Submitted:
28 October 2024
Posted:
29 October 2024
You are already at the latest version
Abstract
Objective To screen and systematically analyze the differential proteins in rats suffer from SCI after treating with HPL and thus explore the therapeutic mechanism of HPL on SCI rats. Method 30 SD rats were selected and randomly divided into three groups: control group, injury group, and treatment group. Allen Test was performed to establish the rat models with injured spinal cord, and2 mL of HPL was intraperitoneally injected into rats in the treatment group every day. After 14 consecutive days of injection, the thoracic spine of rats was sampled and analyzed by proteomics. Result A total of 61 differential proteins were screened in the injury and treatment groups, including 7 up-regulated proteins and 14 down-regulated proteins. The GO enrichment analysis results showed that the differential proteins included cellular components such as ATP dependent chromatin remodeling complex composed of multiple subunits, recycling endosome, extracellular space, and ATP synthase complex. These differential proteins promote serine-type endopeptidase inhibitor activity, myosin-binding protein and peptidase inhibitor molecular functions, and are involved in biological processes including fibrinolysis, sulfur component synthesis and metabolism, cellular modification of amino acid metabolism, and myelination. The KEGG analysis results mainly suggested the therapeutic mechanism of HPL for SCI through nitrogen metabolism, sodium taurocholate hydrate, complement coagulation cascade, and alcoholic liver disease. Conclusion HPL has a good therapeutic effect on SCI via the bioinformatic mechanisms above.
Keywords:
Spinal Cord Injury
; Human Platelet Lysate
; Proteomics
; Therapeutic Mechanism
1. Introduction
SCI most commonly occurs after traumatic events, with high rates of post-traumatic disability and lack of effective treatments. It increases the pain of the patients and brings burden to their families and society. SCI has a complex pathogenesis and stems from many pathophysiological processes. The molecular mechanisms and potential therapeutic targets of SCI can suggest the pathogenesis of SCI from a pathophysiological perspective, which has an important therapeutic significance. Proteomic analysis includes the study of the molecular mechanisms of post-SCI through differential proteins, cellular sites, biological processes and molecular functions of SCI. A proteomic analysis 1 week of post-SCI provides analysis results of different differential proteins, cellular pathways and protein modification methods [1]. Previous analysis results have proven that HPL has a therapeutic effect on SCI cell models and enhances lower limb muscle strength in a SCI rat model. In order to further elucidate its therapeutic mechanism, proteomics technology was adopted to test the rat model with SCI. In addition, the differential proteins after treatment with HPL were screened and then analyzed, and the therapeutic mechanism of HPL on SCI rats was analyzed from a proteomics perspective.
2. Materials and Methods
2.1 Experimental animals
8-week-old male SD rats weighing 180 g to 200 g were provided by the Lab Center of the General Hospital of Northern Theater Command under the animal experiment license of No.: SCXK (Liao) 2020-0001. 8-week-old male SD rats were given sufficient water and food during the experiment to maintain the adaptive growth for one week.
2.2 Drug
HPL Stemulate (research-grade, No: ST-PL-NH100) was purchased from MineBio Life Sciences Ltd.
2.3 Instrument
Allens spinal cord impactor was provided by the Trauma Laboratory of the General Hospital of Northern Theater Command. Brain solid positioner was provided by the Neurosurgery Laboratory of the General Hospital of Northern Theater Command. DiATOME ultra-thin diamond knife (3 mm, 45°) of LeicaLeica UC7 ultra-thin slicer and transmission electron microscope Hitachi-HT7700 were used. Main experimental reagents were obtained from Sinopharm Chemical Reagent Co, Ltd, including No: Servicebio G1102 electron microscope fixative, anhydrous ethanol and acetone, and 812 embedding agent (No: SPI 90529-77-4) was provided by Ted Pella Inc.
2.4. Experimental grouping, modeling and Methods
2.4.1 Experimental grouping
30 rats were randomly divided into 3 groups including Sham-operated Group, SCI group, and treatment group. For the rats in the treatment group, 2 mL HPL was injected intraperitoneally 30 minutes after surgery for 14 continuous days.
2.4.2 Modeling
After adult male SD rats were acclimatized and fed for 7 consecutive days, adult male SD rats were measured (standard body weight range: 200g-220g) and anesthetized by intraperitoneal injection of 1% sodium pentobarbital 40mg/kg. After the disappearance of the corneal reflex, male SD rats were immobilized in the prone position. An incision was made about 3 cm in the center of the T9-T11 spinal cord segment, the fascia was cut, and the muscle above the spinous process was removed. Mosquito forceps were adopted to remove the spinous process to expose the spinal cord completely, and a spinal cord impactor was adopted. An area with a diameter of 2 mm was selected and a 20-g weight was placed in free fall at a height of 5 cm from the spinal cord of male SD rats. After placing the 20g weight on the spinal cord of male SD rats for 5 seconds, it could be noticed that their hind limbs twitched, their tails swung, the tension of the hind limbs and tails disappeared, and they appeared to be reclining, implying the successful modeling process. After hemostasis in the surgical area, the muscles, fascia and skin of male SD rats were sutured sequentially, body temperature was maintained until postoperative recovery, and the bladder was massaged to urinate twice a day until they recovered the function of voluntary urination. During the operation, the rates were administered anesthetic drugs to achieve anesthesia and reduce pain. After the operation, ibuprofen analgesic drugs were given to relieve pain. During tissue collection, the arteries were anesthetized to collect blood and bleeding was continued until death. All animal treatments were in accordance with the Regulations for the Protection and Use of Laboratory Animals in the Northern Theater of Operations of the General Hospital of the Chinese People's Liberation Army (Ethical Approval No. 2023-24).
2.4.3 Main observation indicators
Postoperative daily Basso Beattie Bresnahan (BBB) score and incline plate tests were performed.
2.4.4 Preparation and sampling of specimens
After anesthesia by intraperitoneal injection, the surgical area of SCI in rats was incised, and the spinal cord was gently and quickly removed from the rats, which was then either placed in formalin or washed in PBS and then frozen in a -80°C freezing tube. The spinal cords of three rats were randomly selected from the sham-operated, spinal cord injury and treatment groups, washed in PBS and stored in liquid nitrogen for proteomic analysis.
2.4.5 Immunohistochemical Staining
Step 1: Blocking treatment: treat with endogenous peroxidase blocker for 20 minutes.
Step 2: Soak in double distilled water 3 times for 5 minutes each time.
Step 3: Antigen repair: prepare 400-600 mL of 10 nM citric acid antigen repair solution (pH=6.0), put the slices into a beaker, seal the beaker mouth with cling film to prevent the repair solution from overflowing due to boiling, and then heat it in the microwave at high temperature for 10 minutes.
Step 4: After heating, add sterile double distilled water (DDW) to fill the liquid level, and heat it in a microwave for 40 minutes (medium to low temperature until thawed), and fill the liquid level with double distilled water every 10 minutes.
Step 5: After completing the antigen repair, cool the repair solution naturally at room temperature.
Step 6: After cleaning with DDW for 180 seconds, clean it for 3 times using photospace buffered saline (PBS) with 3 minutes each time.
Step 7: Perform primary antibody incubation, and store it in at 4 ℃, and raise the temperature to 37 ℃ in the next day. Then, perform a 5-minute PBS cleaning.
Step 8: Add reaction enhancer to the beaker, incubated continuously at room temperature for 20 minutes, and then performed PBS cleaning for 3 times with 3 minutes each time.
Step 9: Add goat anti-mouse/rabbit IgG/IgM H&L (HRP polymer), incubated at room temperature for 20 minutes, and then performed a PBS cleaning for 3 times with 3 minutes each time.
Step 10: Add DAB colorimetric solution dropwise (prepared in a 1:19 ratio when using), chose the staining time according to different antibodies, incubate at room temperature for 2 to 10 minutes, and gently rinsed DAB colorimetric solution with tap water after completing this experimental step.
Step 11: Re-dyeing: Stain it with hematoxylin for 50 seconds, cleaned with DDW for 3 minutes, and then performed PBS cleaning for 3 times with 5 minutes each time.
Step 12: Dehydration: Soak the slices in different concentrations of alcohol sequentially: 70 % (0 min), 80 % (1 min), 90 % (1 min), 95 % I (1 min), 95 % II (5 min), 100 % I (5 min), 100 % II (5 min), and 5 min.
Step 13: Transparent treatment: Treat it with xylene I and xylene II for 20 minutes each time.
Step 14: Used neutral resin to seal the slide, seal it with a glass slide (without residual small bubbles in the tissue), and dried it in the air naturally.
2.4.6 Tandem Mass Tag (TMT) Technology (Detected by Hangzhou Lianchuan Biotechnology Co, Ltd.)
TMT technology was employed to detect 9 rat tissue samples using TMT labeled quantitative proteomics technology. The protein composition, expression level differences, and corresponding biological functions within the samples were analyzed. TMT (Tandem mass tag) is an in vitro stable isotope labeling protein quantification technique developed by ThermoFisher. This technology utilizes isotope reagents to simultaneously label up to 18 peptide samples (TMTpro-18plex). After mixing the labeled peptide samples in equal amounts, it can perform liquid chromatography separation and tandem mass spectrometry (MS/MS) detection to obtain primary and secondary mass spectrometry information for each peptide segment. In the secondary mass spectrometry, chemical bond cleavage releases TMT reporter ions, generating 18 reporter ion peaks in the low-quality region of the mass spectrometry, and the intensity of chemical bond cleavage can indicate the relative expression level information of the peptide segment in different samples. In addition, the mass to charge ratio of the peptide fragment ion peak in the secondary mass spectrometry can indicate the sequence information of the peptide segment. By retrieving these raw mass spectrometry data in the database, it can obtain qualitative and relative quantitative information of the protein.
2.4.7 Protein Extraction
(1) An appropriate amount of sample was added into a 2-mL centrifuge tube, and then an appropriate amount of steel beads and lysis buffer (50 mMTris-HCl, 8 M Urea), as well as a final concentration of 1X Roche Cocktail were added, and the mixture was placed on ice block for 5 minutes; (2) A grinder was used to crush and lysed the sample (frequency: 60 HZ, time: 2 min), centrifuged 20,000 g sample at 4°C for 15 min, and then extracted the supernatant; (3) DTT with a final concentration of 10 mM was added and water bath at 37 °C for 1 hour; (4) IAA with a final concentration of 20 mM was added, and placed in the dark for 30 minutes.
2.4.8 Protein Extraction Quality Control
(1) Bradford quantification: Added standard protein (0.2 μg/μL BSA) 0, 2, 4, 6, 8, 10, 12, 14, 16, 18 μL to the 96-well enzyme plate in sequence, then added 20, 18, 16, 14, 12, 10, 8, 6, 4, 2 μL of pure water in sequence, shake and mix, and then added 180 μL of Coomassie Brilliant Blue G-250 quantitative working solution to each well. Used a microplate reader to measure OD595, and created a linear standard curve based on OD595 and protein concentration. Diluted the protein solution to be tested to several times, add 180 μL of quantitative working solution to 20 μL of the protein solution, and read the OD595. Calculated the sample protein concentration based on the standard curve and sample OD595; (2) Used the SDS-PAGE method to sample 10 μg protein solution and add an appropriate amount of loading buffer, mix and heat at 95°C for 5 minutes, centrifuged 20,000g of the sample for 5 minutes, and then extract the supernatant an added it into the spotting holes filling with a 4-12 % SDS polyacrylamide gel, and applied 80V constant voltage electrophoresis for 20 minutes, followed by 120V constant voltage electrophoresis for 60 minutes. After applying electrophoresis, the SDS polyacrylamide gel was stained and destained, then removed and photographed.
2.4.9. Protease Hydrolysis
(1) Extracted 150 μg protein from each sample; (2) Added 3 μg Trypsin enzyme at a ratio of protein: enzyme=50:1, and enzymatically hydrolyzed at 37°C for 14-16 hours; (3) The enzymatically hydrolyzed peptides are desalted using a Waters solid- phase extraction chromatography column and vacuum-dried; (4) The dried peptide samples are reconstituted with pure water and stored at -20°C.
2.4.10. TMT Labeling
(1) Extracted an appropriate amount of peptide fragments from each sample, drain it under vacuum, and redissolve it into 30 μL solvent with 100 mM TEAB; (2) Added 100% anhydrous acetonitrile to the TMT labeling reagent for dissolution. Added each TMT label to each sample according to the ratio of TMT labeling: peptide=5:1, and made it stand for 1-2 hours at room temperature; (3) After labeling, added 5% hydroxylamine to each sample to a final concentration of 0.4 % to terminate labeling, then all the samples were mixed into a tube, shook and mixed well, part of the samples were extracted for labeling quality control, and the rest were drained.
2.4.11. High-pH Reversed-Phase Chromatography
After extracting equal amounts of peptide fragments from all samples, mixing them, diluting them with mobile phase A (5 % ACN, pH 9.8) and selecting samples,
they were separated using the UltiMate™ 3000 binary rapid separation system produced by Thermo Scientific. The model adopted was A 3.5μm 4.6x150mm Agilent ZORBAX 300ExtendC-18 liquid chromatography column was adopted to perform liquid-phase separation of the sample. Elute with a flow rate gradient of 0.3 mL/min: elute from 5% mobile phase B (97 % ACN, pH 9.8) to 21.5 % mobile phase B in 38 min, and elute from 21.5 % to 40 % mobile phase B in 20 min, elute from 40 % to 90 % mobile phase B in 2 min, 90 % mobile phase B for 3 min, and equilibrate with 5 % mobile phase B for 10 min. Monitor the elution peak at a wavelength of 214 nm and collect a component every 1 minute. Aggregate the samples according to the chromatographic elution peak pattern to obtain 10 components, and then freeze and drain the sample.
2.4.12 Mass Spectrometry (nano-flow liquid chromatography tandem mass spectrometry (LC-MS/MS)
We re-solubilized the dried peptide sample with 0. 1 % FA, centrifuged the sample at 20,000 g for 10 minutes, and extracted the supernatant for sample selection. Separation was performed by EASY-nLC™ 1200 system produced by Thermo Scientific. We made the sample enter into the self-loaded liquid chromatography column C18 (100 μm inner diameter, 1.8 μm column particle size, approximately 35 cm column length), and was separated through the following effective gradient at a flow rate of 300 nL/min: 0~48 min, 8 % mobile phase B (98 % ACN, 0. 1 % FA) rises linearly to 32 %; 48~53min, mobile phase B rises from 32 % to 45 %; 53~55 min, mobile phase B rises from 45 % to 90 %; 55~62 min, 90 % mobile phase B. The peptide fragments separated by liquid phase were ionized by the nanoESI source and then entered the mass spectrometer Orbitrap Exploris™ 480 (Thermo Fisher Scientific, San Jose, CA) for DDA (Datadependent Acquisition) mode detection. Main parameter settings: ion source voltage is 2.2KV, primary mass spectrometry scanning range is 350~1,500m/z; resolution is set to 120,000; Normalized AGC Target is set to 300 %; secondary mass spectrometry fragmentation mode is HCD, and fragmentation Energy is set to 32 %; resolution is set to 45,000, and dynamic exclusion time is set to 60s. The starting m/z of the secondary mass spectrum is fixed at 110; the precursor ion screening conditions for secondary fragmentation are: charge 2+ to 6+; the Normalized AGC Target is set to 200 %, and the maximum ion injection time (MIT) is 120 ms.
2.4.13. Protein Identification and Quantification
We adopted MaxQuant software to conduct protein search, identification and quantitative analysis on the TMT-labeled mass spectrum raw data. The main parameters of MaxQuant that should be set are: select the MS2-based reporter
quantitative mode (TMT6plex, TMT10plex, TMT16plex or TMT18plex) , set trypsin to Trypsin/P enzyme, fixed modification to Carbamidomethyl(C), variable modification to Oxidation (M) and Acetyl (protein Nterm), select Match between runs
We extracted the proteins in the sample and then enzymatically hydrolyzed them, and labeled the enzymatically hydrolyzed peptides with TMT reagents and enriched and separated them. The peptides are detected using a high-performance liquid chromatography tandem high-resolution mass spectrometer to obtain a large amount of mass spectrometry data. We adopted MaxQuant (v2.1.4.0) software to identify the proteins in the samples. The identification conditions were: false positive spectrum (PSM FDR) < 0.01, false positive protein (Protein FDR) < 0.05.
2.5 Ethics Statement
This study was approved by the Ethics Committee of the Northern Theater General Hospital: Northern Theater General Hospital Animal Medical Research Ethics Committee Branch(No. 2023-24). This study was performed in accordance with the ARRIVE guidelines.
3. Results
3.1 Effect of HPL on hindlimb movement disorder in rats with SCI
According to the BBB score, the hindlimb movement function of rats in the Sham-operated Group was normal. On the day 1 of treatment, there was no difference between the treatment group, the SCI group and the sham-operated group (P<0.01). There were significant differences between the treatment group, SCI group and sham- operated group on days 3, 7 and 14 (P<0.01). Compared with the SCI group, the hind limb motor functions of the rats in the treatment group were significantly improved on days 3, 7, and 14 (P<0.01; P<0.01; P<0.05) (Figure 1).
3.2 Distribution of Annotation Numbers of Identified Proteins in Major Functional Databases
A total of 254 differentially expressed proteins were screened out from the SCI group and the control group, including 199 up-regulated proteins and 55 down- regulated proteins. Additionally, a total of 61 differentially expressed proteins were screened out from the treatment group and the SCI group, including 7 up-regulated proteins and 14 down-regulated proteins (FC>1.2, P<0.05, Figure 2).
3.3 Comparison of differences between pairwise groups
Screening for differentially expressed proteins in pairwise phenotype groups through univariate analysis. Differentially expressed proteins (Fold change (FC)>1.2, p-value<0.05 in T-test statistical results) were screened out, as shown in Figure 3.
3.4 Differences between SCI Group and Sham-operated Group
3.4.1 Subcellular Localization of Proteins
Localization of differentially expressed proteins is located in the cytoplasm, nucleus, and extracellular space in numericalordination (Figure 4).
3.4.2 GO enrichment
The cellular components of differentially expressed proteins mainly include cytoplasm, extracellular fluid, nucleus, etc, and its molecular functions involve serine carboxypeptidase activity, small molecule binding, purine nucleobase binding, N- acetyl-β-glucosaminidase, etc, and its biological processes include vesicle-mediated transport, regulation of cellular component organization, cellular component composition, catabolism of organic nitrogen compounds, etc (Figure 5).
3.4.3 KEGG enrichment
The signaling pathways are mainly enriched in neomycin, kanamycin, gentamicin biosynthesis, glycosphingolipid biosynthesis, β-alanine metabolism, etc (Figure 6).
3.4.4 Differences between SCI Group and Treatment Group
(1) Subcellular Localization of Proteins
Localization of differentially expressed proteins is located in the extracellular space, cytoplasm and nucleus in numericalordination.A total of 61 differentially expressed proteins were screened out from the SCI group and the treatment group, including 7 up-regulated proteins and 14 down-regulated proteins (FC>1.2, P<0.05), (see Figure 7).
(2) GO enrichment
The cellular components of differentially expressed proteins mainly include ATP-dependent chromatin remodeling complex composed of multi-subunits, recycling endosomes, extracellular space, ATPase complex, etc, and their molecular functions include Serine endopeptidase inhibitor activity, myosin binding, peptidase inhibitors, etc, and its biological processes include fiber dissolution, anabolic process of sulfur components, cell modification amino acid catabolism, myelin assembly, etc (see Figure 8).
(3) KEGG enrichment
The signaling pathways are mainly enriched in nitrogen metabolism, sodium taurocholate hydrate, complement and coagulation cascade, alcoholic liver disease, etc (Figure 9).
3.5 Comparison Results of Differentially Expressed Proteins between 3 Groups The differentially expressed proteins includes Sema7A, CRYM, FBRSL1, MLC1, and CA14 (Figure 10).
3.6 Sema7A immunohistochemistry
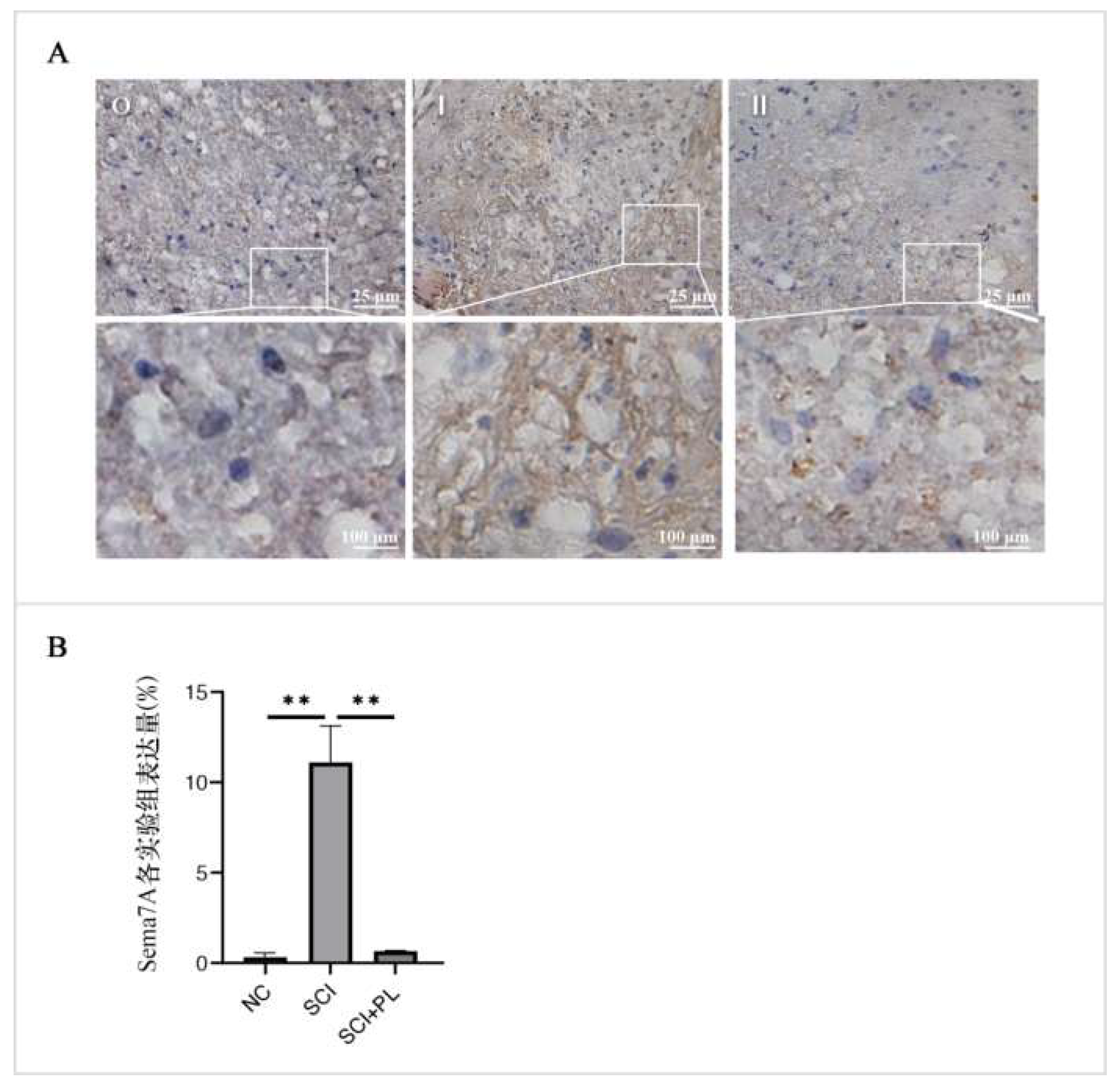
By comparing the expression level of Sema7A in the Sham-operated Group, SCI group, and treatment group on Day 14, there is a significant difference in the expression level of Sema7A in the SCI group, P<0.05, which is with statistically significant compared with the Sham-operated Group (Figure 11).
Figure A: Immunohistochemical results, Figure B: statistical map; O is the normal group, I is the 14 day injury group, II is the 14-day injury group, One-way analysis of variance was used for comparison among all groups, n=3, *P<0.05.
4. Discussion
HPL is a derivative of platelets and contains various biological molecules including growth factors, cytokines and chemical factors. These molecules play important roles in biological processes including cell proliferation, differentiation, migration, and apoptosis. HPL is of great significance in treating and promoting wound healing, nerve repair, angiogenesis, bone regeneration, antibacterial and relieving neuropathic pain [2]. The following are some common growth factors and their functions: platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), insulin-like growth factor (IGF) , fibroblast growth factor (FGF), bone morphogenetic proteins (BMPs), these growth factors play a coordinating role in cell growth, proliferation, differentiation and tissue reconstruction, and can effectively promote wound healing, nerve repair, angiogenesis and bone regeneration, etc[3] .
HPL is a safe alternative with potential applications in the production of serum-free 3-dimensional engineered skin, and can support short-term expansion of fibroblasts and promote epidermal stratification. In addition, the growth factor in the HPL is rich in TGF-β1 and can effectively promote skin maturation. In addition, by measuring the penetration of fluorescent dyes into the skin, the improvement of skin barrier function can be indirectly evaluated [4,5]. HPL can significantly enhance the proliferation of stromal cells and promote cell proliferation by regulating the expression levels of cell cycle-related genes, including TKR, STAT3 and MYC. This may be one of the mechanisms by which HPL promotes cell proliferation [6]. HPL has a positive impact on oral mucosal healing [7], bone regeneration and cell therapy. It can be adopted as a protein source in cell culture media to replace fetal bovine serum [8] and can promote proliferation, differentiation and functional improvement of bone marrow stromal cells [9].
It is found that growth factors and cytokines in HPL can treat SCI by regulating multiple targets and signaling pathways (including Sema7A, CRYM, FBRSL1, MLC1, CA14, etc.).
In summary, HPL plays an important role in cell growth, proliferation, differentiation, and tissue reconstruction, and is considered a safe and effective alternative with potential in the fields of bioengineered skin, oral mucosal healing, bone regeneration, and cell therapy with a potential application prospect. However, a deeper understanding of the therapeutic mechanism of HPL and its effects in different clinical applications is still needed.
Proteomics is a high-throughput, highly automated biotechnology that can screen the molecules responsible for the occurrence and development of the disease under study. The disease can be further elucidated by screening out differential proteins, cell functions and related signaling pathways. The molecular therapeutic mechanism provides reference for subsequent clinical translation research. The results of this experiment show that after the establishment of a rat model of SCI, and treatment with HPL can improve the pathological condition of the rats and enhance the muscle strength of the lower limbs of the rats. The results of the spinal cord proteomics report of this examination show that the differentially expressed proteins are about 5 types, which are all related to the growth and development of the nervous system, transmitter transmission, internal environment stability and other factors.
The studies have shown that Sema7A plays an important role in regulating inflammatory responses, cell survival and apoptosis [10], tissue repair, and neural development. In the pathological process of inflammation, Sema7A can affect the expression levels of inflammatory mediators, regulate the metabolism and polarization state of macrophages, and involve in the induction and resolution of inflammation through multiple signaling pathways [11,12]. Sema7A can affect the levels of inflammatory mediators in intrahepatic cholestasis and non-alcoholic fatty liver disease by regulating the NF-Kβ/P65 signaling pathway [13]. It can also regulate cell adhesion, skeleton and migration phenotypes, and contribute to tissue repair with great significance[14] . In the nervous system, Sema7A is involved in axonal growth, path selection and neural development [15]. In the brain, Sema7A can serve as an immune regulatory factor and is involved in the regulation of epileptic seizure activity [16]. In summary, Sema7A plays a key role in regulating inflammatory responses, cell survival and apoptosis, tissue repair, and neural development. However, further research is still needed to reveal Sema7A’s detailed therapeutic mechanism and potential clinical application value.
CRYM is a protein highly expressed in the nervous system, and its specific function is currently unknown. But there is evidence that CRYM can work in multiple ways. First, studies have shown that CRYM is expressed in the corticospinal tract of rat cortex and that expression levels are reduced in the corticospinal tract of human amyotrophic lateral sclerosis patients. This suggests that CRYM can play a role in axonal degeneration in the corticospinal tract and serve as a marker of this degeneration [17]. Secondly, studies have found that CRYM is expressed in the brain, basal ganglia, hippocampus and corticospinal tract of rats, and is expressed in large pyramidal neurons in the cerebral cortex and pyramidal cells in the CA1 area of the hippocampus. The expression of CRYM is distributed in a mosaic pattern in these areas, that is, CRYM-positive and -negative neurons are distributed alternately. This suggests that CRYM can be expressed in epithelial and medullary cells in the vertebral bodies of the cerebral cortex and hippocampus. In addition, studies also found that CRYM can play an important role in neuroprotection, neurotransmission, cell survival and lysine metabolism through the interaction of T3 or ketoamine. The increased expression level of CRYM in the central nervous system may have a protective effect against neurodegenerative diseases [18,19] . In addition, studies have found that CRYM is an enzyme that can catalyze amino acids with high sulfur content to produce cyclic ketoamines and reduce cyclic ketoamines. These ketoamines can play a potential role in neurotransmitters [20] .
In summary, although the specific functions of CRYM require further research needs to be clarified, there is evidence that it is of great significance in metabolism, neurotransmitter transmission, and cell survival.
The rat tissues adopted in this experiment met the requirements for immunohistochemical examination. According to the protein expression standards, the researchers further verified the expression level of Sema7A in the rat SCI model after treatment with HPL, indicating a high Sema7A expression level in SCI, and after treatment with HPL, indicating a downward trend of Sema7A expression level. The increased expression level of Sema7A led to an increase in the release of inflammatory mediators, triggering subsequent cascade reactions and aggravating spinal cord tissue damage, and Sema7A can cause the changes of ERK in MAPK family through FAK, resulting in a series of changes in the rat’s nervous system. For other therapeutic mechanism of Sema7A, it is needed to conduct further research.
Author Contributions
Conceptualization: Ling Wang, Shuang Wang, Hailong Yu; methodology: Ling Wang, Liangbi Xiang; writing—original draft preparation: Ling Wang; writing—review and editing: Liangbi Xiang. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the application Study on SPEEK Intervertebral Fusion Cage-induced Osteogenic Differentiation of Stem Cells.
Data Availability Statement
Data will be available on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhigang Li, Bingxuan Quan, Xiuyan Li, et al. A proteomic andphosphoproteomic landscape of SCI[J]. Neurosci Lett. 2023,9(25):814:137449. [CrossRef]
- Chiara Puricelli, Elena Boggio, Casimiro Luca Gigliotti, et al. Platelets, Protean Cells with All-Around Functions and Multifaceted Pharmacological Applications[J]. Int. J. Mol. Sci. 2023, 24, 4565. [CrossRef]
- Zhang Binliang, Wang Wenbo, Zhao Zhe. Research Progress on the Treatment of Common Orthopedic Diseases with HPL [J] Medical Review. 2022,28 (10): 1976-1982.
- Banakh,Md. M. Rahman,C. L. Arellano,et al. Platelet lysate can support the development of a 3D-engineered skin for clinical application[J]. Cell and Tissue Research (2023) 391:173–188.doi.org/10. 1007/s00441-022-03698-7.
- Johanka Táborská, Andreu Blanquer, Eduard Brynda.et al. PLCL/PCL Dressings with Platelet Lysate and Growth Factors Embedded in Fibrin for Chronic Wound Regeneration[J]. International Journal of Nanomedicine. 2023:18:595-610. doi.org/10.2147/IJN.S393890.
- Michaela Oeller, Heidi Jaksch-Bogensperger, Markus Templin,et al. Transcription Factors STAT3 and MYC Are Key Players ofHPL-Induced Cell Proliferation[J]. Int. J. Mol. Sci. 2022, 23, 15782.
- Sook-Luan Ng , Nur Ain Azhar , Siti Balkis Budin,et al. Effects of Platelet Lysate Gels Derived from Different Blood Sources on Oral Mucosal Wound Healing: An In Vitro Study[J]. Gels.2023, 9, 343.
- Maria Karadjian, Anne-Sophie Senger, Christopher Essers,et al. HPL Can Replace Fetal Calf Serum as a Protein Source to Promote Expansion and Osteogenic Differentiation of Human Bone-Marrow-Derived Mesenchymal Stromal Cells[J]. Cells.2020, 9, 918. [CrossRef]
- Gabriele Brachtl ,Rodolphe Poupardin, Sarah Hochmann,et al. Batch Effects during Human Bone Marrow Stromal Cell Propagation Prevail Donor Variation and Culture Duration:Impact on Genotype, Phenotype and Function[J]. Cells.2022, 11, 946. [CrossRef]
- Weinan Qi, Dan Zeng, Xiaoshuan Xiong, et al. Knockdown of Sema7A alleviates MPP+-induced apoptosis and inflammation in BV2 microglia via PPAR-γ activation and MAPK inactivationImmunity,inflammation and Disease[J].Immunity,Inflammation and Disease.2022, 12:1–10. [CrossRef]
- Andreas Körnera, Alice Bernarda, Julia C. Fitzgeraldb,et al. Sema7A is crucial for resolution of severe inflammation[J]. Histol Histopathol.2023, 1(18):18624. [CrossRef]
- Arlette L Bruijstens, Christoph Stingl, Coşkun Güzel,et al. Neurodegeneration and humoral response proteins in cerebrospinal fluid associate with pediatric-onset multiple sclerosis and not monophasic demyelinating syndromes in childhood[J].MSJ. 2023, 29(1) 52–62. doi.10. 1177/13524585221125369.
- Xuan Li, Wanlu Xie, Qiong Pan, et al.Semaphorin 7A interacts with nuclear factor NF-kappa-B p105 via integrin β1 and mediates inflammation[J]. Cell Commun Signal.2023,21(1):24.1-16. [CrossRef]
- Ping Hu, Andrew E Miller, Chiuan-Ren Yeh, et al. Sema7A primes integrin α5β1 engagement instructing fibroblast mechanotransduction, phenotype and transcriptional programming[J]. Matrix Biol. 2023,8(121):179-193. [CrossRef]
- Tao Xu, Juan Yang, et al. Sema7A, a brain immune regulator, regulates seizure activity in PTZ-kindled epileptic rats[J].CNS Neurosci Ther. 2020, 26:101–116. [CrossRef]
- Jing Deng, Tao Xu, Juan Yang, et al. Sema7A, a brain immune regulator, regulates seizure activity in PTZ-kindled epileptic rats[J].CNS Neurosci Ther. 2020, 26:101–116.
- Reiji Hommyo, Satoshi O. Suzuki, Nona Abolhassani, et al. Expression of CRYM in different rat organs during development and its decreased expression level in degenerating pyramidal tracts in amyotrophic lateral sclerosis[J].neuropathology.2018.2:1–13. [CrossRef]
- Laetitia Francelle,Laurie Galvan, Marie-Claude Gaillard,et al. Loss of the thyroid hormone-binding protein CRYM renders striatal neurons more vulnerable to mutant huntingtin in Huntington’s disease[J]. Human Molecular Genetics, 2015, 6(24):1563–1573. [CrossRef]
- Robert Chevreau, Hussein Ghazale, Chantal Ripoll, et al .RNA Profiling of Mouse Ependymal Cells of post-SCI Identifies the Oncostatin Pathway as a Potential Key Regulator of Spinal Cord Stem Cell Fate[J]. Cells.2021, 10, 3332:1–24. [CrossRef]
- Kiran Pawara, Brian J. Cummingsa,b,c,d, Aline Thomase,et al. Biomaterial bridges enable regeneration and re-entry of corticospinal tract axons into the caudal spinal cord of post-SCI: association with recovery of forelimb function[J]. Biomaterials. 2015, 65: 1–12. [CrossRef]
Figure 1.
Effect of HPL on hindlimb movement disorder in rats with SCI.
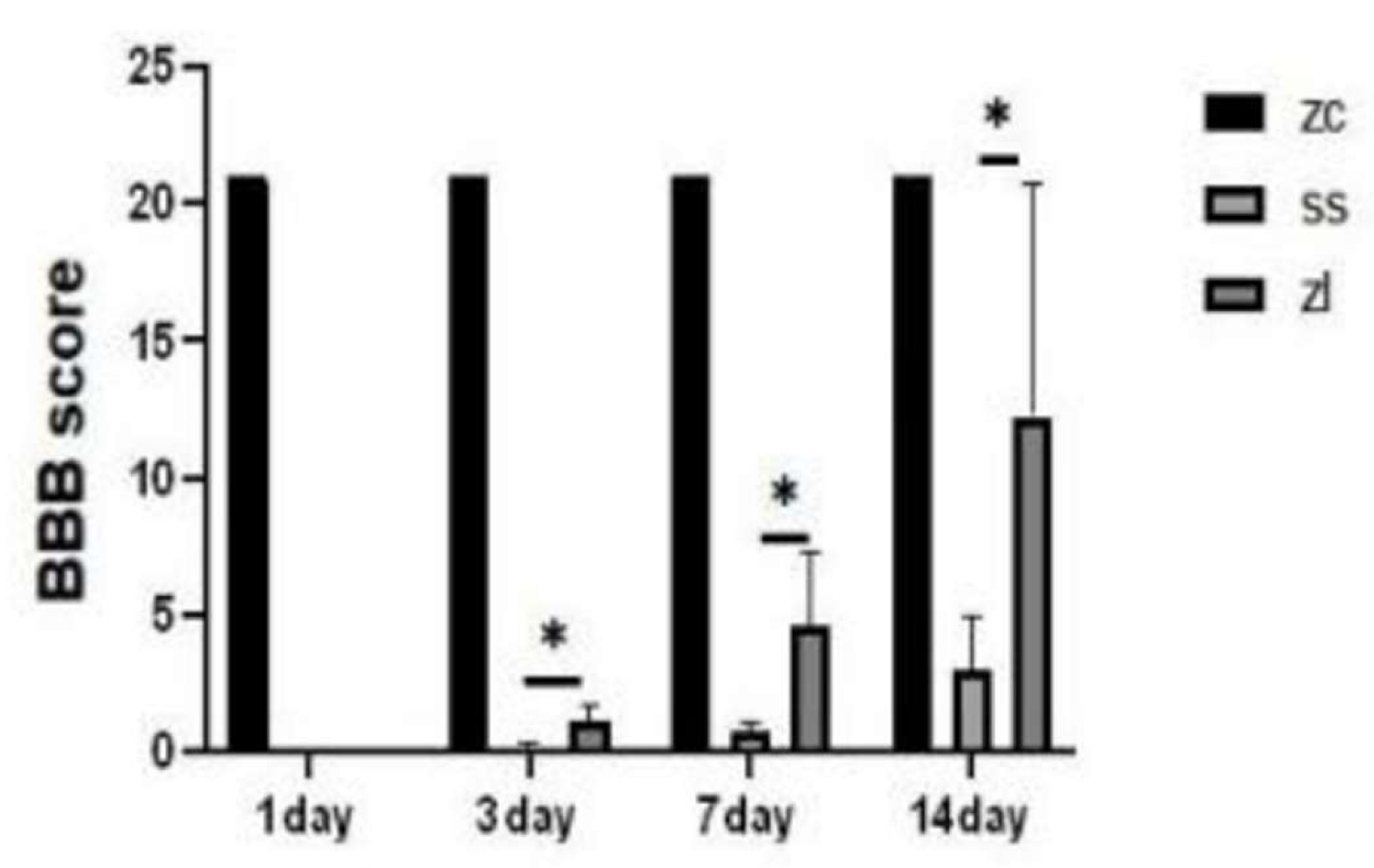
Figure 2.
Distribution of Annotation Numbers.
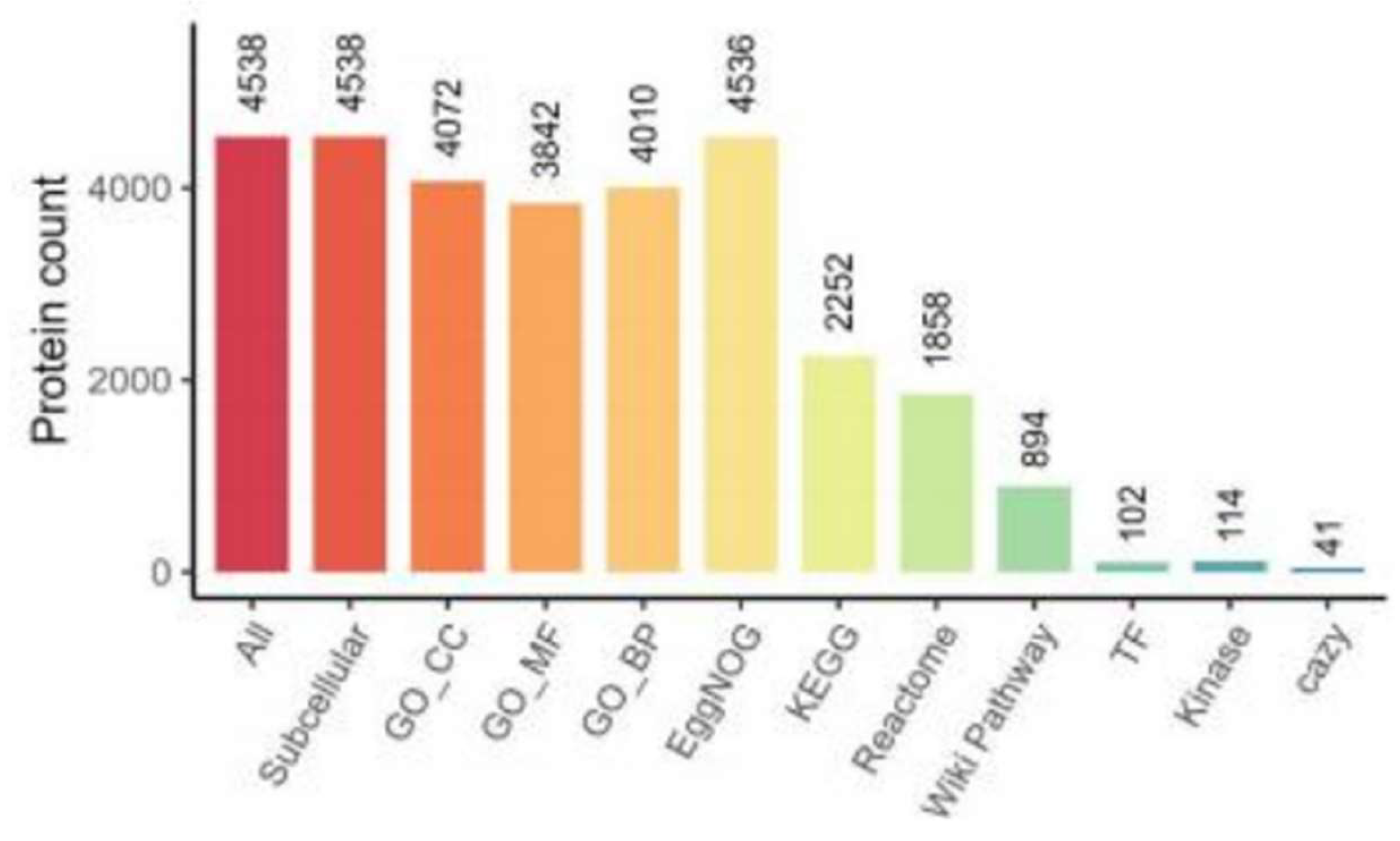
Figure 3.
Comparison of differences between pairwise groups.
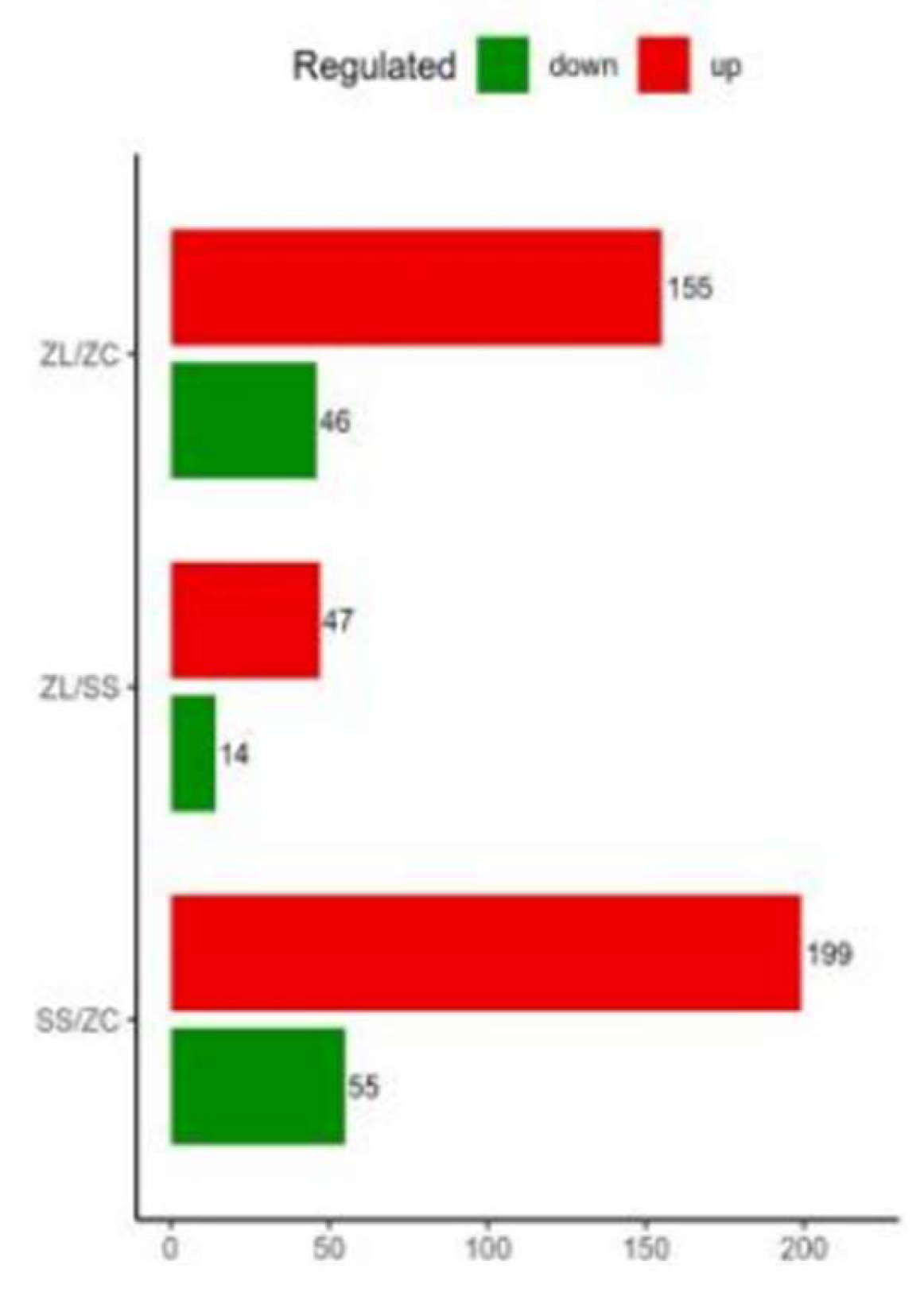
Figure 4.
Subcellular Localization of Proteins.
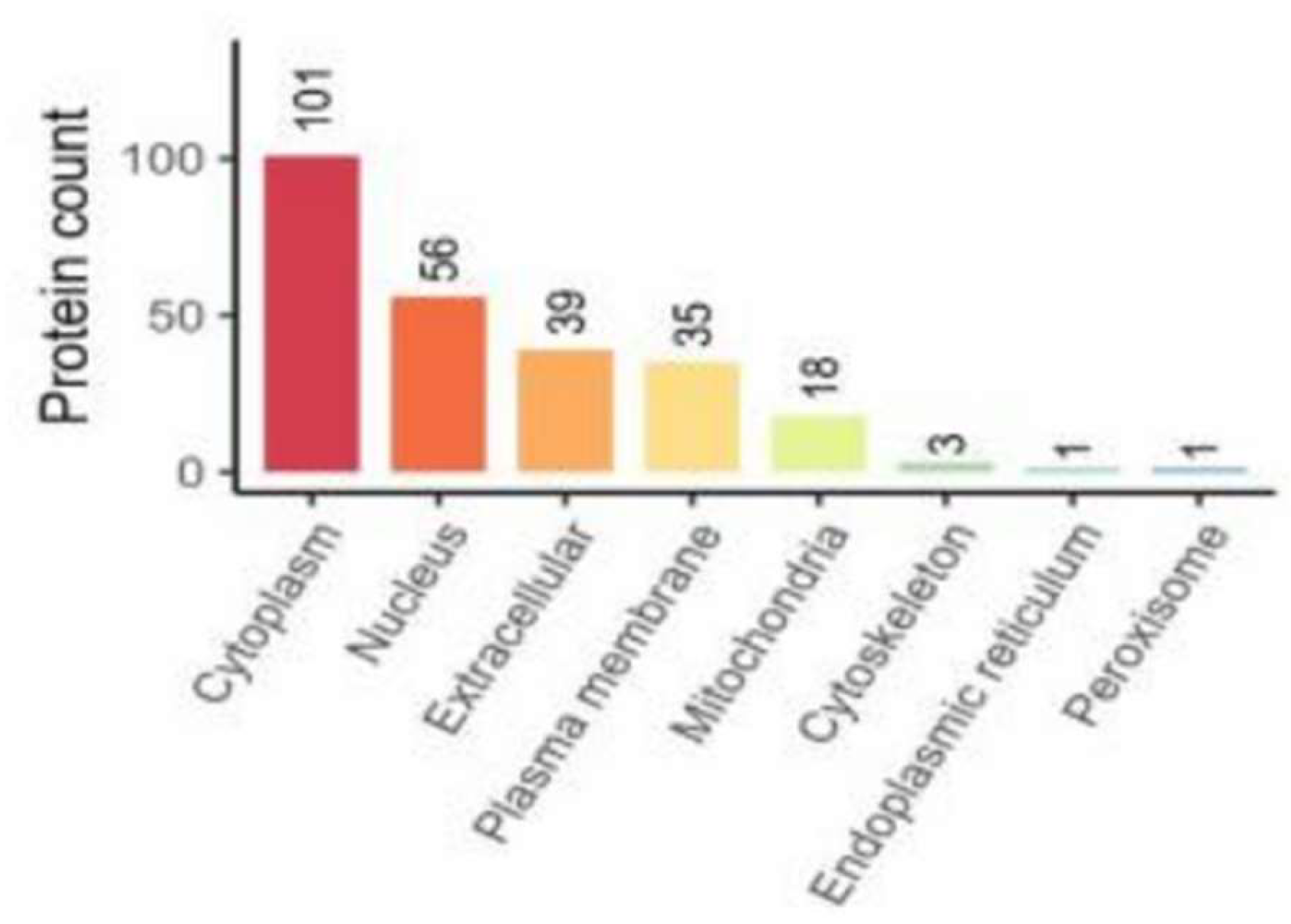
Figure 5.
GO enrichment.
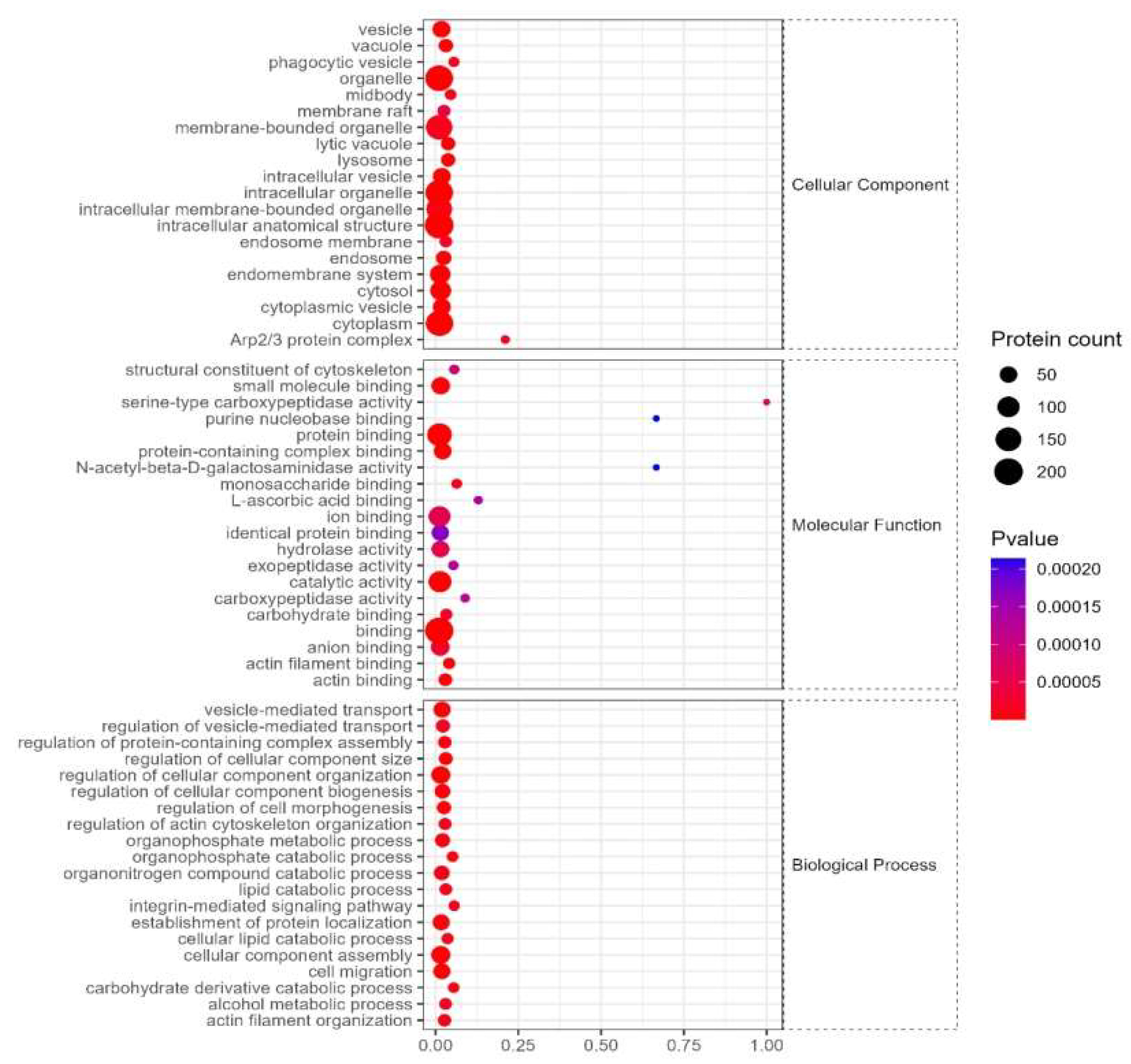
Figure 6.
KEGG.
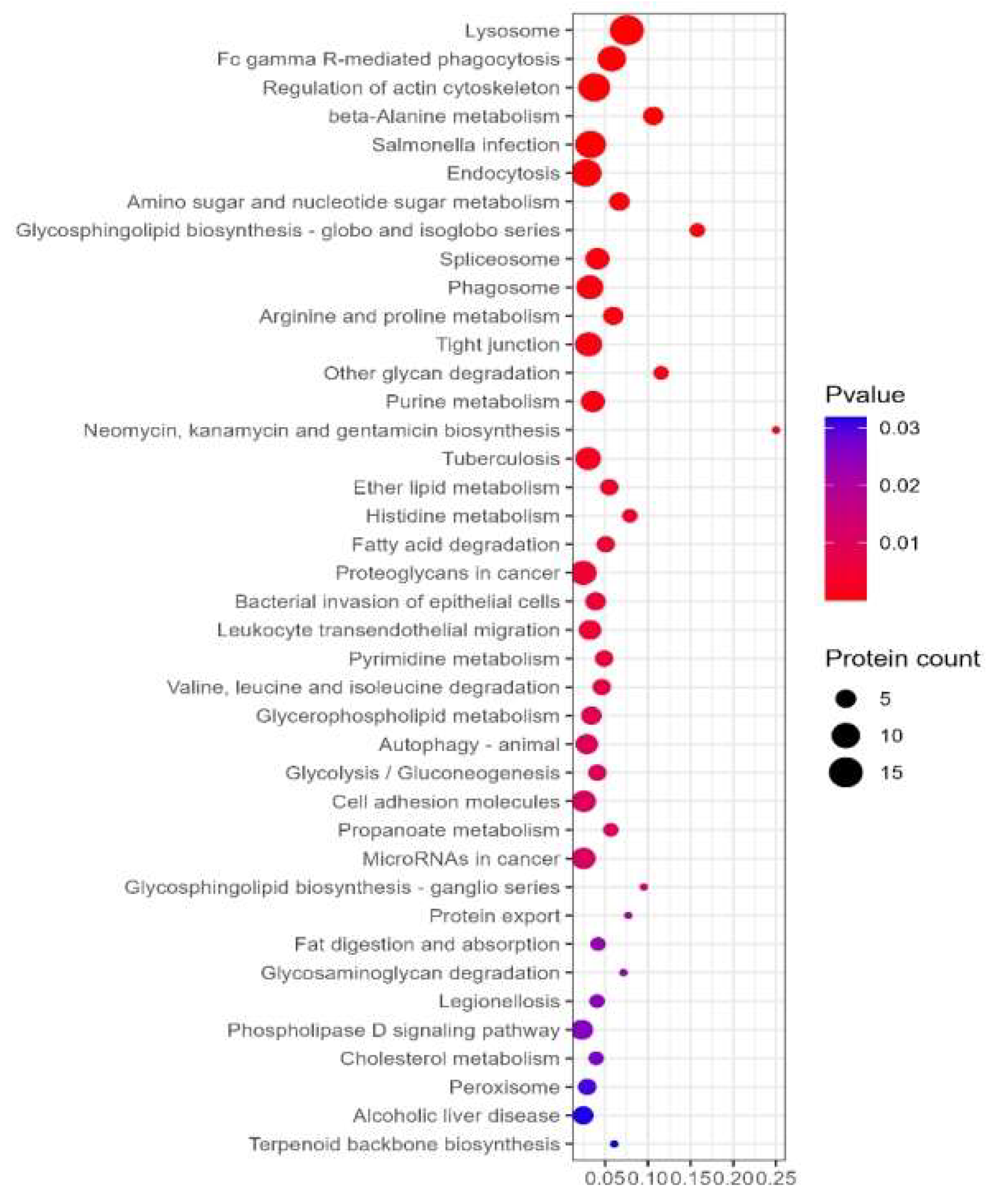
Figure 7.
Subcellular Localization of Proteins.
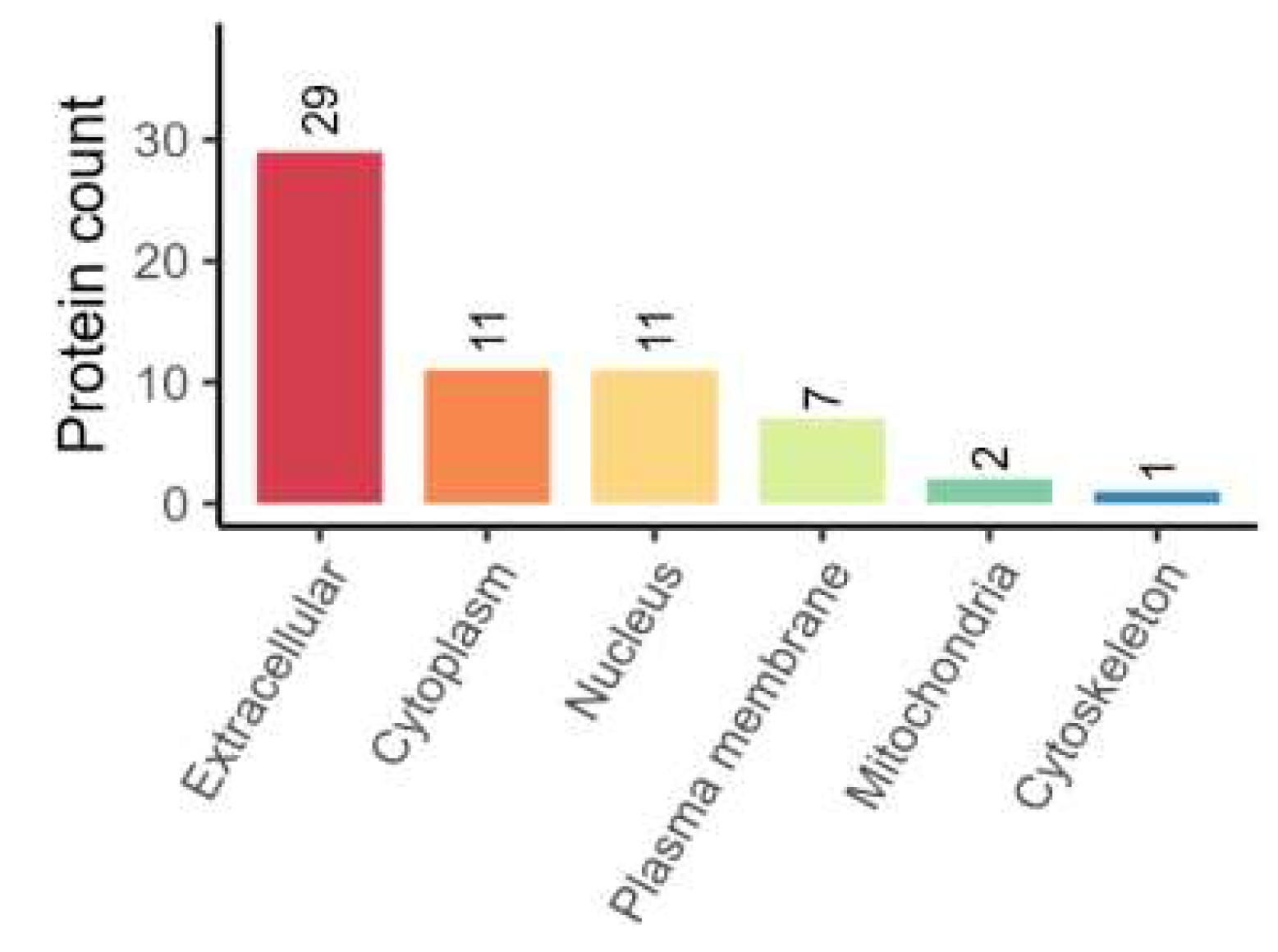
Figure 8.
GO enrichment.
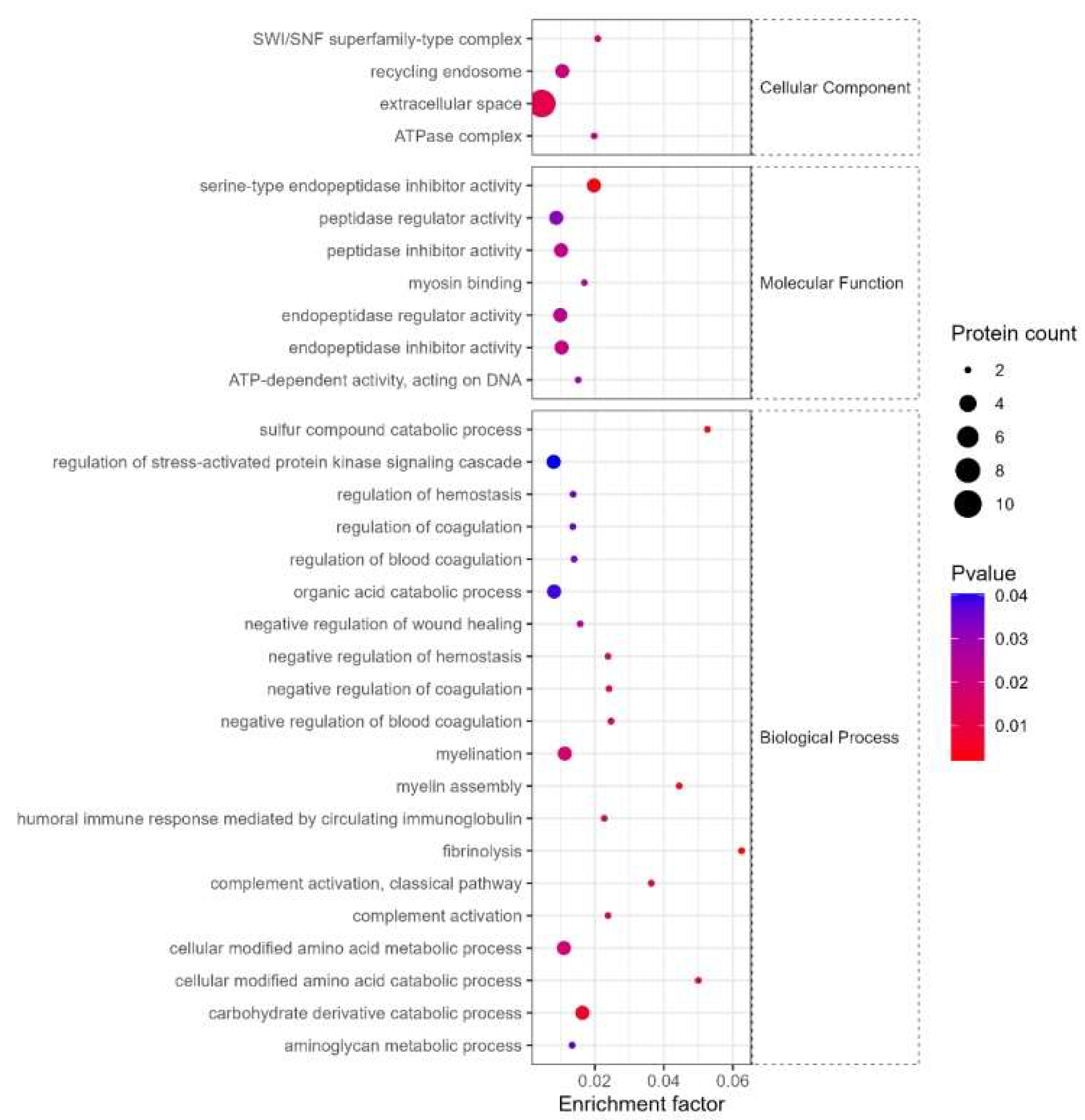
Figure 9.
KEGG.
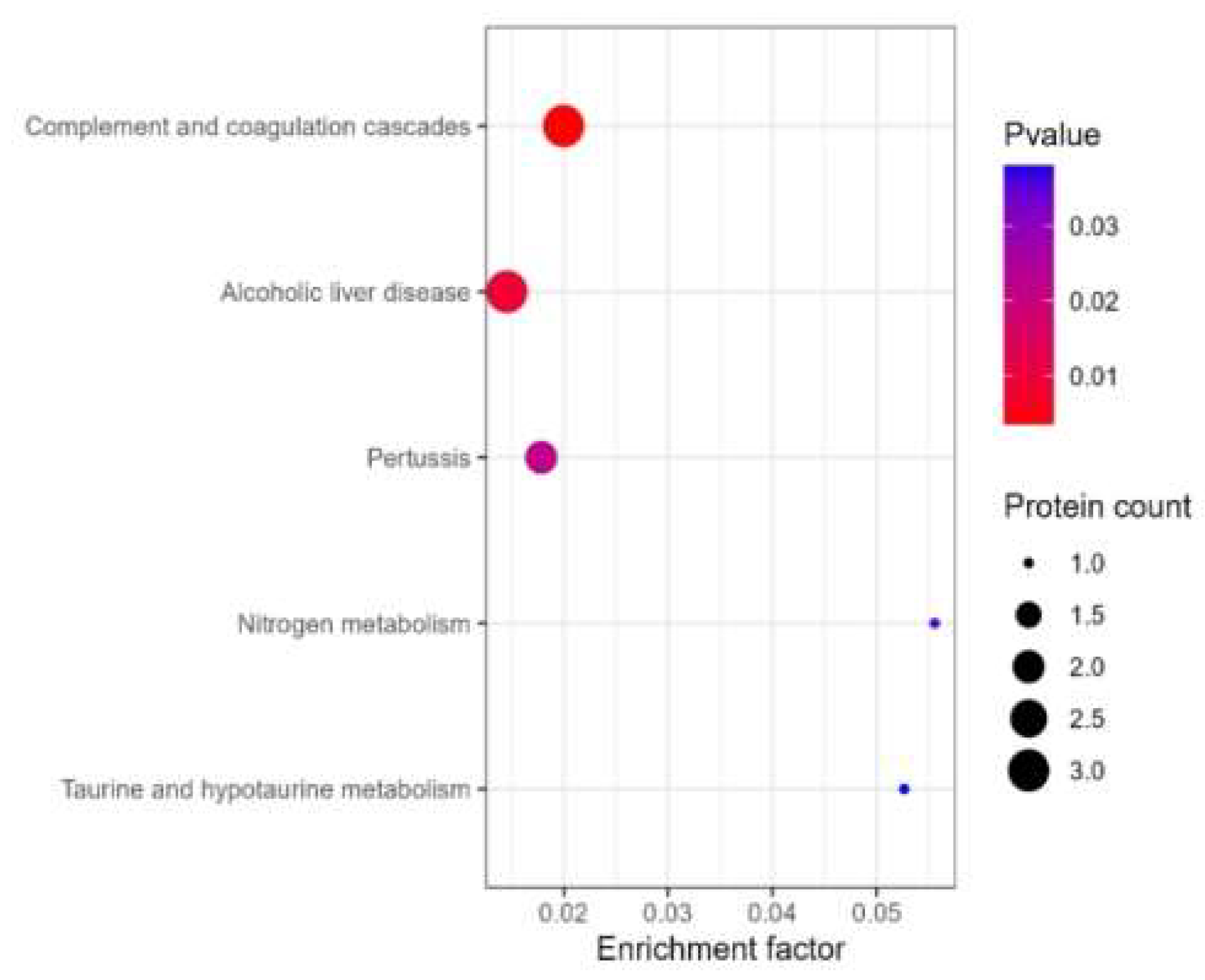
Figure 10.
Comparison Results of Differentially Expressed Proteins.
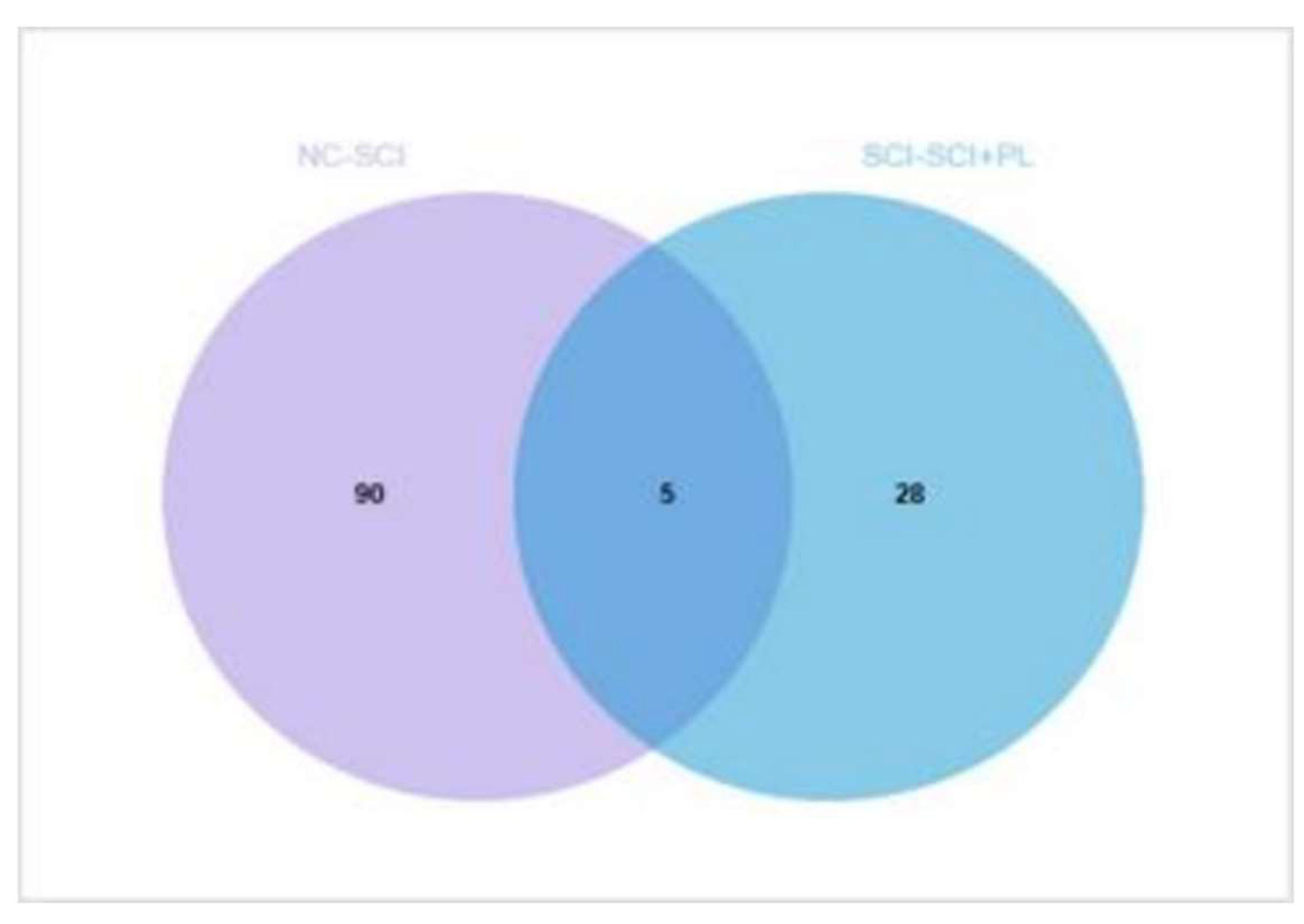
Figure 11.
Sema7A Expression Level.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



