Submitted:
11 October 2024
Posted:
15 October 2024
You are already at the latest version
Abstract
During the past 25 years, vesicular stomatitis virus (VSV) has produced multiple epidemic outbreaks in the US, resulting in the emergence of different viral lineages. Currently, very little is known about the pathogenesis of many of these lineages, thus limiting our understanding of the potential biological factors favoring each lineage in these outbreaks. To determine the potential phenotypical differences between two VSV Indiana (VSIV) serotype epidemic strains using a pig model. These strains are representative of the epidemic lineages that affected the US between 1997-1998 (IN98COE) and 2019-2020 (IN0919WYB2), the latter responsible for one of the most extensive outbreaks in the US. Our initial genome analysis revealed the existence of 121 distinct mutations between both strains, including the presence of a 14-nucleotide insertion in the intergenic region between G and L genes observed in IN0919WYB2. The levels of viral RNA in clinical samples between pigs infected with IN98COE or IN0919WYB2 were compared. Overall, higher and prolonged expression of viral RNA in pigs infected with IN98COE was observed. However, clinically, IN0919WYB2 was slightly more virulent than IN98COE, as well as more efficient to produce infection through contact transmission. Additionally, infectious virus was recovered from more samples when pigs were infected with IN0919WYB2, as revealed by virus isolation in cell culture, indicating the increased ability of this virus to replicate in pigs. Sequence analyses conducted from isolates recovered from both experimental groups showed that IN0919WYB2 produced more variability during the infection, denoting the potential of this strain to evolve rapidly after a single infection-contact transmission event in pigs. Collectively, the results showed that epidemic strains of VSIV may represent disparate phenotypes in terms of virulence/transmissibility for livestock, a situation that may impact the intensity of an epidemic outbreak. This study also highlights the relevance of pathogenesis studies in pigs to characterize phenotypical differences of VSV strains affecting livestock in the field.
Keywords:
Virulence
; vesicular stomatitis virus
; evolution
; pathogenesis
; epidemics
; transmissibility
1. Introduction
Vesicular stomatitis (VS) is a viral disease that annually affects livestock in the Americas [1] and can cause sporadic large epidemic outbreaks in the US [2,3,4,5,6]. VS is an arboviral disease caused by vesicular stomatitis virus (VSV), a negative sense RNA virus belonging to the Rhabdoviridae family, genus Vesiculovirus [7]. The genome of VSV contains five genes encoding five proteins: nucleoprotein (N), phosphoprotein (P), matrix (M), glycoprotein (G), and the large RNA-dependent polymerase (L) [8]. Additionally, diverse nonstructural proteins are encoded by alternative reading frames in the P (proteins C’ and C) [9] and M (M2 and M3 proteins) [10] genes. Two main serotypes have been described: VSV New Jersey virus (VSNJV) and VSV Indiana virus (VSIV) [1]. Pathogenesis studies conducted in pigs have concluded that the VSNJV strains are generally more virulent than VSIV [11]. However, a recent pathogenesis study showed that some VSIV strains could be as virulent as VSNJV ones [12], indicating that more pathogenesis studies are needed to gain insights into the phenotypical differences among VSV strains.
VSV is maintained in well-established endemic settings in Mexico and multiple countries in Central America, where clinical cases are reported annually in livestock [1]. Outbreaks of VSNJV in the US have been documented during 1995-1997 [1], 2004-2006 [4], and 2012-2015 [6,13]. VSIV has been associated with outbreak events occurring in 1997-1998 [5,14] and 2019-2020 [3]. In all cases, these outbreaks have been related to single monophyletic lineages, which display a high level of genome identity with viral strains that circulate in endemic zones of Mexico [6,15].
Very little is known about the molecular factors associated with the emergence of epidemic lineages in the US and the potentially diverse viral phenotypes associated with these viruses. A previous pathogenesis study in pigs suggested that an epidemic strain of VSNJV observed in the US (Strain NJ0612NME6, lineage 1.1) might represent a more virulent phenotype than its endemic relative (Strain NJ0806VCB, lineage 1.2) [16]. This virulent phenotype was associated with an increased number of vesicular lesions produced during the acute stage of the disease; a condition linked to the potential ability of the epidemic strain to evade better the innate host immune response [16].
Based on the current scientific evidence, it can be hypothesized that during epidemics, the maintenance and spread of epidemic lineages is achieved by infections in both livestock and insect vectors [17]. Within affected farms, VSV can be maintained in herd animals by direct contact with infected animals or fomites [17]. In the absence of viremia, the role of livestock as amplifier hosts has been questioned, suggesting that these species are the end hosts in the natural cycle of VSV [1]. However, alternative mechanisms have been experimentally proposed, pointing to livestock as non-conventional amplifier hosts [18,19,20]. These mechanisms imply that livestock is a virus source during vector feeding from active vesicular lesions or infected tissues without them [20]. This observation suggests that VSV strains showing increased levels of virulence in livestock may have potential adaptive advantages, allowing them to survive and thrive in epidemic conditions and thus possibly impacting the intensity of the outbreak events. This advantage might be associated with increased levels of replication in the vertebrate host, providing an optimal condition to produce new viral variants during the infection, a situation poorly explored until now.
The emergence and evolution of an epidemic VSIV lineage in the US was recently observed during the 2019-2020 outbreak [15]. Interestingly, after more than 20 years of absence, this lineage emerged in the US and produced one of the largest outbreaks in the US (Figure 1) [3]. This epidemic lineage was highly related to VSIV strains from endemic zones in Mexico. Throughout the outbreak, it diverged into four distinct phylogenetic groups [15]. A unique feature of this lineage is the presence of a 14-22 nucleotide insertion in the intergenic region between the G and L genes, a characteristic that was observed also in the ancestral endemic relatives of this lineage [15]. Based on the high number of clinical cases reported during the outbreak [3], it is possible to hypothesize that the epidemic VSIV 2019-2020 lineage may represent a virulent phenotype of VSIV in livestock.
This study aimed to determine the phenotypic characteristics of the VSV strain IN0919WYB2, a virus representative of the epidemic VSIV lineage from 2019-2020. For this purpose, a pathogenesis study was conducted using a well-established pig model [16,21,22] as pigs are a natural host of VSV. The VSIV strain 98COE, a virus related to the epidemic US lineage from the 1997-1998 outbreak, was used as a control. Epidemiologically, this lineage produced a considerably lower number of affected premises than lineage 2019-2020 (Figure 1). In this context, based on the epidemiological dynamics of the 2019-2020 VSIV outbreak, we hypothesized that the viral strain IN0919WYB2 represents a more virulent phenotype in pigs than the strain 98COE.
2. Materials and Methods
2.1. Viruses and Cells
The field VSIV strains 98COE (Horse; state of Colorado) and IN0919WYB2 (Bovine; state of Wyoming) were used for this study. Both strains were collected from vesicular lesions produced by natural infections during the epidemic outbreaks of VSIV in the US in 1998 and 2019, respectively. Viral stocks were produced in Vero-76 cells passage 38. Stocks were stored at -70°C until needed.
2.2. Phylogenetic and Evolutionary Analyses
Phylogenetic analyses presented in this study used a maximum likelihood method based on a general time reversible model, with 100 bootstrap replicas as statistical support for the tree topology. Analyses were conducted using the MEGA software version 10.2.5 [23]. Additionally, nonsynonymous and synonymous pairwise distance analyses were performed using Sequence Distances in the SSE software version 1.2 [24].
2.3. Animal Experiments
Pathogenesis experiments in pigs were conducted as previously described [16,21,22]. The studies were conducted at the National Centre for Foreign Animal Disease (NCFAD), a biosafety level 3-agricultural (BSL-3Ag) facility that is part of the Canadian Food Inspection Agency. Two duplicate in vivo experiments were conducted, one with each representative strain. In each experiment, six landrace/duroc pigs (~7 weeks old and 13-15 kg in weight) were housed together in the same animal room. After one week of acclimation, on the inoculation day, three of the pigs (contact group) were moved into a different room to avoid contact with the inoculated pigs for 24 hrs. The three remaining pigs (inoculated group) in the room were sedated using 0.3 ml of Stresnil (40mg/ml Azaperone), administered intramuscularly in the left hamstring. After that, pigs were anesthetized with isoflurane in oxygen. Inoculation was conducted by scarification in the snout via microneedling with a 20G needle up to the bevel of the needle. The snout was pricked 20 times and 107 TCID50 of virus in 50 l of Dulbecco Modified Eagle Medium (DMEM) was placed on the scarified area, and it was pricked again 20 times. The inoculum was allowed to soak for 3 minutes. The remaining inoculum was returned to the lab for back titration. After 24 hours, contact pigs were returned to the room to co-mingle with the inoculated group for the duration of the experiment (21 days) (Figure 2).
2.4. Clinical Evaluation and Sample Collection
Animals were clinically evaluated and sampled daily from day -1 (basal collection) through 10 and again at 16- and 21-days post-infection (dpi). Clinical evaluation consisted of measuring rectal temperature before sample collection and identifying vesicular lesions. The severity and dissemination of vesicular lesions were assessed using a clinical score system previously described [21,22]. Lesions in the feet at each digit contributed two points. An additional point is counted for vesicular lesions observed in the snout of inoculated pigs, and two points are counted for snout lesions in the case of contact animals. Additionally, two points were added for lesions in the carpal/tarsal skin, lips, and oral cavity.
The sampling strategy included collecting blood samples obtained from the jugular vein into EDTA-containing tubes (Vacutainer) or without anticoagulant to obtain serum. Oropharyngeal swabs were collected by targeting the tonsils of the soft palate using a large cotton swab. Nasal swabs were collected by swiping a small cotton swab within the external nares. Additionally, rectal swabs were collected from each pig using a small cotton swab. Swabs were broken off in universal transport media.
Based on animal welfare protocols at CSCHAH, blood, oropharyngeal, and rectal swabs were collected every other day (-1, 2, 4, 6, 8, 10, 16, and 21 dpi). For this purpose, all pigs were sedated and anesthetized as described in the animal experiment subsection. Nasal swabs were collected daily with no requirement for sedation.
2.5. Postmortem Sample Collection
At 21 dpi after being sedated and anesthetized for sample collection, all six pigs were euthanized with 20ml Euthanyl (240 mg/ml sodium pentobarbital), administered intravenously. Necropsies were conducted on all animals, and the following tissues were collected for analysis: tonsil of the soft palate, submandibular lymph node, popliteal lymph node, snout skin, neck skin, liver, spleen, anterior tongue epithelium, gastrohepatic lymph node, parotid lymph node, nasopharyngeal tonsil, and prescapular lymph node. Tissues were placed in individual tubes and frozen at -70 °C until further processing.
2.6. Viral RNA Detection
Viral RNA was extracted using the MagMax™-96 viral RNA isolation kit (AM1836, Applied Biosystems) and a KingFisher automated extraction system (ThermoFisher Scientific) following the manufacturer’s instructions for all clinical samples collected during this study. Specific detection and quantification of 98COE and IN0919WYB2 strains was carried out using a previously published multiplex real-time reverse-transcription polymerase chain reaction assay (RT-qPCR) protocol [25]. The quality of the RNA extractions was evaluated by real-time reverse-transcription polymerase chain reaction assay (RT-qPCR) using the β-actin gene as a target for the reaction by modifying a previously published protocol [26]. Briefly, the primers and probe remained static as per the publication. The master mix and cycling conditions were modified to mimic the VSV RT-qPCR conditions. TaqMan™ Fast Virus 1-Step Master Mix kit (Applied Biosystems #4444434) was used in a final volume of 25 μl. Amplification conditions for this kit are as follows: For reverse transcription, one cycle of 5 min at 50°C followed by 95°C for 20 s, and 45 cycles of 95°C for 15 s and 60°C for 45 s (Data collection). All assays were run on the QuantStudio 7 Pro (Applied Biosystems).
2.7. Detection of Viral Antigen
The detection of viral antigen from clinical samples was conducted using a double antigen-detection sandwich ELISA (DAS ELISA) following a previously described protocol [27]. OD values >0.1 were considered positive for antigen.
2.8. Virus Isolation
Recovery of infectious virus from clinical samples was performed using 24-well plates of Vero-76 cells [28]. Plates were seeded 24 hours before with 2.5x105 cells/well to give 90% confluency. Before infection, plates were washed once with 1 ml of PBS per well, then 300 l of MEM supplemented with glutaMAX and gentamycin (5g/mL) was added per well. Clinical samples were homogenized, clarified by centrifugation, and inoculated (200l sample/well) onto cells. After a one-hour absorption step at 37°C, an additional 500ul of MEM supplemented with glutaMAX, gentamycin (5g/mL), and 4% FBS was added to each well. Plates were incubated for 2 days at 37°C and checked every 24 hours for evidence of cytopathic effect.
2.9. Serum Neutralization Assay
Testing for neutralizing antibodies was conducted as previously described [16]. Briefly, test sera was heat-inactivated at 56°C for 30 min and then two-fold serially diluted in MEM medium (25 µl/well). Two replicates of each dilution were considered for the test. An equal volume (25 μL) of 1000 TCID50 of VSIV (ATCC Indiana Lab strain) was added to each well of the 96-well tissue culture microtiter plate, except for the wells used for serum toxicity testing. The plates were incubated for 1 hr at 37°C followed by the addition of Vero-76 cells (100 µl/well) in MEM medium. The plates were incubated at 37°C in a 5% CO2 incubator, and CPE was scored after 72 hr. Serum titers were defined as the reciprocal of the highest dilution, producing a 100% inhibition of CPE in both replicates. A back titration of the challenge virus and positive and negative serum controls were employed to assess test performance. A titer of ≥1:45 of the final dilution was regarded as positive.
2.10. Sequencing
The presence of the nucleotide insertion in the intergenic G-L region in the strain IN0919WYB2 was confirmed by Sanger sequencing using the following primers: G1206F – 5’-ATTGGACATGGTATGTTGGA – 3’ L193R – 5’-GAGGGAATCGGAAGAGAATT -3’. RT-PCR was performed using a qScript XLT One-Step RT-PCR kit (Quantabio). Amplification conditions were 48°C for 20 min, 94°C for 3 min, followed by 35 cycles of 94°C for 20 s, 52°C for 30 s, and 68°C for 60 s, with a 10-min extension at 68°C. PCR products were purified using the QIAquick PCR purification kit (Qiagen), and Sanger sequencing was carried out using BigDye Terminator V3.1 Cycle Sequencing kit (Applied Biosystems). Sequencing reactions were run on a 3500xL genetic analyzer (Applied Biosystems). Raw data was analyzed using Geneious Prime version 2021.0.3 (Biomatters Ltd.).
Next-generation sequencing (NGS) was conducted to confirm the identity of 98COE and IN0919WYB2 after preparing viral stocks for inoculation. NGS was also used to analyze the isolation positives from the clinical samples. NGS was conducted: DNase treatment (Ambion) was performed on the extracted RNA, followed by cleanup with an RNeasy MinElute kit (Qiagen). cDNA was prepared using SuperScript IV (ThermoFisher) with Endoh random hexamers (IDT) and VSV-specific primers (VSV_junction_S_IND_1_REV - 5’-TTATCCATGATATCTGTTAGTTT-3’ and VSV_junction_S_IND_2_REV - 5’-CTGTCCATGATCTCTGTTAGTTT-3’) (IDT) followed by second strand synthesis with NEBNext Ultra II (NEB) using the manufacturer’s recommended protocol. ds cDNA was purified using AMPure XP Beads (Beckman Coulter). Libraries from the purified cDNA pool were made using Nextera XT DNA Library Preparation Kit (Illumina) and Nextera XT Index Kit v2 (barcodes) (Illumina) following the manufacturer’s protocol using 20 cycles of PCR during Indexing. An Agilent TapeStation 4200 was used for quality control of the Nextera XT Libraries. Once the Library Pool has been analyzed on the Agilent and the concentration has been measured with the Qubit HS DNA kit, the library prep follows the Illumina method to denature the libraries and prepare them to run on the Illumina MiSeq. The resulting fastq files generated by MiSeq were analyzed via the following pipeline.
Preliminary virus detection and assembly were performed with the CFIA-NCFAD/nf-villumina (v2.0.1) [29]. Nextflow pipeline [30]. First, nf-villumina removed Illumina PhiX Sequencing Control V3 reads using BBDuk (v38.96) [31], followed by adaptor removal and quality filtering with fastp [32]. Filtered reads were retained for de novo assembly with Unicycler (v0.4.8) [33], Shovill (v1.1.0) [34], and MEGAHIT (v1.2.9) [35], and the resulting contigs from each assembly were queried against the online NCBI BLAST nucleotide database (accessed May 11-16, 2023; and October 30, 2023) [36]. The fastp filtered reads were mapped to the top VSIV BLAST match for each sample in Geneious Prime (v2023.0.1, https://geneious.com) [37] with the Minimap2 (v2.24) [38] assembler on default settings. A 75% majority consensus sequence was called with a low coverage threshold of 10x coverage. The resulting consensus sequences were aligned to the de novo assembled contigs with MAFFT (v7.490) [39] to manually check for assembly errors and then checked for the presence of complete coding sequences for the polyprotein gene in Geneious Prime (v2023.0.1) with the Find ORFs tool.
3. Results
3.1. Genomic Analysis
A detailed genomic analysis was conducted to determine the genomic differences between the VISV strains 98COE and IN0919WYB2 (Figure 3). Phylogenetic analysis of 98COE and IN0919WYB2, along with strains from distinct geographical origins (sequences obtained from Genbank), showed the close phylogenetic relationship between the two (Figure 3A). Strains 98COE and IN0919WYB2 grouped in the North American cluster (NA) and are phylogenetically distant from the viral strains circulating in Central (CA) and South (SA) America. Pairwise distance analysis comparing the full-length sequence of both strains indicated an overall nucleotide identity of 98.87%. A total of 121 distinct mutations were observed between these strains (Figure 3B–E). Of these, 112 were located in coding regions, with 85 classified as synonymous and 27 as nonsynonymous. Pairwise distance analysis conducted at either synonymous or nonsynonymous sites indicated that most of the phylogenetic differences between both strains could be explained by the genetic distance produced by the accumulation of synonymous mutations, which was ten times higher than the distance created by nonsynonymous mutations (Figure 3B,C). Furthermore, the analysis revealed a contrasting distribution pattern of mutations, showing that the nonsynonymous mutations were confined to genes P, G, and L (Figure 3B,C).
The evolutionary dynamics were tracked using previously published data to identify potentially relevant mutations on the epidemic VSIV lineage affecting the US between 2019-20 [15]. Based on this information, 109 out of 112 distinctive mutations between both strains may be associated with codons evolving under neutral and purifying selection at multiple genes (Figure 3D). Conversely, only three mutations were linked to codons evolving under diversifying selection, indicating that mutations at these codon sites may represent potential phenotypic differences between both strains (Figure 3D). These mutations involve specific codon sites in the P (CGA(R)-239-CTA(L)) and L (CCA(P)-1622-CTA(L)), (CGG(R)-1748-CAG(Q)) genes. Detailed information about all differential mutations between both strains can be found in (Figure S1). Furthermore, no differences were found between both strains in the genome sequences that encode the predicted C’ and C proteins.
A total of ten characteristic mutations were found distributed among all non-coding genome regions of these strains (Figure 3E). No specific additional procedures were undertaken to determine the sequences in the 3’ and 5’ untranslated regions. However, compared with the original sequences of these viruses previously published in the GenBank database, no differences were found between these sequences and the consensus sequences obtained by NGS from the viral stocks used in this study (Figure 3E). These similarities were also consistent during the evaluation of coding regions, indicating that additional passages on Vero-76 cells to produce the viral stocks used in this study did not alter the consensus sequences of these viruses. Moreover, we confirmed the presence of the 14-nucleotide insertion located in the intergenic G-L region of the IN0919WYB2 (Figure 3E), a genetic feature distinctive of the epidemic VSIV lineage from the 2019-20 outbreak [15].
3.2. Clinical Assessment
A pathogenesis study was conducted in pigs to determine potential phenotypical differences between the epidemic VSIV strains 98COE and IN0919WYB2. Overall, clinical results suggested that IN0919WYB2 may represent a sligtly more virulent phenotype for pigs than the 98COE strain. After inoculation by scarification in the snout, two out of three pigs in each group developed vesicular lesions at the inoculation site. In pigs inoculated with IN0919WYB2, lesions were visible in pigs 300 and 302 as early as 2dpi, while in pigs inoculated with 98COE, lesions were observed at 2dpi (pig 308) and 4dpi (pig 307). Fever was only observed in pigs inoculated with IN0919WYB2 (Figure 4A). A temperature of about 40°C was observed in pigs 300 and 302 at 1dpi. This febrile response was prolonged in pig 302 until 3dpi.
Interestingly, another fever spike was detected again in pigs 301 and 302 at 5 dpi, which correlated with the detection of small vesicular lesions in both hind coronary bands of these pigs at 8 dpi. In the contact group, evidence of fever was recorded only in pig 303 at 4dpc, contact of the group inoculated with IN0919WYB2 (Figure 4B). Overall, vesicular lesions were not observed in contact pigs associated with diverse groups.
3.3. Viral Shedding
To evaluate the potential differences in the viral shedding dynamics between 98COE and IN0919WYB2 during infection, RT-qPCR was used to detect the presence of viral RNA in nasal, oral, and rectal swabs. Overall, the results indicated that pigs infected with 98COE shed higher levels of viral RNA for a prolonged period than those infected with IN0919WYB2 (Figure 5).
In nasal swabs, the presence of viral RNA in inoculated animals was detected as soon as 1dpi, being significantly (p-0.0251) higher at 2dpi in pigs inoculated with IN0919WYB2 (Figure 5A). After that, disparate levels of RNA were detected in inoculated animals within groups. In the group infected with 98COE, pig 307 displayed RNA levels between days 3-5dpi that reached 5.6-6.4 predicted TCID50/mL. Similarly, pig 302, inoculated with IN0919WYB2, showed high RNA levels between 5dpi and 7dpi. These levels ranged from 4.8-5.6 predicted TCID/mL (Figure 5A).
In the contact animals (Figure 5B), positive samples were detected at 1dpc, significantly (p-0.0150) higher in contact animals infected with IN0919WYB2. However, after that, significantly higher amounts were detected in pigs infected with 98COE at 2dpc (p-0,0230), 8dpc (p-0.0222), and 9dpc (p-0.0019). At 9dpc, only one (Pig 303) out of 3 contact pigs in the IN0919WYB2 group showed some evidence of viral RNA, indicating that viral shedding was more prolonged in contact pigs infected with 98COE. Viral RNA was not detected beyond 9dpc in either group.
Overall, both groups observed no statistically significant differences in oral shedding (Figure 5CD). However, a prolonged excretion of viral RNA was recorded in pigs inoculated with 98COE (10dpi), compared to pigs inoculated with IN0919WYB2 (8dpi) (Figure 5C). No differences were observed in the duration of oral shedding for contact pigs. Nevertheless, at five and 7dpc increased amounts of viral RNA were detected in pigs 310 and 311, ranging between 3.5-6.0 predicted TCID50/mL.
Finally, viral shedding was assessed in rectal swabs. In inoculated animals, significantly higher levels of RNA were found in pigs inoculated with 98COE at 8 dpi (p-0.0465) and 10 dpi (p-0.03252) (Figure 5E). Similarly, increased RNA levels were observed in 98COE contact animals at 9dpi (p-0.0059) (Figure 5E). In contact animals, we also observed an extended viral shedding in animals infected with 98COE (Figure 5E).
No viral RNA was detected in the blood or serum samples from any pig in either experimental group.
3.4. Detection of Infectious Virus
After assessing differences in RNA shedding by RT-qPCR from clinical samples collected during the experiments, we attempted to understand if the differences in shedding dynamics observed between both groups were consistent with the recovery of infectious virus in Vero-76 cells. Interestingly, despite the increased and prolonged levels of viral RNA shed by pigs infected with 98COE, more infectious VSV was recovered from pigs infected with IN0919WYB2. In particular, viral shedding detected in nasal swabs between 2dpi and 6dpi showed that a higher proportion of pigs infected with IN0919WYB2 excreted more infectious virus than pigs infected with 98COE (Figure 6A). Furthermore, this pattern was also characterized by the recovery of infectious virus from all six pigs infected with IN0919WYB2, contrasting with the detection of infectious particles in just three pigs of the group infected with 98COE (Figure 6A). Overall, despite viral RNA in nasal swabs, infectious virus was not recovered beyond 6dpi (Figure 6B).
In oral swabs, minimal differences were observed between groups. At 4dpi infectious virus was recovered exclusively from two pigs in the group infected with IN0919WYB2, while a single viral isolate was obtained from the group infected with 98COE at 6dpi (Figure 6C). Like the pattern observed in nasal swabs, despite CT values, no infectious virus was recovered beyond 6dpi (Figure 6D). The minimal detection of infectious viral particles in both groups clearly suggested that the shedding of infectious particles from the oral cavity was not an important source of infectious virus induced during the infection with these two strains. No viral isolates were obtained from either group’s animal rectal swabs.
Due to the apparent lack of correlation between the recovery of infectious virus and the presence of CT values, a Pearson r correlation analysis was performed. This analysis indicated a significant negative correlation (p-0.0081, r=-0.8459) between the CT value in the sample and the proportion of viral isolates recovered at specific CT values (Figure 6E). In samples with CT values between 17-19, the percentage of infectious particles recovered was 100%. However, above this range of CT values, there was a decrease in the recovery of infectious viruses (Figure 6E). Interestingly, 73.07% (n=19) of the viral isolates obtained in both groups were associated with CT values between 26 and 34 (Figure 6E).
Additionally, the DAS ELISA was performed to infer if the lack of viral isolates observed in RT-qPCR positive samples was associated with the absence of viral antigens, denoting the presence of viral RNA only in the clinical samples. First, using several ten-fold dilutions of the viral stocks 98COE and IN0919WYB2, we determined the limit of detection of the DAS ELISA for both antigens. RT-qPCR was performed for each dilution. Limits of detection associated with CT values of 21.29 (1x105.43 TCID50/mL) and 22.26 (1x106.00 TCID50/mL) were observed for IN0919WYB2 and 98COE, respectively (Figures S2A and S2B). Overall, all the 9 viral isolates recovered during the experiment used as a control, came positive by DAS ELISA, indicating the ability of this test to detect viral particles recovered from pigs (Figure S2C). In this sense, based on the detection limit predicted for the DAS ELISA, a total of six samples with CT values ranging between 22.75 and 26.49, all negative by viral isolation, were evaluated by DAS ELISA. Positive DAS results were obtained in three (CT values 22.97, 23.46, and 24.76) out of the six samples (Figure S2C), indicating the presence of viral antigens in some of these samples. However, considering the low sensitivity of the DAS ELISA and that most of the isolates obtained during this study were associated with samples with CT values higher than 26, we determined that the results produced by the DAS ELISA must be considered inconclusive.
3.5. Postmortem Sample Evaluation
A total of 12 postmortem tissues were collected from all pigs in each group at 21 dpi to evaluate potential differences in the viral distribution in tissues between pigs infected with either 98COE or IN0919WYB2. Overall, higher levels of viral RNA, based on RT-qPCR CT values, were observed in multiple tissues in pigs infected with IN0919WYB2 than in 98COE (Figure 7). In inoculated pigs, significantly higher levels of viral RNA were found in the tonsil of the soft palate in animals inoculated with IN0919WYB2, contrasting with pigs inoculated with 98COE, where no evidence of viral RNA was found in this tissue (Figure 7A). Furthermore, while no viral RNA was present in the popliteal, parotid, prescapular, gastrohepatic lymph nodes, and snout skin from animals inoculated with 98COE, evidence of viral RNA was found in one of two inoculated animals from IN0919WYB2 group (Figure 7A). Interestingly, evidence of viral RNA was found in popliteal lymph nodes of animals 301 and 302, correlating with the presence of vesicular lesions in both hind coronary bands of these pigs and confirming the ability of IN0919WYB2 to disseminate away from the inoculation site.
In contact animals, viral RNA was undetectable in the tissues of the 98COE group (Figure 7B). In contrast, significantly higher levels of viral RNA were observed in submandibular lymph nodes of contact pigs in the IN0919WYB2 group. In these animals, the presence of viral RNA was also detected in two pigs in the tonsil of the soft palate and one pig in the nasopharyngeal tonsil (Figure 7B).
No viral RNA was detected in either group: neck skin, anterior tongue epithelium, or liver. Furthermore, in all cases, attempts to isolate infectious viruses from tissues collected from both groups produced a negative result. However, based on the potentially high concentrations of predicted viral titers (predicted TCID50/mL based on CT values) (Figure 7A), we decided to look for the presence of viral antigens. Viral antigen was detected in inoculated animals #300 in submandibular and parotid lymph nodes (corrected OD’s 0.227 and 0.183 respectively) and #307 from submandibular lymph node (corrected OD 0.193), showing the presence of viral antigen at 21dpi in both infected groups.
3.6. Neutralizing Antibody Response
Serum samples collected during the experiment were tested for virus neutralization. No differences were observed between inoculated animals from different groups to assess potential differences in the adaptive immune response produced by the infection of pigs with either 98COE or IN0919WYB2 (Figure 8A). Overall, at 6dpi, neutralizing antibodies were recorded in all inoculated animals of both groups. The presence of neutralizing antibodies was consistent until the end of the experiment (Figure 8A). However, a dissimilar pattern was observed in contact animals between groups. While the presence of neutralizing antibodies in contact pigs from the IN0919WYB2 group was evident between 7 and 9 dpc, all contact pigs from the 98COE group failed to develop neutralizing antibodies during the time of the experiment (Figure 8B). Interestingly, this lack of neutralizing antibody response clearly correlates with the absence of viral RNA in multiple tissues collected from contact pigs in the group 98COE (Figure 7B), providing evidence of the absence of contact-transmission for 98COE.
3.7. Evolutionary Dynamics of 98COE and IN0919WYB2 during Infection in Pigs
Finally, we explored potential differences between 98COE and IN0919WYB2 regarding their ability to induce a viral variants during the infection in pigs. Our results showed that IN0919WYB2 had the ability to induce a higher genetic variability during the infection in pigs compared to 98COE (Figure 9). Overall, a total of seven single nucleotide polymorphisms (SNPs) were found distributed in genes N, P, G, and L among viruses recovered from the pigs infected with IN0919WYB2, half of which were associated with nonsynonymous changes (Figure 9A). Additionally, an insertion was found in the intergenic G-L region from pig 302 (Figure 9A), which corresponds to the addition of an extra A in the highly conserved seven adenine polyadenylation signal present in the intergenic regions of VSV [40]. Our sequencing analysis revealed an interesting infection dynamic pattern, where seven viral variants at the consensus level were identified during the infection of pigs with IN0919WYB2 (Figure 9A). The consensus sequence associated with the viral stock (variant #0) was only observed in 2 out of 20 viral isolates recovered during the infection, indicating that multiple viral variants dominated the infection dynamics in this experimental group. Specific changes at codon positions (N/20, P/77, G/352, and L/2066) were observed in different variants, affecting multiple pigs at different time points during the infection. In this context, variant #1 was the most prevalent, identified in three pigs at multiple dpi (Figure 9A). The synonymous mutation GCA-GCC in gene N/codon 20 was the most prevalent mutation during infection and was observed in seven isolates involving variants #1 and #6.
Interestingly, it was the only mutation observed in isolates recovered from all pigs at specific time points during the infection (Figure 9A). Additionally, in pig P304 at four dpi, two different variants (#0 and #7) were recovered from oral and nasal swabs, respectively (Figure 9A). The diverse nature of the mutations observed does not preclude the reversion back to the original state (Figure 9A). Conversely, in pigs infected with 98COE, the infection was dominated by variant #0 (Figure 9B). In this group, two different variants were identified (Figure 9B), one in pig 309 at 4dpi and the other in pig 310 at 4dpi. In pig 310 we observed the same pattern described in the other group, where the original viral phenotype was recovered later during the infection (Figure 9B).
4. Discussion
Recently, the epidemic 2019-20 VSIV lineage IN0919WYB2 emerged in the US, producing one of the largest outbreaks in this country [3]. The epidemiological characteristics of this lineage starkly differ from those of the previous VSIV lineage that impacted the US during 1997-98 [14], suggesting potential phenotypic distinctions between the two. In this scenario, similar to observations in other well-documented arboviruses like the Venezuelan equine encephalitis virus [41], it is uncertain whether the emergence of these epidemic lineages could be associated with the ability of specific viral phenotypes to be more efficient at replicating in either vertebrate hosts or vectors, or even during outbreaks.
In this study, pigs, a natural host of VSV [11,16,21,22], were used to determine phenotypic differences in the vertebrate host between the strains 98COE and IN0919WYB2 associated with the epidemic lineages affecting the US between 1997-98 and 2019-20 respectively. Although these strains were recovered from a horse (98COE) and a bovine (IN0919WYB2) in the field, a previous study showed that pigs represent a neutral host model for pathogenesis studies, being unaffected for the host predilections seen in some VSNJV strains when evaluated in bovine models [42]. In this sense, it is reasonable to suggest that the host origin of these strains did not bias the results obtained in our study.
Multiple genomic differences between 98COE and IN0919WYB2 may be potentially linked to the phenotypic differences observed in this study. Indeed, one of the most remarkable differences between both strains was the 14-nucleotide insertion located in the G-L intergenic region of IN0919WYB2. Originally, it was assumed that the presence of insertions in G-L intergenic region of VSIV, was a hallmark exclusively of VSIV strains from Central American origin [43]. However, our previous study showed that insertions in this region can be also found in strains from North American origin [15]. At this point, we don’t have an explanation regarding the possible role of this insertion in the phenotype displayed by IN0919WYB2 in pigs. Interestingly, our results are consistent with a previous pathogenesis study in pigs [12], showing that VSIV strains from a Central American origin carrying long insertions in the intergenic G-L region appeared more virulent than VSIV strain that originated in North America, suggesting the potential role of this insertion as a promoter of the virulence in VSIV. Future experiments using reverse genetics are needed to get more insights about the possible role of the insertion in the phenotype observed in IN0919WYB2 infections in pigs.
Furthermore, our genomic analysis identified multiple putative mutations between both strains. Among these, three mutations at gene/codons P (CGA(R)-239-CTA(L)) and L (CCA(P)-1622-CTA(L)), (CGG(R)-1748-CAG(Q)) previously found under positive selection on natural populations of VSIV [15], might be considered to explain the phenotypic differences between both viruses. However, since analysis to identify the evolutionary significance of these mutations was conducted in silico [15], future studies using reverse genetics are needed to assess the possible role of these mutations in the pathogenesis of VSIV.
Previously, it was characterized the epidemic VSNJV NJ0612NME6, which resulted in a highly virulent phenotype in pigs when compared with its closet endemic ancestor (NJ0806VCB) [16], suggesting that increased virulence is a hallmark of the epidemic VSV lineages. However, clinically, our results were consistent with previous publications, indicating the overall low virulence displayed by VSIV strains in pigs [11,44,45], even when the strains used in our study were associated with epidemic lineages. In our study, it was impossible to establish any statistically significant difference in terms of virulence between strains based on the development of vesicular lesions. However, based on the development of small secondary vesicular lesions in 2 out of 3 pigs infected with IN0919WYB2, it is possible to hypothesize that this virus may represent a slightly more virulent phenotype for pigs than 98COE. On the other hand, the fact that just 2 out of 3 inoculated pigs at each group developed vesicular lesions in the inoculation site in the snout, strongly suggest that both strains have similar levels of infectivity. Conversely, the augmented capacity of IN0919WYB2 to promote the infection by direct contact, identified transmissibility as distinctive marker between both strains.
Regarding the evaluation of viral RNA in clinical samples, our results contrast with a previous study in pigs, where viral RNA in nasal and tonsil swabs was inconsistently found in infected animals [12]. Conversely, our results were consistent with previous studies on VSNJV using RT-PCR [16,21,22], showing shedding of viral RNA occurring as early as 1dpi and 1dpc in all pigs. A remarkable finding in our study was the lack of correlation between the presence of viral RNA in clinical samples and the recovery of infectious viruses. Despite detecting viral RNA in both groups, live virus, especially from nasal swabs, mainly was recovered from pigs (inoculated and contact) infected with IN0919WYB2. This observation highlights the efficiency of IN0919WYB2 to produce infectious particles during the infection in pigs. In this context, as seen in other viruses like Crimean-Congo hemorrhagic fever virus and yellow fever virus [46], our results make possible to hypothesize that IN0919WYB2 might achieve to orchestrate genome and particle production in an optimal fashion as opposed to 98COE. In terms of transmissibility, this ability might be considered a critical difference between the strains evaluated in our study. Future studies are needed to assess potential differences between both viruses in terms of infectiousness.
Considering the detection of viral antigens by DAS ELISA in few of the clinical samples, a possible explanation for the lack of correlation between viral RNA and infectious virus in the samples evaluated during this study may be the presence of defective interfering virus particles (DIVP). DIVPs are well known for their properties to modulate virulence by triggering the innate immune response and interfering with viral replication [47]. In our study, this lack of correlation was more evident in the group of pigs infected with the 98COE strain, suggesting that increased levels of DIVP might have been produced during the infection of pigs with this strain and, therefore, provide a possible hypothesis for the reduced levels of virulence seen in this experimental group. Interestingly, our results may be consistent with a previous hypothesis which proposed that there was cycling in the production of infectious virus seen in the brain of mice infected with VSV [48], a condition wherein there is alternating dominance between infective and interfering viral particles during the infection. Interestingly, in other arboviruses like the Zika virus, it has been demonstrated that DIVPs are antiviral in vivo, both in mice and mosquito vectors, reducing the transmission in the vector by up to 90% [49]. Further experiments are needed to probe the potential relevance of DIVP during the infection of VSV in pigs. In this sense, the lack of characterization of the specific viral RNA particles detected in the clinical samples (genomic RNA, antigenomic RNA, and messenger RNA) by RT-qPCR must consider a limitation in our study.
A postmortem lymph node evaluation was performed in our study to contrast the ability of both strains to disseminate along the seven lymphatic territories previously described in pigs (parotid, mandibular, dorsal cervical, ventral cervical, subiliac, inguinal, and popliteal) [50]. Overall, in contrast to the group of pigs inoculated with 98COE, the positive results in PTON and SLMN lymph nodes in all three inoculated pigs with IN0919WYB2 suggest the ability of this strain to replicate at higher levels in the parotid and mandibular lymphatics respectively. This observation was consistent with the higher levels of infectious virus recovered from pigs infected with IN0919WYB2, supporting that this strain may represents a more virulent phenotype than 98COE for pigs. Furthermore, considering the inoculation route used in this study and the presence of positive samples in PopLN (popliteal territory) in two out of three pigs inoculated with IN0919WYB2, it can also be suggested that IN0919WYB2 has the potential to disseminate systemically during infection, which may reflect the clamed increase in virulence of this strain. However, since most positive results were found in the parotid (PTON and NTON) and mandibular (SMLN and ParLN) lymphatic territories in both groups, we can state that during the infection in pigs with these two strains, the majority of viral replication takes place locally, close to the primary site of the infection. This situation highly contrasts with our previous results with VSNJV [16], showing the intrinsic differences between both serotypes to produce a systemic infection in pigs. This situation can be correlated with the severity of the clinical outcomes observed during the infection with VSNJV in pigs [16].
Unlike the contact pigs evaluated from the IN0919WYB2 group, no positive results (for nucleic acid or live virus) were obtained from postmortem samples collected from the contact pigs from the 98COE group. This result suggests the lack of transmission between directly inoculated pigs and the contact pigs in the latter group. Interestingly, this fact highly correlates with the lack of neutralizing antibodies observed from contact pigs in the 98COE group, confirming the differences in the ability between these strains to promote transmission by direct contact in pigs. This result is interesting, considering the detection of viral RNA particles in contact pigs from 98COE group. As mentioned above future studies are needed to characterize the presence of DIVP viral gRNA, cRNA, and mRNA in the clinical samples from these pigs. However, the limited number of viral isolations obtained from this contact group (n=1), may reflect the extremely low levels of infectious particles circulating in these pigs, providing a possible explanation not only about the absence of neutralizing antibodies but also the lack of RNA detection in postmortem samples. These results also stress the value of including contact pigs in experimental groups during pathogenesis studies to assess differences in levels of transmissibility among VSIV strains.
Finally, in this study, we evaluated a poorly understood aspect of the evolution of VSV during an in-vivo infection in a natural host. Overall, we consider that the results of this approach generated an interesting perspective, not only due to the possible differences in the evolutionary dynamics between two VSIV epidemic strains but also to increase our insights about the potential role of the vertebrate host during the infectious cycle of VSV in nature. Our results also showed the possible relevance of the vertebrate host in the occurrence of viral diversity during infection, a condition dependent on the intrinsic characteristics of specific viral strains. Based on the above statements, we conclude that the ability of IN0919WYB2 to induce an increased number of viral variants during the infection in pigs might have represented an evolutionary advantage for the VSIV lineage associated with the epidemic outbreak 2019-2020 in the US. This characteristic may be considered an additional factor to help understand the epidemiological differences recorded between the epidemic VSIV lineages from 1997-1998 and 2019-2020 [3].
In this sense, among the multiple mutations reported during our study, the GCT(A)-GTT(V) mutation at codon G/352 associated with a neutralizing epitope [51], was the only mutation found previously reported in VSIV in nature [15]. Interestingly, the report showed that the substitution GCT(A)-GTT(V) was a putative mutation linked to the group of viruses dominating the VSIV outbreak in the US during 2020 [15]. Based on the results of our pathogenesis study, we suggest that the emergence of the mutation GCT(A)-GTT(V) during the outbreak was probably the result of an infection in livestock instead of an insect vector. However, because neither this mutation nor other mutations reported in this study became dominant during the infection in pigs, we speculate that genetic drift instead of natural selection may be responsible for the evolutionary pattern observed in this study, suggesting that mutation GCT(A)-GTT(V) might not be implicated in the adaptation of VSV in livestock. In this context, the fact that the mutation GCT(A)-GTT(V) was fixed and maintained in the sub-lineage dominating the infections in the outbreak during 2020 after multiple transmission events [15] strongly suggests that this mutation might have represented an adaptive advantage for this sub-lineage to replicate in a specific insect vector during the outbreak. More studies are needed to assess the potential role of the multiple mutations described in this study in viral adaptation in livestock and insect vectors.
In conclusion, our study indicates that the VSIV epidemic strains 98COE and IN0919WYB2, which represent the epidemic lineages 1997-1998 and 2019-2020, respectively, constitute two different phenotypes in terms of transmissibility for pigs. Interestingly, this phenotypic difference may be correlated with the epidemiological discrepancies observed between the outbreaks produced by both lineages in the US (3, 14). Interestingly, the recovery of infectious virus from contact pigs was obtained from animals in a subclinical state, suggesting that this situation may be a confounding factor during epidemic outbreaks, favoring the transmission of VSIV by the movement of apparently uninfected animals. In this sense our results make possible to hypothesize a possible scenario where in the absence of clinical disease, other sources of infection beyond the vesicular lesions should be considered as a potential source of the infection of susceptible animals or insect vectors. Finally, our results indicate that protocols for pathogenesis studies of VSV, should be redefined beyond the typical assessment based just on the detection of vesicular lesions.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Differential mutations at multiple gene codon regions between 98COE and IN0919WYB2; Figure S2: Evaluation of clinical samples found positive by RT-qPCR using DAS ELISA.
Author Contributions
Conceptualization, CM, LR, CN, SB LV-S; methodology, KH, CM, CN, SB LV-S.; formal analysis, KH, SB, CM, LV-S; investigation, KH, PS, KHA, MN, OL, ES, SZ, CN, LR, SB, CM, and LV-S. resources, LR, CH, CN, and SB; data curation, KH, PS, MN KHA, OL, ES, SZ, SB and LV-S. ; writing—original draft preparation, KH, SB, CM and LV-S ; writing—review and editing, KH, PS, MN, KHA, OL, ES, SZ, CN, LR, SB, CM, and LV-S visualization, KH, SB, LV-S supervision, KH, SB, LV-S; project administration, SB, CN, LV-S and CM ; funding acquisition, LR, and CH. All authors have read and agreed to the published version of the manuscript.
Funding
This work was conducted under the USDA Research Service CRIS Project "Predicting and Mitigating Vesicular Stomatitis Virus (VSV) in North America" No. 3022-32000-062-000-D.
Institutional Review Board Statement
The project was approved by the Animal Care Committee of the Canadian Science Centre for Human and Animal Health (CSCHAH) under the Animal Use Document (AUD) number C-22-002. Canadian Council for Animal Care (CCAC) guidelines were observed during all procedures with animals.
Data Availability Statement
The raw sequencing data of this project are available in the NCBI Sequence Read Archive (SRA) under the BioProject number
Acknowledgments
The authors thank the Animal Care staff for their valuable help with experiments. We also thank Dr. Peter Simmonds for the use of the SSE program used for pairwise distance analysis.
Conflicts of Interest
The authors declare that the research was conducted without any commercial or financial relationships that could potentially create a conflict of interest.
References
- Rodriguez, L.L., 2002. Emergence and re-emergence of vesicular stomatitis in the United States. Virus Res 85, 211-219. [CrossRef]
- Bertram, M.R., Rodgers, C., Reed, K., Velazquez-Salinas, L., Pelzel-McCluskey, A., Mayo, C., Rodriguez, L., 2023. Vesicular stomatitis Indiana virus near-full-length genome sequences reveal low genetic diversity during the 2019 outbreak in Colorado, USA. Front Vet Sci 10, 1110483. [CrossRef]
- Pelzel-McCluskey, A., Christensen, B., Humphreys, J., Bertram, M., Keener, R., Ewing, R., Cohnstaedt, L.W., Tell, R., Peters, D.P.C., Rodriguez, L., 2021. Review of Vesicular Stomatitis in the United States with Focus on 2019 and 2020 Outbreaks. Pathogens 10. [CrossRef]
- Rainwater-Lovett, K., Pauszek, S.J., Kelley, W.N., Rodriguez, L.L., 2007. Molecular epidemiology of vesicular stomatitis New Jersey virus from the 2004-2005 US outbreak indicates a common origin with Mexican strains. J Gen Virol 88, 2042-2051. [CrossRef]
- Rodriguez, L.L., Bunch, T.A., Fraire, M., Llewellyn, Z.N., 2000. Re-emergence of vesicular stomatitis in the western United States is associated with distinct viral genetic lineages. Virology 271, 171-181. [CrossRef]
- Velazquez-Salinas, L., Pauszek, S.J., Zarate, S., Basurto-Alcantara, F.J., Verdugo-Rodriguez, A., Perez, A.M., Rodriguez, L.L., 2014. Phylogeographic characteristics of vesicular stomatitis New Jersey viruses circulating in Mexico from 2005 to 2011 and their relationship to epidemics in the United States. Virology 449, 17-24. [CrossRef]
- Velazquez-Salinas, L., Zarate, S., Eschbaumer, M., Pereira Lobo, F., Gladue, D.P., Arzt, J., Novella, I.S., Rodriguez, L.L., 2016. Selective Factors Associated with the Evolution of Codon Usage in Natural Populations of Arboviruses. PLoS One 11, e0159943. [CrossRef]
- Dietzgen, R.G., 2012. Morphology. Genome Organization, Transcription and Replication of Rhabdoviruses, in: Kuzmin, R.G.D.a.I.V. (Ed.), Rhabdoviruses, pp. 5-11.
- Spiropoulou, C.F., Nichol, S.T., 1993. A small highly basic protein is encoded in overlapping frame within the P gene of vesicular stomatitis virus. J Virol 67, 3103-3110. [CrossRef]
- Jayakar, H.R., Whitt, M.A., 2002. Identification of two additional translation products from the matrix (M) gene that contribute to vesicular stomatitis virus cytopathology. J Virol 76, 8011-8018. [CrossRef]
- Stallknecht, D.E., Greer, J.B., Murphy, M.D., Mead, D.G., Howerth, E.W., 2004. Effect of strain and serotype of vesicular stomatitis virus on viral shedding, vesicular lesion development, and contact transmission in pigs. Am J Vet Res 65, 1233-1239. [CrossRef]
- Morozov, I., Davis, A.S., Ellsworth, S., Trujillo, J.D., McDowell, C., Shivanna, V., Dasen, E.J., Nichols, R., Martin, B.K., Monath, T.P., Richt, J.A., 2019. Comparative evaluation of pathogenicity of three isolates of vesicular stomatitis virus (Indiana serotype) in pigs. J Gen Virol 100, 1478-1490. [CrossRef]
- Humphreys, J.M., Shults, P.T., Velazquez-Salinas, L., Bertram, M.R., Pelzel-McCluskey, A.M., Pauszek, S.J., Peters, D.P.C., Rodriguez, L.L., 2024. Interrogating Genomes and Geography to Unravel Multiyear Vesicular Stomatitis Epizootics. Viruses 16, 1118. [CrossRef]
- McCluskey, B.J., Hurd, H.S., Mumford, E.L., 1999. Review of the 1997 outbreak of vesicular stomatitis in the western United States. J Am Vet Med Assoc 215, 1259-1262. [CrossRef]
- Zarate, S., Bertram, M.R., Rodgers, C., Reed, K., Pelzel-McCluskey, A., Gomez-Romero, N., Rodriguez, L.L., Mayo, C., Mire, C., Pond, S.L.K., Velazquez-Salinas, L., 2024. Phylogenomic Signatures of a Lineage of Vesicular Stomatitis Indiana Virus Circulating During the 2019–2020 Epidemic in the United States. Preprints 2024, 2024100585. [CrossRef]
- Velazquez-Salinas, L., Pauszek, S.J., Stenfeldt, C., O’Hearn, E.S., Pacheco, J.M., Borca, M.V., Verdugo-Rodriguez, A., Arzt, J., Rodriguez, L.L., 2018. Increased Virulence of an Epidemic Strain of Vesicular Stomatitis Virus Is Associated With Interference of the Innate Response in Pigs. Front Microbiol 9, 1891. [CrossRef]
- Rozo-Lopez, P., Drolet, B.S., Londono-Renteria, B., 2018. Vesicular Stomatitis Virus Transmission: A Comparison of Incriminated Vectors. Insects 9. [CrossRef]
- Mead, D.G., Gray, E.W., Noblet, R., Murphy, M.D., Howerth, E.W., Stallknecht, D.E., 2004. Biological transmission of vesicular stomatitis virus (New Jersey serotype) by Simulium vittatum (Diptera: Simuliidae) to domestic swine (Sus scrofa). J Med Entomol 41, 78-82. [CrossRef]
- Smith, P.F., Howerth, E.W., Carter, D., Gray, E.W., Noblet, R., Mead, D.G., 2009. Mechanical transmission of vesicular stomatitis New Jersey virus by Simulium vittatum (Diptera: Simuliidae) to domestic swine (Sus scrofa). J Med Entomol 46, 1537-1540. [CrossRef]
- Smith, P.F., Howerth, E.W., Carter, D., Gray, E.W., Noblet, R., Smoliga, G., Rodriguez, L.L., Mead, D.G., 2011. Domestic cattle as a non-conventional amplifying host of vesicular stomatitis New Jersey virus. Med Vet Entomol 25, 184-191. [CrossRef]
- Velazquez-Salinas, L., Naik, S., Pauszek, S.J., Peng, K.W., Russell, S.J., Rodriguez, L.L., 2017. Oncolytic Recombinant Vesicular Stomatitis Virus (VSV) Is Nonpathogenic and Nontransmissible in Pigs, a Natural Host of VSV. Hum Gene Ther Clin Dev 28, 108-115. [CrossRef]
- Velazquez-Salinas, L., Pauszek, S.J., Holinka, L.G., Gladue, D.P., Rekant, S.I., Bishop, E.A., Stenfeldt, C., Verdugo-Rodriguez, A., Borca, M.V., Arzt, J., Rodriguez, L.L., 2020. A Single Amino Acid Substitution in the Matrix Protein (M51R) of Vesicular Stomatitis New Jersey Virus Impairs Replication in Cultured Porcine Macrophages and Results in Significant Attenuation in Pigs. Front Microbiol 11, 1123. [CrossRef]
- Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K., 2018. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 35, 1547-1549. [CrossRef]
- Simmonds, P., 2012. SSE: a nucleotide and amino acid sequence analysis platform. BMC Res Notes 5, 50. [CrossRef]
- Hole, K., Nfon, C., Rodriguez, L.L., Velazquez-Salinas, L., 2021. A Multiplex Real-Time Reverse Transcription Polymerase Chain Reaction Assay With Enhanced Capacity to Detect Vesicular Stomatitis Viral Lineages of Central American Origin. Front Vet Sci 8, 783198. [CrossRef]
- Moniwa, M., Clavijo, A., Li, M., Collignon, B., Kitching, P.R., 2007. Performance of a foot-and-mouth disease virus reverse transcription-polymerase chain reaction with amplification controls between three real-time instruments. J Vet Diagn Invest 19, 9-20. [CrossRef]
- Hole K, Velazquez-Salinas L, Clavijo A. Improvement and optimization of a multiplex real-time reverse transcription polymerase chain reaction assay for the detection and typing of Vesicular stomatitis virus. J Vet Diagn Invest. 2010 May;22(3):428-33. [CrossRef]
- Hole, K., Clavijo, A., Pineda, L.A., 2006. Detection and serotype-specific differentiation of vesicular stomatitis virus using a multiplex, real-time, reverse transcription-polymerase chain reaction assay. J Vet Diagn Invest 18, 139-146. [CrossRef]
- Kruczkiewicz, P. CFIA-NCFAD/nf-villumina. (2023).
- Di Tommaso, P., Chatzou, M., Floden, E.W., Barja, P.P., Palumbo, E., Notredame, C., 2017. Nextflow enables reproducible computational workflows. Nat Biotechnol 35, 316-319. [CrossRef]
- Bushnell B. 2022. BBmap. SourceForge. Available from: https://sourceforge.net/projects/bbmap.
- Chen, S., Zhou, Y., Chen, Y., Gu, J., 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884-i890. [CrossRef]
- Wick, R.R., Judd, L.M., Gorrie, C.L., Holt, K.E., 2017. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13, e1005595. [CrossRef]
- Seemann T, Edwards R, Goncalves da Silva A, Kiil K. 2020. Shovill v1.1.0. https://github.com/tseemann/shovill.
- Li, D., Luo, R., Liu, C.M., Leung, C.M., Ting, H.F., Sadakane, K., Yamashita, H., Lam, T.W., 2016. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3-11. [CrossRef]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J., 1990. Basic local alignment search tool. J Mol Biol 215, 403-410. [CrossRef]
- Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S., Duran, C., Thierer, T., Ashton, B., Meintjes, P., Drummond, A., 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647-1649. [CrossRef]
- Li, H., 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094-3100. [CrossRef]
- Katoh, K., Standley, D.M., 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772-780. [CrossRef]
- Stillman EA, Whitt MA. The length and sequence composition of vesicular stomatitis virus intergenic regions affect mRNA levels and the site of transcript initiation. J Virol. 1998 Jul;72(7):5565-72. [CrossRef]
- Weaver, S.C., Ferro, C., Barrera, R., Boshell, J., Navarro, J.-C., 2004. VENEZUELAN EQUINE ENCEPHALITIS*. Annual Review of Entomology 49, 141-174. [CrossRef]
- Smith, P.F., Howerth, E.W., Carter, D., Gray, E.W., Noblet, R., Berghaus, R.D., Stallknecht, D.E., Mead, D.G., 2012. Host predilection and transmissibility of vesicular stomatitis New Jersey virus strains in domestic cattle (Bos taurus) and swine (Sus scrofa). BMC Vet Res 8, 183. [CrossRef]
- Rodriguez, L.L., Pauszek, S.J., Bunch, T.A., Schumann, K.R., 2002. Full-length genome analysis of natural isolates of vesicular stomatitis virus (Indiana 1 serotype) from North, Central and South America. J Gen Virol 83, 2475-2483. [CrossRef]
- Flanagan, E.B., Zamparo, J.M., Ball, L.A., Rodriguez, L.L., Wertz, G.W., 2001. Rearrangement of the genes of vesicular stomatitis virus eliminates clinical disease in the natural host: new strategy for vaccine development. J Virol 75, 6107-6114. [CrossRef]
- Martinez, I., Rodriguez, L.L., Jimenez, C., Pauszek, S.J., Wertz, G.W., 2003. Vesicular stomatitis virus glycoprotein is a determinant of pathogenesis in swine, a natural host. J Virol 77, 8039-8047. [CrossRef]
- Weidmann, M., Sall, A.A., Manuguerra, J.C., Koivogui, L., Adjami, A., Traore, F.F., Hedlund, K.O., Lindegren, G., Mirazimi, A., 2011. Quantitative analysis of particles, genomes and infectious particles in supernatants of haemorrhagic fever virus cell cultures. Virol J 8, 81. [CrossRef]
- Vignuzzi, M., Lopez, C.B., 2019. Defective viral genomes are key drivers of the virus-host interaction. Nat Microbiol 4, 1075-1087. [CrossRef]
- Cave, D.R., Hendrickson, F.M., Huang, A.S., 1985. Defective interfering virus particles modulate virulence. J Virol 55, 366-373. [CrossRef]
- Rezelj, V.V., Carrau, L., Merwaiss, F., Levi, L.I., Erazo, D., Tran, Q.D., Henrion-Lacritick, A., Gausson, V., Suzuki, Y., Shengjuler, D., Meyer, B., Vallet, T., Weger-Lucarelli, J., Bernhauerova, V., Titievsky, A., Sharov, V., Pietropaoli, S., Diaz-Salinas, M.A., Legros, V., Pardigon, N., Barba-Spaeth, G., Brodsky, L., Saleh, M.C., Vignuzzi, M., 2021. Defective viral genomes as therapeutic interfering particles against flavivirus infection in mammalian and mosquito hosts. Nat Commun 12, 2290. [CrossRef]
- Ito, R., Suami, H., 2015. Lymphatic Territories (Lymphosomes) in Swine: An Animal Model for Future Lymphatic Research. Plast Reconstr Surg 136, 297-304. [CrossRef]
- Munis, A.M., Tijani, M., Hassall, M., Mattiuzzo, G., Collins, M.K., Takeuchi, Y., 2018. Characterization of Antibody Interactions with the G Protein of Vesicular Stomatitis Virus Indiana Strain and Other Vesiculovirus G Proteins. J Virol 92. [CrossRef]
Figure 1.
Summary of the epidemiological dynamics of two distinct epidemic VSIV lineages affecting the US during 1997-98 and 2019-20 outbreaks. A) Geographical distribution of the VSIV 1997-98 and VSIV 2019-20 lineages associated with the strains 98COE and IN0919WYB2, respectively. States were abbreviated as Arizona (AZ), Arkansas (AR), Colorado (CO), Kansas (KS), Missouri (MO), Nebraska (NE), New Mexico (NM), Oklahoma (OK), Texas (TX), Utah (UT) and Wyoming (WY). B) Differences between the number of affected premises during the 1997-98 and 2019-20 VSIV outbreaks in the US.
Figure 1.
Summary of the epidemiological dynamics of two distinct epidemic VSIV lineages affecting the US during 1997-98 and 2019-20 outbreaks. A) Geographical distribution of the VSIV 1997-98 and VSIV 2019-20 lineages associated with the strains 98COE and IN0919WYB2, respectively. States were abbreviated as Arizona (AZ), Arkansas (AR), Colorado (CO), Kansas (KS), Missouri (MO), Nebraska (NE), New Mexico (NM), Oklahoma (OK), Texas (TX), Utah (UT) and Wyoming (WY). B) Differences between the number of affected premises during the 1997-98 and 2019-20 VSIV outbreaks in the US.
Figure 2.
General overview of the experiment design. Two groups of pigs were used to evaluate the pathogenesis of the strains 98COE and IN0919WYB2, representative of the epidemic lineages responsible for outbreaks of VSIV in the US during 1997-1998 and 2019-2020. Each experimental group was comprised of six pigs, with three of the pigs in each group being inoculated in the snout. In comparison, the remaining three pigs evaluated the ability of each strain to be transmitted by direct contact. Contact pigs in each room co-mingled with inoculated pigs after 24 hours post-inoculation (hpi). Pig# reflects diverse pigs’ roles during the experiment (inoculated or contact). This figure was created using BioRender.com under the agreement number YH27EC6GAL.
Figure 2.
General overview of the experiment design. Two groups of pigs were used to evaluate the pathogenesis of the strains 98COE and IN0919WYB2, representative of the epidemic lineages responsible for outbreaks of VSIV in the US during 1997-1998 and 2019-2020. Each experimental group was comprised of six pigs, with three of the pigs in each group being inoculated in the snout. In comparison, the remaining three pigs evaluated the ability of each strain to be transmitted by direct contact. Contact pigs in each room co-mingled with inoculated pigs after 24 hours post-inoculation (hpi). Pig# reflects diverse pigs’ roles during the experiment (inoculated or contact). This figure was created using BioRender.com under the agreement number YH27EC6GAL.
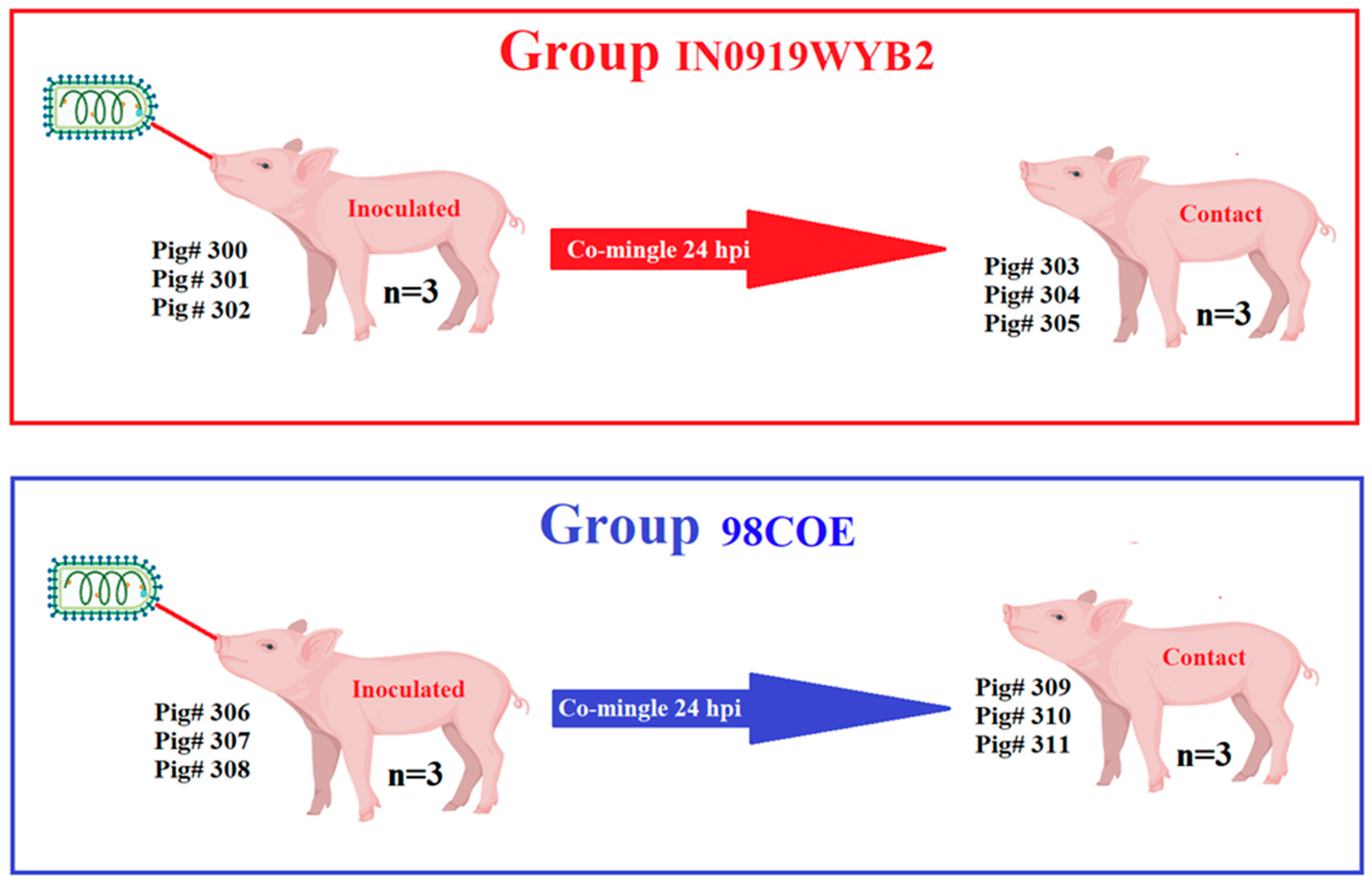
Figure 3.
Genomic characterization between the VSIV epidemic strains 98COE and IN0919WYB2. A) Phylogenetic analysis showing the genetic relationship between 98COE and IN0919WYB2. The analysis was conducted using representative VSIV strains from North America (NA), Central America (CA), and South America (SA) genetic origins. The phylogenetic tree was produced using MEGA version 10.2.5. B) and C) Pairwise distance analysis between 98COE and IN019WYB, showing the genetic distance produced between both strains through the accumulation of synonymous and nonsynonymous mutations at different genes. Bars in the graphics represent pairwise distance using a window of 50 nucleotides, with a step of 25. The graph was produced using SSE version 1.2. D) Summary of the distinctive number of synonymous and nonsynonymous mutations between 98COE and IN019WYB within the different genes of VSIV. Selection class reflects a summary of the evolutionary dynamics at different codon sites in each gene where distinctive mutations were predicted. The selection class at different codons was obtained from previous study assessing the evolution in natural populations of VSIV [15] E) Location of mutations and insertion at non-coding genome regions of VSIV between 98COE and IN019WYB strains. To enhance the analysis, the original sequences of these viruses previously published in the GenBank database were included (AF473864_98COE, and T4337283.2_IN0919WYB2). Nucleotide (nt) boundaries between non-coding regions are based on sequence T4337283.2. Analysis was conducted using the software Jalview version 2.11.1.4.
Figure 3.
Genomic characterization between the VSIV epidemic strains 98COE and IN0919WYB2. A) Phylogenetic analysis showing the genetic relationship between 98COE and IN0919WYB2. The analysis was conducted using representative VSIV strains from North America (NA), Central America (CA), and South America (SA) genetic origins. The phylogenetic tree was produced using MEGA version 10.2.5. B) and C) Pairwise distance analysis between 98COE and IN019WYB, showing the genetic distance produced between both strains through the accumulation of synonymous and nonsynonymous mutations at different genes. Bars in the graphics represent pairwise distance using a window of 50 nucleotides, with a step of 25. The graph was produced using SSE version 1.2. D) Summary of the distinctive number of synonymous and nonsynonymous mutations between 98COE and IN019WYB within the different genes of VSIV. Selection class reflects a summary of the evolutionary dynamics at different codon sites in each gene where distinctive mutations were predicted. The selection class at different codons was obtained from previous study assessing the evolution in natural populations of VSIV [15] E) Location of mutations and insertion at non-coding genome regions of VSIV between 98COE and IN019WYB strains. To enhance the analysis, the original sequences of these viruses previously published in the GenBank database were included (AF473864_98COE, and T4337283.2_IN0919WYB2). Nucleotide (nt) boundaries between non-coding regions are based on sequence T4337283.2. Analysis was conducted using the software Jalview version 2.11.1.4.
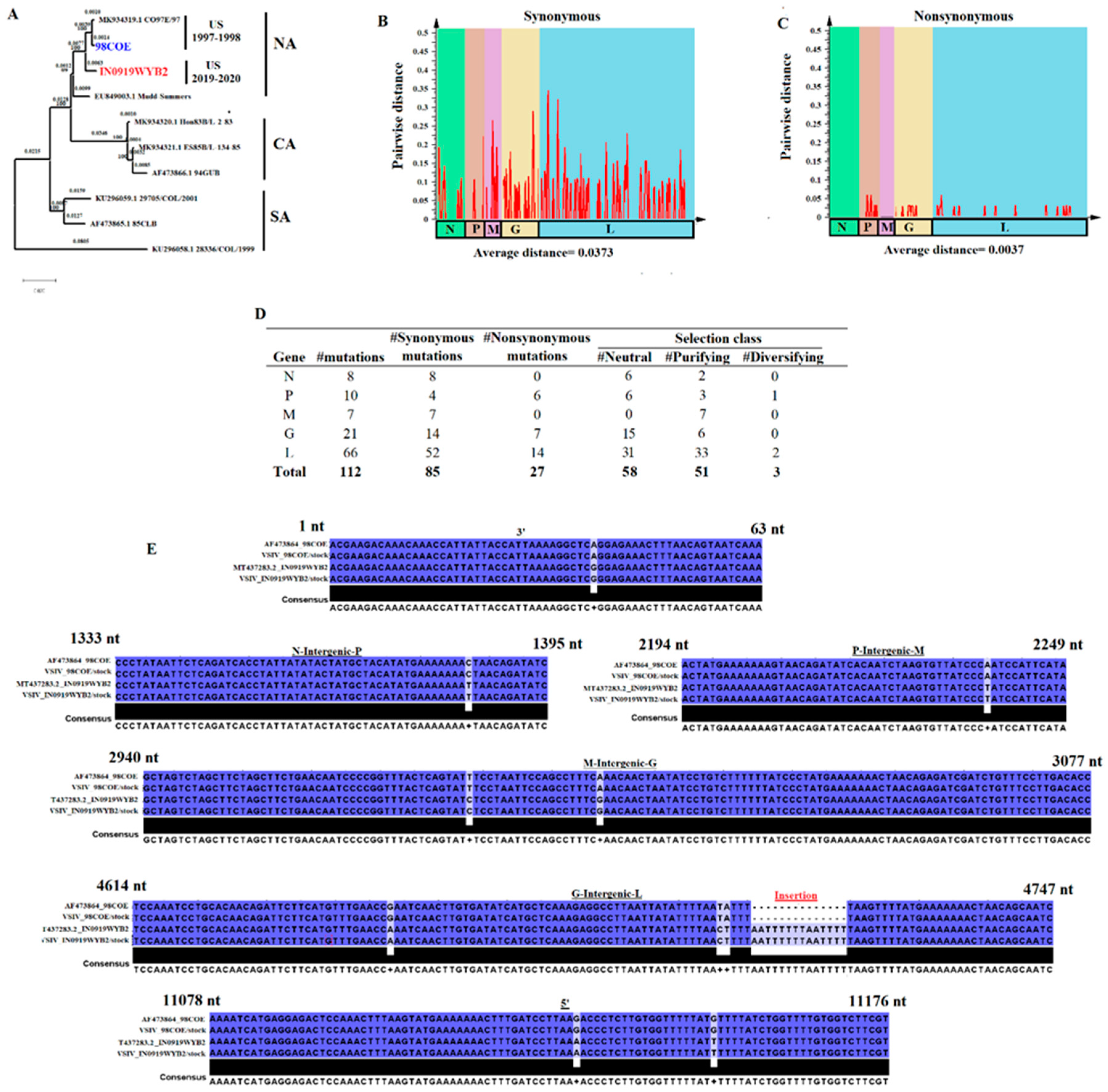
Figure 4.
Comparison of clinical outcomes produced by infection with 98COE and IN0919WYB2. Rectal temperatures were monitored on specific days to detect fever in inoculated (A) and in contact pigs (B) after infection with different viruses. In inoculated pigs, red circles, squares, and triangles denote measurements for pigs 300, 301, and 302, respectively. The same shapes were used in contact pigs to identify pigs 303, 304, and 305 from the IN0919WYB2 group. A similar pattern was followed for blue shapes (98COE group), identifying pigs 306, 307, and 308 (inoculated) and 309, 310, and 311 (contact), respectively. Final clinical scores were recorded from inoculated (C) and contact pigs (D) at each group. Graphics were produced using the software GraphPad version 9.5.0.
Figure 4.
Comparison of clinical outcomes produced by infection with 98COE and IN0919WYB2. Rectal temperatures were monitored on specific days to detect fever in inoculated (A) and in contact pigs (B) after infection with different viruses. In inoculated pigs, red circles, squares, and triangles denote measurements for pigs 300, 301, and 302, respectively. The same shapes were used in contact pigs to identify pigs 303, 304, and 305 from the IN0919WYB2 group. A similar pattern was followed for blue shapes (98COE group), identifying pigs 306, 307, and 308 (inoculated) and 309, 310, and 311 (contact), respectively. Final clinical scores were recorded from inoculated (C) and contact pigs (D) at each group. Graphics were produced using the software GraphPad version 9.5.0.
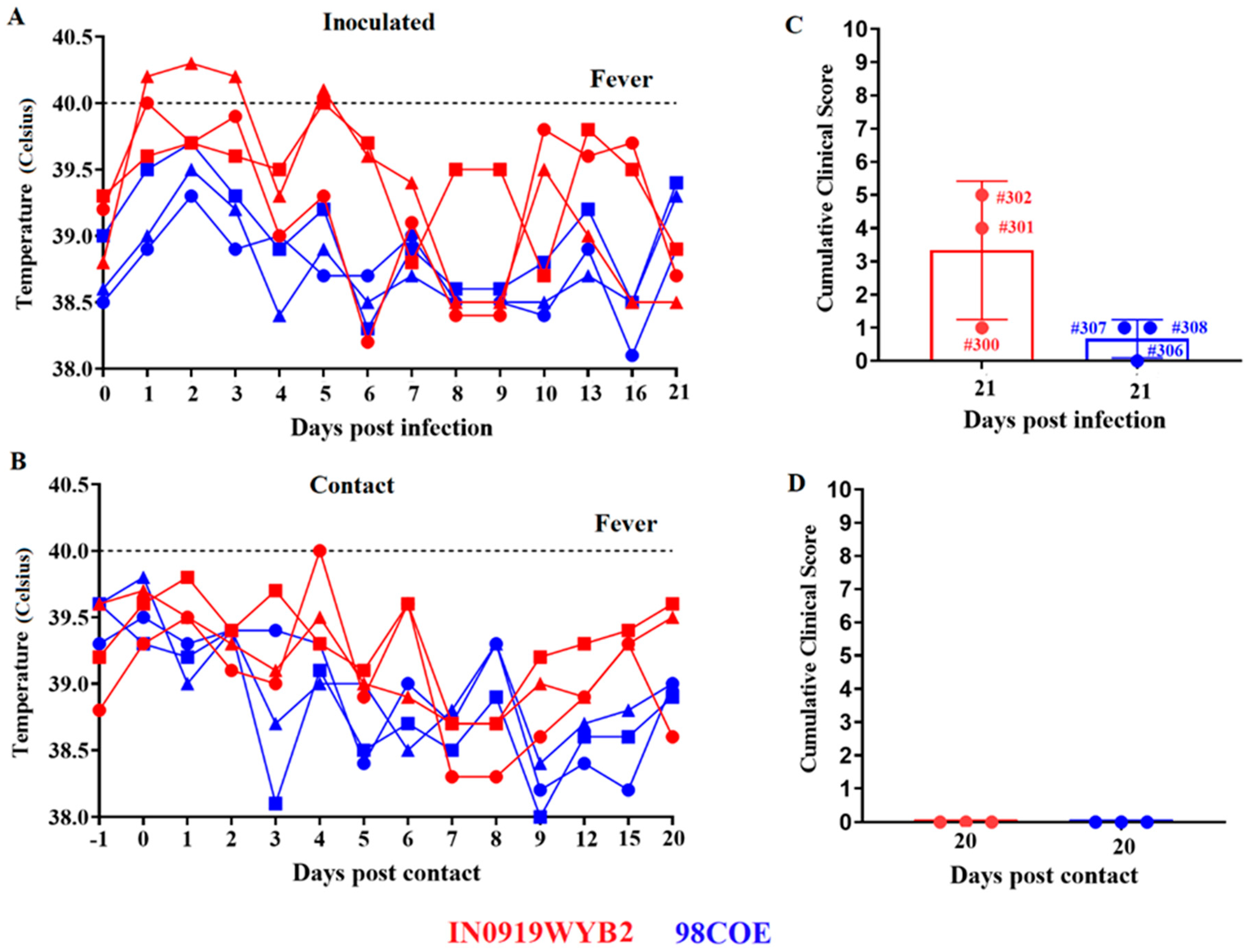
Figure 5.
Shedding dynamics produced by infection with 98COE and IN0919WYB2. RT-qPCR was used to evaluate the shedding of viral RNA during infection in nasal, oral, and rectal swabs collected from inoculated (A, C, and E, respectively) and contact (B, D, and F respectively) animals infected with different viruses. The right Y axis of each graphic represents the predicted TCID50/mL based on the CT value. Predicted levels of infectious virus were considered based on the results of a previous validation (Hole et al., 2021). For these graphs, inoculated pigs (IN0919WYB2 group) are represented by red circles, squares, and triangles for pigs 300, 301, and 302, respectively and to identify contact pigs 303, 304, and 305 (IN0919WYB2 group). Similarly, the same pattern in blue was followed for the 98COE group, identifying pigs 306, 307, and 308 (inoculated) and 309, 310, and 311 (contact), respectively.
Figure 5.
Shedding dynamics produced by infection with 98COE and IN0919WYB2. RT-qPCR was used to evaluate the shedding of viral RNA during infection in nasal, oral, and rectal swabs collected from inoculated (A, C, and E, respectively) and contact (B, D, and F respectively) animals infected with different viruses. The right Y axis of each graphic represents the predicted TCID50/mL based on the CT value. Predicted levels of infectious virus were considered based on the results of a previous validation (Hole et al., 2021). For these graphs, inoculated pigs (IN0919WYB2 group) are represented by red circles, squares, and triangles for pigs 300, 301, and 302, respectively and to identify contact pigs 303, 304, and 305 (IN0919WYB2 group). Similarly, the same pattern in blue was followed for the 98COE group, identifying pigs 306, 307, and 308 (inoculated) and 309, 310, and 311 (contact), respectively.
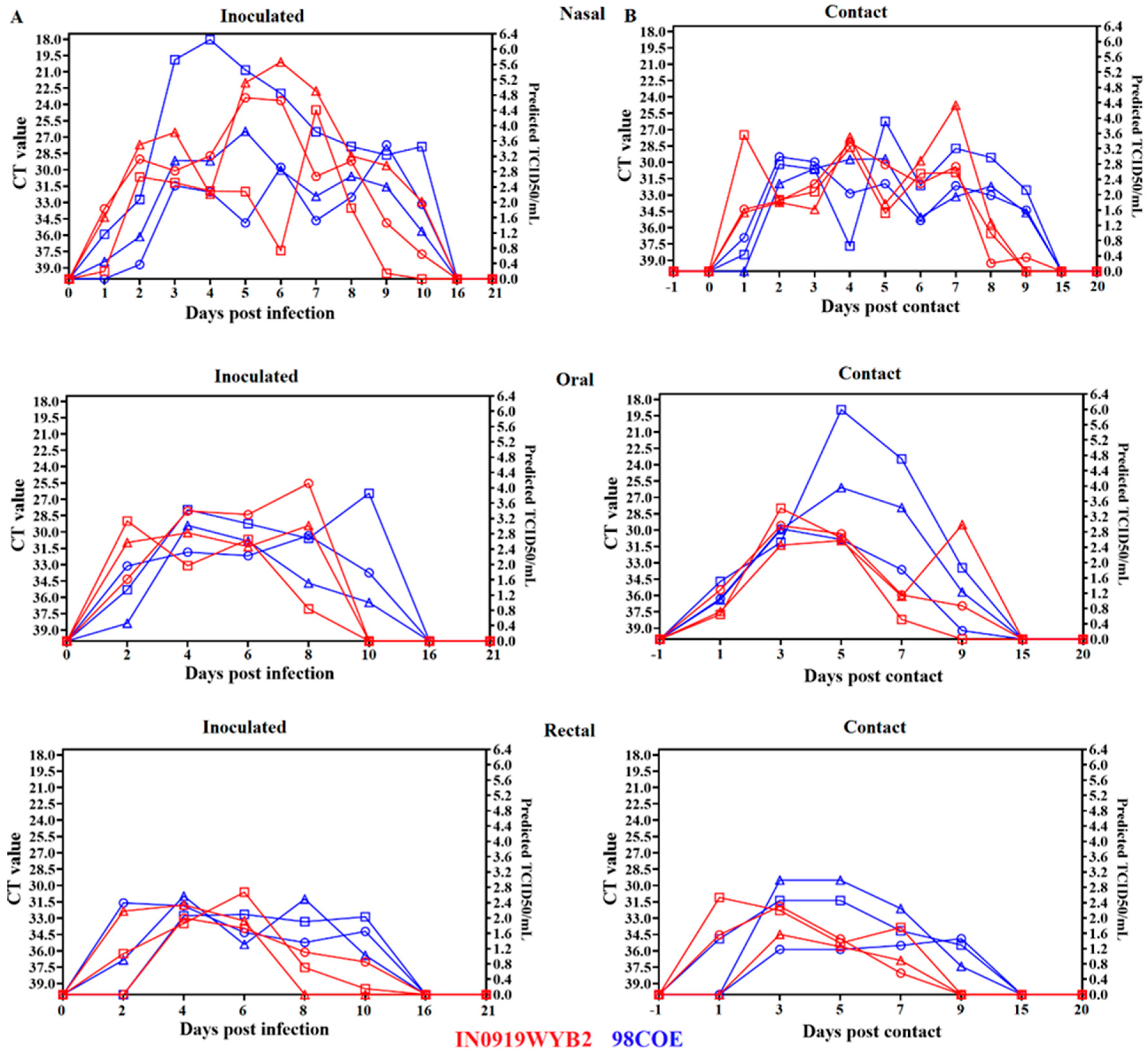
Figure 6.
Analysis of the recovery. Recovery of infectious viral particles from nasal and oral swabs during infection with 98COE and IN0919WYB2. A) Differences in the recovery of infectious virus from nasal swabs between pigs infected with 98COE and IN0919WYB2. B) CT values obtained from the same nasal swabs subjected to viral isolation. C) Differences in the recovery of infectious virus from oral swabs between pigs infected with 98COE and IN0919WYB2. D) CT values obtained from the same oral swabs subjected to viral isolation. Specific days and pigs where viral isolations were obtained were highlighted in yellow in all cases. E) Proportion of positive viral isolates using nasal and oral swabs at specific CT value ranges. The number of isolates obtained for each Ct group is denoted by the numbers (in red) on the top of the bars.
Figure 6.
Analysis of the recovery. Recovery of infectious viral particles from nasal and oral swabs during infection with 98COE and IN0919WYB2. A) Differences in the recovery of infectious virus from nasal swabs between pigs infected with 98COE and IN0919WYB2. B) CT values obtained from the same nasal swabs subjected to viral isolation. C) Differences in the recovery of infectious virus from oral swabs between pigs infected with 98COE and IN0919WYB2. D) CT values obtained from the same oral swabs subjected to viral isolation. Specific days and pigs where viral isolations were obtained were highlighted in yellow in all cases. E) Proportion of positive viral isolates using nasal and oral swabs at specific CT value ranges. The number of isolates obtained for each Ct group is denoted by the numbers (in red) on the top of the bars.
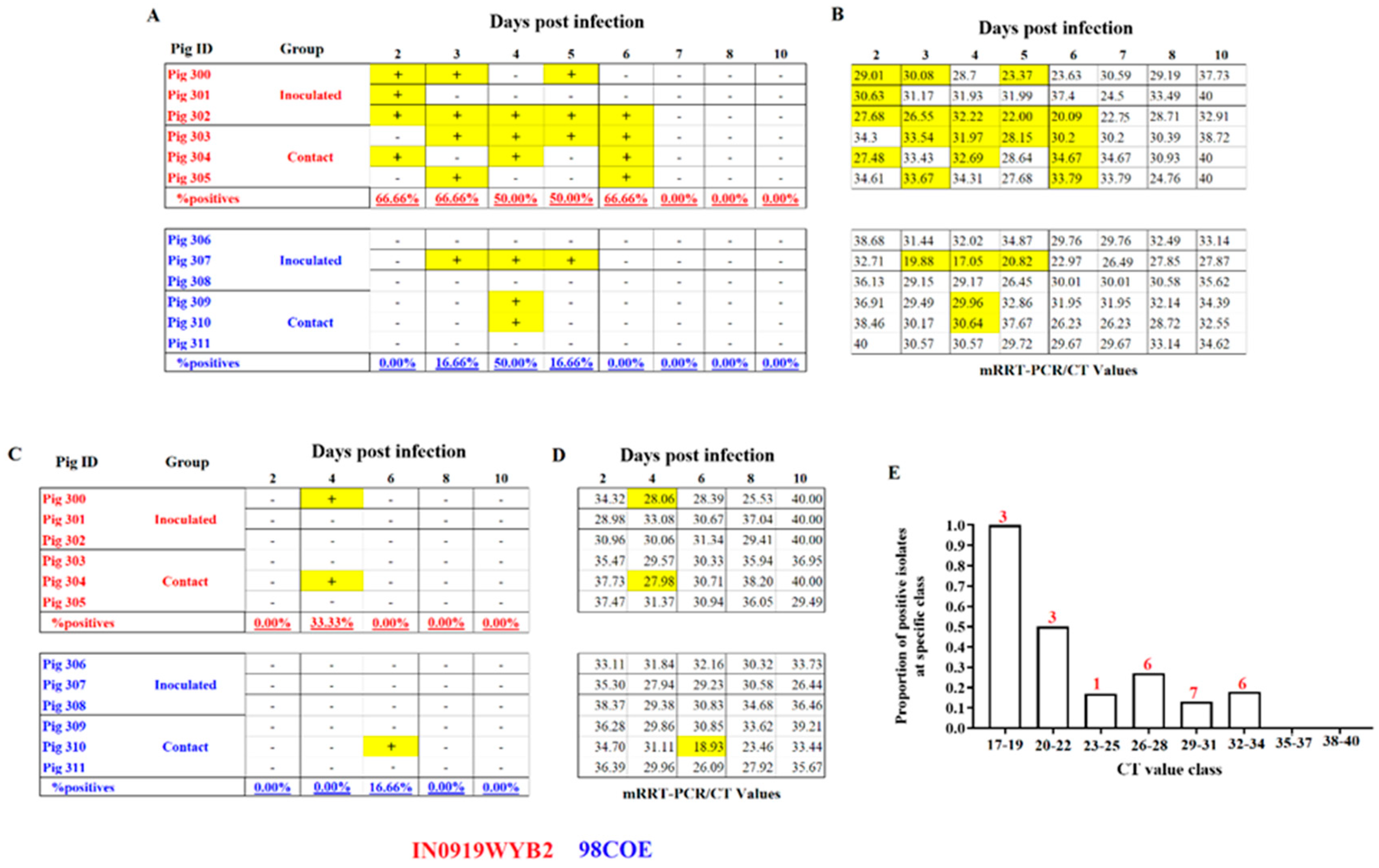
Figure 7.
Postmortem evaluation between 98COE and IN0919WYB2. At 21 dpi, necropsies were conducted in inoculated (A) and contact (B) pigs from each group. Tissues were evaluated by RT-qPCR. The right Y-axis reflects predicted TCID50/mL based on the CT value. Abbreviations indicate Tonsil of the soft palate (PTON), submandibular lymph node (SMLN), popliteal lymph node (R-PopLN), parotid lymph node (ParLN), nasopharyngeal tonsil (NTON) spleen (SPL), prescapular lymph node (PreLN), and gastrohepatic lymph node (GHLN). IN0919WYB2 group pigs identified by red circles, squares, and triangles denote measurements of pigs 300, 301, and 302, respectively. Identical shapes were used in contact pigs to identify pigs 303, 304, and 305. A similar pattern was used to denote the 98COE group, with blue shapes identifying pigs 306, 307, and 308 (inoculated) and 309, 310, and 311 (contact), respectively. .
Figure 7.
Postmortem evaluation between 98COE and IN0919WYB2. At 21 dpi, necropsies were conducted in inoculated (A) and contact (B) pigs from each group. Tissues were evaluated by RT-qPCR. The right Y-axis reflects predicted TCID50/mL based on the CT value. Abbreviations indicate Tonsil of the soft palate (PTON), submandibular lymph node (SMLN), popliteal lymph node (R-PopLN), parotid lymph node (ParLN), nasopharyngeal tonsil (NTON) spleen (SPL), prescapular lymph node (PreLN), and gastrohepatic lymph node (GHLN). IN0919WYB2 group pigs identified by red circles, squares, and triangles denote measurements of pigs 300, 301, and 302, respectively. Identical shapes were used in contact pigs to identify pigs 303, 304, and 305. A similar pattern was used to denote the 98COE group, with blue shapes identifying pigs 306, 307, and 308 (inoculated) and 309, 310, and 311 (contact), respectively. .
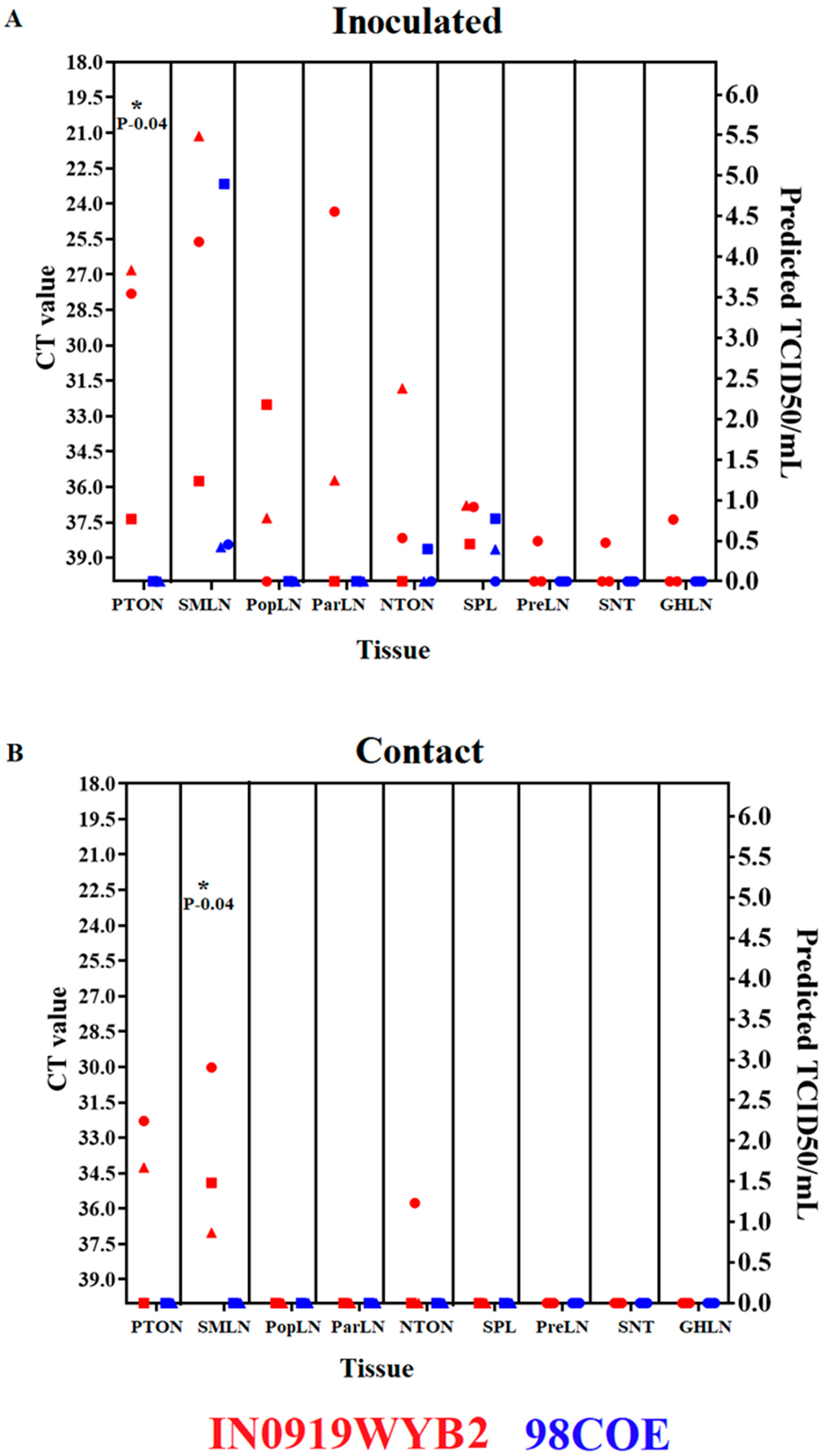
Figure 8.
Differences in the adaptive immune response between 98COE and IN0919WYB2 during the infection in pigs. Graphics show the titer of neutralizing antibodies against VSIV at different times in inoculated (A) and contact (B) pigs infected with 98COE or IN0919WYB2. In inoculated pigs, red circles, squares, and triangles denote measurements from pigs 300, 301, and 302, respectively. The same shapes were used in contact pigs to identify pigs 303, 304, and 305 from IN0919W9YB2 group. Similarly, for blue shapes (98COE group), identifying pigs 306, 307, and 308 (inoculated) and 309, 310, and 311 (contact), respectively.
Figure 8.
Differences in the adaptive immune response between 98COE and IN0919WYB2 during the infection in pigs. Graphics show the titer of neutralizing antibodies against VSIV at different times in inoculated (A) and contact (B) pigs infected with 98COE or IN0919WYB2. In inoculated pigs, red circles, squares, and triangles denote measurements from pigs 300, 301, and 302, respectively. The same shapes were used in contact pigs to identify pigs 303, 304, and 305 from IN0919W9YB2 group. Similarly, for blue shapes (98COE group), identifying pigs 306, 307, and 308 (inoculated) and 309, 310, and 311 (contact), respectively.
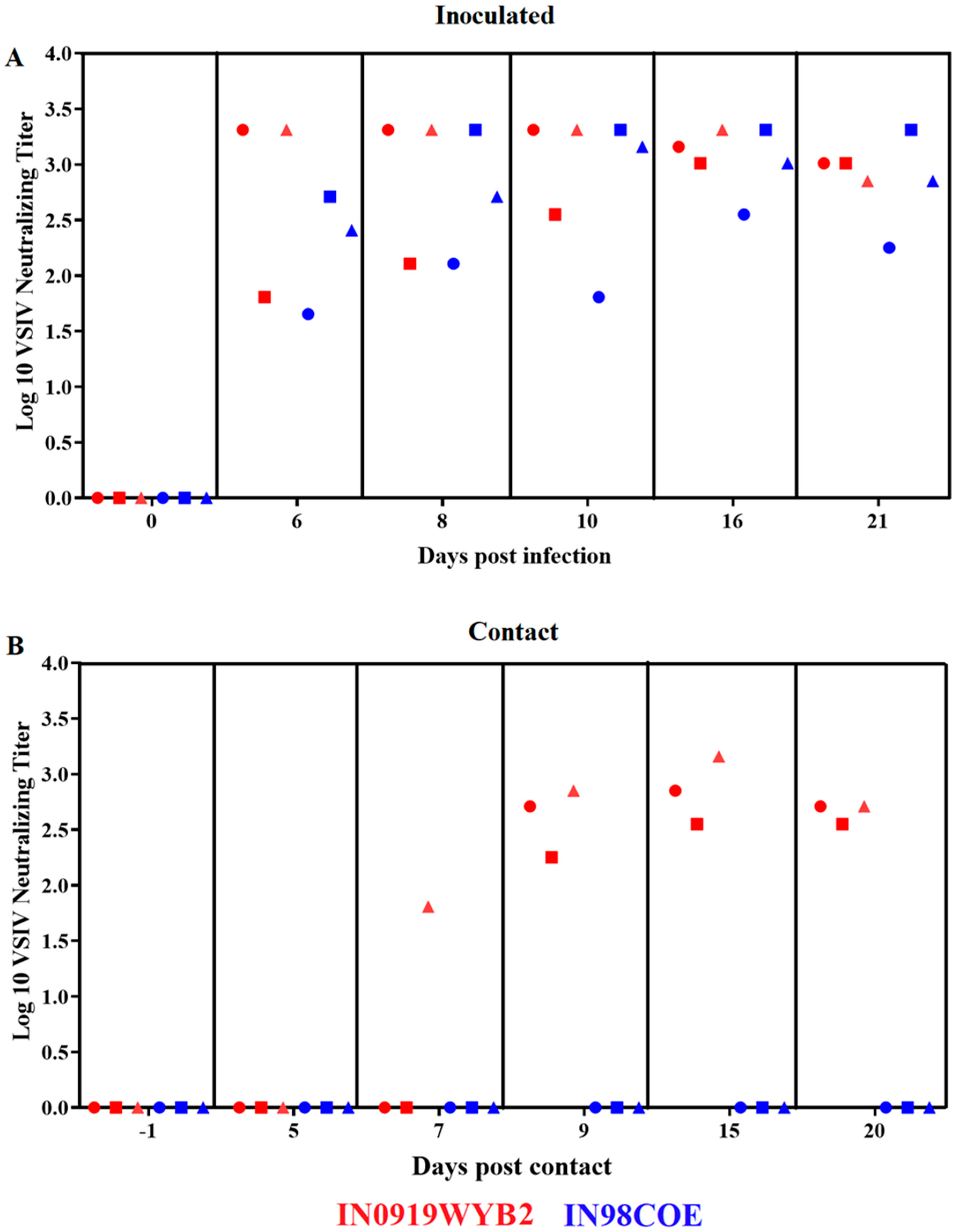
Figure 9.
Genetic characteristics of the viral progeny produced during the infection of IN0919WYB2 and 98COE in pigs. To assess the viral progeny diversity produced during the infection with strains IN0919WYB2 (A) and 98COE (B), viral isolates recovered from pigs infected with each of the strains were subjected to next-generation sequencing (NGS). Mutations at different codons were identified by gene/codon position/ and mutation type (dS= synonymous, or dN= nonsynonymous substitutions). Grey highlights denote substitutions at specific codon positions compared to parental phenotype in the viral stock. Letters in parenthesis reflect the amino acid encoded by different codons.
Figure 9.
Genetic characteristics of the viral progeny produced during the infection of IN0919WYB2 and 98COE in pigs. To assess the viral progeny diversity produced during the infection with strains IN0919WYB2 (A) and 98COE (B), viral isolates recovered from pigs infected with each of the strains were subjected to next-generation sequencing (NGS). Mutations at different codons were identified by gene/codon position/ and mutation type (dS= synonymous, or dN= nonsynonymous substitutions). Grey highlights denote substitutions at specific codon positions compared to parental phenotype in the viral stock. Letters in parenthesis reflect the amino acid encoded by different codons.
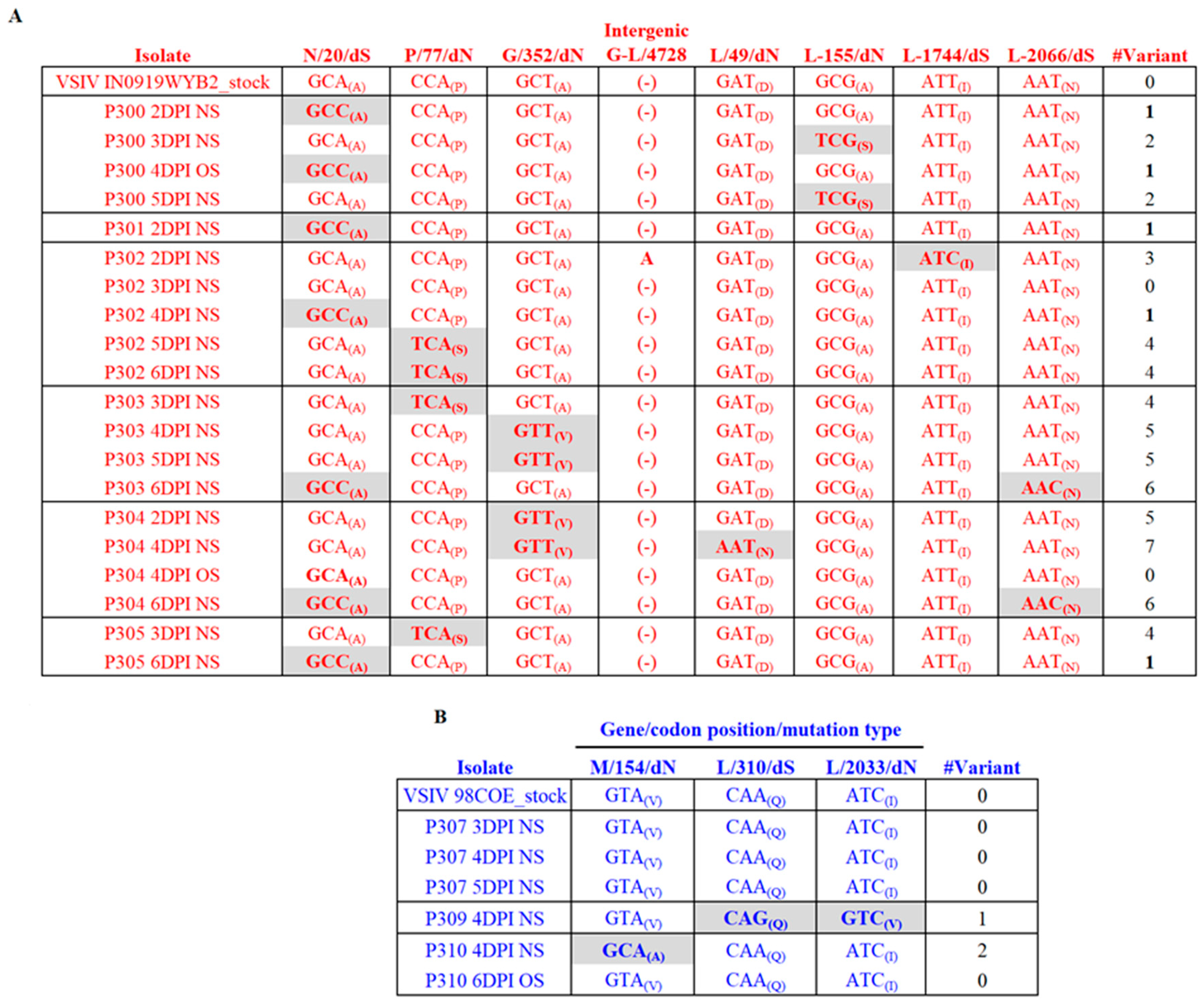
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



