
Submitted:
02 April 2024
Posted:
03 April 2024
You are already at the latest version
Abstract
Glycosidic linkage analysis was conducted on the unfractionated polysaccharides in the alcohol-insoluble residues (AIRs) prepared from six red seaweeds (Gracilariopsis sp., Prionitis sp., Mastocarpus papillatus, Callophyllis sp., Mazzaella splendens, and Palmaria palmata) using GC-MS/FID analysis of partially methylated alditol acetates (PMAAs). The cell walls of P. palmata primarily contained mixed-linkage xylans and small amounts of sulfated galactans and cellulose. In contrast, the unfractionated polysaccharides of the other five species were rich in galactans displaying diverse 3,6-anhydro-galactose and galactose linkages with varied sulfation patterns. Different levels of cellulose were also observed. This glycosidic linkage method offers advantages for cellulose analysis over traditional monosaccharide analysis that is known for underrepresenting glucose in crystalline cellulose. Relative linkage compositions calculated from GC-MS and GC-FID measurements showed that anhydro sugar linkages generated more response in the latter detection method. This improved linkage workflow presents a useful tool for studying polysaccharide structural variations across red seaweed species. Furthermore, for the first time, relative linkage compositions from GC-MS and GC-FID measurements, along with normalized FID and total ion current (TIC) chromatograms without peak assignments, were analyzed using principal component analysis (PCA) as a proof-of-concept demonstration of the technique's potential to differentiate various red seaweed species.
Keywords:
methylation analysis
; reductive hydrolysis
; whole food fiber
; whole cell wall
; anhydro sugar
; agarose
; carrageenan
; sulfated galactan
; mixed linkage xylan
; principal component analysis
1. Introduction
Red algae (Rhodophyta) is the oldest phylogenetic division of eukaryotic algae, comprising approximately 5,000 species that are primarily marine macrophytes known as red seaweeds [1]. Some red seaweed species have been harvested and used as food by humans for over 2,800 years [2]. Red seaweeds contain various polysaccharides, including, but not limited to, Floridean starch, cellulose, xylan, neutral mannan, sulfated mannan, and sulfated galactan [1,3]. Galactans, the major polysaccharide components of majority of red seaweeds, consist of linear chains composed of alternating 3-linked and 4-linked residues and include three types: carrageenan-type, agar-type (e.g., agarose, porphyrans), and dl hybrids of the two types, differing in the following structural features: presence or absence of sulfate substitution, presence or absence of anhydro sugars, and absolute configurations (d, l) of monosaccharides [4]. Sulfated galactans, such as carrageenans, are bioactive carbohydrates and functional additives with many reported health-enhancing properties [5,6,7,8]. These polysaccharides are mass-produced as stabilizers, emulsifiers, homogenizers, and encapsulating materials widely used in the food, pharmaceutical, and other industries [4,9,10]. Two main types of xylans, both homoxylans, have been identified in red algae: the 1,3-β-d-xylans, found as microfibrils in the cell walls of Porphyra umbilicalis (Bangiales), and the mixed-linkage 1,3;1,4-β-d-xylans (MLX), present in the matrix phase of cell walls in the orders Nemaliales and Palmariales [3]. Given the diversity of natural structures and variations in abundance of polysaccharides in red seaweeds, analyzing the chemical composition of these polysaccharides is crucial for understanding their functions and for their better application.
GC-MS/FID-based linkage analysis is a fundamental tool in providing convincing evidences of the types and compositions of monosaccharide linkages in polysaccharides, oligosaccharides, and biomacromolecules that contain carbohydrate moieties [11,12,13,14,15,16,17]. This process involves permethylating the free hydroxyl groups in polysaccharides, followed by hydrolysis to release partially methylated monosaccharides, reduction to C-1 deuterium-labeled alditols via sodium borodeuteride (NaBD4) treatment, then peracetylation to yield partially methylated alditol acetate (PMAA) derivatives for GC-MS/FID analysis [13,18,19]. Conducting linkage analysis on unfractionated cell walls is crucial for understanding the relative compositions of all polysaccharides within the samples, as this approach eliminates the need for fractionation and the potential loss of specific polysaccharide fractions [20]. The procedure has been frequently used for analysis of unfractionated polysaccharides in the form of alcohol-insoluble residues (AIRs) prepared from higher plants and fungi, whose cell walls mainly consist of normal neutral monosaccharide, uronic acid, and amino sugar linkages [21,22,23,24,25,26,27]. However, the procedure cannot be directly applied to the linkage analysis of unfractionated polysaccharides of red seaweeds due to the presence of sulfated and/or anhydro sugar linkages in galactans. This necessitates special treatments to prevent the under-methylation of sulfated galactans due to their poor solubility in DMSO in their inorganic salt forms, the loss of methylated sulfated galactans during product cleanup, or the degradation of 3,6-Anhydro-galactose (AnGal) during acid hydrolysis [19], as detailed in Section 2.1.
This study was aimed to improve the conventional methylation-GC-MS/FID method to enable the reliable determination of glycosidic linkage compositions of unfractionated polysaccharides in six red seaweed species of commercial interests.
2. Results and Discussion
2.1. Adaptation of the Method for Unfracctioned Polysaccharides of Red Seaweed
2.1.1. Permethylation
Sulfated polysaccharides in their inorganic salt forms are poorly dissolved in DMSO and need to be cation-exchanged to their triethylammonium (TEA) salt form for gaining solubility in DMSO. This process is achievable through either dialysis against deionized water solution of triethylammonium chloride (TEAC) or passing cation exchange column (TEA salt form). The former method was found superior to the latter for sulfated galactans [15,19]. However, the dialysis method is not feasible for unfractionated polysaccharides of red seaweeds because, during dialysis, galactans dissolve in water and separate from water-insoluble polysaccharides (e.g., cellulose). This results in the final freeze-dried samples having polysaccharides with varying water solubilities non-homogeneously distributed, making it impossible to aliquot the dialyzed samples for subsequent methylation. Consequently, all samples recovered from the same dialysis tubing must be permethylated together, with the small sample amount constrained by the permethylation process’s capacity. Dialyzing small amounts of samples in separate dialysis tubing increases sample loss and is more inconvenient, labor-intensive, costly, and time-consuming than dialyzing relatively large amounts and then aliquoting for linkage analysis, a common practice for whole cell wall analysis of higher plants [13]. Furthermore, the inability to aliquot effectively and the requirement to use small amounts of starting material in each reaction tube make it difficult to analyze anhydro sugar linkages, which requires aliquoting one portion of the same permethylated sample for TFA hydrolysis and retaining other identical samples from the same tube for reductive hydrolysis. Additionally, there is no evidence to disprove that sulfated polysaccharides may be trapped or encapsulated by insoluble cell wall residues, hindering their dissolution in water and full conversion to TEA salt form during dialysis prior to methylation.
Unfractionated polysaccharides, containing crystalline polysaccharides such as cellulose, need to be well suspended in DMSO for a successful methylation reaction. This is usually achieved by magnetically stirring the sample in heated DMSO (e.g., 60°C) [13,28]. It was observed in this study that finely ball-milled dry AIR powders formed a virtually homogeneous suspension in DMSO at 60 °C with magnetic stirring overnight, in contrast to the observation that the powders of sulfated polysaccharide standards (listed in Section 3.1.3) remained intact without disassembly or disintegration when subjected to the same treatment in DMSO. To the best of our knowledge, this observed difference has not been previously reported. The following mechanisms are proposed for future studies to examine. In dry AIR powder, sulfated galactans are associated with other polysaccharides to form cell wall and extracellular matrix structures [29]. Heated DMSO dissolves non-sulfated polysaccharides from the powder, while sulfated galactans remain in the powder due to their poor solubility in DMSO in their inorganic salt forms. The loss of the non-sulfated polysaccharides weakens the structure and integrity of the powder, causing its breaking into many tiny particles by the physical shearing force of magnetic stirring in heated DMSO. These particles are very small and not easily visible to the naked eye, which results in the appearance of a virtually homogeneous suspension. Additionally, this removal of non-sulfated polysaccharides by DMSO may result in the small pieces from disassembled powder being very porous (with the empty space previously occupied by the non-sulfated polysaccharides), and the porous particles have a relatively large surface area in DMSO, facilitating contact with reactants for methylation. In contrast, powders of purified sulfated polysaccharides, in their inorganic salt forms, form a complex resistant to DMSO extraction due to their poor solubility in DMSO. Consequently, the powder remains unaltered by magnetic stirring, staying visibly intact in DMSO.
Satisfactory methylation was obtained for crystalline cellulose even with only one round of methylation (Figure S1). Following this first methylation, the sample, with sulfated galatans partially methylated, underwent TEAC dialysis during product cleanup, as detailed in Section 3.3.1. An overnight magnetic stirring in DMSO at room temperature then allowed the complete dissolution of the partially methylated galactans (in TEA salt form) and the other polysaccharides for the second round of methylation. During the cleanup step after the second methylation, the product was not subjected to TEAC dialysis. As expected, the methylated sulfated galactans, despite being in their inorganic salt forms, easily dissolved in DMSO with magnetic stirring at room temperature overnight. Apparently, the presence of methyl groups enhanced their solubility in DMSO. An additional round of methylation were conducted to ensure the complete methylation of free hydroxyl groups in the polysaccharides, with the TEAC dialysis step omitted as well. This method was applied to permethylate ~10 mg of polysaccharides in a single glass reaction tube for analytical purposes. The fully methylated polysaccharides dissolved easily in methanol, making it very convenient to aliquot the resulting solution into multiple glass tubes for further experiments.
It is critical to remove methyl iodide during the product cleanup after each methylation to avoid related side reactions during the subsequent acid hydrolysis, reduction, and peracetylation, and most importantly, for the protection of the analysts, as methyl iodide is highly toxic [30]. Stevenson and Furneaux (1991) added water into the permethylated reaction product and then bubbled N2 through the mixture until the visual disappearance of cloudiness caused by the presence of methyl iodide, followed by dialysis of the mixture [19]. The same N2 sparging method was also used to remove methyl iodide from permethylated heparan disaccharide before cleanup with a C18 SPE cartridge [31]. In another report on cleaning up permethylated fucoidan, partitioning between chloroform and acidified deionized water was conducted, followed by the evaporation of the lower chloroform phase containing the methyl iodide to dryness and combining the water phase with the dried lower phase for subsequent dialysis to remove sodium salts and residual DMSO [32]. In the current study, the latter procedure was improved by using dichloromethane (DCM) to replace chloroform for the partitioning, based on the previous report that replacing chloroform with DCM in the extraction of per-O-methylated carbohydrates significantly reduced degradation products to below 0.5%, a reduction that could be further minimized by neutralizing the reaction mixture with acid before partitioning [33]. Furthermore, as detailed in Section 3.3.1, the collected pooled water phases from the multiple partitionings were evaporated to reduce the volume by half under a flow of N2, provided by a generator, to ensure the removal of any minor levels of methyl iodide that may exist in the upper water phase, before pooling the water phase with the dried lower phase for dialysis. It is important to note that both the water phase and the DCM phase from the partitioning need to be collected because permethylated neutral polysaccharides are soluble in DCM and thus stay in the lower phase, while permethylated sulfated galactans are water-soluble and therefore remain in the upper phase.
2.1.2. Depolymerization and Reduction
Permethylated polysaccharides from higher plant cell walls and fungi are typically depolymerized by TFA hydrolysis (e.g., 2 M TFA at 120 °C or 4 M TFA at 100 °C) into monosaccharides that are further reduced by NaBD4 to alditols [13,34]. However, the anhydro sugar AnGal in red seaweed galactans can be destroyed by TFA hydrolysis [19]. Therefore, a reductive hydrolysis process was developed, employing acid-sTable 4-methylmorpholine borane (4-MMB) to reduce the AnGal monosaccharides released by acid hydrolysis to alditols, thus protecting them from degradation [19]. Because 4-MMB was used instead of NaBD4 for the reduction, the resulting alditols were not C-1 deuterium labeled. Consequently, alongside the reductive hydrolysis, it is necessary to perform standard TFA hydrolysis on another aliquot of the same sample, followed by NaBD4 reduction to generate C-1 deuterium-label alditols for non-anhydro sugars, as detailed in Section 3.3.2. This facilitates peak identification based on the electron impact mass spectrometry (EI-MS) ion fragmentation patterns of non-anhydro sugar linkages in unfractionated polysaccharides of red seaweeds. Examples are showed in Figure 1.
Conventional GC-based monosaccharide analysis tends to underestimate crystalline cellulose in unfractionated cell walls due to difficulties in the initial acid hydrolysis stage in quantitatively releasing monosaccharides from the crystalline structure. Crystalline cellulose is resistant to TFA hydrolysis, necessitating the use of stronger acids, such as sulfuric acid, for its effective decrystallization and depolymerization to glucose [20]. Despite the availability of microscale monosaccharide analysis techniques that involve neutralizing and removing sulfuric acid with dioctylamine in chloroform [13], and the Blackeney method for macroscale monosaccharide analysis [35], sulfuric acid hydrolysis remains less convenient and can result in less clean GC chromatograms compared to TFA hydrolysis. Furthermore, the more destructive nature of sulfuric acid compared to TFA can lead to the degradation of monosaccharides released from cell walls, adversely affecting the quantitative quality of the analysis [36]. However, while native crystalline cellulose is resistant to TFA hydrolysis, permethylation before TFA hydrolysis allows the decrystallization of cellulose, facilitating its depolymerization by TFA and enabling reliable GC-MS/FID analysis of released glucose [37,38]. In the current study, crystalline cellulose from the unfractionated polysaccharides of red seaweeds was decrystallized and methylated using improved Ciucanu procedure [18,28], enabling the quantitative release of partially methylated glucose by 4 M TFA hydrolysis for conversion into PMAAs for GC-based quantitation.
2.1.3. Peracetylation and Final Cleanup
Alkaline acetylation, involving the use of base catalysts like pyridine or sodium acetate, became popular for its ability to avoid artifact formation, a common issue with preliminary acid-catalyzed acetylation (e.g., the use of sulfuric acid) [39,40]. Borate, resulting from the reduction with NaBD4, forms stable borate esters with vicinal diol groups under alkaline conditions, inhibiting the acetylation process and necessitating the removal of remaining borates as volatile trimethyl borates through a tedious and time-consuming process of repeated evaporation to dryness with methanolic acetic acid and then with absolute methanol [13]. Voiges et al. (2012) reported that acidic acetylation conditions, utilizing a TFA and acetic anhydride mixture at a 1:5 ratio (v/v) at 60 °C, not only yielded results comparable to alkaline-catalyzed acetylation with high recovery rates and reproducibility but also significantly simplified the process by eliminating the need to remove borate through repeated co-evaporation in methanolic acetic acid and methanol [41]. Therefore, the TFA-catalyzed acetylation was used in this study. After evaporation of the reaction mixture to dryness under N2, the residual TFA was neutralized and removed by partitioning between saturated NaHCO3 and DCM. The residual NaHCO3 was further removed by additional three rounds of partitioning between deionized water and DCM, as detailed in Section 3.3.3. The DCM phase was then filtered through a glass wool-clogged Pasteur pipette packed with sodium sulfate (Na2SO4) to remove residual water, a method that has been proved effective by previous reports [42,43], before evaporation to dryness and reconstitution in ethyl acetate for GC-MS/FID analysis. Ethyl acetate was chosen over DCM as the solvent for the final PMAAs due to having lower toxicity and being a greener chemistry [44,45].
2.1.4. Identification and Quantitation of PMAAs
A 60-m medium polarity SP-2380 column was used for the efficient separation of the PMAAs with enhanced separation capability compared to a 30-m column of the same type. The identification of PMAAs was performed by comparing their retention times and EI-MS ion fragmentation patterns against PMAA references prepared from polysaccharide standards and methyl glycosides. The quantitation of PMAAs was achieved through the combined use of GC-MS and GC-FID. Using EI-MS, pairs of C-1 deuterium-labeled PMAAs derived from 2-Xylp and 4-Xylp, as well as 2,3,6-Galp and 2,4,6-Galp, can be distinguished despite their structural symmetry making them otherwise indistinguishable chromatographically [13]. Moreover, calculating the molar ratio for each pair from their extracted ion chromatograms enables the splitting of their shared peak area, thereby allowing the acquisition of respective TIC or FID peak areas to quantify each PMAA in the pair separately [25]. Without C-1 deuterium labeling, the EI-MS spectra of PMAAs in each pair became indistinguishable, necessitating conventional TFA hydrolysis-NaBD4 reduction alongside reductive hydrolysis, where deuterium labeling is not applicable, on the whole red seaweed cell wall. It is worth mentioning that there has been a preference for GC-MS-based quantitation in the analysis of PMAAs from unfractionated higher plant whole cell walls [24,26,27]. This method often uses relative peak area or, more commonly, relative molar percentages, assuming a direct proportionality between each PMAA’s quantity and ratio of its TIC peak area to its molecular mass, according to a widely accepted protocol [13]. However, red seaweed whole polysaccharides, with fewer and less varied PMAA peaks compared to their higher plant counterparts, allow easier GC-FID peak assignments using comprehensive PMAA standards, as shown in the GC-FID chromatograms of the six seaweed species (Figures 6-11). FID offers superior quantitation capabilities compared to EI-MS, due to its higher linear dynamic range [46]. The GC-FID-based quantitation benefits from utilizing FID response factors calculated based on effective carbon number concept and verified by Sweet et al. (1975) [47]. These FID response factors were applied to all PMAAs except AnGal linkages in the current study. The FID response factor for 4-AnGalp was experimentally determined to be 0.49 from methylation analysis of an agarose standard, applying a 1:1 molar ratio of 4-AnGalp to 3-Galp, with 3-Galp having a response factor of 0.74 [47]. Based on the response factor of 4-AnGalp, the response factor for 2,4-AnGalp was calculated as 0.54, according to the report that substituting a methyl group at the O-2 position with an acetyl group increases the relative response factor by 0.05 [47]. These values were lower than the response factors of 0.64 for 4-AnGalp and 0.72 for 2,4-AnGalp reported by Stevenson and Furneaux (1991) [19]. The discrepancy might be attributed to their use of the Hakomori methylation method, which is milder than the Ciucanu method and better avoids degradation [33,48,49], such as the oxidation of uronic acids [50], even though the Ciucanu method has been proven essential for the permethylation of crystalline polysaccharides in whole cell walls [13,20,28]. However, the greater degradation of AnGal linkages with the Ciucanu methylation compared to the Hakomori methylation is not a significant technical issue, as the degradation margin can be offset by applying lower response factors based on experimental determination.
Significant differences were observed in the TIC and FID responses to PMAAs from AnGal linkages. This discrepancy is highlighted by the notably smaller relative peak areas of AnGal linkages in the GC-TIC chromatogram than in the GC-FID chromatogram of the identical sample solution from same GC vial. Conversely, the relative peak areas of non-anhydro sugar PMAAs appeared similar across both detections. An example of such difference is demonstrated with PMAAs from agarose and corn fiber xylan standards in Figure 2A,B. The observed discrepancy in the response of AnGlap linkages between EI-MS and FID detection cannot be attributed to chromatographic factors, since identical columns were used for PMAA separation in both methods and the quantitatively normal detection of non-anhydro sugar PMAAs was achieved without any chromatographic issues (Figure 2C,D). This pointed to the difference being detector-related. During the GC-MS experiments, the standard ionization energy of 70 eV for EI ionization was used, suggesting that this ionization energy might be suboptimal for ionizing AnGal PMAAs and could lead to low ionization efficiency. It is recommended to investigate the effects of ionization energy on the recovery rate of AnGal PMAAs and to optimize for the best ionization efficiency in future studies. Notably, a recent study calculated the EI-MS response factors for linkages of 4-AnGalp and 2,4-AnGalp relative to their neutral sugar counterparts, based on experimental linkage analysis of κ-carrageenan and ι-carrageenan standards, thereby enabling the reliable determination of seaweed samples containing these linkages by GC-MS [51].
2.2. Results of Linkage Compositions of Unfractionatoed Polysaccharides in Six Red Seaweeds
The linkage analysis of unfractionated polysaccharides in six red seaweed species revealed a total of 38 unique linkages across all species studied. No rhamnose or fucose linkages were detected in any red seaweed, indicating no detectable contamination from green and brown seaweeds. Relative linkage compositions are presented in Figure 3 and Table 1. The relative monosaccharide compositions, calculated by summing up all the linkage values from each monosaccharide, are presented in Figure 4 and Table S1. The polysaccharide compositions, estimated based on the relative linkage compositions, are presented in Figure 5 and Table S2. As discussed in Section 2.1.4, much lower relative compositions of 2-AnGalp and 2,4-AnGlp were observed using GC-MS than with GC-FID. Therefore, all linkage values presented below in Section 2.2.1 to Section 2.2.6 were from GC-FID quantitation. Linkage results calculated based on GC-MS are included in the supplementary document. GC-FID chromatograms of the PMAAs prepared from the six species are shown separately as Figure 6, Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11.
2.2.1. Palmaria palmata
Unlike most red seaweeds, P. palmata, belonging to the order Palmariales, is notable for its rich content of MLX, making it a targeted species for MLX extraction [52]. The AIR of P. palmata was analyzed to reveal compositions of 18.4% 3-Xylp and 54.4% 4-Xylp (Table 1, Figure 6). Xylose accounted for 79.5% of the overall monosaccharide composition (Figure 4, Table S1). The estimated compositions of the AIR included approximately 72.8% MLX, followed by 11.7% galactans, with minor amounts of cellulose and Floridean starch (Figure 5, Table S2), which was in good agreement with previous report [53], indicating that the cell walls of P. palmata were primarily composed of MLX. Minor levels of 2,4-Xylp (0.6%) and 3,4-Xylp (0.5%) were detected. Notably, a low level of 3,4-Xylp has been previously reported in MLX extracted from P. palmata [54]. The identification of 2,4-linked and 3,4-linked xyloses suggested possible branching or substitutions, supported by previous reports that MLX in P. palmata might form covalent bonds with sulfated and/or phosphorylated xylogalactoproteins [55,56]. The 4-Xylp to 3-Xylp ratio in MLX from P. palmata varies from 2 to 4, influenced by extraction methods and harvest season [1,57,58]. A 4-Xylp to 3-Xylp ratio of 3.0 was found for the MLX of P. palmata (Table 1). Galactose linkages accounted for 11.7% of all linkages, including a minor component of 0.1% AnGal linkages (Figure 4, Table S1). There has been no report on the detailed structural elucidation of galactan fractions isolated from P. palmata, probably due to their low abundance in the species. A modest presence of 4-Glcp linkages (4.6%) was noted (Table 1), in good agreement with previous report of 2% - 7% cellulose content in the cell wall of P. palmata [53]. Moreover, minimal levels of arabinose and mannose linkages were also observed (Figure 3, Table 1).
2.2.2. Gracilariopsis sp.
Agarose, an industrially important galactan consisting of alternating 3-Galp and 4-AnGalp units, is frequently extracted from Gracilariaceae family of red seaweeds [59,60]. As expected, the results showed that the AIR of Gracilariopsis sp. contained abundant 3-Galp (39.5%) and 4-AnGalp (27.1%) (Table 1, Figure 7), corresponding to 54.2% of agarose, assuming all 4-AnGalp was attributed to agarose. It is important to note that the ratio of 3-Galp to 4-AnGalp was calculated to be 1.45:1, which exceeded the 1:1 ratio of 3-Galp to 4-AnGalp in agarose, corresponding to a 12.4% composition difference between them. This difference closely aligned with the combined compositions (10.2%) of 4-Galp and its related sulfated forms (2,4-Galp, 3,4-Galp, 4,6-Galp, 2,4,6-Galp, and 2,3,4,6-Galp), suggesting the presence of various minor sulfated galactan components in addition to the major agarose component. Besides, it could not be ruled out that some of the excess 3-Galp might originate from glycoproteins in this specific species, a hypothesis that requires verification by future studies. The presence of the 4,6-Galp linkage, comprising 3.3% of total linkage and being the third most prevalent galactose linkage in the AIR (Table 1, Figure 3), indicated a 4-Galp unit sulfated at the O-6 position, potentially acting as a biological precursor to the 4-AnGalp linkage [61]. The observation of minor levels of xylose, mannose, and arabinose linkages (Table 1, Figure 3) was in good agreement with the previously reported monosaccharide compositions of this species [59,62,63].
2.2.3. Prionitis sp.
Prionitis sp., belonging to the family Halymeniaceae and order Halymeniales, is known for producing complex sulfated galactans [1]. The linkage analysis revealed a variety of galactan-related linkages with 3-Galp (29.8%) emerging as the predominant linkage, followed by notably high levels of 4-AnGalp (11.6%) and 3,4-Galp (10.8%), substantial quantities of 2,4-Galp (6.0%), 4-Galp (5.3%), 3,6-Galp (4.5%), 3,4,6-Galp (4.3%), and 2,3-Galp (3.5%), and lesser amounts of 4,6-Galp (1.8%), 2,3,4,6-Galp (1.5%), 2,4-AnGalp (1.1%), 2,4,6-Galp (0.6%), and 2,3,6-Galp (0.5%) (Table 1, Figure 8). Given the diversity of galactose linkages, various galactans with distinct disaccharide units might exist. For instance, 3-Galp could link to 4-AnGalp to form the disaccharide unit of β-carrageenan, and could link to 4,6-Galp, a biological precursor of 4-AnGalp, to form the disaccharide unit of γ-carrageenan. The presence of 3,4-Galp and 4-AnGalp suggested the potential exitance of κ-carrageenan disaccharide unit. Additionally, the co-exitance of 3,4-Galp and 4,6-Galp suggested the possible presence of disaccharide unit typical of µ-carrageenan. Besides, among all the seaweeds analyzed, Prionitis sp. contained the highest levels of t-Xylp (1.8%) and t-Galp (0.7%), as shown in Table 1, suggesting its AIR sample had more xylosylation and galactosylation than those of the other species.
2.2.4. Callophyllis sp.
Among all the red seaweeds examined, Callophyllis sp. exhibited the greatest variety of linkages, with no single linkage exceeding relative composition of 16%, highlighting the complexity of the galactans present in the seaweed. The most abundant galactan-related linkages in Callophyllis sp. were 4-AnGalp (15.7%) and 3,6-Galp (14.6%), followed by a moderate amount of 2,3-Galp (7.0%), along with many other less abundant linkages (Table 1, Figure 9). Many combinations of monosaccharides could form the disaccharide repeating units of different carrageenans. The abundant 4-AnGalp (15.7%) and 3,6-AnGalp (14.6%) strongly suggested the disaccharide repeating unit of ω-carrageenan [4]. 3-Galp (5.6%) could link with 4-AnGalp (15.7%) to form the disaccharide unit of β-carrageenan, and with 4,6-Galp (1.8%) to form the disaccharide unit of γ-carrageenan. 3,4-Galp (4.4%) could link with 2,4,6-Galp (2.2%) to form ν-carrageenan, with 2,4-AnGalp (2.4%) to form ι-carrageenan, and with 4,6-Galp (1.8%) to form µ-carrageenan. Additionally, 2,3-Galp (7.0%) could link with 2,4,6-Galp (2.2%) to form the disaccharide unit of λ-carrageenan, and with 2,4-AnGalp (2.4%) to form the disaccharide unit of θ-carrageenan. Notably, a galactan fraction isolated from Callophyllis hombroniana was reported to be θ-carrageenan [64]. It was also reported that galactan fractions isolated from Callophyllis variegata were highly sulfated [64]. The disaccharide structures of the galactans in this genus needs to be further investigated in future studies. Besides, Callophyllis sp. contained higher level of cellulose than any other seaweed examined, as evidenced by the high 4-Glcp composition of 14.9% (Table 1) and the estimated cellulose composition of 12.7% (Figure 5, Table S2). The nondetection of terminal linkages t-Xylp and t-Galp (Table 1) supported that the highly acetylated PMAAs were caused by sulfation rather than substitution with xylose and galactose residues, in good agreement with previous reports on this genus [65,66].
2.2.5. Mastocarpus papillatus
M. papillatus is a seaweed belonging to the Phyllophoraceae family within the Gigartinales order. Seaweeds from the Gigartinales order are known to produce carrageenans [65]. Gametophytes from the Phyllophoraceae family are recognized for producing ι- or κ/ι-hybrid carrageenans [67]. The gametophytes of Mastocarpus stellatus was reported to have a carrageenan content of 37.3% (w/w) dry weight, predominantly from the ‘kappa family’ (κ-, ι-, ν-, μ-carrageenans), with κ- and ι-carrageenans accounting for over 80% of the total carrageenan content [68]. Mastocarpus pacificus was reported to contain mainly κ/ι-hybrid carrageenans and low levels of their biological precursors, μ- and ν- carrageenans [69]. As expected, results showed that M. papillatus had the highest levels of AnGal linkages among all examined seaweeds, with 4-AnGalp and 2,4-AnGalp comprising 21.8% and 12.3% of the total linkages, respectively, combining to a total AnGalp linkage of 34.1% (Table 1, Figure 10). The linkage 3,4-Galp, with a high composition level of 46.2%, represented the highest composition value in Table 1. The significant levels of 3,4-Galp, 4-AnGalp, and 2,4-AnGalp indicated the potential presence of disaccharide unit of 3,4-Galp and 4-AnGalp for κ-carrageenan and a disaccharide unit of 3,4-Galp and 2,4-AnGalp for ι-carrageenan. The higher levels of 4-AnGalp compared to 2,4-AnGalp suggested that the gametophyte produces a higher ratio of κ-carrageenan disaccharide unit than ι-carrageenan disaccharide unit. Additionally, some of the extra 3,4-Galp could link to 4,6-Galp (1.3%) and 2,4,6-Galp (1.0%) to form the disaccharide units of µ- and ν-carrageenans, serving as the biological precursors of κ- and ι-carrageenans. Besides, M. papillatus demonstrated lowest cellulose composition among all the seaweeds analyzed (Figure 5, Table S2).
2.2.6. Mazzaella splendins
M. splendens belongs to the family Gigartinaceae that is known to synthesize κ- and κ/ι-hybrid carrageenans from gametophytic plants and λ-carrageenans from sporophytic plants [1]. Results showed that the AIR of M. splendens contained abundant linkages of 3,4-Galp (19.7%), 2,3-Galp (18.4%), 3-Galp (11.5%), 4-AnGalp (9.7%), 2,4,6-Galp (9.9%), and 2,4-AnGalp (8.0%) (Table 1, Figure 11). The sum of 4-AnGalp (9.7%) and 2,4-AnGalp (8.0%), totaling 17.7%, is close to that of 3,4-Galp (19.7%), and the latter could link with 4-AnGalp to form the disaccharide repeating unit of κ-carrageenan and with 2,4-AnGalp to produce the disaccharide unit of ι-carrageenan, possibly from the gametophytic phase of the seaweed. Additionally, some extra 3,4-Galp could link with 4,6-Galp (1.5%) to form µ-carrageenans, the precursor of κ-carrageenan. The presence of λ-carrageenans was supported by the presence of 2,3-Galp (18.4%) and 2,4,6-Galp (9.9%), indicating part of the seaweed being in its diploid macroscopic phase. Some extra 2,3-Galp could possibly form ξ-carrageenan with 2,4-Galp (1.5%) [70]. Moreover, 4-Galp (3.4%) could potentially link to 4-AnGalp to form β-carrageenan. These results were in good agreement with a previous report that the carrageenan fractions isolated from a closely related species, Mazzaella parksii, showed the presence of various carrageenan structures including κ-, ι-, β-, μ-, ν-, κ/ι-hybrid, and κ/β-hybrid carrageenans [71]. Besides, the species demonstrated a relatively low 4-Glcp composition of 5.7% (Table 1) and a low estimated cellulose composition of 3.3% (Figure 5, Table S2).
2.3. Chemometric Analysis by PCA
Principal component analysis (PCA), the most commonly used chemometric technique [72], aims to reduce data dimensionality, enhance interpretability, and minimize information loss by generating new uncorrelated variables that successively maximize variance [73]. In the current study, PCA was used to analyze four kinds of datasets: relative linkage compositions from GC-MS and GC-FID measurements and normalized GC-TIC and GC-FID chromatograms without any peak assignment, as detailed in Section 3.5. The resulting PCA score plots are depicted in Figure 12.
The PCA score plot from GC-FID-derived relative linkage compositions revealed that the first two principal components (PC1 and PC2) accounted for 28.7% and 23.3% (totaling 52.0%) of the total variances (Figure 12A). The PC1 scores distinctly separated P. palmata from the other five species in the PCA plot, a differentiation likely due to its higher abundance of 3-Xylp and 4-Xylp from MLX compared to the other analyzed species. Although the other five species could not be well separated based on PC1 scores, the PC2 scores facilitated the distinct separation of Callophyllis sp., Prionitis sp., and Gracilariopsis sp., while Mastocarpus papillatus and Mazzaella splendens remained unseparated.
The PCA score plot from GC-MS-derived relative linkage compositions showed that the first two principal components (PC1 and PC2) accounted for 32.3% and 21.0% (totaling 53.5%) of the total variances (Figure 12B). As expected, P. palmata demonstrated high positive PC1 scores due to the dominant MLX linkages, allowing its distinct separation from the others. While the other five species could not be well separated based on PC1 scores, they could be partially differentiated based on PC2 scores. The PC2 scores indicated that Prionitis sp. and Gracilariopsis sp. clustered closely, with Callophyllis sp., Mastocarpus papillatus, and Mazzaella splendens clearly differentiated. Notably, despite Callophyllis sp., Mastocarpus papillatus, and Mazzaella splendens belonging to the same order of Gigartinales, they were well separated from each other due to their distinct linkage compositions of galactans (Table 1, Figure 3, Figure 9, Figure 10 and Figure 11).
The PCA score plot from the normalized GC-FID chromatograms showed that PC1 accounted for 95.6% of the total variances (Figure 12C). Spots representing M. papillatus and P. palmata were distinctly separated from the closely clustered spots of the other four species. M. papillatus and P. palmata had similar PC1 scores but distinct PC2 scores, allowing their clear separation.
The PCA score plot from the normalized GC-TIC chromatograms revealed that PC1 accounted for 89.5% of the total variances (Figure 12D). Spots representing M. papillatus and P. palmata were distinctly separated from the other three species that all clustered together. M. papillatus and P. palmata had similar PC1 scores but distinct PC2 scores, allowing their separation. Notably, the spots for M. papillatus and P. palmata appeared in opposite positions compared to their locations on the PCA score plot from the normalized GC-FID chromatograms.
Significantly larger PC1 scores were observed from the PCA score plots of normalized GC-TIC or GC-FID chromatograms than from relative linkage compositions using both detection methods (Figure 12), suggesting that the former methods required fewer principal components to capture the main variances than the latter methods. The normalized chromatograms allowed the distinct separation of M. papillatus from other species. Calculating relative glycosidic linkage compositions from these chromatograms might result in some information loss specific to M. papillatus, potentially compromising its clear distinction from the others by PCA analysis of calculated linkage compositions.
2.4. Further Discussions and Future Considerations
This method offers advantages over conventional monosaccharide analysis by enabling the detection of crystalline cellulose along with other polysaccharides in the whole cell walls and extracellular matrix of red seaweeds. The estimated cellulose contents, ranging from 1.8% to 12.7%, vary among the six red seaweed species (Figure 5, Table S2), indicating the diversity of primary cell wall structures among the species.
In the current study, various linkages of galactose and anhydrogalactose were detected in each of the red seaweed species. The types and abundances of these galactan-related linkages vary among different species (Table 1, Figure 3). However, linkage analysis is fundamentally an analysis of monosaccharide building units. It provides information about the positions of hydroxyl groups involved in the formation of glycosidic linkages and sulfate substitution. It cannot provide information on the sequence of linkages (e.g., disaccharide structure) because sequence information is lost when polysaccharide chains are depolymerized into monosaccharides during PMAA preparation. We suggest the use of tandem mass spectrometry or nuclear magnetic resonance-based methods to investigate the disaccharide structure of galactans from these species in future studies [20].
Our methodology did not account for naturally occurring methyl groups in red seaweed polysaccharides; however, information on methyl groups can be determined through ethylation analysis [15] or deuteromethylation analysis [25] instead of normal methylation analysis. The quantitation of pyruvate from algal polysaccharides can be conducted by the releasing and subsequent derivatization into its dinitrophenylhydrazone form [74]. Confirmation of position of sulfate groups can be achieved by desulfation followed by linkage analysis [1,15,75]. Uronic acid linkages can be determined by reducing the carboxyl group to their corresponding 6,6’-dideuterio neutral sugars before methylation analysis [13]. The current study did not investigate the uronic acid linkages due to their low natural abundance in red seaweed [1].
To the best of our knowledge, this is the first report of PCA being applied to analyze the relative linkage compositions and normalized TIC or FID chromatograms. Limited by the numbers of red seaweed samples, the PCA in this study serves as a preliminary demonstration of its application to linkage analysis data, acting as a proof of concept. Despite such limitations, M. papillatus and P. palmata can be distinctly separated from other species in the PCA score plot using normalized TIC or FID chromatograms, without the necessity for peak assignment. We recommend employing such PCA procedures in future red seaweed linkage analysis studies with larger sample sizes.
3. Materials and Methods
3.1. Materials and Reagents
3.1.1. Seaweed Materials
Dry whole seaweeds of Gracilariopsis sp., Prionitis sp., Mastocarpus papillatus, Callophyllis sp., Mazzaella splendens, and Palmaria palmata were obtained from Canadian Pacifico Seaweeds Ltd. (British Columbia, Canada) in October, 2020. The dry seaweed was ball-milled into fine powder using a Retsch Mixer Mill MM 400 ball mill system (Haan, Germany). The dry powders were sealed in 50 mL tubes and stored at -20 °C before analysis.
3.1.2. Chemicals, Reagents, and Consumables
Dimethyl sulfoxide (DMSO) (Cat. No. 276855), methyl iodide (MeI) (Cat. No. I8507), sodium hydroxide (NaOH) (Cat. No. S5881), methanol (Cat. No. 322415), trifluoroacetic acid (TFA) (Cat. No. 8.08260), glacial acetic acid (Cat. No. 69509), acetic anhydride (Cat. No. 242845), triethylammonium chloride (TEAC) (Cat. No. 8.21135), 4-methylmorpholine borane (4-MMB) (Cat. No. 262323), dichloromethane (DCM) (Cat. No. 1.00668), sodium bicarbonate (NaHCO3) (Cat. No. S8875), 0.5 M methanolic HCl (Cat. No. 07607), sodium sulfate (Na2SO4) (Cat. No. 8.22286), acetonitrile (Cat. No. 271004), ethyl acetate (Cat. No. 1.10972), dimethylformamide (Cat. No. 319937), acetone (Cat. No. 179973), barium oxide (BaO) (Cat. No. 288497), and barium hydroxide octahydrate (Ba(OH)₂·8H₂O) (Cat. No. 217573) were purchased from Sigma-Aldrich Co LLC (Massachusetts, USA). Spectrum Spectra/Por 1 regenerated cellulose (RC) dialysis tubing with molecular weight cut-off (MWCO) of 6,000 – 8,000 Da (Cat. No. 08-670D) and sodium borodeuteride (NaBD4) with an isotopic purity of 99% (Cat. No. 035102.06) were purchased from Thermo Fisher Scientific Inc. (Massachusetts, USA). Ethanol (Cat. No., P016EAAN) was purchased from Commercial Alcohols Inc. (Ontario, Canada). Deionized water was generated from a Barnstead D0809 Nanopure II system (Thermo Fisher Scientific Inc., Massachusetts, USA). All chemical reactions were conducted in Pyrex® disposable glass tubes (thread 15-415, Cat. No. CLS99447161) sealed with Corning® phenolic caps with PTFE liners (Cat. No. CLS999815), and stirred using BRAND® magnetic stirring bars coated with PTFE (8 mm in length and 3 mm in diameter, Cat. No. Z328936), all purchased from Sigma-Aldrich Co LLC (Massachusetts, USA). N2 from a cylinder was used to fill the headspace of the tube, and an Stuart evaporator (Cole-Parmer, Illinois, USA) supplied with N2 from a generator was used for the evaporation of samples to dryness.
3.1.3. Carbohydrate Standards
Methyl β-d-galactopyranoside (Cat. No. M0285), methyl α-d-glucopyranoside (Cat. No. 66940), methyl α-d-mannopyranoside (Cat. No. 67770), ι-carrageenan (Cat. No. C1138), agarose (Cat. No. A9539), beechwood xylan (Cat. No. X4252,), seaweed carrageenan (Cat. No. C1867), curdlan (Cat. No. C7821), maltose monohydrate (Cat. No. 63418), gum arabic (Cat. No. G9752), gellan gum (Cat. No. G1910), xylan oat spelt (Cat. No. X0627), and laminarin (Cat. No. L9634) were from Sigma-Aldrich Co LLC (Massachusetts, USA). Konjac glucomannan (Cat. No. P-GLCML), wheat arabinoxylan (Cat. No. P-WAXYM), soybean rhamnogalacturonan (Cat. No. P-RHAGN), sugar beet debranched arabinan (Cat. No. P-DBAR), potato galactan (Cat. No. P-GALPOT), ivory nut mannan (Cat. No. P-MANIV), lichenan (Cat. No. P-LICHN), guar galactomannan (Cat. No. P-GGM21), pachyman (Cat. No. P-PACHY), carob galactomannan (Cat. No. P-GALMV), arabinogalactan (Larch Wood) (Cat. No. P-ARGAL), xyloglucan (Cat. No. P-XYGLN), and yeast beta glucan (Cat. No. P-BGYST), arabinoxylan (Cat. No. P-RAXY) were purchased from Megazyme (Wicklow, Ireland). Fucoidan (Cat. No. YF157165), κ-carrageenan (Cat. No. YC30039), λ-carrageenan (Cat. No. YC41782), and psyllium gum (Cat. No. YP58645) were purchased from Biosynth International Inc. (Illinois, USA). α-Lactose (Cat. No. L-204) was from Thermo Fisher Scientific Inc. (Massachusetts, USA). Microcrystalline cellulose was purchased from J.T. Baker (New Jersey, USA).
3.2. Preparation of Unfractionated Polysaccharides of Red Seaweeds
AIR samples were prepared according to the literature [26,76], with modifications according to previous report [77]. Briefly, The sample was soaked in 40 mL of 80% ethanol-deionized water solution (v/v) three times (8 h, 16 h, then 8 h), with the tube sealed with PTFE-lined screw cup and rotated on a tube rotator. After each soaking, the sample was centrifuged at 3,000 × g for 30 min, the supernatant was discarded, and the residue was retained for the next round of treatment. The final residue was then subjected to three rounds of washing with acetone (40 mL), followed by three rounds of washing with methanol (40 mL), with the sample being soaked in the solvent for 20 min on the tube rotator for each round. After each wash, the residue was collected by centrifugation at 3,000 × g for 30 min. The residue obtained after the final wash was vacuum-dried using a Savant SPD131DDA SpeedVac concentrator coupled to a Savant RVT5105 refrigerated vapor trap (Thermo Fisher Scientific Inc., Massachusetts, USA). The dry pellets were then ball-milled into fine powder using the aforementioned ball mill system in Section 3.1.1.
3.3. Preparation of PMAA Derivatives from Dry AIR Powder
3.3.1. Permethylation
Dry ball-milled fine power of AIR (~10 mg) was suspended in 2 mL of DMSO by magnetically stirring at 60 °C in a glass tube sealed with PTFE-lined screw cap, with the headspace filled with N2. After cooling the tube to room temperature, approximately 200 mg of freshly ground powder from NaOH pellets were added, followed by magnetic stirring the mixture for 2 h in the sealed tube with headspace filled with N2. Next, 1.2 mL of MeI was added, and the sample tube was then sealed with PTFE-lined screw cup and covered with aluminum foil before being subjected to magnetic stirring for 3 h [14,25,26]. The tube containing the reaction mixture were then placed on ice, followed by the addition of 3 mL of DCM, partitioning twice with 5 mL of 10% acetic acid in deionized water (v/v), then partitioning with deionized water (5 mL) two times. After each partitioning, the upper aqueous phase was carefully collected and pooled into a new tube. Following the final wash, the lower phase was evaporated to dryness under a continuous gentle flow of N2. The pooled upper phase was evaporated under N2 flow to reduce the volume by half and then was transferred back into the original tube containing the dried lower phase. The resulting mixture was carefully transferred into the RC dialysis tubing with a MWCO of 6,000 – 8,000 Da. The original glass tube was washed twice with 1 mL of ethanol and vortexed with cap on after each addition of ethanol. Each ethanol wash was transferred into the same dialysis tubing for dialysis against running water overnight, followed by 24 h of dialysis against 4 L of 0.1 M TEAC, another dialysis for 24 h against 4 L of deionized water, then freeze-drying. The resulting dry samples, in their TEA salt form, were then methylated and cleaned up as previously described, with two exceptions: overnight magnetic stirring in DMSO was conducted at room temperature (instead of at 60 °C), and the dialysis against TEAC was omitted. It was followed by an additional round of overnight magnetic stirring in DMSO at room temperature, permethylation, partitioning, dialysis against running water then deionized water (without TEAC dialysis), and then freeze-drying, as detailed above. A total of three rounds of methylation were conducted.
3.3.2. Hydrolysis and Reduction
The permethylated polysaccharides were redissolved in 8 mL of methanol by magnetically stirring at room temperature in a glass reaction tube. Subsequently, 2 mL of this solution was transferred from the original tube into each of three glass reaction tubes with PTFE-lined screw caps, resulting in a total of four tubes, each containing 2 mL of the methanol solution. After the methanol solution in each tube was evaporated to dryness under N2, approximately one fourth of the total permethylated polysaccharide, corresponding to 2.5 mg of the starting dry AIR, remained dried at the bottom of each tube. Two of these tubes proceeded to the next steps of the derivatization process, while the remaining two were stored at -20 °C, sealed with PTFE-lined screw caps and with the headspace filled with N2, for potential future reanalysis or additional experiments.
3.3.2.1. TFA Hydrolysis-NaBD4 Reduction
The permethylated polysaccharides dried in the bottom of glass reaction tube were magnetically stirred with 2 mL of 4 M TFA for 4 h at 100 °C in a glycerol bath, with the tube sealed with a PTFE-lined cap and the headspace filled with N2. After that, the tube was cooled to room temperate, followed by evaporation to dryness under a continuous gentle flow of N2 provided by a N2 generator. To each tube, 2 mL of freshly prepared NaBD4 in deionized water (10 mg/mL, w/v) was added. The tube was capped and magnetically stirred overnight at room temperature [26]. Subsequently, glacial acetic acid was added dropwise until the fizzing caused by the release of H2 ceased, before the sample was evaporated to dryness under N2 flow.
3.3.2.2. Reductive Hydrolysis
The reductive hydrolysis was conducted according to the literature [19]. To a tube containing the dried methylated polysaccharides, 0.1 mL of freshly prepared 80 mg/mL 4-MMB deionized water solution and 0.4 mL of 3 M TFA were added. The mixture was magnetically stirred at 80 °C for 30 min in a glycerol bath, with the tube sealed by a PTFE-lined cap and tube headspace filled with N2. After cooling the tube to room temperature, an additional 0.1 mL of the 4-MMB solution and 0.6 mL of 2 M TFA were added, followed by gentle magnetic stirring at 120 °C for 2 h, while maintaining an N2 headspace. The tube was then cooled to room temperature again, 0.2 mL of the 4-MMB solution was added, and the sample was immediately subjected to evaporation to dryness under a gentle flow of N2 at 50 °C. After that, 1 mL of acetonitrile was added to each tube, and the sample was evaporated to dryness under N2 without heating.
3.3.3. Peracetylation and Final Cleanup
Peracetylation was performed according to the literature [15,41]. To each tube containing the dry partially methylated alditol sample, 0.25 mL of TFA was added followed by the addition of 1.25 mL of acetic anhydride. The mixture was magnetically stirred at 60 °C for 1 h, with the tube sealed by a PTFE-lined cap and tube headspace filled with N2. After cooling to room temperature, the sample was evaporated to dryness. DCM (3 mL) and a saturated deionized water solution of NaHCO3 (3 mL) were added to the tube, followed by vigorous magnetic stirring with cap loosely attached until the generation of bubbles ceased. After that, the aqueous phase was discarded, and 3 mL of saturated NaHCO3 solution was added. The sample was magnetically stirred for 10 min with cap loosely on, after which the aqueous phase was discarded, 3 mL of deionized water was added, and the tube was sealed with a PTFE-lined screw cap for an additional 20 min of rotation on the tube rotator. The partitioning with deionized water was repeated two more times to ensure the removal of residual NaHCO₃. The final upper phase was discarded, and the lower DCM phase was filtered through a glass wool-clogged Pasteur pipette loaded with anhydrous Na2SO4 powder [42]. The filtrate was evaporated to dryness under N2 flow, redissolved in ethyl acetate, and then transferred to a GC vial for GC-MS/FID analysis.
For each sample, two separate experiments were conducted for Section 3.3.1 to Section 3.3.3.
3.4. Preparation of PMAA Standards
3.4.1. Preparation of PMAA Derivatives from Polysaccharide Standards
Each sulfated polysaccharide standard (~10 mg) was dissolved in 2 mL of deionized water by magnetic stirring at room temperature overnight. The resulting solution was then transferred into dialysis tubing for dialysis against 4 L of 0.1 M TEAC for 24 h, followed by further dialysis with 4 L of deionized water for 24 h, before being freeze-dried. The resulting dry sulfated polysaccharide in its TEA salt form was then dissolved in DMSO by gentle magnetic stirring at room temperature overnight in a sealed glass reaction tube, with the headspace filled with N2. This was followed by permethylation, partitioning, pooling of the concentrated upper phases into the dried lower phase, dialysis of the mixture against running water overnight followed by dialysis against 4 L of deionized water for 24 h, then freeze-drying, as detailed in Section 3.3.1. No additional round of methylation was conducted. Each neutral polysaccharide standard was suspended in DMSO by magnetic stirring at 60 °C overnight, followed by permethylation as described in Section 3.3.1, except that during partitioning, the upper water phases were discarded, and the methylated sample was collected by evaporating the lower DCM phase to dryness, with no need for dialysis or any additional round of methylation. RG-I underwent weak methanolysis (0.5 M methanolic HCl, 80 °C, 20 min) followed by NaBD4 reduction [78,79,80], prior to the aforementioned methylation analysis of neutral polysaccharides.
All the permethylated samples were then converted to their PMAAs by 4 M TFA hydrolysis, NaBD₄ reduction, and peracetylation, as described in Section 3.3.2.1 and Section 3.3.3. For carrageenans and agarose that contain AnGal, an aliquot of the sample was also subjected to reductive hydrolysis, then peracetylation, to generate PMAAs, as described in Section 3.3.2.2 and Section 3.3.3.
3.4.2. Preparation of PMAA Derivatives from Methyl Glycosides
Preparation of PMAA standards based on methyl glycosides was conducted according to the literature [81], with modifications. Each methyl glycoside (~50 mg) was dissolved in DMF (2 mL) in a sealed glass reaction tube by magnetic stirring. Subsequently, BaO (200 mg), Ba(OH)₂·8H₂O (10 mg), and MeI (1 mL) were added to the tube, which was then sealed with a PTFE-lined cap. The tube was covered with aluminum foil to create a dark environment. The mixture was magnetically stirred, and aliquots of 250 μL of the reaction mixture were taken at 15, 30, 45, 60, 90, and 120 min following the start of the reaction. Each aliquot was poured into 5 mL of methanol, followed by evaporation to dryness under a gentle N2 flow at room temperature. After that, the sample underwent peracetylation then was cleaned up by partitioning and filtering through anhydrous Na2SO4 column as described in Section 3.3.3. The purified peracetylated methyl glycoside was then subjected to 4 M TFA hydrolysis, NaBD₄ reduction, and peracetylation for generating PMAAs, as detailed in Section 3.3, except that the 4 M TFA hydrolysis at 100 °C was conducted for 2 h (instead of 4 h).
3.5. GC-MS and GC-FID Analysis of PMAAs
The PMAA derivatives were tested on an Agilent 7890A-5977B GC-MS system (Agilent Technologies, California, USA) installed with a medium-polarity Supelco SP-2380 column (60 m × 0.25 mm × 0.2 µm; Sigma-Aldrich, Massachusetts, USA) with oven temperature programmed to start at 55 °C, followed by increasing at 20 °C/min to 170 °C, at 0.3 °C/min to 190 °C, then at 3 °C/min to 215 °C (hold 20 min). A solvent delay of 12 min was used. The same sample was also tested on an Agilent 7890A GC-FID system (Agilent Technologies, California, USA), equipped with another identical Supelco SP-2380 column, using the same oven program as the GC-MS run. Both GC-FID and GC-MS runs had the same settings of inlet temperature (250 °C) and constant column helium flow of 0.8 mL/min. Splitless injection and split injection (10:1 ratio) were used for the GC-FID and GC-MS, respectively. The ion source temperature was maintained at 230 °C, and an ionization energy of 70 eV was used for electron impact ionization. The quadrupole temperature was set at 150 °C, and the transfer line temperature was set at 280 °C. The mass spectrometer was operated in full scan mode, covering a mass range of 45-350 m/z, with a scan speed of 4.59 scans/s. The FID detector temperature was set to 300 °C, with a dry air flow of 400 mL/min, H2 flow of 30 mL/min, and a make-up N2 flow of 25 mL/min. Data acquisition, peak assignment, and integration were performed through Agilent OpenLab CDS software (Agilent Technologies, California, USA). Identification of the PMAAs was based on the comparison of retention times and EI-MS spectra of the PMAAs with those of reference derivatives generated from carbohydrate standards, and by referring to the literature [82]. Two methods for quantifying PMAAs were compared: one based on the GC-TIC chromatogram, and the other on the GC-FID chromatogram. Quantitation using the TIC chromatogram involved calculating the relative molar composition by dividing the peak area of each PMAA by its molecular mass, following the published protocol [13]. Quantitative analysis via GC-FID chromatography relied on FID response factors derived from the concept of effective carbon number, as detailed in previous study [47], except AnGal linkages. For the 4-AnGalp linkage, the FID response factor was determined experimentally to be 0.49, based on methylation analysis using an agarose standard with a 1:1 molar ratio between 4-AnGalp and 3-Galp, the latter having a response factor of 0.74 [47]. From the established response factor for 4-AnGalp, the response factor for 2,4-AnGalp linkage was calculated to be 0.54, according to the principle that an acetyl group substitution at the O-2 position, in place of a methyl group, results in a 0.05 increase in the relative response factor [47]. Relative polysaccharide compositions were estimated from the relative linkage compositions by assigning glycosidic linkages to corresponding polysaccharide structures and then summing up all the linkage values grouped under each polysaccharide structure [13]. The content of Floridean starch was estimated based on the relative composition of 4,6-Glcp, according to the average degree of branching of 4.8 [83]. The detailed assignment of linkages to relevant polysaccharides is shown in Table S3. All figures related to the linkage analysis results were produced using GraphPad Prism version 8.0.2 (GraphPad Software Inc., California, USA), except that the visually accessible bubble plot depicting the relative linkage composition was generated using R-Studio (Posit PBC, Massachusetts, USA) with the ggplot2 [84] and reshape2 [85] packages. Complete R codes for making the figures are included the supplementary document.
3.6. PCA Analysis
PCA analysis was conducted based on four different types of data: normalized GC-TIC chromatograms of PMAAs, normalized GC-FID chromatograms of PMAAs, relative linkage compositions calculated from GC-MS measurement, and relative linkage compositions calculated from GC-FID measurement. The linkage composition data were already normalized, while the collected raw GC-FID and GC-TIC required normalization before proceeding with PCA analysis. For each raw chromatogram of GC-TIC or GC-FID, signals collected within the range of retention times from 25 to 110 min were normalized in two aspects. First, the absolute retention time (min) for each time point was converted to relative retention time by dividing by the absolute retention time at the highest point of the 4-Glcp peak. Similarly, the absolute abundance reading for each time point was converted to relative abundance by dividing by the absolute abundance at the highest point of the 4-Glcp peak. Each data file was transferred onto an Excel sheet for PCA analysis using R-Studio packages dplyr [86], forcats [87], and ggplot2 [84]. with the script incorporating Z-score standardization to scale the datasets into a standard normal distribution prior to the PCA analysis. Complete R codes for the analysis are included the supplementary document.
4. Conclusions
In conclusion, this improved linkage method has proven to be a useful tool for studying polysaccharide structural variations across red seaweed species. The unfractionated polysaccharides of Palmaria palmata predominantly comprise mixed-linkage xylans, supplemented by smaller quantities of sulfated galactans and cellulose. In contrast, the polysaccharides of the other five species are rich in sulfated galactans, with a variety of linkages in 3,6-anhydro-galactose and galactose with varied sulfation patterns. The study also noted varying cellulose levels among species, highlighting the superiority of the method in cellulose analysis over conventional method of GC-based monosaccharide analysis, which typically underestimates glucose in crystalline cellulose. Furthermore, an enhanced response in anhydro sugar linkages was observed from FID detection compared to EI-MS detection. For the first time, the application of PCA to these linkage composition values and normalized GC-TIC and GC-FID chromatograms without peak assignments has been explored, as proof-of-concept demonstration of the method’s capacity and potential to distinguish various red seaweed species.
Supplementary Materials
The supporting information can be downloaded at: www.mdpi.com/xxx/s1. The supporting documents include supplemental tables for linkage analysis (Table S1-S3), supplemental GC-TIC and GC-FID chromatograms of PMAAs from carbohydrate standards (Figure S1-S11), supplemental EI-MS spectra and ion fragmentation patterns (Figure S12 to S15), supplemental bubble plot of GC-MS-derived linkage compositions (Figure S16), and R Script codes for bubble plot and PCA plot.
Author Contributions
Conceptualization, B.B., X.X., and D.W.A.; methodology, B.B., X.X., and D.W.A.; software, B.B.; validation, B.B., X.X., S.A.T., R.J.G., and D.W.A.; formal analysis, B.B. and X.X.; investigation, B.B. and X.X.; resources, D.W.A.; data curation, B.B.; writing—original draft preparation, B.B. and X.X.; writing—review and editing, D.W.A., S.A.T., and R.J.G.; visualization, B.B. and X.X.; supervision, D.W.A.; project administration, D.W.A.; funding acquisition, D.W.A.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Agriculture and Agri-Food Canada, grant numbers J-002262 and J-003135. In addition, this work was supported by the Canadian government as part of the FACCE-ERA-GAS consortium.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available on request.
Acknowledgments
We thank Canadian Pacifico Seaweeds for providing the red seaweeds.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Usov, A.I. , Polysaccharides of the red algae. In Advances in Carbohydrate Chemistry and Biochemistry, Horton, D., Ed. Academic Press: 2011; Vol. 65, pp 115-217.
- Cunha, L.; Grenha, A. , Sulfated seaweed polysaccharides as multifunctional materials in drug delivery applications. Marine Drugs 2016, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.S.Y.; Harris, P.J. , Xylans of red and green algae: What is known about their structures and how they are synthesised? Polymers 2019, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Ciancia, M.; Matulewicz, M.C.; Tuvikene, R. , Structural diversity in galactans from red seaweeds and its influence on rheological properties. Frontiers in Plant Science 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Tan, H.; Nie, S. , Beneficial effects of seaweed-derived dietary fiber: Highlights of the sulfated polysaccharides. Food Chemistry 2022, 373, 131608. [Google Scholar] [CrossRef] [PubMed]
- McKim, J.M. , Food additive carrageenan: Part I: A critical review of carrageenan in vitro studies, potential pitfalls, and implications for human health and safety. Critical Reviews in Toxicology 2014, 44, 211–243. [Google Scholar] [CrossRef] [PubMed]
- Pangestuti, R.; Kim, S.-K. , Biological activities of carrageenan. In Advances in Food and Nutrition Research, Kim, S.-K., Ed. Academic Press: 2014; Vol. 72, pp 113-124.
- Weiner, M.L. , Food additive carrageenan: Part II: A critical review of carrageenan in vivo safety studies. Critical Reviews in Toxicology 2014, 44, 244–269. [Google Scholar] [CrossRef]
- Renn, D. , Biotechnology and the red seaweed polysaccharide industry: Status, needs and prospects. Trends in Biotechnology 1997, 15, 9–14. [Google Scholar] [CrossRef]
- Li, L.; Ni, R.; Shao, Y.; Mao, S. , Carrageenan and its applications in drug delivery. Carbohydrate Polymers 2014, 103, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-C.; Kao, M.-R.; Saldivar, R.K.; Díaz-Moreno, S.M.; Xing, X.; Furlanetto, V.; Yayo, J.; Divne, C.; Vilaplana, F.; Abbott, D.W.; Hsieh, Y.S.Y. , The Gram-positive bacterium Romboutsia ilealis harbors a polysaccharide synthase that can produce (1,3;1,4)-β-d-glucans. Nature Communications 2023, 14, 4526. [Google Scholar] [CrossRef]
- Jæger, D.; Ndi, C.P.; Crocoll, C.; Simpson, B.S.; Khakimov, B.; Guzman-Genuino, R.M.; Hayball, J.D.; Xing, X.; Bulone, V.; Weinstein, P.; Møller, B.L.; Semple, S.J. , Isolation and structural characterization of echinocystic acid triterpenoid saponins from the Australian medicinal and food plant Acacia ligulata. Journal of Natural Products 2017, 80, 2692–2698. [Google Scholar] [CrossRef]
- Pettolino, F.A.; Walsh, C.; Fincher, G.B.; Bacic, A. , Determining the polysaccharide composition of plant cell walls. Nature Protocols 2012, 7, 1590–1607. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Xing, X.; Tingley, J.P.; Klassen, L.; King, M.L.; Alexander, T.W.; Abbott, D.W. , Analysis of active site architecture and reaction product linkage chemistry reveals a conserved cleavage substrate for an endo-alpha-mannanase within diverse yeast mannans. Journal of Molecular Biology 2020, 432, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Robb, C.S.; Hobbs, J.K.; Pluvinage, B.; Reintjes, G.; Klassen, L.; Monteith, S.; Giljan, G.; Amundsen, C.; Vickers, C.; Hettle, A.G.; Hills, R.; Nitin; Xing, X. ; Montina, T.; Zandberg, W.F.; Abbott, D.W.; Boraston, A.B., Metabolism of a hybrid algal galactan by members of the human gut microbiome. Nature Chemical Biology 2022, 18, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Lahnstein, J.; Hsieh, Y.S.Y.; Xing, X.; Yap, K.; Chaves, A.M.; Scavuzzo-Duggan, T.R.; Dimitroff, G.; Lonsdale, A.; Roberts, E.; Bulone, V.; Fincher, G.B.; Doblin, M.S.; Bacic, A.; Burton, R.A. , Functional characterization of a glycosyltransferase from the moss Physcomitrella patens involved in the biosynthesis of a novel cell wall arabinoglucan. The Plant Cell 2018, 30, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Little, A.; Lahnstein, J.; Jeffery, D.W.; Khor, S.F.; Schwerdt, J.G.; Shirley, N.J.; Hooi, M.; Xing, X.; Burton, R.A.; Bulone, V. , A novel (1,4)-β-Linked glucoxylan is synthesized by members of the cellulose synthase-like F gene family in land plants. ACS Central Science 2019, 5, 73–84. [Google Scholar] [CrossRef]
- Ciucanu, I.; Kerek, F. , A simple and rapid method for the permethylation of carbohydrates. Carbohydrate Research 1984, 131, 209–217. [Google Scholar] [CrossRef]
- Stevenson, T.T.; Furneaux, R.H. , Chemical methods for the analysis of sulphated galactans from red algae. Carbohydr Res 1991, 210, 277–98. [Google Scholar] [CrossRef]
- Tingley, J.P.; Low, K.E.; Xing, X.; Abbott, D.W. , Combined whole cell wall analysis and streamlined in silico carbohydrate-active enzyme discovery to improve biocatalytic conversion of agricultural crop residues. Biotechnology for Biofuels 2021, 14, 16. [Google Scholar]
- Pham, T.A.T.; Kyriacou, B.A.; Schwerdt, J.G.; Shirley, N.J.; Xing, X.; Bulone, V.; Little, A. , Composition and biosynthetic machinery of the Blumeria graminis f. sp. hordei conidia cell wall. The Cell Surface 2019, 5, 100029. [Google Scholar] [CrossRef]
- Pham, T.A.T.; Schwerdt, J.G.; Shirley, N.J.; Xing, X.; Hsieh, Y.S.Y.; Srivastava, V.; Bulone, V.; Little, A. , Analysis of cell wall synthesis and metabolism during early germination of Blumeria graminis f. sp. hordei conidial cells induced in vitro. The Cell Surface 2019, 5, 100030. [Google Scholar] [CrossRef]
- Wood, J.A.; Tan, H.-T.; Collins, H.M.; Yap, K.; Khor, S.F.; Lim, W.L.; Xing, X.; Bulone, V.; Burton, R.A.; Fincher, G.B.; Tucker, M.R. , Genetic and environmental factors contribute to variation in cell wall composition in mature desi chickpea (Cicer arietinum L.) cotyledons. Plant, Cell & Environment 2018, 41, 2195–2208. [Google Scholar]
- Li, J.; Hsiung, S.-Y.; Kao, M.-R.; Xing, X.; Chang, S.-C.; Wang, D.; Hsieh, P.-Y.; Liang, P.-H.; Zhu, Z.; Cheng, T.-J. R.; Shie, J.-J.; Liou, J.-P.; Abbott, D.W.; Kwon, S.W.; Hsieh, Y.S.Y. , Structural compositions and biological activities of cell wall polysaccharides in the rhizome, stem, and leaf of Polygonatum odoratum (Mill.) Druce. Carbohydrate Research 2022, 521, 108662. [Google Scholar] [CrossRef] [PubMed]
- Badhan, A.; Low, K.E.; Jones, D.R.; Xing, X.; Milani, M.R.M.; Polo, R.O.; Klassen, L.; Venketachalam, S.; Hahn, M.G.; Abbott, D.W.; McAllister, T.A. , Mechanistic insights into the digestion of complex dietary fibre by the rumen microbiota using combinatorial high-resolution glycomics and transcriptomic analyses. Computational and Structural Biotechnology Journal 2022, 20, 148–164. [Google Scholar] [CrossRef]
- Low, K.E.; Xing, X.; Moote, P.E.; Inglis, G.D.; Venketachalam, S.; Hahn, M.G.; King, M.L.; Tétard-Jones, C.Y.; Jones, D.R.; Willats, W.G.T.; Slominski, B.A.; Abbott, D.W. , Combinatorial glycomic analyses to direct CAZyme discovery for the tailored degradation of canola meal non-starch dietary polysaccharides. Microorganisms 2020, 8, 1888. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, D.; Xing, X.; Cheng, T.-J. R.; Liang, P.-H.; Bulone, V.; Park, J.H.; Hsieh, Y.S.Y. , Structural analysis and biological activity of cell wall polysaccharides extracted from Panax ginseng marc. International Journal of Biological Macromolecules 2019, 135, 29–37. [Google Scholar] [CrossRef]
- Needs, P.W.; Selvendran, R.R. , An improved methylation procedure for the analysis of complex polysaccharides including resistant starch and a critique of the factors which lead to undermethylation. Phytochemical Analysis 1993, 4, 210–216. [Google Scholar] [CrossRef]
- Synytsya, A.; Čopíková, J.; Kim, W.J.; Park, Y.I. , Cell wall polysaccharides of marine algae. In Springer Handbook of Marine Biotechnology, Kim, S.-K., Ed. Springer Berlin Heidelberg: Berlin, Heidelberg, 2015; pp 543-590.
- Hermouet, C.; Garnier, R.; Efthymiou, M.-L.; Fournier, P.-E. , Methyl iodide poisoning: Report of two cases. American Journal of Industrial Medicine 1996, 30, 759–764. [Google Scholar] [CrossRef]
- Heiss, C.; Wang, Z.; Azadi, P. , Sodium hydroxide permethylation of heparin disaccharides. Rapid Communications in Mass Spectrometry 2011, 25, 774–778. [Google Scholar] [CrossRef]
- Patankar, M.S.; Oehninger, S.; Barnett, T.; Williams, R.L.; Clark, G.F. , A revised structure for fucoidan may explain some of its biological activities. Journal of Biological Chemistry 1993, 268, 21770–21776. [Google Scholar] [CrossRef]
- Ciucanu, I.; Costello, C.E. , Elimination of oxidative degradation during the per-O-methylation of carbohydrates. Journal of the American Chemical Society 2003, 125, 16213–16219. [Google Scholar] [CrossRef]
- Yu, L.; Yakubov, G.E.; Zeng, W.; Xing, X.; Stenson, J.; Bulone, V.; Stokes, J.R. , Multi-layer mucilage of Plantago ovata seeds: Rheological differences arise from variations in arabinoxylan side chains. Carbohydrate Polymers 2017, 165, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Blakeney, A.B.; Harris, P.J.; Henry, R.J.; Stone, B.A. , A simple and rapid preparation of alditol acetates for monosaccharide analysis. Carbohydrate Research 1983, 113, 291–299. [Google Scholar] [CrossRef]
- Emaga, T.; Rabetafika, H.; Blecker, C.; Paquot, M. , Kinetics of the hydrolysis of polysaccharide galacturonic acid and neutral sugars chains from flaxseed mucilage. Biotechnologie Agronomie Société et Environnement 2012, 16, 139–147. [Google Scholar]
- Ndukwe, I.E.; Black, I.; Heiss, C.; Azadi, P. , Evaluating the utility of permethylated polysaccharide solution NMR data for characterization of insoluble plant cell wall polysaccharides. Analytical Chemistry 2020, 92, 13221–13228. [Google Scholar] [CrossRef] [PubMed]
- Black, I.; Heiss, C.; Azadi, P. , Comprehensive monosaccharide composition analysis of insoluble polysaccharides by permethylation to produce methyl alditol derivatives for gas chromatography/mass spectrometry. Analytical Chemistry 2019, 91, 13787–13793. [Google Scholar] [CrossRef] [PubMed]
- Oades, J.M. , Gas-liquid chromatography of alditol acetates and its application to the analysis of sugars in complex hydrolysates. Journal of Chromatography A 1967, 28, 246–252. [Google Scholar] [CrossRef]
- Gunner, S.; Jones, J.; Perry, M. Analysis of sugar mixtures by gas-liquid partition chromatography. Chemistry and Industry 255–256.
- Voiges, K.; Adden, R.; Rinken, M.; Mischnick, P. , Critical re-investigation of the alditol acetate method for analysis of substituent distribution in methyl cellulose. Cellulose 2012, 19, 993–1004. [Google Scholar] [CrossRef]
- Yu, L.; Yakubov, G.E.; Zeng, W.; Xing, X.; Stenson, J.; Bulone, V.; Stokes, J.R. , Multi-layer mucilage of Plantago ovata seeds: Rheological differences arise from variations in arabinoxylan side chains. Carbohydr Polym 2017, 165, 132–141. [Google Scholar] [CrossRef]
- Yang, R.; Li, J.; Jiang, C.; Shi, J. , Preventive and therapeutic effects of an exopolysaccharide produced by Lacticaseibacillus rhamnosus on alcoholic gastric ulcers. International Journal of Biological Macromolecules 2023, 235, 123845. [Google Scholar] [CrossRef]
- Cayot, N.; Lafarge, C.; Bou-Maroun, E.; Cayot, P. , Substitution of carcinogenic solvent dichloromethane for the extraction of volatile compounds in a fat-free model food system. Journal of Chromatography A 2016, 1456, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, B.; van Beek, T.A. , Alternative solvents can make preparative liquid chromatography greener. Green Chemistry 2015, 17, 4073–4081. [Google Scholar] [CrossRef]
- Bartle, K.D.; Myers, P. , History of gas chromatography. TrAC Trends in Analytical Chemistry 2002, 21, 547–557. [Google Scholar] [CrossRef]
- Sweet, D.P.; Shapiro, R.H.; Albersheim, P. , Quantitative analysis by various g.l.c. response-factor theories for partially methylated and partially ethylated alditol acetates. Carbohydrate Research 1975, 40, 217–225. [Google Scholar] [CrossRef]
- Ciucanu, I. , Per-O-methylation reaction for structural analysis of carbohydrates by mass spectrometry. Analytica Chimica Acta 2006, 576, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Jay, A. , The methylation reaction in carbohydrate analysis. Journal of Carbohydrate Chemistry 1996, 15, 897–923. [Google Scholar] [CrossRef]
- Ciucanu, I.; Luca, C. , Avoidance of degradation during the methylation of uronic acids. Carbohydrate Research 1990, 206, 71–77. [Google Scholar] [CrossRef]
- Falshaw, R.; Furneaux, R.H.; Sims, I.M.; Hinkley, S.F.R.; Kidgell, J.T.; Bell, T.J. , Novel 4-O-β-d-xylopyranosyl-3,6-anhydro-l-galactopyranosyl disaccharide units in a polysaccharide from the red alga Pyrophyllon subtumens. Carbohydrate Polymers 2023, 318, 121066. [Google Scholar] [CrossRef] [PubMed]
- Viana, A.G.; Noseda, M.D.; Gonçalves, A.G.; Duarte, M.E.R.; Yokoya, N.; Matulewicz, M.C.; Cerezo, A.S. , β-d-(1→4), β-d-(1→3) ‘mixed linkage’ xylans from red seaweeds of the order Nemaliales and Palmariales. Carbohydrate Research 2011, 346, 1023–1028. [Google Scholar] [CrossRef]
- Lahaye, M.; Michel, C.; Barry, J.L. , Chemical, physicochemical and in-vitro fermentation characteristics of dietary fibres from Palmaria palmata (L.) Kuntze. Food Chemistry 1993, 47, 29–36. [Google Scholar] [CrossRef]
- Turvey, J.R.; Williams, E.L. , The structures of some xylans from red algae. Phytochemistry 1970, 9, 2383–2388. [Google Scholar] [CrossRef]
- Deniaud, E.; Fleurence, J.; Lahaye, M. Preparation and chemical characterization of cell wall fractions enriched in structural proteins from Palmaria palmata (Rhodophyta). 2003, 46, 366–377.
- Deniaud, E.; Quemener, B.; Fleurence, J.; Lahaye, M. Structural studies of the mix-linked β-(1 → 3)/β-(1 → 4)-d-xylans from the cell wall of Palmaria palmata (Rhodophyta). International Journal of Biological Macromolecules 2003, 33, 9–18. [Google Scholar] [CrossRef]
- Painter, T.J. , Algal polysaccharides. In The Polysaccharides, Aspinall, G.O., Ed. Academic Press: 1983; pp 195-285.
- Joubert, Y.; Fleurence, J. , Simultaneous extraction of protein and DNA by an enzymatic treatment of the cell wall of Palmaria palmata (Rhodophyta). Journal of Applied Phycology 2008, 20, 55–61. [Google Scholar] [CrossRef]
- Long, X.; Hu, X.; Liu, S.; Pan, C.; Chen, S.; Li, L.; Qi, B.; Yang, X. , Insights on preparation, structure and activities of Gracilaria lemaneiformis polysaccharide. Food Chem X 2021, 12, 100153. [Google Scholar] [CrossRef]
- Rodríguez Sánchez, R.A.; Canelón, D.J.; Cosenza, V.A.; Fissore, E.N.; Gerschenson, L.N.; Matulewicz, M.C.; Ciancia, M. , Gracilariopsis hommersandii, a red seaweed, source of agar and sulfated polysaccharides with unusual structures. Carbohydrate Polymers 2019, 213, 138–146. [Google Scholar] [CrossRef]
- Rees, D.A. , Enzymic synthesis of 3:6-anhydro-l-galactose within porphyran from l-galactose 6-sulphate units. Biochem J 1961, 81, 347–52. [Google Scholar] [CrossRef]
- Han, R.; Pang, D.; Wen, L.; You, L.; Huang, R.; Kulikouskaya, V. , In vitro digestibility and prebiotic activities of a sulfated polysaccharide from Gracilaria Lemaneiformis. Journal of Functional Foods 2020, 64, 103652. [Google Scholar] [CrossRef]
- Fang, T.; Zhang, X.; Hu, S.; Yu, Y.; Sun, X.; Xu, N. , Enzymatic degradation of Gracilariopsis lemaneiformis polysaccharide and the antioxidant activity of its degradation products. Mar Drugs 2021, 19. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.C.; Merino, E.R.; Pujol, C.A.; Damonte, E.B.; Cerezo, A.S.; Matulewicz, M.C. , Galactans from cystocarpic plants of the red seaweed Callophyllis variegata (Kallymeniaceae, Gigartinales). Carbohydrate Research 2005, 340, 2742–2751. [Google Scholar] [CrossRef]
- Chopin, T.; Kerin, B.F.; Mazerolle, R. , Phycocolloid chemistry as a taxonomic indicator of phylogeny in the Gigartinales, Rhodophyceae: A review and current developments using Fourier transform infrared diffuse reflectance spectroscopy. Phycological Research 1999, 47, 167–188. [Google Scholar] [CrossRef]
- Usov, A.I.; Klochkova, N.G. , Polysaccharides of algae 45. Polysaccharide composition of red seaweeds from Kamchatka coastal waters (Northwestern Pacific) studied by reductive hydrolysis of biomass. Botanica Marina 1992, 35, 371–378. [Google Scholar] [CrossRef]
- McCandless, E.L.; Gretz, M.R. , Biochemical and immunochemical analysis of carrageenans of the Gigartinaceae and Phyllophoraceae. Hydrobiologia 1984, 116, 175–178. [Google Scholar] [CrossRef]
- Tasende, M.G.; Cid, M.; Fraga, M.I. , Qualitative and quantitative analysis of carrageenan content in gametophytes of Mastocarpus stellatus (Stackhouse) Guiry along Galician coast (NW Spain). Journal of Applied Phycology 2013, 25, 587–596. [Google Scholar] [CrossRef]
- Kravchenko, A.O.; Anastyuk, S.D.; Glazunov, V.P.; Sokolova, E.V.; Isakov, V.V.; Yermak, I.M. , Structural characteristics of carrageenans of red alga Mastocarpus pacificus from sea of Japan. Carbohydrate Polymers 2020, 229, 115518. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Viñas, M.; Souto, S.; Flórez-Fernández, N.; Torres, M.D.; Bandín, I.; Domínguez, H. , Antiviral activity of carrageenans and processing implications. Marine Drugs 2021, 19. [Google Scholar] [CrossRef]
- Kravchenko, A.; Anastyuk, S.; Glazunov, V.; Sokolova, E.; Isakov, V.; Yermak, I. , Structural peculiarities of carrageenans from Far Eastern red seaweed Mazzaella parksii (Gigartinaceae). International Journal of Biological Macromolecules 2023, 228, 346–357. [Google Scholar] [CrossRef]
- Kumar, K. , Principal component analysis: Most favourite tool in chemometrics. Resonance 2017, 22, 747–759. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. , Principal component analysis: A review and recent developments. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences 2016, 374, 20150202. [Google Scholar] [CrossRef] [PubMed]
- Sloneker, J.H.; Orentas, D.G. , Pyruvic acid, a unique component of an exocellular bacterial polysaccharide. Nature 1962, 194, 478–479. [Google Scholar] [CrossRef]
- Takano, R. , Desulfation of sulfated polysaccharides. Trends in Glycoscience and Glycotechnology 2002, 14, 343–351. [Google Scholar] [CrossRef]
- Wood, J.; Tan, H.-T.; Collins, H.; Yap, K.; Khor, S.; Lim, W.L.; Xing, X.; Bulone, V.; Burton, R.; Fincher, G.; Tucker, M. , Genetic and environmental factors contribute to variation in cell wall composition in mature desi chickpea (Cicer arietinum L.) cotyledons. Plant, cell & environment 2018, 41. [Google Scholar]
- Klassen, L.; Reintjes, G.; Li, M.; Jin, L.; Amundsen, C.; Xing, X.; Dridi, L.; Castagner, B.; Alexander, T.W.; Abbott, D.W. , Fluorescence activated cell sorting and fermentation analysis to study rumen microbiome responses to administered live microbials and yeast cell wall derived prebiotics. Frontiers in Microbiology 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.H.; Cleary, M.T.; Dokoozlian, N.; Ford, C.M.; Fincher, G.B. , Soluble cell wall carbohydrates and their relationship with sensory attributes in Cabernet Sauvignon wine. Food Chemistry 2019, 298, 124745. [Google Scholar] [CrossRef] [PubMed]
- Muhidinov, Z.K.; Bobokalonov, J.T.; Ismoilov, I.B.; Strahan, G.D.; Chau, H.K.; Hotchkiss, A.T.; Liu, L. , Characterization of two types of polysaccharides from Eremurus hissaricus roots growing in Tajikistan. Food Hydrocolloids 2020, 105, 105768. [Google Scholar] [CrossRef]
- Hosain, N.A.; Ghosh, R.; Bryant, D.L.; Arivett, B.A.; Farone, A.L.; Kline, P.C. , Isolation, structure elucidation, and immunostimulatory activity of polysaccharide fractions from Boswellia carterii frankincense resin. International Journal of Biological Macromolecules 2019, 133, 76–85. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Guo, Y.; Zhang, L.; Huang, L. , A facile method for the synthesis of partially O-methylated alditol acetate standards for GC–MS analysis of galactofuranose-containing structures. Carbohydrate Research 2013, 379, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Carpita, N.C.; Shea, E.M. Linkage structure of carbohydrates by gas chromatography-mass spectrometry (GC-MS) of partially methylated alditol acetates. In Analysis of Carbohydrates by GLC and MS, C.J., B.; McGinnis, G.D., Eds. CRC Press, Inc.: Boca Raton, Florida, 1989; pp 157-216.
- Yu, S.; Blennow, A.; Bojko, M.; Madsen, F.; Olsen, C.E.; Engelsen, S.B. , Physico-chemical characterization of floridean starch of red algae. Starch - Stärke 2002, 54, 66–74. [Google Scholar] [CrossRef]
- Wickham, H. , Ggplot2: Elegant Gaphics for Data Analysis. Springer-Verlag New York: 2016.
- Wickham, H. , Reshaping data with the reshape package. Journal of Statistical Software 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Wickham, H.F.; Henry, L.; Müller, K.; Vaughan, D. Dplyr: a grammar of data manipulation. https://dplyr.tidyverse.org, https://github.com/tidyverse/dplyr.
- Wickham, H. Forcats: tools for working with categorical variables (factors). https://forcats.tidyverse.org/.
Figure 1.
EI-MS spectra and ion fragmentation patterns of PMAAs from (A) 4-AnGalp, (B) 2,4-AnGalp, (C) 3-Galp, and (D) 4-Galp of galactans of Callophyllis sp.
Figure 1.
EI-MS spectra and ion fragmentation patterns of PMAAs from (A) 4-AnGalp, (B) 2,4-AnGalp, (C) 3-Galp, and (D) 4-Galp of galactans of Callophyllis sp.
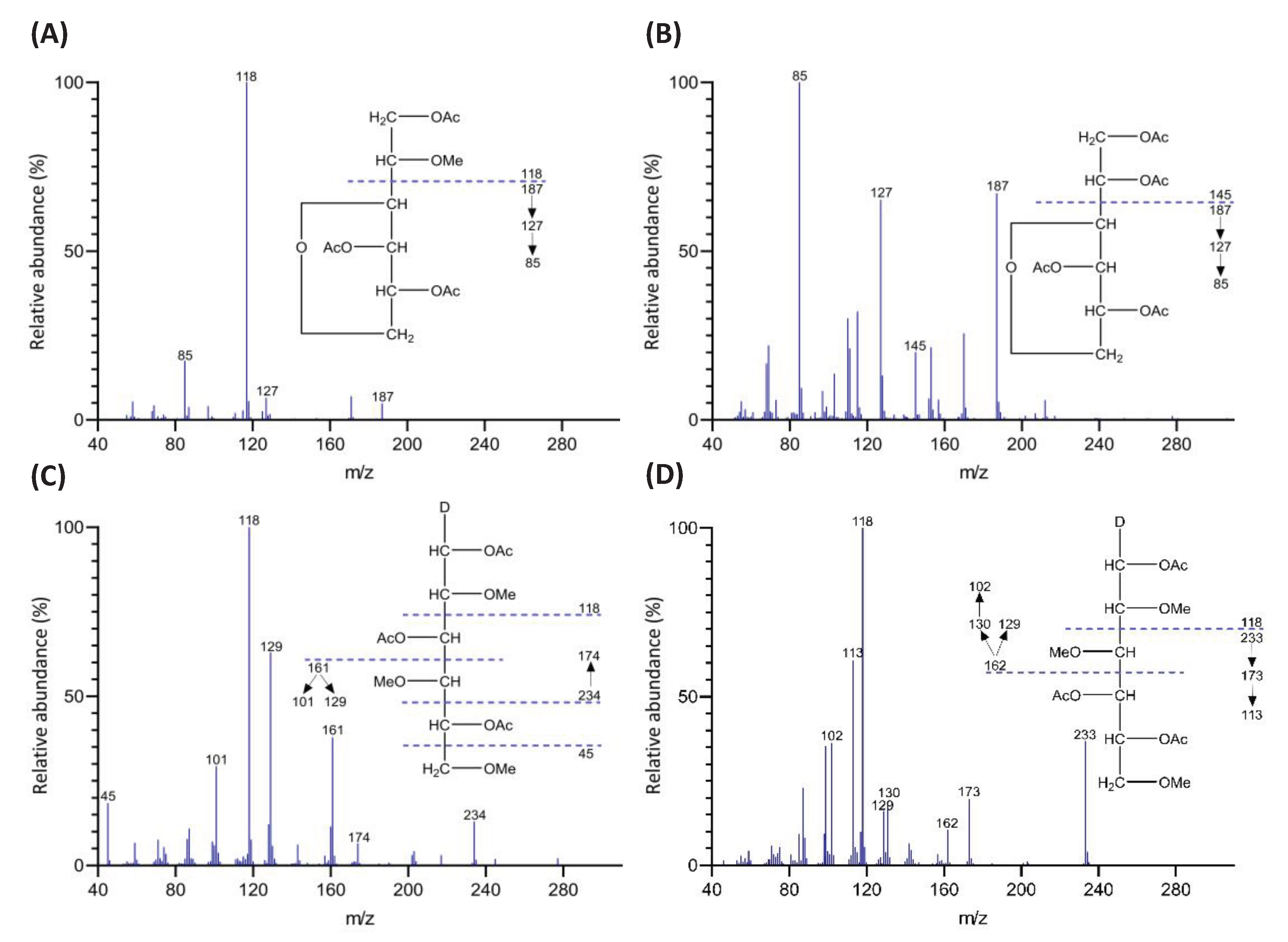
Figure 2.
GC chromatograms of PMAAs prepared from agarose and corn fiber xylan standards, including (A) GC-FID and (B) GC-TIC chromatograms of agarose-derived PMAAs, alongside (C) GC-FID and (D) GC-TIC chromatograms of PMAAs from corn fiber xylan.
Figure 2.
GC chromatograms of PMAAs prepared from agarose and corn fiber xylan standards, including (A) GC-FID and (B) GC-TIC chromatograms of agarose-derived PMAAs, alongside (C) GC-FID and (D) GC-TIC chromatograms of PMAAs from corn fiber xylan.
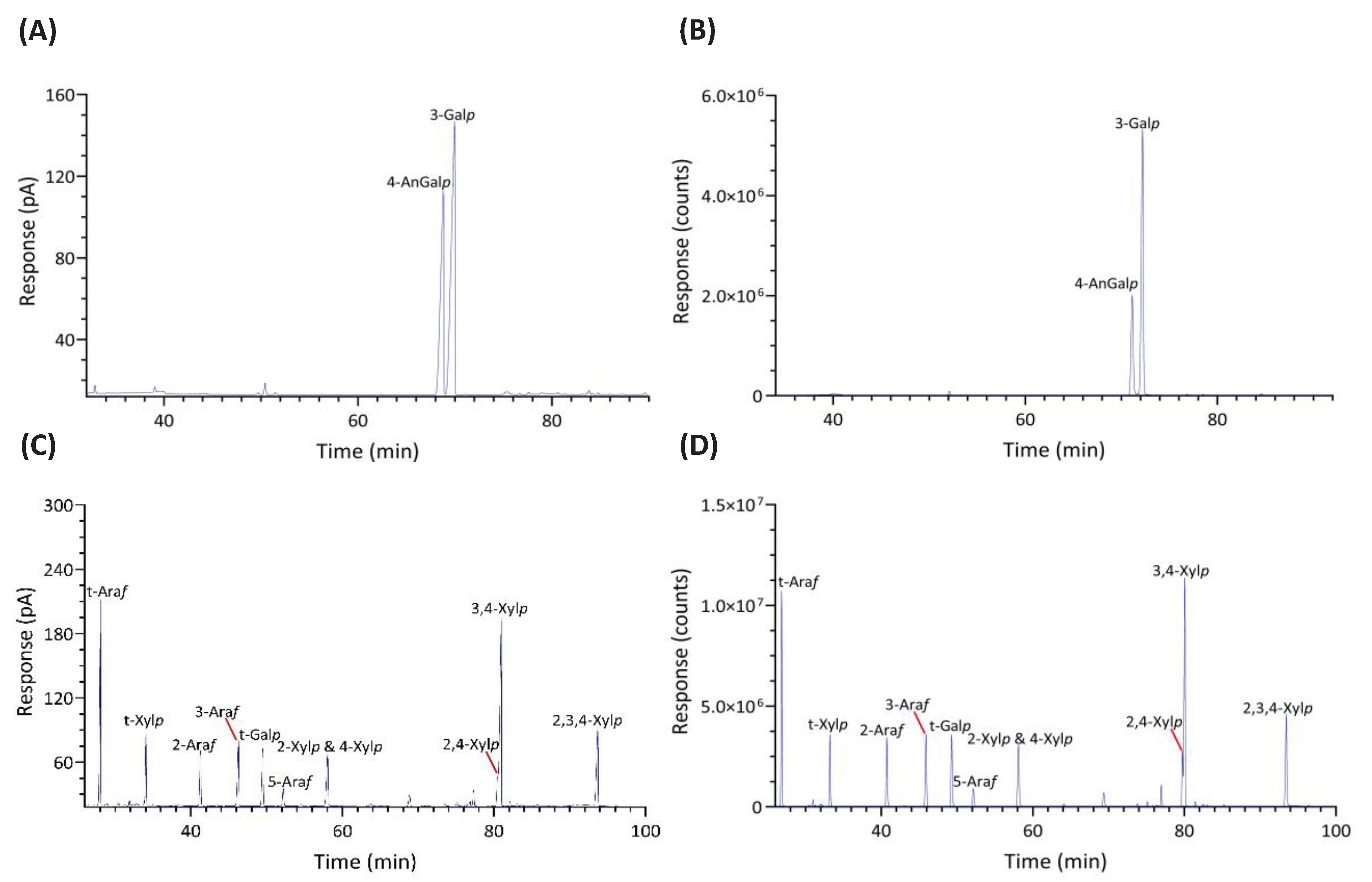
Figure 3.
Bubble plot showing the relative linkage composition (Mol%) quantified by GC-FID from each of the two separate experiments conducted on unfractionated polysaccharides of six red seaweed species.
Figure 3.
Bubble plot showing the relative linkage composition (Mol%) quantified by GC-FID from each of the two separate experiments conducted on unfractionated polysaccharides of six red seaweed species.
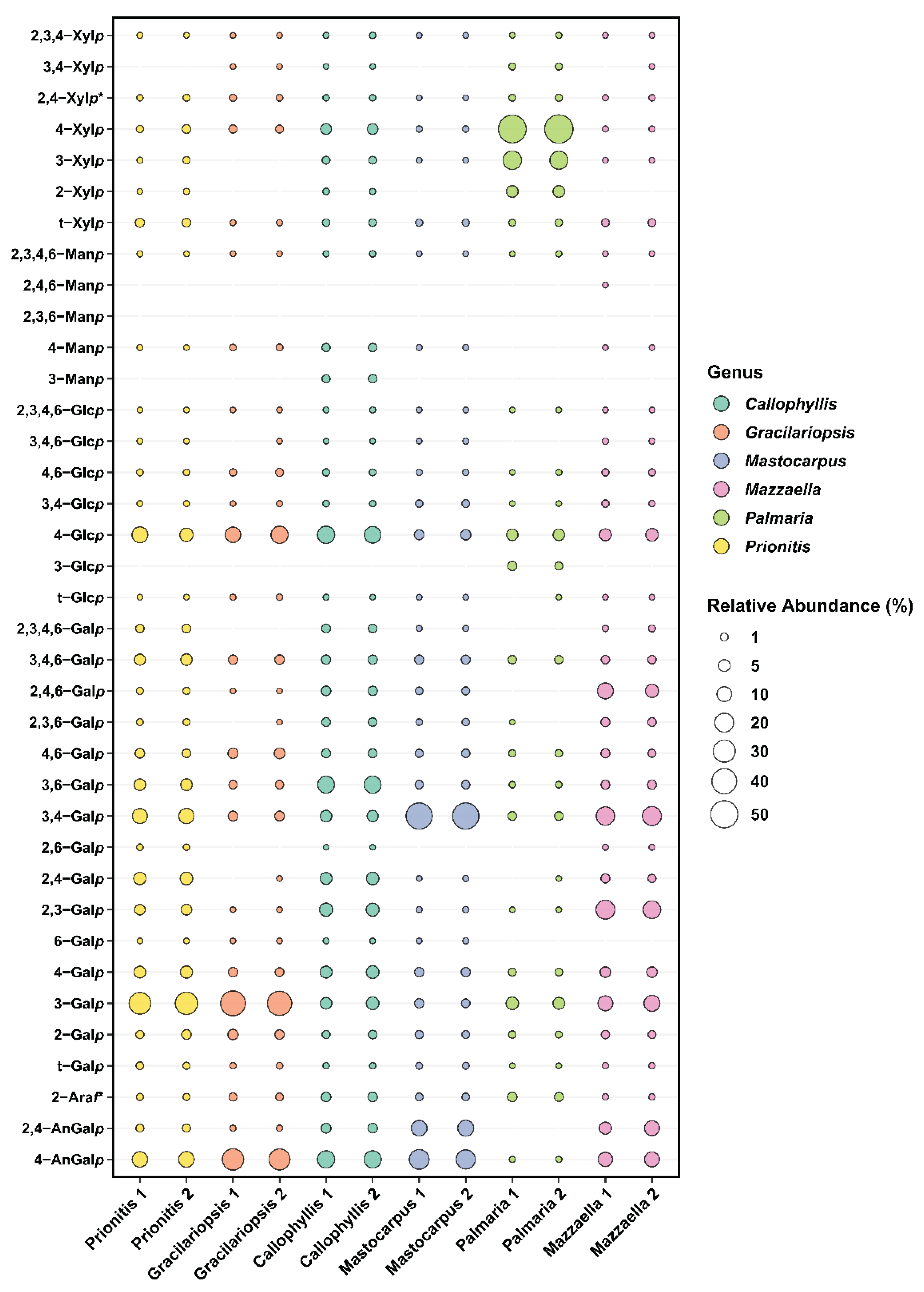
Figure 4.
Relative monosaccharide compositions (Mol%) of unfractionated polysaccharides calculated by summing up all the linkage compositions, quantified by (A) GC-MS and (B) GC-FID, for each monosaccharide of six red seaweed species. AnGal, anhydrogalactose; Ara, arabinose; Gal, galactose; Glc, glucose; Man, mannose; Xyl, xylose. Two separate experiments were conducted.
Figure 4.
Relative monosaccharide compositions (Mol%) of unfractionated polysaccharides calculated by summing up all the linkage compositions, quantified by (A) GC-MS and (B) GC-FID, for each monosaccharide of six red seaweed species. AnGal, anhydrogalactose; Ara, arabinose; Gal, galactose; Glc, glucose; Man, mannose; Xyl, xylose. Two separate experiments were conducted.
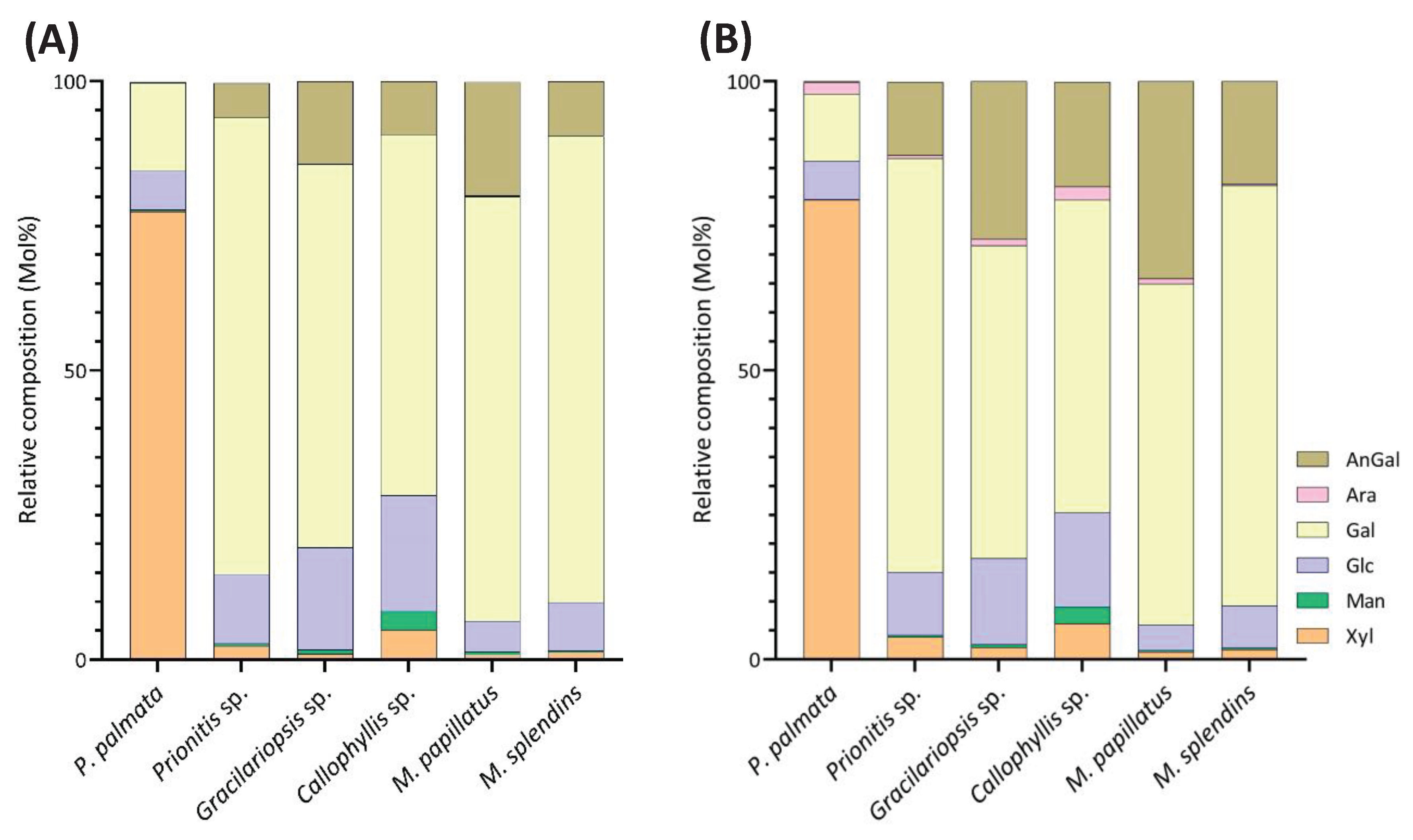
Figure 5.
Estimated polysaccharide compositions (Mol%) calculated by summing up compositions of all the linkages, quantified by (A) GC-MS and (B) GC-FID, assigned to each type of polysaccharide of six red seaweed species. UA: unassigned linkages; XN: xylan; MN: mannan; GN: galactan; CE: cellulose; FS: Floridean starch. Two separate experiments were conducted.
Figure 5.
Estimated polysaccharide compositions (Mol%) calculated by summing up compositions of all the linkages, quantified by (A) GC-MS and (B) GC-FID, assigned to each type of polysaccharide of six red seaweed species. UA: unassigned linkages; XN: xylan; MN: mannan; GN: galactan; CE: cellulose; FS: Floridean starch. Two separate experiments were conducted.
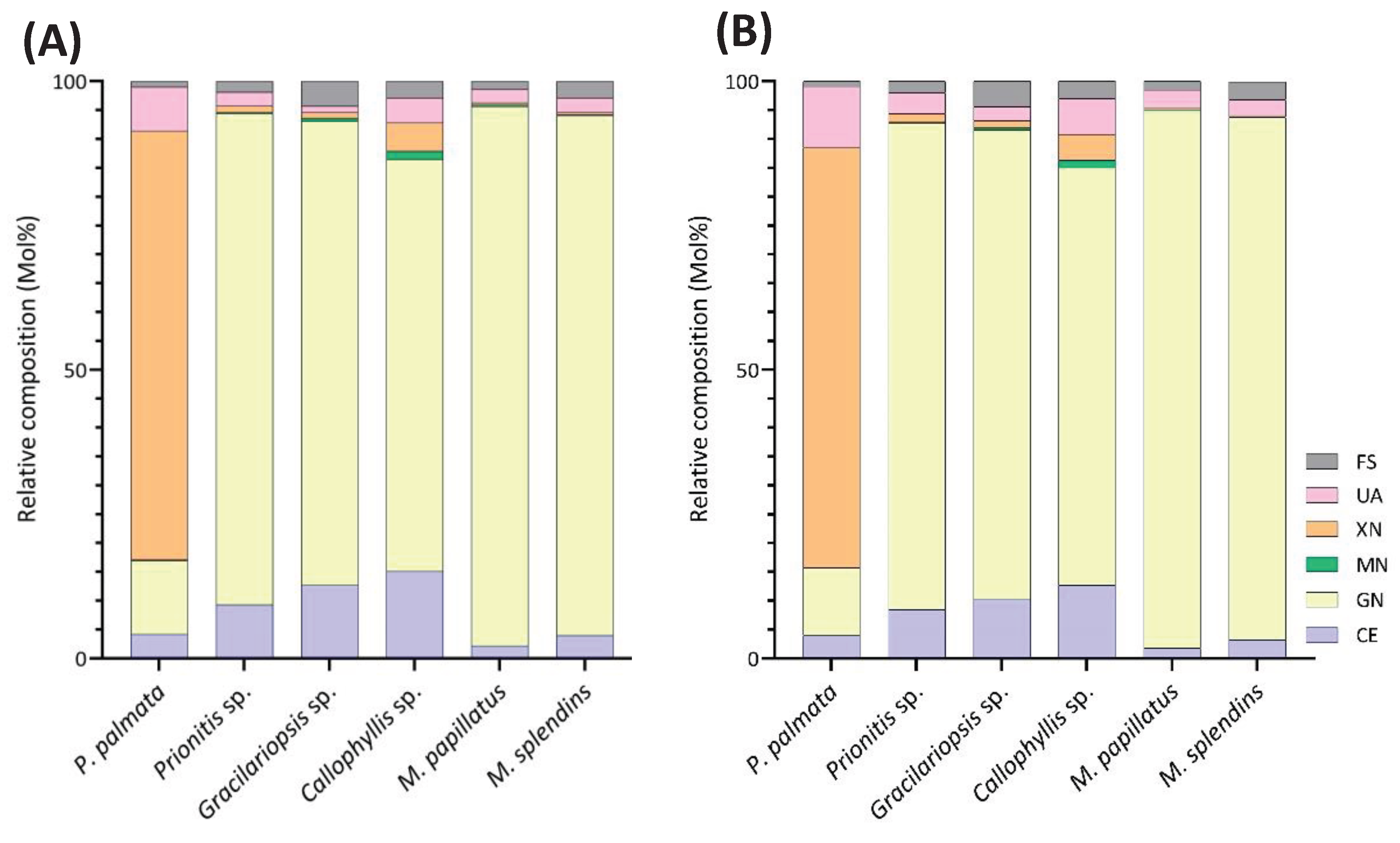
Figure 6.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Palmaria palmata.
Figure 6.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Palmaria palmata.
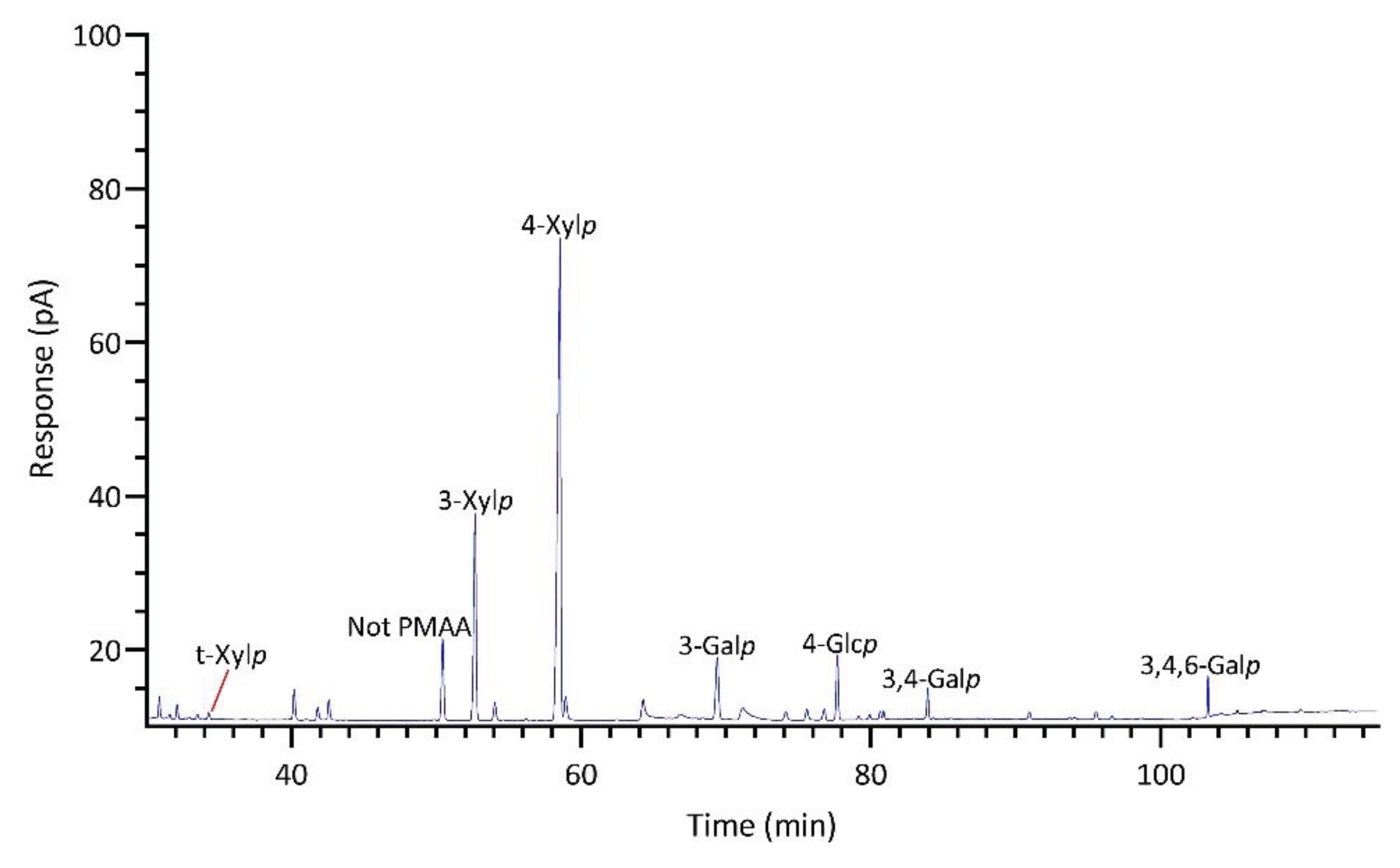
Figure 7.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Gracilariopsis sp.
Figure 7.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Gracilariopsis sp.
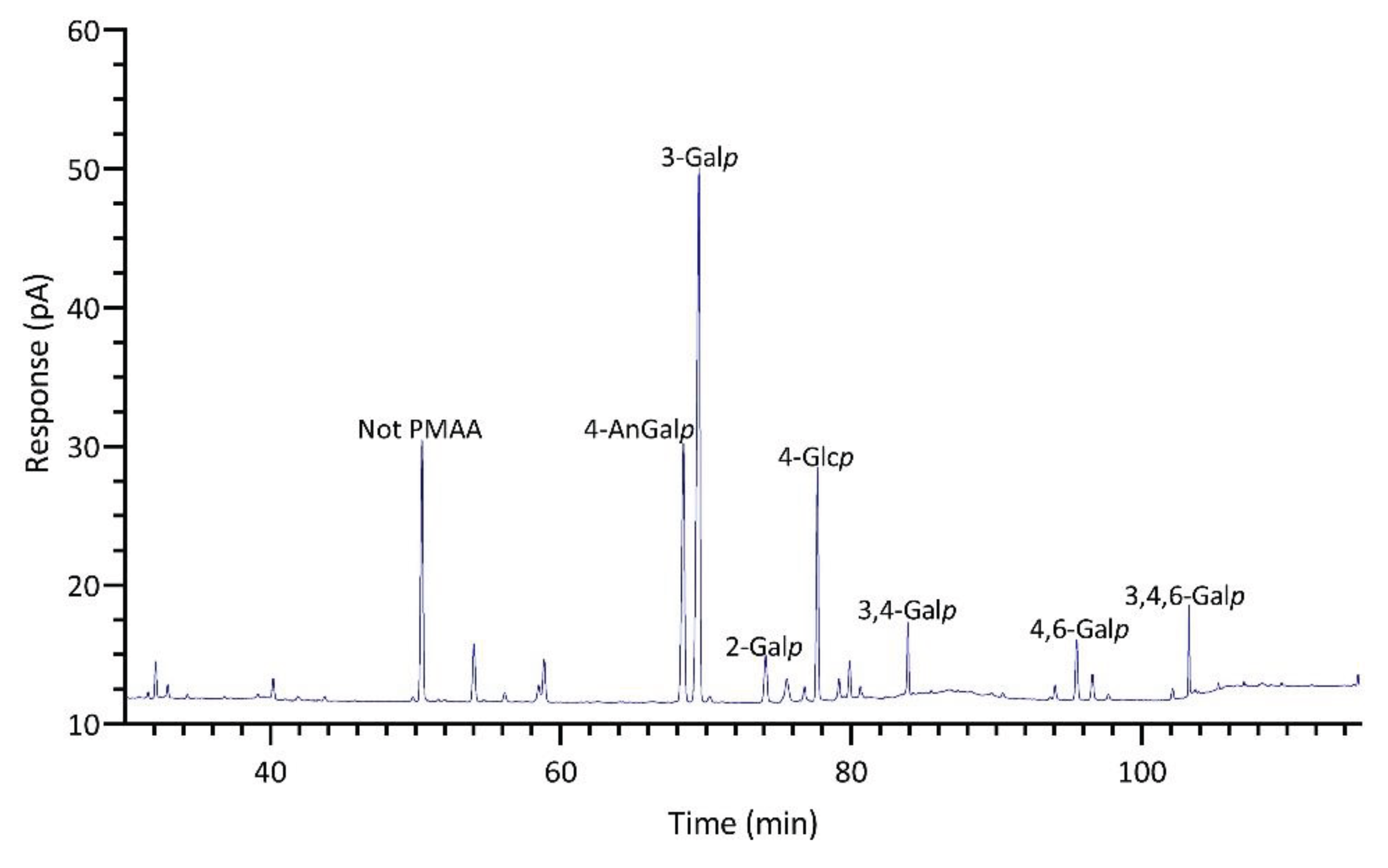
Figure 8.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Prionitis sp.
Figure 8.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Prionitis sp.
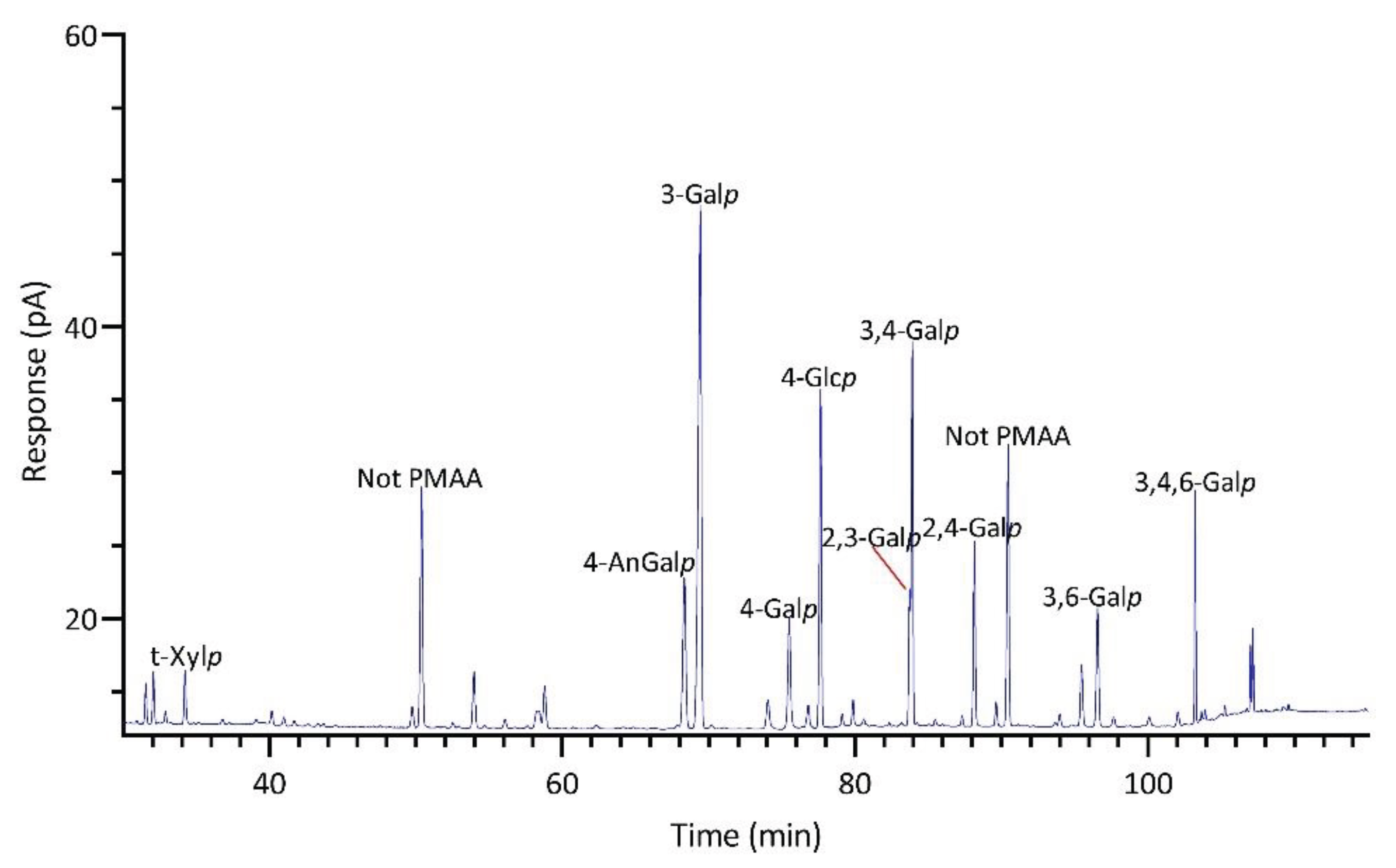
Figure 9.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Callophyllis sp.
Figure 9.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Callophyllis sp.
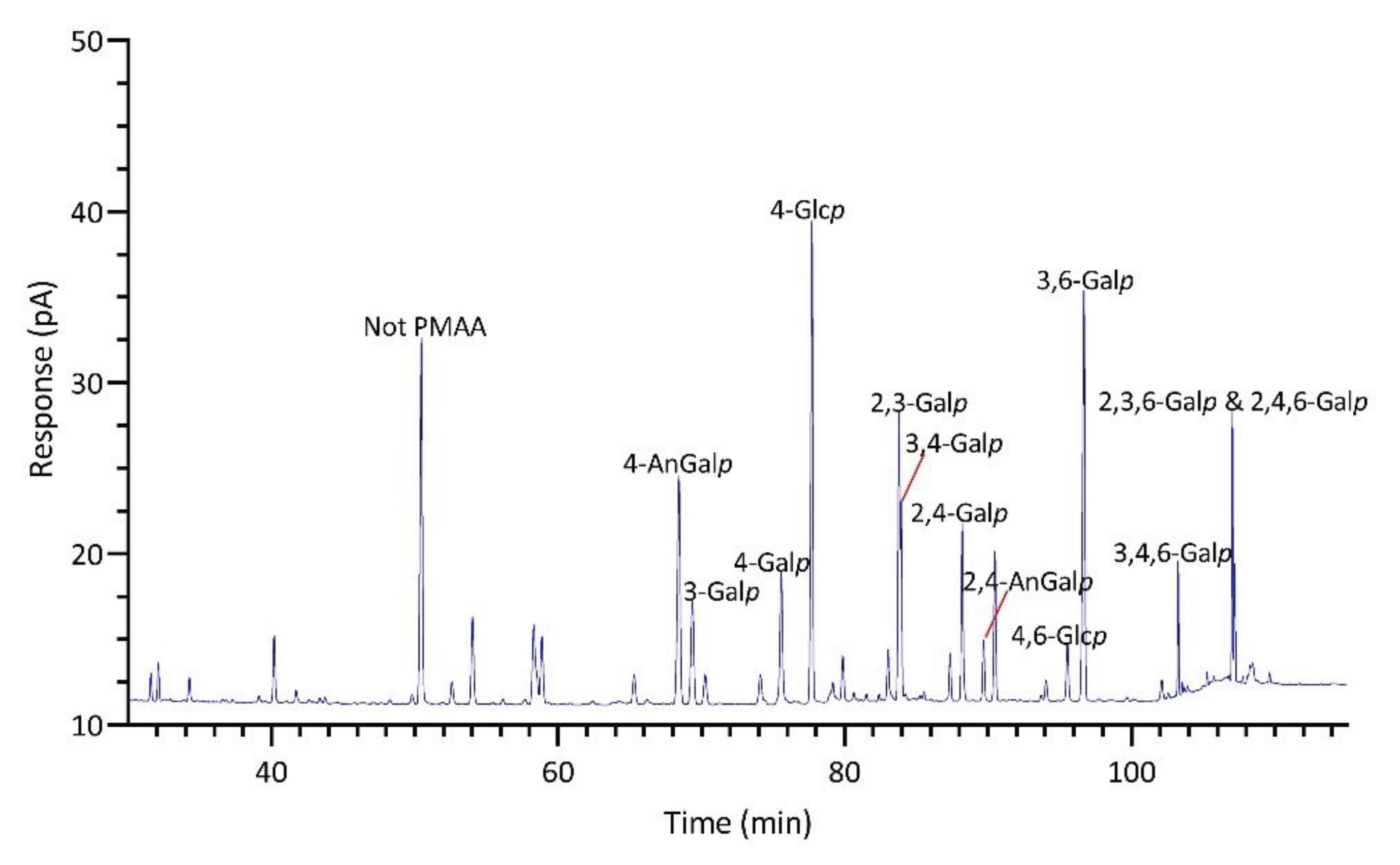
Figure 10.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Mastocarpus papillatus.
Figure 10.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Mastocarpus papillatus.
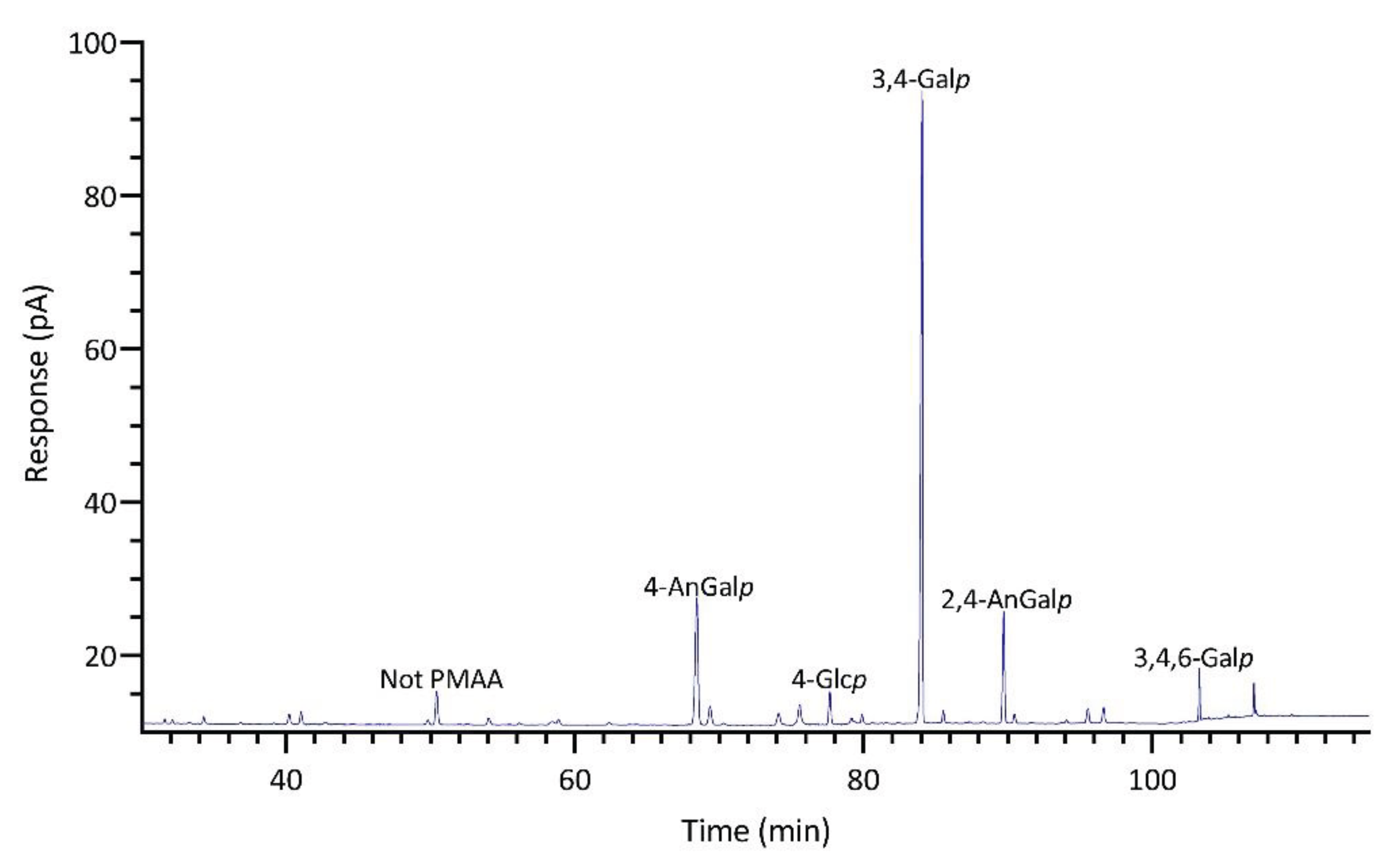
Figure 11.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Mazzaella splendins.
Figure 11.
GC-TIC chromatogram of PMAAs from the glycosidic linkages in unfractionated polysaccharides of Mazzaella splendins.
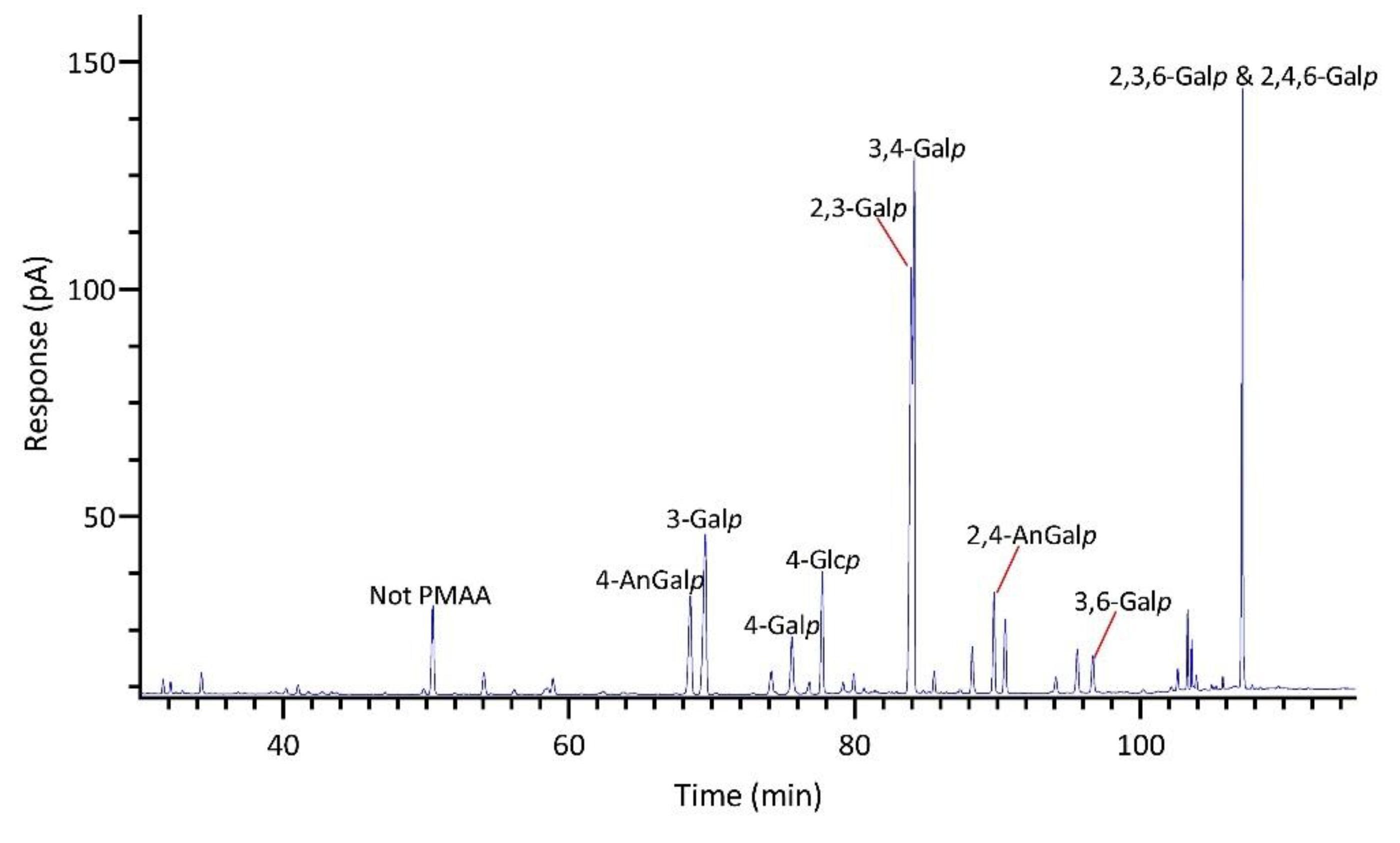
Figure 12.
PCA score plots of (A) relative linkage compositions calculated from GC-FID measurement, (B) relative linkage compositions calculated from GC-MS measurement, (C) normalized GC-FID chromatograms of PMAAs, and (D) normalized GC-TIC chromatograms of PMAAs. Detailed nominalization procedure for raw GC chromatograms is described in Section 3.5.
Figure 12.
PCA score plots of (A) relative linkage compositions calculated from GC-FID measurement, (B) relative linkage compositions calculated from GC-MS measurement, (C) normalized GC-FID chromatograms of PMAAs, and (D) normalized GC-TIC chromatograms of PMAAs. Detailed nominalization procedure for raw GC chromatograms is described in Section 3.5.
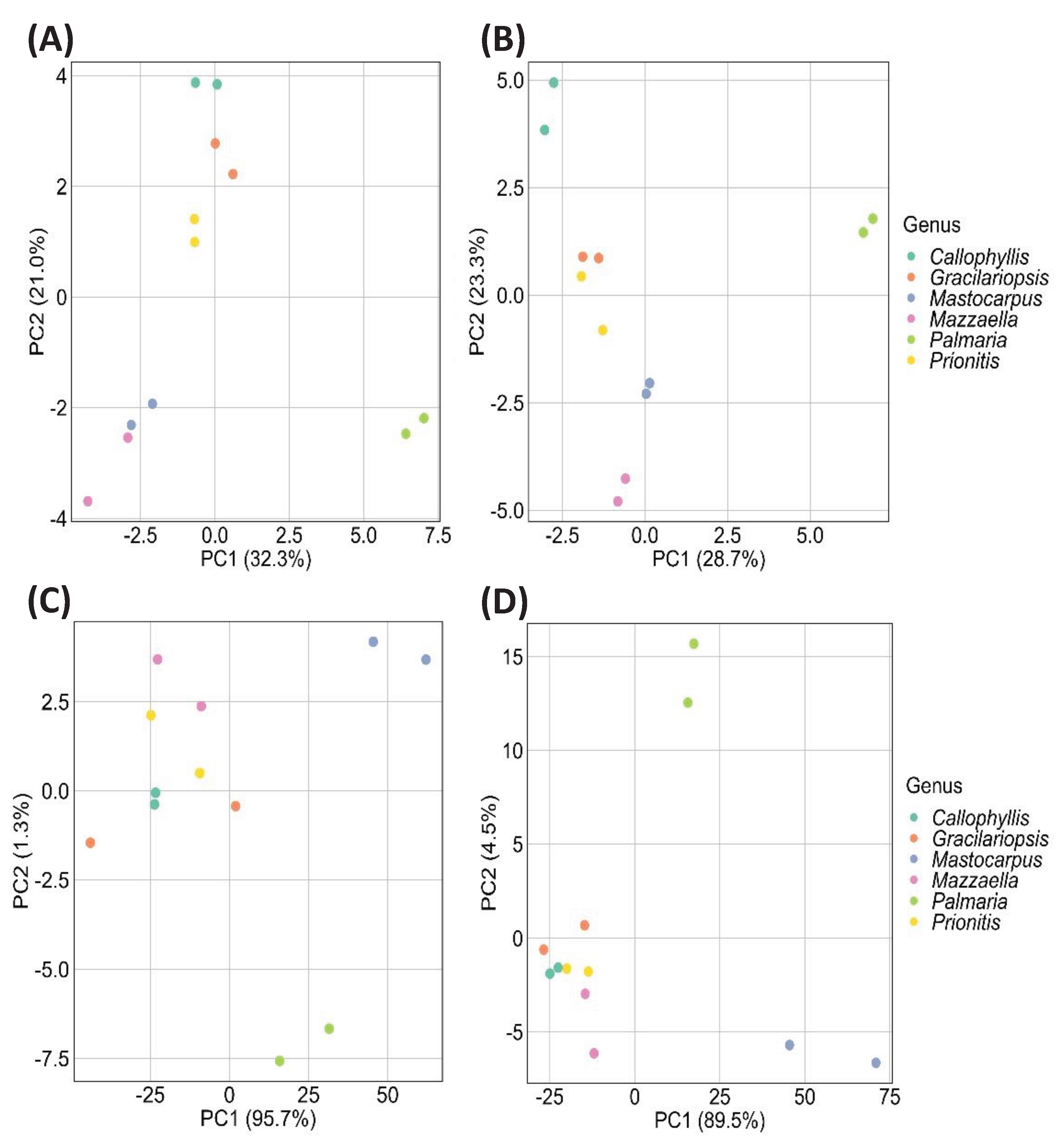

Table 1.
Linkage compositions (Mol%) of unfractionated polysaccharides in red seaweeds determined by GC-FID.
Table 1.
Linkage compositions (Mol%) of unfractionated polysaccharides in red seaweeds determined by GC-FID.
| Linkage | Prionitis | Gracilariopsis | Callophyllis | Mastocarpus | Palmaria | Mazzaella |
|---|---|---|---|---|---|---|
| 4-AnGalp | 11.6 | 27.1 | 15.7 | 21.8 | Tr. | 9.7 |
| 2,4-AnGalp | 1.1 | Tr. | 2.4 | 12.3 | Tr. | 8.0 |
| 2-Araf | 0.5 | 1.1 | 2.3 | 0.9 | 2.0 | Tr. |
| t-Galp | 0.7 | Tr. | Tr. | 0.5 | Tr. | Tr. |
| 2-Galp | 1.8 | 2.7 | 1.3 | 1.1 | 0.7 | 1.3 |
| 3-Galp | 29.8 | 39.5 | 5.6 | 1.9 | 5.6 | 11.5 |
| 4-Galp | 5.3 | 1.9 | 5.8 | 2.1 | 0.9 | 3.4 |
| 6-Galp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| 2,3-Galp | 3.5 | Tr. | 7.0 | Tr. | Tr. | 18.4 |
| 2,4-Galp | 6.0 | Tr. | 5.6 | Tr. | Tr. | 1.5 |
| 2,6-Galp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| 3,4-Galp | 10.8 | 2.5 | 4.4 | 46.2 | 1.6 | 19.4 |
| 3,6-Galp | 4.5 | 1.3 | 14.6 | 1.3 | Tr. | 1.7 |
| 4,6-Galp | 1.8 | 3.3 | 1.8 | 1.3 | 0.7 | 1.5 |
| 2,3,6-Galp | 0.5 | Tr. | 1.6 | 0.6 | Tr. | 1.8 |
| 2,4,6-Galp | 0.6 | Tr. | 2.2 | 1.0 | Tr. | 9.9 |
| 3,4,6-Galp | 4.3 | 2.2 | 2.0 | 2.0 | 1.4 | 1.5 |
| 2,3,4,6-Galp | 1.5 | Tr. | 1.6 | Tr. | Tr. | Tr. |
| t-Glcp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| 3-Glcp | Tr. | Tr. | Tr. | Tr. | 1.6 | Tr. |
| 4-Glcp | 9.9 | 13.4 | 14.9 | 2.9 | 4.6 | 5.7 |
| 3,4-Glcp | Tr. | Tr. | Tr. | 0.9 | Tr. | 0.5 |
| 4,6-Glcp | Tr. | 0.9 | 0.6 | Tr. | Tr. | 0.7 |
| 3,4,6-Glcp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| 2,3,4,6-Glcp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| t-Manp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| 3-Manp | Tr. | Tr. | 1.3 | Tr. | Tr. | Tr. |
| 4-Manp | Tr. | Tr. | 1.3 | Tr. | Tr. | Tr. |
| 2,3,6-Manp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| 2,4,6-Manp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| 2,3,4,6-Manp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
| t-Xylp | 1.8 | Tr. | 0.8 | 0.7 | 0.6 | 1.0 |
| 2-Xylp | Tr. | Tr. | Tr. | Tr. | 4.9 | Tr. |
| 4-Xylp | 1.1 | 1.2 | 3.6 | Tr. | 54.4 | Tr. |
| 3-Xylp | Tr. | Tr. | 0.9 | Tr. | 18.4 | Tr. |
| 2,4-Xylp | Tr. | 0.5 | Tr. | Tr. | 0.6 | Tr. |
| 3,4-Xylp | Tr. | Tr. | Tr. | Tr. | 0.5 | Tr. |
| 2,3,4-Xylp | Tr. | Tr. | Tr. | Tr. | Tr. | Tr. |
Note: Tr. means trace (Mol% < 0.5%). Two separate experiments were conducted to each sample.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



