
Submitted:
07 May 2024
Posted:
10 May 2024
You are already at the latest version
Abstract
BackgroundThe present review summarizes the state-of-the-art of in silico methods and techniques that are the most useful in drug discovery, their relationship with data science, as well as the successful application of data science, machine learning (ML) and artificial intelligence (AI) applications. A meta-analysis of the various technologies available is furthermore proposed as a guideline for the non-expert, reader relative to the several subject areas is also discussed in this article. The scope of this meta-analysis is to rank the enlisted technologies by their field of applications and to depict the latter according to knowledge accessibility, from students to experts.Method The search strategy utilized for this review first produced a general collection of 900 papers without duplications, which were subsequently streamlined and divided into two independent collections: the top 300 most-cited papers of all time (since 2000) and the papers with the highest interest for a systematic review analysis (high-impact exciting papers). Results In Part 1, we discuss the most cited and quality 97 articles in these top 300 papers most relevant to the field of in silico drug discovery. The different disciplines are listed according to their industrial and economic incurred to society, independently from the “metric” results of how many new drug approvals (NDAs) each discipline has generated to date.ConclusionBig data, the ensemble of known items stored in publicly available databases, has improved our understanding of the many fates of a potential drug candidate during its development and even after its commercialization. Moreover, the combination of new screening techniques and “omics” with old drugs has led to a new paradigm in which the unknown knowledge of any biological molecule and its cellular structure, now plays an important role as a target for a series of yet-to-be-developed drugs: the chemical space. Furthermore, leveraging big data, data science, ML, and AI can revolutionize drug discovery by swiftly analyzing massive datasets, predicting efficacy and safety profiles, streamlining development, cutting costs, and boosting success rates for new drugs. AI also speeds up the search for promising drug candidates, advancing innovative therapies.
Keywords:
Data science
; Big data
; Data mining
; Bioinformatics
; Chemometric
; Medicinal chemistry
; Targets
; Knowledge discovery
; Artificial intelligence
; Machine learning
; Deep learning
; Data integration
; Metadata
; Database
; QSAR
; Collaborative drug discovery
; Structure-based drug design
; Ligand-based drug discovery
; Clinical trials
; Product development.
Highlights:
- Advancements in computational methods in in silico drug discovery have become a viable option.
- Artificial intelligence and machine learning improve in silico drug discovery by swiftly analyzing data, predicting interactions, and optimizing candidates with precision.
- The recent technological evolution from 1980 to 2024 of in silico methods is discussed.
- We observed rising trends from big data to chemical space.
- An executive summary is structured according to the most cited articles.
- Milestones are assessed based on their respective timelines.
Executive Summary
| Definition | Drug discovery is a multidisciplinary science mainly devoted to bringing new chemical entities (NCEs) to the market for already-known medical applications. However, this process is very time-consuming (10-12 years) and highly cost-sensitive (in the range of US $ billions). As a result, scientists have developed computational strategies to maximize the results and yield better and larger product portfolios using less time and less money. These computational strategies are referred to in silico methods (as well as computer-assisted methods) in the literature.
Integrating AI, data science, and machine learning into drug discovery streamlines processes, accelerating timelines and optimizing resource allocation. These technologies analyze vast datasets, predict drug efficacy and safety, and enhance accuracy by integrating diverse information sources. This synergy has the potential to revolutionize pharmaceuticals, making drug discovery faster, cost-effective, and more successful in delivering therapeutics to patients. |
| Needs | The origin of in silico methods dates back to the late ’80s. At the time, medicinal chemists were required to produce evidence of structure-activity-relationship (SAR) from molecules within the same pharmacological class of therapeutical agents. This was, for all intents and purposes, essentially an academic exercise. In the ’90s, thanks to the advent of the graphical processor unit (GPU), researchers moved from 2D- to 3D- up to multidimensional SAR. This evolution allowed the boom of in silico methods mainly via two techniques: ligand-based fitting and target-based docking. The growing availability of commercial software as well as the open-approach to developing computer-assisted visualization allowed generating an enormous body of publications in this field from a few hundred per year in the late ’80s to 10,000 per year in the late ’90s. This exponential growth of data prompted the launch of PubMedTM and DrugBankTM in the early 2000s. Following this trend, the largest and most prominent collection of crystallographic structures from Cambridge University has evolved to host several million chemical items and their respective chemical-physical information. Involuntarily, drug discovery entered the big data era. |
| Opportunity | During the study of new chemical entities (NCEs), medicinal chemists have several possibilities at their disposal to bring NCEs to the market as quickly as possible. Sometimes, the success story arrives via serendipity, other times by trial-and- error and, most recently, during clinical investigations via drug repurposing. Drug repurposing is one of those strategies that has improved mostly in the past ten years. A good example of this is the number of new approved drugs (NADs) granted by the USFDA during the COVID-19 pandemic crisis: 90% of these NADs have emerged as a result of drug repurposing. |
| Gap | One of the main forces driving the advance in drug discovery was the search for high selectivity of biological targets, following the principle: one molecule for one specific binding site. However, the inevitable overlap of data stemming from computational visualization, clinical trials, and medical reports has confirmed that this principle of one molecule for one binding site is an excellent theory in a perfect world, but in reality, highly difficult to achieve. The most recent trend in drug discovery is the multi-target drug approach (MTD). In this type of approach, the in silico methods are suitable to discriminate via ab initio the relationship between molecular activity, specific end-user population, and drug-drug (and drug-receptor) interaction upstream of clinical investigations. Moreover, the evolution of databases via web-based server applications has created a brand-new field in the search for novel drugs, called chemical space. |
| Recommendation | For more than a century, the R&D departments of the major pharmaceutical companies were known to be a highly secretive environment with tens of thousands of chemists working on individual and personalized clean benches, and not communicating outside of their close circle of team members. Today’s setting is completely different. The loss of patent protections for the most important blockbusters, and the legal dispute surrounding patents relating to drug repurposing uses has reduced the necessity for this “special environment” in the pharmaceutical industry. Consequently, there has been a large layoff of highly skilled pharmaceutical professionals. Paradoxically, computer-assisted technologies have not produced the expected number of novel solutions, and the emergence of artificial intelligence (AI) now requires a new generation of skilled scientists in this new area going from data science to network pharmacology. Moreover, given the implicit difficulty in finding such multi-task medicinal chemists, the current R&D departments of the major pharmaceutical companies are increasingly becoming incubators/accelerators of start-ups and consortiums of public-private partnerships in which the various stakeholders are invited to interact and collaborate, especially in remote mode. In silico methods have not only brought out the digitalization of pharmaceutical science, but the entire manner in which scientists think about the definition of drug-biology interactions. The next step of in silico methods is the merging of chemical laboratory automation and synthetic tissues (organ-on-chip). At this juncture, even clinical trial investigations will become obsolete, and will run purely via computers: i. e. in silico medicine. |
Introduction
Drug discovery is a multidisciplinary science by which new chemical entities (NCE) are proposed for use as new therapeutical agents and to be administered during clinical trial phases until their final approval as new medications, also commonly known as active pharmaceutical ingredient (API) [1]. Drug design and drug discovery are long-established sciences, predating the emergence of informatics. These fields have developed autonomously and have been exhaustively analyzed and consolidated as comprehensively described in the historical overview by Pina in 2010 [2].
Historically, new drugs were discovered by identifying the active ingredients from traditional remedies such as plants or by serendipitous discovery, e.g. when the identification of the NCE as an API occurred outside of the scope of the main investigation as in the case of penicillin and ViagraTM [3].
Despite advances in technology and a better understanding of cellular pathophysiological signaling downstream, drug discovery remains both time-consuming (up to 10 years of the investigatory program) and capital-intensive (from $ US 800 million up to $ US 4 billion budget for just one new API). It is moreover a highly competitive and regulated sector. Therefore, from a purely financial perspective, drug discovery is an inefficient industrial process [4].
More recently, chemical libraries of small synthetic molecules, natural products, or botanical extracts are being screened via computer-aided systems (also known as virtual screening), allowing new opportunities for understanding receptor docking/fitting and the mechanism of action [5]. One of the results of this approach is to elucidate alternative pharmacological models for new medical indications, classified under the label of investigational new drug (IND). This type of drug discovery process, based on the finding of a new medical indication and subsequently applied to an already-existent API, is commonly called drug repositioning (or repurposing) [6].
Traditional methods of discovering drugs have been effective, although they can take over 12 years and cost approximately USD 1.8 billion from identifying a lead candidate to conducting clinical trials [7]. Moreover, the attrition rate of candidate drugs can reach as high as 96% [7]. In recent times, in silico approaches have gained increasing consideration due to their ability to hasten the drug discovery process in terms of time, labor, and expenses. Owing to novel computational methods, several new drug compounds have been successfully developed.
Lately, an increasing number of scientists are delving into computer-assisted drug discovery (CADD) methods to address the challenges of conventional experimental approaches, such as scale, time, and cost [8].
In silico methods refer to the current practice of drug discovery via the use of specific software, artificial intelligence (AI), together with. IT infrastructures such as high-speed computer systems, web-based servers/databases, and cloud. These methods thus differ from traditional drug discovery approaches performed through biological assays defined as in vitro methods, versus pre-clinical and clinical trials or in vivo methods.
In general, scientists are well-equipped to handle large sets of data comprising several thousand molecules and their properties. Specifically, chemists are trained to re-elaborate large data sets of compounds (and information related to these compounds) for their classification purposes before and after applying statistical regressions, as long as there is an evident rationale: namely, pattern recognition and molecular clustering [9,10,11]. Both of these techniques are crucial in terms of drug delivery, as well as aiding in the design [8], optimization, and understanding of delivery systems [12]. The ensemble of data spanning all possible compounds pertaining to chemistry and its applications is called chemical space [13].
Nevertheless, the ever-increasing amount of data being collected in public repositories/databases, having reached such proportions in terms of volume and velocity, has led in the last decade to the advent of a new science called big data [14,15]. The main difference between large datasets and big data is the total lack in the latter of any apparent homogeneity among the data per se [14,15,16]. In 2018, the number of single chemical entities available for in silico screening overpassed the symbolic threshold of 1 billion compounds. Furthermore, contract research organizations (CROs) offering drug discovery services are already announcing the target of 8 billion chemical entities for the year 2024 [17]. Certain academic teams are even proposing virtual libraries in the range of 11 billion chemical entities [18,19]. The elements of big data are, by definition, a combination of structured, semi-structured, and unstructured datasets. In this review, we discuss how big data is affecting the productivity of chemists working in drug discovery and development, as well as offer an explanatory overview of the rationale and impact of the various selected topics in this field [20].
AI and machine learning (ML), deep learning big data have now become integral components in advancing drug delivery systems [21,22,23]. These technologies offer innovative solutions for designing, optimizing, and personalizing drug delivery, leading to more efficient and effective therapeutic outcomes [14].
This large number of data, generated from genomics, proteomics and metabolomic sciences, combinatorial chemistry, and automated high-throughput screening (HTS), has led to a new trend in drug discovery for data integrations and data mining and its related disciplines [14]. The extremely vast use of public databases from all categories of end-users, ranging from professionals to students, has compelled hosting server providers to release several versions of software capable of data mining and graphical visualization simultaneously, to better serve their users for the many possible applications including virtual screening. The main areas of these applications are known as bioinformatics and cheminformatics [25,26].
In this review, we discuss how big data is affecting the productivity of chemists working in drug discovery and development, as well as offer an explanatory overview of the rationale and impact of the various selected topics in this field. Additionally, we provide an execution summary to facilitate understanding and discuss the large amount of data. To better understand the various trends and global technological evolution of this vast and complex field of study, a meta-analysis of the literature is first proposed, starting from the year 2000. This review contributes as a crucial tool in identifying and examining patterns, themes, and developments that have emerged over the past two decades by delving into the extensive body of literature considering mostly cited papers in the areas.
QSAR: Quantitative-Structure-Activity-Relationship
The main purpose of CADD until the early 2000s was to understand the QSAR of therapeutic agents used in therapy as a means to introduce novel generations of the most selective drugs into the therapeutical arsenal. The result of this gold rush” has translated into more than 30,000 APIs currently available in pharmacies.
CADD involves using computers to identify possible drug targets, screen large chemical libraries to find effective drug candidates, optimize candidate compounds, and assess their potential toxicity. Once these processes are digitally completed, the candidate compounds are then tested in vitro or in vivo to confirm their effectiveness. Consequently, CADD techniques can decrease the number of chemical compounds that need experimental evaluation while increasing the success rate by eliminating inefficient and toxic chemical compounds from consideration [27]. Pharmacophore modelling, QSARs, and AI have been the latest methods used in ligand-based virtual screening methodology [27].
Table 1 depicts the visual summary of the topics (disciplines and sub-disciples) discussed in this review. Each topic is listed as part of big data (pink color) or chemical space (grey color) according to the major relevance of the 300 most cited papers on in silico drug discovery chosen in this review. Key technology areas (blue) such as bioinformatics and cheminformatics are a common background for both big data and chemical space contents. The first result of the analysis of the evolution of big data assumed that one drug can be exclusively selective for one target. However, clinical evidence shows that this exclusivity and selectivity of one molecular target per drug is only theoretical.
Drug repositioning is a growing approach to drug discovery powered by the necessity to achieve greater “approved medical use” for the same API that is on the verge of losing its intellectual property protection status, or already out of the market because it is de facto considered an old drug. In the past, drug repositioning was driven by serendipity as well as by the off-label use of both marketed and generic drugs by physicians in their clinical practice. However, there are ongoing efforts to systematically conduct drug repurposing. The tetracyclines are a prime example of old drugs that are still of interest for new medical purposes. Indeed, for more than 60 years of their history, tetracyclines have been used as antibiotics while in the last 20 years, they have also been used in anticancer treatment as well as for neuroprotection [28,29].
Methodology of the Meta-Analysis
The carried out initial search spanned from January 2023 to May 2023 to identify the most qualified and cited papers from the early 2000s to 2022 related to technological evolution and trends from big data to chemical space for in silico methods in drug discovery. A total of 1475 articles were identified across the three databases, PubMed, Web of Science, and Embase. A comprehensive literature search was conducted to identify relevant articles pertaining to drug discovery, big data in chemical space, QSAR, drug repurposing, orphan drugs, multi-target drugs, and computer-aided drug design (CADD). The search spanned three major databases: PubMed, Web of Science, and Embase. Key terms, including "drug discovery," "big data to chemical space," "QSAR," "drug repurposing," "orphan drugs," "multi-target drugs," and "CADD," were used to retrieve potential articles. Subsequently, duplicates were removed to streamline the dataset, resulting in 900 unique articles. From the second pool of articles, the top 300 most cited papers from the early 2000s to 2022, providing an overview of the most influential works in the field. The second sub-collection aimed at a systematic review analysis and included articles deemed of significant interest for this study. The inclusion criteria for the meta-analysis were meticulously defined to ensure the relevance and reliability of the chosen articles in Figure 1. A total of 97 articles were selected for inclusion in the meta-analysis, adhering to the eligibility criteria outlined in the decision-making strategies presented in the accompanying table.
The article selection process involved multiple stages of review and was conducted independently by the authors involved. Rigorous decision-making strategies were employed to ensure the precision and accuracy of the final selection of articles for inclusion in the meta-analysis. This methodology employed a systematic and comprehensive approach to identify, select, and categorize articles for a meta-analysis, ensuring the robustness and reliability of the findings.
Results
General Consideration: “Quality” of the Data
The definitions of big data and chemical space are primarily based on the sole notion of “quantity” of the data, their storage, and their various fields of application. To date, there are no means to convey a measure of the “quality” of the data that are part of big data and chemical space.
Indeed, the interpretation of the validity (and reliability, accuracy, completeness, and consistency) of data is left to the researchers and the protocols established by their academic institutions [30,31].
While the goal of this review is not to delve into an extensive discussion regarding the “quality” of data, we nonetheless provide some details regarding the different sources of data and the overall relationship between the “quantity” of data and their impact on science and society [32], [33].
Given that the purpose of this review is to measure the “metric relevance” and the type of contribution of the different reported areas of interest, the absolute number of citations per article reported by Google Scholar is taken into consideration.
There is a large body of research as to whether or not Google Scholar can be qualified as a suitable source of scientific information that has been produced from 2017 to 2022 [31,34]. The result of the present search is in support of Google Scholar over other providers [34], particularly in a case such as the current meta-analysis aimed at measuring the impact on the web of each sub-field that comprises the very vast subject of in silico drug discovery.
The mean number of published scientific papers per year in the field of drug discovery has dramatically grown at a pace of tenfold over the decades, passing from the range of hundreds/year in the late ‘80s to a few thousand/year in ’90, and more than tens of thousands per year in the 2000s. AI, ML, and deep learning techniques greatly improve data quality assessment in big data and chemical space. They identify issues like inconsistencies and missing values, automate cleansing, and prioritize data for validation. Deep learning extracts complex features, enhancing accuracy. Leveraging these techniques ensures data reliability, improving scientific research impact in drug discovery. By leveraging these advanced techniques, researchers can ensure the reliability and integrity of data within big data and chemical space, ultimately enhancing the effectiveness and impact of scientific research in drug discovery.
For this review, the absolute number of citations per single article elaborated by Google Scholar is used to establish the impact of each selected article on the web. The dataset of the most cited papers reported herein has been limited to the first 300 papers in the field of drug discovery. The total amount of citations reported for these sole 300 papers corresponds to over 400,000 citations. The dataset chosen for this literature review is extremely significant in understanding the evolution and trends of in silico drug discovery over the last 22 years. It is reasonable to estimate that the chosen dataset is representative of more than 95% of all top-cited articles of the subject area.
Validation of the Proposed Meta-Analysis
The present meta-analysis involves the statistical synthesis of results from multiple studies to obtain a summary estimate of the effect size. In addition to the provided inclusion and exclusion criteria to maintain consistency and relevance in the studies selected, the latter are furthermore supported by other analyses such as VosViever as illustrated in Figure 2 which maps the analysis of the 100 most frequent keywords from over 1,000 articles published in peer-reviewed journals listed in Web of Science (2000-2022). This bibliometric analysis in the form of a network is very useful in gaining a better understanding of the interplay and degree of congruence among the various subject areas.
The distribution of the keywords generated 5 different clusters of articles, depicted in red, green, yellow, blue and (purple, mauve).
The keyword “prediction” emerged as the central point of this bibliometric map. Accordingly, the main purpose of in silico drug discovery is to predict a novel drug in terms of chemical structure and/or in terms of the mechanism of action.
The bibliometric analysis in Figure 3. in the form of a network is highly useful in illustrating the interplay and degree of connection among the different subject areas. This visualization proposes how the information and interest of the articles based on “virtual screening” have shifted from metabolic analysis and pharmacokinetics (2010) to molecular mechanisms, web servers and COVID-19, starting in 2016.
Network Map 2: Focusing on “virtual screening” as one of the main topics in drug discovery.
Figure 3.
Bibliometric analysis illustrating the interplay and degree of congruence of the articles based on “virtual screening” that have migrated from metabolic analysis and pharmacokinetics (2010) to molecular mechanisms, web servers, and COVID-19, starting in2016.
Figure 3.
Bibliometric analysis illustrating the interplay and degree of congruence of the articles based on “virtual screening” that have migrated from metabolic analysis and pharmacokinetics (2010) to molecular mechanisms, web servers, and COVID-19, starting in2016.
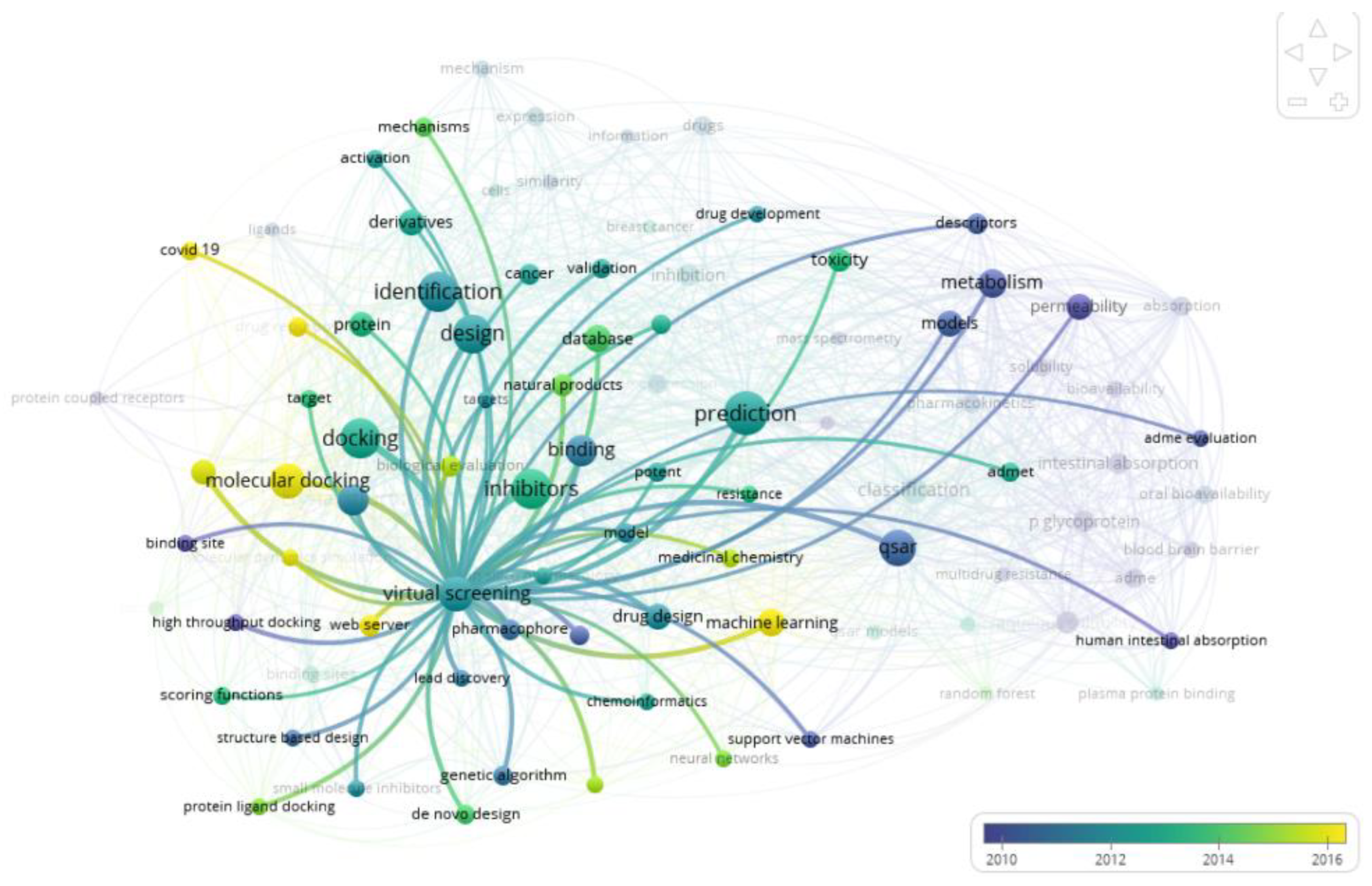
The use of a heatmap is one of the first steps of the rationalization process (cause vs. effect) in which researchers are looking for similarities between very close fields of investigation within the same subject domain, or area.
The use of a color-shade gradient provides a visual interpretation of the degree of trends.
Primary Outcomes of this Meta-Analysis
The primary objective of this meta-analysis was to determine the qualitative and quantitative ranking/classification among the different disciplines of in silico drug discovery: for example, research centers and authors that are involved in the development of software for predictive measure of drugs involved in the development of predictive analytics software for drug monitoring/drug discovery are not necessarily involved in the elaboration of databases or chemical space concepts.
The second objective of this meta-analysis was to understand the relationship and the impact of data science methods in ‘in silico’ drug discovery, if any.
As shown in Figure 4 and Figure 5 below, the findings of the present meta-analysis provide a measure of the impact of the different fields and approach methods within the same subject domain. The most important discriminant is the total number of citations and the ponderal numbers per year, also commonly known as weighted value per year.
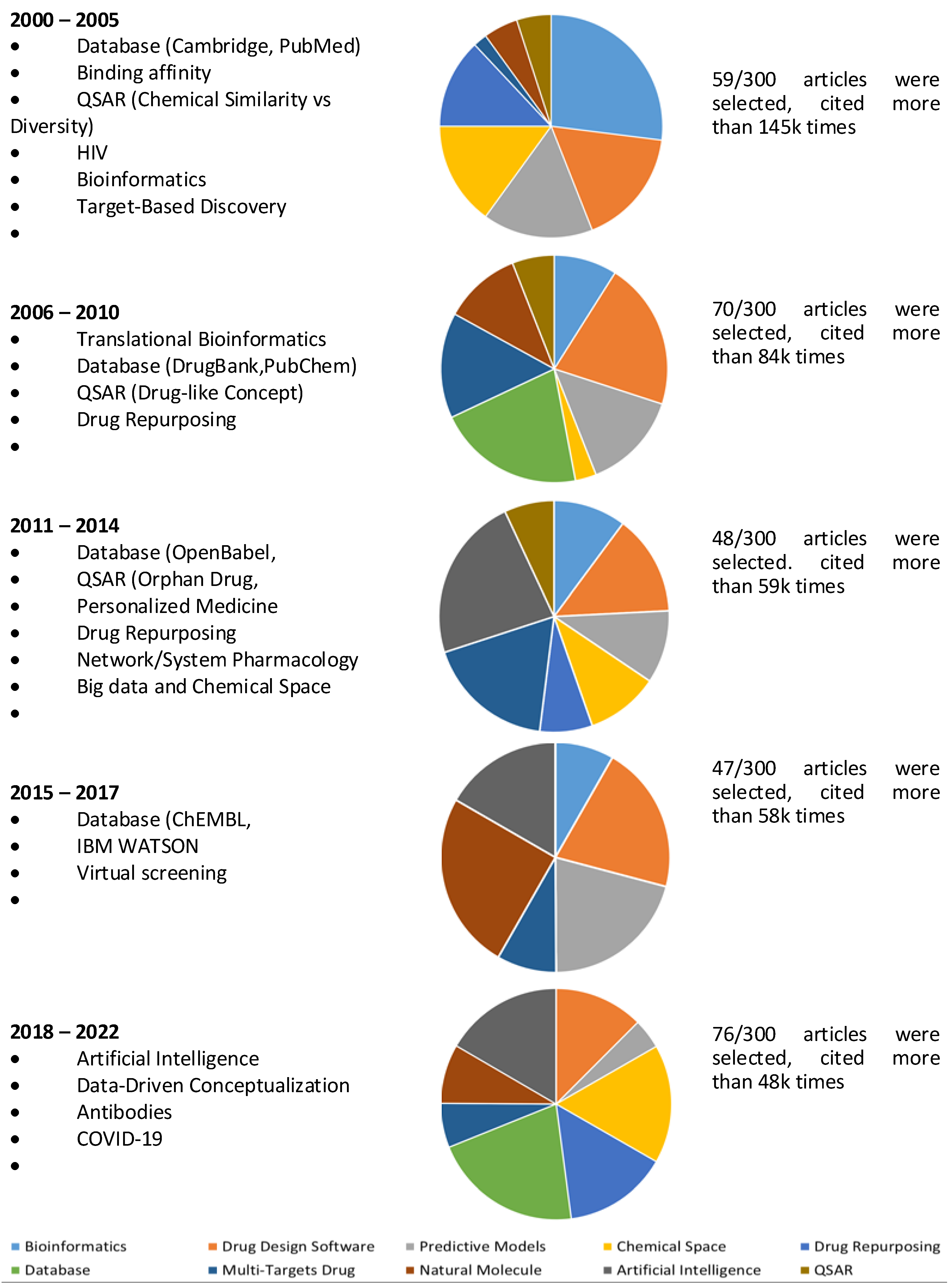
Although the total number of publications reported in PubMed has grown exponentially over the last two decades, with very minimal yearly reduction in pace, analysis of the data depicted in Figure 3 and Figure 4B highlight five very distinct periods unequivocally related to the different trends and having specific impacts on scientific publications. This portrayal of the different trends is a result of the different keywords and topics of the 300 most cited publications as reported from Google Scholar, namely:
Period#1: 2000 – 2005, with a maximum in 2004: 59/300 articles for more than 145 K citations
Period#2: 2006 – 2010, with a maximum in 2008: 64/300 articles for more than 130 K citations
Period#3: 2011 – 2014, with a maximum in 2013: 50/300 articles for more than 100 K citations
Period#4: 2015 – 2017, with a maximum in 2016: 46/300 articles for more than 95 K citations
Period#5: 2018 – 2022, with a maximum in 2021: 79/300 articles for more than 160 K citations
Other extrapolated data from the several technological areas cited so far in this review:
Artificial Intelligence: 8 articles – ca 6,000 citations
Bioinformatics: 33 articles – ca 110,000 citations
Chemiometrics: 28 articles – ca 22,000 citations
Natural Molecule: 11 articles – ca 22,000 citations
Predictive Models : 84 articles – ca 85,000 citations
Drug Design Software: 15 articles – ca 16,000 citations
Chemical Space : 14 articles – ca 14,000 citations
Database: 35 articles – 100,000 citations
Drug Repurposing: 16 articles – ca 33,000 citations
Muti-Target Drugs: 16 articles – ca 8,000 citations
QSAR: 40 articles – ca 35,000 citations
Figure 6.
Variation in topics used in the field of in silico drug discovery methods over time. The relative impacts of each time period over the entirely different subject areas were taken into consideration.
Figure 6.
Variation in topics used in the field of in silico drug discovery methods over time. The relative impacts of each time period over the entirely different subject areas were taken into consideration.
The average number of drugs approved (NDA) by the USFDA per year since 1980 was 43 (series 1) while the average number of new chemical entities (NCE) per year was 14 (series 2) over the same time period (Figure 7, panel A). The difference in terminology NDA vs. NCE lies in the fact that an NDA can be a second-generation drug or any other modification (and/or bioisotere) of a previously approved NCE structure, although NCE is referred to as a chemical moiety of the NDA. The USFDA assesses candidate drugs under final review with two different designations from a panel of experts. Of note, until 2012, the near totality of the NCEs was designated under the category of “Small Molecules”, while after 2012 antibodies, large peptides, and proteins were introduced as NCEs. However, as can be seen in Figure 5B, the very large availability of data did not necessarily lead to an increase in the number of approved drugs nor the variety of chemical structures of the novel approved therapeutics.
Figure 7.
Visualization of (A) the average number of drugs approved (NDA) by the USFDA per year, and (B) big data.
Figure 7.
Visualization of (A) the average number of drugs approved (NDA) by the USFDA per year, and (B) big data.
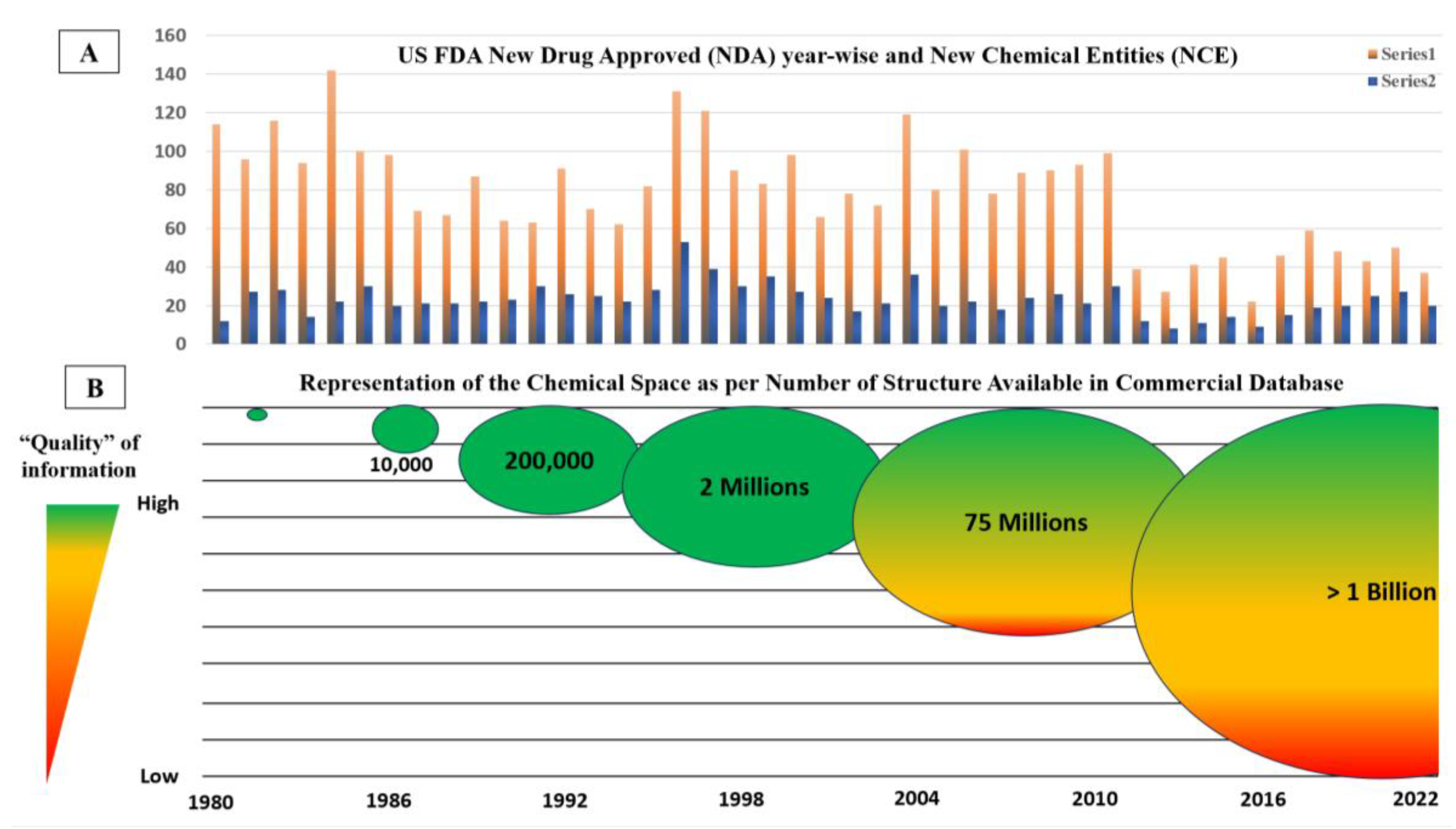

Table 2.
List of the most cited papers encompassing all topics.
| Number | Topic | Title | First author | Year | Citations | Ref |
| 1 | Bioinformatics | Initial sequencing and analysis of the human genome | HG Consortium | 2001 | 28696 | [35] |
| 2 | Database | The Cambridge Structural Database: a quarter of a million crystal structures and rising | FH Allen | 2002 | 14390 | [36] |
| 3 | Bioinformatics | Bioconductor: open software development for computational biology and bioinformatics | RC Gentleman | 2004 | 13462 | [37] |
| 4 | Database | The human genome browser at UCSC | WJ Kent | 2002 | 10583 | [38] |
| 5 | Database | The Cambridge structural database | CR Groom | 2016 | 7366 | [39] |
| 6 | Database | OpenBabel: An open chemical toolbox | NM O Boyle | 2011 | 6735 | [40] |
| 7 | Bioinformatics | A review of feature selection techniques in bioinformatics | Y Saeys | 2007 | 5556 | [41] |
| 8 | Software | CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields | K Vanommeslaeghr | 2010 | 5130 | [42] |
| 9 | Bioinformatics | The Perseus computational platform for comprehensive analysis of omics data | S Tyanova | 2016 | 5068 | [43] |
| 10 | Database | DrugBank 5.0: a major update to the DrugBank database for 2018 | DS Wishart | 2018 | 4951 | [44] |
| 11 | Bioinformatics | Accurate whole human genome sequencing using reversible terminator chemistry | DR Bentley | 2001 | 4691 | [45] |
| 12 | Database | Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening | TA Halgren | 2004 | 4234 | [46] |
| 13 | Model Predict | Lead- and drug-like compounds: the rule-of-five revolution | CA Lipinski | 2004 | 4213 | [47] |
| 14 | Model Predict | Drug-like properties and the causes of poor solubility and poor permeability | CA Lipinski | 2000 | 3912 | [48] |
| 15 | Bioinformatics | Biopython: freely available Python tools for computational molecular biology and bioinformatics | PJA Cock | 2009 | 3885 | [49] |
| 16 | Database | PubChem substance and compound databases | S Kim | 2016 | 3853 | [50] |
| 17 | Bioinformatics | The druggable genome | AL Hopkins | 2002 | 3851 | [51] |
| 18 | Model Predict | Drug discovery: a historical perspective | J Drews | 2000 | 3582 | [52] |
| 19 | Database | New software for searching the Cambridge Structural Database and visualizing crystal structures | IJ Bruno | 2002 | 3581 | [53] |
| 20 | Database | DrugBank: resource in silico drug discovery and exploration | DS Wishart | 2006 | 3544 | [54] |
| 21 | Bioinformatics | From genomics to chemical genomics: new developments in KEGG | M Kaneshisa | 2006 | 3473 | [55] |
| 22 | Chemical Space | Network pharmacology: the next paradigm in drug discovery | AL Hopkins | 2008 | 3438 | [56] |
| 23 | Database | ChEMBL: a large-scale bioactivity database for drug discovery | A Gaulton | 2012 | 3390 | [57] |
| 24 | Database | HMDB 3.0--The Human Metabolome Database in 2013 | DS Wishart | 2012 | 3061 | [58] |
| 25 | QSAR | Random forest: a classification and regression tool for compound classification and QSAR modelingD | V Svetnik | 2003 | 3052 | [59] |
| 26 | Database | HMDB 4.0: the human metabolome database for 2018 | DS Wishart | 2018 | 2928 | [60] |
| 27 | Drug Repositioning | Drug repositioning: identifying and developing new uses for existing drugs | TT Ashburn | 2004 | 2914 | [61] |
| 28 | Database | DrugBank: a knowledgebase of drugs, drug actions, and drug targets | DS Wishart | 2008 | 2785 | [62] |
Figure 8.
Tree map of the most cited papers reported by topics.
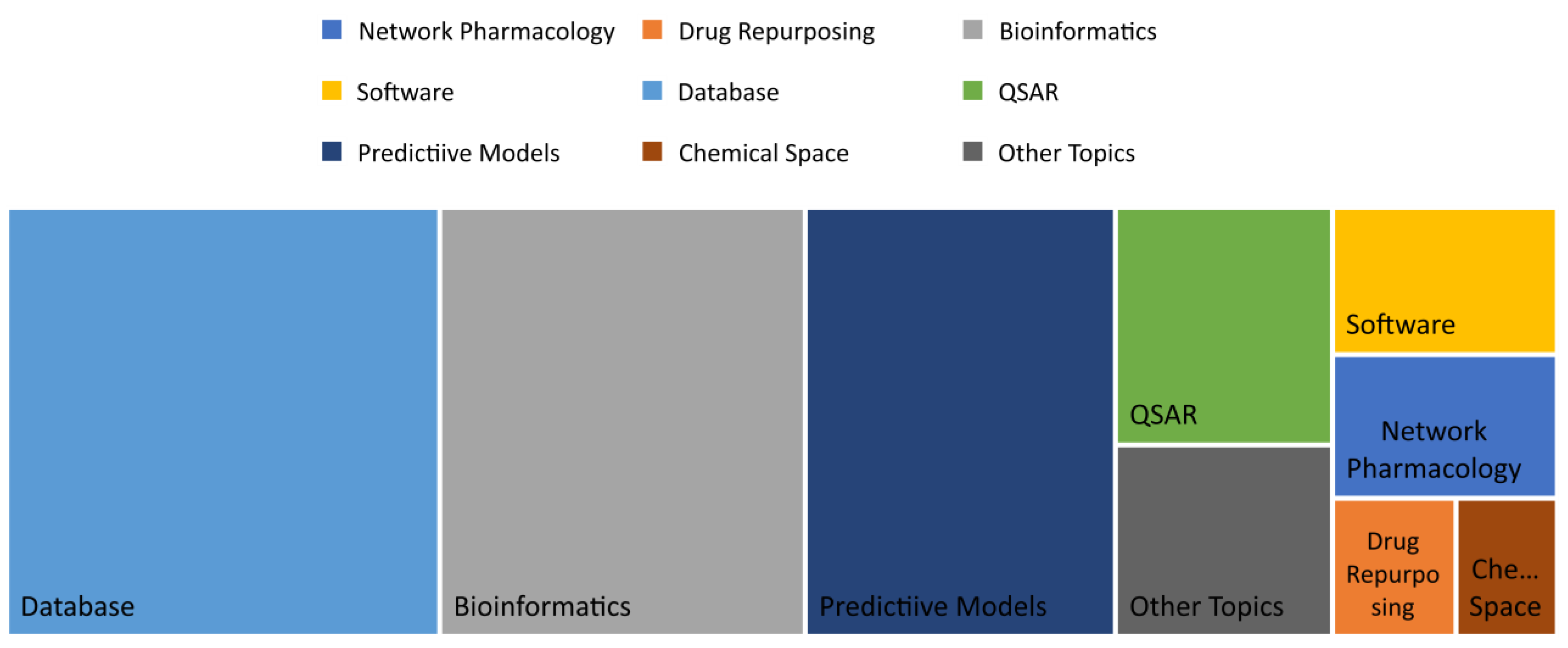
Table 3.
List of the most cited papers in Database and Web-based Library.
| Number | Title | First Author | Year | Citation | Reference |
| 1 | The Cambridge Database: a quarter of a million structure and rising | FH Allen | 2002 | 14390 | [36] |
| 2 | The Human Genome Browser at UCSC | WJ Kent | 2002 | 10583 | [38] |
| 3 | New software for searching the Cambridge Structural Database and visualizing crystal structures | IJ Bruno | 2002 | 3581 | [53] |
| 4 | Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening | TA Halgren | 2004 | 4234 | [46] |
| 5 | DrugBank: a comprehensive resource for in silico drug discovery and exploration | DS Wishart | 2006 | 3544 | [54] |
| 6 | BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities | T Liu | 2007 | 1663 | [63] |
| 7 | ChEBI: a database and ontology for chemical entities of biological interest | K Deglyarenko | 2007 | 1155 | [64] |
| 8 | PubChem: Integrated platform of small molecules and biological actives | EE Bolton | 2008 | 1537 | [65] |
| 9 | DrugBank: a knowledgebase for drugs, drug actions, and drug targets | DS Wishart | 2008 | 2785 | [62] |
| 10 | PubChem: a public information system for analyzing bioactivities of small molecules | Y Wang | 2009 | 1317 | [66] |
| 11 | DrugBank 3.0: a comprehensive resource for ‘OMICS’ research on drugs | C Knox | 2010 | 2034 | [67] |
| 12 | Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database | PCD Hawkins | 2010 | 1367 | [68] |
| 13 | Open Babel: An open chemical toolbox | NM O Boyle | 2011 | 6735 | [40] |
| 14 | ChEMBL: a large-scale bioactivity database for drug discovery | A Gaulton | 2012 | 3390 | [57] |
| 15 | HMDB 3.0—The Human Metabolome Database in 2013 | DS Wishart | 2012 | 3061 | [58] |
| 16 | ZINC: a free tool to discover chemistry for biology | JJ Irwin | 2012 | 2481 | [69] |
| 17 | ChEMBL: a large-scale bioactivity database for drug discovery | A Gaulton | 2012 | 1782 | [57] |
| 18 | DrugBank 4.0: Shedding new light on drug metabolism | V Law | 2014 | 2103 | [70] |
| 19 | The Cambridge structural database | CR Groom | 2016 | 7366 | [39] |
| 20 | PubChem substance and compound databases | S Kim | 2016 | 3853 | [50] |
| 21 | The ChEMBL database in 2017 | A Gaulton | 2017 | 1731 | [71] |
| 22 | DrugBank 5.0: a major update to the DrugBank database for 2018 | DS Wishart | 2018 | 4951 | [44] |
| 23 | HMDB 4.0: the human metabolome database for 2018 | DS Wishart | 2018 | 2928 | [60] |
| 24 | PubChem 2019 update improved access to chemical data | S Kim | 2019 | 2380 | [72] |
| 25 | PubChem in 2021: new data content and improved web interfaces | S Kim | 2021 | 1657 | [73] |
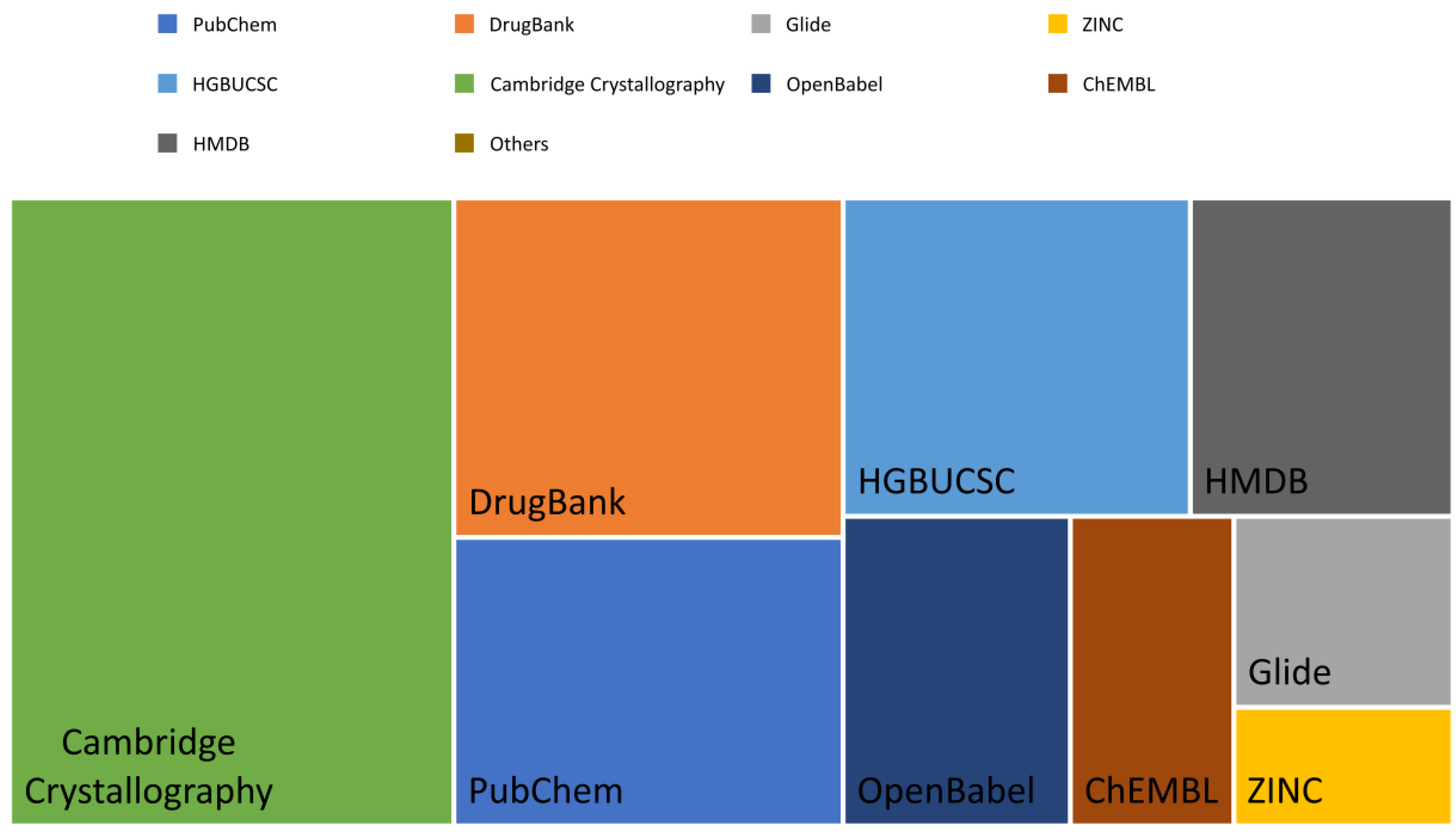
Figure 9.
Tree map of the sub-categories of the main topic: DATABASE/Web-based Library. The list of the most cited papers encompassing all topics for Cambridge Crystallography, DrugBank, PubChem, HGBUCSC, HMDB, OpenBabel, ChEMBL, Glide, and ZINC are shown.
Figure 9.
Tree map of the sub-categories of the main topic: DATABASE/Web-based Library. The list of the most cited papers encompassing all topics for Cambridge Crystallography, DrugBank, PubChem, HGBUCSC, HMDB, OpenBabel, ChEMBL, Glide, and ZINC are shown.
Discussion, Limitations/Uncertainty
The availability of the many compounds documented to date (accounting for active fragments of small and large molecules) has allowed exposure to a myriad of hypotheticals. The most significant result of the “OMICS age” is the growing amount of information introduced by genomics and proteomics in real-time. This wealth of data has provided innumerable targets that could theoretically combine with as many hits for an equal number of active pharmaceutical ingredients [74].
In 2002, Oprea proposed a dataset of one million compounds tested via HTS. However, it would appear that the probability of a compound reaching market status is one in a million [169] .
A mere 10 years later, PubChem had attained 89 million chemical entities in its database such that 89 new drugs could theoretically be obtained as a maximum for this dataset, or one could search for different targets in the same dataset and obtain 89 new drugs every time [75,76].
Millions of scientists from very different domains are familiar with the theory known as the “Lock-Key Model”, the father of which is the 1908 Nobel Prize for Medicine laureate, Sir Paul Ehrlich. The concept that one key can open a lock is at the forefront of molecular pharmacology and, in the last ten years, has evolved into a much more complex paradigm in which a specific key (drug) can unlock a series of locks not necessarily interrelated, exactly as in the case of a master key.
Sir Paul Ehrlich was aware that there is no dogma in science; in fact, he was the first to introduce the concept of Zauberkugel ("magic bullet"). The technology that has been available in the last ten years has shown the full applicability of this paradigm, more than a hundred years after its enunciation, under the terminology of multi-target drugs.
Period #1 – “Testing the reliability of database vs. the selectivity concepts.”
Until the development of web-based library resources, the only force leading the global field of in silico drug discovery was the analysis, via homemade software, of Quantitative-Structure-Activity-Relationship (QSAR) studies, albeit via a limited private library of just a few thousand structures. The principle behind the QSAR analysis was based on the chemical similarity of proven therapeutic agents for the same diseases.
The availability of the first large web-based database, such as PubMed, compelled researchers to introduce new methods in drug discovery, namely data mining and data integration.
In 2004, Pearsons and others [77] designed a good example of data integration among various libraries, from genes vs. a series of bioactive compounds, to link these results to very specific cellular targets or at least to understand whether any correlated pathways existed.
In 2005, Csermely and coll., [78] had already noted that despite the availability of new digital tools in drug discovery, the additional amount of data was not contributing to generating more therapeutical options. On the other hand, they began confirming the new paradigm of multi-target drugs which was the real novelty of this new era of computer-assisted drug design. As stated by the authors:
“At present, the goal is to:
- (i)
- find a target of suitable function.
- (ii)
- identify the ‘best binder’ by high-throughput screening of large combinatorial libraries and/or by rational drug design based on the three-dimensional structure of the target.
- (iii)
- provide a set of proof-of-principle experiments.
- (iv)
- develop a technology platform that predicts potential clinical applications.
However, despite all the careful studies and the considerable drug-development efforts undertaken, the number of successful drugs and novel targets did not increase appreciably during the past decade.”
Period #2 – “Looking for applications of novel drugs inside an old pharmacological space.”
In 2006, a multi-disciplinary team from Pfizer, [79] published a first-ever map of the known pharmacological space at the time. The purpose of this colossal work was to plot the direction or best fitting of more than 275,000 biologically active compounds vs. a library of 1,000 molecular targets. The result of this experience was the introduction of the concepts of chemical promiscuity of therapeutic agents for the same disease and degrees of draggability of cellular targets.
Another interesting novelty of this type of approach is the maturity of bioinformatics and translational bioinformatics.
On another hand, the ensemble of this new evidence of non-pre-determinability of the biological effect, or the obvious lack of exclusive selectivity for the “target”, has induced the purists of the quantitative structure activity-relationship (QSAR) realm to introduce additional dimensions of interactions. The QSAR hence shifted from a mere proprieties of the molecular scaffold (2D) up to induced conformation of the target via environmental forces (3D up to 6D), Lill 2007 [80] and Furches 2010 [81].
Period #3 – “Data-driven drug discovery.”
The most successful result of the early 2010s in the field of drug discovery has been in admitting that one approved drug can have multiple medical indications. This new paradigm based on the concept of drug repurposing produced a record number of 37 designated and marketed orphan drugs in only two years, from 2009-2010, Muthyala [82] .
The easy access to large libraries and real-time collaborative platforms with the major pharmaceutical companies, together with the possibility of exchanging clinical information for new medical indications without incurring legal issues, has fueled the exponential growth of publications and thematic journals. The results of this enormous amount of data unintentionally catapulted drug discovery into the big data era (Lusher 2014 [83]).
Paradoxically, these same digital tools, which were introduced to facilitate in silico drug discovery, also became somewhat of an obstacle requiring the introduction of a new class of researcher to be added to the medicinal chemists’ team, namely the data scientist dedicated to managing and streamlining big data [84].
Period #4 – “Druggability: one drug and many targets”
The growing consensus for muti-target drugs and the search for specific genetic targets dramatically changed the perspective and the scope of the medicinal chemist [85]. The total lack of target selectivity is not due to the chemical moiety per se but rather to the dominant concept of network pharmacology and phenotypic selection [86]. While medicinal chemists usually rely on some version of the “lock and key” paradigm to design novel therapeutics, modern pharmacology recognizes that the mid- and long-term effects of a given drug on a biological system may depend not only on specific ligand-target recognition events but also on the influence of the repeated administration of a drug on cell-gene signature (Talevi 2015 [87], Lavecchia, 2016 [88]).
Period #5 – “Re-discovering the Use of Natural Molecules for Complex Diseases.”
Since 2018, drug repositioning methods have been leading the success of the entire sector of in silico drug discovery, including the search for remedies for COVID-19 treatment. Indeed, COVID-19 is unequivocally a complex disease, consisting not only of a simple viral infection but a complicated framework of symptoms and unpredictable transmissibility (as a result of the variants) with varying outcomes [89] according to the different patient populations (de Oliviera et al. 2018 [90], Ramsay et al. 2018 [91]).
Other fields of interest in this period include neurology [92], with Alzheimer’s disease leading the topics for drug repurposing (Mesiti 2019 et al. (2019) [93]), as well as two other areas related to namely seizures (Geets et al. (2019) [94]) and depression Buenz et al. (2018) [92].
In totally different pathways, cannabinoids (and Cannabis extracts) have resulted in a very interesting natural remedy owing to the success of EpidolexTM in the treatment of seizures in children Namdar et al. (2020) [95] [96].
Finally, following the re-discovering of natural therapy, the latest success in time is the drug repurposing of classical abuse/recreational psychedelics such as psilocybin (and dried “magic mushrooms” powder) for depression and PTDS (Mullowney et al. (2023) [97]).
Conclusion
The significant role of in silico tools has increased exponentially in the last ten years and has markedly revolutionized the field of CADD and brought about technological evolution and recent trends that have galvanized the search for new drugs. All in all, the search approach employed in this review initially yielded a comprehensive set of 900 non-duplicated papers. In Part 1, our focus shifts to the examination of the top 300 papers, identifying the most cited and high-quality 97 articles pertinent to the realm of in silico drug discovery.
A final consideration should also be given as to how these technological trends are affecting the socio-economic aspects of pharmaceutical companies, including:
-
Big Data: Data Science, Data Integration and Data MiningThe result of these disciplines is the continuing improvement in IT infrastructure and software. Incorporating advanced AI and ML techniques can enhance in silico drug discovery by rapidly analyzing vast amounts of data, predicting molecular interactions, and optimizing drug candidates with higher precision and efficiency.
-
Cheminformatics: classification, pattern recognition and clusteringThe result of these disciplines is the improvement in current knowledge regarding the mechanisms of actions of drugs owing to a better understanding of their QSAR.
-
Bioinformatics and Translational BioinformaticsThe result of these disciplines is the seeking of new molecular targets and alternative physio/pathological mechanisms downstream. In particular, these disciplines are pivotal for the understanding of epigenetics and meta-genomics phenomena.
-
Drug RepurposingThe primary scope of this discipline is the life extension of expired patent applications. In practice, the controversy related to patent issues and the ensuing transfer of Drug Master Files (DMF) have accelerated the need for collaborative models among pharmaceutical stakeholders. The result is the shrinking of the required internal R&D workforce.
-
Chemical SpaceThe chemical space is the ensemble of all possible chemical structures, which is believed to contain up to several billion molecules of potential interest for drug discovery as mentioned in this review. One proposed means to explore chemical space is based on the selection of virtual libraries of common scaffold-tree algorithms (grouping) that are overlapped to other “organized maps” of chemical-physical information (pharmacokinetics) and/or chemical interactions (pharmacodynamics). The result of this “spatial analysis” has been used in the last 20 years to generate a new discipline known as Network Medicine or Network Pharmacology. Conversely to drug repurposing strategies in which one molecule (“old API”) is investigated for a new medical indication following the principle of one drug for one targe the results in Network Pharmacology approaches to drug discovery are completely different. Indeed, in terms of the definition of chemical space, there are billions of potential drugs that could virtually match billions of targets. The best matching combinations are hence known as multi-target drugs (MTD), including in Food, Aroma, and other fields.
Author Contributions
A systematic search of the literature was conducted by the first team of the authors (Arife Uzundurukan, Domenico Fuoco) of this review, from January 2023 to May 2023, to identify the most cited articles published from the early 2000s until 2022. The sources of information were combined with Google Scholar and PubMed. This first team of authors came out with the full conceptaualization of the whole paper as well as with data curation; formal analysis; investigation; methodology; visualization; writing – original draft; writing – review and editing, whole project administration. This initial search was validated by a second team of authors (Mark Nelson, Christopher Teske,) of this review using Web of Science and VosViewer. This team has been involved also with the formal analysis; supervision; validation; writing – original draft; writing – review and editing. A third team of authors (Elzagheid Mohamed, John Victor Christy, Mohamed Shahidul Islam) of this review conducted an independent search looking for missing information from the previous two runs. A forth team of authors as assisted with the meta-analysis its rational (Holli-Joi Martin, Eugene Muratov, Samantha Glover). Moreover, this team will play a major role in the upcoing PART II. Meanwhile, this forth team has been involved with theory and rational; methodology; review and editing.
Acknowledgments
The authors are obliged to thank both Prof. Daria Boffito and Prof. Gregory S. Patience for their great support and continuous coaching in leading the year-long journey behind this systematic review of the literature.
References
- Stanley, H. Nusim, Active Pharmaceutical Ingredients, vol. 205. 2005.
- A. S. Pina, A. Hussain, and A. C. A. Roque, “An Historical Overview of Drug Discovery,” in Ligand-Macromolecular Interactions in Drug Discovery: Methods and Protocols, A. C. A. Roque, Ed., Totowa, NJ: Humana Press, 2010, pp. 3–12. [CrossRef]
- Ban, T.A. The role of serendipity in drug discovery. Dialog- Clin. Neurosci. 2006, 8, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Grootendorst, P.; Lexchin, J.; Cunningham, C.; Greyson, D. The cost of drug development: A systematic review. Health Policy 2011, 100, 4–17. [Google Scholar] [CrossRef]
- Xu, C.; Ke, Z.; Liu, C.; Wang, Z.; Liu, D.; Zhang, L.; Wang, J.; He, W.; Xu, Z.; Li, Y.; et al. Systemic In Silico Screening in Drug Discovery for Coronavirus Disease (COVID-19) with an Online Interactive Web Server. J. Chem. Inf. Model. 2020, 60, 5735–5745. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- How to improve R&D productivity: the pharmaceutical industry’s grand challenge | Nature Reviews Drug Discovery.” Accessed: Nov. 28, 2023. [Online]. Available: https://www.nature.com/articles/nrd3078.
- “CADD and Informatics in Drug Discovery | SpringerLink.” Accessed: Nov. 28, 2023. [Online]. Available: https://link.springer.com/book/10.1007/978-981-99-1316-9.
- Nag, S.; Baidya, A.T.K.; Mandal, A.; Mathew, A.T.; Das, B.; Devi, B.; Kumar, R. Deep learning tools for advancing drug discovery and development. 3 Biotech 2022, 12, 1–21. [Google Scholar] [CrossRef]
- Wu, C.; Gudivada, R.C.; Aronow, B.J.; Jegga, A.G. Computational drug repositioning through heterogeneous network clustering. BMC Syst. Biol. 2013, 7, S6–S6. [Google Scholar] [CrossRef]
- Chen, Y.; Kirchmair, J. Cheminformatics in Natural Product-Based Drug Discovery. Mol. Informatics 2020, 39. [Google Scholar] [CrossRef]
- Chen, W.; Liu, X.; Zhang, S.; Chen, S. Artificial intelligence for drug discovery: Resources, methods, and applications. Mol. Ther. - Nucleic Acids 2023, 31, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A. The 'Big Bang' of the chemical universe. Nat. Chem. Biol. 2023, 19, 667–668. [Google Scholar] [CrossRef]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.M.S.-F.; Contrepois, K.; Moneghetti, K.J.; Zhou, W.; Mishra, T.; Mataraso, S.; Dagan-Rosenfeld, O.; Ganz, A.B.; Dunn, J.; Hornburg, D.; et al. A longitudinal big data approach for precision health. Nat. Med. 2019, 25, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Butte, A. Leveraging big data to transform target selection and drug discovery. Clin. Pharmacol. Ther. 2015, 99, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Lerner, R.A. DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information. Annu. Rev. Biochem. 2018, 87, 479–502. [Google Scholar] [CrossRef] [PubMed]
- Sadybekov, A.A.; Sadybekov, A.; Liu, Y.; Iliopoulos-Tsoutsouvas, C.; Huang, X.-P.; Pickett, J.; Houser, B.; Patel, N.; Tran, N.K.; Tong, F.; et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 2021, 601, 452. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Irwin, J.J.; Shoichet, B.K. Modeling the expansion of virtual screening libraries. Nat. Chem. Biol. 2023, 19, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Xie, L. Providing data science support for systems pharmacology and its implications to drug discovery. Expert Opin. Drug Discov. 2016, 11, 241–256. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Thakkar, A.; Mercado, R.; Engkvist, O. Molecular representations in AI-driven drug discovery: a review and practical guide. J. Chemin- 2020, 12, 1–22. [Google Scholar] [CrossRef]
- Vatansever, S.; Schlessinger, A.; Wacker, D.; Kaniskan, H. .; Jin, J.; Zhou, M.; Zhang, B. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: State-of-the-arts and future directions. Med. Res. Rev. 2020, 41, 1427–1473. [Google Scholar] [CrossRef]
- Drug discovery with explainable artificial intelligence | Nature Machine Intelligence.” Accessed: Nov. 28, 2023. [Online]. Available: https://www.nature.com/articles/s42256-020-00236-4.
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef]
- Wooller, S.K.; Benstead-Hume, G.; Chen, X.; Ali, Y.; Pearl, F.M. Bioinformatics in translational drug discovery. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Carracedo-Reboredo, P.; Linares-Blanco, J.; Rodriguez-Fernandez, N.; Cedron, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A review on machine learning approaches and trends in drug discovery. Comput. Struct. Biotechnol. J. 2021, 19, 4538–4558. [Google Scholar] [CrossRef]
- Segall, M.D.; Barber, C. Addressing toxicity risk when designing and selecting compounds in early drug discovery. Drug Discov. Today 2014, 19, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Fuoco, D. Classification Framework and Chemical Biology of Tetracycline-Structure-Based Drugs. Antibiotics 2012, 1, 1–13. [Google Scholar] [CrossRef]
- Subramanian, G.; Mjalli, A.M.M.; Kutz, M.E. Integrated Approaches to Perform In Silico Drug Discovery. Curr. Cancer Drug Targets 2006, 3, 189–197. [Google Scholar] [CrossRef]
- Halevi, G.; Moed, H.; Bar-Ilan, J. Suitability of Google Scholar as a source of scientific information and as a source of data for scientific evaluation—Review of the Literature. J. Inf. 2017, 11, 823–834. [Google Scholar] [CrossRef]
- Radicchi, F.; Weissman, A.; Bollen, J. Quantifying perceived impact of scientific publications. J. Inf. 2017, 11, 704–712. [Google Scholar] [CrossRef]
- Sauvayre, R. Types of Errors Hiding in Google Scholar Data. J. Med Internet Res. 2022, 24, e28354. [Google Scholar] [CrossRef] [PubMed]
- Mayo-Wilson, E.; Li, T.; Fusco, N.; Dickersin, K. ; for the MUDS investigators Practical guidance for using multiple data sources in systematic reviews and meta-analyses (with examples from the MUDS study). Res. Synth. Methods 2017, 9, 2–12. [Google Scholar] [CrossRef]
- Gusenbauer, M.; Haddaway, N.R. Which academic search systems are suitable for systematic reviews or meta-analyses? Evaluating retrieval qualities of Google Scholar, PubMed, and 26 other resources. Res. Synth. Methods 2019, 11, 181–217. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- The druggable genome | Nature Reviews Drug Discovery.” Accessed: Dec. 09, 2023. [Online]. Available: https://www.nature.com/articles/nrd892.
- Drews, J. Drug Discovery: A Historical Perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 389–397. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Kanehisa, M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random Forest: A Classification and Regression Tool for Compound Classification and QSAR Modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Drug repositioning: identifying and developing new uses for existing drugs | Nature Reviews Drug Discovery.” Accessed: Dec. 09, 2023. [Online]. Available: https://www.nature.com/articles/nrd1468.
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Degtyarenko, K.; de Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcantara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2007, 36, D344–D350. [Google Scholar] [CrossRef] [PubMed]
- E. E. Bolton, Y. Wang, P. A. Thiessen, and S. H. Bryant, “Chapter 12 - PubChem: Integrated Platform of Small Molecules and Biological Activities,” in Annual Reports in Computational Chemistry, vol. 4, R. A. Wheeler and D. C. Spellmeyer, Eds., Elsevier, 2008, pp. 217–241. [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37 (Suppl. 2), W623–W633. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: a comprehensive resource for 'Omics' research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2013, 42, D1091–D1097. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.; Hanison, J.; Nirmalan, N. “Omics”-Informed Drug and Biomarker Discovery: Opportunities, Challenges and Future Perspectives. Proteomes 2016, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Kim, S. Getting the most out of PubChem for virtual screening. Expert Opin. Drug Discov. 2016, 11, 843–855. [Google Scholar] [CrossRef] [PubMed]
- D. Fuoco, “Hypothesis for changing models: current pharmaceutical paradigms, trends and approaches in drug discovery,” PeerJ, vol. 3, Feb. 2015. [CrossRef]
- Parsons, A.B.; Brost, R.L.; Ding, H.; Li, Z.; Zhang, C.; Sheikh, B.; Brown, G.W.; Kane, P.M.; Hughes, T.R.; Boone, C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 2003, 22, 62–69. [Google Scholar] [CrossRef]
- Csermely, P.; Agoston, V.; Pongor, S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef]
- Paolini, G.V.; Shapland, R.H.B.; van Hoorn, W.P.; Mason, J.S.; Hopkins, A.L. Global mapping of pharmacological space. Nat. Biotechnol. 2006, 24, 805–815. [Google Scholar] [CrossRef]
- Lill, M.A. Multi-dimensional QSAR in drug discovery. Drug Discov. Today 2007, 12, 1013–1017. [Google Scholar] [CrossRef]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, But Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- Muthyala, R. Orphan/rare drug discovery through drug repositioning. Drug Discov. Today: Ther. Strat. 2011, 8, 71–76. [Google Scholar] [CrossRef]
- Lusher, S.J.; McGuire, R.; van Schaik, R.C.; Nicholson, C.D.; de Vlieg, J. Data-driven medicinal chemistry in the era of big data. Drug Discov. Today 2014, 19, 859–868. [Google Scholar] [CrossRef]
- Fuoco, D. A New Method for Characterization of Natural Zeolites and Organic Nanostructure using Atomic Force Microscopy. Nat. Précéd. [CrossRef]
- D. Fuoco et al., “Identifying nutritional, functional, and quality of life correlates with male hypogonadism in advanced cancer patients,” ecancermedicalscience, vol. 9, 2015, Accessed: Feb. 04, 2024. [Online]. Available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4544574/.
- D. Fuoco, “Cytotoxicity induced by tetracyclines via protein photooxidation,” Advances in Toxicology, vol. 2015, 2015, Accessed: Feb. 04, 2024. [Online]. Available: https://downloads.hindawi.com/archive/2015/787129.pdf.
- Talevi, A. Multi-target pharmacology: possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef]
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef]
- Cheng, F.; Desai, R.J.; Handy, D.E.; Wang, R.; Schneeweiss, S.; Barabási, A.-L.; Loscalzo, J. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Viana, J.d.O.; Félix, M.B.; Maia, M.d.S.; Serafim, V.d.L.; Scotti, L.; Scotti, M.T. Drug discovery and computational strategies in the multitarget drugs era. Braz. J. Pharm. Sci. 2018, 54. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolicb, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Buenz, E.J.; Verpoorte, R.; Bauer, B.A. The Ethnopharmacologic Contribution to Bioprospecting Natural Products. Annu. Rev. Pharmacol. Toxicol. 2017, 58, 509–530. [Google Scholar] [CrossRef]
- Mesiti, F.; Chavarria, D.; Gaspar, A.; Alcaro, S.; Borges, F. The chemistry toolbox of multitarget-directed ligands for Alzheimer's disease. Eur. J. Med. Chem. 2019, 181, 111572. [Google Scholar] [CrossRef]
- Geerts, H.; Wikswo, J.; van der Graaf, P.H.; Bai, J.P.; Gaiteri, C.; Bennett, D.; Swalley, S.E.; Schuck, E.; Kaddurah-Daouk, R.; Tsaioun, K.; et al. Quantitative Systems Pharmacology for Neuroscience Drug Discovery and Development: Current Status, Opportunities, and Challenges. CPT: Pharmacometrics Syst. Pharmacol. 2019, 9, 5–20. [Google Scholar] [CrossRef]
- Namdar, D.; Anis, O.; Poulin, P.; Koltai, H. Chronological Review and Rational and Future Prospects of Cannabis-Based Drug Development. Molecules 2020, 25, 4821. [Google Scholar] [CrossRef]
- Song, Y.-X.; Furtos, A.; Fuoco, D.; Boumghar, Y.; Patience, G.S. Meta-analysis and review of cannabinoids extraction and purification techniques. Can. J. Chem. Eng. 2023, 101, 3108–3131. [Google Scholar] [CrossRef]
- Mullowney, M.W.; Duncan, K.R.; Elsayed, S.S.; Garg, N.; van der Hooft, J.J.J.; Martin, N.I.; Meijer, D.; Terlouw, B.R.; Biermann, F.; Blin, K.; et al. Artificial intelligence for natural product drug discovery. Nat. Rev. Drug Discov. 2023, 22, 895–916. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Article inclusion and exclusion criteria with the numbers.
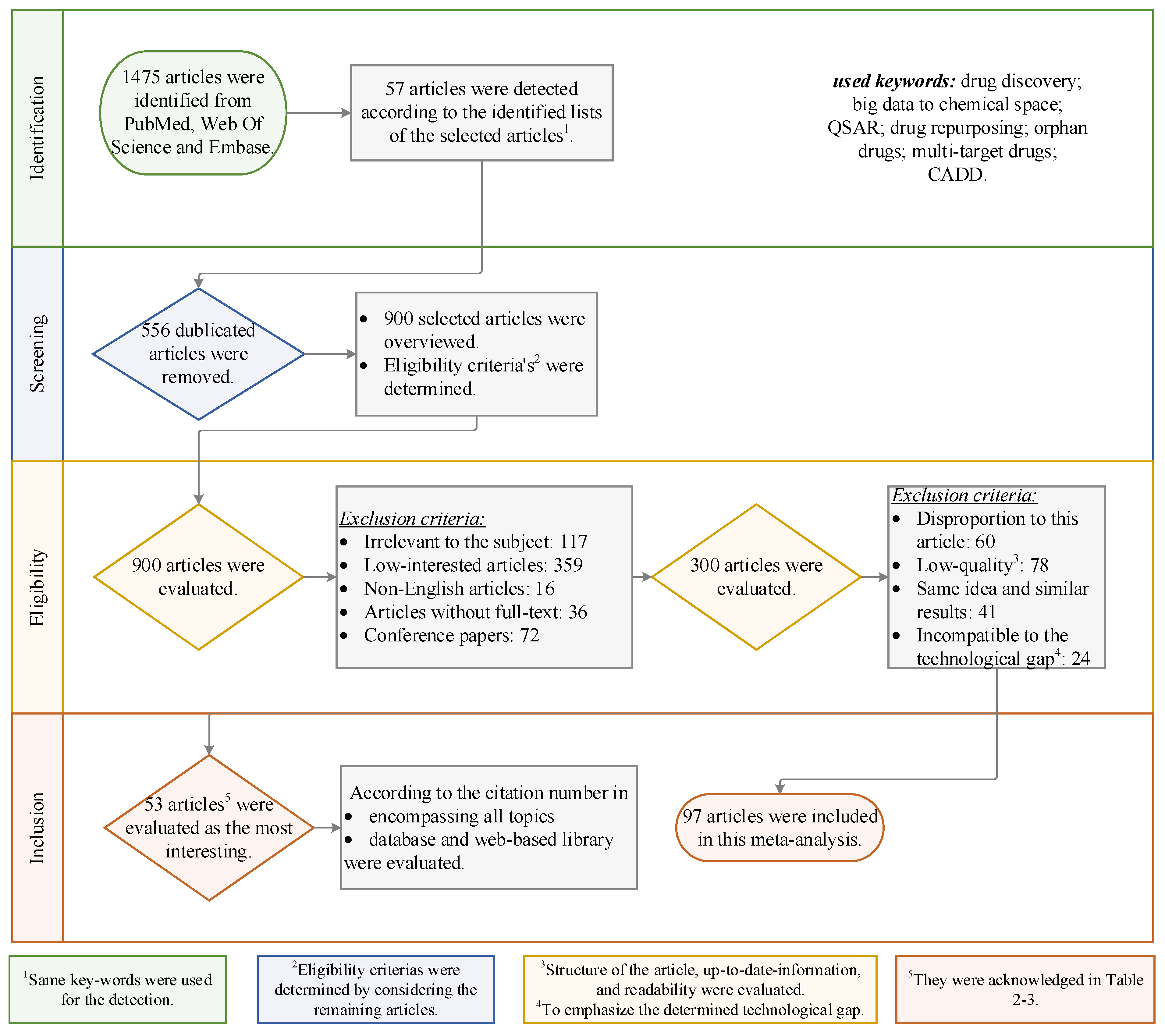
Figure 2.
Bibliometric map derived via the publicly-available software VosViewerTM of the 100 most frequent keywords from over 1,000 articles published in peer-reviewed journals listed in Web of Science (2000-2022).
Figure 2.
Bibliometric map derived via the publicly-available software VosViewerTM of the 100 most frequent keywords from over 1,000 articles published in peer-reviewed journals listed in Web of Science (2000-2022).

Figure 4.
The impact of the different studies involved in in silico drug discovery. This heatmap reports the weighted values, which are extrapolated according to the 300 most cited papers in the respective fields. The full data is available in the Excel file in the supplementary section. Each cell represents the ponderal number per year of absolute citations (Google Scholar Number) of the 300 most cited papers on in silico drug discovery. Additional details regarding the use of the weighted values/year are discussed in the text and the table of reference is reported in an Excel file.
Figure 4.
The impact of the different studies involved in in silico drug discovery. This heatmap reports the weighted values, which are extrapolated according to the 300 most cited papers in the respective fields. The full data is available in the Excel file in the supplementary section. Each cell represents the ponderal number per year of absolute citations (Google Scholar Number) of the 300 most cited papers on in silico drug discovery. Additional details regarding the use of the weighted values/year are discussed in the text and the table of reference is reported in an Excel file.
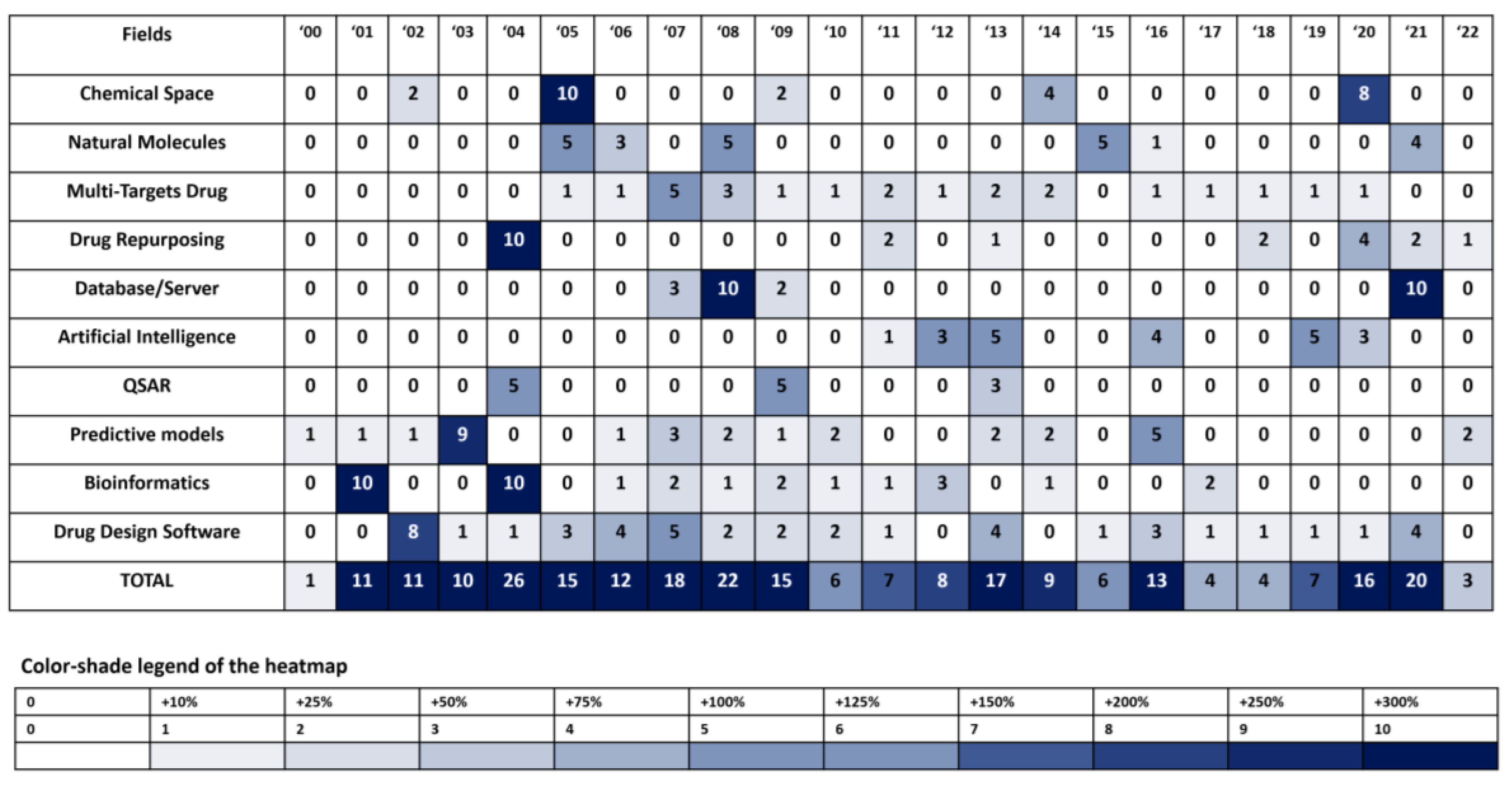
Figure 5.
Visualization of (A) the sum of total publications per year and (B) their impact on the most cited articles over time. Total number of publications reported in PubMed about drug discovery (A). Results of the analysis of trends for the same time span (B). The different periods are depicted by dashed curves. Each color shows one of the five different periods that have emerged from the meta-analysis: purple, red, orange, brown, and green. The blue columns show the relative importance of the 300 selected papers, (on year per year basis). The numbers reported in this lower portion of panel B, are the same as those reported in the heatmap.
Figure 5.
Visualization of (A) the sum of total publications per year and (B) their impact on the most cited articles over time. Total number of publications reported in PubMed about drug discovery (A). Results of the analysis of trends for the same time span (B). The different periods are depicted by dashed curves. Each color shows one of the five different periods that have emerged from the meta-analysis: purple, red, orange, brown, and green. The blue columns show the relative importance of the 300 selected papers, (on year per year basis). The numbers reported in this lower portion of panel B, are the same as those reported in the heatmap.
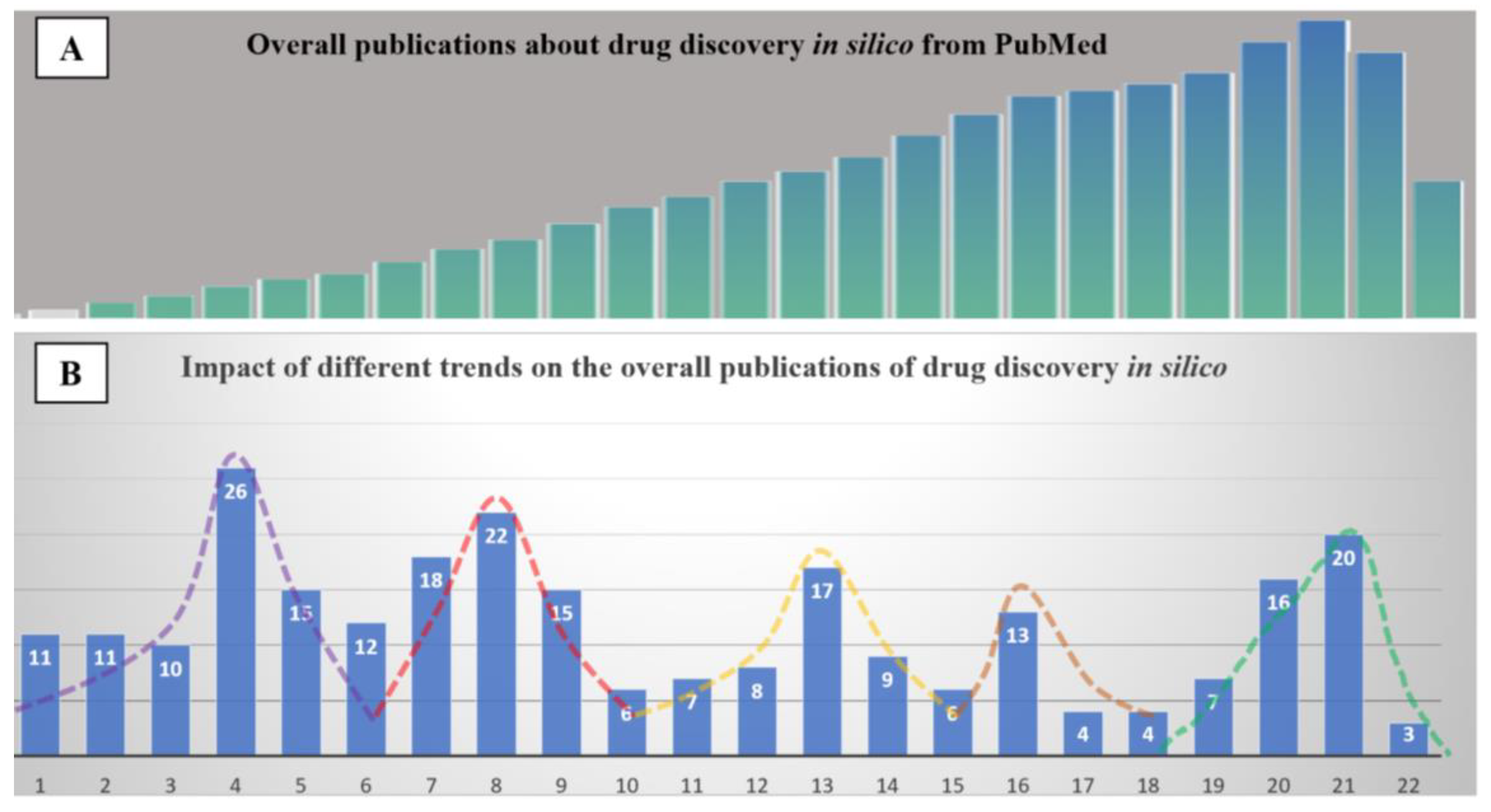
Table 1.
Interplay of current technology and their applications in drug discovery.
| Big Data | |||||||
| Data Source |
Data Collection |
Data Integration |
Data Validation |
Data Application |
Data Visualization |
Data Real-World Evidence |
|
| Disciplines and sub-disciplines by relevance |
in vivo | Diseases | Translational Bioinformatics | Quantitative and Qualitative Structure Activity Relationship (QSAR) |
Active pharmaceutical ingredient (API) “One drug for one molecular target” |
Pharmacophore and molecular target |
Current use of API in clinical practice “One drug for many molecular targets, simultaneously” |
| Clinical outcome | |||||||
| ex vivo | Gene therapy | Bioinformatics | |||||
| Metabolites | |||||||
| in vitro | OMICS | ||||||
| Biochemistry | |||||||
| in silico | Medicinal chemistry | Cheminformatics | Virtual screening and 3D models |
||||
| Chemical physics | |||||||
| Predictive models | |||||||
| Key technologies | DATABASE: • Web-based server • IT-cloud • Extremely fast computing systems |
Data mining coupled with • Machine learning • Deep learning • Artificial intelligence |
Graphical processing unit (GPU): • Dedicated CRO |
Drug repurposing • IND |
Network pharmacology | Multi-target drugs | |
| Chemical Space | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



