
Submitted:
18 March 2024
Posted:
22 March 2024
You are already at the latest version
Abstract
Spherical or hexagonal NaYF4:Yb,Er nanoparticles (UCNPs) with sizes of 25 nm (S-UCNPs) and 120 nm (L-UCNPs) were synthesized by high-temperature coprecipitation and subsequently modified with three kinds of polymers. These included poly(ethylene glycol) (PEG) and poly(N,N-dimethylacrylamide-co-2-aminoethylacrylamide) [P(DMA-AEA)] terminated with an alendronate anchoring group, and poly(methyl vinyl ether-co-maleic acid) (PMVEMA). The internalization of nanoparticles by rat mesenchymal stem cells (rMSCs) and C6 cancer cells was visualized by electron microscopy and the cytotoxicity of the UCNPs and their leaches was measured by the real time proliferation assay. Comet assay was used to determine oxidative damage of UCNPs. In vivo study on mice determined the elimination route and potential accumulation of UCNPs in the body. The results showed that the L- and S-UCNPs were internalized into cells in the lumen of endosomes. Proliferation assay revealed that L-UCNPs were less toxic than S-UCNPs. The viability of rat mesenchymal stem cells (rMSCs) incubated with particles decreased in the order S-UCNP@Ale-(PDMA-AEA) > S-UCNP@Ale-PEG > S-UCNPs > S-UCNP@PMVEMA. Similar results were obtained in C6 cells. Oxidative damage measured by the Comet assay showed that neat L-UCNPs caused oxidative damage to rMSCs more than all coated UCNPs while no difference was observed in C6 cells. An in vivo study indicated that L-UCNPs were eliminated from the body via the hepatobiliary route; L-UCNP@Ale-PEG particles were almost eliminated from the liver 96 h after intravenous application. Pilot fluorescence imaging confirmed the limited in vivo detection capabilities of the nanoparticles.
Keywords:
upconverting nanoparticles
; toxicity
; biological applications
1. Introduction
Since their first development in the early 2000s, lanthanide-doped upconversion nanoparticles (UCNPs) have attracted extensive attention in various cutting-edge bioapplications such as deep tissue bioimaging, biosensing and nanomedicine [1].The ability of UCNPs to convert incident light in the near-infrared (NIR) region into high-energy ultraviolet or visible emission via an anti-Stokes process has been exploited [2,3]. This allows not only relatively deep light penetration and low photodamage effects, but also reduced autofluorescence, light scattering, and phototoxicity, enabling simultaneous applications of luminescent nanomaterials in areas such as precise theranostics, in vivo optogenetics and environmental hazard control [4,5,6].
Important prerequisites for the use of UCNPs in biomedical applications are hydrophilicity and dispersibility in biological buffers, together with chemical and colloidal stability and high signal reproducibility [7]. These properties are exhibited by homogeneous phase-pure particles prepared under specific conditions. Other characteristics of UCNPs influencing their use are biocompatibility and non-toxicity, which are affected by particle size, shape, chemical composition and surface structure [8]. Dissolution of UCNPs in aqueous media can induce cytotoxicity in biological systems, especially under conditions of high dilution in aqueous media [9,10,11,12]. A tendency of Ln fluorides to dissolve in phosphate buffers due to the release of Ln3+ and F- ions, which can lead to a gradual loss of luminescence intensity, uneven particle brightness and even localized cytotoxicity has been recently confirmed [13]. In addition, the release of Y3+ ions can affect geno- and/or cytotoxicity and possible neuronal damage [14], which is a barrier for medical applications [11]. The toxicity assessment of UCNPs is therefore a relevant issue since many researchers are using, them for various applications, such as bioimaging or drug delivery.
There are several methods for producing rare earth-doped UCNPs, e.g., high-temperature coprecipitation [15], thermal decomposition [16], hydro(solvo)thermal [17], microwave [18], or microemulsion synthesis [19]. Among these procedures, the most popular preparation of monodisperse crystalline UCNPs of various sizes and compositions is the aforementioned “user-friendly” coprecipitation using oleic acid as a stabilizer [20]. Recently, some studies have appeared dealing with the protection of UCNPs by introducing a surface layer that provides both their colloidal stability and insolubility in aqueous media, as well as suppresses luminescence quenching and minimizes particle toxicity [12,21]. The advantage of UCNP surface modification is also the possibility of introducing functional groups allowing the attachment of a large number of biomolecules such as peptides, proteins, antibodies, DNAs, drugs, photosensitizers, etc., which facilitate specific targeting and treatment. These functional groups consist of carboxyl, amino, thiol, maleimide, aldehyde, phosphate, bisphosphonate, sulfonate and even o-nitrobenzyl groups [22]. General surface engineering strategies of UCNPs include ligand oxidation, replacement or removal of hydrophobic stabilizers, also silanization, layer-by-layer assembly, coating with amphiphilic polymers, etc. [23]. This allowed the modification of UCNPs with poly(ethylene glycol) (PEG) and its derivatives [24,25], amphiphilic chitosan [26], poly(acrylic acid) [27], polyethyleneimine [28], polyvinylpyrrolidone [29], poly(D,L-lactic-co-glycolic acid) [30], poly(isobutylene-alt-maleic anhydride) [31], etc. The surface design of UCNPs can also be optimized by encapsulating them in polymers via microemulsion polymerization of monomers such as ethylene glycol methyl ether acrylate, 2-hydroxyethyl and glycidyl methacrylate, acrylic acid, or by “grafting-from” and “grafting-on” methods [32,33]. The quality of the surface composition is a key factor affecting the circulation time of UCNPs in the bloodstream and/or passing through cell membranes, which is crucial for their bioapplications. The recommended size for optimal UCNP penetration is less than 100 nm. However, this size may also pose a risk of toxicity due to their potential to penetrate cellular structures and organs via the circulatory system. In addition, UCNPs can generate reactive oxygen species (ROS) that can induce DNA damage, which can lead to damage not only affecting cell growth through protein oxidation, but also impacting mitochondrial respiration [34]. Nanoparticles administered intravenously circulate through the bloodstream, especially to the liver and spleen, kidneys, heart, lungs, bone marrow and brain. Retention in the blood and organs strongly depends on the surface properties of the nanoparticle material. Coatings that promote interactions of NPs with cell membranes promote internalization by different cell types, while the use of biologically inert coatings, i.e., PEG, leads to prolonged circulation of these NPs in the bloodstream. Moreover, the rate and mechanism of uptake and clearance is cell/tissue dependent and varies between NPs of different hydrodynamic size, composition, shape, charge and surface functional groups [35,36].
In this work, we prepared NaYF4:Yb,Er-based UCNPs and investigated the dependence of their morphology, size and type of coating on their cytotoxicity as determined on rMSCs and C6 cells using the xCELLigence real time proliferation assay. We also investigated their internalization in cells and DNA oxidative damage caused by coated and uncoated, small and large UCNPs. As a proof of concept, UCNPs were also tested in experimental animals and imaged in vivo.
2. Results and Discussion
This work is a continuation of our previous papers, in which we investigated the design and properties of small and large surface-engineered UCNPs with emphasis on their colloidal and chemical stability [37,38]. In this report, we compared the two types of UCNPs, small spherical (S-UCNPs) and large hexagonal (L-UCNPs), prepared by high-temperature coprecipitation of the respective lanthanide chlorides depending on the reaction conditions (Figure 1), in terms of morphology, cytotoxicity and detection of DNA damage by the comet assay. While the spherical particles were 25 nm in diameter, their hexagonal counterparts were ~120 nm in size; both types had a relatively narrow particle size distribution (Đ = 1.01), which is essential for biomedical applications as it allows control of particle properties and reproducibility of results. Moreover, three non-toxic biocompatible polymer coatings, namely Ale-PEG, Ale-P(DMA-AEA) and PMVEMA, were selected to ensure the colloidal stability of the particles in media [37]. The properties of these coatings differed; while nonionic Ale-PEG is known for its antifouling properties [39], the positively charged Ale-P(DMA-AEA) coating of UCNPs promoted their engulfment by cells [40]. The latter coating also has the advantage of the presence of reactive amino groups available for prospective attachment of different biomolecules. Both Ale-PEG and Ale-(PDMA-AEA) were terminated with a bisphosphonate group that forms highly stable metal-bisphosphonate complexes firmly anchoring the polymers to the particle surface. In contrast, negatively charged PMVEMA has multiple carboxyl groups that can coordinate with lanthanide surface ions and/or react with various compounds.
Because all polymers did not exhibit phase contrast in the TEM images, the TEM micrographs of the polymer-coated UCNPs did not differ from those of the starting uncoated particles (Figure 1). The hydrodynamic diameter (Dh) of S-UCNPs and S-UCNP@Ale-PEG, S-UCNP@Ale(PDMA-AEA) or S-UCNP@PMVEMA nanoparticles was 101, 68, 102, and 112 nm, respectively. Their ζ-potential, which depends on surface chemistry of the particle, was 30, 8, 27, and -32 mV for neat, Ale-PEG-, Ale(PDMA-AEA)-, and PMVEMA-modified particles, respectively (Table 1). All studied L-UCNPs had comparable ζ-potential as that of S-UCNPs, but due to their larger size (according to TEM) they had larger Dh. The resulting Dh values of L-UCNP, L-UCNP@Ale-PEG, L-UCNP@Ale(PDMA-AEA), and L-UCNP@PMVEMA nanoparticles were 174, 158, 160, and 234 nm, respectively, and the corresponding ζ-potential was 28, 4, 22, and -46 mV, respectively (Table 1). Monitoring the fluoride ions dissolved in media in which the particles were aged showed that their degradation depended on many parameters, including size, coating type, temperature, and the medium used [37,38]. All types of selected coatings were shown to decrease particle degradation in different aqueous media, with the exception of PMVEMA, which slightly increased particles dissolution in water. It was also shown that dissolution of UCNPs was low in water and DMEM, moderate in artificial lysosomal fluid and pronounced in PBS. Increasing temperature directly increased dissolution of particles regardless of their size, coating, and aging medium. Moreover, independently on the type of coating the L-UCNPs dissolved less than the S-UCNPs due to a smaller surface-to-volume ratio. This observation was also confirmed by the ICP-MS, where the amount of Y and Yb was measured in leaches. The results showed that S-UCNPs were more soluble in culture media (Y and Yb within the range of 2-8 µg/ml) compared to the L-UCNPs, where both elements were present at less than 2 µg/ml. Therefore, parameters like particle size, ζ-potential, storage medium can influence particle cytotoxicity due to the interaction of the particles or their degradation products with cells. Our results are in an agreement with other studies in which the stability of NaYF4:Yb3+,Er3+ UCNPs stabilized with phosphonate coatings alendronate and ethylenediamine tetra(methylene phosphonic acid) in PBS or culture medium at room temperature and 37 °C was also investigated. [41,42]
Internalization of UCNPs in Cell Cytoplasm
Both types of nanoparticles (L-UCNPs and S-UCNPs) were internalized into membrane delimited endosomal compartments with noticeable aggregation of nanoparticles into clusters within the lumen of endosomes (Figure 2). Lower magnifications (4,000× and 50,000×) demonstrated position of particle-containing endosomes within the cell, HRTEM images (200 000×) demonstrated crystalline structure of nanoparticles with discernible interplanar spacing (Figure 2, enlarged on the right). Various densities of L-UCNP nanoparticles (an average diameter of 120 nm) and S-UCNP nanoparticles (an average diameter of 25 nm) were found within the section of 60 nm.
Cytotoxicity by xCELLigence Assay
To monitor cell growth dynamics, the viability of C6 and rMSCs incubated with Ale-PEG-, Ale-(PDMA-AEA), and PMVEMA-coated L- and S-UCNPs for 3 days was examined using a real-time proliferation assay. The advantage of this test is the long-term monitoring of cell growth in real time and not only in discrete or terminal values (as MTT assay). The test is performed in wells coated with gold electrodes, where cells growth is expressed as a cell index correlating with an increase in electrical impedance. Cell proliferation curves were determined after incubation with both particle types and their leaches (Figure 3 and Figure 4). Particles were used at a concentration of 20 µg/ml, at which the viability of C6 and rMSCs in previous experiments using Alamar blue or MTT assay reached >80 % [37,38]. The growth of healthy non-tumor rMSCs incubated with any type of UCNPs for 1 day was similar to the control. After 2 days of incubation of rMSCs with S-UCNP@PMVEMA, there was a dramatic decrease in cell proliferation, which continued for the next 24 h (p = 0.031). In the presence of S-UCNP@Ale-(PDMA-AEA) particles, cell growth decreased slightly; then remained unchanged over 72 h (Figure 3a). Cell viability in the presence of S-UCNP@Ale-PEG and neat S-UCNP remained ~80 %. We speculate that uncoated S-UCNPs partially aggregated and thus were not uniformly dispersed in the well. Therefore, the cytotoxicity was lower than that of the S-UCNP@Ale-(PDMA-AEA) particles. In contrast, all types of L-UCNPs had no effect on cell growth, except L-UCNP@PMVEMA, where rMSC proliferation decreased after 48 h of incubation (Figure 3c). In general, S-UCNPs impaired cancer C6 cell viability more significantly than rMSCs. While control cells proliferated rapidly for all 3 days, C6 cells incubated with S-UCNPs for 1 day stopped growing. The viability of C6 cells in the presence of S-UCNP@Ale-PEG particles remained constant but decreased after incubation with neat S-UCNPs for 72 h (p = 0.0142) or their analogues coated with Ale-(PDMA-AEA) (p = 0.0162) or PMVEMA (p = 0.0142; Figure 3 b). Similar results were achieved with L-UCNPs, especially neat ones, which stopped the proliferation of C6 cells that detached from the bottom of the well (Figure 3 d). Prolonged exposure to all polymer-coated L-UCNPs (72 h) at a concentration of 20 µg/ml reduced cell growth (p <0.0001) as did neat L-UCNPs (p <0.0001). Our results are in an agreement with MTS assay studies that reported a 67 % decrease in viability of bone marrow-derived stem cells after 48 h of exposure to UCNPs [43] while shorter incubation times did not affect cell viability. Also, UCNPs coated with polyethylenimine (25 µg/ml) exposed to MSCs for 2 days slightly decreased cell viability to 85 % [44]. Different coatings of UCNPs were also tested on HaCaT keratinocytes by WST-8 assay and similarly, prolonged exposure to UCNPs for 48 hours resulted in a viability drop when compared to 24h [21]. In contrast to nanoparticles, rMSCs incubated with particle leaches from neat S-UCNPs, S-UCNP@PMVEMA, S-UCNP@Ale-(PDMA-AEA), and S-UCNP@Ale-PEG proliferated even faster than in the control group (Figure 4 a), indicating stimulation of cell growth. In the case of C6 cells, their growth was faster after incubation with leaches for 24 h; in later time periods (72 h), leaches from neat S-UCNPs (p = 0.0015), S-UCNP-Ale-PEG (p = 0.0119), S-UCNPs@Ale-(PDMA-AEA) (p = 0.0098) and S-UCNP@PMVEMA (p = 0.0009) were toxic (Figure 4 b). On the other hand, the leaches from L-UCNPs did not affect rMSC proliferation (Figure 4 c); however, C6 growth was slowed down after the incubation with L-UCNP leaches for 72 h (p = 0.0086), L-UCNP@PMVEMA leaches (p = 0.0129) and L-UCNP@Ale-(PDMA-AEA) leaches (p = 0.0086); except for L-UCNP@Ale-PEG leaches, where the cell proliferation decreased only marginally (Figure 4 d).
Thus, it can be concluded that the real time proliferation assay in cells incubated with UCNPs gave similar results to MTT or alamar blue done in previous experiments [37,38], while the leaches, especially from L-UCNPs, were less harmful. In general, L-UCNPs were less toxic than S-UCNPs due to the lower surface-to-volume ratio. L-UCNP nanoparticles almost did not affect the viability of rMSCs, while the viability of C6 significantly decreased to 30% or less. The viability of rMSCs decreased in the order S-UCNP@Ale-(PDMA-AEA) > S-UCNP@Ale-PEG > S-UCNPs > S-UCNP@PMVEMA. The trend was also similar for C6 cells, which readily internalized particles depending on the incubation time. In contrast, rMSCs engulfed particles much less after three days of incubation than after one day [37]. The best biocompatibility was achieved with Ale-(PDMA-AEA) and Ale-PEG coatings, which can be attributed to their hydrophilicity. There are alternative approaches to assess NP cytotoxicity: for example, Das et al. [45] conducted a study on the toxic effects of three types of functionalized UCNPs: oleate ligand-UCNPs, PEG-UCNPs, and bilayered PEG-oleate-UCNPs. They used calcein and propidium iodide viability assay and concluded that bilayer UCNPs exhibit significant toxicity due to functionalization. In another study, Malvindi et al. [46] evaluated the cytotoxicity of silica-coated iron oxide NPs using the WST-8 ([2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] method and the lactate dehydrogenase release assay (LDH assay) to analyze cell viability and cell membrane integrity. The NPs showed good internalization in HeLa cells with no observed toxicity. Meindl et al. [47], on the other hand, assessed the cytotoxicity of UCNPs by measuring intracellular calcium, providing an example of an alternative approach to assess toxicity..
Genotoxicity by Comet Assay
In rMSCs, all nanoparticle treatments were statistically significant to positive control and non-significant to negative control. In addition, when comparing oxidative damage of large vs. small UCNPs and coated vs. uncoated UCNPs on rMSCs, a statistically significant difference was found between L-UCNPs vs. L-UCNP-Ale-PEG (p = 0.021) and S-UCNP@Ale-PMVEMA (p = 0.0158). In C6 cells, no difference was found when comparing different nanoparticle treatments. The difference was found between S-UCNP@Ale-(PDMA-AEA) and negative control (p = 0.0258; Figure 5). In both rMSCs and C6 cells, the negative and positive controls were statistically significant (p <0.001). In general, oxidative damage was ~20 % for rMSCs regardless of nanoparticle treatment, while it was only ~5 % for C6. Thus, C6 cells were less sensitive to oxidative damage than rMSCs under controlled conditions, which can be explained by the reduced sensitivity of the cell lines to higher oxygen levels under normal in vitro incubation conditions than in animal tissues. Similar results have been reported on the UCNPs based on Y2O3/Yb3+,Er3+ where no DNA damage was found on cancer cells in comet assay [48]. On the contrary, lower viability of C6 cells in the presence of UCNPs may be explained by the smaller cell volume relative to the number and size of internalized nanoparticles. These results confirm that functionalized UCNPs can be used without any genotoxic effects for bioimaging.
In Vivo Study
Nanoparticles were investigated in vivo after subcutaneous application in NuNu mice (males, 6-7 weeks old). Upconversion enabled detection of a signal at 535 nm after excitation in near infrared area (980 nm; Figure 6). After systemic (retroorbital) application, in vivo imaging failed to detect the particles. While near infrared excitation light easily penetrates the tissue and may excite, weaker fluorescent light at 535 nm is absorbed. Therefore, the possibility to obtain the fluorescence signal at this wavelength from organs located deep in the body (e.g., the liver) is very limited. The nanoparticles were detected postmortem on excised organs. Fluorescence microscopy proved the presence of L-UCNP@Ale-(PDMA-AEA) in the liver only (Figure 6). L-UCNP@Ale-PEG particles were not detected in any organ. These results were supported by ICP-MS, which confirmed the presence of Yb and Y atoms from the nanoparticles only in the liver. The amount of Yb and Y detected from Ale-PEG-coated UCNPs was significantly lower than that from Ale-(PDMA-AEA)-coated nanoparticles (p <0.0001), where a statistically significant difference was observed compared to the control group. A statistically significant difference between UCNP@Ale-PEG and the control group was not observed; the particles were almost completely eliminated from the body after more than 96 h (Figure 6 a). No Yb and Y was detected by ICP-MS in the kidney. These experiments confirmed fast elimination of the coated UCNPs from the organism and also suggested the hepatobiliary excretion route. Contrary to this, Zhou et al. [49] observed a tendency for NaYF4:Yb,Er@SiO2 to accumulate in the liver after entering the bloodstream. Large amount of NaYF4:Yb,Er@SiO2 was observed in the liver tissue on days 1 and 7 after intravenous administration of these nanoparticles to mice at a dose of 20 mg/kg, suggesting internalization into hepatocytes. However, histology did not reveal any pathological changes in the liver tissue. While fluorescence particle detection in vivo is very difficult, it does not compromise future use of UCNPs in photodynamic therapy in combination with a suitable photosensitizer. If the particles are carefully navigated, they can be excited by NIR light deep in the tissue. Generated fluorescence light can irradiate an immediate vicinity only, so it may excite a suitable photosensitizer locally with no possible harm to other tissues or organs.
4. Materials and Methods
Small (S) spherical and large (L) hexagonal UCNPs were prepared by high-temperature coprecipitation of lanthanide chlorides and their surface was modified with poly(ethylene glycol) (PEG) and poly(N,N-dimethylacrylamide-co-2-aminoethylacrylamide) [P(DMA-AEA)], both terminated with an alendronate (Ale) anchoring group, and poly(methyl vinyl ether-co-maleic acid) (PMVEMA) according to our previous work [37,38]. For cell culturing phosphate-buffered saline (PBS), Dulbecco’s modified Eagle medium (DMEM), supplemented with fetal bovine serum (FBS) (both from Merck; Darmstadt, Germany), a combination of primocin and penicillin - streptomycin (Gibco; Life Technologies, Grand Island, NY, USA) and trypsin (Sigma-Aldrich, St. Luis, MO, USA) was used. For preparation of samples for electron microscopy, we utilized PHEM buffer (pH 7.4) OsO4 solution (Thermo Fisher, Waltham, MA, USA), K3(Fe(CN6)) (VWR International, Radnor, PA, USA), low melting agarose (LMP) (Sigma-Aldrich, St. Luis, MO, USA) , uranyl acetate (Sigma-Aldrich, St. Luis, MO, USA) and Epon HARD complete resin (Electron Microscopis Sciences; Hatfield, USA); for fluorescent microscopy 4′,6-diamidino-2-phenylindole dihydrochloride ( DAPI; Invitrogen; Carlsbad, USA) was obtained.
All other chemicals were purchased from Sigma-Aldrich , or LachNer (Neratovice, Czech Republic).
Distilled and demineralized water (conductivity ˂ 0.1 µS/cm; Millipore; Bedford, MA, USA) was used to prepare all solutions.
Preparation of Leachates
For toxicity analysis of the leachates, these were prepared from dispersions of particles in the culture medium. The nanoparticles were mixed with the medium to a concentration of 20 µl/ml, the mixture was placed in an incubator at 37 °C for three days, the dispersion was centrifuged (160 rcf) for 5 min and the supernatant was analyzed using a real time proliferation assay.
C6 Cell Line
To initiate cell culture, C6 cells were thawed and rinsed in cold PBS. After washing, they were seeded in DMEM, supplemented with FBS and a combination of primocin and penicillin - streptomycin.
Mesenchymal Stem Cells
Rat mesenchymal stem cells (rMSCs) were derived by aspirating bone marrow from the rat’s bone. The harvested bone marrow was subsequently washed twice with PBS. The rinsed bone marrow was then cultured in cell culture flasks containing DMEM. The culture medium was enriched with FBS, along with the addition of primocin and penicillin-streptomycin. The culture flasks were placed in an incubator at 37 °C and 5 % CO2 atmosphere and the culture medium were changed twice a week. Upon reaching approximately 70 % confluence, the cells were passaged using trypsin at 37 °C for 4 min. Trypsin treatment was terminated by FBS. The cells were subsequently washed with PBS and either utilized for the experiment or reseeded for further culture. Both cell cultures were maintained at 37 °C in 5 % CO2 atmosphere. The culture medium was refreshed twice a week to maintain cell viability and growth.
Electron Microscopy
The internalization of the nanoparticles into the C6 and MSCs was investigated by transmission electron microscopy (TEM). The samples were initially centrifuged at 300 g for 5 min at room temperature (RT). They were then fixed with a solution containing 2.5 % glutaraldehyde, 2 % formaldehyde and 0.1 M PHEM buffer (pH 7.4) for 10 min at RT and incubated on ice for 1 h. After fixation, the samples underwent three 5-min ice-cold washes with 0.1 M PHEM buffer (pH 7.4).
Following the washing, the samples were contrasted in 1 % OsO4 solution and 1.5 % K3(Fe(CN6)) in Milli-Q water on ice for 1 h. The samples were washed three times for 5 min each with PHEM buffer (pH 7.4) while kept on ice. Subsequently, the samples were centrifuged at 400 g for 2-5 min at 38 °C, embedded in 2 % LMP at 38 °C, and polymerized for 20 min on ice. Afterward, cubes were cut from the samples, quickly washed with cold Milli-Q water and stained in a 1 % uranyl acetate aqueous solution at RT for 30 min. Dehydration involved washing with a series of cold ethanol solutions (30-50-70-80 %) for 5 min each on ice, followed by RT-warmed 90-100 % ethanol for 5 min at RT. The samples were then transferred into anhydrous acetone at RT, infiltrated and embedded in epoxy resin kit EMBED 812 (EMS#14120) and polymerized at 40 °C for 1 h and at 60 °C for 72 h in an oven. Ultrathin sections were cut on a LEICA UC7 ultramicrotome, collected on formvar carbon coated copper slot grids and post-contrasted with 4 % uranyl acetate and lead citrate. Images were acquired on a JEM 2100 Plus transmission electron microscope (JEOL; Akishima, Japan) operated at 200 kV using TVIPS XF 416 camera.
Real Time Proliferation Assay
Briefly, the culture medium (50 µl) was pipetted into a special E-plate with gold electrodes at the bottom of wells and the background impedance was determined. The cell attachment and proliferation were measured as a change of impedance between electrodes and expressed as a unitless cell index. The appropriate number of cells (5,000 rMSCs or 2,500 C6 cells per well) was added in culture medium (100 µl) and the cells were allowed to attach to the bottom of well for 2 h. This was followed by the addition of nanoparticles or their leaches in medium (50 µl); the final particle concentration was kept at 20 µg/ml per well. The plates were then inserted into the xCELLigence RTCA DP real-time cell analyzer (ACEA Biosciences, now Agilent Technologies; Santa Clara, CA, USA) and the changes in impedance were recorded every 20 min for 3 days. All experiments were done in triplicates and repeated three times.
Determination of Oxidative Damage of DNA by Comet Assay
The comet assay was employed as a highly sensitive and straightforward technique for the assessment of DNA damage at the individual eukaryotic cell level. Initially, 110 μl of normal melting point agarose, preheated to a minimum of 60 °C, was piped onto a glass slide preheated to 50 °C on a hot plate. A glass coverslip was immediately placed over the agarose, and the slide was left on ice to solidify for 5 min. The coverslip was removed and 75 μl of LMP agarose mixed with cells was added. Another glass coverslip was used to cover this layer, and it was allowed to solidify on ice for an additional 5 min. After removing the coverslip, 75 μl of pure LMP agarose was added, and the coverslip was replaced, followed by a 5-min solidification on ice. The coverslip was removed, and the specimen was gently submerged into a cooled lysing solution, taking precautions to shield it from light. It was then placed in the refrigerator for a minimum of 1 h. The samples were taken out of the lysing solution and immersed in an alkaline buffer for electrophoresis for 40 min. The electrophoresis chamber was filled with the alkaline buffer, the voltage was set at 32 V (1.2 V/cm), and a current of 300 mA was applied for 20 min at 4 °C. The specimens underwent three washes with 1 ml of 0.4 M TRIS buffer, each lasting 5 min. Following that, the samples were rinsed twice with distilled water, with each rinse lasting 5 min. The samples were immersed in 70 % ethanol for 15 min and in 99 % ethanol for an additional 15 min. Finally, the samples were air-dried. Lucia Comet Assay software (Laboratory Imaging; Prague, Czech Republic) was used to quantify DNA migration, expressing results as a percentage of DNA in the tail. Both total DNA damage (with enzymes) and DNA strand breaks (DNA-SB; without enzymes) were measured in 100 randomly selected cells (2 sets of 100 cells) per slide, with medians calculated from each group of 50 cells. The level of oxidative DNA damage was assessed by comparing the median of total DNA damage with the median of DNA-SB.
Pilot In Vivo Imaging
As a proof of principle, the large hexagonal nanoparticles (120 nm) were applied to mice and their fluorescence was detected. For in vivo study, the most successful candidates from in vitro investigation were selected, where large nanoparticles with Ale-PEG and Ale-(PDMA-AEA) coating were less toxic.
CD-1® nude mice (Crl:CD1-FOXn1nu), 6 weeks old, were used throughout the experiments. First experiment involved application of nanoparticle dispersions (10 µL of UCNP@Ale-PEG or UCNP@Ale-(PDMA-AEA), concentration 4 mg/mL) subcutaneously to a mouse and scanning using an optical imager Bruker Xtreme (Bruker, Billerica, MA, USA). Excitation of the injection site was performed by a laser diode (980 nm, 100 mW) and a signal at 535 nm was detected (exposition 5 s, field of view 72×72 mm, no binning).
To evaluate biodistribution of the nanoparticles, 100 µL of UCNP@Ale-PEG or UCNP@Ale-(PDMA-AEA) dispersion (concentration 50 µg/mL of mouse blood) was applied retroorbitally to mice (each nanoparticle type to 5 mice). Animals were scanned at similar experimental conditions (an optical imager Bruker Xtreme, excitation using a laser diode at 980 nm, 100 mW, emission at 535 nm, exposition 1 s, field of view 190×190 mm, no binning). Due to a small area irradiated by the diode, it was not possible to excite nanoparticles in the whole animal body. Therefore, only selected organs (the liver, spleen, kidney) were excited in several subsequent measurements. A group of two animals served as a control.
After 96 h, mice were deeply anesthetized with chloralhydrate in a concentration of 400 mg/kg intraperitoneally and then transcardially perfused first with phosphate buffer (PB) and then with 4 % paraformaldehyde in PB. Paraformaldehyde-fixed tissue samples (kidney and liver) were cryopreserved in sucrose solution with gradually increasing concentration (10, 20 and 30 % sucrose in deionized water). The tissue was then embedded in OCT mounting media (VWR; Radnor, USA). Sections were cut on a Cryostar NX70 cryostat (Thermo Fisher Scientific;Waltham, MA, USA) to 30 µm sections (liver and kidney) and stained with DAPI for 10 min. Finally, content of Yb and Y was quantified by inductively coupled plasma mass spectrometry (ICP-MS).
The animal experiments were performed in accordance with national and international guidelines for laboratory animal care and were approved by the Laboratory Animal Care and Use Committee of the First Faculty of Medicine, Charles University, and the Ministry of Education, Youth and Sports of the Czech Republic (MSMT 46304/2020-3).
Laser Scanning Confocal Microscopy
To visualize UCNPs, Carl Zeiss LSM 880 NLO microscope (Oberkochen, Germany), equipped with 40× NA1.1 water immersion objective and a 32 GaAsP array spectral detector covering emission from 410 to 694 nm and operated at single photon counting mode for maximum SNR, was used. Lambda mode at full spectral resolution was used to spectrally prove UCNPs emission (two characteristic distinct and narrow peaks at 544 nm and 660 nm.) and a channel mode was used to combine DAPI (405 nm excitation, 410-500 nm emission, UCNPs (974 nm excitation, 535-565 nm and 650-668 nm emission bands, and transmitted light signals. 974 nm excitation by a TiSa laser (80 MHz, 350 fs laser pulse width at sample plane) provided the highest emission intensity for upconversion and was used for imaging. The 974 nm laser power was kept low at < 50 µW at sample plane. To capture the slow emission of UCNPs (excited state lifetime on the order of hundreds o f microseconds), the scanning speed was the slowest possible, 132 µs per pixel and the pinhole was opened to 300 um (around 4 Airy units). Pixel size was 132 nm, image size 512×512 pixels, bidirectional scan.
Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
A NexION 350D ICP-MS instrument (PerkinElmer; Shelton, USA) equipped with Universal Cell Technology™ for spectral interference elimination was used for ICP-MS measurement. The sample introduction system included an internal peristaltic pump with Tygon® tubing (0.38 mm internal diameter), a polytetrafluorethylene concentric nebulizer, and a glass cyclonic spray chamber with a volume of 100 mL. For measurement of 89Y and 174Yb isotopes, the samples were diluted with 2 % nitric acid and spiked with the internal standard (IS) solution (103Rh).
Calibration Y and Yb solutions and IS solution were prepared from solutions of concentration 1.000±0.002 g/L (Merck, Darmstad, Deutschland).
Statistical Methods
The data are presented as the mean ± standard deviation (SD). Statistical analyses were conducted using the GraphPad Software (GraphPad Prism, version 9), employing a one-way ANOVA test followed by Tukey post hoc test. A significance threshold of p <0.05 was applied to determine statistical significance.
5. Conclusions
In this report, the long-term cyto- and genotoxicity of the particles and/or their extracts was evaluated to better understand the hazards associated with the biological application of UCNPs. In addition, some important information on biodistribution, clearance and accumulation of particles in tissue organs in vivo was obtained. Both large (120 nm according to TEM) and small (25 nm) UCNPs can be internalized in the cell cytoplasm. Real time proliferation assay has confirmed that L-UCNPs were less toxic than the S-UCNPs. The presence of Y and Yb in the leachates confirmed the higher solubility of S-UCNPs in the medium than L-UCNPs, which corresponded to the higher cytotoxicity of the leachates from small nanoparticles, especially in C6 cells. The coating with PMVEMA did not provide sufficient protection against toxicity after incubation with cells. On the contrary, S-UCNPs caused very small oxidative damage, the significantly higher oxidative damage was found after neat L-UCNPs treatment in rMSCs and was diminished after coating with Ale-(PDMA-AEA), PMVEMA and Ale-PEG. Cell line C6 was less sensitive to oxidative damage than primary culture (rMSCs). In vivo study showed that both types of nanoparticles were eliminated from the body via the liver. While L-UCNPs@Ale-PEG particles were almost completely eliminated from liver 96 h after intravenous application, L-UCNPs@Ale-(PDMA-AEA) particles remained in the liver in a significantly higher amount. The polymeric coating can influence the retention of nanoparticles in tissues to some extent. This can be advantageously used in various applications using cell labeling in tissues and organs.
Author Contributions
Lucia Machova Urdzikova was conceptualizing research work, writing publication, evaluated the data, Dana Marekova was responsible for cell culturing, xCell analysis, comet assay and preparation of the EM samples, Vitalii Patsula was preparing UCNPs and writing publication, ,Viktoriia Oleksa, Oleksandr Shapoval and Taras Vasylyshyn were preparing different bathes of UCNPs, Magda Vosmanska was preparing ICP-MS analysis, David Liebl was preparing EM pictures, Aleš Benda was responsible for Laser scanning confocal microscopy, Vit Herynek and Petr Matous conducted in vivo study , Daniel Horák prepare concept of the study and validated data, Pavla Jendelová coordinated research work and writing publication.
Funding
This research was supported by Czech Science Foundation (No. 21-04420S) and by
Institutional Review Board Statement
The animal experiments were performed in accordance with national and international guidelines for laboratory animal care and were approved by the Laboratory Animal Care and Use Committee of the First Faculty of Medicine, Charles University, and the Ministry of Education, Youth and Sports of the Czech Republic (MSMT 46304/2020-3).
Informed Consent Statement
Not applicable
Data Availability Statement
The data will be published at Zenodo repository
Acknowledgments
We acknowledge Imaging Methods Core Facility at BIOCEV, institution supported by the MEYS CR (LM2023050 Czech-BioImaging), for TEM and LSCM data acquisition and Karel Tresnak from IEM CAS for the comet assay preparation.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Loo, J.F.C.; Chien, Y.H.; Yin, F.; Kong, S.K.; Ho, H.P.; Yong, K.T. Upconversion and downconversion nanoparticles for biophotonics and nanomedicine. Coordin Chem Rev 2019, 400. [Google Scholar] [CrossRef]
- Auzel, F. Upconversion and anti-stokes processes with f and d ions in solids. Chem Rev 2004, 104, 139–173. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Xu, J.H.; Wu, Y.M.; Liu, X.G. Energy-Transfer Editing in Lanthanide-Activated Upconversion Nanocrystals: A Toolbox for Emerging Applications. Acs Central Sci 2019, 5, 29–42. [Google Scholar] [CrossRef] [PubMed]
- All, A.H.; Zeng, X.; Teh, D.B.L.; Yi, Z.G.; Prasad, A.; Ishizuka, T.; Thakor, N.; Hiromu, Y.; Liu, X.G. Expanding the Toolbox of Upconversion Nanoparticles for In Vivo Optogenetics and Neuromodulation. Adv Mater 2019, 31. [Google Scholar] [CrossRef]
- Chen, G.Y.; Qju, H.L.; Prasad, P.N.; Chen, X.Y. Upconversion Nanoparticles: Design, Nanochemistry, and Applications in Theranostics. Chem Rev 2014, 114, 5161–5214. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Shikha, S.; Liu, J.L.; Zhang, J.; Mei, Q.S.; Zhang, Y. Upconversion Nanoprobes: Recent Advances in Sensing Applications. Anal Chem 2019, 91, 548–568. [Google Scholar] [CrossRef]
- Duan, C.C.; Liang, L.E.; Li, L.; Zhang, R.; Xu, Z.P. Recent progress in upconversion luminescence nanomaterials for biomedical applications. J Mater Chem B 2018, 6, 192–209. [Google Scholar] [CrossRef]
- Maynard, A.D.; Warheit, D.B.; Philbert, M.A. The New Toxicology of Sophisticated Materials: Nanotoxicology and Beyond. Toxicol Sci 2011, 120, S109–S129. [Google Scholar] [CrossRef]
- Lahtinen, S.; Lyytikäinen, A.; Päkkilä, H.; Hömppi, E.; Perälä, N.; Lastusaari, M.; Soukka, T. Disintegration of Hexagonal NaYF:Yb,Er Upconverting Nanoparticles in Aqueous Media: The Role of Fluoride in Solubility Equilibrium. J Phys Chem C 2017, 121, 656–665. [Google Scholar] [CrossRef]
- Lisjak, D.; Plohl, O.; Ponikvar-Svet, M.; Majaron, B. Dissolution of upconverting fluoride nanoparticles in aqueous suspensions. Rsc Adv 2015, 5, 27393–27397. [Google Scholar] [CrossRef]
- Lisjak, D.; Plohl, O.; Vidmar, J.; Majaron, B.; Ponikvar-Svet, M. Dissolution Mechanism of Upconverting AYF:Yb,Tm (A = Na or K) Nanoparticles in Aqueous Media. Langmuir 2016, 32, 8222–8229. [Google Scholar] [CrossRef]
- Plohl, O.; Kralj, S.; Majaron, B.; Fröhlich, E.; Ponikvar-Svet, M.; Makovec, D.; Lisjak, D. Amphiphilic coatings for the protection of upconverting nanoparticles against dissolution in aqueous media. Dalton T 2017, 46, 6975–6984. [Google Scholar] [CrossRef] [PubMed]
- Dukhno, O.; Przybilla, F.; Muhr, V.; Buchner, M.; Hirsch, T.; Mély, Y. Time-dependent luminescence loss for individual upconversion nanoparticles upon dilution in aqueous solution. Nanoscale 2018, 10, 15904–15910. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.C.; Tian, Y.T.; Zeng, Z.Y.; Shuai, P.; Lan, H.Y.; Zhu, X.S.; Zhong, Y.; Wu, L.H.; Fan, X.N. YCl3 Promotes Neuronal Cell Death by Inducing Apoptotic Pathways in Rats. Biomed Res Int 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.S.; Zhang, Y. Synthesis of Hexagonal-Phase Core-Shell NaYF Nanocrystals with Tunable Upconversion Fluorescence. Langmuir 2008, 24, 12123–12125. [Google Scholar] [CrossRef]
- Boyer, J.C.; Cuccia, L.A.; Capobianco, J.A. Synthesis of colloidal upconverting NaYF: Er/Yb and Tm/Yb monodisperse nanocrystals. Nano Lett 2007, 7, 847–852. [Google Scholar] [CrossRef]
- Zhang, F.; Wan, Y.; Yu, T.; Zhang, F.Q.; Shi, Y.F.; Xie, S.H.; Li, Y.G.; Xu, L.; Tu, B.; Zhao, D.Y. Uniform nanostructured arrays of sodium rare-earth fluorides for highly efficient multicolor upconversion luminescence. Angew Chem Int Edit 2007, 46, 7976–7979. [Google Scholar] [CrossRef]
- Wang, H.Q.; Tilley, R.D.; Nann, T. Size and shape evolution of upconverting nanoparticles using microwave assisted synthesis. Crystengcomm 2010, 12, 1993–1996. [Google Scholar] [CrossRef]
- Shan, S.N.; Wang, X.Y.; Jia, N.Q. Synthesis of NaYF :Yb, Er upconversion nanoparticles in normal microemulsions. Nanoscale Res Lett 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Ohulchanskyy, T.Y.; Liu, S.; Law, W.C.; Wu, F.; Swihart, M.T.; Ågren, H.; Prasad, P.N. Core/shell NaGdF4:Nd3+/NaGdF4 nanocrystals with efficient near-infrared to near-infrared downconversion photoluminescence for bioimaging applications. Acs Nano 2012, 6, 2969–2977. [Google Scholar] [CrossRef] [PubMed]
- Bastos, V.; Oskoei, P.; Andresen, E.; Saleh, M.I.; Rühle, B.; Resch-Genger, U.; Oliveira, H. Stability, dissolution, and cytotoxicity of NaYF-upconversion nanoparticles with different coatings. Sci Rep-Uk 2022, 12. [Google Scholar] [CrossRef]
- Wang, X.T.; Yang, Y.B.; Liu, C.; Guo, H.L.; Chen, Z.F.; Xia, J.Y.; Liao, Y.G.; Tang, C.Y.; Law, W.C. Photo- and pH-responsive drug delivery nanocomposite based on -nitrobenzyl functionalized upconversion nanoparticles. Polymer 2021, 229. [Google Scholar] [CrossRef]
- Sun, L.N.; Wei, R.Y.; Feng, J.; Zhang, H.J. Tailored lanthanide-doped upconversion nanoparticles and their promising bioapplication prospects. Coordin Chem Rev 2018, 364, 10–32. [Google Scholar] [CrossRef]
- Andresen, E.; Resch-Genger, U.; Schäferling, M. Surface Modifications for Photon-Upconversion-Based Energy-Transfer Nanoprobes. Langmuir 2019, 35, 5093–5113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Guo, Z.; Zhang, X.; Gong, L.J.; Dong, X.H.; Fu, Y.Y.; Wang, Q.; Gu, Z.J. Mass production of poly(ethylene glycol) monooleate-modified core-shell structured upconversion nanoparticles for bio-imaging and photodynamic therapy. Sci Rep-Uk 2019, 9. [Google Scholar] [CrossRef]
- Cui, S.S.; Chen, H.Y.; Zhu, H.Y.; Tian, J.M.; Chi, X.M.; Qian, Z.Y.; Achilefu, S.; Gu, Y.Q. Amphiphilic chitosan modified upconversion nanoparticles for photodynamic therapy induced by near-infrared light. J Mater Chem 2012, 22, 4861–4873. [Google Scholar] [CrossRef]
- Xue, Z.L.; Zeng, S.J.; Hao, J.H. Non-invasive through-skull brain vascular imaging and small tumor diagnosis based on NIR-II emissive lanthanide nanoprobes beyond 1500 nm. Biomaterials 2018, 171, 153–163. [Google Scholar] [CrossRef]
- Näreoja, T.; Deguchi, T.; Christ, S.; Peltomaa, R.; Prabhakar, N.; Fazeli, E.; Perälä, N.; Rosenholm, J.M.; Arppe, R.; Soukka, T.; et al. Ratiometric Sensing and Imaging of Intracellular pH Using Polyethylenimine-Coated Photon Upconversion Nanoprobes. Anal Chem 2017, 89, 1501–1508. [Google Scholar] [CrossRef]
- Johnson, N.J.J.; Sangeetha, N.M.; Boyer, J.C.; van Veggel, F.C.J.M. Facile ligand-exchange with polyvinylpyrrolidone and subsequent silica coating of hydrophobic upconverting β-NaYF4:Yb3+/Er3+ nanoparticles. Nanoscale 2010, 2, 771–777. [Google Scholar] [CrossRef]
- Zhao, J.W.; Yang, H.; Li, J.L.; Wang, Y.J.; Wang, X. Fabrication of pH-responsive PLGA(UCNPs/DOX) nanocapsules with upconversion luminescence for drug delivery. Sci Rep-Uk 2017, 7. [Google Scholar] [CrossRef]
- Wilhelm, S.; Kaiser, M.; Würth, C.; Heiland, J.; Carrillo-Carrion, C.; Muhr, V.; Wolfbeis, O.S.; Parak, W.J.; Resch-Genger, U.; Hirsch, T. Water dispersible upconverting nanoparticles: effects of surface modification on their luminescence and colloidal stability. Nanoscale 2015, 7, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.T.T.; Chen, Y.H.; Tawfik, S.A.; Wen, S.H.; Parviz, M.; Shimoni, O.; Jin, D.Y. Systematic investigation of functional ligands for colloidal stable upconversion nanoparticles. Rsc Adv 2018, 8, 4842–4849. [Google Scholar] [CrossRef] [PubMed]
- Sedlmeier, A.; Gorris, H.H. Surface modification and characterization of photon-upconverting nanoparticles for bioanalytical applications. Chem Soc Rev 2015, 44, 1526–1560. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sharma, N.; Maitra, S.S. In vitro and in vivo toxicity assessment of nanoparticles. Int Nano Lett 2017, 7, 243–256. [Google Scholar] [CrossRef]
- Fadeel, B.; Garcia-Bennett, A.E. Better safe than sorry: Understanding the toxicological properties of inorganic nanoparticles manufactured for biomedical applications. Adv Drug Deliver Rev 2010, 62, 362–374. [Google Scholar] [CrossRef]
- Iversen, T.G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Nahorniak, M.; Patsula, V.; Mareková, D.; Matous, P.; Shapoval, O.; Oleksa, V.; Vosmanská, M.; Urdziková, L.M.; Jendelová, P.; Herynek, V.; et al. Chemical and Colloidal Stability of Polymer-Coated NaYF:Yb,Er Nanoparticles in Aqueous Media and Viability of Cells: The Effect of a Protective Coating. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Patsula, V.; Mareková, D.; Jendelová, P.; Nahorniak, M.; Shapoval, O.; Matous, P.; Oleksa, V.; Konefal, R.; Vosmanská, M.; Machová-Urdziková, L.; et al. Polymer-coated hexagonal upconverting nanoparticles: chemical stability and cytotoxicity. Front Chem 2023, 11. [Google Scholar] [CrossRef]
- Lowe, S.; O’Brien-Simpson, N.M.; Connal, L.A. Antibiofouling polymer interfaces: poly(ethylene glycol) and other promising candidates. Polym Chem-Uk 2015, 6, 198–212. [Google Scholar] [CrossRef]
- Oleksa, V.; Macková, H.; Engstová, H.; Patsula, V.; Shapoval, O.; Velychkivska, N.; Jezek, P.; Horák, D. Poly(-dimethylacrylamide)-coated upconverting NaYF:Yb,Er@NaYF:Nd core-shell nanoparticles for fluorescent labeling of carcinoma cells. Sci Rep-Uk 2021, 11. [Google Scholar]
- Andresen, E.; Würth, C.; Prinz, C.; Michaelis, M.; Resch-Genger, U. Time-resolved luminescence spectroscopy for monitoring the stability and dissolution behaviour of upconverting nanocrystals with different surface coatings. Nanoscale 2020, 12, 12589–12601. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.I.; Rühle, B.; Wang, S.; Radnik, J.; You, Y.; Resch-Genger, U. Assessing the protective effects of different surface coatings on NaYF:Yb, Er upconverting nanoparticles in buffer and DMEM. Sci Rep 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Jalil, R.A.; Zhang, Y. Biocompatibility of silica coated NaYF upconversion fluorescent nanocrystals. Biomaterials 2008, 29, 4122–4128. [Google Scholar] [CrossRef] [PubMed]
- Chatteriee, D.K.; Rufalhah, A.J.; Zhang, Y. Upconversion fluorescence imaging of cells and small animals using lanthanide doped nanocrystals. Biomaterials 2008, 29, 937–943. [Google Scholar] [CrossRef]
- Das, G.K.; Stark, D.T.; Kennedy, I.M. Potential Toxicity of Up-Converting Nanoparticles Encapsulated with a Bilayer Formed by Ligand Attraction. Langmuir 2014, 30, 8167–8176. [Google Scholar] [CrossRef]
- Malvindi, M.A.; De Matteis, V.; Galeone, A.; Brunetti, V.; Anyfantis, G.C.; Athanassiou, A.; Cingolani, R.; Pompa, P.P. Toxicity Assessment of Silica Coated Iron Oxide Nanoparticles and Biocompatibility Improvement by Surface Engineering. Plos One 2014, 9. [Google Scholar] [CrossRef]
- Meindl, C.; Kueznik, T.; Bösch, M.; Roblegg, E.; Fröhlich, E. Intracellular calcium levels as screening tool for nanoparticle toxicity. J Appl Toxicol 2015, 35, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, L.; Cuddapah, S.; Costa, M. Oxidative stress under ambient and physiological oxygen tension in tissue culture. Current pharmacology reports 2016, 2, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.Z.; Ge, X.Q.; Ke, D.M.; Tang, H.; Zhang, J.Z.; Calvaresi, M.; Gao, B.; Sun, L.N.; Su, Q.Q.; Wang, H.F. The Bioavailability, Biodistribution, and Toxic Effects of Silica-Coated Upconversion Nanoparticles. Front Chem 2019, 7. [Google Scholar] [CrossRef]
Figure 1.
TEM micrographs of (a-d) small spherical and (e-h) large hexagonal UCNPs; (a, e) non-coated and coated UCNPs with (b, f) Ale-PEG, (c, g) Ale-(PDMA-AEA), and (d, h) PMVEMA.
Figure 1.
TEM micrographs of (a-d) small spherical and (e-h) large hexagonal UCNPs; (a, e) non-coated and coated UCNPs with (b, f) Ale-PEG, (c, g) Ale-(PDMA-AEA), and (d, h) PMVEMA.

Figure 2.
Ultrastructural TEM analysis of nanoparticle intracellular localization. Two representative rMSCs (lower vs. upper panel) are shown for L-UCNPs and S-UCNPs.
Figure 2.
Ultrastructural TEM analysis of nanoparticle intracellular localization. Two representative rMSCs (lower vs. upper panel) are shown for L-UCNPs and S-UCNPs.
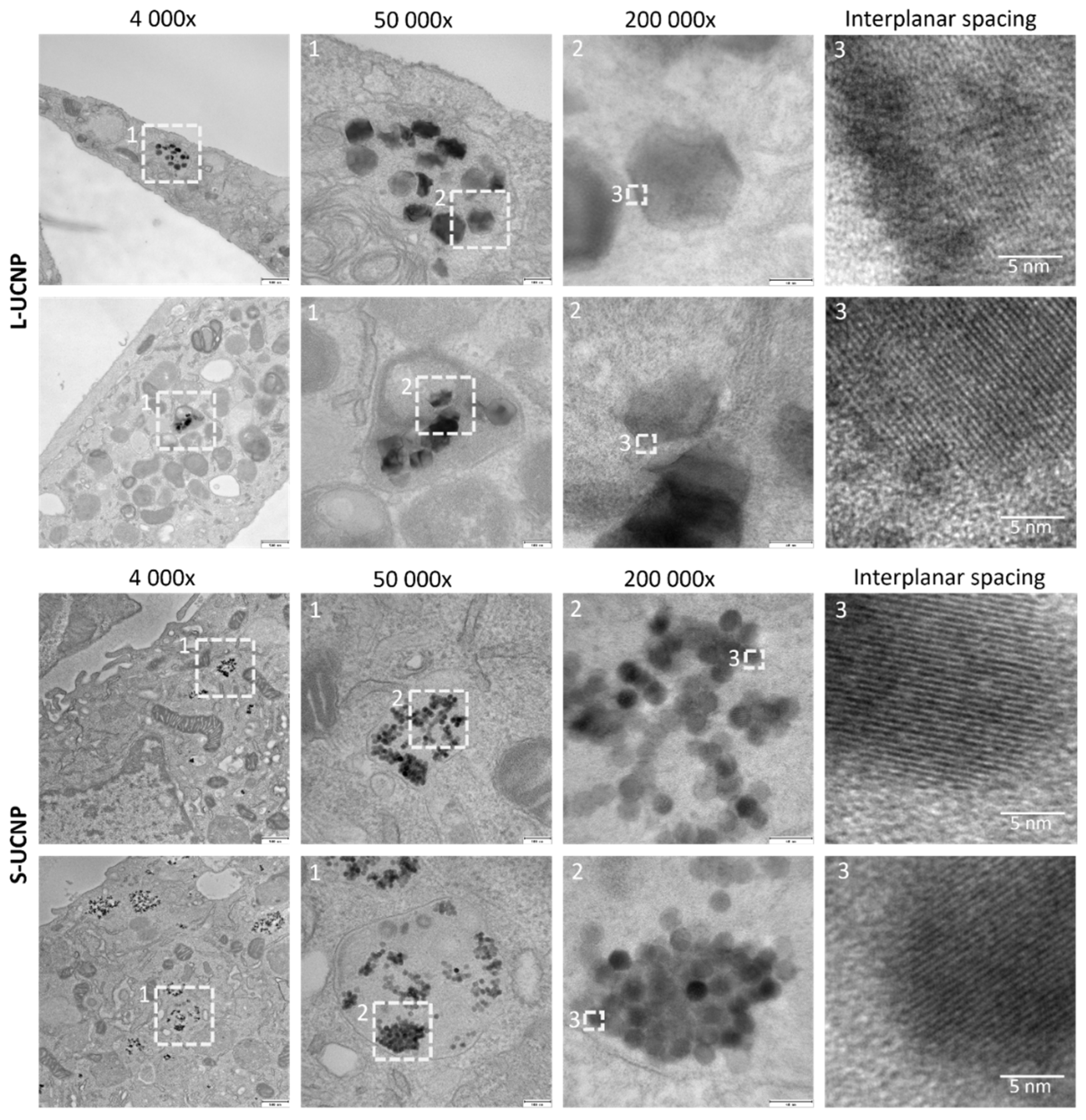
Figure 3.
Proliferation curves of rMSCs and C6 cells incubated with (a, b) small and (c, d) large UCNPs for 7 h.
Figure 3.
Proliferation curves of rMSCs and C6 cells incubated with (a, b) small and (c, d) large UCNPs for 7 h.
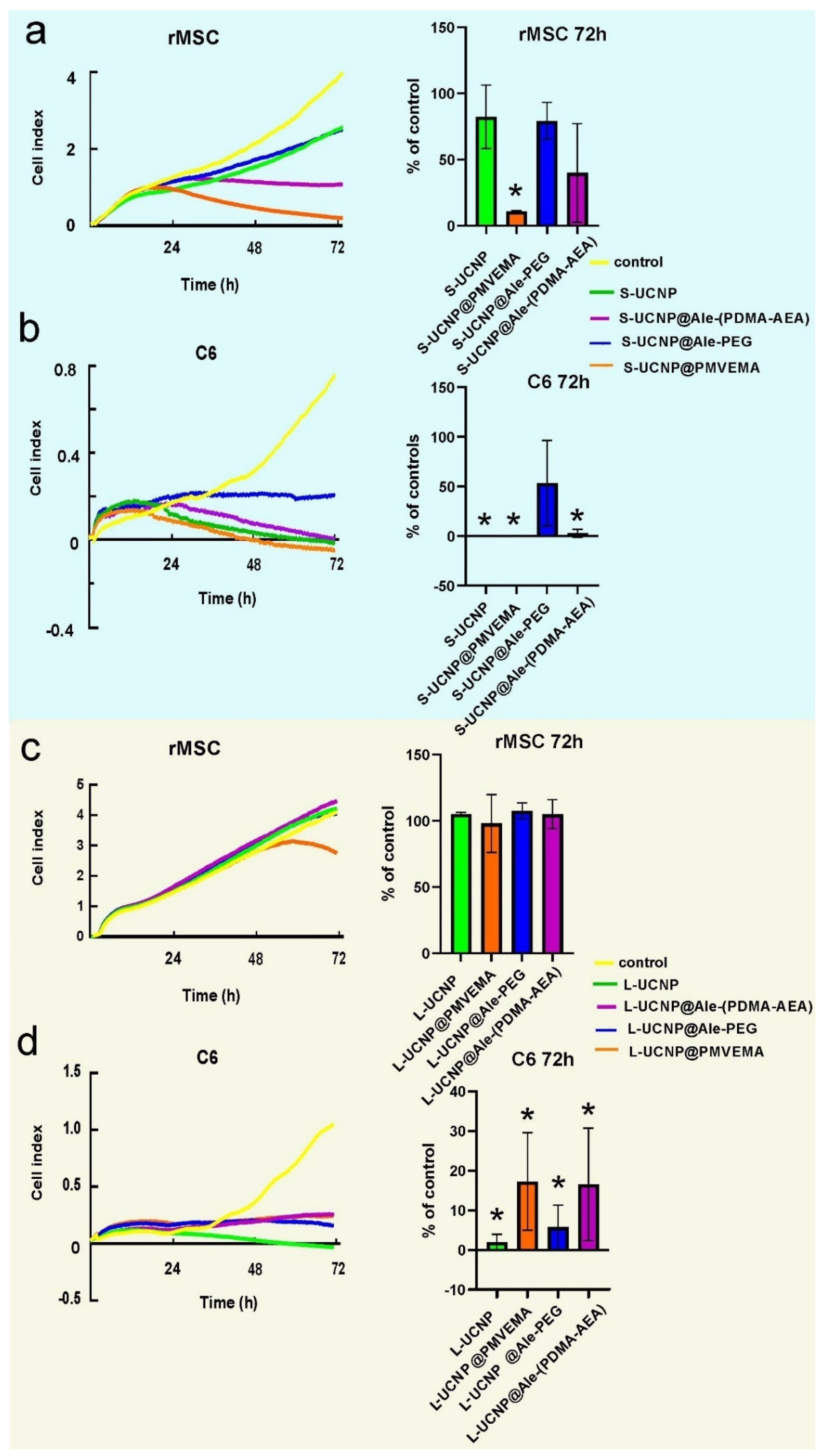
Figure 4.
Proliferation curves of rMSCs a C6 cells incubated with leaches from (a, b) S-UCNPs and (c, d) L-UCNPs for 72 h.
Figure 4.
Proliferation curves of rMSCs a C6 cells incubated with leaches from (a, b) S-UCNPs and (c, d) L-UCNPs for 72 h.
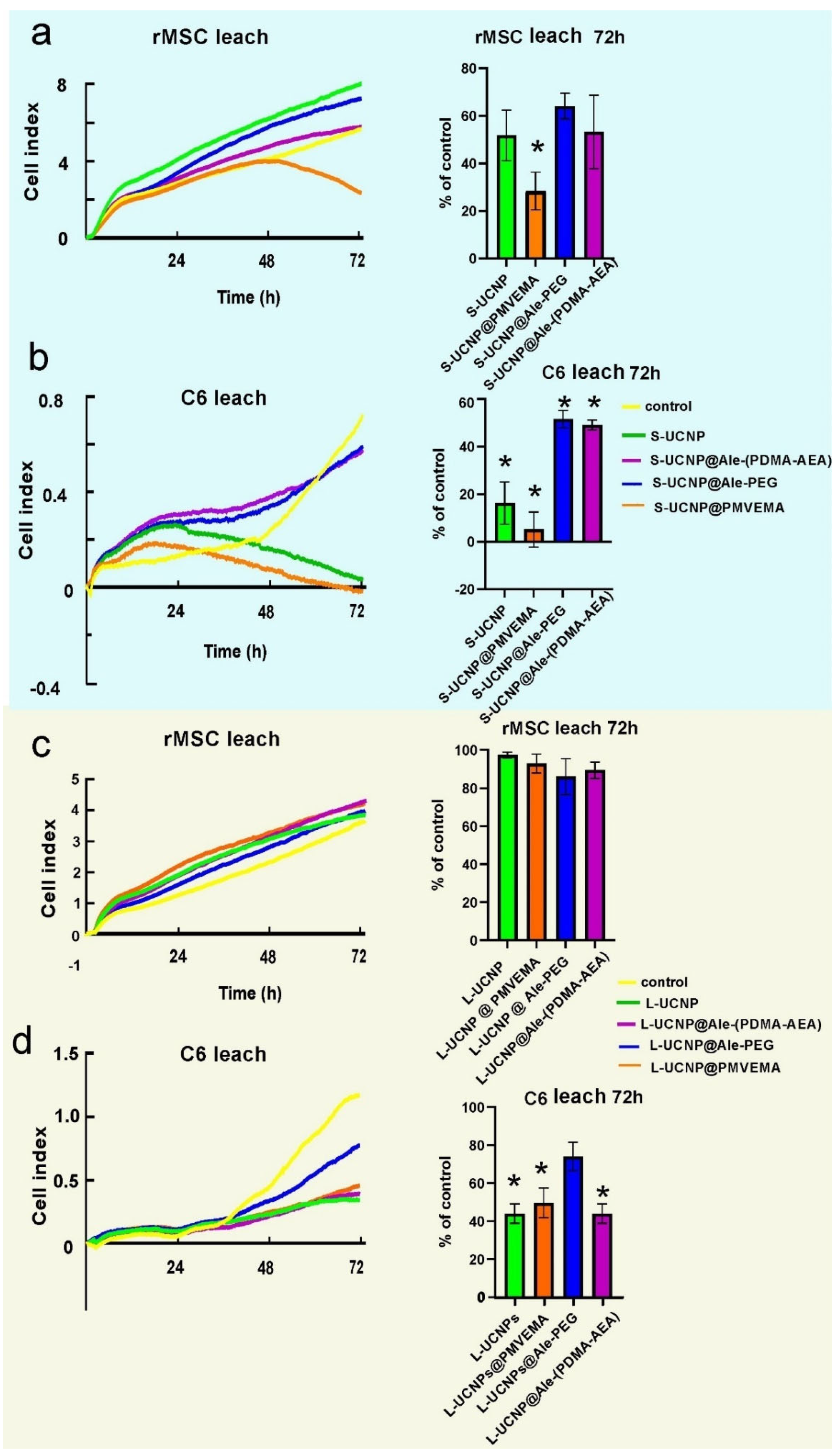
Figure 5.
The oxidative damage of UCNPs expressed as a percentage of tail DNA from rMSCs and C6 line. The statistical difference was found between L-UCNPs vs. L-UCPN@Ale-PEG and S-UCNP@PMVEMA in rMSCs; in C6 cells, no difference was found when comparing different nanoparticle treatments. The difference was found between S-UCNP@Ale-PDMA and negative control.
Figure 5.
The oxidative damage of UCNPs expressed as a percentage of tail DNA from rMSCs and C6 line. The statistical difference was found between L-UCNPs vs. L-UCPN@Ale-PEG and S-UCNP@PMVEMA in rMSCs; in C6 cells, no difference was found when comparing different nanoparticle treatments. The difference was found between S-UCNP@Ale-PDMA and negative control.
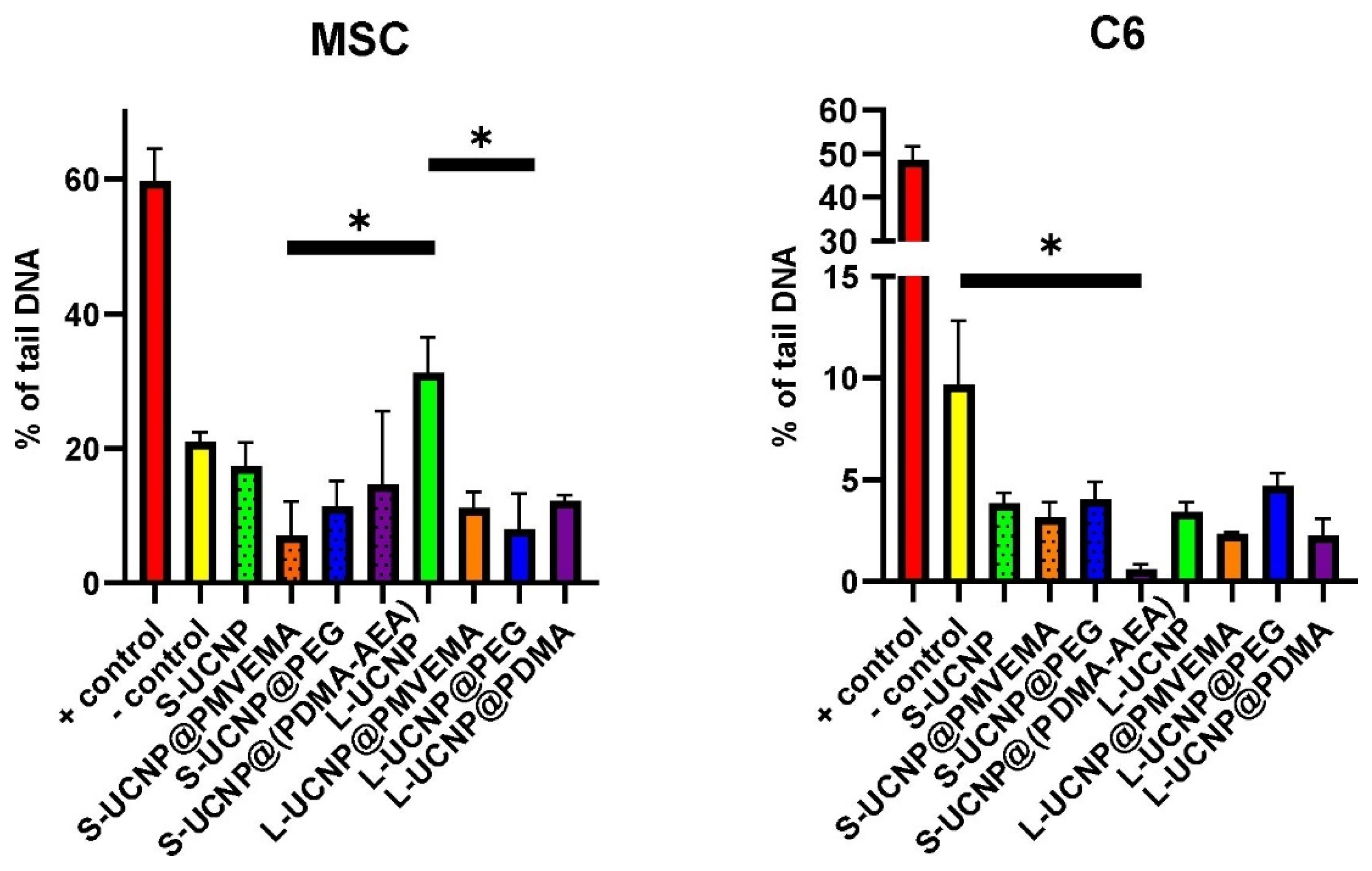
Figure 6.
(a, b) The intravenous application of the L-UCNP@Ale-PEG and L-UCNP@Ale-PDMA particles and saline. (a) L-UCNP@Ale-PEG particles were almost completely eliminated from liver 96 h after application, while L-UCNP@Ale-PDMA particles remained there in significantly higher amount. (b) The L-UCNP@Ale-PDMA particles were detected in liver using two-photon microscopy (see the arrow in (b)). Fluorescence signal at 535 nm of subcutaneously administered (c) L-UCNP@Ale-PEG and (d) L-UCNP@Ale-PDMA particles was detected in vivo in an experimental animal after excitation at 980 nm.
Figure 6.
(a, b) The intravenous application of the L-UCNP@Ale-PEG and L-UCNP@Ale-PDMA particles and saline. (a) L-UCNP@Ale-PEG particles were almost completely eliminated from liver 96 h after application, while L-UCNP@Ale-PDMA particles remained there in significantly higher amount. (b) The L-UCNP@Ale-PDMA particles were detected in liver using two-photon microscopy (see the arrow in (b)). Fluorescence signal at 535 nm of subcutaneously administered (c) L-UCNP@Ale-PEG and (d) L-UCNP@Ale-PDMA particles was detected in vivo in an experimental animal after excitation at 980 nm.
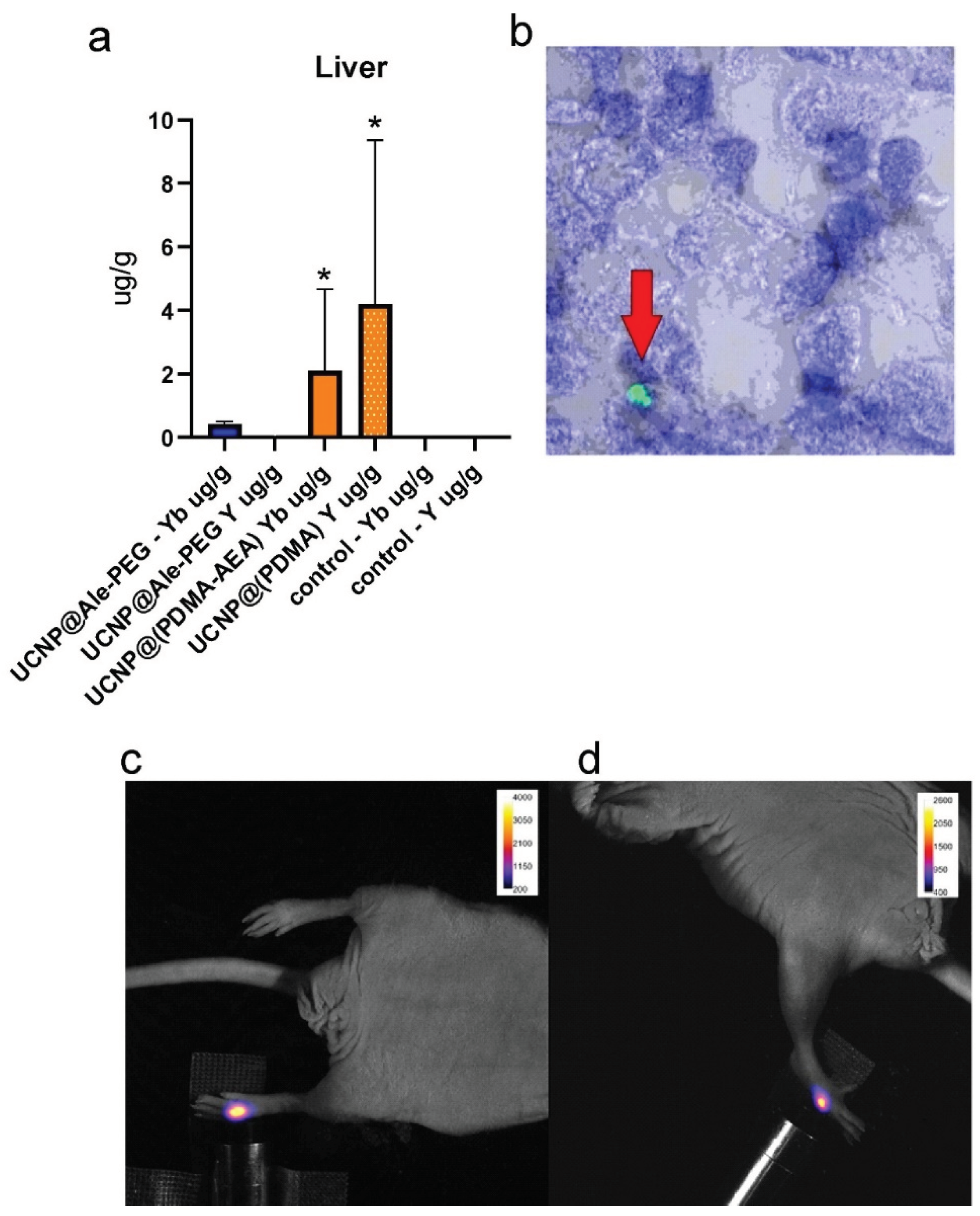

Table 1.
Characterization of neat and polymer-coated UCNPs.
| S-UCNPs | L-UCNPs | |||||||
| Coating |
Dn (nm) |
Đ |
Dh (nm) |
ζ-potential (mV) |
Dn (nm) |
Đ | Dh(nm) | ζ-potential (mV) |
| - | 25 | 1.01 | 101 | 30±5 | 121 | 1.01 | 174 | 28±3 |
| Ale-PEG | 25 | 1.01 | 68 | 8±1 | 119 | 1.02 | 158 | 4±1 |
| Ale-(PDMA-AEA) | 25 | 1.01 | 102 | 27±3 | 122 | 1.01 | 160 | 22±2 |
| PMVEMA | 25 | 1.01 | 112 | -32±2 | 119 | 1.01 | 234 | -46±6 |
UCNPs – upconverting NaYF4:Yb3+,Er3+ nanoparticles; Ale-PEG – poly(ethylene glycol)-alendronate; Ale-(PDMA-AEA) – poly(N,N-dimethylacrylamide-co-2-aminoethylacrylamide)-alendronate; PMVEMA – poly(methyl vinyl ether-co-maleic acid); Dn – number-average diameter (TEM); Ð – dispersity (Dw/Dn; TEM); Dh – hydrodynamic diameter (DLS).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



