Submitted:
11 April 2024
Posted:
12 April 2024
You are already at the latest version
Abstract
Alzheimer’s disease (AD) poses a significant public health challenge due to its irreversible and progressive nature while lack of a cure. As a potential strategy to combat AD, its prevention becomes increasingly attractive. Mild Cognitive Impairment due to AD (MCI-AD) represents a critical transitional stage between normal age-related cognitive decline and more severe AD conditions, occurring just before dementia onset. Unfortunately, there is currently no established animal model that accurately recapitulates MCI-AD characteristics. While many laboratories have traditionally used normally aged wild-type animals as experimental models, this approach falls short in representing the inherently worse state of MCI-AD compared to normal aging. To address this gap, we introduce an animal model—a transgenic mouse line with genetic inactivation of G protein-coupled receptor kinase-5 (GRK5), commonly known as the GRK5 knockout (GRK5KO) mouse. These GRK5KO mice exhibit amnesia, cognitive deficits, increased β-amyloid levels, neurofibrillary tangle (NFT) immunopositive axonopathy, and hippocampal neurodegenerative changes. Importantly, these pathological alterations predominantly impact the entorhinal, transentorhinal, and hippocampal cortices, aligning with human MCI-AD criteria and Braak stage II of human AD progression. Notably, female GRK5KO mice show approximately 2.5 times more NFT-positive axonopathy than males, mirroring the higher prevalence of AD cases in women. Collectively, existing data strongly supports the GRK5KO mouse as a qualified animal model for studying MCI-AD.
Keywords:
Alzheimer’s disease
; Mild cognitive impairment due to Alzheimer’s disease
; aging
; animal model
; GRK5 deficiency
; GRK5 knockout mouse.
1. Introduction
Alzheimer's disease (AD) is the predominant cause of dementia, accounting for 60-80% of dementia cases[1]. It is characterized by progressive decline in memory and other cognitive abilities due to neurodegenerative changes. Currently, there are no known methods for reversing or curing this disease[2]. This makes proactive prevention the most effective strategy to combat AD[3]. In serving such a purpose, a clinical collection of mild cognitive impairment (MCI) due to AD (MCI-AD) was identified, which represents the last chance to intervene before the onset of AD. This article briefly describes the characteristics of MCI-AD in humans and the evidence that G protein-coupled receptor kinase-5 (GRK5) knockout (GRK5KO) mice mirror MCI-AD criteria.
2. Mild Cognitive Impairment Due to Alzheimer's Disease
MCI was initially established to understand the characteristics of individuals in a presumed pre-AD condition, which represents a transitional stage between normal age-related cognitive decline and more severe conditions like AD[4,5]. MCI involves noticeable cognitive decline beyond what is expected for a person’s age but does not meet the criteria for dementia. However, MCI encompasses a heterogeneous group of patients with varying etiologies. Its longitudinal development does not always progress to AD; some individuals develop other dementias, while others remain in the MCI stage or even revert to normal cognition. Only a small portion of MCI cases eventually progress to AD. To better identify the true "pre-AD" population for AD prevention, experts led by the National Institute on Aging (NIA) and the Alzheimer’s Association narrowed down MCI to a subgroup with a much higher tendency to develop AD down the road, terming this subgroup MCI due to AD (MCI-AD)[6,7].
To facilitate both clinical and laboratory research, specific clinical diagnostic criteria and research criteria were established for MCI-AD[6,8]. The clinical diagnostic criteria of MCI-AD were developed to aid clinicians in identifying MCI-AD without relying on invasive and more complex/expensive diagnostic tools such as imaging techniques. This condition is characterized by memory problems and cognitive issues, often serving as early indicators of potential progression to AD. The clinical diagnostic criteria for MCI-AD typically include: (1) Evidence of Cognitive Decline: There should be a noticeable decline in cognitive abilities, particularly in memory, exceeding what is expected for the individual’s age but not severe enough to meet criteria for dementia. (2) Objective Cognitive Impairment: Objective evidence, usually obtained through cognitive assessments and standardized tests, indicates impairment in one or more cognitive domains. (3) Preserved Independence in Functional Abilities: Individuals with MCI generally maintain their independence in daily functioning and do not meet the criteria for dementia, which involves a significant impact on daily activities. These criteria help distinguish MCI from normal age-related cognitive changes and more severe forms of cognitive impairment. The distinction between MCI and AD lies in the severity and impact on daily functioning. In MCI, individuals may experience cognitive challenges, especially in memory, but their overall functional abilities remain largely intact. The cognitive decline is noticeable but not severe enough to significantly interfere with daily activities. This intermediate stage allows healthcare professionals to identify potential early signs of AD or another type of dementia. If there is significant cognitive decline impairing daily functioning, a diagnosis of AD or another form of dementia may be considered.
From a research perspective, MCI-AD requires amnesia and at least another domain of cognitive decline, as well as evidence of concurrent AD pathogenesis[6,7]. Amnesia is essential, but research indicates that having another domain of cognitive impairment significantly increases the odds of developing AD in the future. Individuals with MCI-AD often exhibit pathogenic changes associated with AD, including (1) Beta-Amyloid (Aβ) Plaques: if MCI patients show an accumulation of Aβ or senile plaques (SPs) in the brain, a hallmark characteristic of AD, they are rated as A+ according to the ATN system[9,10]. (2) Tauopathy: Elevated levels of hyperphosphorylated tau (pTau) protein, typically forming neurofibrillary tangles (NFTs). This is another hallmark characteristic of AD and is regarded as T+ in the ATN system. (3) Neurodegeneration: Structural brain changes and neuronal loss may be present in specific regions associated with memory and cognition, indicating neurodegeneration, and is regarded as N+ per the ATN system. These changes contribute to the progression from MCI to AD. However, not everyone with MCI goes on to develop AD, and the rate of progression varies among individuals. Having at least one or two AD characteristic changes in the ATN system is essential for MCI-AD, with more ATN positives indicating a higher likelihood of developing AD. Therefore, the pathogenic subtypes of MCI-AD include A+T-N-, A+T+N-, A-T+N- and A+T+N+, with the latter representing the latest stage, right on the verge of AD onset[11-14].
In MCI-AD, certain brain areas associated with memory and cognitive function are often affected, including the hippocampus, entorhinal cortex, temporal lobes, and prefrontal cortex. The specific areas affected can vary between individuals, and the progression of structural changes may influence the severity and type of cognitive impairment observed. Neuroimaging techniques, such as magnetic resonance imaging (MRI) or positron emission tomography (PET) scans, help researchers and clinicians visualize these changes in the brain. Studies utilizing PET scans have indicated that MCI-AD corresponds to Braak stage II, primarily affecting the entorhinal, transentorhinal, and hippocampal cortices[14].
Furthermore, brain inflammation, or neuroinflammation, is recognized as a significant factor in AD progression. Evidence suggests the involvement of neuroinflammatory processes in MCI-AD, with increased levels of inflammatory markers observed in the brains of affected individuals[6,15]. Microglia, immune cells in the brain, play a role in the inflammatory response. However, the relationship between neuroinflammation and MCI is complex and not fully understood, though it is considered a part of the broader cascade of changes associated with AD progression, with heavier involvement in later stages.
In conclusion, MCI-AD is not a homogeneous condition, and patients may exhibit variability in etiology and clinical presentation. However, commonalities exist among individuals diagnosed with MCI-AD, including underlying Alzheimer’s pathology, memory impairment, neurological changes, and cognitive decline beyond age expectations. Despite these commonalities, considerable heterogeneity exists in MCI cases, emphasizing the need for personalized approaches in understanding and managing this condition.
3. GRK5KO Mice—A Model of GRK5 Deficiency Recapitulates Characteristics of MCI-AD
3.1. GRK5 and Its Relevance to AD
Our comprehension of biological responses to stimuli exceeds that of their diminishing counterpart, desensitization. The latter involves a reduction in responsiveness to persistent stimuli while selectively engaging with new ones. G protein–coupled receptor kinases (GRKs) play a critical role in this process, particularly in signaling mediated by G protein–coupled receptors (GPCRs), which constitute the largest family of signal-transducing proteins[16]. Central to its role in "uncoupling" G proteins from their coupled GPCRs upon the receptor activation for turning off the GPCR signaling[17], this novel GPCR regulatory mechanism earned the Nobel Prize in Chemistry in 2012 for Dr. Robert Lefkowitz[18], who contributed to the discovery of most GRKs and arrestins.
Currently, seven members of GRKs are identified: GRK1 to GRK7 [19,20]. Among them, GRK5 stands out as a ubiquitously distributed member implicated in various physiological functions [19,21]. Its activity has been associated with age-related ailments such as neoplastic, metabolic, neurodegenerative, and cardiovascular diseases, including type 2 diabetes mellitus, cardiac hypertrophy, hypertension, Parkinson’s disease, and AD [22-30]. During aging, dysfunction of the insulinotropic signaling cascade, affecting glucose sensing, uptake, and metabolism, plays a pivotal role in aging across animal models [31]. The insulinotropic/insulin-like growth factor system primarily controls animal longevity, with genes associated mainly with this system {Mathew, 2016, 35-53}. GRK5’s potential role in aging is underscored by its strong expression in adipose tissues, suggesting its impact on the glycoregulatory system [32]. Studies in GRK5KO mice demonstrated significant insulin resistance [33], and GRK5 genetic polymorphisms have been linked to type 2 diabetes mellitus and the efficacy of antidiabetic drugs [34]. Furthermore, studies in GRK5KO mice highlighted its importance in metabolism, showing decreased white adipose tissue mass, reduced adipogenic gene expression, and impaired adipocyte differentiation on a high-fat diet [33,35]. Although human data on GRK5 and metabolism are limited, they suggest an association with apolipoprotein B levels and total LDL-cholesterol, emphasizing its role in cholesterol metabolism. In addition, NF-kB regulates GRK5 expression, indicating a feed-forward loop between these two systems, characteristic of aging [36]. GRK5 emerges as a vital component in energy metabolism and chronic inflammation paradigms [37], both strongly linked to molecular aging pathologies and various human disorders. These insights position GRK5 as a potential therapeutic target for age-related diseases such as cardiovascular disease, neurological disorders, and metabolic disorders [19,21,38].
The relevance of GRK5 to AD research became evident when rapid loss of functional/membrane GRK5 due to Aβ exposure was identified [29]. Subsequently, brain GRK5 deficiency was observed in the prodromal stage of AD in CRND8 AD transgenic mice and human AD autopsy samples [17,29]. GRK5 down-regulation is associated with prolonged platelet-derived growth factor receptor-ß signaling and treatments with gonadotropin-releasing hormone, thyroid stimulating hormone, morphine, or lipopolysaccharide [39-43]. Because plasma membrane-bound GRK5 is considered the functional portion for its GPCR kinase activity, membrane GRK5 deficiency could occur due to reduced membrane-associated GRK5 levels [17,29], in addition to reduced overall GRK5 expression. Physiologically active GRK5 primarily associates with the plasma membrane, where it can be anchored on the inside of the cell membrane by binding to phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphoserine [16,19]. Conversely, in the cytosol, GRK5 tends to associate with Ca2+/Calmodulin and F-actins [16,19]. Therefore, the balance of binding force for GRK5 between membrane (e.g., PIP2) and cytosol (e.g., Ca2+/Calmodulin) may be important to regulate its functionality, transitioning between a GPCR kinase and a signaling molecule, or even a transcriptional regulator in the cell nucleus [17]. For instance, in cultured cells, Aß can cause rapid (within minutes) GRK5 membrane disassociation, leading to membrane GRK5 deficiency, while increased cytosolic GRK5 may participate in mediating Aβ-induced cellular signaling changes that often prime different cells to become more sensitive to many cellular stressors [29,44,45]. From this perspective, potential causes of GRK5 deficiency could be attributed to factors like cellular stress, calcium imbalance, nutritional deficiencies (especially phospholipids), and aging [17,44-48]. The relatively high levels of normal GRK5 distribution in the earliest affected brain regions of AD, particularly the limbic system [49,50], along with Aβ-induced down-regulation of GRK5, implicate involvement of GRK5 deficiency in AD. While relations of GRK dysfunction to Aβ-related AD pathogenesis are still under investigation, another interesting piece of evidence is that a single nucleotide polymorphism associated with a functional GRK5 variant (GRK5-Gln41Leu) appears to decrease GRK5 translocation from the membrane to the cytoplasm, reducing tau hyperphosphorylation, and is associated with a lower risk of late-onset AD [51].
As pointed out by Kohout and Lefkowitz, the regulation of GPCRs by GRKs exhibits both selectivity and redundancy [19]. This redundancy allows GRKs to compensate for each other's loss without significantly impacting the overall efficiency of timely GPCR signaling attenuation. However, studies in individual GRK knockout mice have unequivocally shown selective regulation by GRKs in vivo. For example, mice deficient in GRK2, 3, 5, and 6 display impaired desensitization of adrenergic, odorant, muscarinic, and dopaminergic receptors, respectively [52-55]. Moreover, GRK5 deficiency selectively affects muscarinic receptors, particularly the Gi/o-coupled M2 and M4 receptors, without impacting M1/M3/M5 or non-muscarinic receptors [19,38,52,56]. Importantly, this high selectivity is observed only in vivo, not in vitro, suggesting a potential influence of topographic GRK subtype expression in vivo. Furthermore, this selectivity is apparent only when GRK deficiency occurs without compensation. Specifically, the membrane-associated nature of GRK5 renders its levels in neuronal terminals, such as spines and synapses, highly sensitive to early neurodegenerative changes. This sensitivity arises because GRK5, synthesized in the cytosol, must be transported through neurites to neuronal terminals, while axonal transport deficiency is among the earliest neurodegenerative changes occurring in normal aging and early AD [57]. Consequently, disruptions in neurite transportation, including axonal transport, could negatively impact GRK5 levels in neuronal terminals, including synapses. Notably, brain GRK5 levels sharply decrease during aging in wild-type (WT) mice. If defective axonal transportation occurs during aging, upregulation of GRK5 in an attempt to counteract GRK5 deficiency may not rectify the deficiency in neuronal terminals. Instead, the overexpressed GRK5 could accumulate where it is not deficient, leading to locally upregulated GRK5. Such upregulation could affect a wide range of GPCRs crucial in cardiovascular disorders [16,19,58,59].
3.2. Aged GRK5KO Mice Recaptured MCI-AD Characteristics
MCI-AD represents the last chance to intervene before the onset of AD[4-7]. A good animal model of MCI-AD will help the relevant prevention and therapeutic studies. Currently, many studies have used aged WT animals to model MCI-AD, which we know MCI-AD is a transitional stage between normal aging and AD. This means that a real MCI-AD condition should be significantly worse than normal aging. In another word, normal aging is not a good representation of MCI-AD. In this regard, aged GRK5KO mice mirror the MCI-AD criteria much better than aged WT mice, which will be described in detail below.
Up to now, GRK5KO mice have been studied and analyzed by multiple groups including ours. Given our focus on the brain functionality, the existing data in the GRK5KO mice from our group are summarized below and schematically illustrated in Figure 1:
(1). Behavioral assessment: The earliest indication of behavioral deficits in GRK5KO mice was reported in younger mice, aged 8-12 weeks, displaying impaired social novelty recognition by another group[60]. Our investigations unveiled a pattern of behavioral changes: from 12 months old, GRK5KO mice exhibit reduced travel distance in the open field without impaired sensorimotor function. By 18 months old, they manifest elevated time in the open arm of the elevated plus maze [30], indicating altered alertness and anxiety. These observations coincided with the NFT+ axonopathy in the entorhinal and transentorhinal cortical regions at this age, revealing a connection between impaired behavior and neuropathology. A crucial cognitive deficit related to MCI-AD, amnesia, emerges at 18 months old, affecting spatial working memory in the Radial Arm Water Maze (RAWM) task[30] but not the Y maze and Morris Water Maze (MWM) tasks, an implication of early-stage short memory deficit. Although all the three tasks are able to measure short-term spatial memory[61-63] different complexity of the setups allow them to detect the memory deficit with variable sensitivities (i.e., more complicated 8-arms in RAWM may require more accurate spatial reference memory to succeed, therefore can detect earlier stage deficit). This was indeed supported by the actual data[30]. Overall, the 18-month-old GRK5KO mice exhibit amnesia and other cognitive impairments, fulfilling the behavioral criteria for MCI-AD.
(2). Pathological assessment: The A/T/N system, introduced through the NIA-AA Alzheimer’s Diagnostic Framework, serves as not only an impartial and descriptive classification approach for diagnosing and prognosing AD but also as a valuable tool for comprehending AD pathogenesis[9,10]. This stems from its ability to condense AD pathogenesis into three primary categories of pathological alterations: Aβ, Tau, and Neurodegeneration. This system offers a comprehensive and balanced assessment of pathological changes centered around AD. Hence, we present the pathological data in the GRK5KO mice by aligning them with the ATN system, enhancing clarity, while still able to acknowledge any distinctive features when necessary, such as the NFT+-axonopathy and cholinergic degeneration.
a. T+-related changes: The T+-related alterations in GRK5KO mice, up to 18 months of age, are both significant and represent one of the earliest and most pronounced pathological features. These changes were primarily identified through Campbell-Switzer (CS) Silver staining (which is sensitive to 3-repeats tau) and NFT immunostaining, revealing swollen axonal clusters (SACs)[30]. The characterization of SACs was affirmed through axonal selective protein kinesin heavy chain immunostaining and electron microscopy (as presented in the original article[30]). Interestingly, both KHC and NFT immunostaining demonstrated high sensitivity to the CS Silver positive SACs, reinforcing the T+ nature of these changes. Western Blotting corroborated these histological findings by showing a significant increase in pTau.
Notably, the temporal appearance of NFT+ axonopathy within the brain is particularly striking. It initially emerged in the olfactory region as early as 6-9 months old, subsequently in the piriform/amygdaloid regions, and finally expanded into the hippocampus at mild level around 12 months and continued to accumulate. Remarkably, no such formations were detected in other cortical areas even at 18-months old. These characteristic T+ changes parallel those found in human AD brains at Braak stage I-II and correspond to the clinical stage of MCI-AD. These compelling data offer robust support for the involvement of GRK5 deficiency in MCI-AD.
b. A+-related changes: In 18-month-old GRK5KO mice, the Aβ-related alterations were identified as positive due to the presence of scattered SPs and an increase in SDS-soluble Aβ (sAβ)[30] though the SPs were immature at this stage, being primarily composed of fibrillary Aβ (fAβ) encased by astrocytes without concurrent activation of microglial cells. Additionally, notable inflammation was not globally evident at this point.
c. N+-related changes: In 18-month-old GRK5KO mice, while notable neurodegenerative alterations were observed, they were mild within the ATN system. No significant neuronal loss, either in total or specific to cholinergic neurons, was evidenced by quantification through stereology using NeuN or choline acetyltransferase (ChAT) immunostaining. However, an increase in hippocampal cholinergic axonal swelling (CAS) was noted in the CA3 region, and a decline in cholinergic fiber density (CFD) was observed in the CA3 molecular layer and CA1 stratum oriens across the numerous brain subregions investigated in GRK5KO mice. These findings collectively point to an early-stage cholinergic neurodegeneration.
d. Other pathological changes: The higher incidence of AD in female is a characteristic of AD[64-67]. Previous research from our group reported a 2.5-fold increase of the hippocampal swollen axonal clusters (SACs) in female GRK5KO mice compared to their male counterparts[68], which was aligned with such an AD characteristics, supporting the notion that gender differences in AD-related pathology are reflected in the GRK5KO mouse model. In addition, we previously reported the cognitive evaluation of GRK5KO mice which the data was exclusively from female mice[30,69]. This decision stemmed from our initial characterization of GRK5KO mice, where statistical significance was not achieved from the male or mixed-gender groups although there was a trend23,32. For this reason, unless a study focus specifically requires consideration of gender differences, use of the female mice in experiments may maintain experimental consistency and avoid potential confounding factors related to gender variability.
In summary, GRK5KO mice developed progressive cognitive impairments including MCI-AD characteristic amnesia and other cognitive deficits up to 18 months[30,60]. Pathologically, they exhibited increased Aβ/SPs (A+), prominent CS silver staining and NFT immuno-positive axonopathy with increased pTau (T+), and reduced hippocampal cholinergic fiber density (CFD,→ N+), matching MCI-AD criteria and displaying characteristics of A+T+N+ MCI, a sub-variance of MCI-AD most likely to convert to AD[6,7,14,30]. Notably, NFT distribution in the olfactory, piriform, amygdaloid, and hippocampus of GRK5KO mice[30] aligned with the brain regional locations of the entorhinal (i.e., olfactory) to transentorhinal (i.e., piriform and amygdaloid) and hippocampal locations of the NFTs as defined in the Braak Stage II of human AD[11-14]. The latter coincides with the late stage of MCI-AD according to the recently developed positron emission tomography-based Braak staging[14]. On top of all the evidence, GRK5KO mice also displayed gender difference with the female having worsened pathology, that is an additional coincidence with AD. It is worth noting that all these changes in the GRK5KO mice were age-dependent development (as illustrated in Figure 1) and statistically significant as compared to the age-matched WT control mice. This indicates that GRK5KO mice recapture a disease stage between normal aging and AD and are more suitable to model MCI-AD than the aged WT mice[5,70]. Moreover, these GRK5KO changes replicated human MCI-AD features without altering any known AD genes or mutations, such as β-amyloid precursor protein (APP), Presenilins, tau or ApoE. Therefore, the data collectively suggests that GRK5 deficiency may cause MCI-AD at least in mice.
3.3. GRK5 Deficiency Renders Selective Cholinergic Neuronal Vulnerability-a Potential Mechanism and Therapeutical Implication
The mechanisms underlying selective loss-of-function in GRK5 deficiency remain elusive, potentially tied to the distinct topographic expression patterns of various GRKs in vivo [19,38]. Notably, presynaptic M2 hyperactivity could arise due to insufficient compensation by other GRKs at cholinergic synapses where GRK5 is deficient. Interestingly, this in vivo selectivity between GRKs and GPCRs is absent in vitro, suggesting that beyond chemical binding specificity, the specific spatial arrangement of GRK/GPCR pairs in vivo plays a crucial role. Consequently, upregulating GRK5 as a therapeutic strategy for GRK5 deficiency is ill-suited. Overexpression of GRK5, particularly in cases of impaired axonal transport during aging and AD[71-73], may lead to mislocalization, ineffective compensation, and broad impacts on various GPCRs, potentially elevating cardiovascular disease risks[16,19,58,59].
In the context of potential impacts of GRK5 deficiency in AD, studies have consistently demonstrated that GRK5 deficiency exacerbates AD progression in GRK5-deficient APP (GAP) mice. These impacts manifest as cognitive decline, increased amyloid burden, and heightened inflammation[56,69,74,75]. Notably, in GAP mice, GRK5 deficiency accelerates basal forebrain (BF) cholinergic axonopathy and neuronal loss, leading to an earlier cognitive decline[69]. The underlying mechanism is believed to involve compromised cholinergic neuronal defense due to suppressed cAMP/CREB signaling. This signaling pathway plays a critical role in regulating the pro/anti-apoptotic threshold in cells[76-81]. Consequently, GRK5 deficiency selectively impairs presynaptic M2 receptor desensitization, resulting in presynaptic M2 hyperactivity[48,52,56]. The presynaptic M2 receptor is a Gi-coupled inhibitory receptor. While transient inhibition of M2 only reduces acetylcholine (ACh) release[82-84], prolonged inhibition (from seconds to hours) suppresses the cAMP/CREB signaling pathway[56,69], ultimately lowering the cholinergic neuronal apoptotic threshold and rendering selective vulnerability in cholinergic neurons.
From a therapeutic perspective, directly overexpressing GRK5 may not necessarily compensate for the GRK5 deficiency. Instead, one way to rectify the impacts of GRK5 deficiency is to target the presynaptic M2 hyperactivity. In supporting this therapeutic implication, a recent investigation demonstrated that blocking the M2 receptor with a selective M2 antagonist CN168 in GAP mice effectively prevented BF cholinergic degenerative changes and successfully delayed cognitive decline for 5 months until the end of the experimental observations[85]. This study underscores the utility of this animal model for relevant prevention and therapeutic research.
It is noteworthy that CN168, a selective M2 antagonist, falls under the category of cholinergic modifying drugs, extensively investigated in AD research driven by the cholinergic hypothesis [86-89]. Notably, ChEIs, the most recognized cholinergic modifying drugs for AD, have been disappointing due to their limited efficacy in managing symptoms without modifying the disease course [90-92]. The fundamental difference between CN168 and ChEIs lies in their target mechanisms for addressing the underlying AD pathogenesis (see Table 1 for comparisons between these two therapeutic strategies). ChEIs primarily focus on inhibiting cholinesterase to preserve acetylcholine (ACh) levels in the synaptic cleft, mainly compensating for cholinergic hypofunction and alleviating associated symptoms [90]. In contrast, CN168 targets presynaptic M2 hyperactivity, preventing over-suppression of cAMP/PKA/CREB signaling in cholinergic neurons [38,69,93]. By strengthening the intrinsic defenses of these neurons, CN168 aims to enhance resilience against degenerative insults. Although CN168 may increase ACh release during its use, it was not administered during the cognitive tests in this study. In fact, the drug was withdrawn two weeks before the cognitive tests [85]. This experimental design eliminated any cognitive impact resulting from acute influences on ACh release by the drug. If any, the cognitive tests were conducted during the drug withdrawal period, during which, in the case of ChEI discontinuation or withdrawal, worsening of symptoms might occur [94-97]. Thus, the improvements in cognition observed in this study were likely attributed to the preservation of cholinergic neurons rather than functional compensation for cholinergic hypofunction.
4. Conclusions
GRK5KO mice exhibited amnesia and other cognitive deficits, along with increased AD pathogenesis characterized as A+T+N+[30,38,48,69]. These changes align with the criteria for human MCI-AD[6,7], qualifying for serving as an animal model for MCI-AD. Further exploration and utilization of this model will facilitate the efforts in understanding MCI to AD conversion and prevention.
5. Future Perspectives
The urgent need for effective AD prevention underscores the importance of understanding MCI-AD. GRK5KO mice faithfully replicate MCI-AD characteristics, addressing the unmet need for a suitable animal model. However, the precise roles of GRK5 deficiency in MCI and AD pathogenesis remain to be fully elucidated. The convergence of MCI-AD features across multiple levels cannot be attributed solely to coincidence; hidden logic and phenomena await discovery. Deeper and broader investigations into etiology and epidemiology may yield further evidence.
Immediate utilization of the GRK5KO model can be both mechanistic and therapeutic. By introducing risk factors, we can dissect critical drivers of MCI-to-AD conversion. Targeting known pathways (e.g., M2 blockade) may offer therapeutic or preventive implications. This research avenue has the potential to revolutionize our understanding of AD, shedding light on the mechanisms guiding MCI-to-AD transformation. Ultimately, we aspire to harness this model for effective pharmaceutical AD prevention, instilling hope for millions affected by this condition.
Author Contributions
writing—original draft preparation and review and editing, W.Z.S.
Funding
This work was supported by grants to W.Z.S. from the Alzheimer’s Association (the program of “Novel Pharmacological Strategies to Prevent Alzheimer's Disease” NPSPAD-11-202149), the Medical Research and Development Service, Department of Veterans Affairs (Merit Review 1I01 BX001067-01A2 and 1I01BX004739-01A2).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This review article does not include any new data. The relevant data cited was previously published elsewhere.
Acknowledgments
This work was supported by grants to W.Z.S. from the Alzheimer’s Association (the program of “Novel Pharmacological Strategies to Prevent Alzheimer's Disease” NPSPAD-11-202149), the Medical Research and Development Service, Department of Veterans Affairs (Merit Review 1I01 BX001067-01A2 and 1I01BX004739-01A2), and resources from the Midwest Veterans’ Biomedical Research Foundation.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 2023 Alzheimer's disease facts and figures. Alzheimers Dement 2023, 19, 1598–1695. [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer's disease. Nat Rev Dis Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- McDade, E.; Bateman, R.J. Stop Alzheimer's before it starts. Nature 2017, 547, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Janoutova, J.; Sery, O.; Hosak, L.; Janout, V. Is Mild Cognitive Impairment a Precursor of Alzheimer's Disease? Short Review. Cent Eur J Public Health 2015, 23, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Foidl, B.M.; Humpel, C. Can mouse models mimic sporadic Alzheimer's disease? Neural regeneration research 2020, 15, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Bertens, D.; Vos, S.; Kehoe, P.; Wolf, H.; Nobili, F.; Mendonca, A.; van Rossum, I.; Hort, J.; Molinuevo, J.L.; Heneka, M.; et al. Use of mild cognitive impairment and prodromal AD/MCI due to AD in clinical care: a European survey. Alzheimers Res Ther 2019, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Westbury, J.L.; Peterson, G.M. Rethinking psychotropics in nursing homes. The Medical journal of Australia 2013, 199, 98. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Blazhenets, G.; Frings, L.; Ma, Y.; Sorensen, A.; Eidelberg, D.; Wiltfang, J.; Meyer, P.T.; Alzheimer's Disease Neuroimaging, I. Validation of the Alzheimer Disease Dementia Conversion-Related Pattern as an ATN Biomarker of Neurodegeneration. Neurology 2021, 96, e1358–e1368. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Biel, D.; Brendel, M.; Rubinski, A.; Buerger, K.; Janowitz, D.; Dichgans, M.; Franzmeier, N.; Alzheimer's Disease Neuroimaging, I. Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers Res Ther 2021, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Therriault, J.; Pascoal, T.A.; Lussier, F.Z.; Tissot, C.; Chamoun, M.; Bezgin, G.; Servaes, S.; Benedet, A.L.; Ashton, N.J.; Karikari, T.K.; et al. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nature Aging 2022, 2, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Morgenroth, I.; Michaud, P.; Gasparini, F.; Avrameas, A. Central and Peripheral Inflammation in Mild Cognitive Impairment in the Context of Alzheimer's Disease. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu Rev Biochem 1998, 67, 653–692. [Google Scholar] [CrossRef]
- Suo, W.Z.; Li, L. Dysfunction of G protein-coupled receptor kinases in Alzheimer's disease. TheScientificWorldJournal 2010, 10, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Prize in Chemistry 2012. NobelPrize.org, 2012.
- Kohout, T.A.; Lefkowitz, R.J. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol 2003, 63, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Mayor, F., Jr. Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal 2003, 15, 973–981. [Google Scholar] [CrossRef]
- Hendrickx, J.O.; van Gastel, J.; Leysen, H.; Santos-Otte, P.; Premont, R.T.; Martin, B.; Maudsley, S. GRK5 - A Functional Bridge Between Cardiovascular and Neurodegenerative Disorders. Frontiers in pharmacology 2018, 9, 1484. [Google Scholar] [CrossRef]
- Premont, R.T.; Gainetdinov, R.R. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol 2007, 69, 511–534. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther 2012, 133, 40–69. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gan, W.; Lu, L.; Dong, X.; Han, X.; Hu, C.; Yang, Z.; Sun, L.; Bao, W.; Li, P.; et al. A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes 2013, 62, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.I.; Gao, E.; Shang, X.; Premont, R.T.; Koch, W.J. Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ Res 2012, 111, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.M.; Cohn, H.I.; Pesant, S.; Eckhart, A.D. GPCR signalling in hypertension: role of GRKs. Clinical science 2008, 115, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Arawaka, S.; Wada, M.; Goto, S.; Karube, H.; Sakamoto, M.; Ren, C.H.; Koyama, S.; Nagasawa, H.; Kimura, H.; Kawanami, T.; et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson's disease. J Neurosci 2006, 26, 9227–9238. [Google Scholar] [CrossRef] [PubMed]
- Bychkov, E.R.; Gurevich, V.V.; Joyce, J.N.; Benovic, J.L.; Gurevich, E.V. Arrestins and two receptor kinases are upregulated in Parkinson's disease with dementia. Neurobiol Aging 2008, 29, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Wu, M.; Citron, B.A.; Wong, G.T.; Festoff, B.W. Abnormality of G-protein-coupled receptor kinases at prodromal and early stages of Alzheimer's disease: an association with early beta-amyloid accumulation. J Neurosci 2004, 24, 3444–3452. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Cox, A.A.; Bartelli, N.; Rasul, I.; Festoff, B.W.; Premont, R.T.; Arendash, G.W. GRK5 deficiency leads to early Alzheimer-like pathology and working memory impairment. Neurobiol Aging 2007, 28, 1873–1888. [Google Scholar] [CrossRef]
- Ma, S.; Gladyshev, V.N. Molecular signatures of longevity: Insights from cross-species comparative studies. Seminars in cell & developmental biology 2017, 70, 190–203. [Google Scholar] [CrossRef]
- Diez, J.J.; Iglesias, P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur J Endocrinol 2003, 148, 293–300. [Google Scholar] [CrossRef]
- Wang, F.; Wang, L.; Shen, M.; Ma, L. GRK5 deficiency decreases diet-induced obesity and adipogenesis. Biochem Biophys Res Commun 2012, 421, 312–317. [Google Scholar] [CrossRef]
- Xia, Z.; Yang, T.; Wang, Z.; Dong, J.; Liang, C. GRK5 intronic (CA)n polymorphisms associated with type 2 diabetes in Chinese Hainan Island. PloS one 2014, 9, e90597. [Google Scholar] [CrossRef]
- Wang, L.; Shen, M.; Wang, F.; Ma, L. GRK5 ablation contributes to insulin resistance. Biochem Biophys Res Commun 2012, 429, 99–104. [Google Scholar] [CrossRef]
- Islam, K.N.; Bae, J.W.; Gao, E.; Koch, W.J. Regulation of nuclear factor kappaB (NF-kappaB) in the nucleus of cardiomyocytes by G protein-coupled receptor kinase 5 (GRK5). J Biol Chem 2013, 288, 35683–35689. [Google Scholar] [CrossRef] [PubMed]
- Packiriswamy, N.; Lee, T.; Raghavendra, P.B.; Durairaj, H.; Wang, H.; Parameswaran, N. G-protein-coupled receptor kinase-5 mediates inflammation but does not regulate cellular infiltration or bacterial load in a polymicrobial sepsis model in mice. Journal of innate immunity 2013, 5, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Suo, W.Z. GRK5 Deficiency Causes Mild Cognitive Impairment due to Alzheimer's Disease. J Alzheimers Dis 2022, 85, 1399–1410. [Google Scholar] [CrossRef]
- Wu, J.H.; Goswami, R.; Cai, X.; Exum, S.T.; Huang, X.; Zhang, L.; Brian, L.; Premont, R.T.; Peppel, K.; Freedman, N.J. Regulation of the platelet-derived growth factor receptor-beta by G protein-coupled receptor kinase-5 in vascular smooth muscle cells involves the phosphatase Shp2. J Biol Chem 2006, 281, 37758–37772. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ding, L.; Xu, J.; Williams, R.S.; Chegini, N. Leiomyoma and myometrial gene expression profiles and their responses to gonadotropin-releasing hormone analog therapy. Endocrinology 2005, 146, 1074–1096. [Google Scholar] [CrossRef]
- Nagayama, Y.; Tanaka, K.; Namba, H.; Yamashita, S.; Niwa, M. Expression and regulation of G protein-coupled receptor kinase 5 and beta-arrestin-1 in rat thyroid FRTL5 cells. Thyroid 1996, 6, 627–631. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, J.; Zhang, X.; Yue, W.; Ma, L. Acute and chronic morphine treatments and morphine withdrawal differentially regulate GRK2 and GRK5 gene expression in rat brain. Neuropharmacology 2002, 43, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Malik, A.B. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat Med 2003, 9, 315–321. [Google Scholar] [CrossRef]
- Pitcher, J.A.; Fredericks, Z.L.; Stone, W.C.; Premont, R.T.; Stoffel, R.H.; Koch, W.J.; Lefkowitz, R.J. Phosphatidylinositol 4,5-bisphosphate (PIP2)-enhanced G protein-coupled receptor kinase (GRK) activity. Location, structure, and regulation of the PIP2 binding site distinguishes the GRK subfamilies. J Biol Chem 1996, 271, 24907–24913. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Satpaev, D.K.; Slepak, V.Z.; Benovic, J.L. Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J Biol Chem 1997, 272, 18273–18280. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.L.; De La Cruz, E.M.; Pollard, T.D.; Lefkowitz, R.J.; Pitcher, J.A. Regulation of G protein-coupled receptor kinase 5 (GRK5) by actin. J Biol Chem 1998, 273, 20653–20657. [Google Scholar] [CrossRef] [PubMed]
- Sallese, M.; Iacovelli, L.; Cumashi, A.; Capobianco, L.; Cuomo, L.; De Blasi, A. Regulation of G protein-coupled receptor kinase subtypes by calcium sensor proteins. Biochim Biophys Acta 2000, 1498, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Suo, W.Z. Accelerating Alzheimer’s pathogenesis by GRK5 deficiency via cholinergic dysfunction. Adv Alzheimer’s Dis 2013, 2, 148–160. [Google Scholar] [CrossRef]
- Kunapuli, P.; Benovic, J.L. Cloning and expression of GRK5: a member of the G protein-coupled receptor kinase family. Proc Natl Acad Sci U S A 1993, 90, 5588–5592. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Koch, W.J.; Inglese, J.; Lefkowitz, R.J. Identification, purification, and characterization of GRK5, a member of the family of G protein-coupled receptor kinases. J Biol Chem 1994, 269, 6832–6841. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, J.; Yin, M.; Cai, Y.; Liu, S.; Wang, Y.; Zhang, X.; Cao, H.; Chen, T.; Huang, P.; et al. The influence of two functional genetic variants of GRK5 on tau phosphorylation and their association with Alzheimer's disease risk. Oncotarget 2017, 8, 72714–72726. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Bohn, L.M.; Walker, J.K.; Laporte, S.A.; Macrae, A.D.; Caron, M.G.; Lefkowitz, R.J.; Premont, R.T. Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron 1999, 24, 1029–1036. [Google Scholar] [CrossRef]
- Jaber, M.; Koch, W.J.; Rockman, H.; Smith, B.; Bond, R.A.; Sulik, K.K.; Ross, J., Jr.; Lefkowitz, R.J.; Caron, M.G.; Giros, B. Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc Natl Acad Sci U S A 1996, 93, 12974–12979. [Google Scholar] [CrossRef] [PubMed]
- Peppel, K.; Boekhoff, I.; McDonald, P.; Breer, H.; Caron, M.G.; Lefkowitz, R.J. G protein-coupled receptor kinase 3 (GRK3) gene disruption leads to loss of odorant receptor desensitization. J Biol Chem 1997, 272, 25425–25428. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Bohn, L.M.; Sotnikova, T.D.; Cyr, M.; Laakso, A.; Macrae, A.D.; Torres, G.E.; Kim, K.M.; Lefkowitz, R.J.; Caron, M.G.; et al. Dopaminergic supersensitivity in g protein-coupled receptor kinase 6-deficient mice. Neuron 2003, 38, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rasul, I.; Sun, Y.; Wu, G.; Li, L.; Premont, R.T.; Suo, W.Z. GRK5 deficiency leads to reduced hippocampal acetylcholine level via impaired presynaptic M2/M4 autoreceptor desensitization. J Biol Chem 2009, 284, 19564–19571. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H. Homocysteine, B Vitamins, and Cognitive Impairment. Annu Rev Nutr 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Murga, C.; Ribas, C.; Tutor, A.S.; Peregrin, S.; Mayor, F., Jr. Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res 2006, 69, 46–56. [Google Scholar] [CrossRef]
- Rockman, H.A.; Choi, D.J.; Rahman, N.U.; Akhter, S.A.; Lefkowitz, R.J.; Koch, W.J. Receptor-specific in vivo desensitization by the G protein-coupled receptor kinase-5 in transgenic mice. Proc Natl Acad Sci U S A 1996, 93, 9954–9959. [Google Scholar] [CrossRef]
- Niu, B.; Liu, P.; Shen, M.; Liu, C.; Wang, L.; Wang, F.; Ma, L. GRK5 Regulates Social Behavior Via Suppression of mTORC1 Signaling in Medial Prefrontal Cortex. Cereb Cortex 2018, 28, 421–432. [Google Scholar] [CrossRef]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol Biol 2019, 1916, 105–111. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nature protocols 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Alamed, J.; Wilcock, D.M.; Diamond, D.M.; Gordon, M.N.; Morgan, D. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nature protocols 2006, 1, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Webber, K.M.; Casadesus, G.; Perry, G.; Atwood, C.S.; Bowen, R.; Smith, M.A. Gender differences in Alzheimer disease: the role of luteinizing hormone in disease pathogenesis. Alzheimer Dis Assoc Disord 2005, 19, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Placanica, L.; Zhu, L.; Li, Y.M. Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PloS one 2009, 4, e5088. [Google Scholar] [CrossRef] [PubMed]
- O'Neal, M.A. Women and the risk of Alzheimer's disease. Front Glob Womens Health 2023, 4, 1324522. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.S.; Jiang, Q.W.; Chen, S.D. Sex difference in biological change and mechanism of Alzheimer's disease: From macro- to micro-landscape. Ageing Res Rev 2023, 87, 101918. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rasul, I.; Liu, J.; Zhao, B.; Tang, R.; Premont, R.T.; Suo, W.Z. Augmented axonal defects and synaptic degenerative changes in female GRK5 deficient mice. Brain Res Bull 2009, 78, 145–151. [Google Scholar] [CrossRef]
- He, M.; Singh, P.; Cheng, S.; Zhang, Q.; Peng, W.; Ding, X.; Li, L.; Liu, J.; Premont, R.T.; Morgan, D.; et al. GRK5 Deficiency Leads to Selective Basal Forebrain Cholinergic Neuronal Vulnerability. Scientific reports 2016, 6, 26116. [Google Scholar] [CrossRef] [PubMed]
- Pepeu, G. Mild cognitive impairment: animal models. Dialogues Clin Neurosci 2004, 6, 369–377. [Google Scholar] [CrossRef]
- Frolkis, V.V.; Tanin, S.A.; Gorban, Y.N. Age-related changes in axonal transport. Exp Gerontol 1997, 32, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Vagnoni, A.; Bullock, S.L. A cAMP/PKA/Kinesin-1 Axis Promotes the Axonal Transport of Mitochondria in Aging Drosophila Neurons. Curr Biol 2018, 28, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Carlyle, B.C.; Nairn, A.C.; Wang, M.; Yang, Y.; Jin, L.E.; Simen, A.A.; Ramos, B.P.; Bordner, K.A.; Craft, G.E.; Davies, P.; et al. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci U S A 2014, 111, 5036–5041. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Li, L.; He, S.; Liu, J.; Sun, Y.; He, M.; Grasing, K.; Premont, R.T.; Suo, W.Z. GRK5 deficiency accelerates beta-amyloid accumulation in Tg2576 mice via impaired cholinergic activity. J Biol Chem 2010, 285, 41541–41548. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, J.; Suo, W.Z. GRK5 deficiency exaggerates inflammatory changes in TgAPPsw mice. J Neuroinflammation 2008, 5, 24. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef]
- Fiscus, R.R.; Yuen, J.P.; Chan, S.L.; Kwong, J.H.; Chew, S.B. Nitric oxide and cyclic GMP as pro- and anti-apoptotic agents. J Card Surg 2002, 17, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Eliseev, R.A.; Vanwinkle, B.; Rosier, R.N.; Gunter, T.E. Diazoxide-mediated preconditioning against apoptosis involves activation of cAMP-response element-binding protein (CREB) and NFkappaB. J Biol Chem 2004, 279, 46748–46754. [Google Scholar] [CrossRef]
- Follin-Arbelet, V.; Torgersen, M.L.; Naderi, E.H.; Misund, K.; Sundan, A.; Blomhoff, H.K. Death of multiple myeloma cells induced by cAMP-signaling involves downregulation of Mcl-1 via the JAK/STAT pathway. Cancer letters 2013, 335, 323–331. [Google Scholar] [CrossRef]
- Zhang, W.; Basile, A.S.; Gomeza, J.; Volpicelli, L.A.; Levey, A.I.; Wess, J. Characterization of central inhibitory muscarinic autoreceptors by the use of muscarinic acetylcholine receptor knock-out mice. J Neurosci 2002, 22, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Decossas, M.; Doudnikoff, E.; Bloch, B.; Bernard, V. Aging and subcellular localization of m2 muscarinic autoreceptor in basalocortical neurons in vivo. Neurobiol Aging 2005, 26, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Parnas, H.; Slutsky, I.; Rashkovan, G.; Silman, I.; Wess, J.; Parnas, I. Depolarization initiates phasic acetylcholine release by relief of a tonic block imposed by presynaptic M2 muscarinic receptors. Journal of neurophysiology 2005, 93, 3257–3269. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z. Muscarinic m2 receptor blockade to delay or prevent onset of progressive memory decline. USPTO 2023, US11801241B11801242. [Google Scholar]
- Bartus, R.T.; Dean, R.L., 3rd; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; Esposito, Z.; Koch, G. Beyond the cholinergic hypothesis: do current drugs work in Alzheimer's disease? CNS neuroscience & therapeutics 2010, 16, 235–245. [Google Scholar]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer's disease. Signal Transduct Target Ther 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Beschea Chiriac, S.I.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer's Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, G.T. Cholinesterase inhibitors for the treatment of Alzheimer's disease:: getting on and staying on. Curr Ther Res Clin Exp 2003, 64, 216–235. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer's disease drug development pipeline: 2019. Alzheimers Dement (N Y) 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Buccellato, F.R.; D'Anca, M.; Tartaglia, G.M.; Del Fabbro, M.; Scarpini, E.; Galimberti, D. Treatment of Alzheimer's Disease: Beyond Symptomatic Therapies. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Peng, W.; Zhang, Q.; Ding, X.; Suo, W.Z. GRK5 deficiency leads to susceptibility to intermittent hypoxia-induced cognitive impairment. Behav Brain Res 2016, 302, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, N.; O'Regan, J.; Ruthirakuhan, M.; Kiss, A.; Eryavec, G.; Williams, E.; Lanctot, K.L. A Randomized Placebo-Controlled Discontinuation Study of Cholinesterase Inhibitors in Institutionalized Patients With Moderate to Severe Alzheimer Disease. J Am Med Dir Assoc 2016, 17, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Bidzan, L.; Bidzan, M. Withdrawal syndrome after donepezil cessation in a patient with dementia. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 2012, 33, 1459–1461. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dudley, C. Discontinuation syndrome following donepezil cessation. Int J Geriatr Psychiatry 2003, 18, 282–284. [Google Scholar] [CrossRef]
- Parsons, C.; Lim, W.Y.; Loy, C.; McGuinness, B.; Passmore, P.; Ward, S.A.; Hughes, C. Withdrawal or continuation of cholinesterase inhibitors or memantine or both, in people with dementia. The Cochrane database of systematic reviews 2021, 2, CD009081. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration of Age-dependent changes in GRK5KO mice. The age-dependent changes were illustrated as cholinergic hypofunction and impaired social novelty recognition without pathology at 3 months, impaired alertness/fear and mild axonopathy at 12 months, and amnesia and moderate axonopathy at 18 months in GRK5KO mice. If the trend continues, the age-dependent changes may further develop in more advanced ages.
Figure 1.
Schematic illustration of Age-dependent changes in GRK5KO mice. The age-dependent changes were illustrated as cholinergic hypofunction and impaired social novelty recognition without pathology at 3 months, impaired alertness/fear and mild axonopathy at 12 months, and amnesia and moderate axonopathy at 18 months in GRK5KO mice. If the trend continues, the age-dependent changes may further develop in more advanced ages.
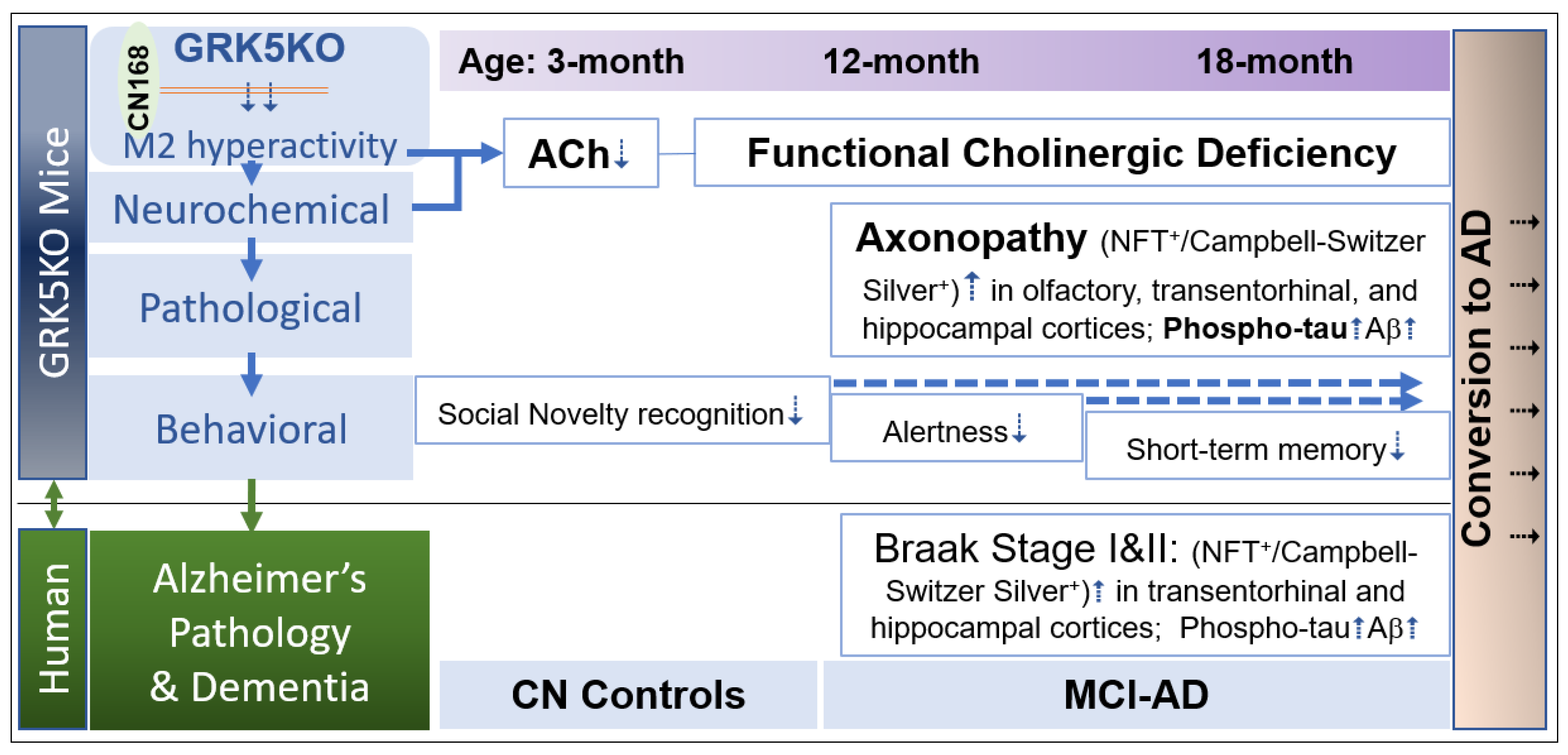

Table 1.
Comparisons between the therapeutic strategies using CN168 versus ChEIs.
| Treatment | CN168 | ChEIs |
|---|---|---|
| Target system | Central cholinergic system | Central cholinergic system |
| Target position | Presynaptic | Postsynaptic |
| Target molecule | M2 autoreceptor | ACh |
| Direct effect | M2 inhibition | Cholinesterase inhibition |
| Acute impacts in the presence of the drug | Increased ACh release and revoked suppression of cAMP/PKA/CREB signaling | Delayed ACh degradation, prolonged ACh effects on all cholinergic receptors including M2 autoreceptor |
| Potential impacts on the cholinergic neurons | Strengthened cholinergic neuronal resilience and survival | Weakened cholinergic neuronal resilience potentially by enhancing the M2 hyperactivity |
| Impacts after the drug withdrawal | Cognitively normal | Potentially worsened symptoms |
| Disease modifying | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



