Submitted:
05 March 2024
Posted:
06 March 2024
You are already at the latest version
Abstract
The reactivity of 3,5-di-(pyridin-4-yl)-1,2,4-thiadiazole (L1) with 1,4-diiodotetrafluorobenzene (1,4-DITFB) was explored and the halogen-bonded 1:1 co-crystal (1) was successfully isolated and structurally characterized.
Keywords:
dipyridyl-1
; 2-4-thiadiazole
; polypyridyl donors
; halogen bonding
; SC-XRD
1. Introduction
1,2,4-Thiadiazoles have been recognized as effective scaffolds in medicinal chemistry, since many derivatives are biologically active and very promising candidates in drug design [1]. Inspired by Cefozopran [2], the first 1,2,4-thiadiazole derivative to enter the market as an antibiotic, extensive synthetic efforts led to the isolation of numerous 1,2,4-thiadiazoles with potential biomedical applications, such as high cytotoxicity against human myeloid leukemia cells [3], inhibitors of Factor XIIIa in the blood coagulation process [4], neuroprotectors [5] and in the treatment of Alzheimer's disease [6]. The synthesis of 1,2,4-thiadiazoles is typically achieved starting from thioamides, whose oxidation is followed by cyclization, and several methods have been reported using a range of oxidants and reaction solvents [7,8]. A valid protocol reported the use of alcoholic thioamide solutions, which can be easily oxidized by molecular dihalogens, leading to the corresponding thiadiazole in good yields [9].
1,2,4-Thiadiazoles featuring pyridyl substituents, such as 3,5-di-(pyridin-4-yl)-1,2,4-thiadiazole (L1) and 3,5-di-(pyridin-3-yl)-1,2,4-thiadiazole (L2) (Scheme 1), have been successfully used as building blocks in supramolecular chemistry by exploring their reactivity towards metal ions in the preparation of coordination polymers and polygons [10,11]. The versatility of donors L1 and L2 as supramolecular synthons became evident when their reactivity towards dihalogens, interhalogens and other halogenated derivatives was investigated [12,13]. In this regard, the reaction of L1 and L2 with dihalogens and interhalogens was previously reported by our research group [12], and the self-assembly outcomes are summarized in Scheme 1. The results showed that donors L1 and L2 can give either Charge-Transfer (CT) adducts or salts with variable degrees of N-protonation (e.g. HL+, H2L2+) depending on the solvent polarity and the experimental setup (Scheme 1). The reaction of L2 with diiodine in CH2Cl2 resulted in the bis-adduct L2·2I2 with a short N···I bond distance (2.505 Å) and a linear N∙∙∙I‒I fragment as typically observed in CT-adducts. Notably, the reaction of L1 with diiodine under the same experimental conditions did not produce a crystalline product and its nature as L1·2I2 was established by microanalytical determinations and Raman spectroscopy [12].
The role of the solvent becomes crucial when considering the products obtained from the reactions between L1 or L2 and I2 or IBr in ethyl alcohol, where the following ionic compounds were obtained: (HL1+)(IBr2−), (HL1+)(I3−), (HL2+)(IBr2−), (HL2+)(I3−), (H2L22+)(I3−)2∙L2, (HL2+)(I5−) (Scheme 1) [12,13]. These structures share cations HL1+ or HL2+ with only one of the two pyridyl nitrogen atoms being protonated, resulting in the formation of head-to-tail polymeric arrays held by NH+···N hydrogen bonds (dN···N distances up to 2.770 Å), whose motif is shaped by the geometrical features of the former donors: wavy chains for cations HL1+ and either helices or zig-zag chains in the case of cations HL2+ [12]. The only exception among these ionic compounds is represented by (H2L22+)(I3−)2∙L2 where the donor L2 appears in both the neutral and the doubly charged HL22+ form.
When acetonitrile was used as a solvent and the donors L1 and L2 were reacted with I2, (H2L12+)(I3−)2∙2H2O and (HL2+)(I−)∙4CH3CN were isolated [13]. Moreover, the reaction of L2 with I2 in an iodoform/acetone mixture produced compound (H2L22+)(I−)2∙L2∙2CHI3 [13]. To further investigate the reactivity of L1 and L2 toward dihalogens, Pennington and coworkers introduced bismuth triiodide as a building block, producing self-assembled salts with formula (H2L12+)2(Bi8I284−)∙4CH3CN, [(H2L12+)(HL1+)](Bi2I93−)∙3H2O, (H2L22+)2(Bi4I164−)∙2CH3CN∙2I2, and (H2L22+)2(Bi6I224−)∙2CH3OH, whose crystal structures show L1 and L2 in their mono- or diprotonated forms along with four unusual polyiodobismuthate counterions [13].
2. Results
The slow evaporation of a chloroform solution of L1 and 1,4-DITFB in 1:1 molar ratio at room temperature afforded colorless crystals, established by means of X-ray diffraction analysis as a 1:1 halogen-bonded co-crystal with formula L2∙1,4-DITFB (compound 1; Figure 1). Compound 1 crystallizes in the triclinic space group P‒1 with two units in the unit cell (see Table S1 for structural data and refinement parameters).
Crystal data for compound 1: C18H8F4I2N4S, (Mr = 642.14 g mol−1) triclinic, P‒1, a = 5.6690(4) Å, b = 12.3300(9) Å, c = 14.1339(9) Å, α = 91.644(6), β = 96.314(6)°, γ = 92.400(6)°, V = 980.54(12) Å3, T = 173(2) K, Z = 2, ρcalc = 2.175 g/cm3, µ(Mo Kα) = 3.363 mm−1. The final R1 was 0.0333 [F2 ≥ 2 σ(F2)], wR2 was 0.0960 (all data) and the GooF = 1.043.
The 1,4-DITFB molecules interact with L1 to form neutral adducts at both N-pyridyl atoms with dN∙∙∙I distances of 2.801(5) and 2.947(4) Å and C–I···N angles of 177.4(2) and 168.3(2)° for N1∙∙∙I1 and N4∙∙∙I2i, respectively (entries a and b in Figure 2; i = 2+x, −1+y, −1+z; Tables S2 and S3). These values are similar to the average N∙∙∙I value of 2.9(2) Å retrieved from the CSD database for the structurally characterized compounds in which 1,4-DITFB interacts with pyridyl-based donors (the search was constrained to N···I distances up to the sum of the atomic van der Waals radii: 3.53 Å).
The resulting (L1∙1,4-DITFB)∞ 1D-chains propagate approximately along the [11] direction and pack into 2D sheets via weak C‒H∙∙∙F interactions (entries c–e in Figure 2 and Table 1) [17]. The FT-IR spectrum (Figure S1) recorded for compound 1 showed a shift towards lower frequency of the ν(C‒I) stretching mode from 760 to 748 cm−1 on passing from free 1,4-DITFB to the co-crystal, as a consequence of the halogen bonding between the two species [14].
3. Materials and Methods
3.1. General
L1 was synthesized according to a literature method [9]. 1,4-DIFTB and chloroform were purchased from Merck and used without any further purification. Elemental analysis determinations were performed with a Perkin Elmer EA CHN elemental analyzer. The FT-IR spectra (4000-400 cm−1) were recorded on KBr pellets on a Thermo Nicolet 5700 spectrometer. Melting point determination was performed on a FALC mod. C apparatus. Single crystal X-ray diffraction data were collected at 173 K on a Rigaku SCX mini diffractometer using graphite monochromated Mo-Kα radiation (0.71073 Å). Data collection and processing were carried out using CrysAlisPro [18]. The structure was solved with the ShelXT [19] solution program using dual methods and the model was refined using full matrix least squares minimization on F2 with ShelXL [20] 2018/3. The crystal was found to be a non-merohedral twin and the model was refined as a two-component twin. Olex2 1.5 [21] was used as the graphical interface.
3.2. Synthesis of (L1)(1,4-DITFB) (1)
L1 (12.0 mg; 5.00 · 10-5 mol) and 1,4-DITFB (20.1 mg; 5.00 · 10-5 mol) were dissolved in chloroform (5 mL) and the mixture was stirred at room temperature for 20 min. The resulting solution was filtered through a PTFE filter and the solvent allowed to evaporate slowly to afford compound 1 as colourless crystals suitable for X-ray diffraction analysis. (10.8 mg; 1.68 · 10-5 mol; 34 %) Elemental analysis calcd (%) for C18H8F4I2N4S: C 32.67, H 1.26, N 8.73. Found: C 31.88, H 0.66, N 8.21. M.p. = 186 °C. FT-IR (KBr, 4000–400 cm–1): 1599m, 1458vs, 1410s, 1335m, 1290m, 1207m, 1124m, 1063m, 995m, 939s, 825ms, 748m, 733ms, 712ms, 677m, 636ms, 505m, 474w, 422w cm–1.
4. Conclusions
The halogen-bonded co-crystal (1) was obtained by the self-assembly of 3,5-di-(pyridin-4-yl)-1,2,4-thiadiazole (L1) and 1,4-diiodotetrafluorobenzene (1,4-DITFB) in chloroform. The crystal structure of 1, determined by means of crystallographic tools, corresponds to the formulation L1∙(1,4-DITFB). A comparison between the FT-IR spectra of 1 and (1,4-DITFB) provided further evidence for the halogen bonding between the two building blocks.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org; Figure S1: FT-IR; Table S1: Crystal data and refinement parameters; Tables S2, S3: bond lengths and angles.
Author Contributions
Conceptualisation and writing (original draft): EP, MCA. MCA, EP, AP were involved in data analysis and the presentation of results. MCA, VL and FI are experts in the field of halogen bonding and extensively investigated the reactivity of L1 towards various halogenated species. AMZS, JDW and CLCW performed the XRD analysis of compound 1. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge Fondazione di Sardegna (FdS Progetti Biennali di Ateneo, annualità 2022).
Data Availability Statement
Crystallographic data were deposited at CCCD (CIF deposition number 2332380).
Acknowledgments
The authors acknowledge the Ministero per l’Ambiente e la Sicurezza Energetica (MASE; formerly Ministero della Transizione Ecologica, MITE) – Direzione generale Economia Circolare for funding (RAEE – Edizione 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Frija, L.M.T.; Pombeiro, A.J.L.; Kopylovich, M.N. Building 1,2,4-Thiadiazole: Ten Years of Progress. European J Org Chem 2017, 2017 (19), 2670–2682. [CrossRef]
- Iizawa, Y.; Okonogi, K.; Hayashi, R.; Iwahi, T.; Yamazaki, T.; Imada, A. Therapeutic Effect of Cefozopran (SCE-2787), a New Parenteral Cephalosporin, against Experimental Infections in Mice. Antimicrob Agents Chemother 1993, 37 (1), 100–105. [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cruz-Lopez, O.; Preti, D.; Tabrizi, M.A.; Fruttarolo, F.; Heilmann, J.; Bermejo, J.; Estévez, F. Hybrid Molecules Containing Benzo[4,5]Imidazo[1,2-d][1,2,4]Thiadiazole and α-Bromoacryloyl Moieties as Potent Apoptosis Inducers on Human Myeloid Leukaemia Cells. Bioorg Med Chem Lett 2007, 17 (10), 2844–2848. [CrossRef]
- Leung-Toung, R.; Tam, T.F.; Wodzinska, J.M.; Zhao, Y.; Lowrie, J.; Simpson, C.D.; Karimian, K.; Spino, M. 3-Substituted Imidazo[1,2-d][1,2,4]-Thiadiazoles: A Novel Class of Factor XIIIa Inhibitors. J Med Chem 2005, 48 (7), 2266–2269. [CrossRef]
- Perlovich, G.L.; Proshin, A.N.; Volkova, T.V.; Petrova, L.N.; Bachurin, S.O. Novel 1,2,4-Thiadiazole Derivatives as Potent Neuroprotectors: Approach to Creation of Bioavailable Drugs. Mol Pharm 2012, 9 (8), 2156–2167. [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, C.; Moreno, F.J. First Non-ATP Competitive Glycogen Synthase Kinase 3 β (GSK-3β) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease. J Med Chem 2002, 45 (6), 1292–1299. [CrossRef]
- Cashman, J.R.; Hanzlik, R.P. Oxidation and Other Reactions of Thiobenzamide Derivatives of Relevance to Their Hepatotoxicity. J.Org. Chem. 1982, 47 (24), 4645–4650. [CrossRef]
- Castro, A.; Castaño, T.; Encinas, A.; Porcal, W.; Gil, C. Advances in the Synthesis and Recent Therapeutic Applications of 1,2,4-Thiadiazole Heterocycles. Bioorg Med Chem 2006, 14 (5), 1644–1652. [CrossRef]
- Meltzer, R.I.; Lewis, A.D.; King, J.A. Antitubercular Substances. IV. Thioamides. J Am Chem Soc 1955, 77 (15), 4062–4066. [CrossRef]
- Aragoni, M.C.; Arca, M.; Coles, S.J.; Crespo Alonso, M.; Coles, S.L.; Davies, R.P.; Hursthouse, M.B.; Isaia, F.; Lai, R.; Lippolis, V. Coordination Polymers and Polygons Using Di-Pyridyl-Thiadiazole Spacers and Substituted Phosphorodithioato Ni II Complexes: Potential and Limitations for Inorganic Crystal Engineering. CrystEngComm 2016, 18 (30), 5620–5629. [CrossRef]
- Podda, E.; Arca, M.; Coles, S.J.; Crespo Alonso, M.; Isaia, F.; Pintus, A.; Lippolis, V.; Aragoni, M.C. Supramolecular Assemblies Tailored by Dipyridyl-1,2-4-Thiadiazoles: Influence of the Building Blocks in the Predictability of the Final Network. Supramol Chem 2020, 32 (4). [CrossRef]
- Aragoni, M.C.; Arca, M.; Caltagirone, C.; Castellano, C.; Demartin, F.; Garau, A.; Isaia, F.; Lippolis, V.; Montis, R.; Pintus, A. Cationic and Anionic 1D Chains Based on NH+⋯N Charge-Assisted Hydrogen Bonds in Bipyridyl Derivatives and Polyiodides. CrystEngComm 2012, 14 (18), 5809–5823. [CrossRef]
- Peloquin, A.J.; McMillen, C.D.; Pennington, W.T. Isolation of Unique Heterocycles Formed from Pyridine-Thiocarboxamides as Diiodine, Iodide, or Polyiodide Salts. CrystEngComm 2022, 24 (35), 6251–6261. [CrossRef]
- Wang, H.; Jin, W.J. Cocrystal Assembled by 1,4-Diiodotetrafluorobenzene and Phenothiazine Based on C - I...π/N/S Halogen Bond and Other Assisting Interactions. Acta Cryst. B 2017, 73 (2), 210–216. [CrossRef]
- Aragoni, M.C.; Podda, E.; Chaudhary, S.; Bhasin, A.K.K.; Bhasin, K.K.; Coles, S.J.; Orton, J.B.; Isaia, F.; Lippolis, V.; Pintus, A.; Slawin, A.M.Z.; Woollins, J.D.; Arca, M. An Experimental and Theoretical Insight into I2/Br2 Oxidation of Bis(Pyridin-2-Yl)Diselane and Ditellane. Chem Asian J 2023, 18 (23), e202300836. [CrossRef]
- Aragoni, M.C.; Podda, E.; Arca, M.; Pintus, A.; Lippolis, V.; Caltagirone, C.; Bartz, R.H.; Lenardão, E.J.; Perin, G.; Schumacher, R.F.; Coles, S.J.; Orton, J.B. An Unprecedented Non-Classical Polyinterhalogen Anion Made of [I2Cl]− and I2 at the 2-(p-Tolyl)Selenopheno[2,3-b]Pyridinium Cation Template. New J. Chem. 2022, 46 (45), 21921–21929. [CrossRef]
- Thalladi, V. R.; Weiss, H.C.; Bläser, D.; Boese, R.; Nangia, A.; Desiraju, G.R. C−H···F Interactions in the Crystal Structures of Some Fluorobenzenes. J. Am. Chem. Soc. 1998, 120, 34, 8702–8710. [CrossRef]
- Rigaku Oxford Diffraction (2015), CrysAlisPro. version 1.171.39.8d. Rigaku Corporation, Tokyo, Japan.
- Sheldrick, G.M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. A 2015, 71 (1), 3–8. [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C 2015, 71 (1), 3–8. [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42 (2), 339–341. [CrossRef]
Scheme 1.
CT adducts and ionic compounds isolated from the reactions between N-donors L1 and L2 and halogenated species. Refcodes are given in parentheses.
Scheme 1.
CT adducts and ionic compounds isolated from the reactions between N-donors L1 and L2 and halogenated species. Refcodes are given in parentheses.
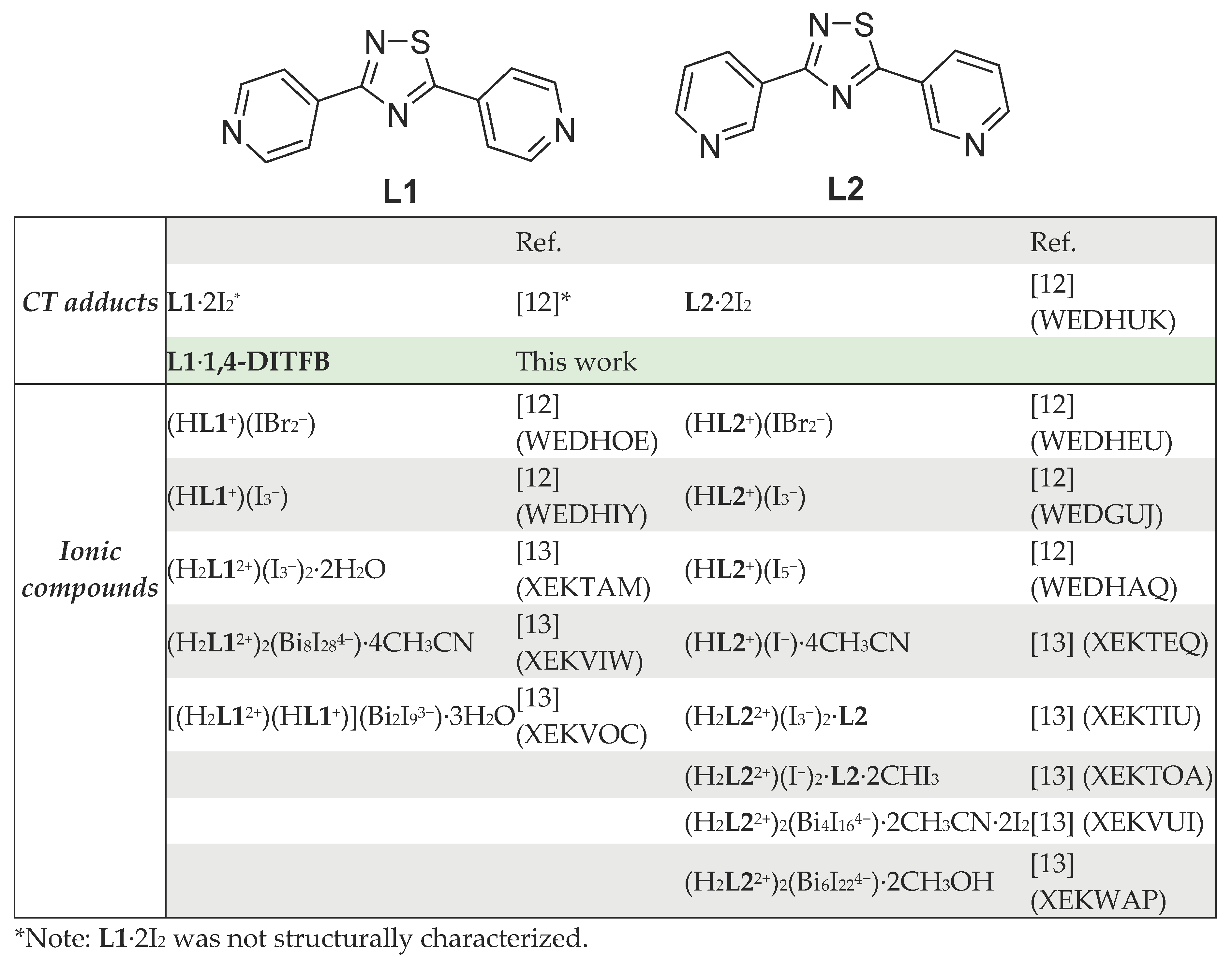
Figure 1.
X-ray crystal structure of compound 1 with the numbering scheme adopted. Displacement ellipsoids were drawn at the 50% probability level.
Figure 1.
X-ray crystal structure of compound 1 with the numbering scheme adopted. Displacement ellipsoids were drawn at the 50% probability level.
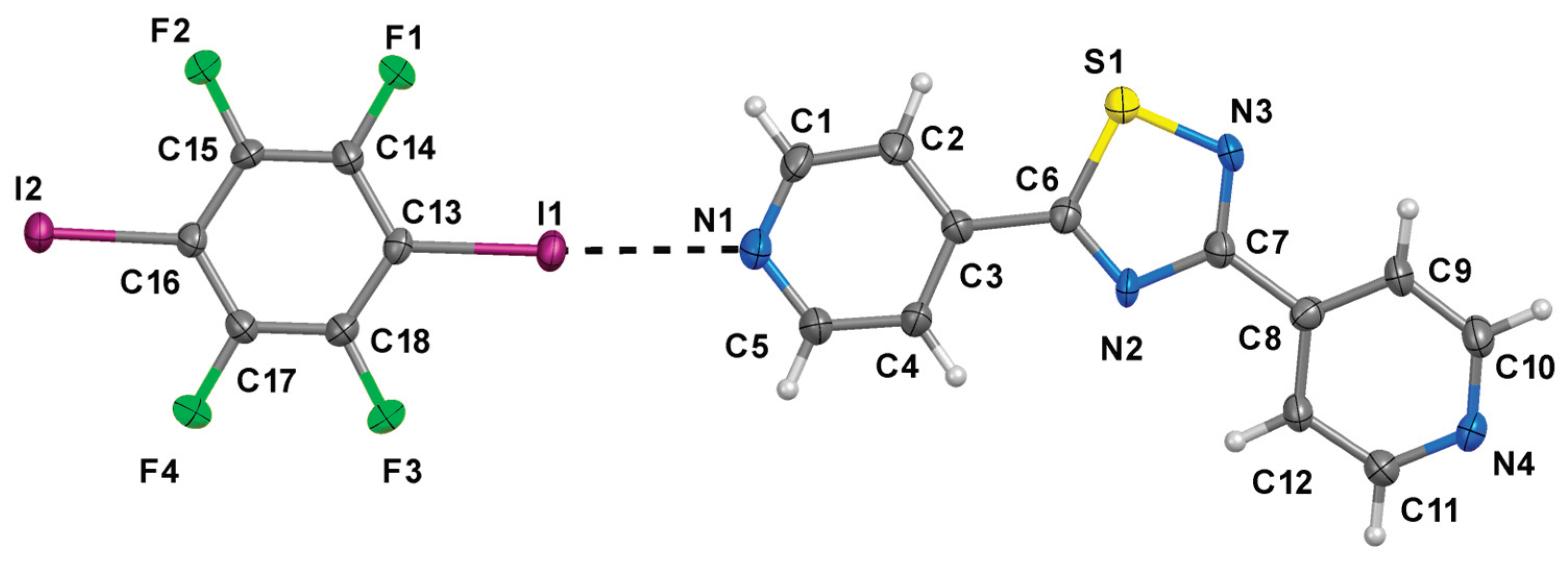
Figure 2.
Partial view of the crystal packing of 1 showing (a) a single layer with the relevant intermolecular interactions labeled according to Table 1, and (b) adjacent layers viewed along the [110] direction.
Figure 2.
Partial view of the crystal packing of 1 showing (a) a single layer with the relevant intermolecular interactions labeled according to Table 1, and (b) adjacent layers viewed along the [110] direction.
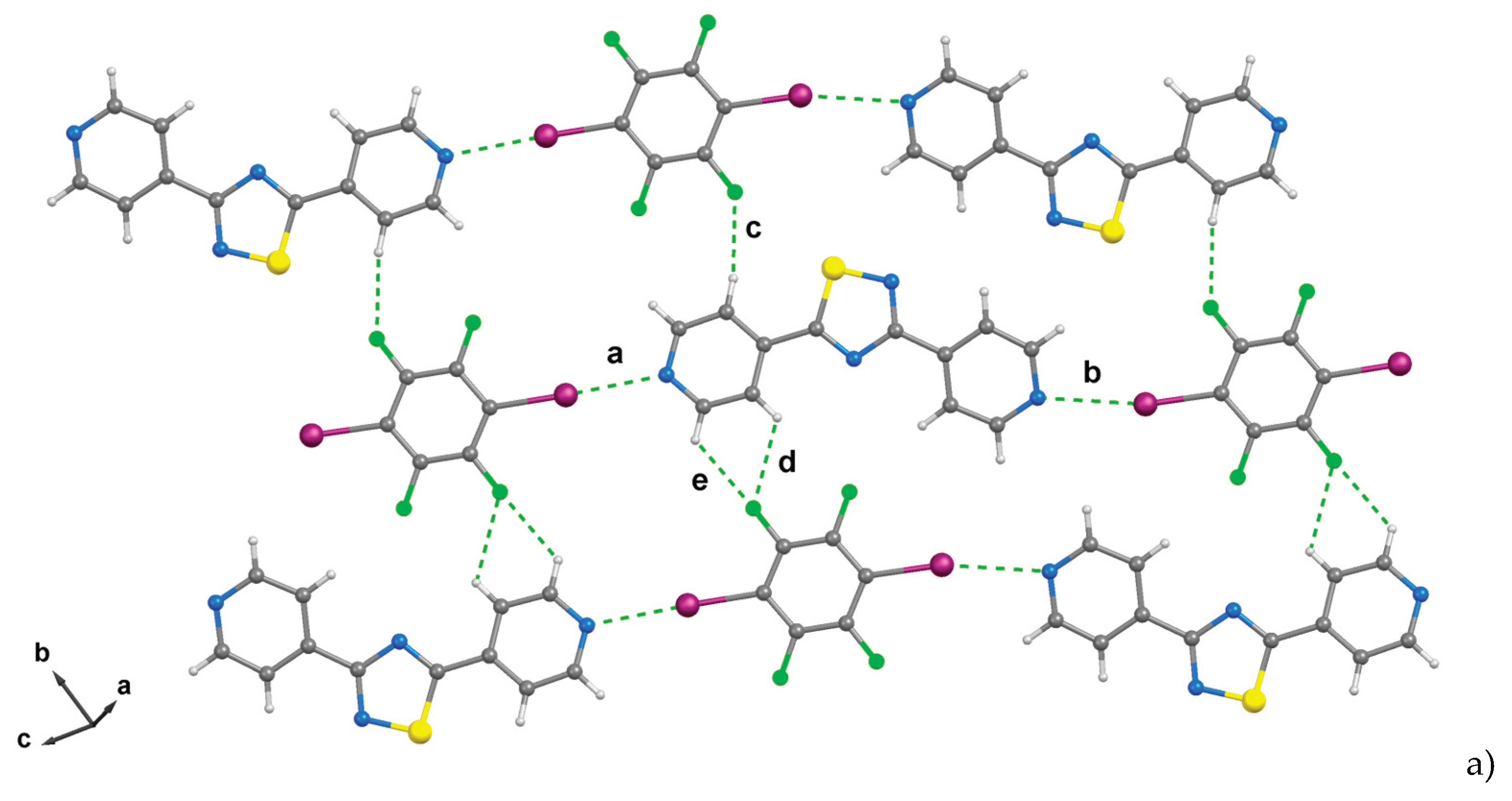

Table 1.
Compound 1 intermolecular interactions.
| C‒I∙∙∙N | dC–I (Å) | dI∙∙∙N (Å) | αC–I···N (°) | ||
| a | C13‒I1∙∙∙N1 | 2.101(5) | 2.801(5) | 177.4(2) | |
| b | C16i‒I2i∙∙∙N4 | 2.092(5) | 2.947(4) | 168.3(2) | |
| C‒H∙∙∙F | dC–H (Å) | dH∙∙∙F (Å) | dC∙∙∙F (Å) | αC–H···F (°) | |
| c | C2‒H2∙∙∙F2ii | 0.95 | 2.450 | 3.307(6) | 150 |
| d | C4‒H4∙∙∙F3iii | 0.95 | 2.607 | 3.142(6) | 122 |
| e | C5‒H5∙∙∙F3iii | 0.95 | 2.505 | 3.111(6) | 116 |
Symmetry codes: i = 2+x, −1+y, −1+z; ii = 1−x, 2−y, 1−z; iii = −x, 1−y, 1−z. .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



