Submitted:
13 May 2024
Posted:
13 May 2024
You are already at the latest version
Abstract
The DNA Damage Response (DDR) network and the mitogen-activated protein kinase (MAPK) signaling pathway are crucial mechanisms for the survival of all living beings. An accumulating body of evidence suggests that there is a crosstalk between these two systems, thus favoring the appropriate functioning of multi-cellular organisms. On the other hand, aberrations within these mechanisms are thought to play a vital role in the onset and progression of several diseases, including cancer, as well as in the emergence of drug resistance. Here, we provide an overview of the current knowledge regarding alterations in the DDR machinery and the MAPK signaling pathway, as well as abnormalities in the DDR-MAPK functional crosstalk in multiple myeloma, the second most common hematologic malignancy. We also present the latest advances in the development of anti-myeloma drugs targeting crucial DDR- and MAPK-associated molecular components. These data could potentially be exploited to discover new therapeutic targets and effective biomarkers, as well as for the design of novel clinical trials. Interestingly, they might provide a new approach to increase the efficacy of anti-myeloma therapy by combining drugs targeting the DDR network and the MAPK signaling pathway.
Keywords:
multiple myeloma (MM)
; DNA damage response (DDR)
; mitogen‑activated protein kinase (MAPK)
; DDR-MAPK interplay
; combination therapy
1. Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by the overproduction of monoclonal immunoglobulins and the clonal proliferation of long-lived plasma cells [1]. Monoclonal gammopathy of undetermined significance (MGUS) is a premalignant condition, which may progress to MM [2]. Three to four percent of the people over the age of fifty have MGUS. Recent data suggest that chronic antigenic stimulation, black race, older age, male sex, diabetes, certain pesticides, family history, inflammatory conditions and obesity are risk factors for developing MGUS [3,4]. Moreover, smoldering MM (SMM), an intermediate more advanced premalignant stage, is clinically identified in certain patients [5]. Annually, ~1% of MGUS cases progress to SMM; for those with SMM, the relevant risk of developing MM is much higher: ~10% per year for the first five years and 3% per year for the subsequent five [6].
Treatment of MM is a multifaceted approach that depends on various factors, including the stage of the disease, the patient's overall health and individualized treatment goals. It involves combinations of drugs with several mechanisms of action, such as corticosteroids, alkylating agents, histone deacetylase inhibitors (HDACi), anthracyclines, proteasome inhibitors (PIs), immunomodulatory drugs (IMID), high-dose chemotherapy followed by autologous stem cell transplantation, monoclonal antibodies (mAbs), chimeric antigen receptor (CAR) T-cell therapy, etc [7,8,9,10]. Despite these advancements, MM remains an incurable disease and the need for new treatment strategies is mandatory.
Using modern biology technologies, molecular characteristics of MGUS and SMM, as well as the progression to active MM are now better understood [11]. Interestingly, several reports identified mutations in genes involved in the DNA Damage Response (DDR) network and the mitogen-activated protein kinase (MAPK) system [12]. Therefore, in this review, we provide data on the current literature regarding aberrations in the DDR network, the MAPK system and their interplay that are involved in the onset and progression of MM, and the development of drug resistance. The latest advances in anti-myeloma drugs targeting critical DDR- and MAPK-related components are also elucidated.
2. The DDR Network
Damage to DNA occurs due to external [ultraviolet (UV) and ionizing radiation, genotoxic drugs] or internal factors (oxidative stress, telomere erosion, replication fork collapse) [13]. To overcome these alterations in the chemical structure of DNA, cells have developed a complex system of pathways, called DDR network that recognize and resolve the damage, thus protecting the integrity of the genome [14]. DDR is triggered following the detection of a DNA lesion. Next, a signal transduction cascade is activated and results in the stimulation of sophisticated mechanisms for genome protection, including DNA repair pathways, cell cycle checkpoints and apoptosis. On the other hand, deregulated DDR may result in mutagenesis and genomic instability [15]. Given that DDR regulates the cellular decision to remove the DNA damage or to activate apoptosis, it is involved in the onset and progression of several diseases, including cancer, as well as in the response to therapeutic interventions.
2.1. The DDR Network in the Onset and Progression of MM
Previous studies have shown that the DNA repair mechanisms are altered in MM, thus allowing the onset and progression of the disease as well as inducing therapy resistance (Figure 1). In fact, deregulation in the Base Excision Repair (BER) pathway plays an important role in MM progression. For example, Liao and colleagues reported that two major BER-related apurinic/apyrimidinic nucleases (APEX1 and APEX2) crosstalk with p73, a transcriptional regulator of RAD51, and results in its transcriptional upregulation, thus increasing the efficiency of Homologous Recombination (HR) and driving genomic instability in MM [16]. Moreover, a study of polymorphisms of BER-associated genes correlates alterations in APEX1 with a reduction in MM patient’s overall survival [17]. In patients’ samples, both APEX1 and APEX2 gene expressions are increased during myelomagenesis [18]. Also, researchers found that high expressions of certain BER genes, such as MPG (N-methylpurine DNA glycosylase) and PARP3 [Poly (ADP-ribose) polymerase 3], are linked to improved overall survival in MM patients who received autologous stem cell transplantation. On the other hand, increased expressions of PARP1 and POLD2 (DNA polymerase delta subunit 2) are associated with worse outcomes in MM, suggesting that targeting BER pathway might improve treatment effectiveness [19,20,21].
Moreover, the gene expression patterns in normal plasma cells and newly diagnosed MM samples reveal that upregulation of the Nucleotide Excision Repair (NER) protein ERCC3 (excision repair cross-complementation group 3) is linked to poorer survival. Additionally, researchers have identified 34 NER-related genes with differential expression in MM plasma cells, along with 23 genes with copy-number variations [22]. Interestingly, polymorphisms of NER have been shown to impact treatment outcomes in MM patients undergoing autologous bone marrow transplantation [23].
It is known that defective Mismatch Repair (MMR) mechanism results in increased mutation rates, particularly in microsatellite DNA regions. This defect, known as microsatellite instability, was observed in many MM patients and becomes more common as the disease progresses and during relapse [24]. Alterations in MMR genes (hMSH2, hMLH1 and hPMS1) have been identified in malignant disorders of B-cell and were associated with aggressive behavior [25,26]. Defective MMR is also implicated in drug resistance [27,28].
The homologous recombination repair (HR/R) mechanism removes DSBs that are formed following therapeutic treatment with several anti-myeloma drugs, such as topoisomerase inhibitors and DNA crosslinking agents. Previous studies have shown elevated expression of HR/R-associated genes, namely RAD50 and RAD51, and increased HR/R activity in MM cell lines and primary MM cells compared with normal plasma cells [29,30]. Since HR/R plays an important role in the recovery of the stalled replication fork and the repair of interstrand cross-links (ICLs), it is of particular importance in drug resistance of the fraction of proliferating MM cells. Indeed, previous studies have shown that following treatment of MM patients with high-dose melphalan (HDM) and autologous stem cell transplantation (ASCT) higher expressions of BRCA1, PRKDC (DNA-PK) and PARP1 genes are linked to poorer outcomes [31]. Moreover, genetic variations in PARP, RAD51, MUTYH, OGG1, PCNA, TPMT and XPC are associated with disease progression [32].
Studies in mice have highlighted the crucial roles of core proteins involved in Non-Homologous End Joining (NHEJ) repair mechanism in preserving genomic stability [33]. In some MM cell lines, such as RPMI-8226, NHEJ activity appears to be compromised, while remains functional in others, including U266 and OPM2 [34]. A study on MM patients treated with thalidomide also revealed that those with specific gene polymorphisms in ERCC1, ERCC5 or XRCC5 (KU80) had higher response rates, with longer overall survival being associated with polymorphisms in ERCC1 and XRCC5. Polymorphisms or abnormal expression of genes such as XRCC4, XRCC6 (KU70), DCLRE1C/Artemis and DNA ligase IV (LIG4) have also been linked to MM risk [30,35,36]. Indeed, increased levels of DCLRE1C/Artemis, DNA–PKcs and XRCC4 proteins were observed in MM cells, while elevated expressions of XRCC5 and DCLRE1C/Artemis genes were linked to poorer prognosis in MM patients [37]. Previous reports have also shown that NSD2 (Nuclear Receptor Binding SET Domain Protein 2), a factor with many biological functions, including DNA repair, plays a crucial role in MM relapse and treatment resistance [38]. In line with these data, loss of NSD2 function reduces the expression of DNA repair genes like RAD51, TP53BP1 and XRCC4 and enhances DNA damage accumulation. On the other hand, overexpression of NSD2 increases DNA repair efficiency, which may contribute to drug resistance, particularly in t(4;14) MM cases [39]. Alternative NHEJ (alt-NHEJ) is a DNA repair pathway, vital for genomic instability and the survival of MM cells. Higher gene expression of LIG3 (component of alt-NHEJ; also involved in NER and BER) in MM patients is linked to shorter survival, especially in advanced disease stages. LIG3 protein levels are elevated in bortezomib-resistant compared to bortezomib-sensitive MM cells; knocking down LIG3 increases DNA damage and inhibits MM cell growth both in vitro and in vivo [40].
Fanconi anemia (FA) is a rare chromosomal instability syndrome, which has been linked to pathogenic variations in at least 22 genes that make up the FA pathway. Interestingly, FA patients’ cells are very sensitive to ICL-inducing drugs, suggesting that FA pathway is implicated in the repair of ICLs [41]. In line with these data, melphalan-resistant myeloma cells express high levels of FANCF (FA Complementation Group F) and RAD51C; depletion of FANCF helps overcome resistance [42].
Gene expression analyses of MM patients treated with HDM and ASCT have revealed the prognostic significance of genes involved in several DNA repair pathways, including NHEJ, HR/R, FA, NER, MMR and BER [43]. Among 84 examined genes, 22 were found to have prognostic value for both event-free and overall survival. These genes included five related to NHEJ [three with negative (NSD2, RIF1, XRCC5/KU80) and two with positive prognostic value (PNKP and POLL)], six to HR/R [five with negative (EXO1, BLM, RPA3, RAD51, MRE11) and one with positive prognostic value (ATM)], three related to FA [all with negative prognostic value (RMI1, FANCI and FANCA)], eight to NER [six with negative (PCNA, RPA3, LIG3, POLD3, ERCC4, POLD1) and two with positive prognostic value (ERCC1, ERCC5)], two involved in MMR [both with negative prognostic value (EXO1 and MSH2)] and one in BER with negative prognostic value (LIG3).
2.2. The DDR Network in the Outcome of Anti-Myeloma Therapy
Extensive observations suggest that the DDR network is implicated in the outcome of genotoxic therapy. Indeed, studies have shown that in vitro resistance to the nitrogen mustard melphalan [44], is linked to increased efficiency of DNA repair mechanisms, including ICL repair [45] and FA/BRCA pathway [42]. In order to elucidate the role of DDR in the outcome of melphalan-treated patients, previous studies reported the formation and repair of DNA damage in peripheral blood mononuclear cells (PBMCs) and bone marrow plasma cells (BMPCs) following in vivo (therapeutic) or ex vivo melphalan treatment [46,47,48,49]. The authors reported that ΜΜ patients, responders to melphalan therapy, are characterized by lower DNA repair capacity and higher accumulation of melphalan-induced DNA damage than non-responders, suggesting that quantification of drug-induced DNA damage formation/repair may help in the selection of patients who may profit from melphalan therapy. Interestingly, they reported that DSB repair (DSB/R) inhibitors, such as the NHEJ inhibitor SCR7, significantly enhanced the cytotoxicity of melphalan against malignant plasma cells, suggesting a promising strategy for the treatment of MM [48].
DSB/R inhibitors are not the only DDR modifiers used in MM therapy. Indeed, previous studies have shown that the combined treatment with inhibitors of ATM (KU-55933) and ATR (VE-821) seriously reduced survival of MM cell lines that exhibited high levels of endogenous DNA damage [50]. Also, PIM-2, a serine/threonine kinase that interacts with DDR and plays a critical role in promoting cell survival and preventing apoptosis, is commonly found upregulated in MM [51,52]. Another study has shown that LT-171-861, a synthetic new PIM-2 inhibitor, induced DNA damage by inhibiting HR/R pathway, activated apoptosis in MM cells and suppressed tumor growth in MM xenograft models [53]. Moreover, the PARP inhibitor olaparib could amplify the anticancer effect of LT-171-861 by reducing the growth of tumors in MM xenografted nude mice.
Panobinostat is an HDACi, which blocks cell cycle progression, induces apoptosome formation and down-regulates the anti-apoptotic Bcl-2 gene [54]. Panobinostat showed synergistic anti-MM effects when combined with genotoxic drugs in vitro [55]. Indeed, in a recent report the authors studied the biological effects of the ex vivo co-treatment of panobinostat and melphalan in BMPCs and PBMCs from MM patients [56]. They found that this combination treatment reduced the efficiency of critical DNA repair mechanisms (NER and DSB/R), augmented the accumulation of cytotoxic DSB lesions and induced apoptosis in BMPCs, but not in PBMCs from the same patients or healthy controls. These data suggest that, compared with melphalan alone, the combined treatment of melphalan and panobinostat showed increased anti-myeloma efficacy and lower side effects.
3. The MAPK System
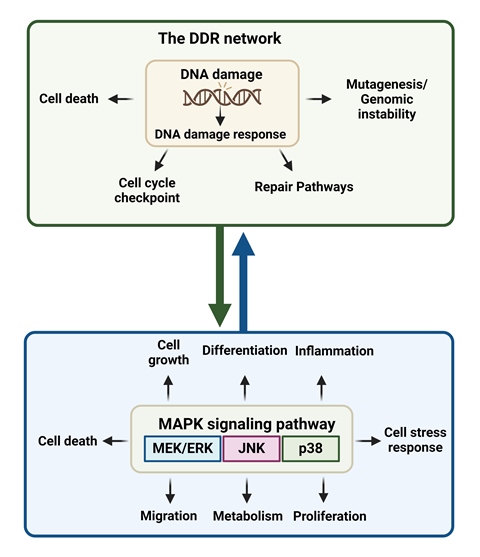
The MAPK signaling pathways organize a network that regulates many cellular processes, including cell growth, differentiation, inflammation, cell stress response, proliferation, metabolism, migration, and apoptosis [57]. In mammals, MAPK consists of three signaling pathways, namely MEK/ERK, c-Jun N-terminal kinase (JNK) and p38 (Figure 2) [57,58]. Interestingly, disruption of the MAPK signaling cascade plays a role in the pathogenesis of different human cancer types [59,60].
3.1. The MAPK Signaling Pathways in the Onset and Progression of MM
Using the next-generation sequencing (NGS) technology, a deeper understanding of the molecular characteristics of MGUS/SMM and the progression toward the active MM has become possible [61]. Numerous mutations were discovered by whole-exome sequencing (WES), with MAPK signaling being one of the most commonly affected pathways. Several reports identified that the NRAS, KRAS and BRAF mutation frequency rises from 24% in SMM [62], to 50% in newly diagnosed MM cases [63,64,65,66,67], and up to 80% in relapsed/refractory MM (RRMM) patients [68,69].
RAS proteins belong to a family of GTP hydrolases (GTPases) that are frequently mutated in human malignancies [70,71]. There are three major isoforms of the RAS gene (NRAS, KRAS and HRAS) that have a significant role in cell proliferation, survival and differentiation. Although in human cancers all three isoforms are commonly mutated oncogenes, most tumors have mutations in the KRAS gene [71]. In MM patients, the mutation incidence is 22–25% for KRAS and 20–25% for NRAS. The most common hotspot mutations are in the codons 12, 13, and 61 of the NRAS and KRAS genes; Q61 mutations (i.e. substitutions of glutamine at amino acid 61 by another amino acid) for NRAS gene are also common in MM [72]. These mutations mostly affect the activity of NRAS and are linked to a more aggressive phenotype and shorter overall survival [73]. Accordingly, in relapsed MM, NRAS mutations are linked to decreased susceptibility to the proteasome inhibitor bortezomib [74].
The development of several cancer types is also associated with mutations in the BRAF proto-oncogene. Among the most frequent mutations observed in the BRAF gene are those encoding the V600E mutant, which results in continuous activation and signal transduction, irrespective of external stimuli. Consequently, cell proliferation and invasion are increased in cancer patients harboring such mutations [75]. The V600E mutation has been specifically associated with melanoma, metastatic colorectal cancer, MM and several other cancer types [76]. The prevalence of the BRAF V600E mutation is high among patients diagnosed with MM, with a frequency ranging from 2% to 4% in newly diagnosed cases and rising to approximately 8% in patients with relapsed/refractory disease or extramedullary involvement [73]. A study of 223 newly diagnosed MM patients exploring gene expression profiles and clinical data detected BRAF mutations in 11% of patients with an unfavorable prognosis [77]. The authors detected both BRAF V600E and non-V600E BRAF mutations, 58% of which were hypoactive or kinase inactive. It is worth noting that 44% of the hypoactive/kinase inactive BRAF patients displayed concurrent mutations in NRAS or KRAS, indicating their involvement in the pathogenesis of the disease by promoting the activation of MAPK via upstream mutated elements.
3.2. The MAPK Signaling Pathway in the Outcome of Anti-Myeloma Therapy
Since the MAPK pathway is mutated in many cancer types, including MM, it is considered to be a major therapeutic target [78,79]. In fact, the FDA has approved four MEK1/2 inhibitors, namely binimetinib, trametinib, cobimetinib and selumetinib [80] and three BRAF inhibitors (vemurafenib, dabrafenib, and encorafenib) for the treatment of several malignancies [78,79].
MEK inhibitors monotherapy in MM has shown mixed results. Indeed, the inhibition of MEK with selumetinib in MM showed a low response in relapsed/refractory MM [81]. On the other hand, trametinib had better response rates in MM patients with MAPK activation [79]. Also, a clinical trial (NCT03312530) evaluating the safety and the efficacy of cobimetinib showed promising results when this MEK inhibitor was administrated alone or in combination with venetoclax (BCL-2 inhibitor) with or without atezolizumab (PD-L1 inhibitor) in t(11;14) MM patients [82,83].
Concerning BRAF inhibition, Andrulis and colleagues examined the mutation status of BRAF V600E in primary tumor samples from 379 MM patients and correlated it with disease outcome [84]. They found that the presence of the BRAF V600E mutation was linked to the development of aggressive extramedullary diseases and shorter overall survival. Moreover, they presented a case study of a MM patient diagnosed with a BRAF V600E mutation and experiencing a relapse of myeloma accompanied by widespread extramedullary involvement; this patient exhibited a rapid and sustained positive response to low doses of vemurafenib.
While most cancer patients show favorable initial responses to BRAF inhibitors, resistance occurs once the ERK pathway is reactivated. To overcome this problem, combinational therapies including BRAF and MEK inhibitors or the use of new second-generation multiple inhibitors, such as the pan-RAF inhibitor tovorafenib (TAK-580; an inhibitor of wildtype BRAF, BRAF V600E, and CRAF) and avutometinib (RO-5126766, also known as CH-5126766; a dual inhibitor of Raf and MEK) were developed [85,86]. Indeed, combined regimes of cobimetinib and vemurafenib showed promising anti-MM results in advanced RRMM [87,88]. Moreover, in a phase II clinical trial (NCT02834364), combined treatment with encorafenib and binimetinib showed positive results in RRMM patients carrying a BRAF V600E mutation [89]. Also, Guo and colleagues reported that the RAF–MEK inhibitor RO-5126766 had antitumor activity against several solid tumors and MM with RAF–RAS–MEK pathway mutations [90]. Interestingly, in the GMMG-BIRMA phase II study (NCT02834364), the combination of binimetinib and encorafenib in RRMM patients with a BRAF V600E or a BRAF V600K mutation showed an 82% overall response rate with 9 out of 11 MM patients having at least partial response [89,91]. In addition, in vitro combination treatment of sorafenib (a RAF and VEGF2 inhibitor) and rapamycin (a potent immunosuppressive drug) showed improved results [92]. Also, a phase I clinical trial using a combination treatment of sorafenib and bortezomib found promising results in several malignancies [93]. However, when investigated in a phase II clinical trial for metastatic or unresectable renal cell carcinoma, the response rates and the progression-free survival were similar to sorafenib monotherapy [94]. In another small study on MM patients, a partial response and a continuous stable disease were observed in 2/11 individuals after sorafenib treatment [95].
The use of p38 MAPK inhibitors, such as talmapimod, plitidepsin and ralimetinib (LY2228820), has shown good preclinical efficacy. Indeed, results from a phase II trial with talmapimod alone or in combination with bortezomib have shown encouraging response rates in the RRMM patients who had previously failed bortezomib monotherapy [96]. Moreover, plitidepsin monotherapy showed 13% response rates in RRMM patients and when it was combined with dexamethasone, response rates reached as high as 22% [97]. However, due to infections, short-lived clinical efficacy and skin damage, to date, there are no FDA-approved drugs against p38 MAPK.
4. The DDR Network and the MAPK System Are Coordinately Activated
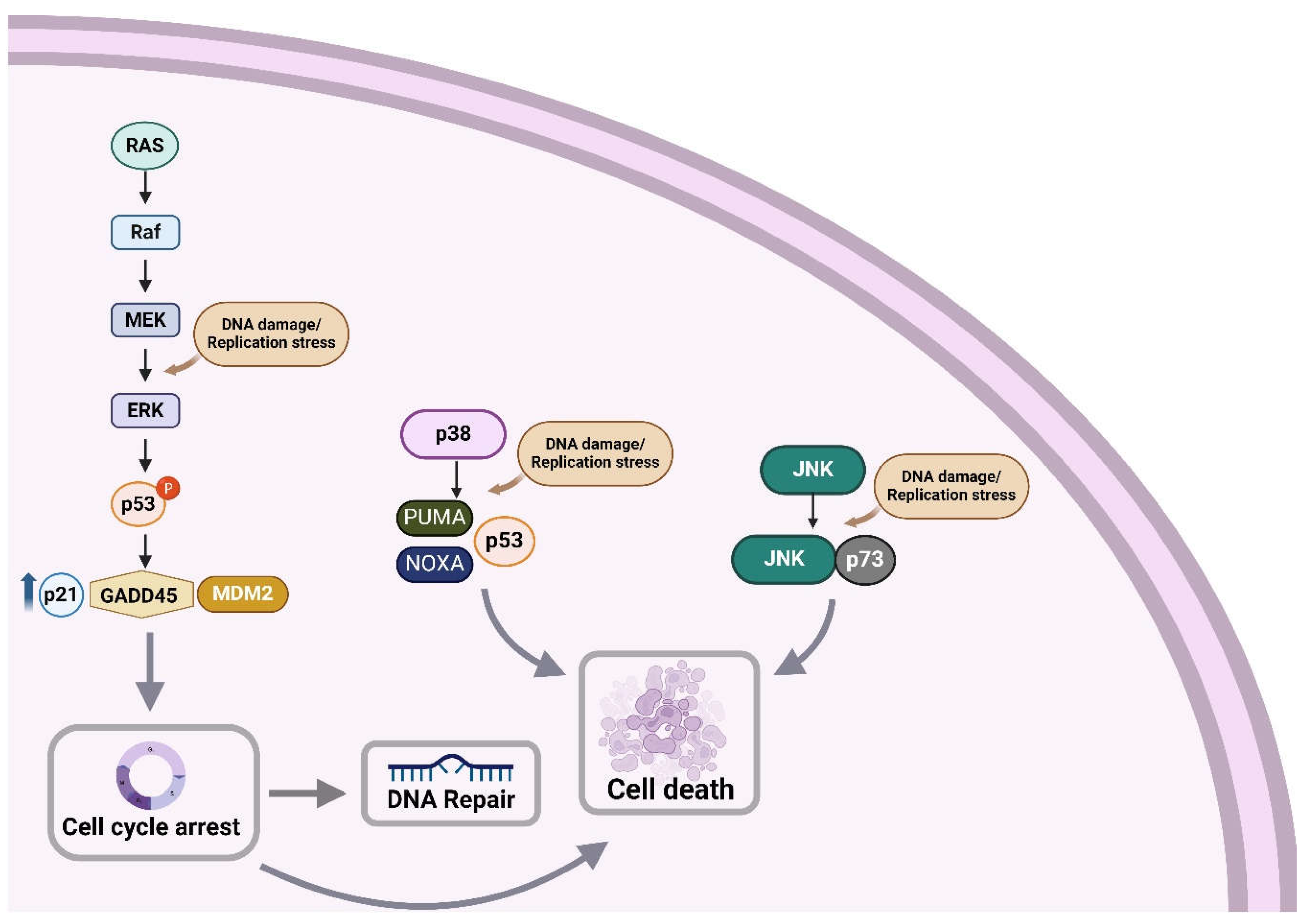
A series of studies have indicated that the DDR network and the MAPK signaling pathway are activated in concert. Indeed, following the activation of DDR, the MEK/ERK pathway is also activated, thus facilitating the proper induction of DDR checkpoints to arrest cell division [98]. Inhibition of the ERK/MAP kinase abrogates cell cycle checkpoint activation, and results in cell proliferation in the presence of DNA lesions, thus triggering the accumulation of mutations and development of tumors [99]. On the other hand, abrogation of checkpoint activation may also induce apoptosis or cell catastrophe, thereby enhancing the efficacy of chemotherapy [100].
4.1. Induction of DDR Activates MAPK
Phosphorylation of the ERK/MAP kinase delivers a survival signal that counteracts pro-apoptotic effects associated with JNK and p38 MAPK activation [101,102,103]. On the other hand, Wang and colleagues have found that in HeLa and A549 cell lines, the activation of ERK/MAP kinase is crucial for the induction of cisplatin-induced apoptosis [104]. Indeed, treatment of HeLa cells with cisplatin caused dose- and time-dependent activation of the MEK/ERK signaling pathway, which ultimately leads to apoptosis (Figure 2). In line with these data, the pretreatment of HeLa cells with TPA (12-O-tetradecanoylphorbol-13-acetate), an activator of the ERK/MAP kinase signaling pathway, enhanced their sensitivity to cisplatin. Moreover, when HeLa cells were pretreated with a MEK1/2 inhibitor (PD98059 or U0126), cisplatin-induced apoptosis was prevented, while cisplatin-resistant HeLa cell variants showed reduced ERK phosphorylation [104]. Together, these data indicate that ERK activation is a fundamental mediator of cisplatin-induced apoptosis that functions upstream of caspase activation to start the apoptosis pathway. However, this is not a universal feature, since Chu and colleagues have found that inhibition of ERK activity in PC-3 prostate cancer cells did not change their sensitivity to cisplatin [105].
Previous studies have shown that cisplatin-associated ERK/MAP kinase activation precedes p53-mediated DDR. Indeed, ERK phosphorylates p53, causing increased expression of the p21CIP1, MDM2 (mouse double minute 2 homolog) and GADD45 (45kd-growth arrest and DNA damage) genes [106]. As such, the activation of ERK may result in cell cycle arrest, providing time for the repair of cisplatin-induced DNA damage via p53. Moreover, p53 affects the sensitivity to apoptosis by activating the transcription of apoptotic genes (BAX) and repressing the transcription of apoptosis-inhibition genes (BCL-2) [107]. On the other hand, inhibition of cisplatin-induced ERK activation increases the sensitivity of cisplatin and decreases the levels of p21CIP1, MDM2 and GADD45 [103].
Other DNA damaging factors, including etoposide, adriamycin (doxorubicin) or UV irradiation also stimulate ERK1/2 MAP kinase in several cell lines [103,104,108]. Interestingly, in response to high or low intensity DNA insults, ERK/MAP kinase activation triggers apoptosis or cell cycle arrest, respectively [109]. Following these results, abrogation of ERK/MAP kinase activation was found when cells pretreated with MEK1/2 inhibitors were exposed to DNA damaging factors, thus verifying the role of MEK1/2 in mediating DNA damage-induced ERK activation [104]. Moreover, treatment with MEK1/2 inhibitors caused inhibition of ERK/MAP kinase and attenuation of p21CIP1 induction, resulting in the release of etoposide-induced G2/M cell cycle arrest. Furthermore, MEK1/2 inhibitors attenuated apoptosis that was induced by high doses of DNA-damaging agents. On the other hand, the excessive expression of the MEK1Q56P gain-of-function variant forced the activation of the ERK1/2 MAP kinase, making cells more susceptible to DNA damage-induced apoptosis [109]. Together, the phosphorylation and activation of the ERK/MAP kinase in the presence of DNA damage contribute to cell cycle arrest and apoptosis, thus explaining why cancer cells with high levels of ERK activation are more sensitive to DNA-damaging agents.
Interestingly, previous research showed that sirtuin 6 (SIRT6; deacetylase involved in DDR) interacts with the ERK signaling-related gene and the ERK-induced transcription factor ELK1 [110]. SIRT6 inhibits the expression of genes involved in the MAPK signaling pathway by interacting with their promoters and deacetylating H3K9 at these locations. In addition, inhibition of ERK2/p90RSK signaling pathway induced by high SIRT6 levels increases the DNA repair by CHK1 (checkpoint kinase 1) and the resistance to DNA damage [111]. In fact, in vitro experiments and human MM xenograft models showed a relationship between SIRT6 and genomic instability of MM cells. That is, they found that MM cells have high amounts of SIRT6, which inhibit the activity of the target genes ELK1, RSK2 (ribosomal S6 Kinase 2) and ERK2 in response to continuous DNA damage and genomic instability. The persistent DNA damage in MM causes SIRT6 to be recruited to DSBs and the downregulation of genes involved in MEK/ERK signaling. On the other hand, depletion of SIRT6 activates several ERK-related genes, including MAPK-activated RSK2 and ELK1-mediated transcriptional activity, thus blocking the G2/M cell cycle checkpoint [111].
In addition to ERK/MAP kinase signaling, JNK pathway is also activated as a result of DNA damage. Indeed, following treatment with cisplatin, DNA damage results in the stabilization and activation of p73, which creates a complex with JNK and triggers drug-induced apoptosis [112]. Moreover, several stress stimuli, including environmental stress, are mediators of cisplatin-induced apoptosis through the activation of p38 MAPK family of signaling proteins. A previous report has shown that cisplatin causes EGFR (epidermal growth factor receptor) internalization through the phosphorylation, and thus activation of the receptor by p38 MAPK [113]. Also, cisplatin induces stabilization of p18 Hamlet, a protein controlled by p38 MAPK, thus increasing the p53's capacity to bind with and activate the pro-apoptotic genes PUMA (p53 upregulated modulator of apoptosis) and PMAIP1 (phorbol-12-myristate-13-acetate-induced protein 1; also known as NOXA) [114]. Together, these results suggest that p38 MAPK pathway plays an important role in the regulation of cisplatin-induced apoptosis.
The involvement of the JNK pathway in the response to cisplatin has been confirmed by the inhibition of the JNK that reduced cisplatin-induced apoptosis in cervical cancer cells [115]. In contrast, blocking the p38 MAPK pathway increased reactive oxygen species levels, activated JNK pathway and made human tumor cells more susceptible to cisplatin-induced cell death [116]. This is also consistent with another study, which found that in epithelial renal tubule cell lines, p38 MAPK inhibition increased cisplatin-induced cell death via glutathione depletion and drug transport alteration [117]. In addition, treatment of several myeloma cell lines (NCI-H929, OPM2, RPMI-8226, U266) with the bifunctional mechlorethamine derivative bendamustine causes cleavage of caspase 3 and induction of apoptosis, while all cell lines experienced G2 cell cycle arrest [118]. Interestingly, the selective p38 MAPK inhibitor SB202190 dramatically boosts bendamustine-induced apoptosis and abrogates G2/M cell cycle arrest, suggesting that the combined treatment with MAPK and DDR modifiers might be used as novel anticancer therapy.
4.2. Induction of MAPK Activates DDR
Progression of the cell cycle from the G0/G1 to the S phase is induced by growth factors and depends on the ERK family of MAP kinases (Figure 2). Interestingly, ERK/MAP kinase activation must be continuous to trigger S phase entry [119,120]. Immediate-early genes and cyclins, among other ERK-dependent upregulated genes, are essential for promoting cell-cycle progression. Therefore, growth factor-stimulated continuous ERK activation could ensure G1 phase progression by upregulating genes that promote proliferation and by downregulating genes that inhibit it. The inactivation of ERK by a MEK inhibitor or a dominant-negative MEK1 at any point before the onset of the S phase decreased the S-phase entry rate [121].
Prior research suggests that BRAF inhibition upregulates the expression of p21CIP1 and p27 and downregulates the expression of retinoblastoma protein (pRb), cyclin D, and cyclin E genes that are implicated in G1 cell cycle progression [122]. For example, vemurafenib promotes cell cycle arrest at the G0/G1 phase and causes apoptosis in melanoma-sensitive, but not in melanoma-resistant cell lines harboring BRAF V600E mutation [123]. Interestingly, in vemurafenib-sensitive cell lines the combination of the HDACi suberoylanilide hydroxamic acid (SAHA) with vemurafenib induced both G0/G1 arrest and apoptosis, while in vemurafenib-resistant cells, the same combination induced G0/G1 and G2/M arrest resulting in dramatic cytostasis. It is noted that in vemurafenib-resistant cells, data from a gene expression study found MAPK hyperactivation and dysregulation of cyclins and CDKs, alterations that are consistent with the cytostatic effects of SAHA.
Even though p38 MAPK is not necessary for the DNA damage-induced G2/M checkpoint activation, it performs a crucial prosurvival role during this cell cycle arrest, through the overexpression of Bcl2 family proteins. In line with these data, inhibition of p38 during G2/M checkpoint arrest results in the simultaneous reduction of Bcl2 protein levels and triggers apoptosis in a p53-independent manner [124]. Another report has shown that p38 MAPK promotes DDB2 degradation and chromatin relaxation, thus stimulating the repair of UV-induced DNA damage by the NER pathway [125]. In fact, following UV irradiation, DDB2 is recruited to the damaged DNA sites, while p38 MAPK rapidly activated and helped DDB2 ubiquitylation. Consequent degradation of DDB2 results in the recruitment of the XPC (Xeroderma pigmentosum complementation group C) protein involved in the recognition of DNA damage through global genome repair (GGR), a critical subpathway of NER. Additionally, p38 MAPK helps to unfold the compacted chromatin by enhancing histone modifications, thus making UV lesions more accessible to NER factors.
CHK1, a serine/threonine protein kinase, is essential for protecting cells from stress and DNA damage during DNA replication [126]. Inhibition of this kinase causes accumulation of DNA damage, possibly due to increased replication stress. Indeed, Dai and colleagues investigated the role of the RAS/MEK/ERK pathway in relation to DNA damage in human MM cells exposed to CHK1 inhibitors and found that RAS/MEK/ERK signaling disruption significantly augmented DNA damage induced by CHK1 inhibitors and increased cells' sensitivity [98].
Moreover, an accumulating body of evidence suggests that MAPK signaling regulates HR/R mechanism in human cells, with JNK and ERK/MAPK pathways being positive, while p38 being negative regulators of HR/R [127]. More specifically, the inhibition of MEK/ERK signaling compromised ATM activity and reduced ATM phosphorylation and localization to foci, suggesting that ERK signaling affects the formation or the stability of repair protein complexes and/or the localization of ATM required for effective HR/R. On the other hand, inhibition of ATM kinase reduced ERK phosphorylation, suggesting that ATM modulates the ERK/MAPK signaling pathway. Therefore, a regulatory feedback loop may control DDR and ERK/MAPK signaling.
Also, using siRNA screening, Köpper and colleagues revealed kinases that contribute to the increased phosphorylation of H2AX at Ser139 (γH2AX) after UV-induced replication stress [128]. They found a dramatic reduction of γH2AX after the knockdown of the MAPK-activated protein kinase 2 (MK2), a kinase implicated in p38 stress signaling and G2 arrest. These results suggest that the cellular response to replication stress and the subsequent accumulation of DNA damage are directly influenced by the p38 MAPK signaling pathway [128,129].
5. Conclusion
Living organisms are protected against endogenous and exogenous hazards by a tightly regulated process that includes the synergistic action of the DDR network and the MAPK system. Aberrations in these networks may contribute to the pathogenesis and progression of several diseases, including MM. Since these alterations may also be involved in the development of drug resistance, they might be exploited as novel therapeutic targets and sensitive/effective biomarkers. Interestingly, these results potentially offer a new approach to enhance the efficacy of anti-myeloma therapy by combining DDR modulators with drugs targeting the MAPK signaling cascade.
Author Contributions
Conceptualization, P.M., C.P. and V.L.S.; writing-review and editing, P.M., C.P, M.G., M.A.D., E.T and V.L.S.; supervision, E.T. and V.L.S. All authors have read and agreed to the published version of the manuscript.
Funding
Funded by the European Union (Project 101097094 - ELMUMY). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or HADEA. Neither the European Union nor the granting authority can be held responsible for them.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in the reference section.
Conflict of Interest
The authors declare no conflicts of interest.
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; Van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple Myeloma. Nat Rev Dis Primers 2017, 3, 1–20. [Google Scholar] [CrossRef]
- Abeykoon, J.P.; Tawfiq, R.K.; Kumar, S.; Ansell, S.M. Monoclonal Gammopathy of Undetermined Significance: Evaluation, Risk Assessment, Management, and Beyond. Fac Rev 2022, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Thordardottir, M.; Lindqvist, E.K.; Lund, S.H.; Costello, R.; Burton, D.; Korde, N.; Mailankody, S.; Eiriksdottir, G.; Launer, L.J.; Gudnason, V.; et al. Obesity and Risk of Monoclonal Gammopathy of Undetermined Significance and Progression to Multiple Myeloma: A Population-Based Study. Blood Adv 2017, 1, 2186. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.M.; Hillengass, J.; Tang, L.; Lesokhin, A.M.; Landgren, O.; Usmani, S.Z.; Moysich, K.B.; McCann, S.E.; Shah, U.A. Dietary Risk Factors for Monoclonal Gammopathy of Undetermined Significance in a Racially Diverse Population. Blood Adv 2024, 8, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Bazou, D.; O’Gorman, P. Smoldering Multiple Myeloma: Prevalence and Current Evidence Guiding Treatment Decisions. Blood Lymphat Cancer 2018, 8, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Tahiru, W.; Izarra Santamaria, A.; Hultdin, J.; Wu, W.Y.Y.; Späth, F. Progression Patterns in Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma Outcome: A Cohort Study in 42 Patients. Exp Hematol Oncol 2022, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Gulla, A.; Anderson, K.C. Multiple Myeloma: The (r)Evolution of Current Therapy and a Glance into Future. Haematologica 2020, 105, 2358–2367. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2022 Update on Diagnosis, Risk-Stratification and Management. Am J Hematol 2022, 97, 1086. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Kumar, S.K. Chimeric Antigen Receptor T-Cells, Bispecific Antibodies, and Antibody-Drug Conjugates for Multiple Myeloma: An Update. Am J Hematol 2022, 97, 99–118. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M. V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Oben, B.; Froyen, G.; Maclachlan, K.H.; Leongamornlert, D.; Abascal, F.; Zheng-Lin, B.; Yellapantula, V.; Derkach, A.; Geerdens, E.; Diamond, B.T.; et al. Whole-Genome Sequencing Reveals Progressive versus Stable Myeloma Precursor Conditions as Two Distinct Entities. Nat Commun 2021, 12, 1861. [Google Scholar] [CrossRef] [PubMed]
- Aksenova, A.Y.; Zhuk, A.S.; Lada, A.G.; Zotova, I. V.; Stepchenkova, E.I.; Kostroma, I.I.; Gritsaev, S. V.; Pavlov, Y.I. Genome Instability in Multiple Myeloma: Facts and Factors. Cancers (Basel) 2021, 13, 5949. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ Mol Mutagen 2017, 58, 235. [Google Scholar] [CrossRef] [PubMed]
- Mavroeidi, D.; Georganta, A.; Panagiotou, E.; Syrigos, K.; Souliotis, V.L. Targeting ATR Pathway in Solid Tumors: Evidence of Improving Therapeutic Outcomes. Int J Mol Sci 2024, 25, 2767. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA Damage Repair in Cancer: From Mechanisms to Applications. Ann Transl Med 2020, 8, 1685. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Talluri, S.; Kumar, S.; Buon, L.; Zhao, J.; Potluri, L.B.; Prabhala, R.; Shammas, M.A.; Munshi, N.C. Base Excision Repair and Homologous Recombination Pathway Intermediates Drive Genomic Instability and Evolution in Myeloma. Blood 2020, 136, 27–28. [Google Scholar] [CrossRef]
- Ushie, C.; Saitoh, T.; Iwasaki, A.; Moriyama, N.; Hattori, H.; Matsumoto, M.; Sawamura, M.; Isoda, J.; Handa, H.; Yokohama, A.; et al. The Polymorphisms of Base Excision Repair Genes Influence the Prognosis of Multiple Myeloma. Blood 2012, 120, 3981. [Google Scholar] [CrossRef]
- Kumar, S.; Talluri, S.; Pal, J.; Yuan, X.; Lu, R.; Nanjappa, P.; Samur, M.K.; Munshi, N.C.; Shammas, M.A. Role of Apurinic/Apyrimidinic Nucleases in the Regulation of Homologous Recombination in Myeloma: Mechanisms and Translational Significance. Blood Cancer J 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524. [Google Scholar] [CrossRef]
- Thomas, M.; Li, J.; King, K.; Persaud, A.K.; Duah, E.; Vangundy, Z.; Hofmeister, C.C.; Lamba, J.K.; Tan, A.C.; Fridley, B.L.; et al. PARP1 and POLD2 as Prognostic Biomarkers for Multiple Myeloma in Autologous Stem Cell Transplant. Haematologica 2023, 108, 2155. [Google Scholar] [CrossRef]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-Induced PARPness to Target Genomic Instability in Multiple Myeloma. Haematologica 2021, 106, 185–195. [Google Scholar] [CrossRef]
- Szalat, R.; Samur, M.K.; Fulciniti, M.; Lopez, M.; Nanjappa, P.; Cleynen, A.; Wen, K.; Kumar, S.; Perini, T.; Calkins, A.S.; et al. Nucleotide Excision Repair Is a Potential Therapeutic Target in Multiple Myeloma. Leukemia 2018, 32, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Vangsted, A.; Gimsing, P.; Klausen, T.W.; Nexø, B.A.; Wallin, H.; Andersen, P.; Hokland, P.; Lillevang, S.T.; Vogel, U. Polymorphisms in the Genes ERCC2, XRCC3 and CD3EAP Influence Treatment Outcome in Multiple Myeloma Patients Undergoing Autologous Bone Marrow Transplantation. Int J Cancer 2007, 120, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Velangi, M.R.; Matheson, E.C.; Morgan, G.J.; Jackson, G.H.; Taylor, P.R.; Hall, A.G.; Irving, J.A.E. DNA Mismatch Repair Pathway Defects in the Pathogenesis and Evolution of Myeloma. Carcinogenesis 2004, 25, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Santón, A.; García-Cosio, M.; Bellas, C. HMLH1 and MGMT Inactivation as a Mechanism of Tumorigenesis in Monoclonal Gammopathies. Mod Pathol 2006, 19, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Kotoula, V.; Hytiroglou, P.; Kaloutsi, V.; Barbanis, S.; Kouidou, S.; Papadimitriou, C.S. Mismatch Repair Gene Expression in Malignant Lymphoproliferative Disorders of B-Cell Origin. Leuk Lymphoma 2002, 43, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Branch, P.; Aquilina, G.; Bignami, M.; Karran, P. Defective Mismatch Binding and a Mutator Phenotype in Cells Tolerant to DNA Damage. Nature 1993, 362, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, K.; Fujii, K.; Suehiro, Y.; Taguchi, K.; Uike, N.; Yoshida, M.A.; Oda, S. Heterochronous Occurrence of Microsatellite Instability in Multiple Myeloma - an Implication for a Role of Defective DNA Mismatch Repair in Myelomagenesis. Leuk Lymphoma 2018, 59, 2454–2459. [Google Scholar] [CrossRef] [PubMed]
- Shammas, M.A.; Reis, R.J.S.; Koley, H.; Batchu, R.B.; Li, C.; Munshi, N.C. Dysfunctional Homologous Recombination Mediates Genomic Instability and Progression in Myeloma. Blood 2009, 113, 2290–2297. [Google Scholar] [CrossRef] [PubMed]
- Roddam, P.L.; Rollinson, S.; O’Driscoll, M.; Jeggo, P.A.; Jack, A.; Morgan, G.J. Genetic Variants of NHEJ DNA Ligase IV Can Affect the Risk of Developing Multiple Myeloma, a TumourCharacterised by Aberrant Class Switch Recombination. J Med Genet 2002, 39, 900–905. [Google Scholar] [CrossRef]
- Patel, P.R.; Senyuk, V.; Sweiss, K.; Calip, G.; Oh, A.; Mahmud, N.; Rondelli, D. Overcoming Melphalan Resistance By Targeting Crucial DNA Repair Pathways in Multiple Myeloma. Biol Blood Marrow Transplant 2020, 26, S224–S225. [Google Scholar] [CrossRef]
- Dumontet, C.; Landi, S.; Reiman, T.; Perry, T.; Plesa, A.; Bellini, I.; Barale, R.; Pilarski, L.M.; Troncy, J.; Tavtigian, S.; et al. Genetic Polymorphisms Associated with Outcome in Multiple Myeloma Patients Receiving High-Dose Melphalan. Bone Marrow Transplant 2010, 45, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ferguson, D.O.; Xie, W.; Manis, J.P.; Sekiguchi, J.A.; Frank, K.M.; Chaudhuri, J.; Horner, J.; DePinho, R.A.; Alt, F.W. Interplay of P53 and DNA-Repair Protein XRCC4 in Tumorigenesis, Genomic Stability and Development. Nature 2000, 404, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Betti, C.; Singh, S.; Toor, A.; Vaughan, A. Impaired NHEJ Function in Multiple Myeloma. Mutat Res 2009, 660, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Roddam, P.L.; Allan, J.M.; Dring, A.M.; Worrillow, L.J.; Davies, F.E.; Morgan, G.J. Non-Homologous End-Joining Gene Profiling Reveals Distinct Expression Patterns Associated with Lymphoma and Multiple Myeloma. Br J Haematol 2010, 149, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Hayden, P.J.; Tewari, P.; Morris, D.W.; Staines, A.; Crowley, D.; Nieters, A.; Becker, N.; De sanjosé, S.; Foretova, L.; Maynadié, M.; et al. Variation in DNA Repair Genes XRCC3, XRCC4, XRCC5 and Susceptibility to Myeloma. Hum Mol Genet 2007, 16, 3117–3127. [Google Scholar] [CrossRef]
- Calimeri, T.; Fulciniti, M.; Lin, J.; Samur, M.K.; Calkins, A.S.; Vahia, A. V; Pal, J.; Cea, M.; Cagnetta, A.; Cottini, F.; et al. Aberrant Non-Homologous End Joining in Multiple Myeloma: A Role in Genomic Instability and As Potential Prognostic Marker. Blood 2012, 120, 2932. [Google Scholar] [CrossRef]
- Liu, J.; Xie, Y.; Guo, J.; Li, X.; Wang, J.; Jiang, H.; Peng, Z.; Wang, J.; Wang, S.; Li, Q.; et al. Targeting NSD2-Mediated SRC-3 Liquid-Liquid Phase Separation Sensitizes Bortezomib Treatment in Multiple Myeloma. Nat Commun 2021, 12, 1022. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Martinez-Garcia, E.; Phillip, J.M.; Chambliss, A.B.; Popovic, R.; Ezponda, T.; Small, E.C.; Will, C.; Phillip, M.P.; Neri, P.; et al. MMSET/WHSC1 Enhances DNA Damage Repair Leading to an Increase in Resistance to Chemotherapeutic Agents. Oncogene 2016, 35, 5905–5915. [Google Scholar] [CrossRef]
- Saitoh, T.; Oda, T. DNA Damage Response in Multiple Myeloma: The Role of the Tumor Microenvironment. Cancers (Basel) 2021, 13, 1–22. [Google Scholar] [CrossRef]
- Peake, J.D.; Noguchi, E. Fanconi Anemia: Current Insights Regarding Epidemiology, Cancer, and DNA Repair. Hum Genet 2022, 141, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Van Der Sluis, P.C.; Boulware, D.; Hazlehurst, L.A.; Dalton, W.S. The FA/BRCA Pathway Is Involved in Melphalan-Induced DNA Interstrand Cross-Link Repair and Accounts for Melphalan Resistance in Multiple Myeloma Cells. Blood 2005, 106, 698–705. [Google Scholar] [CrossRef]
- Kassambara, A.A.; Gourzones-Dmitriev, C.; Sahota, S.; Rème, T.; Moreaux, J.; Goldschmidt, H.; Constantinou, A.; Pasero, P.; Hose, D.; Klein, B. A DNA Repair Pathway Score Predicts Survival in Human Multiple Myeloma: The Potential for Therapeutic Strategy. Oncotarget 2014, 5, 2487. [Google Scholar] [CrossRef] [PubMed]
- Poczta, A.; Rogalska, A.; Marczak, A. Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies—Current Research and Clinical Approaches. J Clin Med 2021, 10, 1841. [Google Scholar] [CrossRef]
- Spanswick, V.J.; Craddock, C.; Sekhar, M.; Mahendra, P.; Shankaranarayana, P.; George Hughes, R.; Hochhauser, D.; Hartley, J.A. Repair of DNA Interstrand Crosslinks as a Mechanism of Clinical Resistance to Melphalan in Multiple Myeloma. Blood 2002, 100, 224–229. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Souliotis, V.L.; Anagnostopoulos, A.; Papadimitriou, C.; Sfikakis, P.P. Extent of Damage and Repair in the P53 Tumor-Suppressor Gene after Treatment of Myeloma Patients with High-Dose Melphalan and Autologous Blood Stem-Cell Transplantation Is Individualized and May Predict Clinical Outcome. J Clin Oncol 2005, 23, 4381–4389. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Souliotis, V.L.; Anagnostopoulos, A.; Bamia, C.; Pouli, A.; Baltadakis, I.; Terpos, E.; Kyrtopoulos, S.A.; Sfikakis, P.P. Melphalan-Induced DNA Damage in Vitro as a Predictor for Clinical Outcome in Multiple Myeloma. Haematologica 2007, 92, 1505–1512. [Google Scholar] [CrossRef]
- Gkotzamanidou, M.; Terpos, E.; Bamia, C.; Munshi, N.C.; Dimopoulos, M.A.; Souliotis, V.L. DNA Repair of Myeloma Plasma Cells Correlates with Clinical Outcome: The Effect of the Nonhomologous End-Joining Inhibitor SCR7. Blood 2016, 128, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Gkotzamanidou, M.; Sfikakis, P.P.; Kyrtopoulos, S.A.; Bamia, C.; Dimopoulos, M.A.; Souliotis, V.L. Chromatin Structure, Transcriptional Activity and DNA Repair Efficiency Affect the Outcome of Chemotherapy in Multiple Myeloma. Br J Cancer 2014, 111, 1293–1304. [Google Scholar] [CrossRef]
- Herrero, A.B.; Gutiérrez, N.C. Targeting Ongoing DNA Damage in Multiple Myeloma: Effects of DNA Damage Response Inhibitors on Plasma Cell Survival. Front Oncol 2017, 7, 98. [Google Scholar] [CrossRef]
- Keane, N.A.; Reidy, M.; Natoni, A.; Raab, M.S.; O’Dwyer, M. Targeting the Pim Kinases in Multiple Myeloma. Blood Cancer J 2015, 5, e325. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, J.; Santo, L.; Siu, K.T.; Panaroni, C.; Raje, N. Pim2 Is Important for Regulating DNA Damage Response in Multiple Myeloma Cells. Blood Cancer J 2016, 6, e462. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yang, D.; Ye, Y.; Chen, Z.; Sun, T.; Zhao, J.; Zhao, K.; Lu, N. Inhibition of Pim-2 Kinase by LT-171-861 Promotes DNA Damage and Exhibits Enhanced Lethal Effects with PARP Inhibitor in Multiple Myeloma. Biochem Pharmacol 2021, 190, 114648. [Google Scholar] [CrossRef] [PubMed]
- Maiso, P.; Carvajal-Vergara, X.; Ocio, E.M.; López-Pérez, R.; Mateo, G.; Gutiérrez, N.; Atadja, P.; Pandiella, A.; San Miguel, J.F. The Histone Deacetylase Inhibitor LBH589 Is a Potent Antimyeloma Agent That Overcomes Drug Resistance. Cancer Res 2006, 66, 5781–5789. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Shen, J.; Steinberg, J.; Li, M.; Wang, C.; Bonavida, B.; Chen, H.; Li, Z.W.; Berenson, J.R. The Histone Deacetylase Inhibitor LBH589 Enhances the Anti-Myeloma Effects of Chemotherapy in Vitro and in Vivo. Leuk Res 2011, 35, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Gkotzamanidou, M.; Terpos, E.; Dimopoulos, M.A.; Souliotis, V.L. The Combination of Panobinostat and Melphalan for the Treatment of Patients with Multiple Myeloma. Int J Mol Sci 2022, 23, 15671. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Li, S.; Westermarck, J. Phosphatase-Mediated Crosstalk between MAPK Signaling Pathways in the Regulation of Cell Survival. FASEB J 2008, 22, 954–965. [Google Scholar] [CrossRef]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF Signaling Pathway in Cancer Therapy. Expert Opin Ther Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; Elsum, I.A.; King, C.L.; Kinross, K.M.; Richardson, H.E.; Humbert, P.O. Loss of Human Scribble Cooperates with H-Ras to Promote Cell Invasion through Deregulation of MAPK Signalling. Oncogene 2008, 27, 5988–6001. [Google Scholar] [CrossRef]
- Majidinia, M.; Sadeghpour, A.; Yousefi, B. The Roles of Signaling Pathways in Bone Repair and Regeneration. J Cell Physiol 2018, 233, 2937–2948. [Google Scholar] [CrossRef]
- Bolli, N.; Genuardi, E.; Ziccheddu, B.; Martello, M.; Oliva, S.; Terragna, C. Next-Generation Sequencing for Clinical Management of Multiple Myeloma: Ready for Prime Time? Front Oncol 2020, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.M.; Deshpande, S.; Tytarenko, R.; Ashby, C.; Wang, Y.; Bauer, M.A.; Johnson, S.K.; Wardell, C.P.; Thanendrarajan, S.; Zangari, M.; et al. The Molecular Make up of Smoldering Myeloma Highlights the Evolutionary Pathways Leading to Multiple Myeloma. Nat Commun 2021, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Rajanna, A.R.; Ziccheddu, B.; Poos, A.M.; Derkach, A.; Maclachlan, K.; Durante, M.; Diamond, B.; Papadimitriou, M.; Davies, F.; et al. Genomic Classification and Individualized Prognosis in Multiple Myeloma. J Clin Oncol 2024, 42, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of Genomic Evolution and Mutational Profiles in Multiple Myeloma. Nat Commun 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Hoang, P.H.; Dobbins, S.E.; Cornish, A.J.; Chubb, D.; Law, P.J.; Kaiser, M.; Houlston, R.S. Whole-Genome Sequencing of Multiple Myeloma Reveals Oncogenic Pathways Are Targeted Somatically through Multiple Mechanisms. Leukemia 2018, 32, 2459–2470. [Google Scholar] [CrossRef] [PubMed]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.X.; Zhu, Y.X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted Sequencing of Refractory Myeloma Reveals a High Incidence of Mutations in CRBN and Ras Pathway Genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The Genetic Architecture of Multiple Myeloma. Nat Rev Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the Untargetable KRAS in Cancer Therapy. Acta Pharm Sin B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Uprety, D.; Adjei, A.A. KRAS: From Undruggable to a Druggable Cancer Target. Cancer Treat Rev 2020, 89, 102070. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Cody Ashby, T.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Pfarr, N.; Endris, V.; Mai, E.K.; Md Hanafiah, N.H.; Lehners, N.; Penzel, R.; Weichert, W.; Ho, A.D.; Schirmacher, P.; et al. Molecular Signaling in Multiple Myeloma: Association of RAS/RAF Mutations and MEK/ERK Pathway Activation. Oncogenesis 2017, 6, e337. [Google Scholar] [CrossRef]
- Mulligan, G.; Lichter, D.I.; Bacco, A. Di; Blakemore, S.J.; Berger, A.; Koenig, E.; Bernard, H.; Trepicchio, W.; Li, B.; Neuwirth, R.; et al. Mutation of NRAS but Not KRAS Significantly Reduces Myeloma Sensitivity to Single-Agent Bortezomib Therapy. Blood 2014, 123, 632–639. [Google Scholar] [CrossRef]
- Haque, E.; Śmiech, M.; Łuczyńska, K.; Bouchard, M.F.; Viger, R.; Kono, H.; Pierzchała, M.; Taniguchi, H. NRF2 DLG Domain Mutations Identified in Japanese Liver Cancer Patients Affect the Transcriptional Activity in HCC Cell Lines. Int J Mol Sci 2021, 22, 5296. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lonardi, S.; Masi, G.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Spadi, R.; Zaniboni, A.; et al. Early Tumor Shrinkage and Depth of Response Predict Long-Term Outcome in Metastatic Colorectal Cancer Patients Treated with First-Line Chemotherapy plus Bevacizumab: Results from Phase III TRIBE Trial by the Gruppo Oncologico Del Nord Ovest. Ann Oncol 2015, 26, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.M.; Ashby, C.; Tytarenko, R.G.; Deshpande, S.; Wang, H.; Wang, Y.; Rosenthal, A.; Sawyer, J.; Tian, E.; Flynt, E.; et al. BRAF and DIS3 Mutations Associate with Adverse Outcome in a Long-Term Follow-up of Patients with Multiple Myeloma. Clin Cancer Res 2020, 26, 2422–2432. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Stamping out RAF and MEK1/2 to Inhibit the ERK1/2 Pathway: An Emerging Threat to Anticancer Therapy. Oncogene 2016, 35, 2547–2561. [Google Scholar] [CrossRef] [PubMed]
- Heuck, C.J.; Jethava, Y.; Khan, R.; Van Rhee, F.; Zangari, M.; Chavan, S.; Robbins, K.; Miller, S.E.; Matin, A.; Mohan, M.; et al. Inhibiting MEK in MAPK Pathway-Activated Myeloma. Leukemia 2016, 30, 976–980. [Google Scholar] [CrossRef]
- Lara, M.S.; Blakely, C.M.; Riess, J.W. Targeting MEK in Non-Small Cell Lung Cancer. Curr Probl Cancer 2024, 101065. [Google Scholar] [CrossRef]
- Holkova, B.; Zingone, A.; Kmieciak, M.; Bose, P.; Badros, A.Z.; Voorhees, P.M.; Baz, R.; Korde, N.; Lin, H.Y.; Chen, J.Q.; et al. A Phase II Trial of AZD6244 (Selumetinib, ARRY-142886), an Oral MEK1/2 Inhibitor, in Relapsed/Refractory Multiple Myeloma. Clin Cancer Res 2016, 22, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Schjesvold, F.; Paiva, B.; Ribrag, V.; Rodriguez-Otero, P.; San-Miguel, J.F.; Robak, P.; Hansson, M.; Onishi, M.; Hamidi, H.; Malhi, V.; et al. Cobimetinib Alone and Plus Venetoclax With/Without Atezolizumab in Patients With Relapsed/Refractory Multiple Myeloma. Clin Lymphoma Myeloma Leuk 2023, 23, e59–e70. [Google Scholar] [CrossRef]
- Schjesvold, F.; Ribrag, V.; Rodriguez-Otero, P.; Robak, P.J.; Hansson, M.; Hajek, R.; Amor, A.A.; Martinez-López, J.; Onishi, M.; Gallo, J.D.; et al. Safety and Preliminary Efficacy Results from a Phase Ib/II Study of CobimetinibAs a Single Agent and in Combination with Venetoclax with or without Atezolizumab in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the BRAF V600E Mutation in Multiple Myeloma. Cancer Discov 2013, 3, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Targeting Oncogenic Raf Protein-Serine/Threonine Kinases in Human Cancers. Pharmacol Res 2018, 135, 239–258. [Google Scholar] [CrossRef]
- Degirmenci, U.; Yap, J.; Sim, Y.R.M.; Qin, S.; Hu, J. Drug Resistance in Targeted Cancer Therapies with RAF Inhibitors. Cancer Drug Resist 2021, 4, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Mey, U.J.M.; Renner, C.; von Moos, R. Vemurafenib in Combination with Cobimetinib in Relapsed and Refractory Extramedullary Multiple Myeloma Harboring the BRAF V600E Mutation. Hematol Oncol 2017, 35, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Otieno, S.B.; Nasir, S.; Weir, A.; Johnson, R. Rapid Response in a Patient with Relapsed/Refractory Multiple Myeloma Treated with BRAF/MEK Inhibitors. Case Rep Hematol 2020, 2020, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Giesen, N.; Chatterjee, M.; Scheid, C.; Poos, A.M.; Besemer, B.; Miah, K.; Benner, A.; Becker, N.; Moehler, T.; Metzler, I.; et al. A Phase 2 Clinical Trial of Combined BRAF/MEK Inhibition for BRAFV600E-Mutated Multiple Myeloma. Blood 2023, 141, 1685–1690. [Google Scholar] [CrossRef]
- Guo, C.; Chénard-Poirier, M.; Roda, D.; de Miguel, M.; Harris, S.J.; Candilejo, I.M.; Sriskandarajah, P.; Xu, W.; Scaranti, M.; Constantinidou, A.; et al. Intermittent Schedules of the Oral RAF-MEK Inhibitor CH5126766/VS-6766 in Patients with RAS/RAF-Mutant Solid Tumours and Multiple Myeloma: A Single-Centre, Open-Label, Phase 1 Dose-Escalation and Basket Dose-Expansion Study. Lancet Oncol 2020, 21, 1478–1488. [Google Scholar] [CrossRef]
- Raab, M.S.; Giesen, N.; Scheid, C.; Besemer, B.; Miah, K.; Benner, A.; Metzler, I.; Khandanpour, C.; Seidel-Glaetzer, A.; Trautmann-Grill, K.; et al. Safety and Preliminary Efficacy Results from a Phase II Study Evaluating Combined BRAF and MEK Inhibition in Relapsed/Refractory Multiple Myeloma (RrMM) Patients with Activating BRAF V600E Mutations: The GMMG-Birma Trial. Blood 2020, 136, 44–45. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Timm, M.; Haug, J.L.; Kimlinger, T.K.; Wellik, L.E.; Witzig, T.E.; Rajkumar, S. V.; Adjei, A.A.; Kumar, S. Sorafenib, a Dual Raf Kinase/Vascular Endothelial Growth Factor Receptor Inhibitor Has Significant Anti-Myeloma Activity and Synergizes with Common Anti-Myeloma Drugs. Oncogene 2010, 29, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Jett, J.; Marks, R.; Richardson, R.; Quevedo, F.; Moynihan, T.; Croghan, G.; Markovic, S.N.; Bible, K.C.; Qin, R.; et al. Phase 1 Study of Sorafenib in Combination with Bortezomib in Patients with Advanced Malignancies. Invest New Drugs 2013, 31, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Lauer, R. Phase II Study of Sorafenib and Bortezomib for First-Line Treatment of Metastatic or Unresectable Renal Cell Carcinoma. Oncologist 2015, 20, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Yordanova, A.; Hose, D.; Neben, K.; Witzens-Harig, M.; Gütgemann, I.; Raab, M.S.; Moehler, T.; Goldschmidt, H.; Schmidt-Wolf, I.G. Sorafenib in Patients with Refractory or Recurrent Multiple Myeloma. Hematol Oncol 2013, 31, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.S.; Krishnan, A.; Lonial, S.; Chatta, G.; Alsina, M.; Jagannath, S.; Richardson, P.; Hohl, R.J.; Lust, J.A.; Bensinger, W.; et al. Phase II Trial of SCIO-469 as Monotherapy (M) or in Combination with Bortezomib (MB) in Relapsed Refractory Multiple Myeloma (MM). Blood 2006, 108, 3580. [Google Scholar] [CrossRef]
- Mateos, M.V.; Cibeira, M.T.; Richardson, P.G.; Prosper, F.; Oriol, A.; De La Rubia, J.; Lahuerta, J.J.; García-Sanz, R.; Extremera, S.; Szyldergemajn, S.; et al. Phase II Clinical and Pharmacokinetic Study of Plitidepsin 3-Hour Infusion Every Two Weeks Alone or with Dexamethasone in Relapsed and Refractory Multiple Myeloma. Clin Cancer Res 2010, 16, 3260–3269. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, S.; Pei, X.Y.; Almenara, J.A.; Kramer, L.B.; Venditti, C.A.; Dent, P.; Grant, S. Interruption of the Ras/MEK/ERK Signaling Cascade Enhances Chk1 Inhibitor-Induced DNA Damage in Vitro and in Vivo in Human Multiple Myeloma Cells. Blood 2008, 112, 2439–2449. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Yan, J.; Tang, D. Extracellular Signal-Regulated Kinases Modulate DNA Damage Response - A Contributing Factor to Using MEK Inhibitors in Cancer Therapy. Curr Med Chem 2011, 18, 5476. [Google Scholar] [CrossRef]
- Rezatabar, S.; Karimian, A.; Rameshknia, V.; Parsian, H.; Majidinia, M.; Kopi, T.A.; Bishayee, A.; Sadeghinia, A.; Yousefi, M.; Monirialamdari, M.; et al. RAS/MAPK Signaling Functions in Oxidative Stress, DNA Damage Response and Cancer Progression. J Cell Physiol 2019, 234, 14951–14965. [Google Scholar] [CrossRef]
- Wang, X.; Martindale, J.L.; Liu, Y.; Holbrook, N.J. The Cellular Response to Oxidative Stress: Influences of Mitogen-Activated Protein Kinase Signalling Pathways on Cell Survival. Biochem J 1998, 333, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Koppenhoefer, U.; Weinstock, C.; Linderkamp, O.; Lang, F.; Gulbins, E. Fas- or Ceramide-Induced Apoptosis Is Mediated by a Rac1-Regulated Activation of Jun N-Terminal Kinase/P38 Kinases and GADD153. J Biol Chem 1997, 272, 22173–22181. [Google Scholar] [CrossRef] [PubMed]
- Persons, D.L.; Yazlovitskaya, E.M.; Cui, W.; Pelling, J.C. Cisplatin-Induced Activation of Mitogen-Activated Protein Kinases in Ovarian Carcinoma Cells: Inhibition of Extracellular Signal-Regulated Kinase Activity Increases Sensitivity to Cisplatin. Clin Cancer Res 1999, 5, 1007–1014. [Google Scholar] [PubMed]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK Activation in Cisplatin-Induced Apoptosis. J Biol Chem 2000, 275, 39435–39443. [Google Scholar] [CrossRef] [PubMed]
- Chu, G. Cellular Responses to Cisplatin. The Roles of DNA-Binding Proteins and DNA Repair. J Biol Chem 1994, 269, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Dehaan, R.D.; Yazlovitskaya, E.M.; Persons, D.L. Regulation of P53 Target Gene Expression by Cisplatin-Induced Extracellular Signal-Regulated Kinase. Cancer Chemother Pharmacol 2001, 48, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of P53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Fang, L.; Igarashi, M.; Ouchi, T.; Lu, K.P.; Aaronson, S.A. Sustained Activation of Ras/Raf/Mitogen-Activated Protein Kinase Cascade by the Tumor Suppressor P53. Proc Natl Acad Sci U S A 2000, 97, 8302–8305. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Wu, D.; Hirao, A.; Lahti, J.M.; Liu, L.; Mazza, B.; Kidd, V.J.; Mak, T.W.; Ingram, A.J. ERK Activation Mediates Cell Cycle Arrest and Apoptosis after DNA Damage Independently of P53. J Biol Chem 2002, 277, 12710–12717. [Google Scholar] [CrossRef]
- Bosch-Presegué, L.; Vaquero, A. Sirtuins in Stress Response: Guardians of the Genome. Oncogene 2014, 33, 3764–3775. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Adamia, S.; Acharya, C.; Tai, Y.T.; Fulciniti, M.; Ohguchi, H.; Munshi, A.; Acharya, P.; Bhasin, M.K.; et al. Evidence for a Role of the Histone Deacetylase SIRT6 in DNA Damage Response of Multiple Myeloma Cells. Blood 2015, 127, 1138–1150. [Google Scholar] [CrossRef]
- Jones, E. V.; Dickman, M.J.; Whitmarsh, A.J. Regulation of P73-Mediated Apoptosis by c-Jun N-Terminal Kinase. Biochem J 2007, 405, 617. [Google Scholar] [CrossRef] [PubMed]
- Winograd-Katz, S.E.; Levitzki, A. Cisplatin Induces PKB/Akt Activation and P38(MAPK) Phosphorylation of the EGF Receptor. Oncogene 2006, 25, 7381–7390. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, V.; Cuadrado, A.; Nebreda, A.R. P18(Hamlet) Mediates Different P53-Dependent Responses to DNA-Damage Inducing Agents. Cell Cycle 2007, 6, 2319–2322. [Google Scholar] [CrossRef]
- Brozovic, A.; Fritz, G.; Christmann, M.; Zisowsky, J.; Jaehde, U.; Osmak, M.; Kaina, B. Long-Term Activation of SAPK/JNK, P38 Kinase and Fas-L Expression by Cisplatin Is Attenuated in Human Carcinoma Cells That Acquired Drug Resistance. Int J Cancer 2004, 112, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Igea, A.; Canovas, B.; Dolado, I.; Nebreda, A.R. Inhibition of P38 MAPK Sensitizes Tumour Cells to Cisplatin-Induced Apoptosis Mediated by Reactive Oxygen Species and JNK. EMBO Mol Med 2013, 5, 1759–1774. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-García, M.E.; Quiroga, A.G.; Castro, J.; Ortiz, A.; Aller, P.; Mata, F. Inhibition of P38-MAPK Potentiates Cisplatin-Induced Apoptosis via GSH Depletion and Increases Intracellular Drug Accumulation in Growth-Arrested Kidney Tubular Epithelial Cells. Toxicol Sci 2009, 111, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Gaul, L.; Mandl-Weber, S.; Baumann, P.; Emmerich, B.; Schmidmaier, R. Bendamustine Induces G2 Cell Cycle Arrest and Apoptosis in Myeloma Cells: The Role of ATM-Chk2-Cdc25A and ATM-P53-P21-Pathways. J Cancer Res Clin Oncol 2008, 134, 245–253. [Google Scholar] [CrossRef]
- Weber, J.D.; Raben, D.M.; Phillips, P.J.; Baldassare, J.J. Sustained Activation of Extracellular-Signal-Regulated Kinase 1 (ERK1) Is Required for the Continued Expression of Cyclin D1 in G1 Phase. Biochem J 1997, 326 (Pt 1), 61–68. [Google Scholar] [CrossRef]
- Meloche, S.; Seuwen, K.; Pagès, G.; Pouysségur, J. Biphasic and Synergistic Activation of P44mapk (ERK1) by Growth Factors: Correlation between Late Phase Activation and Mitogenicity. Mol Endocrinol 1992, 6, 845–854. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK Activation Downregulates Antiproliferative Genes throughout G1 Phase to Allow Cell-Cycle Progression. Curr Biol 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.W.; Pratilas, C.A.; Poulikakos, P.I.; Tadi, M.; Wang, W.; Taylor, B.S.; Halilovic, E.; Persaud, Y.; Xing, F.; Viale, A.; et al. The RAF Inhibitor PLX4032 Inhibits ERK Signaling and Tumor Cell Proliferation in a V600E BRAF-Selective Manner. Proc Natl Acad Sci U S A 2010, 107, 14903–14908. [Google Scholar] [CrossRef] [PubMed]
- Toress-Collado, A.X.; Nazarian, R.; Jazirehi, A.R. Rescue of Cell Cycle Progression in BRAFV600E Inhibitor-Resistant Human Melanoma by a Chromatin Modifier. Tumour Biol 2017, 39, 1010428317721620. [Google Scholar] [CrossRef] [PubMed]
- Phong, M.S.; Horn, R.D. Van; Li, S.; Tucker-Kellogg, G.; Surana, U.; Ye, X.S. P38 Mitogen-Activated Protein Kinase Promotes Cell Survival in Response to DNA Damage but Is Not Required for the G2 DNA Damage Checkpoint in Human Cancer Cells. Mol Cell Biol 2010, 30, 3816. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Barakat, B.M.; Qin, S.; Ray, A.; El-Mahdy, M.A.; Wani, G.; Arafa, E.S.; Mir, S.N.; Wang, Q.E.; Wani, A.A. The P38 Mitogen-Activated Protein Kinase Augments Nucleotide Excision Repair by Mediating DDB2 Degradation and Chromatin Relaxation. J Biol Chem 2008, 283, 32553–32561. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A.M. Small-Molecule Cyclin-Dependent Kinase Modulators. Oncogene 2003, 22, 6609–6620. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Neill, S.; Dent, P.; Povirk, L.F.; Valerie, K. Extracellular Signal-Related Kinase Positively Regulates Ataxia Telangiectasia Mutated, Homologous Recombination Repair, and the DNA Damage Response. Cancer Res 2007, 67, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Köpper, F.; Bierwirth, C.; Schön, M.; Kunze, M.; Elvers, I.; Kranz, D.; Saini, P.; Menon, M.B.; Walter, D.; Sørensen, C.S.; et al. Damage-Induced DNA Replication Stalling Relies on MAPK-Activated Protein Kinase 2 Activity. Proc Natl Acad Sci U S A 2013, 110, 16856–16861. [Google Scholar] [CrossRef]
- Gaestel, M. MAPKAP Kinases - MKs - Two’s Company, Three’s a Crowd. Nat Rev Mol Cell Biol 2006, 7, 120–130. [Google Scholar] [CrossRef]
Figure 1.
Deregulated DNA repair pathways involved in myelomagenesis. The most commonly altered DNA repair genes are shown (Figure was created with BioRender.com).
Figure 1.
Deregulated DNA repair pathways involved in myelomagenesis. The most commonly altered DNA repair genes are shown (Figure was created with BioRender.com).
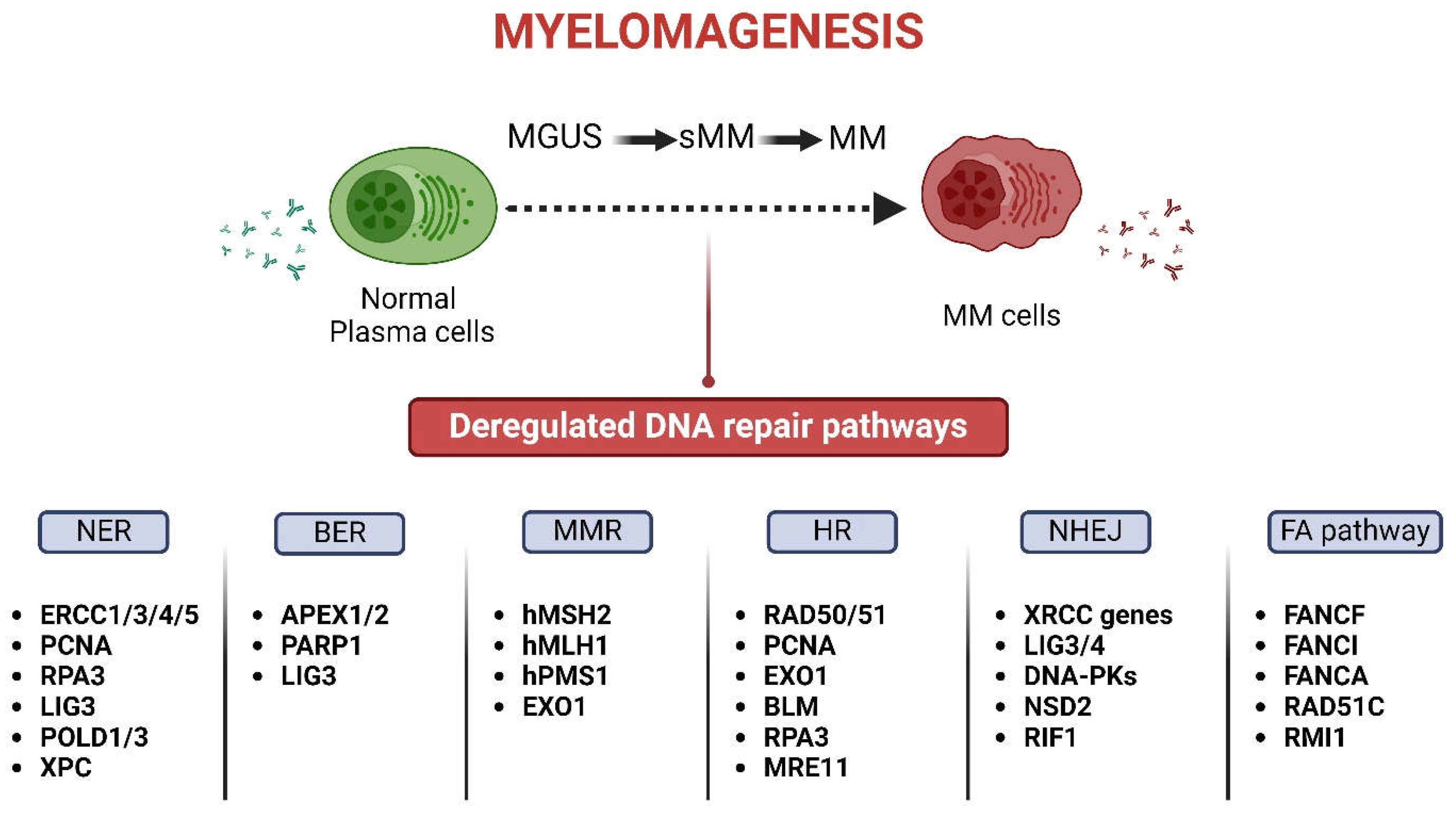
Figure 2.
Schematic diagram displaying the three MAPK signaling pathways and their coordinated action with the DDR network (Figure was created with BioRender.com).
Figure 2.
Schematic diagram displaying the three MAPK signaling pathways and their coordinated action with the DDR network (Figure was created with BioRender.com).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



