
Submitted:
18 May 2024
Posted:
20 May 2024
You are already at the latest version
Abstract
Methylation is a biochemical process involving the addition of a methyl group (-CH3) to various chemical compounds. It plays a crucial role in maintaining the homeostasis of the endothelium, which lines the interior surface of blood vessels and has been linked, among other conditions, to coronary artery disease (CAD). Despite significant progress in CAD diagnosis and treatment, intensive research continues into genotypic and phenotypic CAD biomarkers. This review explores the significance of the methylation pathway and folate metabolism in CAD pathogenesis, with a focus on endothelial dysfunction resulting from deficiency in the active form of folate (5-MTHF). We discuss emerging areas of research into CAD biomarkers and factors influencing the methylation process. By highlighting genetically determined methylation disorders, particularly the MTHFR polymorphism, we propose the potential use of the active form of folic acid (5-MTHF) as a novel CAD biomarker and personalized pharmaceutical for selected patient groups. Our aim is to improve the identification of individuals at high risk of CAD and enhance their prognosis.
Keywords:
Methylation
; Folate
; 5-MTHF
; Coronary artery disease
; biomarker
; personalized medicine
1. Introduction
In cardiovascular diseases (CVD), significant progress has been made in novel diagnostic and treatment methods. These include interventions in cardiology and the management of post-hospitalization patients through hybrid comprehensive cardiac rehabilitation using telemedicine. This approach has been shown to enhance the quality of life for patients with heart failure (The Telerehabilitation in Heart Failure Patients trial, TELE-REH-HF) [1].
The primary objective of innovation in cardiology is to reduce the incidence of CVD and Major Adverse Cardiovascular Events (MACE), often defined as myocardial infarction, cardiac arrest, coronary revascularization procedure, ischemic stroke, or death from cardiovascular causes. Despite extensive research, including multi-omics data (including genomics, epigenomics, transcriptomics, proteomics, and metabolomics) along with artificial intelligence (AI), CVDs are widespread in the general population [2].
The identification of prognostic biomarkers in primary CAD prevention and the management of MACE in CAD patients during secondary prevention are crucial for enhancing patient management and outcomes. Presently, CAD prevention strategies primarily target traditional risk factors such as obesity, diabetes, hypertension, and dyslipidemia. However, there exists a notable gap in addressing additional risks posed by non-traditional factors [3].
Impaired methylation pathway, which involves folate and methyltetrahydrofolate (5-MTHF) metabolism disorders, has been gaining interest with regard to CAD caused by genetic polymorphism of the methyltetrahydrofolate reductase gene (MTHFR). The field of pharmacogenomics, which focuses on individual drug selection based on the genetic polymorphisms and mutations of each patient, is rapidly advancing. Patients with genet-ic methylation disorders may be suitable candidates for personalized therapy utilizing the active form of folate.
The aim of this review is to clarify the physiological importance of the methylation pathway and folate metabolism in CAD patients, as this area is not yet widely under-stood. We aim to explore how this knowledge could be utilized to uncover novel, person-alized treatment approaches for patients.
2. Overview of the Methylation Pathway
Methylation is a biochemical process in which a methyl group (-CH3) is attached to chemical compounds such as neurotransmitters, lipids, proteins, and DNA [4].
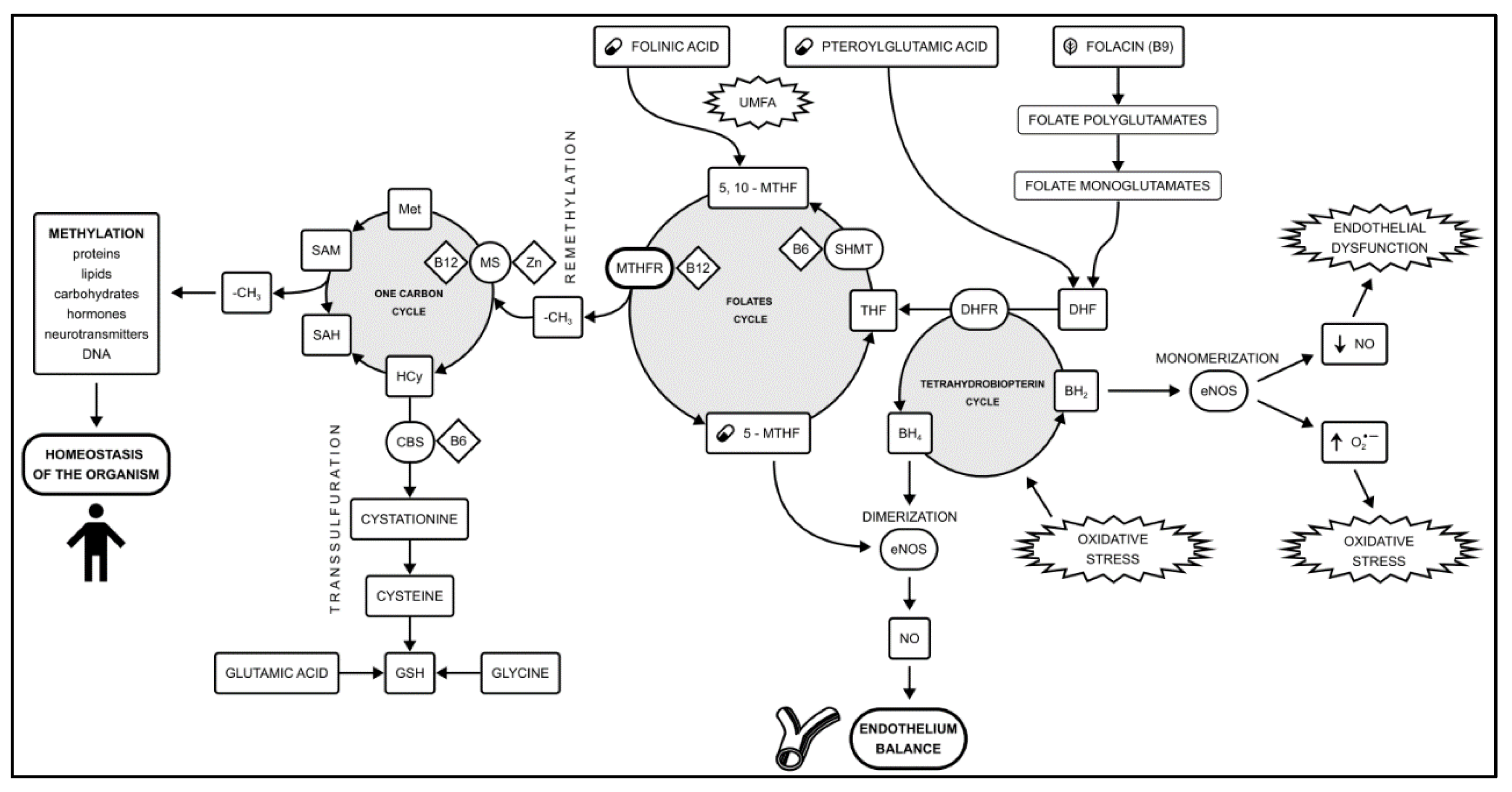
The main metabolic pathways involving methylation are the folate cycle and the one-carbon group cycle [5]. These pathways are essential for generating methyl groups to regulate all methylation-related processes, associated with metabolic processes governing cell division and tissue development. An additional role in this pathway is played by the transsulfuration and tetrahydrobiopterin (BH4) cycle. The connections between the cycles forming the methylation pathway are shown in Figure 1.
The universal donor of the methyl group is S-adenosylmethionine (SAM) - a derivative of adenosine and methionine, which is the most important substrate in methylation processes. SAM is necessary for regulating the functions of nucleic acids and many pro-teins important for proper prenatal development, as well as throughout life after birth. Af-ter delivering the methyl group to target sites (e.g., DNA), S-adenosylhomocysteine (SAH) is formed, which is then transformed into homocysteine and adenosine via the enzyme adenosylhomocysteinase [6]. The SAM to SAH ratio has been used as a marker of "cellular methylation capacity" [7], and it has a documented role in the pathogenesis of CAD [8].
Methylation plays role in the gene expression, functioning of multiple enzymes, synthesis of catecholamines as well as in the DNA repair. DNA and histone methylation occur during cell division, epigenesis, and imprinting. Maintaining the appropriate balance between substrates, products, and cofactors of the methylation pathway is essential for the homeostasis of the body. Methylation is crucial for maintaining the proper function of the vascular endothelium, the maturation and development of the central nervous system, and in the regulation of gametogenesis and embryo development [9]. Moreover, recently, the folate cycle and the one-carbon cycle have been identified as regulators of the aging process [10].
Impairment of the methylation pathway leads to the development of neurological disorders such as Alzheimer's disease, autism, Down syndrome, multiple sclerosis; gynecological conditions such as impaired fertility, intrauterine death; as well as immune disorders, cancer, thrombophilia; and cardiovascular diseases such as CAD and ischemic stroke [11,12,13,14,15,16,17,18,19,20,21,22].
Figure 1.
The methylation pathway formed by the one-carbon cycle and the folates cycle. An additional role is played by the transsulfuration pathway and the tetrahydrobiopterin cycle. Methylation disorders caused by the MTHFR polymorphism play a role in the development of vascular endothelial dysfunction. The 5-MTHF availability plays a key role in the amount of circulating NO. Shifting the balance between NO production and oxidative stress in endothelial cells is a key step in the CAD development. Based on [23,24,25]. Abbreviations used: (O2•−) Superoxide anion radical; 5,10-MTHF - 5,10-methylenetetrahydrofolate; 5-MTHF - methylated folic acid, levomefolic acid; BH2 - Dihydrobiopterin; BH4 - Tetrahydrobiopterin; CAD - coronary artery disease; CBS - cystathionine β-synthase; DHF - dihydrofolate; DHFR - dihydrofolate reductase; eNOS - endothelial nitric oxide synthase; GSH - reduced glutathione; Hcy - Homocysteine; Met - Methionine; MS - Methionine synthase; MTHFR - methyltetrahydrofolate reductase; NO - nitric oxide; SAH - S-adenosylhomocysteine; SAM - S-adenosylmethionine; SHMT - serine hydroxymethyltransferase; THF - tetrahydrofolate; UMFA - Unmetabolized Folic Acid syndrome.
Figure 1.
The methylation pathway formed by the one-carbon cycle and the folates cycle. An additional role is played by the transsulfuration pathway and the tetrahydrobiopterin cycle. Methylation disorders caused by the MTHFR polymorphism play a role in the development of vascular endothelial dysfunction. The 5-MTHF availability plays a key role in the amount of circulating NO. Shifting the balance between NO production and oxidative stress in endothelial cells is a key step in the CAD development. Based on [23,24,25]. Abbreviations used: (O2•−) Superoxide anion radical; 5,10-MTHF - 5,10-methylenetetrahydrofolate; 5-MTHF - methylated folic acid, levomefolic acid; BH2 - Dihydrobiopterin; BH4 - Tetrahydrobiopterin; CAD - coronary artery disease; CBS - cystathionine β-synthase; DHF - dihydrofolate; DHFR - dihydrofolate reductase; eNOS - endothelial nitric oxide synthase; GSH - reduced glutathione; Hcy - Homocysteine; Met - Methionine; MS - Methionine synthase; MTHFR - methyltetrahydrofolate reductase; NO - nitric oxide; SAH - S-adenosylhomocysteine; SAM - S-adenosylmethionine; SHMT - serine hydroxymethyltransferase; THF - tetrahydrofolate; UMFA - Unmetabolized Folic Acid syndrome.
3. Folate Metabolism
Folates are essential to life, serving as components and catalysts in biochemical reactions, particularly through their central role in the methylation cycle. They participate in processes crucial for the growth, development, and reproduction of all body cells, as well as in the synthesis of nucleic acids, purines, and pyrimidines. Folates also contribute to the hydroxylation of long-chain fatty acids and the proper functioning of the hematopoetic, nervous, and cardiovascular systems [26].
- Folates, which are chemically salts of folic acid, represent a group of pterin derivatives. Research has identified approximately 150 different forms of folic acid, with about 20 found in nature [27]. Humans cannot synthesize folate, and because it is water-soluble, it can be stored to limited extent. For this reason, folate must be included in the diet or externally supplemented. Forms of folates currently available as pharmaceuticals have been presented in Table 2. The variations among individual folate compounds lie in the oxidation level of the pyridine ring and the number of glutamic acid residues. The main folates include:
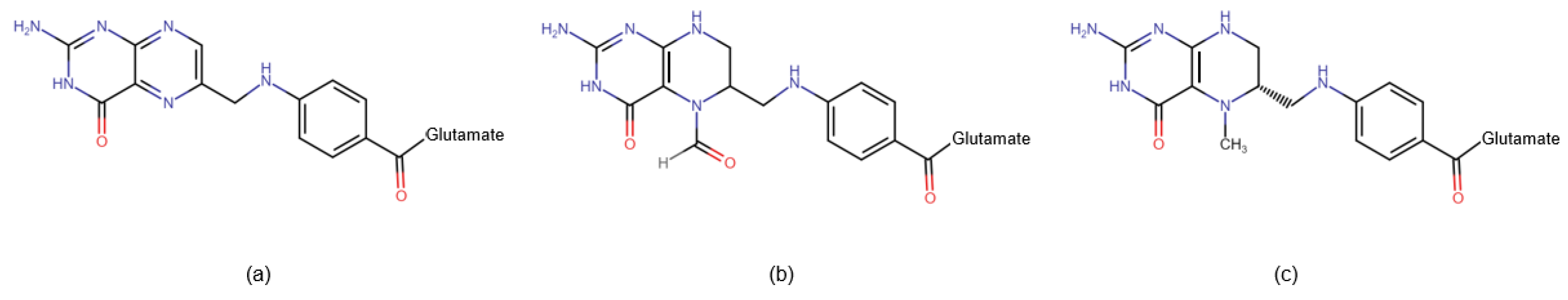
- Folic acid (also known as folacin or vitamin B9): This naturally occurring water-soluble molecule is predominantly found in green leafy vegetables such as asparagus, spinach, lettuce, and broccoli. In food, folic acid exists as complex compounds known as polyglutamine conjugates. These compounds are broken down in the small intestine into monoglutamates, which are absorbed into enterocytes. Within the cell, folic acid is converted to dihydrofolate (DHF), and then to tetrahydrofolate (THF) by the enzyme dihydrofolate reductase (DHFR). Subsequently, the enzyme serine hydroxymethyltransferase (SHMT) transfers the methylene group from the serine side chain to THF, resulting in the production of 5,10-methylenetetrahydrofolate (5,10-MTHF) and glycine. Methylenetetrahydrofolate reductase (MTHFR) then facilitates the formation of 5-methyltetrahydrofolate (5-MTHF), the biologically active form of folic acid.
- Pteroylglutamic acid: This synthetically produced molecule consists of a pteroyl residue and 2 to 7 glutamine residues. It is used as a dietary supplement and for food fortification. Before entering the folate cycle, it must be reduced by DHFR to DHF and then to THF, ultimately being converted to biologically active 5-MTHF. Supplementation with synthetic folic acid may lead to a syndrome known as Unmetabolized Folic Acid (UMFA) syndrome.
- Folinic acid (leucovorin): This synthetic molecule is a 5-formyl derivative of THF, which is converted into 5,10-MTHF without the need for DHFR. MTHFR is required for its conversion to 5-MTHF. Folinic acid is used to mitigate the toxic effects of chemotherapy agents that disrupt folate metabolism by inhibiting DHFR (e.g., methotrexate).
- Levomefolic acid, methyltetrahydrofolate (5-MTHF): This represents the predominant physiological form of folic acid found in the blood. The availability of 5-MTHF facilitates the conversion of methionine to S-adenosylmethionine (SAM), a universal methylation effector. After the release of the methyl group, S-adenosylhomocysteine (SAH) and homocysteine are produced, exerting feedback that inhibits methylation.
Figure 2.
Chemical structure of folate pharmaceuticals. (a) Pteroylglutamic acid – the most commonly used form of folic acid, requires conversion by two enzymes: DHFR and MTHR; (b) Folinic acid – used in antifolates (ex. methotrexate) overdose, requires conversion by MTHFR; (c) 5-MTHF – biologically active form of folic acid, does not require conversion by the enzymes. Abbreviations used: DHFR - dihydrofolate reductase; MTHFR - methyltetrahydrofolate reductase.
Figure 2.
Chemical structure of folate pharmaceuticals. (a) Pteroylglutamic acid – the most commonly used form of folic acid, requires conversion by two enzymes: DHFR and MTHR; (b) Folinic acid – used in antifolates (ex. methotrexate) overdose, requires conversion by MTHFR; (c) 5-MTHF – biologically active form of folic acid, does not require conversion by the enzymes. Abbreviations used: DHFR - dihydrofolate reductase; MTHFR - methyltetrahydrofolate reductase.
2.1. Differences between Folates – Diagnostic Insights
Limited awareness of the differences among individual compounds within the folate group and of the variations in tests used for detecting folate levels in the blood, contributes to substantial misinterpretation of test results [28]. Fluorescence-based tests, commonly employed in diagnostics, evaluate levels of all folates containing a pteroyl ring. However, using such tests to monitor folic acid therapy does not accurately reflect the level of the biologically active form of folic acid. In contrast, tests utilizing Liquid Chromatography - Mass Spectrometry (LC-MS) have the capability to differentiate between 5-MTHF, pteroylglutamic and folinic acid. Therefore, these tests, despite not being widely utilized yet, should be employed in assessing folate status, as they solely enable the evaluation of the concentration of the metabolically active methyl donor and provide a genuine reflection of the folate status in patients with genetic methylation disorders [29].
4. Pathogenesis of CAD and its Risk Factors
CAD is usually the first and most frequent manifestation of cardiovascular disease, and it remains the leading cause of mortality and morbidity worldwide [3]. It is caused by impairment in coronary blood flow, leading to chronic or acute ischemia of the heart muscle. Coronary blood flow impairment is typically associated with atherosclerosis, which affects the epicardial coronary arteries. However, there are many additional mechanisms leading to myocardial ischemia, such as thrombosis, coronary vasospasm, inflammation, and microcirculatory dysfunction [30].
Atherosclerosis, a chronic arterial disease characterized by typical lesions due to lipid accumulation and fibrosis, is primarily driven by oxidized low-density lipoprotein particles (oxyLDL), which are involved in nearly all stages of its progression. Lipoproteins, high-molecular complexes comprising a hydrophobic lipid core containing cholesterol esters and triglycerides, along with a polar shell consisting of phospholipids, free cholesterol, and apolipoproteins, play a crucial role in lipid metabolism and transport.
While the inflammatory process within vessel walls is currently recognized as the primary cause of atherosclerosis [31], endothelial dysfunction contributes to CAD pathogenesis in its earliest stages [30]. Contrary to previous beliefs that atherosclerosis was an irreversible, age-associated degenerative process, it is now understood to develop episodically, potentially subsiding, with lifestyle modifications and medications capable of modulating its course [32].
The traditional cardiovascular risk factors were identified in the 1960s. According to the European Society of Cardiology, they are dyslipidemia, hypertension, smoking, diabetes and obesity. The risk of CAD increases after the age of 40. These risk factors have been included in the Systemic Coronary Risk Estimation 2 (SCORE2), SCORE2-Older Persons (SCORE2-OP) and SCORE2-Diabetes cardiovascular risk classification systems [33,34]. Despite the implementation of therapy for traditional cardiovascular risk factors, such as recommendations to lose weight, stop smoking, implement antihypertensive, lipid-lowering and antidiabetic drugs, the therapeutic goals are not achieved by the patients from the high cardiovascular risk group [35].
In addition to lifestyle-related risk factors, numerous genetic, biochemical, and vascular wall characteristics contribute to heightened cardiovascular risk. Physicians may consider non-traditional risk factors including circulating biomarkers, genetic factors, psychosocial and socioeconomic factors, air pollution exposure, ethnicity, frailty, and family history. Imaging test results such as coronary artery calcium scoring, contrast computed tomography angiography (CCTA), and carotid ultrasound findings should also be factored in. Only a multidimensional approach holds promise for precise risk prediction [36].
The current approach to CAD risk factors divides them into three main categories: genetic susceptibility, environmental exposure and the lifestyle factors [37]. The individual risk of developing CAD is modulated by the interplay of genetic and lifestyle factors. The environmental factors modulate gene expression by the epigenetic mechanisms [38]. Personalized risk prediction tools such as coronary artery calcium score, polygenic risk score, and metabolic risk score may improve cardiovascular risk assessment.
4.1. Genetics and Genomics—A Novel Approach to Understanding the Pathogenesis of CAD
CAD results from a combination of environmental and genetic factors. A comprehensive understanding of the genetic components contributing to CAD is a key objective of modern medicine, with genetic factors estimated to contribute between 40-60% to CAD risk [37]. The genetic underpinnings of CAD may be indicated by various factors, including early onset of CAD (before 55 years in men and 65 years in women), extensive angiographic progression, familial clustering of CAD cases, presence of multiple risk factors in family history, and the absence of classic risk factors in affected family members [39].
The rapid growth of genetics results in complex terminology: The term genetics refers to the study of the way that certain conditions are passed down from one generation to another. Genomics describes the sequence of genetic material and map the whole patients’ genome and determines all relationships and interactions within the genome. Epigenetics deals with the study of changes in gene expression that do not result from sequence modifications in the deoxyribonucleotide (DNA) strand.
The human genome responds dynamically to changes in the environment through-out a person's life, influencing the phenotypic expression of diseases. The small noncoding parts of the RNA (MicroRNAs) are recognized as major regulatory gene families playing role in epigenetic regulation [40]. For example, the loss of nitric oxide synthesis which is an initial step of atherogenesis may be detected by the microRNA [41]. MicroRNAs have been proved to discriminate unstable CAD patients from stable ones [42]. Epigenetics may play a key role in the development of CAD because the environment influences epigenetic patterns and can modulate gene expression [38].
Understanding the etiology of CAD requires determining which genes are involved in the development or modification of the phenotype and detecting potentially harmful mutations, as well as determining the mutual interactions of individual genes and genes and the environment.
4.1.1. Large Scale Research or Candidate Genes Approach?
In recent years, there has been a surge in research focusing on the genetic predisposition to CAD, particularly through large-scale studies such as genomics, transcriptomics, proteomics, and metabolomics. Genomics involves mapping the entire genome, while transcriptomics examines gene activity states, proteomics studies protein interactions, and metabolomics analyzes cellular pathway metabolites.
Genome-Wide Association Studies (GWAS) utilize an approach that scans the entire genome for associations between Single Nucleotide Polymorphisms (SNPs) and traits of interest. DNA microarray technology allows for the simultaneous identification of numerous SNPs, which can predict the risk of lifestyle diseases. Molecular diagnostics in this context involve determining an individual's SNP pattern and comparing it with known patterns associated with specific diseases. SNP analysis results may potentially serve as the basis for creating genetic profiles in the future [43].
However, the contribution of genes identified through GWAS to CAD pathogenesis is often unclear [44]. While initially, 55 CAD-related loci were identified, by 2018, the number had increased to 163 genetic variants deemed important in CAD development [45]. Further studies have led to the identification of over 321 CAD susceptibility loci, with on-going efforts to identify new variants [44,46]. Despite this, these genes explain less than 20% of CAD heritability [47]. Notably, most genetic loci associated with CAD are not linked to traditional risk factors [48], and the biological functions of many gene variants remain poorly understood. However, the total risk summarized in one number called genetic risk score (GRS) play a role in identifying individuals at high CAD risk, allowing for early preventive strategies [47,49], and have shown promise in reducing major adverse cardiovascular events (MACE) by 40% to 50% in clinical trials [37].
In contrast, the candidate gene approach in genetic testing focuses on the association between genetic variation within specific genes and disease phenotypes, rather than at-tempting to identify all SNPs associated with the disease [50].
The formation and progression of atherosclerotic plaques involve biochemical processes involving numerous enzymes, receptors and their ligands, molecules encoded by various genes that interact with environmental factors. In the development of CAD, the mechanisms responsible for the inflammatory response, endothelial function, platelet function, lipid and apolipoprotein metabolism, folate metabolism, development of thrombosis, insulin sensitivity and mechanisms of blood pressure regulation should be taken into account [39].
Studies show that polymorphisms in genes encoding the folate cycle significantly contribute to atherogenesis and endothelial dysfunction [51]. For instance, in a me-ta-analysis conducted on over 87,000 participants it was found that the T allele of the 665C>T MTHFR polymorphism is a risk factor for CAD, which is partially mediated by abnormal lipid levels [52]. Recently selected polymorphisms of the ADAMTS7 (15q24.2)gene encoding metalloproteinases (MMPs) with proteolytic activity against extracellular substrates was proved to increase the risk of CAD together with total cholesterol and LDL concentrations abnormalities in serum [53].
Selected genes responsible for the development of CAD with known pathomecha-nism and taking into account the type of inheritance are presented in Table 1.
Identifying individuals at high risk of CAD through genetic diagnosis enables early preventive measures and personalized therapy, thereby improving prognosis. As ad-vancements in this field continue, there is potential for genetics to play a more prominent role in prevention strategies. Furthermore, the insights gleaned from individual genetic information have the potential to drive the development of new therapeutic agents [55].
5. CAD Biomarkers
Biomarkers are objectively measurable biological parameters that reflect specific physiological states of the body. These may result from physical tests, laboratory tests, or imaging tests [56]. Biomarkers are crucial in the early detection of chronic diseases.
CAD biomarkers are divided into those causally related to the development of atherosclerosis and those that indicate early damage to the vessel wall [57]. Recent studies show that up to 25% of patients hospitalized for first-time ACS do not have any of the four main risk factors (dyslipidemia, hypertension, cigarette smoking, diabetes) [58], high-lighting the need to discover new biomarkers for the early identification of CAD patients and the development of new targeted therapies.
5.1. Patomechanisms at the Early Stage of Atherosclerosis and Related Biomarkers
An increasingly better understanding of the pathomechanisms leading to the development of CAD and the constant improvement of research techniques result in an in-creasing list of new biomarkers, such as various lipoproteins and exosomes playing a role in lipid metabolism, and inflammatory markers [59].
Some biomarkers may have a causal relationship with the development of atherosclerosis (e.g., lipoprotein (a) reflecting the pathogenic lipid fraction) or may explain its pathogenic mechanism (e.g., C-reactive protein indicating inflammation or inflammatory hematological ratios [60]). Others may be markers of early heart damage (e.g., high-sensitivity cardiac troponin) or heart failure (e.g., natriuretic peptides). These biomarkers are useful in identifying patients with established cardiovascular disease, but their role is partially limited due to individual differences and the lack of reflection of intracellular processes at the early stage of CAD development.
Currently, there is a lack of blood biomarkers that reflect the early stage of the atherosclerosis. Oxidative stress leads to the modification of endothelial nitric oxide synthase (eNOS), which, by uncoupling BH4, causes endothelial cell dysfunction and initiates the development of atherosclerotic plaque [58]. The central role of oxidative signaling in cardiovascular pathophysiology positions measurements of redox state parameters as excellent markers for research and clinical applications. For example, mitochondrial superoxide levels were recently proposed as a marker of CAD [61]. It seems crucial to identify a biomarker that correlates with the redox state and may lead to early detection of CAD. However, despite this, no redox biomarker is currently widely used clinically, mainly due to analytical problems, including the relative instability of reactive oxygen species [62].
5.2. Public Health Perspective
According to the current ESC guidelines, in order to include a potential new marker in the assessment of cardiovascular risk, it must: improve risk prediction; have an impact on public health; be feasible in everyday practice; not only its impact on increasing the cardiovascular risk, but also on reducing cardiovascular risk must be known; and the literature on the topic cannot be biased. Currently, only a few of the markers meet all of the required criteria at the same time.
For example, the increased concentration of Lipoprotein (a), which is genetically determined and twice more common than familial hypercholesterolemia (FH), is associated with the significant risk of atherosclerotic diseases, including CAD [63]. Lp(a) is recognized as an independent cardiovascular risk factor, and from 2019, it is recommended to measure its concentration at least once in a lifetime in every adult and in selected people with a family history of premature cardiovascular disease [64].
Routine measurement of the non-classical biomarkers concentration in blood or urine is not recommended according to the current state of knowledge [57]. However, in a specific group of patients, especially in patients with a history of myocardial infarction at a young age, without classic risk factors or with a strong family history of CAD, additional tests may be justified [65].
This approach is used to prioritize public health interventions for primary and secondary prevention programs but does not encompass all etiological factors of CAD.
6. The Role of Methylation Pathway in Endothelial Dysfunction
The endothelium is a single layer of cells that lines the inner walls of vessels. It regulates the tension of blood vessel walls and blood flow through the vessels thanks to the controlled release of vasodilators such as nitric oxide (NO), prostacyclin (PGI2), hyperpolarizing factor, and vasoconstrictors like endothelins, platelet-activating factor (PAF), thromboxane A2, and prostaglandin H2 [66].
The distribution of atherosclerotic lesions in the arteries is heterogeneous. Lesions appear most quickly or progress most rapidly in places of turbulent blood flow, such as divisions of the arteries. Laminar blood flow protects against the development of atherosclerosis by stimulating mechanoreceptors on the surface of endothelial cells, which results in increased expression and activation of endothelial nitric oxide synthase (eNOS).
Endothelial homeostasis is a strict balance between vasodilation and vasoconstriction, pro- and antithrombotic, pro- and anti-inflammatory, and pro- and antiproliferative processes. The endothelium has a vasoprotective function by producing and metabolizing numerous endothelial transmitters released in response to chemical and mechanical stimuli (e.g., shear forces). The actions of endothelial NO are believed to play a major role in endothelial function [67].
Nitric oxide is produced from L-arginine via an enzyme called nitric oxide synthase (Endothelial Nitric oxide synthase, eNOS). The release of NO occurs due to constant stimulation of endothelial cells by vascular shear forces. NO activates soluble guanylate cyclase and causes an increase in cyclic guanosine monophosphate (cGMP) concentration in target cells, causing vasodilation. NO additionally reduces platelet aggregation and adhesion, prevents smooth muscle hyperplasia, inhibits leukocyte adhesion and the expression of pro-inflammatory cytokine genes, and prevents the oxidation of low-density lipoproteins (LDL) [68]. Studies have shown that patients with cardiovascular risk factors and CAD exhibit significantly lower NO concentration and a higher concentration of endothelin compared to the control group [69].
A shift in the balance between NO production and oxidative stress in endothelial cells leads to endothelial dysfunction (ED), [68], which is considered an early phase of the development of atherosclerotic plaque [70,71]. ED is a state when the endothelium has a limited ability to produce substances with a vasodilatory effect, resulting in an insufficient vascular response to stimuli increasing the oxygen demand of the myocardium. Moreover, ED leads to increased penetrability, which exacerbates the penetration of proteins, including lipoproteins, into the vessel wall.
The synthesis of endothelial NO is catalyzed by eNOS, and its cofactors are tetrahydrobiopterin (BH4), reduced nicotinamide adenine dinucleotide phosphate (NADPH), and the active forms of vitamin B2 - flavin adenine dinucleotide (FAD) and flavin mono-nucleotide (FMN). Reduced availability of NO produced from L-arginine by eNOS occurs in the case of: decreased eNOS expression or activity; deficiency of the substrate - L-arginine; presence of an eNOS inhibitor in the circulation - asymmetric dimethylarginine (ADMA), which is produced as a result of the methylation of arginine; increased elimination of NO from the body - due to increased production of superoxide anion by NADPH oxidases and xanthine oxidases, producing peroxynitrite (ONOO-) and eNOS uncoupling [72,73].
Low levels of the active form of folic acid, 5-methyltetrahydrofolate (5-MTHF), have been shown to play a key role in inducing BH4 deficiency, which results in eNOS monomerisation and the production of free oxygen radicals instead of NO (oxidative stress) [68]. The reduced availability of NO due to decreased levels of methylated folic acid can initiate endothelial dysfunction (ED), contributing to the development of CAD. The availability of 5-MTHF plays a key role in the amount of circulating nitric oxide, and this is a mechanism independent of homocysteine concentration [68,74,75]. In endothelial cells of blood vessels, homocysteine is transformed by remethylation. The remethylation cycle of homo-cysteine to methionine requires the supply of methyltetrahydrofolate (5-MTHF) as a me-thyl donor and the presence of methionine synthase and its cofactor, vitamin B12. 5-MTHF, which is a methyl donor, cannot donate a methyl group in the case of vitamin B12 (cobalamin) deficiency. During the folic acid cycle, one molecule of tetrahydrobiopterin (BH4) is also regenerated from the form of BH2. BH4 is a cofactor for nitric oxide synthase (eNOS).
When the methylation cycle is impaired, the production of BH4 is significantly weakened, and as a result, the amount of nitric oxide (NO) produced is reduced. This ex-plains the dysfunction of the vascular endothelium in metabolic disorders of the cellular methylation pathway, which is the stage initiating the development of atherosclerosis in the pathogenesis of coronary artery disease (Figure 1).
7. Methylation Disorders
Methylation is influenced by various environmental (epigenetic) and genetic factors [76]. Externally, the proper course of methylation mainly depends on the supply of micronutrients that are donors of methyl groups (-CH3): choline, betaine, folacin, methionine, and the supply of cofactors such as vitamin B12, vitamin B6, vitamin C, iron, and zinc [77]. However, the low stability of folates results in their limited bioavailability.
The body's ability to properly carry out methylation reactions is also influenced by abnormal functioning of the digestive system, such as intestinal malabsorption syndromes, abnormal pH in the intestine, and abnormal hepatocyte function [78]. Alcohol and cigarettes are among the products that negatively affect folate absorption.
The increased need for folates (e.g., during pregnancy, hemolytic anemia) and drug interactions (anti-inflammatory drugs, antiepileptic drugs, contraceptives, antimalarials, and barbiturates) are the causes of impaired methylation cycles [79]. Methotrexate therapy, inhibiting the conversion of dihydrofolate to tetrahydrofolate, requires folate supple-mentation to prevent methylation disorders [80].
7.1. Genetically Determined Methylation Disorders
The ability to methylate is genetically determined. Genetic changes in genes encoding key enzymes in the methylation process affect their activity. The proper functioning of the metabolic pathways of the folic acid cycle and one-carbon groups depends on identified single nucleotide polymorphisms (SNPs), which alter the activity of proteins participating in the cycle, thus impairing specific physiological functions and contributing to the pathogenesis of infertility, neurological disorders, and cardiovascular diseases [11].
For instance, thymidylate synthase (TS), which catalyzes the conversion of 5,10-MTHF to DHF and deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), has recently been associated with CAD susceptibility [81].
Methylation disorders may also arise from polymorphisms responsible for the formation of enzymatic proteins that carry out these reactions, such as the enzyme methyl-tetrahydrofolate reductase (MTHFR). Polymorphisms identified in the field of folate metabolism include:
- absorption of folate from food: enzyme encoding glutamine carboxypeptidase II - polymorphism 475H>Y (rs61886492)
- folate transport - polymorphism (rs1051266) of the RFC1 gene
- activity of folate receptors - genes encoding FOLR1, FOLR2
- folate metabolism - polymorphisms encoding the genes dihydrofolate reductase (DHFR), methylenetetrahydrofolate reductase (MTHFR), methionine synthase (rs1805087).
7.1.1. Methylenetetrahydrofolate Reductase Gene Polymorphisms
The MTHFR gene is responsible for the production of 5,10-methylenetetrahydrofolate reductase (MTHFR), a key enzyme involved in folate metabolism. This enzyme catalyzes the reduction of 5,10-methylenetetrahydrofolate to the active form of folic acid (5-MTHF, levomefolic acid), which is fundamental in the process of homocysteine remethylation [80]. Single nucleotide polymorphisms (SNPs) in the MTHFR gene result in many variants of the enzyme, reducing its ability to generate 5-MTHF. MTHFR polymorphisms are associated with cardiovascular diseases, cancer, neurological diseases, diabetes, and psoriasis.
The MTHFR locus is located on chromosome 1 at the end of the short arm (1p36.6) [82]. Many variants of the MTHFR gene polymorphism have been described, but the most attention is paid to the c.665C>T and c.1286A>C polymorphisms. These polymorphisms can be found in heterozygous (polymorphism in only one allele), homozygous (polymorphism in both alleles), and also in the form of compound heterozygotes (one of the above-mentioned polymorphisms in one allele and the other in the second allele). Recently, the nomenclature for the most common MTHFR polymorphisms have changed (Table 2).
In carriers of the c.665C>T polymorphism (the terms C677T or C665T are no longer recommended; rs1801133 according to the Single Nucleotide Polymorphism Database), there is a transition from cytosine (C) to thymine (T) at nucleotide position 665 in exon 4 of the MTHFR gene, and consequently, the change of alanine to valine in amino acid 222 of this protein (p.Ala222Val).
In carriers of the c.1286A>C polymorphism (the term A1298C is no longer recommended; rs1801131 according to the Single Nucleotide Polymorphism Database), there is a transversion of adenine (A) to cytosine (C) at position 1286 of the MTHFR gene and, consequently, the replacement of glutamine with alanine at amino acid 429 of this protein (p.Glu429Ala) [83].

| Current nomenclature | Alleles | Past nomenclature | Genetic “raw data” | |
|---|---|---|---|---|
|
c.665C>T MTHFR polymorphism |
C677T MTHFR polymorphism |
|||
| Genotypes | c.[665C=]c;[665C=] | Both “wild type” alleles | C677C | G/G |
| c.[665C>T];[665C=] | One polymorphic allele: c.665 C>T heterozygote |
C677T | A/G | |
| c.[665C>T];[665C>T] | Both polymorphic alleles: c.665 C>T homozygote |
T677T | A/A | |
|
c.1286A>C MTHFR polymorphism |
A1298C MTHFR polymorphism |
|||
| Genotypes | c.[1286A=];[1286A=] | Both “wild type” alleles | A1298A | T/T |
| c.[1286A>C];[1286A=] | One polymorphic allele: c.1286 A>C heterozygote |
A1298C | G/T | |
| c.[1286A>C];[1286A>C] | Both polymorphic alleles: c.1286 A>C homozygote |
C1298C | G/G | |
The frequency of polymorphisms varies depending on geographical location and ethnicity. They are most common in the heterozygous variant. According to the Genome Aggregation Database (gnomAD), in the general population, the polymorphic allele c.665C>T affects 30.85% of the population, while the c.1286A>C allele affects 28.58% of the population [86]. MTHFR gene polymorphisms are particularly common in Caucasians, slightly less common in Yellows, and are least common in Blacks [87].
Heterozygous c.665C>T occurs in 20% to 40% of the white population and 1% to 4% of most other ethnic groups. Homozygous c.665C>T occurs in approximately 10% of the general population in Europe and may affect up to 25% of individuals in some populations (Iran, China, Turkey, Spain, southern Italy). The heterozygous variant c.1286A>C is found in 20% of Europeans, 8-15% of the general white population, and 1-4% of Asians; the homozygous variant is found in 9% of the general population [88].
In a multicenter study conducted in Poland in 2011, the genotype frequency of the c.665C>T polymorphism of the MTHFR gene was determined. For the variants found c.[665C=];[665C=], c.[665C>T];[665C=], c.[665C>T];[665C>T], in men it was 47%, 43%, and 10%, and in women 49%, 42%, and 9% [86]. In a 2015 study conducted on the population of Polish women, the frequency of the above c.665C>T polymorphisms was 50.60%, 39.88%, and 9.52%, respectively. However, the genotypes of the c.1286A>C c.[1286A=];[1286A=], c.[1286A>C];[1286A=], c.[1286A>C];[1286A>C] polymorphism occurred with a frequency of 42.75%, 47.88%, and 9.37%[87].
The combination of two variants as heterozygotes is common (>20%). Combined homozygosity for one variant and heterozygosity for another is rare, but does exist: 0.4%. No double homozygosity was detected; it is probably a lethal combination [11].
The presence of MTHFR polymorphisms causes a loss of 40 to 70% of enzyme function in the case of the c.665C>T variant and from 30 to 50% in the case of the c.1298A>C variant [83]. The remaining activity of the MTHFR enzyme in the case of particular genotypes have been presented in the Table 3.
A decrease in activity in terms of 5-MTHF formation leads to a decrease in the con-centration of folic acid in the blood [89]. The availability of 5-MTHF plays a key role in the amount of circulating nitric oxide, and this is a mechanism independent of homocysteine concentration [68,74,75]. A shift in the balance between NO production and oxidative stress in endothelial cells leads to vascular endothelial dysfunction (ED), which is a key step in the initiation of CAD development.
8. Practical Aspects of Genetics and Pharmacogenomics in CAD Patients
Despite the increased availability of genetic diagnostics, it is currently not widely used, and there are no recommendations for the routine use of genetic markers in the assessment of cardiovascular risk [57]. Genetic testing remains at the stage of scientific re-search. This is related to treatment options, as the majority of the results do not translate into clinical practice due to the lack of targeted pharmacotherapy.
Presently, the only exception in genetic diagnostics for cardiovascular diseases is in the case of monogenic inherited familial hypercholesterolemia (FH). FH is caused by mutations in the genes for the LDL receptor (LDLR - the most common cause), apolipoprotein B (apoB) and protein convertase subtilisin/kexin type 9 (PCSK9). FH may be homozygous (incidence 1:160,000–1,000,000) or heterozygous (1:200–500). Currently, genetic testing for FH is recommended only in individuals who meet any of the following criteria: serum total cholesterol (TC) ≥ 310 mg/dl (≥ 8 mmol/l) in an adult patient or family member; premature coronary heart disease in the patient or a member of his family (men < 55 years of age, women < 60 years of age); tendon xanthomas in you or a member of your family; sudden cardiac death of a family member at a young age [90]. According to current recommendations, although genetic testing may facilitate and accelerate identification of patients, is not required to make the diagnosis. In The Dutch Lipid Clinic Network scoring system containing diagnostic criteria for FH, genetic testing is not necessary [91].
In the case of FH, pharmacogenomics can be utilized, which matches the drug to the patient's genotype. PCSK9 inhibitors were first included in the ESC guidelines for the treatment of lipid disorders in 2019 [54]. Proprotein subtilisin/kexin convertase type 9 is a protein involved in the metabolism of LDL receptors (LDLRs) by binding to them and stimulating endocytosis of the LDLR-PCSK9 complex and LDLR degradation in the lysosome. On the other hand, PCSK9 inhibitors are monoclonal antibodies to PCSK9 that lead to a reduction in LDLR degradation and cause a decrease in LDL-C concentration by an average of 60% regardless of other lipid-lowering treatment carried out simultaneously [92]. PCSK9 inhibitors available in Poland are alirocumab and evolocumab, not reimbursed or available as part of the drug program conducted in selected Metabolic Disorders Clinics. Due to too narrow program criteria and the high cost of treatment when it is not reimbursed, access to effective personalized therapy remains limited.
Therapeutic decisions regarding the initiation of pharmacological treatment hinge on the overall cardiovascular risk assessment, which is informed by classical risk factors and risk-modifying factors [33]. Despite the identification of elevated serum levels of various biomarkers associated with CAD risk, regrettably, there are no specific treatments directly targeting these biomarkers to reduce CAD risk, thus limiting their clinical utility. Additionally, while gene editing preclinical studies are ongoing, the effectiveness and safety of this approach in humans require further validation [93].
9. Translating the Pathomechanism into Clinical Practice - New CAD Therapies
According to current knowledge, ED is a reversible process [94]. So far, all drugs in-spired by genetic discoveries in CAD concern lipid metabolism. Although the clinical results are promising, they do not represent all new pathophysiological concepts. Mechanisms leading to uncoupling of eNOS are considered a promising therapeutic target. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) commonly used in atherosclerotic cardiovascular diseases among many beneficial effects on endothelial physiology also include the prevention of eNOS uncoupling [95].
Reports suggest the use of antioxidants such as vitamins C and E, N-acetylcysteine, glutathione, as well as BH4 and folic acid, to enhance vascular endothelial function [68,96,97,98]. However, direct interventions aiming to administer tetrahydrobiopterin analogues (BH4 - eNOS cofactor) or supplementing with L-Arginine (eNOS substrate) did not yield a clearly beneficial effect [95].
9.1. Folic Acid versus 5-MTHF in CAD Therapy
Pharmacological interventions in CAD typically target classic risk factors such as hypertension and dyslipidemia. According to current standards, studies on supplementation of B vitamins (B6, B12), folic acid, and vitamins C and D in patients with CAD have not demonstrated beneficial effects. However, there have been no studies conducted on the methylated forms of folic acid [57].
The recommended intake of folic acid in the adult diet is 200 μg per day, primarily from green leafy vegetables. Folic acid supplementation is well-established in gynecology, recommended before conception and during pregnancy to prevent neural tube defects (NTDs). Over 80 countries, including the USA, have implemented mandatory food enrichment of flour with synthetic folic acid for NTD prevention [99]. In countries without food fortification policies, such as Poland, women planning to conceive are advised to take a preventive dose of folic acid 400 μg daily or its active metabolites.
Currently, the choice of the form of folic acid does not depend on the genotype. To prevent defects of the central nervous system in women who have previously given birth to a child with such a defect, a higher dose of folic acid of 4–5 mg should be used [84]. However, too high doses of folic acid (>400 ug per day) may be potentially harmful. Supplementation with synthetic folic acid may lead to the syndrome recognized as unmetabolized folic acid syndrome (Unmetabolized folic acid Syndrome, UMFA) [100]. For in-stance, high folate concentration was linked to gestational diabetes mellitus [101,102].
Clinical trials investigating the use of folic acid in the treatment of CVD have produced conflicting results, despite promising experimental data. These trials have largely failed to demonstrate a beneficial effect of folic acid supplementation on the cardiovascular-lar system. Folic acid can enhance the expression and activation of certain genes, particularly in vascular smooth muscles, which may promote cell division and proliferation. Interestingly, there are reports indicating a significantly higher incidence of restenosis in stents among patients who used high doses (>1 mg) of folic acid following coronary artery revascularization procedures [103]. A meta-analysis of clinical trial data has corroborated these findings, suggesting that folic acid supplementation is not effective in preventing cardiovascular events in individuals with pre-existing vascular disease [104].
Given the increasing consumption of large doses (>1 mg) of folic acid through fortified foods and dietary supplements, there is a pressing need to monitor its effects closely [105]. The perspective on folic acid supplementation needs to shift. Instead of simply in-creasing the dosage, factors such as the bioavailability of folic acid and the patient's genotype should be carefully considered initially. This approach may offer a more personalized and potentially effective strategy for managing cardiovascular health.
Folic acid remains inactive in the human body and requires conversion in the liver into its active form, 5-methyltetrahydrofolate (5-MTHF). This active molecule serves as a crucial methyl group donor (-CH3) in various metabolic reactions, including the biosynthesis of glycine from serine, the conversion of homocysteine to methionine, and the formation of purines and pyrimidines essential for DNA synthesis. It is fundamental to growth in the embryonic and fetal phases. In the adult human cardiovascular system, endothelial cells retain the ability to divide and move cells and require 5-MTHF for proliferation [106]. Because 5-MTHF does not require activation, it does not accumulate in the blood like folic acid in cases of reduced hepatic metabolism [107].
5-MTHF plays a key role in regulating the level of circulating nitric oxide [68,74,75]. It is synthesized by the enzyme methylenetetrahydrofolate reductase (MTHFR) from 5,10-methylenetetrahydrofolate (5,10-MTHF) sourced from the gastrointestinal tract. However, it's noteworthy that in about 30% of the European population, the function of this enzyme is impaired [108].
To determine how the MTHFR gene polymorphism affects methylation disorders, it seems reasonable to check its activity by measuring the 5-MTHF concentration. It has been reported that 5-MTHF levels in red blood cells represent long-term folate status and may be a more reliable marker of cellular methylation pathway disorders than homocysteine, and its deficiency is associated with CAD [109]. The level of the active folic acid metabolite 5-MTHF plays a critical role in circulating nitric oxide levels [68]. Low concentrations of 5-MTHF in red blood cells are found in patients with hypertension [110]. In a prospective cohort study conducted in 2011-2014 on 10,661 participants, an increased risk of death from any cause was found in people with low serum 5-MTHF levels (<23.9 nmol/l) [111].
The effect of 5-MTHF supplementation on endothelial function is independent of lowering homocysteine concentration. Folates directly interact with eNOS and improve the binding affinity of BH4 to eNOS, chemically stabilize BH4 and enhance the regeneration of BH4 from BH2 [25]. Carriers of polymorphisms in genes related to folic acid metabolism or absorption may benefit from 5-MTHF instead of folic acid [112].
The use of 5-MTHF in several diseases has been studied in animal models. It has been shown that 5-MTHF administered intravenously in rats with acute kidney injury reduced oxidative stress by increasing glutathione concentrations and reducing renal lipid peroxidation [113]. Rats with developed aortic atherosclerosis treated with 5-MTHF showed a decrease in homocysteine levels, an improvement in the serum lipid profile, an increase in the expression of NO and NOS, an enhancement of the antioxidant properties of GSH activity and a decrease in the expression of inflammatory factors: TNF-α, IL-6, IL -1β and endothelial cell damage factors: ET-1 and sICAM-1. 5-MTHF has been shown to have anti-inflammatory and antioxidant effects and reduce the occurrence of aortic dam-age [114].
There are single experimental studies on humans using 5-MTHF. In couples diagnosed with infertility and confirmed c.665C>T polymorphism in both partners, treatment with 5-MTHF resulted in a decrease in homocysteine concentration in the blood [115]. 5-MTHF infusion improved impaired endothelium-dependent vasodilation in patients with familial hypercholesterolemia [116] and CAD [117]. The active circulating form of folic acid has been shown to increase the vascular BH4/BH2 ratio, reversing eNOS uncoupling and restoring endothelial function in coronary artery bypass grafting patients [75].
The most recent guidelines on food fortification now incorporate 5-MTHF into their standards [118]. Considering the 5-MTHFS ability to restore the endothelial cells function, is seems crucial to verify its medical utility in the clinical trials with the CAD patients.
10. Conclusions and Future Directions
Currently, CAD prevention is tailored to the lifestyle, but not to the individual genotype of the patient. The current recommendations do not relate to the frequent methylation disorders caused by the methyltetrahydrofolate reductase gene polymorphism in CAD subjects and to the usage of folates in this group of patients. Folates encompass a range of chemical compounds. However, routine clinical tests assess the total folate concentration without differentiating them or reflecting the level of the biologically active form of folic acid (5-MTHF). Deficiency in 5-MTHF is significant in the development of coronary heart disease, but diagnostic tests for 5-MTHF blood concentration are currently limited to scientific research. Identifying CAD patients with methylated folic acid deficiency may lead to the use of 5-MTHF in pharmacotherapy, but further research on this topic is necessary.
In summary, information derived from the determination of folate concentrations, including its methylated form, should not translate into supplementation and increasing the dosage of folic acid. Instead, it should focus on its bioavailability and consider the patient's genotype in the development of personalized medicine.
Additionally, knowledge of the genetic CAD basis combined with the assessment of the phenotype based on new biomarkers may contribute to the development of pharmacogenomics, focusing on the individual selection of drugs depending on the genetic polymorphisms and mutations of a specific patient.
Future research is needed on the use of the active (methylated) form of folic acid (5-MTHF) in CAD patients with methylation disorders caused by MTHFR gene polymorphisms.
Author Contributions
Conceptualization, R.P., A.P.-R. and R.I.; validation R.I.; Resources, R.I.; Data curation, A.P.-R. and R.P.; Writing—original draft preparation, A.P.-R.; Writing—review and editing, R.P.; Visualization, A.P.-R.; Supervision, R.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Irzmański, R.; Glowczynska, R.; Banach, M.; Szalewska, D.; Piotrowicz, R.; Kowalik, I.; et al. Prognostic Impact of Hybrid Comprehensive Telerehabilitation Regarding Diastolic Dysfunction in Patients with Heart Failure with Reduced Ejection Fraction-Subanalysis of the TELEREH-HF Randomized Clinical Trial. J Clin Med 2022, 11, 1844. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Health Estimates: Life expectancy and leading causes of death and disability. 2019. Available online: www.who.int/data/gho/data/themes/mortality-and-global-health-estimates.
- Byrne, R.A.; Rossello, X.; Coughlan, J.J.; Barbato, E.; Berry, C.; Chieffo, A.; et al. 2023 ESC Guidelines for the management of acute coronary syndromes. Eur Heart J 2023, 44, 3720–3826. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, R. Methylation. Brittanica 2018. [Google Scholar]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate's role. Adv Nutr 2012, 3, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Menezo, Y.; Clement, P.; Clement, A.; Elder, K. Methylation: An Ineluctable Biochemical and Physiological Process Essential to the Transmission of Life. Int J Mol Sci 2020, 21, 9311. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr 2002, 132 (Suppl. 8), 2361s–2366s. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liao, R.; Dai, X.; Guo, H.; Wang, D.; Xia, M.; et al. Association between plasma S-adenosylmethionine and risk of mortality in patients with coronary artery disease: A cohort study. Am J Clin Nutr 2021, 114, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Karthika, C.L.; Ahalya, S.; Radhakrishnan, N.; Kartha, C.C.; Sumi, S. Hemodynamics mediated epigenetic regulators in the pathogenesis of vascular diseases. Mol Cell Biochem 2021, 476, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Fardous, A.M.; Heydari, A.R. Uncovering the Hidden Dangers and Molecular Mechanisms of Excess Folate: A Narrative Review. Nutrients 2023, 15, 4699. [Google Scholar] [CrossRef]
- Menezo, Y.; Elder, K.; Clement, A.; Clement, P. Folic Acid, Folinic Acid, 5 Methyl TetraHydroFolate Supplementation for Mutations That Affect Epigenesis through the Folate and One-Carbon Cycles. Biomolecules 2022, 12, 197. [Google Scholar] [CrossRef]
- Costello, J.F.; Plass, C. Methylation matters. J Med Genet 2001, 38, 285–303. [Google Scholar] [CrossRef] [PubMed]
- Cosar, A.; Ipcioglu, O.M.; Ozcan, O.; Gultepe, M. Folate and homocysteine metabolisms and their roles in the biochemical basis of neuropsychiatry. Turk J Med Sci 2014, 44, 1–9. [Google Scholar] [CrossRef]
- Ziolkowska, K.; Tobola-Wrobel, K.; Pietryga, M.; Kasprzak, G.; Jamsheer, A.; Wysocka, E. An Assessment of Selected Molecular and Biochemical Markers of the Folate Pathway as Potential Risk Factors for Fetal Trisomy 21 during the First Trimester of Pregnancy in the Polish Population. J Clin Med 2022, 11, 1190. [Google Scholar] [CrossRef]
- Czechowicz, P.; Malodobra-Mazur, M.; Lebioda, A.; Jonkisz, A.; Dobosz, T.; Smigiel, R. Polymorphisms of the MTHFR gene in mothers of children with trisomy 21 (Down syndrome) in a Polish population. Adv Clin Exp Med 2020, 29, 251–256. [Google Scholar] [CrossRef]
- Chorazy, M.; Wawrusiewicz-Kurylonek, N.; Goscik, J.; Posmyk, R.; Czarnowska, A.; Wiesik, M.; et al. Association between polymorphisms of a folate - homocysteine - methionine - SAM metabolising enzyme gene and multiple sclerosis in a Polish population. Neurol Neurochir Pol 2019, 53, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Zur-Wyrozumska, K.; Pera, J.; Dziubek, A.; Sado, M.; Golenia, A.; Slowik, A.; et al. Association between C677T polymorphism of MTHFR gene and risk of amyotrophic lateral sclerosis: Polish population study and a meta-analysis. Neurol Neurochir Pol 2017, 51, 135–139. [Google Scholar] [CrossRef]
- Goracy, I.; Cyrylowski, L.; Kaczmarczyk, M.; Fabian, A.; Koziarska, D.; Goracy, J.; et al. C677T polymorphism of the methylenetetrahydrofolate reductase gene and the risk of ischemic stroke in Polish subjects. J Appl Genet 2009, 50, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Wolski, H.; Kurzawinska, G.; Drews, K.; Barlik, M.; Kadziolka, P.; Malewski, Z.; et al. MTHFR genetic polymorphism and the risk of intrauterine fetal death in Polish women. Ginekol Pol 2019, 90, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Nowak, I.; Bylinska, A.; Wilczynska, K.; Wisniewski, A.; Malinowski, A.; Wilczynski, J.R.; et al. The methylenetetrahydrofolate reductase c.c.677 C>T and c.c.1298 A>C polymorphisms in reproductive failures: Experience from an RSA and RIF study on a Polish population. PLoS One 2017, 12, e0186022. [Google Scholar] [CrossRef] [PubMed]
- Stanislawska-Sachadyn, A.; Borzyszkowska, J.; Krzeminski, M.; Janowicz, A.; Dziadziuszko, R.; Jassem, J.; et al. Folate/homocysteine metabolism and lung cancer risk among smokers. PLoS One 2019, 14, e0214462. [Google Scholar] [CrossRef]
- Wolski, H.; Barlik, M.; Drews, K.; Klejewski, A.; Kurzawinska, G.; Ozarowski, M.; et al. Contribution of inherited thrombophilia to recurrent miscarriage in the Polish population. Ginekol Pol 2017, 88, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, M.; Kagawa, Y. Genetic polymorphisms and folate status. Congenit Anom 2017, 57, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Hale, A.B.; Channon, K.M. Dihydrofolate reductase protects endothelial nitric oxide synthase from uncoupling in tetrahydrobiopterin deficiency. Free Radic Biol Med 2011, 50, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Moens, A.L.; Vrints, C.J.; Claeys, M.J.; Timmermans, J.-P.; Champion, H.C.; Kass, D.A. Mechanisms and potential therapeutic targets for folic acid in cardiovascular disease. American Journal of Physiology-Heart and Circulatory Physiology 2008, 294, H1971–H1977. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J.; Narayanan, S.; Brand, G.M.; Pirie, L.; Grant, G. Impact of Folate Deficiency on DNA Stability. The Journal of Nutrition 2002, 132, 2444S–2449S. [Google Scholar] [CrossRef] [PubMed]
- Magri, V.R.; Rocha, M.A.; de Matos, C.S.; Petersen, P.A.D.; Leroux, F.; Petrilli, H.M.; et al. Folic acid and sodium folate salts: Thermal behavior and spectroscopic (IR, Raman, and solid-state (13)C NMR) characterization. Spectrochim Acta A Mol Biomol Spectrosc 2022, 273, 120981. [Google Scholar] [CrossRef] [PubMed]
- Sobczyńska-Malefora, A.; Harrington, D.J. Laboratory assessment of folate (vitamin B(9)) status. J Clin Pathol 2018, 71, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Finer, S.; Saravanan, P.; Hitman, G.; Yajnik, C. The role of the one-carbon cycle in the developmental origins of Type 2 diabetes and obesity. Diabet Med 2014, 31, 263–272. [Google Scholar] [CrossRef]
- Marzilli, M.; Merz, C.N.; Boden, W.E.; Bonow, R.O.; Capozza, P.G.; Chilian, W.M.; et al. Obstructive coronary atherosclerosis and ischemic heart disease: an elusive link! J Am Coll Cardiol 2012, 60, 951–956. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-Garcia, O.; Dominguez-Perez, M.; Gonzalez-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int J Mol Sci 2021, 22, 3850. [Google Scholar] [CrossRef]
- Dawson, L.P.; Lum, M.; Nerleker, N.; Nicholls, S.J.; Layland, J. Coronary Atherosclerotic Plaque Regression: JACC State-of-the-Art Review. J Am Coll Cardiol 2022, 79, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Visseren, F.L.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; Cosyns, B. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). European Heart Journal 2021, 42, 3227–3337. [Google Scholar] [CrossRef] [PubMed]
- Marx, N.; Federici, M.; Schutt, K.; Muller-Wieland, D.; Ajjan, R.A.; Antunes, M.J.; et al. 2023 ESC Guidelines for the management of cardiovascular disease in patients with diabetes. Eur Heart J 2023, 44, 4043–4140. [Google Scholar] [CrossRef] [PubMed]
- Kotseva, K.; De Backer, G.; De Bacquer, D.; Rydén, L.; Hoes, A.; Grobbee, D.; et al. Primary prevention efforts are poorly developed in people at high cardiovascular risk: A report from the European Society of Cardiology EURObservational Research Programme EUROASPIRE V survey in 16 European countries. Eur J Prev Cardiol 2021, 28, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Verma, K.P.; Inouye, M.; Meikle, P.J.; Nicholls, S.J.; Carrington, M.J.; Marwick, T.H. New Cardiovascular Risk Assessment Techniques for Primary Prevention: JACC Review Topic of the Week. J Am Coll Cardiol 2022, 80, 373–387. [Google Scholar] [CrossRef]
- Roberts, R.; Chang, C.C.; Hadley, T. Genetic Risk Stratification: A Paradigm Shift in Prevention of Coronary Artery Disease. JACC Basic Transl Sci 2021, 6, 287–304. [Google Scholar] [CrossRef]
- Rizzacasa, B.; Amati, F.; Romeo, F.; Novelli, G.; Mehta, J.L. Epigenetic Modification in Coronary Atherosclerosis: JACC Review Topic of the Week. J Am Coll Cardiol 2019, 74, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, M.T. Genetic evaluation for coronary artery disease. Genet Med 2003, 5, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr Opin Chem Biol 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Torres-Paz, Y.E.; Gamboa, R.; Fuentevilla-Alvarez, G.; Soto, M.E.; Gonzalez-Moyotl, N.; Martinez-Alvarado, R.; et al. Overexpression of microRNA-21-5p and microRNA-221-5p in Monocytes Increases the Risk of Developing Coronary Artery Disease. Int J Mol Sci 2023, 24, 8641. [Google Scholar] [CrossRef]
- Barbalata, T.; Niculescu, L.S.; Stancu, C.S.; Pinet, F.; Sima, A.V. Elevated Levels of Circulating lncRNAs LIPCAR and MALAT1 Predict an Unfavorable Outcome in Acute Coronary Syndrome Patients. Int J Mol Sci 2023, 24, 12076. [Google Scholar] [CrossRef]
- Thorisson, G.A.; Stein, L.D. The SNP Consortium website: past, present and future. Nucleic Acids Res 2003, 31, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Schunkert, H. Coronary Artery Disease Genetics Enlightened by Genome-Wide Association Studies. JACC Basic Transl Sci 2021, 6, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, J.; Kessler, T.; Munoz Venegas, L.; Schunkert, H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res 2018, 114, 1241–1257. [Google Scholar] [CrossRef]
- Koyama, S.; Ito, K.; Terao, C.; Akiyama, M.; Horikoshi, M.; Momozawa, Y.; et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet 2020, 52, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wiernek, S.; Evans, J.P.; Runge, M.S. Genetics of coronary artery disease and myocardial infarction. World J Cardiol 2016, 8, 1–23. [Google Scholar] [CrossRef]
- Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013, 45, 25–33. [Google Scholar] [CrossRef]
- Pechlivanis, S.; Lehmann, N.; Hoffmann, P.; Nöthen, M.M.; Jöckel, K.H.; Erbel, R.; et al. Risk prediction for coronary heart disease by a genetic risk score - results from the Heinz Nixdorf Recall study. BMC Med Genet 2020, 21, 178. [Google Scholar] [CrossRef]
- Moore, S.R. Commentary: What is the case for candidate gene approaches in the era of high-throughput genomics? A response to Border and Keller (2017). J Child Psychol Psychiatry 2017, 58, 331–334. [Google Scholar] [CrossRef]
- Timizheva, K.B.; Ahmed, A.A.M.; Ait Aissa, A.; Aghajanyan, A.V.; Tskhovrebova, L.V.; Azova, M.M. Association of the DNA Methyltransferase and Folate Cycle Enzymes' Gene Polymorphisms with Coronary Restenosis. Life 2022, 12, 245. [Google Scholar] [CrossRef]
- Luo, Z.; Lu, Z.; Muhammad, I.; Chen, Y.; Chen, Q.; Zhang, J.; et al. Associations of the MTHFR rs1801133 polymorphism with coronary artery disease and lipid levels: a systematic review and updated meta-analysis. Lipids Health Dis 2018, 17, 191. [Google Scholar] [CrossRef] [PubMed]
- Iwanicka, J.; Balcerzyk-Matic, A.; Iwanicki, T.; Mizia-Stec, K.; Banka, P.; Filipecki, A.; et al. The Association of ADAMTS7 Gene Polymorphisms with the Risk of Coronary Artery Disease Occurrence and Cardiovascular Survival in the Polish Population: A Case-Control and a Prospective Cohort Study. Int J Mol Sci 2024, 25, 2274. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). European Heart Journal 2019, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Assimes, T.L.; Roberts, R. Genetics: Implications for Prevention and Management of Coronary Artery Disease. J Am Coll Cardiol 2016, 68, 2797–2818. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D. biomarker. Encyclopedia Britannica.
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Back, M.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Vernon, S.T.; Coffey, S.; Bhindi, R.; Soo Hoo, S.Y.; Nelson, G.I.; Ward, M.R.; et al. Increasing proportion of ST elevation myocardial infarction patients with coronary atherosclerosis poorly explained by standard modifiable risk factors. Eur J Prev Cardiol 2017, 24, 1824–1830. [Google Scholar] [CrossRef]
- Della Corte, V.; Todaro, F.; Cataldi, M.; Tuttolomondo, A. Atherosclerosis and Its Related Laboratory Biomarkers. Int J Mol Sci 2023, 24, 15546. [Google Scholar] [CrossRef]
- Tudurachi, B.S.; Anghel, L.; Tudurachi, A.; Sascau, R.A.; Statescu, C. Assessment of Inflammatory Hematological Ratios (NLR, PLR, MLR, LMR and Monocyte/HDL-Cholesterol Ratio) in Acute Myocardial Infarction and Particularities in Young Patients. Int J Mol Sci 2023, 24, 14378. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.E.; Besnier, M.; Genetzakis, E.; Tang, O.; Kott, K.A.; Vernon, S.T.; et al. High-Throughput Measure of Mitochondrial Superoxide Levels as a Marker of Coronary Artery Disease to Accelerate Drug Translation in Patient-Derived Endothelial Cells Using Opera Phenix((R)) Technology. Int J Mol Sci 2023, 25, 22. [Google Scholar] [CrossRef]
- Karimi Galougahi, K.; Antoniades, C.; Nicholls, S.J.; Channon, K.M.; Figtree, G.A. Redox biomarkers in cardiovascular medicine. Eur Heart J 2015, 36, 1576–1582. [Google Scholar] [CrossRef]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol 2018, 3, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J. New cardiovascular risk factors exist, but are they clinically useful? Eur Heart J 2008, 29, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Irzmański, R.; Serwa-Stepień, E.; Barylski, M.; Banach, M.; Kowalski, J.; Pawlicki, L. [Endothelial dysfunction in hypertension. The role of natriuretic peptides and endothelin]. Kardiol Pol 2005, 63, S457–S461. [Google Scholar] [PubMed]
- Della Roca Domenico, P.C. Czynność sródbłonka i niepomyślne rokowanie chorób układu krążenia. Kardiologia po Dyplomie 2011, 10, 14–26. [Google Scholar]
- Yuyun, M.F.; Ng, L.L.; Ng, G.A. Endothelial dysfunction, endothelial nitric oxide bioavailability, tetrahydrobiopterin, and 5-methyltetrahydrofolate in cardiovascular disease. Where are we with therapy? Microvasc Res 2018, 119, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Goch, A.; Banach, M.; Mikhailidis, D.P.; Rysz, J.; Goch, J.H. Endothelial dysfunction in patients with noncomplicated and complicated hypertension. Clin Exp Hypertens 2009, 31, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Zeiher, A.M. Endothelial function: cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Siekierzycka, A.; Płoska, A.; Dobrucki, I.T.; Kalinowski, L. Endothelial Dysfunction Driven by Hypoxia-The Influence of Oxygen Deficiency on NO Bioavailability. Biomolecules 2021, 11, 982. [Google Scholar] [CrossRef]
- Sullivan, J.C.; Pollock, J.S. Coupled and uncoupled NOS: separate but equal? Uncoupled NOS in endothelial cells is a critical pathway for intracellular signaling. Circ Res 2006, 98, 717–719. [Google Scholar] [CrossRef]
- Forstermann, U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch 2010, 459, 923–939. [Google Scholar] [CrossRef] [PubMed]
- Doshi, S.N.; McDowell, I.F.; Moat, S.J.; Payne, N.; Durrant, H.J.; Lewis, M.J.; et al. Folic acid improves endothelial function in coronary artery disease via mechanisms largely independent of homocysteine lowering. Circulation 2002, 105, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Baarholm, O.A.; Van-Assche, T.; Cunnington, C.; et al. MTHFR 677 C>T Polymorphism reveals functional importance for 5-methyltetrahydrofolate, not homocysteine, in regulation of vascular redox state and endothelial function in human atherosclerosis. Circulation 2009, 119, 2507–2515. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B(12), folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J Inherit Metab Dis 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A. Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection. Int J Mol Sci 2023, 24, 2346. [Google Scholar] [CrossRef] [PubMed]
- Kadayifci, F.Z.; Zheng, S.; Pan, Y.X. Molecular Mechanisms Underlying the Link between Diet and DNA Methylation. Int J Mol Sci 2018, 19, 4055. [Google Scholar] [CrossRef]
- Kim, D.J.; Venkataraman, A.; Jain, P.C.; Wiesler, E.P.; DeBlasio, M.; Klein, J.; et al. Vitamin B12 and folic acid alleviate symptoms of nutritional deficiency by antagonizing aryl hydrocarbon receptor. Proc Natl Acad Sci USA 2020, 117, 15837–15845. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, F.; Panzavolta, G. Folate, folic acid and 5-methyltetrahydrofolate are not the same thing. Xenobiotica 2014, 44, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.O.; Ryu, C.S.; Lee, J.Y.; Ko, E.J.; Ha, Y.H.; Sung, J.H.; et al. Association of Thymidylate Synthase (TS) Gene Polymorphisms with Incidence and Prognosis of Coronary Artery Disease. Int J Mol Sci 2023, 24, 12591. [Google Scholar] [CrossRef]
- Liew, S.C.; Gupta, E.D. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: epidemiology, metabolism and the associated diseases. Eur J Med Genet 2015, 58, 1–10. [Google Scholar] [CrossRef]
- Undas, A.; Chojnowski, K.; Klukowska, A.; Letowska, M.; Mital, A.; Mlynarski, W.; et al. Determination and interpretation of MTHFR gene mutations in gynecology and internal medicine. Pol Arch Intern Med 2019, 129, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Moczulska, H.; Pesz, K.; Gach, A.; Borowiec, M.; Sieroszewski, P.; Sąsiadek, M.; et al. Stanowisko ekspertów Polskiego Towarzystwa Genetyki Człowieka i Polskiego Towarzystwa Ginekologów i Położników w sprawie zlecania i interpretacji wyników badań pod kątem wariantów genetycznych w genie MTHFR. Ginekologia i Perinatologia Praktyczna 2017, 2, 234–238. [Google Scholar]
- MTHFR Mutation: What is it? How to check your raw data.
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Wolski, H.; Kocięcka, M.; Mrozikiewicz, A.E.; Barlik, M.; Kurzawińska, G. Coexistence of the 677C>T and 1298A>C MTHFR polymorphisms and its significance in the population of Polish women. Ginekologia Polska 2015, 86. [Google Scholar] [CrossRef]
- Botto, L.D.; Yang, Q. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol 2000, 151, 862–877. [Google Scholar] [CrossRef] [PubMed]
- Tsang, B.L.; Devine, O.J.; Cordero, A.M.; Marchetta, C.M.; Mulinare, J.; Mersereau, P.; et al. Assessing the association between the methylenetetrahydrofolate reductase (MTHFR) 677C>T polymorphism and blood folate concentrations: a systematic review and meta-analysis of trials and observational studies. Am J Clin Nutr 2015, 101, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, F.M.; Barylski, M.; Cybulska, B.; Wozakowska-Kaplon, B.; Krasinski, Z.; Mamcarz, A.; et al. Recommendation for the management of dyslipidemia in Poland - Third Declaration of Sopot. Interdisciplinary Expert Position Statement endorsed by the Polish Cardiac Society Working Group on Cardiovascular Pharmacotherapy. Cardiol J 2018, 25, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. European Heart Journal 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Navarese, E.P.; Kolodziejczak, M.; Kereiakes, D.J.; Tantry, U.S.; O'Connor, C.; Gurbel, P.A. Proprotein Convertase Subtilisin/Kexin Type 9 Monoclonal Antibodies for Acute Coronary Syndrome: A Narrative Review. Ann Intern Med 2016, 164, 600–607. [Google Scholar] [CrossRef]
- Mak, M.C.E.; Gurung, R.; Foo, R.S.Y. Applications of Genome Editing Technologies in CAD Research and Therapy with a Focus on Atherosclerosis. Int J Mol Sci 2023, 24, 14057. [Google Scholar] [CrossRef]
- Dobarro, D.; Gomez-Rubin, M.C.; Sanchez-Recalde, A.; Moreno, R.; Galeote, G.; Jimenez-Valero, S.; et al. Current pharmacological approach to restore endothelial dysfunction. Cardiovasc Hematol Agents Med Chem 2009, 7, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Janaszak-Jasiecka, A.; Płoska, A.; Wierońska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial dysfunction due to eNOS uncoupling: molecular mechanisms as potential therapeutic targets. Cell Mol Biol Lett 2023, 28, 21. [Google Scholar] [CrossRef] [PubMed]
- Paulo, M.; Costa, D.; Bonaventura, D.; Lunardi, C.N.; Bendhack, L.M. Nitric Oxide Donors as Potential Drugs for the Treatment of Vascular Diseases Due to Endothelium Dysfunction. Curr Pharm Des 2020, 26, 3748–3759. [Google Scholar] [CrossRef] [PubMed]
- Varadharaj, S.; Kelly, O.J.; Khayat, R.N.; Kumar, P.S.; Ahmed, N.; Zweier, J.L. Role of Dietary Antioxidants in the Preservation of Vascular Function and the Modulation of Health and Disease. Front Cardiovasc Med 2017, 4, 64. [Google Scholar] [CrossRef]
- Settergren, M.; Böhm, F.; Malmström, R.E.; Channon, K.M.; Pernow, J. L-arginine and tetrahydrobiopterin protects against ischemia/reperfusion-induced endothelial dysfunction in patients with type 2 diabetes mellitus and coronary artery disease. Atherosclerosis 2009, 204, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Centeno Tablante, E.; Pachon, H.; Guetterman, H.M.; Finkelstein, J.L. Fortification of wheat and maize flour with folic acid for population health outcomes. Cochrane Database Syst Rev 2019, 7, CD012150. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.W.; Ayling, J.E. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc Natl Acad Sci USA 2009, 106, 15424–15429. [Google Scholar] [CrossRef]
- Xie, K.; Xu, P.; Fu, Z.; Gu, X.; Li, H.; Cui, X.; et al. Association of maternal folate status in the second trimester of pregnancy with the risk of gestational diabetes mellitus. Food Sci Nutr 2019, 7, 3759–3765. [Google Scholar] [CrossRef]
- Zhu, B.; Ge, X.; Huang, K.; Mao, L.; Yan, S.; Xu, Y.; et al. Folic Acid Supplement Intake in Early Pregnancy Increases Risk of Gestational Diabetes Mellitus: Evidence From a Prospective Cohort Study. Diabetes Care 2016, 39, e36–e37. [Google Scholar] [CrossRef]
- Lange, H.; Suryapranata, H.; De Luca, G.; Borner, C.; Dille, J.; Kallmayer, K.; et al. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med 2004, 350, 2673–2681. [Google Scholar] [CrossRef]
- Bazzano, L.A.; Reynolds, K.; Holder, K.N.; He, J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA 2006, 296, 2720–2726. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, B.; Sahyoun, N.R. Application of the Key Events Dose-response Framework to Folate Metabolism. Crit Rev Food Sci Nutr 2016, 56, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.J.A.; Lewis, J.; et al. Molecular Biology of the Cell. 4th edition. Science NYG, editor2002.
- Ferrazzi, E.; Tiso, G.; Di Martino, D. Folic acid versus 5-methyl tetrahydrofolate supplementation in pregnancy. Eur J Obstet Gynecol Reprod Biol 2020, 253, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Golbahar, J.; Rezaian, G.; Fathi, Z.; Aminzadeh, M.A. Association of low red blood cell folate concentrations with coronary artery disease in Iranians: a matched case-control study. J Vasc Res 2005, 42, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Golbahar, J.; Mostafavi, E. Association between low red blood cell 5-methyltetrahydrofolate and hyperhomocysteinaemia with hypertension: a cross-sectional study. High Blood Press Cardiovasc Prev 2012, 19, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, Z.; Zhou, C.; Li, Q.; He, P.; Zhang, Y.; et al. Relationship of several serum folate forms with the risk of mortality: A prospective cohort study. Clin Nutr 2021, 40, 4255–4262. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Holzgreve, W.; Pietrzik, K. Is 5-methyltetrahydrofolate an alternative to folic acid for the prevention of neural tube defects? J Perinat Med 2013, 41, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Wijerathne, C.U.B.; Au-Yeung, K.K.W.; Siow, Y.L.; O, K. 5-Methyltetrahydrofolate Attenuates Oxidative Stress and Improves Kidney Function in Acute Kidney Injury through Activation of Nrf2 and Antioxidant Defense. Antioxidants 2022, 11, 1046. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Z.; Wang, Y.; Zhang, T.; Qi, S.; Tang, Y.; et al. Investigation into the Properties of L-5-Methyltetrahydrofolate and Seal Oil as a Potential Atherosclerosis Intervention in Rats. J Nutr Sci Vitaminol 2022, 68, 87–96. [Google Scholar] [CrossRef]
- Clément, A.; Menezo, Y.; Cohen, M.; Cornet, D.; Clément, P. 5-Methyltetrahydrofolate reduces blood homocysteine level significantly in C677T methyltetrahydrofolate reductase single-nucleotide polymorphism carriers consulting for infertility. J Gynecol Obstet Hum Reprod 2020, 49, 101622. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, M.C.; Wever, R.M.; Kastelein, J.J.; van Dam, T.; Koomans, H.A.; Rabelink, T.J. 5-methyltetrahydrofolate, the active form of folic acid, restores endothelial function in familial hypercholesterolemia. Circulation 1998, 97, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Warrick, N.; Cai, S.; de Bono, J.; Lee, J.; et al. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 2006, 114, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Efsa Panel on Nutrition, N.F.; Food, A.; Turck, D.; Bohn, T.; Castenmiller, J.; De Henauw, S.; et al. Safety of monosodium salt of l-5-methyltetrahydrofolic acid as a novel food pursuant to Regulation (EU) 2015/2283 and the bioavailability of folate from this source in the context of Directive 2002/46/EC, Regulation (EU) No 609/2013 and Regulation (EC) No 1925/2006. EFSA J 2023, 21, e8417. [Google Scholar] [CrossRef]
Table 1.
Selected genes and their polymorphic variants participating in the development of coronary artery disease [39,47,54].
| Type of inheritance | Genes, enzymes, receptors and ligands | Clinical implications |
|---|---|---|
|
Monogenic inheritance. Among monogenic mutations, most are involved in lipid metabolism. |
Genetic causes of elevated LDL cholesterol | |
| LDLR, APOB, PCSK9 | Familial hyperlipidemia | |
| USF1 | Familial combined hyperlipidemia | |
| Genetic causes of reduced HDL cholesterol levels | ||
| APOA1 | Primary hypoalphalipoproteinemia | |
| ABCA1 | Tangier disease | |
| LCAT | Norum disease, Fish eye disease | |
| ABCG5/8 | Sitosterolemia | |
| Genetic causes of hypertriglyceridemia | ||
| LPL, APOC2, APOAV, GPIHBP1, LMF1 | Familial chylomicronemia syndrome | |
| APOA1/C3/A4/A5 | Familial combined hyperlipidemia and familial hypertriglyceridemia | |
| ATHS | Atherogenic lipoprotein phenotype | |
|
Polygenic inheritance. Mutations and polymorphisms of genes encoding enzymes, receptors and ligands play a role in the development of atherosclerosis. |
APOE, APOB, LPL, OLR1 (LOX1), SORT1, TRIB1 | Lipid and apolipoprotein metabolism |
| E-selectin, P-selectin, Interleukin 6, Paroxonase data | Inflammatory response | |
| Connexin 37, eNOS, metalloproteinase 9, stomelysin 1 | Endothelium function | |
| GP Ia/II receptor, GP IIIa receptor | Platelet function | |
| Factor V Leiden, prothrombin 20210A, protein C deficiency, protein S non-deficiency, anti-thrombin deficiency | Thrombosis and fibrinolysis | |
| MTHFR | Folate metabolism | |
| ACE, AGTR1 | Blood pressure regulation | |
ABCA - ATP-binding cassette transporter; ABCG5/8 - ATP-binding cassette sub-family G member 5/8; ACE - angiotensin-converting enzyme; AGTR1 - angiotensin-II receptor 1; APOA1 - apolipoproteine AI, APOA1/C3/A4/A5 Gene Cluster; APOA5 - apolipoproteine A-V; APOB - apolipoproteine B; APOB - apolipoproteine B; APOC2 - apolipoproteine C-II; APOE - apolipoproteine E; ATHS - atherosclerosis susceptibility; eNOS - endothelial nitric oxide synthase; GPIHBP1 - Glycosylphosphatidylinositol Anchored High Density Lipoprotein Binding Protein 1; LCAT - Lecithin–cholesterol acyltransferase; LDLR - low-density lipoprotein receptor; LMF1 - Lipase Maturation Factor 1; LPL - lipoprothein lipase; MTHFR - Methylenetetrahydrofolate reductase; OLR1 (LOX1) - Oxidized low-density lipoprotein receptor 1; PCSK9 - proprotein subtylisine/keksine convertase; SORT1 - Sortilin 1; TRIB1 - Tribbles Pseudokinase 1; USF1 - Upstream stimulatory factor 1.
Table 3.
Activity (%) of methyltetrahydrofolate reductase (MTHFR) depending on the presence of c.665C>T and c.1286A>C polymorphisms [87].
Table 3.
Activity (%) of methyltetrahydrofolate reductase (MTHFR) depending on the presence of c.665C>T and c.1286A>C polymorphisms [87].
| Genotype | MTHFR c.665 C>T | |||
| c.[665C=];[665C=] | c.[665C>T];[665C=] | c.[665C>T];[665C>T] | ||
| MTHFR c.1286A>C | c.[1286A=];[1286A=] | 100 | 60-70 | 30-40 |
| c.[1286A>C];[1286A=] | 70-80 | 50-60 | - | |
| c.[1286A>C];[1286A>C] | 50-60 | - | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



