Submitted:
11 May 2024
Posted:
13 May 2024
You are already at the latest version
Abstract
New furan, thiophene and triazole oximes were synthesized through several-step reaction paths to investigate their potential for development of central nervous systems (CNS)-active and cholinesterase-targeted therapeutics in organophosphorus compound (OP) poisonings. Acute OP poisoning is still a challenge in treating patients despite the development of a large number of oxime compounds that should have the capacity to reactivate acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). The activity of these two enzymes, crucial for neurotransmission, are blocked by the OP, which has the consequence of disturbing normal cholinergic nerve signal transduction in the peripheral and CNS, leading to cholinergic crisis. Oximes in use have one or two pyridinium rings and cross the brain–blood barrier poorly due to the quaternary nitrogen. Following our recent study on 2-thienostilbene oximes, in this paper we described the synthesis of 63 heterostilbene derivatives out of which 26 oximes were tested as inhibitors and reactivators of AChE and BChE inhibited by OP nerve agents – sarin and cyclosarin. While the majority of oximes were potent inhibitors of both enzymes in micromolar range, we identified several oximes as BChE or AChE selective inhibitors with a potential for drug development. Moreover, even oximes were poor reactivators of AChE, four heterocyclic derivatives reactivated the cyclosarin-inhibited BChE up to 70%, and cis,trans-5 [2-((Z)-2-(5-((E)-(hydroxyimino)methyl)thiophen-2-yl)vinyl)benzonitrile] had comparable reactivation efficacy to the standard oxime HI-6. In silico analysis and molecular docking studies connected the kinetic data to the structural features of these oximes, and confirmed their productive interactions with the active site of the cyclosarin-inhibited BChE. Based on inhibition and reactivation and their ADMET properties regarding lipophilicity, CNS activity, and hepatotoxicity, these compounds could be considered for further development of CNS-active reactivators in OP poisoning as well as cholinesterase-targeted therapeutics in neurodegenerative diseases like Alzheimer’s and Parkinson’s.
Keywords:
cholinergic
; HI-6
; nerve agents
; reactivators
; Vilsmeier
; Wittig reaction
1. Introduction
The current therapy for acute exposure to organophosphates (OP) consists of a combination of active substances that includes an anticholinergic, a cholinesterase reactivator, and an anticonvulsant [1,2]. Oximes, reactivators of enzyme acetylcholinesterase (AChE), possess the C=N-OH group that, by a nucleophilic attack on the electrophilic phosphorus atom, breaks the bond between phosphorus and the catalytic serine of AChE, resulting in a free enzyme and a phosphorylated oxime. It should be emphasized that although oximes are of great importance in adequate therapy for poisoning with OP compounds, the oximes in today's use have a limited effect, which is defined by their structure and the structure of the OP-cholinesterase conjugate. Reference or standard oximes are usually specific to individual OP compounds and are not broad-spectrum [3,4,5,6,7]. In addition to the lack of universal access, another problem with reference oximes is their pharmacological properties, which are defined by their rapid dissociation in the body into ionized form [8]. Hydrophilic oximes with a charged nitrogen atom hardly pass through the blood-brain barrier (BBB) to central synapses. The catalytic serine of BChE undergoes the same phosphylation reactions as AChE in the presence of OP compounds. However, different kinetic properties of cholinesterases, due to the different structure of the active sites, affect the ability and speed of inhibition of cholinesterase by certain OP compounds. Previous research of therapy for poisoning with OP compounds was based on finding effective reactivators of inhibited AChE, and oxime structures were designed to achieve the best possible interaction with the phosphorylated active site of AChE, i.e. its reactivation. Consequently, most oximes developed to restore the activity of OP-AChE conjugates have been shown to be nearly ineffective in restoring OP-BChE activity. Moreover, although all of the reactivators tested so far showed a somewhat successful recovery of AChE catalytic activity inhibited by individual OP compounds [9,10], no oxime was an universal reactivator of various OP-AChE conjugates.
To find new oximes that would retain excellent biochemical properties, great attention was given to trihydroxy stilbene resveratrol, and at the same time to heterocyclic compounds in general. Trans-resveratrol is known for a very wide range of biological activities [11], among which its participation in mechanisms in the pathology of Alzheimer's disease (AD) should be emphasized in this context [12,13]. On the other hand, most therapeutics possess at least one heterocyclic ring in their structure [14,15,16]. Applying rational design principles, we strategically incorporated the furan, thiophene or triazole ring into the resveratrol scaffold and explored different combinations of substituents and their positions to enhance their potential for development of CNS-active and cholinesterase-targeted therapeutics. The existence of a double bond on the (hetero)stilbene subunit opens up the possibility of different configurational isomers, which together with diverse configuration of the oxime group result in different properties.
In our previous research [17], uncharged thienostilbene oximes were synthesized, and characterized as reactivators of AChE and BChE inhibited by OP nerve agents. Four trans-derivatives reactivated cyclosarin-inhibited BChE up to 70% within two hours of reactivation. Docking studies confirmed their constructive interactions with the active site of cyclosarin-inhibited BChE, while in silico evaluated ADME properties pointed to a new class of oximes with the potential for further development of CNS-active therapeutics in OP poisoning. Building on those promising results, in this study, a number of new uncharged oxime derivatives were synthesized by a series of reactions in order to examine more deeply the influence of the heterocyclic nucleus, the position and type of substituents on the nucleus, and the position and configuration of the oxime group on their cholinesterase reactivation potential.
The general skeleton of new compounds is shown on Figure 1. Further on, we evaluated their physical-chemical properties important for BBB penetration in silico and cytotoxicity in vitro. Oximes were tested as inhibitors of human AChE and BChE, and as reactivators of their activities upon inhibition with OP nerve agents - sarin and cyclosarin. In order to rationalize the positive interaction between the tested compounds and cholinesterases (ChE), we performed molecular docking of reversible enzyme-ligand complex and modelling of the near-attack conformation for the most potent reactivators of BChE inhibited by cyclosarin.
2. Results and Discussion
2.1. Synthesis of New Uncharged Oximes 1–21
Following the previously synthesized uncharged thienostilbene oximes as potential cholinesterase reactivators [17], a number of new oximes 1–21 were synthesized starting from the prepared phosphonium salts and corresponding aldehydes. The new oximes 1–3 (Figure 2) have the oxime group directly attached to the triazole ring and without the presence of the central double bond in the skeleton, while the other derivatives belong to heterostilbenes with an oxime group on the thiophene (4, 5, 7, 8, 18–21) or furan (6, 9–17) ring (Figure 3).
Triazole oximes 1–3 were obtained (in 8-50% of isolated yields) from the triazole nitro aldehyde 24 (Scheme 1) either by direct conversion into an oxime (compound 3) or by conversion of nitro aldehyde in a reaction with amine into new substituted triazole aldehydes 22 and 23, and then by their conversion into an oxime (compounds 1 and 2). This synthesis involves the corresponding starting amines in reaction with triazole nitro aldehyde 24 [18,19] to produce new triazole aldehydes 22, 23 and 25 (see Experimental Section) as starting substrates for the Wittig reaction.
Scheme 1, Scheme 2, Scheme 3 and Scheme 4 show the synthetic route to the new uncharged oximes 4–21. The preparation of the desired heterostilbene oximes 4–21 was achieved according to the three-step reaction path initiated by forming a double bond using the Wittig reaction to yield the starting compounds 45–63 as mixtures of cis- and trans-isomers (11–97%) except in the case of 45, 45’, 47, 50 and 55 where only a trans-isomer was formed, or for the heterostilbene 49 when only a cis-isomer was obtained. The Wittig reaction was performed using commercially available carbaldehydes, and the selected phosphonium salts prepared in our laboratory. In the second step, the obtained heterostilbenes 45–63 represented the basic building blocks subjected to the Vilsmeier formylation reaction, providing the corresponding aldehydes 25–44 (7–98%) as mixtures of isomers. In this reaction, POCl3 and DMF initially formed a chloriminium ion, a Vilsmeier reagent, which then reacted with the heterostilbene molecule, and an iminium ion was formed and hydrolysed to the final aldehydes 25–44. The trans-isomers of 45–63 reacted more successfully in the formylation carried out for the partially unreacted cis-isomers, and thus the proportion of trans-isomers of aldehydes 25–44 was usually higher in comparison to those for the cis-isomers in the reaction mixture after formylation. For aldehyde trans-30, the configuration inversion was happened in the formylation reaction starting from cis-49.
In the third step, formyl derivatives were transformed to oximes 4–21 as the targeted structures in a very wide range of isolated yields (2–98%) and with different proportions of individual configurational isomers. Mixtures of the geometrical isomers of oximes 4–21 were separated to obtain pure compounds by repeated column chromatography. The reaction of converting the synthesized aldehydes 25–44 into the corresponding oximes 4–21 involved the use of NH2OH × HCl and a mixture of ethanol and water as solvents and was based on the reaction described in the literature on simpler heteroaromatic systems [20]. According to 1H NMR spectroscopy, the nature and position of the substituent and the oxime group, as well as the nature of the heterocyclic ring, directed the ratio of configuration both on the double C=C bond and the configuration of the C=N bond of heterostilbene oximes 4–21 (Figure 3). The configuration on the double bonds is based exclusively on the proton chemical shift.
After successive column chromatography, the trans,anti-, trans,syn-, cis,anti- and cis,syn-isomers of individual oxime derivatives 4–21 isolated at a sufficient quantity were subjected to further examination as potential cholinesterase inhibitors and reactivators. All isolated oximes 1–21 were successfully spectroscopically characterized (See Experimental Section 4.1. and Supporting Data). Pure isolated isomers of a single oxime or a mixture of isomers were tested if there were too few pure isomers of a single oxime separated (as in the case of oximes 1–3, 7, 8 and 13). For some of the isolated configurational isomers, there was enough mass only for spectroscopic characterization. In the case when it was not possible to separate all of the isomers, data for individual isomers (if they were formed and clearly seen in the 1H NMR spectra) were listed from the spectra of oxime mixtures.
2.2. Prediction of ADME Properties
Chemical absorption, distribution, metabolism, excretion (ADME) including toxicity play key roles in drug discovery and development. A high-quality drug candidate should not only have sufficient efficacy against the therapeutic target, but also show appropriate ADME properties at a therapeutic dose. For potential AChE and BChE targeted therapeutics, it is important to have good absorption and high potential to cross the BBB to achieve its biological activity on AChE and BChE in brain [21,22].
Therefore, for the 26 prepared oxime compounds we predicted lipophilicity (AlogP) and polar surface area (PSA) based on molecular weight (MW), the number of hydrogen bond donors and acceptors (HBDs and HBAs), and number of rotating bonds (RBs) estimated their BBB-penetration ability and compared to recommended values of CNS-active drugs (Figure 4) [23]. An examination of the radar and AlogP and 2D PSA correlation plot reveals a high potential of new uncharged oximes to cross the BBB. An exception are the ADME properties of compounds 3 and trans-8 that seem to have larger PSAs than the recommended value for a CNS-active drug. However, as Lipinski’s rule allows one exception [24], having in mind their high absorption, these compounds can be considered potentially orally CNS-active compounds.
A high lipophilicity implied low solubility in preferential phosphate buffers for cholinesterases, so additional attention was given to the impact of the solvent on cholinesterase activity [25]. In our recent study we reported natural deep eutectic solvent (NADES) as an alternative solvent for preparation of CNS-active oximes [26], which is capable not only increase the solubility, but also improve oxime properties in interactions with AChE. Indeed, whether a molecule is going to provide a satisfying therapeutic effect depends on how it interacts with the molecular target (pharmacodynamic aspect), but also on how it is processed by the body (pharmacokinetic aspect). Therefore, the search for molecules with better pharmacodynamics and pharmacokinetic characteristics is still an ongoing pursuit with focus on the CNS-active cholinesterase-targeted therapeutics [27]. It is worth mentioning that CNS-active AChE reactivators - zwitterionic oxime RS194B [28,29] and 3-hydroxy-2-pyridine reactivator JR595 [30,31], showed an improvement in BBB penetration compared to standard oximes. However, the challenge seems to be a quick elimination, which could be solved with multiple oral application. Therefore, it is important to note that in silico prediction tests showed that our new compounds possess high bioavailability and are suitable for multiple oral administration to increase and maintain a therapeutic dose of compounds, as well as increasing their residency in the brain.
2.3. Cytotoxicity of New Uncharged Oximes
To estimate the impact of the prepared compounds on cellular homeostasis, we evaluated 24-hour cytotoxicity by measuring the succinate dehydrogenase mitochondrial activity in an exposed liver cell line (HepG2), one of the standard cell-lines for metabolism and drug-safety evaluations (Figure S299). It is worth highlighting that these results also show the effects of the degradation products of compounds, which are expected to form in a 24 h-long assay. The cytotoxic effect of the new uncharged oximes in terms of IC50 is summarized in Table 1. Out of the 26 tested compounds, the IC50 values of 17 oximes were estimated to be higher than 400 μM categorizing those oximes as likely non-hepatotoxic compounds. Although nine oximes exhibited a toxic effect within the tested concentration range, only for compounds cis,syn-16, trans,syn-10, cis,syn-21 and cis,syn-17 IC50 values were equal to or lower than 100 µM demonstrating a relatively high hepatotoxicity.
It seems there is a prevalence of cis-derivatives over trans-derivatives, since 9 out of 10 cis-oximes exhibited hepatoxicity. In accordance with recent studies the mechanism of toxicity depended on the structure of oximes and variations in the structure significantly altered the toxic effect on the cells [32,33,34]. It is important to mention that these studies on various cell types representing the liver, kidney, neurons and muscles revealed that cytotoxicity was not dependent on cell type and was induced by different mechanisms/types of the regulated cell death activation by caspase-dependent apoptosis or cell death [32,33,34]. More precisely, pyridinium oximes (IC50 10–300 µM) induced apoptosis by an intrinsic mitochondria-dependent pathway through the activation of specific caspase 9, while imidazolium oximes (IC50 120–320 µM) triggered necrotic events characterized by elevated levels of reactive oxygen species, loss of mitochondrial potential and uncontrolled intracellular lactate dehydrogenase leakage accompanied by cell burst [33]. Biological response and cell death signalling pathways modulated by uncharged 3-hydroxy-2-pyridine aldoximes with tetrahydroisoquinoline moieties (IC50 8–120 µM) showed time-dependent effects and stimulated mitochondria-mediated activation of the intrinsic apoptosis pathway through ERK1/2 and p38-MAPK signalling and subsequent activation of initiator caspase 9 and executive caspase 3 accompanied with DNA damage, as observed already after 4 h of exposure. Mitochondria and fatty acid metabolism were also likely targets of 3-hydroxy-2-pyridine aldoximes with tetrahydroisoquinoline moiety, due to increased phosphorylation of acetyl-CoA carboxylase [34].
2.3. Reversible Inhibition of Cholinesterase by New Uncharged Oximes
We can generally emphasize that new heterocyclic oximes bind to both AChE and BChE in a reversible inhibitor manner in the micromolar range. The inhibition potency in terms of inhibition dissociation constants (Ki) is given in Table 2. Due to poor solubility and poor enzyme binding affinity, Ki constants could not be determined for 8 compounds, while only 3 of them – trans-13, trans,anti-15, and cys,syn-19’, were poor inhibitors of both human AChE and BChE. BChE showed the highest affinity (1/Ki) for 2,5-substituted furan oximes trans,anti-4 and cis,syn-5, and triazole oxime 2. Interestingly, in the case of AChE, heterostilbenes trans,syn-18, trans,syn-17, and cis,syn-12 – with the syn configuration on the oxime group, and the substituent in para-position, were the most potent inhibitors, although about 10-fold lower than the most potent BChE inhibitor, trans,anti-4.
Moreover, the most potent AChE inhibitors were poor inhibitors of BChE and vica versa (with the exception of cys,syn-5 and trans,syn-10, which were potent inhibitors of both enzymes). Therefore, we can discuss these results in terms of inhibition selectivity as the Ki ratio between the two enzymes. In addition, BChE inhibitor trans,anti-4, which had the lowest Ki, was identify as a potent and selective inhibitor of BChE. These results should encourage further tests of these compounds as potential therapeutics of neurological disorders, as both non-selective or BChE-selective inhibitors are highlighted as potential drugs in AD and other neuromuscular disorders [35,36,37,38]. More precisely, both AChE and BChE are targets in drug-development for symptomatic treatment of Alzheimer’s disease (AD), e.g. galantamine and donepezil, which are currently in use, are AChE inhibitors [36]. However, since BChE activity in certain brain regions increased by AD progression, BChE inhibitors became additional targets in the development of drugs for AD and related dementias [35].
While in our previous paper eight 2-thienostilbene oxime derivatives were reported as reversible inhibitors of human AChE and BChE with Ki constants also in micromolar range, seven of the oximes were more potent inhibitors of AChE (4–80 µM) than of BChE (44–573 µM) [17]. In addition, inhibition of AChE was uncompetitive, while all of the compounds exhibited competitive binding for BChE, which is quite opposite for the studied heterocyclic oximes. Along with somewhat mixed findings – out of the 26 compounds, nine and eleven oximes were competitive, while six and five were uncompetitive inhibitors in case of AChE and BChE, respectively – all BChE potent inhibitors showed an uncompetitive character of inhibition meaning that potency of inhibition did not depend on the concentration of substrate acetylthiocholine (Figure S300). It is also important to highlight that these uncharged heterostilbenes identified as the most potent inhibitors of AChE and BChE possess binding affinity within the range of charged oximes [31,39,40,41,42,43,44]. Therefore, our results confirmed a previous study on (thiophen-2-yl)-aldoximes that reported that the hetero atom, due to its potential for polarization, helps to stabilize the negative charge of its anionic form, similarly to the pyridinium ring of the standard oxime 2-PAM [20].
It is also very interesting to recapitulate our previous results on analogues of the well-known bioactive molecule resveratrol that underwent evaluation for antioxidant activity along with their potential to inhibit non-human AChE and BChE [45]. The biological tests have shown that the derivative with trans-configuration, similar to trans,anti-4, exhibited significant antioxidative and BChE inhibitory potential, as evidenced by lower IC50 values compared to the established standards, trans-resveratrol and galantamine, respectively.
2.4. Molecular Modelling of Heterostilbene Oximes−ChE Complex
To rationalise interactions between oxime molecule and amino acids lining the active site gorge, we performed docking to visualise possible binding interactions (Figure 5 and Figure S301). Several modes of binding in the ChE active site were observed. Due to the hydrophobic nature of the active site the main interactions for the stabilisation came from aromatic residues Phe, Tyr or Trp. Moreover, the active site of AChE prefers elongated ligands, which can simultaneously bind to the choline binding site and peripheral site resulting in higher inhibition potency [46]. In this study compounds with trans-configuration of double bond trans,syn-18 and trans,syn-17 were the most potent inhibitors of AChE (Table 2, Figure S301C,D). Molecular modelling showed stabilisation of substituted phenyl group via multiple hydrophobic interactions (Phe295, Phe297, Tyr337, Phe338 and His447) including the acyl binding site, while the oxime group attached to heteroaromatic ring was stabilised via H-bonds from the peripheral site Asp74 and Tyr341 or Thr83, Asn87 and Tyr341.
Compounds with a cis-configuration of the double bond cis,syn-5 and cys,syn-12 in human AChE showed similar stabilisation of the oxime group via multiple H-bonds from Asp74 and Tyr341 or Ser125 (Figure S301). Due to the bent shape of these molecules, the phenyl group is stabilised in the choline binding site away from the acyl binding site. Multiple hydrophobic interactions were included in the stabilisation of the cyano or chlorine substituted phenyl group, primarily from Trp86 and additionally Tyr337, and His447.
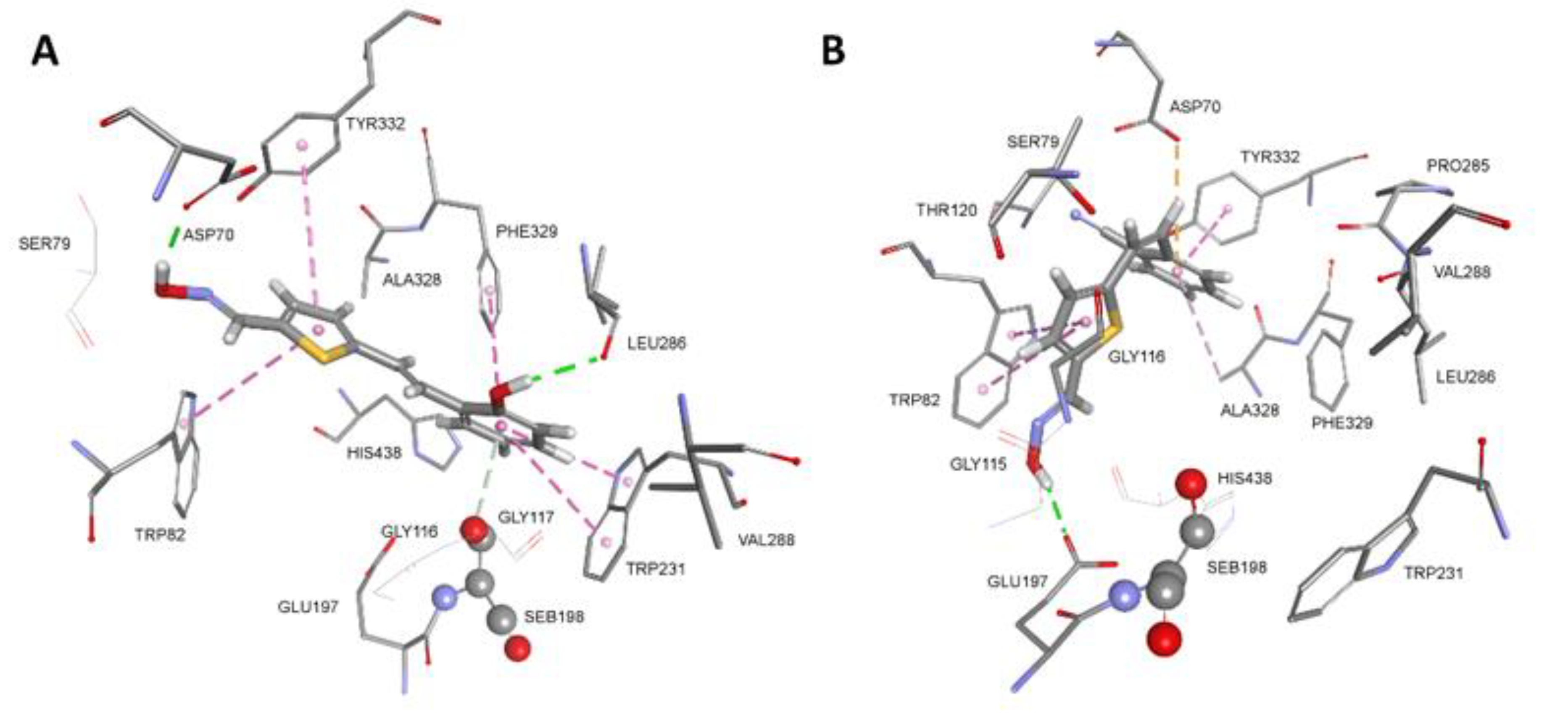
In human BChE, both thiostilbenes trans,anti-4 and cis,syn-5 seemed to bind simultaneously to the choline binding site (Trp82) and the peripheral binding site (Asp70, Trp332) (Figure 5). Strong stabilisation of trans,anti-4 occurs due to the formation of multiple hydrophobic interactions with Trp82, Trp231 and Phe329. Additional stabilisation came from the H-bond between Asp70 and the oxime group, and Leu286 and the hydroxyl group. Catalytic Ser198 also stabilises trans,anti-4 via electron-π interaction from the oxygen (Oγ) of Ser198. The thiostilbene ring of cis,syn-5 forms a π-π sandwich with the indole ring of Trp82 with additional stabilisation in the form of an H-bond between Glu197 and the oxime group. It is worth mentioning that the formation of a π-π sandwich between thiostilbene and the indole ring of Trp82 is analogous to the crystal structure of the triazole compound III bound in the human BChE active site (PDB code 6T9P) where the imidazole ring forms a π-π sandwich with Trp82 [47]. The cyano group at the phenyl ring is stabilised in the BChE peripheral binding site creating hydrophobic interactions with Tyr332 and Ala328 and an additional electrostatic π-anion interaction from Asp70. A similar binding mode was determined for triazole compound 2 that binds simultaneously to the choline binding site and acyl binding site of BChE (Figure S301E). The triazole ring bearing an oxime group is stabilised by Trp82 via hydrophobic interactions and with an additional H-bond from Gly115, a member of the oxyanion hole. Chlorine at the phenyl ring is stabilised via multiple hydrophobic interactions formed with Val288, Trp231, Phe332 including electrostatic electron-π interaction from the oxygen (Oγ) of Ser198. Our results on docking are in agreement with crystal structures of ligands showing that potent inhibitors of BChE simultaneously bind to the choline binding site and acyl pocket or peripheral binding site [48].
Figure 5.
Conformation of complex between native human BChE and trans,anti-4 (A) and cis,syn-5 (B). Interactions with amino acid residues are represented as dashed lines: hydrophobic (purple), hydrogen bonds (green) and electrostatic (orange). Crystal structure of human BChE was used (PDB code 2PM8) [49].
Figure 5.
Conformation of complex between native human BChE and trans,anti-4 (A) and cis,syn-5 (B). Interactions with amino acid residues are represented as dashed lines: hydrophobic (purple), hydrogen bonds (green) and electrostatic (orange). Crystal structure of human BChE was used (PDB code 2PM8) [49].
2.5. Oxime-Assisted Reactivation of AChE and BChE
The new heterostilbene and triazole were initially tested for reactivation of sarin- and cyclosarin-inhibited AChE and BChE at the given oxime concentration of 0.1 mM. Reactivation of AChE did not exceed 20% within 5 h in case of both sarin and cyclosarin. While for the reactivation of the sarin-inhibited BChE no oxime had the capability to return its activity, cyclosarin-inhibited BChE was promptly reactivated, higher than 20%, with 11 uncharged oximes and results were sorted in terms of the observed first-order reactivation rate (kobs) (Figure 6). Oxime cis,syn-16 exhibited the highest reactivation capabilities up to 80%, while cis,anti-21 was also able to conceive a high reactivation percentage of 75%, but with a slow reactivation rate. Evidently, the standard oxime HI-6 demonstrated the highest observed first-order reactivation rate, but not significantly higher than cis,syn-12, which reactivated with the highest kobs of all uncharged oximes, but the maximal reactivation was low (Figure 6). Along with cis,syn-16, oximes trans,anti-4, cis,syn-5, and trans,syn-10 reactivated BChE activity above 50% with moderately high reactivation rates about 4-fold higher than kobs determined for the 2-thiostilbenes previously [17]. It is worth mentioning that the inhibition of BChE with oximes which was uncounted in the reactivation assay corresponded to the affinity described above (cf. Table 2).
Based on initial screening, out of the 26 uncharged oximes, four oximes two 2,5-thienstilbenes trans,anti-4 and cis,syn-5, 2,3-furostilbene cis,syn-16, and 2,5-furanstilbene trans,syn-10 were singled out for further detailed kinetics reactivation of BChE inhibited by cyclosarin. The reactivation kinetic parameters were determined in a limiting concentration range (0.01–0.2 mM) due to poor solubility, and compared to standard reactivator HI-6 (Table 3). The reactivation of the cyclosarin-inhibited BChE with all of the selected oximes reached maximal reactivation in up to 140 minutes, where the highest reactivation of 80% was achieved in the presence of chlorinated 2,5-furanstilbene oxime, cis,syn-16. The thienstilbene compound containing a cyano group, cis,syn-5, exhibited the highest reactivation efficiency as a result of primarily relatively high binding affinity, even surpassing that of standard HI-6 [30]. Indeed, cyclosarin-conjugated BChE probably had low binding affinity for the other tested oximes, indicated with a linear relationship between kobs and oxime concentration enabling us to determine the overall reactivation constant only.
Nevertheless, our reactivation study confirms that although both AChE and BChE share the same mechanism of reaction, the structure of OP moiety conjugated at the catalytic serine and difference in residues in the active site direct the specificities of AChE and BChE reactivation, and therefore, no single oxime, reported in the literature, is equally efficient against a variety of OP and potent for both AChE and BChE [30,50]. It is worth mentioning here that, with design of these uncharged oximes, we joined the endeavour undertaken by several research teams to develop a new generation of CNS-active reactivators [9,51,52,53,54,55,56,57,58,59,60,61,62,63,64].
2.6. Modelling of a Complex between an Oxime and Cyclosarin-Inhibited BChE
Since molecular docking fails in modelling of a near-attack conformation of inhibited ChE by OPs, we performed an alternative approach using the structure of a reactivation product, a phosphorylated oxime [65]. The complex between oxime cis,syn-5 or cis,syn-16 and cyclosarin-inhibited BChE representing the near-attack conformation is given in Figure 7. The following van der Waals distances were obtained from the oxime group oxygen to the phosphorus atom of the phosphoester conjugate: 3.8 Å and 3.3 Å for cis,syn-5 and cis,syn-16, respectively. It is very obvious that the near-attack conformation was stabilised in different binding pose with less interactions from BChE active site residues than the reversible complex representing inhibition (cf. Figure 5B and S2). Compound cis,syn-5 was stabilised via electron-π interaction involving sulphur and Phe329 and an additional H-bond from Tyr332. The presence of the cyclohexyl ring in the active site blocked the interaction of cis,syn-5 with Trp82 but stabilised position of the cyano substituted phenyl ring (cf. Figure 5B). Multiple hydrophobic interactions were formed between the para-chlorinated phenyl group and mainly aromatic residues of active site (Phe329, Tyr332, Trp430) and Ala328 and Met437. This type of binding and chlorine interactions were analogues to the crystal structure of a chlorinated tacrine derivate, a potent cholinesterase inhibitor, bound to BChE (PDB code 6I0B) [66]. Stronger stabilisation of cis,syn-16 may cause lower reactivation potency compared to cis,syn-5 due to less freedom for the movement required for the nucleophilic attack of the oxime group.
3. Materials and Methods
3.1. Chemistry
All nuclear magnetic resonance (NMR) spectroscopic data for 1H and 13C were recorded in deuterated chloroform, CDCl3 and deuterated methanol CD3OD using tetramethylsilane as a standard at room temperature on Bruker Avance 300 and 600 MHz spectrometers. For the full characterization of the targeted oximes, the additional techniques 2D-CH correlation (HSQC) and 2D-HH-COSY were used. The following abbreviations were used in the NMR spectra: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets) and m (multiplet). Chemical shifts were reported in parts per million (ppm). High-resolution mass spectrometry (HRMS) analyses were carried out on a mass spectrometer (MALDI TOF/TOF analyzer) equipped with an Nd:YAG laser operating at 355 nm with a fitting rate of 200 Hz in the positive (H+) or negative (-H) ion reflector mode. All of the compounds tested for reversible inhibition and oxime-assisted reactivation of cholinesterases were >95% pure by High Resolution Mass Spectrometry (HRMS) or HPLC analyses (see Supplemental Information). The results of HRMS and HPLC analyses are included in the Supporting Information. All used solvents for the synthesis were purified by distillation and were commercially available. Anhydrous magnesium sulfate, MgSO4, was used for drying organic layers after extractions. The column chromatography was performed on columns with silica gel (Fluka 0.063–0.2 nm and Fluka 60 Å, technical grade) using the appropriate solvent system. Abbreviations used in experimental procedures were ACN – acetonitrile, EtOAc – ethyl acetate, PE – petroleum ether, E – diethylether, EtOH – ethanol, DCM – dichloromethane, DMF – dimethylformamide, POCl3 – phosphoryl chloride, NaOH – sodium hydroxide. All solvents were removed from the solutions by rotary evaporator under reduced pressure. The initial phosphonium salts were prepared in the laboratory from the corresponding bromides, while the other starting compounds used were purchased chemicals.
Heterostilbenes 45–63 were obtained as mixtures of cis- and trans-isomers ((11–97%) except in the case of 45, 45’, 47, 50 and 55 where only a trans-isomer was formed, or for the heterostilbene 49 when only a cis-isomer was obtained) using Wittig reaction. The reaction apparatus was purged with N2 for 15 min before adding the reactants. The reactions were carried out in three-necked flasks (100 mL) equipped with a chlorine-calcium tube and an N2 balloon connected. Phosphonium salts (5 mmol) were added to the 40 mL of EtOH, and mixtures were stirred with a magnetic stirrer. Solution of sodium ethoxide (5 mmol, 1.1 eq of Na dissolved in 10 mL of absolute ethanol) was added in strictly anhydrous conditions under N2 dropwise. The corresponding aldehydes (5 mmol) were then added to the reaction mixtures, which were allowed to stir for 24 h at room temperature. The reaction mixtures were evaporated on a vacuum evaporator and dissolved in toluene. Products were then extracted with toluene (3 × 15 mL). The organic layers were dried under anhydrous MgSO4. Products 45–63 were isolated by repeated column chromatography on silica gel using PE/E, PE/DCM, and E/EtOAc solvent systems. The first isomer to eluate was cis-isomer (but in some cases was not isolated), and the trans-isomer was isolated in the last fractions. The spectroscopic characterization of new isolated heterostilbenes is given below.
Compound trans-45 was converted to the aldehyde by Vielsmeier formylation, however, it did not give the product. After that, protection of its OH group was carried out in a round flask (25 mL) using acetic anhydride (1.5 mL) at room temperature overnight in pyridine. After that, a mixture of water, toluene and acetone (1:3:3) was added, and the reaction mixture was evaporated under reduced pressure, and a yellow solid remained in the flask (trans-45').
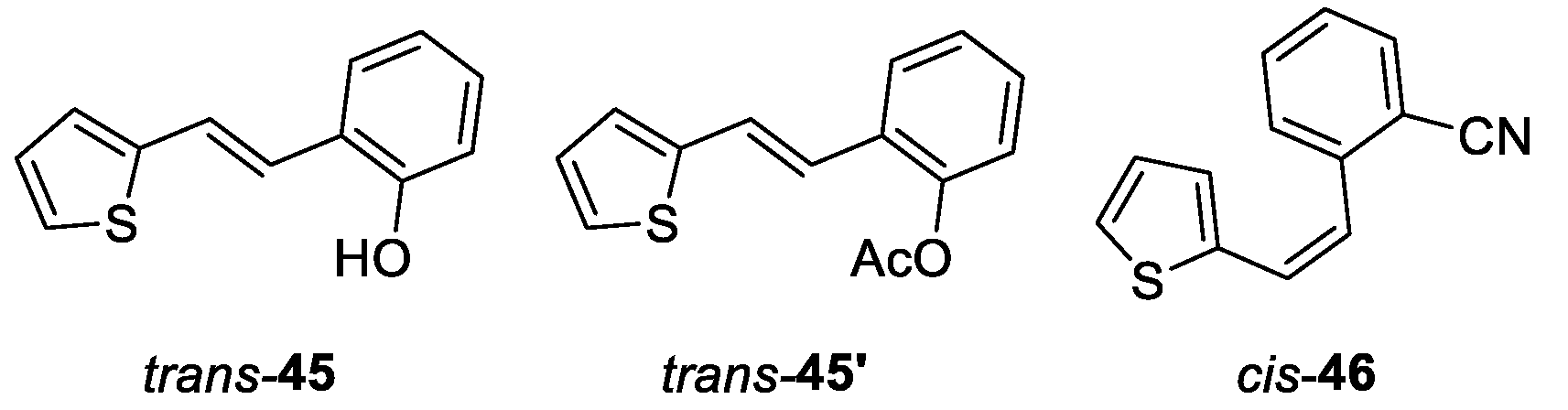
(E)-2-(2-(thiophen-2-yl)vinyl)phenol (trans-45) [45] 495 mg, 82% isolated yield; white powder; Rf (PE/DCM (50%)) = 0.33; UV (ACN) λmax/nm (ε/dm3mol-1cm-1) 335 (27412); 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.46 (dd, J = 7.7, 1.6 Hz, 1H), 7.28 (d, J = 16.1 Hz, 1H), 7.20 – 7.16 (m, 2H), 7.10 (t, J = 7.4 Hz, 1H), 7.07 (d, J = 3.7 Hz, 1H), 6.99 (dd, J = 5.1, 3.5 Hz, 1H), 6.93 (t, J = 7.5 Hz, 1H), 6.78 (dd, J = 8.1, 1.2 Hz, 1H), 4.97 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 152.9, 143.3, 128.6, 127.6, 127.2, 126.0, 124.4, 124.3, 123.3, 122.7, 121.2, 115.9; MS (ESI) (m/z) (%, fragment): 202 (25), 105 (100).
(E)-2-(2-(thiophen-2-yl)vinyl)phenyl acetate (trans-45’): 286 mg, 95% isolated yield; colorless oil; Rf (DCM) = 0.75; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.62 (dd, J = 7.6, 1.5 Hz, 1H), 7.28 – 7.20 (m, 4H), 7.08 – 7.06 (m, 2H), 7.00 (dd, J = 5.2, 3.7 Hz, 1H), 6.94 (d, J = 16.1 Hz, 1H), 2.37 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 169.3, 148.1, 142.7, 129.6, 128.4, 127.6, 126.6, 126.3, 124.7, 124.0, 122.8, 122.8, 121.5, 20.9.
(Z)-2-(2-(thiophen-2-yl)vinyl)benzonitrile (cis-46): 350 mg, 50% isolated yield; colorless oil; Rf (DCM) = 0.65; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.71 (d, J = 7.7 Hz, 1H), 7.57 – 7.53 (m, 2H), 7.42 – 7.39 (m, 1H), 7.12 (d, J = 5.8 Hz, 1H), 6.96 (d, J = 3.6 Hz, 1H), 6.93 (d, J = 12.1 Hz, 1H), 6.90 (dd, J = 4.9, 3.6 Hz, 1H), 6.62 (d, J = 12.1 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 182.9, 147.6, 143.5, 140.0, 135.9, 133.5, 133.1, 130.3, 129.7, 128.9, 128.8, 126.4, 117.4, 112.3.
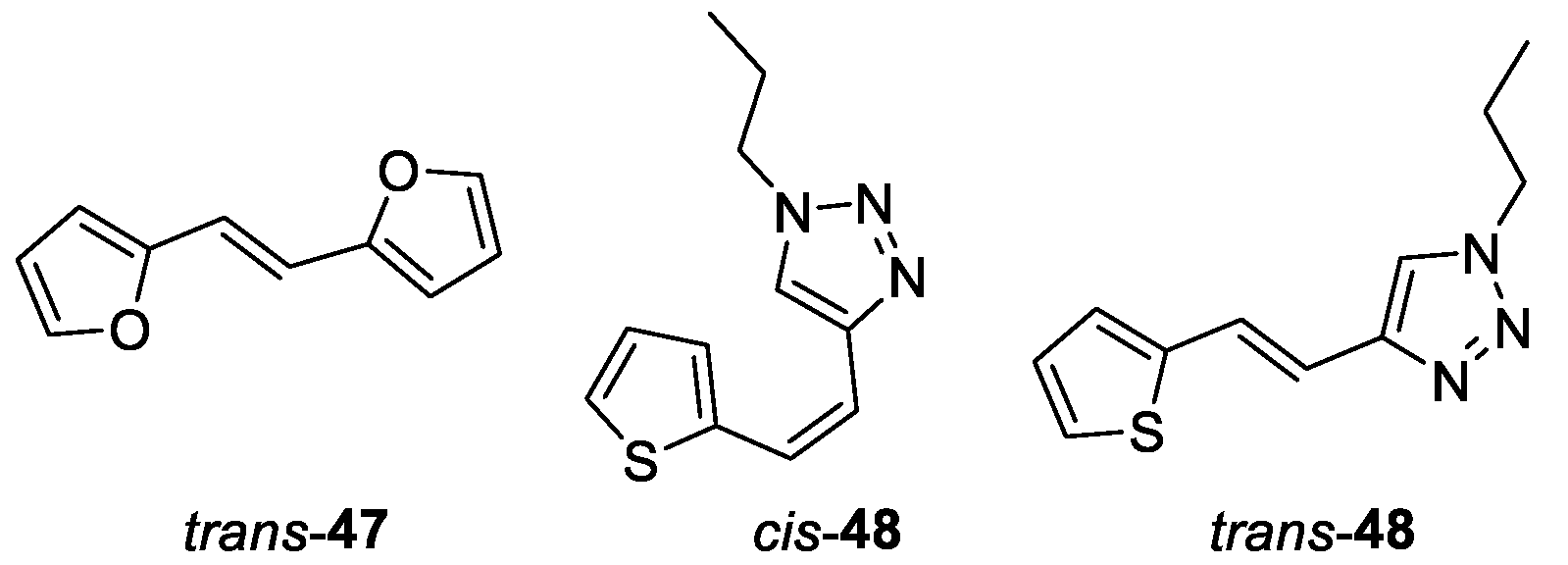
(E)-1,2-di(furan-2-yl)ethene (trans-47): 93 mg, 20% isolated yield; colorless powder; Rf (PE) = 0.58; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.42 (d, J = 17.7 Hz, 1H), 7.34 (d, J = 17.7 Hz, 1H), 6.81 (t, J = 19.4 Hz, 2H), 6.42 – 6.40 (m, 2H), 6.32 (d, J = 3.2 Hz, 2H).
(Z)-1-propyl-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole ( cis-48) [68]: 110 mg, 48% isolated, colorless oil; Rf (PE/E (40%)) = 0.34; UV (ethanol, 96%) λmax/nm (ε/dm3mol-1cm-1) 296 (16418), 310 (14455, sh); 1H NMR (CDCl3, 300 MHz) δ /ppm: 7.53 (s, 1H), 7.25 (d, J = 4.7 Hz, 1H), 7.22 (d, J = 3.7 Hz, 1H), 6.99 (dd, J = 4.9, 3.4 Hz, 1H), 6.70 (d, J = 12.1 Hz, 1H), 6.54 (d, J = 12.1 Hz, 1H), 4.27 (t, J = 14.2 Hz, 3H), 1.93 – 1.87 (m, 2H), 0.94 (t, J = 14.6 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ /ppm: 143.8, 139.5, 128.5, 127.1, 126.0, 123.8, 122.1, 118.3, 51.9, 23.74, 11.1.
(E)-1-propyl-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole ( trans-48) [69]: 20 mg, 10% isolated; white powder; Rf (PE/E (40%)) = 0.29; UV (ethanol, 96%) λmax/nm (ε/dm3mol-1cm-1) 314 (30210); 1H NMR (CDCl3, 600 MHz) δ /ppm: 7.51 (s, 1H), 7,44 (d, J = 16.2 Hz, 1H), 7.20 (d, J = 8.2 Hz, 1H), 7.07 (d, J = 3.5 Hz, 1H), 7.00 (dd, J = 5.1, 3.6 Hz, 1H), 6.89 (d, J = 16.2 Hz, 1H), 4.33 (t, J = 7.2 Hz, 2H), 1.95 (m, J = 7.3 Hz, 2H), 0.98 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ /ppm: 145.2, 141.7, 127.1, 125.9, 124.2, 123.1, 119.6, 115.7, 51.4, 23.2, 10.6.
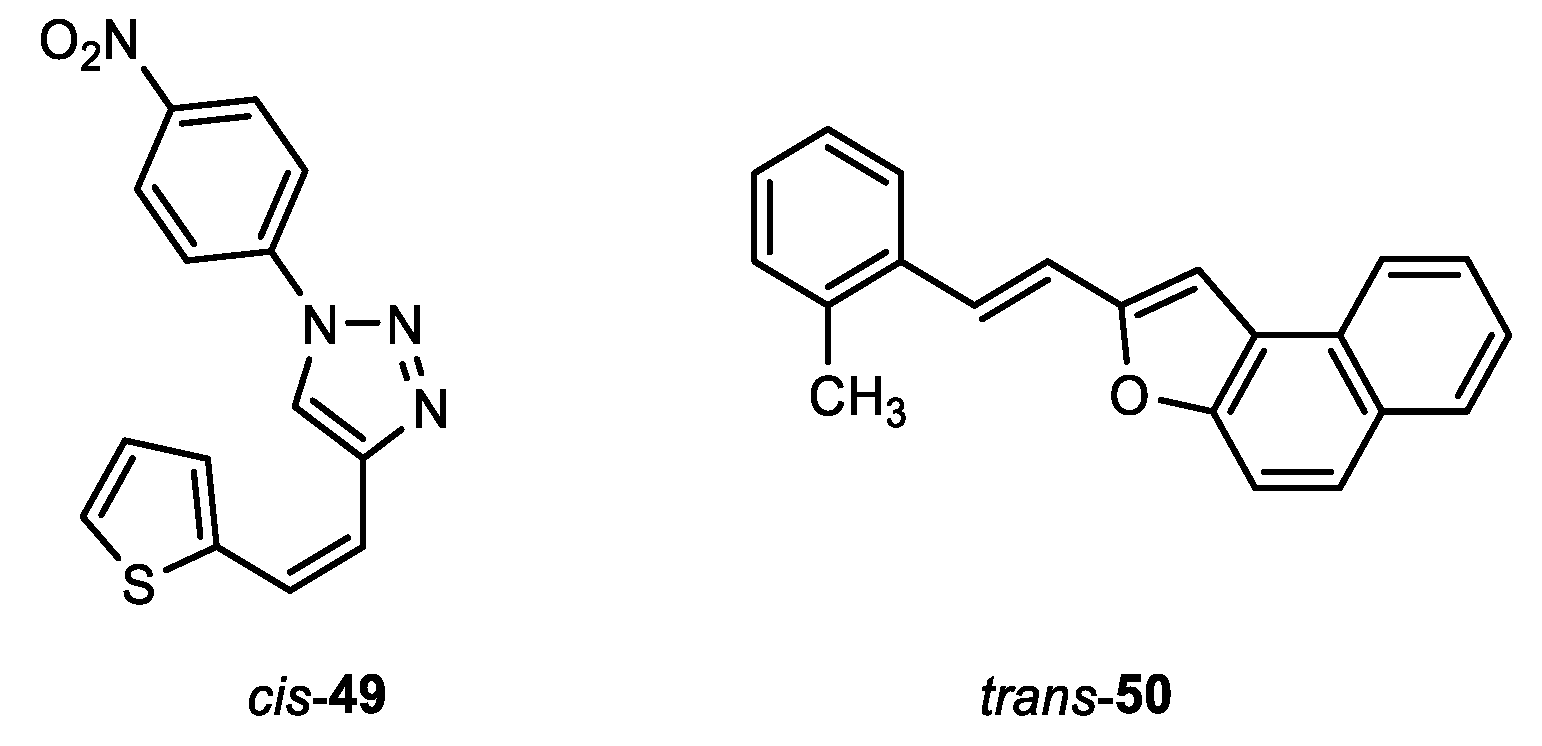
(Z)-1-(4-nitrophenyl)-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole (cis-49): 85 mg, 42% isolated yield; yellow oil; Rf (E) = 0.85; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.41 (d, J = 8.7 Hz, 2H), 8.10 (s, 1H), 7.94 (d, J = 8.9 Hz, 2H), 7.32 (d, J = 5.5 Hz, 1H), 7.28 (d, J = 3.3 Hz, 1H), 7.06 (t, J = 4.1 Hz, 1H), 6.86 (d, J = 12.2 Hz, 1H), 6.59 (d, J = 12.2 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 147.2, 145.3, 141.5, 138.9, 129.2, 127.3, 126.6, 125.7, 125.6, 120.4, 119.8, 116.7.
(E)-2-(2-methylstyryl)naphtho[2,1-b]furan (trans-50): 210 mg, 95% isolated yield; yellow powder; Rf (PE/E (50%)) = 0.95; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.12 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.74 – 7.55 (m, 5H), 7.48 (t, J = 7.7 Hz, 1H), 7.25 – 7.18 (m, 4H), 7.01 (d, J = 16.2 Hz, 1H), 2.50 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 154.9, 152.4, 136.2, 135.6, 130.6, 130.4, 128.8, 127.9, 127.5, 127.1, 126.2, 126.2, 125.6, 125.5, 125.0, 124.6, 124.5, 123.5, 117.5, 112.1, 104.2, 19.9.
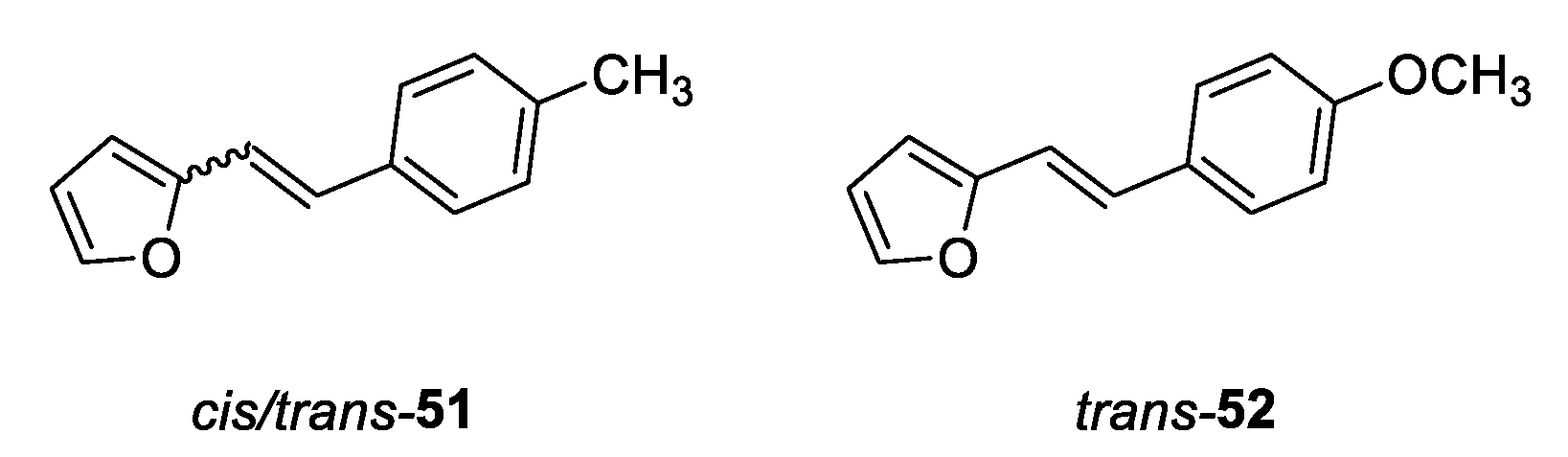
(E)-2-(4-methoxystyryl)furan (trans-52): 750 mg, 36% isolated yield; white powder; Rf (PE/E (2%)) = 0.31; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.39 (d, J = 9.7 Hz, 2H), 6.99 (d, J = 15.9 Hz, 1H), 6.87 (d, J = 8.9 Hz, 2H), 6.75 (d, J = 15.9 Hz, 1H), 6.41 – 6.39 (m, 1H), 6.29 (d, J = 2.4 Hz, 1H), 3.81 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 159.4, 153.7, 141.7, 129.8, 127.5, 126.7, 114.6, 114.5, 111.5, 107.7, 55.3.
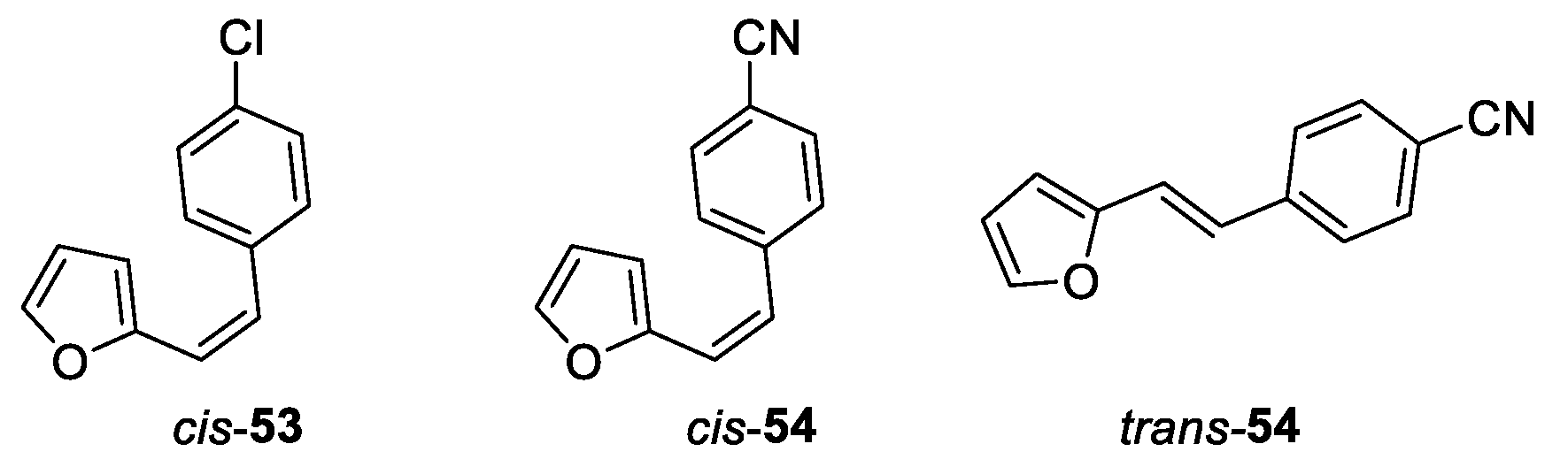
(Z)-2-(4-chlorostyryl)furan (cis-53): 240 mg, 23% isolated yield; colorless oil; Rf (PE/E (2%)) = 0.95; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.38 (d, J = 8.5 Hz, 2H), 7.28 (d, J = 1.8 Hz, 1H), 7.29 (d, J = 8.5 Hz, 2H), 6.40 (d, J = 12.5 Hz, 1H), 6.35 (d, J = 12.5 Hz, 1H), 6.35 – 6.32 (m, 1H), 6.26 (d, J = 3.2 Hz, 1H).
(Z)-4-(2-(furan-2-yl)vinyl)benzonitrile (cis-54): 170 mg, 9% isolated yield; colorless oil; Rf (PE/E (30%)) = 0.19; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.55 (d, J = 8.7 Hz, 2H), 7.50 (d, J = 8.6 Hz, 2H), 7.30 (d, J = 1.9 Hz, 1H), 6.44 (d, J = 12.4 Hz, 1H), 6.40 (d, J = 12.5 Hz, 1H), 6.37 – 6.35 (m, 1H), 6.31 (d, J = 3.7 Hz, 1H).
(E)-4-(2-(furan-2-yl)vinyl)benzonitrile (trans-54): 150 mg, 8% isolated yield; white powder; Rf (PE/E (30%)) = 0.22; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.54 (d, J = 8.7 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 7.44 (d, J = 1.9 Hz, 1H), 7.01 (d, J = 16.3 Hz, 1H), 6.97 (d, J = 16.3 Hz, 1H), 6.45 (d, J = 3.3 Hz, 1H), 6.36 (dd, J = 1.9, 3.3 Hz, 1H).
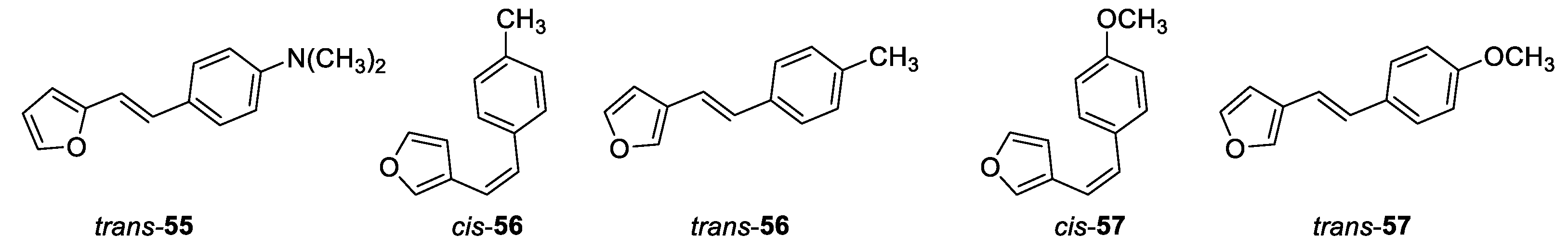
(E)-4-(2-(furan-2-yl)vinyl)-N,N-dimethylaniline (trans-55): 79 mg, 11% isolated yield; yellow powder; Rf (PE/E (5%)) = 0.36; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.44 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 1.6 Hz, 1H), 6.73 – 6.68 (m, 2H), 6.42 (d, J = 12.9 Hz, 1H), 6.33 (d, J = 2.5 Hz, 2H), 6.18 (d, J = 12.9 Hz, 1H), 2.98 (s, 6H).
(Z)-3-(4-methylstyryl)furan (cis-56): 214 mg, 40% isolated yield; yellow oil; Rf (PE/E (10%)) = 0.86; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.36 (s, 1H), 7.22 (d, J = 7.9 Hz, 2H), 7.23 (d, J = 1.4 Hz, 1H), 7.11 (d, J = 7.9 Hz, 2H), 6.51 (d, J = 12.2 Hz, 1H), 6.33 (d, J = 12.2 Hz, 1H), 6.16 (d, J = 1.4 Hz, 1H), 2.35 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 142.5, 141.9, 136.9, 134.9, 129.5, 128.9, 128.6, 122.4, 119.5, 110.3, 21.2.
(E)-3-(4-methylstyryl)furan (trans-56): 219 mg, 41% isolated yield; white powder; Rf (PE/E (10%)) = 0.81; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.51 (s, 1H), 7.40 (t, J = 1.7 Hz, 1H), 7.34 (d, J = 8.1 Hz, 2H), 7.14 (d, J = 8.1 Hz, 2H), 6.92 (d, J = 16.2 Hz, 1H), 6.78 (d, J = 16.2 Hz, 1H), 6.65 (d, J = 1.7 Hz, 1H), 2.34 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 143.6, 140.7, 137.2, 134.6, 129.3, 128.4, 126.0, 124.7, 117.4, 104.4, 21.2.
(Z)-3-(4-methoxylstyryl)furan (cis-57): 160 mg, 50% isolated yield; white oil; Rf (PE) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.36 (s, 1H), 7.27 (d, J = 8.8 Hz, 2H), , 6.85 (d, J = 8.8 Hz, 2H), 6.48 (d, J = 12.8 Hz, 1H), 6.31 (d, J = 12.8 Hz, 1H), 6.18 (s, 1H), 3.81 (s, 3H).
(E)-3-(4-methoxylstyryl)furan (trans-57): 217 mg, 30% isolated yield; white powder; Rf (PE) = 0.75; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.50 (s, 1H), 7.38 (d, J = 9.1 Hz, 3H), 6.88 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 16.1 Hz, 1H), 6.76 (d, J = 16.1 Hz, 1H), 6.64 (s, 1H), 3.82 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 143.6, 140.5, 131.3, 130.2, 127.9, 127.3, 124.7, 116.4, 114.2, 107.4, 55.3.
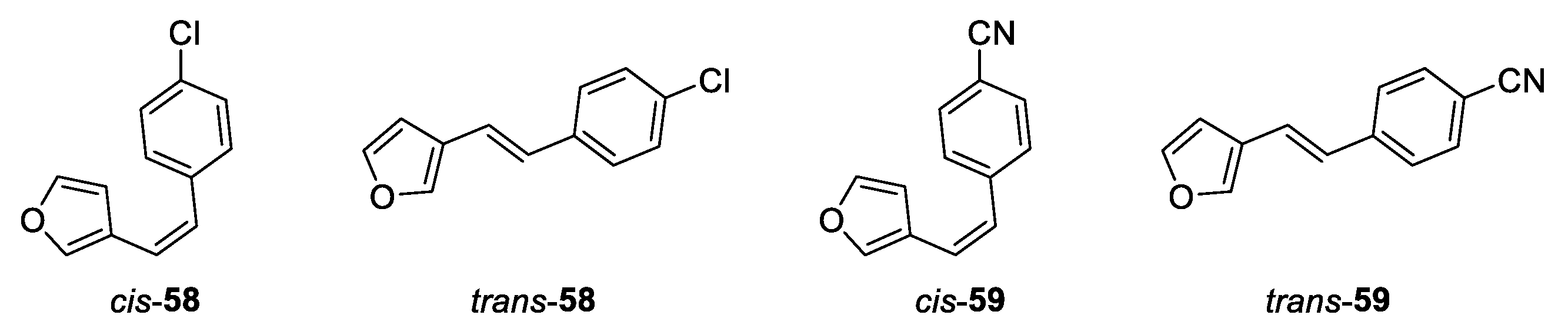
(Z)-3-(4-chlorostyryl)furan (cis-58): 256 mg, 63% isolated yield; colorless oil; Rf (PE) = 0.64; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.36 (s, 1H), 7.30 – 7.26 (m, 4H), 7.25 (d, J = 1.3 Hz, 1H), 6.47 (d, J = 12.1 Hz, 1H), 6.39 (d, J = 12.1 Hz, 1H), 6.11 (d, J = 1.3 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 142.7, 142.2, 136.3, 132.9, 130.0, 128.4, 128.1, 121.9, 120.8, 110.0.
(E)-3-(4-chlorostyryl)furan (trans-58): 95 mg, 21% isolated yield; white powder; Rf (PE) = 0.43; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.54 (s, 1H), 7.41 (d, J = 1.1 Hz, 1H), 7.37 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 16.3 Hz, 1H), 6.75 (d, J = 16.3 Hz, 1H), 6.64 (d, J = 1.1 Hz, 1H).
(Z)-4-(2-(furan-3-yl)vinyl)benzonitrile (cis-59): 278 mg, 61% isolated yield; colorless oil; Rf (PE/E (5%)) = 0.54; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.60 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 1.2 Hz, 1H), 7.27 (s, 1H), 6.53 (d, J = 12.4 Hz, 1H), 6.48 (d, J = 12.4 Hz, 1H), 6.06 (d, J = 1.2 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 143.1, 142.7, 132.5, 132.1, 129.5, 127.4, 126.5, 124.0, 122.7, 121.6, 109.8.
(E)-4-(2-(furan-3-yl)vinyl)benzonitrile (trans-59): 134 mg, 30% isolated yield; white powder; Rf (PE/E (5%)) = 0.33; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.61 (d, J = 8.5 Hz, 2H), 7.51 (d, J = 8.5 Hz, 2H), 7.60 (s, 1H), 7.44 (d, J = 1.3 Hz, 1H), 7.09 (d, J = 16.2 Hz, 1H), 6.79 (d, J = 16.2 Hz, 1H), 6.66 (d, J = 1.3 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.1, 142.1, 141.9, 134.5, 126.5, 124.0, 122.2, 119.1, 110.3, 107.2.
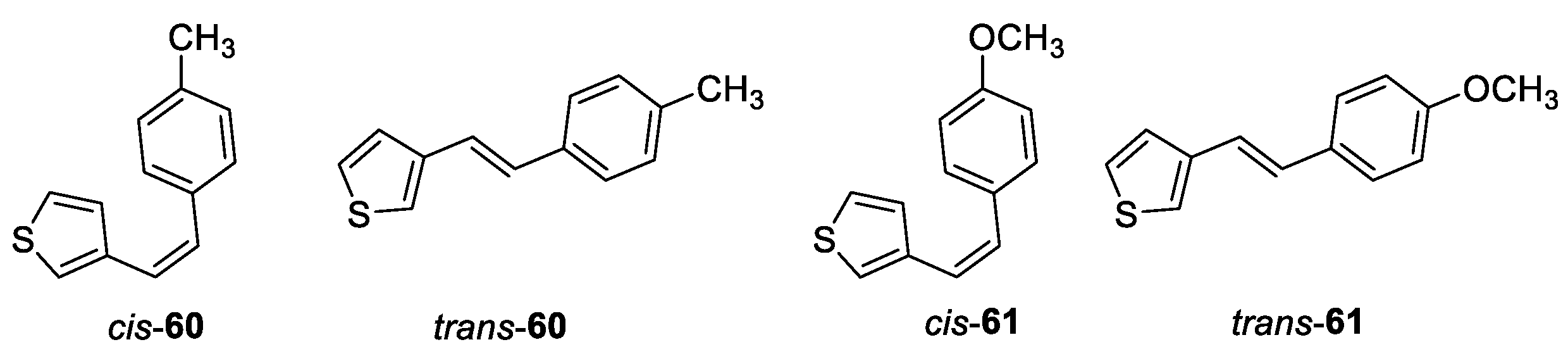
(Z)-3-(4-methylstyryl)thiophene (cis-60): 377 mg, 44% isolated yield; colorless oil; Rf (PE) = 0.32; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.19 (d, J = 8.1 Hz, 2H), 7.14 – 7.11 (m, 2H), 7.09 (d, J = 8.1 Hz, 2H), 6.89 (d, J = 4.8 Hz, 1H), 6.54 (d, J = 12.3 Hz, 1H), 6.49 (d, J = 12.3 Hz, 1H), 2.34 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 138.4, 136.9, 134.8, 129.5, 129.0, 128.6, 128.1, 124.8, 123.8, 123.8, 21.3.
(E)-3-(4-methylstyryl)thiophene (trans-60): 289 mg, 33% isolated yield; white powder; Rf (PE) = 0.21; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.37 (d, J = 8.1 Hz, 2H), 7.33 – 7.29 (m, 2H), 7.24 – 7.22 (m, 1H), 7.15 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 16.3 Hz, 1H), 6.92 (d, J = 16.3 Hz, 1H), 2.35 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 140.3, 137.3, 134.6, 129.4, 128.6, 126.2, 126.1, 124.9, 122.0, 121.9, 21.2.
(Z)-3-(4-methoxystyryl)thiophene (cis-61): 244 mg, 40% isolated yield; colorless oil; Rf (PE) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.37 (s, 1H), 7.26 (d, J = 9.2 Hz, 2H), 7.25 (t, J = 1.7 Hz, 1H), 6.85 (d, 1H, J = 8.8 Hz, 2H), 6.48 (d, J = 11.8 Hz, 1H), 6.31 (d, J = 11.8 Hz, 1H), 6.18 (d, J = 5.1 Hz, 1H), 3.81 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 158.7, 142.5, 141.8, 130.3, 129.8, 129.2, 122.4, 118.9, 113.6, 110.3, 100.0, 55.2.
(E)-3-(4-methoxystyryl)thiophene (trans-61): 248 mg, 41% isolated yield; white powder; Rf (PE) = 0.76; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.41 (d, J = 8.7 Hz, 2H), 7.34 – 7.29 (m, 2H), 7.22 – 7.20 (m, 1H), 7.00 (d, J = 16.2 Hz, 1H), 6.91 (d, J = 16.2 Hz, 1H), 6.89 (d, J = 8.7 Hz, 2H), 3.82 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 159.2, 140.4, 130.2, 128.2, 127.5, 126.1, 124.9, 121.5, 120.9, 114.2, 55.3.
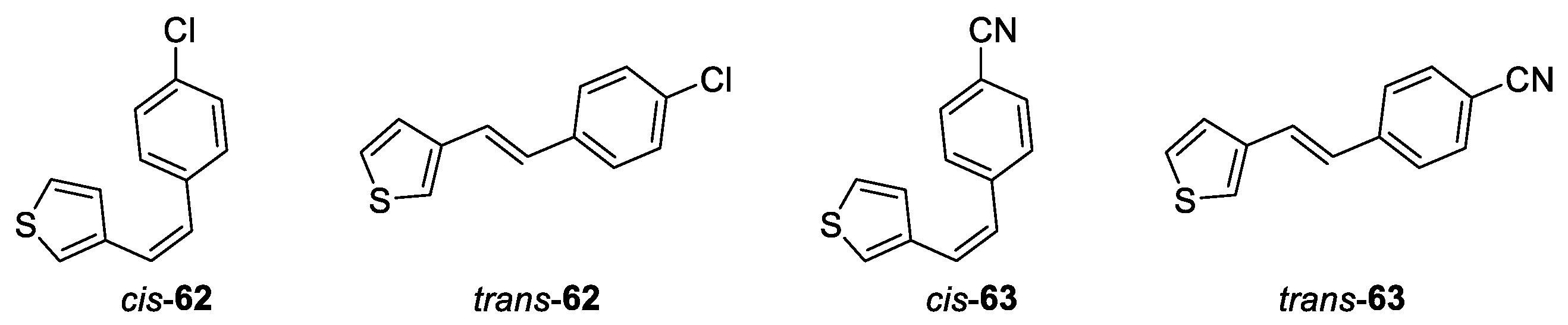
(Z)-3-(4-chlorostyryl)thiophene (cis-62): 222 mg, 41% isolated yield; colorless oil; Rf (PE) = 0.64; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.23 (d, J = 8.7 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 7.15 (dd, J = 5.1, 2.9 Hz, 1H), 7.11 (d, J = 2.9 Hz, 1H), 6.85 (d, J = 5.1 Hz, 1H), 6.56 (d, J = 12.1 Hz, 1H), 6.48 (d, J = 12.1 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 137.9, 136.2, 132.9, 130.1, 128.5, 128.2, 127.8, 125.2, 125.1, 124.3.
(E)-3-(4-chlorostyryl)thiophene (trans-62): 186.1 mg, 34% isolated yield; white powder; Rf (PE) = 0.49; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.40 (d, J = 8.7 Hz, 2H), 7.33 – 7.27 (m, 5H), 7.09 (d, J = 16.3 Hz, 1H), 6.89 (d, J = 16.3 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 139.8, 135.9, 133.0, 128.8, 127.4, 127.3, 126.3, 124.8, 123.5, 122.7.
(Z)-4-(2-(thiophen-3-yl)vinyl)benzonitrile (cis-63): 474 mg, 73% isolated yield; colorless oil; Rf (PE/E (15%)) = 0.69; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.56 (d, J = 8.3 Hz, 2H), 7.39 (d, J = 8.1 Hz, 2H), 7.18 (dd, J = 4.9, 3.0 Hz, 1H), 7.13 (d, J = 2.9 Hz, 1H), 6.81 (d, J = 5.0 Hz, 1H), 6.68 (d, J = 12.1 Hz, 1H), 5.52 (d, J = 12.1 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 142.6, 137.3, 132.0, 132.1, 129.5, 127.5, 127.1, 125.6, 125.0, 118.9, 110.6.
(E)-4-(2-(thiophen-3-yl)vinyl)benzonitrile (trans-63): 158 mg, 23% isolated yield; white powder; Rf (PE/E (15%)) = 0.48; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.62 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.36 (s, 2H), 7.26 (s, 1H), 7.23 (d, J = 16.3 Hz, 1H), 6.93 (d, J = 16.3 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 142.0, 139.2, 132.5, 126.7, 126.6, 126.6, 126.4, 124.8, 124.3, 119.1, 110.4.
Corresponding amines (1.2 eq) were added to a solution of triazole nitro aldehyde 24 [18,19] (1 eq) in dry dioxane and purged with argon, Ar. Reaction mixtures were stirred at 130 °C. After overnight, reaction mixtures were cooled to room temperature and evaporated till dryness to obtain crude products 22, 23 and 25. Crude products were purified by a column chromatography using E/EtOAc solvent system.
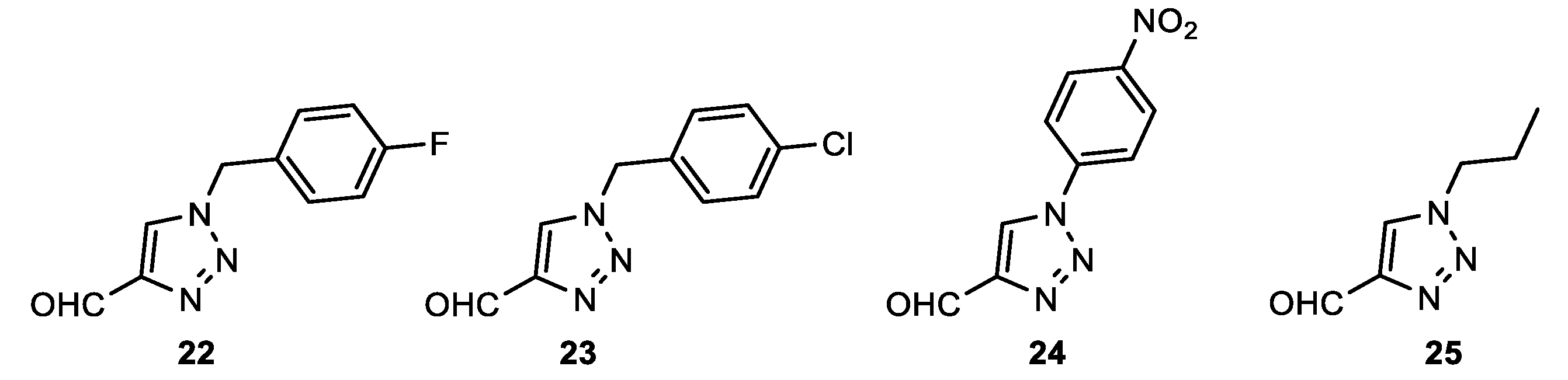
1-(4-fluorobenzyl)-1H-1,2,3-triazole-4-carbaldehyde (22): 128 mg, 78% isolated yield; yellow oil; Rf (DCM/EtOAc (2%)) = 0.53; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.13 (s, 1H), 7.99 (s, 1H), 7.33 – 7.30 (m, 2H), 7.13 – 7.08 (m, 2H), 5.57 (s, 2H).
1-(4-chlorobenzyl)-1H-1,2,3-triazole-4-carbaldehyde (23): 85 mg, 68% isolated yield; yellow oil; Rf (DCM/EtOAc (2%)) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.13 (s, 1H), 8.01 (s, 1H), 7.41 – 7.38 (m, 4H), 5.57 (s, 2H).
1-(4-nitrophenyl)-1H-1,2,3-triazole-4-carbaldehyde (24) [12,70]: 720 mg, 72% isolated yield; yellow powder; Rf (DCM/EtOAc (2%)) = 0.52; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.20 (s, 1H), 8.66 (s, 1H), 8.49 (d, J = 8.8 Hz, 2H), 8.04 (d, J = 9.0 Hz, 2H).
1-propyl-1H-1,2,3-triazole-4-carbaldehyde (25): 100 mg, 40% isolated yield; yellow oil; Rf (DCM/EtOAc (2%)) = 0.55; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.14 (s, 1H), 8.15 (s, 1H), 4.95 – 4.88 (m, 1H), 1.65 (s, 3H), 1.64 (s, 3H).
The obtained heterostilbenes 45–63 were subjected to Vilsmeier formylation reaction. The selected heterostilbenes, as mixtures of isomers, were dissolved in 2 mL of DMF and stirred for 10 min at 10 °C. This temperature was achieved using a water bath with a few ice cubes and monitored with a thermometer. The weighed amount of POCl3 was slowly added dropwise. After 30 min, the water bath was removed, and the reaction mixture was allowed to stir. Upon completion of the reaction, the reaction mixture was neutralized using 10% NaOH solution. When neutralization was achieved, extraction was carried using E and water. The combined organic layer was washed with water and dried over MgSO4, filtered, and the solvent was evaporated. The dry reaction mixture was purified by column chromatography on silica gel using a PE/E or PE/DCM variable polarity eluent. In the first fractions, the unreacted substrates were isolated (as cis-isomers), while in the last fractions, the desired formyl derivatives 26–44 were obtained (mainly as trans-isomers). They were further used in the preparation of oximes 4–21.
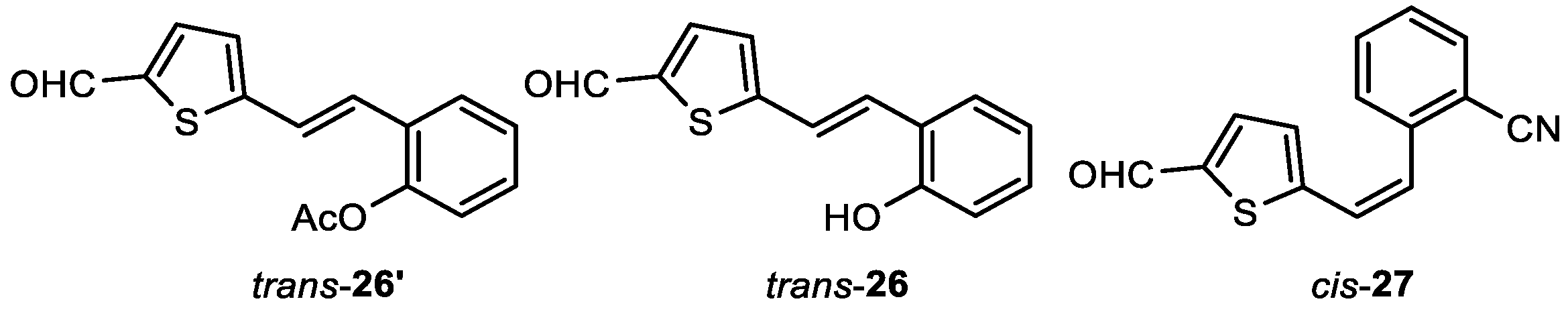
(E)-2-(2-(5-formylthiophen-2-yl)vinyl)phenyl acetate (trans-26'): 128 mg, 78% isolated yield; yellow oil; Rf (DCM) = 0.36; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.86 (s, 1H), 7.65 – 7.64 (m, 2H), 7.33 (t, J = 7.9 Hz, 1H), 7.27 – 7.24 (m, 1H), 7.21 (d, J = 16.1 Hz, 1H), 7.17 (d, J = 16.1 Hz, 1H), 7.14 (d, J = 3.9 Hz, 1H), 7.11 (d, J = 8.0 Hz, 1H), 2.39 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 182.7, 169.2, 152.1, 148.5, 141.9, 137.2, 129.6, 128.5, 127.2, 126.4, 125.9, 123.0, 122.9, 99.9, 20.9.
(E)-5-(2-hydroxystyryl)thiophene-2-carbaldehyde (trans-26): 60 mg, 50% isolated yield; yellow powder; Rf (E) = 0.26; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.85 (s, 1H), 7.67 (d, J = 3.7 Hz, 1H), 7.50 (dd, J = 7.7, 1.4 Hz, 1H), 7.47 (d, J = 16.2 Hz, 1H), 7.31 (d, J = 16.2 Hz, 1H), 7.19 (t, J = 7.6 Hz, 1H), 7.16 (d, J = 3.8 Hz, 1H), 6.96 (t, J = 7.4 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 5.38 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 182.8, 153.7, 153.5, 141.3, 137.5, 129.8, 128.0, 127.7, 126.4, 123.3, 121.8, 121.2, 116.2.
(Z)-2-(2-(5-formylthiophen-2-yl)vinyl)benzonitrile (cis-27): 10 mg, 20% isolated yield; yellow oil; Rf (DCM) = 0.40; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.79 (s, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.61 (t, J = 8.1 Hz, 1H), 7.57 (d, J = 4.6 Hz, 1H), 7.51 – 7.47 (m, 2H), 7.05 (d, J = 4.1 Hz, 1H), 6.96 (d, J = 11.9 Hz, 1H), 6.87 (d, J = 11.9 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 182.9, 147.6, 143.5, 140.1, 135.9, 133.5, 130.3, 129.6, 128.8, 126.4, 117.4, 112.3.
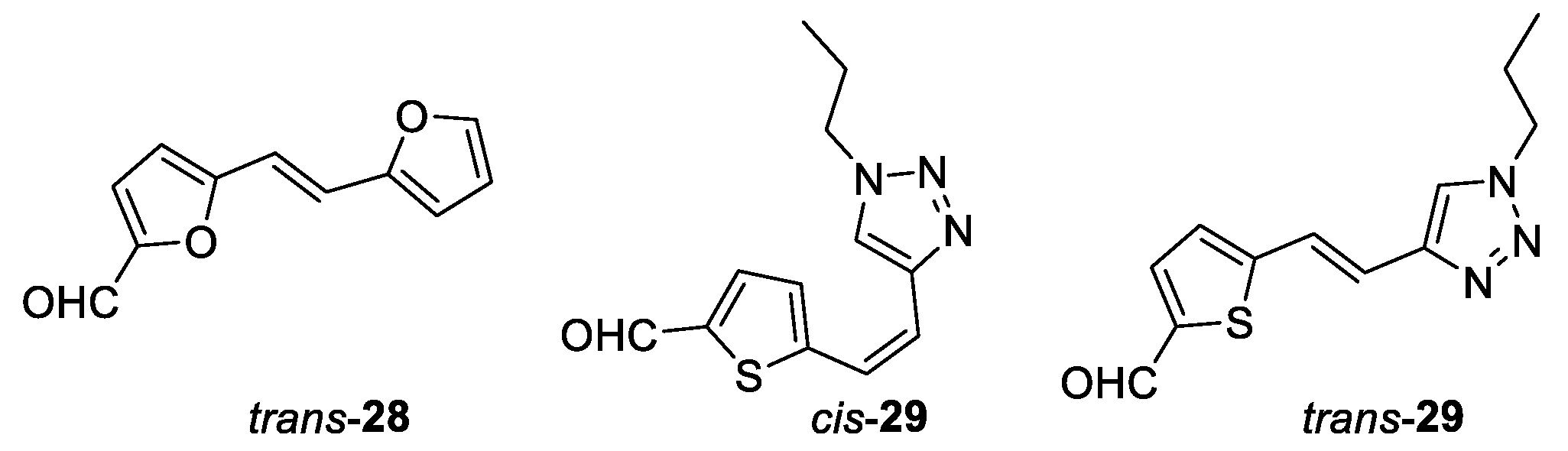
(E)-5-(2-(furan-2-yl)vinyl)furan-2-carbaldehyde (trans-28): 74 mg, 60% isolated yield; red oil; Rf (PE/E (20%)) = 0.85; 1H NMR (CDCl3, 300 MHz) δ/ppm: 9.57 (s, 1H), 7.44 (d, J = 1.2 Hz, 1H), 7.26 – 7.23 (m, 1H), 7.16 (d, J = 16.2 Hz, 1H), 6.82 (d, J = 16.2 Hz, 1H), 6.49 – 6.45 (m, 3H).
(Z)-5-(2-(1-propyl-1H-1,2,3-triazol-4-yl)vinyl)thiophene-2-carbaldehyde (cis-29): 25 mg, 25% isolated yield; yellow oil; Rf (PE/E (9%)) = 0.25; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.89 (s, 1H), 7.69 (d, J = 4.1 Hz, 1H), 7.63 (s, 1H), 7.51 (d, J = 4.1 Hz, 1H), 6.72 (d, J = 12.4 Hz, 1H), 6.69 (d, J = 12.4 Hz, 1H), 4.36 – 4.33 (m, 2H), 2.00 – 1.93 (m, 2H), 1.00 – 0.96 (m, 3H).
(E)-5-(2-(1-propyl-1H-1,2,3-triazol-4-yl)vinyl)thiophene-2-carbaldehyde (trans-29): 25 mg, 25% isolated yield; yellow oil; Rf (PE/E (90%)) = 0.23; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.87 (s, 1H), 7.67 (d, J = 4.1 Hz, 1H), 7.57 (s, 1H), 7.51 (d, J = 15.9 Hz, 1H), 7.17 (d, J = 4.8 Hz, 1H), 7.11 (d, J = 15.9 Hz, 1H), 4.36 – 4.33 (m, 2H), 2.00 – 1.93 (m, 2H), 1.00 – 0.96 (m, 3H).
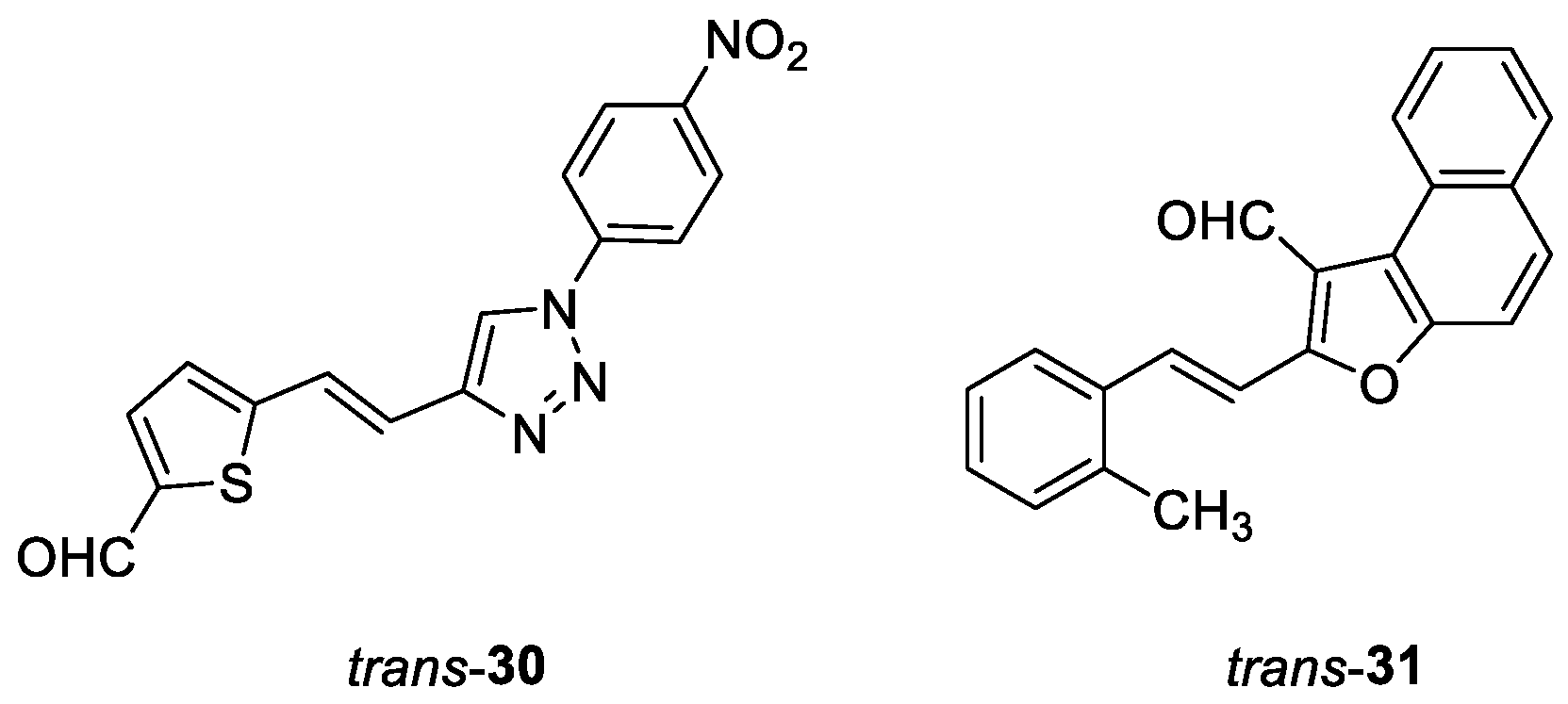
(E)-5-(2-(1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)vinyl)thiophene-2-carbaldehyde (trans-30): 12 mg, 10% isolated yield; yellow oil; Rf (DCM) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.82 (s, 1H), 8.45 (d, J = 9.1 Hz, 2H), 8.11 (s, 1H), 8.00 (d, J = 9.6 Hz, 2H), 7.69 (d, J = 1.7 Hz, 1H), 7.68 (d, J = 14.1 Hz, 1H), 7.23 (d, J = 3.6 Hz, 1H), 7.68 (d, J = 14.1 Hz, 1H).
(E)-2-(2-methylstyryl)naphtho[2,1-b]furan-1-carbaldehyde (trans-31): 75 mg, 75% isolated yield; yellow powder; Rf (PE/E (50%)) = 0.75; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.67 (s, 1H), 9.30 (d, J = 8.3 Hz, 1H), 7.97 (d, J = 15.9 Hz, 1H), 7.95 (d, J = 5.9 Hz, 1H), 7.86 (d, J = 9.3 Hz, 1H), 7.75 – 7.63 (m, 4H), 7.58 – 7.54 (m, 2H), 7.31 – 7.29 (m, 2H), 2.55 (s, 3H).
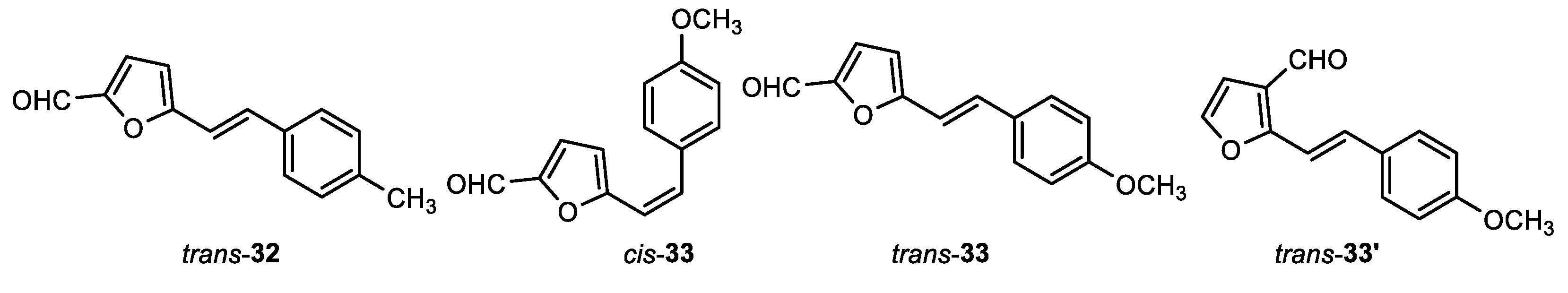
(E)-5-(4-methylstyryl)furan-2-carbaldehyde (trans-32): 693 m, 76% isolated yield; white powder; Rf (PE/E (2%)) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.58 (s, 1H), 7.40 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 16.5 Hz, 1H), 7.24 (d, J = 3.8 Hz, 1H), 7.18 (d, J = 7.7 Hz, 2H), 6.88 (d, J = 16.5 Hz, 1H), 6.50 (d, J = 3.8 Hz, 1H), 2.36 (s, 3H).
(Z)-5-(4-methylstyryl)furan-2-carbaldehyde (cis-33): 17 mg, 4% isolated yield; colorless oil; Rf (PE/E (1%)) = 0.27; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.54 (s, 1H), 7.42 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 3.8 Hz, 1H), 6.89 (d, J = 8.7 Hz, 2H), 6.73 (d, J = 12.4 Hz, 1H), 6.41 (d, J = 3.8 Hz, 1H), 6.36 (d, J = 12.4 Hz, 1H), 3.84 (s, 3H) ; 13C NMR (CDCl3, 600 MHz) δ/ppm: 177.4, 159.7, 157.8, 151.1, 134.3, 130.2, 128.7, 115.9, 113.9, 111.6, 55.3.
(E)-5-(4-methoxystyryl)furan-2-carbaldehyde (trans-33): 90 mg, 17% isolated yield; white powder; Rf (PE/E (1%)) = 0.22; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.56 (s, 1H), 7.44 (d, J = 8.6 Hz, 2H), 7.34 (d, J = 16.1 Hz, 1H), 7.24 (d, J = 4.0 Hz, 1H), 6.90 (d, J = 9.1 Hz, 2H), 6.78 (d, J = 16.1 Hz, 1H), 6.47 (d, J = 4.0 Hz, 1H), 3.82 (s, 3H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 176.7, 160.3, 159.3, 151.4, 133.1, 133.2, 128.4, 114.3, 112.9, 109.9, 55.4.
(E)-2-(4-methoxystyryl)furan-3-carbaldehyde (trans-33'): 60 mg, 26% isolated yield; white powder; Rf (PE/E (1%)) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.91 (s, 1H), 7.57 (d, J = 1.9 Hz, 1H), 7.49 (d, J = 9.1 Hz, 2H), 7.47 (d, J = 16.9 Hz, 1H), 7.09 ( d, J = 16.9 Hz, 1H), 6.91 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 1.5 HZ, 1H), 3.84 (s, 3H).
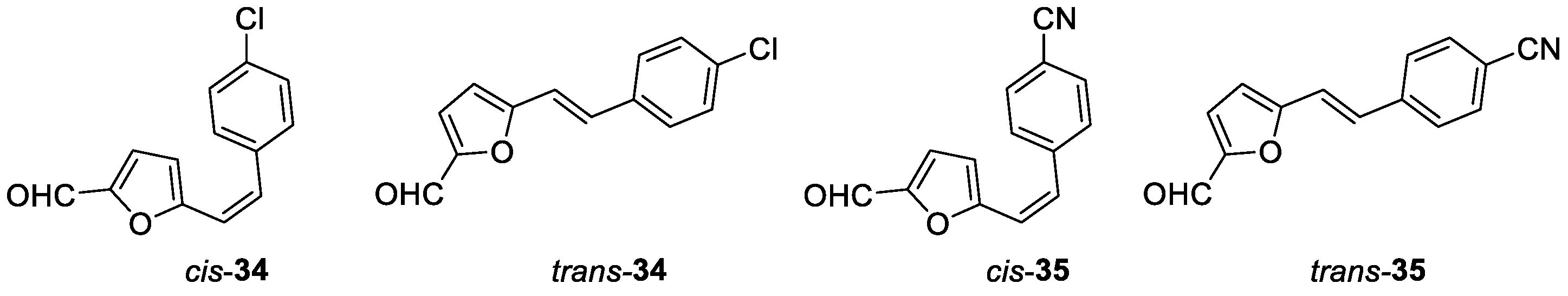
(Z)-5-(4-chlorostyryl)furan-2-carbaldehyde (cis-34): 128 mg, 21% isolated yield; yellow oil; Rf (PE/E (1%)) = 0.23; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.54 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 3.6 Hz, 1H), 6.73 (d, J = 13.1 Hz, 1H), 6.47 (d, J = 13.1 Hz, 1H), 6.34 (d, J = 3.6 Hz, 1H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 177.3, 156.9, 151.4, 134.7, 134.2, 132.9, 129.9, 128.7, 117.9, 112.3.
(E)-5-(4-chlorostyryl)furan-2-carbaldehyde (trans-34): 62.7 mg, 11% isolated yield; white powder; Rf (PE/E (1%)) = 0.18; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.57 (s, 1H), 7.41 (d, J = 8.7 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 15.9 Hz, 1H), 7.24 (d, J = 3.5 Hz, 1H), 6.88 (d, J = 15.9 Hz, 1H), 6.53 (d, J = 3.7 Hz, 1H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 176.9, 158.2, 151.8, 134.6, 134.3, 131.8, 129.1, 128.1, 115.5, 111.0.
(Z)-4-(2-(5-formylfuran-2-yl)vinyl)benzonitrile (cis-35): 21 mg, 3% isolated yield; white oil; Rf (PE/E (20%)) = 0.33; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.54 (s, 1H), 7.67 (d, J = 7.7 Hz, 2H), 7.55 (d, J = 7.8 Hz, 2H), 7.16 (d, J = 3.8 Hz, 1H), 6.76 (d, J = 12.3 Hz, 1H), 6.56 (d, J = 12.3 Hz, 1H), 6.37 (d, J = 3.8 Hz, 1H).
(E)-4-(2-(5-formylfuran-2-yl)vinyl)benzonitrile (trans-35): 62 mg, 9% isolated yield; white powder; Rf (PE/E (20%)) = 0.23; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.64 (s, 1H), 7.67 (d, J = 7.9 Hz, 2H), 7.57 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 16.5 Hz, 1H), 7.28 (d, J = 3.8 Hz, 1H), 7.03 (d, J = 16.5 Hz, 1H), 6.64 (d, J = 3.8 Hz, 1H).
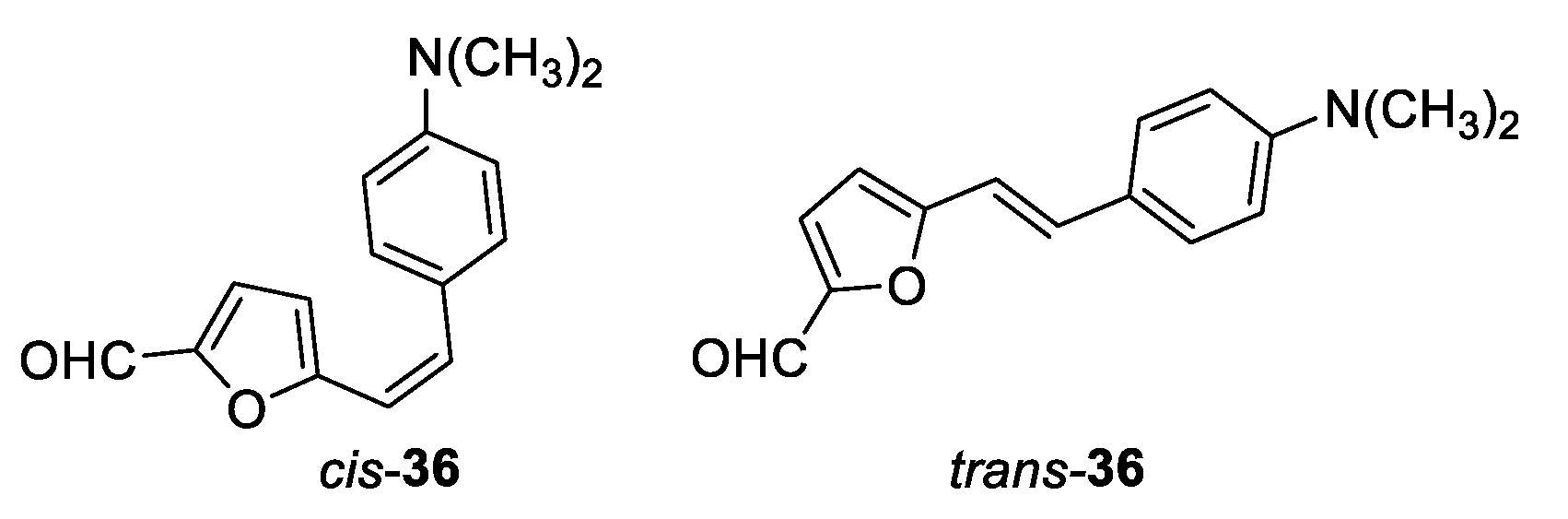
(Z)-5-(4-(dimethylamino)styryl)furan-2-carbaldehyde (cis-36): 10 mg; 2% isolated yield; yellow oil; Rf (PE/E (5%)) = 0.16; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.55 (s, 1H), 7.45 (d, J = 8.5 Hz, 2H), 7.17 (d, J = 3.7 Hz, 1H), 6.70 (d, J = 8.7 Hz, 2H), 6.69 (d, J = 12.4 Hz, 1H), 6.52 (d, J = 3.6 Hz, 1H), 6.23 (d, J = 12.4 Hz, 1H), 3.01 (s, 6H).
(E)-5-(4-(dimethylamino)styryl)furan-2-carbaldehyde (trans-36): 5 mg; 5% isolated yield; yellow powder; Rf (PE/E (5%)) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.53 (s, 1H), 7.41 (d, J = 8.6 Hz, 2H), 7.33 (d, J = 16.2 Hz, 1H), 7.24 (d, J = 3.6 Hz, 1H), 7.20 (d, J = 16.2 Hz, 1H), 6.70 (d, J = 8.7 Hz, 2H), 6.43 (d, J = 3.7 Hz, 1H), 3.01 (s, 6H).
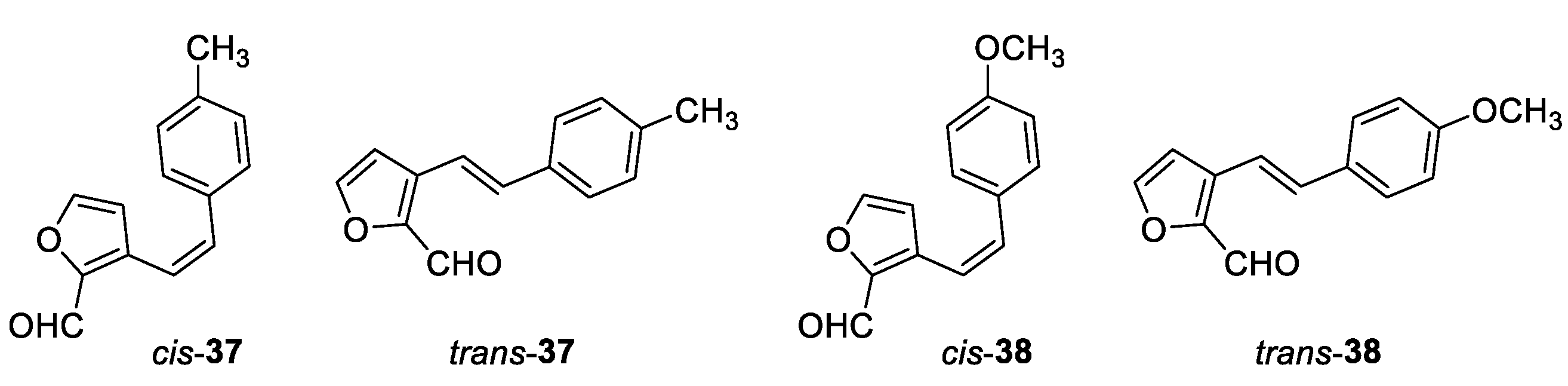
(Z)-3-(4-methylstyryl)furan-2-carbaldehyde (cis-37): 151 mg, 43% isolated yield; orange oil; Rf (PE/E (5%)) = 0.40; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.81 (s, 1H), 7.43 (d, J = 1.2 Hz, 1H), 7.18 (d, J = 8.1 Hz, 2H), 7.11 (d, J = 8.1 Hz, 2H), 6.85 (d, J = 12.4 Hz, 1H), 6.85 (d, J = 12.4 Hz, 1H), 6.27 (d, J = 1.2 Hz, 1H), 2.35 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 178.2, 148.5, 146.8, 138.0, 135.7, 133.6, 129.1, 128.7, 127.0, 117.4, 112.9, 21.3.
(E)-3-(4-methylstyryl)furan-2-carbaldehyde (trans-37): 103 mg, 51% isolated yield; yellow powder; Rf (PE/E (5%)) = 0.26; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.91 (s, 1H), 7.58 (d, J = 1.3 Hz, 1H), 7.50 (d, J = 16.2 Hz, 1H), 7.45 (d, J = 7.8 Hz, 2H), 7.19 (d, J = 7.8 Hz, 2H), 7.11 (d, J = 16.2 Hz, 1H), 6.85 (d, J = 1.3 Hz, 1H), 2.37 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 178.5, 147.7, 147.5, 139.0, 135.3, 133.4, 129.5, 129.1, 128.7, 127.1, 109.8, 21.3.
(Z)-3-(4-methoxystyryl)furan-2-carbaldehyde (cis-38): 90 mg, 30% isolated yield; yellow oil; Rf (PE/DCM (30%)) = 0.30; 1H NMR (CDCl3, 300 MHz) δ/ppm: 9.81 (s, 1H), 7.44 (s, 1H), 7.26 – 7.21 (m, 2H), 6.85 – 6.76 (m, 4H), 6.31 (d, J = 1.6 Hz, 1H), 3.82 (s, 3H).
(E)-3-(4-methoxystyryl)furan-2-carbaldehyde (trans-38): 135 mg, 56% isolated yield; yellow powder; Rf (PE/DCM (30%)) = 0.25; 1H NMR (CDCl3, 300 MHz) δ/ppm: 9.90 (s, 1H), 7.57 – 7.44 (m, 4H), 7.09 (d, J = 15.0 Hz, 1H), 6.93 – 6.83 (m, 3H), 3.84 (s, 3H).
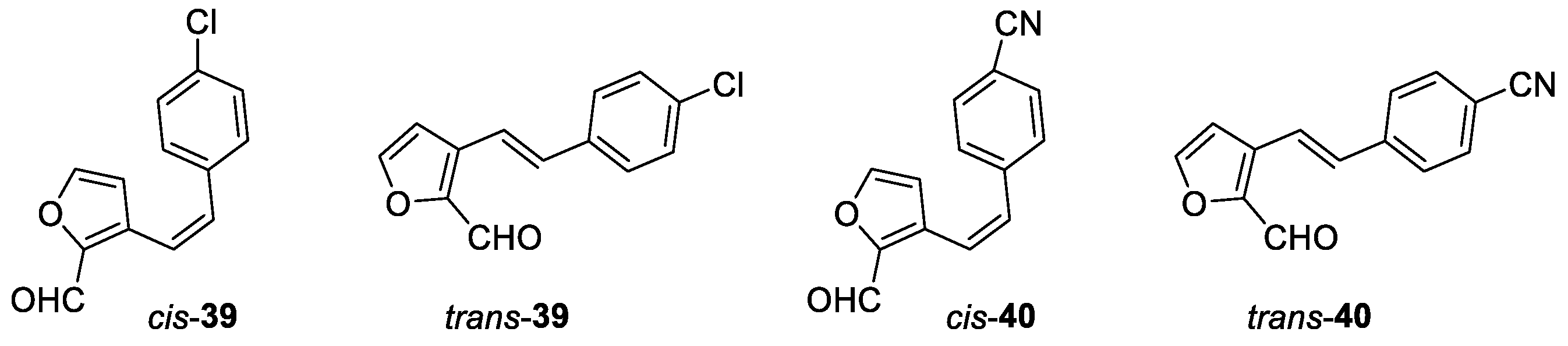
(Z)-3-(4-chlorostyryl)furan-2-carbaldehyde (cis-39): 187 mg, 67% isolated yield; orange oil; Rf (PE/E (10%)) = 0.30; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.83 (s, 1H), 7.44 (d, J = 1.6 Hz, 1H), 7.29 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 8.6 Hz, 2H), 6.94 (d, J = 12.0 Hz, 1H), 7.82 (d, J = 12.0 Hz, 1H), 6.22 (d, J = 1.6 Hz, 1H).
(E)-3-(4-chlorostyryl)furan-2-carbaldehyde (trans-39): 58 mg, 23% isolated yield; yellow powder; Rf (PE/E (5%)) = 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.92 (s, 1H), 7.62 (d, J = 16.3 Hz, 1H), 7.59 (d, J = 1.5 Hz, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.35 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 16.3 Hz, 1H), 6.85 (d, J = 1.5 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 178.5, 147.6, 146.9, 134.2, 133.8, 130.1, 129.0, 128.7, 128.2, 112.8, 109.9.
(Z)-4-(2-(2-formylfuran-3-yl)vinyl)benzonitrile (cis-40): 175 mg, 68% isolated yield; yellow oil; Rf (PE/E (20%)) = 0.55; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.85 (s, 1H), 7.61 (d, J = 8.3 Hz, 2H), 7.47 (d, J = 1.6 Hz, 1H), 7.40 (d, J = 8.3 Hz, 2H), 7.07 (d, J = 12.3 Hz, 1H), 6.85 (d, J = 12.3 Hz, 1H), 6.15 (d, J = 1.6 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 178.7, 148.9, 147.0, 133.2, 132.6, 132.3, 129.5, 127.4, 121.1, 112.6, 111.6, 109.9.
(E)-4-(2-(2-formylfuran-3-yl)vinyl)benzonitrile (trans-40): 119 mg, 45% isolated yield; yellow powder; Rf (PE/E (20%)) = 0.46; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.94 (s, 1H), 7.76 (d, J = 16.5 Hz, 1H), 7.66 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 1.9 Hz, 1H), 7.12 (d, J = 16.5 Hz, 1H), 6.87 (d, J = 1.9 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 179.4, 148.4, 147.6, 140.6, 132.9, 132.6, 127.4, 120.6, 118.8, 111.7, 109.9.
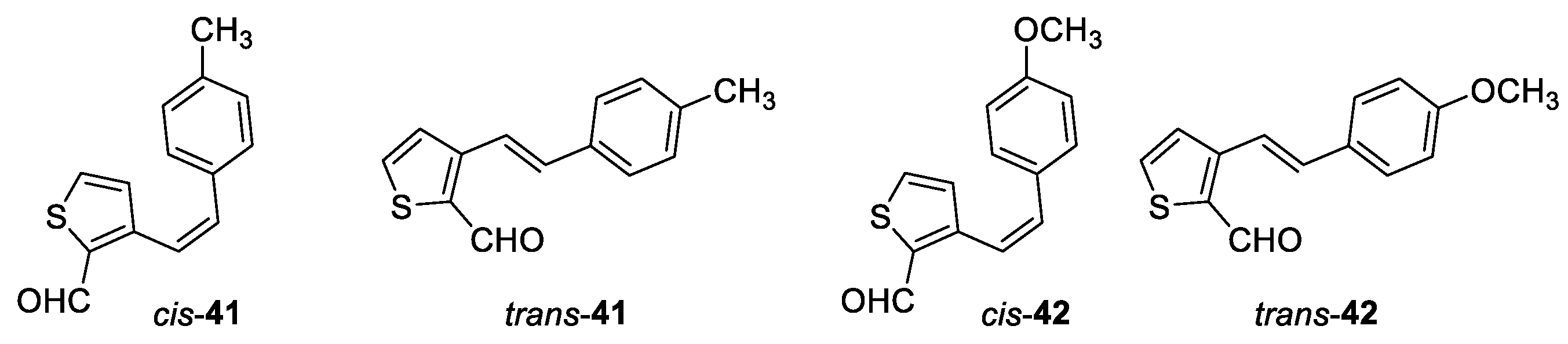
(Z)-3-(4-methylstyryl)thiophene-2-carbaldehyde (cis-41): 110 mg, 27% isolated yield; Rf (PE/E (10%)) = 0.57; 1H NMR (CDCl3, 300 MHz) δ/ppm: 9.99 (s, 1H), 7.56 (d, J = 4.9 Hz, 1H), 7.06 (t, J = 9.4 Hz, 4H), 6.92 (d, J = 5.0 Hz, 1H), 6.84 (d, J = 12.1 Hz, 1H), 6.78 (d, J = 12.1 Hz, 1H), 2.31 (s, 3H).
(E)-3-(4-methylstyryl)thiophene-2-carbaldehyde (trans-41): 186 mg, 64% isolated yield; white powder; Rf (PE/E (10%)) = 0.42; 1H NMR (CDCl3, 300 MHz) δ/ppm: 10.21 (s, 1H), 7.65 (d, J = 5.6 Hz, 1H), 7.63 (d, J = 16.2 Hz, 1H), 7.43 (d, J = 6.6 Hz, 3H), 7.20 (d, J = 8.0 Hz, 2H), 7.14 (d, J = 16.2 Hz, 1H), 2.37 (s, 3H).
(Z)-3-(4-methoxystyryl)thiophene-2-carbaldehyde (cis-42): 45 mg, 20% isolated yield; white oil; Rf (PE/DCM (60%)) = 0.55; 1H NMR (CDCl3, 300 MHz) δ/ppm: 9.99 (s, 1H), 7.57 (d, J = 5.2 Hz, 1H), 7.12 (d, J = 9.1 Hz, 2H), 6.94 (d, J = 4.6 Hz, 1H), 6.82 – 6.71 (m, 4H), 3.78 (s, 3H).
(E)-3-(4-methoxystyryl)thiophene-2-carbaldehyde (trans-42): 130 mg, 52% isolated yield; white powder; Rf (PE/DCM (60%)) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 10.21 (s, 1H), 7.66 (d, J = 5.2 Hz, 1H), 7.59 – 7.42 (m, 7H), 3.85 (s, 3H).
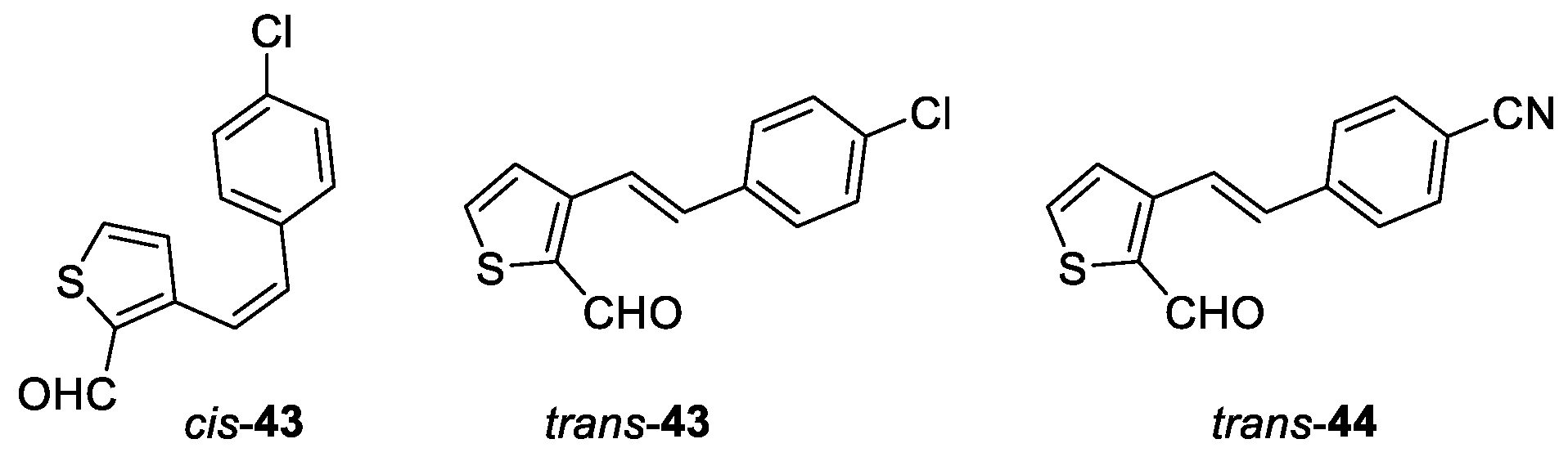
(Z)-3-(4-chlorostyryl)thiophene-2-carbaldehyde (cis-43): 98 mg, 41% isolated yield; white oil; Rf (PE/E (10%)) = 0.19; 1H NMR (CDCl3, 300 MHz) δ/ppm: 9.99 (s, 1H), 7.58 (d, J = 4.9 Hz, 1H), 7.22 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 6.90 – 6.87 (m, 3H), 6.86 (d, J = 5.0 Hz, 1H), 6.80 (d, J = 12.2 Hz, 1H).
(E)-3-(4-chlorostyryl)thiophene-2-carbaldehyde (trans-43): 96 mg, 40% isolated yield; white powder; Rf (PE/E (10%)) = 0.15; 1H NMR (CDCl3, 300 MHz) δ/ppm: 10.19 (s, 1H), 7.85 (d, J = 16.5 Hz, 1H), 7.72 – 7.60 (m, 5H), 7.48 (d, J = 4.7 Hz, 1H), 7.15 (d, J = 16.5 Hz, 1H).
(E)-4-(2-(2-formylthiophen-3-yl)vinyl)benzonitrile (trans-44): 31 mg, 86% isolated yield; white powder; Rf (PE/E (10%)) = 0.24; 1H NMR (CDCl3, 300 MHz) δ/ppm: 10.19 (s, 1H), 7.86 (d, J = 16.2 Hz, 1H), 7.71 (d, J = 5.2 Hz, 1H), 7.68 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 5.2 Hz, 1H), 7.16 (d, J = 16.2 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 181.9, 149.7, 140.8, 134.5, 139.5, 133.0, 132.6, 132.3, 129.5, 127.3, 123.1, 118.7.
The obtained aldehydes 23–25 were converted into oximes 1–3 while aldehydes 26–44 produced the corresponding oximes 4–21 according to the literature [20]. Crystals of NH2OH × HCl were dissolved in a prepared mixture of 10 mL of EtOH and 3 mL of distilled water. After a homogeneous solution was obtained, the corresponding prepared heterostilbene aldehydes 26–44 were added. The reaction mixture was stirred at room temperature for 24 h. When the reaction was completed, the solvent was evaporated on a rotavapor under reduced pressure. The reaction mixture was purified by repeated column chromatography on silica gel using PE/DCM and DCM/methanol variable polarity eluents. The targeted oximes 1–21 were isolated and the spectroscopic data and yields of their pure isomers are given below.
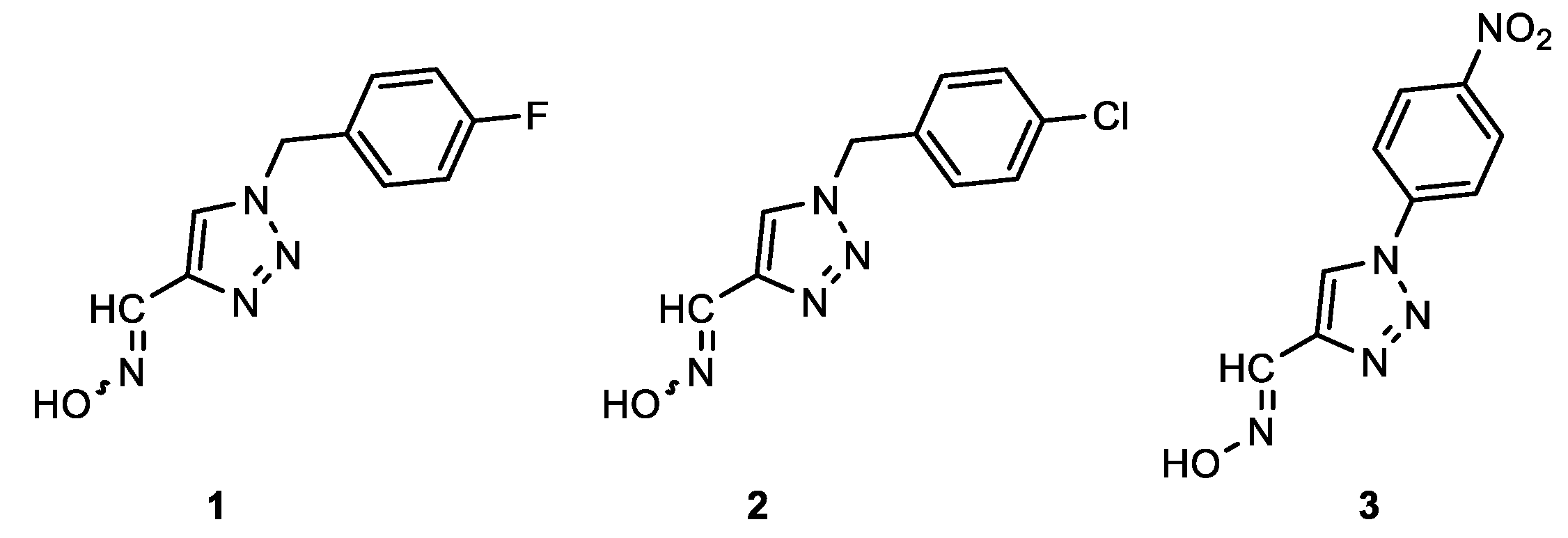
1-(4-fluorobenzyl)-1H-1,2,3-triazole-4-carbaldehyde oxime (1): 30 mg, 16% isolated yield; yellow oil; Rf (DCM/MeOH (30%)) = 0.22; 1H NMR (CD3OD, 600 MHz) δ/ppm: 8.48 (s, 1H), 8.44 (s, 1H), 8.12 (d, J = 9.3 Hz, 2H), 6.87 (d, J = 8.2 Hz, 2H), 5.68 (s, 2H). In the mixture, the ratio of the two isomers is approximately 1:1.
1-(4-chlorobenzyl)-1H-1,2,3-triazole-4-carbaldehyde oxime (2): 30 mg, 8% isolated yield; yellow oil; Rf (DCM/MeOH (30%)) = 0.21; 1H NMR (CD3OD, 600 MHz) δ/ppm: 8.11 (s, 1H), 8.10 (s, 1H), 6.86 (d, J = 8.7 Hz, 2H), 6.60 (d, J = 8.7 Hz, 2H), 4.81 (s, 2H). In the mixture, the ratio of the syn- and anti-isomers is approximately 2:1.
1-(4-nitrophenyl)-1H-1,2,3-triazole-4-carbaldehyde oxime (3): 70 mg, 50% isolated yield; yellow oil; Rf (DCM/MeOH (30%)) = 0.25; 1H NMR (CD3OD, 600 MHz) δ/ppm: 9.29 (s, 1H), 8.48 (d, J = 9.4 Hz, 2H), 8.24 (d, J = 9.2 Hz, 2H), 7.75 (s, 1H). The majority syn-isomer is formed. MS (ESI) m/z (%, fragment): 234 (5); 121 (100); HRMS (m/z) for C9H7N5O3: [M + H]+calcd = 233.0548, and [M + H]+measured = 233.0549.
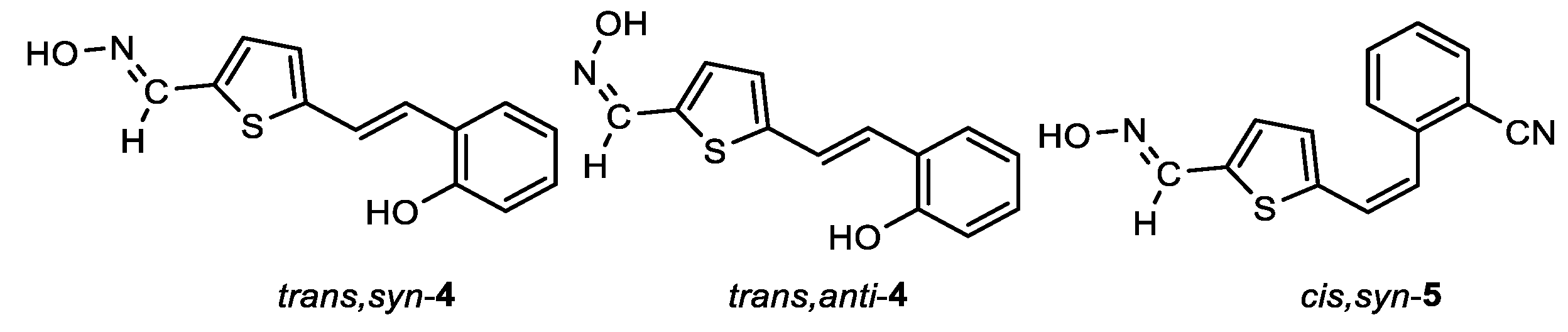
(E)-5-((E)-2-hydroxystyryl)thiophene-2-carbaldehyde oxime (trans,syn-4): 18 mg, 25%; Rf (DCM/MeOH (10%)) = 0.52; 1H NMR (CDCl3, CD3OD, 600 MHz) δ/ppm: 8.20 (s, 1H), 7.46 (d, J = 7.2 Hz, 1H), 7.29 (d, J = 16.1 Hz, 2H), 7.25 (d, J = 16.1 Hz, 1H), 7.11 (t, J = 7.3 Hz, 1H), 7.05 (d, J = 3.8 Hz, 1H), 6.97 (d, J = 4.2 Hz, 1H), 6.87 (t, J = 7.8 Hz, 1H), 6.81 (d, J = 8.2 Hz, 1H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 153.9, 149.1, 145.6, 133.9, 132.4, 130.2, 128.9, 127.3, 125.9, 125.1, 122.2, 120.7, 116.0.
(Z)-5-((E)-2-hydroxystyryl)thiophene-2-carbaldehyde oxime (trans,anti-4): 41 mg, 52% isolated yield; white powder; m.p. = 173 – 175˚C; Rf (DCM/MeOH (10%)) = 0.45; 1H NMR (CDCl3, CD3OD, 600 MHz) δ/ppm: 7.61 (s, 1H), 7.47 (dd, J = 7.6, 1.5 Hz, 1H), 7.38 (d, J = 16.2 Hz, 1H), 7.30 (d, J = 16.2 Hz, 1H), 7.24 (t, J = 3.7 Hz, 1H), 7.13 – 7.10 (m, 1H), 7.04 (d, J = 4.4 Hz, 1H), 6.88 (t, J = 7.5 Hz, 1H), 6.81 (d, J = 8.1 Hz, 1H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 158.6, 152.9, 144.8, 135.9, 133.1, 132.8, 130.8, 129.3, 128.5, 127.8, 125.5, 123.8, 119.6.
MS (ESI) m/z (%, fragment): 242 (100); 200 (30); HRMS (m/z) for C13H9NO2S: [M + H]+calcd = 243.0354, and [M + H]+measured = 243.0355.
2-((Z)-2-(5-((E)-(hydroxyimino)methyl)thiophen-2-yl)vinyl)benzonitrile (cis,syn-5): 38 mg, 33% isolated yield; white powder; m.p. = 171 – 173˚C; Rf (DCM/MeOH (5%)) = 0.65; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.12 (s, 1H), 7.72 (d, J = 7.9 Hz, 1H), 7.60 – 7.55 (m, 2H), 7.43 (t, J = 7.1 Hz, 1H), 7.09 (s, 1H), 6.99 (d, J = 4.1 Hz, 1H), 6.91 (d, J = 4.1 Hz, 1H), 6.88 (d, J = 12.1 Hz, 1H), 6.69 (d, J = 12.1 Hz, 1H); MS (ESI) m/z (%, fragment): 255 (100); HRMS (m/z) for C14H10N2OS: [M + H]+calcd = 254.0513, and [M + H]+measured = 254.0514.
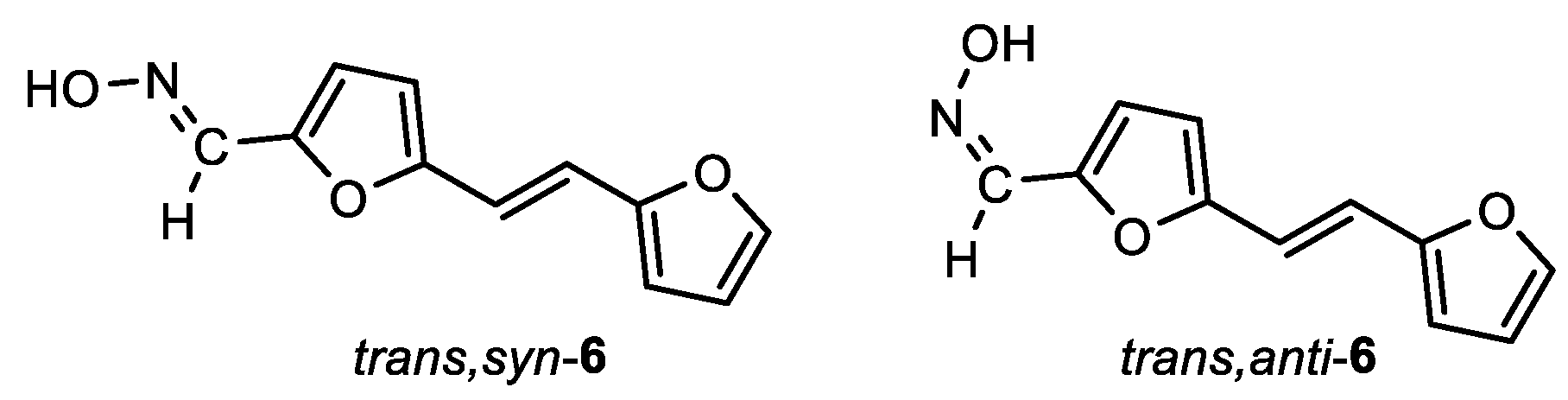
(E)-5-((E)-2-(furan-2-yl)vinyl)furan-2-carbaldehyde oxime (trans,syn-6): 15 mg, 23%; Rf (DCM/MeOH (10%)) = 0.56; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.97 (s, 1H), 7.45 (s, 1H), 6.97 (d, J = 16.2 Hz, 1H), 6.77 (d, J = 16.2 Hz, 1H), 6.63 (d, J = 3.6 Hz, 1H), 6.42 – 6.41 (m, 1H), 6.38 (d, J = 3.7 Hz, 1H), 6.37 (d, J = 3.2 Hz, 1H).
(Z)-5-((E)-2-(furan-2-yl)vinyl)furan-2-carbaldehyde oxime (trans,anti-6): 34 mg, 48% isolated yield; white powder; m.p. = 150 – 153˚C; Rf (DCM/MeOH (10%)) = 0.53; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.51 (s, 1H), 7.41 (d, J = 1.8 Hz, 1H), 7.31 (d, J = 3.7 Hz, 1H), 6.93 (d, J = 16.2 Hz, 1H), 6.79 (d, J = 16.2 Hz, 1H), 6.46 (d, J = 3.6 Hz, 1H), 6.44 – 6.43 (m, 1H), 6.40 (d, J = 3.4 Hz, 1H).
MS (ESI) m/z (%, fragment): 204 (100); HRMS (m/z) for C11H10NO3: [M + H]+calcd = 203.0582, and [M + H]+measured = 203.0586.
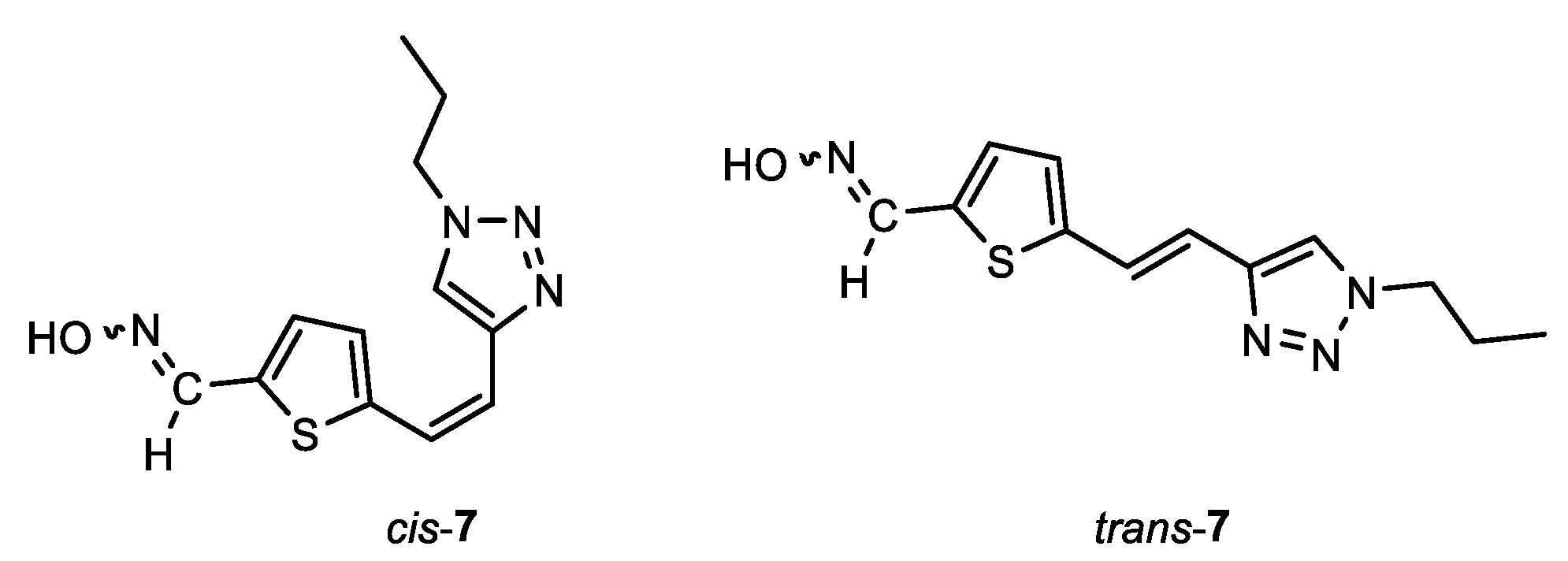
(E)-5-((Z)-2-(1-propyl-1H-1,2,3-triazol-4-yl)vinyl)thiophene-2-carbaldehyde oxime (cis,syn-7): 17 mg; 9%; Rf (DCM/MeOH (5%)) = 0.22; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.21 (s, 1H), 7.53 (s, 1H), 7.08 – 7.06 (m, 2H), 6.69 (d, J = 12.4 Hz, 1H), 6.63 (d, J = 12.4 Hz, 1H), 4.35 – 4.29 (m, 2H), 1.98 – 1.91 (m, 2H), 0.99 – 0.95 (m, 3H).
(Z)-5-((Z)-2-(1-propyl-1H-1,2,3-triazol-4-yl)vinyl)thiophene-2-carbaldehyde oxime (cis,anti-7): 20 mg; 11%; Rf (DCM/MeOH (5%)) = 0.22; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.64 (s, 1H), 7.63 (s, 1H), 7.10 (d, J = 3.2 Hz, 1H), 7.08 – 7.06 (m, 1H), 6.65 (d, J = 12.1 Hz, 1H), 6.58 (d, J = 12.1 Hz, 1H), 4.35 – 4.29 (m, 2H), 1.98 – 1.91 (m, 2H), 0.99 – 0.95 (m, 3H).
(E)-5-((E)-2-(1-propyl-1H-1,2,3-triazol-4-yl)vinyl)thiophene-2-carbaldehyde oxime (trans,syn-7): 22 mg; 12%; Rf (DCM/MeOH (5%)) = 0.21; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.22 (s, 1H), 7.53 (s, 1H), 7.47 (d, J = 15.6 Hz, 1H), 7.31 (d, J = 3.8 Hz, 1H), 7.29 (d, J = 3.8 Hz, 1H), 7.01 (d, J = 15.6 Hz, 1H), 4.35 – 4.29 (m, 2H), 1.98 – 1.91 (m, 2H), 0.99 – 0.95 (m, 3H).
(Z)-5-((E)-2-(1-propyl-1H-1,2,3-triazol-4-yl)vinyl)thiophene-2-carbaldehyde oxime (trans,anti-7): 19 mg; 10%; Rf (DCM/MeOH (5%)) = 0.21; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.65 (s, 1H), 7.61 (s, 1H), 7.42 (d, J = 15.4 Hz, 1H), 7.08 (d, J = 3.7 Hz, 1H), 7.08 (d, J = 4.2 Hz, 1H), 6.92 (d, J = 15.4 Hz, 1H), 4.35 – 4.29 (m, 2H), 1.98 – 1.91 (m, 2H), 0.99 – 0.95 (m, 3H).
MS (ESI) m/z (%, fragment): 263 (100); HRMS (m/z) for C12H15N4OS: [M + H]+calcd = 262.0888, and [M + H]+measured = 262.0890.
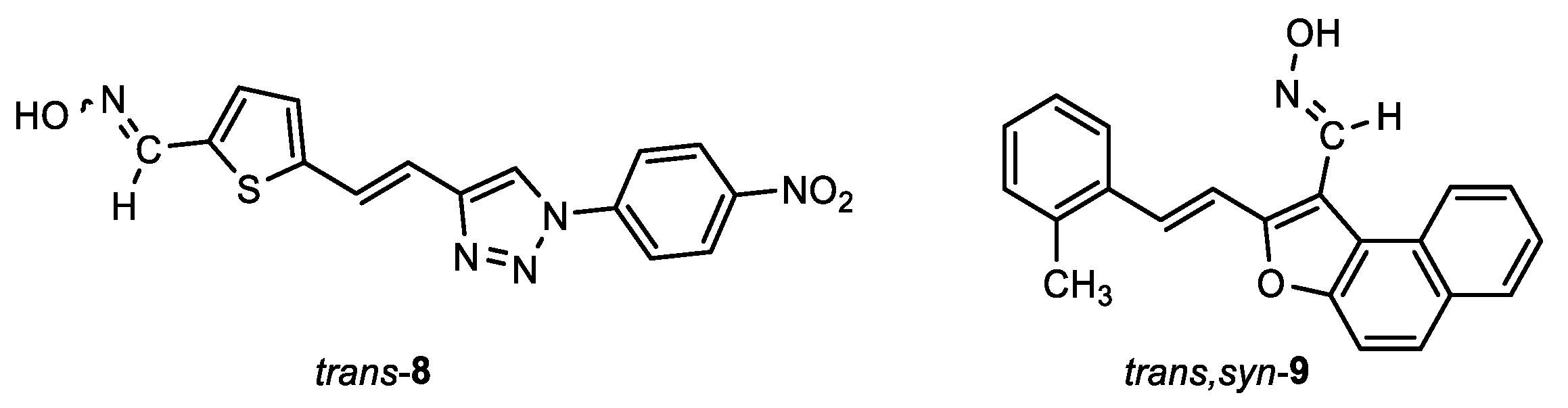
5-(2-(1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)vinyl)thiophene-2-carbaldehyde oxime (trans-8): 6 mg, 2%; Rf (DCM/MeOH (5%)) = 0.12; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.82 (s, 1H), 8.79 (s, 1H), 8.48 (d, J = 9.8 Hz, 2H), 8.19 (d, J = 9.6 Hz, 2H), 7.67 (d, J = 16.6 Hz, 1H), 7.65 (d, J = 4.7 Hz, 1H), 7.63 (d, J = 5.1 Hz, 1H), 7.61 (d, J = 16.6 Hz, 1H); MS (ESI) m/z (%, fragment): 342 (30); 121 (100); HRMS (m/z) for C15H11N5O3S: [M + H]+calcd = 341.0583, and [M + H]+measured = 341.0574.
(E)-2-((E)-2-methylstyryl)naphtho[2,1-b]furan-1-carbaldehyde oxime (trans,syn-9): 38 mg, 40% isolated yield; yellow powder; m.p. = 148 – 152˚C; Rf (DCM) = 0.35; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.60 (s, 1H), 7.50 – 7.45 (m, 1H), 7.40 (d, J = 16.6 Hz, 1H), 7.30 (d, J = 16.6 Hz, 1H) 7.25 (d, J = 3.7 Hz, 1H), 7.11 (t, J = 7.9 Hz, 1H), 7.04 (d, J = 3.7 Hz, 1H), 6.87 (t, J = 7.9 Hz, 1H), 6.83 (d, J = 8.5 Hz, 1H), 3.22 (s, 3H); MS (ESI) m/z (%, fragment): 328 (100); 121 (30); HRMS (m/z) for C22H17NO2: [M + H]+calcd = 327.1259, and [M + H]+measured = 327.1254.
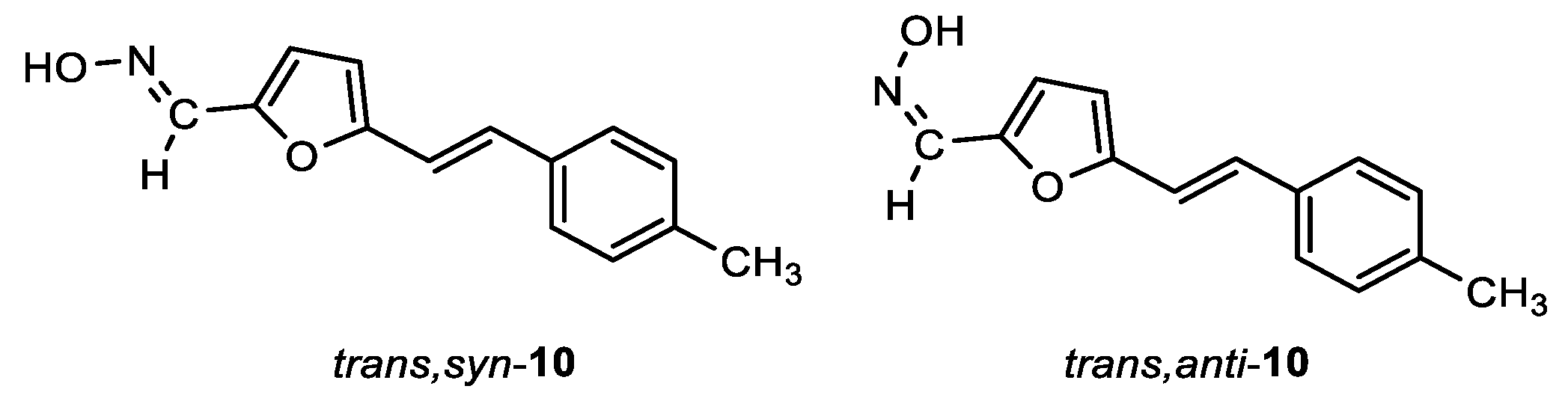
(E)-5-((E)-4-methylstyryl)furan-2-carbaldehyde oxime (trans,syn-10): 203 mg, 28% isolated yield; white powder; m.p. = 153 – 154˚C; Rf (DCM/MeOH (1%)) = 0.35; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.99 (s, 1H), 7.59 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.17 – 7.15 (m, 3H), 6.83 (d, J = 16.3 Hz, 1H), 6.68 (d, J = 3.5 Hz, 1H), 6.38 (d, J = 3.5 Hz, 1H), 2.35 (s, 3H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 155.3, 146.2, 140.2, 138.1, 133.8, 129.5, 129.5, 126.5, 115.4, 114.7, 109.9, 21.3; MS (ESI) m/z (%, fragment): 228 (100); HRMS (m/z) for C14H13NO2: [M + H]+calcd = 227.0946, and [M + H]+measured = 227.0947.
(Z)-5-((E)-4-methylstyryl)furan-2-carbaldehyde oxime (trans,anti-10): 168 mg, 23% isolated yield; yellow oil; Rf (DCM/MeOH (1%)) = 0.26; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.53 (s, 1H), 7.38 (d, J = 7.1 Hz, 2H), 7.31 (d, J = 3.1 Hz, 1H), 7.16 (d, J = 7.7 Hz, 2H), 7.15 (d, J = 16.5 Hz, 1H), 6.84 (d, J = 16.5 Hz, 1H), 6.46 (d, J = 3.3 Hz, 1H), 2.36 (s, 3H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 153.9, 144.0, 138.2, 137.4, 133.7, 129.7, 126.5, 120.3, 114.9, 110.8, 21.3.
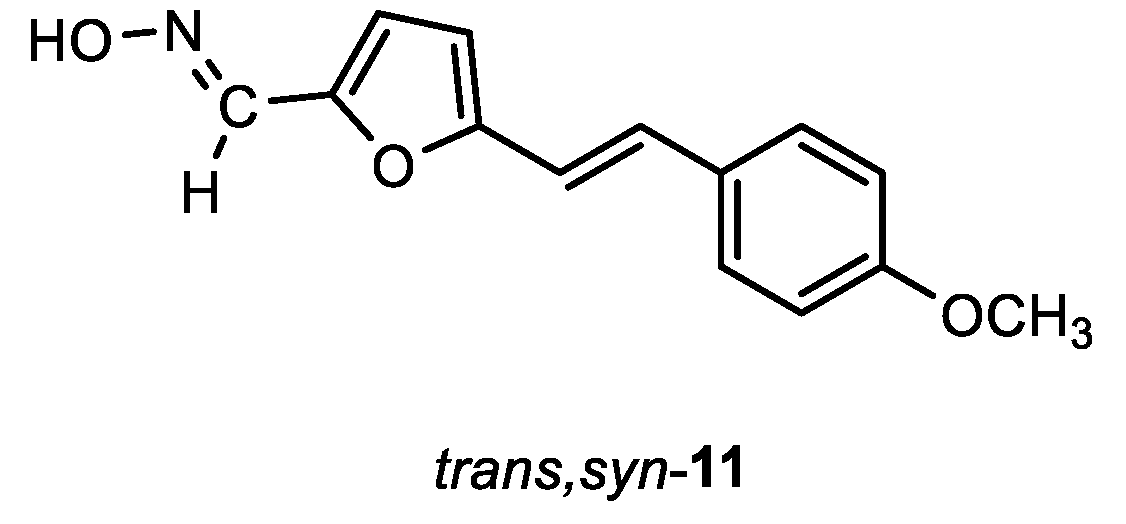
(E)-5-((E)-4-methoxystyryl)furan-2-carbaldehyde oxime (trans,syn-11): 7 mg, 6% isolated yield; yellow powder; m.p. = 160 – 162˚C; Rf (DCM/E (1%)) = 0.26; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.20 (s, 1H), 7.861 (s, 1H), 7.43 – 7.41 (m, 3H), 7.03 (d, J = 15.8 Hz, 1H), 6.91 – 6.84 (m, 3H), 6.71 (d, J = 1.2 Hz, 1H), 3.82 (s, 3H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 159.1, 143.9, 142.4, 139.2, 130.3, 129.1, 127.3, 126.2, 114.4, 113.7, 108.4, 54.8.
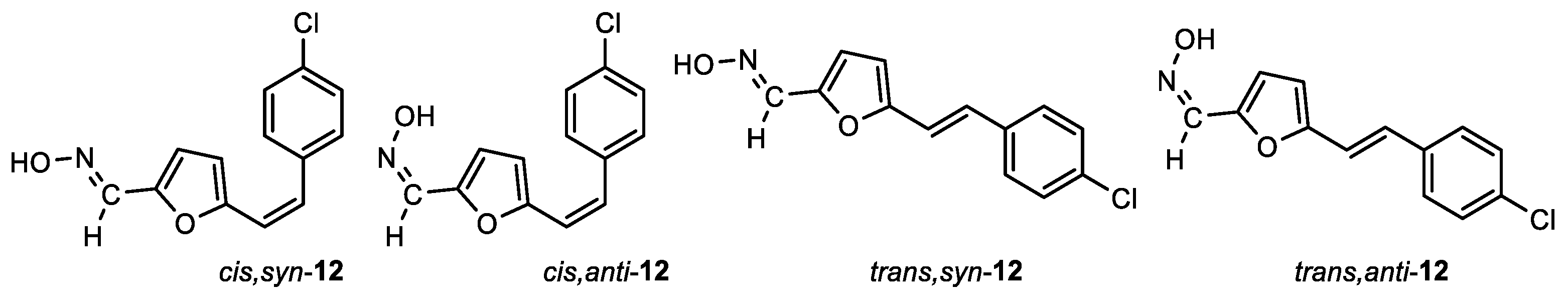
(E)-5-((Z)-4-chlorostyryl)furan-2-carbaldehyde oxime (cis,syn-12): 39 mg, 42% isolated yield; white powder; m.p. = 178 – 180˚C; Rf (DCM/MeOH (1%)) = 0.73; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.0 (s, 1H), 7.91 (s, 1H), 7.40 (d, J = 7.6 Hz, 2H), 7.31 (d, J = 7.6 Hz, 2H), 6.56 (d, J = 3.4 Hz, 1H), 6.50 (d, J = 12.6 Hz, 1H), 6.38 (d, J = 12.6 Hz, 1H), 6.28 (d, J = 3.5 Hz, 1H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 153.4, 146.0, 140.1, 135.3, 133.5, 130.0, 128.9, 128.5, 118.0, 114.2, 111.9; MS (ESI) m/z (%, fragment): 248 (100); HRMS (m/z) for C13H10ClNO2: [M + H]+calcd = 247.0400, and [M + H]+measured = 247.0401.
(Z)-5-((Z)-4-chlorostyryl)furan-2-carbaldehyde oxime (cis,anti-12): 15 mg, 14%; Rf (DCM/MeOH (1%)) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.63 (s, 1H), 7.39 (s, 1H), 7.38 – 7.28 (m, 4H), 7.23 (d, J = 3.2 Hz, 1H), 6.54 (d, J = 12.8 Hz, 1H), 6.38 (d, J = 3.4 Hz, 1H), 6.37 (d, J = 12.8 Hz, 1H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 152.2, 143.8, 137.0, 135.3, 133.5, 130.1 129.3, 128.3, 119.7, 117.8, 113.0.
(E)-5-((E)-4-chlorostyryl)furan-2-carbaldehyde oxime (trans,syn-12): 19 mg, 20%; Rf (DCM/MeOH (1%)) = 0.44; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.98 (s, 1H), 7.49 (s, 1H), 7.39 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 16.3 Hz, 1H), 6.84 (d, J = 16.2 Hz, 1H), 6.65 (d, J = 3.5 Hz, 1H), 6.42 (d, J = 3.5 Hz, 1H); 13C NMR (CDCl3, 600 MHz) δ/ppm: 154.7, 146.6, 140.2, 135.1, 133.7, 128.9, 128.0, 127.6, 116.1, 115.3, 110.7.
(Z)-5-((E)-4-chlorostyryl)furan-2-carbaldehyde oxime (trans,anti-12): 5 mg, 5%; Rf (DCM/MeOH (1%)) = 0.34; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.10 (s, 1H), 7.99 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.9 Hz, 2H), 7.14 (d, J = 16.1 Hz, 1H), 6.83 (d, J = 16.1 Hz, 1H), 6.65 (d, J = 3.1 Hz, 1H), 6.42 (d, J = 3.4 Hz, 1H).
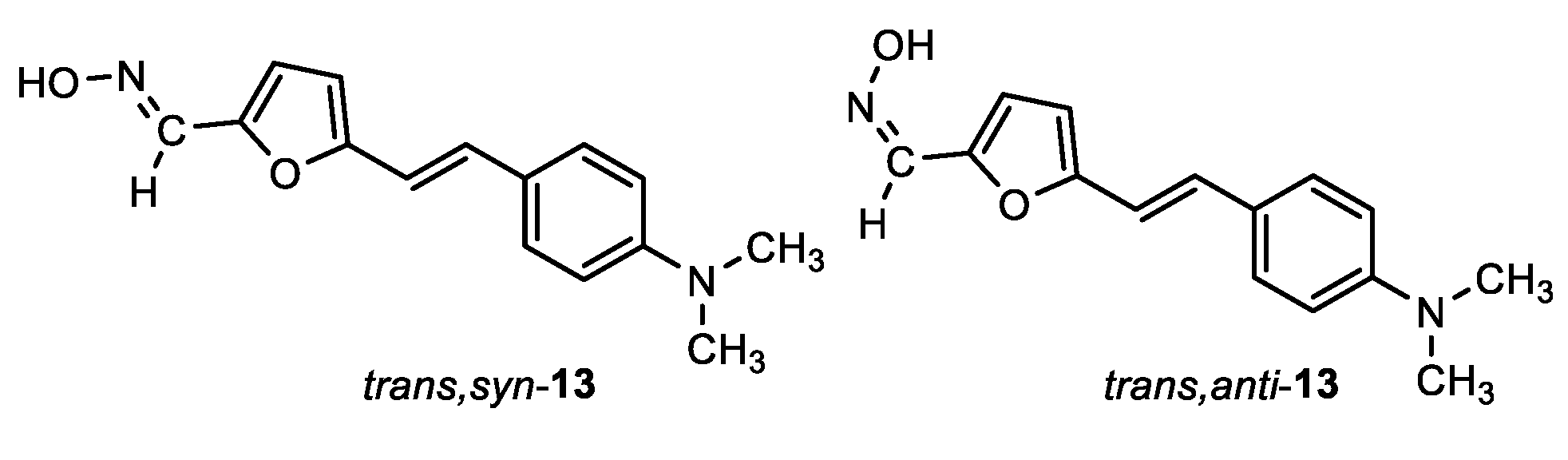
(E)-5-((E)-4-(dimethylamino)styryl)furan-2-carbaldehyde oxime (trans,syn-13): 10 mg, 5%; Rf (DCM/MeOH (5%)) = 0.65; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.96 (s, 1H), 7.38 (d, J = 8.8 Hz, 2H), 7.37 (d, J = 8.8 Hz, 2H), 7.09 (d, J = 16.5 Hz, 1H), 6.60 (d, J = 16.5 Hz, 1H), 6.62 (d, J = 3.5 Hz, 1H), 6.31 (d, J = 3.5 Hz, 1H), 2.99 (s, 6H).
(Z)-5-((E)-4-(dimethylamino)styryl)furan-2-carbaldehyde oxime (trans,anti-13): 11 mg, 7%; Rf (DCM/MeOH (5%)) = 0.59; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.51 (s, 1H), 7.38 (d, J = 8.8 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 3.7 Hz, 1H), 7.13 (d, J = 16.4 Hz, 1H), 7.09 (d, J = 16.4 Hz, 1H), 6.39 (d, J = 3.6 Hz, 1H), 2.99 (s, 6H).
Data for the isomers written from the 1H NMR of the mixture of isomers. MS (ESI) m/z (%, fragment): 239 (100); HRMS (m/z) for C14H10N2O2: [M + H]+calcd = 238.0742, and [M + H]+measured = 238.0741.
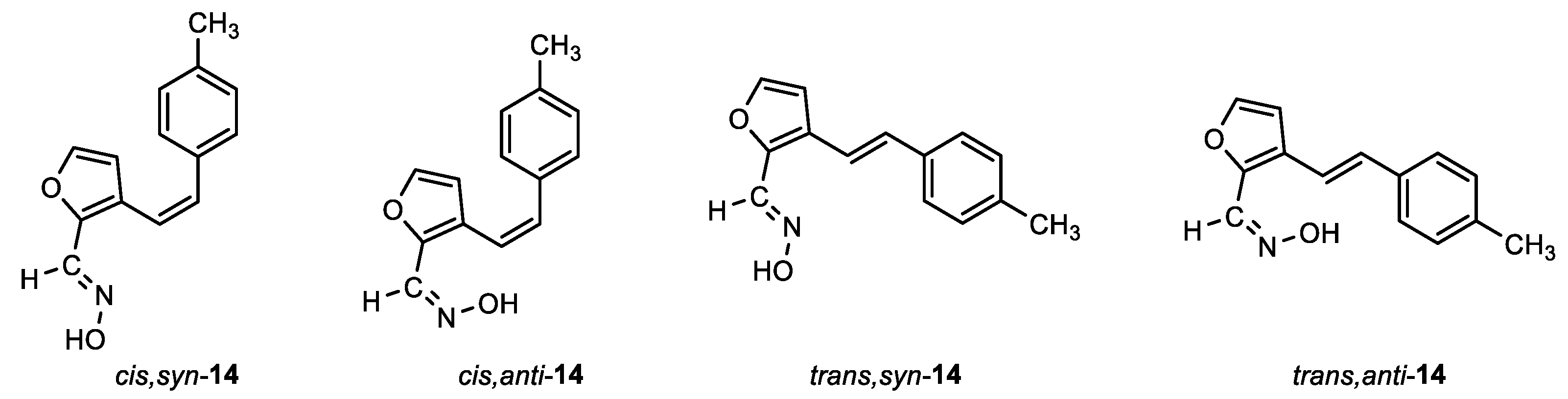
(E)-3-((Z)-4-methylstyryl)furan-2-carbaldehyde oxime (cis,syn-14): 113 mg, 38% isolated yield; white powder; m.p. = 145 – 148˚C; Rf (PE/E (15%)) = 0.27; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.08 (s, 1H), 8.11 (s, 1H), 7.27 (d, J = 1.6 Hz, 1H), 7.19 (d, J = 8.0, 2H), 7.09 (d, J = 8.0, 2H), 6.65 (d, J = 12.2 Hz, 1H), 6.44 (d, J = 12.2 Hz, 1H), 6.17 (d, J = 1.6 Hz, 1H), 2.33 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.1, 143.6, 139.4, 137.5, 134.0, 132.4, 129.0, 128.8, 124.3, 117.9, 111.9, 21.3.
(Z)-3-((Z)-4-methylstyryl)furan-2-carbaldehyde oxime (cis,anti-14): 2 mg, 1%; Rf (PE/E (15%)) = 0.22; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.43 (d, J = 1.7 Hz, 1H), 7.26 (s, 1H), 7.18 (d, J = 7.9 Hz, 2H), 7.09 (d, J = 7.9 Hz, 2H), 6.70 (d, J = 11.8 Hz, 1H), 6.45 (d, J = 11.8 Hz, 1H), 6.22 (d, J = 1.7 Hz, 1H), 2.34 (s, 3H).
(E)-3-((E)-4-methylstyryl)furan-2-carbaldehyde oxime (trans,syn-14): 70 mg, 34% isolated yield; white powder; m.p. = 146 – 147˚C; Rf (PE/E (10%)) = 0.17; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.23 (s, 1H), 7.45 (s, 1H), 7.43 (d, J = 1.9 Hz, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 16.2 Hz, 1H), 6.89 (d, J = 16.2 Hz, 1H), 6.72 (d, J = 1.9 Hz, 1H), 2.36 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.4, 143.2, 140.1, 138.0, 134.1, 131.2, 129.5, 126.5, 126.4, 116.1, 109.0, 21.3.
(Z)-3-((E)-4-methylstyryl)furan-2-carbaldehyde oxime (trans,anti-14): 5 mg, 2%; Rf (PE/E (10%)) = 0.10; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.49 (d, J = 1.7 Hz, 1H), 7.44 (s, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.19 (s, 1H), 7.10 (d, J = 8.0 Hz, 2H), 7.05 (d, J = 16.1 Hz, 1H), 6.84 (d, J = 16.1 Hz, 1H), 6.67 (d, J = 1.7 Hz, 1H), 2.29 (s, 3H).
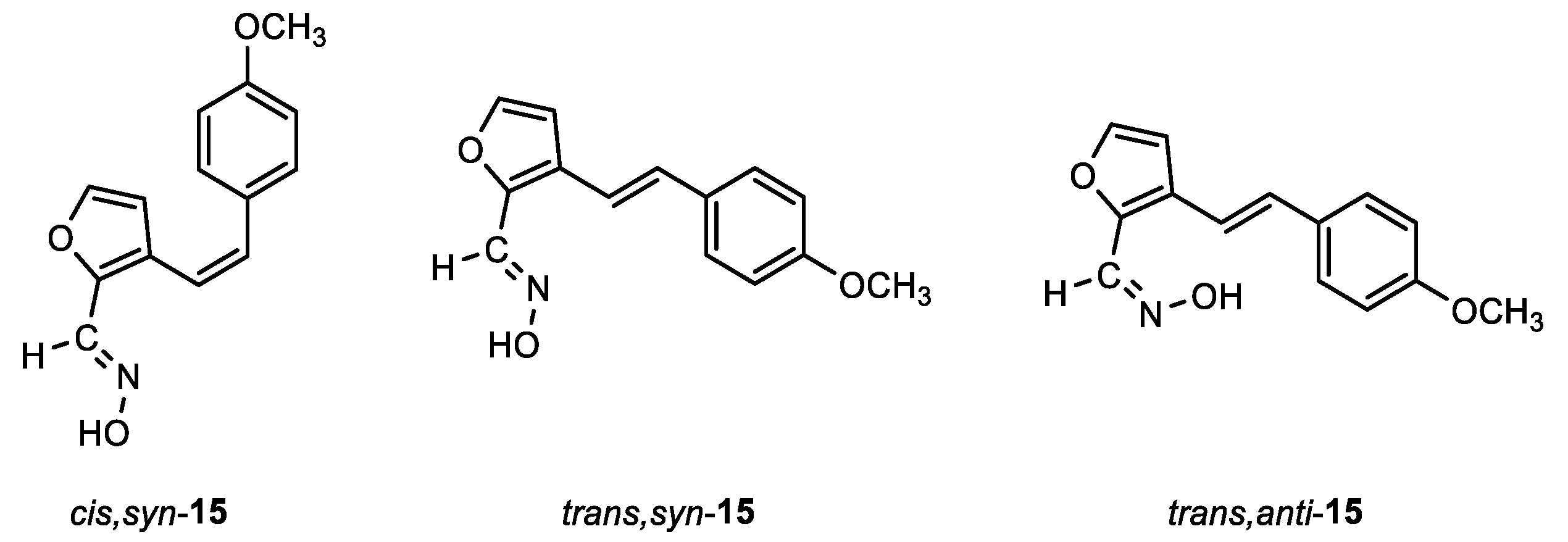
(E)-3-((Z)-4-methoxystyryl)furan-2-carbaldehyde oxime (cis,syn-15): 26 mg, 15%; Rf (PE/DCM (60%)) = 0.25; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.10 (s, 1H), 7.55 (s, 1H), 7.30 (d, J = 1.8 Hz, 1H), 7.24 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 9.2 Hz, 2H), 6.63 (d, J = 12.3 Hz, 1H), 6.42 (d, J = 12.3 Hz, 1H), 6.21 (d, J = 1.9 Hz, 1H), 3.81 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 159.1, 143.9, 143.6, 140.1, 132.0, 130.2, 129.4, 124.8, 117.2, 113.7, 111.9, 55.2.
(E)-3-((E)-4-methoxystyryl)furan-2-carbaldehyde oxime (trans,syn-15): 36 mg, 15%; Rf (PE/DCM (60%)) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.23 (s, 1H), 7.84 (s, 1H), 7.42 (d, J = 8.6 Hz, 3H), 7.03 (d, J = 16.6 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 16.6 Hz, 1H), 6.71 (d, J = 2.1 Hz, 1H), 3.83 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 159.6, 144.4, 142.9, 139.9, 130.8, 129.7, 127.8, 126.7, 114.9, 114.2, 108.9, 55.3; MS (ESI) m/z (%, fragment): 244 (100); HRMS (m/z) for C14H13NO3: [M + H]+calcd = 243.0895, and [M + H]+measured = 243.0901.
(Z)-3-((E)-4-methoxystyryl)furan-2-carbaldehyde oxime (trans,anti-15): 70 mg, 26% isolated yield; white powder; m.p. = 158 – 164˚C; Rf (PE/DCM (60%)) = 0.13; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.23 (s, 1H), 7.93 (s, 1H), 7.42 (d, J = 9.1 Hz, 3H), 7.03 (d, J = 15.9 Hz, 1H), 6.89 (d, J = 9.1 Hz, 2H), 6.87 (d, J = 15.9 Hz, 1H), 6.71 (d, J = 1.9 Hz, 1H), 3.83 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 159.6, 144.4, 142.9, 139.9, 130.8, 129.7, 127.8, 126.7, 114.9, 114.2, 108.9, 55.3.
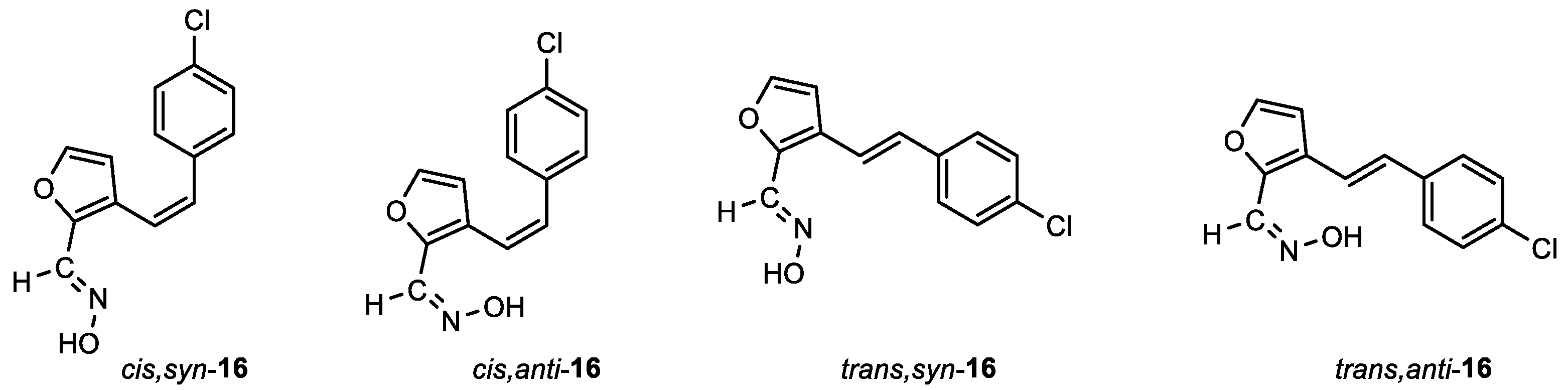
(E)-3-((Z)-4-chlorostyryl)furan-2-carbaldehyde oxime (cis,syn-16): 114 mg, 24% isolated yield; white powder; m.p. = 189 – 191˚C; Rf (PE/E (15%)) = 0.47; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.10 (s, 1H), 7.99 (s, 1H), 7.28 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.5 Hz, 2H), 6.63 (d, J = 12.0 Hz, 1H), 6.55 (d, J = 12.0 Hz, 1H), 6.12 (d, J = 1.2 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.4, 143.7, 139.8, 135.5, 133.4, 130.9, 130.2, 128.5, 124.0, 119.4, 111.7; MS (ESI) m/z (%, fragment): 248 (100); HRMS (m/z) for C13H10ClNO2: [M + H]+calcd = 247.0400, and [M + H]+measured = 247.0405.
(Z)-3-((Z)-4-chlorostyryl)furan-2-carbaldehyde oxime (cis,anti-16): 5 mg, 1%; Rf (PE/E (15%)) = 0.28; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.44 (d, J = 1.6 Hz, 1H), 7.35 (s, 1H), 7.26 (d, J = 8.6 Hz, 2H), 7.21 (d, J = 8.6 Hz, 2H), 6.66 (d, J = 16.2 Hz, 2H), 6.53 (d, J = 16.2 Hz, 2H), 6.16 (d, J = 1.8 Hz, 1H).
(E)-3-((E)-4-chlorostyryl)furan-2-carbaldehyde oxime (trans,syn-16): 48 mg, 20% isolated yield; white powder; m.p. = 181 – 183˚C; Rf (PE/E (15%)) = 0.40; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.22 (s, 1H), 7.48 (s, 1H), 7.48 (s, 1H), 7.44 (d, J = 1.6 Hz, 1H), 7.41 (d, J = 8.5 Hz, 2H), 7.32 (d, J = 8.5 Hz, 1H), 7.19 (d, J = 16.1 Hz, 1H), 6.86 (d, J = 16.1 Hz, 1H), 6.72 (d, J = 1.7 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.4, 143.7, 140.2, 135.4, 133.5, 129.8, 128.9, 127.6, 125.8, 118.0, 108.9.
(Z)-3-((E)-4-chlorostyryl)furan-2-carbaldehyde oxime (trans,anti-16): 12 mg, 3%; Rf (PE/E (15%)) = 0.25; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.61 (d, J = 1.6 Hz, 1H), 7.50 (s, 1H), 7.42 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.5 Hz, 2H), 7.17 (d, J = 16.1 Hz, 1H), 6.88 (d, J = 16.1 Hz, 1H), 6.74 (d, J = 1.6 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.9, 144.5, 140.1, 135.4, 134.3, 130.3, 129.8, 129.0, 127.9, 117.9, 108.7.
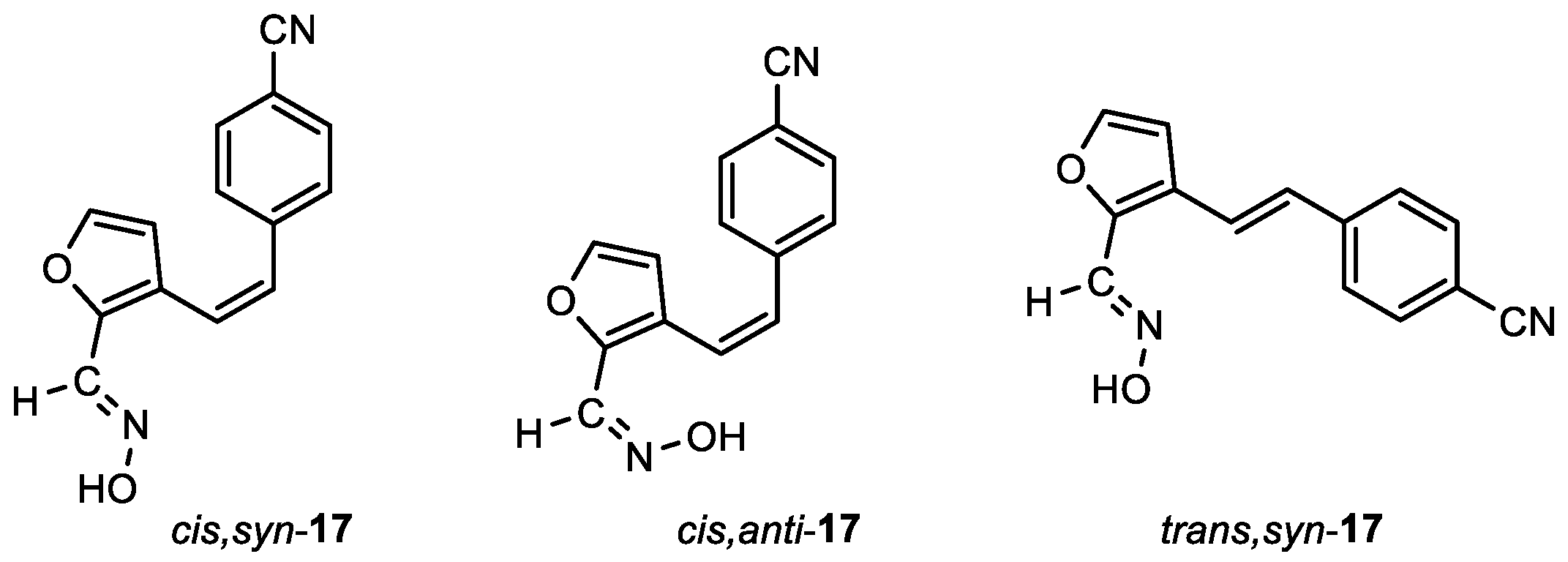
4-((Z)-2-(2-((E)-(hydroxyimino)methyl)furan-3-yl)vinyl)benzonitrile (cis,syn-17): 141 mg, 43% isolated yield; white powder; m.p. = 198 – 201˚C; Rf (PE/E (20%)) = 0.56; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.10 (s, 1H), 7.59 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 1.9 Hz, 1H), 6.71 (d, J =12.1 Hz, 1H), 6.64 (d, J =12.1 Hz, 1H), 6.07 (d, J = 1.9 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 143.9, 140.2, 132.5, 132.2, 130.0, 129.9, 129.5, 126.8, 123.1, 121.7, 117.7, 111.6; MS (ESI) m/z (%, fragment): 239 (100); HRMS (m/z) for C14H10N2O2: [M + H]+calcd = 247.0400, and [M + H]+measured = 247.0405.
4-((Z)-2-(2-((Z)-(hydroxyimino)methyl)furan-3-yl)vinyl)benzonitrile (cis,anti-17): 17 mg, 6%; Rf (PE/E (20%)) = 0.34; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.58 (d, J = 8.3 Hz, 2H), 7.45 (d, J = 1.9 Hz, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.33 (s, 1H), 6.69 (d, J = 12.3 Hz, 1H), 6.67 (d, J = 12.3 Hz, 1H), 6.11 (d, J = 1.9 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.5, 141.4, 134.2, 132.1, 130.7, 129.5, 125.5, 121.9, 118.7, 111.5, 111.1, 103.0.
4-((E)-2-(2-((E)-(hydroxyimino)methyl)furan-3-yl)vinyl)benzonitrile (trans,syn-17): 47 mg, 16% isolated yield; white powder; m.p. = 193 – 195˚C; Rf (PE/E (30%)) = 0.53; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.23 (s, 1H), 7.76 (s, 1H), 7.63 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.2 Hz, 2H), 7.47 (d, J = 1.8 Hz, 1H), 7.37 (d, J = 16.3 Hz, 1H), 6.90 (d, J = 16.3 Hz, 1H), 6.74 (d, J = 1.8 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.6, 144.5, 141.4, 140.3, 132.6, 132.5, 126.9, 125.0, 121.2, 118.9, 110.8, 108.9.
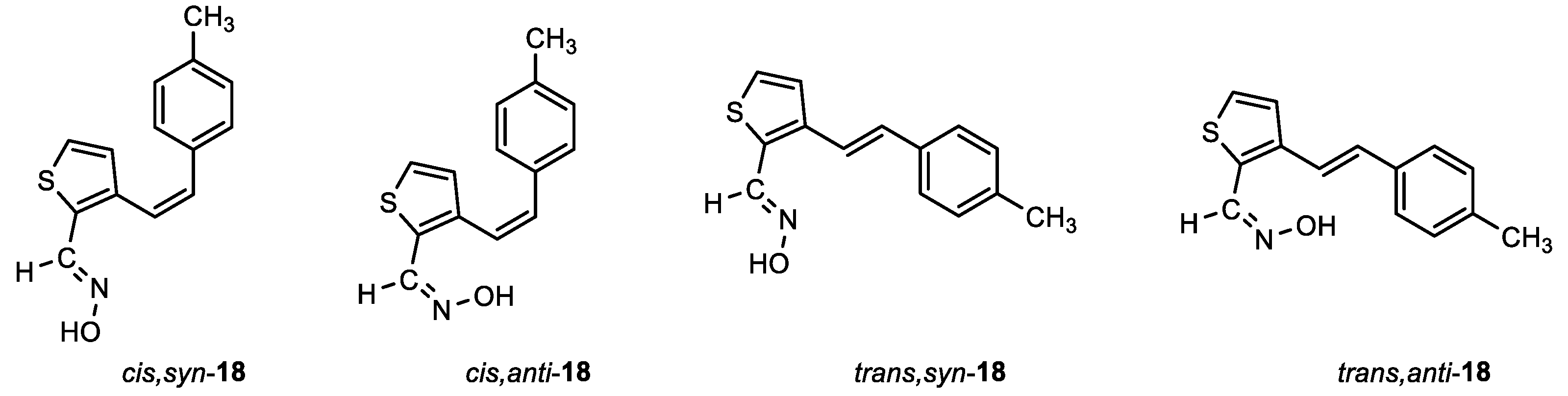
(E)-3-((Z)-4-methylstyryl)thiophene-2-carbaldehyde oxime (cis,syn-18): 72 mg, 28% isolated yield; white powder; m.p. = 158 – 160˚C; Rf (PE/E (10%)) = 0.53; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.32 (s, 1H), 7.15 (d, J = 5.2 Hz, 2H), 7.09 (d, J = 8.2 Hz, 2H), 7.04 (d, J = 8.2 Hz, 2H), 6.79 (d, J = 5.3 Hz, 1H), 6.68 (d, J = 12.1 Hz, 1H), 6.50 (d, J = 12.1 Hz, 1H), 2.31 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.5, 140.0, 137.6, 132.8, 130.4, 129.0, 128.9, 128.8, 126.4, 121.6, 121.1, 21.2; MS (ESI) m/z (%, fragment): 244 (100); HRMS (m/z) for C14H13NOS: [M + H]+calcd = 243.0718, and [M + H]+measured = 243.0717.
(Z)-3-((Z)-4-methylstyryl)thiophene-2-carbaldehyde oxime (cis,anti-18): 58 mg, 23% isolated yield; white powder; m.p. = 155 – 156˚C; Rf (PE/E (10%)) = 0.34; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H), 7.42 (d, J = 5.1 Hz, 1H), 7.05 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 5.2 Hz, 1H), 6.73 (d, J = 12.1 Hz, 1H), 6.58 (d, J = 12.1 Hz, 1H), 2.29 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 141.5, 140.1, 137.6, 133.5, 133.5, 130.4, 129.0, 128.9, 127.6, 125.8, 121.5, 99.9, 21.2.
(E)-3-((E)-4-methylstyryl)thiophene-2-carbaldehyde oxime (trans,syn-18): 87 mg, 34% isolated yield; white powder; m.p. = 151 – 153˚C; Rf (PE/E (10%)) = 0.40; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.55 (s, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.34 (s, 1H), 7.30 (k, 2H), 7.18 (d, J = 6.6 Hz, 3H), 6.98 (d, J = 16.1 Hz, 1H), 2.37 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.0, 140.6, 138.2, 134.1, 131.4, 129.8, 129.5, 127.2, 126.5, 125.6, 118.9, 21.3.
(Z)-3-((E)-4-methylstyryl)thiophene-2-carbaldehyde oxime (trans,anti-18): 43 mg, 13%; Rf (PE/E (10%)) = 0.23; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.02 (s, 1H), 7.52 (d, J = 5.4 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 5.4 Hz, 1H), 7.29 (d, J = 8.7 Hz, 1H), 7.19 (d, J = 8.1 Hz, 2H), 7.04 (d, J = 16.1 Hz, 1H), 2.37 (s, 3H).
(E)-3-((Z)-4-methoxystyryl)thiophene-2-carbaldehyde oxime (cis,syn-19): 7 mg, 6%; Rf (DCM) = 0.55; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.32 (s, 1H), 7.69 (s, 1H), 7.16 (d, J = 4.3 Hz, 1H), 7.13 (d, J = 8.6 Hz, 2H), 6.81 (d, J = 5.1 Hz, 1H), 6.80 (d, J = 8.8 Hz, 2H), 6.64 (d, J = 11.8 Hz, 1H), 6.45 (d, J = 11.8 Hz, 1H), 3.78 (s, 3H).
(Z)-3-((Z)-4-methoxystyryl)thiophene-2-carbaldehyde oxime (cis,anti-19): 16 mg, 13%; Rf (DCM) = 0.45; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.81 (s, 1H), 7.44 (d, J = 5.2 Hz, 1H), 7.08 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 5.2 Hz, 1H), 6.74 (d, J = 8.7 Hz, 2H), 6.69 (d, J = 11.9 Hz, 1H), 6.52 (d, J = 11.9 Hz, 1H), 3.77 (s, 3H).
(E)-4-((Z)-4-methoxystyryl)thiophene-2-carbaldehyde oxime (cis,syn-19'): 100 mg, 78% isolated yield; white powder; m.p. = 159 – 160 ˚C; Rf (DCM) = 0.60; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.26 (s, 1H), 8.11 (s, 1H), 7.30 (d, J = 1.7 Hz, 1H), 7.23 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 9.2 Hz, 2H), 6.61 (d, J = 12.0 Hz, 1H), 6.39 (d, J = 12.0 Hz, 1H), 6.19 (d, J = 1.8 Hz, 1H), 3.79 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 159.2, 144.7, 143.1, 138.7, 131.4, 129.8, 123.7, 117.3, 113.7, 111.4, 54.3.
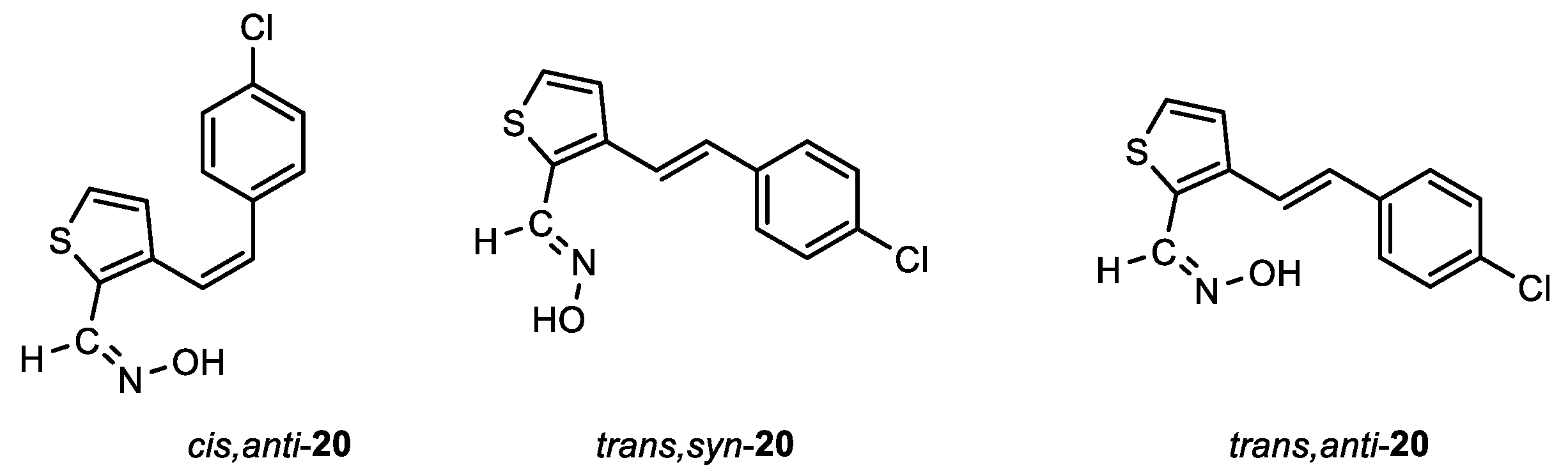
(Z)-3-((Z)-4-chlorostyryl)thiophene-2-carbaldehyde oxime (cis,anti-20): 8 mg, 29%; Rf (PE/E (20%)) = 0.35; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.29 (s, 1H), 7.21 (d, J = 8.5 Hz, 2H), 7.17 (d, J = 5.3 Hz, 1H), 7.12 (d, J = 8.5 Hz, 2H), 6.74 (d, J = 5.3 Hz, 1H), 6.65 (d, J = 12.1 Hz, 1H), 6.58 (d, J = 12.1 Hz, 1H).
(E)-3-((E)-4-chlorostyryl)thiophene-2-carbaldehyde oxime (trans,syn-20): 6 mg, 21%; Rf (PE/E (20%)) = 0.62; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.54 (s, 1H), 7.42 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.6 Hz, 2H), 7.30 (s, 2H) 7.22 (d, J = 16.1 Hz, 1H), 6.95 (d, J = 16.1 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 143.8, 139.9, 135.4, 133.7, 130.0, 129.0, 127.7, 127.4, 125.5, 124.3, 120.5.
(Z)-3-((E)-4-chlorostyryl)thiophene-2-carbaldehyde oxime (trans,anti-20): 8 mg, 29%; Rf (PE/E (20%)) = 0.27; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.93 (s, 1H), 7.53 (s, 2H), 7.42 – 7.38 (m, 2H), 7.35 (d, J = 16.1 Hz, 1H), 7.30 (t, J = 8.4 Hz, 1H), 7.24 – 7.17 (m, 2H), 6.96 (d, J = 16.1 Hz, 1H).
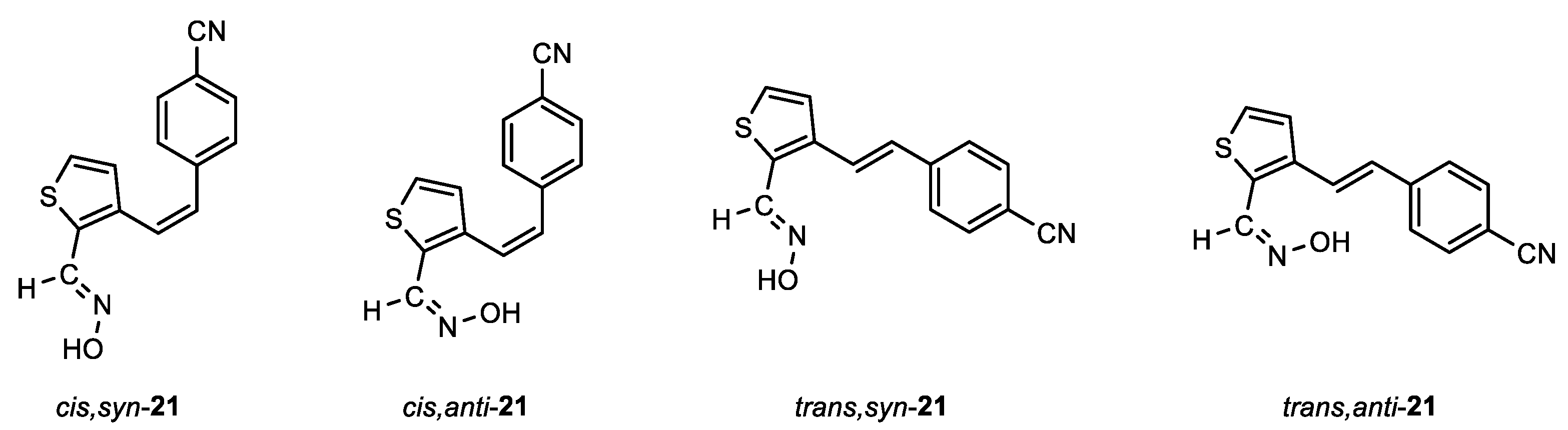
4-((Z)-2-(2-((E)-(hydroxyimino)methyl)thiophen-3-yl)vinyl)benzonitrile (cis,syn-21): 62 mg, 34% isolated yield; yellow powder; m.p. = 169 – 171˚C; Rf (PE/E (25%)) = 0.58; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.25 (s, 1H), 7.53 (d, J = 8.3 Hz, 2H), 7.34 (s, 1H), 7.29 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 5.2 Hz, 1H), 6.74 – 6.66 (m, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 138.6, 136.0, 133.0, 126.9, 126.7, 125.5, 124.2, 123.0, 121.9, 119.9, 113.5, 105.8; MS (ESI) m/z (%, fragment): 255 (100); HRMS (m/z) for C14H10N2OS: [M + H]+calcd = 254.0514, and [M + H]+measured = 254.0512.
4-((Z)-2-(2-((Z)-(hydroxyimino)methyl)thiophen-3-yl)vinyl)benzonitrile (cis,anti-21): 52 mg, 27% isolated yield; yellow powder; m.p. = 170 – 171˚C; Rf (PE/E (25%)) = 0.24; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.75 (s, 1H), 7.50 (d, J = 8.3 Hz, 2H), 7.47 (d, J = 5.4 Hz, 1H), 7.24 (d, J = 8.3 Hz, 2H), 6.83 (d, J = 12.2 Hz, 1H), 6.79 (d, J = 5.3 Hz, 1H), 6.75 (d, J = 12.2 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 143.8, 141.1, 139.8, 138.3, 132.1, 130.8, 129.5, 128.3, 127.1, 125.7, 118.7, 111.0.
4-((E)-2-(2-((E)-(hydroxyimino)methyl)thiophen-3-yl)vinyl)benzonitrile (trans,syn-21): 11 mg, 5%; Rf (PE/E (25%)) = 0.47; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.53 (s, 1H), 7.65 (d, J = 8.3 Hz, 2H), 7.57 (d, J = 8.3 Hz, 2H), 7.36 – 7.35 (m, 4H), 6.98 (d, J = 16.1 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 148.7, 143.6, 141.4, 132.6, 129.2, 127.6, 126.9, 125.5, 123.4, 121.2, 118.9, 111.1.
4-((E)-2-(2-((Z)-(hydroxyimino)methyl)thiophen-3-yl)vinyl)benzonitrile (trans,anti-21): 15 mg, 7%; Rf (PE/E (25%)) = 0.22; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.05 (s, 1H), 7.72 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.64 (s, 1H), 7.60 (s, 1H), 7.49 (d, J = 16.0 Hz, 1H), 7.46 (d, J = 5.4 Hz, 1H), 7.14 (d, J = 16.0 Hz, 1H).
4.2. Oxime ADME Properties and ChE Docking
Physical-chemical properties were calculated using the Calculate molecule properties protocol in BioVia Discovery Studio v21.1 (BioVia, San Diego, CA, USA) and ADMET descriptors calculation to determine the probability of crossing the BBB. Molecular docking of ligands in the active site of AChE and BChE was performed using CDOCKER protocol in BioVia Discovery Studio v21.1 (BioVia, San Diego, CA, USA). The protocol based on a CHARMm force field generated 20 binding poses for each ligand and scoring function CDOCKER Energy was used to rank the obtained binding poses [71,72]. After an evaluation of poses and comparison to known crystal structures of AChE or BChE inhibitor complexes, we selected the final ones [73]. The crystal structure of human BChE and human AChE deposited in the Protein Data Bank, PDB code 2PM8 [47] and 4PQE [74], respectively, were used. The detailed ligand docking protocol used was described previously [5]. Modelling of the near-attack conformation of oxime reactivators in the cyclosarin-inhibited BChE was performed using the structure of phosphorylated cis,syn-5 and cis,syn-12 as described previously [65]. The structure of the cyclosarin-inhibited BChE was generated from the crystal structure of the tabun-inhibited BChE (PDB code 3DJY) [67] using molecular replacement of matching cyclosarin moiety. The final minimised structure of the near-attack conformation of oxime in the cyclosarin-inhibited BChE was submitted to full minimisation using a CHARMm force field with RMS gradient 1.0 kcal/(mol⋅Å).
4.3. AChE and BChE Activity Assays
Stock solutions of oximes (100 mM) were dissolved in dimethyl sulphide solvent (DMSO, Kemika, Zagreb, Croatia), stored at 4 °C and diluted just before use. Acetylthiocholine iodide (ATCh), 5,5′ -dithiobis(2-nitrobenzoic acid) (DTNB), and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Stock solutions (5000 μg/mL) of sarin and cyclosarin purchased from NC Laboratory (Spiez, Switzerland) were made in isopropyl alcohol, and diluted in water just before use. Recombinant human AChE was a generous gift from Dr Zoran Radić, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California at San Diego, La Jolla, USA, while human BChE derived from purified plasma was a generous gift from Dr Xavier Brazzolotto and Dr Florian Nachon, Institut de Recherche Biomédicale des Armées, Bretigny-sur-Orge, France. Enzyme activity was measured using the Ellman method [75] at 25 °C. Enzyme inhibition was assessed using various oxime concentrations ranging from 50 to 150 μM for AChE and 5 to 250 μM for BChE in the presence of ATCh (0.02-0.7 mM) and DTNB (0.3 mM) diluted in 0.1 mM sodium phosphate buffer, pH 7.4 in 96-well plates. The tested concentration of DMSO did not significantly inhibit enzymes. The enzyme-oxime dissociation constant of complex (Ki) was derived using the Hunter-Downs equation as previously described [42]. Measurements were conducted on a Tecan Infinite M200PRO microplate reader at 25 °C for 5 minutes.
For reactivation assays the enzyme was inhibited almost completely (95–100%) during the incubation with a 10-fold excess of the OP for 60 min and the inhibited enzyme was separated from the excess of unconjugated OP by filtration using ProbeQuantTM G-50 Micro Columns (Cytiva, Buckinghamshire, UK). For assessing the recovery of enzyme activity, the inhibited enzyme was added to a reactivation mixture containing an oxime (100 μM in screenings and 10–200 μM in detailed kinetics). Aliquots of the reactivation mixture were diluted at designated intervals in sodium phosphate buffer containing DTNB (0.3 mM) and enzyme activity was measured upon the addition of ATCh (1 mM) on a CARY 300 spectrophotometer (Varian Inc., Mulgrave, Australia) at 25 °C for 2 minutes. An equivalent prepared sample of uninhibited enzyme, whose activity was measured in the presence of oxime, served as a 100% activity control. Spontaneous reactivation was not observed. If the maximal percentage of reactivation (Reactmax) was higher than 40%, the first-order reactivation rate constant (kobs) was determined from the one phase exponential curve of reactivation% over time. Selected oximes were then evaluated in a wide concentration range (10–200 μM) in order to determine the maximal reactivation rate constant (k2), dissociation constant (KOX), and overall second-order reactivation rate constant (kr) as described previously [76]. Results were analysed with Prism 9 software (GraphPad by Dotmatics, Boston, MA, USA).
4.4. Cytotoxicity Assay
Certified human liver cell culture, HepG2 (ECACC 85011430; European Collection of Authenticated Cell Cultures, England) were cultured as adherent cultures under controlled conditions (5% pCO2 and a temperature of 37 °C) in Eagle’s Minimum Essential Medium supplemented with a 1% solution of penicillin-streptomycin (Pen-Strep, Sigma-Aldrich, Germany), 2 mM of glutamine, 10% (v/v) FBS, and 1% non-essential amino acids. The MTS detection reagent assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega, Madison, WI, USA) was used to evaluate in vitro cytotoxicity.
HepG2 cells were subjected to concentrations of the tested oximes spanning from 3 to 400 μM in durations of 24 hours. For utilization of the commercially procured MTS detection reagent assay, cells were cultivated in 96-well plates at a density of 20,000 cells/well and treated with oxime compounds, as in a previously established protocol [40]. After incubation of 24 h at 37 °C in an environment infused with 5% CO2, the cells underwent a PBS buffer rinse and each well received 120 μL of the MTS reagent (diluted with PBS at a 1:6 ratio). In intervals of 0.5 to 3-hour colour incubation, optical density readings were captured at 492 nm using an Infinite M200PRO plate reader (Tecan Austria GmbH, Salzburg, Austria). Inhibitory concentration (IC50) values represent the oxime concentration responsible for a 50% cell mortality rate. The DMSO content within the assay remained consistent at 0.4% and posed no perceptible impact on cell vitality. Data was evaluated using predefined equations from Prism 9 software (GraphPad by Dotmatics, Boston, MA, USA).
4. Conclusions
This paper presents the design and synthesis of uncharged furan, thiophene and triazole oximes by an economical synthesis using the Wittig reaction and Vilsmeier formylation in high yields. The expected targeted products, 26 compounds, were pure cis- and trans-isomers of syn- and anti-oximes containing different substituents bound in the para- or ortho-position of the benzene ring. Based on their binding affinity, reactivation potency, estimated ADME and hepatotoxicity properties, several compounds should be highlighted as potential CNS-active and cholinesterase-targeted therapeutics (Figure 8). The list is headed by two selective BChE inhibitors – trans,anti-4 and 2, three selective AChE inhibitors – trans,syn-18, trans,syn-17, and cys,syn-12, and two non-selective inhibitors of ChE – cis,syn-5 and trans,syn-10. These compounds present a new scaffold for drug development because the current therapy in AD, which basically increase acetylcholine levels in synapses, are selective AChE inhibitors. In Parkinson’s and all stages of AD, non-selective inhibitors of ChE are preferentially in use [21], while selective BChE inhibitors are promising therapeutics for later stages of AD, since BChE activity in certain brain regions increases up to 120% of its normal values during AD progression [35,68]. Even more importantly, in this study we identified CNS-active BChE reactivators – among which cis-derivatives cis,syn-5 and cis,syn-16 showed an exceptional potential for reactivation of cyclosarin-inhibited BChE. The productive and energy-efficient positioning in the active site reported with the near attack modelling, along with an optimal geometry of reactivation transition state, return BChE activity within a short amount of time. Furthermore, as uncharged ligands, these leading compounds are predicted to efficiently cross the BBB and therefore they could potentially achieve significant concentrations both at the neuromuscular junction and in the brain and as such could result in an overall improved therapeutic outcome after OP poisonings or for use in neuromuscular diseases and neurodegenerative disorders.
Graphical Abstract
Figure 8.
New uncharged heterocyclic oximes as potent human AChE (grey) and human BChE (blue) inhibitors and reactivators of human BChE inhibited by cyclosarin (yellow).
Figure 8.
New uncharged heterocyclic oximes as potent human AChE (grey) and human BChE (blue) inhibitors and reactivators of human BChE inhibited by cyclosarin (yellow).
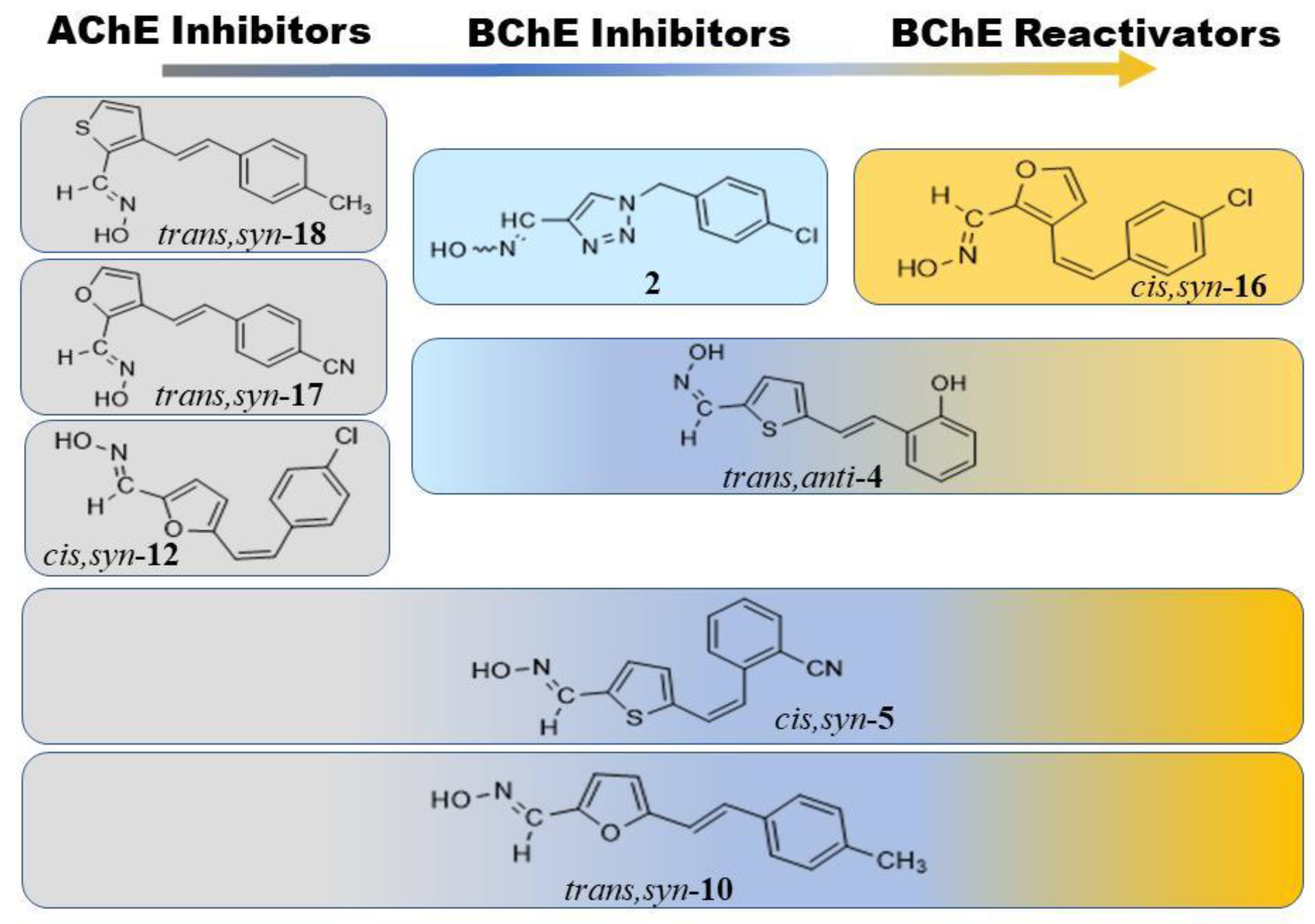
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, NMR spectra (Figures S1–S282), mass spectra and HRMS analyses (Figures S283–S298), evaluation of cytotoxicity (Figure S299), inhibition of AChE and BChE with selected oximes (Figure S300), molecular docking of human AChE and BChE (Figure S301).
Author Contributions
Conceptualization, I.S. and Z.K.; methodology, M.M., T.C.; formal analysis, G.S.; investigation, M.M., T.C. and G.S.; resources, I.S. and Z.K.; writing—original draft preparation, M.M. and T.C.; writing—review and editing, M.M., T.C., G.S., I.S. and Z.K.; supervision, I.S. and Z.K.; funding acquisition, I.S. and Z.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the University of Zagreb for short-term scientific support for 2023 under the title Novel styryl-heterocyclic systems: synthesis, biological activity and computational studies.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy.
Acknowledgments
The authors thank Zlata Lasić from the Pliva pharmaceutical company for MS and HRMS analyses, and the NMR Centre at Ruđer Bošković Institute for recording all the NMR spectra. This research was supported by the University of Zagreb (short-term scientific support for 2022 under the title Experimental and computational studies of new heterocyclic o-divinylbenzenes), Croatian Science Foundation (IP-2018-01-7683 and IP-2022-10-6685), European Regional Development Fund (KK.01.1.1.02.0007) and European Union – Next Generation EU (Class: 643-02/23-01/00016, Reg. no. 533-03-23-0006).
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
References
- B.J. Lukey, J.A.J. Romano, H. Salem, Chemical warfare agents: Chemistry, pharmacology, toxicology, and therapeutics, 2nd Ed, CRC Press, Boca Roton, 2007.
- C.M. Timperley, J.E. Forman, M. Abdollahi, A.S. Al-Amri, A. Baulig, D. Benachour, V. Borrett, F.A. Cariño, M. Geist, D. Gonzalez, W. Kane, Z. Kovarik, R. Martínez-Álvarez, N.M.F. Mourão, S. Neffe, S.K. Raza, V. Rubaylo, A.G. Suárez, K. Takeuchi, C. Tang, F. Trifirò, F.M. van Straten, P.S. Vanninen, S. Vučinić, V. Zaitsev, M. Zafar-Uz-Zaman, M.S. Zina, S. Holen, Advice on assistance and protection provided by the Scientific Advisory Board of the Organisation for the Prohibition of Chemical Weapons: Part 1. On medical care and treatment of injuries from nerve agents, Toxicology. 415 (2019) 56–69. [CrossRef]
- N. Amend, K. V. Niessen, T. Seeger, T. Wille, F. Worek, H. Thiermann, Diagnostics and treatment of nerve agent poisoning—current status and future developments, Ann. N. Y. Acad. Sci. 1479 (2020) 13–28. [CrossRef]
- F. Worek, H. Thiermann, The value of novel oximes for treatment of poisoning by organophosphorus compounds, Pharmacol. Ther. 139 (2013) 249–259. [CrossRef]
- T. Čadež, D. Kolić, G. Šinko, Z. Kovarik, Assessment of four organophosphorus pesticides as inhibitors of human acetylcholinesterase and butyrylcholinesterase, Sci. Rep. 11 (2021) 21486. [CrossRef]
- D.E. Lorke, G.A. Petroianu, Treatment of organophosphate poisoning with experimental oximes: A review, Curr. Org. Chem. 23 (2019) 628–639. [CrossRef]
- K. Kuča, J. Cabal, J. Kassa, A comparison of the efficacy of a bispyridinium oxime - 1,4-bis-(2-hydroxyiminomethylpyridinium) butane dibromide and currently used oximes to reactivate sarin, tabun or cyclosarin-inhibited acetylcholinesterase by in vitro methods, Pharmazie. 59 (2004) 795–798.
- H. Thiermann, F. Worek, Pro: Oximes should be used routinely in organophosphate poisoning, Br. J. Clin. Pharmacol. 88 (2022) 5064–5069. [CrossRef]
- R.K. Sit, Z. Radić, V. Gerardi, L. Zhang, E. Garcia, M. Katalinić, G. Amitai, Z. Kovarik, V. V. Fokin, K.B. Sharpless, P. Taylor, New structural scaffolds for centrally acting oxime reactivators of phosphylated cholinesterases, J. Biol. Chem. 286 (2011) 19422–19430. [CrossRef]
- K. Musilek, D. Malinak, E. Nepovimova, R. Andrys, A. Skarka, K. Kuča, Novel cholinesterase reactivators, in: Handb. Toxicol. Chem. Warf. Agents, 3rd Ed, Academic Press, 2020: pp. 1161–1177. [CrossRef]
- T. Belwal, S.M. Nabavi, S.F. Nabavi, A.R. Dehpour, S. Shirooie, Naturally occurring chemicals against Alzheimer’s disease, Academic, Elsevier, London, 2021. [CrossRef]
- T.S. Anekonda, Resveratrol-A boon for treating Alzheimer’s disease? Brain Res. Rev. 52 (2006) 316–326. [CrossRef]
- D. Martelli, M.J. McKinley, R.M. McAllen, The cholinergic anti-inflammatory pathway: A critical review, Auton. Neurosci. Basic Clin. 182 (2014) 65–69. [CrossRef]
- I. Lee, Y.S. Choe, J.Y. Choi, K.H. Lee, B.T. Kim, Synthesis and evaluation of 18F-labeled styryltriazole and resveratrol derivatives for β-amyloid plaque imaging, J. Med. Chem. 55 (2012) 883–892. [CrossRef]
- G. Wang, Z. Peng, J. Wang, X. Li, J. Li, Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors, Eur. J. Med. Chem. 125 (2017) 423–429. [CrossRef]
- A. Carneiro, M.J. Matos, E. Uriarte, L. Santana, Trending topics on coumarin and its derivatives in 2020, Molecules. 26 (2021) 501. [CrossRef]
- M. Mlakić, T. Čadež, D. Barić, I. Puček, A. Ratković, Ž. Marinić, K. Lasić, Z. Kovarik, I. Škorić, New uncharged 2-thienostilbene oximes as reactivators of organophosphate-inhibited cholinesterases, Pharmaceuticals. 14 (2021) 1147. [CrossRef]
- A. Ratković, M. Mlakić, W. Dehaen, T. Opsomer, D. Barić, I. Škorić, Synthesis and photochemistry of novel 1,2,3-triazole di-heterostilbenes. An experimental and computational study, Spectrochim. Acta - Part A Mol. Biomol. Spectrosc. 261 (2021) 120056. [CrossRef]
- T. Opsomer, K. Valkeneers, A. Ratković, W. Dehaen, 1-(4-nitrophenyl)-1H-1,2,3-triazole-4-carbaldehyde: scalable synthesis and its use in the preparation of 1-alkyl-4-formyl-1,2,3-triazoles, Organics. 2 (2021) 404–414. [CrossRef]
- T.S. Ribeiro, A. Prates, S.R. Alves, J.J. Oliveira-Silva, C.A.S. Riehl, J.D. Figueroa-Villar, The effect of neutral oximes on the reactivation of human acetylcholinesterase inhibited with paraoxon, J. Braz. Chem. Soc. 23 (2012) 1216–1225. [CrossRef]
- A. Matošević, D.M. Opsenica, M. Spasić, N. Maraković, A. Zandona, S. Žunec, M. Bartolić, Z. Kovarik, A. Bosak, Evaluation of 4-aminoquinoline derivatives with an n-octylamino spacer as potential multi-targeting ligands for the treatment of Alzheimer’s disease, Chem. Biol. Interact. 382 (2023). [CrossRef]
- T. Zorbaz, P. Mišetić, N. Probst, S. Žunec, A. Zandona, G. Mendaš, V. Micek, N. Maček Hrvat, M. Katalinić, A. Braïki, L. Jean, P.Y. Renard, V. Gabelica Marković, Z. Kovarik, Pharmacokinetic evaluation of brain penetrating morpholine-3-hydroxy-2-pyridine oxime as an antidote for nerve agent poisoning, ACS Chem. Neurosci. 11 (2020) 1072–1084. [CrossRef]
- H. Pajouhesh, G.R. Lenz, Medicinal chemical properties of successful central nervous system drugs, NeuroRx. 2 (2005) 541–553. [CrossRef]
- C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev. 23 (1997) 3–25. [CrossRef]
- N. Maček Hrvat, T. Zorbaz, G. Šinko, Z. Kovarik, The estimation of oxime efficiency is affected by the experimental design of phosphylated acetylcholinesterase reactivation, Toxicol. Lett. 293 (2018) 222–228. [CrossRef]
- M. Panić, N. Maček Hrvat, M. Štokić, I. Radojčić Redovniković, Z. Kovarik, K. Radošević, Natural deep eutectic solvents improve the solubility of acetylcholinesterase reactivator RS194B, Sustain. Chem. Pharm. 27 (2022) 100654. [CrossRef]
- T. Zorbaz, Z. Kovarik, Neuropharmacology: Oxime antidotes for organophosphate pesticide and nerve agent poisoning, Period. Biol. 121–122 (2020) 35–54. [CrossRef]
- R.K. Sit, Z. Kovarik, N. Maček Hrvat, S. Zunec, C. Green, V. V. Fokin, K.B. Sharpless, Z. Radic, P. Taylor, S. Žunec, C. Green, V. V. Fokin, K.B. Sharpless, Z. Radic, P. Taylor, Pharmacology, pharmacokinetics, and tissue disposition of zwitterionic hydroxyiminoacetamido alkylamines as reactivating antidotes for organophosphate exposure, J. Pharmacol. Exp. Ther. 367 (2018) 363–372. [CrossRef]
- P. Taylor, Y.J. Shyong, N. Samskey, K.Y. Ho, Z. Radić, W. Fenical, K.B. Sharpless, Z. Kovarik, G.A. Camacho-Hernandez, Ligand design for human acetylcholinesterase and nicotinic acetylcholine receptors, extending beyond the conventional and canonical, J. Neurochem. (2021) 1–6. [CrossRef]
- T. Zorbaz, A. Braiki, N. Maraković, J. Renou, E. de la Mora, N. Maček Hrvat, M. Katalinić, I. Silman, J.L. Sussman, G. Mercey, C. Gomez, R. Mougeot, B. Pérez, R. Baati, F. Nachon, M. Weik, L. Jean, Z. Kovarik, P.Y. Renard, Potent 3-hydroxy-2-pyridine aldoxime reactivators of organophosphate-inhibited cholinesterases with predicted blood–brain barrier penetration, Chem. - A Eur. J. 24 (2018) 9675–9691. [CrossRef]
- T. Zorbaz, D. Malinak, T. Hofmanova, N. Maraković, S. Žunec, N. Maček Hrvat, R. Andrys, M. Psotka, A. Zandona, J. Svobodova, L. Prchal, S. Fingler, M. Katalinić, Z. Kovarik, K. Musilek, Halogen substituents enhance oxime nucleophilicity for reactivation of cholinesterases inhibited by nerve agents, Eur. J. Med. Chem. 238 (2022) 114377. [CrossRef]
- T. Zorbaz, D. Malinak, N. Maraković, N. Maček Hrvat, A. Zandona, M. Novotny, A. Skarka, R. Andrys, M. Benkova, O. Soukup, M. Katalinić, K. Kuča, Z. Kovarik, K. Musilek, Pyridinium oximes with ortho-positioned chlorine moiety exhibit improved physicochemical properties and efficient reactivation of human acetylcholinesterase inhibited by several nerve agents, J. Med. Chem. 61 (2018) 10753–10766. [CrossRef]
- A. Zandona, N. Maraković, P. Mišetić, J. Madunić, K. Miš, J. Padovan, S. Pirkmajer, M. Katalinić, Activation of (un)regulated cell death as a new perspective for bispyridinium and imidazolium oximes, Arch. Toxicol. 95 (2021) 2737–2754. [CrossRef]
- Zandona, J. Madunić, K. Miš, N. Maraković, P. Dubois-Geoffroy, M. Cavaco, P. Mišetić, J. Padovan, M. Castanho, L. Jean, P.Y. Renard, S. Pirkmajer, V. Neves, M. Katalinić, Biological response and cell death signaling pathways modulated by tetrahydroisoquinoline-based aldoximes in human cells, Toxicology. 494 (2023) 153588. [CrossRef]
- E. Giacobini, Butyrylcholinesterase: Its function and inhibitors, 1st Ed, Martin Dunitz, Taylor & Francis Group plc, London, 2003.
- E. Giacobini, G. Pepeu, The brain cholinergic system, CRC Press, London, 2006. [CrossRef]
- N.H. Greig, T. Utsuki, D.K. Ingram, Y. Wang, G. Pepeu, C. Scali, Q.S. Yu, J. Mamczarz, H.W. Holloway, T. Giordano, D. Chen, K. Furukawa, K. Sambamurti, A. Brossi, D.K. Lahiri, Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent, Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 17213–17218. [CrossRef]
- K. Chen, X. Jiang, M. Wu, X. Cao, W. Bao, L.Q. Zhu, Ferroptosis, a potential therapeutic target in Alzheimer’s disease, Front. Cell Dev. Biol. 9 (2021) 704298. [CrossRef]
- M. Katalinić, N. Maček Hrvat, K. Baumann, S. Morasi Piperčić, S. Makarić, S. Tomić, O. Jović, T. Hrenar, A. Miličević, D. Jelić, S. Žunec, I. Primožič, Z. Kovarik, A comprehensive evaluation of novel oximes in creation of butyrylcholinesterase-based nerve agent bioscavengers, Toxicol. Appl. Pharmacol. 310 (2016) 195–204. [CrossRef]
- M. Katalinić, A. Zandona, A. Ramić, T. Zorbaz, I. Primožič, Z. Kovarik, New cinchona oximes evaluated as reactivators of acetylcholinesterase and butyrylcholinesterase inhibited by organophosphorus compounds, Molecules. 22 (2017). [CrossRef]
- A. Zandona, M. Katalinić, G. Šinko, A. Radman Kastelic, I. Primožič, Z. Kovarik, Targeting organophosphorus compounds poisoning by novel quinuclidine-3 oximes: development of butyrylcholinesterase-based bioscavengers, Arch. Toxicol. 94 (2020) 3157–3171. [CrossRef]
- Z. Kovarik, N. Ciban, Z. Radić, V. Simeon-Rudolf, P. Taylor, Active site mutant acetylcholinesterase interactions with 2-PAM, HI-6, and DDVP, Biochem. Biophys. Res. Commun. 342 (2006) 973–978. [CrossRef]
- Z. Kovarik, J. Kalisiak, N. Maček Hrvat, M. Katalinić, T. Zorbaz, S. Žunec, C. Green, Z. Radić, V. V. Fokin, K.B. Sharpless, P. Taylor, Reversal of tabun toxicity enabled by a triazole-annulated oxime library—reactivators of acetylcholinesterase, Chem. - A Eur. J. 25 (2019) 4100–4114. [CrossRef]
- D. Gašo Sokač, A. Zandona, S. Roca, D. Vikić-Topić, G. Lihtar, N. Maraković, V. Bušić, Z. Kovarik, M. Katalinić, Potential of vitamin B6 dioxime analogues to act as cholinesterase ligands, Int. J. Mol. Sci. 23 (2022) 13388. [CrossRef]
- M. Mlakić, I. Odak, D. Barić, S. Talić, I. Šagud, Z. Štefanić, K. Molčanov, Z. Lasić, B. Kovačević, I. Škorić, New resveratrol analogs as improved biologically active structures: Design, synthesis and computational modeling, Bioorg. Chem. 143 (2024) 106965. [CrossRef]
- G. Šinko, Assessment of scoring functions and in silico parameters for AChE-ligand interactions as a tool for predicting inhibition potency, Chem. Biol. Interact. 308 (2019) 216–223. [CrossRef]
- N. Maraković, A. Knežević, I. Rončević, X. Brazzolotto, Z. Kovarik, G. Šinko, Enantioseparation, in vitro testing, and structural characterization of triple-binding reactivators of organophosphate-inhibited cholinesterases, Biochem. J. 477 (2020) 2771–2790. [CrossRef]
- T.L. Rosenberry, X. Brazzolotto, I.R. MacDonald, M. Wandhammer, M. Trovaslet-Leroy, S. Darvesh, F. Nachon, Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study, Molecules. 22 (2017) 2098. [CrossRef]
- M.N. Ngamelue, K. Homma, O. Lockridge, O.A. Asojo, Crystallization and X-ray structure of full-length recombinant human butyrylcholinesterase, Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63 (2007) 723–727. [CrossRef]
- Z. Kovarik, G. Moshitzky, N. Maček Hrvat, H. Soreq, Recent advances in cholinergic mechanisms as reactions to toxicity, stress, and neuroimmune insults, J. Neurochem. (2023) 1–15. [CrossRef]
- M.C. de Koning, M. van Grol, D. Noort, Peripheral site ligand conjugation to a non-quaternary oxime enhances reactivation of nerve agent-inhibited human acetylcholinesterase, Toxicol. Lett. 206 (2011) 54–59. [CrossRef]
- N. Maraković, A. Knežević, V. Vinković, Z. Kovarik, G. Šinko, Design and synthesis of N-substituted-2-hydroxyiminoacetamides and interactions with cholinesterases, Chem. Biol. Interact. 259 (2016) 122–132. [CrossRef]
- G. Mercey, T. Verdelet, G. Saint-André, E. Gillon, A. Wagner, R. Baati, L. Jean, F. Nachon, P.Y. Renard, First efficient uncharged reactivators for the dephosphylation of poisoned human acetylcholinesterase, Chem. Commun. 47 (2011) 5295–5297. [CrossRef]
- G. Mercey, T. Verdelet, J. Renou, M. Kliachyna, R. Baati, F. Nachon, L. Jean, P.Y. Renard, Reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents, Acc. Chem. Res. 45 (2012) 756–766. [CrossRef]
- Z. Radić, R.K. Sit, E. Garcia, L. Zhang, S. Berend, Z. Kovarik, G. Amitai, V. V. Fokin, K.B. Sharpless, P. Taylor, Mechanism of interaction of novel uncharged, centrally active reactivators with OP-hAChE conjugates, Chem. Biol. Interact. 203 (2013) 67–71. [CrossRef]
- J. Renou, J. Dias, G. Mercey, T. Verdelet, C. Rousseau, A.J. Gastellier, M. Arboléas, M. Touvrey-Loiodice, R. Baati, L. Jean, F. Nachon, P.Y. Renard, Synthesis and in vitro evaluation of donepezil-based reactivators and analogues for nerve agent-inhibited human acetylcholinesterase, RSC Adv. 6 (2016) 17929–17940. [CrossRef]
- J. Renou, M. Loiodice, M. Arboléas, R. Baati, L. Jean, F. Nachon, P.Y. Renard, Tryptoline-3-hydroxypyridinaldoxime conjugates as efficient reactivators of phosphylated human acetyl and butyrylcholinesterases, Chem. Commun. 50 (2014) 3947–3950. [CrossRef]
- J. Renou, G. Mercey, T. Verdelet, E. Pǎunescu, E. Gillon, M. Arboléas, M. Loiodice, M. Kliachyna, R. Baati, F. Nachon, L. Jean, P.Y. Renard, Syntheses and in vitro evaluations of uncharged reactivators for human acetylcholinesterase inhibited by organophosphorus nerve agents, Chem. Biol. Interact. 203 (2013) 81–84. [CrossRef]
- R.K. Sit, V. V. Fokin, G. Amitai, K.B. Sharpless, P. Taylor, Z. Radić, Imidazole aldoximes effective in assisting butyrylcholinesterase catalysis of organophosphate detoxification, J. Med. Chem. 57 (2014) 1378–1389. [CrossRef]
- Z. Radić, R.K. Sit, Z. Kovarik, S. Berend, E. Garcia, L. Zhang, G. Amitai, C. Green, B. Radić, V. V. Fokin, K.B. Sharpless, P. Taylor, Refinement of structural leads for centrally acting oxime reactivators of phosphylated cholinesterases, J. Biol. Chem. 287 (2012) 11798–11809. [CrossRef]
- Z. Radić, T. Dale, Z. Kovarik, S. Berend, E. Garcia, L. Zhang, G. Amitai, C. Green, B. Radić, B.M. Duggan, D. Ajami, J. Rebek, P. Taylor, Catalytic detoxification of nerve agent and pesticide organophosphates by butyrylcholinesterase assisted with nonpyridinium oximes, Biochem. J. 450 (2013) 231–242. [CrossRef]
- J. Kalisiak, E.C. Ralph, J.R. Cashman, Nonquaternary reactivators for organophosphate-inhibited cholinesterases, J. Med. Chem. 55 (2012) 465–474. [CrossRef]
- J. Kalisiak, E.C. Ralph, J. Zhang, J.R. Cashman, Amidine-Oximes: Reactivators for organophosphate exposure, J. Med. Chem. 54 (2011) 3319–3330. [CrossRef]
- Z. Kovarik, N. Maček, R.K. Sit, Z. Radić, V. V. Fokin, K.B. Sharpless, P. Taylor, Centrally acting oximes in reactivation of tabun-phosphoramidated AChE, Chem. Biol. Interact. 203 (2013) 77–80. [CrossRef]
- G. Šinko, Modeling of a near-attack conformation of oxime in phosphorylated acetylcholinesterase via a reactivation product, a phosphorylated oxime, Chem. Biol. Interact. 383 (2023). [CrossRef]
- K. Chalupova, J. Korabecny, M. Bartolini, B. Monti, D. Lamba, R. Caliandro, A. Pesaresi, X. Brazzolotto, A.J. Gastellier, F. Nachon, J. Pejchal, M. Jarosova, V. Hepnarova, D. Jun, M. Hrabinova, R. Dolezal, J. Zdarova Karasova, M. Mzik, Z. Kristofikova, J. Misik, L. Muckova, P. Jost, O. Soukup, M. Benkova, V. Setnicka, L. Habartova, M. Chvojkova, L. Kleteckova, K. Vales, E. Mezeiova, E. Uliassi, M. Valis, E. Nepovimova, M.L. Bolognesi, K. Kuca, Novel tacrine-tryptophan hybrids: Multi-target directed ligands as potential treatment for Alzheimer’s disease, Eur. J. Med. Chem. 168 (2019) 491–514. [CrossRef]
- E. Carletti, H. Li, B. Li, F. Ekström, Y. Nicolet, M. Loiodice, E. Gillon, M.T. Froment, O. Lockridge, L.M. Schopfer, P. Masson, F. Nachon, Aging of cholinesterases phosphylated by tabun proceeds through O-dealkylation, J. Am. Chem. Soc. 130 (2008) 16011–16020. [CrossRef]
- A. Matošević, A. Knežević, A. Zandona, N. Maraković, Z. Kovarik, A. Bosak, Design, synthesis and biological evaluation of biscarbamates as potential selective butyrylcholinesterase inhibitors for the treatment of Alzheimer’s disease, Pharmaceuticals. 15 (2022) 1220. [CrossRef]
- M. Mlakić, I. Odak, I. Faraho, S. Talić, M. Bosnar, K. Lasić, D. Barić, I. Škorić, New naphtho/thienobenzo-triazoles with interconnected anti-inflammatory and cholinesterase inhibitory activity, Eur. J. Med. Chem. 241 (2022) 114616. [CrossRef]
- B.R. Brooks, R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, M. Karplus, CHARMM: A program for macromolecular energy, minimization, and dynamics calculations, J. Comp. Chem. 4 (1983) 187–217.
- G. Wu, D.H. Robertson, C.L. Brooks, M. Vieth, Detailed analysis of grid-based molecular docking: A case study of CDOCKER - A CHARMm-based MD docking algorithm, J. Comput. Chem. 24 (2003) 1549–1562. [CrossRef]
- A. Miličević, G. Šinko, Evaluation of the key structural features of various butyrylcholinesterase inhibitors using simple molecular descriptors, Molecules. 27 (2022) 6894. [CrossRef]
- Dym, T. Unger, L. Toker, I. Silman, J. Sussman, Israel Structural Proteomics Center (ISPC) - Crystal structure of human acetylcholinesterase, (2015). [CrossRef]
- G.L. Ellman, K.D. Courtney, V. Andres, R.M. Featherstone, A new and rapid colorimetric determination of acetylcholinesterase activity, Biochem. Pharmacol. 7 (1961) 88–95. [CrossRef]
- Z. Kovarik, Z. Radić, H.A. Berman, V. Simeon-Rudolf, E. Reiner, P. Taylor, Mutant cholinesterases possessing enhanced capacity for reactivation of their phosphonylated conjugates, Biochemistry. 43 (2004) 3222–3229. [CrossRef]
Figure 1.
The general structures of the new oxime derivatives.
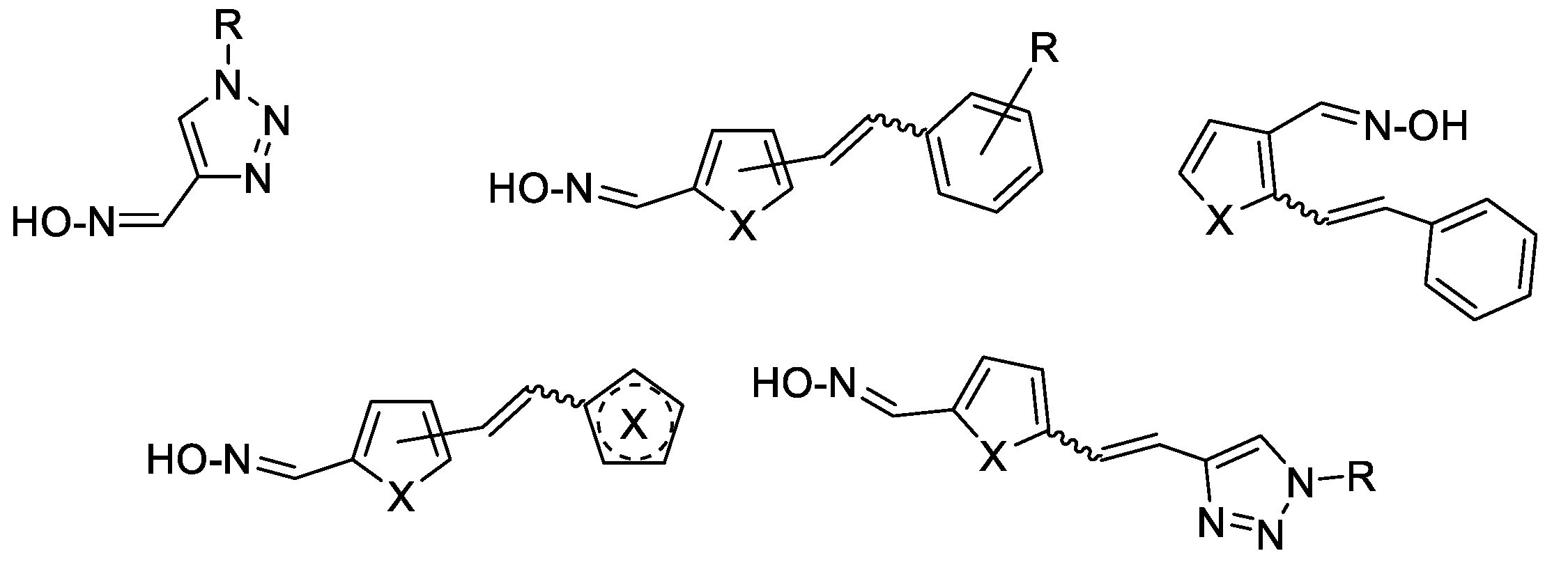
Figure 2.
New triazole oximes 1–3 as the only non-stilbene type oximes.
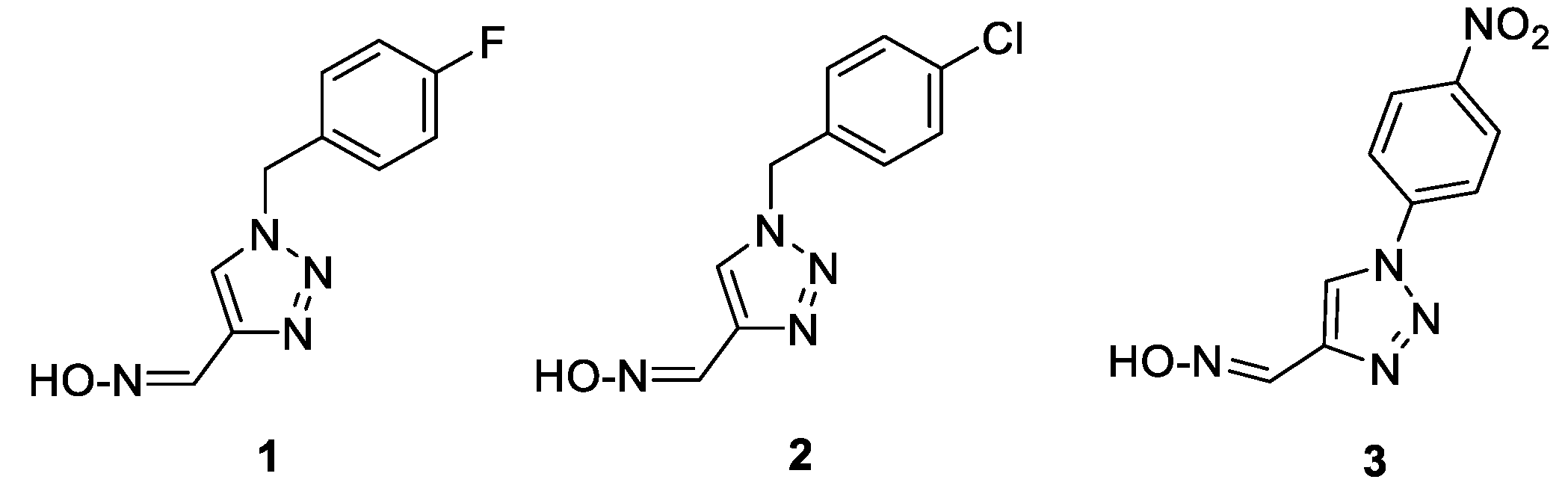
Figure 3.
New oxime derivatives 4–21 with a heterostilbene skeleton.
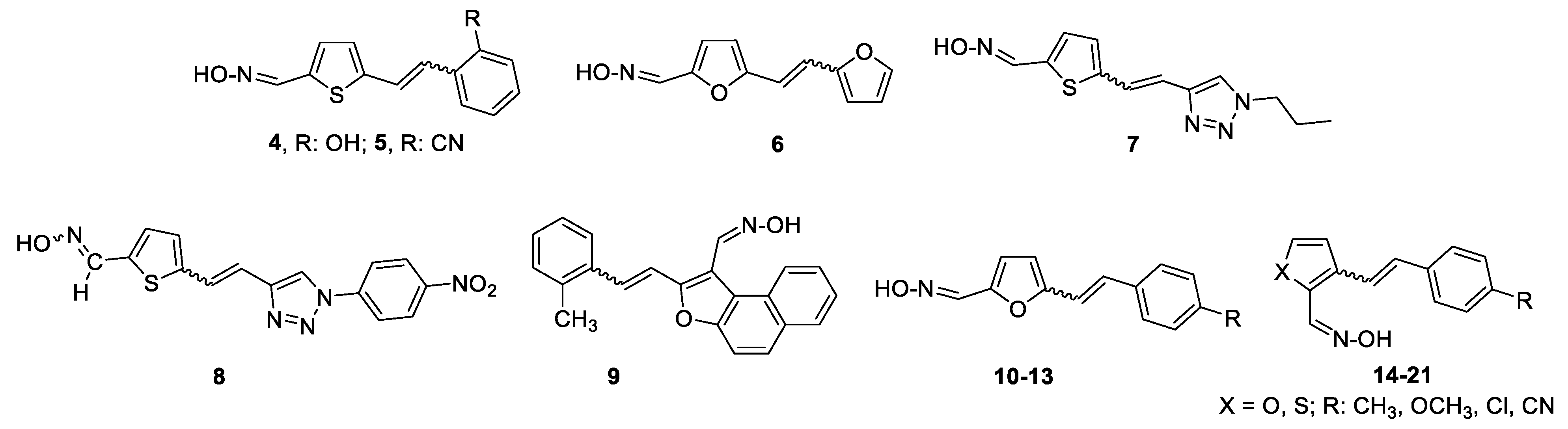
Scheme 1.
First synthetic steps towards uncharged oximes 4–21.
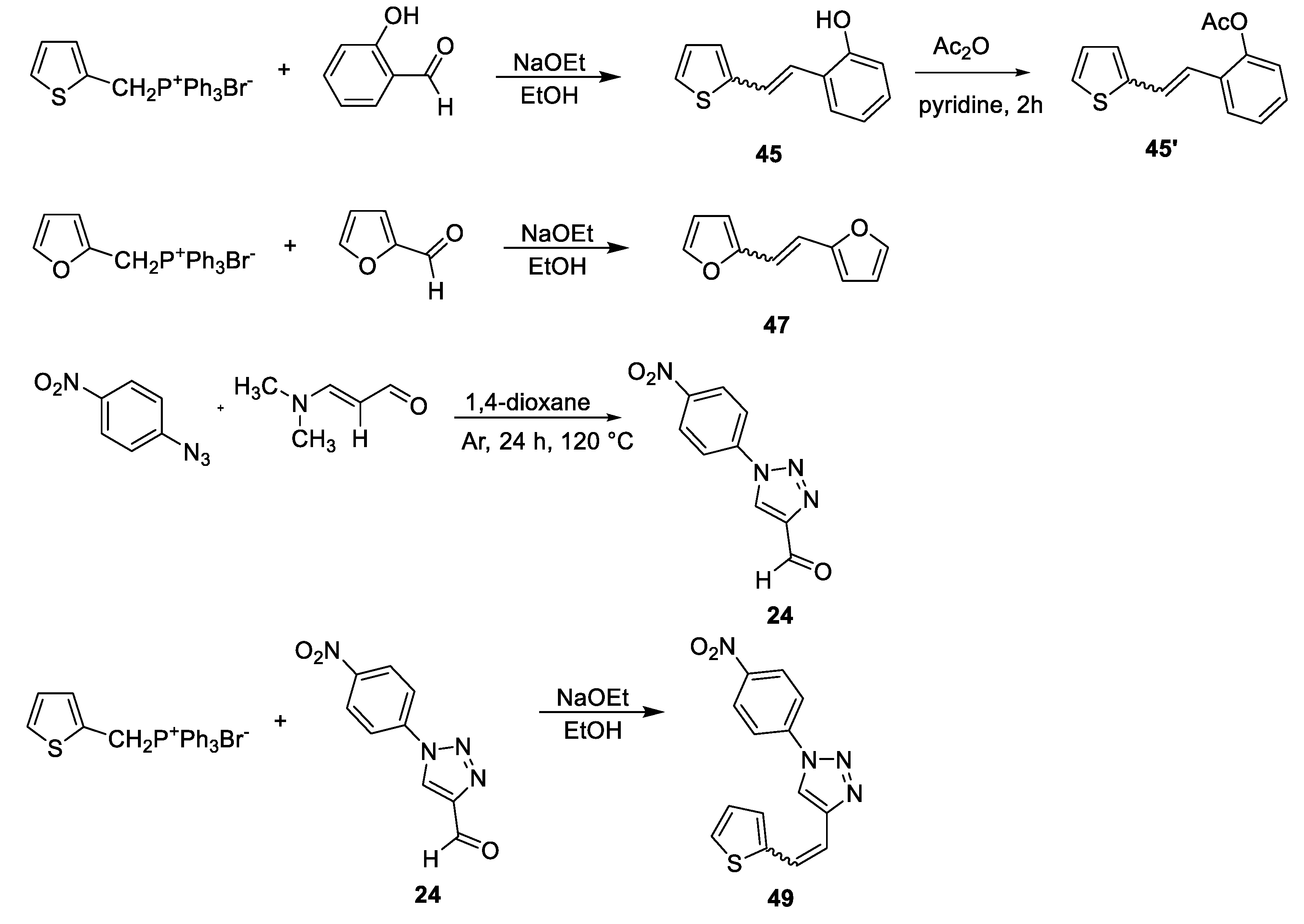
Scheme 2.
Vilsmeier formylation of new heterostilbenes and their conversion to uncharged oximes 4–9.
Scheme 2.
Vilsmeier formylation of new heterostilbenes and their conversion to uncharged oximes 4–9.
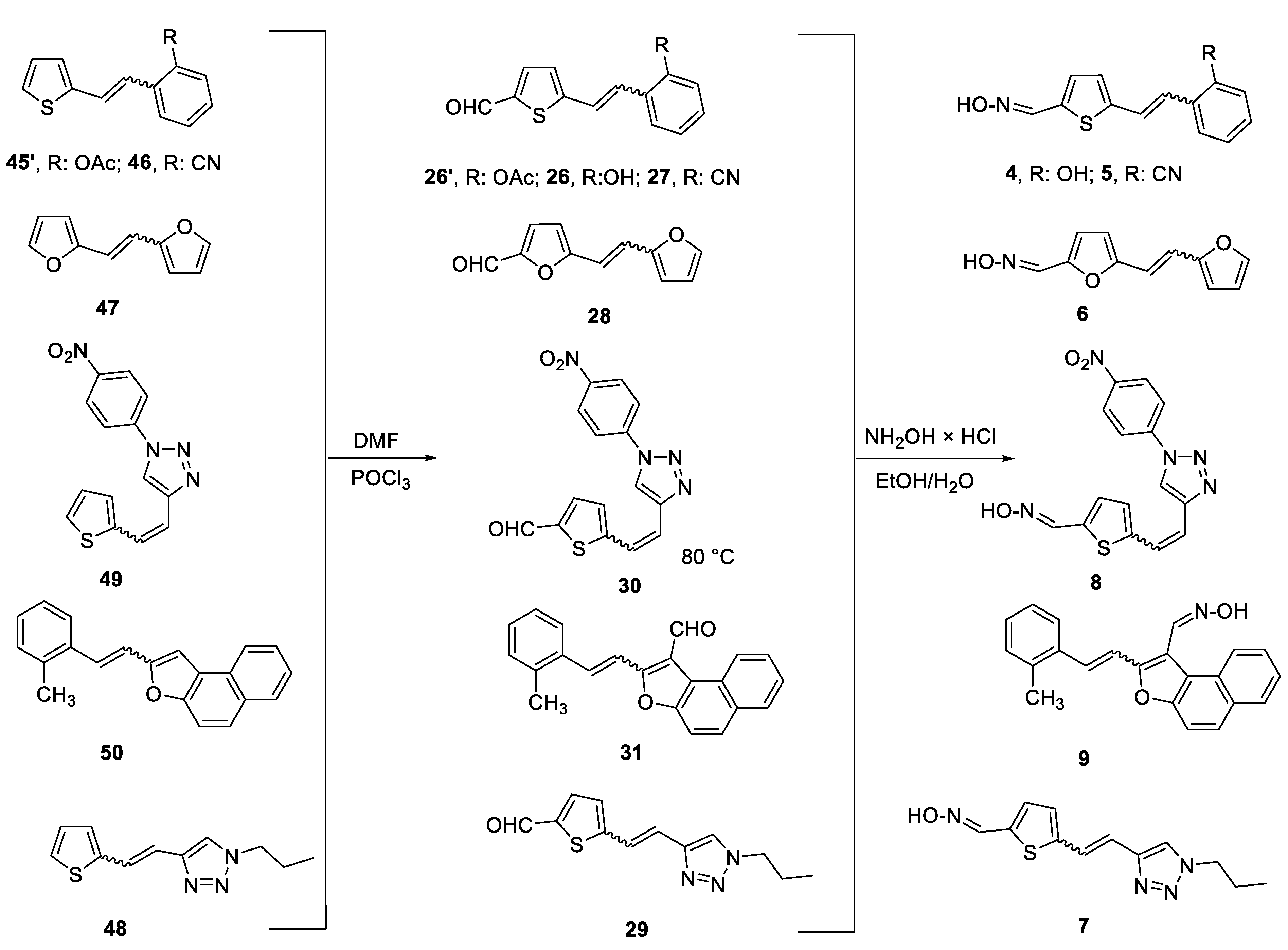
Scheme 3.
Synthetic pathway of new uncharged oximes 10–13 (R = CH3, OCH3, Cl, CN, N(CH3)2).
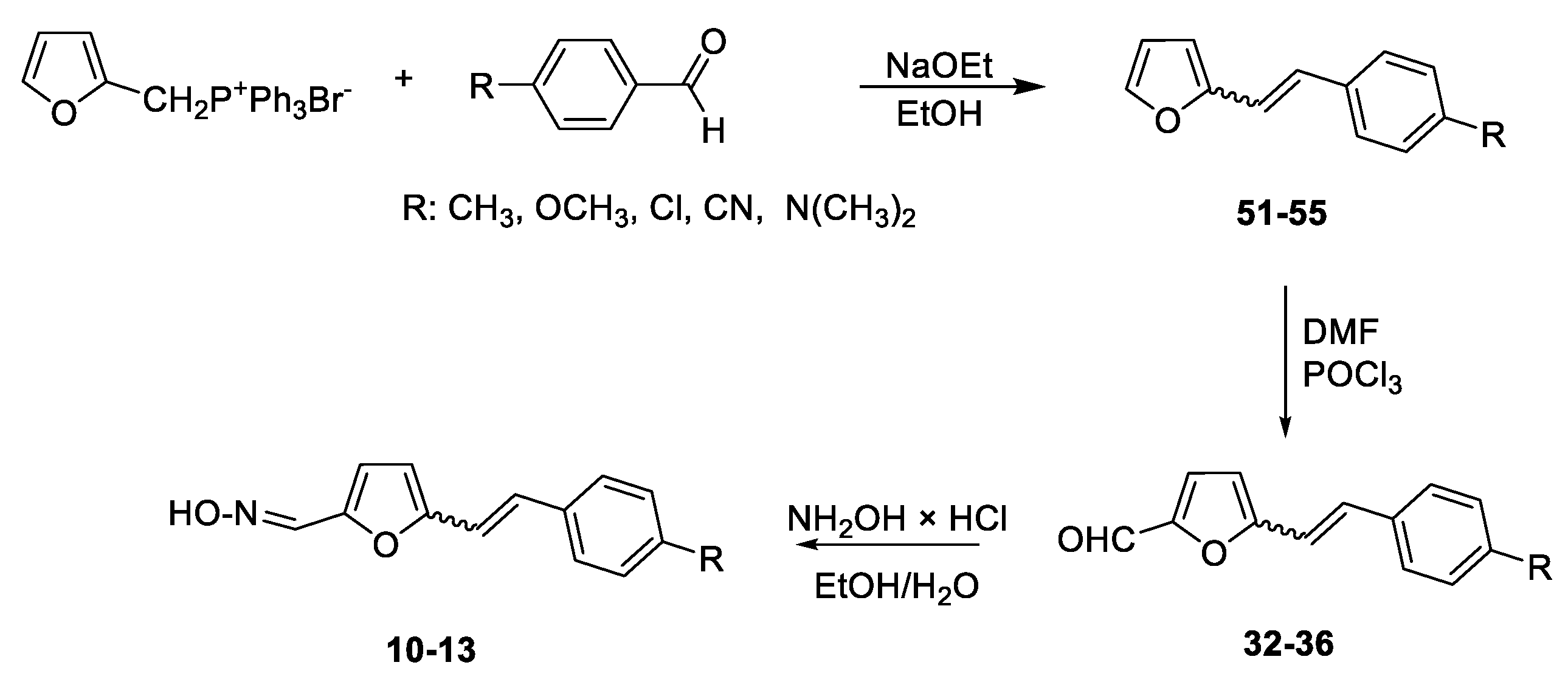
Scheme 4.
Synthetic route of new uncharged oximes 14–21 (R = CH3, OCH3, Cl, CN; X = O, S).
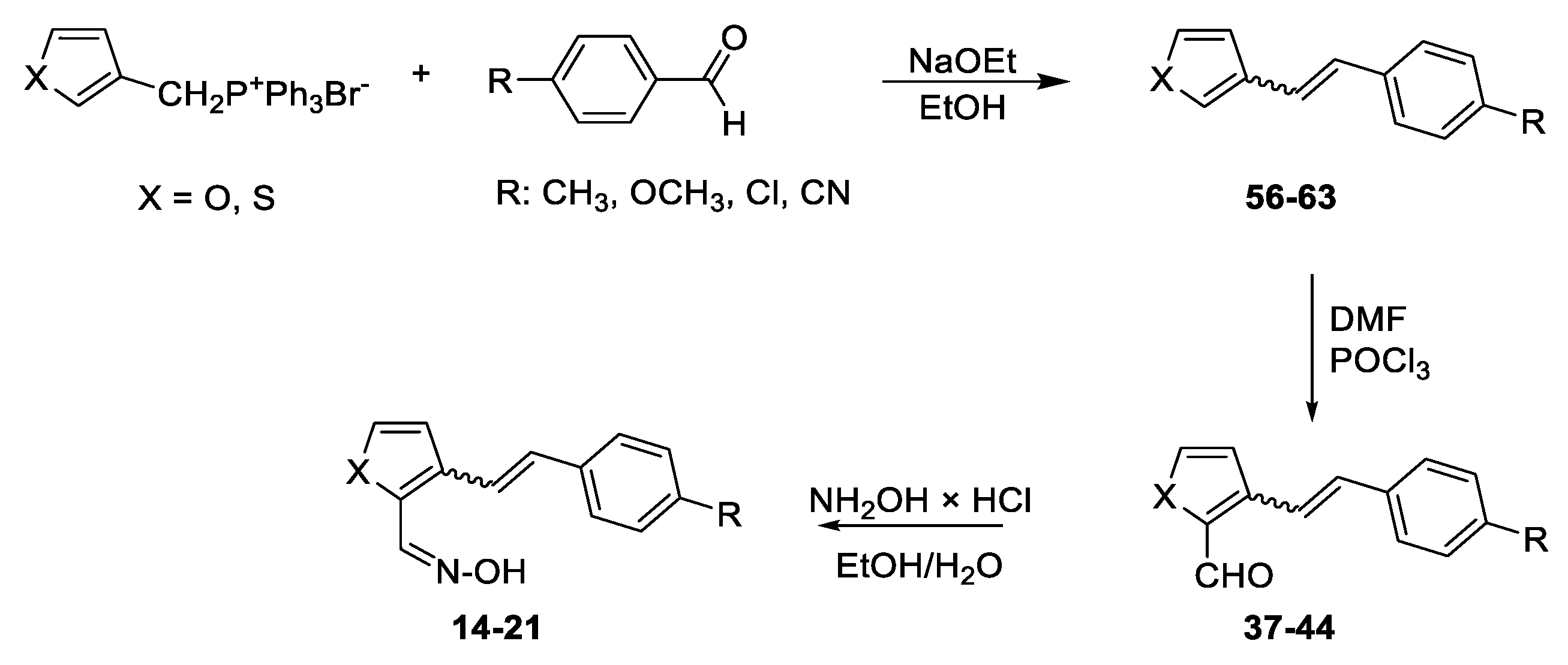
Figure 4.
(A) Physical-chemical properties - molecular weight (MW), number of hydrogen bond donors and acceptors (HBDs and HBAs), number of rotating bonds (RBs), lipophilicity (AlogP), and polar surface area (PSA) of uncharged oximes in relation to the recommended values (red line). (B) Correlation plot for prediction of BBB penetration. Recommended values of CNS-active drugs are given by Pajouhesh and Lenz [23].
Figure 4.
(A) Physical-chemical properties - molecular weight (MW), number of hydrogen bond donors and acceptors (HBDs and HBAs), number of rotating bonds (RBs), lipophilicity (AlogP), and polar surface area (PSA) of uncharged oximes in relation to the recommended values (red line). (B) Correlation plot for prediction of BBB penetration. Recommended values of CNS-active drugs are given by Pajouhesh and Lenz [23].

Figure 6.
Reactivation of cyclosarin-inhibited BChE by 100 µM oximes in terms of the reactivation rate constants, kobs (grey columns), maximal reactivation in 240 min (purple columns), and inhibition of BChE by oximes (green columns). The presented values are means of at least two experiments measured at 25 °C.
Figure 6.
Reactivation of cyclosarin-inhibited BChE by 100 µM oximes in terms of the reactivation rate constants, kobs (grey columns), maximal reactivation in 240 min (purple columns), and inhibition of BChE by oximes (green columns). The presented values are means of at least two experiments measured at 25 °C.
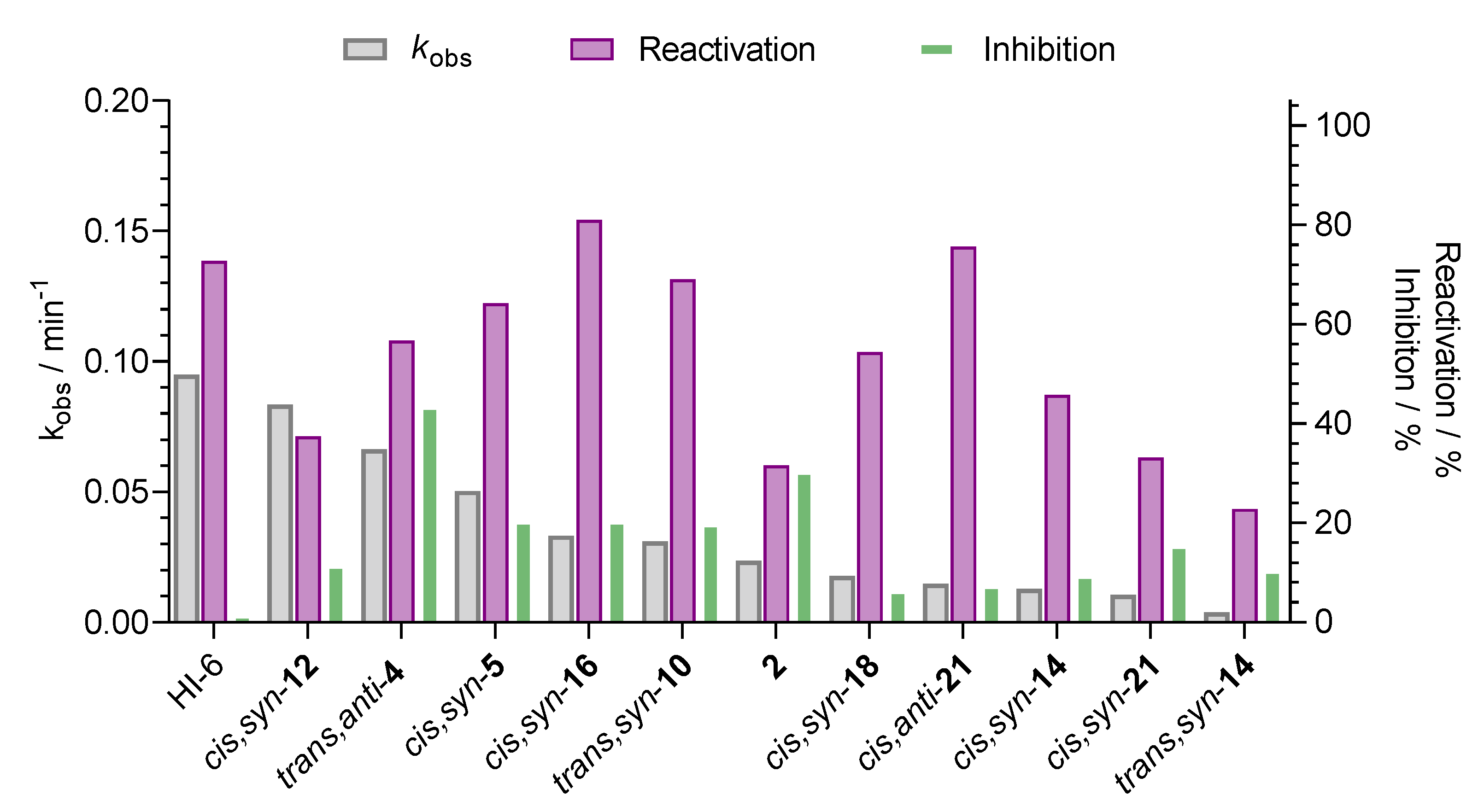
Figure 7.
Minimised conformation of a complex in near-attack conformation between cyclosarin-inhibited BChE and cis,syn-5 (A) and cis,syn-16 (B). The Van der Waals distance of the oxime group from the oxygen (Oγ) of catalytic serine (Ser198) is red. Oxime interactions with active site residues are represented as dashed lines: hydrophobic (purple and yellow), and hydrogen bonds (green). Crystal structure of human BChE inhibited by tabun (PDB code 3DJY) [67] was used for the modelling of cyclosarin-inhibited BChE [65].
Figure 7.
Minimised conformation of a complex in near-attack conformation between cyclosarin-inhibited BChE and cis,syn-5 (A) and cis,syn-16 (B). The Van der Waals distance of the oxime group from the oxygen (Oγ) of catalytic serine (Ser198) is red. Oxime interactions with active site residues are represented as dashed lines: hydrophobic (purple and yellow), and hydrogen bonds (green). Crystal structure of human BChE inhibited by tabun (PDB code 3DJY) [67] was used for the modelling of cyclosarin-inhibited BChE [65].
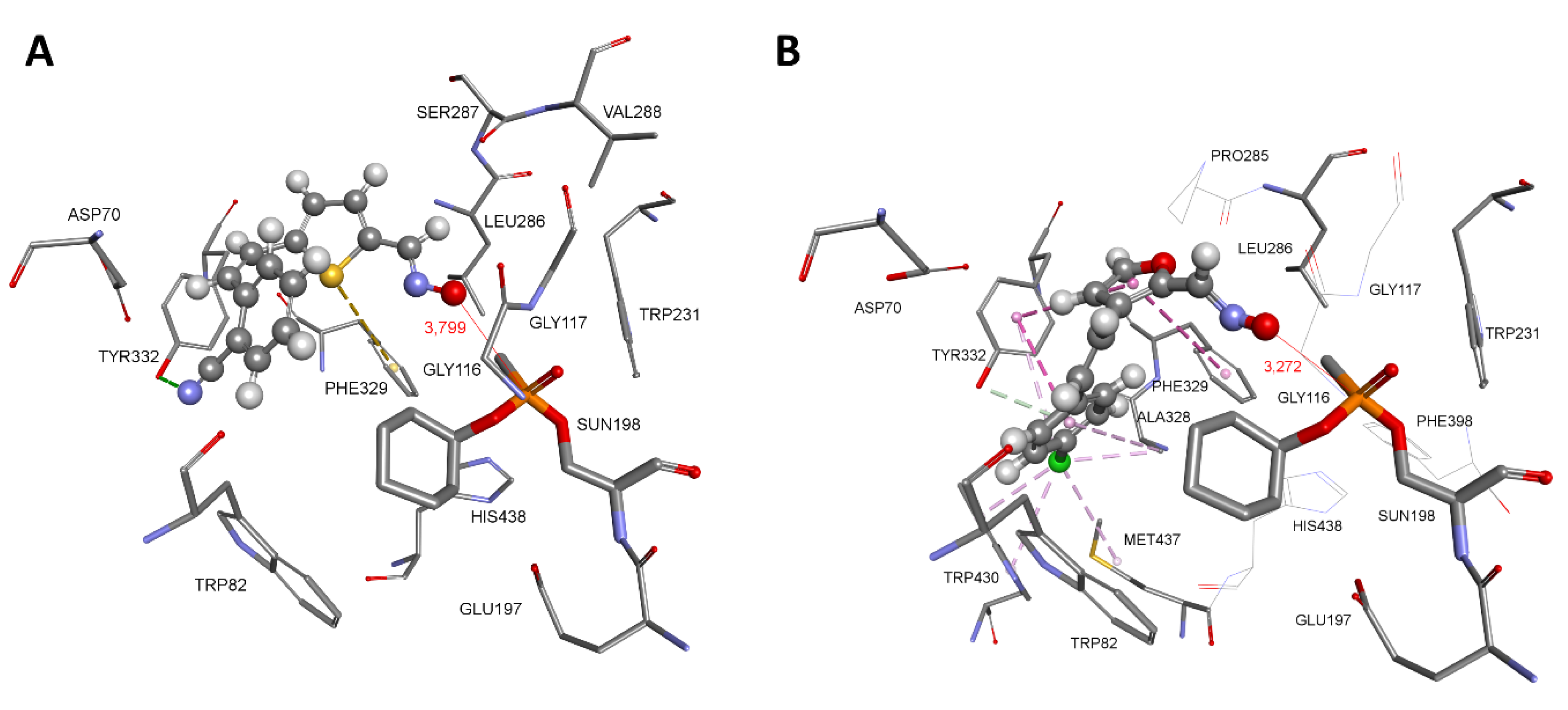

Table 1.
The cytotoxicity of new uncharged oximes 1–21 evaluated by MTS assay after 24-hour liver cell line (HepG2) exposure. Data was calculated from at least three experiments.
Table 1.
The cytotoxicity of new uncharged oximes 1–21 evaluated by MTS assay after 24-hour liver cell line (HepG2) exposure. Data was calculated from at least three experiments.
| Oxime | IC50 / μM |
|---|---|
| HepG2 | |
| 1 a | > 400 |
| 2 a | > 400 |
| 3 a | > 400 |
| trans,anti-4 | 191 ± 21 |
| cis,syn-5 | ≥ 400 |
| trans,anti-6 | ≥ 400 |
| trans-7 a | > 400 |
| trans-8 a | > 400 |
| trans,syn-9 | > 400 |
| trans,syn-10 | 105 ± 4 |
| trans,anti-10 | > 400 |
| cis,syn-12 | 226 ± 10 |
| 13 a | > 400 |
| cis,syn-14 | 182 ± 32 |
| trans,syn-14 | > 400 |
| trans,anti-15 | > 400 |
| cis,syn-16 | 65 ± 13 |
| trans,syn-16 | > 400 |
| cis,syn-17 | 120 ± 6 |
| trans,syn-17 | > 400 |
| cis,syn-18 | 269 ± 38 |
| cis,anti-18 | > 400 |
| trans,syn-18 | > 400 |
| cis,syn-19’ | > 400 |
| cis,syn-21 | 118 ± 46 |
| cis,anti-21 | 182 ± 13 |
| a mixture of configurational isomers | |
Table 2.
Dissociation constants (Ki ± S.E) for reversible inhibition of human AChE and BChE with uncharged oximes at 25 °C from at least three experiments.
Table 2.
Dissociation constants (Ki ± S.E) for reversible inhibition of human AChE and BChE with uncharged oximes at 25 °C from at least three experiments.
| Oxime | Ki / μM | |
|---|---|---|
| AChE | BChE | |
| 1 a | 271 ± 29 | 503 ± 32 |
| 2 a | 267 ± 92 | 24 ± 2.9 |
| 3 a | 280 ± 30 | 303 ± 66 |
| trans,anti-4 | 258 ± 90 | 6.5 ± 0.5 |
| cis,syn-5 | 112 ± 21 | 17 ± 2.0 |
| trans,anti-6 | nd b | 123 ± 23 |
| trans-7 a | 147 ± 43 | 49 ± 11 |
| trans-8 a | 199 ± 38 | 143 ± 22 |
| trans,syn-9 | 172 ± 29 | 303 ± 65 |
| trans,syn-10 | 111 ± 45 | 38 ± 9.8 |
| trans,anti-10 | 180 ± 40 | nd |
| cis,syn-12 | 68 ± 37 | 147 ± 37 |
| trans-13 a | nd | nd |
| cis,syn-14 | 627 ± 245 | 93 ± 30 |
| trans,syn-14 | nd | 122 ± 47 |
| trans,anti-15 | nd | nd |
| cis,syn-16 | nd | 119 ± 44 |
| trans,syn-16 | 217 ± 160 | nd |
| cis,syn-17 | 544 ± 105 | nd |
| trans,syn-17 | 66 ± 24 | nd |
| cis,syn-18 | nd | 86 ± 32 |
| cis,anti-18 | nd | 760 ± 122 |
| trans,syn-18 | 59 ± 39 | nd |
| cis,syn-19’ | nd | nd |
| cis,syn-21 | 171 ± 68 | 59 ± 8.0 |
| cis,anti-21 | 187 ± 54 | 53 ± 10 |
| a mixture of configurational isomers; b nd – not determine due to enzyme activity was higher than 80% at the highest oxime concentration | ||
Table 3.
Reactivation of cyclosarin-inhibited BChE by selected uncharged oximes. Determined parameters (mean ± S.E.) - maximal reactivation rate constant (k2), dissociation constant (KOX), overall reactivation rate constant (kr), maximal percentage of reactivation (Reactmax) and time (t) in which maximal reactivation was achieved were determined from at least three experiments at 25 °C.
Table 3.
Reactivation of cyclosarin-inhibited BChE by selected uncharged oximes. Determined parameters (mean ± S.E.) - maximal reactivation rate constant (k2), dissociation constant (KOX), overall reactivation rate constant (kr), maximal percentage of reactivation (Reactmax) and time (t) in which maximal reactivation was achieved were determined from at least three experiments at 25 °C.
| Oxime | k2 / min-1 | KOX / µM | kr / M-1 min-1 | Reactmax /% | t / min |
|---|---|---|---|---|---|
| trans,anti-4 | -a | - | 592 ± 79 | 50 | 50 |
| cis,syn-5 | 0.11 ± 0.02 | 105.2 ± 47.3 | 1,082 ± 270 | 70 | 60 |
| trans,syn-10 | - | - | 240 ± 14 | 70 | 140 |
| cis,syn-16 | - | - | 424 ± 39 | 80 | 105 |
| HI-6b | - | - | 780 ± 30 | 90 | 30 |
a Not evaluated due to a linear dependence of kobs and oxime concentration (0.01 – 0.2 mM selected oximes and 0.1 – 1 mM HI-6). b from [30].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



