
Submitted:
02 August 2024
Posted:
06 August 2024
You are already at the latest version
Abstract
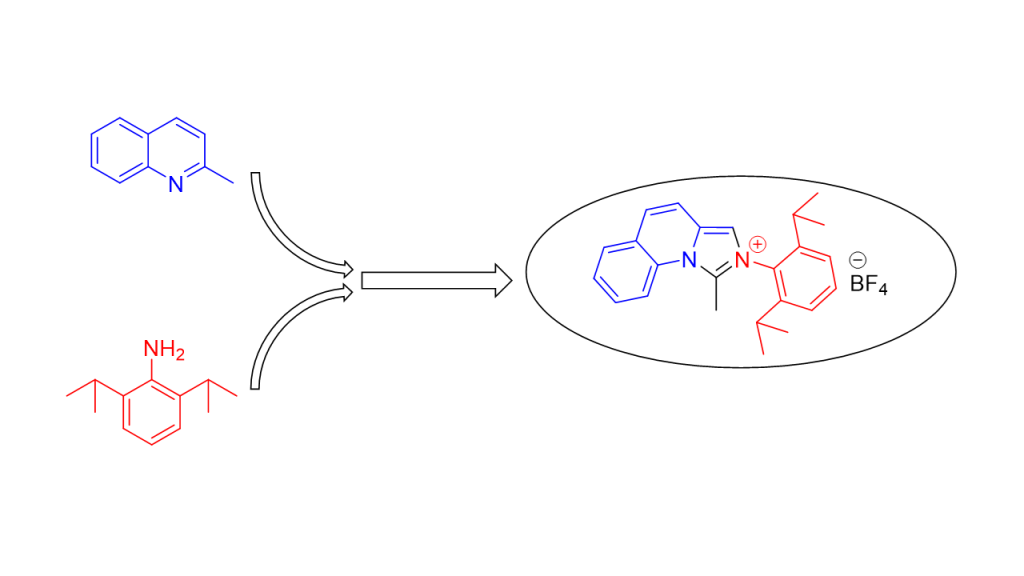
A new heterocyclic compound, 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate 1 was obtained from its precursor, N-(2,6-diisopropylphenyl)-N-(quinolin-2-ylmethyl)acetamide 2, by POCl3-mediated cyclization. For the first time – the tertiary acetamide 2, precursor of 1, was synthesized by using commercially available starting materials. The structure of 1 was unquestionably confirmed by 1H-, 13C-, 2D-NMR-, IR- spectroscopy and mass-spectrometry. Its optical properties were also studied.
Keywords:
heterocyclic compounds
; imidazolium salts
; abnormal N-heterocyclic carbenes
; cationic heterocycles
1. Introduction
In recent years the interest in new imidazolium salts has been growing rapidly. This is mostly as a result of the fact that they are precursors for the synthesis of N-heterocyclic carbenes [1]. The free N-heterocyclic carbene is generated after deprotonation of the imidazolium salt with a proper base. The formed carbene can serve as organocatalyst [2] or as transition metal ligand [3,4,5,6,7]. Such NHC-based metal complexes are used as homogeneous catalysts [4,5,6,7], for instance the PEPPSI-catalyst [8]. When the carbon atom, located between the two nitrogen atoms is substituted, deprotonation occurs in other position in the imidazolium ring and thus abnormal N-heterocyclic carbenes are generated [9]. Their respective metal complexes are also reported [10,11].
On the other hand, imidazolium salts bearing suitable substituents and counterions are also useful ionic liquids [12,13]. They are mainly used as green recyclable solvents due to their stability, low vapor pressure and low toxicity.
There are two main routes for the synthesis of imidazolium salts: a) building an imidazole ring [14] and b) alkylating an imidazole ring [14]. The first approach is applicable when phenyl substituted imidazolium salt is desired. The second approach is preferred when imidazolium salt with two alkyl substituents is needed.
Imidazolium salts with imidazole ring, condensed with aromatic system, are also reported in the literature [15,16,17,18,19]. However, imidazoquinolinium salts are relatively rare [20,21,22,23,24,25]. Their synthesis can also follow the mentioned methods for preparation of imidazolium salts. Representatives of this class of imidazolium salts have been prepared by cyclization of tertiary formamides [20,21], cyclization of aldimines [22,23,24,25] and by alkylation of imidazoquinolines [24,25].
Despite the reported imidazoquinolinium salts, salts bearing a substituent at the carbon atom located between the two nitrogen atoms are not described in the literature.
Herein we report for the first time a synthetic procedure for obtaining such type of substituted imidazoquinolinium salt, which emits in the UV-region of the light spectrum. This compound could later serve as a precursor for abnormal N-heterocyclic carbene.
On the other hand, the new compound possesses a methyl group next to a positively charged quaternary nitrogen atom. It is known that such type of heterocycles can participate in aldol type condensation, making this methyl-substituted imidazoquinoinium salt an attractive building block for cationic hemicyanine and styryl dyes [26,27].
2. Results and Discussion
2.1. Synthesis of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate 1
The synthesis of the title compound, 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate 1, was achieved in several steps, using adapted previously reported [20,21] procedure (Scheme 1 and Scheme 2).
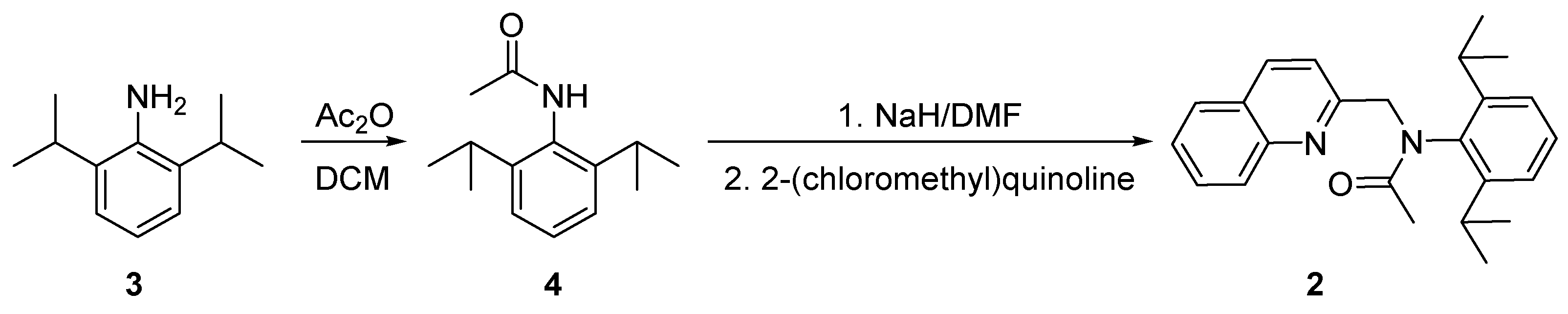
N-(2,6-diisopropylphenyl)-N-(quinolin-2-ylmethyl)acetamide 2, was prepared by acylation of the respective aniline 3 with acetic anhydride at room temperature (Scheme 1). The amide 4 was isolated with good yield. It is interesting to note that our first attempt for acylation, which includes boiling for 8 hours of 2,6-diisopropylaniline 3 with a large excess of acetic acid, was unsuccessful because of the sterically hindered NH2-group.
The second step of the procedure includes alkylation of the nitrogen atom of the resulting primary amide 4 by 2-(chloromethyl)quinoline [28] in the presence of NaH in anhydrous conditions (Scheme 1). The product, N-(2,6-diisopropylphenyl)-N-(quinolin-2-yl methyl)acetamide 2, was found to be unstable on silica. Fortunately, it was successfully purified by recrystallization. This tertiary amide 2 was applied as a substrate in a POCl3-mediated cyclization to the desired 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium salt 1 (Scheme 2). In order to increase its solubility in organic solvents, the product of the cyclization was treated with an aqueous solution of NH4BF4.
2.2. NMR Studies
1D (1H, 13C, DEPT-135) and 2D (COSY, HSQC, HMBC) NMR-techniques were used to confirm the structure of all compounds. See Supplementary Materials for copies of the NMR-spectra. N-(2,6-diisopropylphenyl)-N-(quinolin-2-ylmethyl)acetamide 4 was studied in two deuterated solvents - chloroform and dimethyl sulfoxide (Figures S1-S6). In the 1H-NMR-spectrum in chloroform were observed two sets of signals, assigned as major and minor form, in ratio 1:0.57. This behavior in solution is particularly discussed previously [29]. In dimethyl sulfoxide the compound demonstrates virtually only one form. This is the reason why 2D-NMR studies were carried out in this solvent. Using these 2D-NMR-experiments each signal was assigned to the respective atom. In contrast to the secondary amide, tertiary amid N-(2,6-diisopropylphenyl)-N-(quinolin-2-ylmethyl)acetamide 2 demonstrates only one form in solution (Figures S6-S17). In its 1H spectrum clearly could be seen signals corresponding to the quinoline and 2,6-diisopropylphenyl systems, bonded by CH2-group. In the HMBC spectrum of the compound interactions between methylene protons (4.98 ppm) and carbon atoms from both systems were observed.
All the spectra of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate 1 were recorded in deuterated dimethyl sulfoxide and methanol in order to avoid signal overlap (Figures S19-S42). Unfortunately, this wasn’t avoided completely: in the proton spectra, recorded in DMSO three aromatic protons were overlapped. When methanol was used as a solvent, signals for only two protons were overlapped, however, the signal for one carbon atom could not be detected. Thus, deuterated DMSO was preferred. In the 1H-spectrum, the singlet, corresponding to the imidazolium CH3-group is shifted downfield (3.03 ppm) in comparison with the signal in the tertiary amid spectrum. Instead of a singlet for the protons of CH2-group at 4.98 ppm, in the proton spectrum of compound 1 a signal for one proton (H3 from imidazole ring) at 8.51 ppm was observed. The signal of the carbon atom from the C=O group in the spectrum of N-(2,6-diisopropylphenyl)-N-(quinolin-2-ylmethyl)acetamide 2 in the spectrum of the imidazolium tetrafluoroborate 1 is shifted upfield.
2.2. Optical Properties
The photophysical properties of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate 1 were determined by UV-VIS and fluorescent spectroscopy. The absorption and fluorescence spectra were recorded in methanol (MeOH) and acetonitrile (MeCN), Figure 1 and Table 1. In the UV spectrum in MeOH are observed maxima at 246, 256, 264, 300, 311, and 325 nm corresponding to the typical for the benzene and quinoline π-π* electronic transitions (see Figure S43 for the whole spectrum). The same applies to the spectrum in MeCN. The molar absorptivities are almost identical – 10 444 M-1.cm-1 (y = 10443x - 0.0366; R2=0.9990) and 10 106 M-1.cm-1 (y = 10106x + 0.0013; R2=0.9998) in MeOH and MeCN, respectively (Table 1 and Figure S45). The emission spectra are also very similar as a band with two maxima at 348 nm and 358 nm is observed in both solvents. The only difference is the ratio of the maxima – in MeOH the maximum at 358 nm is slightly less intensive compared to that at 348 nm, while in the case of MeCN, they are with equal intensity. Since 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate emits at shorter wavelengths than visible light, it doesn’t possess fluorescent properties.
4. Materials and Methods
Melting points were determined on an SRS MPA120 EZ-Melt apparatus. The IR spectra were recorded with a Shimadzu FTIR-8400S spectrophotometer. 1H and 13C NMR spectra were recorded on a Bruker AVNEO 400 spectrometer (at 400 MHz for 1H and 100.6 MHz for 13C respectively. Chemical shifts are given in ppm. The NMR-spectra of all synthesized compounds could be found in the Supplementary Materials. Liquid chromatography mass spectrometry analysis (LC-HRAM) was carried out on Q Exactive® hybrid quadrupole-Orbitrap® mass spectrometer (ThermoScientific Co, Waltham, MA, USA) equipped with a HESI® (heated electrospray ionization) module, TurboFlow® Ultra High Performance Liquid Chromatography (UHPLC) system (ThermoScientific Co, Waltham, MA, USA) and HTC PAL® autosampler (CTC Analytics, Zwingen, Switzerland). The chromatographic separations of the analyzed compounds were achieved on Nucleoshell C18 (100 × 2.1 mm, 2.7 µm) analytical column (Macherey-Nagel, Düren, Germany). Full-scan mass spectra over the m/z range 100–600 were acquired in positive ion mode at resolution settings of 140,000. The used mass spectrometer operating parameters were: spray voltage—4.0 kV; capillary temperature—320 °C; probe heater temperature—300 °C; sheath gas flow rate 40 units; auxiliary gas flow 12 units; sweep gas 2 units (units refer to arbitrary values set by the Q Exactive Tune software); and S-Lens RF level of 50.00. Nitrogen was used for sample nebulization and collision gas in the HCD cell. All derivatives were quantified using 5 ppm mass tolerance filters to their theoretically calculated m/z values. Data acquisition and processing were carried out with XCalibur® ver 2.4 software package (ThermoScientific Co, Waltham, MA, USA). UV-Vis spectra were carried out on a Shimadzu UV-1800 spectrophotometer. Fluorescence spectra were recorded at room temperature on a PerkinElmer LS45. Reactions were monitored by TLC on silica gel 60 F254.
Trichloroisocyanuric acid and 2,6-diisopropylaniline were purchased from Acros Organics. 2-Methylquinoline and toluene were purchased from Honeywell. All the other solvents and reagents were purchased from local suppliers. Toluene was dried by refluxing with sodium and benzophenone under argon atmosphere and distilled. N,N-Dimethylformamide (DMF) was dried by storing for one week over CaH2 and used without distillation.
Synthesis of N-(2,6-diisopropylphenyl)acetamide 4
2,6-Diisopropylaniline 3 (1.078 g, 6.08 mmol) was dissolved in 10 mL dichloromethane by stirring at room temperature. Acetic anhydride (1.24 g, 12.16 mmol) was added to the solution in one portion. The mixture was stirred at room temperature for 8 hours. Dichloromethane was removed under reduced pressure and the crystalline residue was dried under a vacuum (water aspirator) at room temperature for 3 hours. The light pink crystalline residue was dissolved in dichloromethane and transferred in a separatory funnel. Concentrated aqueous solution of Na2CO3 was added and the funnel was shaken vigorously. The water layer was extracted with dichloromethane and the combined organic layers were dried with anhydrous Na2SO4. After removal of the solvent (rotavapor) the product (1.3 g, 98%) was additionally purified by recrystallization from toluene to afford snow white crystals, m.p. = 189.0–189.5 °C.
1H NMR (500 MHz, DMSO) δ 9.19 (s, 1H, NHCO), 7.24 (t, J = 7.8 Hz, 1H, H4), 7.14 (d, J = 7.7 Hz, 2H, H3, H5), 3.05 (hept, J = 6.9 Hz, 2H, CH-isopropyl), 2.05 (s, 3H, CH3CO), 1.13 (s, 12H, CH3-isopropyl). 1H NMR (500 MHz, CDCl3), Major form: δ 7.31 (d, J = 7.7 Hz, 1H, H4), 7.19 (d, J = 7.7 Hz, 2H, H3, H5), 6.86 (s, 1H, NHCO), 3.10 (d, J = 6.9 Hz, 2H, CH-isopropyl), 2.23 (s, J = 3.6 Hz, 3H, CH3CO), 1.22 (d, J = 6.9 Hz, 12H, CH3-isopropyl). Minor form: δ 7.36 (t, J = 7.7 Hz, 1H), 7.22 (d, J = 7.7 Hz, 2H, H3, H5), 7.04 (s, 1H, H4), 3.22 (d, J = 6.9 Hz, 2H, CH-isopropyl), 1.76 (s, J = 3.2 Hz, 3H, CH3CO), 1.27 (d, J = 6.9 Hz, 6H, CH3-isopropyl), 1.18 (d, J = 6.8 Hz, 6H, CH3-isopropyl). Major to minor form ratio = 1:0.57. 13C NMR (126 MHz, DMSO) δ 169.47 (CH3CO), 146.40 (C2, C6), 133.22 (4C1), 127.81 (C4), 123.23 (C3, C5), 28.46 (CH-isopropyl), 24.18 (CH3-isopropyl), 23.66 (CH3-isopropyl), 22.97 (CH3CO). HRMS (ESI) m/z calculated for [M+H]+ 220.16993, found 220.16959 (ppm: 1.54).
Synthesis of N-(2,6-diisopropylphenyl)-N-(quinolin-2-ylmethyl)acetamide 2
N-(2,6-diisopropylphenyl)acetamide 4 (0.329 g, 1.5 mmol) was dissolved in 5 mL dry DMF. The system was purged with argon and cooled in an ice bath for 15 minutes. To the transparent colorless solution NaH (0.108 g, 2.2 mmol, 50% in mineral oil) was added. Moderate effervescence was observed. The resulting white suspension was stirred for one hour in an ice bath. After that, 2-(chloromethyl)quinoline (0.266 g, 1.5 mmol) was added and the reaction mixture was stirred at room temperature for 12 hours. DMF was removed under reduced pressure and 5 mL of water were added to the crude residue. To the suspension ethyl acetate was added, and the aqueous layer was extracted with the same solvent. The combined organic layers were dried with Na2SO4 and the volatiles were removed under reduced pressure (rotavapor), leaving yellow crystals (0.540 g, quantitative yield). The recrystallization from cyclohexane gave 0.250 g (46%) yellow crystals, m.p. = 124.0–125.0 °C.
1H NMR (500 MHz, CDCl3) δ 8.07 (d, J = 8.5 Hz, 1H, H4), 7.86 (d, J = 8.5 Hz, 1H, H8), 7.81 (d, J = 8.5 Hz, 1H, H3), 7.72 (d, J = 8.0 Hz, 1H, H5), 7.57 (t, J = 7.1 Hz, 1H, H7), 7.43 (t, J = 7.5 Hz, 1H, H6), 7.24 (t, J = 7.7 Hz, 1H, H4-2,6-diisopropylphenyl), 7.07 (d, J = 7.7 Hz, 2H, H3, H5-2,6-diisopropylphenyl), 4.98 (s, 2H, CH2N), 2.85 (hept, J = 6.8 Hz, 2H, CH-isopropyl), 1.80 (s, 3H, CH3CO), 1.06 (d, J = 6.9 Hz, 6H, CH3-isopropyl), 0.72 (d, J = 6.8 Hz, 6H, CH3-isopropyl). 13C NMR (126 MHz, CDCl3) δ 172.50 (CH3CO), 158.05 (4C2), 147.32 (4C8a), 146.29 (4C2, 4C6 - 2,6-diisopropylphenyl), 138.80 (4C1-2,6-diisopropylphenyl), 136.48 (C4), 129.33 (C7), 129.09 (C4-2,6-diisopropylphenyl), 129.04 (C8), 127.45 (C5), 127.42 (4C4a),126.32 (C6), 124.86 (C3, C5 - 2,6-diisopropylphenyl), 122.25 (C3), 57.30 (CH2N), 28.32 (CH-isopropyl), 24.36 (CH3-isopropyl), 24.19 (CH3-isopropyl), 22.28 (CH3CO). HRMS (ESI) m/z calculated for [M+H]+ 361.22806, found 361.22744 (ppm: 1.72).
Synthesis of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate 1
N-(2,6-diisopropylphenyl)-N-(quinolin-2-ylmethyl)acetamide 2 (0.170 g, 0.47 mmol) was dissolved in 30 mL dry toluene. The resulting solution was heated to 100oC and POCl3 (0.2 ml, 0.328 g, 2.14 mmol) was added. The temperature was elevated to 105 oC and the reaction mixture was heated for 48 hours. Volatiles were distilled under reduced pressure, leaving dark solid. The crude product was dissolved in minimal amount of boiling water and the solution was filtered. To the yellow hot filtrate concentrated aqueous NH4BF4 (0.065 g, 6.2 mmol) was added, resulting in the formation of a white precipitate. After cooling to room temperature, the precipitate was filtered and air dried. Yield: 0.180 g (89%) white solid, m.p. > 230 °C.
1H NMR (500 MHz, DMSO) δ 8.57 (d, J = 8.5 Hz, 1H, H9), 8.51 (s, 1H, H3), 8.10 (dd, J = 7.7, 1.4 Hz, 1H, H6), 7.86 (t, J = 7.9 Hz, 1H, H8), 7.80 (t, J = 7.3 Hz, 1H, H7), 7.76 – 7.67 (m, 3H, H4, H5, H4-2,6-diisopropylphenyl), 7.56 (d, J = 7.9 Hz, 2H, H3,H5-2,6-diisopropylphenyl), 3.03 (s, 3H, CH3-imidazole), 2.28 (hept, J = 6.8 Hz, 2H, CH-isopropyl), 1.13 (d, J = 6.8 Hz, 6H, CH3-isopropyl), 1.11 (d, J = 6.9 Hz, 6H, CH3-isopropyl). 1H NMR (500 MHz, MeOD) δ 8.63 (d, J = 8.6 Hz, 1H, H9), 8.30 (s, 1H, H3), 8.05 (d, J = 7.7 Hz, 1H, H6), 7.89 (t, J = 7.7 Hz, 1H, H8), 7.80 (t, J = 7.5 Hz, 1H, H7), 7.77 – 7.69 (m, 2H, H5, H4-2,6-diisopropylphenyl), 7.66 (d, J = 9.6 Hz, 1H, H4), 7.57 (d, J = 7.8 Hz, 2H, H3,H5-2,6-diisopropylphenyl), 3.14 (s, 3H, CH3-imidazole), 2.28 (d, J = 6.8 Hz, 2H, CH-isopropyl), 1.24 (d, J = 6.8 Hz, 6H, CH3-isopropyl), 1.21 (d, J = 6.8 Hz, 6H, CH3-isopropyl). 13C NMR (126 MHz, DMSO) δ 145.96 (4C2, 4C6 - 2,6-diisopropylphenyl), 140.86 (4C1-CH3), 132.48 (C4-2,6-diisopropylphenyl), 132.04 (4C9a), 130.20 (4C1-2,6-diisopropylphenyl), 130.17 (C8), 130.06 (C6), 129.47 (4C3a), 128.77 (C7), 127.32 (C5), 126.22 (4C5a), 125.48 (C3,C5-2,6-diisopropylphenyl), 119.55 (C9), 116.38 (C3), 115.95 (C4), 28.08 (CH-isopropyl), 25.01 (CH3-isopropyl), 23.57 (CH3-isopropyl), 15.16 (CH3-imidazole). HRMS (ESI) m/z calculated for [M-BF4+H]+ 343.21739, found 343.21633 (ppm: 3.10). IR (nujol): ν = 3120, 1645, 1610, 1597, 1568, 1423, 1049, 1030 cm-1 (see Figure S46).
5. Conclusions
A new methyl-substituted imidazoquinolinium salt was prepared and was studied via 1H-, 13C-, 2D-NMR-, IR-, UV- and fluorescent spectroscopies and mass-spectrometry. Its optical properties in methanol and acetonitrile are very similar as in both solvents the longest wavelength maxima in the UV spectrum is at 315 nm. Although the synthesized imidazoquinolinium salt possesses planar conjugated fragments, it emits in the UV region at 358 nm, and thus it is non-fluorescent.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figures S1–S46. 1H, 13C, COSY, HSQC, HMBC NMR spectra of all compounds, UV, fluorescent and IR spectra of compound 1.
Author Contributions
Conceptualization, R.L. and L.B.; methodology, R.L., L.B., I.K., M.I. and V.L.; validation, L.B., I.K., V.L., M.I. and R.L.; investigation, L.B., I.K., V.L., M.I and R.L.; data curation, R.L., L.B. and I.K.; writing—original draft preparation, R.L. and I.K.; writing—review and editing, L.B., I.K., V.L. and M.I.; visualization, R.L., L.B. and I.K.; supervision, R.L.; project administration, R.L.; funding acquisition, R.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Sofia University research fund, grant number 80-10-7 from 29.3.2024.
Data Availability Statement
Additional research data can be obtained from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Arduengo III, A. J.; Harlow, R. L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 1, 361–363. [CrossRef]
- Flanigan, D.; Romanov-Michailidis, F.; White, N.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 17, 9307–9387. [CrossRef]
- Herrmann, W.; Köcher, C. N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. Engl. 1997, 36, 1047-1054. [CrossRef]
- Kantchev, E.; O’Brien, C.; Organ, M. Palladium Complexes of N-Heterocyclic Carbenes as Catalysts for Cross-Coupling Reactions—A Synthetic Chemist’s Perspective. Angew. Chem. Int. Ed. 2007, 46, 2768–2813. [CrossRef]
- Herrmann, W.A. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [CrossRef]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic Carbenes: Synthesis and Coordination Chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [CrossRef]
- Herrmann, W.; Weskamp, T.; Böhm, V. Metal Complexes of Stable Carbenes. Adv. Organomet. Chem. 2001, 48, 1-69.
- O’Brien, C.J.; Kantchev, E.A.B.; Valente, C.; Hadei, N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Michael, G.; Organ, M.G. Easily Prepared Air- and Moisture-Stable Pd–NHC (NHC=N-Heterocyclic Carbene) Complexes: A Reliable, User-Friendly, Highly Active Palladium Precatalyst for the Suzuki–Miyaura Reaction. Chem. A Eur. J. 2006, 12, 4743- 4748. [CrossRef]
- Aldeco-Perez, E.; Rosenthal, A.; Donnadieu, B.; Parameswaran, P.; Frenking, G.; Bertrand, G. Isolation of a C5-Deprotonated Imidazolium, a Crystalline “Abnormal” N-Heterocyclic Carbene, Science 2009, 326, 556-559. [CrossRef]
- Arnold, P.L.; Pearson, S. Abnormal N-heterocyclic carbenes. Co-ord. Chem. Rev. 2007, 251, 596–609. [CrossRef]
- Schuster, O.; Yang, L.; Raubenheimer, H.G.; Albrecht, M. Beyond ConventionalN-Heterocyclic Carbenes: Abnormal, Remote, and Other Classes of NHC Ligands with Reduced Heteroatom Stabilization. Chem. Rev. 2009, 109, 3445–3478. [CrossRef]
- Vekariya, R.L. A review of ionic liquids: Applications towards catalytic organic transformations. J. Mol. Liq. 2017, 227, 44–60. [CrossRef]
- Ali, M.; Praveen, M.; Ghouse, A.; Fatima, F. A Brief Review of Ionic Liquids: Synthesis and Applications. Int. J. Biomed. Res. 2021, 1, 2, 11-19.
- Benhamou, L.; Chardon, E.; Lavigne, G.; Bellemin-Laponnaz, S.; César, V. Synthetic Routes to N-Heterocyclic Carbene Precursors. Chem. Rev. 2011, 111, 4, 2705–2733. [CrossRef]
- Sivaram, H.; Tan, J.; Huynh, H.V. Syntheses, Characterizations, and a Preliminary Comparative Cytotoxicity Study of Gold(I) and Gold(III) Complexes Bearing Benzimidazole- and Pyrazole-Derived N-Heterocyclic Carbenes. Organometallics 2012, 31, 5875–5883. [CrossRef]
- Gillen, J.H.; Moore, C.A.; Vuong, M.; Shajahan, J.; Anstey, M.R.; Alston, J.R.; Bejger, C.M. Synthesis and disassembly of an organometallic polymer comprising redox-active Co4S4 clusters and Janus biscarbene linkers. Chem. Commun. 2022, 58, 4885–4888. [CrossRef]
- Wang, H.; Xia, Y.; Lv, S.; Xu, J.; Sun, Z. Facial and practical synthesis of benzimidazole-based N-heterocyclic carbenes. Tetrahedron Lett. 2013, 54, 2124–2127. [CrossRef]
- Tronnier, A.; Pöthig, A.; Metz, S.; Wagenblast, G.; Münster, I.; Strassner, T. Enlarging the π System of Phosphorescent (C^C*) Cyclometalated Platinum(II) NHC Complexes. Inorg. Chem. 2014, 53, 6346–6356. [CrossRef]
- Ullah, F.; Kindermann, M.; Jones, P.; Heinicke, J. Annulated N-Heterocyclic Carbenes: 1,3-Ditolylphenanthreno[9,10-d]imidazol-2-ylidene and Transition Metal Complexes Thereof, Organometallics, 2009, 28, 8, 2441–2449. [CrossRef]
- Alcarazo, M.; Roseblade, S.J.; Cowley, A.R.; Fernández, R.; Brown, J.M.; Lassaletta, J.M. Imidazo[1,5-a]pyridine: A Versatile Architecture for Stable N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2005, 127, 3290–3291. [CrossRef]
- Lyapchev, R.; Petrov, P.; Dangalov, M.; Vassilev, N.G. Synthesis and structure elucidation of allyl Pd(II) complexes of NHC ligands derived from substituted imidazo[1,5-a]quinolin-1(2H)-ylidene. J. Organomet. Chem. 2017, 851, 194–209. [CrossRef]
- Kriechbaum, M.; Winterleitner, G.; Gerisch, A.; List, M.; Monkowius, U. Synthesis, Characterization and Luminescence of Gold Complexes Bearing an NHC Ligand Based on the Imidazo[1,5-a]quinolinol Scaffold. Eur. J. Inorg. Chem. 2013, 2013, 5567–5575. [CrossRef]
- Tao, W.; Nakano, R.; Ito, S.; Nozaki, K. Copolymerization of Ethylene and Polar Monomers by Using Ni/IzQO Catalysts. Angew. Chem. Int. Ed. 2016, 55, 2835–2839. [CrossRef]
- Konwar, M.; Hazarika, N.; Sarmah, B.; Das, A. Ruthenium(II)-Catalyzed Oxidative Annulation of Imidazo[1,5-a]quinolin-2-iums Salts and Internal Alkynes via C−H Bond Activation. Chem. Eur. J. 2024, 30, e202401133. [CrossRef]
- Konwar, M.; Hazarika, N.; Sarmah, B.; Das, A. Ru/O2-Catalyzed Oxidative C–H Activation/Alkyne Annulation Using Quinoline-Functionalized NHC as a Directing and Functionalizable Group. Org. Lett. 2024, 26, 2965–2970. [CrossRef]
- Deligeorgiev, T.; Vasilev, A.; Kaloyanova, S.; Vaquero, J.J. Styryl dyes – synthesis and applications during the last 15 years. Color. Technol. 2010, 126, 55–80. [CrossRef]
- Said, A.; Kandinska, M.; Vasilev, A.; Grabchev, I. Styryl hemicyanine-DNA assembly for selective Hg2+ sensing and molecular computing. J. Photochem. Photobiol., A 2024, 452, 115590- 115600. [CrossRef]
- Jeromin, G.E.; Orth, W.; Rapp, B.; Weiß, W. Seitenkettenchlorierungen von N-Heterocyclen mit Trichlorisocyanursäure (TCC). Chem. Ber. 1987, 120, 649–651. [CrossRef]
- Kessler, H. Nachweis innermolekularer beweglichkeit durch nmr-spektrometrie-III: Magnetische nichtäquivalenz geminaler gruppen durch rotationshinderung in achiralen molekülen. Tetrahedron 1968, 24, 1857-1867. [CrossRef]
Scheme 1.
Synthesis of N-(2,6-diisopropylphenyl)-N-(quinolin-2-ylmethyl)acetamide 2.
Scheme 2.
Synthesis of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate 1.
Scheme 2.
Synthesis of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate 1.
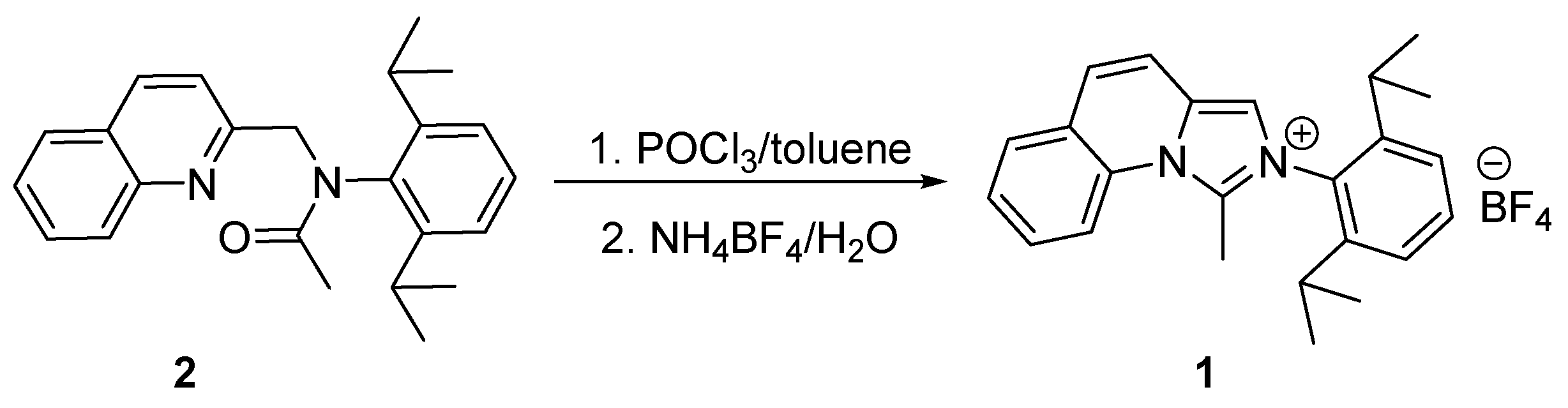
Figure 1.
Normalized UV-VIS and fluorescent spectra of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate: (a) UV-VIS (red line) and fluorescent spectra (blue line) in MeOH; (b) Comparison between the spectra in MeOH and MeCN (dotted lines: UV-VIS – cyan and fluorescent spectra – black line). The UV spectrum below 270 nm is shown on Figure S43.
Figure 1.
Normalized UV-VIS and fluorescent spectra of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate: (a) UV-VIS (red line) and fluorescent spectra (blue line) in MeOH; (b) Comparison between the spectra in MeOH and MeCN (dotted lines: UV-VIS – cyan and fluorescent spectra – black line). The UV spectrum below 270 nm is shown on Figure S43.
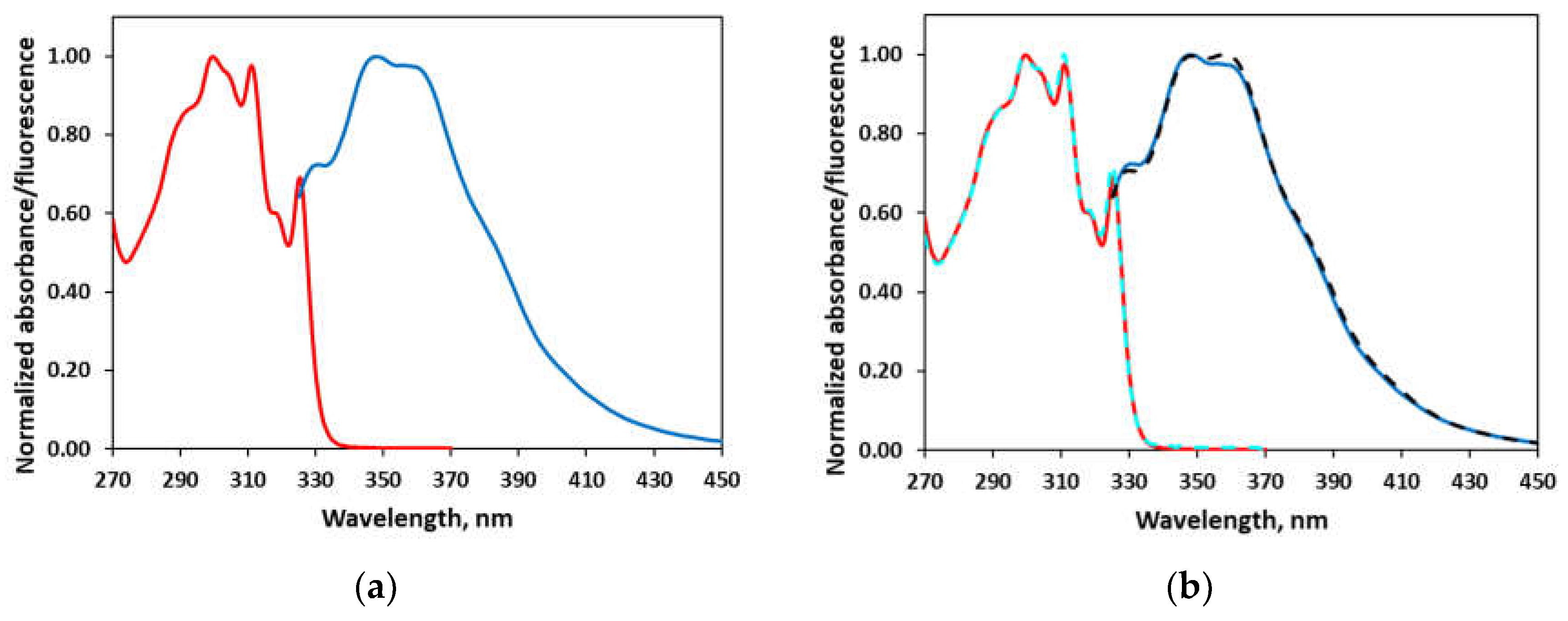

Table 1.
Summary of the photophysical data of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate.
Table 1.
Summary of the photophysical data of 2-(2,6-diisopropylphenyl)-1-methylimidazo[1,5-a]quinolin-2-ium tetrafluoroborate.
| Solvent | ε, M-1.cm-1 | λabs, nm 1 | λem, nm 2 | Stokes Shift, cm-1 |
|---|---|---|---|---|
| MeOH | 10444 | 325 | 348; 358 | 2034; 2863 |
| MeCN | 10106 | 325 | 348; 358 | 2034; 2863 |
1 The longest wavelength maximum. 2 Excitation at 325 nm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



