
Submitted:
12 September 2024
Posted:
12 September 2024
You are already at the latest version
Abstract
This study investigates the correlation between gut microbiota diversity and various hematological and meat quality parameters in different cattle breeds, including Angus, Simmental, Braunvieh, Brahman and F1(SMxBR). Using 16S rRNA and 18S rRNA gene sequencing, microbial communities were profiled to discern breed-specific impacts on gut microbiota. Significant correlations were identified between gut microbiota diversity and health parameters, emphasizing the role of microbial diversity in influencing skin condition, fat deposition, and muscle quality. Key bacterial genera such as Blautia, Christensenellaceae_R-7_group, and Coprococcus exhibited significant correlations with meat quality traits. In fungi, genera like Rhodotorula exhibited positive correlations with muscle quality, while protist genera such as Cryptosporidium negatively correlated with meat quality parameters. These findings suggest that host genetics significantly shape the gut microbiota and highlight the potential for targeted microbiota management to optimize gut health, enhance feed efficiency, and improve meat quality in livestock.
Keywords:
gut microbiota
; cattle breeds
; microbial diversity
; meat quality
; hematological parameters
; host genetics
; 16S/18S rRNA
1. Introduction
The normal intestine contains more than 100 trillion microorganisms, mostly bacteria (98%), as well as fungi, viruses, protists, and archaea, establishing a symbiotic relationship with the host that is essential for nutrient absorption, immune system regulation, and the maintenance of intestinal barrier integrity [1,2]. The gut microbiota, in addition to producing beneficial substances such as antimicrobial peptides, vitamins, and enzymes, is influenced by the host species and their health status [3]. Recent studies have indicated that host genetics can influence the composition of the rumen microbiota, which is related to rumen metabolites, feed efficiency, and milk quality [4,5,6,7].
Intestinal microbial communities play a fundamental role in nutrient digestion, energy harvesting, immune system regulation, and disease development [8]. Variation in the composition and function of the gut microbiota has been associated with the health and productive performance of farm animals [9]. Both environmental factors (such as diet, medication, and hygiene conditions) and host genetic factors (including hereditary genetics, sex, and age) can shape these microbial communities [10]. Evidence has highlighted the diversity of the gut microbiome in different animal breeds fed under identical conditions, identifying breed-specific biomarkers in pigs and broiler chickens [11,12,13]. Additionally, research has explored rumen microbial differences driven by various breeds and crossbreeding for cattle combinations, indicating significant impacts on microbiota and metabolites [14,15,16,17]. These findings underscore the complex interplay between host genetics, gut microbiota, and phenotypic traits, which is crucial for the livestock industry.
The quality of beef is crucial for the livestock industry, as it directly affects consumer satisfaction and market value. Genetic and environmental factors play a significant role in determining essential attributes such as marbling, tenderness, and flavor, which can optimize breeding practices and improve production efficiency [18]. Enhancing beef quality also promotes sustainable management by enabling efficient resource use and improving animal welfare [19]. Characteristics such as backfat thickness, intramuscular fat, and tenderness are related to lipid metabolism and fat deposition [20,21]. Sensory quality, which includes attributes like color, flavor, pH, drip loss, marbling, tenderness, and juiciness, is essential for consumer satisfaction [22]. Additionally, beef quality encompasses processing quality, nutritional value, and hygiene standards [23].
Cattle farming in Peru has advanced significantly due to the introduction of various breeds with valuable attributes for production. In 1993, the Peruvian government created a herd that included the Brahman, Braunvieh, Gyr, and Simmental breeds. The purpose was to promote research in reproductive technologies such as artificial insemination and embryo transfer [24]. Currently, the Estación Experimental Agraria Donoso (EEA Donoso) of INIA in Huaral, under the supervision of the Ministry of Agriculture and Irrigation, is dedicated to the production of in vivo embryos of the Brahman, Gir, Fleckvieh, and Braunvieh breeds with the aim of promoting agricultural development [25]. The Angus breed, recognized worldwide for its high-quality marbled meat, is distinguished by its tenderness and flavor, making it highly valued in the meat industry [26]. The Simmental breed, originating from Switzerland, is appreciated for its robustness and high milk production, also contributing to the quality of its meat [27]. Similarly, the Braunvieh breed, also from Switzerland, is known for its dual purpose, providing both milk and high-quality beef, with a balanced distribution of fat and tenderness [28]. Finally, the Brahman breed, originating from India, is highly resistant to heat and parasites, making it ideal for tropical climates [29]; its crossbreeding with Simmental enhances these traits along with meat quality.
The 16S rRNA gene is a key molecular marker used to explore the ruminal microbiota, offering valuable insights into the structure, taxonomy, and diversity of microbial communities [30]. Similarly, the 18S rRNA marker is vital for studying the intestinal microbiota of fungi and protists due to its conserved nature among eukaryotes, enabling accurate identification and classification [31]. When combined with high-throughput sequencing technologies such as Illumina, this marker facilitates detailed profiling of microbial communities, including those with low abundance [32]. This comprehensive methodology is essential for linking microbial diversity to host health, thereby supporting the development of targeted probiotic therapies to enhance animal health and productivity [33].
This study aims to elucidate the alterations in gut microbiota across various cattle breeds within a genetic nucleus and their associations with beef quality. By analyzing breeds such as Angus, Simmental, Braunvieh, Brahman, and a Simmental-Brahman crossbreed, we intend to discern how breed-specific microbial communities’ impact critical beef quality attributes. This research will employ molecular markers, specifically the 16S rRNA and 18S rRNA genes, alongside high-throughput sequencing technologies to comprehensively profile microbial communities. The insights derived from this study are anticipated to inform the development of targeted probiotic interventions, thereby enhancing animal health and productivity.
2. Materials and Methods
2.1. Animal Experiment and Sample Collection
In the Huaral region of Lima, situated at an elevation of 128 meters above sea level (coordinates 11°31’18” S and 77°14’06” W), a total of 23 fecal samples were collected from healthy cattle (13 females and 10 males) of 4 different breeds (7 Angus, 5 Braunvieh, 4 Brahman, and 3 Simmental) and 4 crossbreeds F1(SMxBR) from the EEA Donoso. All the cattle were one year and six months old and had lived under the same conditions.
The diet at the EEA Donoso mainly consists of fresh forages, supplemented with specific additives (Table 1). Fecal samples were collected from the rectum of each animal using disposable gloves. These samples were promptly transported to the laboratory in liquid nitrogen and stored at -80°C until DNA extraction. The veterinary team at the EEA Donoso Genetic Center in Huaral regularly monitored the animals, conducting parasitological examinations that confirmed the absence of cysts, oocysts, or larvae. Routine veterinary evaluations, including physical exams, medical history assessments, and laboratory tests, were performed to ensure high health standards. As a result, there were no sick animals in the genetic nucleus. This study was carried out in accordance with Peruvian National Law No. 30407: “Animal Protection and Welfare.”
2.2. Meat Quality Traits Detection
To acquire the ultrasound images, the cattle were immobilized and secured by the head in a squeeze chute. The imaging sites were identified through physical palpation to ensure precise determination of the scanning locations. The animals were manually restrained, ensuring that no abnormal conditions arose that could cause stress. Ultrasound scanning was performed only when the animals were in a relaxed posture, allowing for accurate measurements. Ultrasound imaging was performed to assess the Loin skin thickness (GPL), loin fat thickness (GGL), loin thickness (GL), hip skin thickness (GPC), hip fat thickness (GGC), marbled beef ultrasonography right buttock (NMG1), marbled beef ultrasonography right buttock (NMG2), and Loin area (AL), were measure in vivo using an ESAOTE (Esaote Pie Medical, Aquila Vet model, with a 6 MHz linear transducer, Maastrich, The Netherlands) ultrasound machine equipped with an APS 3.5 MHz transducer. To enhance image clarity, the measurement site was shaved, cleaned, and lubricated with vegetable oil, and a soft material guide was employed to improve contact between the transducer and the animal’s curved body surfaces. The weight of the animal was measured at the same time of the ultrasonography.
2.3. Analyses of Blood Parameters
Blood samples were collected from all cattle via jugular venipuncture. A total of 23 samples were analyzed for red blood cell count, white blood cell count, and platelets using the Dx® hematology analyzer (IDEXX Laboratories, Westbrook, MA, USA). Additionally, 23 plasma samples were analyzed for triglyceride levels using the Beckman-CX4 automatic biochemical analyzer (Beckman Coulter, Inc., Brea, CA, USA). Detailed information on the variables analyzed is provided in Supplementary Table S1
2.4. DNA Extraction and Sequencing
The genomic DNA from each fecal sample was extracted utilizing the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA, USA). The DNA concentration and purity were assessed with a NanoDrop 2000 spectrophotometer, and the 260/280 absorbance ratio was measured at the same time (Thermo Fisher Scientific, USA). Additionally, the integrity of the DNA was confirmed by 0.8% agarose gel electrophoresis. To construct the Illumina amplicon sequencing library, approximately 10 ng of DNA from each sample were used for PCR amplification with primers 515F/806R for 16S rRNA and 528F/706R for 18S rRNA. The PCR conditions for 16S rRNA amplification protocol consisted of an initial denaturation at 94 °C for 1 minute, followed by 30 cycles of denaturation at 94 °C for 20 seconds, annealing at 54 °C for 30 seconds, and elongation at 72 °C for 30 seconds, with a final extension at 72 °C for 5 minutes. For 18S rRNA, the protocol began with an initial denaturation at 94°C for 2 minutes, followed by an initial set of 5 cycles consisting of denaturation at 94°C for 45 seconds, annealing at 52/54°C for 45 seconds each, and elongation at 72°C for 1 minute. This was followed by 35 additional cycles with a reduced annealing temperature of 50/52°C, and concluded with a final elongation step at 72°C for 10 minutes. Sequencing libraries were prepared using the Illumina TruSeq DNA PCR-Free Library Preparation Kit (Illumina, USA) as per the protocol, which included the addition of index sequences. The quality of the libraries was assessed with a Qubit 2.0 fluorometer (Thermo Scientific). The validated libraries were then sequenced by the sequencing service of Novogene (USA) on the Illumina NovaSeq 6000 platform with 250 bp paired-end reads (Illumina Inc., San Diego, CA, USA) following the manufacturer’s instructions.
2.5. Bioinformatics Analysis
In the QIIME2 analysis, the data underwent trimming and quality filtering. Paired-end reads, demultiplexed by Illumina, were then used to create an Amplicon Sequence Variants (ASVs) table using the qiime2-dada2 plugin. To reduce the number of false positive ASVs, sequences with fewer than 10 reads in total across all samples were removed. Additionally, sequences of plant origin were excluded. Taxonomic classification of the ASVs was conducted using the SILVA v138.1 database, which provided the basis for identifying bacteria through 16S sequences and fungi and protists through 18S sequences. The high-quality filtered sequences were subsequently aligned using the integrated MAFFT aligner. Using the QIIME2 phylogenetic module, both rooted and unrooted phylogenetic trees were constructed for bacteria, fungi, and protists with the FastTree algorithm.
2.6. Statistics Analysis
The dataset was analyzed statistically using R software v.4.1.1 [34] with the Phyloseq [35], Microeco [36] and MicrobiotaProcess [37] packages. Initially, rarefaction curves were generated for each sample to assess sequencing depth. Various alpha diversity indices, including Observed, Chao1, ACE, Pielou, Simpson, and Shannon, were calculated to evaluate the diversity of intestinal bacteria, fungi, and protists. Beta diversity was assessed using the Jaccard and Unweighted Unifrac methods, and the results were visualized through Principal Coordinate Analysis (PCoA). To examine differences in microbial communities among groups, a two-way PERMANOVA test with 9999 permutations was conducted. A linear discriminant analysis (LDA) with LEfSe, an algorithm for identifying biomarkers of significant statistical and biological importance, was conducted. Additionally, Spearman rank correlation analyses were performed to explore relationships between blood and meat quality parameters and alpha diversity indices. Finally, Mantel tests with 999 permutations were used to investigate the association between variables and community composition.
3. Results
In the bacteria, 32,670,654 high-quality filtered reads were obtained. On average, 142,046 high-quality reads were recorded per sample, reaching a maximum of 2,159,093 and a minimum of 775,522 high-quality reads. In fungi, 6,720,444 high-quality filtered reads were obtained. On average, 29,219 high-quality reads were recorded per sample, reaching a maximum of 849,583 and a minimum of 77,722 high-quality reads. In protists, 20,393,204 high-quality filtered reads were obtained. On average, 88,666 high-quality reads were recorded per sample, reaching a maximum of 1,466,373 and a minimum of 151,082 high-quality reads. The rarefaction curves (Fig. S1) indicated that the sampling depth was adequate to capture the biodiversity within the dataset.
3.1. Analysis of Effect of Breed on the Gut Microbiota Diversity and Composition
The ACE, Chao1, Observed, Pielou, Shannon, and Simpson diversity indices were calculated for bacteria, fungi (Figure 1), and protists (Fig. S2) in the communities of different cattle breeds, revealing clear differences. In bacterial communities (Figure 1A), significant differences were identified between Angus and Brahman for ACE (p = 0.042), Shannon (p = 0.042), and Simpson (p = 0.042). Significant differences were detected between Angus and Braunvieh in ACE (p = 0.0025), Chao1 (p = 0.0025), Observed (p = 0.0057), Pielou (p = 0.0025), Shannon (p = 0.0025), and Simpson (p = 0.0025). Angus and F1(SMxBR) exhibited significant differences in Pielou (p = 0.012), Shannon (p = 0.024), and Simpson (p = 0.042). Significant differences were recorded between Angus and Simmental in ACE (p = 0.017), Chao1 (p = 0.017), Observed (p = 0.022), Pielou (p = 0.033), Shannon (p = 0.017), and Simpson (p = 0.033). Differences in ACE (p = 0.032), Chao1 (p = 0.032), Observed (p = 0.032), and Shannon (p = 0.032) were significant between Brahman and Braunvieh.
Regarding fungi (Figure 1B), significant differences were identified between Angus and Brahman in ACE (p = 0.012), Chao1 (p = 0.0061), and Observed (p = 0.0061). Significant differences were detected between Angus and Braunvieh in ACE (p = 0.0025), Chao1 (p = 0.0057), and Observed (p = 0.0056). Significant differences were recorded between Brahman and Braunvieh in ACE (p = 0.029), Chao1 (p = 0.029), and Observed (p = 0.029). Differences in ACE (p = 0.036), Chao1 (p = 0.036), and Observed (p = 0.035) were significant between Braunvieh and Simmental. Finally, significant differences were identified between Braunvieh and F1(SMxBR) in ACE (p = 0.016), Chao1 (p = 0.019), and Observed (p = 0.019).
In protists (Fig. S2), significant differences were identified between Angus and Brahman in Chao1 (p = 0.042), Observed (p = 0.012), Pielou (p = 0.0061), Shannon (p = 0.0061), and Simpson (p = 0.0061). Significant differences were detected between Angus and Braunvieh in Pielou (p = 0.01), Shannon (p = 0.0025), and Simpson (p = 0.01). Significant differences were recorded between Brahman and F1(SMxBR) in Observed (p = 0.029), Pielou (p = 0.029), Shannon (p = 0.029), and Simpson (p = 0.029). Finally, significant differences were identified between Braunvieh and F1(SMxBR) in Pielou (p = 0.016), Shannon (p = 0.016), and Simpson (p = 0.016).
In beta diversity analysis of gut microbiota communities in cattle, using unweighted UniFrac and Jaccard metrics, a PCoA plot demonstrated that the composition of microbial communities was influenced by breed and sex (Figure 2). For both UniFrac and Jaccard unweighted metrics in bacteria have been influenced by race and sex (Figure 2A,B), but only in Jaccard, community composition was significantly affected by the interaction of race and sex (p = 0.045), as verified by PERMANOVA (Table 2). In fungal composition, the Unweighted UniFrac metric (Figure 2C) revealed a significant effect of sex (p = 0.027), while the Jaccard metric (Figure 2D) indicated significant effects of both breed (p = 0.001) and sex (p = 0.036). Lastly, in the protist composition, the Unweighted UniFrac (p = 0.004) and Jaccard (p = 0.003) PCoA (Figure 2E,F) plots indicated that the community was significantly influenced by breed.
Venn diagrams illustrate ASVs of bacteria, fungi and protists, both unique and shared, among cattle of different breeds (Fig. S3). For bacterial ASVs (Fig. S3A), 1483 are common to all breeds, while 1569 are unique to Braunvieh, 1092 to Brahman, 1686 to Angus, 722 to Simmental and 784 to F1(SMxBR). In the fungal ASVs (Fig. S3B), 43 are common to all breeds, with 36 unique to Braunvieh, 30 to Brahman, 97 to Angus, 39 to Simmental, and 49 to F1(SMxBR). For protist ASVs (Fig. S3C), 35 are common to all breeds, while 93 are unique to Braunvieh, 40 to Brahman, 109 to Angus, 42 to Simmental, and 61 to F1(SMxBR).
3.2. Effect of Breed on the Gut Microbiota Taxonomy
At the phylum level, Firmicutes were the dominant bacterial phylum (Figure 4A), comprising 69% in Angus, 67% in Brahman, 67% in Braunvieh, 65% in Simmental, and 65% in F1(SMxBR). Bacteroidota accounted for 26%, 32%, 31%, 33%, and 31% in Angus, Brahman, Braunvieh, Simmental, and F1(SMxBR), respectively. All other bacterial taxa collectively represented 5%. The dominant fungal phylum (Figure 4C) was Ascomycota, with representations of 95% in Angus, 97% in Brahman, 93% in Braunvieh, 95% in Simmental, and 88% in F1(SMxBR). All other fungal phyla collectively represented 5%. Among protists (Fig. S4A), the dominant phyla were Ciliophora and Incertae Sedis. Ciliophora constituted 46% in Angus, 67% in Brahman, 85% in Braunvieh, 4% in Simmental, and 22% in F1(SMxBR). Incertae Sedis were represented by 36% in Angus, 27% in Brahman, 12% in Braunvieh, 84% in Simmental, and 55% in F1(SMxBR). Chlorophyta represented 7%, 3%, 2%, 7%, and 12% in Angus, Brahman, Braunvieh, Simmental, and F1(SMxBR), respectively. Apicomplexa accounted for 9%, 2%, 1%, 3%, and 7% in Angus, Brahman, Braunvieh, Simmental, and F1(SMxBR), respectively. All other protist phyla collectively represented 5%.
Figure 3.
Unweighted UniFrac and Jaccard analysis of beta diversity in the different breeds of cattle. A) Unweighted Unifrac of bacteria B) Jaccard of bacteria C) Unweighted Unifrac of fungi. D) Fungi Jaccard E) Protist Unweighted Unifrac F) Protist Jaccard.
Figure 3.
Unweighted UniFrac and Jaccard analysis of beta diversity in the different breeds of cattle. A) Unweighted Unifrac of bacteria B) Jaccard of bacteria C) Unweighted Unifrac of fungi. D) Fungi Jaccard E) Protist Unweighted Unifrac F) Protist Jaccard.
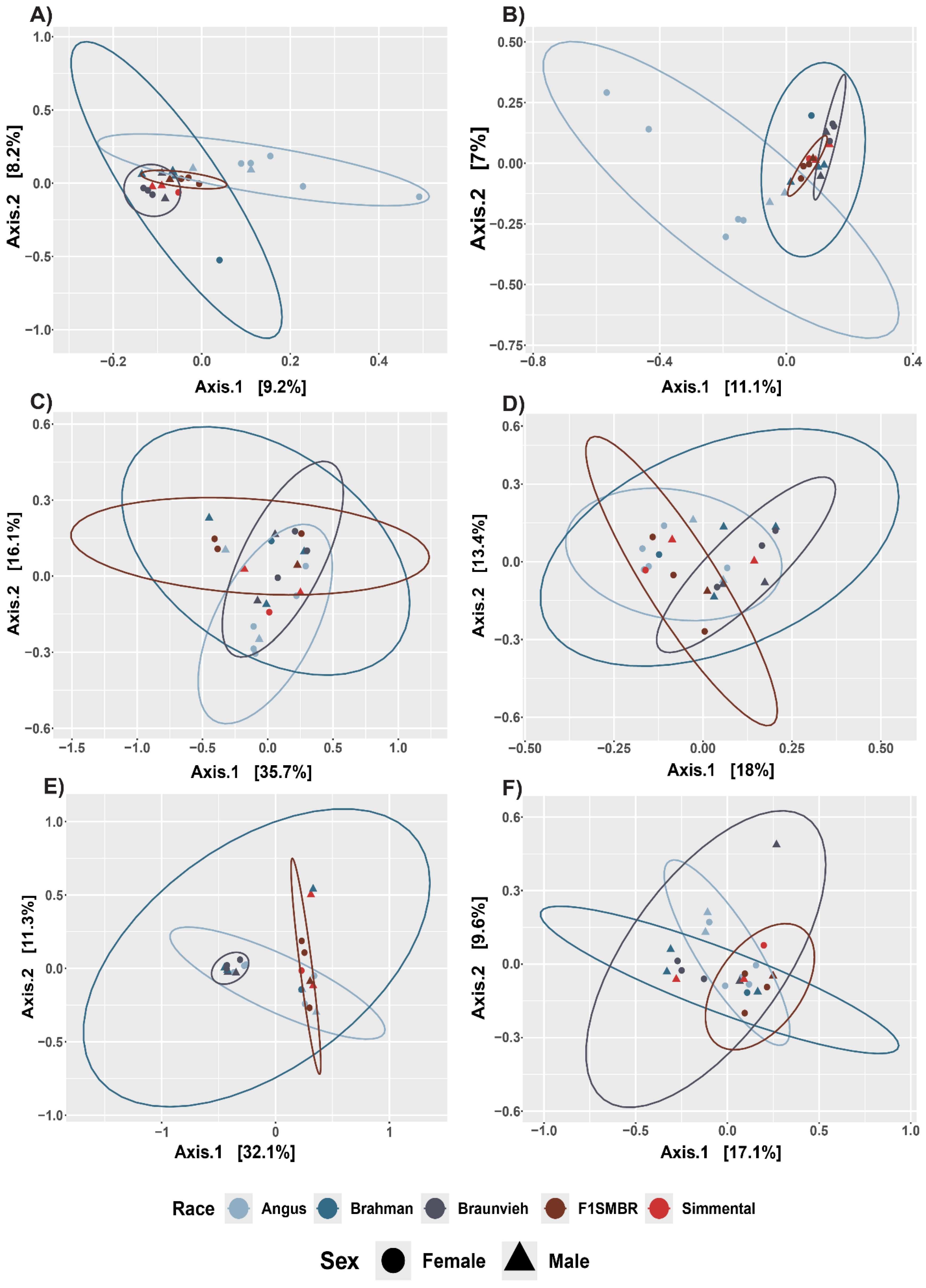
Figure 4.
Relative abundances in the gut microbiota at the phylum and genus level in different cattle breeds. A) Bar graph analysis illustrates the abundance of bacterial phyla in each breed. B) Heat map with the main bacterial abundances of the 20 genera in each breed. C) Bar graph analysis illustrates the abundance of fungal phyla in each breed; D) Heat map with the main abundances of fungi of 20 genera in each breed.
Figure 4.
Relative abundances in the gut microbiota at the phylum and genus level in different cattle breeds. A) Bar graph analysis illustrates the abundance of bacterial phyla in each breed. B) Heat map with the main bacterial abundances of the 20 genera in each breed. C) Bar graph analysis illustrates the abundance of fungal phyla in each breed; D) Heat map with the main abundances of fungi of 20 genera in each breed.
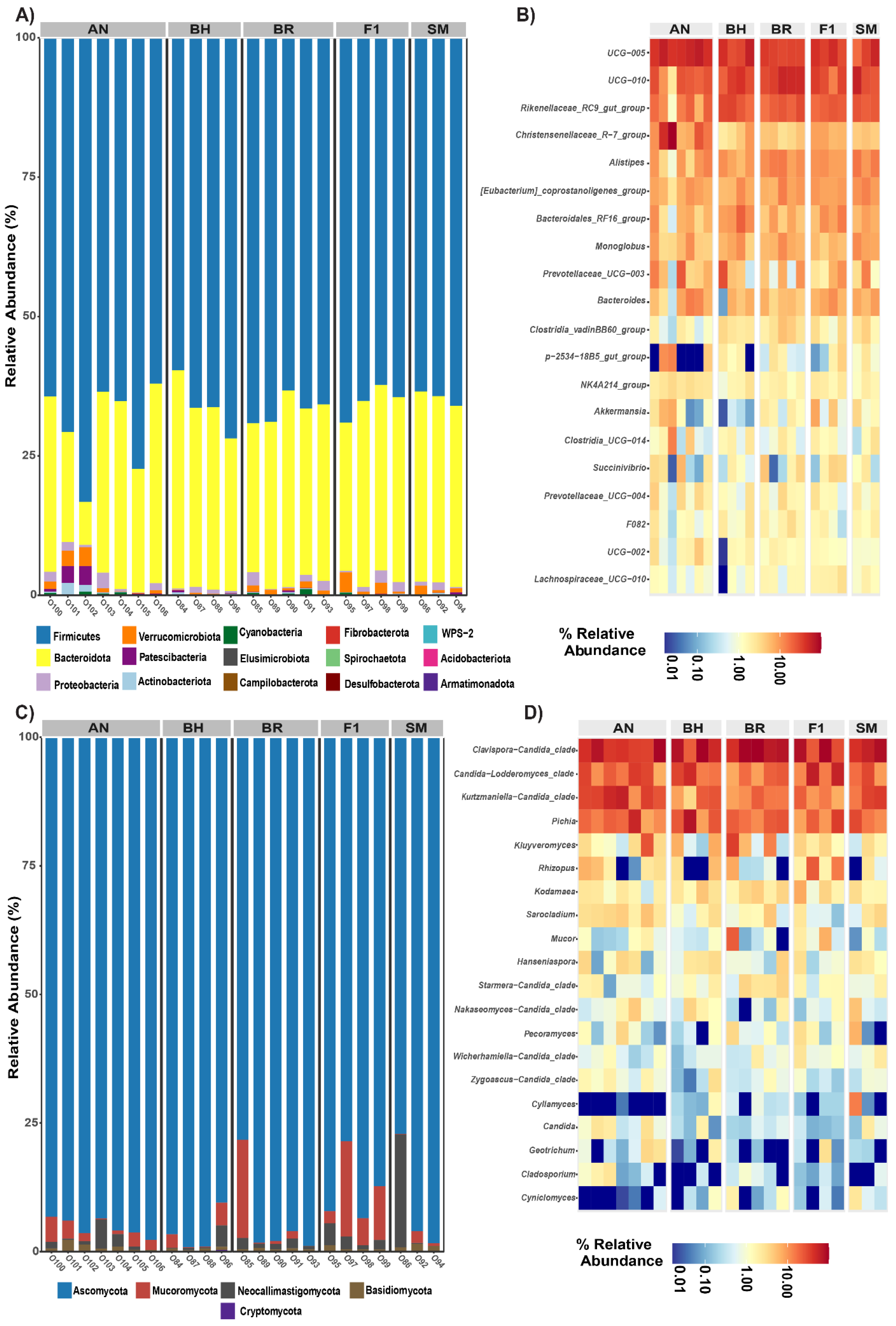
The heatmap illustrates the relative abundance of various genera across different breeds (Figure 4B,D). The 20 most abundant genera were observed. In the analysis of bacterial composition at the genus level (Figure 4B), the most abundant genera observed were UCG-005, UCG-010, Rikenellaceae_RC9_gut_group, Christensenellaceae_R-7_group, and Alistipes. Regarding fungal composition (Figure 4D), the predominant genera were Clavispora-Candida_clade, Candida-Lodderomyces_clade, Kurtzmaniella-Candida_clade, and Pichia. In the protist composition (Fig. S4B), the most abundant genera were Trichostomatia, Blastocystis, Trebouxiophyceae, and Gregarina.
3.3. Relationship between Gut Microbiota and Beef Quality Variables
The Spearman correlation analysis between meat quality variables and the relative abundances of intestinal microbiota genera revealed several significant associations (Figure 5). Among the bacterial genera (Figure 5A), NGM1 and NGM2 exhibited significant positive correlations with NK4A214_group and Christensenellaceae_R-7_group, and significant negative correlations with Odoribacter, Clostridia_vadinBB60_group, Coprococcus, gir-aah93h0, and Agathobacter. Additionally, NGM2 was significantly negatively correlated with [Clostridium]_methylpentosum_group. GPL demonstrated a significant negative correlation with Bifidobacterium and a positive correlation with Hydrogenoanaerobacterium. GGL and GGC were negatively correlated with Negativibacillus and Lachnospiraceae_UCG-010, with GGC also exhibiting a significant negative correlation with Veillonellaceae_UCG-001. GPC was negatively correlated with Blautia, Christensenellaceae_R-7_group, and Veillonellaceae_UCG-001. Body weight exhibited significant negative correlations with Lachnospiraceae_UCG-010 and Veillonellaceae_UCG-001. GL was negatively correlated with Lachnospiraceae_UCG-010. Finally, AL demonstrated significant negative correlations with the Christensenellaceae_R-7_group and Lachnospiraceae_UCG-010.
Regarding fungal genera (Figure 5B), only Rhodotorula exhibited a significant positive correlation with NGM1 and NGM2 and a significant negative correlation with GPL. Among protist genera (Figure 5C), NGM1 and NGM2 had significant negative correlations with Pseudoperkinsidae and Colpodida. GL was significantly negatively correlated with Cryptosporidium, Colpodella, Tetramitia, Trebouxiophyceae, Gregarina, Colpodida, Ochromonas, Blastocystis, and Cercomonas, while demonstrating a significant positive correlation with Trichostomatia. AL exhibited significant negative correlations with Cryptosporidium and Colpodella. Lastly, GPC had significant negative correlations with Cryptosporidium, Colpodella, Tetramitia, Blastocystis, and Cercomonas.
3.4. Biomarkers Identification for Different Breed
To identify specific bacterial taxa associated with different breeds, a comparative analysis of gut microbiota compositions was performed. This analysis was performed using the linear discriminant analysis effect size method (LEfSe). The most significant differentiation in taxa, from phylum to genus level, was determined through an LDA score (Figure 6).
In Angus cattle, the following bacterial taxa were identified (Figure 6A) one phylum (Patescibacteria), two orders (Christensenellales and Saccharimonadales), one class (Saccharimonadia), two families (Christensenellaceae and Saccharimonadaceae), and two genera (Christensenellaceae_R-7_group and Candidatus_Saccharimonas). In Braunvieh, one genus (Dorea) was identified. In F1(SMxBR), one order (Lachnospirales), one family (Lachnospiraceae), and two genera (Lachnospiraceae_UCG-010 and Coprococcus) were identified. In Simmental, one family ([Eubacterium]_coprostanoligenes_group) and one genus ([Eubacterium]_coprostanoligenes_group) were identified. Regarding fungal biomarkers (Figure 6B), in Angus, one genus (Candida) and one species (Candida cylindracea) were identified. In Simmental, one genus (Kurtzmaniella-Candida_clade) was identified. In Braunvieh, one genus (Cyllamyces) was identified.
For protist biomarkers (Figure 6C), in Brahman, one phylum (Ciliophora), one order (Litostomatea), one class (Intramacronucleata), one family (Trichostomatia), one genus (Trichostomatia), and one species (Buxtonella sulcata) were identified. In F1(SMxBR), two phyla (Apicomplexa and Chlorophyta), one order (Trebouxiophyceae), two classes (Conoidasida and Trebouxiophyceae), one family (Trebouxiophyceae), one genus (Trebouxiophyceae), and one species (Prototheca zopfii) were identified. In Simmental, one genus (Blastocystis) and one species (Blastocystis sp.) were identified.
3.5. Breed Relationship of Alpha/Beta Diversity with Variables
The variables, hematological and quality meat, were analyzed using the Kruskal-Wallis test, with breed as the variable of interest (Table S1). Only significant parameters for breeds included GL, GPC, GGC, NGM1, and NGM2 (Table S2). Subsequently, Bonferroni post-hoc analysis was conducted. For GL, the comparison between Braunvieh and F1(SMxBR) yielded a significance of 0.0175. For GPC, the comparison between Brahman and F1(SMxBR) indicated a significance of 0.0457. In the case of GGC, the comparison between Brahman and Braunvieh had a significance of 0.0445, and Brahman versus F1(SMxBR) had a significance of 0.0061. For NGM1, the comparison between Angus and F1(SMxBR) was significant at 0.005. Finally, for NGM2, the comparison between Angus and F1(SMxBR) was significant at 0.0084 (Table S3).
A Spearman correlation analysis was conducted to examine the relationship between variables and alpha diversity indices for bacteria, fungi, and protists (Figure 7). For bacteria (Figure 7A), the Shannon index exhibited a significant positive correlation with GPL. The Simpson index was significantly positively correlated with GPC and significantly negatively correlated with NGM2. The Pielou index demonstrated significant negative correlations with NGM1 and NGM2.
Regarding fungi (Figure 7B), the Observed index had significant positive correlations with MON% and significant negative correlations with EOS, GL, and GPC. The Chao index was significantly positively correlated with MON%, MON, and PLT, and significantly negatively correlated with EOS, GL, AL, and GPC. The ACE index exhibited significant positive correlations with MON% and significant negative correlations with EOS, GL, AL, and GPC. The Shannon and Pielou indices were significantly positively correlated with NGM2. Finally, the Simpson index exhibited significant positive correlations with NGM1 and NGM2.
For protists (Figure 7C), the Observed, Chao1, ACE, Shannon, Simpson, and Pielou indices were significantly positively correlated with PLT. These indices were also significantly negatively correlated with WG, GL, AL, and GPC. Shannon, Simpson, and Pielou indices were significantly positively correlated with MON% and significantly negatively correlated with GGL and GGC. The Observed index was significantly negatively correlated with HGB and GGC. The Chao1 index was significantly negatively correlated with HCT and GPL. Finally, the Simpson index was significantly negatively correlated with EOS.
In the correlation analysis of variables with beta diversity for fungi and protists, several significant results were identified. No significant correlations were observed for bacteria (Table 3). In fungi, the Jaccard index in the Mantel test exhibited significant correlations for NGM1 (p = 0.016) and NGM2 (p = 0.033). The Partial Mantel test also revealed significant correlations for NGM1 (p = 0.017) and NGM2 (p = 0.036). For the Unweighted Unifrac index in the Mantel test, significant correlations were identified for WBC (p = 0.019), Weight (p = 0.015), RBC (p = 0.031), MCV (p = 0.019), MCHC (p = 0.025), NEU (p = 0.038), and SEG (p = 0.039). In the Partial Mantel test, WBC (p = 0.041) and Weight (p = 0.044) were also significantly correlated.
Regarding protists, the Jaccard index in the Mantel test exhibited significant correlations for PLT (p = 0.021) and NEU% (p = 0.031). The Partial Mantel test also identified a significant correlation for PLT (p = 0.024). For the Unweighted Unifrac index in the Mantel test, significant correlations were identified for MON (p = 0.013), PLT (p = 0.026), GL (p = 0.01), and AL (p = 0.033). The Partial Mantel test revealed significant correlations for MON (p = 0.008), PLT (p = 0.026), GL (p = 0.006), and AL (p = 0.044).
4. Discussion
In this study, alpha diversity of the intestinal microbiota exhibited significant correlations with various hematological and meat quality parameters across different cattle breeds. Angus cattle exhibited higher alpha diversity indices compared to Brahman, Braunvieh, Simmental, and F1(SMxBR) crosses, with significant differences observed in the ACE, Chao1, Observed, Pielou, Shannon, and Simpson indices. Additionally, significant differences were identified in the taxonomic composition of bacteria, fungi, and protists among the various breeds. These findings underscore the importance of considering genetic and environmental factors, including breed, in shaping the gut microbiota. Therefore, this study was conducted to further understand these interactions and develop strategies to optimize gut health, enhance feed efficiency, and improve meat quality in livestock production.
Clinical studies were carried out that indicated normal hematological values in cattle, indicating that the animals were healthy [38]. Additionally, meat quality analyses demonstrated that parameters such as marbling and fat content were within acceptable ranges [39].
The race exhibited a significant correlation with alpha diversity of the intestinal microbiota, consistent with previous findings in bacteria [3,40], fungi [2,41], and protists [42,43,44]. These studies underscore the crucial role of genetic and environmental factors in shaping the microbial communities within the gut. The observed differences in alpha diversity among various races suggest that host genetics significantly influence the richness and evenness of gut microbiota. This aligns with evidence from other animal studies, where specific breeds demonstrate distinct microbial profiles due to inherent genetic traits and adaptive responses to their environments [2,3]. In pigs, alpha diversity of gut eukaryotic communities, including fungi and protists, exhibited low but notable heritability, indicating a limited genetic influence on the diversity of these communities [44]. Similarly, in cattle, different breeds exhibit significant variations in rumen bacterial diversity, which can affect nutrient digestion and energy harvest [45,46].
In this study, beta diversity analysis revealed significant variations in the gut microbiota among different breeds. Both breed and the interaction of breed and gender influenced the microbiota composition. These findings are consistent with previous research, highlighting the importance of considering breed and gender in microbiota studies [47,48]. Changes related to breed in the microbiota of cattle are well documented, with specific breeds demonstrating distinct microbial communities [3,46]. For instance, studies have indicated that bacterial diversity in the rumen varies between breeds, with some breeds exhibiting higher diversity associated with lower nutrient utilization efficiency [7]. Similarly, the influence of breed and the interplay between breed and sex has been observed in dogs [49] and pigs [50]. Similarly, the influence of breed on protist and fungal communities was observed, akin to findings in bulls [43], pigs [44], and humans [42]. Additionally, the effect of sex on fungal communities was noted in goats [41] and dogs [51].
These studies underscore the crucial role of breed and sex in shaping gut microbiota, as they highlight significant variations in microbiota composition linked to these biological factors, suggesting their profound impact on the configuration of microbial communities across different species. Understanding these correlations is essential for developing targeted strategies to manipulate gut microbiota for improving health and disease management in different breeds.
All the analyzed breeds exhibited the same predominant bacterial phyla, Firmicutes and Bacteroidota, which is consistent with previous studies in cattle and other ruminants, such as goats [4,52], alpacas [53,54], and cattle [3,55]. Firmicutes, known for their role in fermenting complex carbohydrates and producing short-chain fatty acids, are crucial for the digestive efficiency of ruminants [56]. Bacteroidota, on the other hand, are essential for breaking down plant polysaccharides and producing volatile fatty acids [57]. Similarly, all breeds exhibited Ascomycota as the predominant fungal phylum, followed by Mucoromycota, corroborating previous studies in cattle [47,58], alpacas [53], and goats [41]. Ascomycota includes many fungi that decompose organic matter and contribute to the nutrient cycle in the rumen ecosystem [3]. Mucoromycota also plays a role in organic material decomposition and is important in the structure and function of the ruminal microbiome [3]. Additionally, the analysis of protists revealed that the dominant phyla were Ciliophora and Incertae Sedis, which have also been reported in primates [59] and humans [42,60]. Ciliophora, characterized by the presence of cilia, are important in predating bacteria and maintaining microbial balance [61].
The increase in butyrate-producing bacteria, particularly UCG-010 and UCG-005, across different breeds highlights their role in enhancing butyrate production, crucial for intestinal health. These bacteria improve digestion and intestinal maturation by converting lactate into beneficial compounds, thus providing energy and reducing inflammation [62,63]. The presence of Rikenellaceae RC9 in breeds underscores its importance in breaking down fibrous plant materials, facilitating the degradation of polysaccharides such as starch, cellulose, and lignin in the hindgut [64,65,66]. Additionally, the genus Alistipes was abundantly present across all breeds, playing a significant role in the intestinal microbiome. Alistipes is associated with protective effects against conditions like liver fibrosis and colitis, but also with pathogenic roles in colorectal cancer and mental health disorders [67]. This widespread presence across breeds indicates a fundamental impact on intestinal health, influenced by genetic and environmental factors.
Detection of Clavispora and Blastocystis across all breeds with healthy subjects suggests their potential role as stable components of the gut microbiota, likely influenced by environmental factors [68]. Known for stimulating mucus production through IL-22 and promoting bacterial diversity, Blastocystis may have beneficial impacts on intestinal health and immune function [69]. Pichia emerged as one of the most abundant genera in healthy cattle breeds, suggesting its beneficial role in their gut microbiota. Although Pichia has been associated with higher body mass index (BMI) in humans [68], its presence in healthy cattle and alpacas points to a species-specific role in supporting intestinal health without adverse effects [70]. Additionally, Trichostomatia was identified as a prevalent genus in cattle, pigs [44], and other bovines [43], underscoring its significant role in the intestinal microbiota. This aligns with findings of Trichostomatia as a diverse and notable component of the gut microbiota in patients with healthcare-associated diarrhea [71]. These consistent observations across different species and health conditions suggest that Trichostomatia plays a crucial role in maintaining a varied and resilient intestinal ecosystem.
A significant negative correlation was observed between the genus Blautia and hip skin thickness (GPC). Similarly, in a study on cattle, Blautia wexlerae exhibited a negative correlation with backfat thickness (BFT) [45]. Additionally, decreased abundance of Blautia wexlerae was associated with reduced levels of acetic and butyric acids, essential for lipid metabolism and adiposity traits [72,73]. In the same study, Coprococcus also demonstrated a negative correlation with meat quality parameters, similar to this study where a negative correlation was observed with the gluteus medius muscle area (NGM) in both NGM1 and NGM2. Likewise, Christensenellaceae R-7 presented significant negative correlations [74] with GPC and loin area (AL). These consistent correlations across different studies [74,75] underscore the potential role of these genera in lipid metabolism and adiposity traits in cattle, suggesting they could influence fat deposition and meat quality. This knowledge is crucial for developing targeted strategies to improve meat quality through modulation of gut microbiota composition. Dietary supplementation with Rhodotorula mucilaginosa has been demonstrated to significantly improve all color parameters of chicken meat, highlighting its potential to optimize the visual quality of meat products [76]. In this study, Rhodotorula exhibited a positive correlation with the gluteus medius muscle area (NGM) in both NGM1 and NGM2, and a negative correlation with the thickness of the loin skin, suggesting a role in reducing adiposity and improving muscle composition. The presence of Rhodotorula sp. in the gut could positively influence meat quality by producing beneficial nutrients such as proteins, lipids, folate, and carotenoids [77]. Additionally, its probiotic effect, which regulates the multiplication of pathogenic bacteria and neutralizes their toxins [78], could improve intestinal health and, consequently, meat quality.
Cryptosporidium, a genus of protists, has been linked to significant negative correlations with several meat quality traits in cattle, including AL, GL, and GPC. This relationship may be due to its impact on the intestinal microbiota, which is crucial for nutrient absorption and metabolism. Studies have demonstrated that the composition of the gut microbiota significantly influences meat quality by affecting metabolic processes and nutrient utilization [79,80]. Therefore, the presence of Cryptosporidium could negatively affect these processes, leading to poorer meat quality characteristics, such as reduced muscle mass and increased fat deposition [81,82]. Understanding these interactions is vital to developing strategies to mitigate the negative effects of Cryptosporidium on meat quality through targeted microbiota management and improved animal health practices.
In Angus cattle, Christensenellaceae has been identified as a biomarker associated with lean body mass and metabolic health, suggesting its influence on fat deposition and overall meat quality [83]. In Braunvieh cattle, the genus Dorea was identified, known for its ability to regulate intestinal health and nutrient absorption, playing a crucial role in the gut microbiome [84,85]. In Simmental cattle, Eubacterium coprostanoligenes was identified. Members of this genus are known for their butyrate production, which plays a critical role in energy homeostasis, colonic motility, immunomodulation, and suppression of gut inflammation. Additionally, Eubacterium is involved in bile acid and cholesterol transformation, contributing to their homeostasis [86,87]. Gut dysbiosis and altered representation of Eubacterium in the gut have been associated with various disease states in humans [88].
The Spearman correlation analysis reveals intricate relationships between various alpha diversity indices and hematological and meat quality parameters, highlighting the multifaceted interactions within the gut microbiota and host physiological traits. For bacterial alpha diversity, significant positive correlations were observed between the Shannon index and GPL, suggesting that greater bacterial diversity might be associated with better skin condition [89]. Conversely, negative correlations of the Simpson and Pielou indices with NGM2 marbling indicate a potential link between bacterial evenness and marbling traits. These findings align with previous studies emphasizing the influence of gut microbiota diversity on animal health and meat quality [45,84].
Significant correlations between the observed fungal index and blood parameters such as MON% and EOS suggest an interaction between fungal diversity and immune cell populations. Negative correlations with GL, AL and GPC also indicate that greater fungal diversity could influence fat deposition and distribution. This is consistent with previous studies highlighting the role of fungal communities in nutrient metabolism and immune modulation [90]. Furthermore, hematological health can affect the intestinal mycobiota, crucial for understanding how variations in hematological parameters influence the diversity and functionality of the intestinal mycobiota [91]. For protists, positive correlations of alpha diversity indices with PLT and negative correlations with growth and loin parameters (WG, GL, AL, GPC) underscore the complex role of protists in intestinal health and animal growth [92,93].
The correlation analysis indicated that several meat quality parameters are significantly correlated with the beta diversity of fungi and protists. These correlations align with recent studies demonstrating that the gut microbiota is influenced by host genetic factors [94]. For example, the composition of the rumen microbiota and the function of certain microorganisms are determined by host genetics and can influence feed efficiency and meat quality [74]. Studies have identified genomic regions associated with the abundance of certain rumen bacteria, suggesting that genetic selection could be used to improve ruminal function and feed efficiency in cattle [95].
The approach to control for age-related variations limited the sample size and the proportion of sexes of Simmental, Brahman, and crossbred cattle available for the study. The cattle’s age was uniform to eliminate the influence of this factor in group comparisons.
5. Conclusions
In this study, significant correlations were observed between gut microbiota diversity and various hematological and meat quality parameters across different cattle breeds. These results emphasize the intricate relationship between the gut microbiome and host physiological traits, suggesting that microbial diversity plays a vital role in influencing health and production traits. The findings indicate that specific microbial communities within the gut are linked to better skin condition, fat deposition, and muscle quality, highlighting the potential for targeted microbiota management. Notably, a limitation of the study was the control for age-related variations, which reduced the sample size and the proportion of sexes of Simmental, Brahman, and crossbred cattle available for analysis. The study underscores the importance of considering genetic and environmental factors, including breed, in shaping the gut microbiota. By understanding these interactions, strategies can be developed to optimize gut health, enhance feed efficiency, and improve meat quality in livestock. Future research should further explore these relationships to refine cattle management and breeding practices, ultimately contributing to the advancement of livestock production systems.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Rarefaction curves of species richness show the sequencing depth of data obtained from the gut samples. Figure S2: Alpha diversity metrics of protists were evaluated in cattle in five breeds. Diversity indices exhibited include ACE, Chao1, Observe, Pielou, Shannon and Simpson. Figure S3. The Venn diagram based on ASVs of cattle fecal microbiota from the five breeds. Figure S4. Relative abundances in the gut microbiota at the phylum and genus level in different cattle breeds. Table S1: List of abbreviations. Table S2: Variables of five breeds groups. Table S3: Kruskal Wallis results for clinical parameters in cattle by breed. Table S4: Posthoc Bonferroni results for variables in cattle by breed.
Author Contributions
Conceptualization, C.Q., R.E. and Y.R.; Methodology, C.Q. R.E. and D.R.; Software, R.E., D.R. and Y.R.; Validation, C.Q., R.E. and Y.R.; Formal analysis, C.Q., R.E. and D.R.; Investigation, C.Q., Y.R. and D.R.; Resources, D.R., R.D.H.-Q. and M.A.; Data curation, R.E., Y.R. and D.R.; Writing—original draft preparation, C.Q., Y.R. and D.R.; Writing—review and editing, R.E., C.A. and J.L.M.; Visualization, R.E., W.G. and J.L.M.; Supervision, R.M., C.A., J.L.M. and H.V.V.; Project administration, R.M., C.A., R.E. and H.V.V.; Funding acquisition, R.M., R.D.H.-Q., M.A. and W.G.; H.V.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the following research project: “Mejoramiento de la disponibilidad de material genético de ganado bovino con alto valor a nivel nacional 7 departamentos” of the Ministry of Agrarian Development and Irrigation (MIDAGRI) of the Peruvian Government, with grant number CUI 2432072.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Ethics Committee of the Universidad Nacional Toribio Rodríguez de Mendoza de Amazonas (CIEI-N°0072, 20 June 2024).
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
We acknowledge the “Promeg Nacional” team for supporting the logistic activities. C.I.A. thanks Vicerrectorado de Investigación of UNTRM.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, J.; Wang, H.; Lin, L.; Miao, C.; Zhang, Y.; Zhou, B. Intestinal Barrier Damage Involved in Intestinal Microflora Changes in Fluoride-Induced Mice. Chemosphere 2019, 234, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Liu, B.; Li, F.; He, Y.; Wang, L.; Fakhar-e-Alam Kulyar, M.; Li, H.; Fu, Y.; Zhu, H.; Wang, Y.; et al. Integrated Bacterial and Fungal Diversity Analysis Reveals the Gut Microbial Alterations in Diarrheic Giraffes. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, D.; Zhang, Y.; Li, K.; Wang, M.; Ma, J. Dynamic Distribution of Gut Microbiota in Cattle at Different Breeds and Health States. Front. Microbiol. 2023, 14, 1113730. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Chen, Y.; Liu, J.; Zhang, C.; Irving, B.; Fitzsimmons, C.; Plastow, G.; Guan, L.L. Host Genetics Influence the Rumen Microbiota and Heritable Rumen Microbial Features Associate with Feed Efficiency in Cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef]
- Abbas, W.; Howard, J.T.; Paz, H.A.; Hales, K.E.; Wells, J.E.; Kuehn, L.A.; Erickson, G.E.; Spangler, M.L.; Fernando, S.C. Influence of Host Genetics in Shaping the Rumen Bacterial Community in Beef Cattle. Sci. Rep. 2020, 10, 15101. [Google Scholar] [CrossRef]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A Heritable Subset of the Core Rumen Microbiome Dictates Dairy Cow Productivity and Emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef]
- Sasson, G.; Kruger Ben-Shabat, S.; Seroussi, E.; Doron-Faigenboim, A.; Shterzer, N.; Yaacoby, S.; Berg Miller, M.E.; White, B.A.; Halperin, E.; Mizrahi, I. Heritable Bovine Rumen Bacteria Are Phylogenetically Related and Correlated with the Cow’s Capacity To Harvest Energy from Its Feed. mBio 2017, 8, 10.1128/mbio.00703-17. [Google Scholar] [CrossRef]
- Zmora, N.; Suez, J.; Elinav, E. You Are What You Eat: Diet, Health and the Gut Microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Canibe, N.; O’Dea, M.; Abraham, S. Potential Relevance of Pig Gut Content Transplantation for Production and Research. J. Anim. Sci. Biotechnol. 2019, 10, 55. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.R.; Waters, J.L.; Clark, A.G.; Ley, R.E. Cross-Species Comparisons of Host Genetic Associations with the Microbiome. Science 2016, 352, 532–535. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, K.; Xiang, Y.; Zhou, W.; Gui, G.; Yang, H. The Fecal Microbiota Composition of Boar Duroc, Yorkshire, Landrace and Hampshire Pigs. Asian-Australas. J. Anim. Sci. 2017, 30, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Pandit, R.J.; Hinsu, A.T.; Patel, N.V.; Koringa, P.G.; Jakhesara, S.J.; Thakkar, J.R.; Shah, T.M.; Limon, G.; Psifidi, A.; Guitian, J.; et al. Microbial Diversity and Community Composition of Caecal Microbiota in Commercial and Indigenous Indian Chickens Determined Using 16s rDNA Amplicon Sequencing. Microbiome 2018, 6, 115. [Google Scholar] [CrossRef]
- Cheng, P.; Wang, Y.; Liang, J.; Wu, Y.; Wright, A.; Liao, X. Exploratory Analysis of the Microbiological Potential for Efficient Utilization of Fiber Between Lantang and Duroc Pigs. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wright, A.-D.G.; Si, H.; Wang, X.; Qian, W.; Zhang, Z.; Li, G. Changes in the Rumen Microbiome and Metabolites Reveal the Effect of Host Genetics on Hybrid Crosses. Environ. Microbiol. Rep. 2016, 8, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, M.L.; Cersosimo, L.M.; Wright, A.-D.G.; Kraft, J. Rumen Bacterial Communities Shift across a Lactation in Holstein, Jersey and Holstein × Jersey Dairy Cows and Correlate to Rumen Function, Bacterial Fatty Acid Composition and Production Parameters. FEMS Microbiol. Ecol. 2016, 92, fiw059. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Sanabria, E.; Goonewardene, L.A.; Wang, Z.; Zhou, M.; Moore, S.S.; Guan, L.L. Influence of Sire Breed on the Interplay among Rumen Microbial Populations Inhabiting the Rumen Liquid of the Progeny in Beef Cattle. PLOS ONE 2013, 8, e58461. [Google Scholar] [CrossRef]
- Roehe, R.; Dewhurst, R.J.; Duthie, C.-A.; Rooke, J.A.; McKain, N.; Ross, D.W.; Hyslop, J.J.; Waterhouse, A.; Freeman, T.C.; Watson, M.; et al. Bovine Host Genetic Variation Influences Rumen Microbial Methane Production with Best Selection Criterion for Low Methane Emitting and Efficiently Feed Converting Hosts Based on Metagenomic Gene Abundance. PLOS Genet. 2016, 12, e1005846. [Google Scholar] [CrossRef]
- Gagaoua, M.; Picard, B. Current Advances in Meat Nutritional, Sensory and Physical Quality Improvement. Foods 2020, 9, 321. [Google Scholar] [CrossRef]
- Sakowski, T.; Grodkowski, G.; Gołebiewski, M.; Slósarz, J.; Kostusiak, P.; Solarczyk, P.; Puppel, K. Genetic and Environmental Determinants of Beef Quality—A Review. Front. Vet. Sci. 2022, 9. [Google Scholar] [CrossRef]
- Petracci, M.; Soglia, F.; Berri, C. Chapter 3 - Muscle Metabolism and Meat Quality Abnormalities. In Poultry Quality Evaluation; Petracci, M., Berri, C., Eds.; Woodhead Publishing Series in Food Science, Technology and Nutrition; Woodhead Publishing, 2017; pp. 51–75 ISBN 978-0-08-100763-1.
- Gerber, P.J.; Mottet, A.; Opio, C.I.; Falcucci, A.; Teillard, F. Environmental Impacts of Beef Production: Review of Challenges and Perspectives for Durability. Meat Sci. 2015, 109, 2–12. [Google Scholar] [CrossRef]
- Thorslund, C.A.H.; Sandøe, P.; Aaslyng, M.D.; Lassen, J. A Good Taste in the Meat, a Good Taste in the Mouth – Animal Welfare as an Aspect of Pork Quality in Three European Countries. Livest. Sci. 2016, 193, 58–65. [Google Scholar] [CrossRef]
- Joo, S.T.; Kim, G.D.; Hwang, Y.H.; Ryu, Y.C. Control of Fresh Meat Quality through Manipulation of Muscle Fiber Characteristics. Meat Sci. 2013, 95, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Corredor, F.-A.; Figueroa, D.; Estrada, R.; Salazar, W.; Quilcate, C.; Vásquez, H.V.; Gonzales, J.; Maicelo, J.L.; Medina, P.; Arbizu, C.I. Genetic Diversity and Population Structure of a Peruvian Cattle Herd Using SNP Data. Front. Genet. 2023, 14. [Google Scholar] [CrossRef]
- Marín, G.Y.G.; Torre-Trinidad, L.L.; Farje-Muguruza, P.; Vargas, J.F.G. Desarrollo y calidad embrionaria de un protocolo de superovulación en vacas Brahman en el distrito de Codo del Pozuzo, Huánuco, Perú. Braz. J. Anim. Environ. Res. 2024, 7, e70052–e70052. [Google Scholar] [CrossRef]
- Chen, D.; Wang, X.; Guo, Q.; Deng, H.; Luo, J.; Yi, K.; Sun, A.; Chen, K.; Shen, Q. Muscle Fatty Acids, Meat Flavor Compounds and Sensory Characteristics of Xiangxi Yellow Cattle in Comparison to Aberdeen Angus. Animals 2022, 12, 1161. [Google Scholar] [CrossRef] [PubMed]
- Conanec, A.; Campo, M.; Richardson, I.; Ertbjerg, P.; Failla, S.; Panea, B.; Chavent, M.; Saracco, J.; Williams, J.L.; Ellies-Oury, M.-P.; et al. Has Breed Any Effect on Beef Sensory Quality? Livest. Sci. 2021, 250, 104548. [Google Scholar] [CrossRef]
- Cziszter, L.-T.; Ilie, D.-E.; Neamt, R.-I.; Neciu, F.-C.; Saplacan, S.-I.; Gavojdian, D. Comparative Study on Production, Reproduction and Functional Traits between Fleckvieh and Braunvieh Cattle. Asian-Australas. J. Anim. Sci. 2017, 30, 666–671. [Google Scholar] [CrossRef]
- Burrow, H.M. Genetic Aspects of Cattle Adaptation in the Tropics. Genet. Cattle 2015, 571–597. [Google Scholar] [CrossRef]
- Anderson, C.J.; Koester, L.R.; Schmitz-Esser, S. Rumen Epithelial Communities Share a Core Bacterial Microbiota: A Meta-Analysis of 16S rRNA Gene Illumina MiSeq Sequencing Datasets. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Vaulot, D.; Geisen, S.; Mahé, F.; Bass, D. Pr2-Primers: An 18S rRNA Primer Database for Protists. Mol. Ecol. Resour. 2022, 22, 168–179. [Google Scholar] [CrossRef]
- Mishra, P.; Tulsani, N.J.; Jakhesara, S.J.; Dafale, N.A.; Patil, N.V.; Purohit, H.J.; Koringa, P.G.; Joshi, C.G. Exploring the Eukaryotic Diversity in Rumen of Indian Camel (Camelus Dromedarius) Using 18S rRNA Amplicon Sequencing. Arch. Microbiol. 2020, 202, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Bukin, Y.S.; Mikhailov, I.S.; Petrova, D.P.; Galachyants, Y.P.; Zakharova, Y.R.; Likhoshway, Y.V. The Effect of Metabarcoding 18S rRNA Region Choice on Diversity of Microeukaryotes Including Phytoplankton. World J. Microbiol. Biotechnol. 2023, 39, 229. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PloS One 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. Microeco: An R Package for Data Mining in Microbial Community Ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Xu, S.; Zhan, L.; Tang, W.; Wang, Q.; Dai, Z.; Zhou, L.; Feng, T.; Chen, M.; Wu, T.; Hu, E.; et al. MicrobiotaProcess: A Comprehensive R Package for Deep Mining Microbiome. The Innovation 2023, 4, 100388. [Google Scholar] [CrossRef]
- Motta, G.A.; Neto, P.S.M.; Nociti, R.P.; Santana, Á.E. Hematological Normality, Serum Biochemistry, and Acute Phase Proteins in Healthy Beef Calves in the Brazilian Savannah. Animals 2023, 13, 2398. [Google Scholar] [CrossRef]
- Geletu, U.S.; Usmael, M.A.; Mummed, Y.Y.; Ibrahim, A.M. Quality of Cattle Meat and Its Compositional Constituents. Vet. Med. Int. 2021, 2021, 7340495. [Google Scholar] [CrossRef]
- Fan, P.; Nelson, C.D.; Driver, J.D.; Elzo, M.A.; Jeong, K.C. Animal Breed Composition Is Associated With the Hindgut Microbiota Structure and β-Lactam Resistance in the Multibreed Angus-Brahman Herd. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Lv, Q.-B.; Meng, J.-X.; Ma, H.; Liu, R.; Qin, Y.; Qin, Y.-F.; Geng, H.-L.; Ni, H.-B.; Zhang, X.-X. Description of Gut Mycobiota Composition and Diversity of Caprinae Animals. Microbiol. Spectr. 2023, 11, e0242422. [Google Scholar] [CrossRef]
- Parfrey, L.W.; Walters, W.A.; Lauber, C.L.; Clemente, J.C.; Berg-Lyons, D.; Teiling, C.; Kodira, C.; Mohiuddin, M.; Brunelle, J.; Driscoll, M.; et al. Communities of Microbial Eukaryotes in the Mammalian Gut within the Context of Environmental Eukaryotic Diversity. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Chen, Y.; Wu, H.; Meng, Q.; Guan, L.L. Metatranscriptomic Profiling Reveals the Effect of Breed on Active Rumen Eukaryotic Composition in Beef Cattle With Varied Feed Efficiency. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Prenafeta-Boldú, F.; Zingaretti, L.M.; Gonzalez-Rodriguez, O.; Dalmau, A.; Quintanilla, R.; Ballester, M. Gut Eukaryotic Communities in Pigs: Diversity, Composition and Host Genetics Contribution. Anim. Microbiome 2020, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Sun, Y.; Chen, L.; Zhang, Y.; Wang, J.; Li, H.; Yan, X.; Xia, L.; Yao, G. Differences in Meat Quality between Angus Cattle and Xinjiang Brown Cattle in Association with Gut Microbiota and Its Lipid Metabolism. Front. Microbiol. 2022, 13. [Google Scholar] [CrossRef]
- Daghio, M.; Ciucci, F.; Buccioni, A.; Cappucci, A.; Casarosa, L.; Serra, A.; Conte, G.; Viti, C.; McAmmond, B.M.; Van Hamme, J.D.; et al. Correlation of Breed, Growth Performance, and Rumen Microbiota in Two Rustic Cattle Breeds Reared Under Different Conditions. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Nan, S.; Li, J.; Kuang, Y.; Feng, J.; Wang, H.; Niu, J.; Wu, Y.; Zhang, W.; Nie, C. Differential Microbial Composition and Interkingdom Interactions in the Intestinal Microbiota of Holstein and German Simmental × Holstein Cross F1 Calves: A Comprehensive Analysis of Bacterial and Fungal Diversity. Microorganisms 2024, 12, 486. [Google Scholar] [CrossRef]
- Zhu, D.; Lu, L.; Zhang, Z.; Qi, D.; Zhang, M.; O’Connor, P.; Wei, F.; Zhu, Y.-G. Insights into the Roles of Fungi and Protist in the Giant Panda Gut Microbiome and Antibiotic Resistome. Environ. Int. 2021, 155, 106703. [Google Scholar] [CrossRef]
- You, I.; Kim, M.J. Comparison of Gut Microbiota of 96 Healthy Dogs by Individual Traits: Breed, Age, and Body Condition Score. Animals 2021, 11, 2432. [Google Scholar] [CrossRef]
- Wang, C.; Wei, S.; Chen, N.; Xiang, Y.; Wang, Y.; Jin, M. Characteristics of Gut Microbiota in Pigs with Different Breeds, Growth Periods and Genders. Microb. Biotechnol. 2022, 15, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Meineri, G.; Martello, E.; Atuahene, D.; Miretti, S.; Stefanon, B.; Sandri, M.; Biasato, I.; Corvaglia, M.R.; Ferrocino, I.; Cocolin, L.S. Effects of Saccharomyces Boulardii Supplementation on Nutritional Status, Fecal Parameters, Microbiota, and Mycobiota in Breeding Adult Dogs. Vet. Sci. 2022, 9, 389. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Zhu, L.; Xu, Y.; Liu, N.; Sun, X.; Hu, L.; Huang, H.; Wei, K.; Zhu, R. Dynamic Distribution of Gut Microbiota in Goats at Different Ages and Health States. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Zapata, C.; Estrada, R.; Oros, O.; Sánchez, D.; Maicelo, J.L.; Arbizu, C.I.; Coila, P. Alterations in the Gut Microbial Composition and Diversity Associated with Diarrhea in Neonatal Peruvian Alpacas. Small Rumin. Res. 2024, 235, 107273. [Google Scholar] [CrossRef]
- Carroll, C.; Olsen, K.D.; Ricks, N.J.; Dill-McFarland, K.A.; Suen, G.; Robinson, T.F.; Chaston, J.M. Bacterial Communities in the Alpaca Gastrointestinal Tract Vary With Diet and Body Site. Front. Microbiol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Estrada, R.; Romero, Y.; Figueroa, D.; Coila, P.; Hañari-Quispe, R.D.; Aliaga, M.; Galindo, W.; Alvarado, W.; Casanova, D.; Quilcate, C. Effects of Age in Fecal Microbiota and Correlations with Blood Parameters in Genetic Nucleus of Cattle. Microorganisms 2024, 12, 1331. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, P.; Liu, X.; Zhang, C.; Lu, Q.; Xi, D.; Yang, R.; Wang, S.; Bai, W.; Yang, Z.; et al. The Composition of Fungal Communities in the Rumen of Gayals (Bos Frontalis), Yaks (Bos Grunniens), and Yunnan and Tibetan Yellow Cattle (Bos Taurs). Pol. J. Microbiol. 2019, 68, 505–514. [Google Scholar] [CrossRef]
- Zhao, G.; Niu, Y.; Wang, H.; Qin, S.; Zhang, R.; Wu, Y.; Xiao, X.; Xu, Y.; Yang, C. Effects of Three Different Plant-Derived Polysaccharides on Growth Performance, Immunity, Antioxidant Function, and Cecal Microbiota of Broilers. J. Sci. Food Agric. 2024, 104, 1020–1029. [Google Scholar] [CrossRef]
- Zhu, Y.; Cidan, Y.; Sun, G.; Li, X.; Shahid, M.A.; Luosang, Z.; Suolang, Z.; Suo, L.; Basang, W. Comparative Analysis of Gut Fungal Composition and Structure of the Yaks under Different Feeding Models. Front. Vet. Sci. 2023, 10. [Google Scholar] [CrossRef]
- Mann, A.E.; Mazel, F.; Lemay, M.A.; Morien, E.; Billy, V.; Kowalewski, M.; Di Fiore, A.; Link, A.; Goldberg, T.L.; Tecot, S.; et al. Biodiversity of Protists and Nematodes in the Wild Nonhuman Primate Gut. ISME J. 2020, 14, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Liu, Y.; Xu, W.; Li, G.; Xue, B.; Feng, Y.; Tang, S.; Wei, W.; Yuan, H. Eukaryotes May Play an Important Ecological Role in the Gut Microbiome of Graves’ Disease. Front. Immunol. 2024, 15. [Google Scholar] [CrossRef]
- Dubik, M.; Pilecki, B.; Moeller, J.B. Commensal Intestinal Protozoa—Underestimated Members of the Gut Microbial Community. Biology 2022, 11, 1742. [Google Scholar] [CrossRef]
- Yang, C.; Tsedan, G.; Liu, Y.; Hou, F. Shrub Coverage Alters the Rumen Bacterial Community of Yaks (Bos Grunniens) Grazing in Alpine Meadows. J. Anim. Sci. Technol. 2020, 62, 504–520. [Google Scholar] [CrossRef]
- Yao, L.; Wang, B.; Wang, Y.; Bai, J.; Gao, Y.; Ru, X.; Bi, C.; Li, J.; Shan, A. Effects of Sex on Fat Deposition through Gut Microbiota and Short-Chain Fatty Acids in Weaned Pigs. Anim. Nutr. 2024, 17, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Morrison, M.; Yu, Z. Evaluation of Different Partial 16S rRNA Gene Sequence Regions for Phylogenetic Analysis of Microbiomes. J. Microbiol. Methods 2011, 84, 81–87. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial Degradation of Complex Carbohydrates in the Gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, M.R.; Dawson, J.A.; Stevenson, D.M.; Cunningham, A.C.; Bramhacharya, S.; Weimer, P.J.; Kendziorski, C.; Suen, G. Unique Aspects of Fiber Degradation by the Ruminal Ethanologen Ruminococcus Albus 7 Revealed by Physiological and Transcriptomic Analysis. BMC Genomics 2014, 15, 1066. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Nourrisson, C.; Scanzi, J.; Brunet, J.; Delbac, F.; Dapoigny, M.; Poirier, P. Prokaryotic and Eukaryotic Fecal Microbiota in Irritable Bowel Syndrome Patients and Healthy Individuals Colonized With Blastocystis. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Rajamanikam, A.; Isa, M.N.M.; Samudi, C.; Devaraj, S.; Govind, S.K. Gut Bacteria Influence Blastocystis Sp. Phenotypes and May Trigger Pathogenicity. PLoS Negl. Trop. Dis. 2023, 17, e0011170. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.; Zapata, C.; Romero, Y.; Flores-Huarco, N.H.; Oros, O.; Alvarado, W.; Quilcate, C.; Guevara-Alvarado, H.M.; Estrada, R.; Coila, P. Parasitism-Induced Changes in Microbial Eukaryotes of Peruvian Alpaca Gastrointestinal Tract. Life 2024, 14, 187. [Google Scholar] [CrossRef]
- Herrera, G.; Vega, L.; Patarroyo, M.A.; Ramírez, J.D.; Muñoz, M. Gut Microbiota Composition in Health-Care Facility-and Community-Onset Diarrheic Patients with Clostridioides Difficile Infection. Sci. Rep. 2021, 11, 10849. [Google Scholar] [CrossRef]
- Jang, L.-G.; Choi, G.; Kim, S.-W.; Kim, B.-Y.; Lee, S.; Park, H. The Combination of Sport and Sport-Specific Diet Is Associated with Characteristics of Gut Microbiota: An Observational Study. J. Int. Soc. Sports Nutr. 2019, 16, 21. [Google Scholar] [CrossRef]
- Benítez-Páez, A.; Gómez del Pugar, E.M.; López-Almela, I.; Moya-Pérez, Á.; Codoñer-Franch, P.; Sanz, Y. Depletion of Blautia Species in the Microbiota of Obese Children Relates to Intestinal Inflammation and Metabolic Phenotype Worsening. mSystems 2020, 5, 10.1128/msystems.00857-19. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, J.; Wang, X.; Han, L.; Yang, Y.; Wang, Q.; Yu, Q. Metagenomic and Transcriptomic Analyses Reveal the Differences and Associations Between the Gut Microbiome and Muscular Genes in Angus and Chinese Simmental Cattle. Front. Microbiol. 2022, 13. [Google Scholar] [CrossRef]
- Chen, X.; An, M.; Zhang, W.; Li, K.; Kulyar, M.F.-E.-A.; Duan, K.; Zhou, H.; Wu, Y.; Wan, X.; Li, J.; et al. Integrated Bacteria-Fungi Diversity Analysis Reveals the Gut Microbial Changes in Buffalo With Mastitis. Front. Vet. Sci. 2022, 9, 918541. [Google Scholar] [CrossRef]
- Grigore, D.-M.; Mironeasa, S.; Ciurescu, G.; Ungureanu-Iuga, M.; Batariuc, A.; Babeanu, N.E. Carcass Yield and Meat Quality of Broiler Chicks Supplemented with Yeasts Bioproducts. Appl. Sci. 2023, 13, 1607. [Google Scholar] [CrossRef]
- Hof, H. Rhodotorula Spp. in the Gut – Foe or Friend? GMS Infect. Dis. 2019, 7, Doc02. [Google Scholar] [CrossRef]
- Ge, Y.; Huang, K.; Xie, W.; Xu, C.; Yao, Q.; Liu, Y. Effects of Rhodotorula Mucilaginosa on the Immune Function and Gut Microbiota of Mice. Front. Fungal Biol. 2021, 2. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa-Seki, M.; Fereig, R.M.; Masatani, T.; Kinami, A.; Takahashi, Y.; Kida, K.; Nishikawa, Y. Development of CpGP15 Recombinant Antigen of Cryptosporidium Parvum for Detection of the Specific Antibodies in Cattle. Parasitol. Int. 2019, 69, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zou, W.; Chen, X.; Sun, G.; Cidan, Y.; Almutairi, M.H.; Dunzhu, L.; Nazar, M.; Mehmood, K.; Zhu, Y.; et al. Effects of Cryptosporidium Parvum Infection on Intestinal Fungal Microbiota in Yaks (Bos Grunniens). Microb. Pathog. 2023, 183, 106322. [Google Scholar] [CrossRef]
- Kooh, P.; Thébault, A.; Cadavez, V.; Gonzales-Barron, U.; Villena, I. Risk Factors for Sporadic Cryptosporidiosis: A Systematic Review and Meta-Analysis. Microb. Risk Anal. 2021, 17, 100116. [Google Scholar] [CrossRef]
- Ryan, U.; Power, M. Cryptosporidium Species in Australian Wildlife and Domestic Animals. Parasitology 2012, 139, 1673–1688. [Google Scholar] [CrossRef]
- Holman, D.B.; Gzyl, K.E.; Scott, H.; Prieto, N.; López-Campos, Ó. Cara Service Associations between the Rumen Microbiota and Carcass Merit and Meat Quality in Beef Cattle. Appl. Microbiol. Biotechnol. 2024, 108, 287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.; Wu, C. Modulation of Gut Microbiota and Immune System by Probiotics, Pre-Biotics, and Post-Biotics. Front. Nutr. 2022, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Z.; Li, B.; Hao, W.; Yin, W.; Ai, S.; Han, J.; Wang, R.; Duan, Z. Depicting Fecal Microbiota Characteristic in Yak, Cattle, Yak-Cattle Hybrid and Tibetan Sheep in Different Eco-Regions of Qinghai-Tibetan Plateau. Microbiol. Spectr. 2022, 10, e00021-22. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, H.; Cheng, Y.; Xu, F.; Ruan, G.; Ying, S.; Tang, W.; Chen, L.; Chen, M.; Lv, L.; et al. Fecal Microbiota Transplantation Relieves Gastrointestinal and Autism Symptoms by Improving the Gut Microbiota in an Open-Label Study. Front. Cell. Infect. Microbiol. 2021, 11. [Google Scholar] [CrossRef]
- Hu, J.; Guo, P.; Mao, R.; Ren, Z.; Wen, J.; Yang, Q.; Yan, T.; Yu, J.; Zhang, T.; Liu, Y. Gut Microbiota Signature of Obese Adults Across Different Classifications. Diabetes Metab. Syndr. Obes. 2022, 15, 3933–3947. [Google Scholar] [CrossRef]
- Mukherjee, A.; Lordan, C.; Ross, R.P.; Cotter, P.D. Gut Microbes from the Phylogenetically Diverse Genus Eubacterium and Their Various Contributions to Gut Health. Gut Microbes 2020, 12, 1802866. [Google Scholar] [CrossRef] [PubMed]
- Bay, V.; Gillespie, A.; Ganda, E.; Evans, N.J.; Carter, S.D.; Lenzi, L.; Lucaci, A.; Haldenby, S.; Barden, M.; Griffiths, B.E.; et al. The Bovine Foot Skin Microbiota Is Associated with Host Genotype and the Development of Infectious Digital Dermatitis Lesions. Microbiome 2023, 11, 4. [Google Scholar] [CrossRef]
- Zhang, L.; Zhan, H.; Xu, W.; Yan, S.; Ng, S.C. The Role of Gut Mycobiome in Health and Diseases. Ther. Adv. Gastroenterol. 2021, 14, 17562848211047130. [Google Scholar] [CrossRef]
- Zhang, F.; Aschenbrenner, D.; Yoo, J.Y.; Zuo, T. The Gut Mycobiome in Health, Disease, and Clinical Applications in Association with the Gut Bacterial Microbiome Assembly. Lancet Microbe 2022, 3, e969–e983. [Google Scholar] [CrossRef]
- Lukeš, J.; Stensvold, C.R.; Jirků-Pomajbíková, K.; Parfrey, L.W. Are Human Intestinal Eukaryotes Beneficial or Commensals? PLOS Pathog. 2015, 11, e1005039. [Google Scholar] [CrossRef]
- Laforest-Lapointe, I.; Arrieta, M.-C. Microbial Eukaryotes: A Missing Link in Gut Microbiome Studies. mSystems 2018, 3, 10.1128/msystems.00201-17. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, Y.; Guan, L.L. Translational Gut Microbiome Research for Strategies to Improve Beef Cattle Production Sustainability and Meat Quality. Anim. Biosci. 2024, 37, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Teng, J.; Wang, X.; Xu, B.; Niu, Y.; Ma, L.; Yan, X. Multi-Omics Analysis Reveals Gut Microbiota-Induced Intramuscular Fat Deposition via Regulating Expression of Lipogenesis-Associated Genes. Anim. Nutr. 2022, 9, 84–99. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Alpha diversity metrics of bacteria and fungi were evaluated in cattle in five breeds. Diversity indices exhibited include ACE, Chao1, Observe, Pielou, Shannon and Simpson. A) Alpha diversity of bacteria B) Alpha diversity of fungi.
Figure 2.
Alpha diversity metrics of bacteria and fungi were evaluated in cattle in five breeds. Diversity indices exhibited include ACE, Chao1, Observe, Pielou, Shannon and Simpson. A) Alpha diversity of bacteria B) Alpha diversity of fungi.
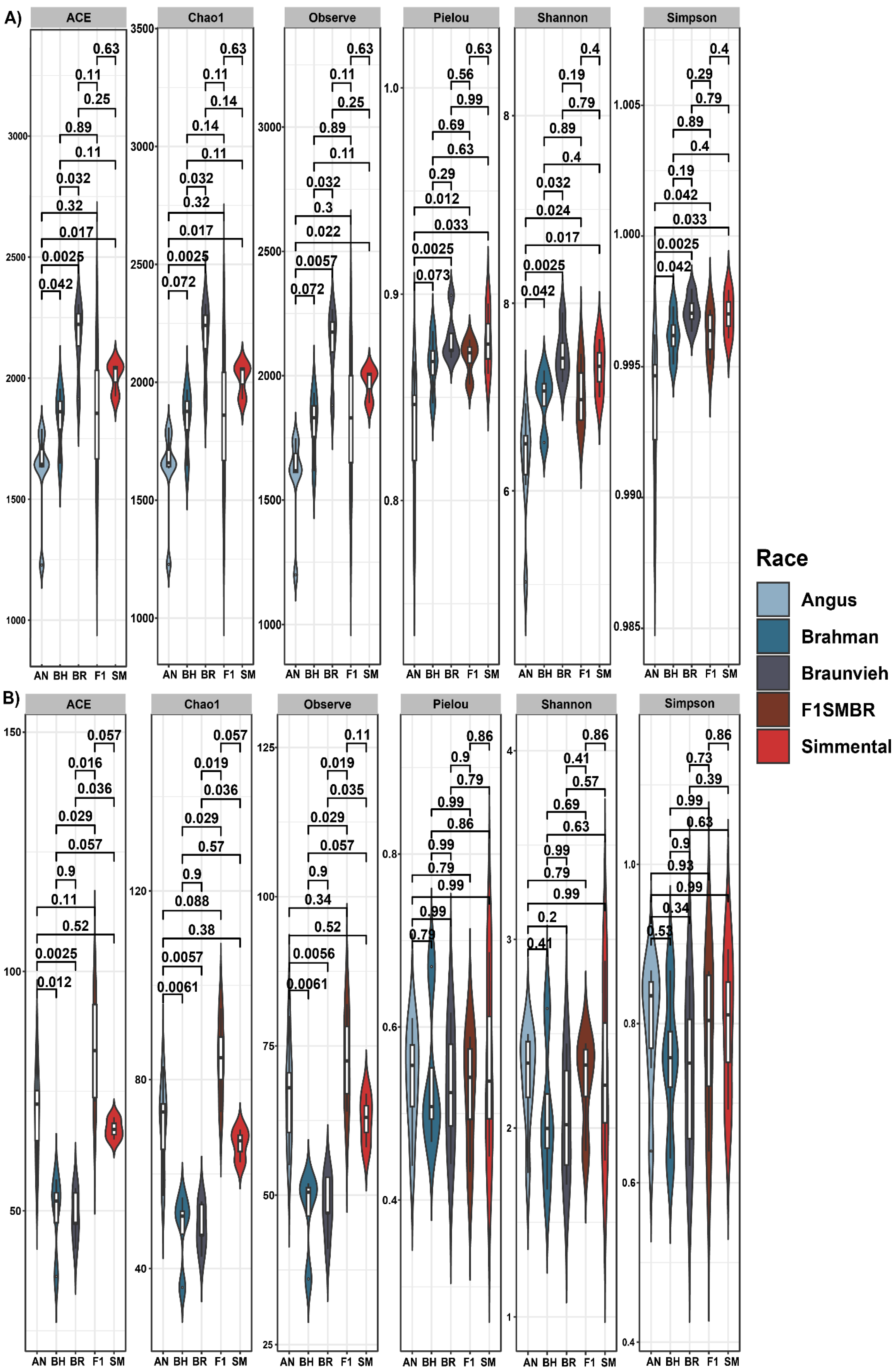
Figure 5.
Heat map of Spearman correlation analysis between the genera of intestinal microbes and meat variables. *p < 0.05; **p<0.01; ***p<0.001. A) Heat map of bacterial genera. B) Heat map of fungi genera. C) Heat map of protist genera.
Figure 5.
Heat map of Spearman correlation analysis between the genera of intestinal microbes and meat variables. *p < 0.05; **p<0.01; ***p<0.001. A) Heat map of bacterial genera. B) Heat map of fungi genera. C) Heat map of protist genera.
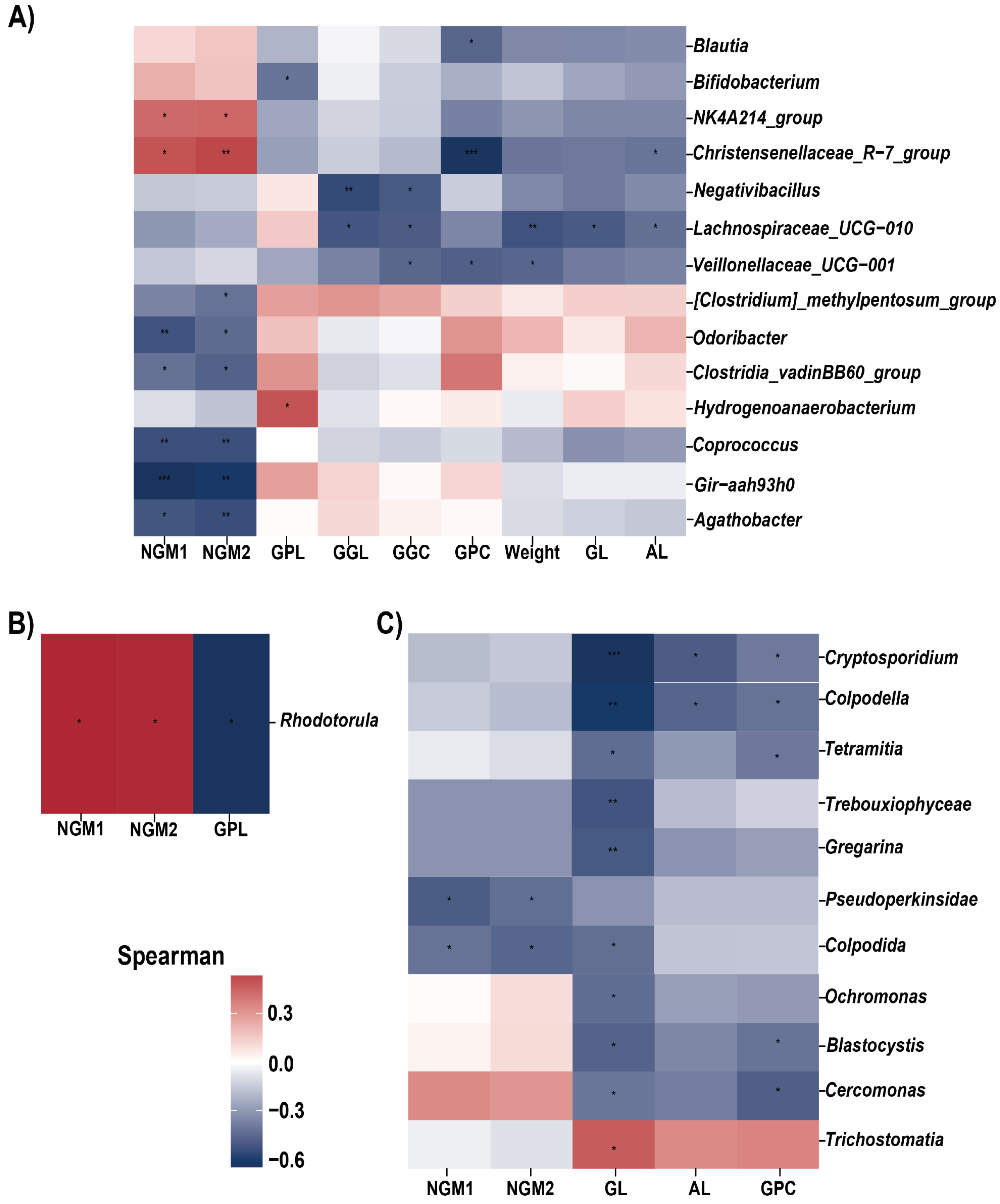
Figure 6.
LEfSe analysis of the intestinal microbiome in different breeds. A) LEfSe of bacteria. B) LEfSe of fungi. C) LEfSe of protist.
Figure 6.
LEfSe analysis of the intestinal microbiome in different breeds. A) LEfSe of bacteria. B) LEfSe of fungi. C) LEfSe of protist.
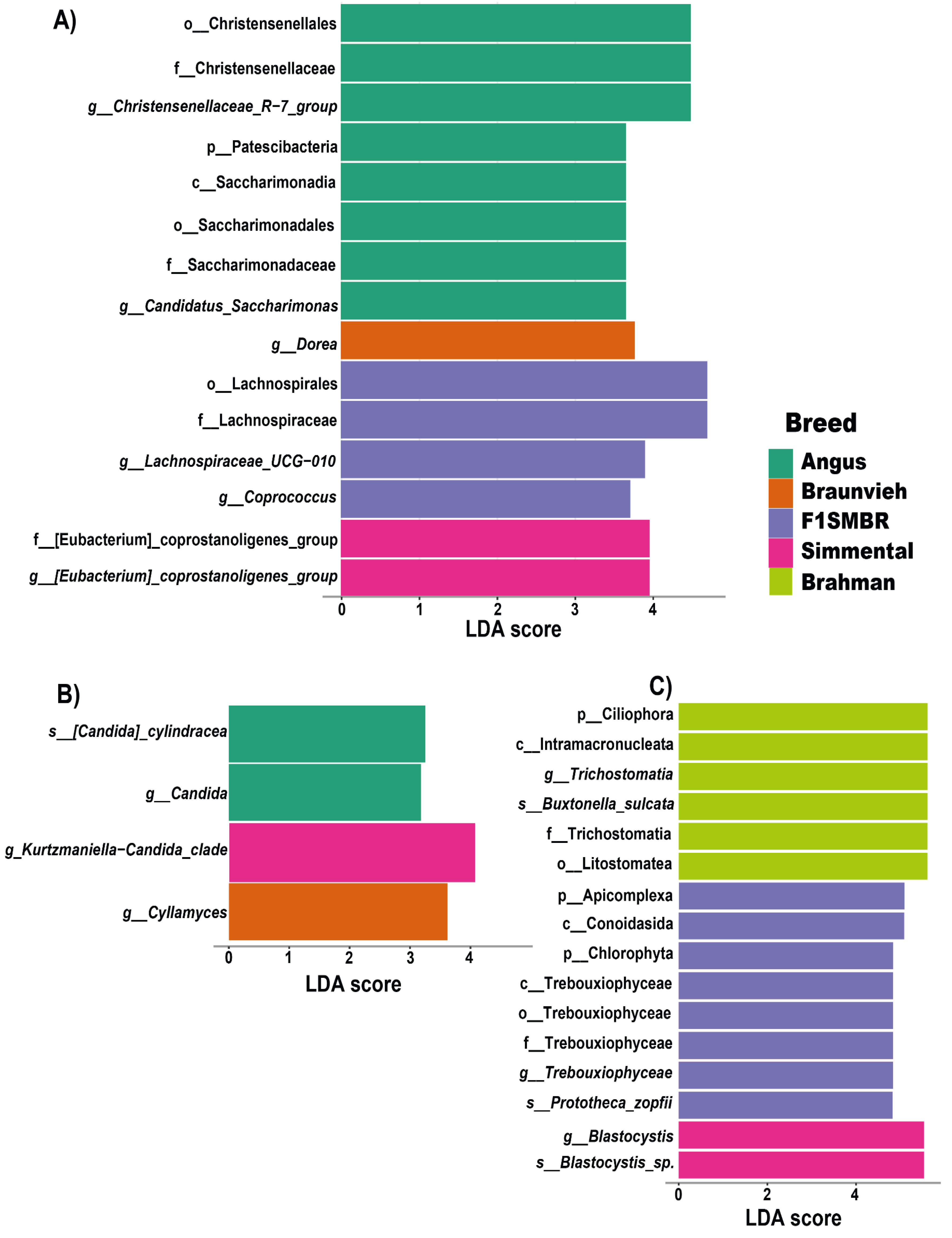
Figure 7.
Correlations between the alpha diversity of the intestinal microbiota and hematological and meat variables. A) Spearman correlation of alpha diversity of bacteria. B) Spearman correlation of fungal alpha diversity. C) Spearman correlation of protist alpha diversity.
Figure 7.
Correlations between the alpha diversity of the intestinal microbiota and hematological and meat variables. A) Spearman correlation of alpha diversity of bacteria. B) Spearman correlation of fungal alpha diversity. C) Spearman correlation of protist alpha diversity.
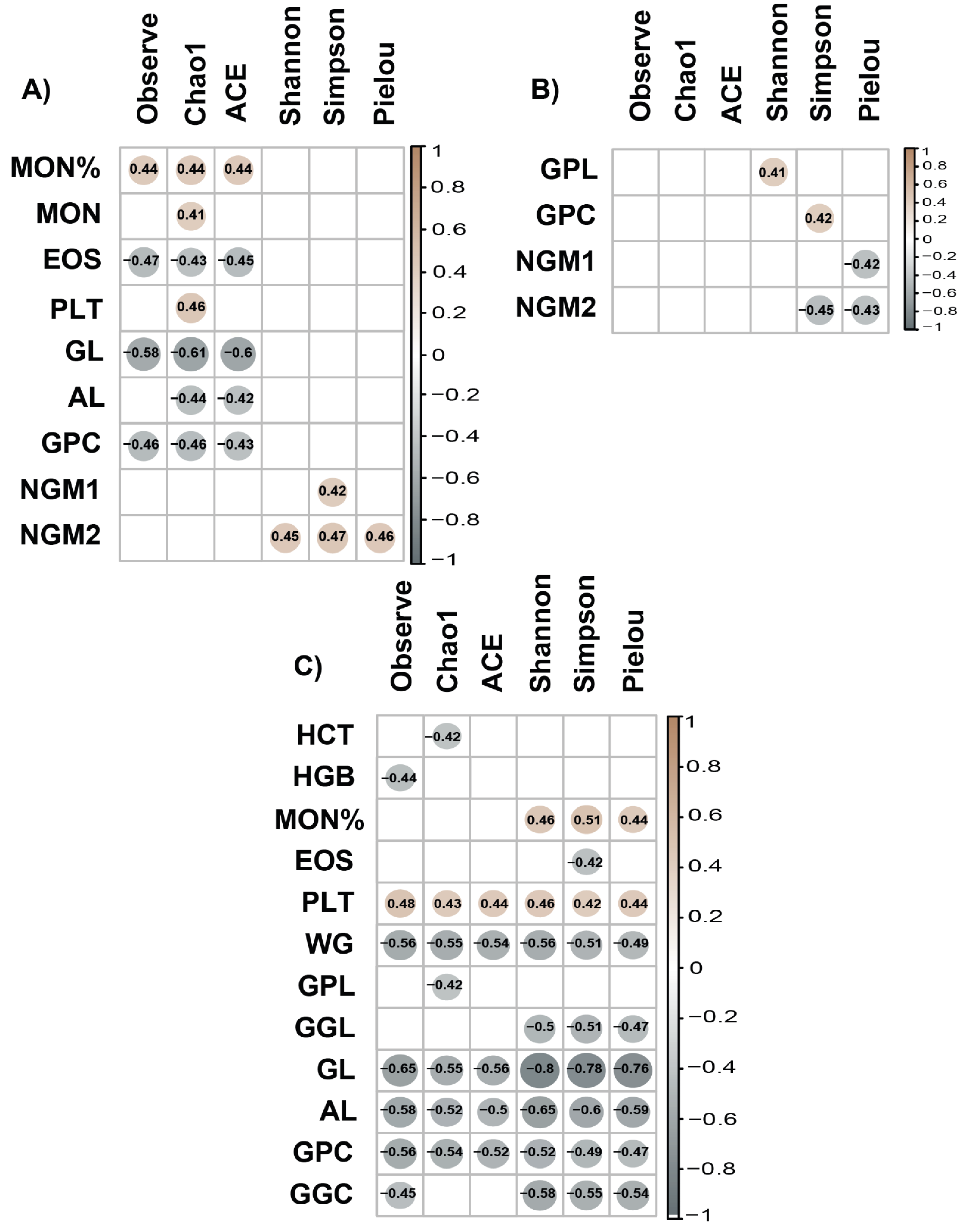

Table 1.
Diet of Cattle at EEA Donoso.
| Feed Type | Amount | Dry Matter | Moisture |
|---|---|---|---|
| Corn Silage | 3% of body weight | 24%-26% | 74%-76% |
| Balanced Feed | 2 kg | 90% | 10% |
| Crude Protein | 14% |
Table 2.
PERMANOVA of Unweighted Unifrac and Jaccard methods.
| Bacteria | Fungi | Protists | ||||||||||
| Unweighted unifrac | Jaccard | Unweighted unifrac | Jaccard | Unweighted unifrac | Jaccard | |||||||
| Items | R2 | p | R2 | p | R2 | p | R2 | p | R2 | p | R2 | p |
| Race | 0.22 | 0.001** | 0.22 | 0.001** | 0.21 | 0.058 | 0.22 | 0.001** | 0.27 | 0.004** | 0.23 | 0.003** |
| Sex | 0.05 | 0.022* | 0.05 | 0.035* | 0.07 | 0.027* | 0.05 | 0.036* | 0.05 | 0.225 | 0.05 | 0.058 |
| Race:Sex | 0.19 | 0.083 | 0.19 | 0.045* | 0.15 | 0.691 | 0.17 | 0.438 | 0.2 | 0.1 | 0.17 | 0.082 |
Table 3.
Correlation of variables with Beta diversity (Jaccard and Unweighted Unifrac) using Mantel and Partial Mantel Tests. Only Significant Variables Presented.
Table 3.
Correlation of variables with Beta diversity (Jaccard and Unweighted Unifrac) using Mantel and Partial Mantel Tests. Only Significant Variables Presented.
| Fungi | Jaccard | Unweighted Unifrac | ||||||
|---|---|---|---|---|---|---|---|---|
| Mantel test | Partial Mantel test | Mantel test | Partial Mantel test | |||||
| Variables | r | p | r | p | r | p | r | p |
| NGM1 | 0.22821 | 0.016 | 0.23833 | 0.017 | - | - | - | - |
| NGM2 | 0.19406 | 0.033 | 0.20089 | 0.036 | - | - | - | - |
| WBC | - | - | - | - | 0.22348 | 0.019 | 0.1771 | 0.041 |
| WEIGHT | - | - | - | - | 0.28379 | 0.015 | 0.22588 | 0.044 |
| RBC | - | - | - | - | 0.26543 | 0.031 | - | - |
| MCV | - | - | - | - | 0.27068 | 0.019 | - | - |
| MCHC | - | - | - | - | 0.25847 | 0.025 | - | - |
| NEU | - | - | - | - | 0.19148 | 0.038 | - | - |
| SEG | - | - | - | - | 0.19148 | 0.039 | - | - |
| Protists | r | p | r | p | r | p | r | p |
| MON | - | - | - | - | 0.19531 | 0.013 | 0.20835 | 0.008 |
| PLT | 0.22961 | 0.021 | 0.2268 | 0.024 | 0.16086 | 0.026 | 0.1609 | 0.026 |
| GL | - | - | - | - | 0.25734 | 0.01 | 0.27912 | 0.006 |
| AL | - | - | - | - | 0.13362 | 0.033 | 0.1335 | 0.044 |
| NEU% | 0.21743 | 0.031 | - | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



