I. Introduction
In recent decades, the field of genomics has witnessed considerable advancement, particularly with the advent and rapid evolution of high-throughput sequencing technology. This has empowered researchers to collect and analyse genetic data on an unprecedented scale. Genome sequencing plays a pivotal role in the identification of gene mutations associated with rare genetic disorders. Additionally, it is a valuable tool in the investigation of polygenic diseases, such as complex traits and cancer. However, the advent of new sequencing technologies has resulted in a significant increase in the volume of data generated, with a single sequencing event yielding tens of terabytes of data [
1].
This has led to the necessity for repeated calculations and analyses to draw scientific conclusions. To illustrate, whole genome sequencing (WGS) necessitates the storage of hundreds of gigabytes of data and multiple rounds of data processing, including quality control, sequence alignment, variant detection, and functional annotation [
2]. The aforementioned processing steps are not only computationally intensive but also time-sensitive, which often presents a challenge in meeting the demands of existing local computing resources.
The investigation of genetic diseases is a highly intricate undertaking that necessitates the processing and analysis of vast quantities of genomic data in order to ascertain the correlations between genetic mutations and disease phenotypes. In the context of genomic data processing, the task of gene sequence alignment is one that is particularly computationally complex. In order to achieve an accurate alignment, it is necessary to perform a series of alignment and similarity calculations for each pair of sequences [
3]. Given that a typical genome comprises millions of nucleotides, the alignment of each locus necessitates the utilisation of a sophisticated alignment algorithm.
As the number of samples in a study increases, so too does the amount of computation required. In fact, the amount of computation increases exponentially as the number of samples rises. Furthermore, in the process of data analysis, such as mutation detection and functional annotation, each genomic sample must process a substantial amount of mutation data in a step-by-step manner. This further amplifies the data volume and computational requirements [
4]. Traditional single-machine or small computing clusters are unable to accommodate such large data loads, resulting in significant limitations in computing efficiency and processing speed. Consequently, a single experiment may take weeks or even months to complete.
In recent years, cloud computing platforms have emerged as the preferred architectural choice for data-intensive applications. In comparison with traditional computing resources, the cloud computing platform is distinguished by its capacity for elastic expansion and on-demand allocation, which enables a flexible adjustment of resources in accordance with the specific requirements of the computing task [
5]. The analysis of genetic data is characterised by volatility and high load, and the elastic scalability of cloud computing platforms makes them an ideal choice to meet this demand. Furthermore, the distributed storage system provided by the cloud platform can facilitate efficient data access and reading, and distribute large-scale genetic data to different nodes for parallel processing. This distributed storage architecture not only enhances the parallelism of data access, but also effectively mitigates the data transmission bottleneck inherent to centralized storage, thereby enhancing overall computing efficiency.
Although cloud computing offers significant advantages in terms of scalability and resource management, it still presents a number of technical challenges when applied to the analysis of genetic data [
6]. The block storage and distribution of genetic data represents a crucial step in enhancing computing efficiency. The challenge of balancing the load and reducing data transfer between cloud nodes has emerged as a pivotal issue. Furthermore, the absence of resource scheduling and load balancing mechanisms may result in the inefficient execution of tasks.
To address these challenges, this paper proposes a multi-layer parallel computing framework for a cloud platform designed specifically for genetic disease research. This framework combines data blocking, containerisation technology and a dynamic resource scheduling mechanism to achieve efficient and flexible parallel processing capabilities. Additionally, this paper presents a resource scheduling algorithm based on reinforcement learning, which can optimise resource allocation in real time and improve computing efficiency in a multi-node cloud environment. This algorithm is designed to cope with the fluctuations of different task loads.
III. Methodologies
A. Containerization and Virtualization Layers
On cloud platforms, data distribution is critical to efficiency. We treat the raw genetic data
as a matrix
, where each elemental
represents mutation information at a gene location. In order to achieve chunk optimization, we use a chunking algorithm to divide
into
sub-chunks to maximize read efficiency and reduce inter-node transfer costs. The objective of block optimisation can be expressed as a cost function, as demonstrated in Equation 1.
where
represents the size of the block
,
denotes the bandwidth of the node that processes the block
, and
signifies the data read delay. The Lagrange multiplier method is employed to optimise the objective function, thereby identifying the optimal block distribution scheme that maximises read efficiency, as illustrated in Equation 2.
where
represents the Lagrange multiplier, whereas
denotes the total amount of data. By determining the partial derivative of each
, it is possible to identify the optimal distribution scheme.
The utilisation of containerisation technology facilitates the dynamic expansion of parallel computing in the cloud. Furthermore, the model proposed in this paper represents a further refinement of the container allocation strategy. In this context,
represents the resource requirements of a single container, while
represents the available resources of node
. The objective is to achieve the optimal resource allocation while avoiding resource contention. The model should be optimised in accordance with the specifications set out in Equation 3.
where
, this signifies that container
is deployed on node
. Conversely, if
, then
. Additionally, the resource dependencies between containers and nodes are constrained in accordance with the specifications set forth in Equation 4.
Constrainted optimisation algorithms, such as dynamic programming, are employed to achieve the aforementioned goal, thereby ensuring that the resource allocation of each node is optimised. Furthermore, in order to account for fluctuations in load, we introduce the Resource Resilience Factor
, which is expressed as Equation 5.
Once the value reaches a predetermined threshold (e.g. ), the system initiates a resource scaling or migration mechanism to guarantee container stability during periods of peak load.
B. Load Balancing Optimization
The
model plays a pivotal role in data processing. We put forward an enhanced
framework based on dynamic load balancing, with the objective of optimising gene alignment tasks. In the context of the gene sequence alignment task
, the sequence set is represented by
, while the query sequence is represented by
. The objective is to minimise the overall time overhead of sequence alignment
, as illustrated in Equation 6.
where
represents the load weight of the node
and the sequence
. The
denotes the alignment distance of the sequence
and
. Finally,
is the complexity factor. The objective is to optimise the distribution of
in order to ensure that the alignment tasks are distributed evenly among the nodes, as shown in Equation 7.
Furthermore, in order to enhance the degree of parallelism, the computationally demanding steps involved in the gene alignment process were divided into discrete segments, and the recursive computation method of segmented parallelism was employed. In the event that the calculation step is divided into
segments, the formula for the
segment is given by Equation 8.
where the functions
and
represent the weight calculation in different comparison modes. The recursive solution of the aforementioned formulae effectively distributes the computational burden among the constituent nodes.
In order to enhance the efficiency with which resources are utilised, an adaptive scheduling algorithm based on reinforcement learning was proposed in the resource scheduling layer. The algorithm employs a dynamic approach to resource allocation, with the objective of minimising system idle time and transmission latency. In this context, let
denote the idle time and
denote the transmission time. The objective function for optimisation is given by Equation 9.
Note that,
and
are regulatory factors. A reinforcement learning strategy based on
is introduced, whereby
represents the benefit of taking action
in state
. The formula should be updated in accordance with the instructions set out in Equation 10.
where
represents the learning rate, γ denotes the discount factor, and
signifies the immediate reward for the current state. By means of a continuous updating of the
value, the scheduler is enabled to learn and select the optimal scheduling strategy during operation, thereby achieving optimal resource allocation and dynamic load balancing.
IV. Experiments
A. Experimental Setups
The UK Biobank dataset provides a comprehensive repository of data pertaining to the correlation between the human genome and health status. It encompasses a wealth of information on genetic variation and phenotypic characteristics associated with health and disease. Approximately 500 genomic samples, encompassing both autosomal and sex chromosome sequences, were selected for comprehensive evaluation of the framework's suitability for diverse genetic data analysis. With regard to the block size, the original gene dataset has been allocated 1 MB sub-blocks, thus optimising the distributed storage performance of the cloud. The number of containers is subject to dynamic adjustment in accordance with the resource configuration of each node, with a maximum of 20 containers to facilitate high-density parallel computing.
B. Experimental Analysis
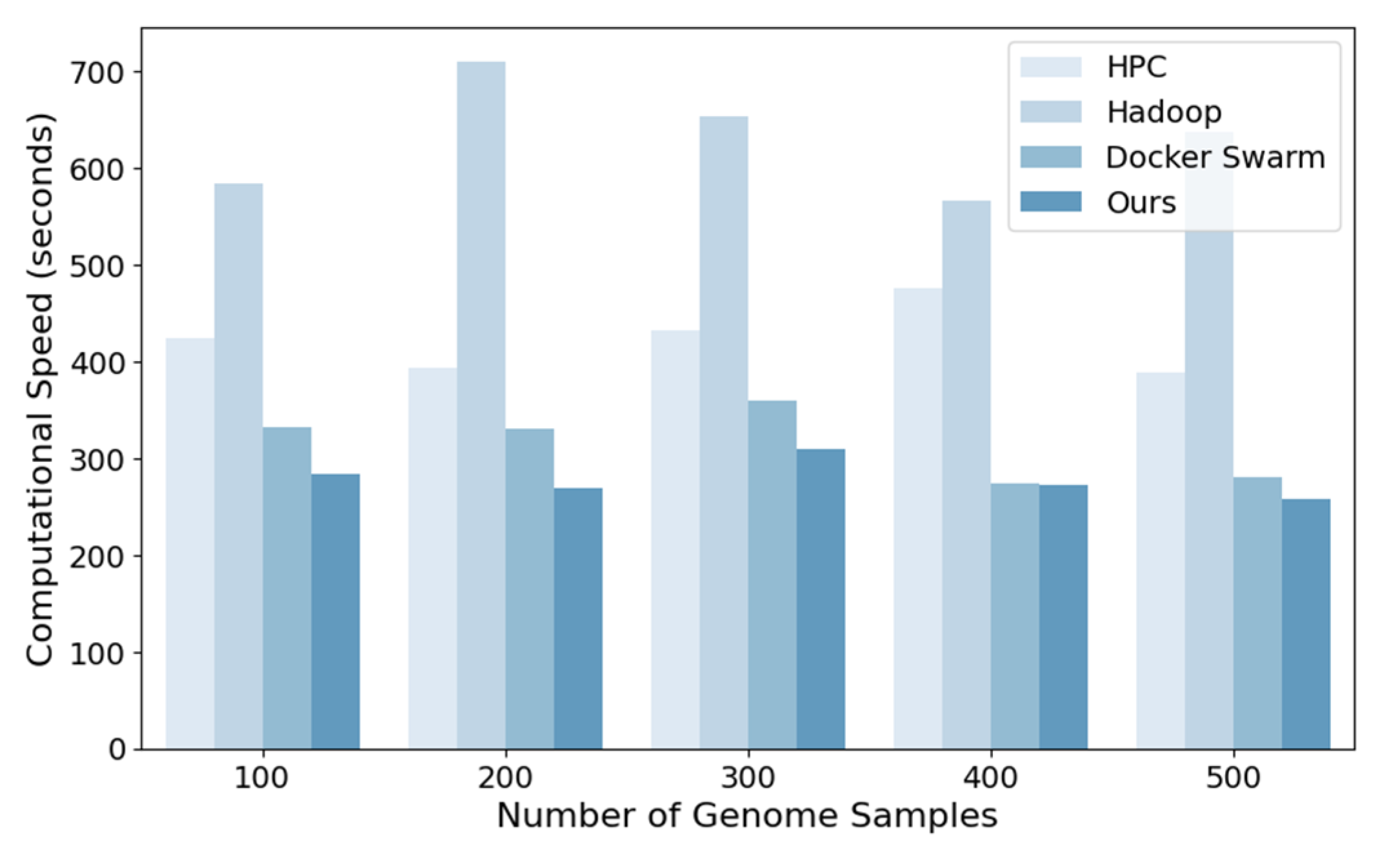
In order to verify the large-scale genetic data processing of the proposed cloud computing parallel framework, a series of comparison methods were established, including traditional high-performance computing clusters (HPC), MapReduce (Hadoop), containerisation-based local clusters (Docker Swarm), and Spark-based cloud platform processing frameworks. The computational speed of a parallel framework is an important indicator of its efficiency. It is used to measure the time taken for the framework to complete genetic data processing tasks under different data volumes and nodes.
Figure 1 illustrates the comparison of the computing speed of the aforementioned methods: HPC, Hadoop, Docker Swarm, and the proposed method. As the number of genomic samples increases, the computational speed of the various methods can be compared.
Figure 1 demonstrates the notable disparity in computational efficiency between the methods at varying sample sizes, with the proposed method exhibiting a substantial advantage in computational speed.
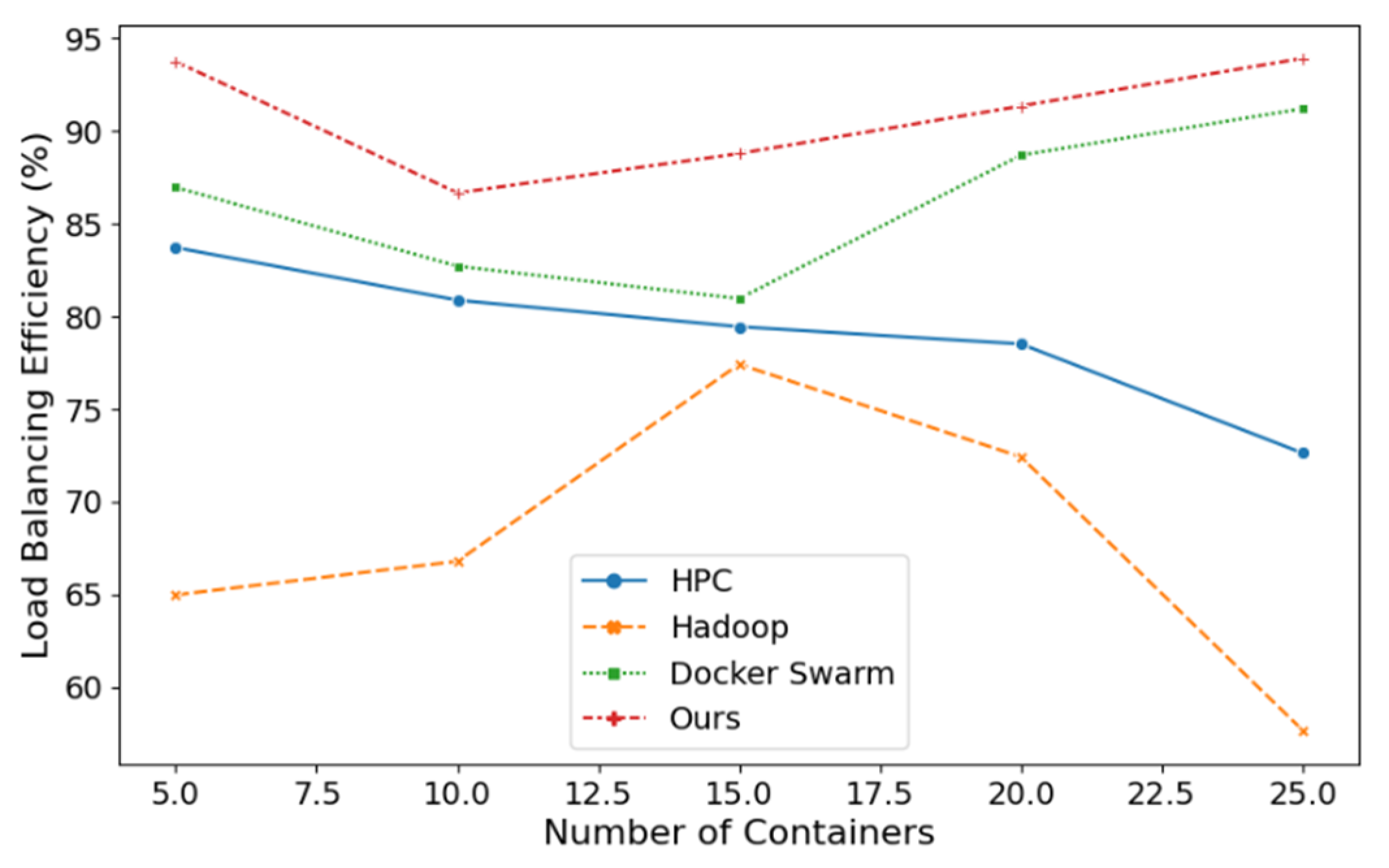
Load balancing is employed to ascertain the distribution of workload among compute nodes. A higher degree of load balancing within a compute node signifies a more uniform distribution of tasks across the nodes.
Figure 2 presents a comparative analysis of load balancing efficiency between diverse computing methodologies, including HPC, Hadoop, Docker Swarm, and our proposed approach, across a spectrum of container configurations. The load balancing efficiency of the various methods is to be compared through the growth of the number of containers. As is evident from
Figure 2, our method demonstrates superior load balancing efficiency with a narrow margin of error for each container configuration, indicating greater stability in its efficiency.
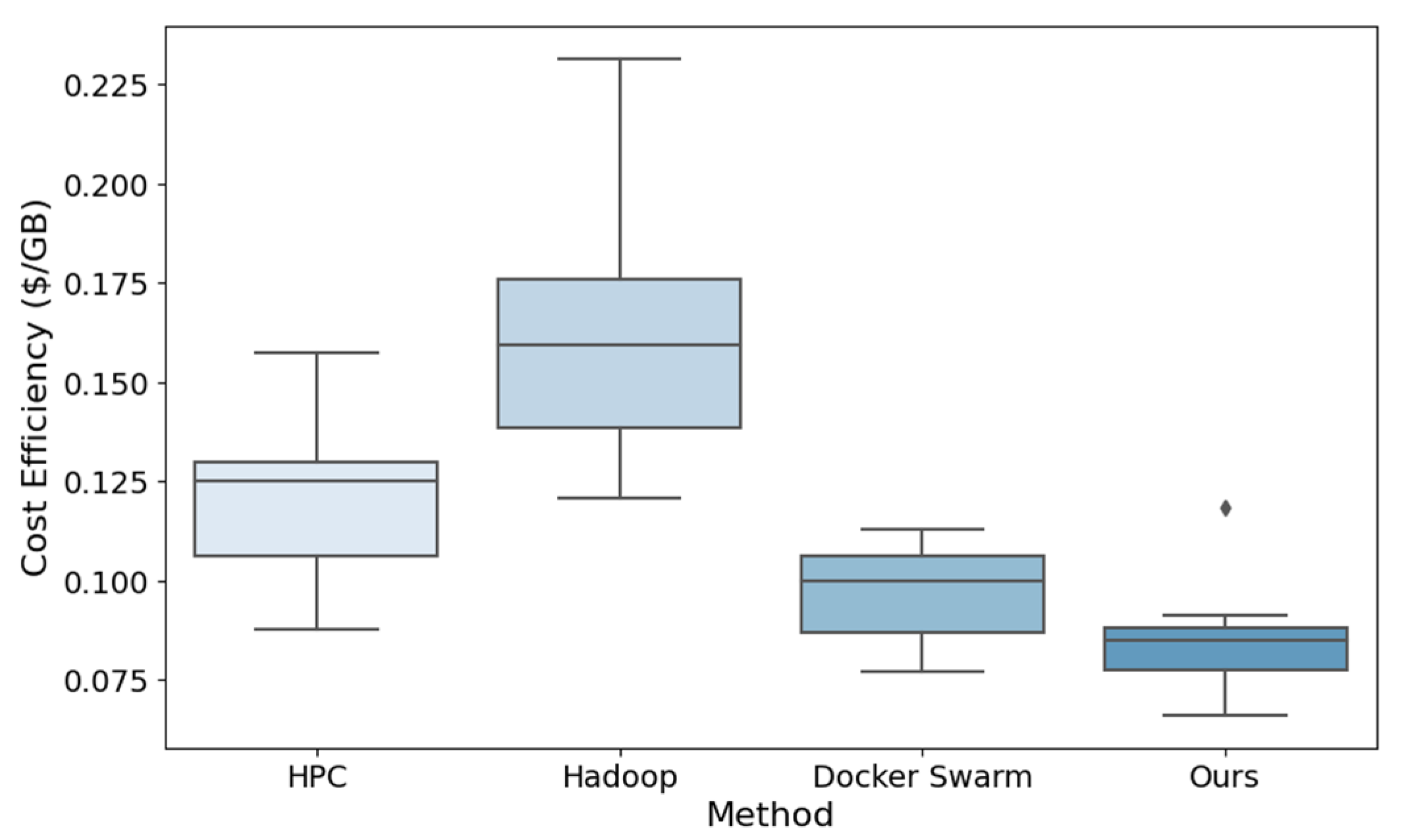
In a cloud computing environment, cost is a significant factor. The framework's cost efficiency is evaluated based on the cost per unit of data to be processed.
Figure 3 depicts the efficiency of various calculation methods in terms of processing cost per gigabyte of data. The median and data distribution in
Figure 3 clearly illustrate the cost efficiency of each method. Our method demonstrates lower processing costs and a narrower distribution range, indicating that it is cost-efficient and stable.
V. Conclusions
In conclusion, the objective of this study is to propose a cloud-based parallel computing framework designed for large-scale genomic data analysis in genetic disease research. A comparison of our approach with comprehensive experiments in traditional HPC clusters, Hadoop, and Docker Swarm environments reveals significant advantages in multiple metrics, including compute speed, load balancing efficiency, and cost efficiency. The framework demonstrates superior performance in terms of load balancing stability and unit data processing cost compared to the comparison methods, indicating its suitability for large-scale, cost-effective genomic analysis. Future research could focus on further optimising the computational efficiency and resource management of the framework to support larger-scale genomic data analysis and a broader range of genetic disease research.
References
- Sharma, Sheelesh Kumar, and Ram Jee Dixit. "Applications of Parallel Data Processing for Biomedical Imaging." Applications of Parallel Data Processing for Biomedical Imaging. IGI Global, 2024. 1-24.
- Rai, Shivangi, et al. "Translational Bioinformatics Ontology In Healthcare With Cloud Computing." 2023 International Conference on Innovations in Engineering and Technology (ICIET). IEEE, 2023.
- Kaliappan, Seeniappan, et al. "Enhancing the Efficiency of Computational Genetic Epidemiology using HPC-Driven AI Models." 2023 IEEE International Conference on ICT in Business Industry & Government (ICTBIG). IEEE, 2023.
- Raghul, S. , and G. Jeyakumar. "Parallel and distributed computing approaches for evolutionary algorithms—a review." Soft Computing: Theories and Applications: Proceedings of SoCTA 2020, Volume 1 (2022): 433-445.
- Gao, Yuan, et al. "Stability analysis of a deep and large open pit based on fine geological modeling and large-scale parallel computing: a case study of Fushun West Open-pit Mine." Geomatics, Natural Hazards and Risk 14.1 (2023): 2266663. [CrossRef]
- Johansson, Lennart F., et al. "A unified data infrastructure to support large-scale rare disease research." medRxiv (2023): 2023-12.
- Langmead, Ben, and Abhinav Nellore. "Cloud computing for genomic data analysis and collaboration." Nature Reviews Genetics 19.4 (2018): 208-219. [CrossRef]
- Grossman, Robert L. "Data lakes, clouds, and commons: a review of platforms for analyzing and sharing genomic data." Trends in Genetics 35.3 (2019): 223-234. [CrossRef]
- Molnár-Gábor, Fruzsina, et al. "Computing patient data in the cloud: practical and legal considerations for genetics and genomics research in Europe and internationally." Genome Medicine 9 (2017): 1-12. [CrossRef]
- Navale, Vivek, and Philip E. Bourne. "Cloud computing applications for biomedical science: A perspective." PLoS computational biology 14.6 (2018): e1006144. [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).