
Submitted:
07 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
(R)-2-Amino-1-hydroxyethylphosphonic acid 2 was prepared by hydrolytic kinetic resolution of rac-diethyl oxiran-2-ylphosphonate followed by reaction with benzylamine, acid hydrolysis, catalytic hydrogenolysis and anion exchange chromatography. Recrystallization from water-ethanol gave 2, which was characterized by IR, 1H NMR, 13C NMR, 31P NMR, polarimetry, elemental microanalysis, high-resolution mass spectrometry and single-crystal X-ray diffraction. The acid 2 crystallized in the orthorhombic chiral space group P212121 with cell constants a = 6.303(2) Å, b = 7.104(2) Å, c = 11.627(3) Å. The X-ray crystal structure confirmed the (R)-configuration of 2 and revealed that 2 is zwitterionic in the solid state, with extensive intermolecular hydrogen bonding between the hydroxyl, ammonium cation and phosphonate anion groups.
Keywords:
X-ray structure
; amino phosphonic acid
; hydrogen bonding
1. Introduction
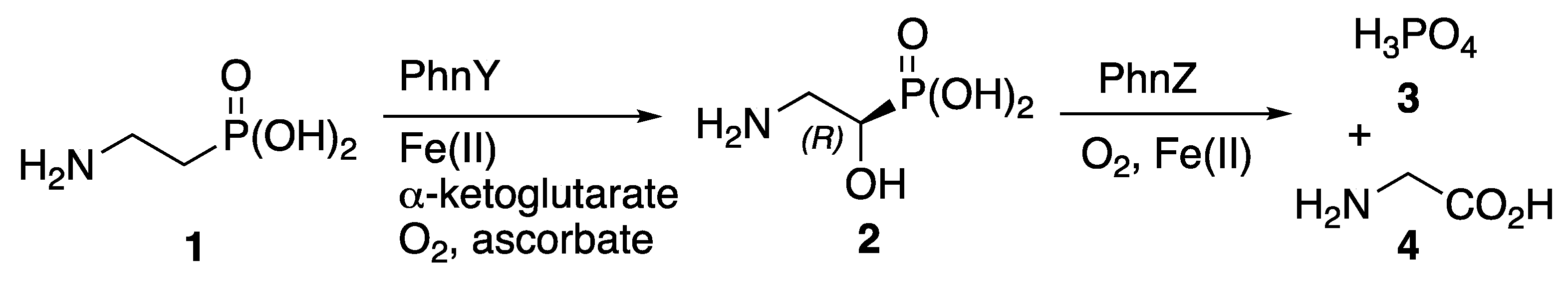
Phosphorus is essential for all life on Earth, but the low bioavailability of this vital element in many marine environments places stress on biological systems [1]. Microorganisms can use phosphonic acid derivatives to manage scarce phosphorus resources. Thus, in phytoplankton, oxidation of 2-aminoethylphosphonic acid 1 (Scheme 1), mediated by the iron-dependent enzyme PhnY, leads to stereoselective formation of (R)-2-amino-1-hydroxyethylphosphonic acid 2. This catabolic intermediate 2 is then used a reservoir for supplying inorganic phosphate 3, which is released alongside glycine 4 following further stereoselective oxidation, mediated by a second iron-dependent enzyme, PhnZ [2]. A 2.1 Å resolution X-ray crystal structure of the PhnZ enzyme bound to its substrate 2 has been reported [3].
We have shown [4] that enantioselective chemical synthesis of the natural product 2 can be achieved using hydrolytic kinetic resolution of racemic epoxyphosphonate 5 in the presence of the Jacobsen [5] cobalt catalyst 6 (Scheme 2). The unchanged (R)-enantiomer of the epoxide 5 was ring-opened by benzylamine to give the intermediate 7, then all protecting groups were removed to afford (R)-2, with physical data consistent with those of material obtained from natural sources [6], by preparation of diastereoisomeric salts [6] and by kinetic resolution of an ester precursor using a lipase [7]. Recently the formation of 2 by borane reduction of a nitrile precursor was used to determine the absolute configuration of the natural product (–)-hydroxynitralaphos [8].
There have been only a few crystal structures reported for aminophosphonic acids. These include compounds 1 [9] and 8 - 11 (Figure 1) [10,11,12,13]. In this article we provide experimental details and characterization for the chemical synthesis of the acid 2 and we report a high resolution (0.7 Å) single crystal X-ray structure determination of 2, thus defining the hydrogen bond network that exists in the solid state for pure (R)-2, a small but highly functionalized chiral molecule.
2. Results
The synthesis of 2 proceeded as previously outlined [4]; product 2 was recrystallized from aqueous ethanol, then dried under vacuum. Physical data for 2, including NMR spectra, melting point and optical rotation matched existing literature. High resolution mass spectrometry confirmed the molecular formula and elemental analysis demonstrated that the crystals did not contain residual solvent.
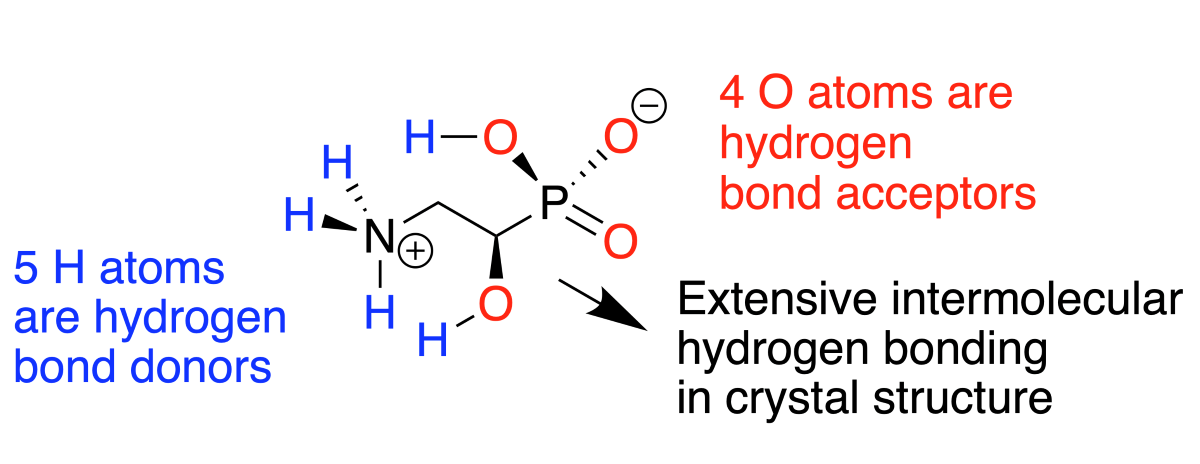
Single-crystal X-ray diffraction (Figure 2) revealed the structure of 2, which exists in the chiral space group P212121. The presence of the relatively heavy phosphorus atom within the compound allowed the absolute configuration to be confirmed with confidence: a Flack [14] absolute structure parameter of 0.023(13) was calculated for the (R)-enantiomer. Molecules of 2 exist as zwitterions with transfer of one proton from each phosphonic acid group to the amino group generating phosphonate anions and ammonium cations. The conformation of each molecule is such that the phosphorus and nitrogen atoms are antiperiplanar to one another, with P–C(1)-C(2)-N torsion angles of 173.8°, similar to the value of 174.2° found in the crystal structure for 2 that is bound to the PhnZ enzyme [3] and to the conformations of compounds 1 and 8 observed in previous reports [9, 10]. The polar functional groups of 2 engage in complex networks of hydrogen bonds between neighboring molecules of 2; no intramolecular hydrogen bonds are observed and, in accord with the microanalysis results, no solvent molecules are retained within the crystals. This absence of solvation contrasts with compounds rac-10b, which crystallized as a hydrate, and rac-11, which contained ethanol of crystallization.
For each molecule of acid 2 in the crystal, there were four N–H…O and two O–H…O type intermolecular hydrogen bonds to adjacent molecules of 2 (Table 1). Figure 3 illustrates packing of molecules of 2 within the crystal and the associated network of hydrogen bonds. The ammonium and phosphonate groups adopt similar hydrogen bond-forming roles to those seen in crystals of compounds 1 and 8 - 11. All three N–H groups are hydrogen bond donors, with bifurcation at H4. The three oxygen atoms of the phosphonate group are all hydrogen bond acceptors, with O2 and O3 each linking to two H atoms in neighboring molecules. Additionally, the phosphonate O1-H7 and the alcohol O4–H8 groups act as both hydrogen bond donors and acceptors.
To conclude, we have established the crystal structure of enantiomerically pure 2, prepared according to our previously outlined method, for which experimental details are now provided. Notably, this highly polar substance crystallizes from aqueous ethanol without retaining solvent. In common with other amino phosphonic acids, 2 is zwitterionic. All the functional groups of 2 contribute to intermolecular hydrogen bonding within the crystal.
3. Experimental
3.1. General Experimental Details
Unless otherwise stated, all commercially available solvents and reagents were used without further purification. Melting points were measured using a hot-stage microscope (Reichert). IR spectra were obtained by attenuated total reflection (ATR) using a Perkin-Elmer Spectrum 65 FT-IR spectrometer. NMR spectra were recorded using Bruker AM250, Jeol JNM-EX270 and Bruker AVIII 400 spectrometers. Elemental microanalysis was performed by Medac Ltd., Chobham, Surrey, UK. Mass spectra were obtained by the EPSRC NMSF, Swansea.
3.2. (R)-Diethyl oxiran-2-ylphosphonate (R)-5
AcOH (7 μL, 0.12 mmol) was added to a solution of (R,R)-(–)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminocobalt(II) 6 (38.0 mg, 0.06 mmol) in dry toluene (1.5 mL) and stirred in air at 20 °C for 1 h. The acetic acid was evaporated under vaccuum and rac-diethyl oxiran-2-ylphosphonate 5 [15] (1.261 g, 7.00 mmol) was added, followed by water (101 μL, 5.60 mmol). Stirring was continued for 4 d, after which purification by flash chromatography (CH2Cl2-EtOAc, 3:1) gave (R)-diethyl oxiran-2-ylphosphonate (R)-5 (520.1 mg, 2.71 mmol) [ee > 95%, based on 31P integration of the product of epoxide opening with neat (S)-α-methylbenzylamine (5 equivalents, 16 h, 20 °C)][4].
3.3. (R)-Diethyl (2-benzylamino-1-hydroxyethyl)phosphonate 7
A mixture of (R)-diethyl oxiran-2-ylphosphonate 5 (455.3 mg, 2.53 mmol) and benzylamine (1.35 g, 12.6 mmol) was kept at 20 °C for 16 h, after which the excess benzylamine was evaporated in vacuo (70 °C, 0.5 mmHg, 1 h). The residue was subjected to flash chromatography (gradient from EtOAc-CH2Cl2 1:1 to EtOH-EtOAc 3:7). Appropriate fractions were pooled, evaporated and the residue was taken up in Et2O, filtered through celite to remove silica gel residues, and re-evaporated to give (R)-diethyl (2-benzylamino-1-hydroxyethyl)phosphonate 7 (557.5 mg, 77%) as a liquid [α]D30 –7.6 (c = 1.67, MeOH); δH (270 MHz, CDCl3) 7.37-7.20 (5H, m), 4.22-4.09 (4H, m), 4.00 (1H, dd, J 11.2, 6.8 Hz), 3.80 (2H, s), 3.38 (2H, br s), 3.03-2.86 (2H, m), 1.35-1.28 (6H, m).
3.4. (R)-2-Amino-1-hydroxyethylphosphonic Acid 2
(R)-Diethyl (2-benzylamino-1-hydroxyethyl)phosphonate 7 (272.4 mg, 0.95 mmol) was refluxed gently for 16 h with 12 M hydrochloric acid (20 mL) then evaporated in vacuo. Water (16 mL), acetic acid (8 mL) and palladium black (120 mg) were added and the mixture was stirred under a balloon of hydrogen at room temperature for 48 h. The mixture was filtered through Celite and the solvent was evaporated in vacuo. The residue was re-evaporated from water, then redissolved in water and applied to a column of Amberlite IR400 (OH), which was eluted first with water and then with 10% aqueous AcOH. The ninhydrin-positive fractions were combined and evaporated. The residue was recrystallized from water-ethanol to give the title compound 2 (109.3 mg, 82%) as colorless needles, mp 264-265 °C (lit. [6] 244-246 °C); [α]D27 –31.9 (c = 0.525, H2O) (lit. [6] [α]D20 –30.5). Found: C, 17.10; H, 5.73; N, 9.57 calcd for C2H8NO4P: C, 17.03; H, 5.72; N, 9.92; νmax/cm–1 (KBr) 3458, 3157, 2936, 2640, 1663, 1539, 1259, 1159, 1096, 937, 781; δH (400 MHz, D2O) 3.90 (1H, td, J = 9.9, 2.7 Hz), 3.30 (1H, ddd, J = 13.2, 6.4, 3.2 Hz), 3.11 (1H, ddd, J = 13.3, 9.8, 6.2 Hz); δc (62.9 MHz, D2O) 62.7 (d, 1JCP = 155 Hz), 44.4 (d, 2JCP = 8 Hz); δP (162 MHz, D2O) 15.1; m/z (ESI) found: 142.0262; calcd for C2H9NO4P (M+H+): 142.0264.
3.5. X-ray Structure Determination of 2
Single-crystal X-ray diffraction was carried out at the Queen Mary University of London using the KAPPA APEX ii DUO diffractometer, with MoKα radiation (λ = 0.71073 Å). X-ray crystal structures were solved and refined using the Bruker SHELXTL software package.
A translucent colorless block-like specimen of C2H8NO4P, approximate dimensions 0.100 mm × 0.120 mm × 0.200 mm, was used for the X-ray crystallographic analysis. The X-ray intensity data were measured. A total of 2253 frames were collected. The total exposure time was 6.26 h. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The integration of the data using an orthorhombic unit cell yielded a total of 11622 reflections to a maximum θ angle of 30.56° (0.70 Å resolution), of which 1582 were independent (average redundancy 7.346, completeness = 98.9%, Rint = 1.45%) and 1559 (98.55%) were greater than 2σ(F2). The final cell constants of a = 6.303(2) Å, b = 7.104(2) Å, c = 11.627(3) Å, α = β = γ = 90° and volume = 520.6(2) Å3 are based upon the refinement of the XYZ-centroids of 9792 reflections above 20 σ(I) with 6.721° < 2θ < 61.11°. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.927. The calculated minimum and maximum transmission coefficients (based on crystal size) were 0.9150 and 0.9560, respectively.
The final anisotropic full-matrix least-squares refinement on F2 with 76 variables converged at R1 = 1.61%, for the observed data and wR2 = 4.64% for all data. The goodness of fit was 1.167. The largest peak in the final difference electron density synthesis was 0.400 e−/Å3, and the largest hole was −0.159 e−/Å3, with an RMS deviation of 0.052 e−/Å3. On the basis of the final model, the calculated density was 1.800 g/cm3 and F(000), 296 e−.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, CIF report for the crystal structure of 2, Figure S1: 1H NMR spectrum of 2 (400 MHz, D2O); Figure S2: 1H {31P} NMR spectrum of 2 (400 MHz, D2O); Figure S3: 13C NMR spectrum of 2 (62.9 MHz, D2O); Figure S4: 31P {1H} NMR spectrum of 2 (162 MHz, D2O).
Author Contributions
Conceptualization, P.B.W.; methodology, P.B.W., M.M., P.B. and I.A.; investigation, P.B.W., M.M., P.B., C.E.W., and I.A.; writing—original draft preparation, P.B.W.; writing—review and editing, P.B.W., and I.A.; visualization, P.B.W., M.M., and I.A.; supervision, P.B.W.; project administration, P.B.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Crystallographic data (excluding structure factors) for the acid 2 are available in the Supplementary Materials of this paper and were deposited at the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2381816. Copies of the data can be obtained, free of charge, upon application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgments
We are grateful to the EPSRC NMSF at Swansea University for mass spectrometry and to the European Union for enabling P.B. to participate in the ERASMUS student exchange programme.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Duhamel, S.; Diaz, J.M.; Adams, J.C.; Djaoudi, K.; Steck, V.; Waggoner, E.M. Phosphorus as an integral component of global marine geochemistry. Nature Geoscience 2021, 14, 359-368. [CrossRef]
- McSorley, F. R.; Wyatt, P. B.; Martinez, A.; DeLong, E. F.; Hove-Jensen, B.; Zechel, D.L. PhnY and PhnZ comprise a new oxidative pathway for enzymatic cleavage of a carbon-phosphorus bond. J. Am. Chem. Soc. 2012, 134, 8364-8367. [CrossRef]
- Van Staalduinen, L.M.; McSorley, F.R.; Schiessl, K.; Séguin, J.; Wyatt, P.B.; Hammerschmidt, F.; Zechel, D.L.; Jia, Z. Crystal structure of Phnz in complex with substrate reveals a di-iron oxygenase mechanism for catabolism of organophosphates. Proc. Natl. Acad. Sci. USA 2014, 111, 5171-5176. [CrossRef]
- Wyatt, P.B.; Blakskjær, P. An enantioselective synthesis of (R)-2-amino-1-hydroxyethylphosphonic acid by hydrolytic kinetic resolution of (±)-diethyl oxiranephosphonate. Tetrahedron Lett. 1999, 40, 6481-6483. [CrossRef]
- Tokunaga, M.; Larrow, J.F.; Kakiuchi, F.; Jacobsen, E.N. Asymmetric catalysis with water: efficient kinetic resolution of terminal epoxides by means of catalytic hydrolysis. Science, 1997, 277, 936-938. [CrossRef]
- Hammerschmidt, F.; Völlenkle, H. Absolute Konfiguration der (2-Amino-1-hydroxyethyl)phosphonsäure aus Ancanthamoeba castellanii (Neff) – Darstellung der Phosphonsaure-Analoga von (+)- und (–)- Serin.Liebigs Ann. Chem. 1989, 577-583. [CrossRef]
- Hammerschmidt, F.; Lindner, W.; Wuggenig, F.; Zarble, E. Enzymes in organic chemistry. Part 10: Chemo-enzymatic synthesis of L-phosphaserine and L-phosphaisoserine and enantioseparation of amino-hydroxyethylphosphonic acids by non-aqueous capillary electrophoresis with quinine carbamate as chiral pair agent. Tetrahedron: Asymmetry 2000, 11, 2955-2964. [CrossRef]
- Pallitsch, K.; Happl, B.; Stieger, C. Determination of the absolute configuration of (–)-hydroxynitralaphos and related biosynthetic questions. Chem. Eur. J. 2017, 23, 15655-15665.
- Okaya, Y. Crystal structure of the stable modification of 2-aminoethylphosphonic acid, β-ciliatine. Acta Cryst. 1966, 20, 712-715. [CrossRef]
- Crawczyk, H.; Albrecht, L.; Wojciechowski, J.; Wolf, W. M. Synthesis and crystal structure of 1-(aminomethyl)vinylphosphonic acid. Tetrahedron 2008, 64, 5051-5054. [CrossRef]
- Chen, S-P.; Zhang, Y-Q.; Hu, L.; He, H-Z.; Yuan, L-J. Hydrogen-bonded assembly of aminophosphonic anions: different 1D, 2D and 3D supramolecular architectures. CrystEngComm. 2010, 12, 3327-3336. [CrossRef]
- Bogomilova, A.; Hägele, G.; Troev, K.; Wagner, E.; Günther, M. Hydrogen bonding in α-aminophosphonic acids, Phosphorus, Sulfur and Silicon, 2012, 187, 165-180. [CrossRef]
- Wanat, W.; Dziuk, B.; Kafarski, P. New crystal structures of α-aminophosphonic acid analogues of phenylglycine. Structural Chemistry, 2020, 31, 1197-1209. [CrossRef]
- Flack, H. D.; On enantiomorph-polarity estimation. Acta. Crystallogr. 1983, A39, 876-881. [CrossRef]
- Sturtz, G.; Pondaven-Raphalen, A. Etude de la réaction de cyclisation des halohydrines-1,2-éthyl phosphonates de diéthyle pour l’obtention de l’époxy-1,2 éthyl phosphonate de diéthyle. Phosphorus and Sulfur 1984, 20, 35-47. [CrossRef]
Scheme 1.
Catabolic pathway in which (R)-2-amino-1-hydroxyethylphosphonic acid 2 acts as a reservoir supplying inorganic phosphate 3.
Scheme 1.
Catabolic pathway in which (R)-2-amino-1-hydroxyethylphosphonic acid 2 acts as a reservoir supplying inorganic phosphate 3.
Scheme 2.
Chemical synthesis of (R)-2-amino-1-hydroxyethylphosphonic acid 2 from racemic epoxyphosphonate 5 using an enantioselective hydrolytic kinetic resolution [4].
Scheme 2.
Chemical synthesis of (R)-2-amino-1-hydroxyethylphosphonic acid 2 from racemic epoxyphosphonate 5 using an enantioselective hydrolytic kinetic resolution [4].
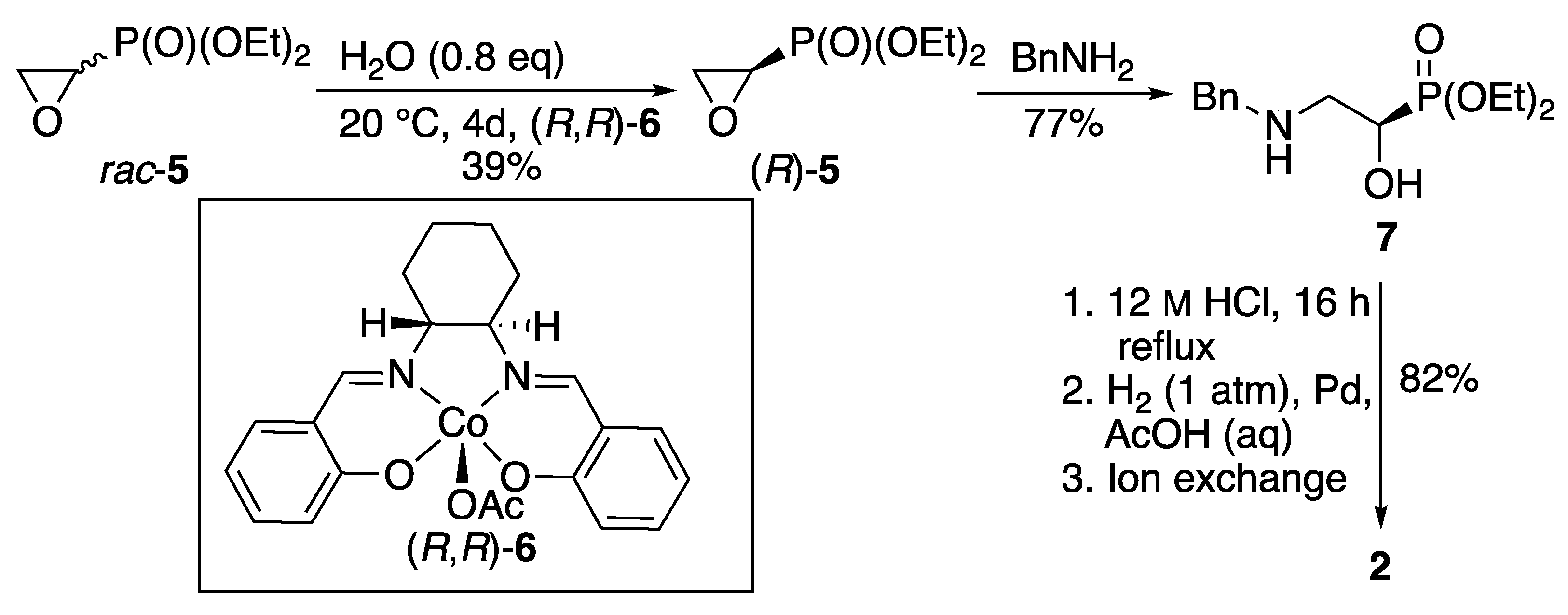
Figure 1.
Aminophosphonic acids 8 - 11 that have been characterized by X-ray crystallography. .
Figure 2.
Ball and stick representation of the X-ray crystal structure of the acid 2 showing the atom numbering scheme.
Figure 2.
Ball and stick representation of the X-ray crystal structure of the acid 2 showing the atom numbering scheme.
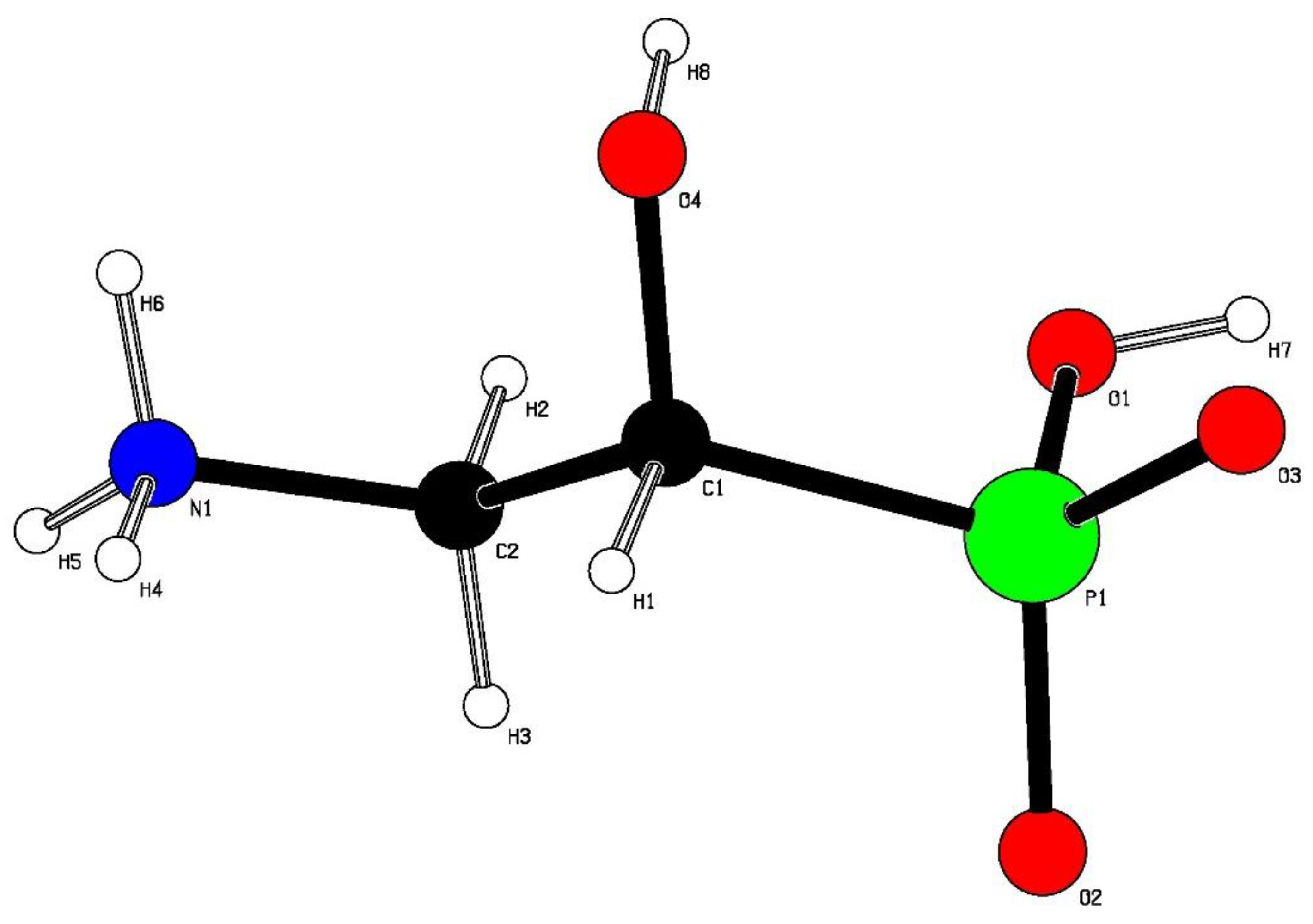
Figure 3.
Ball and stick representation showing hydrogen bonding (dashed lines) within the crystal structure of acid 2. Key: C black, H white, N blue, O red, P green.
Figure 3.
Ball and stick representation showing hydrogen bonding (dashed lines) within the crystal structure of acid 2. Key: C black, H white, N blue, O red, P green.
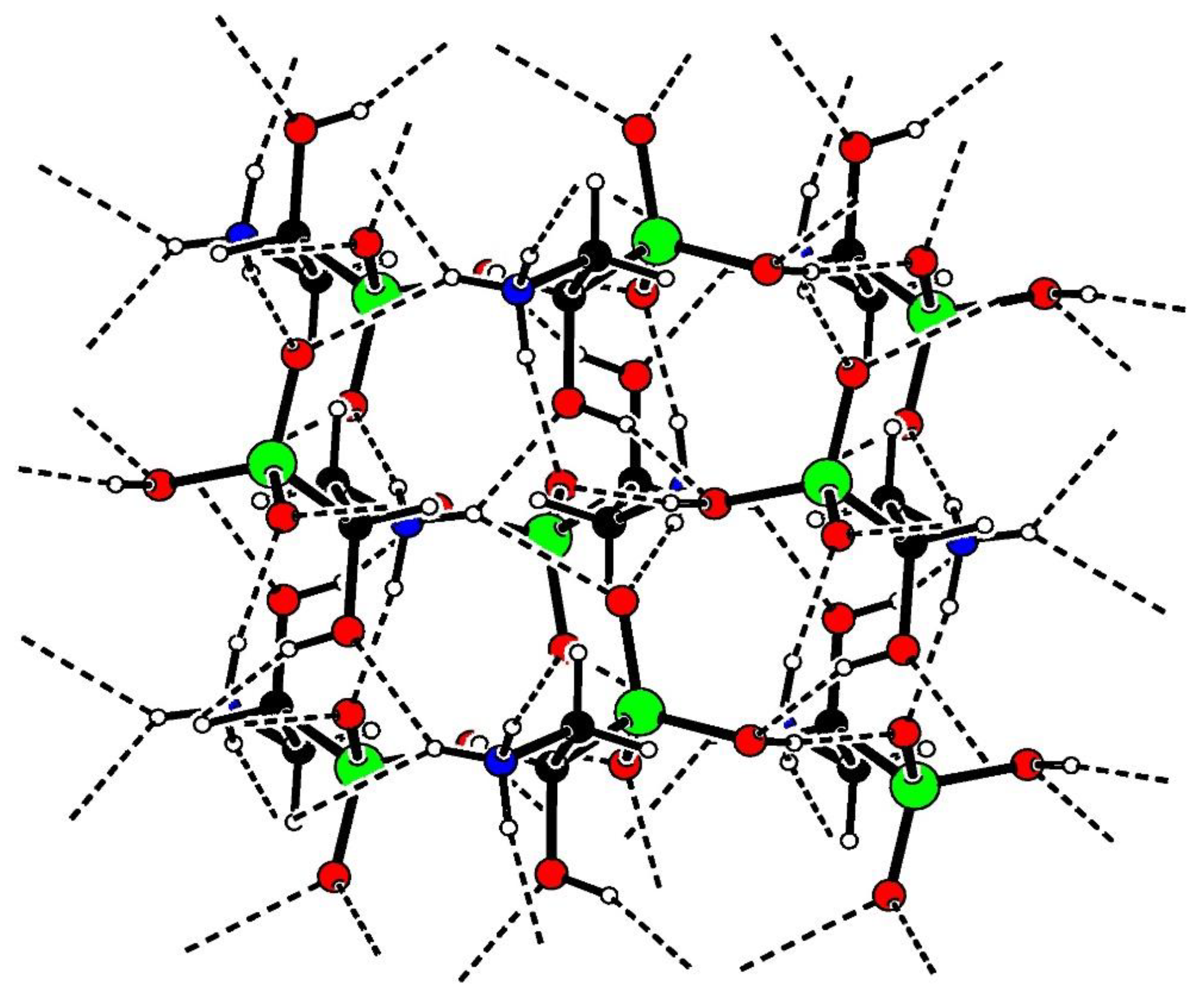

Table 1.
Hydrogen bond distances (Å) and angles (°) for acid 2.
| Donor-H | Acceptor-H | Donor-Acceptor | Angle | |
|---|---|---|---|---|
| N1-H4...O2 | 0.91 | 2.23 | 2.951(2) | 135.5 |
| N1-H4...O4 | 0.91 | 2.20 | 2.945(1) | 139.1 |
| N1-H5...O2 | 0.91 | 1.84 | 2.730(1) | 165.6 |
| N1-H6...O3 | 0.91 | 1.90 | 2.795(1) | 165.9 |
| O1-H7...O3 | 0.84 | 1.69 | 2.503(1) | 163.6 |
| O4-H8...O1 | 0.84 | 2.12 | 2.880(1) | 150.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



