Submitted:
22 March 2024
Posted:
22 March 2024
You are already at the latest version
Abstract
As the global arid climate intensifies and the groundwater becomes increasingly scarce, drought stress has become a major challenge to plant growth and development worldwide, especially causing yield reduction and extinction of cultivated crops. In a large number of drought resistance studies, DNA methylation has been found dynamically associated with response to drought stress in plants by regulating gene expression and developments in plants, but the specific mechanisms are not yet clear. In this article, the current advances of research have been summarized on DNA methylation response to drought stress in various plant species, in terms of methylation detection methods, mechanisms of pattern changes (DNA de novo methylation, DNA maintenance methylation, and DNA demethylation), and its response to drought stress. With Most of the studies showed significant changes in genome or gene promoter methylation levels in plants subjected to drought stress, but the identification of the genes and pathways involved is still in the minority. The aim is to provide references for in-depth research on the response of plants to drought stress through epigenetics, and to uncover drought resistance genes regulated by DNA methylation, specific signaling pathways, and molecular mechanisms of action.
Keywords:
plant
; epigenetics
; DNA methylation
; drought stress
; mechanism
; research advances
1. Introduction
Epigenetics has become a popular area of study in recent years because it regulates gene activity without altering the DNA nucleotide sequence. This allows genes to change their function and expression, which can then be passed down to offspring in a stable manner in response to environmental changes [1]. This preserves organisms' normal growth and development and enables them to adapt to stress brought on by unstable environmental factors. Epigenetic modifications contain DNA methylation, histone modifications, chromatin remodeling, and non-coding RNA (ncRNA) etc., which affect the structure and accessibility of chromatin, thus dynamically regulate gene expression [2].
DNA methylation has become one of the most thoroughly investigated research field in categories of epigenetic modification, with the rapid advancement in high-throughput sequencing technologies for genome DNA methylation. As a conserved epigenetic modification, DNA methylation is a widespread covalent modification in biological genomes, which can regulate the corresponding functions of genomes without changing the primary structure of DNA molecules. It plays a vital role in gene regulation and genome stability [3]. DNA methylation is a form of chemical modification of DNA, occurs by the transfer and covalent binding of methyl groups (CH3-) from S-adenosylmethionine (SAM) to sites on the DNA sequence where methylation can occur, such as adenine N-6, guanine G-7, and cytosine C-5 [4]. The process is catalyzed by various DNA methyltransferases (DNMT) after DNA replication. The DNA is further modified by DNA methylation to form N6-methyl purine (N6-mA), 7-methyl guanine (7-mG), and 5-methyl cytosine (5-mC) [5]. Among them, 5-mC occurs most frequently in eukaryotic organisms in the methylation site enriched region and has a role in regulating the transcription of biological genes [6]. It also allows organisms to respond to the environmental changes by maintaining genome stability, regulating gene expression, and altering genetic phenotype without altering DNA sequences.
The United Nations Intergovernmental Panel on Climate Change (IPCC) reports that the average temperature will rise by 1.8-4.0°C by 2100 and that many regions of the world will face the environmental change problem due to increased drought. Under the circumstances of external water stress, coupled with the low water table, drought has become a major threat to crop cultivation worldwide [7]. The impact of drought has been exacerbated by climate change in recent years, as drought stress has led to stunted crop growth, devastating effects on crops, and severe yield losses [8]. In recent years, DNA methylation has often been found in plants in response to biotic and abiotic stresses, e.g., in biotic stress, the total DNA methylation level of rice was decreased in response to bacterial infection [9]; in abiotic stress, increased genome-wide level of rice under high salt stress [10]; under heat stress, different levels of heat stress induced different levels of DNA methylation in Arabidopsis thaliana [11]; cold stress caused rice's demethylation of the promoter region of cold tolerance genes [12]; and UV stress induces demethylation of the promoter region of key factors in Arabidopsis [13], leading to differential DNA methylation in plants in response to different patterns of climate change; drought stress caused an increase in the overall methylation level of linseed [14], the research progress of DNA methylation on its drought stress is collated to provide an important idea for the improvement of crop breeding for drought resistance.
2. Characteristics of DNA Methylation Modifications
In 1925, 5-mC was first discovered in the hydrolysis product of tuberculinic acid from the nucleic acid of the tubercle bacillus [15]. In subsequent studies, higher levels of DNA methylation modifications of 5-mC were also found in plants [16].
DNA methylation is based on the nature of the target gene and its function is reflected in the regulation of gene expression, transposon silencing, chromosomal interactions, and genetic characteristics [4].
DNA methylation is also crucial for maintaining genomic stability [17]. DNA methylation in plants can maintain genomic stability by inhibiting transposon and exogenous gene transcription to reduce genome disruptions such as translocation and recombination. Recent studies have revealed that transposons show high methylation levels after amplification, which can indicate that transposons in a highly active state can be regulated by DNA methylation and further act as a repressor.
DNA methylation modification of genes in general is often found to exhibit a negative correlation with gene transcription[18,19], i.e., the higher the frequency and level of DNA methylation, the lower the level of gene transcription, which in turn affects the expression of the corresponding gene, this further increases the phenotypic differences between organisms. This is the reason why the same organism with the same whole genome sequence has different traits that are adapted to environmental changes [20]. This is in contrast to the methylation that occurs on transposons, where CG methylation within the gene region allows for moderate expression of the corresponding gene, mostly at different tissue sites and does not silence the gene[21,22].
In most eukaryotes, the frequent DNA methylation is usually occurs in the cytosine base, which contains CG, CHG and CHH (H for A, T or C) in total three types [18]. Among these three types, the CG methylation level is significantly higher than the CHG or CHH methylation types [23]. CG methylation can occur in the promoter region and part of the pre-transcriptional region, as well as in the 3' end and part of the post-transcriptional region and may inhibit gene expression. There are two theories that explain this repression of expression: first, methylation that occurs in the promoter and enhancer regions prevents the binding of transcription factors required for gene transcription, affecting gene transcription and thereby inhibiting or even preventing gene expression; Second, cytosine sites that have undergone methylation can attract proteins bound to them, causing histone deacetylases and chromatin remodeling proteins to be attracted, and chromatin to be compressed. This structural change results in the inability to transcribe, thereby inhibiting gene expression [19]. Although mostly occurring promoter DNA methylation inhibits gene transcription, it also acts as a promoter in a small number of cases [3].
Compared with mammals, the proportion of 5-mC in plant genomes is relatively high, and the genome-wide cytosine methylation level varies from species to species, ranging from 6- 25% in different species [24]. For example, in the model plant Arabidopsis, 5% of cytosine is methylated [25], 24.3% of cytosine is methylated in young spikes of wild and cultivated rice and in wheat it is over 20%. This significant difference is caused by the enrichment of repetitive sequences. The level of cytosine methylation also varies significantly in different regions of the same genome, and several studies have demonstrated that cytosine methylation levels are tissue-, organ-, and developmental stage-specific [18]. DNA methylation of transposable elements (TEs) is strikingly similar across species and DNA repeat sequences, with 50% methylation differences between ecotypes [4]. Methylation polymorphisms were found to occur most frequently in the upstream or downstream regions of genes, after repressing the transcription of related genes, making its level negatively correlated with gene expression levels. Although the effect is not significant, studies on plants have mostly proven that DNA methylation also occurs in the gene body. However, it has been discovered in the study of poplar that methylation in the gene body significantly inhibits gene transcription more than methylation in the promoter region [26].
The first genome-wide methylation map of plants was published in 2016 by Zhang et al. [27] using the model plant Arabidopsis thaliana as a guide. The map's findings revealed that more than one-third of the expressed genes in the genome were methylated. There is a significant difference in that only 5% of the genes are methylated in the promoter region. Based on this genome-wide methylation map, it was found that Arabidopsis genes that were methylated in the gene region were expressed and could reach high expression levels. Subsequently, Inagaki et al. [28] found that gene region methylation was higher in transcribed regions than in non-transcribed regions, however, the mechanism of this association is unclear. Graaf et al. [29] evaluated the mutation rate per CpG site per haploid per generation in Arabidopsis and found that the forward mutation rate (i.e., methylation gain rate) was about 2.56 × 10-4 and the reverse mutation rate (i.e., methylation loss rate) was about 6.30 × 10-4 in Arabidopsis, and these methylation mutation rates are about five times higher than the mutation rates found by Ossowski et al. [30].
3. Methylation Detection Methods
3.1. Methylation Sensitive Amplified Polymorphism (MSAP)
MSAP is based on the amplified fragment length polymorphism(AFLP)method [31]. Methylation-specific isoschizomer HpaII and MSPI with a restriction endonuclease and genomic DNA, instead of AFLP functional enzymes and target bands, double digestion to obtain DNA fragments of different sizes, then join the enzymatically cleaved DNA fragments with the corresponding restriction endonuclease as a junction, then Design the primers according to the junction. Due to the high methylation specificity stemming from the coordinated activity of two methyltransferase enzymes, gDNA methylation levels can be further refined to distinguish between holo- and hemi-methylation states [32]. This assay is mostly used in early DNA methylation studies.
3.2. High Performance Liquid Chromatography (HPLC)
HPLC is divided into normal-phase HPLC and reversed-phase HPLC, which, in the study of calf thymus and salmon sperm, Kuo et al. [33] first detected DNA methylation using reversed-phase HPLC (RP-HPLC) [34], suggesting that HPLC can be a reliable method for detecting gDNA methylation levels. On this basis, the deformed HPLC was linked with PCR to form the DHPLC-PCR method system by Baumer, and this method makes DNA methylation detection more convenient and efficient [35]. As the research progressed, it was updated to high performance liquid chromatography-mass spectrometry (HPLC-MS) [36], which further improved the method technique for 5-mC detection.
3.3. Methylated DNA Immunoprecipitation-Sequencing (MeDIP-seq)
MeDIP is a technique that uses monoclonal antibodies or DNA methylation-binding proteins that bind specifically to methylation sites to quantitatively capture enriched methylated DNA against 5-mC in the sample [37]. Highly methylated regions of gDNA can be identified, but not at the level of single base methylation.
3.4. Amplified Fragment Single Nucleotide Polymorphism and Methylation (AFSM)
The AFSM test employs restriction endonucleases with varying sensitivity to methylation to double cleave the genome and produce DNA fragments of various sizes for the effective detection of DNA methylation [38]. This assay is based on the lower cost and higher accuracy of second-generation sequencing technology[39,40]. AFSM is currently the only method in the world that can simultaneously detect single-nucleotide polymorphisms (SNPs) by high-throughput, insertion and deletion (inDels) and methylation sites in the whole genome with high throughput.
3.5. Methylation Sensitive Restriction Endonuclease (MSREs)
MSREs are a class of restriction endonucleases that are methylation-sensitive at the recognition site [41]. The fragment is obtained by cleaving the CpG sequence using its isozyme, which is insensitive to methylation, and then analyzed by Southern Blot. The MSREs method is a method that combines the sensitivity of methylation and the specificity of restriction enzymes to identify the methylation status of CpG sequences. It is convenient because it does not require detailed information about the sequence of the entire gDNA and the primary structure of DNA, but its application is more restricted because it needs a great deal of DNA with a high relative molecular mass and can only detect methylated alleles with a high copy number ratio.
3.6. Bisulfite Sequencing PCR (BSP)
BSP was first proposed by Frommer et al [42] to be applied to 5-mC detection, where gDNA was first treated with hydrosulfite to react unmethylated cytosine C into uridine U [43]. In a subsequent study by Bianchessi et al [44] to detect methylation in the mitochondrial DNA, non-coding region of endothelial cells, it was found that 5-mC was not randomly scattered but aggregated within the DNA coding region. Many other studies have shown that the BSP is still the most often used assay because it is accurate and dependable and can identify the methylation status of individual CpG sites despite the BSP's limitations in detecting DNA methylation, such as the high cost and complexity of the method [45].
3.7. High-Performance Capillary Electrophoresis (HPCE)
It is a kind of product separation using the principle that narrow pore fused silica capillaries [46]. It can separate different chemical components from the complex to achieve quantitative detection of modified DNA by using the different charge properties, structure size and chemical properties of DNA hydrolysis products in the background of the strong electric field.
3.8. TET Enzyme-Assisted Pyridineborane Sequencing (TAPS) and Enzymatic Methyl-seq (EM-seq)
These both techniques use enzymatic and chemical reactions to complete sequencing in order to prevent degradation by bisulfite stimulation of most DNA. TAPS uses TET1 oxidase to oxidize 5-mC and 5-hmC to 5-caC, which is chemically converted to DHU by the reducing agent pyridine borane, which is then used as a template to be recognized by the corresponding DNA polymerase for U base, producing a C to T conversion by PCR amplification products [47]. Similarly, EM-seq uses TET2 and oxidation enhancer to oxidize 5-mC and 5-hmC to 5-caC, and then deaminates cytosine with APOBEC3A to deaminate the unmodified C to U, which is then recognized [48].
4. Mechanisms of Methylation Change Patterns
4.1. Mechanism of Methylation Action
DNA methylation is closely related to genome maintenance, parental imprint formation, and transcriptional regulation, and it is important to clarify the molecular mechanism for further research. It is known that the dynamic changes of DNA methylation include three processes of de novo methylation, maintenance methylation, and demethylation.
4.1.1. De Novo Methylation
De novo methylation is an asymmetric type CHH fully dependent methylation type occurring after completion of replication and refers to the methylation of cytosines that have not been modified by methylation in the presence of an associated methyltransferase. De novo methylation is mediated in plants by RNA, i.e. there is small interfering RNA (siRNA), scaffold RNA and the corresponding protein DNA methylation pathway [49], the de novo methylation can be divided into classical RNA-directed DNA methylation (RdDM) pathway (Figure 1) and non-classical RdDM pathway. As shown in Figure 1, classical RdDM is divided into two major steps, the first step is the synthesis of siRNA precursor RNA, i.e. Pol IV-dependent RNA (P4RNA). The SAWADEE HOMEODOMAIN HOMOLOG 1 (SHH1) protein binds to the H3K9 histone modified by methylation of the lysine at the ninth position of the tail through the Tudor-like fold structure of the SAWADEE structural domain, and then recruits RNA polymerase Pol IV to the specific site to synthesize single strand RNA (ssRNA)[50,51]. It was found that in Arabidopsis, this ssRNA, called P4RNA, is a precursor of 24nt siRNA. ssRNA is synthesized into double-stranded RNA (dsRNA) by RNA-dependent RNA polymerase 2 (RDR2) [52,53,54,55]. It is further cleaved by the cleavage-like enzyme Dicer-like protein 3 (DCL3) into 24nt siRNA, which requires the action of RNA methyltransferase HUA ENHANCER 1 (HEN1) to prevent degradation by other nucleases and maintain stability. The mature 24nt siRNA is loaded onto the effector protein Argonaute 4 (AGO4), mainly AGO4, and degrades the new strand generated by the action of RDR2 for pairing. In the second step, transcription to produce scaffold RNA. While the stable 24nt siRNA is loaded onto AGO4 [53], the scaffold RNA is transcribed from the DNA damage repair (DDR) protein complex (DRD1/ DMS3/ RDM1), which interacts with the suppressor of Variegation Homologous2/9 (SUVH2/9) protein to attract Pol V to the specific site[56,57]. After complementary pairing of siRNA bases [58], it recruits domain rearranged methylase 2 (DRM2) to complete the de novo methylation through protein catalysis of multiple RdDM pathways. The main difference between non-classical RdDM and classical RdDM is the small RNAs (sRNAs) that mediate methylation [59], i.e., sRNAs other than the 24-nt heterochromatic siRNA (hetsiRNA) can also mediate DNA methylation to occur [60,61,62,63]. In addition, a few scaffold RNAs can also be obtained by Pol II transcription [64], AGOs other than AGO4 have also been partially found to mediate DNA methylation[65,66].
4.1.2. Maintenance of Methylation
Maintenance methylation is performed based on semi-conserved replication, i.e., when the parental chain originally contains a methylation site, the methylation modification occurs on the new synthetic chain paired with it. By this semi-conserved replication, maintenance methylation allows methylation to occur at two symmetric sites, CG and CHG, but maintenance of methylation at CHH asymmetric sites can only occur by de novo methylation [67]. Further, CG-type methylation is thought to be maintained by a simple replication mechanism that allows methylation to occur [49], whereas CHG-type methylation is more complex and requires maintenance of methylation through a combination of H3K9-containing and SRA-containing proteins [49,68,69] But the two are not independent of each other, they influence each other, for example, CG methylation can act to maintain CHG methylation, while the specific site of CHG methylation can determine CG methylation [70]. The maintenance of DNA methylation in plants is associated with cytosine sequences and is regulated by different mechanisms catalyzed by DNA methylation transferases (Figure 2). One of the first homologous mammalian proteins catalyzing CG site methylation in plants, methyltransferase 1 (MET1) was identified (Figure 2A) [71]. Chromomethylase (CMT)-specific transferases that maintain CHG methylation are endemic in plants [72]. Specifically, it was demonstrated that the chromethylase3 (CMT3) with or without the loss of SUVH4 influenced whether DNA methylation levels were significantly lower or higher [73]. This suggests that CMT3 and SUVH4 are associated with CHG methylation. The histone methyltransferase SUVH4 structural domain binds to the hemi methylated site through its SRA structure, causing histone H3K9 in this site region to undergo and methylation to produce H3K9me2, thereby recruiting CMT3 to interact with the specific site, causing CMT3 to bind to nucleosomes, hemi methylation to become fully methylated and maintain the original DNA methylation[56,74](Figure 2B). It has been shown that the active state of the chromatin remodeling factor decreased and methylation 1 (DDM1) is important for the maintenance of MET1 and CMT3, two methylation transferases [75]. DRM2 and CMT2 maintain the methylation of asymmetric CHH through different pathways, with DRM2 maintaining the methylation status of the RdDM target region through the de novo methylation pathway (Figure 2C) [76]. While CMT2 catalyzes the methylation of CHH containing histone H1 and heterochromatin [74]. In addition, it has also been shown that MET1 and CMT3 are involved in the maintenance of asymmetrically methylated CHH[57,77]. However, overall, the RdDM pathway plays a crucial role in the maintenance of CHH methylation.
4.1.3. Demethylation
DNA methylation can inhibit biological gene regulation, and there must be a mechanism in the organism that dynamically balances with methylation to maintain a stable state, and this mechanism is demethylation. Demethylation refers to the change of reverting a site originally modified with methylation to cytosine. It may be classified into two types based on the mechanism: passive demethylation and active demethylation [78].
Passive demethylation
Passive demethylation inhibits the maintenance of de novo methylation and symmetric site methylation [79]. After passive DNA demethylation acts on DNA replication, when the DNA strand with methylation modification is replicated semi-conservatively, the methylation-dependent DNA transferase activity decreases or the concentration does not reach the required level, and the corresponding site where the methylation modification occurs is still cytosine, resulting in the loss of methylation of the newly synthesized strand [80]. The final level of DNA methylation in the organism is reduced [3]. However, this passive demethylation based on semi-conserved replication is far from meeting the need to inhibit DNA methylation and prevent gene silencing, so active demethylation is still the main way to cope with environmental changes.
Active demethylation
Active demethylation is a specific enzymatic reaction involving DNA glycosylases, cleavage enzymes such as repressor of silencing1 (ROS1), demeter (DME), demeter-like2 (DML2), and demeter-like3 (DML3) enzymes[81,82]. In this process, the E3 ligase enhances the stability of ROS1 to advance the reaction [83], recognizes the 5-mC at the site of DNA methylation and then hydrolyzes it to break the glycosidic bond and remove the methylated cytosine from the DNA backbone [84]. In combination with the base excision repair (BER) mechanism, the synthesized unmethylated cytosine is used to complete the repair by completing the gap to achieve active demethylation[80,85]. This is also consistent with Ikeda et al [86] and Zhu’ s findings [87]. As shown in Figure 3 , there are currently three different pathways to achieve active demethylation, the first of which is mediated by the first DNA active demethylation complex identified in plants, where the structural domain protein neuronal pentraxin 1(NPX1) and the methyl-CpG-binding domain protein 9 (MBD9) can preferentially recognize acetylated histone marks established by increased DNA methylation 1 (IDM1) on CG-type methylation, thereby recruiting the INO80 chromatin remodeling complex SWR1 to specific chromatin to deposit variant H2A of histone H2A.Z, which further recruits the ROS1 to specific target sites to complete demethylation [88]. The second one is mediated by RWD40, the first DNA demethylation complex containing DNA demethylase ROS1 found in Arabidopsis. ROS1 recruits structural domain protein RWD40 to specific sites and interacts with DNA methylation binding protein RMB1, zinc finger, and structural domain protein RHD1 to form the RWD40 complex, which regulates endogenous site methylation of all methylation types. It can also regulate the gene expression level of ROS1 through the ROS1 gene promoter, thus completing the active DNA demethylation to regulate DNA methylation level [89]. Third, the AGENET domain-containing protein 3 (AGDP3) identified by forward genetic screening recognizes and binds to the methylated histone H3K9me2 on the one hand, on the other hand, recruits ROS1 to target the genomic site, and causes the methylated site to be demethylated by base excision repair [90].
4.2. Pattern Variation and Genetic Characteristics
Factors that cause changes in DNA methylation are classified as endogenous and exogenous [91]. Exogenous factors are environmental changes that have a significant impact on an organism's ability to grow and are categorized as biotic and abiotic stresses. Endogenous factors are methylation changes brought on by changes at the genetic level, such as transposon insertions and deletions, chromosome rearrangements, and mutations in methylation-related factors. However, it has been suggested that exogenous factors are more influential than endogenous factors for heritable DNA methylation changes from a long evolutionary perspective [92]. The level and status of DNA methylation is not fixed in an organism, and there is a distinction between "transient" and "long-lasting". The majority of cases are "transient" and are dynamically regulated to meet the needs of the organism in response to changes in DNA methylation through the involvement of a series of related enzymes that target specific sites through different pathways [67]. The state and level of a dynamic balance of DNA methylation, which can be passed on through DNA replication as a relatively stable imprint of epistatic modifications, is generally in a relatively stable state in the organism, but can be changed when stimulated and passed on to offspring, called stress memory, which is a "long-lasting" situation [93]. Depending on the duration of the memory, it can be divided into short-term somatic memory, which is caused by physiology and metabolism for a few days or weeks, and long-term intergenerational stress memory, which is inherited through mitosis and meiosis[49,94].
In the study of Williams [95], Arabidopsis thaliana regulates 5-mC glycosylases through corresponding methylation genes as a way to respond to stress and to inherit this memory across generations, giving offspring the ability to stably cope with the associated resistance.
5. Effect of DNA Methylation on Plant Response to Drought Stress
Since plants are rooted to a single point and they cannot change position to escape environmental challenges. This makes abiotic stresses such as drought, one of the major limiting factors, for the growth and development and production of most plants worldwide [96]. Drought stress can disrupt the relatively stable equilibrium built up in plants, causing disruptions at the molecular level, resulting in physiological disorders, further hindering growth and development, affecting key indicators such as yield, and even causing plant death [67]. In contrast, plants themselves can also protect themselves against external damage by re-establishing the regulatory mechanisms of cellular homeostasis. Studies have demonstrated that drought stress can induce significant changes in DNA methylation levels, activate related signaling pathways, alter the expression of corresponding drought genes, and affect plant growth and development[97,98]. Among the gene families responding to drought stress, methylation and demethylation were found in genes in a CG background [99],therefore, DNA methylation is thought to be involved in the regulation of plant drought stress response, and most DNA methylation variants can be transmitted from generation to generation, altering the physiological and ecological changes of plant growth and development to adapt to the environment[3,4].
5.1. Effect of DNA Methylation on Plant Growth and Development and Stress Resistance
DNA methylation is prevalent in the whole genome of plants and acts in different tissue regions with different methylation patterns at different developmental periods, which in turn specifically regulates different developmental stages, allowing specific genes to be expressed in specific developmental stages or suppressing the expression of specific genes, thus ensuring cell differentiation and normal plant growth and development [67]. Candaele et al. found that DNA methylation transferase in maize leaves, after the cells undergo a gradual movement of spatial gradients, makes CG and CHG methylation of different backgrounds occur in the division , transition, extension, and maturation zone , proving that DNA methylation plays an important role in the growth and development of maize leaves [100].
In recent studies, it has been found that biological clocks are closely associated with DNA methylation. The biological clock is a complex regulatory system derived by plants themselves over a long period of evolution, by which organisms can sense the changing patterns of their surroundings and adapt to environmental changes to survive [101] . DNA methylation, however, can directly affect the biological clock of plants and participate in the regulatory signaling network at the molecular level to meet and respond to the needs of plant growth and development and stress, etc. [102]. It has been evaluated in many studies that DNA methylation can regulate many key developmental stages of plants, such as flowering, immunity, maturation, etc., through the biological clock [103,104,105]. And in tomato [106] , strawberry [107] , sweet orange [108] and pepper [109], it has been found that DNA methylation changes dynamically with the growth and development of the organism, and the corresponding changes in methylation levels can in turn act on the organism to promote or inhibit its maturation. It has also been found that inhibition of DNA methylation transferase can significantly prolong the biological clock in Arabidopsis [110] .
Application of zebularine demethylation altered the development of drought-stressed parental polygonum seedlings, with significant changes in leaf area, root length, and biomass [111] ; application of methylation inhibitor to spring wheat resulted in significant phenotypic changes, reduced malondialdehyde (MDA) content, increased proline and soluble sugar content, activated superoxide dismutase (SOD), peroxidase (POD) and catalase (CAT) enzyme activities under salt stress, and caused plant dwarfing, and significantly improved antioxidant capacity of wheat leaves under salt stress [112]. The exogenous application of the DNA methylation inhibitor 5-azadC to potato significantly inhibited the growth and development of potato, and the phenotype was significantly different from that of the control, with a significant decrease in plant dry weight, plant height, number of leaves and total root length, and a significant increase in SOD and POD activity to alleviate the abiotic stress [113]. Meanwhile, transcriptomic studies revealed that genes of the MAPK signaling pathway, glutathione metabolism, glycolysis/gluconeogenesis, phosphatidylinositol metabolism and phytohormone signaling pathways in potato plants responded to both drought stress and demethylation treatments [114].
5.2. Progress of DNA Methylation Involved in Drought Stress Response
DNA methylation is closely related to plant growth and development. Under abiotic stresses such as drought, it regulates the expression of related genes and activates related pathways or signals through dynamic patterns and changes in levels [4]. Under abiotic stresses such as drought, DNA methylation can be used to regulate the expression of related genes, activate related pathways or signals through dynamic patterns and level changes, and cause physiological and morphological changes in plant growth and development to cope with the damage caused by stress. It has been shown that the polymorphic changes in DNA methylation sites induced by drought stress accounted for 12.1% of the genome-wide DNA methylation sites, thus affecting growth and development in response to drought stress, and 29% of the methylation was retained even after the subsequent stress was removed [115]. Therefore, DNA methylation is very important for plant response to drought stress. Table 1 shows the current studies on DNA methylation changes in different plants subjected to drought stress.
Based on previous studies, we found that plants have DNA methylation level changes in response to drought stress, which is directly reflected in a more pronounced proportion of methylation changes compared with control material treated with regular moisture. In particular, most of the increase in DNA methylation levels occurred genome-wide, specifically, two studies in Table 1 showed a genome-wide decrease in DNA methylation levels, and seven studies showed an increase in DNA methylation levels; whereas the increase or decrease in methylation levels of the internal part of the promoter was specifically determined by the positive or negative regulation of the gene under drought stress; the proportion of changes mostly ranged from 10%-20%, but some showed only slight changes, which may be related to tissue specificity; In contrast, the proportions and trends of methylation sites in the three contexts were different, with the trends of symmetric methylation sites such as CG and CHG remaining the same, whereas most of the CHH asymmetric methylation sites were opposite to the symmetric methylation sites, which might be more sensitive to drought environment.
6. Conclusions and Outlook
Abiotic stresses such as drought have a huge impact on plants and can affect a wide range of plant growth and development, morphological indicators, yield, etc. Coupled with the fact that drought stress, an environmental problem, is a global issue, research related to drought tolerance and drought resistance in plants has been one of the key focuses of researchers. With the research of epigenetics becoming a new topical issue in recent years and making great research progress, it is able to produce intergenerationally transmissible changes in phenotypic traits without altering the gene sequences. As one of the important modification modes, DNA methylation has been found to respond to a variety of crops in the face of drought and other abiotic stresses through changing patterns and levels, mainly through the increase of methylation throughout the genome, so that the expression of the corresponding genes is down-regulated for regulation, and through a variety of pathways, such as biological clocks, regulating the plant's growth and development, and altering the morphology and physiology of plants as a means of adapting to the changes in the environmental conditions or to survive in the extreme conditions.
However, more specific studies, such as the molecular mechanisms of DNA methylation in response to drought stress, including the transmission of signaling molecules, the activation of related pathways and the investigation of transcription factors, have not been fully investigated; and how DNA methylation occurs, in response to environmental stress changes from short-term genetic effects to long-term genetic effects have not been clearly studied [6]. The study of DNA methylation in response to environmental stress has not yet been clarified. In lieu of the global problem of drought and the challenge of food security, the combination of gene editing, molecular marker-assisted selection and other modern molecular breeding tools are of great significance to the research for the development of drought-resistant varieties of plants and crops and germplasm innovation, which can enrich new strategies for plant adaptation to adverse environments to solve the problem of food security in the changing climate of the planet [79].
Author Contributions
Conceptualization, Y.F. and C.S.; writing—original draft preparation, Y.F. and C.S.; writing—review and editing, K.Y., I.H., E.M.G., P.K., Z.B. and P.Y.; visualization, P.L. and Z.L.; supervision, J.B. and Y.L.; funding acquisition, C.S. and J.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (32060502), the Gansu Science and Technology fund (21JR7RA804 and 23JRRA1414), the State Key Laboratory of Aridland Crop Science of China (GSCS-2022-Z01).
Acknowledgments
We thank Kazim Ali (National Institute for Genomics and Advanced Biotechnology, National Agricultural Research Centre, Park Road, Islamabad 45500, Pakistan) for linguistic and grammatical corrections to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A role for epigenetic regulation in the adaptation and stress responses of non-model plants. Front. Plant Sci. 2019, 10, 246. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.P.; Serra, T.; Figueiredo, D.D.; Barros, P.; Lourenco, T.; Chander, S.; Oliveira, M.M.; Saibo, N.J. Transcription regulation of abiotic stress responses in rice: A combined action of transcription factors and epigenetic mechanisms. OMICS 2011, 15, 839–857. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Zhang, S.; Niu, Y.; Tang, Q.; Wei, D.; Wang, Z. Advances in research on the mechanism of dna methylation in plants. Chinese Journal of Biotechnology 2020, 36, 838–848. [Google Scholar] [PubMed]
- Hongyang, G.; Danyun, X.; Liangyun, Z.; Bi, L.; Quan, Y. Advances in dna methylation in plants. Hubei Forestry Science and Technology 2020, 49, 6. [Google Scholar]
- Lijun, D. The analysis of dna methylation in microrna promoters and the biological fuction of atwrky30 transcription factor in arabidopsis thaliana. 硕士 Thesis, Shandong Agricultural University, 2012.
- Meiling, Z.; Zhiqiang, X.; Wenquan, W. Research progress in dna methylation in plant population. Chinese Journal of Tropical Agriculture 2021, 41, 49–59. [Google Scholar]
- Jaleel, C.A.; Manivannan, P.; Kishorekumar, A.; Sankar, B.; Gopi, R. , Somasundaram, R., & Panneerselvam, R. Alterations in osmoregulation, antioxidant enzymes and indole alkaloid levels in catharanthus roseus exposed to water deficit. Colloids and Surfaces, B. Biointerfaces 2007, 59, 150–157. [Google Scholar]
- Paramasivam, M.; Cheruth, A.J.; Ramamurthy, S.; Rajaram, P. Osmoregulation and antioxidant metabolism in drought-stressed helianthus annuus under triadimefon drenching. Comptes rendus - Biologies, 2008; 331, 418–425. [Google Scholar]
- Atighi, M.R.; Verstraeten, B.; De Meyer, T.; Kyndt, T. Genome-wide dna hypomethylation shapes nematode pattern-triggered immunity in plants. New Phytol. 2020, 227, 545–558. [Google Scholar] [CrossRef]
- Rajkumar, M.S.; Shankar, R.; Garg, R.; Jain, M. Bisulphite sequencing reveals dynamic dna methylation under desiccation and salinity stresses in rice cultivars. Genomics 2020, 112, 3537–3548. [Google Scholar] [CrossRef]
- Malabarba, J.; Windels, D.; Xu, W.; Verdier, J. Regulation of dna (de)methylation positively impacts seed germination during seed development under heat stress. Genes 2021, 12, 457. [Google Scholar] [CrossRef]
- Guo, H.; Wu, T.; Li, S.; He, Q.; Yang, Z.; Zhang, W.; Gan, Y.; Sun, P.; Xiang, G.; Zhang, H.; Deng, H. The methylation patterns and transcriptional responses to chilling stress at the seedling stage in rice. Int. J. Mol. Sci. 2019, 20, 5089. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, J.; Sanders, D.; Qian, S.; Ren, W.; Song, J.; Liu, F.; Zhong, X. Uvr8 interacts with de novo dna methyltransferase and suppresses dna methylation in arabidopsis. Nat. Plants 2021, 7, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Tan, M.; Wang, L.; Zhao, W.; You, J.; Wang, L.; Yan, X.; Wang, W. The pattern of alternative splicing and dna methylation alteration and their interaction in linseed (linum usitatissimum l.) Response to repeated drought stresses. Biol. Res. 2023, 56, 12. [Google Scholar] [CrossRef]
- Johnson, T.B.; Coghill, R.D. Researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacillus1. J. Am. Chem. Soc. 1925, 47, 2838–2844. [Google Scholar] [CrossRef]
- Bewick, A.J.; Schmitz, R.J. Gene body dna methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Zhu, J.K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Boris, F.V.; Vasili, V.A. Dna methylation in higher plants: Past, present and future. BBA - Gene Regulatory Mechanisms 2011, 1809, 360–368. [Google Scholar]
- Gallusci, P.; Hodgman, C.; Teyssier, E.; Seymour, G.B. Dna methylation and chromatin regulation during fleshy fruit development and ripening. Front. Plant Sci. 2016, 7, 807. [Google Scholar] [CrossRef] [PubMed]
- Ian, R.H.; Steven, E.J. Epigenetic inheritance in plants. Nature: International weekly journal of science 2007, 447, 418–424. [Google Scholar]
- Bird, A. Dna methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of arabidopsis thaliana dna methylation uncovers an interdependence between methylation and transcription. Nature Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef]
- Cao, Q.; Huang, L.; Li, J.; Qu, P.; Tao, P.; Crabbe, M.; Zhang, T.; Qiao, Q. Integrated transcriptome and methylome analyses reveal the molecular regulation of drought stress in wild strawberry (fragaria nilgerrensis). BMC Plant Biol. 2022, 22, 613. [Google Scholar] [CrossRef] [PubMed]
- Steward, N.; Kusano, T.; Sano, H. Expression of zmmet1, a gene encoding a dna methyltransferase from maize, is associated not only with dna replication in actively proliferating cells, but also with altered dna methylation status in cold-stressed quiescent cells. Nucleic Acids Res. 2000, 28, 3250–3259. [Google Scholar] [CrossRef] [PubMed]
- Dhar, M.K.; Vishal, P.; Sharma, R.; Kaul, S. Epigenetic dynamics: Role of epimarks and underlying machinery in plants exposed to abiotic stress. Int. J. Genomics 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yun-Lei, Z.H.; Wang, Y.W.; Fan, J.; Bao, X.; Yan, S. L. Review of dna methylation and plant stress-tolerance. Acta Botanica Boreali-Occidentalia Sinica 2009, 29, 1479–1489. [Google Scholar]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; Ecker, J.R. Genome-wide high-resolution mapping and functional analysis of dna methylation in arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Kakutani, T. What triggers differential dna methylation of genes and tes: Contribution of body methylation? Cold Spring Harb Symp Quant Biol 2012, 77, 155–160. [Google Scholar] [CrossRef] [PubMed]
- van der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colome-Tatche, M.; Johannes, F. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6676–6681. [Google Scholar] [CrossRef]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledo, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Reyna-Lopez, G.E.; Simpson, J.; Ruiz-Herrera, J. Differences in dna methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol Gen Genet 1997, 253, 703–710. [Google Scholar] [CrossRef]
- Chwialkowska, K.; Korotko, U.; Kosinska, J.; Szarejko, I.; Kwasniewski, M. Methylation sensitive amplification polymorphism sequencing (msap-seq)-a method for high-throughput analysis of differentially methylated ccgg sites in plants with large genomes. Front. Plant Sci. 2017, 8, 2056. [Google Scholar] [CrossRef]
- Kuo, K.C.; Mccune, R.A.; Gehrke, C.W.; Midgett, R.; Ehrlich, M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in dna. Nucleic Acids Res. 1980, 8, 4763–4776. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.W.; Harding, K.; Bremner, D.H.; Souch, G.; Green, J.; Lynch, P.T.; Grout, B.; Benson, E.E. Hplc analysis of plant dna methylation: A study of critical methodological factors. Plant Physiol. Biochem. 2005, 43, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Alessandra, B. Analysis of the methylation status of imprinted genes based on methylation-specific polymerase chain reaction combined with denaturing high-performance liquid chromatography. Methods 2002, 27, 139–143. [Google Scholar]
- Zhang, L.; Zhang, L.; Zhou, K.; Ye, X.; Zhang, J.; Xie, A.; Chen, L.; Kang, J.X.; Cai, C. Simultaneous determination of global dna methylation and hydroxymethylation levels by hydrophilic interaction liquid chromatography-tandem mass spectrometry. J Biomol Screen 2012, 17, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, X.; Cao, H.; Xu, H.; Xu, Q.; Deng, X. Dynamic changes in methylome and transcriptome patterns in response to methyltransferase inhibitor 5-azacytidine treatment in citrus. DNA Res. 2017, 24, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Zou, M.; Zhang, S.; Feng, B.; Wang, W. Afsm sequencing approach: A simple and rapid method for genome-wide snp and methylation site discovery and genetic mapping. Sci Rep 2014, 4, 7300. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Lu, C.; Zhang, S.; Chen, Q.; Sun, X.; Ma, P.; Hu, M.; Peng, M.; Ma, Z.; Chen, X.; Zhou, X.; Wang, H.; Feng, S.; Fang, K.; Xie, H.; Li, Z.; Liu, K.; Qin, Q.; Pei, J.; Wang, S.; Pan, K.; Hu, W.; Feng, B.; Fan, D.; Zhou, B.; Wu, C.; Su, M.; Xia, Z.; Li, K.; Wang, W. Epigenetic map and genetic map basis of complex traits in cassava population. Sci Rep 2017, 7, 41232. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Zhang, S.; Wen, M.; Lu, C.; Sun, Y.; Zou, M.; Wang, W. Construction of an ultrahigh-density genetic linkage map for jatropha curcas l. And identification of qtl for fruit yield. Biotechnol. Biofuels 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Rein, T.; Depamphilis, M.L.; Zorbas, H. Identifying 5-methylcytosine and related modifications in dna genomes. Nucleic Acids Res. 1998, 26, 2255–2264. [Google Scholar] [CrossRef]
- Frommer, M.; Mcdonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual dna strands. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Platt, A.; Gugger, P.F.; Pellegrini, M.; Sork, V.L. Genome-wide signature of local adaptation linked to variable cpg methylation in oak populations. Mol. Ecol. 2015, 24, 3823–3830. [Google Scholar] [CrossRef]
- Bianchessi, V.; Vinci, M.C.; Nigro, P.; Rizzi, V.; Farina, F.; Capogrossi, M.C.; Pompilio, G.; Gualdi, V.; Lauri, A. Methylation profiling by bisulfite sequencing analysis of the mtdna non-coding region in replicative and senescent endothelial cells. Mitochondrion 2016, 27, 40–47. [Google Scholar] [CrossRef]
- Liu, T.; Li, Y.; Duan, W.; Huang, F.; Hou, X. Cold acclimation alters dna methylation patterns and confers tolerance to heat and increases growth rate in brassica rapa. J. Exp. Bot. 2017, 68, 1213–1224. [Google Scholar] [CrossRef]
- Karger, B.L. High-performance capillary electrophoresis. Nature 1989, 339, 641–642. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Siejka-Zielińska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Böckler, B.; Song, C. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Vaisvila, R.; Ponnaluri, V.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; Samaranayake, M.; Samuelson, J.C.; Church, H.E.; Tamanaha, E.; Correa, I.J.; Pradhan, S.; Dimalanta, E.T.; Evans, T.J.; Williams, L.; Davis, T.B. Enzymatic methyl sequencing detects dna methylation at single-base resolution from picograms of dna. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef]
- Kawashima, T.; Berger, F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014, 15, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Mosher, R.A. Rna-directed dna methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Law, J.A.; Vashisht, A.A.; Wohlschlegel, J.A.; Jacobsen, S.E. Shh1, a homeodomain protein required for dna methylation, as well as rdr2, rdm4, and chromatin remodeling factors, associate with rna polymerase iv. PLoS Genet. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Smith, L.M.; Pontes, O.; Searle, I.; Yelina, N.; Yousafzai, F.K.; Herr, A.J.; Pikaard, C.S.; Baulcombe, D.C. An snf2 protein associated with nuclear rna silencing and the spread of a silencing signal between cells in arabidopsis. Plant Cell 2007, 19, 1507–1521. [Google Scholar] [CrossRef]
- Zhai, J.; Bischof, S.; Wang, H.; Feng, S.; Lee, T.F.; Teng, C.; Chen, X.; Park, S.Y.; Liu, L.; Gallego-Bartolome, J.; Liu, W.; Henderson, I.R.; Meyers, B.C.; Ausin, I.; Jacobsen, S.E. A one precursor one sirna model for pol iv-dependent sirna biogenesis. Cell 2015, 163, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ebright, Y.W.; Yu, B.; Chen, X. Hen1 recognizes 21-24 nt small rna duplexes and deposits a methyl group onto the 2' oh of the 3' terminal nucleotide. Nucleic Acids Res. 2006, 34, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ji, L.; Huang, Q.; Vassylyev, D.G.; Chen, X.; Ma, J.B. Structural insights into mechanisms of the small rna methyltransferase hen1. Nature 2009, 461, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Ausin, I.; Johnson, L.M.; Vashisht, A.A.; Zhu, J.K.; Wohlschlegel, J.A.; Jacobsen, S.E. A protein complex required for polymerase v transcripts and rna- directed dna methylation in arabidopsis. Curr. Biol. 2010, 20, 951–956. [Google Scholar] [CrossRef]
- Johnson, L.M.; Du, J.; Hale, C.J.; Bischof, S.; Feng, S.; Chodavarapu, R.K.; Zhong, X.; Marson, G.; Pellegrini, M.; Segal, D.J.; Patel, D.J.; Jacobsen, S.E. Sra- and set-domain-containing proteins link rna polymerase v occupancy to dna methylation. Nature 2014, 507, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Ream, T.S.; Haag, J.R.; Pikaard, C.S. Rna polymerase v transcription guides argonaute4 to chromatin. Nature Genet. 2009, 41, 630–634. [Google Scholar] [CrossRef] [PubMed]
- G, H.; Q, X. Mechanism of de novo dna methylation in plants. Chin Sci Bull 2021, 66, 1821–1834. [Google Scholar]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. Dna methylation mediated by a microrna pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Panda, K.; Mccue, A.D.; Slotkin, R.K. Arabidopsis rna polymerase iv generates 21-22 nucleotide small rnas that can participate in rna-directed dna methylation and may regulate genes. Philos. Trans. R. Soc. B-Biol. Sci. 2020, 375, 20190417. [Google Scholar] [CrossRef]
- Wu, L.; Mao, L.; Qi, Y. Roles of dicer-like and argonaute proteins in tas-derived small interfering rna-triggered dna methylation. Plant Physiol. 2012, 160, 990–999. [Google Scholar] [CrossRef]
- Ye, R.; Chen, Z.; Lian, B.; Rowley, M.J.; Xia, N.; Chai, J.; Li, Y.; He, X.J.; Wierzbicki, A.T.; Qi, Y. A dicer-independent route for biogenesis of sirnas that direct dna methylation in arabidopsis. Mol. Cell 2016, 61, 222–235. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, Z.; Li, S.; Yu, B.; Liu, J.; Chen, X. Intergenic transcription by rna polymerase ii coordinates pol iv and pol v in sirna-directed transcriptional gene silencing in arabidopsis. Genes Dev. 2009, 23, 2850–2860. [Google Scholar] [CrossRef]
- Mccue, A.D.; Panda, K.; Nuthikattu, S.; Choudury, S.G.; Thomas, E.N.; Slotkin, R.K. Argonaute 6 bridges transposable element mrna-derived sirnas to the establishment of dna methylation. Embo J. 2015, 34, 20–35. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Guo, X.; Wang, X.J.; Zhang, X. Arabidopsis ago3 predominantly recruits 24-nt small rnas to regulate epigenetic silencing. Nat. Plants 2016, 2, 16049. [Google Scholar] [CrossRef] [PubMed]
- Hui-Wen, H.; Ying, F. Review of plant development and epigenetics. Modern Agricultural Science and Technology 2018, 47–50. [Google Scholar]
- Bernatavichute, Y.V.; Zhang, X.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide association of histone h3 lysine nine methylation with chg dna methylation in arabidopsis thaliana. PLoS ONE 2008, 3, e3156. [Google Scholar] [CrossRef] [PubMed]
- Lianna, M.J.; Magnolia, B.; Xiaoyu, Z.; Edward, K.; Ian, H.; Judy, C.; Steven, E.J. The sra methyl-cytosine-binding domain links dna and histone methylation. Curr. Biol. 2007, 17, 379–384. [Google Scholar]
- Hume, S.; Maxim, V.C.G.; Suhua, F.; Yana, V.B.; Steven, E.J. Comprehensive analysis of silencing mutants reveals complex regulation of the arabidopsis methylome. Cell 2015, 161, 352–364. [Google Scholar]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis met1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [CrossRef]
- Lindroth, A.M.; Saarikoski, P.; Flygh, G.; Clapham, D.; Grönroos, R.; Thelander, M.; Ronne, H.; von Arnold, S. Two s-adenosylmethionine synthetase-encoding genes differentially expressed during adventitious root development in pinus contorta. Plant Mol.Biol. 2001, 46, 335–346. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Kovac, K.A. Plant dna methyltransferases. Plant Mol.Biol. 2000, 43, 189–201. [Google Scholar] [CrossRef]
- Jiamu, D.; Lianna, M.J.; Martin, G.; Suhua, F.; Christopher, J.H.; Sisi, L.; Ajay, A.V.; Javier, G.; James, A.W.; Dinshaw, J.P.; Steven, E.J. Mechanism of dna methylation-directed histone methylation by kryptonite. Mol. Cell 2014, 55, 495–504. [Google Scholar]
- Zemach, A.; Kim, M.Y.; Hsieh, P.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The arabidopsis nucleosome remodeler ddm1 allows dna methyltransferases to access h1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang-Wei, L.; Chang-Rong, S.; Cui-Jun, Z.; Jin-Xing, Z.; Su-Wei, Z.; Lin, L.; She, C.; Huan-Wei, H.; Tao, C.; Xin-Jian, H. The set domain proteins suvh2 and suvh9 are required for pol v occupancy at rna-directed dna methylation loci. PLoS Genet. 2014, 10, e1003948. [Google Scholar]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-cg methylation patterns shape the epigenetic landscape in arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef]
- Christof, N. Active dna demethylation and dna repair. Differentiation 2008, 77, 1–11. [Google Scholar]
- Manman, Z.; Qingsong, Z.; Xia, L.; Mingpu, T. Research progress of dna methylation in plant response to stress. Plant Physiology Journal 2021, 57, 780–792. [Google Scholar]
- Wei, C.; Yingzeng, Y.; Feng, C.; Wenguan, Z.; Kai, S. Epigenetic modification-mediated memory for plant stress. Chinese Bulletin of Botany 2019, 54, 779–785. [Google Scholar]
- Gehring, M.; Huh, J.H.; Hsieh, T.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. Demeter dna glycosylase establishes medea polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef]
- Morales-Ruiz, T.; Ortega-Galisteo, A.P.; Ponferrada-Marín, M.I.; Martínez-Macías, M.I.; Ariza, R.R.; Roldán-Arjona, T. Demeter and repressor of silencing 1 encode 5-methylcytosine dna glycosylases. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 6853–6858. [Google Scholar] [CrossRef]
- Xiangfeng, K.; Yechun, H.; Yi-Feng, H.; Huan, H.; Xue, L.; Zhe, S.; Jian-Kang, Z. Siz1-mediated sumoylation of ros1 enhances its stability and positively regulates active dna demethylation in arabidopsis. Mol. Plant. 2020, 13, 1816–1824. [Google Scholar]
- Zhizhong, G.; Teresa, M.; Rafael, R.A.; Teresa, R.; Lisa, D.; Jian-Kang, Z. Ros1, a repressor of transcriptional gene silencing in arabidopsis, encodes a dna glycosylase/lyase. Cell 2002, 111, 803–814. [Google Scholar]
- Chun-Yu, L.; Yu, W.; Li-Nan, X. Progress on the active dna demethylation pathways and their regulation mechanisms in animals and plants. Life Science Research 2020, 24, 415–424. [Google Scholar]
- Ikeda, Y.; Kinoshita, T. Dna demethylation: A lesson from the garden. Chromosoma 2009, 118. [Google Scholar] [CrossRef]
- Jian-Kang, Z. Active dna demethylation mediated by dna glycosylases. Annu. Rev. Genet. 2009, 43. [Google Scholar]
- Nie, W.; Lei, M.; Zhang, M.; Tang, K.; Huang, H.; Zhang, C.; Miki, D.; Liu, P.; Yang, Y.; Wang, X.; Zhang, H.; Lang, Z.; Liu, N.; Xu, X.; Yelagandula, R.; Zhang, H.; Wang, Z.; Chai, X.; Andreucci, A.; Yu, J.; Berger, F.; Lozano-Duran, R.; Zhu, J. Histone acetylation recruits the swr1 complex to regulate active dna demethylation in arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 2019, 116. [Google Scholar] [CrossRef]
- Liu, P.; Nie, W.F.; Xiong, X.; Wang, Y.; Jiang, Y.; Huang, P.; Lin, X.; Qin, G.; Huang, H.; Niu, Q.; Du, J.; Lang, Z.; Lozano-Duran, R.; Zhu, J.K. A novel protein complex that regulates active dna demethylation in arabidopsis. J. Integr. Plant Biol. 2021, 63, 772–786. [Google Scholar] [CrossRef]
- Zhou, X.; Wei, M.; Nie, W.; Xi, Y.; Peng, L.; Zheng, Q.; Tang, K.; Satheesh, V.; Wang, Y.; Luo, J.; Du, X.; Liu, R.; Yang, Z.; La, H.; Zhong, Y.; Yang, Y.; Zhu, J.K.; Du, J.; Lei, M. The h3k9me2-binding protein agdp3 limits dna methylation and transcriptional gene silencing in arabidopsis. J. Integr. Plant Biol. 2022, 64, 2385–2395. [Google Scholar] [CrossRef]
- Ales, P.; Ahmed, A.; Giang, T.H.V. Hidden genetic nature of epigenetic natural variation in plants. Trends Plant Sci. 2013, 18. [Google Scholar]
- Jörg, H.; Claude, B.; Jonas, M.; Oliver, S.; Rhonda, C.M.; George, W.; Korbinian, S.; Joffrey, F.; Thomas, A.; Joy, B.; Karsten, B.; Detlef, W. Century-scale methylome stability in a recently diverged arabidopsis thaliana lineage. PLoS Genet. 2015, 11. [Google Scholar]
- Sun, C.; Ali, K.; Yan, K.; Fiaz, S.; Dormatey, R.; Bi, Z.; Bai, J. Exploration of epigenetics for improvement of drought and other stress resistance in crops: A review. Plants 2021, 10. [Google Scholar] [CrossRef]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18. [Google Scholar] [CrossRef]
- Williams, B.P.; Gehring, M. Stable transgenerational epigenetic inheritance requires a dna methylation-sensing circuit. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Hilker, M.; Schmülling, T. Stress priming, memory, and signalling in plants. Plant, cell & environment 2019, 42. [Google Scholar]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.K. Critical roles of dna demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E4511–E4519. [Google Scholar] [CrossRef]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; Di Pierro, E.A.; Gouzy, J.; Rees, D.; Guerif, P.; Muranty, H.; Durel, C.E.; Laurens, F.; Lespinasse, Y.; Gaillard, S.; Aubourg, S.; Quesneville, H.; Weigel, D.; van de Weg, E.; Troggio, M.; Bucher, E. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nature Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Ai, P.; Xue, J.; Shi, Z.; Liu, Y.; Li, Z.; Li, T.; Zhao, W.; Khan, M.A.; Kang, D.; Wang, K.; Wang, Z. Genome-wide characterization and expression analysis of myb transcription factors in chrysanthemum nankingense. BMC Plant Biol. 2023, 23, 140. [Google Scholar] [CrossRef]
- Candaele, J.; Demuynck, K.; Mosoti, D.; Beemster, G.T.S.; Inzé, D.; Nelissen, H. Differential methylation during maize leaf growth targets developmentally regulated genes. Plant Physiol. 2014, 164. [Google Scholar] [CrossRef]
- Hua, W.; Yan, W.; Baogu, L.; Lei, W. Advances in research on plant biological clocks and their regulation of growth and development. Chinese Bulletin of Botany 2018, 53, 456–467. [Google Scholar]
- Jiashuo, Z.; Hua, W.; Lei, W. The molecular networks of circadian clock-regulated plant growth and development. Plant Physiology Journal 2022, 58, 3–12. [Google Scholar]
- Wim, J.J.S.; Steven, E.J.; Carlos, A.; James, P.J.; Tetsuji, K.; Maarten, K.; Anton, J.M.P. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell 2000, 6. [Google Scholar]
- He, L.; Wu, W.; Zinta, G.; Yang, L.; Wang, D.; Liu, R.; Zhang, H.; Zheng, Z.; Huang, H.; Zhang, Q.; Zhu, J. A naturally occurring epiallele associates with leaf senescence and local climate adaptation in arabidopsis accessions. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic dna methylation in response to biotic stress. Proc. Natl. Acad. Sci. U. S. A. 2012, 109. [Google Scholar] [CrossRef]
- Zhong, S.; Fei, Z.; Chen, Y.; Zheng, Y.; Huang, M.; Vrebalov, J.; Mcquinn, R.; Gapper, N.; Liu, B.; Xiang, J.; Shao, Y.; Giovannoni, J.J. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31. [Google Scholar] [CrossRef]
- Cheng, J.; Niu, Q.; Zhang, B.; Chen, K.; Yang, R.; Zhu, J.; Zhang, Y.; Lang, Z. Downregulation of rddm during strawberry fruit ripening. Genome Biol. 2018, 19. [Google Scholar] [CrossRef]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.; Lang, Z. Global increase in dna methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. U. S. A. 2019, 116. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Chen, J.; He, Q.; Wang, Y.; Shen, H.; Sun, L. Dna methylation is involved in the regulation of pepper fruit ripening and interacts with phytohormones. J. Exp. Bot. 2020, 71. [Google Scholar] [CrossRef]
- Tian, W.; Wang, R.; Bo, C.; Yu, Y.; Zhang, Y.; Shin, G.I.; Kim, W.Y.; Wang, L. Sdc mediates dna methylation-controlled clock pace by interacting with ztl in arabidopsis. Nucleic Acids Res. 2021, 49, 3764–3780. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.J.; Sultan, S.E. Dna methylation mediates genetic variation for adaptive transgenerational plasticity. Proc. R. Soc. B-Biol. Sci. 2016, 283. [Google Scholar] [CrossRef]
- Youfang, L.; Jianguo, J.; Jingwu, Z.; Weihua, L. Preliminary studies on the involvement of dna methylation in regulating salt stress response in wheat. In 第十届全国小麦基因组学及分子育种大会: Yantai, Shandong, China, 2019, p. 1.
- Peng-Cheng, L.; Zhen-Zhen, B.; Wen-Jun, L.; Chao, S.; Jun-Lian, Z.; Jiang-Ping, B. Dna methylation involved in regulating drought stress response of potato. ACTA AGRONOMICA SINICA 2019, 45, 1595–1603. [Google Scholar]
- Peng-Cheng, L.; Zhen-Zhen, B.; Chao, S.; Tian-Yuan, Q.; Wen-Jun, L.; Yi-Hao, W.; De-Rong, X.; Yu-Hui, L.; Jun-Lian, Z.; Jiang-Ping, B. Key genes mining of dna methylation involved in regulating drought stress response in potato. ACTA AGRONOMICA SINICA 2021, 47, 599–612. [Google Scholar]
- Wang, W.; Pan, Y.; Zhao, X.; Dwivedi, D.; Zhu, L.; Ali, J.; Fu, B.; Li, Z. Drought-induced site-specific dna methylation and its association with drought tolerance in rice (oryza sativa l.). J. Exp. Bot. 2011, 62. [Google Scholar] [CrossRef]
- Alex, B.; Todd, B.; Youli, Y.; Andrey, G.; Andriy, B.; Yaroslav, I.; Jens, H.; Frederick, M.; Igor, K. Correction: Transgenerational adaptation of arabidopsis to stress requires dna methylation and the function of dicer-like proteins. PLoS ONE 2010, 5. [Google Scholar]
- Dawei, M.; Yue, W.; Peixuan, L.; Yuwei, Z.; Yao, Z.; Yu, H.; Chenjing, L.; Taicheng, J.; Liping, Y. Drought-introduced dna demethylation of atgstf14 gene. Molecular Plant Breeding 2020, 18, 6108–6113. [Google Scholar]
- Liang, D.; Zhang, Z.; Wu, H.; Huang, C.; Shuai, P.; Ye, C.; Tang, S.; Wang, Y.; Yang, L.; Wang, J.; Yin, W.; Xia, X. Single-base-resolution methylomes of populus trichocarpa reveal the association between dna methylation and drought stress. BMC Genet. 2014; 15. [Google Scholar]
- Zhou, J.; Xiao, L.; Huang, R.; Song, F.; Li, L.; Li, P.; Fang, Y.; Lu, W.; Lv, C.; Quan, M.; Zhang, D.; Du, Q. Local diversity of drought resistance and resilience in populus tomentosa correlates with the variation of dna methylation. Plant Cell Environ. 2023, 46, 479–497. [Google Scholar] [CrossRef]
- González, R.M.; Ricardi, M.M.; Iusem, N.D. Atypical epigenetic mark in an atypical location: Cytosine methylation at asymmetric (cnn) sites within the body of a non-repetitive tomato gene. BMC Plant Biol. 2011, 11. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Wei, J.; Pan, Y.; Su, C.; Zhang, X. Sppke1, a multiple stress-responsive gene confers salt tolerance in tomato and tobacco. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Garg, R.; Narayana, C.V.; Shankar, R.; Jain, M. Divergent dna methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci Rep 2015, 5. [Google Scholar] [CrossRef]
- Li, P.; Yang, H.; Wang, L.; Liu, H.; Huo, H.; Zhang, C.; Liu, A.; Zhu, A.; Hu, J.; Lin, Y.; Liu, L. Physiological and transcriptome analyses reveal short-term responses and formation of memory under drought stress in rice. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Ahmad, F.; Farman, K.; Waseem, M.; Rana, R.M.; Nawaz, M.A.; Rehman, H.M.; Abbas, T.; Baloch, F.S.; Akrem, A.; Huang, J.; Zhang, H. Genome-wide identification, classification, expression profiling and dna methylation (5mc) analysis of stress-responsive zfp transcription factors in rice (oryza sativa l.). Gene 2019, 718, 144018. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, X.; Zhang, Q.; Zheng, Q.; Yao, H.; Gu, X.; Liu, D.; Tian, X.; Wang, X.; Li, Y.; Zhu, Z. H3k36 demethylase jmj710 negatively regulates drought tolerance by suppressing myb48-1 expression in rice. Plant Physiol. 2022, 189, 1050–1064. [Google Scholar] [CrossRef]
- Sallam, N.; Moussa, M. Dna methylation changes stimulated by drought stress in aba-deficient maize mutant vp10. Plant Physiol. Biochem. 2021, 160, 218–224. [Google Scholar] [CrossRef]
- Mao, H.; Wang, H.; Liu, S.; Li, Z.; Yang, X.; Yan, J.; Li, J.; Tran, L.P.; Qin, F. A transposable element in a nac gene is associated with drought tolerance in maize seedlings. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Nehal, S.; Mounir, M.; Mohamed, Y.; Huda, M.S. Detection of dna methylation in dbf1 gene of maize inbred w64a and mutant vp 14 exposed to drought stress. Cereal Res. Commun. 2021. [Google Scholar]
- Rehman, Z. Drought stress induces differential dna methylation shift at symmetric and asymmetric cytosine sites in the promoter region of zmexpb2 gene in maize. International journal of agriculture and biology 2021, 25, 319–326. [Google Scholar] [CrossRef]
- Chwialkowska, K.; Nowakowska, U.; Mroziewicz, A.; Szarejko, I.; Kwasniewski, M. Water-deficiency conditions differently modulate the methylome of roots and leaves in barley (hordeum vulgare l.). J. Exp. Bot. 2016, 67. [Google Scholar] [CrossRef]
- Drosou, V.; Kapazoglou, A.; Letsiou, S.; Tsaftaris, A.S.; Argiriou, A. Drought induces variation in the dna methylation status of the barley hvdme promoter. J. Plant Res. 2021, 134, 1351–1362. [Google Scholar] [CrossRef]
- Moglia, A.; Gianoglio, S.; Acquadro, A.; Valentino, D.; Milani, A.M.; Lanteri, S.; Comino, C. Identification of dna methyltransferases and demethylases in solanum melongena l., And their transcription dynamics during fruit development and after salt and drought stresses. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Sharma, R.; Vishal, P.; Kaul, S.; Dhar, M.K. Epiallelic changes in known stress-responsive genes under extreme drought conditions in brassica juncea (l.) Czern. Plant Cell Reports 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Ackah, M.; Guo, L.; Li, S.; Jin, X.; Asakiya, C.; Aboagye, E.T.; Yuan, F.; Wu, M.; Essoh, L.G.; Adjibolosoo, D.; Attaribo, T.; Zhang, Q.; Qiu, C.; Lin, Q.; Zhao, W. Dna methylation changes and its associated genes in mulberry (morus alba l.) Yu-711 response to drought stress using methylrad sequencing. Plants 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Jiang, L.; Cao, F.; Liu, C.; Guo, J.; Zhang, Z.; Yue, Q.; Hou, N.; Liu, Z.; Li, X.; Tahir, M.M.; He, J.; Li, Z.; Li, C.; Ma, F.; Guan, Q. Methylation of a mite insertion in the mdrfnr1-1 promoter is positively associated with its allelic expression in apple in response to drought stress. Plant Cell 2022, 34, 3983–4006. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, P.; Jin, L.; Yv, X.; Wen, M.; Wu, S.; Liu, F.; Xu, J. Methylome and transcriptome analysis of flowering branches building of citrus plants induced by drought stress. Gene 2023, 880, 147595. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, X.; Sun, Z.; Wu, Y.; Malkodslo, M.M.; Ge, J.; Jing, Z.; Zhou, Q.; Cai, J.; Zhong, Y.; Huang, M.; Jiang, D. Dna methylation levels of tap5cs and tabadh are associated with enhanced tolerance to peg-induced drought stress triggered by drought priming in wheat. Plant Physiol. Biochem. 2023, 200, 107769. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Classical RdDM pathway-mediated de novo methylation pattern.
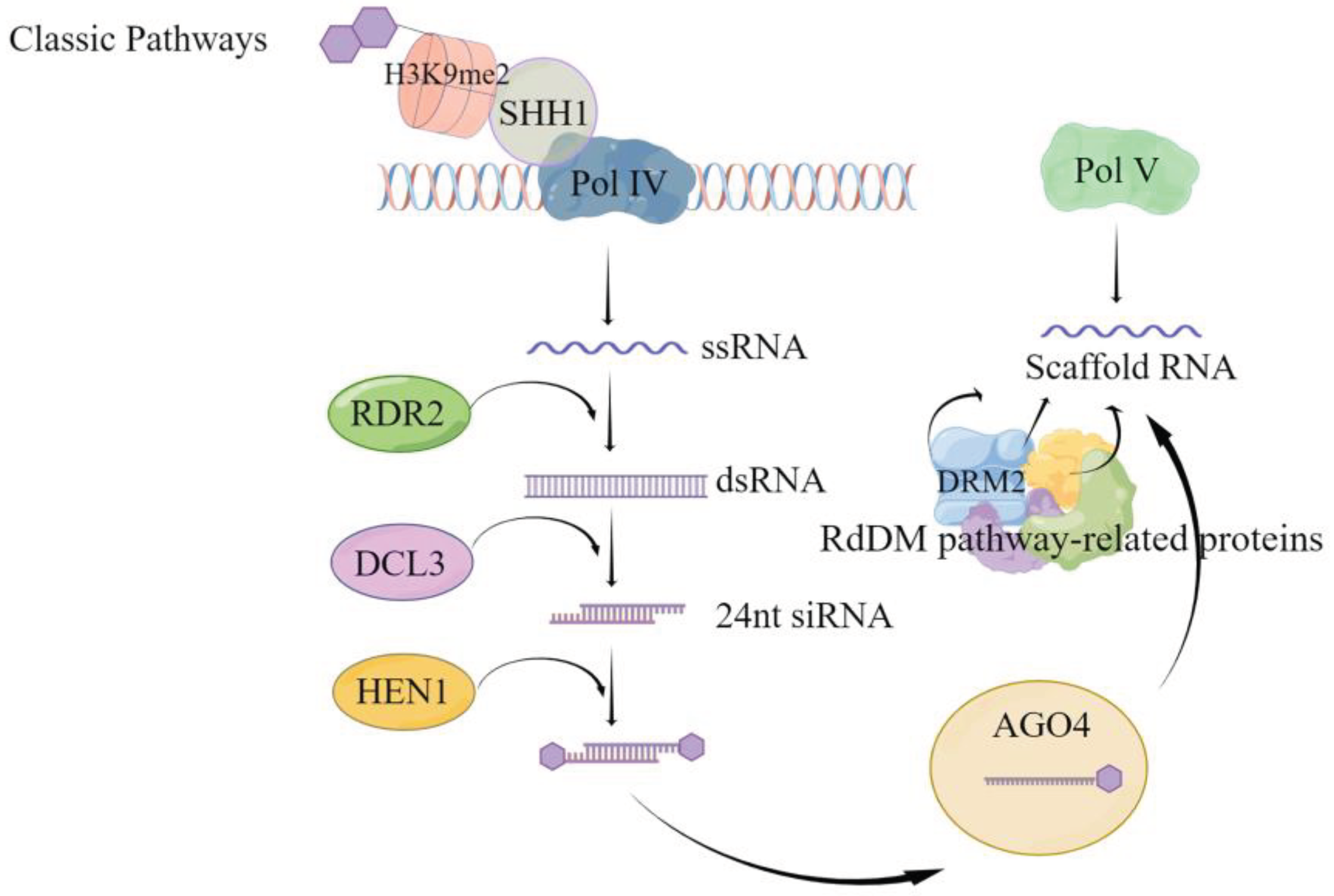
Figure 2.
Mode of action of DNA methylation transferases involved in maintaining methylation. (a) CG maintains methylation; (b) CHG maintains methylation; (c) CHH maintains methylation.
Figure 2.
Mode of action of DNA methylation transferases involved in maintaining methylation. (a) CG maintains methylation; (b) CHG maintains methylation; (c) CHH maintains methylation.
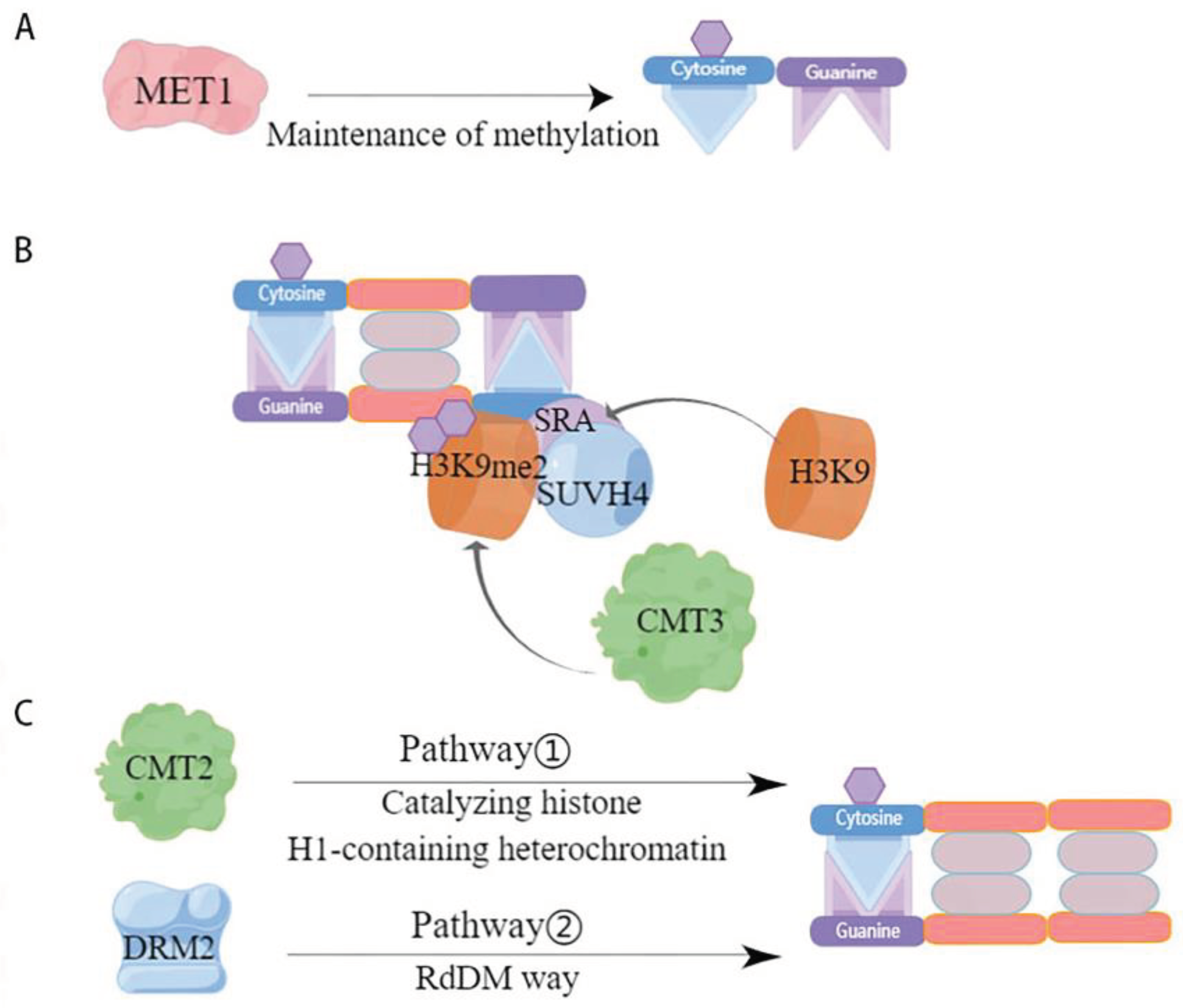
Figure 3.
Active demethylation mode of action of three different pathways.
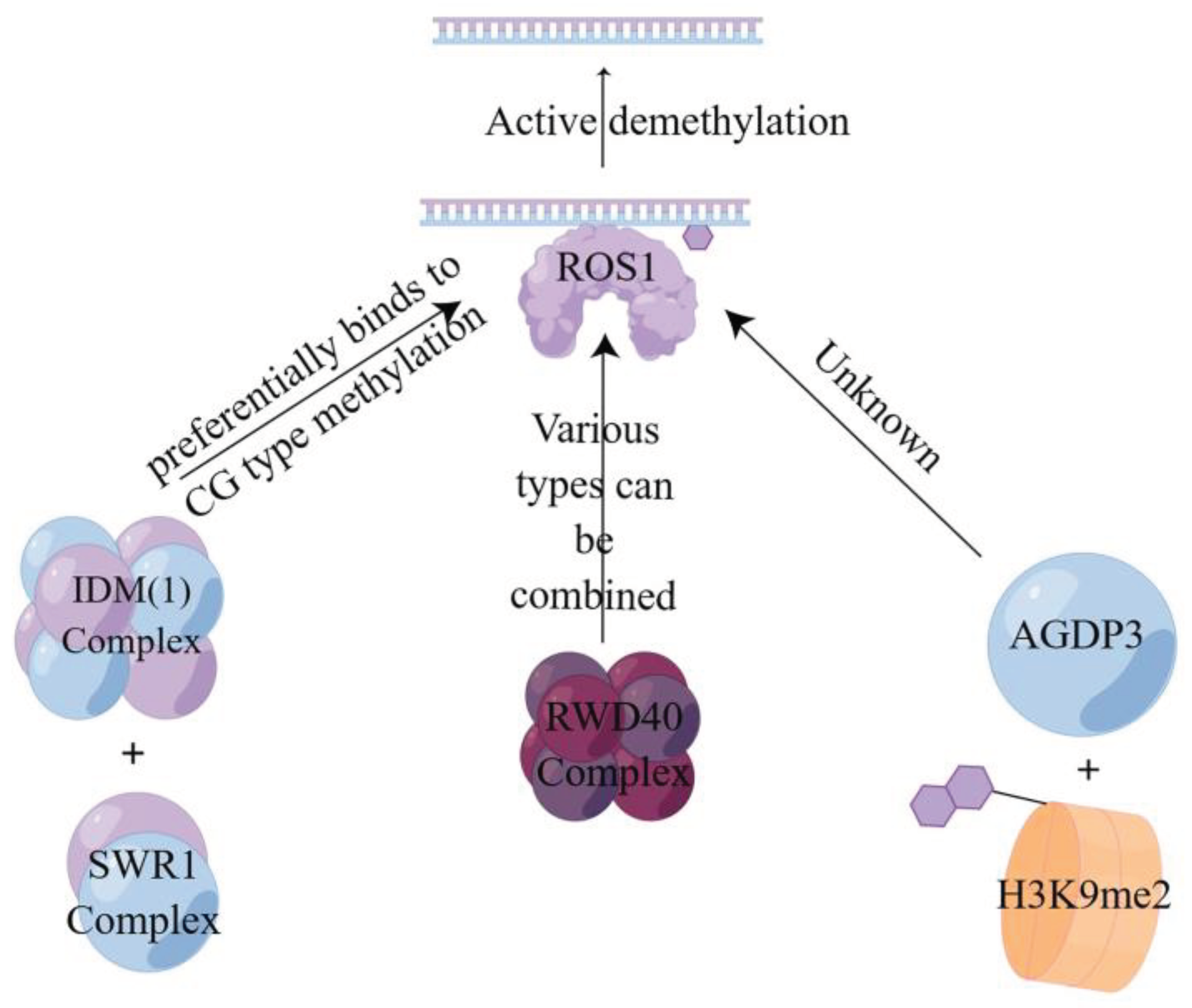

Table 1.
DNA methylation changes in different plants under drought stress.
| Species | Processing | DNA methylation changes | Related genes or access |
Associated phenotypes | References |
|---|---|---|---|---|---|
| Arabidopsis thaliana (L.) Heynh. | After 7-30dpg growth, stop water treatment for 20 days | A significant decrease in the 5-meC content | Related to DCL2/DCL3 pathway | Decreased homologous recombination frequency (Increased generally HFR, DNA hypermethylation, and higher stress tolerance) | [116] |
| After 4 weeks of growth, the treatment group stopped water for 20 days | DNA methylation levels in the promoter region of AtGSTF14 were significantly reduced |
AtGSTF14 | N.A. | [117] | |
| Populus trichocarpa | After 2 months of growth, the soil moisture content is controlled at about 10% | Significantly higher methylation levels of methylated cytosine, upstream 2kp, downstream 2kb and repetitive sequences | C2C2, WRKY, MYB, EIL gene family | N.A. | [118] |
| Populus tomentosa | After 2 months of growth, soil moisture content was controlled at 20%-25% under 37 days | Significant reduction in genomic DNA methylation levels | GATA9, LECRK-VIII.2 | Ceases leaf photosynthetic activity;Accumulation of ABA, osmolytes such as glycine betaine (BETA), proline (PRO) and osmotic regulator (ORS) | [119] |
|
Solanum lycopersicum |
Grow for 3 weeks to clean the roots and place on blotting paper under incandescent light until wilting occurs | Elevated CG methylation level in exon 1 of Asr1 and loss of methyl markers at CNN sites (mainly intron regions) | Asr1 | N.A. | [120] |
| Solanum pennellii | Seedlings are removed from the soil and placed on filter paper | The DNA of the PKE1 promoter was highly methylated in fruit and leaf | PKE1 | N.A. | [121] |
|
Oryza sativa |
Different tolerant species | Elevated levels of genomic methylation | smRNA pathway | N.A. | [122] |
| 28 ℃ air dry 80 minutes, rehydration 22h after the cycle of treatment 2 rounds | DNA methylation regulates the expression of stress memory transcripts | ABA Access Road | Relative water content was sharply dropped;the endogenous contents of these phytohormones (ABA, JA) content was increased | [123] | |
| After 2 weeks of growth, treatment with 20% PEG6000 for 12h | Genome & ZFP promoter and CDS region are highly methylated | ZFP | N.A. | [124] | |
| 1/ 2 MS medium with 20% (w/v) PEG6000 | JMJ710 demethylated H3K36me2 both in vivo and in vitro | JMJ710 | The survival rates and water loss experiment with detached leaves are higher than CK | [125] | |
| Zea mays L. | Growing for 1 month, drought treatment for 9 days | Reduced total methylation levels in the maize mutant vp10 | ABA pathway | The rapid decrease in the leaf relative water content | [126] |
| Seedlings were not watered until they had three true leaves and were re-watered for six days when significant wilting was observed. | Nearest the MITE insertion, were hypermethylated in ZmNAC111 promoter | ZmNAC111 | The leaf photosynthesis rates (PS), stomatal conductance (SC) and transpiration rates (TR) were significantly smaller than CK | [127] | |
| Growing for 1 month, drought treatment for 9 days | DNA methylation in the upstream region of the DBF1 gene | DBF1 | The average relative water content were highly significant | [128] | |
| Stop watering for 15d when growth reaches the 5-leaf stage | DNA hypermethylation at CG and CHG sites and DNA hypermethylation at CHH site in the middle of ZmEXPB2 gene promoter | ZmEXPB2 | Significant decrease in fresh weight of whole plant, 6th leaf length, stunted secondary root growth, and increased primary root length | [129] | |
| Hordeum vulgare L. | After germination, water deficit treatment for 10d | high overall DNA methylation level | HvDRM | N.A. | [130] |
| After 7d of growth, stop hydroponics for 10d | Methylation and demethylation of different regions of the HvDME promoter | HvDME | N.A. | [131] | |
| Solanum melongena L. | After 3 weeks of growth, water was stopped for 2d | Upregulation of demethylase expression | SmelMET1, SmelCMT, SmelDRM | N.A. | [132] |
| Brassica juncea | Watering was stopped for 15 d after seed germination until the leaves were yellow and curled. | Gene body methylation is increased for all genes, while promoter methylation is gene function dependent | BAX inhibitor 1, metacaspase 4, B3, DIE2/ALG10, F-box, Bcl2 |
N.A. | [133] |
| Morus alba | Growth in February (new leaves appear ah), 14d water stop | Increased genomic DNA methylation | Phenylpropanoid biosynthesis and other multi-pathways | Relative water content (RWC) was decreased, leaf lengths remained shorter | [134] |
| Malus pumila Mill. | Growing for 4 months, incubated with Hoagland solution containing 20% PEG8000 for 6h (short-term) or 15d (long-term) | Increased DNA methylation level of MdRFNR1-1 promoter | MdRFNR1-1 | The fresh weights of all calli decreased;POD and CAT activities were lower in MdRFNR1 RNAi lines than in GL-3 plants | [135] |
| Citrus | Around 18–20% soil moisture content | High global DNA methylation level | FLC、BFT | A significant increase in the flowering branches, whereas an apparent decrease in vegetative branches | [136] |
| Triticum aestivum L. | Drought primed for 24 h via the addition of 10% (-0.36 Mpa), 15% (-0.58 Mpa) and 20% PEG 6000 (-0.91 Mpa), applied with 20% PEG 6000 for 72 h at the six-leaf stage | The CG and CHG methylation rates were decreased of TaP5CS、TaBADH promoter | TaP5CS、TaBADH | Plant dry weight and leaf area were significantly reduced, significantly inhibits ΦPSII and increased ΦNPQ, higher photosynthetic rate and stomatal conductance | [137] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



