Submitted:
23 August 2024
Posted:
26 August 2024
Read the latest preprint version here
Abstract
The Rose grain aphid, a notable agricultural pest, releases saliva while feeding. Yet, there is a need for a comprehensive understanding of the specific identity and role of secretory proteins released during probing and feeding. Therefore, a combined transcriptomic and proteomic approach was employed to identify putative secretory proteins. The transcriptomic sequencing result led to the assembly of 18030 unigenes out of 31344 transcripts. Among these, 705 potential secretory proteins were predicted and functionally annotated against publicly accessible protein databases. Notably, a substantial proportion of secretory genes (71.5%, 69.08%, and 60.85%) were predicted to encode known proteins in Nr, Pfam, and Swiss-Prot databases, respectively. Conversely, 27.37% and 0.99% of gene transcripts were predicted to encode known proteins with unspecified functions in the Nr and Swiss-Prot databases, respectively. Meanwhile, the proteomic analysis result discovered, 30 salivary proteins. Interestingly, most salivary proteins (53.3% proteins) showed close similarity to A. craccivora, while 36.67% to A. pisum and A. glycines. However, to verify the expression of these secretory genes and characterize the biological function of salivary proteins further intervention should be geared towards gene expression and functional analysis.
Keywords:
Metopolophium dirhodum
; unigene
; transcriptome
; proteome
; salivary protein
1. Introduction
Aphids (Hemiptera: Aphididae) are voraciously phytophagous insects that ingest plant phloem sap by their needle-like stylets (mouth parts). They are unusual herbivores because their feeding site is a single phloem cell in the sieve element buried deep within plant tissues; yet, they represent one of the most important insect pests in temperate agriculture Gupta [2,3]. Globally, there are approximately 5558 aphid species belonging to 703 genera and 30 subfamilies [4]. Among the recorded aphid species, around 450 of them feed on crop plants, and 250 aphid species are considered economically important agricultural pests causing significant economic losses [5,6]. Four aphid species cause serious damage to wheat farms in China. The major aphid species infesting wheat in China are the grain aphid (Sitobion avenae Fabricius), green bug (Schizaphis graminum Rondani), bird cherry oat aphid (Rhopalosiphum padi Linnaeus) and rose grain aphid (Metopolophium dirhodum Walker) [7]. Some Phloem feeding insects, such as pea aphids (Acyrthosiphon pisum Harris) have a wide host range often colonizing many leguminous plant species. Unlike the other types of aphids, M. dirhodum are primarily restricted to crops that belong to the Rosaceae (mainly cherries and peaches), but are a secondary pest of cereal crops (wheat and other cereal crops).
Certain plants have the ability to withstand aphid feeding without negative consequences, either through inherent genetic resistance or by adjusting their interactions with aphids. This natural defense mechanism has proven effective in controlling aphids in various crops. The interactions between plants and aphids can be seen as a process of coevolution, where both parties adapt to each other [8]. Aphids extract nutrients from host plants by piercing the phloem to access sap with their stylets, releasing saliva from salivary glands that contain effector proteins to counter plant defenses [9,10]. The composition of watery saliva, containing a diverse blend of enzymes and other substances, varies significantly among aphid species and even within the same species depending on their diet [11,12,13]. The ability of aphids to adapt to different host plants is closely tied to the diversity in watery saliva content. This generally involves the secretion of secretory molecules specifically effectors to target host molecules [14]. Many studies have shown that insects, including aphids, produce and secrete effectors that suppress or induce plant defense responses [15,16,17,18]. These aphid secretory proteins are thought to be produced predominantly inside the head and are secreted along with saliva during probing and feeding [19,20,21]. Nonetheless, limited research has delved into the connection between feeding patterns and the genes expressed in aphid salivary glands during shifts in host preferences.
The recent availability of data on the genome and transcriptome sequences of aphids has enabled the development of an approach to identify potential secretory protein of aphids [9,15,19]. Bioinformatics pipelines to identify putative secreted proteins have been developed and applied to several aphid species [17,22]. In addition, saliva collection methods based on artificial feeding systems combined with mass spectrometry allow for the detection of many proteins in the saliva of several types of aphids released during probing and feeding [15,23]. Significant progress has been made in the identification and functional annotation of potential effectors of economically important aphid species of cereals, such as bird cherry-oat aphids, and green bug aphids, through proteomic and transcriptomic analysis. However, limited gene and/or proteomic data for rose-grain aphids have been documented, despite the fact that rose-grain aphids are the fourth most dominant and destructive aphids affecting wheat production in many temperate regions, including China. Furthermore, the molecular mechanisms underlying these differences have not been documented because there is a lack of sufficient genetic data for rose grain aphids in public databases. Consequently, this study employed a combined transcriptomic and proteomic approach to mark an initial step in identifying candidate secretory proteins involved in plant infestation and environmental adaptation.
2. Materials and Methods
2.1. Aphid Handling and RNA Preparation
Both apterous and alate adult rose grain aphids from a clonal lineage were raised on susceptible wheat (Triticum aestivum L. Var. Zhongmai-175) in a controlled laboratory setting. Aphid heads along with salivary glands were dissected under a stereomicroscope and temporarily stored in liquid nitrogen. RNA was extracted using TRIzol reagent and then purified using a Tianmobio total RNA extraction kit. The degradation and contamination of RNA were examined using a 1% agarose gel. RNA purity was measured with a Nano-drop spectrophotometer (IMPLEN, CA, USA). The quantity and integrity of RNA were assessed using the RNA Nano 6000 Assay Kit (Agilent Technologies, CA, USA).
2.2. cDNA Library Preparation, Cluster Generation and Sequencing
The process of constructing the cDNA library commenced with a 1.5 µg RNA sample. The synthesis of the cDNA library involved fragmenting mRNA using divalent cations in the presence of random hexamer primers and M-MuLV Reverse transcriptase. DNA Polymerase I and RNase H were employed to construct the first and second cDNA strands. The overhangs were then converted into blunt ends through exonuclease/polymerase activity. Three microliters of enzyme (NEB, USA) were used with size-selected, adaptor-ligated cDNA at 37 °C for 15 minutes, followed by 5 minutes at 95 °C. PCR amplification was carried out using Phusion High-Fidelity DNA polymerase, universal PCR primers, and Index Primer. The PCR products were purified using the AMPure XP system (Beckman Coulter, Beverly, USA), the library quality and insert size was assessed on the Agilent Bioanalyzer 2100 system. Furthermore, the library's quantity was measured using a Qubit 2.0 Fluorometer. A paired-end library of 250-300bp size was prepared according to Illumina's protocol/instructions. Subsequently, the samples were clustered with the aid of cBot Cluster Generation System along with the TruSeq PE Cluster Kit v3-cBot-HS from Illumina (Illumina, China).
2.3. Quality Control, Transcriptome Assembly, and Gene Annotation
The first stage of the analysis involved processing of the raw data (raw reads) in FASTQ format through custom Perl scripts. Subsequently, clean reads were acquired by removing intact adapter reads (the proportion of reads with connectors or linkers), ambiguous reads (The proportion of reads with unspecified base information), and low-quality reads (Q Phred ≤ 20 is the number of bases in the total read length of 50% or more of the reads) from the raw data. At the same time, the clean data was evaluated for Q20 (The percentage of bases with a QPhred value greater than 20 to the total bases, where QPhred=-10log10(e)), Q30 (The percentage of bases with a QPhred value greater than 30 to the total bases), GC content, and sequence duplication level. The transcriptome was assembled using Trinity software with min_kmer_cov set to 2, and all other parameters with default settings [24]. Following the assembly, reads were aligned back to the transcriptome assembly using the align_and_estimate_abundance.pl script, with RNA-seq by Expectation Maximization (RSEM) method and Bowtie to estimate abundance and mapping the reads. Transcripts with fewer than 0.5 transcripts per million mapped reads (TPM) or representing less than 10% of the expression value of the dominant isoform for each unigene were excluded from the transcriptome assembly. Protein coding regions were identified using Trans-decoder v.3.0.1, retaining the highest scoring open reading frame (ORF) for each transcript with the single_best_orf option. Finally, transcripts without open reading frames were removed from the assembly.
Functional annotation of the derived genes was done using the Basic Local Alignment Search Tool (BLAST2go) against public databases, including the National Center for Biotechnology Information (NCBI), Non-redundant Nucleotide Acid Database, Non-redundant Protein Database, and BLASTx against the Swiss-Prot Database. The best matched alignment results were considered to annotate all unigenes; if the alignment results among these databases differed, preference was given to the results from the NR database. All genes that matched the Swiss-Prot database entries were further classified according to Gene Ontology terms (matched genes with an E-value < 10-6). Furthermore, all genes that matched the Swiss-Prot database were classified based on the Eukaryotic Orthologous Groups (KOG)/Clusters of Orthologous Groups of proteins (COG) (matched genes with an e-value < 10−3), and the pathways were annotated using the Kyoto Encyclopedia of Genes and Genomes (KEGG). Automatic Annotation Server (KAAS) with a cut-off E-value < 1.0x10−1. Gene homology was established by searching public databases, such as non-redundant proteins (Nr, with genes having an e-value < 10−5), nucleotide sequences (Nt, with genes having an e-value < 10−5), Pfam (with genes having an e-value < 0.01), and Swiss-Prot (with genes having an e-value < 1.0x10−5), for all genes. Gene Ontology enrichment analysis of the differentially expressed genes (DEGs) was conducted using the GOseq R package. The KOBAS software [25] was utilized to assess the statistical enrichment of the differentially expressed genes in the KEGG pathways.
2.4. Prediction of Secretory Proteins
To discover potential effectors, the coding sequences of every unigene were translated into peptide sequences and subsequently analyzed using a web-based tool for predicting signal peptides (https://services.healthtech.dtu.dk/services/SignalP-6.0/). Out of 705 peptide sequences, clusters containing a signal peptide at the N-terminal of the polypeptide chain were identified. These sequences with signal peptides were then assessed using a transmembrane domain prediction web-based tool (https://dtu.biolib.com/DeepTMHMM) to identify any transmembrane domains within the protein sequence. Ultimately, protein sequences featuring a signal peptide at the N-terminal and those with one or devoid of transmembrane domains were classified as candidate secretory proteins.
2.5. Saliva Collection, Extraction and Protein Identification
2.5.1. Saliva Collection and Extraction
The rose grain aphids were dislodged from wheat plants by shaking and then placed on sterile diets (consisting of 15 % sucrose, 100 mM L-serine, 100 mM L-methionine, and 100 mM L-aspartic acid, pH 7.2 (KOH)) [21]. The diet was prepared under aseptic conditions and filtered using a 0.22μm syringe filter (Millipore, MA, USA). Approximately 250 aphids were confined in each glass tube, with 1 mL of the diet provided between two layers of Parafilm (Neenah, WI, USA). After 72 hours of feeding, the diets were collected from the space between the two layers of Parafilm using a pipette. Ultrafiltration was performed with a 3-kDa molecular-weight cutoff Amicon Ultra-4 Centrifugal Filter Device (Millipore) at 5000g at 4 °C for 30 minutes. The concentrated samples were then precipitated using a trichloroacetic acid protein precipitation kit (Sangon, Shanghai, China). The pellets were solubilized in 200µl of SDT buffer and Bicinchoninic Acid (BCA) protein determination assay (Bio-Rad, USA). The proteins were then separated on a gel using SDS-PAGE and stained with Coomassie Blue R-250 dye to visualize protein bands. Gel slices were processed further by destaining, reduction, alkylation, digestion with trypsin, and peptide extraction. The resulting peptides were dehydrated and reconstituted in 0.1% formic acid for downstream analysis.
Figure 1.
Aphid feeding on artificial diet and saliva collection.
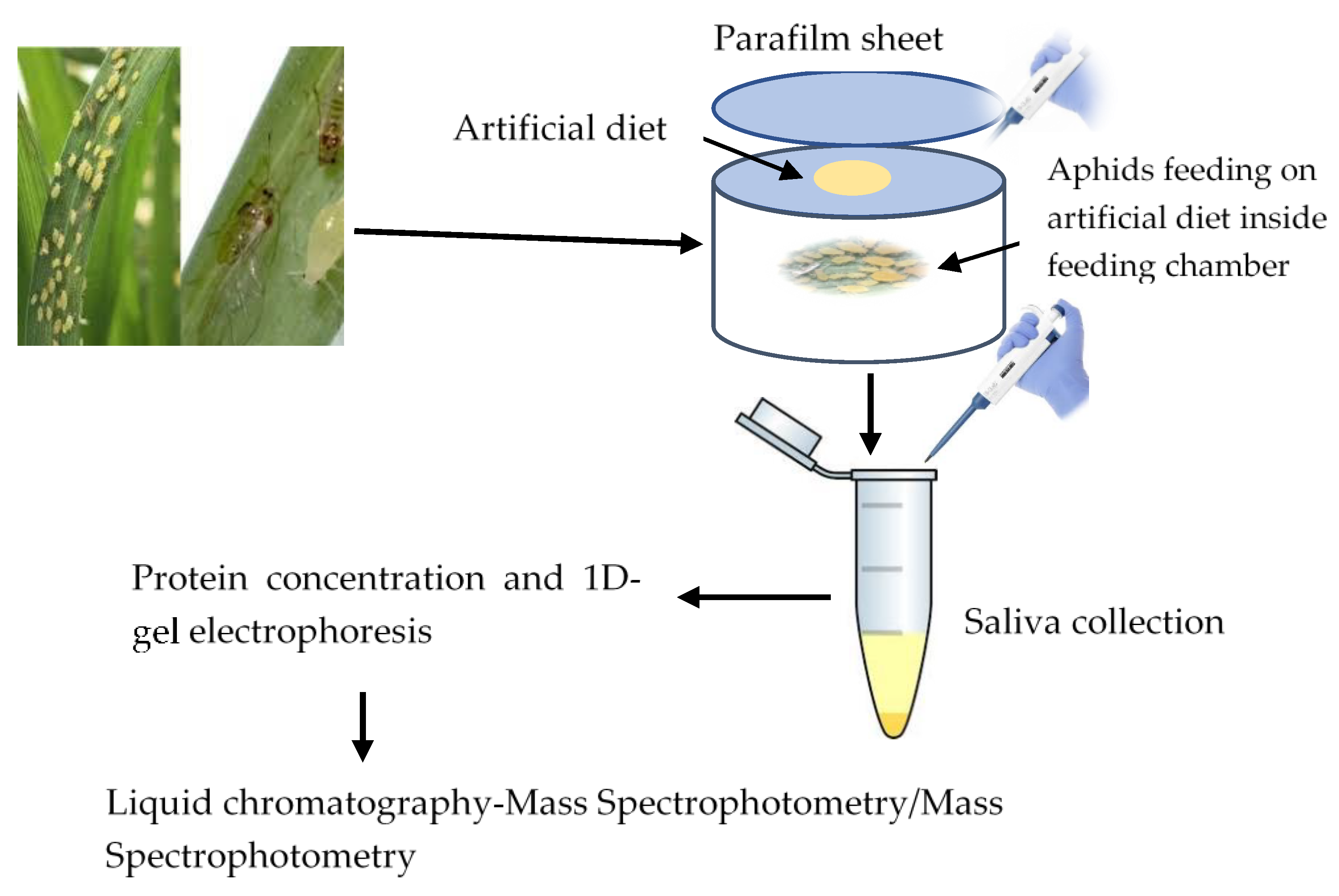
2.5.2. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
LC-MS/MS analysis was performed on a Q Exactive mass spectrometer (Thermo Scientific) coupled with Easy nano-liquid chromatography (Thermo Fisher Scientific). While the sample eluted from the LC column, thousands of mass spectra were acquired, and subsequently, the mass spectrum of all peptides was measured at that time point. The mass analyzer separated the peptides based on their mass-to-charge ratio, and the detector detected precursor peptide ions. The most abundant precursor peptide ion (300-1800 m/z) was further fragmented by collision with neutral gas molecules after passing it through the filter unit of the mass spectrometer.
2.5.3. Proteomic Data Analysis and Similarity Search
The acquired mass spectrometry (MS) data were analyzed using the MASCOT engine (Matrix Science, London, UK; version 2.4). MS data were searched against the UniProtKB Escherichia coli database. The search followed an enzymatic cleavage rule for trypsin, and allowed a maximum of two missed cleavage sites and a peptide mass tolerance of 20ppm for fragment ions. The cutoff global false discovery rate (FDR) for peptide and protein identification was set to 0.01 or (FDR≤0.01).
BLAST score ratio (BSR) tests were employed to compare the proteome similarities among various aphid species, including M. dirhodum, Aphis craccivora, Aphis glycines, Acyrthosiphon pisum, Aphis gossypii, Schizaphis graminum, Sitobion avenae, Rhopalosiphum padi, Myzus persicae, Melanaphis sacchari, and the yellow sugar cane aphid, Sipha flava. The reference comparisons were conducted using the BLAST software available at http://blast.ncbi.nlm.nih.gov/Blast.cgi. The BSR index was determined by dividing the BLAST query score by the reference score and normalized the result to a scale from 0 to 1. The reference library included A. craccivora, A. glycines, A. pisum, Aphis gossypii, Schizaphis graminum, S. avenae, R. padi, M. persicae, M. sacchari, and S. flava.
3. Results
Altogether, 47,565,328 bp of raw reads were retrieved from the sequencing of the established library. After removing low-quality reads, ambiguous reads, and those related to adapters, 46,238,772 clean reads were produced using Illumina paired-end RNA-seq technology, achieving a Q20 value of 97.61% in the transcriptomic assembly. Approximately 39,293,370 reads (84.98% of the total clean reads) were successfully aligned to the reference sequence (longest transcript unigene). After filtering out adapter-related reads (609272), ambiguous reads (18194), and low-quality reads (35812), clean reads were retained. The transcriptome of the rose grain aphid head was then de novo assembled using the short-read assembling program (Trinity), resulted in clustering of 31344 transcripts and 18030 genes (Table 1). The transcripts varied in length from 301 to over 2000 base pairs, with an average size of 1532 base pairs. Out of the transcripts, 7830 (24.98%) were in the range of 301 to 500 base pairs, 6510 (20.77%) were between 501 to 1000 base pairs, 8229 (26.25%) ranged from 1001 to 2000 base pairs, and 8775 (28%) were longer than 2000 base pairs. Furthermore, among the genes that were assembled, 4949 (27.45%) had ranged between 301 to 500 base pairs, and 4261 (23.63%) fell within the range of 501 to 1000 base pairs. Among the genes analyzed, 4377 (24.27%) fell within the range of 1001 to 2000 base pairs, while 4443 (24.64%) surpassed 2000 base pair mark (Figure 2). The mean size of the genes derived from the transcripts was 1413 base pairs. In comparison, the transcripts produced by Trinity had an average length of 1532 base pairs (N50=2335bp), demonstrating longer transcripts than the average gene length of 1413 base pairs (N50=2205 bp). All unigene assembled in this study are illustrated in supplementary table 1. The raw sequencing data have been submitted to the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) database with the accession number PRJNA1134911 (http://www.ncbi.nlm.nih.gov/bioproject/ PRJNA1134911).
3.1. Functional Annotation of Gene Transcripts
Head tissue libraries were annotated utilizing BLASTX against the NCBI database, and all unigenes underwent homology searches. The functional annotation of all unigenes of M. dirhodum were subjected to BLAST search against seven databases including NCBI's non-redundant protein (Nr) and nucleotide (Nt) sequences, protein family (Pfam), the manually annotated and reviewed Swiss-Prot protein sequence database, Eukaryotic Clusters of Orthologous Groups of proteins (KOG/COG), the Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog database, and Gene Ontology (GO). This comprehensive approach was employed to annotate the functions of each gene transcript. Of the total assembled unigenes (18030), 12589 (69.82%), 14253 (79.05%), 5587 (30.98%), 9467 (52.5%), 9314 (51.65%), 9314 (51.65%), and 5850 (32.44%) genes were matched to known proteins in the non-redundant protein sequences, non-redundant nucleotide sequences (Nt), KEGG Orthology (KO), Swiss-Prot, Protein family (Pfam), Gene Ontology (GO), and Eukaryotic Orthologous Groups (KOG) databases, respectively (Table 2). The alignment result of the total unigenes were not similar across all databases and hence, the NR database results were preferentially employed to annotate all unigenes.
The newly de novo assembled transcriptomic sequences, were examined using a BLAST search against the NCBI database to deduce the functional and structural characteristics of the proteins encoded by these transcripts. The comparison involved matching all coding sequences of M. dirhodum genes with established transcriptomic sequences from various aphid species found in recognized protein databases such as Nr, Swiss-Prot, and Pfam. Consequently, the de novo assembly of M. dirodum transcripts resulted in the prediction of 12589, 9467, and 9314 functionally known and unknown gene products (proteins) in the Nr, Swiss-Prot, and Pfam genome databases, respectively. The E-value distribution for the top matches against the nr database indicated that 17.18% of the sequences exhibit strong homology (E-value < 1.0E-60), with most E-values falling between 0 and 1.0E-100 (Figure 3A). Conversely, the similarity distribution revealed that 97.18% of the unique sequences with top matches have a similarity greater than 60%, whereas only 2.82% of the hits showed a similarity below 60% (Figure 3B). On top of that, the sequences of assembled unigenes were aligned to gene sequences of other organisms accessible in prominent databases to look for conserved genes (homologous sequence search). Most of the sequences of unigenes (68.2%) were aligned to A. pisum sequences, followed with M. persicae (18%), for which the genome sequences were made available in the database (Figure 3C). However, some gene transcripts (13.7%) revealed less semblance to other economically important aphid species.
3.2. Gene Ontology and Eukaryotic Orthologous Groups classification (KOG)
The gene transcripts characterized based on the Nr and Swiss-Prot databases were further annotated against Gene ontology to determine the number of genes associated with each functional category. Of the 18030 assembled gene clusters, 9314 were assigned to three functional groups. The three most abundant functional categories were biological processes, cellular components, and molecular function with 4493 (48.24%), 2762 (29.65%) and 2059 (22.1%) unigenes associated with each functional category, respectively. Accordingly, a wide distribution and assignment of unigenes were implicated in biological process. Based on the GO enrichment analysis results, the top 54 over-represented genes were cellular processes (5212), metabolic processes (4516), single organism processes (4165), biological regulation (2113), regulation of biological process (1992), and localization (1823) respectively. On top of that among the genes associated with molecular functions, 5389, 3902, and 848 unigenes were involved in binding, catalytic and transport activities respectively. In light of cellular components, most unigenes were associated in cells (2777), cell parts (2777), organelles (1922), and membranes (1914) (Figure 4).
Eukaryotic Orthologous Groups (KOG) were extrapolated from Clusters of Orthologous Groups (COG) to specifically predict the sequence of unknown genes from previously identified and stored in known databases of evolutionarily related sequences of different organisms. Based on KOG, 6575 genes were annotated into known (6126 genes) and unknown (449 genes) functional ortholog groups. Among the known functional groups, the highest percentage of genes (914,13.9%) was annotated to the general function prediction, followed by signal transduction mechanisms (800, 12.17%), post-translational modifications, protein turnover, and chaperones (605, 9.2%) (Figure 5).
3.3. Metabolic Pathway Analysis by Kyoto Encyclopedia of Genes and Genomes (KEGG)
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway was used to describe the structural elements of the gene products and metabolic pathways in cell. Based on KEGG, 5587 gene transcripts were assembled from the transcriptomic sequences of the heads of M. dirhodum aphids. The transcripts of these genes were annotated to 228 functional pathways.
Based on KEGG pathway annotations, the transcripts of genes associated with the KEGG pathway were grouped into five functional categories: Cellular Processes, Environmental Information Processing, Genetic Information Processing, Metabolism and Organismal Systems. In the cellular process category, the highest number of unigenes were associated with the transport and catabolism (347, 5.74%) and cellular (262, 4.33%) functional pathways, respectively. In the environmental information processing category, significant number of unigenes were implicated in signal transduction (718, 11.88%) while extremely few numbers of genes involved in membrane transport (55), signal molecules and interaction (118). Of these genes associated to genetic information processing category, the highest number of genes (410, 6.78%) were involved in Translation and folding, followed by sorting, and degradation (330, 5.46%) KEGG pathways. In the metabolism group, carbohydrate metabolism pathway was significantly enriched with 296 (4.89%) unigenes. In the organismal systems category, the number of the transcripts of genes associated with endocrine system, digestive system, immune system and nervous system were 402 (6.65%) and 253 (4.18%), 230 (3.8%) and 230 (3.8%) respectively (Figure 6). These findings offer significant insight into the environmental information processing pathways within the head tissue of M. dirhodum. They may serve as a foundation for future research aimed at identifying and characterizing the genes and transcripts involved in key signal transduction pathways.
3.4. Prediction of Secretory Proteins
Salivary proteins are believed to be involved in plant interactions only when they are secreted during aphid probing and feeding. All genes were compared with the priority order of non-redundant protein sequences (NR protein), Protein family (Pfam) and the Swiss-Prot protein library to analyze the presence of signal peptides. Therefore, in this study, 705 putative secretory genes were predicted of all unigenes assembled from the transcriptome of the head tissue. These proteins which had signal peptides associated to the N-terminal of amino-acid sequence along with one and /or zero transmembrane domains, indicating their secretory nature (supplementary files Table S2). Of all clusters of transcripts of secretory genes, 504(71.5%), 487 (69.08%), and 429 (60.85%) genes were characterized as encoding known secretory proteins with known functions in the Nr, Pfam, and Swiss-Prot databases, respectively. However, 193 (27.37%), and 7 (0.99%) transcripts of genes were predicted to encode known proteins with uncharacterized functions in the Nr, and Swiss-Prot databases, respectively. In contrast, the type and function of gene transcripts of some secretory genes were not known in the NR (8 gene), Swiss-Prot (269 genes) and Pfam (218 genes) databases. Most functionally annotated products of putative secretory genes were highly conserved (orthologous) among different aphid species, including pea aphids. Thus, they had similar biological and molecular functions in plant-aphid interactions during feeding.
3.5. Salivary Proteins and Sequence Similarity among Aphid Species
The release of aphid salivary proteins plays indispensable role during aphid-plant interaction. A thorough examination of the saliva from M. dirhodum uncovered the existence of 30 salivary proteins (Table 3). However only 16.67% of the salivary proteins (Glucose dehydrogenase like-protein 2, Heat shock protein, Protein slit, Putative sheath protein, and Vitellogenin domain-containing protein) had signal peptides associated to the N-terminal of amino-acid sequences of each protein. It is not always true that all salivary proteins originate in the head tissue of sap sucking insects. As hemolymph is continually circulated through the salivary glands, it is likely that various biological molecules, including proteins originate from different tissues, are transported into the salivary glands along with the hemolymph or through other, currently unknown, mechanisms.
The identity of the majority of these proteins were determined based on sequence homology search (38.8%) followed by transcript expression levels (13.3%) and sequence prediction (20%). Among the salivary proteins actin had depicted high similarities with 4066 insects and micro-organisms. The sequence of salivary proteins like Elongation factor 1-alpha (1691), 60S acidic ribosomal protein P (1611), Tubulin beta chain (391), Heat shock protein (331) and ACYPI010077 protein (288) had also shown close resemblance with protein sequence of considerable insects and micro-organisms at 50% similarity level (Table 3). The biochemical activity and property of these salivary protein comprises like enzymes, binding proteins, putative effectors, and regulatory proteins (mainly transcription factors). The presence of salivary proteins like glucose dehydrogenase, elongation factors, putative sheath, protein slit, heat shock protein, actin, histone and odorant receptor proteins in the saliva of M. dirhodum, which was widely found in other phloem feeding insects and plant pathogenic micro-organisms, indicating us the significance of salivary proteins in determination of behavioral and functional interaction of aphids with crop plants. Overall, we observed a significant level of similarity across species, typically exceeding 50% identity between the rose grain aphid and the rest of phloem-feeding insects and plant pathogens. This allowed us to anticipate conserved salivary proteins and contribute to a deeper comprehension of the molecular processes involved in the interaction between aphids and host.
The discovery of diverse salivary proteins in the liquid saliva of sap-sucking insects led to the exploration of homologous proteins through sequence comparisons among aphid species. The BLAST score ratio analysis was employed to visualize the extent of salivary protein similarity among different aphid species. The findings revealed that 30% of the salivary proteins were conserved among the three or more aphid species (Table 4). Using the predicted salivary proteins of the rose grain aphid as a query sequence, it was found that 53.3% of the total salivary proteins exhibited close similarity with proteins in the cowpea aphid, Aphis craccivora, while 36.67% of these proteins showed high similarity to those in A. pisum and the cotton aphid, Aphis gossypii. Nevertheless, only 33.3% and 30% of the total salivary proteins exhibited close similarities with sugar cane aphids (Melanaphis sacchari and Sipha flava). Meanwhile, only few numbers of salivary proteins of rose grain aphid, 3.3% and 6.67% displayed close similarity with peach aphid, Myzus persicae and oat aphid, Rhopalosiphum padi respectively.
In general, of the total salivary proteins discovered in the saliva of M. dirhodum, 90% had shown close resemblance to aphids that primarily depend on pulse crops as source food and sustenance, (Aphis craccivora, Aphis glycines, A. pisum, and A. gossypii). In contrast, only 50% of the salivary protein sequences of the rose grain aphid displayed close similarities with cereal aphids, including S. graminum, S. avenae, R. padi, M. persicae, Melanaphis sacchari, and the yellow sugar cane aphid, Sipha flava (Table 4). The close similarity in the biological makeup and composition of salivary proteins between rose grain aphids and pulse crop aphids may indicate that they employ comparable functional and/or behavioral adaptation strategies, setting them apart from cereal aphids.
4. Discussion
In this research, we employed a multidisciplinary strategy to characterize the transcriptome sequence of rose grain aphid, M. dirhodum, and to identify potential secretory proteins. The study of the rose grain aphid's head transcriptome and salivary proteome revealed four distinct trends: (1) a significant number of head transcriptome secretory proteins possess signal peptide sequences, (2) some portion of the head transcriptome proteins associated with secretory proteins have unknown functions, (3) with the exception of five proteins most saliva proteins had no signal peptides associated to their sequence, and (4) Except five proteins, most secretory proteins predicted in the transcriptome of the head tissue could not detected in the saliva of rose grain aphid. This inconsistency is likely attributable to the highly specialized role of aphid saliva to adapt to the diverse and evolving defense mechanisms of plants.
Some of the inconsistencies between the salivary gland transcriptome and proteome might stem from the differences in expression levels between gene transcripts and proteins. Expression profiles of mRNA are highly dynamic, and there is often a lack of direct correlation between mRNA expression and protein levels [26]. Therefore, discrepancies between the expressed transcript and the resulting protein are likely to be common. Nonetheless, the goal of our study was to provide an initial catalogue of potential secreted proteins from the head tissue, utilizing both transcriptomic and proteomic data. The integration of these data sets marks a significant preliminary step in identifying candidate secretory proteins for future aphid research.
4.1. Putative Effectors
At present, there is no functional data to clarify the roles of the aphid salivary proteins identified in this study. Nevertheless, possible effector functions for many of these candidate proteins can be inferred from their homology or similarity to effector proteins involved in pathogenesis or parasitism, as observed in other sap-sucking insects and plant pathogens. Host plants meticulously detect and respond to incoming injuries caused by the release of secretory proteins and mechanical damage from herbivorous insects. However, plant-feeding insects subsequently suppress the immune responses triggered by these elicitors. Consistent with the finding of Huang, et al. [27] putative sheath proteins (encoded after the gene shp-1) were predicted in both saliva and transcriptomic sequence of rose grain aphid heads. According to Huang, et al. [27] putative sheath protein (LsSP1) binds to the salivary sheath via mucin-like protein (LsMLP1) when released into plants during feeding and probing [28]. The protein specifically synthesized in the head tissue of sap sucking insects and release into plant to suppress plant defense system.
Secretory proteins like glucose dehydrogenase protein-1 (secretory proteins encoded after the gene gld-1) and elongation factor 1 alpha (regarded as functional product of EF1A_0 gene) were also detected in the secretome and transcriptome of rose grain aphid heads. Glucose dehydrogenase like protein 1 facilitates oxidation-reduction process [29]. Ubiquitous plant defense responses against invading plant pathogens and insect pests may be degraded by proteins involved in oxidoreductase activity [30]. Thus, it is a potential effector that deters plant defense responses against aphid infestation. Glucose dehydrogenase like protein 1 also plays indispensable role in insect development and suppressing plant defense mechanisms.
Lipid-binding and transporting proteins (apolipophorins), (Cluster-3959.1281), was predicted in the transcripts of secretory genes. This protein is homologous among some aphid species like potato aphid Myzus euphorbiae and M. persicae. The fecundity rate of M. persicae was increased following the overexpression of Me10 in N. benthamiana, indicating species specific ability to deter plant defense responses [31]. Beta-glucosidase transcript (Cluster-9628.0) were predicted from the transcriptome of M. dirhodum head tissue. It is a typical enzyme in termites (Neotermes koshunensis) that aids in degrading hemicellulose [32]. On top of that it may also serve as a signaling pathway that triggers plant defense responses against herbivores. Treatment of cabbage leaves with beta-glucosidase induces the release of volatile compounds like salicylic acid (SA), ethylene, and H2O2 [33].
Gene transcripts specifically, Cluster-3959.2598, Cluster-3959.726, Cluster-1501.0, Cluster-45.0, Cluster-671.0, and Cluster-4581.0 were predicted encoding of glucose dehydrogenase. By the same token, glucose dehydrogenase was also detected in the saliva of rose grain aphid (glucose dehydrogenase-like protein 1 and glucose dehydrogenase-like protein 2). These proteins were reported in the saliva and salivary glands of some aphid species like M. persicae [6], S. avenae [30,34], and A. craccivora [35]. Glucose dehydrogenase is a potential effector that deters pattern triggered immune responses against aphid infestation.
The transcripts of lipase (Cluster-7728.1) and phospholipases (Cluster-3959.2585, Cluster-10963.0 and Cluster-7682.0) were extrapolated from the transcriptomic sequence of M. dirhodum heads with salivary glands. Lipase and phospholipase hydrolyze the linkages of triacylglycerols and phospholipids, respectively [36]. Unlike phospholipase, lipid processing, food digestion and transport has been enhanced by the activity of lipase [33]. The transcript of secretory proteins (Cluster-3959.2585, Cluster-12716.1, Cluster-10963.0, & Cluster-7682.0), phospholipase often involve in lipid biosynthesis, acyltransferase activity (catalysis of the transfer of an acyl group to an oxygen atom), calcium ion and receptor binding activities. The calcium binding activity of the phospholipase protein subsequently led to prevention of calcium-mediated occlusion and obliteration of phloem sieve elements against aphid feeding. Characterization of putative effectors of pea aphid using proteome mass spectrometry indicated both metalloprotease and calcium-binding proteins inactivate plant defense responses and inhibit calcium-mediated blockade of plant sieve elements, respectively [9]. However, both lipase and phospholipase have versatile roles in various biological and industrial applications. The biproduct of phospholipase, phosphatidic acid, plays an important role in signal transduction cascades and lipid metabolic pathways, thus determining plant response to stress. When plants are exposed to stress such as mechanical stress (wounding and frost) and biological stress (pathogen and insect attack), the expression and activity of the phospholipase D protein increases rapidly [37]. Based on the findings of [38], rice infection with Xanthomonas oryzae increased phospholipase accumulation in its plasma membrane. Phospholipases also trigger plant defense responses (release of abscisic acid, ethylene, and nicotinamide adenine dinucleotide phosphate ion (NADPH)) against herbivorous insects and diseases [37]. The transcript of trehalose (Cluster-608.0) facilitates the expression of trehalose transporter Tret1-2 homolog isoform X2, trehalose-phosphatase, and alpha-trehalose-phosphate synthase. In plants, trehalose plays a regulatory role in sugar metabolism, growth, development, and responses to stress induced by biotic and abiotic factors [39]. The overexpression of trehalose biosynthetic gene enacts plant and microbes to receive exogenous trehalose, thereby increase tolerance to stress [40,41]. Trehalose has also been reported in the salivary glands of B. tabaci and S. avenae [42,43]. Based on the findings of [43], trehalose plays an important role in deterring the accumulation of excess trehalose in plant cells to suppress immune response.
4.2. Detoxifying Secretory Proteins
When plants are attacked by herbivorous insects, they release secondary toxic metabolic products (mainly protease inhibitors). Nonetheless, herbivorous insects secrete detoxifying proteins to degrade the toxins produced upon feeding, thus paving the way for adaptation to adverse conditions. Esterase is one of the detoxifying proteins released by aphids manipulate plant secondary metabolites and insecticides [44]. The transcripts of secretory genes (Cluster-6992.0, and Cluster-6189.0) were predicted to encode secretory protein, esterase E4. This protein breaks down the effect of chemical pesticides when farmers sprayed on farm to manage aphids. A prevalent strategy utilized by Myzus persicae aphid to build resistance against insecticide involves boosting the expression of genes that encode for Esterase E4 [45].
Secretory proteins such as glutathione S-transferase 6 and CAAX prenyl protease 1 homolog (ortholog metalloproteases) were also predicted in the transcriptome assembly of the head tissue after gene transcripts of Cluster-11681.0 and Cluster-3959.1466. Glutathione S-transferases allow insects to survive under chemical stress through metabolism of xenobiotics or by providing protection against oxidative stress [46]. Although there is insufficient evidence to support our findings, a report [47] indicated, many types of metalloproteinases are involved in detoxifying plant secondary metabolites.
Clusters of gene transcripts (Cluster-7473.0, Cluster-477.0, Cluster-3959.111, Cluster-3959.1431, Cluster-3959.824, and Cluster-3959.2743) were also predicted encoding secretory proteins, specifically peroxidase and oxidoreductase. These proteins are essential for basic biological functions, such as response against stress and defense mechanisms. Previously, the secretory proteins were discovered in some aphid species, mainly in S. avenae [43], A. pisum [9], Mayetiola destructor [48] and Megoura viciae [47]. Unlike other species of aphids, only superoxide dismutase (Cluster-3959.779) was predicted in the transcriptomic sequences of M. dirhodum. Research conducted on parasitoid wasp (Leptopilina heterotoma), revealed that extracellular superoxidase dismutase was synthesized and released along with the fluid and act as virulence factors to counteract the immune response of Drosophila species [49]. It is also used by pathogenic bacteria, fungi, and pathogenic protozoan parasites as virulence factors against the resistant gene of the host at early infection. This protein initiates plant defense responses when aphids begin to probe and feed on plants. Thus, it is considered to be a signaling pathway in the plant defense system. Peroxidase transcripts (Cluster-3959.111, Cluster-3959.824, and Cluster-7473.0) were predicted in transcriptomic sequences of secretory proteins from M. dirhodum aphids. Based on the findings of [43], the presence of peroxidase in aphid saliva serves as an antioxidant involved in the scavenging of hydrogen peroxides and thus plays significant role as a detoxifying enzyme and in suppressing reactive oxygen species-induced plant defense responses.
4.3. Digestive Secretory Proteins
Probing and feeding activities of aphids are also enhanced by digestive enzyme release into the host. The transcript of serine protease (Cluster-12396.0) was assembled from the transcriptomic sequences of rose-grain aphids. Serine protease is a digestive enzyme secreted by the midguts of phloem-feeding insects to facilitate the digestion of food assimilates while ingesting phloem saps from the plant [50]. Serine protease also acts against host-related serine protease inhibitors and phenol oxidase defense response. Secretory gene transcripts, namely Cluster-1194.0, Cluster-1892.0, Cluster-2182.0, Cluster-2350.0, Cluster-3666.0, Cluster-3959.1850, Cluster-3959.2179, Cluster-3959.964, Cluster-6237.0, & Cluster-7233.0 were assembled and predicted encoding digestive secretory proteins, most likely trypsin. Trypsin is secreted by phloem feeding aphids to facilitate the break down and oral digestion of plant phloem constituents [44].
4.4. Ca2+ Binding Secretory Proteins
Putative calcium-binding protein (regucalcin) was predicted in the transcriptional product of secretory gene (Cluster-11867.0) assembled from the transcriptomic sequence of M. dirhodum heads with salivary glands. Similarly, in this experiment we had predicted sarcalumenin (SAR) from secretory gene transcript, Cluster-3959.1170. Sarcalumenin is associated with calumenin calcium buffer proteins to modulate the uptake of calcium ions and release the excitation and contraction of muscle fibers. It also plays an important role in other physiological functions such as muscle resistance against fatigue, muscle development, sarco-endoplasmic reticulum calcium ATPase stabilization, and store-operated calcium entry mechanisms [51]. Plants release calcium ions into the sieve element lumen to block the outflow of phloem saps during mechanical damage, feeding, and probing. In response, sap-sucking insects release water saliva consisting of calcium ion-binding proteins into the lumen of the sieve element to prevent plugging and clogging of feeding sites for insects [52]. With the aid of transcriptomic and proteomic approaches, this protein was detected in the saliva and salivary glands of some homopterous insects such as A. pisum and Nephotettix cincticeps. These results revealed that the synthesis and secretion of calcium-binding proteins could be a means of suppressing plant defense responses against all phloem sap-sucking insects [53].
4.5. Zn-Binding Secretory Proteins
The transcripts of aminopeptidase N (Cluster-8442.0, Cluster-11978.0, and Cluster-7461.0) were detected in the transcriptomic sequences of M. dirhodum secretory proteins. This protein is implicated in binding zinc ions and also known for metallopeptidase activity during aphid-host interactions. Generally, Aminopeptidase N is a multifunctional protein and expressed at different organs and cells of many insects [54]. This protein catalyzes the breakdown of the amide group of amino acids from the N terminal of proteins or peptides. Other versatile types of aminopeptidases have also been identified in M. dirhodum aphid transcripts. Endoplasmic reticulum aminopeptidase 2 was predicted to be synthesized from the transcripts of secretory proteins (Cluster-3959.627) and was found to play important molecular functions, such as metallopeptidase, zinc binding, and hydrolysis. The protein acts against ester bonds of the amino acid, thereby facilitate the disintegration of the disulfide bonds of polypeptides and/or proteins. The functional product of the transcript of gene (Cluster-3959.1656), that is, thyrotropin-releasing hormone-degrading ectoenzyme, is also an integral part of the aminopeptidase protein typically secreted in the salivary glands of insects and functions as the binding of zinc ions (to the active site of the protein, aminopeptidase) and other metallopeptidase activities (penetration of plant cell wall and membrane while probing their stylet to the layers of a cell’s phloem sieve element) at a specific amino acid location of the protein. Clusters of unigenes transcripts, Cluster-3959.1011, Cluster-3959.1012, and Cluster-7461.0 were assembled from the transcriptomic sequences of aphid heads and predicted to encode metallopeptidase and lysosomal alpha-mannosidase proteins which in turn bind zinc. Zinc, a crucial micronutrient vital for the growth, development, and defense of all living things, plays a significant role in the interaction between hosts and pests in plant systems, where competition for Zn can influence the outcome [55].
4.6. Reproduction and Development
Vitellogenin (encoded after gene vit-6) was detected in the proteome and transcriptome (Cluster-1741.0) of M. dirhodum head tissue. It is synthesized in the fat body, and plays multiple roles in insect reproduction, mainly during oocytes and embryo development. Vitellogenin receptors (VgR) present on the surface of oocytes, are responsible for vitellogenin transportation from haemolymph to oocytes [56]. The vitellogenin genes have been extensively investigated in root-knot nematodes, Caenorhabditis elegans [57]. Clusters of secretory gene transcripts, Cluster-3959.2780, & Cluster-4827.0, were also predicted to encoding secretory proteins (protein slit). On top of that it was detected in the secretome of rose grain aphid. This secretory protein plays essential role as midline repellents, stopping longitudinal axons from crossing the central nervous system's midline in a range of insects, nematodes, and planarians [58].
4.7. Protein Synthesis and Secretion
Disulfide isomerase was identified through transcriptomic analysis of head tissues, but it was not notably present in the proteomic profile of salivary proteins. Protein disulfide isomerases are widespread and versatile enzymes found in eukaryotic organisms. These enzymes belong to the thioredoxin superfamily. Classical disulfide isomerases are highly conserved and include an N-terminal signal peptide [59]. This enzyme is believed to play a role in protein folding regulation. Additionally, disulfide isomerase is found in the salivary secretions of plant-parasitic nematodes and has been linked to an increased production of salivary proteins [60,61].
4.8. Odorant Binding and Chemosensory Proteins
In addition to visual signals, aphids largely depend on certain metabolic products as cues to identify their host within complex and spatially diverse environments [62]. In line with the information of Shih, et al. [62], chemosensory protein was predicted among the secretary genes (Cluster-3959.2546) assembled from the transcriptomic sequence of M. dirhodum aphids. Previously, chemosensory proteins were reported in the transcriptome of salivary glands of N. lugens, M. persicae, M. cerasi, and bird cherry-oat aphid R. padi [63]. According to the review of Shih, et al. [62], multiple gene products typically chemosensory proteins, odorant binding proteins, gustatory receptors, and olfactory receptors are important to locate and exploit host resources following the release of phenolic compounds from the plant. In insects, the transportation of organic compounds that convey specific chemical signals (semio-chemicals) is performed with the help of chemosensory proteins. Aphids communicate with each other and locate their food sources and ovipository sites with the aid of odorant binding and chemosensory proteins expressed and released through their antennae and mouth [64]. However, odorant binding and chemosensory proteins may also be located in other parts of insect tissues or organs, mainly the leg, head, thorax, abdomen, and salivary glands of the head. The function of these proteins may differ depending on the location of their expression, in which they may aid in insect development, regeneration of insect legs, host interaction, and immune responses [65,66]. The product of unigene transcripts (Cluster-3959.2546), i.e., chemosensory proteins, predicted from the transcriptome sequence of M. dirhodum showed high similarity to proteins reported in S. avenae and M. persicae aphid species. According to a functional assay carried out on Nicotiana benthamiana, the overexpression of Mp10 protein in N. benthamiana suppressed bacteria-associated molecular patterns and local cell death that impede the fecundity of M. persicae [67]. The odorant binding and chemosensory proteins of Spodoptera frugiperda recognize chemical, behavioral and/or physiological responses of plants [68].
5. Conclusion and Recommendation
The successful colonization of plants by invading pathogen hinges on a variety of secretory proteins released into the host to either suppress or modulate specific innate defenses. This study aimed to determine whether the rose grain aphid possesses a similar array of potential secretory proteins as other phloem-feeding insects. The analysis of the rose grain aphid head tissue secretome revealed three key characteristics: (1) a significant portion of head tissue proteins possess a peptide secretion signal; (2) six salivary proteins identified in the saliva of rose grain aphid consisting of signal peptides associated to the N-terminal of amino-acid sequences; and (3) there have been notable similarities in secretory protein composition between rose grain aphid head tissue and other aphid species. A transcriptomic analysis of M. dirhodum aphids revealed 18,030 genes from 31,344 transcripts in the head tissue along with salivary glands, with 705 genes encoding secretory proteins. Notably, 28.5% of the total gene transcripts were deem of unknown functions in the Nr database. The proteomic analysis unveiled the discovery 30 salivary proteins, including enzymes, binding proteins, putative effectors, and regulatory proteins, highlighting their role in aphid-plant interactions. Despite lacking signal peptides, salivary proteins such as TTF-type domain-containing protein and ATP synthase subunit were found in the watery saliva, suggesting unknown secretion mechanisms. 30% of these proteins are conserved across three or more aphid species. Furthermore, 53.3% of the total salivary protein showed close similarity to Aphis craccivora, followed by 36.67% of these proteins had close similarity with A. pisum and Aphis gossypii. However, only 3.3% and 6.67% of these proteins were similar to those of Myzus persicae and Rhopalosiphum padi, respectively. 90% of the salivary proteins of rose grain aphid closely matched to aphids targeting leguminous crops. Hence, rose grain aphid may share functional and/or behavioral adaptive strategies with pulse aphids. The study underscores the need for further research on gene expression profiling and functional analyses of salivary proteins to elucidate the nature, expression and their role in aphid-plant interactions.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supporting information, List of unigenes assembled from the transcriptomic sequence of rose grain aphid and List of secretory genes and gene products predicted from total unigenes. Supplementary file of salivary proteins, Sample one and Sample two. These materials are made available free of charges via the Internet portal at https://zenodo.org/uploads/11634547.
Funding
This research was supported by Beijing Natural Science Foundation of China (6242026), National Key R&D Plan in China (2023YFD1400800) and China’s Donation to the CABI Development Fund (IVM10051).
References
- Stoletzki, N.; Eyre-Walker, A. The positive correlation between dN/dS and dS in mammals is due to runs of adjacent substitutions. Mol Biol Evol 2011, 28, 1371–1380. [Google Scholar] [CrossRef]
- Gupta, V. Aphids on the world's crops. An identification and information guide. 2001.
- International Aphid Genomics Consortium. Genome Sequence of the Pea Aphid Acyrthosiphon pisum. PLoS Biology 2010, 8, e1000313. [Google Scholar] [CrossRef]
- Favret, C.; Normandin, É.; Cloutier, L. The Ouellet-Robert Entomological Collection: new electronic resources and perspectives. The Canadian Entomologist 2019, 151, 423–431. [Google Scholar] [CrossRef]
- Blackman, R.; Eastop, V. Aphids on the world herbaceous plants and shrubs: An identification and information guide. 2006.
- Harmel, N.; Létocart, E.; Cherqui, A.; Giordanengo, P.; Mazzucchelli, G.; Guillonneau, F.; De Pauw, E.; Haubruge, E.; Francis, F. Identification of aphid salivary proteins: a proteomic investigation of Myzus persicae. Insect molecular biology 2008, 17, 165–174. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, R.; Liu, Q.; Lu, J.; Qiao, G.; Huang, X. Transcriptomic and proteomic analyses provide insights into host adaptation of a bamboo-feeding aphid. Frontiers in Plant Science 2023, 13. [Google Scholar] [CrossRef]
- Goggin, F.L. Plant–aphid interactions: molecular and ecological perspectives. Current opinion in plant biology 2007, 10, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Carolan, J.C.; Caragea, D.; Reardon, K.T.; Mutti, N.S.; Dittmer, N.; Pappan, K.; Cui, F.; Castaneto, M.; Poulain, J.; Dossat, C. Predicted effector molecules in the salivary secretome of the pea aphid (Acyrthosiphon pisum): a dual transcriptomic/proteomic approach. Journal of proteome research 2011, 10, 1505–1518. [Google Scholar] [CrossRef]
- Hogenhout, S.A.; Bos, J.I. Effector proteins that modulate plant--insect interactions. Curr Opin Plant Biol 2011, 14, 422–428. [Google Scholar] [CrossRef]
- Guo, K.; Wang, W.; Luo, L.; Chen, J.; Guo, Y.; Cui, F. Characterization of an aphid-specific, cysteine-rich protein enriched in salivary glands. Biophys Chem 2014, 189, 25–32. [Google Scholar] [CrossRef]
- Wang, W.; Dai, H.; Zhang, Y.; Chandrasekar, R.; Luo, L.; Hiromasa, Y.; Sheng, C.; Peng, G.; Chen, S.; Tomich, J.M.; et al. Armet is an effector protein mediating aphid-plant interactions. Faseb j 2015, 29, 2032–2045. [Google Scholar] [CrossRef]
- Wang, W.; Luo, L.; Lu, H.; Chen, S.; Kang, L.; Cui, F. Angiotensin-converting enzymes modulate aphid-plant interactions. Sci Rep 2015, 5, 8885. [Google Scholar] [CrossRef]
- Sharma, A.; Khan, A.; Subrahmanyam, S.; Raman, A.; Taylor, G.; Fletcher, M. Salivary proteins of plant-feeding hemipteroids–implication in phytophagy. Bulletin of entomological research 2014, 104, 117–136. [Google Scholar] [CrossRef]
- Atamian, H.S.; Chaudhary, R.; Cin, V.D.; Bao, E.; Girke, T.; Kaloshian, I. In planta expression or delivery of potato aphid Macrosiphum euphorbiae effectors Me10 and Me23 enhances aphid fecundity. Molecular Plant-Microbe Interactions 2013, 26, 67–74. [Google Scholar] [CrossRef]
- Bos, J.I.; Prince, D.; Pitino, M.; Maffei, M.E.; Win, J.; Hogenhout, S.A. A functional genomics approach identifies candidate effectors from the aphid species Myzus persicae (green peach aphid). PLoS genetics 2010, 6, e1001216. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, D.A.; De Vos, M.; Jander, G. Suppression of plant defenses by a Myzus persicae (green peach aphid) salivary effector protein. Molecular Plant-Microbe Interactions 2014, 27, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Pitino, M.; Hogenhout, S.A. Aphid protein effectors promote aphid colonization in a plant species-specific manner. Molecular Plant-Microbe Interactions 2013, 26, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.I.; Armstrong, M.R.; Gilroy, E.M.; Boevink, P.C.; Hein, I.; Taylor, R.M.; Zhendong, T.; Engelhardt, S.; Vetukuri, R.R.; Harrower, B.; et al. Phytophthora infestans effector AVR3a is essential for virulence and manipulates plant immunity by stabilizing host E3 ligase CMPG1. Proc Natl Acad Sci U S A 2010, 107, 9909–9914. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Garzo, E.; Fernandez-Mata, G.; Kassem, M.; Aranda, M.; Fereres, A. Aphids secrete watery saliva into plant tissues from the onset of stylet penetration. Entomologia Experimentalis et Applicata 2011, 139, 145–153. [Google Scholar] [CrossRef]
- Will, T.; Tjallingii, W.F.; Thönnessen, A.; van Bel, A.J. Molecular sabotage of plant defense by aphid saliva. Proceedings of the National Academy of Sciences 2007, 104, 10536–10541. [Google Scholar] [CrossRef]
- Cock, P.J.; Grüning, B.A.; Paszkiewicz, K.; Pritchard, L. Galaxy tools and workflows for sequence analysis with applications in molecular plant pathology. PeerJ 2013, 1, e167. [Google Scholar] [CrossRef]
- Cooper, W.R.; Dillwith, J.W.; Puterka, G.J. Salivary proteins of Russian wheat aphid (Hemiptera: Aphididae). Environmental entomology 2010, 39, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Wu, G.; Zhang, W. Correlation between mRNA and protein abundance in Desulfovibrio vulgaris: a multiple regression to identify sources of variations. Biochemical and biophysical research communications 2006, 339, 603–610. [Google Scholar] [CrossRef]
- Huang, H.J.; Wang, Y.Z.; Li, L.L.; Lu, H.B.; Lu, J.B.; Wang, X.; Ye, Z.X.; Zhang, Z.L.; He, Y.J.; Lu, G.; et al. Planthopper salivary sheath protein LsSP1 contributes to manipulation of rice plant defenses. Nat Commun 2023, 14, 737. [Google Scholar] [CrossRef]
- Huang, H.-J.; Wang, Y.-Z.; Li, L.-L.; Lu, H.-B.; Lu, J.-B.; Wang, X.; Ye, Z.-X.; Zhang, Z.-L.; He, Y.-J.; Lu, G. Planthopper salivary sheath protein LsSP1 contributes to manipulation of rice plant defenses. Nature Communications 2023, 14, 737. [Google Scholar] [CrossRef]
- Kunieda, T.; Fujiyuki, T.; Kucharski, R.; Foret, S.; Ament, S.; Toth, A.; Ohashi, K.; Takeuchi, H.; Kamikouchi, A.; Kage, E. Carbohydrate metabolism genes and pathways in insects: insights from the honey bee genome. Insect molecular biology 2006, 15, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.A.; Carolan, J.C.; Wilkinson, T.L. Proteomic profiling of cereal aphid saliva reveals both ubiquitous and adaptive secreted proteins. PloS one 2013, 8, e57413. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Atamian, H.S.; Shen, Z.; Briggs, S.P.; Kaloshian, I. Potato aphid salivary proteome: enhanced salivation using resorcinol and identification of aphid phosphoproteins. Journal of Proteome Research 2015, 14, 1762–1778. [Google Scholar] [CrossRef]
- Tokuda, G.; Saito, H.; Watanabe, H. A digestive β-glucosidase from the salivary glands of the termite, Neotermes koshunensis (Shiraki): distribution, characterization and isolation of its precursor cDNA by 5′-and 3′-RACE amplifications with degenerate primers. Insect Biochemistry and Molecular Biology 2002, 32, 1681–1689. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, G.; Xiang, C.; Du, M.; Cheng, J.; Liu, S.; Lou, Y. β-Glucosidase treatment and infestation by the rice brown planthopper Nilaparvata lugens elicit similar signaling pathways in rice plants. Chinese Science Bulletin 2008, 53, 53–57. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, Y.; Francis, F.; Liu, X.; Chen, J. Insight into watery saliva proteomes of the grain aphid, Sitobion avenae. Archives of Insect Biochemistry and Physiology 2021, 106, e21752. [Google Scholar] [CrossRef] [PubMed]
- Pavithran, S.; Murugan, M.; Mannu, J.; Yogendra, K.; Balasubramani, V.; Sanivarapu, H.; Harish, S.; Natesan, S. Identification of salivary proteins of the cowpea aphid Aphis craccivora by transcriptome and LC-MS/MS analyses. Insect Biochemistry and Molecular Biology 2024, 165, 104060. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, G.M.; Trono, D. Recombinant lipases and phospholipases and their use as biocatalysts for industrial applications. International journal of molecular sciences 2015, 16, 20774–20840. [Google Scholar] [CrossRef]
- Bargmann, B.O.; Munnik, T. The role of phospholipase D in plant stress responses. Current opinion in plant biology 2006, 9, 515–522. [Google Scholar] [CrossRef]
- Young, S.A.; Wang, X.; Leach, J.E. Changes in the plasma membrane distribution of rice phospholipase D during resistant interactions with Xanthomonas oryzae pv oryzae. The Plant Cell 1996, 8, 1079–1090. [Google Scholar] [CrossRef]
- Grennan, A.K. The role of trehalose biosynthesis in plants. Plant Physiology 2007, 144, 3–5. [Google Scholar] [CrossRef]
- Almeida, A.M.; Villalobos, E.; Araújo, S.S.; Leyman, B.; Van Dijck, P.; Alfaro-Cardoso, L.; Fevereiro, P.S.; Torné, J.M.; Santos, D.M. Transformation of tobacco with an Arabidopsis thaliana gene involved in trehalose biosynthesis increases tolerance to several abiotic stresses. Euphytica 2005, 146, 165–176. [Google Scholar] [CrossRef]
- Iordachescu, M.; Imai, R. Trehalose biosynthesis in response to abiotic stresses. Journal of integrative plant biology 2008, 50, 1223–1229. [Google Scholar] [CrossRef]
- Su YunLin, S.Y.; Li JunMin, L.J.; Li Meng, L.M.; Luan JunBo, L.J.; Ye XiaoDong, Y.X.; Wang XiaoWei, W.X.; Liu ShuSheng, L.S. Transcriptomic analysis of the salivary glands of an invasive whitefly. 2012.
- Zhang, Y.; Fan, J.; Sun, J.; Francis, F.; Chen, J. Transcriptome analysis of the salivary glands of the grain aphid, Sitobion avenae. Scientific Reports 2017, 7, 15911. [Google Scholar] [CrossRef]
- Hayashi, H.; Chino, M. Collection of pure phloem sap from wheat and its chemical composition. Plant and Cell Physiology 1986, 27, 1387–1393. [Google Scholar] [CrossRef]
- Field, L.M.; Devonshire, A.L. Evidence that the E4 and FE4 esterase genes responsible for insecticide resistance in the aphid Myzus persicae (Sulzer) are part of a gene family. Biochem J 1998, 330 ( Pt 1) Pt 1, 169–173. [Google Scholar] [CrossRef]
- BK, S.K.; Moural, T.; Zhu, F. Functional and structural diversity of insect glutathione S-transferases in xenobiotic adaptation. International Journal of Biological Sciences 2022, 18, 5713. [Google Scholar] [CrossRef]
- Vandermoten, S.; Harmel, N.; Mazzucchelli, G.; De Pauw, E.; Haubruge, E.; Francis, F. Comparative analyses of salivary proteins from three aphid species. Insect Molecular Biology 2014, 23, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-S.; Zhao, H.-X.; Zhu, Y.C.; Scheffler, B.; Liu, X.; Liu, X.; Hulbert, S.; Stuart, J.J. Analysis of transcripts and proteins expressed in the salivary glands of Hessian fly (Mayetiola destructor) larvae. Journal of insect physiology 2008, 54, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Belghazi, M. Extracellular superoxide dismutase in insects: characterization, function and interspecific variation in parasitoid wasps' venom. 2011.
- Shao, E.; Song, Y.; Wang, Y.; Liao, Y.; Luo, Y.; Liu, S.; Guan, X.; Huang, Z. Transcriptomic and proteomic analysis of putative digestive proteases in the salivary gland and gut of Empoasca (Matsumurasca) onukii Matsuda. BMC genomics 2021, 22, 1–17. [Google Scholar] [CrossRef]
- Conte, E.; Dinoi, G.; Imbrici, P.; De Luca, A.; Liantonio, A. Sarcoplasmic reticulum Ca2+ buffer proteins: A focus on the yet-to-Be-explored role of sarcalumenin in skeletal muscle health and disease. Cells 2023, 12, 715. [Google Scholar] [CrossRef]
- Xue WenXin, X.W.; Fan Jia, F.J.; Zhang Yong, Z.Y.; Xu QingXuan, X.Q.; Han ZongLi, H.Z.; Sun JingRui, S.J.; Chen JuLian, C.J. Identification and expression analysis of candidate odorant-binding protein and chemosensory protein genes by antennal transcriptome of Sitobion avenae. 2016.
- Carolan, J.C.; Fitzroy, C.I.; Ashton, P.D.; Douglas, A.E.; Wilkinson, T.L. The secreted salivary proteome of the pea aphid Acyrthosiphon pisum characterised by mass spectrometry. Proteomics 2009, 9, 2457–2467. [Google Scholar] [CrossRef]
- Jakhar, R.; Gakhar, S. Study and comparison of mosquito (Diptera) aminopeptidase N protein with other order of insects. International journal of Mosquito Research 2019. [Google Scholar]
- Cabot, C.; Martos, S.; Llugany, M.; Gallego, B.; Tolrà, R.; Poschenrieder, C. A role for zinc in plant defense against pathogens and herbivores. Frontiers in plant science 2019, 10, 448458. [Google Scholar] [CrossRef]
- Upadhyay, S.K.; Singh, H.; Dixit, S.; Mendu, V.; Verma, P.C. Molecular characterization of vitellogenin and vitellogenin receptor of Bemisia tabaci. PloS one 2016, 11, e0155306. [Google Scholar] [CrossRef] [PubMed]
- Wileman, H.J.; Perry, R.N.; Davies, K.G. Comparative phylogenetic analysis of vitellogenin in species of cyst and root-knot nematodes. Nematology 2023, 25, 467–476. [Google Scholar] [CrossRef]
- Brown, H.E.; Reichert, M.C.; Evans, T.A. In vivo functional analysis of drosophila Robo1 fibronectin type-III repeats. G3: Genes, Genomes, Genetics 2018, 8, 621–630. [Google Scholar] [CrossRef]
- Fu, J.; Shi, Y.; Wang, L.; Zhang, H.; Li, J.; Fang, J.; Ji, R. Planthopper-Secreted Salivary Disulfide Isomerase Activates Immune Responses in Plants. Front Plant Sci 2020, 11, 622513. [Google Scholar] [CrossRef]
- Darby, N.J.; Penka, E.; Vincentelli, R. The multi-domain structure of protein disulfide isomerase is essential for high catalytic efficiency. Journal of Molecular biology 1998, 276, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Schönbrunner, E.R.; Mayer, S.; Tropschug, M.; Fischer, G.; Takahashi, N.; Schmid, F.X. Catalysis of protein folding by cyclophilins from different species. Journal of Biological Chemistry 1991, 266, 3630–3635. [Google Scholar] [CrossRef]
- Shih, P.Y.; Sugio, A.; Simon, J.C. Molecular Mechanisms Underlying Host Plant Specificity in Aphids. Annu Rev Entomol 2023, 68, 431–450. [Google Scholar] [CrossRef]
- Thorpe, P.; Cock, P.J.; Bos, J. Comparative transcriptomics and proteomics of three different aphid species identifies core and diverse effector sets. BMC genomics 2016, 17, 1–18. [Google Scholar] [CrossRef]
- Missbach, C.; Vogel, H.; Hansson, B.S.; Groβe-Wilde, E. Identification of odorant binding proteins and chemosensory proteins in antennal transcriptomes of the jumping bristletail Lepismachilis y-signata and the firebrat Thermobia domestica: evidence for an independent OBP–OR origin. Chemical senses 2015, 40, 615–626. [Google Scholar] [CrossRef]
- Dippel, S.; Oberhofer, G.; Kahnt, J.; Gerischer, L.; Opitz, L.; Schachtner, J.; Stanke, M.; Schütz, S.; Wimmer, E.A.; Angeli, S. Tissue-specific transcriptomics, chromosomal localization, and phylogeny of chemosensory and odorant binding proteins from the red flour beetle Tribolium castaneum reveal subgroup specificities for olfaction or more general functions. BMC genomics 2014, 15, 1–14. [Google Scholar] [CrossRef]
- Stathopoulos, A.; Van Drenth, M.; Erives, A.; Markstein, M.; Levine, M. Whole-genome analysis of dorsal-ventral patterning in the Drosophila embryo. Cell 2002, 111, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.A.; Stam, R.; Warbroek, T.; Bos, J.I. Mp10 and Mp42 from the aphid species Myzus persicae trigger plant defenses in Nicotiana benthamiana through different activities. Molecular Plant-Microbe Interactions 2014, 27, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Mohamed, A.; Cattaneo, A.M.; Huang, X.; Keyhani, N.O.; Gu, M.; Zang, L.; Zhang, W. Odorant-binding proteins and chemosensory proteins in Spodoptera frugiperda: From genome-wide identification and developmental stage-related expression analysis to the perception of host plant odors, sex pheromones, and insecticides. International Journal of Molecular Sciences 2023, 24, 5595. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Sequence length distribution of genes and transcripts of the trinity generated with de novo assembly driven out of the raw reads of rose grain aphid transcriptome.
Figure 2.
Sequence length distribution of genes and transcripts of the trinity generated with de novo assembly driven out of the raw reads of rose grain aphid transcriptome.
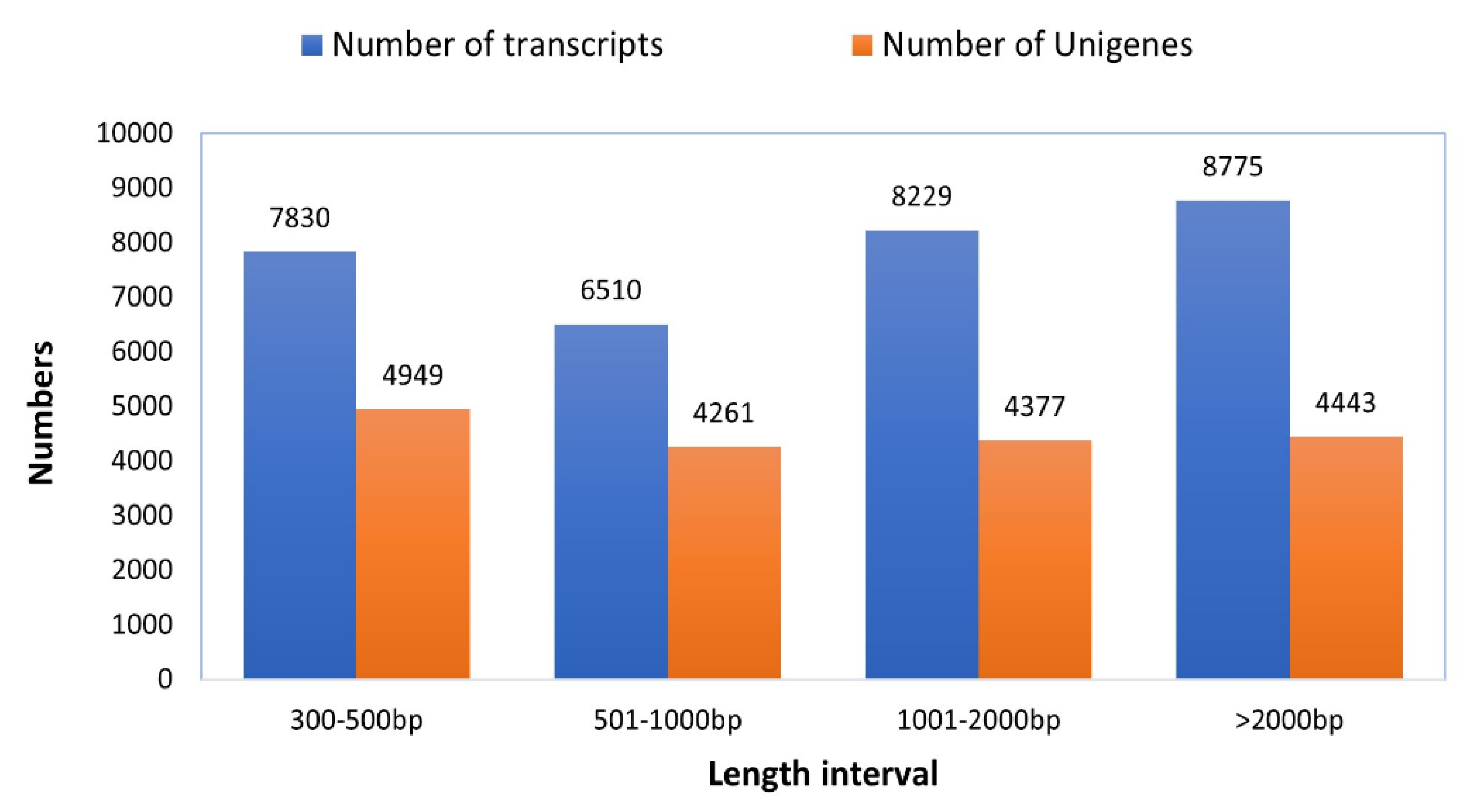
Figure 3.
Homology analysis of M. dirhodum genes against non-redundant protein sequence database. (A) Expected threshold value(E-value) in which the number BLAST hits of each gene appear by chance (E-value <1.0E−5), (B) Similarity distribution, (C) Species classification based on sequence similarity of genes among different organisms.
Figure 3.
Homology analysis of M. dirhodum genes against non-redundant protein sequence database. (A) Expected threshold value(E-value) in which the number BLAST hits of each gene appear by chance (E-value <1.0E−5), (B) Similarity distribution, (C) Species classification based on sequence similarity of genes among different organisms.
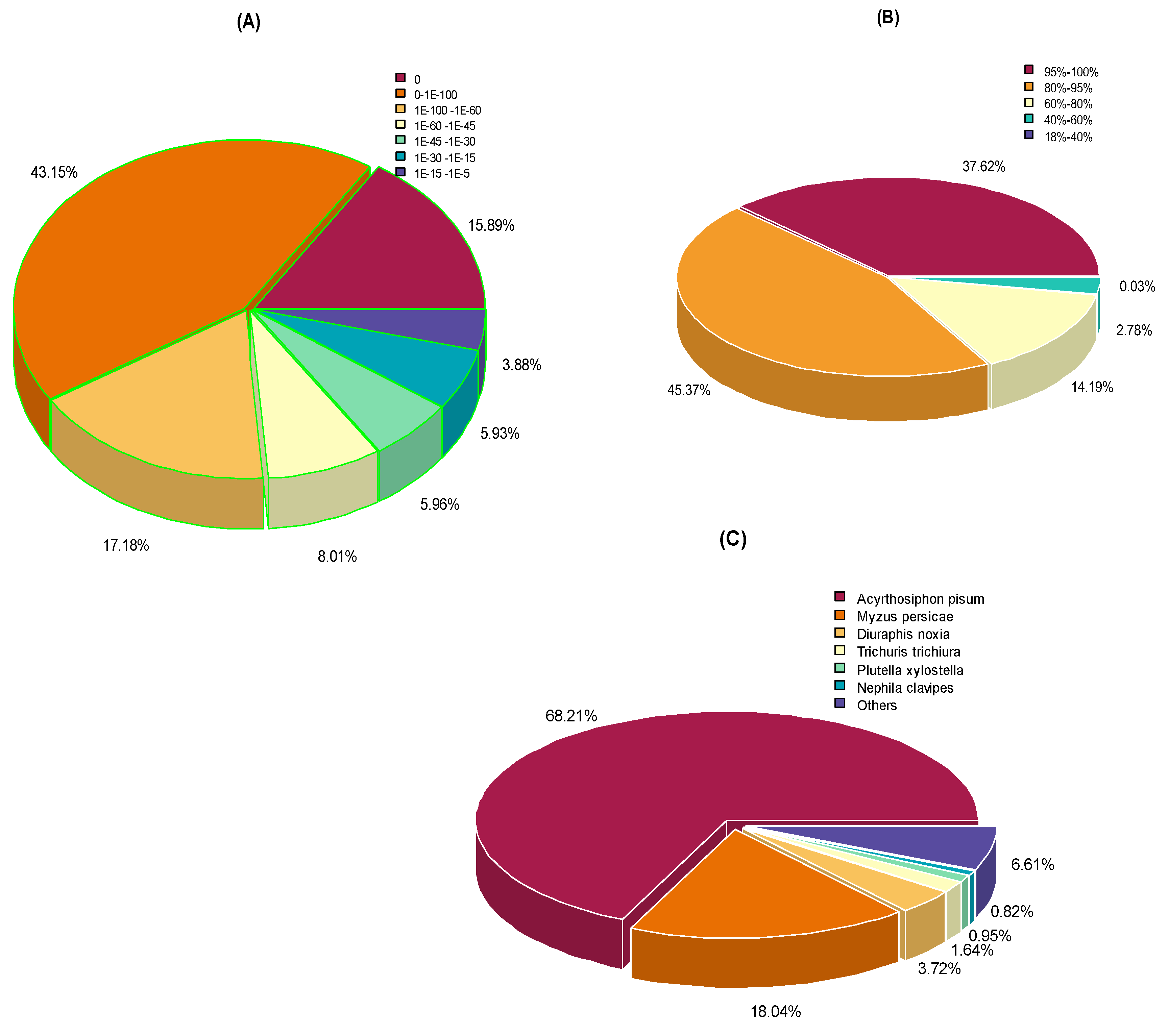
Figure 4.
The biological, molecular and cellular classification of genes associated with each functional category.
Figure 4.
The biological, molecular and cellular classification of genes associated with each functional category.

Figure 5.
Genes annotated based on Eukaryotic Orthologous Groups classification (KOG).
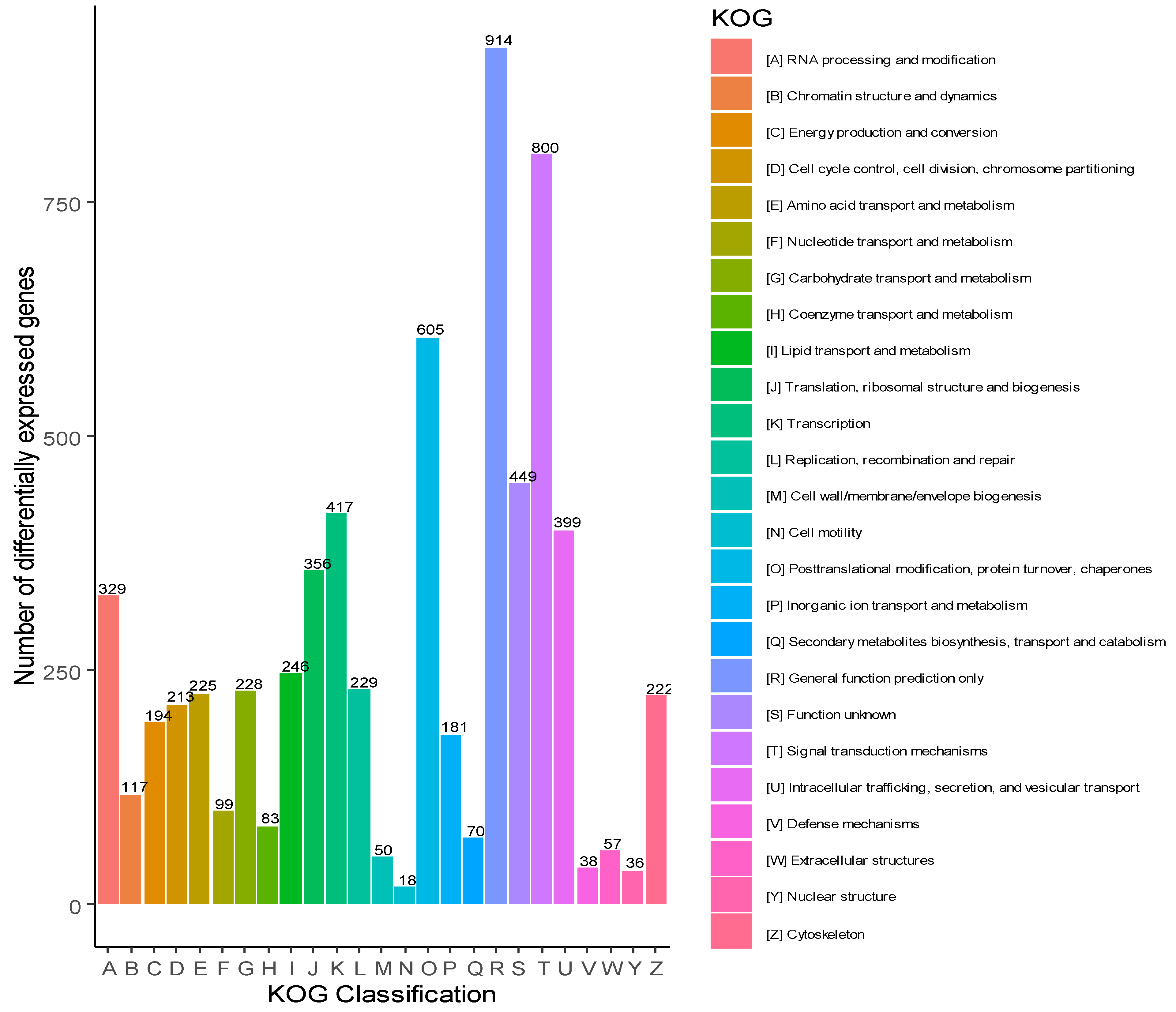
Figure 6.
Functional classification of unigenes based on KEGG pathways.
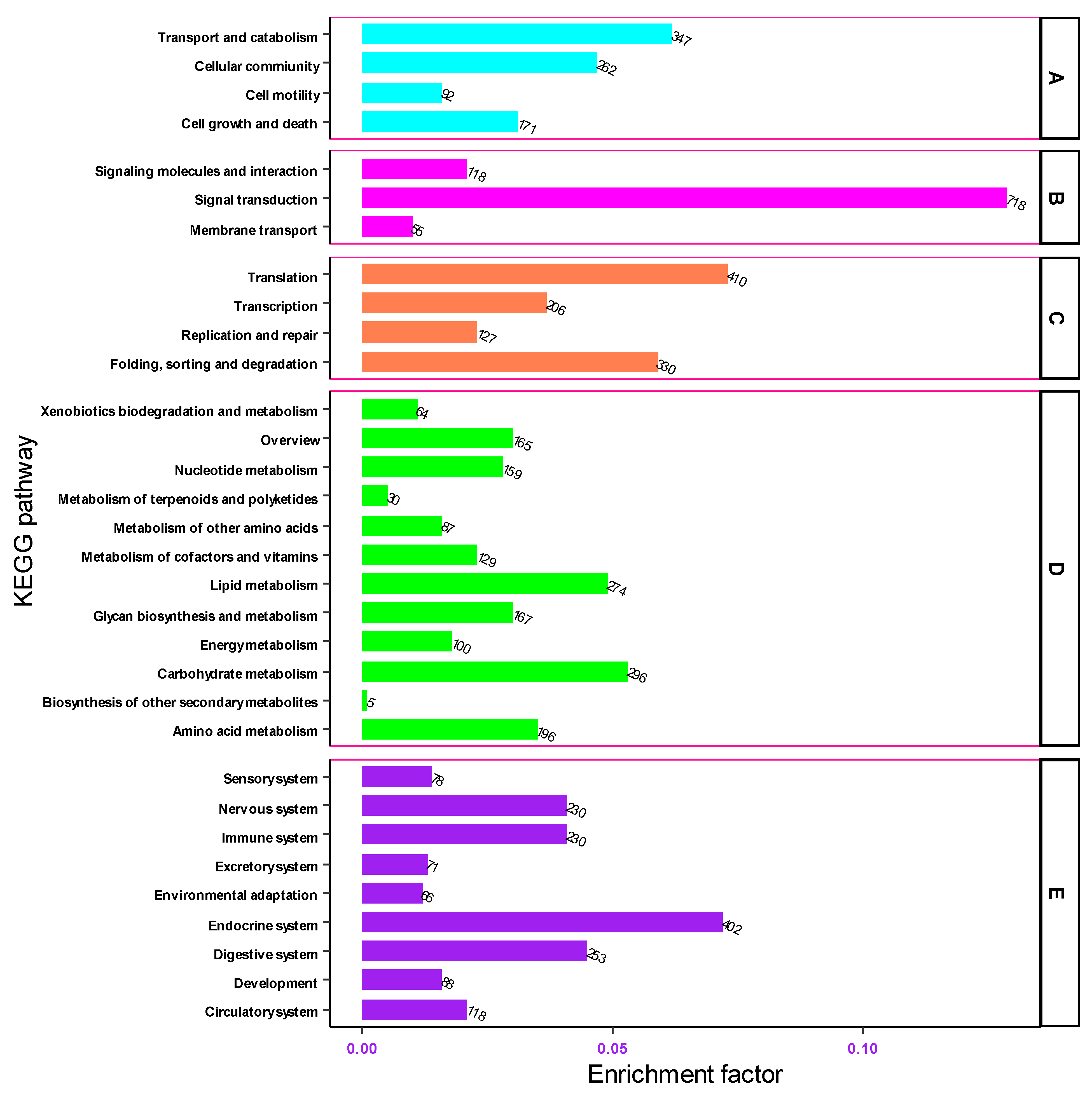

Table 1.
The quality of rose grain aphid head gene sequence and assembly.
| S/N | Statistic | Read |
| 1 | Raw Reads (bp) | 47565328 |
| 2 | Clean Reads (bp) | 46238772 |
| 3 | Clean Bases (Gb) | 6.94Gb |
| 4 | Error (%) | 0.03 |
| 5 | Q20(%) | 97.61 |
| 6 | Q30(%) | 93.04 |
| 7 | N percentage (%) | 0 |
| 8 | Total length of transcripts | 48023560 |
| 9 | Total length of genes | 25480502 |
| 10 | Number of Transcripts | 31344 |
| 11 | Number of genes | 18030 |
| 12 | Mean Length of Transcript | 1532 |
| 13 | Mean Length of gene | 1413 |
| 14 | N50 Transcript | 2335 |
| 15 | N50 Genes | 2205 |
| 16 | GC Content (%) | 42.47 |
Table 2.
Number and percent of transcripts of genes annotated against all known data bases.
| s/n | Data base | Number of Genes | Percentage of Genes (%) |
|---|---|---|---|
| 1 | Nr | 12589 | 69.82 |
| 2 | Nt | 14253 | 79.05 |
| 3 | KO | 5587 | 30.98 |
| 4 | Swiss-Prot | 9467 | 52.5 |
| 5 | Pfam | 9314 | 51.65 |
| 6 | GO | 9314 | 51.65 |
| 7 | KOG | 5850 | 32.44 |
| 8 | All data bases | 3670 | 20.35 |
| 9 | At least one data base | 14887 | 82.56 |
| 10 | Total Genes | 18030 | 100 |
Table 3.
Sequence similarity of salivary proteins among different organisms.
|
Proteins identified in the saliva of M. dirhodum |
Entry | Secretory nature | Similarity level | |||
| Yes | No | 100% | 90% | 50% | ||
| 60S acidic ribosomal protein P0 (Fragment) | A0A2H8TGI4 | √ | x | 6 | 1611 | |
| 6-phosphogluconate dehydrogenase, decarboxylating |
A0A9P0J6A7 | √ | x | x | 6 | |
| Actin related protein 1 isoform X1 | A0A6G0YHT2 | √ | 182 | 190 | 4066 | |
| ACYPI010077 protein | C4WWE5 | √ | 6 | 12 | 288 | |
| ATP synthase subunit alpha, mitochondrial | A0A6G0YU19 | √ | x | x | 6 | |
| DNA-dependent protein kinase catalytic subunit CC3 domain-containing protein |
A0A8R2JNU0 | √ | x | 3 | 4 | |
| Elongation factor 1-alpha | A0A2H8TTF1 | √ | 13 | 373 | 1691 | |
| Exoribonuclease II (Fragment) | A0A6G0WGD2 | √ | 2 | 2 | 2 | |
| Genome assembly, chromosome: | A0A9P0JBE8 | √ | 2 | 2 | 2 | |
| Genome assembly, chromosome: | A0A9P0NDC0 | √ | 2 | 3 | 4 | |
| Glucose dehydrogenase like-protein 1 | K0DCK7 | √ | x | 2 | 2 | |
| Glucose dehydrogenase like-protein 2 | K0D9J0 | √ | x | x | 2 | |
| Glycine hydroxy methyltransferase | C4WVD4 | √ | x | x | x | |
| Heat shock protein 70KD | A0A5E4MRQ3 | √ | √ | x | 14 | 331 |
| Histone H2A / H2B / H4 (Fragment) | A0A2S2NHS9 | √ | 4 | 4 | 5 | |
| Odorant receptor 49b-like | A0A6G0YXA8 | √ | x | x | x | |
| Peroxiredoxin 1 | A0A2S2NYR8 | √ | x | x | 4 | |
| PHD domain-containing protein | A0A6G0VQ84 | √ | x | x | x | |
| Protein slit | A0A2S2NEH5 | √ | x | x | 2 | |
| Putative sheath protein (Fragment) | K0D9J4 | √ | x | x | 2 | |
| RNA-directed DNA polymerase (Fragment) | A0A6G0VWQ4 | √ | x | x | x | |
| TTF-type domain-containing protein | A0A8R2H9Y1 | √ | x | 2 | 2 | |
| Tubulin beta chain | A0A6G0ZHT7 | √ | 6 | 39 | 391 | |
| Uncharacterized protein | A0A6G0U7P0 | √ | x | x | x | |
| Uncharacterized protein | A0A6G0YI97 | √ | x | x | x | |
| Uncharacterized protein | A0A8R2HBC2 | √ | x | x | x | |
| Uncharacterized protein | A0A8R2NN35 | √ | x | 8 | 14 | |
| Uncharacterized protein | A0A8R2JL05 | √ | x | 4 | 45 | |
| Uncharacterized protein (Fragment) | A0A2S2P8E1 | √ | x | x | x | |
| Vitellogenin domain-containing protein | A0A8R2F8V9 | √ | x | x | 9 | |
Table 4.
Sequence similarity of salivary proteins among different aphid species based on BLAST score ratio (BSR).
Table 4.
Sequence similarity of salivary proteins among different aphid species based on BLAST score ratio (BSR).
|
Protein names identified from the saliva of M. dirhodum |
Accession number | Pulse Crop Aphids | Cereal Crop Aphids | ||||||||
| A. craccivora | A. glycines | A. pisum | A. gossypii | S. graminum | S. avenae | R. padi | M. persicae | M. sacchari | Sipha flava | ||
| 60S acidic ribosomal protein P0 | A0A2H8TGI4 | √ | √ | √ | √ | x | x | x | x | √ | √ |
| 6-phosphogluconate dehydrogenase, | A0A9P0J6A7 | x | x | x | √ | x | x | x | x | x | x |
| Actin related protein 1 isoform X1 | A0A6G0YHT2 | √ | √ | √ | √ | x | √ | √ | x | √ | √ |
| ACYPI010077 protein | C4WWE5 | √ | √ | √ | √ | √ | x | x | x | √ | √ |
| ATP synthase subunit alpha, mitochondrial | A0A6G0YU19 | √ | x | x | x | x | x | x | x | x | x |
| DNA-dependent protein kinase catalytic subunit CC3 domain-containing protein |
A0A8R2JNU0 | √ | x | √ | × | x | x | x | x | x | x |
| Elongation factor 1-alpha | A0A2H8TTF1 | x | x | x | x | √ | x | x | x | √ | x |
| exoribonuclease II (Fragment) | A0A6G0WGD2 | √ | x | x | x | x | x | x | x | x | x |
| Genome assembly, chromosome: | A0A9P0JBE8 | x | x | x | √ | x | x | x | x | √ | x |
| Genome assembly, chromosome: | A0A9P0NDC0 | x | x | x | √ | x | x | x | x | √ | x |
| Glucose dehydrogenase like-protein 1 | K0DCK7 | x | x | x | x | x | √ | x | x | x | x |
| Glucose dehydrogenase like-protein 2 | K0D9J0 | x | x | x | x | x | √ | x | x | x | x |
| Glycine hydroxy methyltransferase | C4WVD4 | x | x | √ | x | x | x | x | x | x | x |
| Heat shock protein 70KD | A0A5E4MRQ3 | √ | √ | √ | √ | x | x | √ | √ | √ | √ |
| Histone H2A / H2B / H4 (Fragment) | A0A2S2NHS9 | √ | √ | x | x | √ | x | x | x | x | x |
| Odorant receptor 49b-like | A0A6G0YXA8 | √ | x | x | x | x | x | x | x | x | x |
| Peroxiredoxin 1 | A0A2S2NYR8 | x | x | x | x | √ | x | x | x | x | √ |
| PHD domain-containing protein | A0A6G0VQ84 | √ | x | x | x | x | x | x | x | x | x |
| Protein slit | A0A2S2NEH5 | x | x | x | x | √ | x | x | x | x | x |
| Putative sheath protein | K0D9J4 | x | x | x | x | x | √ | x | x | x | x |
| RNA-directed DNA polymerase | A0A6G0VWQ4 | √ | x | x | x | x | x | x | x | x | x |
| TTF-type domain-containing protein | A0A8R2H9Y1 | x | x | √ | x | x | x | x | x | x | x |
| Tubulin beta chain | A0A6G0ZHT7 | √ | √ | x | √ | x | x | x | x | √ | √ |
| Uncharacterized protein | A0A6G0U7P0 | x | √ | x | x | x | x | x | x | x | x |
| Uncharacterized protein | A0A6G0YI97 | √ | x | x | x | x | x | x | x | x | x |
| Uncharacterized protein | A0A8R2HBC2 | x | x | √ | x | x | x | x | x | x | x |
| Uncharacterized protein | A0A8R2NN35 | √ | √ | √ | √ | √ | x | x | x | √ | √ |
| Uncharacterized protein | A0A8R2JL05 | √ | √ | √ | √ | x | x | x | x | x | √ |
| Uncharacterized protein | A0A2S2P8E1 | x | x | x | x | √ | x | x | x | x | x |
| Vitellogenin domain-containing protein | A0A8R2F8V9 | √ | √ | √ | √ | x | x | x | x | √ | √ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



