Submitted:
26 August 2024
Posted:
27 August 2024
You are already at the latest version
Abstract
PARP inhibitors are used in tumor therapy exploiting their inhibitory role in DNA repair. Radiation therapy leads to DNA damage in the affected region comprising both tumor tissue and adjacent normal tissue. In addition, radiotherapy has long-standing effects on DNA integrity. The mechanisms for this are not well understood. We studied normal tissue damage of the lung after application of ionizing radiation (IR) in two well established rat models comprising both external (eIR) and internal (iIR) ionizing radiation. We found a nearly complete (~350-fold) loss of PARP-1 in the irradiated tissue in both models but no change in PARylated protein. This may be explained by the strong decrease in both ARH3 and PARG, both deparylating enzymes. We demonstrate, that of all these proteins are expressed in type II pneumocytes, but also in type I pneumocytes and the bronchiolar epithelium. The data suggest a severe disturbance of PARylation in these cells which may affect their major functions such as maintenance of the integrity of the alveolar wall. It is likely that the application of PARP inhibitors could have cumulative effects in radiation-damaged normal tissue.
Keywords:
PARP1
; PARP2
; ARH3
; PARG
; TARG1
; PARylation
; radiation damage
1. Introduction
The addition of poly-ADP-ribose molecules to proteins (PARylation) is a post-translational modification in response to DNA damage [1]. PARylation is carried out by members of the PAR polymerase (PARP) enzyme family [2]. Removal of ADP-ribose (ADPR) from PARylated proteins is carried out by enzymes such as PAR glycohydrolase (PARG) [3], ADP ribosylhydrolase 3 (ARH3) [4,5] as well as the macro domain (MacroD) containing terminal ADPR protein glycohydrolase 1 (TARG1) [6].
PARylation plays a role in several DNA repair pathways, possibly by providing a landing point or scaffold for multiple DNA repair factors [7]. PARP-1 binds to DNA via its zinc-finger domain [7]. During double strand break repair, PARP-1 is involved in both homologous recombination repair (HR) repair and in non-homologous end-joining (NHEJ), the latter both in classical and alternative pathways [1]. After single strand breaks, PARP-1 is involved in base excision repair (BER), both in short patch repair and in long patch repair. During short-patch repair, PARP-1 binds to the BRCT (BRCA1 C-terminal) domain of XRCC1 (X-ray repair cross-complementing 1) protein [7]. Via a second BRCT domain, XRCC1 protein is also bound to DNA ligase III [7].
PARylation is also a central factor in a special form of cell death named parthanatos [8]. During parthanatos, an early transient hyperactivation of PARylation occurs [9], which leads to NAD+ and ADP depletion and collapse of the mitochondrial membrane potential. After that, PARP-1 is cleaved by caspases which occurs immediately after caspase activation [10]. Caspase cleavage leads to 24- and 89 kDa fragments of PARP-1 [11]. The 24 kDa fragment remains bound to DNA lesions whereas the 89 kDa fragment is attached to PAR and – via PAR – to apoptosis inducing factor (AIF) [11]. In addition to the functions described, PARP-1 can also regulate inflammatory mediators, cellular energetics, gene transcription, mitosis and cellular signaling such as sex hormone and ERK (extracellular signal regulated kinase) signaling [12].
Due to their role in DNA repair, PARP inhibitors are successfully used in tumor therapy, e.g. in the treatment of breast, ovarian, pancreatic and prostate cancers [13]. However, a synthetically lethal interaction with the mutation in another DNA repair protein is required, namely mutations in BRCA1/2 (breast cancer growth suppressor protein 1 or 2) which are required for HR [13]. PARP inhibitors exert their action by PARP trapping at sites of DNA damage with subsequent collapse of the replication forks, disrupted processing of Okazaki fragments, up-regulation of p53 but also increased replication fork speed [13]. Due to the biologic synergy of PARPi and IR, PARPi act as radiosensitizers [14,15].
IR induces and double strand breaks and PARP family members are among the first proteins arriving at the site of damage by binding to DNA via its zinc finger domain [16]. By acting as a scaffold, PARP attracts other DNA repair proteins. In addition, a number of immediate-early genes are induced as a stress response [17,18,19]. These processes occur during tumor therapy both in tumor and in normal tissue. The affected tissue displays a sterile inflammation which – e.g. in the lung - is called radiation pneumonitis (RP) [20]. After the acute damage has been repaired, there is usually a gap period where nearly no macroscopic and microscopic changes occur in the tissue. This gap period can take several weeks to years. Then, the phase of chronic radiation damage starts involving destruction of organ-typic cells, endothelial cells and formation of scar tissue. In the lung, this process is called fibrosing alveolitis (FA) with the final result being pulmonary fibrosis (PF), also called radiation fibrosis (RF) [21]. Several inflammatory mediators, cytokines and growth factors are involved in the lung injury, such as TNFalpha [22], TGFbeta1 [23,24,25]. Impaired regeneration due to growth factor deficiency has been attempted to be treated with the administration of exogenous growth factors, such as KGF (keratinocyte growth factor) [26]. Although the gap period shows little changes in lung architecture, signaling pathways seem to deteriorate. In a well-characterized model of IR induced FA, we found a continuous increase in NF-kappaB DNA binding activity reaching its highest level at 12 weeks after IR. When fibrosing alveolitis starts, which takes place around 2 months after irradiation, NF-kappaB activity is sub-maximal [27]. In contrast, the transcription factors Sp1 and AP-1 lose their DNA binding activity after IR [28,29]. At 4.5 weeks after IR, AP-1 starts to lose its DNA binding activity which is completely lost at 5.5 weeks after IR. Thus, 8 weeks after IR, with early FA and submaximal NF-kappaB activity and 12 weeks (3 months) after IR when NF-kappaB levels reach its maximum are critical time points to study molecules that might influence the process of FA.
As mentioned earlier, radiosensitization can be exerted by PARP-1 inhibitors. In this study, we wanted to find out, whether IR may sensitize normal tissue to PARP-inhibition. In the above-mentioned rat model of IR, we found a nearly complete loss of PARP-1 in radiation-damaged lung tissue, a condition that may aggravate tissue damage caused by PARP inhibitors.
2. Materials and Methods
Animal models. For the external irradiation (eIR), the right lungs of female Fischer rats were irradiated with a single dose of 20 Gy IR as described [29]. These animal experiments have been approved by the animal ethics committee of the State of Saxony (approval number AZ 24-9168.11-1/2014-1). For internal irradiation (iIR), 188Re-microspheres were injected intravenously into adult Wistar rats as described [30]. These animal experiments were approved by the Gouvernment of Saxony (Approval No. AZ 24-9168.11-1-2004-8). In these experiments, doses up to 54 Gy were applied. Tissue was fixed in 4 % neutrally buffered formaldehyde overnight and subsequently embedded in paraffin or was snap frozen and stored at -80 °C.
Western blotting (WB). Nuclear and crude cytoplasmic protein extracts were prepared as described [31]. Membrane and organelle (M/O) fractions were prepared by pelleting the crude cytoplasmic fraction at 20,817 x g (14000 rpm) and resuspending the pellet in nuclear extraction buffer. Proteins were separated with denaturing polyacrylamide gel electrophoresis [32] and blotted using a semi-dry apparatus (TE77XP, Hoefer, Bridgewater, USA) onto a polyvinylidene difluoride (PVDF) membrane. Primary antibodies and their dilutions are listed in the supplementary informations (Table S1). Secondary antibodies were labeled with horseradish peroxidase (HRP) at a 10-fold dilution compared to the primary antibodies. Luminescence was induced by incubation with Super Signal West Femto substrate (Thermo Fisher Scientific, Erlangen, Germany). Signals were detected on a ChemoCam Imager (INTAS, Göttingen, Germany). Bands were quantified with the programs Labimage 1D (INTAS, Göttingen, Germany) or with ImageJ [33]. Ponceau S stained PVDF membranes served as loading controls. Molecular weights (MW) of fragments were calculated with Microsoft Excel or Labimage 1D.
Immunohistochemistry (IH) and Immunofluorescence (IF) stainings. Both IH and IF stainings were performed on sections from paraffin-embedded material. The dilutions of the primary antibodies are given in Table S1. Horseradish peroxidase (HRP) conjugated secondary antibodies were used at a concentration of about 5 µg/ml. IH was developed with the Vectastain Elite Kit (Biozol, Eching, Germany) and IF was developed with TSA (Tyramide signal ampflification) Superboost 488 or 594 kits (Thermo Fisher Scientific, Erlangen, Germany), both according to the manufacturers instructions. IH pictures were obtained with an Axioskop 2 (Zeiss, Oberkochen, Germany) using the ZEN blue software of the same company. IF pictures were obtained on a Leica SP5 laserscanning microscope (Leica, Wetzlar, Germany).
3. Results
3.1. Down-regulation of PARP1 proteins
In the eIR experiments full-length PARP-1 (FL-PARP-1) could be detected in the membrane/organelle (M/O) fraction and in the nuclear (Nuc) protein fraction of control (C) lungs (Figure 1A). In the cytoplasmic fraction, nearly no FL PARP-1 could be detected. At 2 and 3 months after iIR, also nearly no FL PARP-1 could be detected.
Quantification of the FL PARP-1 bands in the nuclear fraction (Figure 1C and Figure S2) shows that the nuclear FL PARP-1 protein levels at 2 and 3 months after eIR, were only 3.22 and 0.28 % of the control level respectively which was a highly significant difference. In addition to the FL PARP-1, there were also other bands visible in the WB gels of eIR lungs, which presumably represented PARP-1 fragments. In Figure 1A, the major fragments that could be observed in all compartments had apparent MWs of 72 and 55 kDa, the latter being most prominent in the nuclear fraction of control lungs and at 3 months after IR. The 72 kDa band was present all fractions from control lungs as well as in the cytoplasmic and nuclear compartments of the 3 months time point. In order to detect other possible fragments in the nuclei, gel electrophoresis with a wider molecular weight spectrum was performed (Figure 1B). In these gels, many additional bands in the nuclear protein extracts from both control and irradiated lungs down to 17 kDa could be detected. We do not see a 89 kDa fragment. There are bands that have decreased intensity such as bands at 100, 76, 72, 39 and 21 kDa. On the other hand, band at 55, ~42 and 26 kDa are increased in intensity in the irradiated lung. Of note, in this gel, there is a FL PARP-1 band at 2 months after IR which indicates that at this time there is a switching point of PARP-1 integrity. After iIR, which had been applied using different dose ranges, there was a strong decrease of FL PARP-1 at doses of 28 Gy or above pointing at a stronger decrease with stronger IR damage (Figure 1D).
3.2. Levels of PARP-2, PAR, and deparylation enzymes
PARP-2 levels after eIR fluctuated in parallel with the ponceau S stained bands (Figure 2A). The calculations revealed no significant differences in band intensities (Figure 2B and Figure S3). After iIR, there was a tendency of decreased band intensities with higher doses, but no significance in the differences (Figure 2C and Figure S4). PAR levels remained quite constant at the different time points after eIR (Figure 2D). Also in rat lungs after iIR, there was nearly no change in PARylation (Figure 2E).
The deparylation enzyme ARH3 was only detectable in the cytoplasmic fraction of control lungs (Figure 2F). At 2 and 3 months after eIR, there was nearly no ARH3 detectable. Another deparylating enzyme, PARG, was detectable in all cellular fractions in the control lungs (Figure 2F). However, at 2 and 3 months after eIR, nearly no PARG was detectable. The deparylating enzyme TARG1 was also detectable in all 3 cellular fractions (Figure 2G). At 2 months after eIR, the TARG1 level decreased massively in all cellular fractions, but at 3 months after IR, TARG1 levels were restored (3 months) or even exceeded control values.
3.3. Cellular distribution of PARylation related proteins
All PARylation-related proteins included in this study including the PARylation itself could be found in both type I and type II pneumocytes as well as in the bronchiolar epithelium, with the exception of TARG1 that could not be seen in type I pneumocytes (Table 1). All these cell types together represented the lung-specific epithelium. Alveolar macrophages, members of the hematopoietic lineage, mostly showed a strong staining with PARP-1. Concerning endothelial cells, PARG was detectable in capillary endothelial cells but not the other proteins studied. In contrast, all proteins studied except ARH3 could be found in endothelial cells of arterioles and venules. In the connective tissue, all proteins studied, except ARH3, could be found. In smooth muscle cells around bronchioles PARP-2 was stained but not PARP-1, ARH3 and TARG. Mast cells contained no PARP-1, PARP-2 and PARylated proteins.
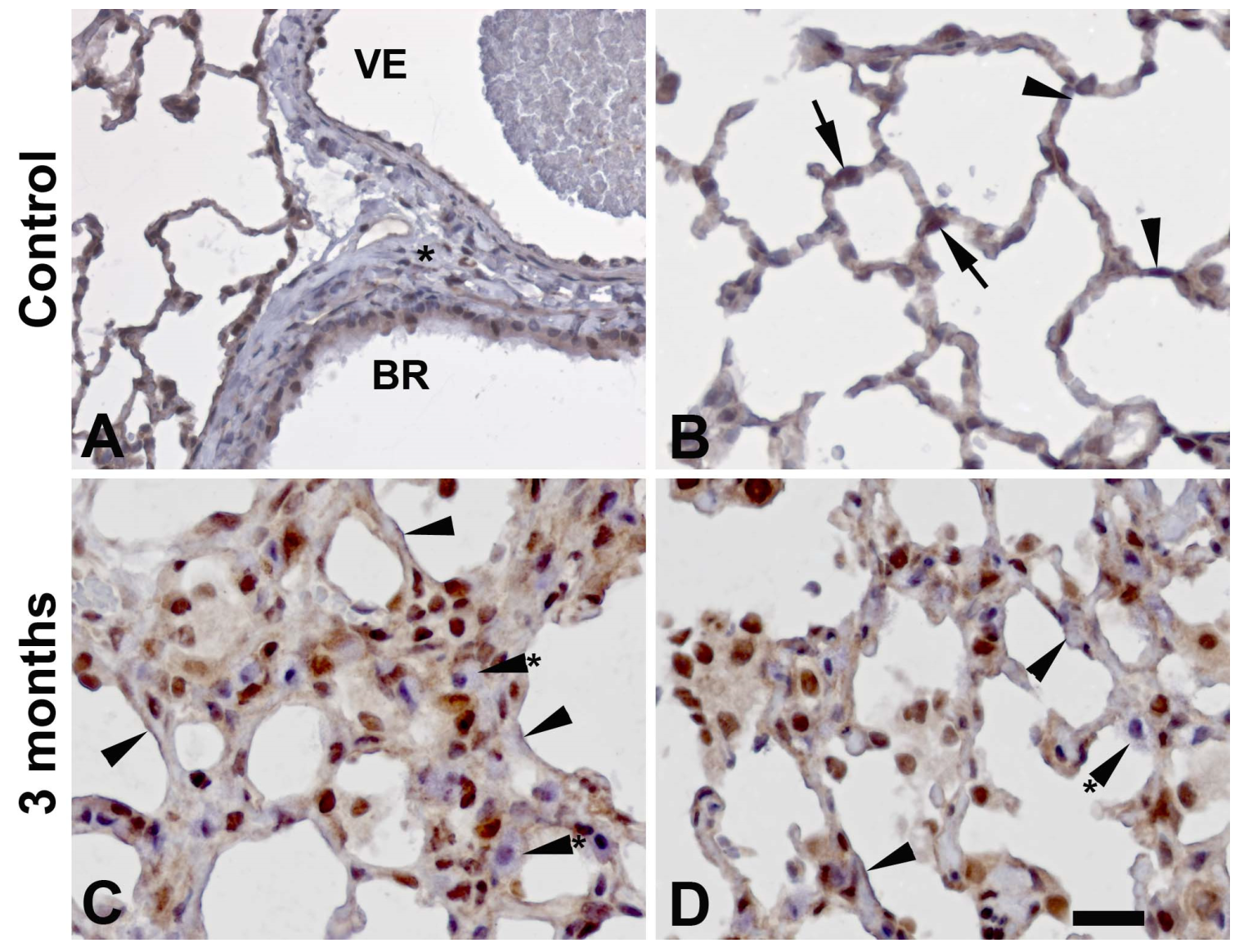
In Figure 3A, PARP-1 positive bronchial epithelial (BR) cells can be seen, as well as a weak positivity in the endothelium of a venule (VE). Examples of PARP-1 positive type II pneumocytes (arrows) are demonstrated in Figure 3B. Arrowheads point at type I pneumocytes which are slightly positive, also in the irradiated lung (Figure 3C and 3D). Mast cells were negative for PARP-1 (Figure 3C,D, arrowheads with asterix). Moderate to strong positivity of PARP-2 could be demonstrated in bronchial epithelial cells lining a bronchiolus (BR, Figure 4A), whereas endothelial cells, in this case lining a venule (VE) display a very weak staining.Positivity of PARP-2 in type II pneumocytes (arrows) and type I pneumocytes (arrowheads) are shown in Figure 4B. In the irradiated lung, alveolar macrophages were increased in number, and they were positive for PARP-2 (Figure 4C,D, arrowheads with asterix).
Positivity of type I pneumocytes (arrwoheads) could also be demonstrated in these two figures. The nature of the very strongly stained cells in Figure 4C,D could not be determined.Immunohistochemistry Figure 5A-F very clearly demonstrate PARylated proteins in type I pneumocytes (arrowheads) and also type II pneumocytes (arrow). In addition, bronchiolar epithelial cells lining a bronchiole (BR) are positive for PARylated proteins (Figure 5C). Endothelial cells lining a venule (VE) were clearly positive for PARylated proteins (Figure 5D). Macrophages were mostly strongly stained for PARylation (Figure 5E,F, arrowheads with asterix).
In order to prove these descriptions, immunofluorescence double stainings were performed. Pro-surfactant protein C, a marker for type II pneumocytes, could be found in the cytoplasm of cells that contain PARP-1 in the nuclei (arrow), confirming the positivity of type II pneumocytes for PARP-1 (Figure 6A-D). However, also PARP-1 negative pneumocytes of type II could be found (arrowhead in the same figures). PARylation could be demonstrated in some but not many type II pneumocytes (Figure 6D-F, arrows). The cell in the center indicated with an arrow demonstrates PARylation at the surface. The PARylation postitivity of type I pneumocytes can be demonstrated by a co-staining with aquaporin 5 (Aqp5) which is a marker for type I pneumocytes (Figure 7A-C). However, the staining of both targets was mutually exclusive as seen by an alternative staining pattern changing from PAR staining (arrowheads) to Aqp5 staining (arrowheads with asterix). A marker for macrophages, CD68, demonstrated that many macrophages displayed only a very weak positivity or a negativity for PAR (Figure 7D-F). The macrophage indicated with an arrow had very low cytoplasmic positivity for PAR whereas the macrophage labelled with an arrowhead was negative for PAR or showed a slight staining of its cell membrane.
4. Discussion
A Due to the important role of PARP-1 in DNA repair, PARP-1 inhibitors are used therapeutically in malignant tumors [14]. A combination of both PARP-1 inhibition and IR renders tumor cells more susceptible to cell death since unrepaired SSB are converted to DSB when repair is inhibited, e.g. by mutations in BRCA proteins [14]. In addition, BRCA proteins are down-regulated after a combination of PARP-1 inhibition with Olaparib and radiation [34]. Yet, PARP-1 inhibition works best in BRCA deficient tumors providing also a selectivity of cell damage in tumors compared to normal tissue. In this study, we asked whether there may be sensitization of irradiated tissue to PARP-1 inhibition in the long run. We used two well-characterized rat models of radiation-induced lung damage [29,30], namely eIR and iIR. In both models, there was a strong decrease in FL PARP-1 protein levels after 2 and 3 months (eIR) or about 2.5 months (iIR). The effect was especially strong after eIR reaching a decrease to 0.28 % of the control level corresponding to a more than 350-fold decrease in FL PARP-1 (Figure 1). In contrast, PARP-2 levels remained quite constant after eIR and decreased slightly but not significantly after iIR (Figure 2A,C) which suggests that PARP-2, a protein similar in function and sequence [2], is unlikely to compensate for the loss of PARP-1 function. However, PARylation remains constant in all cellular fractions and in both models (Figure 2D,E). These PAR results were in contrast to the expectations. Therefore, we also analyzed the levels of deparylation enzymes. Both ARH3 and PARG are strongly down-regulated in the lung 2 and 3 months after eIR (Figure 2F) whereas TARG1 is transiently but strongly down-regulated at 2 months after IR (Figure 2G). Thus, both PARylation and de-PARylation enzymes are down-regulated and thereby explain why total PARylation remains constant. This also means that PARylation looses much of its flexibility. Cellular processes like DNA repair require a quick flexibility to direct PARylation to sites of DNA damage. These data suggest that this “frozen” state of PARylation leads to a severe disturbance of DNA repair, but also to the other functions of PARP mentioned above including inflammation, cellular energetics and mitosis [12]. Therefore, these data predict the accumulation of DNA damage also months after IR. Since the proteins studied are present in all indigenous lung cells, namely type I and type II pneumocytes and bronchiolar epithelial cells, the data suggest that this disturbance of PARylation takes place in all these cells.
Western blotting revealed that not only FL PARP-1, but also a number of fragments are present in the cells (Figure 1B). It had been shown that some of these fragments occur under specific conditions and have distinct functions. 85 and 24/25 kDa fragments have been detected in various types of cell death [35], the latter obviously corresponding to the 26 kDa fragments in our study. During apoptosis, PARP is cleaved by caspase 3 directly [9]. A 24 kDA N-terminal fragment has been described to bind tightly to DNA, and it has been supposed that this would impair DNA repair [36]. Indeed, it has been shown that this N-terminal fragment strongly inhibits PARP-1 activity [36,37]. In our study, this ~24-26 kDa fragment is increased at 3 months after eIR (Figure 1B) which is likely to result in even more reduction in PARP-1 activity (and in more reduction in DNA repair). PARP-1 can also be cleaved during necrosis, producing a 50 kDa fragment by the activity of cathepsins [38]. We see a ~55 kDa band which may correspond to the 50 kDa fragment mentioned which is somewhat increased at 3 months after IR (Figure 1B). Thus, the increased ~55 kDa band 3 months after IR may be caused by an increased activity of cathepsins.
Taken together, we demonstrate a strong decrease in PARylation and dePARylation enzymes in the rat lung at 2 and 3 months after irradiation, a condition which should correspond to a severe disturbance in all PARylation dependent processes, most importantly DNA repair. Thus, radiation damage of normal tissue may interfere with side effects of therapies with PARP-1 inhibitors.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Table S1: Antibodies used for the study; Table S2. Quantification of PARP-1 band staining intensity of rat lung western blots after eIR; Table S3: Quantification of PARP-2 band staining intensity of rat lung western blots after eIR; Table S4: Quantification of PARP-2 band staining intensity of western blots of rat lung after iIR; Figure S1. Immunofluorescence double-labelling of rat lungs at 3 months after irradiation with PARP-2 (A, C, D, F, G, I) and CD68 (B, C, E, F, H, I).
Author Contributions
Felicitas Mayer performed experiments, prepared figures and was involved in writing the manuscript. Ivonne Kemnitz performed experiments. Peter Geyer was involved in animal experiments with eIR. Jörg Kotzerke was involved in animal experiments with iIR. Guido Fitze was involved in designing the study and in Manuscript preparation. Michael Haase designed and supervised the study and wrote the manuscript.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, MU 1299/1-1) and the BMBF (01ZZ9604, 03ZIK041) as well as a grant from the Faculty of Medicine of the Dresden University of Technology to MGH.
Institutional Review Board Statement
The animal experiments have been approved by the animal ethics committee of the State of Saxony (approval numbers AZ 24-9168.11-1/2014-1 and. AZ 24-9168.11-1-2004-8).
Informed Consent Statement
Not applicable
Acknowledgments
The authors thank Anke Klawitter for excellent technical assistance. We also thank Diana Faulhaber and Roland Jung for their help in the iIR experiments. We thank Birgit Henzel for her help with eIR.
Conflicts of Interest
The authors declare no conflicts of interest
References
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Ame, J.C.; Heberle, E.; Camuzeaux, B.; Dantzer, F.; Schreiber, V. Purification of Recombinant Human PARG and Activity Assays. Methods Mol. Biol. 2017, 1608, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine ADP-ribosylation reversal by the hydrolase ARH3. Elife 2017, 6. [Google Scholar] [CrossRef]
- Abplanalp, J.; Leutert, M.; Frugier, E.; Nowak, K.; Feurer, R.; Kato, J.; Kistemaker, H.V.A.; Filippov, D.V.; Moss, J.; Caflisch, A.; et al. Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase. Nat. Commun. 2017, 8, 2055. [Google Scholar] [CrossRef]
- Žaja, R.; Aydin, G.; Lippok, B.E.; Feederle, R.; Lüscher, B.; Feijs, K.L.H. Comparative analysis of MACROD1, MACROD2 and TARG1 expression, localisation and interactome. Sci. Rep. 2020, 10, 8286. [Google Scholar] [CrossRef]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell Biol. 1998, 18, 3563–3571. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Mayer, F.; Kemnitz, I.; Fitze, G.; Haase, M.G. Dynamics of caspase activation upon UV induced genotoxic injury. International Journal of Radiation Biology 2020, 1–7. [Google Scholar] [CrossRef]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Yamada, S.; Negi, S.; Bu, X.; Kato, J.; et al. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef]
- Weaver, A.N.; Yang, E.S. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol. 2013, 3, 290. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O'Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Angel, M.; Zarba, M.; Sade, J.P. PARP inhibitors as a radiosensitizer: a future promising approach in prostate cancer? Ecancermedicalscience 2021, 15, ed118. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Chu, A.; Song, R.; Liu, S.; Chai, T.; Wang, X.; Liu, Z. PARP inhibitors combined with radiotherapy: are we ready? Front. Pharmacol. 2023, 14, 1234973. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair (Amst) 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Usenius, T.; Tenhunen, M.; Koistinaho, J. Ionizing radiation induces expression of immediate early genes in the rat brain. Neuroreport 1996, 7, 2559–2563. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Hallahan, D.; Fuks, Z.; Kufe, D. Radiation induction of immediate early genes: effectors of the radiation-stress response. Int. J. Radiat. Oncol. Biol. Phys. 1994, 30, 229–234. [Google Scholar] [CrossRef]
- Hagan, M.; Wang, L.; Hanley, J.R.; Park, J.S.; Dent, P. Ionizing radiation-induced mitogen-activated protein (MAP) kinase activation in DU145 prostate carcinoma cells: MAP kinase inhibition enhances radiation-induced cell killing and G2/M-phase arrest. Radiat. Res. 2000, 153, 371–383. [Google Scholar] [CrossRef]
- Tucker, S.L.; Travis, E.L. Time course for the hazard of radiation-induced pneumonitis death in mice. Int. J. Radiat. Biol. 1992, 62, 627–639. [Google Scholar] [CrossRef]
- Arroyo-Hernández, M.; Maldonado, F.; Lozano-Ruiz, F.; Muñoz-Montaño, W.; Nuñez-Baez, M.; Arrieta, O. Radiation-induced lung injury: current evidence. BMC Pulm. Med. 2021, 21, 9. [Google Scholar] [CrossRef]
- Przybyszewska, M.; Miloszewska, J.; Rzonca, S.; Trembacz, H.; Pysniak, K.; Kotlarz, A.; Swoboda, P.; Zalewska, M.; Malecki, M. Soluble TNF-alpha receptor I encoded on plasmid vector and its application in experimental gene therapy of radiation-induced lung fibrosis. Archivum Immunologiae et Therapiae Experimentalis (Warszawa) 2011, 59, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sheldon, K.; Chen, M.; Yin, M.S.; Hayman, J.A.; Kalemkerian, G.P.; Arenberg, D.; Lyons, S.E.; Curtis, J.L.; Davis, M.; et al. The predictive role of plasma TGF-beta1 during radiation therapy for radiation-induced lung toxicity deserves further study in patients with non-small cell lung cancer. Lung Cancer 2008, 59, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Rube, C.E.; Uthe, D.; Schmid, K.W.; Richter, K.D.; Wessel, J.; Schuck, A.; Willich, N.; Rube, C. Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2000, 47, 1033–1042. [Google Scholar] [CrossRef]
- Martin, M.; Lefaix, J.; Delanian, S. TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int. J. Radiat. Oncol. Biol. Phys. 2000, 47, 277–290. [Google Scholar] [CrossRef]
- Yi, E.S.; Williams, S.T.; Lee, H.; Malicki, D.M.; Chin, E.M.; Yin, S.; Tarpley, J.; Ulich, T.R. Keratinocyte growth factor ameliorates radiation- and bleomycin-induced lung injury and mortality. Am. J. Pathol. 1996, 149, 1963–1970. [Google Scholar]
- Haase, M.G.; Klawitter, A.; Geyer, P.; Alheit, H.; Baumann, M.; Kriegel, T.M.; Kasper, M.; Baretton, G.B. Sustained elevation of NF-kappaB DNA binding activity in radiation-induced lung damage in rats. Int. J. Radiat. Biol. 2003, 79, 863–877. [Google Scholar] [CrossRef]
- Haase, M.; Geyer, P.; Appold, S.; Schuh, D.; Kasper, M.; Muller, M. Down-regulation of Sp1 DNA binding activity in the process of radiation-induced pulmonary fibrosis. Int. J. Radiat. Biol. 2000, 76, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Haase, M.G.; Klawitter, A.; Bierhaus, A.; Yokoyama, K.K.; Kasper, M.; Geyer, P.; Baumann, M.; Baretton, G.B. Inactivation of AP1 proteins by a nuclear serine protease precedes the onset of radiation-induced fibrosing alveolitis. Radiat. Res. 2008, 169, 531–542. [Google Scholar] [CrossRef]
- Liepe, K.; Faulhaber, D.; Wunderlich, G.; Andreeff, M.; Haase, M.; Jung, R.; Oehme, L.; Dorr, W.; Kotzerke, J. Radiation pneumopathy in the rat after intravenous application of (188)Re-labeled microspheres. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 529–536. [Google Scholar] [CrossRef]
- Schreiber, E.; Matthias, P.; Muller, M.M.; Schaffner, W. Rapid detection of octamer binding proteins with 'mini-extracts', prepared from a small number of cells. Nucleic Acids Res. 1989, 17, 6419. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Molecular Cloning. A Laboratory Manual., 4 ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, 2012. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nature Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bourton, E.C.; Ahorner, P.A.; Plowman, P.N.; Zahir, S.A.; Al-Ali, H.; Parris, C.N. The PARP-1 inhibitor Olaparib suppresses BRCA1 protein levels, increases apoptosis and causes radiation hypersensitivity in BRCA1(+/-) lymphoblastoid cells. J. Cancer 2017, 8, 4048–4056. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar] [PubMed]
- Smulson, M.E.; Pang, D.; Jung, M.; Dimtchev, A.; Chasovskikh, S.; Spoonde, A.; Simbulan-Rosenthal, C.; Rosenthal, D.; Yakovlev, A.; Dritschilo, A. Irreversible binding of poly(ADP)ribose polymerase cleavage product to DNA ends revealed by atomic force microscopy: possible role in apoptosis. Cancer Res. 1998, 58, 3495–3498. [Google Scholar]
- D'Amours, D.; Sallmann, F.R.; Dixit, V.M.; Poirier, G.G. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J. Cell Sci. 2001, 114, 3771–3778. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.; Boucher, C.C.; Nadeau, D.; Poirier, G.G. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 2001, 8, 588–594. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Western blots showing loss of PARP-1 in lung after eIR and iIR. A: Representative experiment showing the expression of full length (FL) PARP-1 protein as well as some of its fragments in different cellular compartments of the lung after external irradiation (eIR) at 2 and 3 months after IR (M/O, membrane/organelles; C, cytoplasm; N, nucleus). B: Blot of nuclear PARP-1 showing fragments down to 15 kDa size in lung tissue after eIR. C: Quantification of nuclear PARP-1 of eight independent experiments of eIR. D: Repesentative western blot showing loss of PARP-1-FL after iIR at or above doses of 28 Gy.
Figure 1.
Western blots showing loss of PARP-1 in lung after eIR and iIR. A: Representative experiment showing the expression of full length (FL) PARP-1 protein as well as some of its fragments in different cellular compartments of the lung after external irradiation (eIR) at 2 and 3 months after IR (M/O, membrane/organelles; C, cytoplasm; N, nucleus). B: Blot of nuclear PARP-1 showing fragments down to 15 kDa size in lung tissue after eIR. C: Quantification of nuclear PARP-1 of eight independent experiments of eIR. D: Repesentative western blot showing loss of PARP-1-FL after iIR at or above doses of 28 Gy.
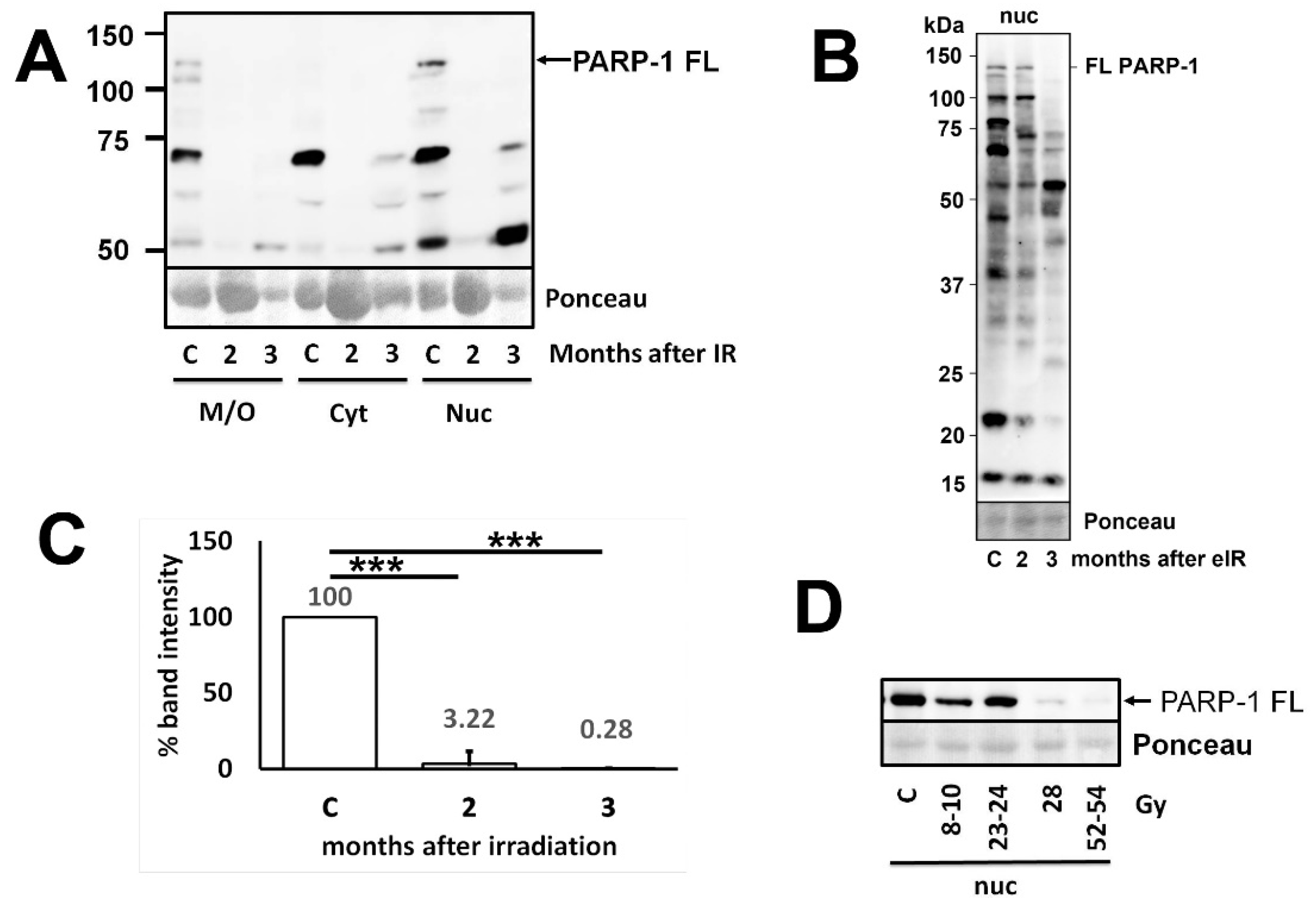
Figure 2.
Western blots of lung tissue showing PARP-2 and PAR as well as different deparylating enzymes after IR. A: Expression of PARP2 protein in different cellular fractions of the lung after eIR together with the quantification diagram (B) demonstrating no significant (n.s.) changes. C: Slight but not significant reduction of PARP2 after iIR. D, E: No major changes in PARylated protein both after eIR (D) and iIR (E). F: Strong reduction in the deparylating enzymes ARH3 and PARG after eIR. G: Transient reduction of the deparylating enzyme TARG1 after eIR.
Figure 2.
Western blots of lung tissue showing PARP-2 and PAR as well as different deparylating enzymes after IR. A: Expression of PARP2 protein in different cellular fractions of the lung after eIR together with the quantification diagram (B) demonstrating no significant (n.s.) changes. C: Slight but not significant reduction of PARP2 after iIR. D, E: No major changes in PARylated protein both after eIR (D) and iIR (E). F: Strong reduction in the deparylating enzymes ARH3 and PARG after eIR. G: Transient reduction of the deparylating enzyme TARG1 after eIR.
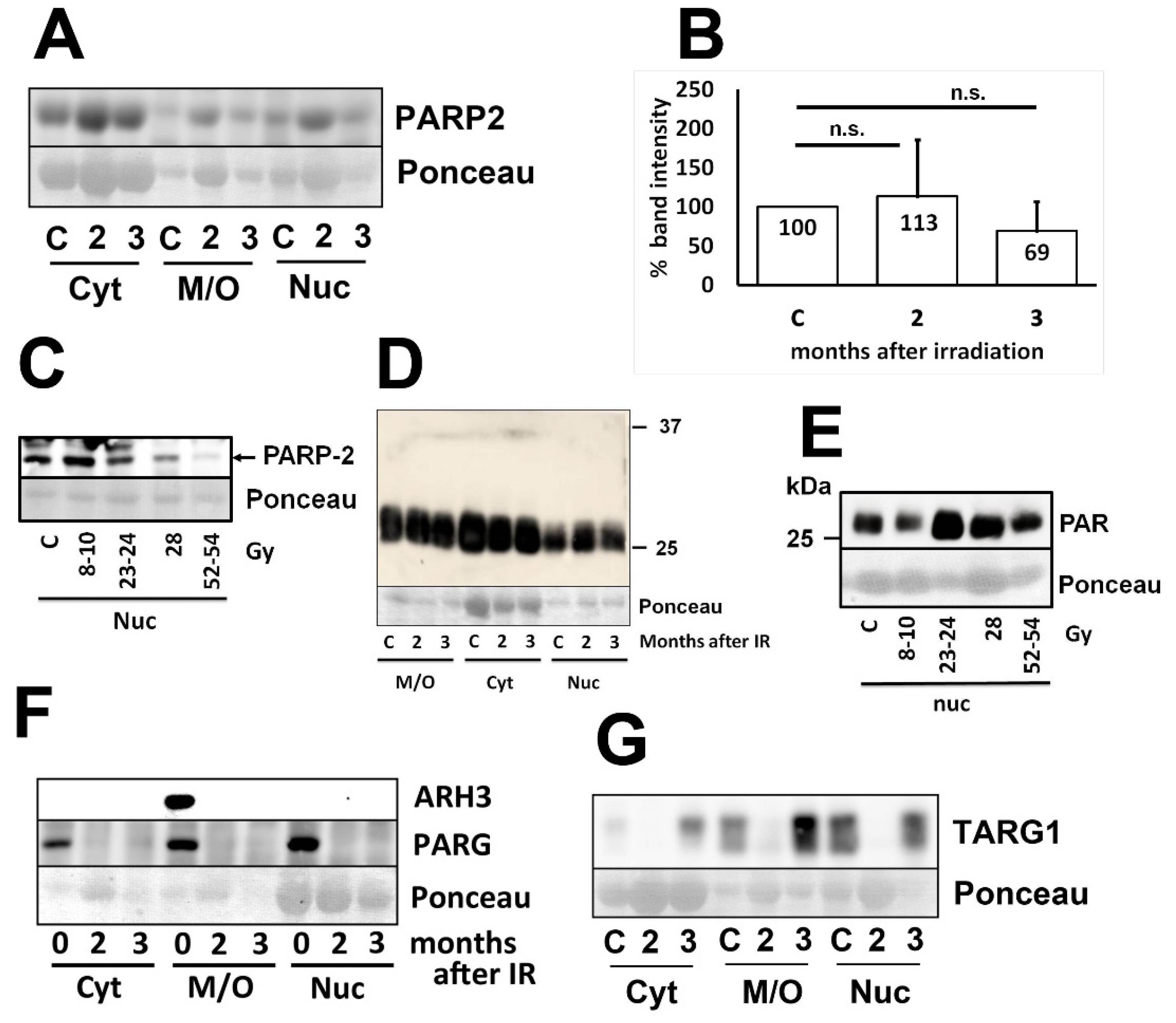
Figure 3.
Immunohistochemistry stainings demonstrating the distribution of PARP-1 protein in the control rat lung (A, B) and in rat lung 3 months after eIR (C, D). Staining is predominantly nuclear, especially in the irradiated lungs. Type II pneumocytes display strong to very strong staining (arrows) whereas staining in type I pneumocytes (arrwoheads) is weak to moderate. Mast cells (arrowheads with asterix) are usually negative. Venules (VE) show strong or very strong staining (cells are very flat), whereas bronchioles show a weak to moderate staining intensity. In the connective surrounding vessels or bronchioles, only few cells are weakly positive. Bar is 20 µm in A, B and 10 µm in C, D.
Figure 3.
Immunohistochemistry stainings demonstrating the distribution of PARP-1 protein in the control rat lung (A, B) and in rat lung 3 months after eIR (C, D). Staining is predominantly nuclear, especially in the irradiated lungs. Type II pneumocytes display strong to very strong staining (arrows) whereas staining in type I pneumocytes (arrwoheads) is weak to moderate. Mast cells (arrowheads with asterix) are usually negative. Venules (VE) show strong or very strong staining (cells are very flat), whereas bronchioles show a weak to moderate staining intensity. In the connective surrounding vessels or bronchioles, only few cells are weakly positive. Bar is 20 µm in A, B and 10 µm in C, D.
Figure 4.
Immunohistochemistry stainings demonstrating the distribution of PARP-2 protein in the control rat lung (A, B) and in rat lung 3 months after eIR (C, D). Arrows point at positive type II pneumocytes and arrowheads to type I pneumocytes. Arrowheads with asterix point at alveolar macrophages which are moderately to strongly stained. Bronchiolar epithelium (BR) is also moderately to strongly stained whereas endothelial cells of venules (VE) are weakly stained. Bar is 20 µm in A, B and 10 µm in C, D.
Figure 4.
Immunohistochemistry stainings demonstrating the distribution of PARP-2 protein in the control rat lung (A, B) and in rat lung 3 months after eIR (C, D). Arrows point at positive type II pneumocytes and arrowheads to type I pneumocytes. Arrowheads with asterix point at alveolar macrophages which are moderately to strongly stained. Bronchiolar epithelium (BR) is also moderately to strongly stained whereas endothelial cells of venules (VE) are weakly stained. Bar is 20 µm in A, B and 10 µm in C, D.
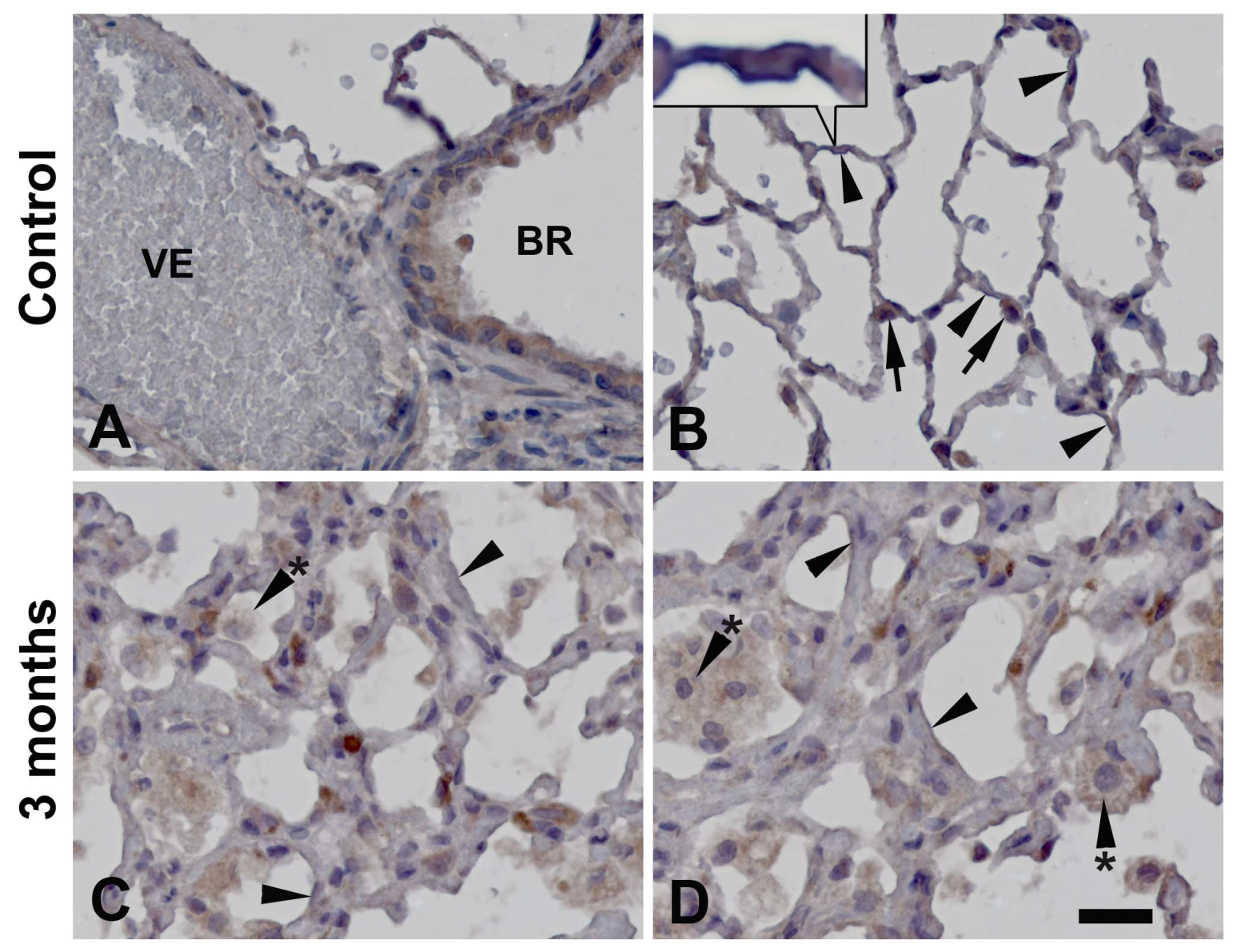
Figure 5.
Immunohistochemistry stainings demonstrating the distribution of PAR in the control rat lung (A, B) and in rat lung 3 months after eIR (C-F). Arrows point at type II pneumocytes, arrowheads at type I pneumocytes. Bronchioli (BR) are moderately to strongly stained. The endothelium of blood vessels such as venules (VE) is also moderately stained. Cells in the alveolar walls containing granular or foamy cytoplasm (arrowheads with asterix) probably represent tissue residing macrophages. Bar is 20 µm in A, B and 10 µm in C-F.
Figure 5.
Immunohistochemistry stainings demonstrating the distribution of PAR in the control rat lung (A, B) and in rat lung 3 months after eIR (C-F). Arrows point at type II pneumocytes, arrowheads at type I pneumocytes. Bronchioli (BR) are moderately to strongly stained. The endothelium of blood vessels such as venules (VE) is also moderately stained. Cells in the alveolar walls containing granular or foamy cytoplasm (arrowheads with asterix) probably represent tissue residing macrophages. Bar is 20 µm in A, B and 10 µm in C-F.
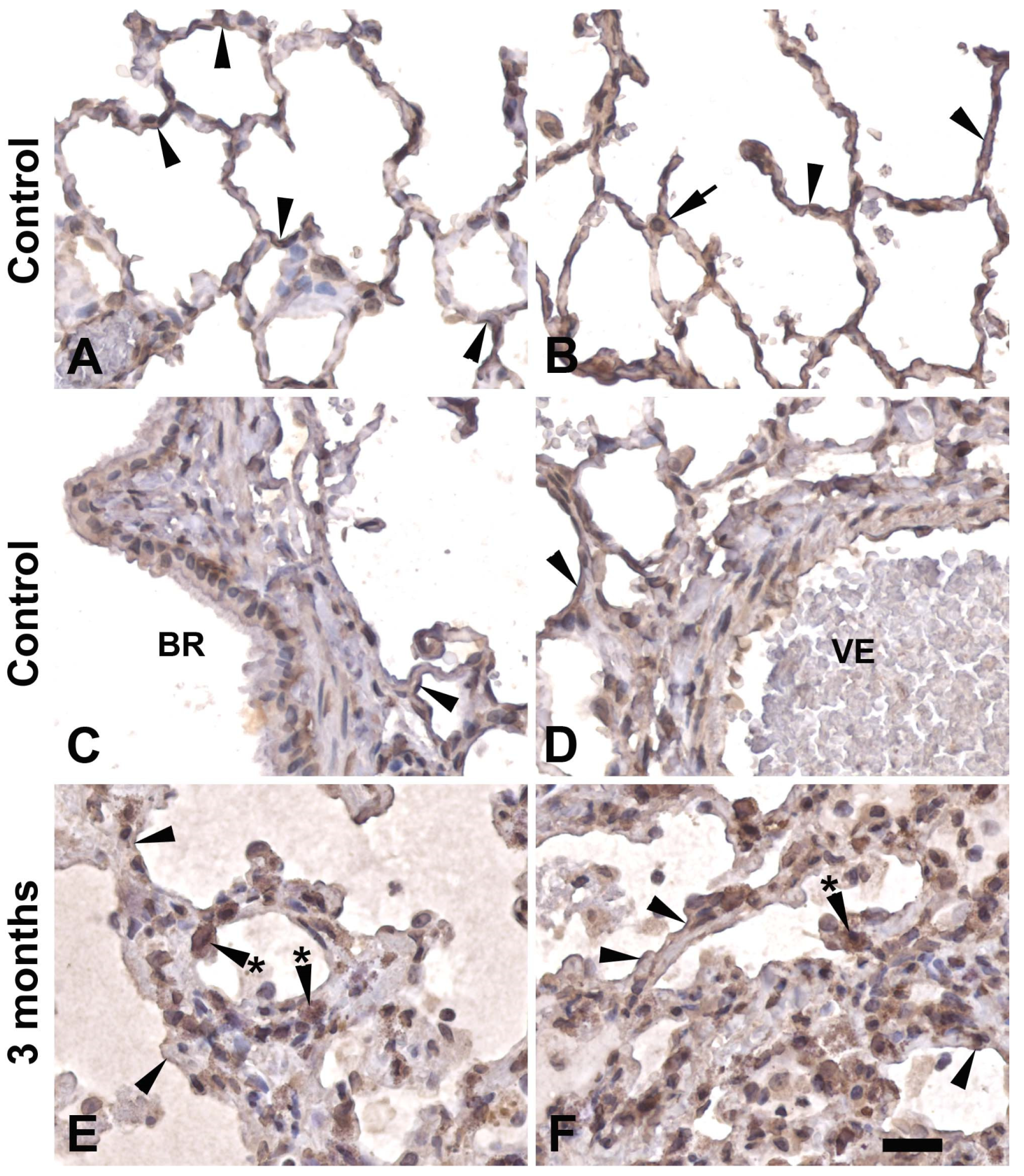
Figure 6.
Immunofluorescence double-labelling of SFTPC, a marker of type II pneumocytes, and PARP-1 (A-C) as well as PAR (D-F) in rat lungs. A-C: Most of the type II pneumocytes contain PARP-1 (arrwos), but there are some type II pneumocytes which are PARP-1 negative (arrowheads). PAR is expressed as a thin line on the surface of alveoli, but also at the surface of type II pneumoytes (arrows, such the cell in the center). Bar corresponds to 50 µm.
Figure 6.
Immunofluorescence double-labelling of SFTPC, a marker of type II pneumocytes, and PARP-1 (A-C) as well as PAR (D-F) in rat lungs. A-C: Most of the type II pneumocytes contain PARP-1 (arrwos), but there are some type II pneumocytes which are PARP-1 negative (arrowheads). PAR is expressed as a thin line on the surface of alveoli, but also at the surface of type II pneumoytes (arrows, such the cell in the center). Bar corresponds to 50 µm.
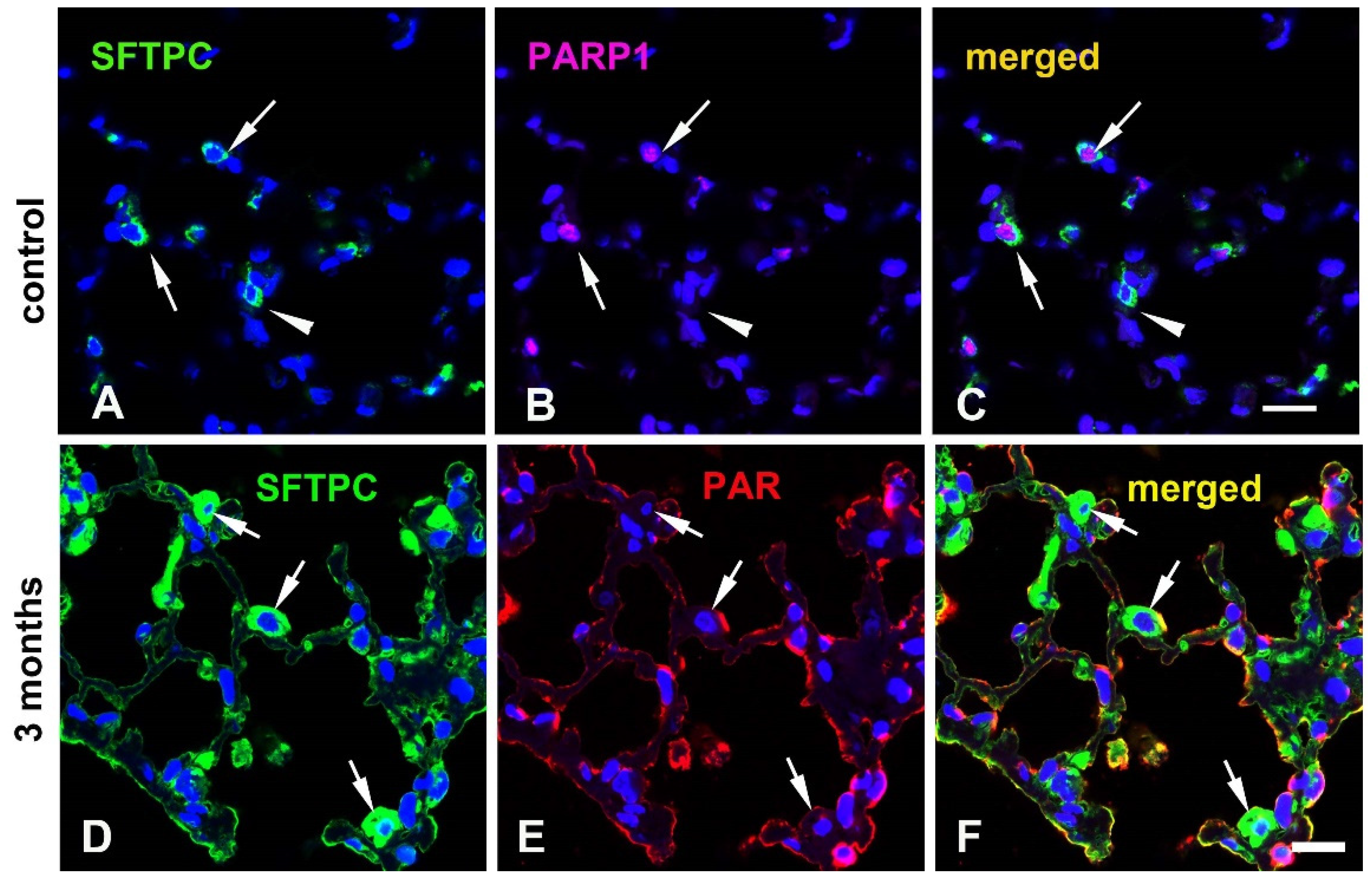
Figure 7.
Immunofluorescence double-labelling of PAR with aquaporin 5 (Aqp5), a marker of type I pneumocytes (A-C) and with CD68, a marker of macrophages (D-F). A-C: The surface of the alveoli is labelled by both PAR (arrowheads) and Aqp5 (arrowheads with asterix), but in a mutually exclusive way. D-F: Macrophages (arrows) are mostly PAR negative or very weakly positive (arrowheads), but there are also some PAR positive macrophages as for example the cell above the cell labelled with an arrow. Bar corresponds to 50 µm.
Figure 7.
Immunofluorescence double-labelling of PAR with aquaporin 5 (Aqp5), a marker of type I pneumocytes (A-C) and with CD68, a marker of macrophages (D-F). A-C: The surface of the alveoli is labelled by both PAR (arrowheads) and Aqp5 (arrowheads with asterix), but in a mutually exclusive way. D-F: Macrophages (arrows) are mostly PAR negative or very weakly positive (arrowheads), but there are also some PAR positive macrophages as for example the cell above the cell labelled with an arrow. Bar corresponds to 50 µm.
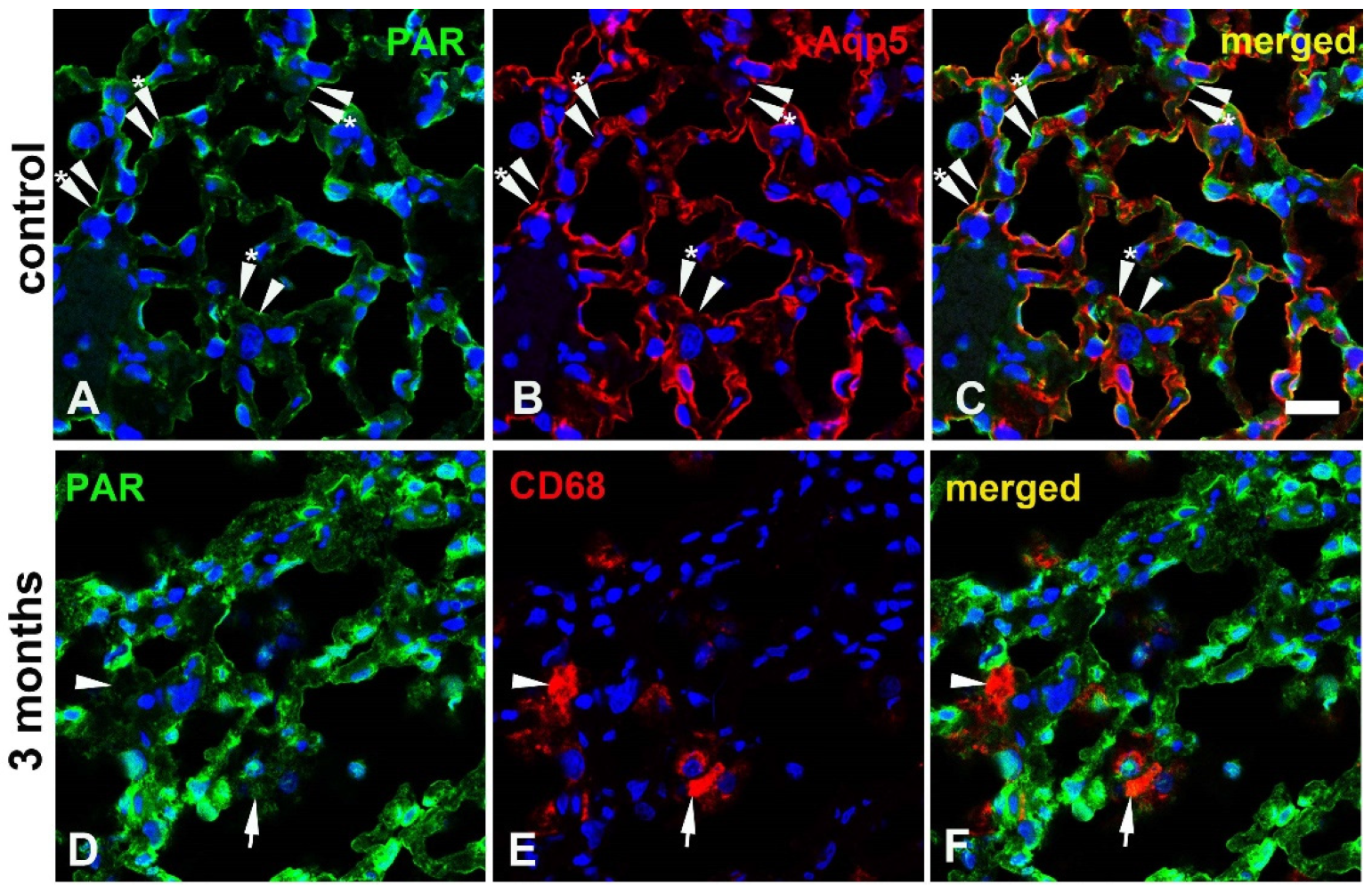
Table 1.
Expression pattern of PARylation related proteins in the rat lung.
Compartments: N, nuclear, C, cytoplasmic, M, membrane or organelles; °, retronuclear; #, apical. Staining intensity: 0, not detectable; 1, weak or rare staining; 2, medium staining; 3, strong staining; 4, very strong staining. Abbreviations for the cell types: art, arteriole; bron, bronchiolar; cap, capillary; con, connective; endo, endothelium; m, muscle; pneu, pneumocytes; s.m., smooth muscle; tiss, tissue; ven, venule.
Table 1.
Expression pattern of PARylation related proteins in the rat lung.
Compartments: N, nuclear, C, cytoplasmic, M, membrane or organelles; °, retronuclear; #, apical. Staining intensity: 0, not detectable; 1, weak or rare staining; 2, medium staining; 3, strong staining; 4, very strong staining. Abbreviations for the cell types: art, arteriole; bron, bronchiolar; cap, capillary; con, connective; endo, endothelium; m, muscle; pneu, pneumocytes; s.m., smooth muscle; tiss, tissue; ven, venule.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



