
Submitted:
31 July 2024
Posted:
02 August 2024
You are already at the latest version
Abstract
Sarcoglycanopathies are among the most frequent and severe autosomal recessive forms of limb-girdle muscular dystrophies /LGMD/ with childhood onset. Mutations in , , and sarcoglycan genes lead to various kinds of LGMD. We present the clinical variability of LGMD 2C/R5 among a genetically homogeneous group of 56 patients, belonging to 35 pedigrees. Molecular genetic analysis showed thаt all 57 patients were homozygous for C283Y variant. The muscles of the pelvic girdle and the trunk were early and more severely affected, followed by the the shoulder girdle. Macroglossia, hypertrophy of the calves, scapular winging and lumbar hyperlordosis were common in the ambulatory phase. A great variability in the clinical presentation of LGMD 2C/R5 was observed both intra and interfamilial, despite the same underlying molecular defect. Females demonstrated a relatively milder clinical course, compared to males. Mean CK levels were 20 times above normal values. Muscle CT of MRI showed earlier and more severe involvement of the flexor proximal limb muscles in comparison to the extensor ones.
Keywords:
LGMD 2C/R5
; gamma-sarcoglycanopathy
; Bulgarian Muslim Roma
1. Introduction
Sarcoglycanopathies are among the most frequent and severe autosomal recessive limb-girdle muscular dystrophies /LGMD/ with childhood onset. Mutations in α, β, γ and δ sarcoglycan genes lead to various kinds of LGMD. More than 5 different mutations in the gamma sarcoglycan gene have been identified to date resulting in LGMD 2C/R5 [1].
In 1983, Ben Hamida et al. [4] were the first to describe a severe autosomal recessive limb-girdle muscular dystrophy that was exceptionally common in Tunisia. Later, it was found that the disease in three Tunisian families was linked to the pericentromeric region of chromosome 13q [5].
Piccolo et al. [2] studied a severe autosomal recessive muscular dystrophy with sarcoglycan deficiency in seven large, unrelated Roma families from various Western European countries (France, Spain, and Italy). Genetic linkage analysis showed that the disease was linked to the gamma-sarcoglycan locus on chromosome 13q12. The variant p.C283Y (G>A, affecting codon 283), which leads to the replacement of the conserved cysteine with tyrosine at position 283 in the protein, was identified. All patients were homozygous for the variant, while their parents were heterozygous.
Following the description of the private Gypsy mutation p.C283Y in the γ-sarcoglycan gene in LGMD2C/R5 patients in Western Europe, we have reasoned that, similar to other single gene disorders of the Gypsies, LGMD2C/R5 should be expected to have a pan-European distribution, resulting from the historical migration routes of this population. This rationale led us to conduct an extensive search for similarly affected individuals of Gypsy ethnicity in Bulgaria. Both hospital records and extensive field work were used as a source of information, leading to the identification of 57 living patients with the LGMD2C/R5 phenotype, homozygous for the mutation p.C283Y, and an additional 17 deceased affected siblings. LGMD2C/R5 thus appears to be the most common myopathy among the Romani minority of Bulgaria, with a prevalence higher than that of Duchenne muscular dystrophy [6].
LGMD2C is more restricted and represent the founding and expansion of the Gypsy migrational categories 20+ generations ago. LGMD2C/R5 was fully confined to the Balkan and Western European Gypsies and was not present in Vlax Gypsies. The mean age of p.C283Y mutation is ~600 (525–775) years [7]. L. Kalaydjieva was the first to genetically verify the disease in Bulgaria and Todorova et al. introduced routine molecular-genetic analysis for the presence of the C283Y variant in Bulgaria. [8,9].
The aim of our study is to investigate the phenotypic variability of the largest group of genetically homogenous LGMD 2C/R5 patients in Bulgaria, belonging to the group of Bulgarian Roma.
2. Materials and Methods
Fifty-seven patients (29 men and 28 women) from 35 pedigrees were identified. Catamnestic data were also collected for 17 deceased patients with gamma-sarcoglycanopathy belonging to the affected families.
All the patients underwent standart clinical and neurological examination, including Walton functional clinical scale, manual muscle testing using the MRC scale.
The patients were categorized in three groups, according to age at loss of ambulation. Loss of the ability to walk independently ambulation before age 13 defines the cate-gory of severe progression that characterizes the Duchenne phenotype; disability after age 16 characterizes the Becker phenotype; and disability between ages 13 and 16 defines the intermediate phenotype [10].
Laboratory tests, including creatine phosphokinase and its isoenzymes, nerve conduction studies (NCS) and electromyography (EMG), conduction studies (NCS) and electromyography (EMG) were done in all patients. ECG and echocardiography were performed in 20 patients, and respiratory function assessment were provided in 6 patients. Computer tomography (CT) or magnetic resonance imaging (MRI) of the major muscles were conducted on 10 patients. Muscle morphological changes were graded based on the relative proportion of muscle areas with decreased and normal density: mild (hypodense areas smaller than 50% of normally appearing muscle areas), moderate (hypodense areas from 1/2 to 2/3 of normally appearing muscle areas), and severe (muscles are atrophic and more than 2/3 are replaced by low-density tissues). A molecular-genetic analysis for the presence of the variant p.C283Y was performed on all patients. Blood for DNA extraction, with informed consent for genetic testing, was collected from all patients, their parents, siblings, and other relatives. The molecular-genetic testing was conducted in the National Genetic Laboratory at the University Hospital “Maichin Dom,” Medical University- Sofia; and the Centre for Human Genetics, Edith Cowan University, Perth. Immunohistochemical studies of muscles were conducted on 7 patients. An ethnological study was also conducted to identify the groups among which the disease is prevalent.
3. Results
3.1. Epidemiological Data
The clinical cohort included 73 patients with genetically verified gamma-sarcoglycanopathy. The age of the patients at the time of the study ranged from 7 to 40 years. Thirty-five pedigrees with the disease were identified. All patients belong to the group of settled Muslim Roma – Millet. This is a long-settled and long-established group in our lands. Figure 1 presents a pedigree with patients with LGMD 2C/R5.
The disease has been identified in 13 regions – 11 in Eastern Bulgaria: Targovishte, Razgrad, Ruse, Silistra, Dobrich, Varna, Shumen, Veliko Tarnovo, Sliven, Yambol, Stara Zagora, and 2 in Western Bulgaria: Sofia and Pernik. To a large extent, the distribution of the disease outlines the path of migration. Despite extensive field studies of the disease throughout the country, no cases of LGMD 2C/R5 have been identified in other regions or among other Roma groups. Figure 2 shows the geographic distribution of LGMD 2C/R5 in Bulgaria.
3.2. History
The disease usually begins with complaints of difficulty walking, frequent falls, walking on tiptoes, difficulty climbing stairs, running and rising from a squatting position. Summary data on the onset, duration of the disease and average age are presented in Table 1.
The age at onset in later in women than in men. Males lose ambulation earlier than female patients. Life expectancy is longer in women. The earlier the onset of the disease, the earlier the disability, respectively the shorter the duration of walking.
3.3. Clinical and Paraclinical Data
- The initial muscle weakness involved the pelvic girdle, followed by the shoulder girdle. At more advanced stages, muscle weakness becomes diffuse, but the mm. glutei, psoas, sacrospinalis, periscapularis and trapezius are most severely affected. In the proximal limb muscles, there is early selective involvement of the flexors (m. biceps bachii and m. flexor femoris) with relatively good preservation of the extensors (m. triceps brachii and m. quadriceps). Figurе 3.
- The axial muscles are affected after proximal muscles with occurrence of lumbar hyperlordosis. Calf pseudohypertrophy and macroglossia are typical features of the disease. Macroglossia was found in 21 of 57 patients (8/28 women and 13/29 men). Distal muscle strength was preserved even in the more advanced stages. No patient was found to have facial or bulbar muscle weakness. Pseudohypertrophy of the hamstrings, scapulae alate and lumbar hyperlordosis were seen in most patients at the earlier stages.. Progressive scoliosis with significant neck stiffness was found in 18 patients, all of whom were disabled(10 men and 8 women). Ankle joint contractures are found after the age of 8 years in 17/29 males and 15/28 females.
- Although there is no difference between the sexes in the type of muscle damage and the degree of severity, in men the disease has an earlier onset and more rapid progression. Table 2 shows the percentage of all patients and their gender distribution according to the type of disease.
- The mean CK value was 20 times higher than normal in all patients.
- Intellectual functions were preserved in all patients.
- Cardiac functions were examined in 20 patients aged 10 to 40 years and one patient was described to have cardiac involvement with dilated cardiomyopathy and marked systolic dysfunction requiring medical therapy.
- Respiratory function was disturbed in 6 patients, and 4 patients were found to have a mild restrictive type of respiratory disorder. Two were found to have severe respiratory failure, one of whom required treatment with invasive pulmonary ventilation from 35 yr of age.
- EMG was consistent with mixed myogenic and neurogenic changes. Spontaneous activity (fibrillations, positive sharp waves) is found predominantly in mm. biceps brachii, rectus femoris and tibialis anterior. The duration of action potentials is shortened. In the automatic analysis, the values were 2.0-3.8 ms for m. rectus femoris, 2.5-3.3 ms for m. tibialis anterior, 2.0-3.3 ms for m. biceps brachii and 4.5-8.6 ms for triceps brachii. The amplitudes of the action potentials were in the region of the lower limit of normal in the automatic analysis. For m.rectus femoris and m.triceps brachii the amplitudes were < 0.9mV /0.7-0.9/, for m.tibialis anterior < 0.7mV /0.3-0.7/ and for m.biceps brachii < 0.4mV /0.2-0.4/. Conduction velocities along motor and sensory fibres were within normal limits.
- CT and MRI of lower limb muscles were performed in 12 patients. The degree of muscle involvement correlated with age and stage of disease in the respective patients. There was symmetrical involvement of the lumbar musculature, with changes progressing distally-more distal muscle groups persisting longer in the course of the disease. Mm. glutei (medius and minimus) are minimally affected in the two younger patients who can independently climb stairs; moderately affected in 10- to 12-year-old patients who are still walking independently; severely affected in patients who are unable to walk; completely replaced by adipose tissue in the oldest and most severely debilitated patients. M. subscapularis and m. trapezius are minimally affected in younger patients, in whom m. del-toideus is preserved; severely affected between the ages of 12 and 14, when m. deltoideus is minimally to moderately affected; and completely atrophic in the oldest patients in whom only traces of m. deltoideus were found. Selectively affected muscles in all patients studied were mm. glutei, mm. adductori, flexor femoris, abdominalis, spinalis, supraspinatus, infraspinatus, subscapularis and soleus. In the earlier stages, relatively more severe involvement of flexor than extensor muscle groups were found in all four limbs. In the stage of complete debilitation, most muscles are atrophic or completely replaced by adipose tissue, whereas mm. deltoideus, quadriceps, sartorius, gracilis, and gastrocnemius are relatively preserved.
- Тhe immunohistochemical characterization of sarcoglycan and dystroglycan components was studied in seven patients with LGMD 2C/R5. Similar changes were found in all patients studied: persistent absence of α- dystroglycan and well-preserved β- dystroglycan. By using different domain-specific antibodies against α- sarcoglycan, we found relative preservation of its intramembrane domain and absence of its C- and N-terminal parts. β-sarcoglycan was severely reduced or absent. The γ- sarcoglycan is also completely absent in all patients. δ- sarcoglycan is relatively best preserved and is expressed to a significantly greater extent in LGMD 2C/R5 than in Duchenne-type PMD.
- Significant inter- and intra-familial variations in the course of the disease were found. In three families with patients of both sexes (1F+2M; 1F+1M; 1F+2M) the disease course was significantly milder and more delayed in women than in men. In fourth family with LGMD 2C/R5 (1F+2M) had a more severe course in one brother and sister, than in the other brother. In two families in which the patients were only female (2F; 2F), phenotypic variations were also found- in the one family the disease had the same onset in both sisters, but with earlier disability in the older sister; in the other family, the course was more severe (with earlier onset and earlier disability) in the younger sister. In two other families in which the patients were only male (4M; 2M), the phenotypic variations were significantly less pronounced.
- Statistically significant sex differences were found during LGMD 2C/R5. Mathematical and statistical analyses were performed to assess the significance of various disease-related variables and their interactions. The disease starts later in women than in men (p<0.01); men stop walking earlier (p<0.05) than women. The effect of gender on age of disability is also evident from Levene’s test, which shows statistical significance at equal variances – p<0.01. The results of the analysis of variance (ANOVA) also demonstrate statistical significance (p<0.01) of the age at onset X sex and age of disability X sex interactions. Life expectancy was longer in females. There was a statistically significant correlation between age of onset and age of disability (p<0.01). The earlier the onset of the disease, the earlier the disability, i.e., the shorter the duration of walking. The severity of patients’ condition correlated with age at the time of examination (p<0.001).
3.4. Genetical Findings
The mutation p.C283Y was found in all patients.
35 pedigrees with the disease were identified. All patients’ parents shared no complaints and had normal neurological status. Two parental pairs were also tested for CK values, which were normal. LGMD 2C/R5 has an autosomal recessive type of inheritance. A detailed pedigree analysis was performed in each family, one is presented in Figure 1. Only three families were found to have consanguineous marriages, two of which were fifth-degree and one seventh-degree related.
4. Discussion
LGMD 2C/R5 is the most common myopathy among Roma in Bulgaria, although it has a regional prevalence in the eastern part of the country. All patients belong to the group of settled Muslim Roma - Millet. No cases of LGMD 2C/R5 have been identified in other Roma groups. The study of affected families included a genetically homogeneous group of 57 patients with LGMD 2C/R5, all confirmed homozygous for the C283Y mutation. The disease has been identified in only 13 regions, 11 of them of Eastern Bulgaria.
Pseudohypertrophy of the lower limbs, scapular winging, lumbar hyperlordosis and macroglossia were typically observed in the earlier stages. Clinical examination and muscle imaging show early selective involvement of mm. glutei, adductors, flexor femoris, abdominalis, spinalis, subscapularis and soleus and relative sparing of m. quadriceps. Flexor muscle groups are more severely affected than extensor ones. Our clinical observations in LGMD 2C/R5 patients are similar to the findings of the joint European study [10] regarding the natural history of the disease, the rate of progression of the disability, the pattern of muscle involvement and the absence of cognitive impairment and cardiomyopathy. Restrictive type respiratory failure usually is a common finding [11], but only one patient with severe respiratory failure requiring invasive pulmonary ventilation was found in the Bulgarian population.
The described Duchenne-like phenotype was seen in the largest percentage of patients, with the remainder distributed almost equally between those classified with moderate disease severity and those with a milder phenotype-similar to Becker-type MD, despite all patients having the same mutation in the homozygous state. Differences were observed in some of our affected pedigrees- two families presented with all three phenotypes in three affected children in one family and five additional families with manifestations of two phenotypes in them. These differences may be related to the degree of involvement of other sarcoglycan components or may be due to the influence of modifying genes or environmental factors. Sarcoglycan deficiency leads to changes in the entire dystrophin-glycoprotein complex with α-dystroglycan and β-sarcoglycan being the most severely affected.
Males were found to have an earlier onset and more rapid progression. . Although no sex-related differences were observed with respect to muscle involvement, loss of independent gait occurred earlier in men. Milder phenotypes were significantly more common among female patients, with the exception of one representative in whom severe respiratory failure with the need for IVF occurred. Gender differences with more severe male involvement were also reported by G. Leal and E. Da Silva [12] in a Brazilian family with gamma-sarcoglycanopathy caused by the del521T variant. The authors found sex differences in only one of the four families studied. Ben Hamida et al. [4] described significant inter- and intra-familial variation but found no sex differences. Such variations were also observed in the families we studied with patients of even the same sex.
In LGMD 2C/R5, compared to Duchenne muscular dystrophy, scoliosis develops later and to a lesser degree. Due to this fact, respiratory complications occur most often after the age of 30. It is known that life expectancy in Duchenne MD is severely limited and only a minority of patients survive more than 25 years without mechanical ventilation [13]. In gamma-sarcoglycanopathy with the variant p.C283Y, 25% of patients survive to this age. The higher life expectancy is most likely due to the absence of early respiratory disorders and heart failure.
Gamma-sarcoglycanopathy with the pathogenic variant p.C283Y has a more severe course than that caused by the del521T variant seen in North Africa [4]. Only 20% of Tunisian patients lose the ability to walk independently by the age of 15 years, compared with 81.5% of patients with the “private “ Gypsy mutation. The phenotype in Roma and Tunisians is similar, except that macroglossia is not found in the latter and cardiomyopathy is more commonly reported.
5. Conclusions
LGMD 2C/R5 in Bulgaria is described in a large group of settled Muslim Roma - Millets, inhabiting mainly the eastern part of the country. All are carriers of the same pathogenic variant p.C283Y in a homozygous state, described as a “private” Gypsy mutation. The clinical phenotype follows the natural history of the disease, with a great phenotypic variability observed both intra- and inter-familial, despite the same underlying molecular defect. Statistically significant sex differences in the course of the disease were found. The disease is more severe in men than in women. Although there is no difference between the sexes in the type of muscle damage and the degree of severity, the disease has an earlier onset in men and leads to more rapid disability. Females demonstrated a relatively milder clinical course of the disease.
Author Contributions
Conceptualization, A.T., Al. T., L.K., I.T.; methodology, V.G., T. C., T.V., S.B., T.T., M.G., R. P; O.A., L.A., V.M; formal analysis, A.T., I.T.; investigation, A.T., V.G., V.B., T.C., T.V., S.B., T. T., M.G., R.P., T.T., Al. T., L. K., I.T.; resources, I. T., A.T., L. K.; data curation, I.T.; writing—A. T.; writing—review and editing, A.T; I.T.; visualization, A.T.; supervision, I.T.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received financial support from the Bulgarian National Science Fund, project B02/240, and European Union-NextGeneration EU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project No BG-RRP-2.004-0004-C01.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Angelini, C.; Fanin, M. Pathogenesis, clinical features and diagnosis of sarcoglycanopathies. Exp. Opin. on Orph. Dr., 2016, 4, 1239–1251. [Google Scholar] [CrossRef]
- Piccolo, F.; Jeanpierre, M.; Leturcq, F.; et al. A founder mutation in the γ-sarcoglycan gene of Gypsies possibly predating their migration out of India. Hum. Mol. Genet., 1996, 5, 2019–2022. [Google Scholar] [CrossRef] [PubMed]
- Tournev, I.; Aneva, L.; Kamenov, O.; Ishpekova, B.; Katsarova, M.; Gergelcheva, V.; Angelicheva, D.; Kalaydjieva, L. Gamma sarcoglycan deficiency in Bulgarian Gypsies. Muscle & Nerve, 1998, Suppl. 7, 136.
- Ben Hamida, M.; Fardeau, M.; and Attia, N. Severe childhood muscular dystrophy affecting both sexes and frequent in Tunisia. Muscle & Nerve 1983, 6, 469–480. [Google Scholar] [CrossRef]
- Ben Othmane, K.; Ben Hamida, M.; Pericak-Vance, M.A.; et al. Linkage of Tunisian autosomal recessive Duchenne-like muscular dystrophy to the pericentromeric region of chromosome 13q. Nat. Genet. 1992, 2, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Tournev, I. Clinical, genetic and epidemiological study of six novel hereditary diseases in Roma (Gypsy) in Bulgaria. Doctoral thesis, Medical University Sofia, Bulgaria, 2001.
- Morar, B.; Gresham, D.; Angelicheva, D.; Tournev, I.; Gooding, R.; Guergueltcheva, V.; Schmidt, C.; Abicht, S.; Lochmuller, H.; Tordai, A.; et al. Mutation history of the Roma /Gypsies/. Am.J.Hum.Gen, 2004, 75, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Todorova, A.; Ashikov, A.; Beltcheva, O.; Tournev, I.; Kremenski, I. C283Y Mutation and other C-Terminal Nucleotide Changes in the γ-Sarcoglycan Gene in the Bulgarian Gypsy Population. Hum. Mutat. 1999, 14, 40–44. [Google Scholar] [CrossRef]
- Todorova, A.; Tournev, I.; Ninova, N.; Georgieva, V.; Kremensky, I. Screening for C283Y gamma-sarcoglycan mutation in high risk group of Bulgarian Gypsies: Evidence for geographical localization and non-random distribution among Gypsy subgroups. Comm. Genetics, 2002, 5, 217–221. [Google Scholar] [CrossRef]
- Merlini, L.; Barois, A.; Monte, A.; et al. Homogeneous phenotype of the Gypsy Limb-girdle Muscular Dystrophy with the gamma-sarcoglycan C283Y mutation. Neurology 2000, 54, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Politano, L.; Nigro, V.; Passamano, L.; et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromusc. Disord. 2001, 11, 178–85. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.F.; Da-Silva, E.O. Limb-girdle muscular dystrophy with apparently different clinical courses within sexes in a large inbred kindred. J. Med. Genet. 1999, 36, 714–718. [Google Scholar] [PubMed]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromusc. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pedigree with LGMD2C/R5 from the villages of Aprilovo, Golyamo Novo, Elenovo in the Targovishte region, and the villages of Sinya Voda and Rakovski in the Razgrad region.
Figure 1.
Pedigree with LGMD2C/R5 from the villages of Aprilovo, Golyamo Novo, Elenovo in the Targovishte region, and the villages of Sinya Voda and Rakovski in the Razgrad region.
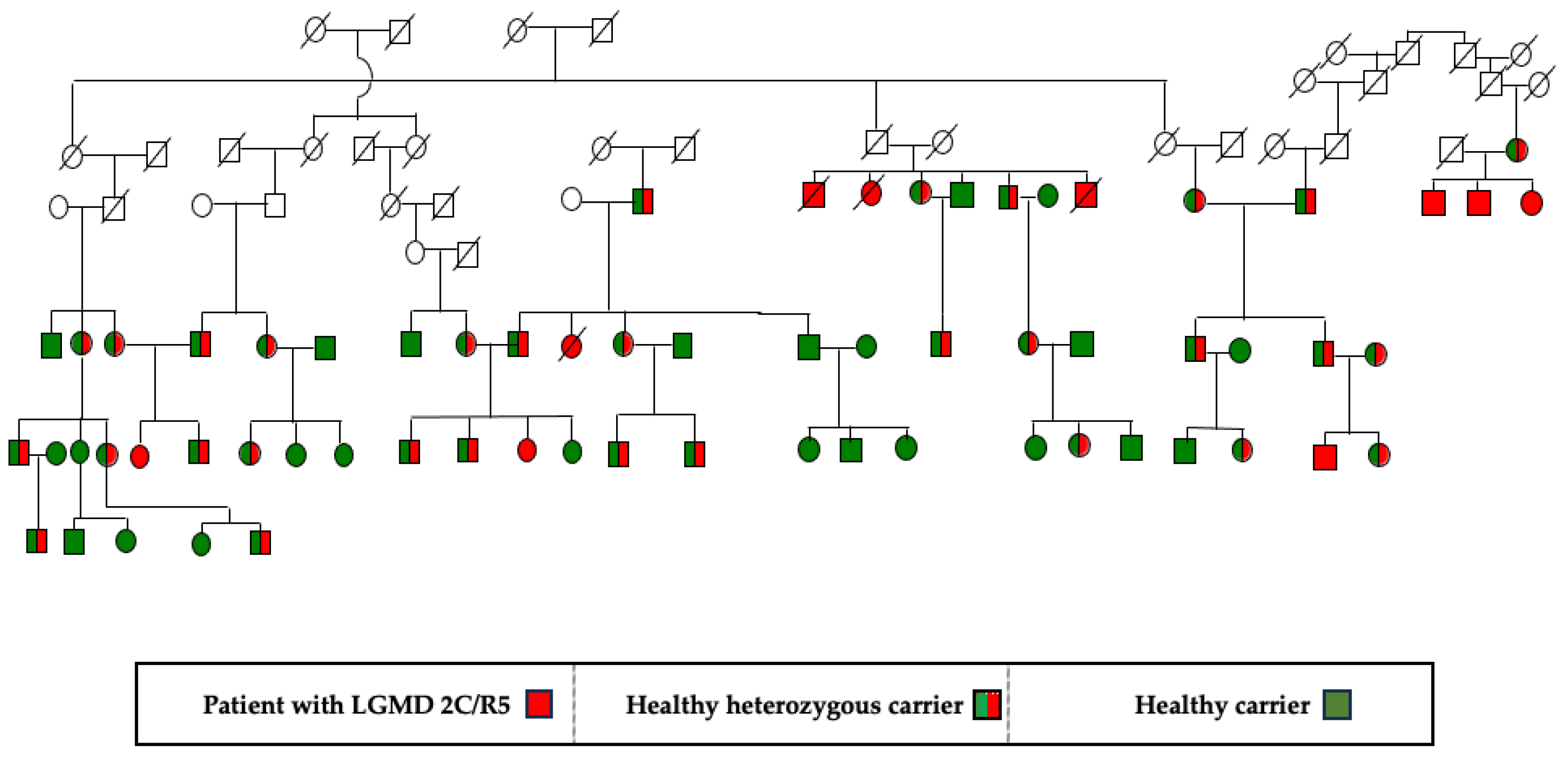
Figure 2.
Distribution of LGMD 2C/R5 among Roma in Bulgaria by regions.
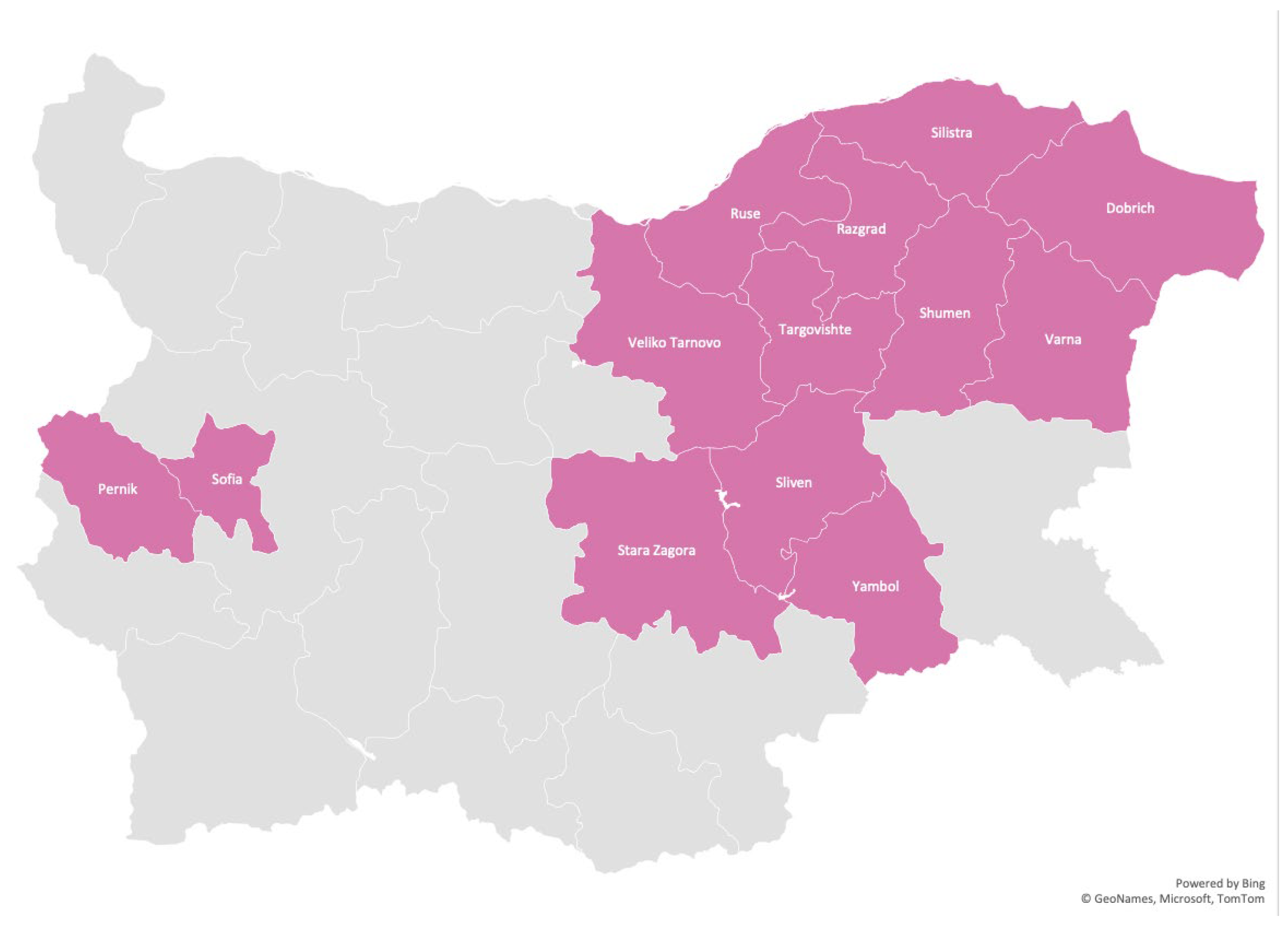
Figure 3.
Boy with LGMD 2C/R5 with typical phenotype, 13y., Maysko village, Elena.
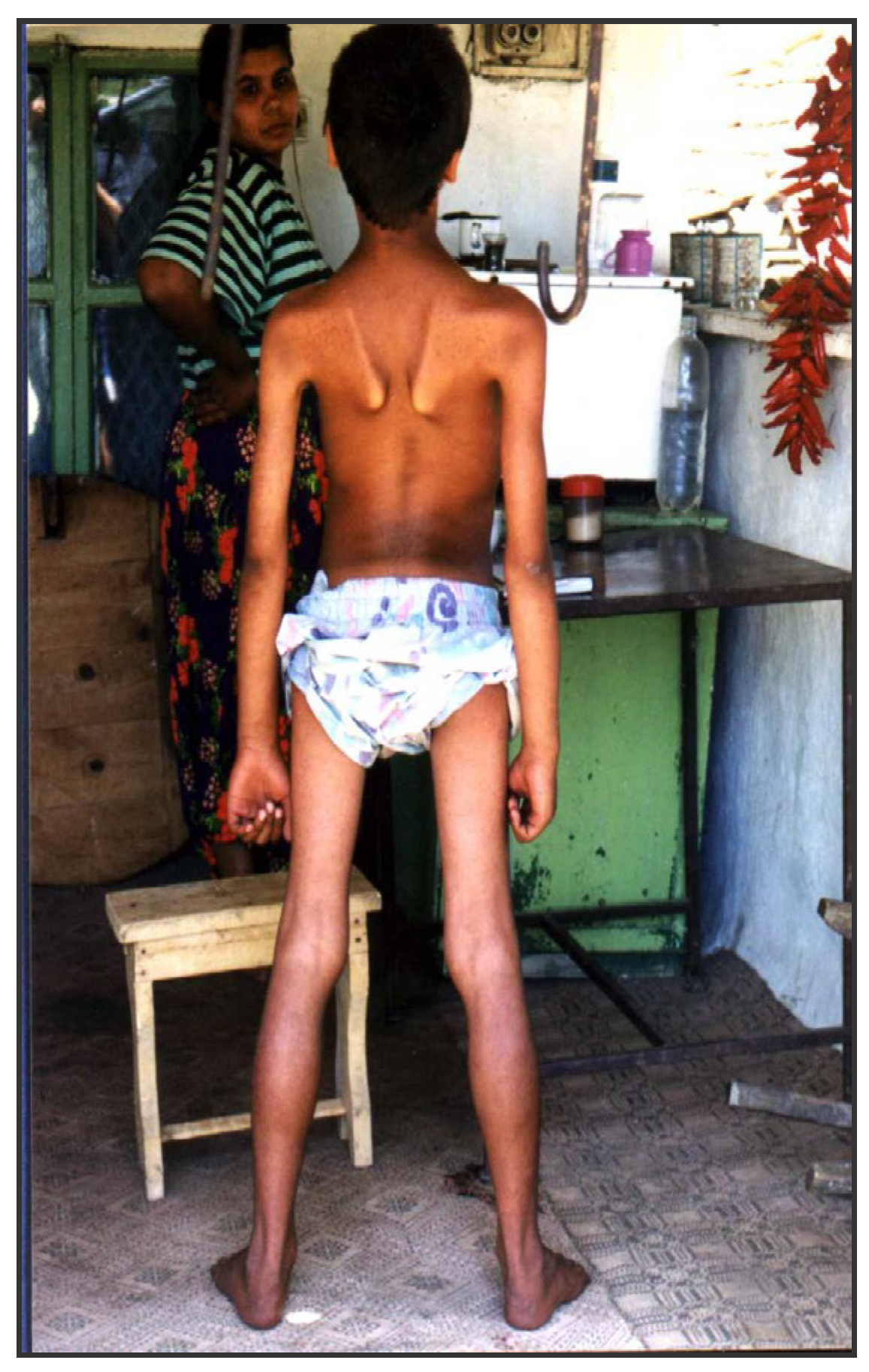

Table 1.
Distribution by sex, age of onset, duration of disease, gait loss and death of the cohort of patients with LGMD 2C/ R5.
Table 1.
Distribution by sex, age of onset, duration of disease, gait loss and death of the cohort of patients with LGMD 2C/ R5.
| All patients | Female | Male | |
|---|---|---|---|
|
Mean age at the time of the study |
21.9 ± 7.8 | 22.9 ± 8.7 | 20.9 ± 7.0 |
| Mean age of onset the disease (between 2 and 13 yrs. of age) | 6.7 ± 2.5 г | 7.8± 2.5 | 5.7 ± 2.0 |
| Mean age of lost ambulation | 13.6 г.±3.2 | 14.9 ± 3.7 | 12.4 ± 2.1 |
| Average duration of ability to walk after disease onset | 6.9±3.1 | 7.1.±3.8 | 6.7.±2.3 |
| Mean age of death | 28.13 ± 3.5 | 32.6 ± 3.1 | 25.9 ± 3.8 |
Table 2.
Percentage of patients in relation to the clinical course of LGMD 2C/R5.
| % All patients | % Female | % Male | |
|---|---|---|---|
| Duchenne- like phenotype | 52.5 | 36,8 | 66.7 |
| Intermediate phenotype | 27.5 | 26.3 | 28.6 |
| Becker-like phenotype | 20 | 36.8 | 4.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



