
Submitted:
17 June 2024
Posted:
18 June 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has acquired multiple mutations since its emergence. Analyses of the SARS-CoV-2 genomes from infected patients exhibit a bias toward C-to-U mutations, which are suggested to be caused by the apolipoprotein B mRNA editing enzyme polypeptide-like 3 (APOBEC3, A3) cytosine deaminase proteins. However, the role of A3 enzymes in SARS-CoV-2 replication remains unclear. To address this question, we investigated the effect of A3 family proteins on SARS-CoV-2 replication in THP-1 cells lacking A3A to A3G genes. The Wuhan, BA.1, and BA.5 variants had comparable viral replication in parent and A3A-to-A3G-null THP-1-ACE2 cells. On the other hand, the replication and infectivity of these variants were abolished in A3A-to-A3G-null THP-1-ACE2 cells in a series of passage experiments over 20 days. In contrast to previous reports, we observed no evidence for A3-induced SARS-CoV-2 mutagenesis in the passage experiments. Furthermore, our analysis of a large number of publicly available SARS-CoV-2 genomes did not reveal conclusive evidence for A3-induced mutagenesis. Taken together, our studies suggest that A3 family proteins can positively contribute to SARS-CoV-2 replication, however this effect is deaminase-independent.
Keywords:
APOBEC3 family proteins
; SARS-CoV-2
; Deaminase-independent mechanism
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for coronavirus disease 2019 (COVID-19). Since the first cases of novel coronavirus infection were detected in Wuhan, Hubei Province, China, in December 2019 [1,2], it spread rapidly worldwide. The World Health Organization (WHO) declared a Public Health Emergency of International Concern (PHEIC) as a COVID-19 pandemic on January 30, 2020 [3] and announced the termination of PHEIC on May 5, 2023 [4]. However, the COVID-19 pandemic has continued.
The apolipoprotein B mRNA editing enzyme polypeptide-like 3 (APOBEC3, A3) family of proteins are composed of seven DNA cytosine deaminases (A3A, A3B, A3C, A3D, A3F, A3G, and A3H proteins) in humans (reviewed in [5,6,7,8]). These A3 proteins are involved in an innate host defense mechanism against parasitic DNA-based elements (reviewed in [8,9,10,11]). Retroviruses are susceptible to cytosine to uracil (C-to-U) deamination caused by A3 family proteins because they produce single-stranded cDNA intermediates that act as the substrate for these enzymes (reviewed in [5,6,7,8,12]). Notably, human immunodeficiency virus type 1 (HIV-1) is the best characterized substrate for A3 family proteins. In primary CD4+ T cells, at least four A3 enzymes (A3D, A3F, A3G, and only stable A3H) restrict HIV-1 replication by deaminating viral cDNA intermediates and physically blocking reverse transcription [13,14,15,16,17,18,19,20,21,22]. A3 enzymes recognize specific dinucleotide motifs for deamination, such as 5′-CC for A3G or 5′-TC for other A3 enzymes at target cytosine bases, which appear as 5′-AG or 5′-AA mutations in the genomic strand [15,17,23,24]. A3 protein-mediated mutations were observed in the genome of single-stranded DNA virus [Transfusion-transmitted virus (TTV)] (reviewed in [8,12]) and various double-stranded DNA viruses [Epstein-Barr virus (EBV), herpes simplex virus type 1 (HSV-1), alpha human papillomaviruses (α-HPV), and BK polyomavirus (BK PyV)] (reviewed in [8,11,12]). However, due to deaminase-independent mechanisms, the antiviral activity of A3 family proteins is not simply associated with its enzymatic activity [25,26,27,28].
Although A3 family proteins generally prefer single-stranded DNA for the deamination, several reports demonstrated that certain A3 proteins induce C-to-U mutations in single-stranded RNA substrates [29,30,31,32,33,34]. In addition, A3 protein-mediated mutations have been reported in the human coronavirus NL63 (HCoV-NL63) genome [35]. However, whether A3-induced mutations are associated with antiviral activity against RNA viruses remains unclear [35,36,37].
SARS-CoV-2 is continually evolving since its emergence in late 2019 [38,39,40,41,42,43,44]. C-to-U mutations are among the most frequent mutations accumulated in the SARS-CoV-2 genome [39,40,41,43,44]. This has been speculated to be due to the deaminase activity of APOBEC proteins. These mutations are thought to produce viruses that are more infectious and evade adaptive immunity. Recently, it has been reported that A3A protein is the potential source of C-to-U mutations in the SARS-CoV-2 genome [40]. Another in vitro study focusing on A1, A3A and A3G proteins suggested that A1 and A3A proteins (and to a much lesser extent A3G protein) have the capacity to mutate the SARS-CoV-2 genome, however these mutations do not impact viral replication. Unexpectedly, the expression of these editing enzymes promoted SARS-CoV-2 replication and propagation [45]. The authors suggested that APOBEC-induced mutations may provide a fitness advantage. Despite these illuminating studies, the role of A3 family proteins in SARS-CoV-2 replication is yet to be fully determined.
We have previously reported that A3H encoded in THP-1 is an unstable haplotype and it is not involved in the restriction to HIV-1 [21]. Further, we created THP-1 cells lacking A3A, A3B, A3C, A3D, A3F, and A3G genes (A3A-to-A3G-null) and characterized the susceptibility of A3A-to-A3G-null THP-1 cells to HIV-1 infection [21]. Importantly, these cell lines completely abolished restriction activity against Vif-deficient HIV-1 [21]. Therefore, we exploited the A3A-to-A3G-null THP-1 cells to create a version stably expressing ACE2 protein and examined the effect of A3 family proteins on SARS-CoV-2 replication.
2. Results
2.1. Creation of THP-1 and A3A-to-A3G-null THP-1 Cells Stably Expressing ACE2 Protein
It has been reported previously that A3C, A3F, and A3H proteins can inhibit HCoV-NL63 infection [35]. More recently, it has been shown that A3A protein and, to a significantly lesser extent, A3G protein are capable of inflicting C-to-U mutations in the SARS-CoV-2 genome. These studies have led to the speculation that A3 enzymes may be the potential source of the observed C-to-T mutations in the SARS-CoV-2 clinical isolates [40,45]. To address whether A3 family proteins are associated with SARS-CoV-2 mutation and restriction, we introduced the ACE2 gene into THP-1 parent cells and their derivatives lacking the expression of A3A to A3G proteins (THP-1#11-4).
As shown in Figure 1A, ACE2 protein expression was not detected in THP-1 parent and THP-1#11-4 cells. However, 36% of THP-1 parental cells and 43% of THP-1#11-4 cells showed ACE2 protein expression after transduction. Next, A3 mRNA expression levels in these ACE2-transduce THP-1 cells were quantified by RT-qPCR (Figure 1B). Except for a minor difference in the A3H mRNA expression, A3A to A3G mRNA expression levels remain consistent with the previous observation [21]. These data indicate that THP-1#11-4 cells stably express ACE2 protein and still fail to express the functional versions of any of these proteins.
2.2. Effect of A3 Family Proteins on SARS-CoV-2 Replication
We next investigated viral replication in THP-1 parent and THP-1#11-4 cells stably expressing ACE2 protein. We did not observe viral (BA.1 and BA.5) replication in cells lacking ACE2 protein expression, indicating that ACE2 is required for viral replication in THP-1 cells (Figure 2A). However, the viral replication of these variant between THP-1 parent and THP-1#11-4 cells was comparable during 96 hours of time course (Figure 2A). We repeated the time course of viral replication assay until passage 5 (~20 days). In THP-1 parent, the Wuhan, BA.1, and BA.5 variants showed continuous viral replication until passage 5 (Figure 2B). Surprisingly, the lack of A3A to A3G genes in THP-1 diminished the viral replication of all variants and their viral RNAs were undetectable until passage 5 (Figure 2B). These data suggest that A3A to A3G proteins may be associated with long-term (~20 days) SARS-CoV-2 replication in THP-1 cells.
2.3. Effect of A3 Family Proteins on SARS-CoV-2 Infectivity
As mentioned above, the viral RNA of all variants tested became undetectable by 20 days postinfection, in contrast to that observed in THP-1 parent cells (Figure 2B). To know whether the Wuhan variant obtained from each passage was infectious, we performed plaque assay in VeroE6/TMPRSS2 (Figure 3). Consistent with viral replication results (Figure 2B), the number of PFU obtained from the Wuhan variant infection in THP-1 parent cells was increased during the passage experiments (Figure 3). However, A3A-to-A3G gene disruption caused a decrease in viral infectivity and it finally became undetectable (Figure 3). These data suggest that A3A to A3G proteins may contribute positively to the production of SARS-CoV-2 infectious particles from THP-1 cells.
2.4. Effect of A3 Proteins on SARS-CoV-2 Mutagenesis
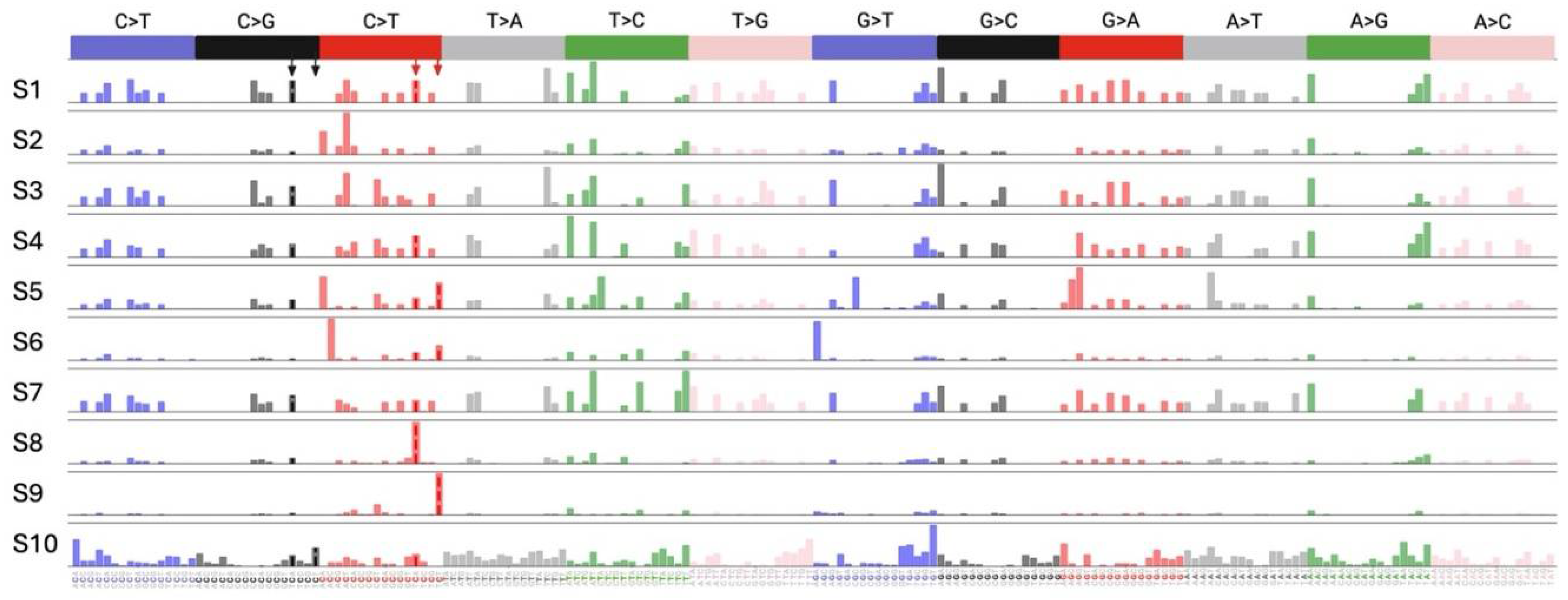
Finally, we asked whether A3 proteins contribute to C-to-U mutations in the SARS-CoV-2 genome. To address this question, we performed WGS analysis for viral RNA isolated from each passage. Our analysis revealed mutations in seven positions only two of which were C-to-T mutations, but none were in the TCA or TCT contexts that are known as APOBEC targets (Figure 4). Additionally, there was no mutational burden difference between the cells with and without A3A to A3G genes (Figure 4). These results indicate that A3 proteins do not play a role in SARS-CoV-2 mutagenesis. To investigate whether lack of A3-induced mutagenesis in SARS-CoV-2 is specific to THP-1 cells or it is a general feature of SARS-CoV-2 infection, we conducted a bioinformatics analysis of 40,000 whole genome SARS-CoV-2 sequences from NCBI. Our analysis involved the quantification of all possible 192 types of mutations followed by mutational signature deconvolution by NMF. We investigated models with up to 20 mutational signatures and could not find any mutational signatures closely resembling SBS2 [represented by TC(T/A)-to-TT(T/A)] or SBS13 [represented by TC(T/A)-to-TG(T/A)] as shown in Figure 5. However, we noted two signatures (S8 and S9), each represented by only one of the main SBS2 peaks (TCA-to-TTA in S8 and TCT-to-TTT in S9). Since such signatures containing only a single dominant peak have not been previously reported for any A3 enzymes, further studies are needed to provide evidence that these signatures are footprint of A3 enzymes.
3. Discussion
The role of A3 enzymes in SARS-CoV-2 mutations have been implicated in several studies [39,40,41,43,44], however little is known about the functional relevance of A3 proteins in SARS-CoV-2 infection. In this study, we examined the effect of A3 proteins on SARS-CoV-2 replication by conducting infection in THP-1 cells lacking A3 enzymes. We showed that the Wuhan, BA.1, and BA.5 variants had comparable viral RNA production in THP-1-ACE2 parent and THP-1 cells lacking the expression of A3A to A3G proteins during 96 hours of the time course (Figure 2A). However, A3 family proteins affected SAR-CoV-2 RNA production in the passage experiments for up to 20 days (Figure 2B). Further, the results of plaque assay showed that A3 family proteins might contribute to the production of infectious virus particles in THP-1 cells (Figure 3). Notably, the effect of A3 family proteins on SARS-CoV-2 replication is independent of C-to-U mutations (Figure 4). Taken together, our findings suggest that A3 family proteins may influence SARS-CoV-2 replication in a deaminase-independent manner.
C-to-U deamination by A3 family proteins is required to restrict HIV-1 [13,21,22,23,46,47,48,49,50]. However, deaminase-independent mechanisms also contribute to the anti-HIV-1 activity of A3 family proteins [14,21,48,49,51,52,53,54,55]. Indeed, deaminase-independent mechanisms are reported to be the predominant method of viral restriction against many viruses [35,37,56,57,58,59]. Several mechanisms have been proposed for the deaminase-independent action. First, the binding of A3F and A3G proteins to HIV-1 genomic RNA blocks the elongation of reverse transcription directly [14,49,50,51,55]. Second, a direct interaction between the A3G protein and HIV-1 Reverse transcriptase causes the disruption of cDNA synthesis [20,60]. Third, an interesting notion is that the A3B protein promotes stress granule formation through a protein kinase R signaling pathway that mediates translational shutdown in cells infected with diverse RNA viruses, such as Sendai virus, Polio virus, and Sindbis virus [61]. These findings indicate that deaminase-independent mechanisms mediate the interaction of the A3 proteins with viral and non-viral proteins. Therefore, the range of innate defense mechanisms by A3 family proteins may be broadly applied to numerous viruses more than those that have been reported so far.
THP-1-ACE2 cells mainly express A3B, A3C, A3F, and A3G mRNAs under normal cell culture conditions (Figure 1B). These proteins have the capacity to form high molecular mass ribonucleoprotein (HMM RNP) complexes [62,63,64,65,66,67,68]. Since HMM RNP complexes are composed of A3G-binding RNAs, A3G-binding proteins, and numerous cellular RNA-binding proteins [62,63,64,66,67,68,69,70], an essential negative factor(s) that can support SARS-CoV-2 replication may be sequestered in the HMM RNP complexes mediated by A3 proteins. Meanwhile, the myeloid cell line, THP-1 used in this study is a non-natural target cell of SARS-CoV-2 because this cell line does not express ACE2 and SARS-CoV-2 cannot replicate in this cell line (Figure 1A and 2A). As an alternative idea, the phenomenon observed in this study may be limited in THP-1 cells where A3 protein-mediated HMM RNP complexes may include an essential negative protein(s) for SARS-CoV-2 replication that is not expressed in non-natural target cells and the sequestration of this protein could support viral replication. Further investigation revealing the underlying mechanisms will be needed for understanding the role of A3 proteins in SARS-CoV-2 replication.
In summary, we found that A3 family proteins support SARS-CoV-2 replication in THP-1 independently of their enzymatic activity. Understanding the underlying mechanism may provide new insight into the interaction between A3 family proteins and coronaviruses. Further, A3 family proteins may be a potential therapeutic target for drug development to alleviate disease severity in respiratory diseases caused by coronaviruses.
4. Materials and Methods
4.1. Cell lines and Culture Conditions
THP-1 cells were provided by Dr. Andrea Cimarelli (INSERM, France) [71]. The generation and characterization of THP-1 ΔA3A-to-A3G#11-4 has been reported previously [21]. THP-1 cells and their derivatives were maintained in RPMI (Thermo Fisher Scientific, Cat# C11875500BT) with 10% fetal bovine serum (FBS) (NICHIREI, Cat#175012) and 1% penicillin/streptomycin (P/S) (Wako, Cat# 168-23191). GP2-293 cells (HEK293 expressing Moloney murine leukemia virus gag/pol protein; TAKARA, Cat# 631530) were maintained in high glucose Dulbecco’s Modified Eagle Medium (DMEM, Wako, Cat# 044-29765) containing 10% FBS and 1% P/S. VeroE6/TMPRSS2 cells (VeroE6 cells stably expressing human TMPRSS2 protein; JCRB Cell Bank, JCRB1819) [72] were maintained in low glucose Dulbecco’s Modified Eagle Medium (DMEM, Wako, Cat# 041-29775) containing 10% FBS, G418 (1 mg/ml; Wako, Cat#070-06803) and 1% P/S (Wako, Cat# 168-23191). All cells were maintained at 37°C with 5% CO2.
4.2. Virus Preparation
SARS-CoV-2 Wuhan variant (strain SARS-CoV-2/Hu/DP/Kng/19-020 strain, Genbank accession no. LC528232) [73,74] was provided by Drs. Tomohiko Takasaki and Jun-Ichi Sakuragi (Kanagawa Prefectural Institute of Public Health). SARS-CoV-2 Omicron BA.1 (strain TY38-873, GISAID ID: EPI_ISL_7418017) [74,75] variant was obtained from National Institute of Infectious Diseases. BA.5 (strain TKYS14631; GISAID ID: EPI_ISL_12812500) [76,77,78] variants were provided by Tokyo Metropolitan Institute of Public Health.
Virus propagation was performed as previously described [73,74,79,80]. Briefly, VeroE6/TMPRSS2 cells (5 × 106 cells) were seeded in a T-75 flask the day before infection. Virus was diluted in virus dilution buffer [1M HEPES, DMEM (low glucose), Non- essential Amino acid (gibco, Cat# 11140-050), 1% P/S] and the dilution buffer containing virus was added to the flask after removing the initial medium. After 1 hour of incubation at 37°C, the supernatant was replaced with 15 ml of 2% FBS/DMEM (low glucose) and cell culture was continued to incubate at 37°C until visible cytopathic effect (CPE) was clearly observed. Then, cell culture supernatant was collected, centrifuged at 300 × g for 10 minutes and frozen at –80°C as working virus stock. The titer of the prepared working virus was determined as the 50% tissue culture infectious dose (TCID50) [76,80,81]. The day before infection, VeroE6/TMPRSS2 cells (10,000 cells) were seeded in a 96-well plate and infected with serially diluted working virus stocks. The infected cells were incubated at 37°C for 4 days and the appearance of CPEs in the infected cells was observed by a microscope. The value of TCID50/ml was calculated by the Reed-Muench method [82].
4.3. A3 mRNA Quantification
Cells were harvested and washed with PBS twice. Then, total RNA was isolated by RNA Premium Kit (NIPPON Genetics, Cat# FG-81250) and cDNA was synthesized by Transcriptor Reverse Transcriptase (Roche, Cat# 03531287001) with random hexamer. RT-qPCR was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Cat# 4367659). Primers for each A3 mRNA have been reported previously [83,84]. A3A forward: (5’-GAG AAG GGA CAA GCA CAT GG) and A3A reverse: (5’-TGG ATC CAT CAA GTG TCT GG). A3B forward: (5’-GAC CCT TTG GTC CTT CGA C) and A3B reverse: (5’-GCA CAG CCC CAG GAG AAG). A3C forward: (5’-AGC GCT TCA GAA AAG AGT GG) and A3C reverse: (5’-AAG TTT CGT TCC GAT CGT TG). A3D forward: (5’-ACC CAA ACG TCA GTC GAA TC) and A3D reverse: (5’-CAC ATT TCT GCG TGG TTC TC). A3F forward: (5’-CCG TTT GGA CGC AAA GAT) and A3F reverse: (5’-CCA GGT GAT CTG GAA ACA CTT). A3G forward: (5’-CCG AGG ACC CGA AGG TTA C) and A3G reverse: (5’-TCC AAC AGT GCT GAA ATT CG). A3H forward: (5’-AGC TGT GGC CAG AAG CAC) and A3H reverse: (5’-CGG AAT GTT TCG GCT GTT). TATA-binding protein (TBP) forward: (5’-CCC ATG ACT CCC ATG ACC) and TBP reverse: (5’-TTT ACA ACC AAG ATT CAC TGT GG). Fluorescent signals from resulting PCR products were acquired using a Thermal Cycler Dice Real Time System III (Takara). Finally, each A3 mRNA expression level was represented as values normalized by TBP mRNA expression levels (Figure 1B).
4.4. ACE2 Transduction
pLV-EF1a-human ACE2-IRES-Puro [85] was used as a template to amplify human ACE2 gene using primers, forward: (5’-NNN NNG TTA ACA CCA TGT CAA GCT CTT CCT GGC TCC TTC) and reverse: (5’-NNN NNC TCG AGC TAA AAG GAG GTC TGA ACA TCA TCA GTG). Then, the amplified human ACE2 gene was inserted into pMSCVneo retroviral vector (Takara, Cat# 634401) at HpaI and XhoI site. The inserted human ACE2 gene was Sanger sequenced (AZENTA) and the data were analyzed by Sequencher DNA sequence analysis software v5.5.6 (Gene Codes Corporation).
Retroviral transduction was performed as previously described in lentivirus [46,48,85]. VSV-G-pseudotyped virus expressing human ACE2 protein was generated by transfecting 4 µg of pMSCV-ACE2-neo plasmid and pVSV-G expression vector (Addgene, cat# 138479) using TransIT-LT1 reagent (Takara, Cat# MIR2306) into 293gp cells (3 × 106 cells). Forty-eight hours later, supernatants were harvested, filtered (0.45 μm filters, Merck, Cat# SLHVR33RB), and subjected to ultracentrifugation at 22,000 × g at 4˚C for 2 hours. After resolving the viral pellets with 10% FBS/RPMI, the concentrated retrovirus was inoclulate into THP-1 cells and its derivatives (1.5 × 105 cells) and incubated at 37˚C. At 72 hours posttransduction, the cells were selected by 1 mg/ml G418 (Wako, Cat#070-06803). G418-selected cells with relatively higher ACE2 expression were sorted by a FACS Aria II (BD Biosciences) and expanded. After expansion, the expression level of surface ACE2 was verified by a FACS Canto II (BD Biosciences). A goat anti-ACE2 polyclonal antibody (R&D Systems, Cat# AF933, 1:50) and an APC-conjugated donkey anti-goat IgG (R&D Systems, Cat# F0108, 1:50) were used for surface ACE2 staining (Figure 1A). Normal goat IgG (R&D Systems, Cat# AB-108-C, 1:100) was used as the negative control for this assay.
4.5. SARS-CoV-2 Infection
5 × 105 parent and A3A-to-A3G-null THP-1 cells were seeded into a 24-well plate, inoculated with SARS-CoV-2 (5,000 TCID50) and incubated at 37°C for 1 hour. After washing with phosphate buffered saline (PBS), 1 ml of fresh cell culture medium was added. 15 μl of cell culture supernatant was harvested at the indicated timepoints and used for RT-qPCR to quantify the viral RNA copy number (see “RT-qPCR” section) (Figure 2A and B). For passage experiments, 10 µl of the cell culture supernatant at 96 hours postinfection was transferred to the next target cells. The passage of the cell culture supernatant was repeated until passage 5.
4.6. RT-qPCR for SARS-CoV-2 RNA
RT-qPCR was performed as previously described [81,85,86,87,88,89]. Briefly, 5 μl of culture supernatant was mixed with 5 μl of 2 × RNA lysis buffer [2% Triton X-100 (Nacalai Tesque, Cat#12969-25), 50 mM KCl, 100mM Tris-HCl (pH 7.4), 40% glycerol, 0.8 U/μl recombinant RNase inhibitor (Takara, Cat# 2313A)] and incubated at room temperature for 10 minutes. 90 μl of RNase free water was added and then 2.5 μl of diluted sample was used for real-time RT-PCR according to the manufacturer’s protocol with One step TB green PrimeScript PLUS RT-PCR Kit (Takara, Cat# RR096A) and primers for Nucleocapsid (N) gene; Forward N, 5’-AGC CTC TTC TCG TTC CTC ATC-3’ and Reverse N, 5’-CCG CCA TTG CCA GCC ATT C-3’. The viral RNA copy number was standardized using a SARS-CoV-2 direct detection RT-qPCR kit (Takara, Cat# RC300A). Fluorescent signals from resulting PCR products were acquired using a Thermal Cycler Dice Real Time System III (Takara).
4.7. Plaque Assay
Plaque assay was performed as previously described [79,87,90,91]. One day before infection, 1 × 105 VeroE6/TMPRSS2 cells were seeded into 24 well plate and infected with serial dilution of cell culture supernatants including SARS-CoV-2 (10, 100, 1000, and 10000-fold dilution, respectively) at 37◦C for 1 hour. 3% FBS and 1.5% carboxymethyl cellulose (Wako, Cat# 039-1335) containing mounting solution was overlaid, followed by incubation at 37◦C. At 3 days postinfection, the cell culture medium was removed, and the cells were washed with PBS three times and fixed with 4% paraformaldehyde phosphate (Nacalai Tesque, Cat# 09154-85). The fixed cells were washed with tap water, dried, and stained with 0.1% methylene blue (Nacalai Tesque, Cat# 22412-14) in water for 30 minutes. The stained cells were washed with tap water and dried. Finally, the number of plaques were counted and indicated as plaque forming unit (PFU)/ml (Figure 3).
4.8. SARS-CoV-2 Whole Genome Sequencing (WGS)
The WGS of SARS-CoV-2 RNA genome was performed as previously described [92]. Briefly, cDNA synthesis, viral sequence enrichment, library amplification, and indexing were performed using the QIAseq DIRECT SARC-CoV-2 kit (QIAgen, Cat# 333891) according to the manufacturer's protocol. After multiplexing with QIASeq DIRECT UDI Set-A (QIAgen), 25 µl library was prepared from each sample. The quality of the enriched libraries was evaluated by electrophoresis using TapeStation 4150 system (Agilent Technologies). The prepared libraries were subjected to sequencing using MiSeq reagent Micro and Nano Kits (Version 2, 300 cycles) in the MiSeq desktop sequencing system (Illumina). The data analysis was done as previously performed [92].
4.9. SARS-CoV-2 Mutational Signature Analysis
We downloaded 40,000 whole genome SARS-CoV-2 sequences from NCBI. These sequences were sampled between the years 2019 and 2022 and were highly diverse in terms of geographic distribution. 38,830 of these sequences that had less than 5% insertion/deletion and/or non-A/C/G/T, were selected and aligned to the most common ancestor of SARS-CoV-2 as described by Kumar et al. [93]. For each sequence, the frequencies of all of the possible 192 mutation types (NnN-to-NmN where n mutates to m and N:A/C/G/T) were quantified. These mutation counts were organized in a data matric of (38,830 by 192) and used as input for analysis using the Non-negative Matrix Factorization (NMF) method. NMF is a matrix decomposition method used routinely to deconstruct mutational signatures in cancer and viral genomes [94,95,96,97]. Here, NMF was used to investigate if deconstruction of all SARS-CoV-2 sequences provides evidence for the existence of an APOBEC mutational signature similar to the known SBS2 and SBS13 signatures [94]. We built NMF models with up to 20 components to investigate the presence of SBS2 and SB13.
4.10. Statistical Analyses
GraphPad Prism software v8.4.3 was used for statistical analysis including two-tailed t tests (Figure 1B).
5. Conclusion
Our results suggest that A3 family proteins may contribute to SARS-CoV-2 replication in THP-1 cells. This effect of A3 family proteins on SARS-CoV-2 replication is independent of deaminase activity.
Funding
This study was supported in part by AMED Research Program (JP23fk0410052 and JP23wm0325068 to YS, JP21fk0108494, JP22fk0108511, JP22fk0108534, and JP22fk0410055, to TI); JSPS KAKENHI Grant-in-Aid for Scientific Research C (22K07103, to Terumasa Ikeda); JSPS Leading Initiative for Excellent Young Researchers (LEADER) (to TI); Takeda Science Foundation (to TI); Mochida Memorial Foundation for Medical and Pharmaceutical Research (to TI); The Naito Foundation (to TI); Waksman Foundation of Japan (to TI); The Uehara Memorial Foundation (to TI); an intramural grant from Kumamoto University COVID-19 Research Projects (AMABIE) (to TI); International Joint Research Project of the Institute of Medical Science, the University of Tokyo (to TI).
Acknowledgements
We would like to thank all Ikeda lab members. We also thank Drs. Tomohiko Takasaki and Jun-Ichi Sakuragi (Kanagawa Prefectural Institute of Public Health) for providing SARS-CoV-2 Wuhan variant (strain SARS-CoV-2/Hu/DP/Kng/19-020, Genbank accession no. LC528232) and National Institute of Infectious Diseases for providing SARS-CoV-2 Omicron BA.1 (strain TY38-873, GISAID ID: EPI_ISL_7418017).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- WHO. COVID-19 Public Health Emergency of International Concern (PHEIC) Global research and innovation forum (Feb 12, 2020), https://www.who.int/publications/m/item/covid-19-public-health-emergency-of-international-concern-(pheic)-global-research-and-innovation-forum.
- WHO. Statement on the fifteenth meeting of the IHR (2005) Emergency Committee on the COVID-19 pandemic (May 5, 2023), https://www.who.int/news/item/05-05-2023-statement-on-the-fifteenth-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-coronavirus-disease-(covid-19)-pandemic.
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J Mol Biol 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479-480C, 131–145. [Google Scholar] [CrossRef]
- Ikeda, T.; Yue, Y.; Shimizu, R.; Nasser, H. Potential utilization of APOBEC3-mediated mutagenesis for an HIV-1 functional cure. Front Microbiol 2021, 12, 686357. [Google Scholar] [CrossRef] [PubMed]
- Jonathan, M.; Ikeda, T. APOBEC3 family proteins as drivers of virus evolution. Frontiers in Virology 2023, 3. [Google Scholar] [CrossRef]
- Holmes, R.K.; Malim, M.H.; Bishop, K.N. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci 2007, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Koito, A.; Ikeda, T. Apolipoprotein B mRNA-editing, catalytic polypeptide cytidine deaminases and retroviral restriction. Wiley interdisciplinary reviews. RNA 2012, 3, 529–541. [Google Scholar] [CrossRef]
- Cheng, A.Z.; Moraes, S.N.; Shaban, N.M.; Fanunza, E.; Bierle, C.J.; Southern, P.J.; Bresnahan, W.A.; Rice, S.A.; Harris, R.S. APOBECs and herpesviruses. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Willems, L.; Gillet, N.A. APOBEC3 Interference during Replication of Viral Genomes. Viruses 2015, 7, 2999–3018. [Google Scholar] [CrossRef]
- Newman, E.N.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Current biology : CB 2005, 15, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.K.; Koning, F.A.; Bishop, K.N.; Malim, M.H. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem 2007, 282, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol 2011, 85, 11220–11234. [Google Scholar] [CrossRef]
- Wang, X.; Abudu, A.; Son, S.; Dang, Y.; Venta, P.J.; Zheng, Y.H. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J Virol 2011, 85, 3142–3152. [Google Scholar] [CrossRef]
- Refsland, E.W.; Hultquist, J.F.; Harris, R.S. Endogenous origins of HIV-1 G to A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog 2012, 8, e1002800. [Google Scholar] [CrossRef] [PubMed]
- Ooms, M.; Brayton, B.; Letko, M.; Maio, S.M.; Pilcher, C.D.; Hecht, F.M.; Barbour, J.D.; Simon, V. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 2013, 14, 411–421. [Google Scholar] [CrossRef]
- Refsland, E.W.; Hultquist, J.F.; Luengas, E.M.; Ikeda, T.; Shaban, N.M.; Law, E.K.; Brown, W.L.; Reilly, C.; Emerman, M.; Harris, R.S. Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS genetics 2014, 10, e1004761. [Google Scholar] [CrossRef] [PubMed]
- Pollpeter, D.; Parsons, M.; Sobala, A.E.; Coxhead, S.; Lang, R.D.; Bruns, A.M.; Papaioannou, S.; McDonnell, J.M.; Apolonia, L.; Chowdhury, J.A.; et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat Microbiol 2018, 3, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Shimizu, R.; Nasser, H.; Carpenter, M.A.; Cheng, A.Z.; Brown, W.L.; Sauter, D.; Harris, R.S. APOBEC3 degradation is the primary function of HIV-1 Vif determining virion infectivity in the myeloid cell line THP-1. mBio 2023, e0078223. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, E.; Opi, S.; Takeuchi, H.; Khan, M.; Goila-Gaur, R.; Kao, S.; Strebel, K. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J Virol 2007, 81, 13346–13353. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.; Carpenter, M.A.; Demir, O.; Ikeda, T.; Li, M.; Shaban, N.M.; Law, E.K.; Anokhin, D.; Brown, W.L.; Amaro, R.E.; et al. The local dinucleotide preference of APOBEC3G can be altered from 5'-CC to 5'-TC by a single amino acid substitution. J Mol Biol 2013, 425, 4442–4454. [Google Scholar] [CrossRef] [PubMed]
- Poulain, F.; Lejeune, N.; Willemart, K.; Gillet, N.A. Footprint of the host restriction factors APOBEC3 on the genome of human viruses. PLoS Pathog 2020, 16, e1008718. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Verhalen, B.; Starrett, G.J.; Harris, R.S.; Jiang, M. Functional Upregulation of the DNA Cytosine Deaminase APOBEC3B by Polyomaviruses. J Virol 2016, 90, 6379–6386. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Van Doorslaer, K.; Pandey, A.; Espinosa, J.M.; Pyeon, D. Role of the host restriction factor APOBEC3 on papillomavirus evolution. Virus Evol 2015, 1, vev015. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat Commun 2015, 6, 6881. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Baysal, B.E. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci Rep 2016, 6, 39100. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Baysal, B.E. Stem-loop structure preference for site-specific RNA editing by APOBEC3A and APOBEC3G. PeerJ 2017, 5, e4136. [Google Scholar] [CrossRef]
- Sharma, S.; Patnaik, S.K.; Kemer, Z.; Baysal, B.E. Transient overexpression of exogenous APOBEC3A causes C-to-U RNA editing of thousands of genes. RNA Biol 2017, 14, 603–610. [Google Scholar] [CrossRef]
- Sharma, S.; Wang, J.; Alqassim, E.; Portwood, S.; Cortes Gomez, E.; Maguire, O.; Basse, P.H.; Wang, E.S.; Segal, B.H.; Baysal, B.E. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol 2019, 20, 37. [Google Scholar] [CrossRef]
- Alqassim, E.Y.; Sharma, S.; Khan, A.; Emmons, T.R.; Cortes Gomez, E.; Alahmari, A.; Singel, K.L.; Mark, J.; Davidson, B.A.; Robert McGray, A.J.; et al. RNA editing enzyme APOBEC3A promotes pro-inflammatory M1 macrophage polarization. Commun Biol 2021, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-mediated restriction of RNA virus replication. Sci Rep 2018, 8, 5960. [Google Scholar] [CrossRef] [PubMed]
- Pauli, E.K.; Schmolke, M.; Hofmann, H.; Ehrhardt, C.; Flory, E.; Munk, C.; Ludwig, S. High level expression of the anti-retroviral protein APOBEC3G is induced by influenza A virus but does not confer antiviral activity. Retrovirology 2009, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Fehrholz, M.; Kendl, S.; Prifert, C.; Weissbrich, B.; Lemon, K.; Rennick, L.; Duprex, P.W.; Rima, B.K.; Koning, F.A.; Holmes, R.K.; et al. The innate antiviral factor APOBEC3G targets replication of measles, mumps and respiratory syncytial viruses. J Gen Virol 2012, 93, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.W. Mutations on COVID-19 diagnostic targets. Genomics 2020. [Google Scholar] [CrossRef]
- Simmonds, P. Rampant C-->U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Nakata, Y.; Ode, H.; Kubota, M.; Kasahara, T.; Matsuoka, K.; Sugimoto, A.; Imahashi, M.; Yokomaku, Y.; Iwatani, Y. Cellular APOBEC3A deaminase drives mutations in the SARS-CoV-2 genome. Nucleic Acids Res 2023, 51, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci Adv 2020, 6, eabb5813. [Google Scholar] [CrossRef]
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. SARS-CoV-2 within-host diversity and transmission. Science 2021, 372. [Google Scholar] [CrossRef]
- Kosuge, M.; Furusawa-Nishii, E.; Ito, K.; Saito, Y.; Ogasawara, K. Point mutation bias in SARS-CoV-2 variants results in increased ability to stimulate inflammatory responses. Sci Rep 2020, 10, 17766. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.D.; Goes, L.R.; Alves, B.M.; de Carvalho, P.S.; Cicala, C.; Arthos, J.; Viola, J.P.B.; de Melo, A.C.; Soares, M.A. SARS-CoV-2 genomic analyses in cancer patients reveal elevated intrahost genetic diversity. Virus Evol 2021, 7, veab013. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Calabrese, P.; Wang, S.; Qin, C.; Rao, Y.; Feng, P.; Chen, X.S. The roles of APOBEC-mediated RNA editing in SARS-CoV-2 mutations, replication and fitness. Sci Rep 2022, 12, 14972. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Molan, A.M.; Jarvis, M.C.; Carpenter, M.A.; Salamango, D.J.; Brown, W.L.; Harris, R.S. HIV-1 restriction by endogenous APOBEC3G in the myeloid cell line THP-1. J Gen Virol 2019, 100, 1140–1152. [Google Scholar] [CrossRef]
- Browne, E.P.; Allers, C.; Landau, N.R. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology 2009, 387, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.D.; Ikeda, T.; Moghadasi, S.A.; Martin, A.S.; Brown, W.L.; Harris, R.S. Natural APOBEC3C variants can elicit differential HIV-1 restriction activity. Retrovirology 2018, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Gillick, K.; Pollpeter, D.; Phalora, P.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol 2013, 87, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Symeonides, M.; Albin, J.S.; Li, M.; Thali, M.; Harris, R.S. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog 2018, 14, e1007010. [Google Scholar] [CrossRef]
- Bishop, K.N.; Verma, M.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog 2008, 4, e1000231. [Google Scholar] [CrossRef]
- Shaban, N.M.; Shi, K.; Lauer, K.V.; Carpenter, M.A.; Richards, C.M.; Salamango, D.; Wang, J.; Lopresti, M.W.; Banerjee, S.; Levin-Klein, R.; et al. The Antiviral and Cancer Genomic DNA Deaminase APOBEC3H Is Regulated by an RNA-Mediated Dimerization Mechanism. Mol Cell 2018, 69, 75–86. [Google Scholar] [CrossRef]
- Iwatani, Y.; Chan, D.S.; Wang, F.; Stewart-Maynard, K.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res 2007, 35, 7096–7108. [Google Scholar] [CrossRef]
- Li, X.Y.; Guo, F.; Zhang, L.; Kleiman, L.; Cen, S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem 2007, 282, 32065–32074. [Google Scholar] [CrossRef]
- Belanger, K.; Savoie, M.; Rosales Gerpe, M.C.; Couture, J.F.; Langlois, M.A. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res 2013, 41, 7438–7452. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol 2007, 81, 4465–4472. [Google Scholar] [CrossRef]
- Narvaiza, I.; Linfesty, D.C.; Greener, B.N.; Hakata, Y.; Pintel, D.J.; Logue, E.; Landau, N.R.; Weitzman, M.D. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog 2009, 5, e1000439. [Google Scholar] [CrossRef]
- Stavrou, S.; Zhao, W.; Blouch, K.; Ross, S.R. Deaminase-Dead Mouse APOBEC3 Is an In Vivo Retroviral Restriction Factor. J Virol 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Boi, S.; Kolokithas, A.; Shepard, J.; Linwood, R.; Rosenke, K.; Van Dis, E.; Malik, F.; Evans, L.H. Incorporation of mouse APOBEC3 into murine leukemia virus virions decreases the activity and fidelity of reverse transcriptase. J Virol 2014, 88, 7659–7662. [Google Scholar] [CrossRef]
- Wang, X.; Ao, Z.; Chen, L.; Kobinger, G.; Peng, J.; Yao, X. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J Virol 2012, 86, 3777–3786. [Google Scholar] [CrossRef]
- Manjunath, L.; Oh, S.; Ortega, P.; Bouin, A.; Bournique, E.; Sanchez, A.; Martensen, P.M.; Auerbach, A.A.; Becker, J.T.; Seldin, M.; et al. APOBEC3B drives PKR-mediated translation shutdown and protects stress granules in response to viral infection. Nat Commun 2023, 14, 820. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Witkowska, H.E.; Hall, S.C.; Santiago, M.; Soros, V.B.; Esnault, C.; Heidmann, T.; Greene, W.C. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci U S A 2006, 103, 15588–15593. [Google Scholar] [CrossRef]
- Kozak, S.L.; Marin, M.; Rose, K.M.; Bystrom, C.; Kabat, D. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J Biol Chem 2006, 281, 29105–29119. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.M.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol 2007, 81, 2165–2178. [Google Scholar] [CrossRef]
- Niewiadomska, A.M.; Tian, C.; Tan, L.; Wang, T.; Sarkis, P.T.; Yu, X.F. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J Virol 2007, 81, 9577–9583. [Google Scholar] [CrossRef]
- Soros, V.B.; Yonemoto, W.; Greene, W.C. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog 2007, 3, e15. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Holmes, R.K.; Swanson, C.M.; Fernandez-Ocana, M.; Byers, H.L.; Ward, M.A.; Malim, M.H. Comparison of cellular ribonucleoprotein complexes associated with the APOBEC3F and APOBEC3G antiviral proteins. J Virol 2008, 82, 5636–5642. [Google Scholar] [CrossRef]
- Xiao, X.; Yang, H.; Arutiunian, V.; Fang, Y.; Besse, G.; Morimoto, C.; Zirkle, B.; Chen, X.S. Structural determinants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res 2017, 45, 7494–7506. [Google Scholar] [CrossRef]
- Shirakawa, K.; Takaori-Kondo, A.; Yokoyama, M.; Izumi, T.; Matsui, M.; Io, K.; Sato, T.; Sato, H.; Uchiyama, T. Phosphorylation of APOBEC3G by protein kinase A regulates its interaction with HIV-1 Vif. Nat Struct Mol Biol 2008, 15, 1184–1191. [Google Scholar] [CrossRef]
- Maeda, K.; Almofty, S.A.; Singh, S.K.; Eid, M.M.; Shimoda, M.; Ikeda, T.; Koito, A.; Pham, P.; Goodman, M.F.; Sakaguchi, N. GANP Interacts with APOBEC3G and Facilitates Its Encapsidation into the Virions To Reduce HIV-1 Infectivity. J Immunol 2013, 191, 6030–6039. [Google Scholar] [CrossRef]
- Berger, G.; Durand, S.; Fargier, G.; Nguyen, X.N.; Cordeil, S.; Bouaziz, S.; Muriaux, D.; Darlix, J.L.; Cimarelli, A. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog 2011, 7, e1002221. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci U S A 2020, 117, 7001–7003. [Google Scholar] [CrossRef]
- Fukuda, M.; Islam, M.S.; Shimizu, R.; Nasser, H.; Rabin, N.N.; Takahashi, Y.; Sekine, Y.; Lindoy, L.F.; Fukuda, T.; Ikeda, T.; et al. Lethal Interactions of SARS-CoV-2 with Graphene Oxide: Implications for COVID-19 Treatment. Acs Appl Nano Mater 2021, 4, 11881–11887. [Google Scholar] [CrossRef]
- Begum, M.M.; Ichihara, K.; Takahashi, O.; Nasser, H.; Jonathan, M.; Tokunaga, K.; Yoshida, I.; Nagashima, M.; Sadamasu, K.; Yoshimura, K.; et al. Virological characteristics correlating with SARS-CoV-2 spike protein fusogenicity. Frontiers in Virology 2024, 4. [Google Scholar] [CrossRef]
- Suzuki, R.; Yamasoba, D.; Kimura, I.; Wang, L.; Kishimoto, M.; Ito, J.; Morioka, Y.; Nao, N.; Nasser, H.; Uriu, K.; et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 2022, 603, 700–705. [Google Scholar] [CrossRef]
- Kimura, I.; Yamasoba, D.; Tamura, T.; Nao, N.; Suzuki, T.; Oda, Y.; Mitoma, S.; Ito, J.; Nasser, H.; Zahradnik, J.; et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 subvariants, including BA.4 and BA.5. Cell 2022, 185, 3992–4007. [Google Scholar] [CrossRef]
- Saito, A.; Tamura, T.; Zahradnik, J.; Deguchi, S.; Tabata, K.; Anraku, Y.; Kimura, I.; Ito, J.; Yamasoba, D.; Nasser, H.; et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2.75 variant. Cell Host Microbe 2022. [Google Scholar] [CrossRef]
- Islam, M.S.; Rabin, N.N.; Begum, M.M.; Goto, N.; Tagawa, R.; Nagashima, M.; Sadamasu, K.; Yoshimura, K.; Sekine, Y.; Ikeda, T.; et al. SARS-CoV-2 inactivation: Assessing the efficacy of GO-anchored filters versus various commercial masks. RSC Appl Interfaces 2024. [Google Scholar] [CrossRef]
- Islam, M.S.; Fukuda, M.; Hossain, M.J.; Rabin, N.N.; Tagawa, R.; Nagashima, M.; Sadamasu, K.; Yoshimura, K.; Sekine, Y.; Ikeda, T.; et al. SARS-CoV-2 suppression depending on the pH of graphene oxide nanosheets. Nanoscale Adv 2023, 5, 2413–2417. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat Commun 2023, 14, 2800. [Google Scholar] [CrossRef]
- Ito, J.; Suzuki, R.; Uriu, K.; Itakura, Y.; Zahradnik, J.; Kimura, K.T.; Deguchi, S.; Wang, L.; Lytras, S.; Tamura, T.; et al. Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant. Nat Commun 2023, 14, 2671. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endopoints. Am J Epidemiol 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Refsland, E.W.; Stenglein, M.D.; Shindo, K.; Albin, J.S.; Brown, W.L.; Harris, R.S. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res 2010, 38, 4274–4284. [Google Scholar] [CrossRef]
- 10.1093/nar/gkq174.
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Kimura, I.; Yamasoba, D.; Nasser, H.; Ito, H.; Zahradnik, J.; Wu, J.; Fujita, S.; Uriu, K.; Sasaki, J.; Tamura, T.; et al. Multiple mutations of SARS-CoV-2 Omicron BA.2 variant orchestrate its virological characteristics. J Virol 2023. [Google Scholar] [CrossRef]
- Tamura, T.; Irie, T.; Deguchi, S.; Yajima, H.; Tsuda, M.; Nasser, H.; Mizuma, K.; Plianchaisuk, A.; Suzuki, S.; Uriu, K.; et al. Virological characteristics of the SARS-CoV-2 Omicron XBB.1.5 variant. Nat Commun 2024, 15, 1176. [Google Scholar] [CrossRef]
- Tamura, T.; Mizuma, K.; Nasser, H.; Deguchi, S.; Padilla-Blanco, M.; Oda, Y.; Uriu, K.; Tolentino, J.E.M.; Tsujino, S.; Suzuki, R.; et al. Virological characteristics of the SARS-CoV-2 BA.2.86 variant. Cell Host Microbe 2024, 32, 170–180. [Google Scholar] [CrossRef]
- Kimura, I.; Yamasoba, D.; Nasser, H.; Zahradnik, J.; Kosugi, Y.; Wu, J.; Nagata, K.; Uriu, K.; Tanaka, Y.L.; Ito, J.; et al. The SARS-CoV-2 spike S375F mutation characterizes the Omicron BA.1 variant. iScience 2022, 25, 105720. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Rajib, S.A.; Ogi, Y.; Hossain, M.B.; Ikeda, T.; Tanaka, E.; Kawaguchi, T.; Satou, Y. A SARS-CoV-2 Delta variant containing mutation in the probe binding region used for RT-qPCR test in Japan exhibited atypical PCR amplification and might induce false negative result. J Infect Chemother 2022, 28, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tao, Q.; Weaver, S.; Sanderford, M.; Caraballo-Ortiz, M.A.; Sharma, S.; Pond, S.L.K.; Miura, S. An Evolutionary Portrait of the Progenitor SARS-CoV-2 and Its Dominant Offshoots in COVID-19 Pandemic. Mol Biol Evol 2021, 38, 3046–3059. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Lamb, K.D.; Luka, M.M.; Saathoff, M.; Orton, R.J.; Phan, M.V.T.; Cotten, M.; Yuan, K.; Robertson, D.L. Mutational signature dynamics indicate SARS-CoV-2's evolutionary capacity is driven by host antiviral molecules. PLoS Comput Biol 2024, 20, e1011795. [Google Scholar] [CrossRef]
- Zhu, B.; Xiao, Y.; Yeager, M.; Clifford, G.; Wentzensen, N.; Cullen, M.; Boland, J.F.; Bass, S.; Steinberg, M.K.; Raine-Bennett, T.; et al. Mutations in the HPV16 genome induced by APOBEC3 are associated with viral clearance. Nat Commun 2020, 11, 886. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
Figure 1.
Validation of expression levels for ACE2 protein and A3 mRNAs in THP-1 parent and A3A-to-A3G-null THP-1 cells. (A) ACE2 protein expression levels on the surface of THP-1 parent and #11-4 (A3A-to-A3G-null THP-1) cells. The ACE2 gene was introduced by lentiviral vector and the expression levels of the surface ACE2 protein were detected by an anti-ACE2 polyclonal antibody (red). The number in each graph shows the percentage of ACE2+ cells compared to those stained by isotype control (gray). (B) RT-qPCR data. A3 mRNA expression levels were quantified by RT-qPCR and are normalized to TBP mRNA levels. Each bar represents the average of three independent experiments with Standard deviation (SD). Statistical significance was determined using the two-sided unpaired t test. *, P < 0.05 compared to THP-1 parent cells.
Figure 1.
Validation of expression levels for ACE2 protein and A3 mRNAs in THP-1 parent and A3A-to-A3G-null THP-1 cells. (A) ACE2 protein expression levels on the surface of THP-1 parent and #11-4 (A3A-to-A3G-null THP-1) cells. The ACE2 gene was introduced by lentiviral vector and the expression levels of the surface ACE2 protein were detected by an anti-ACE2 polyclonal antibody (red). The number in each graph shows the percentage of ACE2+ cells compared to those stained by isotype control (gray). (B) RT-qPCR data. A3 mRNA expression levels were quantified by RT-qPCR and are normalized to TBP mRNA levels. Each bar represents the average of three independent experiments with Standard deviation (SD). Statistical significance was determined using the two-sided unpaired t test. *, P < 0.05 compared to THP-1 parent cells.
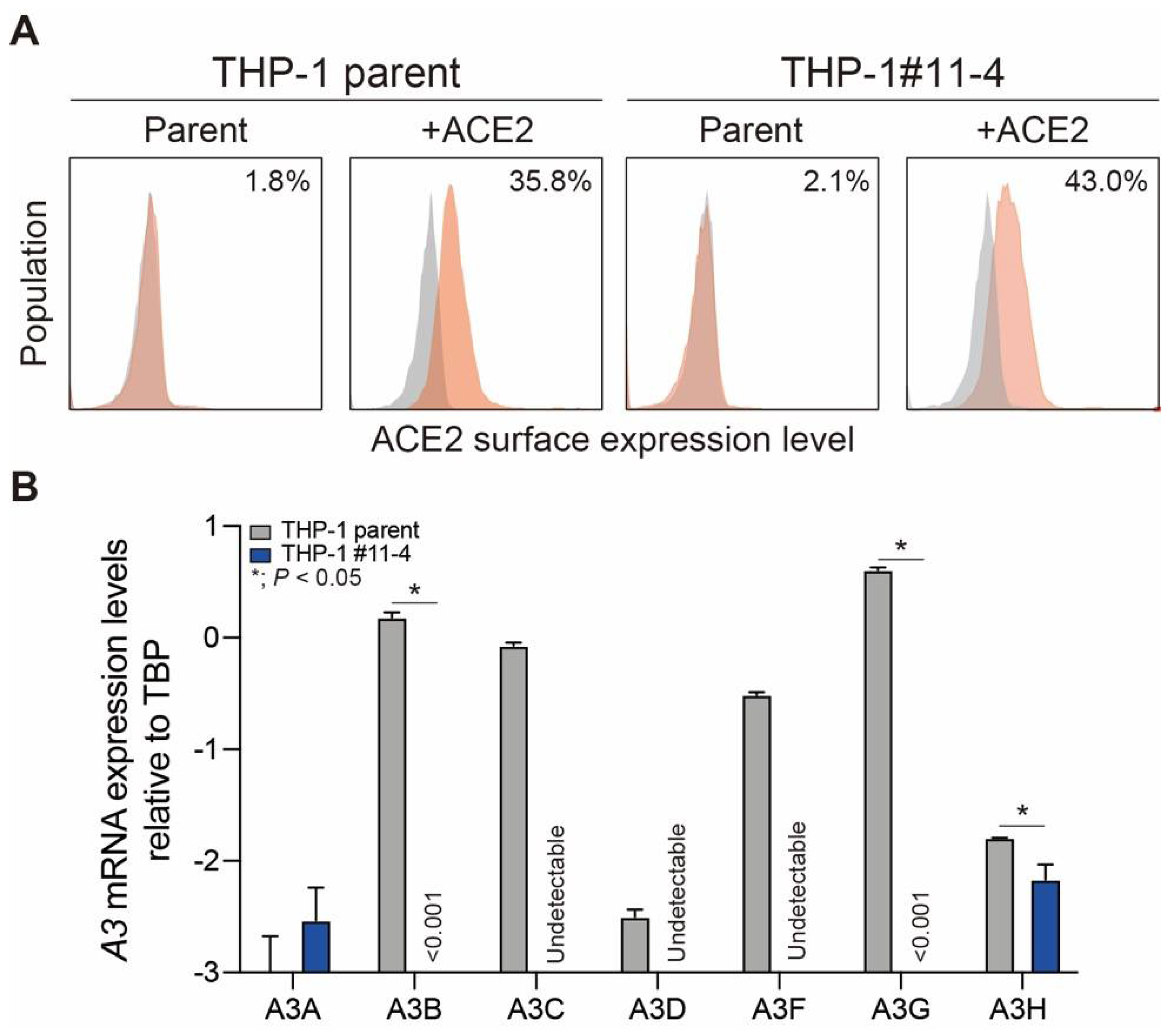
Figure 2.
SARS-CoV-2 replication in THP-1 parent and A3A-to-A3G-null THP-1 cells. (A) Replication kinetics of the Wuhan, BA.1, and BA.5 variants produced from THP-1 parent and A3A-to-A3G-null THP-1 (A3A-to-A3G-null THP-1) cells without (blue line) or with (red line) ACE2 protein expression. The SARS-CoV-2 N gene was quantified by RT-qPCR to monitor the viral RNA copy number across the indicated timepoints. Each timepoint represents the average of four independent experiments with SD. (B) Passage experiments. The SARS-CoV-2 N gene in the cell culture supernatants produced from THP-1 parent or A3A-to-A3G-null THP-1 (A3A-to-A3G-null THP-1) cells at 96 hours postinfection of each passage was quantified by RT-qPCR to monitor the viral RNA copy number of the Wuhan (gray), BA.1 (blue), and BA.5 (red) variants. Each bar represents the average of three independent experiments with SD.
Figure 2.
SARS-CoV-2 replication in THP-1 parent and A3A-to-A3G-null THP-1 cells. (A) Replication kinetics of the Wuhan, BA.1, and BA.5 variants produced from THP-1 parent and A3A-to-A3G-null THP-1 (A3A-to-A3G-null THP-1) cells without (blue line) or with (red line) ACE2 protein expression. The SARS-CoV-2 N gene was quantified by RT-qPCR to monitor the viral RNA copy number across the indicated timepoints. Each timepoint represents the average of four independent experiments with SD. (B) Passage experiments. The SARS-CoV-2 N gene in the cell culture supernatants produced from THP-1 parent or A3A-to-A3G-null THP-1 (A3A-to-A3G-null THP-1) cells at 96 hours postinfection of each passage was quantified by RT-qPCR to monitor the viral RNA copy number of the Wuhan (gray), BA.1 (blue), and BA.5 (red) variants. Each bar represents the average of three independent experiments with SD.
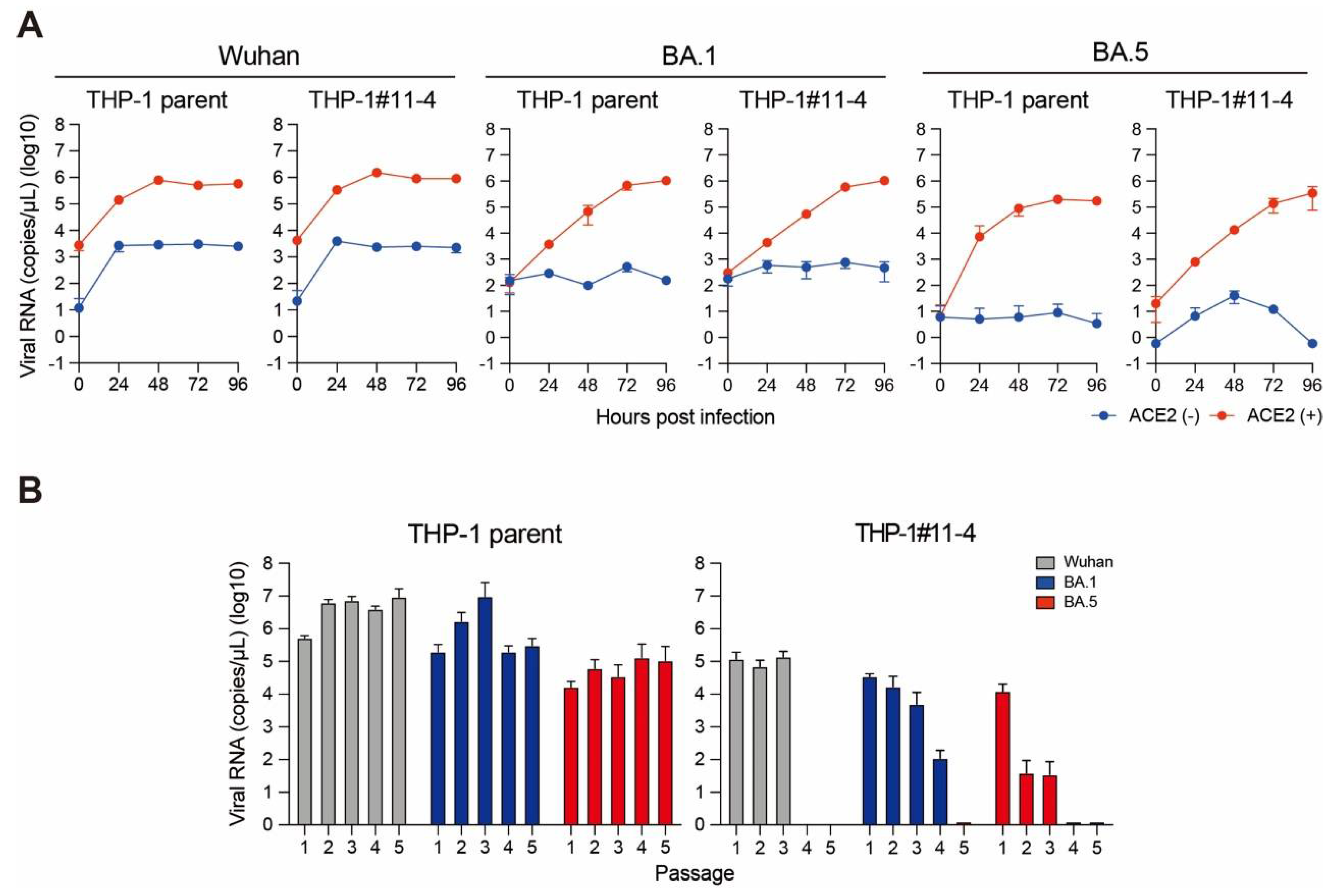
Figure 3.
SARS-CoV-2 infectivity produced from THP-1 parent and A3A-to-A3G-null THP-1 cells during passage experiments.(A) Representative pictures of plaque assay. Cell culture supernatants obtained from the passage experiments for the Wuhan variant were also used for plaque assay with serial 10-times dilution. (B) PFU/ml of the Wuhan variant produced from THP-1 parent (gray) or A3A-to-A3G-null THP-1 (A3A-to-A3G-null THP-1) (blue) cells at 96 hours postinfection during passage experiments.
Figure 3.
SARS-CoV-2 infectivity produced from THP-1 parent and A3A-to-A3G-null THP-1 cells during passage experiments.(A) Representative pictures of plaque assay. Cell culture supernatants obtained from the passage experiments for the Wuhan variant were also used for plaque assay with serial 10-times dilution. (B) PFU/ml of the Wuhan variant produced from THP-1 parent (gray) or A3A-to-A3G-null THP-1 (A3A-to-A3G-null THP-1) (blue) cells at 96 hours postinfection during passage experiments.
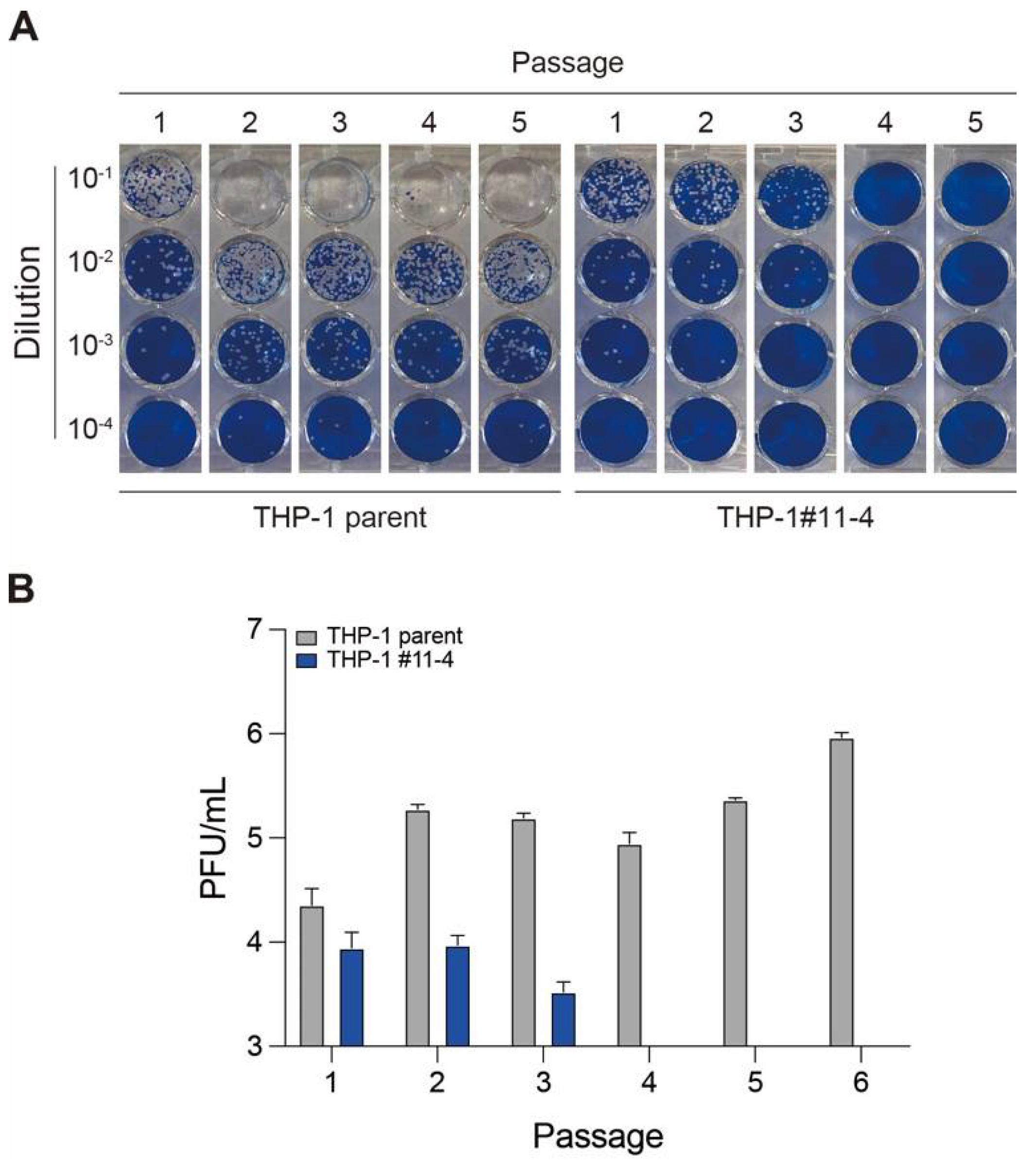
Figure 4.
Analysis of mutations in SARS-CoV-2 genomes produced from THP-1 parent and A3A-to-A3G-null THP-1 cells during passage experiments. SARS-CoV-2 genomic RNA was isolated and subjected to WGS. Sample 1 to 4: Wuhan variant from passage 1 of THP-1 parent cells. Sample 5 to 8: Wuhan variant from passage 5 of THP-1 parent cells. Sample 9 to 12: Wuhan variant from passage 1 of A3A-to-A3G-null THP-1 cells. Sample 13 and 14: Wuhan variant from passage 3 of A3A-to-A3G-null THP-1 cells. Sample 15 and 16: Wuhan variant from passage 4 and passage 5 of A3A-to-A3G-null THP-1 cells. Green boxes on the top show each SARS-CoV-2 ORF gene with nucleotide position.
Figure 4.
Analysis of mutations in SARS-CoV-2 genomes produced from THP-1 parent and A3A-to-A3G-null THP-1 cells during passage experiments. SARS-CoV-2 genomic RNA was isolated and subjected to WGS. Sample 1 to 4: Wuhan variant from passage 1 of THP-1 parent cells. Sample 5 to 8: Wuhan variant from passage 5 of THP-1 parent cells. Sample 9 to 12: Wuhan variant from passage 1 of A3A-to-A3G-null THP-1 cells. Sample 13 and 14: Wuhan variant from passage 3 of A3A-to-A3G-null THP-1 cells. Sample 15 and 16: Wuhan variant from passage 4 and passage 5 of A3A-to-A3G-null THP-1 cells. Green boxes on the top show each SARS-CoV-2 ORF gene with nucleotide position.

Figure 5.
Analysis of mutational signatures in publicly available SARS-CoV-2 genomes. All possible 192 mutation types (NnN-to-NmN where n mutates to m and N:A/C/G/T) were quantified in a total of 38,830 whole genome SARS-CoV-2 sequences sampled between the years 2019 and 2022 and reprted in NCBI. These mutation counts were organized in a data matric of (38,830 by 192) and used as input for analysis using the Non-negative Matrix Factorization (NMF) method. Models with up to 20 components were built. Only a model with 10 component is shown for simplicity. None of the models showed signatures closely related to SBS2 and SBS13. Positions of the four major C>T and C>G peaks in SBS2 and SBS13 are shown by arrows.
Figure 5.
Analysis of mutational signatures in publicly available SARS-CoV-2 genomes. All possible 192 mutation types (NnN-to-NmN where n mutates to m and N:A/C/G/T) were quantified in a total of 38,830 whole genome SARS-CoV-2 sequences sampled between the years 2019 and 2022 and reprted in NCBI. These mutation counts were organized in a data matric of (38,830 by 192) and used as input for analysis using the Non-negative Matrix Factorization (NMF) method. Models with up to 20 components were built. Only a model with 10 component is shown for simplicity. None of the models showed signatures closely related to SBS2 and SBS13. Positions of the four major C>T and C>G peaks in SBS2 and SBS13 are shown by arrows.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



