Submitted:
03 June 2024
Posted:
04 June 2024
Read the latest preprint version here
Abstract
Magnesium monofluoride is a polar molecule amenable to laser cooling which has caused renewed interest in its spectroscopy.
In this work we consider the case of three low-lying electronic excitations, namely $\rm X \, {}^{2}\Sigma \to A \, {}^{2}\Pi, \ \to B \, {}^{2}\Sigma, \ \to C \,{}^{2}\Sigma$,
using well developed quantum chemistry approaches, i.e., without reference to the spin-orbit splitting of the $\rm A \, {}^{2}\Pi$ states.
Accurate experimental data for these transitions have been available for over 50 years. Here we explore the linear response method at the level of CC2 theory, as well as
equation of motion methods at the level of CCSD and CC3, using two families of basis sets.
Excellent agreement is obtained for the first three transitions when using the correlation-consistent basis sets and extrapolation to the complete basis limit within EOM-CC3 (at a relative precision of $10^{-4}$), and qualitative agreement for the other two methods.
The purpose of this paper is to serve as a guide in how to approach the accurate calculation of excitations in polar diatomic molecules.
Keywords:
polar molecules
; electronic excitation energies
; coupled-cluster methods
; quantum chemistry
1. Introduction
The spectroscopy of polar diatomic molecules, such as magnesium monofluoride (MgF) has a long history, and has led in the late 1960ies to results with remarkable accuracy. Barrow and Beale [1] resolved the rotational structure of gaseous MgF and carried out a detailed analysis resulting in the determination of molecular constants for the ground and first three electronic excited states with precise values based on the two lowest vibrational states (). Higher excitations were measured and reported in 1971 by Novikov and Gurvich[2]. The detailed structure of the transitions including spin-orbit effects (the doublet is split by about 35 ) focused on the nature of the -type doubling, and it was concluded that the doublet was inverted (negative A parameter). A recent study [3] does, however, conclude that the doublet is normal, i.e., not inverted. Hyperfine-resolved measurements are also reported in Ref. [4].
The interest in laser cooling of MgF has resulted in a detailed study of potential energy curves and spectroscopic constants at the level of multi-reference configuration interaction (MRCI) [5,6]. These results can serve the present study as a theoretical benchmark, in addition to the experimental results [7]. The ground-state potential energy curve can be determined also from infrared spectroscopy [8] which leads to an extended Morse oscillator description of this curve. This allows us to focus directly on the calculation of electronic excitations. These studies (and the present work) ignore spin-orbit effects. Such effects can be calculated in some standard packages, e.g., using two-component density functional theory [9] as implemented in TURBOMOLE [10].
Our interest in studying the accuracy of excited-state calculations within coupled-cluster (CC) theory relates to experimental work on matrix isolated barium monofluoride (BaF) [11]. In this context earlier work on matrix isolated atomic barium [12,13] was extended to the case of BaF in argon [14,15] and neon [16]. The matrix isolation work is motivated by performing measurements of the electron electric dipole moment. While the BaF molecule is much more complicated than MgF, the use of effective core potentials makes the computational effort comparable for the two cases. The purpose of the present work is to establish expectations on accuracy with respect to methodology and basis sets in order to understand how reliable the CC methods may be in the matrix isolation environment.
The current status of describing the electronic excitations of gas-phase BaF (and its lighter homologs CaF and SrF) can be described as follows. Results from scalar relativistic CC and MRCI calculations are reported in Ref. [17]. The all-electron Fock-space CC results for the excitation energies (which include spin-orbit splittings on the order of 400-600 ) agree with experiment at the level of about 200 . Better agreement (down to about 10 ) was obtained in Ref. [18] by including QED effects and basis set extrapolation of correlation effects calculated at the level of CCSDT(Q). A similar approach based on four-component CC Dirac theory resulted in accurate ionization energies [19]. Hyperfine constants for ground and excited-state BaF molecules were obtained in Ref. [20] and provide some confidence in the quality of the Fock-space CC wavefunctions.
To return to the topic of the present paper we note that in Ref. [21] radiative decay rates and branching fractions of the first excited state of 24 were reported in a comparative experimental-theoretical work. While the theory is the relativistic Fock-space CC method, comparison can be made with the present non-relativistic work due to the smallness of spin-orbit effects in MgF. The experimental data presented in Ref. [21] deviate somewhat (at the level) from the earlier work [1]. Studies which investigate equation-of-motion (EOM) coupled cluster methods have been carried out and are typically benchmarked against full configuration interaction calculations, but the goal of this work is to only compare the theoretical spectroscopic parameters to experimental determinations.
The layout of the paper is as follows. We begin in Section 2 with a short summary of how to use CC methods for electronic excitations. In Section 3 we present our results for excitation energies of MgF using three methods and two basis set families, and compare them with experimentally determined values, and with the relativistic Fock-space CC results of Ref. [20]. We draw some conclusions in Section 4.
2. Theory
Electronic excitation energies for molecules can be calculated in many quantum chemistry packages with a selection of methods. For the purpose of the present work the accuracy of time-dependent density functional theory methods is not sufficient, since we would like to reach percent-level agreement with measured values or better. This requires the inclusion of electron correlation with a systematic treatment as provided by configuration interaction or coupled cluster methodology.
A fast method in this realm is the CC2 method coupled with response theory [22]. It is, e.g., implemented in TURBOMOLE [10], alongside with a faster version based on Møller-Plesset perturbation theory, namely the ADC(2) method [23]. These allow for fast calculations based on the resolution-of-identity methods, and efficient basis sets of triple- and quadruple-zeta quality [24], (e.g, def2-QZVPPD). Transition moments and other excited-state first-order properties are calculated quickly even for larger molecules [25]. We will refer to this method as CC2-RPA. It is mostly restricted to not use the full symmetry, i.e., it is running with C1 symmetry by default in TURBOMOLE. One can use the properties of the excited states, such as transition moments to get some idea of which excited states have been generated.
If one requires higher accuracy, one has available a general excited-state method at the level of CC theory including single and double excitations, named EOM-CCSD (EOM stands for equation of motion) [26,27]. Since the class of polar molecules to which MgF belongs is dominated by single excitations, it should provide reasonable accuracy. One can ask for a number of excitations for each irreducible representation. We used Psi4 [28] to perform the calculations. The CCEOM package in Psi4 also allows to perform higher-accuracy calculations at the level of EOM-CC3 theory. These are demanding calculations, but are considered the best there is as a general-purpose CC method for excited states [29]. The EOM-CC3 method (as implemented in Psi4) first calculates the CC3 ground state, then performs an EOM-CCSD for multiple roots (which in our work has the trend of being higher in energy than the usual EOM-CCSD roots), and then one has to pick one root at a time to calculate the EOM-CC3 value. Thus, the method is more cumbersome to use. Faster implementations of EOM-CC3 than generally available have been developed [30].
In addition to the mentioned methods one can also attempt so-called -CC methods [31]. One complements a standard ground-state CC calculation with a run where the SCF calculation is carried our for an excited state obtained by switching two orbitals (e.g., highest occupied and lowest unoccupied) to seed an SCF calculation for a Slater determinant which represents the excited state. This step may or may not work, as the SCF calculation may collapse back to the ground state. When it works, the resulting orbitals can be used in a correlated calculation at the CCSD or CCSD(T) level or higher. For the MgF molecule we were not able to carry the -CC approach, since the step failed, but it is interesting to note that in cases where the method works the excitation energies from -CCSD and EOM-CCSD in general do not agree with each other [32]. The agreement may become better at the level of the CC3 model. Such results for the barium monofluoride (BaF) molecule using a pseudopotential for the inner electrons of barium for the , , and states will be reported in a separate publication.
In terms of basis sets we used the def2-family at triple- and quadruple-zeta level and made a guess at the complete basis set (CBS) limit by using an extrapolation of the form
where and are the energies obtained with def2-TZVPPD and def2-QZVPPD respectively.
We also used the correlation-consistent augmented basis sets aug-cc-PVnZ with [33,34]. From the calculated energies the CBS limit can be obtained using
which follows from Equation (2) in Ref. [35].
For the MgF molecule the known bond lengths for the ground and excited states in question are very close (Å). Thus, we restrict our calculations here to a single Mg-F separation, since we are interested in comparing values of , i.e., for excitation energies the difference in the minimal energies between the potential energy curves. Detailed calculations of such curves at the level of four-component (relativistic) Fock space CC theory are given, e.g., in Figure 1 of Ref. [21] and can be used for justification of this shortcut. For other molecules, such as, e.g., CaF, SrF and BaF one has to calculate the potential energy curves, i.e., energies as a function of R, e.g., as given in Ref. [17] in order to determine accurate adiabatic excitation energies. From these curves one can then determine the spectroscopic parameters and , which follow from the nuclear vibrational excitation energies in accord with
In the present work, however, the focus is entirely on the determination of accurate values of for a few excited states relative to the ground state.
A final general comment for this section can be made: each of the theory models and for each of the chosen basis sets the ground-state is calculated at some level of precision. All CC calculations start from a common self-consistent field result, which for our calculations is based on spin-unrestricted Hartree-Fock theory. The changes in ground-state energy with basis set at this level are not too significant. The differences between these values from these calculations are appreciable, and they arise mainly for two reasons: (i) the correlation contributions included in CC2, CCSD, and CC3 are different, and (ii) the truncated basis set introduces a significant error when computing correlation effects. Excitation energies are calculated relative to ground states which are numerically quite different. Nevertheless, they can be compared directly to each other when looking for excited-state values.
3. Results
3.1. Mg()
The first excitation is to the states. The present non-relativistic calculation can be compared to the experimental analysis of Ref. [1] which lists not but the transition energies to the lowest two vibrational states . The doublet has a separation of which will be of the order of the accuracy goal for calculating this excitation. Figure 1 shows this doublet as a pair of solid lines, and the relativistic calculation from Ref. [21] as a pair of dashed lines at the right. This calculation gives results which are higher by about , but it should be noted that Ref. [21] includes an analysis of the data of Ref. [7] which results in higher values for the excitation energies than the original data of Barrow and Beale [1] by about . Such a difference exceeds the accuracy of the original experimental analysis, and can be related to the use of the effective Hamiltonian involved in the analysis of the data. An example of such a discrepancy was reported in Ref. [36]. For our purposes one may then argue that the best nonrelativistic result would fall not in the middle between the two black lines, but closer to the shown energy.
On the left in Figure 1 we observe that the extrapolated CC2-RPA results underestimate the transition energy: the extrapolated result using the def2 basis sets (blue square) disagrees by about . The aug-cc basis set calculations fare a bit better, reducing the discrepancy to about . The open symbols show that the results from the EOM-CCSD method overestimate the excitation energy. The relationship between the two basis set families remains similar to the results from the CC2-RPA method. Finally, the EOM-CC3 results (solid circles) turn out to be lower than EOM-CCSD, with the aug-cc basis set results leading to an extrapolated value that falls within of the results from Ref. [1], or Ref. [7] as re-analyzed in Ref. [21]. The EOM-CC3 method with the aug-cc basis set family can be considered a great success in reaching better than per mille relative accuracy for the transition energy. It would be interesting to find out whether the relativistic Fock-space CC method of Ref. [21] can be pushed to higher order to reach this level of precision on a system which involves 21 electrons.
3.2. Mg()
Excitations to higher electronic states can be expected to come out with less accuracy, particularly since they are in the same symmetry group. Figure 2 shows that this is clearly the case: the ordinate spans a wider range. All methods approach the experimental excitation energy from above, and exhibit qualitatively a similar trend when comparing the def2 and aug-cc basis sets. The differences are larger (on the order of ), and the aug-cc basis set calculations clearly do better. Remarkably the EOM-CC3 result for the aug-cc basis set falls right onto the experimental value when extrapolated to the CBS limit. The Fock-space CC results of Ref. [21] are more than too high.
3.3. Mg()
The next-higher state was considered by Barrow and Beale [1] to be a Rydberg state. The energy scale in Figure 3 is expanded to cover . Interestingly, this range is required mostly to show the difference between results from the two basis set families. The EOM-CC3 method gives an excellent result in the CBS limit. In order to assess whether this is a lucky coincidence, we report in Table 1 the actual EOM-CC3 data for the aug-cc basis sets. These data allow one to observe some interesting trends, particularly as to how the extrapolation works.
For the excitation to the the progression of numbers approaches the CBS from below in a systematic way. The extrapolation to a value which is about above the answer is based on the fact that the quadruple- and quintuple-zeta basis result differ by almost .
For the excitation the progression of results is not monotonic. It follows, however, from Equation (2) that the estimated CBS limit result is governed by the increase in excitation energy between the results. The variation of the excitation energy with basis set quality is at the level. This is an interesting observation in the context of the relativistic calculation of Ref. [21] which overestimates experiment by about twice this amount. It leads to the conclusion that the EOM-CC3 method includes the important correlation contributions almost independent of the value of n when using the aug-cc-pVnZ basis. Given that the def2 basis result was less successful (cf. Figure 2), one can argue that the augmentation of the basis set is important to achieve the reported accuracy.
For the case of excitation the sequence of excitation energies for is systematic and again approaches the extrapolated result from below. The data for all three excited states presented in Table 1 support the notion that the triple-zeta (n=3) calculations can be used to obtain a quick overview of the situation accurate at the few-hundred level.
4. Conclusions
Electronically excited states of magnesium fluoride, a basic polar diatomic molecule were calculated at few levels of coupled-cluster theory. The very fast CC2-RPA method as implemented in TURBOMOLE yields good first estimates of the excitation spectrum, particularly in combination with the aug-cc basis set family.
The EOM-CCSD method which is a turnkey method to obtain the spectrum tends to overestimate the excitation energies. While the def2 basis works reasonably well for the lower two excitations, it leads to overestimated energies for the state both in the CC2-RPA and the EOM-CCSD methods.
The EOM-CC3 method requires more effort (both operationally and in terms of computing time), but represents the most accurate of these toolsets. For molecules where the methodology works, i.e., higher roots than for the ground state can be found computations should represent an economical alternative to it.
Acknowledgments
I thank Eric Hessels, Gregory Koyanagi and Rene Fournier for many discussions. Financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC) (RGPIN-2017-05655 and RGPIN-2019-06305) is gratefully acknowledged.
References
- Barrow, R.F.; Beale, J.R. Rotational analysis of electronic bands of gaseous MgF. Proceedings of the Physical Society 1967, 91, 483. [Google Scholar] [CrossRef]
- Novikov, M.M.; Gurvich, L.V. The electronic spectra of MgF and MgF+. Journal of Applied Spectroscopy 1971, 14, 820–822. [Google Scholar] [CrossRef]
- Xu, S.; Xia, M.; Yin, Y.; Gu, R.; Xia, Y.; Yin, J. Determination of the normal A2∏ state in MgF with application to direct laser cooling of molecules. The Journal of Chemical Physics 2019, 150, 084302. [Google Scholar] [CrossRef] [PubMed]
- Doppelbauer, M.; Wright, S.C.; Hofsäss, S.; Sartakov, B.G.; Meijer, G.; Truppe, S. Hyperfine-resolved optical spectroscopy of the A2∏ ← X2∑+ transition in MgF. The Journal of Chemical Physics 2022, 156, 134301. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Gao, Y.; Kuang, F.; Gao, T.; Du, J.; Jiang, G. Theoretical study of laser cooling of magnesium monofluoride using ab initio methods. Phys. Rev. A 2015, 91, 042511. [Google Scholar] [CrossRef]
- Kang, S.; Gao, Y.; Kuang, F.; Gao, T.; Du, J.; Jiang, G. Erratum: Theoretical study of laser cooling of magnesium monofluoride using ab initio methods [Phys. Rev. A 91, 042511 (2015)]. Phys. Rev. A 2015, 92, 069902. [Google Scholar] [CrossRef]
- Gu, J.; Xiao, Z.; Yu, C.; Zhang, Q.; Chen, Y.; Zhao, D. High resolution laser excitation spectra and Franck-Condon factors of A2∏–X2∑+ electronic transition of MgF. Chinese Journal of Chemical Physics 2022, 35, 58–68. [Google Scholar] [CrossRef]
- Hou, S.; Bernath, P.F. Line list for the MgF ground state. Journal of Quantitative Spectroscopy and Radiative Transfer 2017, 203, 511–516. [Google Scholar] [CrossRef]
- Bruder, F.; Franzke, Y.J.; Weigend, F. Paramagnetic NMR Shielding Tensors Based on Scalar Exact Two-Component and Spin–Orbit Perturbation Theory. The Journal of Physical Chemistry A 2022, 126, 5050–5069. [Google Scholar] [CrossRef] [PubMed]
- TURBOMOLE V7.8 2024, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available fromhttp://www.turbomole.com.
- Vutha, A.C.; Horbatsch, M.; Hessels, E.A. Oriented Polar Molecules in a Solid Inert-Gas Matrix: A Proposed Method for Measuring the Electric Dipole Moment of the Electron. Atoms 2018, 6. [Google Scholar] [CrossRef]
- Buchachenko, A.A.; Viehland, L.A. Interaction potentials and transport properties of Ba, Ba+, and Ba2+ in rare gases from He to Xe. The Journal of Chemical Physics 2018, 148, 154304. [Google Scholar] [CrossRef] [PubMed]
- Kleshchina, N.N.; Kalinina, I.S.; Leibin, I.V.; Bezrukov, D.S.; Buchachenko, A.A. Stable axially symmetric atomic impurity in an fcc solid—Ba in rare gases. The Journal of Chemical Physics 2019, 151, 121104. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, G.; Lambo, R.; Ragyanszki, A.; Fournier, R.; Horbatsch, M.; Hessels, E. Accurate calculation of the interaction of a barium monofluoride molecule with an argon atom: A step towards using matrix isolation of BaF for determining the electron electric dipole moment. Journal of Molecular Spectroscopy 2023, 391, 111736. [Google Scholar] [CrossRef]
- Lambo, R.L.; Koyanagi, G.K.; Ragyanszki, A.; Horbatsch, M.; Fournier, R.; Hessels, E.A. Calculation of the local environment of a barium monofluoride molecule in an argon matrix: A step towards using matrix-isolated BaF for determining the electron electric dipole moment. Molecular Physics 2023, 121, e2198044. [Google Scholar] [CrossRef]
- Lambo, R.L.; Koyanagi, G.K.; Horbatsch, M.; Fournier, R.; Hessels, E.A. Calculation of the local environment of a barium monofluoride molecule in a neon matrix. Molecular Physics 2023, 121, e2232051. [Google Scholar] [CrossRef]
- Hao, Y.; Pašteka, L.F.; Visscher, L.; Aggarwal, P.; Bethlem, H.L.; Boeschoten, A.; Borschevsky, A.; Denis, M.; Esajas, K.; Hoekstra, S.; Jungmann, K.; Marshall, V.R.; Meijknecht, T.B.; Mooij, M.C.; Timmermans, R.G.E.; Touwen, A.; Ubachs, W.; Willmann, L.; Yin, Y.; Zapara, A.; eEDM Collaboration), N. High accuracy theoretical investigations of CaF, SrF, and BaF and implications for laser-cooling. The Journal of Chemical Physics 2019, 151, 034302. [Google Scholar] [CrossRef]
- Skripnikov, L.V.; Chubukov, D.V.; Shakhova, V.M. The role of QED effects in transition energies of heavy-atom alkaline earth monofluoride molecules: A theoretical study of Ba+, BaF, RaF, and E120F. The Journal of Chemical Physics 2021, 155, 144103. [Google Scholar] [CrossRef]
- Kyuberis, A.A.; Pašteka, L.F.; Eliav, E.; Perrett, H.A.; Sunaga, A.; Udrescu, S.M.; Wilkins, S.G.; Garcia Ruiz, R.F.; Borschevsky, A. Theoretical determination of the ionization potentials of CaF, SrF, and BaF. Phys. Rev. A 2024, 109, 022813. [Google Scholar] [CrossRef]
- Denis, M.; Haase, P.A.B.; Mooij, M.C.; Chamorro, Y.; Aggarwal, P.; Bethlem, H.L.; Boeschoten, A.; Borschevsky, A.; Esajas, K.; Hao, Y.; Hoekstra, S.; van Hofslot, J.W.F.; Marshall, V.R.; Meijknecht, T.B.; Timmermans, R.G.E.; Touwen, A.; Ubachs, W.; Willmann, L.; Yin, Y. Benchmarking of the Fock-space coupled-cluster method and uncertainty estimation: Magnetic hyperfine interaction in the excited state of BaF. Phys. Rev. A 2022, 105, 052811. [Google Scholar] [CrossRef]
- Norrgard, E.B.; Chamorro, Y.; Cooksey, C.C.; Eckel, S.P.; Pilgram, N.H.; Rodriguez, K.J.; Yoon, H.W.; Pašteka, L.c.v.F.; Borschevsky, A. Radiative decay rate and branching fractions of MgF. Phys. Rev. A 2023, 108, 032809. [Google Scholar] [CrossRef]
- Hättig, C.; Weigend, F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. The Journal of Chemical Physics 2000, 113, 5154–5161. [Google Scholar] [CrossRef]
- Hättig, C. Structure Optimizations for Excited States with Correlated Second-Order Methods: CC2 and ADC(2). In Response Theory and Molecular Properties (A Tribute to Jan Linderberg and Poul Jørgensen); Jensen, H., Ed.; Academic Press, 2005; Vol. 50, Advances in Quantum Chemistry, pp. 37–60. [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Hättig, C.; Köhn, A. Transition moments and excited-state first-order properties in the coupled-cluster model CC2 using the resolution-of-the-identity approximation. The Journal of Chemical Physics 2002, 117, 6939–6951. [Google Scholar] [CrossRef]
- Gauss, J. Equation-of-Motion Coupled-Cluster Theory, 2017. https://www.esqc.org/lectures/gauss_EOM_lecture_handout.pdf [Accessed: , 2024]. 31 May.
- Crawford, T.D.; Schaefer III, H.F. , An Introduction to Coupled Cluster Theory for Computational Chemists. In Reviews in Computational Chemistry; John Wiley & Sons, Ltd, 2000; pp. 33–136. [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; James, A.M.; Lehtola, S.; Misiewicz, J.P.; Scheurer, M.; Shaw, R.A.; Schriber, J.B.; Xie, Y.; Glick, Z.L.; Sirianni, D.A.; O’Brien, J.S.; Waldrop, J.M.; Kumar, A.; Hohenstein, E.G.; Pritchard, B.P.; Brooks, B.R.; Schaefer, Henry F. , I.; Sokolov, A.Y.; Patkowski, K.; DePrince, A. Eugene, I.; Bozkaya, U.; King, R.A.; Evangelista, F.A.; Turney, J.M.; Crawford, T.D.; Sherrill, C.D. PSI4 1.4: Open-source software for high-throughput quantum chemistry. The Journal of Chemical Physics 2020, 152, 184108. [Google Scholar] [CrossRef]
- Damour, Y.; Quintero-Monsebaiz, R.; Caffarel, M.; Jacquemin, D.; Kossoski, F.; Scemama, A.; Loos, P.F. Ground- and Excited-State Dipole Moments and Oscillator Strengths of Full Configuration Interaction Quality. Journal of Chemical Theory and Computation 2023, 19, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.C.; Myhre, R.H.; Koch, H. New and Efficient Implementation of CC3. Journal of Chemical Theory and Computation 2021, 17, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Damour, Y.; Scemama, A.; Jacquemin, D.; Kossoski, F.; Loos, P.F. State-Specific Coupled-Cluster Methods for Excited States. 2024; arXiv:physics.chem-ph/2401.05048. [Google Scholar]
- Lee, J.; Small, D.W.; Head-Gordon, M. Excited states via coupled cluster theory without equation-of-motion methods: Seeking higher roots with application to doubly excited states and double core hole states. The Journal of Chemical Physics 2019, 151, 214103. [Google Scholar] [CrossRef] [PubMed]
- Prascher, B.P.; Woon, D.E.; Peterson, K.A.; Dunning, T.H.; Wilson, A.K. Gaussian basis sets for use in correlated molecular calculations. VII. Valence, core-valence, and scalar relativistic basis sets for Li, Be, Na, and Mg. Theoretical Chemistry Accounts 2011, 128, 69–82. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, Thom H. , J.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. The Journal of Chemical Physics 1992, 96, 6796–6806, https://pubs.aip.org/aip/jcp/article-pdf/96/9/6796/18998924/6796_1_online.pdf. [Google Scholar] [CrossRef]
- Peterson, K.A.; Woon, D.E.; Dunning, Thom H. , J. Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H+H2→H2+H reaction. The Journal of Chemical Physics 1994, 100, 7410–7415. [Google Scholar] [CrossRef]
- Field, R.W.; Bergeman, T.H. Radio-Frequency Spectroscopy and Perturbation Analysis in CS A 1 ∏ (theta = 0). The Journal of Chemical Physics 1971, 54, 2936–2948. [Google Scholar] [CrossRef]
Figure 1.
Excitation energies in wavenumbers () for the electronic transition . Solid lines: experimental value extracted from the analysis of rotational and vibrational () excitations for the and states based on data given in Ref. [1]. The short dashed lines show the Fock-space CC results from Ref. [21]. The present theoretical results are without spin-orbit coupling and should fall in between the black solid lines. Squares: results from the CC2-RPA method, open circles: from EOM-CCSD, and solid circles from EOM-CC3. For each method basis sets of increasing accuracy were applied and extrapolated to a complete basis set limit: on the left (in blue) for the def2-TZVPPD and def2-QZVPPD basis sets, and on the right (in red) for the aug-cc-pVnZ family with n=3, 4, 5.
Figure 1.
Excitation energies in wavenumbers () for the electronic transition . Solid lines: experimental value extracted from the analysis of rotational and vibrational () excitations for the and states based on data given in Ref. [1]. The short dashed lines show the Fock-space CC results from Ref. [21]. The present theoretical results are without spin-orbit coupling and should fall in between the black solid lines. Squares: results from the CC2-RPA method, open circles: from EOM-CCSD, and solid circles from EOM-CC3. For each method basis sets of increasing accuracy were applied and extrapolated to a complete basis set limit: on the left (in blue) for the def2-TZVPPD and def2-QZVPPD basis sets, and on the right (in red) for the aug-cc-pVnZ family with n=3, 4, 5.
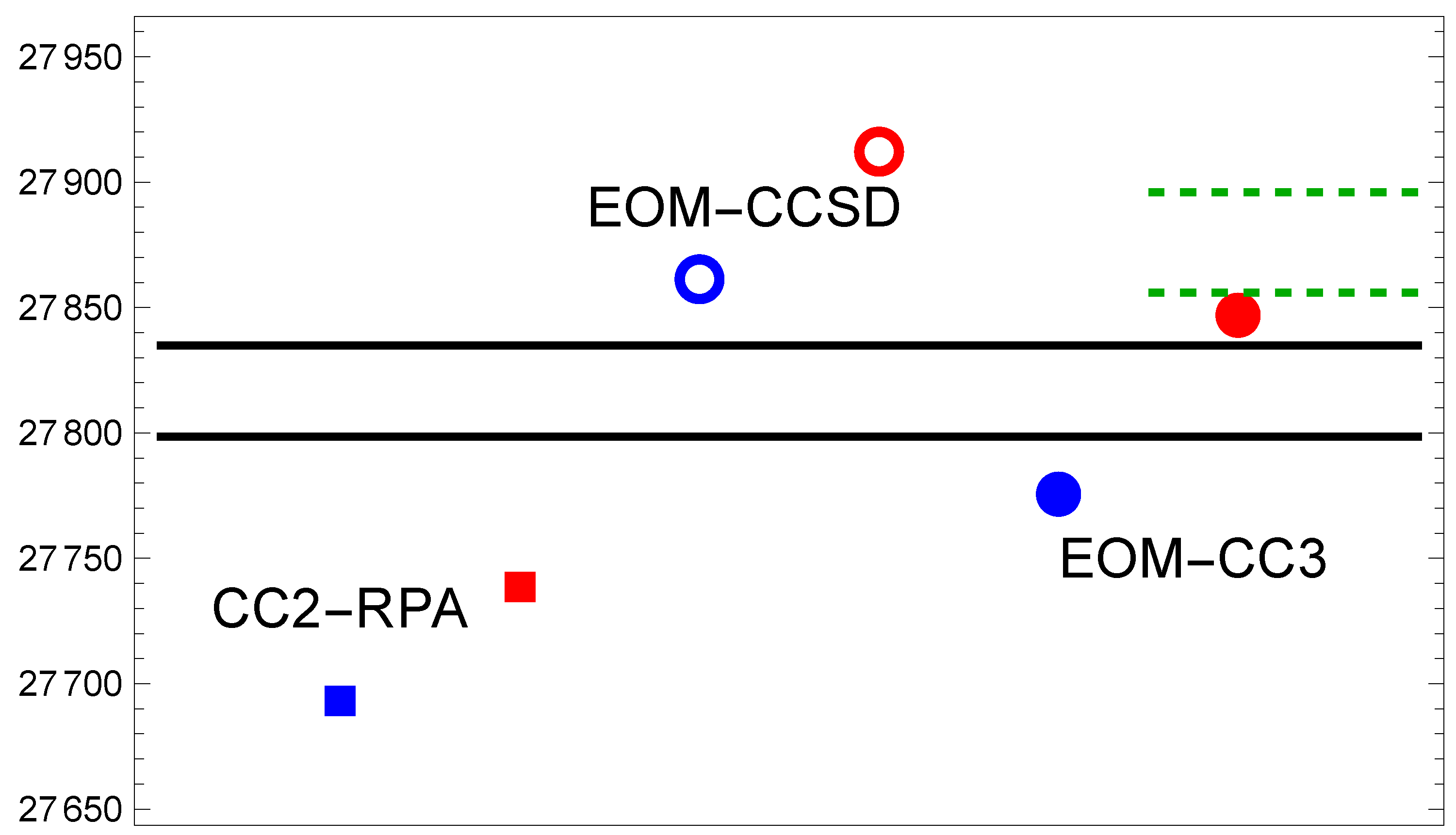
Figure 2.
Same as in Figure 1, but for the transition.
Figure 2.
Same as in Figure 1, but for the transition.
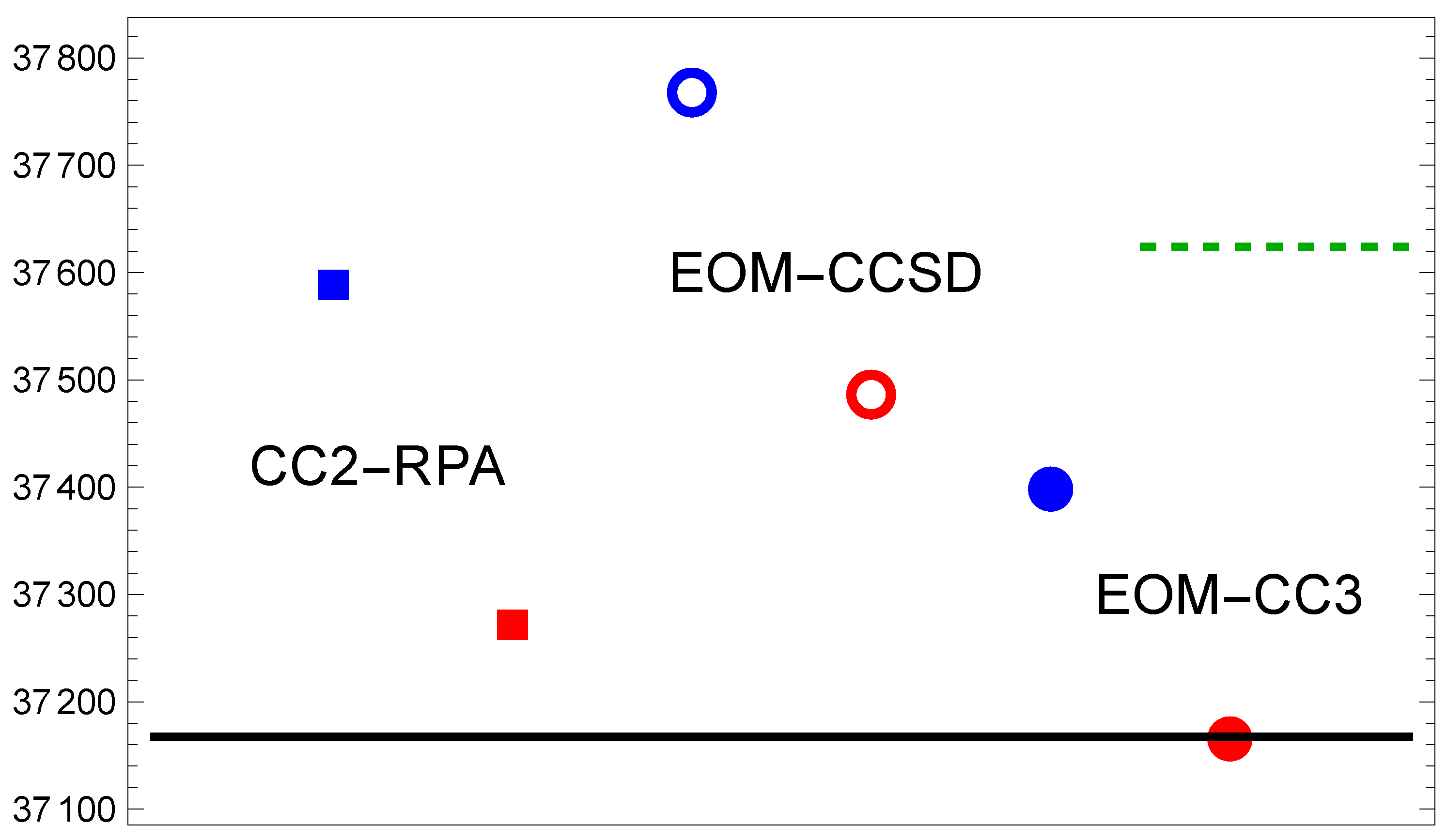
Figure 3.
Same as in Figure 1, but for the transition.
Figure 3.
Same as in Figure 1, but for the transition.
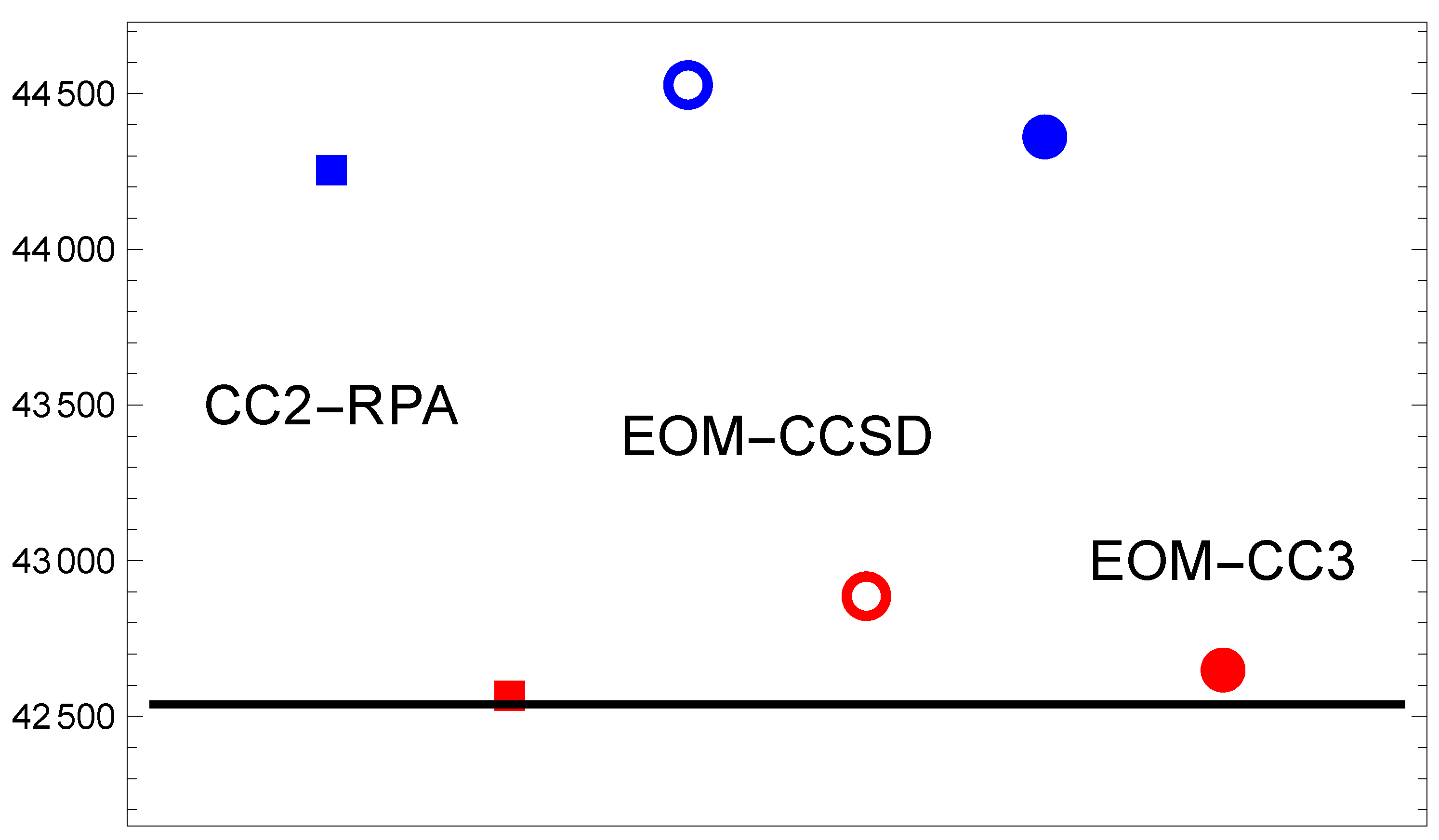

Table 1.
Excitation energies in using the aug-cc-pVnZ basis and the EOM-CC3 method. Experimental data in the last column are from Ref. [1]; for the excited state the number marked with † is based on the measurement of Ref. [7] as re-analyzed and reported in Ref. [21].
| CBS | Expt | ||||
|---|---|---|---|---|---|
| 27 507 | 27 556 | 27 744 | 27 849 | 27 817 27 834† | |
| 36 966 | 36 895 | 37 067 | 37 169 | 37 167 | |
| 42 257 | 42 316 | 42 534 | 42 663 | 42 590 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



