
Submitted:
17 May 2024
Posted:
20 May 2024
You are already at the latest version
Abstract
Mutations in the SACS gene are associated with autosomal recessive spastic ataxia of Charlevoix-Saguenay disease (ARSACS) or complex clinical phenotypes of Charcot-Marie-Tooth disease (CMT). This study aimed to identify SACS mutations in a Korean CMT cohort with cerebellar ataxia and spasticity. As a result, eight pathogenic SACS mutations in four families were identified as the underlying causes of these complex phenotypes. The prevalence of CMT families with SACS mutations was determined to be 0.3%. All the patients showed sensory, motor, and gait disturbances with increased deep tendon reflexes. Lower limb magnetic resonance imaging (MRI) was done in four patients, and all had fatty replacements. Of note, they all had similar fatty infiltrations between the proximal and distal lower limb muscles, different from the neuromuscular imaging feature in most CMT patients without SACS mutations who had distal dominant fatty involvement. Therefore, these findings were considered a characteristic feature in CMT patients with SACS mutations. Although further studies with more cases are needed, our results highlight lower extremity MRI findings in CMT patients with SACS mutations and broaden the clinical spectrum. We suggest screening for SACS in recessive CMT patients with complex phenotypes of ataxia and spasticity.
Keywords:
autosomal recessive spastic ataxia of Charlevoix-Saguenay disease (ARSACS)
; cerebellar ataxia
; Charcot-Marie-Tooth disease (CMT)
; Korean
; SACS
1. Introduction
Charcot-Marie-Tooth disease (CMT), also called hereditary motor and sensory neuropathy (HMSN), is a genetically heterogeneous group of peripheral neuropathic disorders characterized by progressive muscle weakness, atrophy, and sensory loss, primarily in the distal extremities. The genetic underpinnings of CMT have been extensively studied, leading to the identification of more than 130 causative genes [1]. CMT is traditionally considered a rare, simple Mendelian genetic disease but has a loose genotype-phenotype correlation.
CMT is sometimes accompanied by additional symptoms such as central nervous abnormality, myopathy, nephropathy, optic atrophy, hearing loss, ataxia, and spasticity [2], which are believed to occur due to the multi-organ pleiotropic nature of many CMT-relevant genes. SACS (MIM 604490), generally known to cause autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS; MIM 270550), is one of the CMT-related genes showing multi-organic complex phenotypes [3,4]. CMT patients with homozygous or compound heterozygous mutations in the SACS gene have complex phenotypes, including peripheral polyneuropathy, spinocerebellar ataxia, and spasticity [5,6]. Besides SACS mutations, mutations in some genes including MFN2, REEP1, B4GALNT1, and C12ORF65 have been reported to be related with the complex phenotypes of CMT and spasticity [7,8,9].
Since ARSACS was first documented in the 1970s in Quebec [10], linkage analysis mapped its genetic locus to chromosome 13q11 [11]. Then, mutations in SACS located within the linked disequilibrium region were identified as the underlying causes of ARSACS [3,4]. Thus far, several hundred homozygous or compound heterozygous mutations have been reported to be the genetic cause of ARSACS [12,13]. In the public databases of ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and OMIM (https://www.omim.org/), more than 600 SACS variants have been registered as pathogenic or likely pathogenic variants in several recessive disorders, such as spastic ataxia with a pyramidal sign and peripheral neuropathy. Most SACS variants occur within short sequences, while several long deletion or insertion variants (copy number variants) have also been reported [14].
The SACS gene encodes a SACSIN protein, which integrates with the ubiquitin-proteasome system and HSP70 chaperone machinery [15]. Besides its chaperone activity, the SACSIN protein is believed to have important roles in several cellular processes, including cytoskeletal organization [16,17], mitochondrial homeostasis [18], and cell migration [3,18]. Notably, the regulation of mitochondrial functions by SACSIN may share common mechanisms with many neurodegenerative diseases [16,18,19].
When ARSACS was first reported in Quebec, the affected individuals revealed similar phenotypes characterized by early onset cerebellar ataxia, spasticity, and peripheral neuropathy [3,11]. After that, many cases have shown atypical phenotypes in other ethnic groups. Late-onset patients have been frequently found [20,21], and some Japanese patients showed no spasticity [22,23,24]. ARSACS cases have been reported in East Asian countries, including Japan [22,23,24] and China [14,25,26]. However, limited cases of SACS mutations have been reported in Korean patients [27,28,29].
The primary purpose of this study was to identify pathogenic SACS mutations in a Korean CMT cohort. As a result, compound heterozygous SACS mutations from four CMT families were identified with concurrent ataxia and spasticity. This study also characterized clinical phenotypes, including lower extremity MRI features, and analyzed the genotype-phenotype correlation in the affected individuals with the SACS mutations.
2. Results
2.1. Identification of Compound Heterozygous SACS Mutations
This study identified trans-arrayed compound heterozygous mutations of SACS in four CMT families presenting concurrent cerebellar ataxia and spasticity (Table 1). The eight observed mutations were evaluated to be pathogenic (P) or likely pathogenic (LP) by the ACMG/AMP criteria (Table S1). All the mutations were completely cosegregated with the affected individual(s) in each family with the recessive mode.
In the family FC591, c.1966_1967insT (p.S656Ffs*1) and c.4138C>G (p.P1380A) were identified in two affected siblings. Insertion and missense mutations were transmitted from their unaffected father and mother, respectively (Figure 1A). In the family FC937, two heterozygous mutations of c.2439_2440delAT (p.V815Gfs*2) and c.10897T>G (p.F3633V) were in a sporadically affected male. The p.F3633V missense mutation was observed in his father; thus, the other 2-bp deletion mutation seemed to be transmitted from his non-examined mother (Figure 1B). The c.2439_2440delAT homozygous mutation has been reported several times in affected individuals with early-onset cerebellar ataxia and peripheral neuropathy from consanguineous families [30,31]. In addition, compound heterozygous SACS mutations of c.2439_2440delAT and c.434C>G were observed in a patient with leg spasticity and sensorimotor axonal polyneuropathy [32]. In the FC1157 family, two deletion mutations of c.2903_2906delACAG (p.D968Vfs*13) and c.13217delC (p.T4406Rfs*45) were observed in an affected sister and brother. Their unaffected father and mother had the p.D968Vfs*13 and p.T4406Rfs*45, respectively (Figure 1C). Homozygous mutation of the c.2903_2906delACAG was once reported in a patient with axonal CMT, ataxic gait, and cerebellar phenotype [33]. In the FC1176 family, two stop-gain mutations of c.1596T>A (p.Y532X) and c.3159_3160delCT (p.F1054X) were identified in an affected female. Two mutations were transmitted from each unaffected parent (Figure 1D).
The c.2439_2440delAT and c.2903_2906delACAG have been reported as pathogenic mutations in the homozygous or heterozygous states [30,31,32,33]. In contrast, the other six mutations were not reported in public human genome databases, including IGSR, gnomAD, and KRGDB (Table 2). In addition, four pairs of heterozygous mutations found in the analyzed families have not yet been reported as an underlying cause of CMT or ARSACS.
Figure 2A,B show sequencing chromatograms and mutation locations on the SACSIN protein, respectively. Two missense mutation sites and their surrounding sequences are highly conserved among vertebrate species, from fish to mammals (Figure 2C). Several in silico analyses by Mutation Taster, REVEL, MUpro, and PolyPhen-2 predicted that all the mutations were pathogenic (Table 2).
2.2. Prediction of Conformational Changes by Missense Mutations
Considerable conformational changes were predicted in the mutant proteins due to missense mutations (Figure 3). In the wild protein, the F3633 residue was predicted to connect with the R3636 residue through a cation-π interaction, and the F3666 residue was predicted to have hydrophobic contact with the L3640 and F3633 residues. However, in the p.F3633V mutant, the cation-π interaction with the R3636 residue was replaced by a weak hydrogen bond, and all hydrophobic contacts with the F3666 residue are predicted to be broken. As a result, the distance between surrounding α-helices was predicted to be distant (Figure 3A). In the wild type, the P1380 residue was not connected with the L1360, D1406, D1402, and H1383 residues, but due to the p.P1380A mutation, a hydrogen bond was predictively created between the P1380A and the H1383 residues. The H1383 residue was linked with the D1406 residue by a hydrogen bond and with the D1402 residue by an ionic interaction. It was also predicted that the distance between the two α-helices will become closer. Additionally, it was predicted that a hydrophobic contact would be created between the D1406 and L1360 residues because the α-helix would be closed. As a result, the distance between the surrounding α-helices was predicted to become closer (Figure 3B). When we applied the PremPS, MAESTROweb, and DynaMut2 programs using the predicted PDB, it was predicted that the two missense mutations significantly decreased the protein stability (Table 3).
2.3. Clinical Manifestation
We examined the clinical features in the affected individuals with SACS mutations from the four families (Table 4). The mean age at which neuropathy symptoms first appeared was 9.83 ± 6.46 years (early-to-mid onset ranging from 3 to 17 years), and the age at the time of examination at the hospital was 28.0 ± 4.98 years (22 to 27 years). Table 3 shows the clinical features of six patients with compound heterozygous SACS mutations.
They showed a similar muscle weakness between the proximal and distal limb muscles. Therefore, their muscle weakness differed from most other CMT patients with length-dependent axonal degeneration. Sensory disturbance was seen in all patients, and the vibration sense was worse than the pain sense. Deep tendon reflexes were hyperactive in all patients. Cerebellar ataxia, spasticity, foot deformity, hearing loss, and nystagmus were observed in all patients. Disability scores were severe (FDS > 3, CMTNSv2 > 21) in three patients, and moderate (FDS = 2, 10 < CMTNSv2 ≤ 20) in the other patients.
2.4. Electrophysiological Findings
Table S2 shows the results of six patients who underwent nerve conduction studies. These six patients were compatible with HMSN. The upper and lower extremity MNCVs were slow, and the CMAPs were decreased. The sensory nerve damage was more remarkable than the motor nerve damage. In four patients, the upper and lower limb SNAPs were not evoked. Hence, the level of sensory neuropathy manifests earlier and exhibits greater severity when contrasted with the degree of motor neuropathy. BAEP was performed on three patients, and all showed a prolonged I-III and III-V interpeak latency, indicating abnormal findings.
2.5. MRI Features of the Brain and Lower Extremity
The lower extremity MRI findings for the four patients are summarized in Table S3. These four patients had minimal to mild fatty infiltration (Goutallier grade 1 or 2) in the lower extremity muscles: II-1 in FC937 (Figure 4A,B), II-2 in FC1157 (Figure 4C,D), II-4 in FC1157 (Figure 4E,F), and II-1 in FC1176 (Figure 4G,H) [31]. Of note, there were no cases in which normal muscle exists without fatty infiltration. Considering that the age at which the MRI was taken was from 22 to 27 years old, the fatty replacement on the lower extremity MRI was very unusual. Moreover, no clear predominance of fatty involvement was observed at the distal levels or specific compartment muscles. Both the proximal and distal muscles appeared to be similarly involved, a characteristic finding that differed from the more severe damage to the distal part in the other CMT subtypes.
Brain MRI scans were performed on three affected individuals, II-2 and II-4 in FC1157 and II-1 in FC1176 (Figure S1). There were no notable findings of significant atrophy or white matter changes in the cerebrum. However, all three individuals showed atrophy, particularly in the superior cerebellar vermis. A bilateral linear hypointensity in the brainstem was observed in the pontine region on the T2 and FLAIR images.
3. Discussion
This is the first cohort study on SACS mutations in Korean CMT patients. Thus far, only three sporadic cases have been reported in the Korean population [27,28,29]. In this study, we identified eight heterozygous SACS mutations in four recessive early-to-mid onset CMT families with cerebellar spastic ataxia. Among them, six were unreported novel mutations, and the compound heterozygous mutations shown in the examined families were all unreported new combinations of heterozygous mutation pairs. We identified these pairs as the underlying causes of the complex phenotypes observed in polyneuropathy and cerebellar spastic ataxia. This result was based on several key factors: complete cosegregation with affected individuals, genotype-phenotype correlation, unreported or very low frequent mutant alleles, well conservation of mutation sites, in silico prediction of pathogenicity, and likely pathogenic evaluation by the ACMG/AMP criteria.
The prevalence of patients with SACS mutations in the Korean CMT cohort was calculated to be 0.3% of the total examined families; while 0.4% of the families were negative for PMP22 duplication. As a genetic feature, no homozygous SACS mutation was found in this study, which is believed to be partly related to the strong prohibition of marriage between relatives. In countries with frequent consanguineous marriages, recessive genetic diseases in many patients have been caused by homozygous mutations [34,35,36], whereas in Korea, recessive conditions in the majority of patients have been reported to be caused by compound heterozygous mutations [37,38].
Mutations in the SACS gene are known to affect the expression of the SACSIN protein, leading to mitochondrial dysfunction, influencing the HSP70 chaperone pathway, and ultimately contributing to neurodegeneration. The findings of cerebellar atrophy and bilateral pontine linear hypointensity in this study are commonly observed in other SACS mutations. They are associated with cerebellar ataxia and upper motor neuron signs. Other studies have reported the presence of demyelinating or mixed-type polyneuropathy [4,5,26]. However, the cases in this study showed five mixed-type polyneuropathy and one axonal polyneuropathy; thus, no demyelinating polyneuropathy was found.
Sensorimotor polyneuropathy in patients with SACS gene mutations is well known. Additionally, it is well known that patients with sensorimotor polyneuropathy show abnormal fatty involvement on lower extremity MRI. Therefore, patients with SACS mutations may have lower extremity muscle damage and may be expected to show abnormal fatty infiltration findings on lower extremity MRI. However, lower extremity MRI findings in patients with SACS mutations have not been reported. Therefore, this study performed lower extremity MRI on four SACS mutation patients with typical clinical features. To our knowledge, this is the first report on lower extremity MRI in patients with SACS mutations. All four patients had minimal to mild fatty infiltration in the thigh and calf muscles of the lower extremities. In general, CMT patients show characteristics of length-dependent axonal degeneration; thus, the damage to the calf muscles, the distal part of the lower extremity, is more severe than to the thigh muscles, the proximal part. As the disease gradually worsens, signs of fatty infiltration appear in the proximal thigh muscles. However, in the patients with the SACS mutations, the degree of fatty infiltration in the thigh and calf muscles was similar. This was equally observed in all four patients with SACS mutations. Thus, these findings require further study with a larger number of SACS patients; however, these findings are considered characteristic findings of lower extremity MRI examinations in SACS patients.
This study was the first attempt to investigate SACS variants thoroughly from a CMT genetic cohort. As a result, biallelic SACS variants could be identified as the genetic causes of four recessive CMT families. Because some ARSACS patients showed atypical phenotypes, such as the absence of spasticity and mid-to-late onset [22,23], we suggest that SACS variants should be considered as the genetic causes of recessive CMT with complex phenotypes.
4. Materials and Methods
4.1. Patients
This study recruited a cohort consisting of 1,889 CMT patients from 1,363 unrelated Korean families between April 2005 and May 2023. Then, 413 CMT type 1A (CMT1A) families with the PMP22 duplication were excluded. Thus, the remaining 950 families were screened for SACS mutations. Written informed consent was obtained from all participants by a protocol approved by the Institutional Review Boards of Sungkyunkwan University, Samsung Medical Center (2014-08-057-002) and Kongju National University (KNU_IRB_2018-62). Parents provided written consent for the minors involved in this study.
4.2. Molecular Genetic Analysis
Genomic DNA was purified from whole blood using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The affected individuals, who tested negative for PMP22 duplication, underwent whole exome sequencing (WES). The exomes were captured using the SureSelect Human All Exon 50M Kit (Agilent Technologies, Santa Clara, CA, USA), and sequencing was performed using the HiSeq 2500 Genome Analyzer (Illumina, San Diego, CA, USA).
The human genome assembly hg19 (GRCh37) was used as the reference sequence (http://genome.ucsc.edu). VCF files were generated from the FastQ files of the WES low data using the GATK and SAMtools algorithms. Functionally significant variants (missense, nonsense, exonic indel, and splicing site variants) were first selected from the VCF files. Then, unreported or rare variants with allele frequencies of ≤ 0.01 were subsequently chosen from CMT- and spastic ataxia-related genes. Minor allele frequencies (MAF) were cross-referenced with various public databases, including dbSNP (http://www.ncbi.nlm.nih.gov), the International Genome Sample Resource (IGSR; https://www.internationalgenome.org), the Genome Aggregation Database (gnomeAD; https://gnomad.broadinstitute.org), and the Korean Reference Genome Database (KRGDB; http://coda.nih.go.kr/coda/KRGDB). The identified variants were confirmed through Sanger sequencing using the SeqStudio Genetic Analyzer (Life Technologies-Thermo Fisher Scientific, Foster City, CA, USA).
4.3. Conservation, Conformational Change, and In Silico Prediction of Mutant Proteins
Conservation analysis of the mutation sites was conducted using MEGA6, ver. 6.0 (http://www.megasoftware.net). Genomic evolutionary rate profiling (GERP) scores were determined using the GERP++ program (http://mendel.stanford.edu/SidowLab/downloads/gerp/). In silico prediction of mutations was performed using the Mutation taster (https://www.genecascade.org/MutationTaster2021), MUpro (http://www.ics.uci.edu/~baldig/mutation), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), and REVEL (https://sites.google.com/site/revelgenomics/). Pathogenicity was evaluated using the guidelines established by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) criteria (http://wintervar.wglab.org/). Three-dimensional conformational changes of mutant proteins were predicted using I-TASSER (https://seq2fun.dcmb.med.umich.edu//I-TASSER/) and were visualized through the Mol*features of the Protein Data Bank (https://www.rcsb.org/3d-view). Based on the generation of the Protein Data Bank File (PDB), the effects of the mutations on the protein stability were evaluated using the MAESTROweb (https://biwww.che.sbg.ac.at/maestro/web), PremPS (https://lilab.jysw.suda.edu.cn/research/PremPS/), and DynaMut2 (http://biosig.unimelb.edu.au/dynamut2) programs.
4.4. Clinical Assessment
Clinical data were collected in a standardized manner, including evaluations of motor and sensory impairments, deep tendon reflexes, and muscle atrophy. The strength of the flexor and extensor muscles was assessed manually using the widely accepted Medical Research Council (MRC) scale [39]. CMT neuropathy score version 2 (CMTNSv2) and functional disability scale (FDS) were used to quantify physical disability. Sensory impairments were evaluated based on the level and severity of pain, temperature perception, vibration sense, and positional awareness. The ages of onset were determined through patient reports, specifically by inquiring about the timing and age at which initial symptoms, such as distal muscle weakness, foot deformities, or sensory changes, first manifested.
4.5. Electrophysiological Examination
Motor and sensory conduction velocities of the median, ulnar, peroneal, tibial, and sural nerves were assessed using the standard methods of surface stimulation and recording electrodes. Motor nerve conduction velocities (MNCVs) and compound muscle action potentials (CMAPs) of the median and ulnar nerves were measured by stimulating at both the elbow and wrist. Similarly, MNCVs and CMAPs of the peroneal and tibial nerves were determined by stimulation at the knee and ankle. CMAPs were quantified by measuring the difference between the baseline and negative peak values. Sensory nerve conduction velocities (SNCVs) were evaluated over a finger-wrist segment for the median and ulnar nerves using orthodromic scoring, and recordings were also obtained for the sural nerves. Sensory nerve action potentials (SNAPs) were determined by measuring the amplitude between the positive and negative peaks. Brainstem auditory evoked potential (BAEP) was obtained by monitoring the two channels placed in the bilateral mastoid processes (A1 and A2, left and right, respectively) referenced to the Cz position when 43.9 Hz auditory stimuli were given by 1.5 ms/div using an in-ear microphone. Bilateral latencies and amplitudes from wave I-V were recorded at baseline.
4.6. Brain and Lower Extremity MRI
MRI scans were conducted in a supine position using the Ingenia 3.0T CX MRI system (Philips Healthcare, Best, Netherlands). T1-weighted axial and sagittal images and T2-weighted axial fluid-attenuated inversion recovery (FLAIR) and susceptibility weighted images (SWIs) were sequentially obtained to evaluate the brain involvement. Comprehensive MRI scans were obtained in the pelvic girdle, bilateral thigh, and lower legs for lower extremity images. Fatty infiltration in the thigh and lower leg muscles was assessed on axial T1-weighted turbo spin-echo images using a five-point semiquantitative scale [40]. Thigh and lower leg muscles were evaluated bilaterally at two levels (proximal and distal), respectively.
5. Conclusions
This study identified eight SACS mutations in four recessive CMT families as the underlying cause of the complex phenotypes. Six were unreported novel mutations, and the compound heterozygous mutations shown in the examined families were all unreported new combinations of mutation pairs. The prevalence of CMT families with SACS mutations was determined to be 0.3% in the Korean CMT cohort. This study provides new insights into SACS mutations in a Korean CMT cohort study, revealing their genetic patterns, prevalence, and clinical and neuroimaging manifestations. All the patients showed similar fatty infiltrations between the proximal and distal lower limb muscles, which differed from the CMT patients without SACS mutations who had distal dominant fatty involvement. The consistent clinical presentation aligns with established knowledge, and the distinctive lower extremity MRI findings broaden the clinical spectrum of CMT patients with SACS mutations. This study emphasizes the testing of the SACS gene for recessive CMT patients showing complex symptoms of cerebellar ataxia and spasticity.
Supplementary Materials
The following supporting information can be downloaded at Preprints.org, Figure S1: Brain MRIs of three CMT patients with SACS mutations, Table S1: Evaluation of the SACS variants by the ACMG/AMP criteria, Table S2: Electrophysiological features in CMT patients with SACS mutations, Table S3: Thigh and calf MRI features of CMT patients with SACS mutations.
Author Contributions
Conceptualization, K.W.C. and B.-O.C.; methodology, B.K.P., Y.H.C. H.S.K., S.H.N. and D.E.N.; validation, S.H.N., H.S.K. and S.B.K.; investigation, B.K.P., H.S.K., A.J.L. and D.E.N.; resources, Y.H.C., H.J.P., S.B.K. and B.O.C.; writing-original draft preparation, B.K.P. and Y.H.C.; writing-review and editing, K.W.C.; funding acquisition, B.-O.C.; supervision, B.-O.C and K.W.C. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by grants from the National Research Foundation (2021R1A4A2001389 and 2022R1I1A2068995), the Korean Health Technology R&D Project, Ministry of Health and Welfare (HR22C1363 and HX23C1756), the Korean Fund for Regenerative Medicine, Ministry of Science and ICT, and Ministry of Health and Welfare (23C0115L1), and Future Medicine 2030 Project of the Samsung Medical Center (SMO1240041), Korea.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and The Institutional Review Boards of Kongju National University (KNU_IRB_2018-62) and Sungkyunkwan University, Samsung Medical Center (2018-05-102-002) approved this study.
Informed Consent Statement
All participants provided written informed consent by a protocol approved by the Institutional Review Boards. For minors under the age of 18 years, written consent was provided by their parents.
Data Availability Statement
Data available on request from the authors.
Acknowledgments
We appreciate all the patients and their family members for providing consent, samples, and clinical information.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: opportunities and challenges. Nat Rev Neurol 2019, 15, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Scaioli, V.; Laura, M. Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease. Neuromolecular Med 2006, 8, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Engert, J.C.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 2000, 24, 120–125. [Google Scholar] [CrossRef] [PubMed]
- El Euch-Fayache, G.; Lalani, I.; Amouri, R.; Turki, I.; Ouahchi, K.; Hung, W.Y.; Belal, S.; Siddique, T.; Hentati, F. Phenotypic features and genetic findings in SACSin-related autosomal recessive ataxia in Tunisia. Arch Neurol 2003, 60, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.V.S.; Bortholin, T.; Naylor, F.G.M.; Pinto, W.B.V.R.; Oliveira, A.S.B. Early-onset axonal Charcot-Marie-Tooth disease due to SACS mutation. Neuromuscul Disord 2018, 28, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Zaman, Q.; Khan, M.A.; Sahar, K.; Rehman, G.; Khan, H.; Rehman, M.; Najumuddin; Ahmad, I. ; Tariq, M.; Muthaffar, O.Y.; et al. Novel variants in MPV17, PRX, GJB1, and SACS cause Charcot-Marie-Tooth and spastic ataxia of Charlevoix-Saguenay type diseases. Genes 2023, 14, 328. [Google Scholar] [CrossRef] [PubMed]
- Feely, S.M.; Laura, M.; Siskind, C.E.; Sottile, S; Davis, M. ; Gibbons, V.S.; Reilly, M.M.; Shy, M.E. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 2011, 76, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Blair, N.F.; Sue, C.M. An update on the hereditary spastic paraplegias: new genes and new disease models. Mov Disord Clin 2015, Pract 2, 213–223. [Google Scholar] [CrossRef]
- Toft, A.; Birk, S.; Ballegaard, M.; Duno, M.; Hjermind, L.E.; Nielsen, J.E.; Svenstrup, K. Peripheral neuropathy in hereditary spastic paraplegia caused by REEP1 variants. J Neurol 2019, 266, 735–744. [Google Scholar] [CrossRef]
- Bouchard, J.P.; Barbeau, A.; Bouchard, R.; Bouchard, R.W. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci 1978, 5, 61–69. [Google Scholar] [CrossRef]
- Engert, J.C.; Dore, C.; Mercier, J.; Ge, B.; Betard, C.; Rioux, J.D.; Owen, C.; Berube, P.; Devon, K.; Birren, B.; et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): high-resolution physical and transcript map of the candidate region in chromosome region 13q11. Genomics 1999, 62, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Aly, K.A.; Moutaoufik, M.T.; Zilocchi, M.; Phanse, S.; Babu, M. Insights into SACS pathological attributes in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Curr Opin Chem Biol 2022, 71, 102211. [Google Scholar] [CrossRef] [PubMed]
- Bagaria, J.; Bagyinszky, E.; An, S.S.A. Genetics of autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) and role of SACSin in neurodegeneration. Int J Mol Sci, 2022, 23, 552. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.B.; Zi, X.H.; Shen, L.; Hu, ZhM. ; Huang, ShX.; Yu, D.L.; Li, H.B.; Xia, K.; Tang, B.S.; et al. A novel hemizygous SACS mutation identified by whole exome sequencing and SNP array analysis in a Chinese ARSACS patient. J Neurol Sci 2016, 362, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein SACSin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum Mol Genet 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Lariviere, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. SACS knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum Mol Genet 2015, 24, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Murtinheira, F.; Migueis, M.; Letra-Vilela, R.; Diallo, M.; Quezada, A.; Valente, C.A.; Oliva, A.; Rodriguez, C.; Martin, V.; Herrera, F. SACSin deletion induces aggregation of glial intermediate filaments. Cells 2022, 11, 299. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Lariviere, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.; Duchen, M.R.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc Natl Acad Sci USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gehring, K. Structural studies of parkin and SACSin: Mitochondrial dynamics in neurodegenerative diseases. Mov Disord 2015, 30, 1610–1619. [Google Scholar] [CrossRef]
- Baets, J.; Deconinck, T.; Smets, K.; Goossens, D.; Van den Bergh, P.; Dahan, K.; Schmedding, E.; Santens, P.; Rasic, V.M.; Van Damme, P.; et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 2010, 75, 1181–1188. [Google Scholar] [CrossRef]
- Synofzik, M.; Soehn, A.S.; Gburek-Augustat, J.; Schicks, J.; Karle, K.N.; Schule, R.; Haack, T.B.; Schoning, M.; Biskup, S.; Rudnik-Schoneborn, S.; et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis 2013, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Miyake, N.; Doi, H.; Saitsu, H.; Ogata, K.; Kawai, M.; Matsumoto, N. A novel SACS mutation in an atypical case with autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Intern Med 2012, 51, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Doi, H.; Kunii, M. Autosomal recessive spinocerebellar ataxias in Japan. Rinsho Shinkeigaku 2016, 56, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Aida, I.; Ozawa, T.; Fujinaka, H.; Goto, K.; Ohta, K.; Nakajima, T. Autosomal recessive spastic ataxia of Charlevoix-Saguenay without spasticity. Intern Med 2021, 60, 3963–3967. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Tang, J.G.; Yang, Y.F.; Tan, Z.P.; Tan, J.Q. A novel homozygous SACS mutation identified by whole-exome sequencing in a consanguineous family with autosomal recessive spastic ataxia of Charlevoix-Saguenay. Cytogenet Genome Res 2017, 152, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, X.; Jin, Y.; Li, D.; Ye, X.; Tao, C.; Zhou, M.; Jiang, H.; Yu, H. A novel SACS variant identified in a Chinese patient: case report and review of the literature. Front Neurol 2022, 13, 845318. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.Y.; Huh, K.; Eun, B.L.; Yoo, H.W.; Kamsteeg, E.J.; Scheffer, H.; Koh, S.B. A probable Korean case of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci 2015, 42, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Lyoo, C.H.; Park, S.E.; Seo, Y.; Han, S.H.; Han, J. Optical coherence tomography findings facilitate the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Korean J Ophthalmol 2021, 35, 330–331. [Google Scholar] [CrossRef] [PubMed]
- Bong, J.B.; Kim, S.W.; Lee, S.-T.; Choi, J.R.; Shin, H.Y. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. J Korean Neurol Assoc 2019, 37, 69–72. [Google Scholar] [CrossRef]
- Hammer, M.B.; Eleuch-Fayache, G.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Sassi, C.; Bouhlal, Y.; Hentati, F.; Amouri, R.; Singleton, A.B. Exome sequencing: an efficient diagnostic tool for complex neurodegenerative disorders. Eur J Neurol 2013, 20, 486–492. [Google Scholar] [CrossRef]
- Shakya, S.; Kumari, R.; Suroliya, V.; Tyagi, N.; Joshi, A.; Garg, A.; Singh, I.; Kalikavil Puthanveedu, D.; Cherian, A.; Mukerji, M.; et al. Whole exome and targeted gene sequencing to detect pathogenic recessive variants in early onset cerebellar ataxia. Clin Genet 2019, 96, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Panwala, T.F.; Garcia-Santibanez, R.; Vizcarra, J.A.; Garcia, A.G.; Verma, S. Childhood-onset hereditary spastic paraplegia (HSP): a case series and review of literature. Pediatr Neurol 2022, 130, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, M.; Storbeck, M.; Strathmann, E.A.; Delle Vedove, A.; Holker, I.; Altmueller, J.; Naghiyeva, L.; Schmitz-Steinkruger, L.; Vezyroglou, K.; Motameny, S.; et al. Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum Mutat 2018, 39, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Imtiaz, A.; Mujtaba, G.; Maqsood, A.; Bashir, R.; Bukhari, I.; Khan, M.R.; Ramzan, M.; Fatima, A.; Rehman, A.U.; et al. Genetic causes of moderate to severe hearing loss point to modifiers. Clin Genet 2017, 91, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, S.; Choi, Y.J.; Lim, S.O.; Choi, H.J.; Park, J.H.; Nuzhat, R.; Khan, A.; Perveen, S.; Choi, B.O.; Chung, K.W. Novel homozygous mutations in Pakistani families with Charcot-Marie-Tooth disease. BMC Med Genomics 2021, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kanwal, S.; Hameed, R.; Tamanna, N.; Perveen, S.; Mahreen, H.; Son, W.; Lee, K.S.; Chung, K.W. Biallelic mutations in pakistani families with autosomal recessive prelingual nonsyndromic hearing loss. Genes Genomics 2023, 45, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, S.C.; Hong, Y.B.; Yoo, J.H.; Koo, H.; Lee, J.H.; Hong, H.D.; Kim, S.B.; Chung, K.W.; Choi, B.O. Recessive optic atrophy, sensorimotor neuropathy and cataract associated with novel compound heterozygous mutations in OPA1. Mol Med Rep 2016, 14, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Nam, S.H.; Park, J.M.; Kanwal, S.; Choi, Y.J.; Lee, H.J.; Lee, K.S.; Lee, J.E.; Park, J.S.; Choi, B.O.; et al. ; Compound heterozygous mutations of SH3TC2 in Charcot-Marie-Tooth disease type 4C patients. J Hum Genet 2019, 64, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Paternostro-Sluga, T.; Grim-Stieger, M.; Posch, M.; Schuhfried, O.; Vacariu, G.; Mittermaier, C.; Bittner, C.; Fialka-Moser, V. Reliability and validity of the Medical Research Council (MRC) scale and a modified scale for testing muscle strength in patients with radial palsy. J Rehabil Med 2008, 40, 665–671. [Google Scholar] [CrossRef]
- Goutallier, D.; Postel, J.M.; Bernageau, J.; Lavau, L.; Voisin, M.C. Fatty muscle degeneration in cuff ruptures: pre- and postoperative evaluation by CT scan. Clin Orthop Relat Res 1994, 304, 78–83. [Google Scholar] [CrossRef]
Figure 1.
Pedigrees of autosomal recessive CMT families with spastic ataxia. Compound heterozygous mutations in SACS were found in all the affected individuals. Haplotypes of two mutation pairs in each family are indicated at the bottom of all the examined individuals with brief chromosomal signs. Black and white symbols represent affected and unaffected individuals, respectively. The probands are indicated by an arrow (□: male, and ○: female). Mutations transmitted from the father and mother are shown in red and blue, respectively, and common (wild) alleles are indicated by “+”. (A) FC591, (B) FC937, (C) FC1157, and (D) FC1176.
Figure 1.
Pedigrees of autosomal recessive CMT families with spastic ataxia. Compound heterozygous mutations in SACS were found in all the affected individuals. Haplotypes of two mutation pairs in each family are indicated at the bottom of all the examined individuals with brief chromosomal signs. Black and white symbols represent affected and unaffected individuals, respectively. The probands are indicated by an arrow (□: male, and ○: female). Mutations transmitted from the father and mother are shown in red and blue, respectively, and common (wild) alleles are indicated by “+”. (A) FC591, (B) FC937, (C) FC1157, and (D) FC1176.
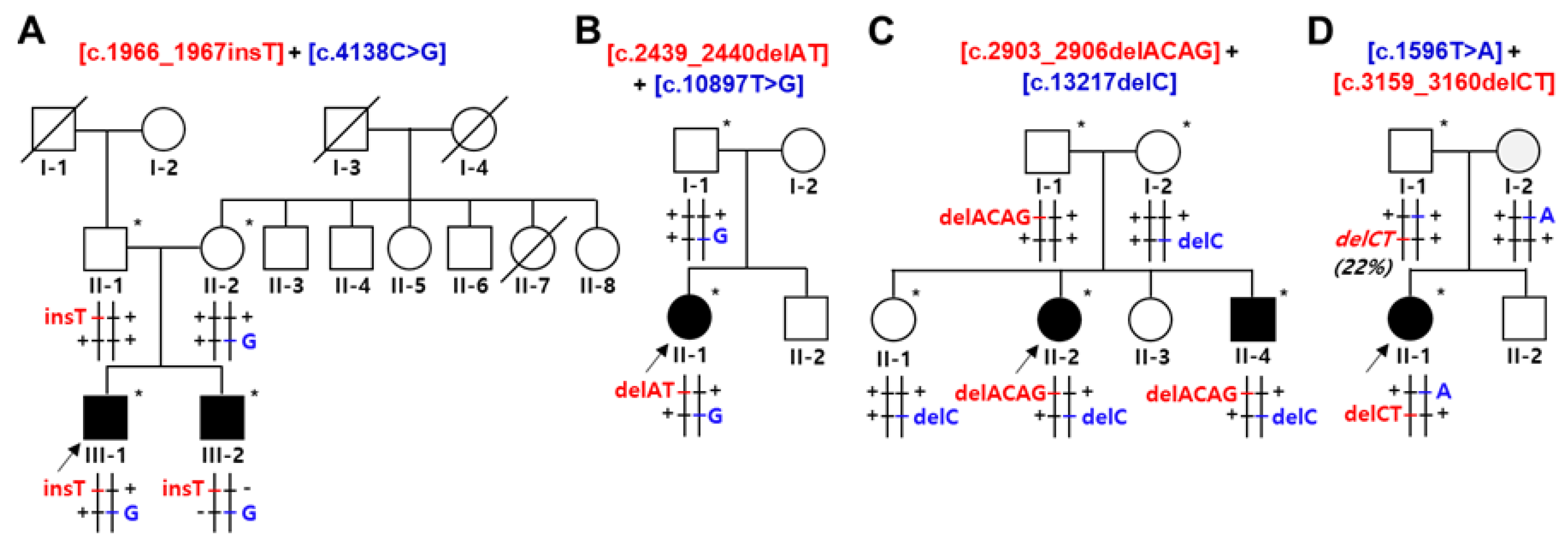
Figure 2.
Identification and conservation of SACS mutations. (A) Sequencing chromatograms of the SACS mutations. The mutation sites are indicated by arrows. (B) Schematic structure of SACSIN protein. Mutations identified in this study are indicated by red arrows. Blue, green, and pink arrows indicate previously reported frameshift, nonsense, and missense mutations, respectively (DNAJ: DnaJ molecular chaperone homology domain; HEPN: higher eukaryotes and prokaryotes nucleotide-binding domain; SRR: SACSIN repeating region; UbL: ubiquitin-Like domain; XPCB: xeroderma pigmentosum complementation group C binding domain). (C) Conservation of missense mutation sites. Two missense mutations are located near SRR2 (p.P1380A) and XPCB (p.F3633V), and their sites were well conserved among vertebrate species from fish to mammals.
Figure 2.
Identification and conservation of SACS mutations. (A) Sequencing chromatograms of the SACS mutations. The mutation sites are indicated by arrows. (B) Schematic structure of SACSIN protein. Mutations identified in this study are indicated by red arrows. Blue, green, and pink arrows indicate previously reported frameshift, nonsense, and missense mutations, respectively (DNAJ: DnaJ molecular chaperone homology domain; HEPN: higher eukaryotes and prokaryotes nucleotide-binding domain; SRR: SACSIN repeating region; UbL: ubiquitin-Like domain; XPCB: xeroderma pigmentosum complementation group C binding domain). (C) Conservation of missense mutation sites. Two missense mutations are located near SRR2 (p.P1380A) and XPCB (p.F3633V), and their sites were well conserved among vertebrate species from fish to mammals.
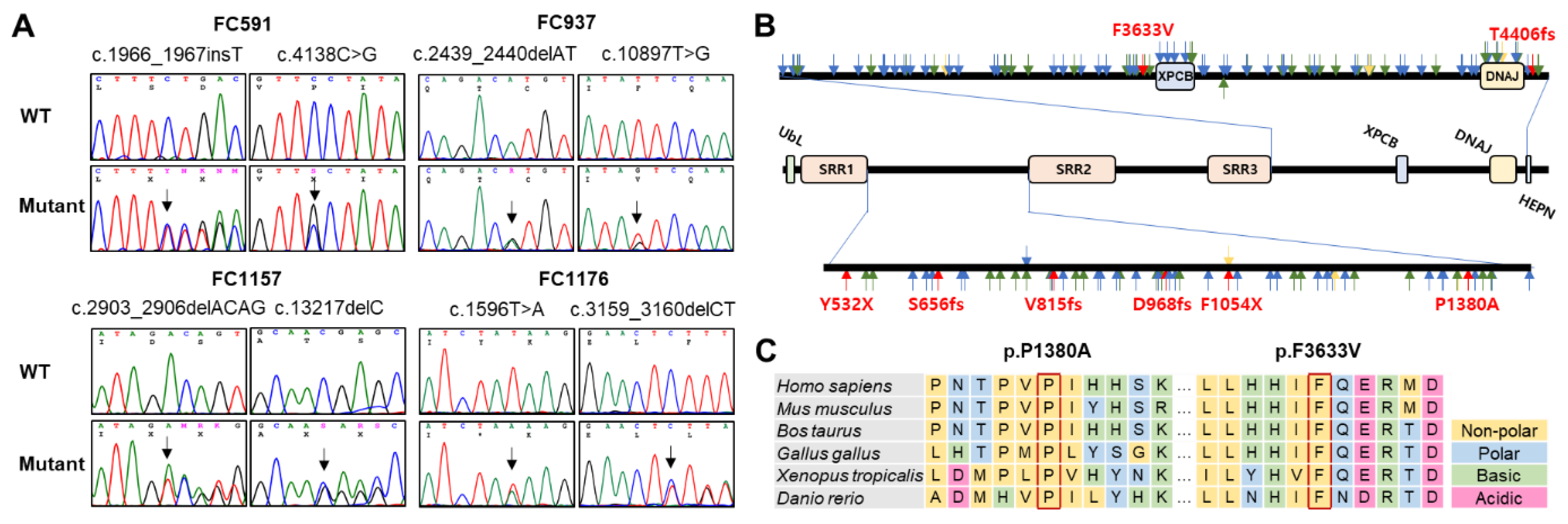
Figure 3.
Prediction of 3D structural changes due to missense mutations. Mutated residues are shown in blue, and surrounding residues predicted to undergo structural changes due to the mutations are shown in yellow, pink, red, and green. (A) p.F3633V, (B) p.P1380A.
Figure 3.
Prediction of 3D structural changes due to missense mutations. Mutated residues are shown in blue, and surrounding residues predicted to undergo structural changes due to the mutations are shown in yellow, pink, red, and green. (A) p.F3633V, (B) p.P1380A.
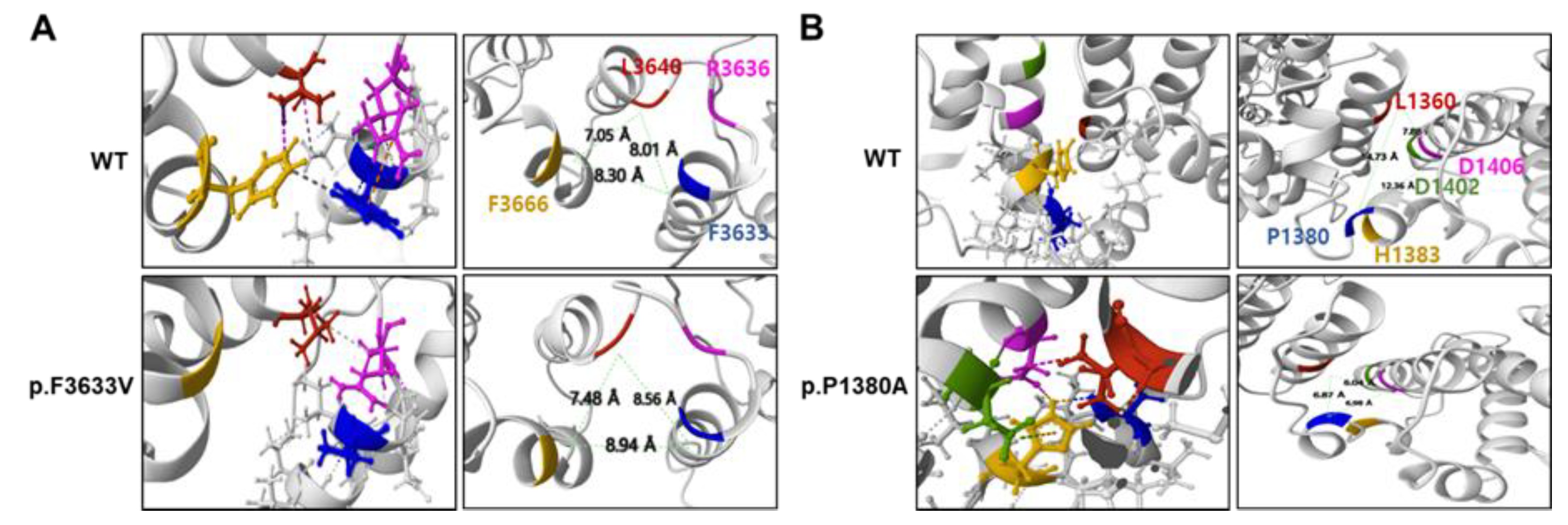
Figure 4.
T1-weighted lower limb MR images of four patients with SACS mutations: II-1 (F/27 years) in FC937 (A, B), II-2 (F/26 years) in FC1157 (C, D), II-4 (M/25 years) in FC1157 (E, F), and II-1 (M/22 years) in FC1176 (G, H). MRIs were prepared at the thigh level (A, C, E, G) or calf level (B, D, F, H). Notably, no clear predominance of fatty changes was observed at the distal levels or in specific compartment muscles.
Figure 4.
T1-weighted lower limb MR images of four patients with SACS mutations: II-1 (F/27 years) in FC937 (A, B), II-2 (F/26 years) in FC1157 (C, D), II-4 (M/25 years) in FC1157 (E, F), and II-1 (M/22 years) in FC1176 (G, H). MRIs were prepared at the thigh level (A, C, E, G) or calf level (B, D, F, H). Notably, no clear predominance of fatty changes was observed at the distal levels or in specific compartment muscles.

Table 1.
Compound heterozygous mutations of SACS in CMT patients with cerebellar ataxia.
| Family ID | Mutations | Clinical phenotype | ACMG/AMP | |
|---|---|---|---|---|
| Nucleotide 1 | Amino acid 1 | |||
| FC591 | [c.1966_1967insT] + [c.4138C>G] | [p.S656Ffs*1] + [p.P1380A] | CMT, cerebellar ataxia, spasticity, and HL | P p |
| FC937 | [c.2439_2440delAT] + [c.10897T>G] | [p.V815Gfs*2] + [p.F3633V] | CMT, cerebellar ataxia, and spasticity, HL | P LP |
| FC1157 | [c.2903_2906delACAG] + [c.13217delC] | [p.D968Vfs*13] + [p.T4406Rfs*45] | CMT, cerebellar ataxia, spasticity, and HL | P P |
| FC1176 | [c.1596T>A] + [c.3159_3160delCT] | [p.Y532X] + [p.F1054X] | CMT, cerebellar ataxia, spasticity, and HL | P P |
1 Tables Abbreviations: ACMG/AMP: American College of Medical Genetics and Genomics and Association for Molecular Pathology, CMT: Charcot-Marie-Tooth disease, HL: hearing loss, LP: likely pathogenic, P: pathogenic. 1 Reference sequences of nucleotides: SACS: NM_014363.6 and amino acids: NP_055178.3.
Table 2.
Allele frequencies and in silico prediction of the SACS variants.
| Variants 1 | dbSNP Acc. No. | Mutant allele frequencies | GERP | In silico analyses 2 | References | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| IGSR | gnomAD | KRGDB | MutT | REVEL | PP2 | MU | ||||
| p.Y532X | rs2137720760 | - | - | - | -2.77 | 200/0* | - | - | - | |
| p.S656Ffs*1 | - | - | - | - | 5.14 | 200/0* | - | - | - | |
| p.V815Gfs*2 | rs775059063 | - | 0.0000142 | - | -1.5 | 200/0* | - | - | - | 30-32 |
| p.D968Vfs*13 | rs1259615333 | - | 0.0000099 | - | 3.71 | 199/1* | - | - | - | 33 |
| p.F1054X | rs2137637877 | - | - | - | 5.09 | 200/0* | - | - | - | |
| p.P1380A | - | - | - | - | 6.06 | 66/34* | 0.637* | 0.965* | -0.757* | |
| p.F3633V | rs1382541188 | - | 0.000013 | - | 6.04 | 72/28* | 0.626* | 0.999* | -0.292* | |
| p.T4406Rfs*45 | - | - | - | - | 5.85 | 198/2* | - | - | - | |
Abbreviations: GERP: genomic evolutionary rate profiling score, gnomAD: Genome Aggregation Database, IGSR: International Genome Sample Resource, KRGDB: Korean Reference Genome Database, LP: likely pathogenic, P: pathogenic, UR: unreported. 1 Reference amino acid sequence: NP_055178.3. 2 In silico scores of Mutation taster (MutT)> 0.5, Rare Exome Variant Ensemble Learner (REVEL) > 0.5, PROVEAN (PRO) < -2.5, and MUpro (MU) < 0 indicate pathogenic prediction (* denotes a pathogenic prediction).
Table 3.
Prediction of protein stabilities by the missense mutations.
| Mutation | Protein stability prediction tools 1 | ||
|---|---|---|---|
| PremPS | MAESTROweb | DynaMut2 | |
| p.F3633V | 0.68* | 0.283* | -0.73* |
| p.P1380A | 0.91* | 0.121* | -1.10* |
1 Protein stability scores of PremPS > 0, MAESTROweb > 0, and DynaMut2 < 0 indicate instability mutations (* denotes an instability prediction).
Table 4.
Clinical manifestations of the CMT patients with SACS mutations.
| Family ID | FC591 | FC937 | FC1157 | FC1176 | ||
|---|---|---|---|---|---|---|
| Patients | III-1 | III-2 | II-1 | II-2 | II-4 | II-1 |
| Mutations | p.S656Ffs*1 + p.P1380A |
p.V815Gfs*2 + p.F3633V |
p.D968Vfs*13 + p.T4406Rfs*45 |
p.Y532X+ p.F1054X | ||
| Sex | M | M | F | F | M | M |
| Examined age (yrs) | 35 | 33 | 27 | 26 | 25 | 22 |
| Onset age (yrs) | 4 | 5 | 15 | 17 | 15 | 3 |
| Muscle weakness | ||||||
| Upper limb (MRC) | ||||||
| Proximal (Rt/Lt) 1 | 4+/4+ | 4+/4+ | 4/4 | 4+/4+ | 4+/4+ | 4/4 |
| Distal (Rt/Lt) 2 | 4+/4+ | 4+/4+ | 4/4 | 4+/4+ | 4+/4+ | 4/4 |
| Lower limb (MRC) | ||||||
| Proximal (Rt/Lt) 3 | 4/4 | 4+/4+ | 4-/4- | 4+/4+ | 4/4 | 4/4 |
| Distal (Rt/Lt) 4 | 4/4 | 4+/4+ | 4-/4- | 4+/4+ | 4/4 | 4/4 |
| Muscle atrophy | Mild | Minimal | Moderate | Minimal | Mild | Mild |
| Sensory disturbance | Yes | Yes | Yes | Yes | Yes | Yes |
| DTR | ||||||
| Biceps jerk reflex | ++ | ++ | +++ | +++ | +++ | +++ |
| Knee jerk reflex | +++ | +++ | +++ | +++ | +++ | +++ |
| Disability score | ||||||
| FDS | 2 | 2 | 4 | 2 | 4 | 4 |
| CMTNSv2 | 13 | 11 | 24 | 17 | 22 | 23 |
| Pyramidal sign | Yes | Yes | Yes | Yes | Yes | Yes |
| Spasticity | Yes | Yes | Yes | Yes | Yes | Yes |
| Cerebellar ataxia | Yes | Yes | Yes | Yes | Yes | Yes |
| Dysarthria | No | No | Yes | Yes | No | Yes |
| Nystagmus | Yes | Yes | Yes | Yes | Yes | Yes |
| Foot deformity | Yes | Yes | Yes | Yes | Yes | Yes |
| Intellectual disability | No | No | No | No | No | No |
| Hearing loss | Yes | Yes | Yes | Yes | Yes | Yes |
| Electrophysiology | ||||||
| Nerve conduction | HMSN | HMSN | HMSN | HMSN | HMSN | HMSN |
| BAEP | NA | NA | Abnormal | Abnormal | NA | Abnormal |
| Brain MRI | NA | NA | NA | Cerebellar atrophy | Cerebellar atrophy | Cerebellar atrophy |
Abbreviations: BAEP: brainstem auditory evoked potential, CMT: Charcot-Marie-Tooth disease, CMTNSv2: CMT neuropathy score version 2, DTR: deep tendon reflex, FDS: functional disability scale, HMSN: hereditary motor and sensory polyneuropathy (axonal polyneuropathy), Lt: left side, NA: not available, MRC: Medical Research Council scale, Rt: right side. 1 Proximal (upper limb): elbow flexion MRC grade, 2 distal (upper limb): finger abduction MRC grade, 3 proximal (lower limb): knee flexion MRC grade, 4 distal (lower limb): ankle dorsiflexion MRC grade.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



