
Submitted:
04 March 2024
Posted:
05 March 2024
You are already at the latest version
Abstract
Rarely has a chemical elicited as much controversy as has dichloroacetate (DCA). DCA was initially considered a dangerous toxic industrial waste product. Then it was a potential treatment for lactic acidosis. However, the main controversies started in 2008 when DCA was found to have anti-cancer effects in experimental animals. These publications showed contradictory results in vivo and in vitro, so that a thorough consideration of this compound in cancer is merited. Despite 50 years of experimentation, DCA’s future in therapeutics is uncertain. Without adequate clinical trials and lack of health authorities’ approval, DCA has been introduced in off -label cancer treatments in alternative medicine clinics in Canada, Germany, and other European countries. The lack of well-planned clinical trials and its use by people without medical training has discouraged consideration by the scientific community. There are limited thorough clinical studies of DCA, and many publications are individual case reports. Case
reports of DCA benefits against cancer have been increasing recently. Furthermore, it has been shown that DCA synergizes with conventional treatments and other repurposable drugs. Beyond the classic DCA target, pyruvate dehydrogenase kinase, new target molecules have also been recently discovered. These findings have renewed interest in DCA.
This paper explores whether existing evidence justifies further research on DCA for cancer treatment and it explores the role DCA may play in oncology treatment.
Keywords:
dichloroacetate
; cancer
; lactic acidosis
; glioma
; pyruvate dehydrogenase kinase
; pyruvate dehydrogenase
; pyruvate dehydrogenase phosphatase
1. Introduction
1.1. Lactate and Cancer
Before understanding a putative role for DCA in cancer treatment, it is important to understand some basics about cancer and lactic acid metabolism.
Cancer cells differ from normal cells in many ways, including their metabolism as was described by Warburg [1,2]. Cancer metabolism is bound to the disease’s development and progression in such an intricate way that we may assume that there is no malignancy without the appropriate metabolic changes. Originally it was thought that the metabolic alterations mainly affected the carbohydrate catabolic pathways, but nowadays there is abundant and clear evidence that cancer’s metabolic alterations extend well beyond carbohydrates [3,4,5].
Among the many metabolic alterations in cancer, glycolytic as opposed to oxidative metabolism, is one of the hallmarks of cancer [6]. Cancer “needs” a glycolytic phenotype to avoid cell death [7]. Apoptosis “needs” an oxidative phenotype [8]. Cells that depend primarily on the glycolytic pathway are therefore resistant to cell death [9,10,11], and cancer cells are particularly resistant to death.
The reversion of the glycolytic phenotype decreases proliferation and increases apoptosis of malignant cells as we shall further analyze in this review.
The main drivers of this metabolic transformation are the expressions of oncogenic genes and hypoxia. Very early in a tumor’s development these switch the preferential form of metabolism from oxidative to glycolytic. We describe the process as “preferential”, because not all the cells in the tumor adopt the glycolytic metabolism, some remain oxidative. Furthermore, some non-malignant stromal cells are “enslaved” by the tumor and also become glycolytic [12]. These produce lactic acid that is taken up and utilized by malignant cells that retain some oxidative properties as an energy source. It is important to note that even glycolytic cancer cells are not fully glycolytic and a certain percentage of glucose and lactate are metabolized through the mitochondrial oxidative pathway [13]. This is because glycolysis and oxidative metabolism are not mutually exclusive, but to a certain extent they are cooperative [14].
It is also important to understand lactic acid metabolism in cancer cells. Warburg found that the high production of lactic acid in cancer (60 times more than in normal cells, expressed in cubic millimeters per hour) is characteristic of cancer glycolytic metabolism [15,16]. The extrusion of this lactic acid from the cell is a vital activity for cell survival because if it were to remain inside, the intracellular pH would become very acidic, and the cell would undergo acidic stress. Malignant cells need a slightly alkaline intracellular milieu for proliferation and biosynthesis [17,18] therefore, it is essential for them to export this excess lactic acid. This is done by specialized lactic acid transporters (MCTs). Of these MCT1 and MCT4 are mainly responsible for lactate extrusion. Lactate transport is coupled with proton translocation, so export of lactate also implies an equimolecular number of exported protons. The extruded lactic acid is thus partly responsible for the extracellular acidity. As noted above, cancer cells can also import lactate and MCT1 is the main lactate importer in malignant oxidative cells.
Cancer cells also have very important alterations in their intracellular and extracellular pH. With the generation of excess intracellular acid from lactate and other sources, there is simultaneously a very active extrusion of protons mediated by NHE1 (sodium-hydrogen exchanger-1) and other membrane ion channels [19]. These transporters prevent intracellular pH acidification. Additionally, there is a particular pH condition that is generated in cancer where the intracellular pH becomes slightly alkaline while extracellular pH is quite acid. This situation is exactly the opposite of that found in normal tissues and is called “inversion of the pH gradient” [20].
This inversion of the pH gradient is not exclusively a consequence of high lactic acid production and its extrusion. Lactic acid is an important factor in the equation, and in normal blood the lactate concentration does not go beyond 1 mM, while in the tumor microenvironment it can rise to levels of 30-40 mM [21]. However, hypoxia and elevated proton extrusion are also strong mediators of extracellular acidity in the tumor microenvironment. In this regard, Newell [22] and Yamagata [23] have found that lactic acid is not essential for the creation of a highly acid extracellular matrix (ECM) in tumors.
Increased production of lactic acid and its extrusion are the direct consequences of six phenomena:
- 1)
- Malignant cells’ increased glucose uptake and metabolism.
- 2)
- Glycolytic metabolism which occurs even in the presence of oxygen (Warburg effect).
- 3)
- Decreased activity of the pyruvate dehydrogenase (PDH) complex.
- 4)
- Increased activity of pyruvate dehydrogenase kinases (PDK)
- 5)
- Increased expression and activity of the lactate extrusion proteins MCT1 and MCT4.
- 6)
- Increased expression and activity of the glycolytic enzymes.
To summarize these causes, we may say that increased lactate is the result of increased glycolytic flux.
Not surprisingly, there has been study of methods to block MCT transport as a form of chemotherapy [24,25,26,27,28] however, as yet there is no clinically approved drug for this purpose. Lipophilic statins (atorvastatin, simvastatin) have shown MCT4 inhibitory effects at the experimental level, while the hydrophilic statins had negligible effects [29]. However, statins are not yet used clinically for MCT inhibition.
Developing strong clinically useful inhibitors of MCTs will not be easy, because normal cells need MCT activity. For example, muscle, retina, and other tissues use lactates as an energy source. This probably means that MCT inhibitors will show an elevated toxicity towards normal cells. This is the case with AZD3965, an MCT1 inhibitor in an advanced state of research that has undergone phase I and II clinical trials [30].
Another more clinically useful approach could be to reduce lactic acid production which would push the cell towards oxidative phosphorylation: this would be the supposed function of dichloroacetate (DCA).
The importance of lactic acid in cancer must be emphasized. It is true that many publications suggest that lactic acid is an end product or a “waste product” of glycolytic carbohydrate metabolism. However, lactates or lactic acid are not end products or waste products.
Lactic acid serves as a fuel for oxidative malignant cells, as we have described above. The lactate shuttle, that carries lactate from glycolytic cells to oxidative cells (referred to as lactophagic cells) means that lactate is not an end product of metabolism but can be further used by the appropriate cell types. Evidence that lactic acid is not a waste product with physiological or pathological roles such as pro-tumoral effects comes from the following studies which show that
- 1)
- 2)
- 3)
- 4)
- 5)
- 6)
- 7)
- Lactate activates HIF-1α in non-glycolytic cells (but not in glycolytic cells) which in turn further stimulates glycolysis and angiogenesis [50].
- 8)
- 9)
- Lactate has a positive correlation with radioresistance [53].
- 10)
- It increases hyaluronan production involved in migration and growth [54].
- 11)
- Lactate modulates the tumor microenvironment [55].
- 12)
- 13)
- Lactylation has also been found to be an important mechanism of post-translational modification of proteins associated with poor prognosis in cancer progression [59].
- 14)
- Lactate is an important source of energy for oxidative lactophagic cells [60].
- 15)
- The pyruvate to lactate conversion by lactate dehydrogenase regenerates nicotinamide adenine dinucleotide (NAD+) in the cytoplasm [61] and NAD+ is an important metabolite for redox processes.
(For a review on lactate in cancer see Kocianova et al. [62]).
All the above effects of lactate clearly show that it is not an innocent bystander, but an active tumor promoter. Some authors have called it an "oncogenic lacthormone" [63].
Therefore, the reduction of lactate production, or its extrusion from cells becomes another important weapon in the fight against cancer. This is where DCA may have a contribution to make in the fight against cancer.
1.2. Dichloroacetate (DCA)
1.2.1. Therapeutic History of Dichloroacetate
DCA is an organic compound with the formula Cl2CHCO2H that is an analogue of acetic acid. DCA has been used for the treatment of lactic acidosis in acutely affected children and adults [64,65] and in congenital cases in children [66] since the 1970s [67]. The efficacy of DCA for treatment of lactic acidosis was examined in a randomized placebo-controlled study of DCA by Stacpoole et al. They did not find clear advantages of the treatment, although it decreased arterial blood lactic acid concentrations by 20% or more and increased arterial blood pH [68].
Another study examined children with lactic acidosis caused by malaria. Their treatment with DCA showed better results than in congenital cases [69]. A third study on DCA examined congenital lactic acidosis. This is a disease caused by mutations of the pyruvate dehydrogenase complex (PDH) or it can be caused by mutations of enzymes of the mitochondrial respiratory chain. Stacpoole et al. [70] examined over 200 pediatric and adult patients with congenital lactic acidosis, and DCA lowered levels of lactic acid in blood. They fell at least 20% within 6 hours of administration, although this did not improve clinical outcomes. DCA still remains a drug being investigated for the treatment of congenital lactic acidosis.
1.2.2. DCA Enters the Oncology Terrain
In 2007 a group headed by Drs. Bonnet and Michelakis [71] called attention to DCA`s putative usefulness in cancer. They studied three highly glycolytic cell lines that are: glioblastoma (MO59K), lung adenocarcinoma (A549) and breast cancer cells (MCF7). The three cell types had a hyperpolarized mitochondrial membrane potential (ΔΨm) which was reversed to normal by incubation with DCA in a dose-dependent manner. DCA also increased voltage gated potassium channel (Kv1.5) expression which was decreased in the malignant cells. The combination of hyperpolarized ΔΨm and low expression of Kv1.5 is a consequence of high glycolytic flux metabolism and induces resistance to apoptosis. They found that DCA induced apoptosis and decreased proliferation by restoring mitochondrial oxidative metabolism, without toxicity to normal cells. Figure 1.
Since then, many clinics, usually called “DCA Clinics” have opened, mainly in Canada and Germany. These clinics provide other naturopathic treatments for cancer, but the main therapy given is based on DCA. However, due to lack of FDA approval, DCA has not entered mainstream of oncology practice. Although DCA use was born as an alternative cancer treatment, unfortunately its real benefits in cancer are still unproven, although there is much evidence that supports the need for more testing of the drug. The fact that in many cases it has been co-opted by medically unqualified people raises doubts as to if it will ever be validated or accepted as an effective cancer treatment. (Figure 2).
The objectives of this review are:
- 1)
- To summarize and review published data on DCA use in cancer.
- 2)
- To establish an impartial view of benefits/harm that DCA may cause.
- 3)
- To determine the role that DCA should or should not have in cancer treatment.
- 4)
- To evaluate the opportunity and convenience of further research.
- 5)
- To find out if DCA deserves to be rescued from the hands of unqualified people and introduced as a complementary treatment for cancer.
1.2.3. Chemistry and Pharmacology of DCA
DCA is an investigational drug used to treat lactic acidosis, pulmonary hypertension, familial hyperlipidemia, and more recently cancer. Its use is unofficially accepted for the treatment of lactic acidosis in children with malaria in some African countries [73].
Humans are exposed to small amounts of DCA through drinking water. Chlorinated acetic acids, including DCA, are formed from organic material during water chlorination [74]. According to the US Environmental Protection Agency DCA concentrations in drinking water range from 1 to 99 µg/l.
The structure of DCA is shown in Figure 3. DCA is a small molecule and an organochlorine compound of acetic acid that is sometimes sold as a sodium salt.
DCA is an orally available molecule that is quickly and almost completely absorbed by the digestive system [77]. It is initially distributed to the liver and muscles, and afterwards to other tissues [78]. DCA is metabolized by GSTZ1 (a glutathione transferase isoform) [79]. (Figure 3). 20% of DCA is bound to plasma proteins [80]. An example of how it is distributed throughout tissues was shown by James et al. In young adult rats given a single oral dose of 50 mg of sodium dichloroacetate per kg of body weight, the tissue distribution was: muscle (11.9%), liver (6.19%), gastrointestinal tract (3.74%), fat (3.87%), and kidneys (0.53%), the rest went to other tissues [81].
Effects in humans were examined with DCA administered to 16 healthy individuals. With 1 to 50 mg/kg IV infusion, concentrations of plasma glucose, lactate and alanine were examined. Plasma levels linearly followed those of the administered dose up to 30 mg/kg. 35 mg/kg was considered the most effective dose regarding lactic acid, which fell to 75% below baseline concentrations within 2 hours of the infusion [82]. Alanine concentrations were also reduced while blood glucose was not affected in these healthy individuals but was reduced in diabetic patients through stimulation of peripheral glucose utilization and inhibition of gluconeogenesis [83]. DCA also inhibited lipogenesis and cholesterol synthesis.
In diabetic patients with slightly elevated lactic acid, daily administration of oral DCA 50 mg/kg reduced alanine and lactic acid in plasma [84].
It should be noted that experimental administration of a single dose of DCA may yield misleading effects on dose and metabolism, compared with effects of repeated dose treatment that could be used in patient treatment. Gonzalez-Leon et al. [85] have shown that DCA drug has a surprising feature when repeatedly administered to rats: it inhibits its own metabolism. This means that the chronic administration of DCA differs in its plasma concentration, toxicology and metabolism from the metabolism that occurs with single doses.
In humans, the half life of DCA was examined using IV infusion. With a 10 mg/kg infusion the maximum plasma concentration achieved was between 19.9 μg/ml and 24.7 μg/ml with a half life of only 20 minutes. If the infused dose was increased to 20 mg/kg the plasma concentration was between 57.3 and 74.9 μg/ml with a half life of 36 minutes. In dogs and rats the half life was much longer; the half life of a 100 mg/kg dose was 4 hours in rats and 24 hours for dogs [86]. These marked differences among species call attention to:
- 1)
- doubts about the possibility of translating findings in other animals to humans.
- 2)
- the importance of the inter-species differences of the clearance mechanism.
Confirming these inter-species differences, Maissenbacher et al. have shown that dogs have a slower DCA metabolism and a slower clearance than humans and rats due to greater inhibition of GSTZ1 [87].
The first dose is more rapidly cleared from plasma than subsequent doses, as Gonzalez-Leon et al. have shown [85]. This is probably due to GSTZ1 inhibition by DCA and is similar in all species. The decrease in DCA clearance by multiple doses is not a minor issue, because the initial clearance may be reduced to less than 25% of the initial one in successive doses [88].
The metabolic pathway of DCA starts with dehalogenation to monochloroacetate and then glyoxylate. After the dechlorination, degradation continues to glycine, and the final products are oxalate and carbon dioxide [89].
1.2.4. Mechanism of Action
The best known and main mechanism of action of DCA is the inhibition of pyruvate dehydrogenase kinase (PDK) and its isoforms. This inhibition increases the flux of pyruvate into the mitochondria, promoting glucose oxidation instead of glycolysis [90].
Pyruvate dehydrogenase kinase (PDK) is a kinase that inactivates the pyruvate dehydrogenase enzyme (PDH) complex through phosphorylation. By downregulating the activity of this complex, PDK decreases the oxidation of pyruvate in mitochondria and increases the conversion of pyruvate to lactate in the cytosol.
PDH is regulated by two systems:
- a)
- an inhibitory system represented by the four isoforms of PDK and
- b)
- a "disinhibitory" system represented by the two PDH phosphatases (see below)
Other mechanisms of action of DCA in cancer can be found in the medical literature. Stockwin et al. [91] found that cytotoxicity of DCA was only achieved in those cells that suffered mitochondrial DNA mutations that “condemn” them exclusively to the glycolytic pathway. (This would explain the synergy between DCA and metformin, see below).
2.
2.1. PDH Complex
The PDH complex is the gate for pyruvate to enter the oxidative metabolic pathway. It is prudent to review the complex and its regulation. This enzymatic complex is formed by three enzymes. The first, pyruvate dehydrogenase, decarboxylates pyruvate forming hydroxyethyl thiamin. Thiamin acts as a coenzyme (Figure 4). The second enzyme, dihydrolipoyl transacetylase transfers the acetyl group to lipoamide and then to coenzyme A, forming acetyl-CoA that enters the Krebs Cycle. The third enzyme, dihydrolipoyl dehydrogenase, captures two protons that are transferred to FAD. Figure 4. The PDH complex is regulated by phosphorylation through pyruvate dehydrogenase kinases and dephosphorylation by pyruvate dehydrogenase phosphatases. Insulin is a strong activator of the PDH complex [92,93].
2.2. PDK Family of Enzymes
The PDK enzymatic complex has been called the gatekeeper of mitochondria [94].
PDK was cloned and sequenced by Popov et al. in 1992 [95] and it also functions as a histidine protein kinase. In humans, at least, four isoforms were identified: PDK1, PDK2, PDK3 and PDK4 [96,97]. PDK1 is under HIF-1α modulation and cMyc [98,99] as part of cell adaptation to hypoxic conditions. PDK2, PDK3 and PDK4 seem to be regulated by the peroxisome proliferators activation receptor β/δ (PPAR β/δ) [100,101].
p53 negatively regulates PDK2 [102], thus, promoting oxidative metabolism.
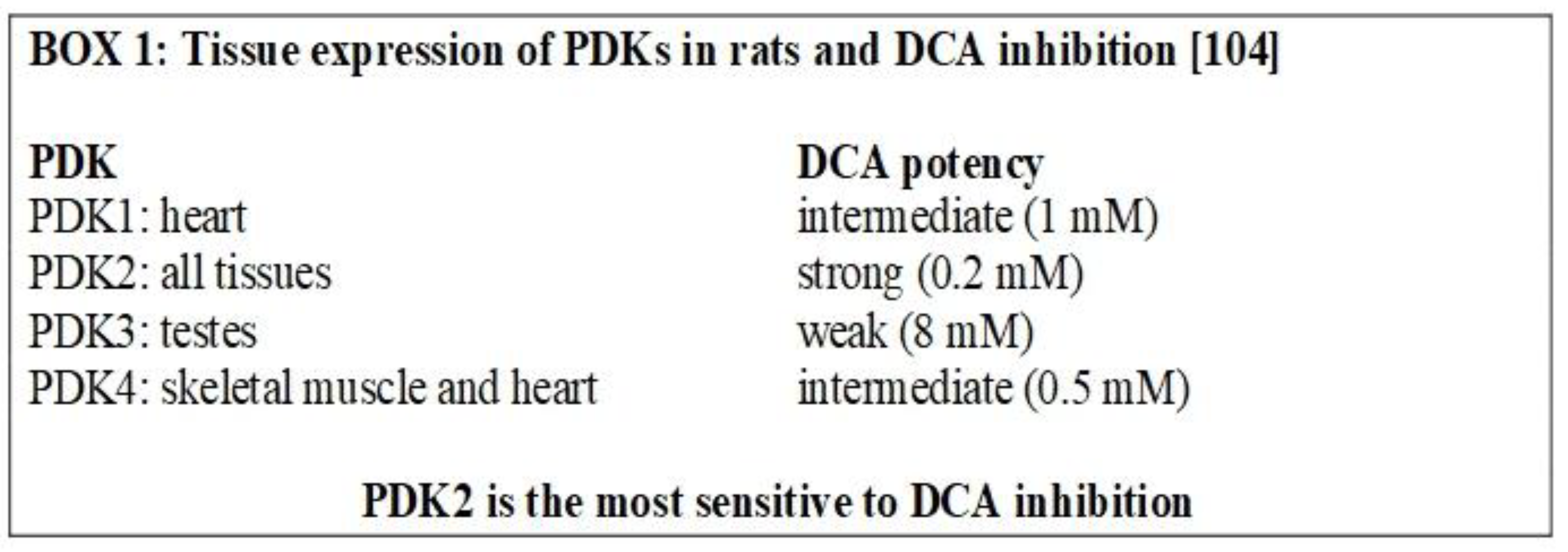
The mechanism by which PDKs inhibit PDH is a serine phosphorylation at site 1 (there are three possible sites, but only site 1 is responsible for full deactivation of the PDH complex [103]). All the PDKs can phosphorylate PDH, but PDK2 has the greatest phosphorylation activity in vivo [104] (Box 1). P53 is another negative regulator of PDK2 [105].
2.3. Inhibition of PDKs
PDK1 and PDK3 can be inhibited by different compounds. The mechanism of action of the investigational drug AZD7545 is through binding the lipoyl-binding pocket of the enzyme while DCA binds the interior of the helix bundle in the N-terminal domain, promoting conformational changes. Radicicol, another inhibitor, binds to the ATP binding pocket [106].
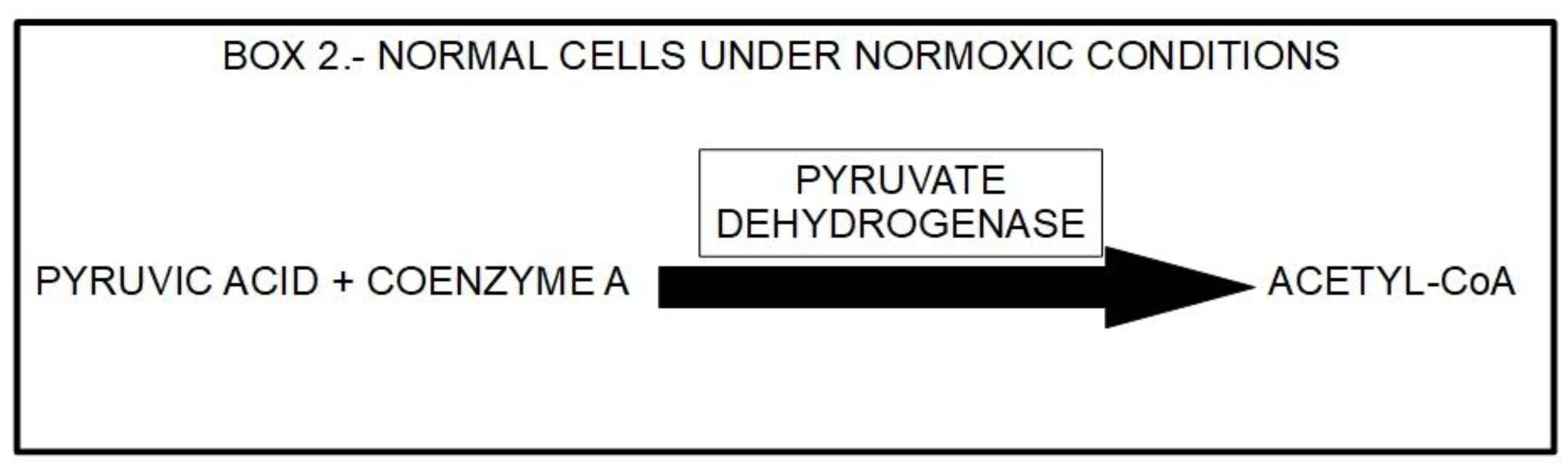
The downstream mechanism of DCA inhibition of PDK, is to “force” the cancer cell to abandon its preferred metabolic process (aerobic glycolysis). This allows the PDH complex to drive pyruvic acid towards the mitochondrial tricarboxylic cycle. Box 2.
Box 2.
Pathway of pyruvic acid in normal cells during normoxia.
In cancer cells with elevated glycolytic metabolism, PDH is inhibited through phosphorylation performed by PDK. Thus, the chemical reaction shown above (Box 2) does not take place, and pyruvate is transformed into lactic acid through the enzymatic activity of lactic dehydrogenase (LDH). This means that the pyruvate, that normally goes into mitochondria as acetyl-coenzyme A and proceeds to the Krebs cycle, does not do so in cancer cells. Instead, it remains in the cytosol and is converted into lactic acid. The lactic acid is then extruded from the cell through the activity of monocarboxylate transporters (MCTs).
Normal cells may also use this same mechanism when oxygen levels are very low. In this case it is called anaerobic glycolysis. When oxygen levels improve, the handling of pyruvic acid returns to the normal pathway (Box 2). The interesting phenomenon is that cancer cells use this mechanism even when oxygen levels have improved. This was shown to occur almost a 100 years ago in yeast by Crabtree [107] and in mammalian cancer cells by Warburg. This phenomenon has been called aerobic glycolysis or the Warburg effect. Not only does this occur in cancer cells, but the malignant cells are also highly dependent on this aerobic glycolysis. Box 3 illustrates the pathway leading to production of lactic acid in cancer cells and Figure 5 illustrates the major metabolic pathways in normal versus malignant cells.
Box 3 Carbohydrate metabolism of cancer cells. (HIF, Hypoxia-Inducible Factor) PDK is under control of the HIF complex. Up-regulation of PDK results in PDH inhibition and elevated aerobic glycolysis and lactic acid (See also Section 2.2.).

2.4. Pyruvate Dehydrogenase Phosphatases (PDP)
PDH is not permanently inactivated by phosphorylation. The enzyme is reactivated via the action of two PDPs (PDP1 and PDP2) which dephosphorylate PDH. The PDPs are regulated by magnesium and calcium [108].
Figure 5.
Glucose metabolism in normal cells (oxidative metabolism, left side) and in cancer cells (glycolytic metabolism, right side). In normal cells, the chemical end products are CO2 and H2O. In cancer cells the end product is lactic acid, that is extruded from the cell through MCTs. The activity of PDH is the cornerstone that defines which way metabolism will go. In this case, mitochondrial PDH irreversibly decarboxylates pyruvate to acetyl coenzyme A, linking glycolysis to the tricarboxylic acid cycle (left panel). This is a critical step in carbohydrate metabolism where a decision point is reached: glycolytic or oxidative pathway. If PDH is inhibited by phosphorylation by PDK (right panel), pyruvate will be taken care of by the glycolytic pathway. An important fact is that malignant cells show 2-to-20-fold higher glucose intake which means an average 60-fold increase in lactate production. When this lactate is extruded, it has an impact on extracellular acidity. However, lactate does not seem to be the main cause of extracellular acidity. Figure 6.
Figure 5.
Glucose metabolism in normal cells (oxidative metabolism, left side) and in cancer cells (glycolytic metabolism, right side). In normal cells, the chemical end products are CO2 and H2O. In cancer cells the end product is lactic acid, that is extruded from the cell through MCTs. The activity of PDH is the cornerstone that defines which way metabolism will go. In this case, mitochondrial PDH irreversibly decarboxylates pyruvate to acetyl coenzyme A, linking glycolysis to the tricarboxylic acid cycle (left panel). This is a critical step in carbohydrate metabolism where a decision point is reached: glycolytic or oxidative pathway. If PDH is inhibited by phosphorylation by PDK (right panel), pyruvate will be taken care of by the glycolytic pathway. An important fact is that malignant cells show 2-to-20-fold higher glucose intake which means an average 60-fold increase in lactate production. When this lactate is extruded, it has an impact on extracellular acidity. However, lactate does not seem to be the main cause of extracellular acidity. Figure 6.
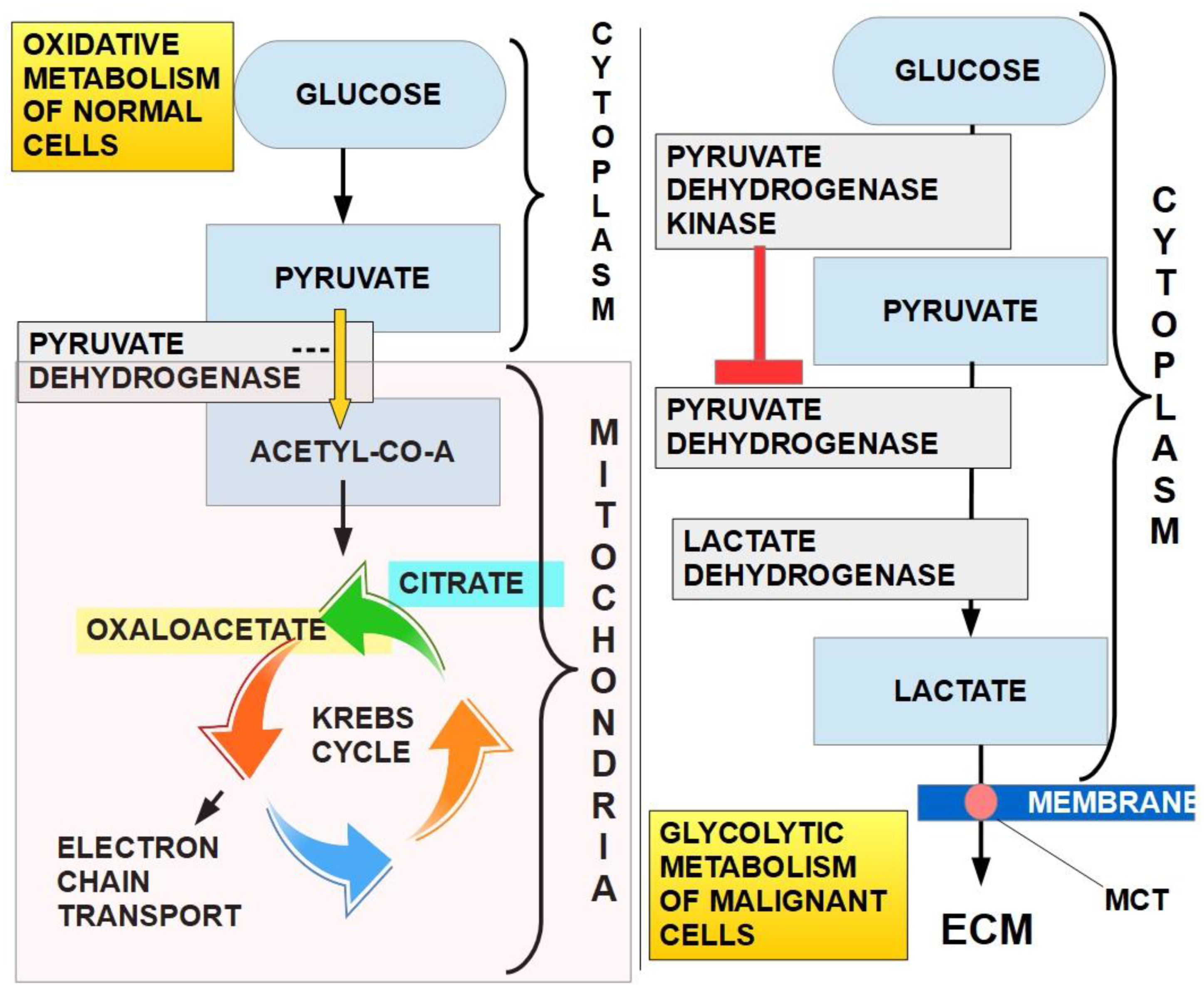
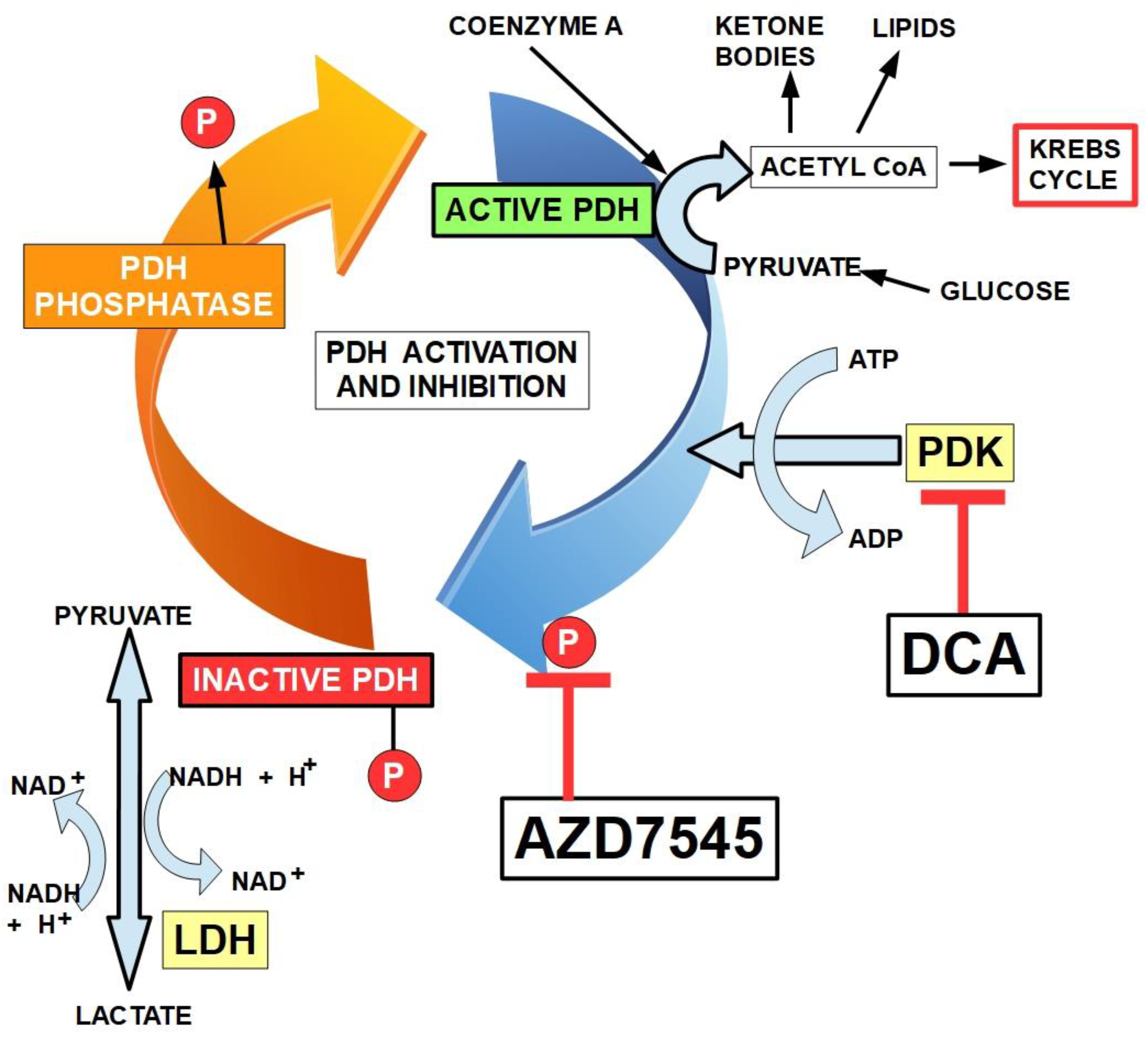
Figure 6.
Mechanism of regulation of PDH (pyruvate dehydrogenase) by PDK (pyruvate dehydrogenase kinase). PDK phosphorylates PDH and inactivates it. PDH phosphatase de-phosphorylates PDH and activates the enzyme. Two different mechanisms of PDK inhibition are shown: DCA and AZD7545 [109]. DCA binding to PDK1 induces local conformational changes that inactivate the kinase activity. Radicicol, another PDK inhibitor (not shown) binds to the ATP-binding pocket of PDK3. Inactivated PDH may have up to three phosphorylated sites. There is only one shown in the diagram.
Figure 6.
Mechanism of regulation of PDH (pyruvate dehydrogenase) by PDK (pyruvate dehydrogenase kinase). PDK phosphorylates PDH and inactivates it. PDH phosphatase de-phosphorylates PDH and activates the enzyme. Two different mechanisms of PDK inhibition are shown: DCA and AZD7545 [109]. DCA binding to PDK1 induces local conformational changes that inactivate the kinase activity. Radicicol, another PDK inhibitor (not shown) binds to the ATP-binding pocket of PDK3. Inactivated PDH may have up to three phosphorylated sites. There is only one shown in the diagram.
2.5. Mechanism of Action of DCA
The main mechanism of anti-cancer activity by DCA is the inhibition of mitochondrial pyruvate dehydrogenase kinase. This prevents inhibition of pyruvate dehydrogenase and therefore facilitates the switch from a glycolytic to an oxidative metabolism. The inhibition by DCA is noncompetitive with ATP [110]. An important consequence of this conversion to an oxidative metabolism by DCA, is an increase in caspase-mediated apoptosis, preventing the increase in cell survival that a glycolytic metabolism enhances [111]. DCA was first recognized as a PDK inhibitor by Whitehouse and Randle fifty years ago [112]. They wrote: "Evidence is given that dichloroacetate may facilitate the conversion of pyruvate dehydrogenase from an inactive (phosphorylated) form into an active (dephosphorylated) form". Their research on DCA was prompted by a previous report in 1962 by Lorini and Ciman that showed that diisopropylammonium dichloroacetate increased oxygen consumption in diabetic rats [113].
Another possible mechanism of action of DCA involved in anti-tumor activity is the modification of the intracellular pH that paradoxically has been shown to decrease in glioblastoma cells in mice following intravenous injection of DCA [114]. And here we underline the paradoxical issue, since DCA produces a decrease in lactic acid production, an increase of intracellular pH (pHi) would be expected. This does not happen. On the contrary, pHi decreases (at least temporarily). This means that there must be mechanisms other than lactic acid production, that affect pHi. Details of these are not yet fully known.
A different study by Anemone et al. [115] also treated mice with DCA though in this case they examined extracellular pH in breast tumor bearing mice in vivo. They found that there was an increase in tumor extracellular pH 3 days after treatment. However, after 15 days there was resistance to DCA treatment and extracellular pH and lactic acid levels returned to the original levels. To date the results of these last two studies have not been reconciled with one another, though it is tempting to suggest that the more acute effects in these two studies were due to decreased acid extrusion by unknown mechanisms.
In support of this suggestion is a study which showed that the decrease of pHi produced by a single dose of DCA is approximately one third of the pHi decrease produced by one injection of cariporide [116] in a glioblastoma model. Cariporide is a Na+/H+ exchanger (NHE1) inhibitor and its inhibition would decrease extrusion of protons by this antiporter.
Several other effects of DCA are known. DCA increases the expression of COX2 and the latter increases tumor resistance to DCA. Therefore, a possible integrated treatment could be COX2 inhibitor co-administered with DCA [117].
Other actions of DCA are inhibition of lipogenesis and endogenous cholesterol production. DCA also shows anti HIF-1α activity. DCA suppresses HIF-1α activity and angiogenesis through the inhibition of PDK-II [118].
Figure 7 summarizes DCA`s pharmacologic effects.
Lactylation and DCA
In 2019, Zhang et al. [128] identified histone lysine lactylation (lactyl-amide conjugation) and protein lactylation as an effect of lactate accumulation able to introduce epigenetic changes and modify post-translational characteristics of some proteins. Since then, it was found that lactylation participates in many pro-tumoral hallmarks [129,130,131,132,133,134,135,136,137,138,139]. Interestingly, DCA is one of the drugs that can decrease/inhibit lactylation. Wang et al. [140] found that DCA reduced protein lactylation in microglia. Decreased protein and histone lactylation induced by DCA was also found in reparative genes after myocardial infarction [141], neuroendocrine type lung and prostate tumors [142], colon cancer cells [143], and lung adenocarcinoma cells [144]. This rapidly accumulating evidence indicates that one of the anti-cancer mechanisms of DCA is the reduction of lactylation. Figure 8.
- 1)
- 2)
- can external lactate that enters the cell by MCT1 activity be integrated into lactylation, or is it required that it be converted into pyruvate? If external lactate (from the lactate shuttle) participates significantly in lactylation, DCA would be ineffective in preventing it. Thus far, there is only evidence of lactylation that originates from lactate metabolically produced by the cell [149]. However, for now there is no evidence that excludes lactate imported into cells from participating in lactylation.
3. Experimental Evidence on DCA Activity in Cancer
Evidence of DCA’s effects in different tumors will be discussed in sections 3.1 and 3.13.
3.1. Breast Cancer
Breast cancer is a predominantly glycolytic malignancy [150] involved in tumor progression [151] and resistance to therapy [152]. Supporting these observations are the experiments of Godoy et al. [153] who found that 90% of breast cancers had a high or very high expression of the glucose transporter 1 and 2 (GLUT1, GLUT2) which means that they have a high glycolytic flux. GLUT5 which is a fructose transporter was over-expressed in 88% of breast cancers. Furthermore, breast cancer stem cells are also glycolytic [154]. Evidence for the importance of glycolysis in breast cancer was also provided by a study that showed that inhibition of aerobic glycolysis decreased the AKT/mTOR/HIF-1alpha axis signaling and could restore sensitivity to tamoxifen in resistant cells [155]. Additionally, most breast malignancies have been shown to be glycolysis-addicted [156]. Fluorodeoxylucose PET scanning confirmed this addiction to glycolytic metabolism [157]. Therefore, since DCA promotes glucose oxidation over glycolysis, it is expected to be inhibitory to these tumors.
Breast cancer has perhaps been the type of cancer in which the effects of DCA have been studied most extensively. Experiments in different models examined effects of DCA alone and in combination with other treatments. In studies with DCA treatment alone, a variety of beneficial effects have been found. For example, Sun et al. [158] found that in an in vivo rat model with mammary adenocarcinoma cell injection, DCA administration greatly (58%) reduced lung metastasis. Additional experiments showed that growth of breast cancer cell lines was inhibited by DCA in vitro, while non-cancerous control cells were unaffected. The results with cell lines were similar to those of Gang et al. [159] whereby DCA treatment (1-5 mM) of various breast cancer cell lines decreased cell viability markedly but had little effect on non-cancerous cells. In this study DCA was also shown to reduce extracellular lactate concentrations indicating a reversal of the Warburg effect.
Some other studies used DCA alone to treat breast cancer cells. Harting et al. [160] treated canine mammary carcinoma and mammary gland cell lines with DCA and found that cell proliferation was inhibited, without inducing apoptosis. However, this study (and others) used a high dose of DCA (10 mM) that is not clinically achievable. De Preter et al. [161] also used DCA alone and showed that DCA decreased the pentose phosphate pathway, the glycolytic pathway and reduced proliferation in different breast and cervical cancer cell lines. Effects of DCA alone were also confirmed by Sun et al. [158]. DCA treatment of breast cancer cell lines reversed the glycolytic phenotype and inhibited proliferation of several breast cancer cell lines. In vivo, in that study it was also shown that in rats, DCA inhibited lung metastases. In a similar in vivo study in mice, Blackburn et al. [162] showed that in a murine mammary adenocarcinoma model, DCA treatment reduced tumor growth and numbers, and complete regression of one tumor occurred.
Another approach to the use of DCA in cancer, has been to combine it with other compounds to facilitate either their effects, or to facilitate DCA’s therapeutic effect. This approach has been used in several studies. For example, Gang et al. [159] showed that DCA can enhance apoptosis induced by PENAO (phenylarsonous acid) in breast cancer cells, but DCA alone did not cause apoptosis or inhibit growth. Similarly, Wang et al. [163] showed that DCA markedly enhanced doxorubicin-induced breast cancer cell death and had anti-proliferation effects in vitro. DCA could inhibit doxorubicin induced autophagy. DCA combined therapy could also inhibit tumor growth in vivo and prolong mouse survival times. Other compounds that DCA has been successfully combined with include: tamoxifen [164] where it sensitized MCF7 breast cancer cells to tamoxifen- induced cell death by downregulating epidermal growth factor expression (EGF); Metformin [165], where DCA increased oxidative damage and apoptosis in MCF7 and T47D breast cancer cells with a synergistic effect specific to cancer cells; arsenic trioxide [166] which when combined with DCA, synergistically inhibited breast cancer cell proliferation and induced apoptosis in a panel of cell lines representing various subtypes of breast cancer; and finally pegylated human arginase [167], which when combined with DCA showed synergistic anti-proliferative effects in triple negative breast cancer cells.
Robey et al. [168] did not find significant advantages by co-administering bicarbonate with DCA in a MDA-MB231 metastatic breast cancer model in tumor-bearing mice.
The above studies were examples whereby DCA may enhance effectiveness of some other compounds. A closely related approach is studies in which another compound enhanced DCA’s effects on cancer cells. For example, Hong et al. [169] showed that knockdown or inhibition of the S6 Kinase 1 (S6K1) enhances DCA cell death in various breast cancer cell lines. It should be noted however that here again, high concentrations of DCA, up to 20 mM were used.
Of note is the observation that some of these types of effects may vary with different breast cancer subtypes. One study [170] compared effects of different glycolytic metabolic inhibitors and DCA against two breast cancer cell lines. DCA showed higher potency against MCF-7 cell growth relative to MDA-MB-231 cells. Some other metabolic inhibitors of glycolysis also had this trend, though not all of them. These results suggest that this general approach to breast cancer treatment, may be very dependent on the cancer subtype and that any future treatments that use these approaches, may have to be tailored to the cancer subtype.
A few studies have delved into some more details of the mechanisms of action of DCA including some of its downstream effects. Lefort et al. [171] examined different breast cell lines (MDA-MB-231 and MCF-7 and a normal control cell line, MCF-10A) that were treated with allopurinol in combination with DCA. Treatments of breast cancer cells with DCA and allopurinol resulted in larger changes in metabolites found in extracellular medium compared with the changes found in intracellular pools. DCA had no metabolic effects on normal breast cells. Apoptosis was low in the three cell lines. Treatment with low doses of DCA did not lead to high level of apoptosis in MDA-MB-231 cells. Allopurinol decreased the metabolic effects of DCA. DCA resulted in reduced influx of glucose into MCF7 cells and higher glucose in the extracellular medium. This study confirmed that DCA has metabolic effects specific to cancer cells, including reduced glucose uptake. A different and more recent study [172] confirmed some of the suspected modes of action of DCA in two triple negative breast cancer cell lines. DCA decreased phosphorylation of pyruvate dehydrogenase and lowered lactate production. DCA also increased reactive oxygen species production 15-fold in hypoxic cancer cells but not in aerobic cells. Additionally, DCA made hypoxic tumor cells more sensitive to radiotherapy. A very recent study [173] also demonstrated that DCA treatment can also affect the expression of anti-apoptotic genes and oncogenic MiRNAs. These were reduced in MDA-MB231 triple negative breast cancer cells treated with 50 mM DCA. DCA also reduced cell viability in a dose-dependent manner. However, again, these concentrations of DCA are very high.
3.2. Prostate Cancer
Prostate cancer has a peculiar metabolism that changes during its progression [174]. Initially prostate cancer is not a glycolytic type of tumor, rather it adopts a lipogenic phenotype.
Two differences in prostate cancer metabolism compared to other tumors are that:
- 1)
- in prostate cancer cells Krebs cycle oxaloacetic acid is not regenerated but produced from imported aspartate [175].
- 2)
- prostate cancer is initially not a glycolytic type of tumor, rather it adopts a lipogenic phenotype until some critical time later, during its progression when it becomes glycolytic. This last peculiarity in metabolism contrasts with breast cancer where glycolysis is the predominant metabolic feature from very early stages. This also explains the reasons why fluorodeoxyglucose positron emission scans are of little help at initial stages of prostate cancer [176] and become useful in the advanced stages [177].
Despite the unusual metabolic features, DCA has shown therapeutic potential in prostate cancer. An early observation [178] tested the effect of DCA on human prostate cancer cells and DCA alone had significant cytotoxic effects and was associated with G1 cell cycle arrest and increased apoptosis. The combination of DCA with irradiation sensitized cells to radiation's cytotoxic effects. A study soon after that one [179] demonstrated that effects of DCA can vary with the prostate cancer cell type. PC3 and DU145 prostate cancer cell lines were unresponsive to DCA suggesting that their glycolytic phenotype was due to mitochondrial disfunction rather than inhibition of PDH. LNCaP prostate cancer cells were more oxidative than the other two cell types and were the only cell type to respond to DCA treatment with increased oxygen consumption. Much later, Harting et al. [180] examined six canine prostate adenocarcinoma and transitional cell carcinoma cell lines after their exposure to 10 mM DCA. DCA decreased proliferation of all but one cell line and lead to reduced lactate release. Survivin (an inhibitor of apoptosis) expression was decreased and miR-375 (a microRNA which acts as a tumor suppressor) levels were increased.
Similar to studies on breast cancer cells, several studies have also examined DCA effects on prostate cancer cells in combination with other chemotherapy agents. Zeng et al. [181] showed that DCA dramatically enhances the anti-tumor effect of cisplatin in PC3 and DU145 prostate cancer cells. A similar effect was also shown in other tumor cell types by Olszewski et al. [182]
One report [183] examined the effect of DCA on African American cell lines, since African Americans are known to respond poorly to therapy compared with Caucasian American patients. They found that DCA inhibited cell proliferation in both African American and Caucasian prostate cancer cells. DCA increased taxol cell death in Caucasian cells (only) and sensitized African American cells to doxorubicin. (African American cells were more aggressive and metastatic than Caucasian derived cells.)
A very recent report [184] used a prostate cancer cell line (PC3) and showed that DCA can inhibit the cancer stem cell like characteristics of the cells and strongly influenced the metabolic pathway of the cells causing a shift from glycolysis to oxidative phosphorylation. Their conclusion was that by inhibiting the Warburg effect, DCA may be useful in preventing future cancer relapses caused by cancer stem cells.
Overall, it is clear that like observations seen in breast cancer, there is significant potential for beneficial effects of DCA either alone or in combination with other therapies for prostate cancer.
3.3. Colon Cancer
The glycolytic pathway is activated in colon cancer cells [185] and it has been shown that most colon cancers (76%), but not all, show over-expression of glycolytic genes indicating enhancement of glycolytic pathways. Experiments in which SW480 and SW460 (colorectal cancer cells from patients) were stimulated with glucose, resulted in a proliferative response and the inhibitor of glycolysis 2- deoxyglucose, had the opposite effect [186]. Therefore, there is a large group of colon cancer patients that may benefit from inhibiting glycolysis.
In this regard, Madhok et al. [187] found that while DCA did not reduce growth of non-cancerous cells it caused significant decreases in cancer cell proliferation, which was associated with apoptosis and G2 phase cell-cycle arrest. It was also found that DCA induced autophagy in human colon cancer cells with ROS production and mTOR inhibition, and reduced lactate excretion. These effects reversed upon cessation of treatment [188]. Delaney et al. [189] reported that DCA did not increase apoptosis in colon cancer cells, despite decreasing levels of anti-apoptotic protein Mcl-1, but DCA treatment correlated with a decrease in proliferation. DCA reduced stemness markers in colorectal cancers and importantly, it was able to induce ferroptosis by sequestering iron in the lysosomes [190].
There are also several reports examining synergistic effects of DCA in colon cancer. DCA was found to act synergistically with 5-fluorouracil (5-FU) [191] and restored colorectal cancer chemosensitivity in 5-FU [192] and oxaliplatin [193] resistant cells. Olszewski et al. [182] did a study in which they examined the effects of DCA in combination with platinum compounds on several cell types including COLO205 (colon cancer epithelial) cells. DCA only very slightly increased in vitro cytotoxicity of carboplatin and satraplatin on these cells and was more efficacious in other cell types.
Notably, there is also a case report of a 57-years-old female with stage IV colon cancer treated with oral DCA with tumor stabilization for a period of nearly 4 years, without serious toxicity [194].
3.4. Melanoma
Melanoma is a tumor with a high glycolytic activity [195]. Several studies have shown that glycolytic pathways are increased in melanoma. For example, immunocytochemical analysis of lymph node metastasis of patients with melanoma showed increased expression of glycolytic genes [196].
Additionally, the BRAF V600E is a common mutation initiating melanomas and mutant melanoma with this mutation had a particularly high expression level of GLUT1 [197].
Like studies with breast cancer and prostate cancer, there have been studies attempting to use DCA to treat melanoma, or to improve other treatments for melanoma. Franco-Molina et al. [198] tested the effect of DCA on several different tumor cell lines including breast cancer, prostate cancer and melanoma and they found that murine melanoma cancer cells were the most susceptible cell line to DCA treatment in vitro. DCA induced apoptosis, inhibited invasion, and angiogenesis. In mouse experiments in vivo with the melanoma allografts, DCA reduced volume and weight of tumors.
Similarly, another study showed that blocking lactate generation with DCA or oxamate (a lactate dehydrogenase inhibitor) in metformin-treated melanoma cells, decreased cell proliferation and tumor progression in mice. Inhibition of complex I alone did not induce apoptosis, whereas inhibiting complex I and lactate generation caused metabolic catastrophe induced apoptosis [199]. Three other studies confirmed positive effects of DCA treatment on melanomas and have novel insights into mechanisms involved. Pópulo et al. [200] showed that melanomas overexpress PDKs which made them a candidate for DCA treatment. In their study DCA produced modifications of melanoma cell metabolism: decreased glucose uptake and lactic acid production, downregulating proliferation. Also, Abildgaard et al. [201] tested the effect of DCA on melanoma cells in combination with BRAFV600E inhibition. DCA alone reduced glycolytic activity and intracellular ATP levels and inhibited cellular growth in melanoma cells. Co-treatment of BRAFV600E-mutant melanoma cells with DCA and the BRAF V600E inhibitor vemurafenib induced a greater reduction in intracellular ATP levels and cellular growth than either compound alone. DCA was also effective in vitro against cells with acquired resistance to vemurafenib. These results suggested that DCA potentiates effects of BRAFV600E inhibition through cooperative inhibition of energy production. A third study found that DCA increases the anti-tumoral effects of the chemotherapy agent capecitabine in a model study using mouse melanoma allograft [202].
Finally, there has been one case report of a 32-year-old male with advanced BRAF positive metastatic melanoma treated with only DCA (500 mg three times a day). He achieved stable disease and shrinkage of the tumor mass for over 4 years. The recurrence after 4 years occurred following failure to comply with the medication [203]. This was an isolated case report, but clearly gives some weight to the suggestion that DCA might have a role in treating malignant melanoma.
3.5. Glioblastoma
Glioblastoma is a glycolytic tumor. Studies have shown that there is an elevation of glycolysis that is facilitated by hypoxia inducible factor [204]. Some evidence that supports the critical role or glycolysis was provided by Zhou et al. [205]. They isolated a highly tumorigenic group of cells, resistant to chemotherapy, that can be considered human glioblastoma stem cells. These cells had a 100-fold increase in tumor initiating ability, and were very dependent on glycolysis, had a preference for hypoxia and had decreased mitochondrial respiration. The glycolytic dependency was demonstrated by glyxolytic inhibition with a derivative of 3 bromopyruvate which caused a significant decrease in viability of the cells. Both, stem cells and non stem cells were very sensitive to glycolytic inhibition. Other evidence for the importance of glycolysis in this cell type was provided by Sanzey et al. [206] who showed that interference with the expression of glycolytic enzymes in glioblastoma decreased tumor growth in hypoxic and non-hypoxic tumors. While another study demonstrated that high expression of glycolytic genes in patients with glioblastoma correlates with poor survival [207]. Conversely, inhibition of glycolysis with DCA and fatty acid oxidation with ranolazine improved survival in mice [208]. DCA effects in glioblastoma are summarized in Table 1.
The studies included in Table 1 showed DCA positive effects with inhibition of tumor growth or synergistic effects with other chemotherapeutic compounds. However, there was not a uniform response in all the tumors. The lack of a uniform response of gliomas to DCA is probably due to the metabolic heterogeneity of gliomas, as has been shown by Duraj et al. [227], and other authors [228,229]. Glycolytic areas may coexist with oxidative areas. Oxidative areas would not be responsive to DCA. Experiments with different glioma cell lines show different metabolic profiles which may account for different efficacy of treatments. The metabolic heterogeneity of tumors in general, is probably related to DCA failures in other cancers as well.
3.6. Hematopoietic Tumors
Most hematological malignancies suffer similar metabolic alterations to those seen in solid tumors including elevated glucose uptake, elevated glycolysis, and elevated lactate production [230]. Suppressing glycolysis has shown potential benefits in hematological cancers [231,232,233,234]. Herst et al. [235] proposed the extent of glycolysis in myeloblasts in acute myeloblastic leukemia as a marker of pretreatment prognosis.
3.6.1. Myeloma
Myeloma is a glycolytic tumor [236,237], and therefore a potential target for DCA. Sanchez et al. [238] found that DCA inhibited glycolytic metabolism and increased sensitivity to the proteosome inhibitor bortezomib in myeloma. The combined treatment increased survival in mice. A test of several glycolysis inhibitors, including DCA, also induced apoptosis in myeloma cells [239]. The combination of DCA with bortezomib showed higher cytotoxicity than each drug alone [240]. However, the concentrations used in these experiments were above those achievable at the bedside: concentration of DCA 10 mM or higher, and increased ROS production required 40 mM DCA.
In a clinical trial with seven myeloma patients in partial remission, one patient showed a complete response and two had partial response. The patient with complete response had higher DCA blood concentrations due to lower GSTZ1 activity and developed severe neuropathy [241]. We may assume that toxic concentrations of DCA are necessary for significant therapeutic results.
3.6.2. Lymphoma
The combination of metformin and DCA showed synergistic apoptotic cell death in B-Cell lymphocytic leukemia with down regulation of the anti-apoptotic Mcl-1 protein [242]. DCA-dependent tumor regression and chemosensitization to cisplatin was found in Dalton lymphoma [243].
There are a few case reports regarding DCA in lymphoma. Flavin et al. [244] reported the case of a 48-year-old male patient, with Stage 4 Non-Hodgkin's Follicular Lymphoma (NHL), treated for 3 months with conventional chemotherapy resulting in a complete remission. Almost one year later tumors returned in the nasopharynx and neck lymph glands. The patient began self-administering DCA 900 mg daily with a PET scan showing complete remission four months later, remaining tumor-free after 1 year. Another report is the case of a man with documented relapse after state-of-the-art chemotherapy for non-Hodgkin’s lymphoma, a significant response to DCA was documented with a complete remission, ongoing after 4 years [245].
3.6.3. Leukemia
DCA decreased the viability of several types of leukemia cells in vitro in one study [246], and in another study decreased viability of primary B-chronic lymphocytic leukemia cells lines [247] while up-regulating the tumor suppressor p53. It also exhibited anti-leukemic activity in p53 mutated B-chronic lymphocytic leukemia cells [248]. DCA potentiated the cytotoxicity of arsenic trioxide against acute myeloid leukemia cells [249].
3.7. Ovarian, Cervical and Uterine Cancer
High grade serous ovarian cancers are glycolytic while other types of ovarian tumors may be of the non glycolytic phenotype. Advanced stages are more glycolysis-dependent [250]. Evidence for a putative role of DCA in treatment was provided by a study which showed that DCA increased apoptosis in ovarian cancer cells [251,252]. DCA derivatives were also screened in ovarian carcinoma cells, and they were shown to be more cytotoxic than cisplatin in vitro [253].
Cervical cancer is also glycolytic and over-expresses genes involved in glucose metabolism. This includes HPV-16 positive cervical cells [254]. In HeLa cells, which originated from a cervical cancer tissue sample, DCA shifted the metabolism from aerobic glycolysis to oxidation. The change in metabolism led to a drop in mitochondrial membrane potential and an increase in the level of apoptotic proteins. Combined DCA and cisplatin chemotherapy exhibited a significant synergy in inhibition of the proliferation of HeLa cells [255].
Endometrial cancer is also associated with elevated glycolysis and with expression of genes associated with glycolysis that have prognostic value in the disease [256]. DCA treatment was used in several experiments with endometrial cancer cells. Apoptosis could be induced in five low to moderately invasive endometrial cancer cell lines treated with DCA. The treatment had no effect on non-cancerous cells. Two highly invasive endometrial adenocarcinoma cell lines were resistant to DCA-induced apoptosis [257]. A later study also showed that DCA could inhibit cell proliferation and induce apoptosis in three endometrial cell lines [258].
3.8. Lung Cancer
3.8.1. Non Small Cell Lung Cancer (NSCLC)
Lung cancers are glycolytic in general, with high glycolytic flux and elevated levels of glycolytic enzymes [259]. However, some metabolic differences can be found between the different lung cancer subtypes. For example, PCK2 (phosphoenolpyruvate carboxykinase 2) is over-expressed in lung adenocarcinoma and GLUT1 expression was increased in squamous cell carcinoma [260]. The common point in both cases is that glycolysis is a potential target that was stimulated [261].
Several studies have examined the effect of DCA on various types of lung cancer cells. Oylumlu, et al. [262] found that DCA induced significant apoptosis in A549 lung cancer cells (adenocarcinoma cells) after 48 hous of incubation. It decreased the expression of PDK1 and increased the expression of PDH1. DCA was also found to interrupt the Warburg effect and decreased proliferation. Another study showed that DCA increases cytotoxicity of radiation treatment in A549 and H1299 lung cancer cells [263].
DCA has also been shown to have synergistic effects in NSCLC cells. It was shown to enhance the apoptotic effect of chemotherapeutics through down regulation of autophagy in vivo and in vitro in NSCLC cells [264]. Additionally, DCA re-sensitized human lung cancer cells that were resistant to paclitaxel. There was a synergistic effect between the two drugs [265]. Furthermore, DCA showed synergistic effects with EGFR tyrosine kinase inhibitors in vitro in NSCLC cells [266].
DCA was used against two different lines of non-small lung cancer cells, A549 and LNM35. In both lines DCA reduced cell viability in a dose-dependent manner and also reduced growth of tumor xenografts and angiogenesis. However, it had no effect on migration and invasion. It also showed additive (LNM35)/synergistic (A549) effects with gefitinib and erlotinib [267].
A DCA derivative, dichloroacetophenone biphenylsulfone, a powerful PDK1 inhibitor, induced apoptosis in NSCLC cells at sub-micromolar concentrations [268].
Regarding lung carcinoid cells, DCA re-sensitized cells resistant to carboplatin [269]. In Lewis lung carcinoma DCA decreased slightly the proliferation of the primary tumor (30% less) and decreased markedly the number and volume of metastasis [270].
Feng et al. [271] used integrated metabolomics and transcriptomics studies to elucidate some of the mechanisms of action of DCA in mice with lung cancer xenografts. The comparative metabolomic study between mice with lung cancer treated with, and without DCA, showed significant differences in the citric acid cycle (Krebs cycle). This indicated that these two groups had different metabolic profiles. Specific genetic analysis showed important expression differences in four genes: MIF (macrophage migration inhibitory factor), CLEC3B, FCN3, and EMCN. The DCA treated group showed considerably smaller tumors, elevated citric acid and reduction of MIF expression compared to controls. Importantly, Feng et al. [271] confirmed that DCA treatment was able to shift malignant cells away from aerobic glycolysis and increase oxidative phosphorylation. They also postulated an interaction between the lowering of the levels of the cytokine MIF and the modification of the malignant metabolic phenotype. This interaction has been confirmed by other authors [272,273,274,275]. Figure 9.
3.8.2. Small Cell Lung Cancer (SCLC)
SCLC is a highly malignant and metastasizing tumor that represents approximately 15% of lung cancers [276] and they are usually glycolytic tumors [277]. On the other hand, in this cancer type the cancer stem cells seem to be predominantly oxidative [278]. Lactate transport in SCLC has been targeted in some experiments [279]. However, we found no publications on DCA treatment in SCLC.
3.9. Head and Neck Squamous Cell Carcinoma (HNSCC)
Glycolytic tumor propagating cells drive squamous cell carcinoma and glycolysis is a main provider of energy in at least some types of the disease [280]. The most frequently mutated/overexpressed driver genes in HNSCC are EGFR, TP53, NOTCH and PI3K [281,282,283]. These genes favor glycolytic metabolism by different pathways. Figure 10.
All the above (Figure 10) confirms that HNSCCs are glycolytic tumors and potential candidates for treatment with DCA. However, a distinction must be made between HPV negative and HPV positive oropharyngeal tumors. The former are more glycolysis-dependent and over-express PDKs, while the HPV positive mainly utilizes mitochondrial respiration and PDK expression is much lower. DCA sensitized HPV negative cells to irradiation [289].
Other studies testing the effect of DCA on this type of tumor include that of Ruggieri et al. [290]. They found that in oral squamous tumors DCA treatment of the three oral squamous cell carcinoma (OSCC) cell lines at pharmacological concentrations, resulted in stimulation of the oxidative metabolism and caused a remarkably distinctive pro-apoptotic/cytostatic effect on HSC-2 and HSC-3. It also enhanced production of ROS. Under hypoxic conditions, paclitaxel-resistant OSCC cells were re-sensitized through paclitaxel-DCA synergy. This was not observed in paclitaxel-sensitive cells under normoxia [291]. DCA also achieved reversal of cisplatin resistance in vitro and in vivo with a concentration of 30 mM [292]. These studies led to a phase II clinical trial, adding DCA to chemoradiotherapy in HNSCC patients significantly improved end of treatment response rate but did not modify survival (NCT01386632) [293]. Metformin and DCA acted synergistically reducing the viability of oral squamous cell carcinoma cells [294].
Overall, HNSCC is clearly another glycolytic tumor type, and DCA has shown beneficial effects and beneficial potential in treatment of the disease.
3.10. Renal Tumors
A study has shown that the Warburg effect is quite pronounced in clear cell renal carcinomas (CCRC) [295], and glycolysis-related gene expression is a potential prognostic marker in CCRC [296].
Most renal cancers (75%) are clear cell renal carcinomas (CCRC). Many CCRCs show a characteristic mutation in VHL (Von Hippel Lindau gene). The VHL protein binds to the transcription factor hypoxia inducible factor alpha 1 (HFI-α1) and drives it to degradation in the proteosome [297]. Mutations of VHL impede HFI-α1 degradation and create a pseudohypoxic condition. Active HFI-α1 translocates to the nucleus where in association with HIF-β1 it acts as a transcription factor activating the expression for more than 300 different genes (hypoxia inducible genes). Importantly, glucose transporters 1 and 4 and nine of the ten glycolytic enzymes genes are under this HIF regulation. Therefore, CCRCs are glycolytic-dependent tumors that are often dependent on this mechanism [298,299].
Several studies have examined effects of DCA on CCRC. Nunez-Xavier et al. [300] found that DCA significantly reduced the viability of CCRC cells but at the same time increased AKT expression. Kinnaird et al. [301] reported that PDK activity was elevated in the tumor when compared with normal kidney tissue of the same patient. DCA reduced HIF expression, and increased mitochondrial activity with increased ROS, increased p53 activity, elevated apoptosis, and decreased proliferation. DCA also reduced viability in a different type of renal cancer cells (G401), Wilms’ tumor, promoting apoptosis and inhibiting migration and invasion. It reduced the expression of glycolysis-related genes [302]. It also reduced cell viability of renal cell adenocarcinoma cells (ACHN) in a concentration-dependent manner inducing G1 arrest and apoptosis [303]. In summary, in CCRC DCA again had beneficial effects on this glycolytic tumor type.
3.11. Pancreatic Cancer
One of the hallmarks of pancreatic ductal adenocarcinoma (PDAC) is aerobic respiration typical of the Warburg effect [304]. Pancreatic tumors also over-express glucose transporters (GLUTs) like glycolytic tumors. However, PDAC frequently also over-expresses SGLT2 a glucose transporter found in kidneys. This transporter is inhibited by canagliflozin, a drug in clinical use for the treatment of diabetes type 2. Inhibition of SGLT2 with canagliflozin has reduced pancreatic cancer growth in mice [305].
Several studies have shown potentially beneficial effects of DCA in pancreatic cancer. In one, when different metabolic modifiers were tested against human pancreatic cancer xenografts in mice, the most effective regarding tumor growth inhibition was phenformin (5 out 12 individuals) and DCA (2 out of 6) [306]. DCA and other metabolic modifiers such as 2-deoxyglucose and phenformin increased cytotoxicity of paclitaxel albumin nanoparticles (nab paclitaxel) on pancreatic cancer cells [307]. Another study demonstrated that radioresistant PDAC cells over-expressed PDK and this resistance was reversed by DCA. Importantly, radioresistant cells showed increased glycolysis and high flux towards the pentose phosphate pathway [308]. As expected, in pancreatic cancer cell lines, DCA was shown to increase mitochondrial oxidative metabolism and could decrease glycolysis and non-essential amino acid synthesis [309]. However, according to Tataranni et al. [310] DCA showed cytostatic rather than cytotoxic effects on pancreatic cancer cell lines. DCA also reduced the expression of stemness markers in some cell lines but not in others. Again, to summarize, in these cases of pancreatic cancer, DCA shows beneficial effects, often promoting the effects of other therapeutic treatments.
3.12. Hepatocarcinoma
It was found that DCA enhanced adryamicin cytotoxicity through increased ROS production in hepatocarcinomas [311]. Similar results were obtained with pirarubicin [312]. The co-application of metformin and DCA suppressed human liver cancer cell proliferation inducing apoptosis through inhibition of mTORC1 and increased ROS in vitro and in vivo [313].
3.13. Other Tumors
Table 2 summarizes the findings in other tumors not considered above. Generally, the results are similar to those found above with the other cancers and effects of DCA include, cytotoxicity, glycolysis inhibition, enhancing cytotoxicity of other chemo/immunotherapeutic agents. There is variability in the responses, in some cases DCA did not suppress tumor growth, in others it did. In some cases, DCA accentuated effect of therapeutic type of treatments and in others it did not. As noted above, this variability in effect may have to do with the specific characteristics of the tumors, some being very glycolytic and others being oxidative. Other characteristics of the tumors might also affect the efficacy of treatments such as multidrug resistance transporters. In these cases, most of the studies have not looked at these other factors and how they affect effectiveness of DCA treatment.
4. Resistance to DCA
There is little published on this issue. Anemone et al. [115], commented above, when measuring extracellular pH in vivo found that an initial increase in extracellular pH of tumors in mice when treated with DCA. After 15 days of treatment, extracellular pH returned to pre-treatment levels. Is there a resistance mechanism to DCA?
On a speculative basis we should presume that non-glycolytic tumors should not respond to DCA (primary resistance). A similar lack of response would be found in the oxidative cells that are also present in metabolic heterogeneous tumors.
The appearance of resistance after 15 days of treatment with initial response (secondary resistance) should involve a metabolic switch in which the tumors adopt oxidative metabolism and/or a glutaminergic phenotype [326]. To avoid the development of this type of metabolic resistance DCA should be given simultaneously with other metabolic drugs such as metformin [327] or 2 deoxyglucose. See below.
SLC5A8 (solute carrier gene family 5a, member 8), (also known as SMCT1 or sodium-coupled monocarboxylate transporter 1), is a Na(+)-coupled high-affinity transporter membrane transporter that is considered a tumor suppressor by most authors [328,329]. It facilitates the penetration of DCA into the cell (Figure 11). It mainly transports monocarboxylates, short chain fatty acids (importantly, butyrate), ketone bodies, pyruvate, acetate, and lactate. SLC5A8 has 610 amino acid residues, and its predicted structure is that of a protein that spans the cell membrane 13 times with a long intracytoplasmic C-terminal and a short extracellular N-terminal [330]. Figure 11.
It has been shown [331] that cancer cells can epigenetically inhibit the expression of SLC5A8 and authors have proposed the co-administration of a DNA methylation inhibitor to enhance DCA effects. This transporter has been shown to be silenced by promoter methylation of the gene in colon cancer [332], papillary thyroid cancers [333], gliomas [334], pancreatic [335] and gastric [336] cancers. Anti-inflammatories in general, decrease the transport activity of SCL5A8, thus they can reduce DCA penetration in the cell.
5. DCA and Some Interesting Associations
5.1. DCA and Metformin
As noted above, DCA in combination with metformin has shown beneficial effects in a number of cell types including in breast cancer cells [165], melanoma cells [199], glioma cells [223,224], lymphoma [242], oral squamous cells [294], and Hela cells [320]. The effect of this combination of compounds was also examined in some detail by Li et al. [337] who noted that the simultaneous use of DCA that decreases lactate production in the cells, and metformin that increases lactate production, have interesting effects: they synergistically suppressed ovarian cancer cells growth and increased apoptosis, in vivo and in vitro. Ward et al. [338] searched for an answer to this paradox. They found that in glioblastoma cells, complex I inhibition increased DCA cytotoxicity, whether produced by metformin or other complex I inhibitors like rotenone, through increased oxidative stress. This last finding seemed to explain DCA’s cytotoxic activity. It is achieved through an increase in oxidative metabolism that increases oxidative stress, which is enhanced by complex I inhibition. This pathway is shown in Figure 12, that fully illustrates DCA’s mechanism of action, although there are controversies on this issue.
5.2. DCA and COX2 Inhibitors
As noted above Li et al. [117] have demonstrated that inhibition of COX2 enhances the actions of DCA in cervical cancer cells. They also noted that DCA induces COX2 expression, so that DCA could be co-administered with a COX2 inhibitor to achieve better results. On the other hand, COX2 inhibitors may reduce DCA entrance into the cell. This issue is based on indirect evidence: inhibition of SCL5A8 by NSAIDs.
5.3. DCA and Lipoic Acid
Alpha lipoic acid (ALA) is a cofactor of PDH [346] and inhibits PDK [347]. Increased activity of PDH (as produced by inhibition of PDK) should require increased lipoic acid. Alpha lipoic acid decreases lactate and pyruvate levels in diabetes type II patients [348], but at the same time, ALA is a strong antioxidant and antiapoptotic compound [349]. This last issue may bring some doubts about the convenience of co-administering ALA with DCA treatment. On the other hand, there are publications showing anti-tumoral effects of ALA [350], and elevated ROS generation through increased oxidative metabolism [351,352,353]. Feuerecker et al. [354] found that ALA was more effective against proliferation, decreasing lactate production and inducing apoptosis than DCA.
We think, even lacking all the necessary experimental evidence, that DCA for cancer treatment could be co-administered with lipoic acid and a COX2 inhibitor like celecoxib.
Further experiments on this hypothesis are suggested.
5.4. DCA and 2D-Deoxy Glucose (2DG)
As noted above, 2-deoxyglucose, the inhibitor of glycolysis, has shown beneficial effects in experiments with colon cancer [186], pancreatic cancer cells [307]. In another case, 2DG markedly increased DCA anti-cancer effects in Lewis lung carcinoma. DCA alone showed a reduction of 60% of metastasis in this model and a decrease of 90% of metastasis volume. DCA and 2DG did not affect primary tumor growth, but the co-administration decreased its volume [355].
5.5. DCA and Bicarbonate
Robey and Martin [168, [356] found that the chronic co-administration of DCA with sodium bicarbonate to tumor bearing mice prolonged survival, and reduced tumor volume of re-ocurring lesions in lung of mice with resected tumors. DCA alone did not achieve any of these results. According to the authors, the reasons for these results are related to a decrease in extracellular acidity. As noted above, Ishiguro et al. [316] used omeprazole, a H+, K+-ATPase inhibitor that reduces acid secretion, in combination with DCA. They found a synergistic effect on tumors, and this seems to be more than a coincidence.
5.6. DCA and Sulindac
The co-administration of this anti-inflammatory with DCA increased tumor cell killing through increased ROS production [357] .
5.7. Mitaplatin
As noted above, a number of studies have shown that DCA in combination with various platinum derivatives, can have synergistic effects in a variety of cancer types including cholangiocarcinoma [325], head and neck cancer [292], lung cancer cells [269], Hela cells [291], Dalton lymphoma [243], retinoblastoma [215], colorectal cancer [192] and some other cell types. The large number of positive results with this combination therapy appears to have led to the development of Mitaplatin. Mitaplatin is a fusion compound formed by the association of two molecules of DCA and one of a platinum compound. This allows simultaneous targeting of DNA and mitochondria. The cytotoxicity of mitaplatin equals that of cisplatin in a variety of cancer cell lines with the advantage of very low toxicity or no toxicity for normal cells [358,359]. However, the production of this drug has been discontinued by all the major chemical companies, apparently due to lack of beneficial effects.
5.8. Thiamin
High doses of thiamin (vitamin B1) have effects similar to those of DCA: reduced PDH phosphorylation, reduced lactate production and increased caspase 3 activity with reduced proliferation in colon cancer cells [360]. This should raise the question: can vitamine B1 replace DCA as a nontoxic PDK inhibitor? Further experimentation with animal models would seem to be warrented.
5.9. DCA and Betulinic Acid
Similar to the approach with DCA incorporation into Mitaplatin, Saha et al. [361] synthesized a compound whereby DCA was bound to betulinic acid (they called it Bet-CA). They found that this new drug had potent anti-cancer activity against a wide range of malignant cell lines. It was also tested in vivo in a syngeneic mouse melanoma model and the resulting tumors were much smaller in the treated group than in the control group. Bet-Ca is not being further developed by the pharmaceutical industry.
5.10. DCA and Rapamycin
Chen et al.[362] found that rapamycin, an mTORC1 inhibitor, inactivates PDH thus limiting its activity on cancer cells. However, DCA co-administration with rapamycin increases cell susceptibility to mTORC1 inhibitors in vitro and in vivo.
5.11. DCA and Vemurafenib
5.12. DCA and Ivermectin
Three patients (one with metastatic breast cancer, a second with femur osteosarcoma and a third with lung adenocarcinoma) were treated with co-administration of DCA, omeprazole, tamoxifen and ivermectin. All three patients showed dramatic improvement in all symptoms when ivermectin was added to the scheme [364].
5.13. DCA and TRAIL Liposomes
DCA administered with TNF-related apoptosis-inducing ligand (TRAIL) nanoliposomes synergistically increased apoptosis in lung adenocarcinoma, colorectal, and breast cancer cells. The mechanism involved seems to be an increased expression of death receptor 5 (DR5) produced by the metabolic shift triggered by DCA [365].
5.14. DCA and 5-Fluorouracil (5-FU)
A novel derivative of 5-FU and DCA in the form of a codrug showed higher cytotoxicity in diverse malignant cells [366].
5.15. DCA and Chemotherapeutic Drugs in General
5.16. DCA and Salinomycin
Synergy between DCA and salinomycin has been found in colon cancer cell lines [370]. We believe this synergy is, in part, related to additive cytoplasmic acidification. However, the authors of this publication do not agree with this criterion. They say “we demonstrate that the synergistic effect of compounds may be related to the inhibitory effect of dichloroacetate on multidrug resistance proteins, and in contrast, it is not related to dichloroacetate-induced reduction of intracellular pH.”
They also suggested “our experiments replacing DCA by ACE show that reduced pHi is not a sufficient factor for potentiation of salinomycin effect”.
5.17. DCA and Propranolol
The beta blocker propranolol has been identified as a cancer-preventive and anti-tumoral drug [371,372,373,374]. The mechanisms involved are multiple [375]. Propranolol reduction of norepinephrine levels reduces invasion and causes a limitation of oxidative metabolism. This causes malignant cells to be dependent on the glycolytic pathway. When DCA is co-administered with propranolol, the glycolytic escape is blocked and there is a resultant synergistic cytotoxicity [376].
5.18. DCA and All-Transretinoic Acid (ATRA)
ATRA induces growth inhibition in many tumors [377,378]. In melanoma, one of multiple mechanisms involved is reduction of oxidative metabolism with a concomitant major dependence on glycolysis, in a similar fashion as propranolol. When DCA is associated with ATRA the produced metabolic stress produced increased cytotoxicity [379].
5.19. DCA and Radiotherapy
5.20. DCA and Omeprazol
Toledo et al. [382] found synergic reduction of cell viability of human and canine melanoma cells with this association.
5.21. DCA and 2-Methoxiestradiol
This association under hypoxic conditions inhibited growth and migration of lung adenocarcinoma cells (A549) [383].
5.22. DCA and Sirtinol
Sirtinol is a SIRT2 inhibitor. DCA showed synergistic anti-tumor effects with sirtinol in non-small cell lung cancer cells, in vitro and in vivo [384].
5.23. DCA and EGFR Tyrosine Kinase Inhibitors
6. DCA and T Cells
DCA improved T cell function in tumors by reducing lactate in the microenvironment, thus, allowing T cell activation. DCA improved the performance of adoptive T cell therapies when used during the in vitro expansion phase [387]. However, DCA effects on immunity are not that clear. It can induce regulatory T-cell (Tregs) differentiation in human lymphocytes. Effector T-cells depend on aerobic glycolysis while regulatory T-cells do not. Therefore, DCA by inhibiting glycolysis, can deviate lymphocyte maturation towards Tregs [388,389].
7. Side Effects, Toxicity and Doses
Initially, DCA was hailed in magazines as almost a miracle “cheap and safe” drug that kills multiple cancers. Studies with animals and cell lines provided promising results and with administration to rats (75mg/L in drinking water), tumor growth was reversed without apparent toxicity [71]. This led to DCA being proposed for many cancer therapies including glioblastoma. However, like most drugs, the specificity seems to decline with further use (and further investigation). In several cases neurological problems have been reported and this is of course closely related to dose and method of administration.
Patients who receive doses above 25 mg/Kg/day may show a mild sedative effect or drowsiness. The most serious published side effect is reversible peripheral neuropathy [390] that was found in a double-blind placebo-controlled study of 30 patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke like episodes (MELAS) that were administered 25 mg/kg/day [391]. No renal, cardiac or bone marrow toxicity have been found though no benefit in the disease was shown. In another study, Brandsma et al. [392] describe a case of severe encephalopathy due to DCA in a 46-year -old male patient with melanoma treated with 15 mg/kg/day.
The incidence of polyneuropathy in children with mitochondrial disease receiving 12.5 mg/Kg twice a day is 10% [393] and in adults it reaches to 86%.
The mechanism producing neuropathy has not been fully elucidated. It is known that DCA can produce a dose-dependent demyelination through reversible inhibition of myelin-related proteins [394]. Based on some evidence we speculate that DCA inhibition of glycolysis is the cause, and studies have shown that glycolysis is required by myelinating Schwann cells [395,396,397,398]. In theory, it may be possible to reduce DCA neurotoxicity by administering antioxidants [399] because oxidative stress is also one of DCA`s effects. However, this requires experimental confirmation. Importantly, peripheral neuropathy is a reversible phenomenon which improves after a few days of drug wash out. As noted above (section 1.2.3) glutathione transferase zeta 1 (GSTZ1) is important in the metabolism of DCA and polymormorhisms in humans of the GSTZ1 gene may enhance DCA toxicity [400].
Other side effects of DCA may be related to unintentional cell death. DCA treatment affects predominantly cancer cells, but non cancer cells may also be affected, although in a less significant way [401]. However, at high doses cellular damage is probably not restricted to cancer cells. There is also a report of DCA induction of liver carcinogenesis and lipoperoxidation in rats and mice [402,403]. Here the doses of DCA in drinking water were up to 2g/L which are much higher than others have used, and oral administration was up to 100 mg/kg doses.
There is a fatal case reported of bone marrow and liver toxicity with the artesunate-DCA association in a patient with glioblastomas [404]. In this case an unknown amount of DCA was given by an alternative practitioner so a very high dose may have been administered. There is also experimental evidence of potential liver damage by DCA in mice [405,406,407,408]. though these studies have used up to 2 g/L in drinking water or 300 mg/kg acutely and the high doses may account for some of their results. In some cases, the results also may be due to co-administration of trichloroacetic acid with DCA.
8. DCA Dosage
10 to 50 mg/kg body weight/day has been found to be a safe dose. However, single nucleotide polymorphisms (SNPs) in the gene of the enzyme GSTZ1 cause difficulties in establishing a universal dose [409] as noted above.
9. DCA Concentrations in Humans and Animals: A Key Issue
With the unpleasant risk of being repetitive, Table 3 shows the maximum concentrations achieved in humans after an IV injection of DCA [86]. These concentrations are in the level of µg or µM. The reason for this repetition is that DCA concentration in tumors is the key issue to understanding whether DCA has or may have any therapeutic effect at the bedside.
The concentrations reached in humans are enough for inhibiting PDK2 that requires 0.2 mM (see Box 1) [104]. PDK2 is a widely distributed isoform of the enzyme and the main player in cancer.
Another study showed completely different results. A 25 mg/Kg infusion achieved an average level of 129 ± 73 µg/ml the first day of the infusion and this incresead to 163 ± 84 µg after the fifth infusion [89]. A 50 mg/Kg dose achieved a maximum concentration range between 150 and 250 µg/ml [82].
Most of the in vitro studies with DCA were performed at the millimolar or milligram level instead of the micromolar or microgram level. For example, in first study that highlighted DCA’s anti-tumor activity, by Bonnet et al. [71], the lowest level of DCA concentration was 0.5 mM. The experiments of Sun et al. [158] with breast cancer cells in vitro used DCA at a concentration of 1 to 5 mM. The in vivo experiments carried out in rats had one group that received very high doses (200 mg/Kg). According to the estimates of the authors’ the plasma concentrations were in the range of 0.5-1 mM with oral administration and 1.5-3.0 mM for intraperitoneal injection. These data are well beyond the maximum concentration peak of 0.15 mM that can be achieved in humans. Furthermore, this is a peak concentration that decreases swiftly after the injection.
The experiments of others also used very high concentrations in vitro. Gang et al. [159] used concentrations between 1 to 5 mM on breast cancer cells. Harting et al. [160] treated canine mammary carcinoma cells with DCA concentrations of 10 mM. At this very high concentration they found a decreased proliferation but no apoptosis. Duan et al. [210] treated C6 glioblastoma cells with DCA concentrations of 5, 10, and 25 mM. They found that apoptosis required an IC50 of 27.0 ± 3.0 mM. At 5 mM there was no reduction of cell proliferation or apoptosis. Kolesnik et al. [213] showed that under hypoxia, DCA’s cytotoxicity in glioblastoma C6 cells was increased compared with normoxia (IC50 18.2 ± 3.9 mM vs. 51.2 ± 8.1 mM). But these concentrations are still at levels not achievable at the bedside.
Two other studies used DCA to induce apoptosis in cells. Oylumlu et al. [262] used lung adenocarcinoma A549 cells and found apoptosis at a concentration of 100 mM. Wong et al. [257] studied apoptosis in different endometrial cancer cell lines. Significant apoptosis was only detected with concentrations above 10 mM. Again, both studies used high concentrations of DCA.
A study by Korsakova et al. [219] used somewhat lower concentrations for part of their study on DCA. They found cytotoxicity in glioma cells with a combination of metformin and DCA. The problem is that there were no effects with concentrations of 5 mM DCA and 2.5 mM metformin. The cytotoxic effects were only evident with the higher concentrations of 10 mM DCA and 5 mM metformin. Cytotoxic concentrations could not be reached with non-toxic doses of both compounds. It is of note that even the 5 mM DCA and 2.5 mM metformin concentrations that have no cytotoxic effects are well beyond levels achievable at the bedside.
Sanchez et al. [238] found that DCA inhibited aerobic glycolysis in multiple myeloma cells at concentrations between 5 and 10 mM, but it required a concentration between 10-25 mM to establish increased sensitivity to bortezomib.
Prokhorova et al. [223] demonstrated metformin-DCA synergy in glioma tumors in rats. They administered doses of 1.1 g/Kg DCA, and 2.6 g/Kg metformin which are considered in the therapeutic range in rats. However, as stated above, this treatment cannot necessarily be extrapolated to humans. The problem is that DCA concentrations tolerated in rats, mice, and dogs, are very different from those in humans. Therefore, animal experiment results cannot directly be translated to humans. Humans have a lower tolerance to DCA than animals so that DCA concentrations tolerated by dogs may be toxic in humans.
Other studies that also explored synergistic effects of chemotherapeutics used concentrations closer to those safely achievable at the bedside. For example, Xie et al. [255] used concentrations of 2, 4, 8, and 16 mM DCA alone. At 2 and 4 mM very little cytotoxic activity was found. Only at 16 mM did cell viability decrease to 75% and more significant synergistic effects were only seen with the highest concentrations of DCA.
As to DCA concentration-cancer effects, it is necessary to further discuss the research by Stockwin et al. [91] who examined non-cancer cells (normal human dermal fibroblasts and human umbilical vein endothelial cells) and some tumor cell lines (transformed fibroblasts MRC5 and a T lymphoblast cell line rho(0) MOLT4 cells). They found that DCA is inactive at concentrations of 17 mM and it only induces apoptosis at concentrations above 25 mM, and at this concentration was not cancer-selective, meaning that it was also toxic for normal cells. DCA was cancer selective when mitochondria were not functional such as in the rho(0) cells. This explains its synergy with mitochondrial poisons such as metformin, phenformin, and also with cisplatin and topotecan that both damage mitochondrial DNA [410,411].
10. DCA Derivatives
DCA has been used as the basic part of other chemical structures designed to be more efficient PDK inhibitors. For example, diisopropylamine DCA showed stronger anti-cancer effects than DCA in a xenografted breast cancer model [412] Figure 13.
In another study, She et al. [413] (Figure 14) synthesized a variety of DCA derivatives combined with arsenicals. Of these compound “1f” showed powerful PDK inhibition. The new 1f DCA compound showed high efficacy at a very low concentration (IC50 = 2.0 µM ). Furthermore, the authors loaded 1f DCA into nanoparticles which when administered to tumor bearing rats, caused up to 90% tumor shrinkage. Trapella et al. [414] also developed a novel DCA ompound they called compound 10 which showed 30-fold increased in vitro anti-tumor activity. This molecule is the result of three DCA moieties bound to a nitrogenated scaffold. DCA compound 10 showed 30-fold cytotoxic increase in vitro [414].
Another DCA derivative is N-iodophenyl dichloroacetamide. This compound has shown high cytotoxicity in tumors in mice with low general toxicity [416]. While a different DCA derivative, DCA-oxaliplatin, overcomes platinum compound resistance and increased cytotoxicity. It activated an autophagic response in colon cancer cells [417].
The best known DCA derivative is diisopropylamine dicholoroacetate (DADA, Figure 13). DADA is a compound of DCA and diisopropylamine that is sold over the counter for the prevention and treatment of hepatic fibrosis and cirrhosis. It inhibits activation and proliferation of hepatic stellate cells [418]. It has been proposed as a PDK4 inhibitor for the treatment of cytokine storm and multiorgan failure. [419]. Importantly, DADA has been found to have a superior anti-tumor activity when compared with DCA in a xenotrasplanted model of breast cancer cells [420]. It requires lower concentrations than DCA to achieve growth inhibition.
11. Clinical Cases
DCA has been tested in cancer patients with some promising results. Some of these results have been mentioned above, these and others are summarized below.
Kahn et al. [421] studied effects of DCA intravenous treatment in different kinds of patients with advanced cancers. In one patient with colon cancer there appeared to be an improvement as indicated by a decline in serum enzymes. Another case with metastatic angiosarcoma had mixed responses, some positive, while in a third case DCA treatment may have stabilized the tumor. The authors concluded there was some activity in this neuroendocrine pancreatic carcinoma. A different study by this group suggested that DCA could stabilize stage 4 colon cancer [194]. And a third study by the same group showed stabilization of a patient with metastatic melanoma [203]. The group headed by Khan and working in a DCA clinic, reviewed their clinical experience with DCA in patients in a recent publication [422].
There is also one case in which a patient with Non-Hodgkin’s follicular lymphoma had returning tumors that were self-administered with 900 mg DCA daily [244]. There was a complete remission in this case, though of course a single such case is not sound and strong evidence of cause and effect. A similar result was shown by Strum et al [245].
In another study seven patients with myeloma were treated with DCA and one patient had a full remission and two had remissions [241].
As outlined above [209] the Michelakis group treated 5 patients who had glioblastoma with oral DCA (12.5-25 mg/kg twice daily) for up to 15 months. Three patients showed evidence of tumor regression.
Some other individual cases include one where Ishiguro et al. [423] used DCA together with omeprazol and tamoxifen in a 51-year-old female patient with cholangiocarcinoma in whom no previous standard treatment was effective. They found the DCA combination therapy blocked further tumor development. Lemmo et al. [424] also described a case of prolonged survival (over a year) after DCA treatment of 500-750 mg/day of leptomeningeal carcinomatosis that originated from a NSCLC.
11.1. Clinical Trials
NCT01111097 [425]: Was a phase I study to evaluate the safety of DCA in 15 patients with recurrent malignant brain tumors (glioblastoma and brain metastasis of other tumors). Patients were separated into two groups according to genotype: low metabolizers of DCA and quick metabolizers of DCA, with different dose schedules for each group. The starting dose 8 mg/Kg twice daily was increased to 12.5 mg/Kg twice daily for the quick metabolizers. No patients suffered toxicities, despite the relatively high dose. The result of the trial showed “clinical and radiological evidence of disease stabilization through the first 4 weeks of DCA administration in all 8 evaluable patients”.
NCT00566410: is a Canadian phase I trial of DCA in 23 patients with solid tumors. No results were published. They established the initial dose at 12.5 mg/Kg/day.
NCT05120284: is a multicenter, Phase IIA trial of oral DCA in surgical patients with recurrent glioblastoma who have clinically indicated debulking surgery planned. It is ongoing and patients will be randomized to receive DCA (N=20) or no DCA (N=20) for one week prior to surgery. Completion is expected in 2025.
Clinical Trials with Poor Results
Not all case studies and clinical trials resulted in positive results. There are in fact several that had poor outcomes. These include several clinical trials. One, NCT01029925 (a USA clinical trials) [426] was a phase I study to evaluate safety of DCA in metastatic breast cancer and lung cancer phase II, with seven cases. Only three patients completed 28 days of treatment and only two completed 56 days. Two patients suffered sudden death and another 2 patients had pulmonary embolism. Due to these serious adverse events, the study was terminated prematurely. The dose used in this clinical trial was 6.25 mg/Kg twice a day. The authors concluded that that “patients with previously treated advanced NSCLC did not benefit from oral DCA”.
Another clinical trial NCT01386632 [427] was a study of DCA in combination with cisplatin and radiation for squamous cell carcinoma of the head and neck. 25 patients received placebo and 25 received DCA. Concomitant in both cohorts they received radio and chemotherapy. Two-year survival and progression free were similar in both groups. Two patients in the DCA group experienced delirium, but no other serious side events were observed.
There are other three phase I clinical trials with DCA for cancer treatment, but none have posted results.
12. Negative Results with DCA
There are also several studies that have had more negative results with DCA treatment. For example, Hesche et al. [428] evaluated DCA in pediatric tumor cells and a non- malignant cell line and they found that DCA only moderately decreased cancer cell growth. DCA also decreased the cytotoxicity of cisplatin and doxorubicin. It did not activate caspases and decreased caspase activation by cisplatin.
Shahrzad et al. [429] found that DCA had a cytoprotective effect, decreasing apoptosis, in certain cases of colon carcinoma under hypoxic conditions. It also increased the growth of xenografts of SW480 tumors. While there is no clear explanation for this result it is noted that the effects of DCA may be cell type specific, and another report has found that DCA can promote survival of some hypoxic cells [430]. This seems counterintuitive as DCA inhibits glycolytic metabolism and promotes oxidative flux. It was suggested that in this case it might support development of cell clones with more tolerance to hypoxia.
A few other studies found results consistent with these. One study showed that DCA induced tumor readiosensitivity in vitro, but in vivo, DCA decreased radiation induced growth delays in tumors [431]. In another study in xenotransplanted nude mice neuroblastoma, progression and proliferation was observed [432]. Anemone et al. [115] followed the effects of DCA in vivo in xerotransplanted mice with breast cancer. They found that initially lactic acid production was decreased but after 15 days of continuous administration of the drug, lactic acid production and extracellular acidification returned to pre-treatment levels. Thus, the effect of DCA can be ephemeral.
There is also an important question about DCA effects on the immune system. Many cells of the immune system utilize glycolysis under normal conditions. Can DCA induce an immunosuppressive condition by inhibiting the glycolytic pathway? Dendritic cells are glycolytic, natural killer cells are glycolytic and oxidative, T effector cells are highly glycolytic and weakly oxidative, while T regulatory cells are oxidative [433]. Therefore, in theory, eliminating glycolysis may be detrimental to some of these cell types and may cause decreased anti-tumor immune activity. Specifically, for example, aerobic glycolysis is necessary for CD4+ T-cell proliferation and differentiation into effector T-cells. DCA can induce apoptosis in these cells or may induce their differentiation towards T regulatory cells [434]. Therefore, we must conclude that DCA can induce immunosuppression. At the same time, by reducing lactate in the extracellular milieu, DCA can counteract lactate immunosuppression improving T-cell functions [435]. Clearly the net benefit or harm of the effects of DCA on the immune system should be further studied.
13. Discussion
There is ample evidence showing that DCA is a metabolic modifier which, by inhibiting PDK, restricts glycolysis and enhances mitochondrial oxidative metabolism. This basic mechanism of action has been repeatedly confirmed in different tumors and circumstances as shown above and in additional references [436,437,438,439,440]. There is evidence that DCA also has other anti-tumoral effects summarized in Figure 7. Importantly, all these effects can be achieved with oral administration with low toxicity and few side effects.
However, the DCA concentration required to reach cytotoxic effects on tumor cells is well beyond those that can be achieved with non-toxic doses in humans. Most of the results of experiments described above were performed in animal models or cell cultures, where high concentrations can be used. Here, we must insist that these results cannot be extrapolated to humans. Considering the findings of Stockwin et al. [91], the only way to make DCA a useful antineoplasic drug is by simultaneously targeting mitochondria. This could be with topotecan, cisplatin or by inactivating the electron transfer chain with metformin, phenformin, atovaquone [441,442,443] or some other mitochondrial poison. Furthermore, Robey et al. [168] found that DCA concentrations under 20 mM increased cell viability, and 40 mM or higher concentrations were required to decrease cell viability in vitro. Olszewski et al. [182] found that co-administration of DCA (10 mM) with certain platinum compounds could increase cytotoxicity. However, when the cells were pre-incubated with DCA alone and subsequently treated with platinum-based drugs, the effect was exactly the opposite. Therefore, when administered with other drugs, the effects of DCA can vary greatly according to the administration schedule.
The question now is: can the interruption of the glycolytic metabolism stop tumor growth and induce apoptosis?
To answer this question, we must consider that tumors are formed not only by glycolytic cells but also by oxidative cells. The latter should not be affected by the elimination of the glycolytic pathway. If the tumor is predominantly composed of glycolytic cells, the growth of these cells will be hampered while the oxidative cells will continue to thrive. However, it must be remembered that even if DCA can stop proliferation of glycolytic cells, it is unable to induce their apoptosis, unless it is associated with other cytotoxic drugs.
Abundant examples showing that DCA can indeed stop tumor proliferation in different cancer cells, in vitro, and in vivo have been discussed above. However, these effects are temporary, do not occur in all cancer types, and at very high concentrations not achievable in the clinical setting, probably due to some of the following causes:
- a)
- cells that survive can switch to another type of metabolism such as mitochondrial oxidative metabolism;
- b)
- there may be a persistence of oxidative cells that are not affected by DCA;
- c)
- DCA is cytostatic rather than cytotoxic, and both require very high concentrations;
- d)
- the tumor is predominantly oxidative.
The tumor heterogeneity implied above, is an important point. As noted, it may account for the wide diversity of results discussed above, where sometimes DCA seems remarkably effective and other times it seems totally ineffective or even harmful.
The in vivo achievable concentration at the tumor site, is of course relevant to the clinical effectiveness of DCA. In this regard, Stockwin et al. [91] have shown that DCA requires very high concentrations in the tumor (above 25 mM) to induce apoptosis, concentrations that cannot be reached without intolerable toxicity for normal cells. Therefore, we must conclude that its activity at tolerable doses may sometimes only be cytostatic and may not achieve cytotoxicity. Stockwin et al. [91] also showed that DCA can induce apoptosis in those cells that are unable to switch into mitochondrial oxidative metabolism. Therefore, clearly the nature and type of the tumor affects the efficacy of DCA. Once again, the tumor type and its heterogeneity will affect DCA efficacy. We suggest that in the rare case studies when DCA administration alone appears to have strong clinically positive effects, this may usually be because it was used in tumors that were highly glycolytic, and possibly with small or negligible components of oxidative tumor cells. There may also be a serendipidous component to some of the results in individual case studies.
A first conclusion is that DCA is not a stand-alone drug because its effects are cytostatic and seem to be temporary, and the tumor cells are not eliminated. Confirming the research by Stockwin et al. [91] is the fact that DCA becomes more efficient when it is co-administered with an inhibitor of the oxidative metabolism such as phenformin or metformin. In this association, the main escape mechanism, that is, switching to mitochondrial oxidative metabolism, is blocked. Another confirmation of the cytostatic rather than cytotoxic effects of DCA is the remarkable synergy between DCA and cytotoxics such as cisplatin. Importantly, Kumar et al. [211] showed the synergistic antitumoral effects of DCA with antiangiogenic drugs such as bevacizumab. We did not find any follow up of this significant finding.
DCA also has other anti-tumor effects such as decreasing cell stemness and inhibiting MIF. However, it is not clear if these experimental findings can be utilized without intolerable toxicity at the bedside. Further, we do not know how important these effects are regarding tumor progression. DCA also can decrease mitochondrial uncoupling protein 2 (UCP2) expression, thus increasing ROS production in oxidative cells [444]. Similarly, it is not clear if these effects are exploitable clinically.
We remind the reader of differences between normal and tumor cells. Under normoxia (normal oxygen conditions), non-cancer cells catabolize glucose to pyruvate. Then pyruvate is taken up by the mitochondria where it is further catabolized. As part of this, electrons from the Krebs cycle are taken up by oxygen in the respiratory electron transfer chain. However, tumors are highly hypoxic, therefore, there is very little oxygen for electron uptake, so that this process does not occur or occurs at a reduced level compared with non-tumor cells. The alternative used by tumor cells is anaerobic glycolysis, where pyruvate is not taken up by the mitochondria and instead is converted to lactic acid. The great conductor of this process is Hypoxia Inducible Factor 1 alpha (HIF-1α). HIF-1α is a transcription factor responsible for the overexpression of more than 300 genes (HIF responsive genes). PDK overexpression is regulated by HIF-1α [445] and is an adaptation to hypoxic conditions. Evidence for this regulation has been demonstrated. Under hypoxic conditions, when HIF-1α is down-regulated in mouse embryos, there is no activation of PDK1. The cell also undergoes apoptosis due to an increase in ROS. By re-expressing HIF-1α, apoptosis is avoided, and ROS generation is decreased [99].
To summarize these effects Papandreou et al. [99] state that “The HIF-1 transcription factor drives hypoxic gene expression changes that are thought to be adaptive for cells exposed to a reduced-oxygen environment. For example, HIF-1 induces the expression of glycolytic genes”.
The increased activity of glycolytic genes and enzymes in tumors represents an important resistance factor against apoptosis [446]. DCA anti-tumor effects are significantly increased when used in association with PX-478, a HIF-1α inhibitor [447].
PDH activity represents a key metabolic hub where a “decision” is made as to whether glycolytic metabolism or oxidative metabolism is the next step. PDK is the modulator of this “decision”. In malignant glycolytic cells, the activity of PDH is restrained by over-expression of PDK. The blocking of pyruvate oxidative mechanism by PDK implies a “functional mitochondrial shutdown”. This is advantageous for malignant cells, because mitochondria produce reactive oxygen species (ROS) which may create oxidative stress. Mitochondrial shutdown avoids this danger. DCA is an inhibitor of PDK, and probably this is its main mechanism of action in cancer.
There is abundant evidence showing that PDK inhibition allows malignant cells to return to oxidative metabolism. In many cases this return is accompanied by inhibition of proliferation and even apoptosis. Malignant glycolytic cells are highly dependent on glycolytic metabolism, to the point that its reversal to oxidative metabolism causes apoptosis in many, but not all cases.
As a proof of concept, Li et al. [448] used epigallocatechin 3 gallate (EGCG) (found in green tea leaves) on glycolytic hepatocarcinoma cells reversing the glycolytic metabolism and inducing apoptosis. EGCG acted as an inhibitor of PDK. In the same research, they found that PDK knockdown with siRNA also induced apoptosis.
Furthermore, DCA concentrations that can be reached in vivo (0.5 mM), seem sufficient to decrease glycolysis and lactate production [449]. But these concentrations are insufficient for inducing apoptosis. Figure 15.
However, as we said above, DCA alone seems not to fulfill all the requirements needed to be considered a fully chemotherapeutic drug, but when it is used in combination with other specific drugs, the results seem to change.
There is strong evidence showing that the association of metformin and DCA has significant cytotoxic effects. However, this requires concentrations that are higher than those achievable at the bedside. To this approach we must add a third compound: a COX2 inhibitor like celecoxib to decrease COX2 expression induced by DCA. We note that, the triple association of DCA, metformin and celecoxib, which has never been experimentally tested in patients, deserves well planned phase II clinical trials.
While, even if DCA is far from achieving the cytotoxicity expected from a chemotherapeutic drug, it seems to be an anti-metastatic compound that has properties that can be clinically useful. For example, DCA enhances the cytotoxic activity of the commonly used chemotherapeutic drugs cisplatin, paclitaxel, adryamicin, 5FU, capecitabine, bevacizumab, sorafenib, and doxorubicin and in many cases reverses acquired resistance [450,451]. Therefore, DCA should also be considered as a complement to standard chemotherapeutic protocols, especially if chemoresistance has developed.
The intravenous administration of DCA in a once a week or once every two weeks schedule as performed in many DCA clinics does not make therapeutic sense. According to its mechanism of action and pharmacodynamic features, (short half life), DCA needs a metronomic scheme (which can use the oral route).
The membrane transporter SLC5A8 seems to play an important role in transporting DCA to the interior of the cell. This transporter shows usually very low expression in malignant cells. But those tumors that have a normal or increased expression of SLC5A8 show increased response to DCA treatment [452], possibly due to increased uptake of DCA through this transporter.
Even if it is slightly farfetched, we believe that the glycolytic phenotype in cancer may be due to two different conditions, according to metabolic response to DCA:
- 1)
- DCA responders: the glycolytic phenotype is due to up-regulation of PDK.
- 2)
- DCA non responders: the glycolytic phenotype is not dependent on PDK up-regulation.
Again, we note that DCA has sometimes fleeting positive effects. In prostate cancer cell lines Kailavasan et al. [453] showed a major metabolic shift produced by DCA in highly metastatic glycolytic cells but no changes were found in cell lines with low metastatic potential. The responding cells lacked LDH-B whereas non-responders expressed both LDH-A and LDH-B. However, the concentrations of DCA used (50 mM), were well above those that can be reached with non-toxic doses in patients. Another example of the elusiveness of the positive effects of DCA is the study of Zwicker et al. [431]. They found that while DCA induced radiosensitization in vitro it showed no such effects in vivo and in fact showed a slight but significant enhancement of tumor growth in their model in vivo.
The elusiveness of positive DCA effects in these examples above should also be remembered when proposing DCA for many other diseases such as diabetes [454,455], pulmonary hypertension [456], ulcerative colitis [457], hyperlipemia [458], prevention of platelet aggregation and thrombosis [459], atherosclerosis [460], endometriosis [461], and amyotrophic lateral sclerosis [462].
Finally, it is important to mention that patients may show variable responses to DCA due to the type of cancer and presence of other diseases. For example, patients with hepatic cirrhosis have a significant decrease of DCA clearance [463] but does not alter the metabolic response to DCA administration [464]. GSTZ1 is down-regulated in hepatocellular carcinoma while it is up-regulated in breast cancer [465]. Age also decreases DCA clearance [79].
14. Future Perspectives
DCA will never become a stand-alone chemotherapeutic compound. The fundamental reason for this statement is that the drug can only reach micromolar blood concentrations without toxicity and requires millimolar levels to be cytotoxic. It therefore has no chance of becoming a therapeutic alternative. Even when associated with metformin, it still requires clinically unreachable concentrations. However, its synergy with conventional chemotherapeutic drugs opens a window of opportunity which may be exploitable for the benefit of a significant number of patients. Another alternative would be DCA association with other PDK inhibitors which are under development, such as cryptotanshinone [466,467], oxindole-base-derivatives [468], BX795 [469], amino-triazine derivatives [470], stearic acid [471], shikonon derivatives [472]. Finally, the toxicity limitation of DCA will probably be improved by novel delivery systems such as the antibody-drug conjugates (ADCs). These can release a high dose of a potent cytotoxic agent directly totumors without affecting normal cells [473]. For example, natural products such as albiziabioside A can be conjugated with DCA enhancing its effects [474].
ADCs are probably the best way to transform DCA into a real cytotoxic drug because novel and more powerful PDK inhibitors would not be able to circumvent the toxicity involved in PDK inhibition. Some important normal cells depend on a functional glycolyric metabolism (e.g. Schwann’s cells, T4 lymphocytes).
15. General Conclusions
DCA, when used in toxic doses, decreases tumor growth through inhibition of PDK and glycolysis and via increased oxidative metabolism with elevated ROS production.
DCA decreases the number and size of metastases in vitro in the laboratory setting.
DCA activity is particularly evident in highly hypoxic tumors.
DCA metabolic effects are short lived in vivo.
DCA is not a stand-alone drug and must be used in association with conventional chemotherapeutic compounds or others such as metformin, omeprazol, and bicarbonate. These treatment profiles have not yet been defined.
DCA has synergic effects with most standard chemotherapeutic drugs.
DCA does not induce apoptosis, thus the need for its association with other chemotherapeutics.
DCA should not be used in association with allopurinol, NSAIDs, or flavonoids because they reduce cellular DCA uptake.
To achieve important metabolic changes in cancer cells, DCA must be used for extended periods, at high doses, and on a daily basis. However, there is no evidence of lasting effects.
DCA is not devoid of toxicity, particularly when it is used in adults for a long time and at high doses.
DCA is useful to reverse resistance to chemotherapeutic drugs in highly hypoxic tumors.
Experiments performed in rats or dogs cannot be translated to humans due to important pharmacodynamic differences.
DCA is well absorbed when given orally, therefore intravenous administration is not necessary.
Cancer cells in which the glutaminergic pathway is up-regulated, will not be susceptible to DCA treatment.
DCA will probably fail as a stand-alone treatment of tumors with a high proportion of oxidative malignant cells.
DCA, either alone or in association with other compounds has never been tested in phase II clinical trials and we strongly discourage the use of DCA as a compassionate treatment until well planned and supervised studies are conducted. On the other hand, we believe that DCA deserves more efforts to elucidate its precise role in the treatment of cancer.
Presently, although there is abundant basic experimental evidence using DCA at high concentrations, this is not sufficient on clinical grounds to support its use due to toxicity at high doses and short-lived effects.
16. Main Conclusion
DCA alone, reduces glycolytic metabolism in tumors and has a cytostatic effect. However, this is not sufficient to induce apoptosis at achievable non-toxic concentrations in humans.
17. Declarations
No human nor animal experiments were performed.
English spelling and syntax revised by Ms. Julia H. Weiss.
All the figures are original.
Author Contributions
Both authors contributed equally to the manuscript.
Funding
No financial support has been received for this manuscript.
Conflicts of Interest
The authors declare no conflicts of interests.
References
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. The Journal of General Physiology 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell metabolism 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: fatty acid oxidation in the limelight. Nature reviews Cancer 2013, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C.Y.; Krauss, S.; Ranger, A.M.; Datta, S.R.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; Gygi, S.P. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003, 424, 952. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. Journal of the National Cancer Institute 2006, 98, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Dey, R.; Moraes, C.T. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-xL in osteosarcoma cells. Journal of Biological Chemistry 2000, 275, 7087–7094. [Google Scholar] [CrossRef]
- Park, S.Y.; Chang, I.; Kim, J.Y.; Kang, S.W.; Park, S.H.; Singh, K.; Lee, M.S. Resistance of mitochondrial DNA-depleted cells against cell death: role of mitochondrial superoxide dismutase. Journal of Biological Chemistry. 2004, 279, 7512–7520. [Google Scholar] [CrossRef]
- Harris, M.H.; Vander Heiden, M.G.; Kron, S.J.; Thompson, C.B. Role of oxidative phosphorylation in Bax toxicity. Molecular and cellular biology 2000, 20, 3590–3596. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; Pestell, R.G.; Sotgia, F.; Philp, N.J.; Lisanti, M.P. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.I. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation. Oncology letters 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. The Journal of physiology 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg's contributions to current concepts of cancer metabolism. Nature Reviews Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; T Supuran, C.; O Alfarouk, K. The Warburg effect and the Hallmarks of Cancer. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents) 2017, 17, 164–170. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.B.; Elhassan, G.O.; Ibrahim, M.E.; Orozco, J.D.; Cardone, R.A.; Reshkin, S.J.; Harguindey, S. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777. [Google Scholar] [CrossRef]
- Harguindey, S.; Reshkin, S.J. The new pH-centric anticancer paradigm in Oncology and Medicine"; SCB, 2017. In Seminars in cancer biology 2017 Apr (Vol. 43, pp. 1–4). [CrossRef]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: mirror and motor of tumor malignancy. InSeminars in radiation oncology 2004 Jul 1 (Vol. 14, No. 3, 267-274). WB Saunders. [CrossRef]
- Newell, K.; Franchi, A.; Pouyssegur, J.; Tannock, I. Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proceedings of the National Academy of Sciences 1993, 90, 1127–1131. [Google Scholar] [CrossRef]
- Yamagata, M.; Hasuda, K.; Stamato, T.; Tannock, I.F. The contribution of lactic acid to acidification of tumours: studies of variant cells lacking lactate dehydrogenase. British journal of cancer 1998, 77, 1726. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Meredith, D.; Fox, J.E.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) Is the Target for Organomercurial Inhibition of Monocarboxylate Transporter Isoforms 1 and 4 THE ANCILLARY PROTEIN FOR THE INSENSITIVE MCT2 IS EMBIGIN (gp70). Journal of Biological Chemistry 2005, 280, 27213–27221. [Google Scholar] [CrossRef] [PubMed]
- Polański, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; Smith, P.D. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clinical cancer research 2013, 20, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Bueno, V.; Binet, I.; Steger, U.; Bundick, R.; Ferguson, D.; Murray, C.; Donald, D.; Wood, K. The specific monocarboxylate transporter (MCT1) inhibitor, AR-C117977, a novel immunosuppressant, prolongs allograft survival in the mouse. Transplantation 2007, 84, 1204–1207. [Google Scholar] [CrossRef] [PubMed]
- Ovens, M.J.; Davies, A.J.; Wilson, M.C.; Murray, C.M.; Halestrap, A.P. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10. Biochemical Journal 2010, 425, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.M.; Hutchinson, R.; Bantick, J.R.; Belfield, G.P.; Benjamin, A.D.; Brazma, D.; Bundick, R.V.; Cook, I.D.; Craggs, R.I.; Edwards, S.; Evans, L.R. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nature chemical biology 2005, 1, 371. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Otsuka, Y.; Itagaki, S.; Hirano, T.; Iseki, K. Inhibitory effects of statins on human monocarboxylate transporter 4. International journal of pharmaceutics 2006, 317, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In vivo anticancer activity of AZD3965: a systematic review. Molecules 2021, 27, 181. [Google Scholar] [CrossRef] [PubMed]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. International journal of oncology 2011, 39, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; Bosserhoff, A.K. Lactate promotes glioma migration by TGF-β2–dependent regulation of matrix metalloproteinase-2. Neuro-oncology 2009, 11, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Hussien, R.; Oommen, S.; Gohil, K.; Brooks, G.A. Lactate sensitive transcription factor network in L6 cells: activation of MCT1 and mitochondrial biogenesis. The FASEB Journal 2007, 21, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor–macrophage interplay to promote breast cancer metastasis. Proceedings of the National Academy of Sciences 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? . The Journal of pathology 2013, 230, 350–355. [Google Scholar] [CrossRef]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunology, Immunotherapy. 2016, 65, 821–33. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Puig-Kröger, A.; Muniz-Pello, O.; Selgas, R.; Criado, G.; Bajo, M.A.; Sánchez-Tomero, J.A.; Alvarez, V.; del Peso, G.; Sánchez-Mateos, P.; Holmes, C.; Faict, D. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: effect of lactate and glucose-degradation products. Journal of Leucocyte Biology 2003, 73, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; Renner, K.; Timischl, B.; Mackensen, A.; Kunz-Schughart, L.; Andreesen, R.; Krause, S.W.; Kreutz, M:. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Constant, J.S.; Feng, J.J.; Zabel, D.D.; Yuan, H.; Suh, D.Y.; Scheuenstuhl, H.; Hunt, T.K.; Hussain, M.Z. Lactate elicits vascular endothelial growth factor from macrophages: a possible alternative to hypoxia. Wound Repair Regen 2000, 8, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Milovanova, T.N.; Bhopale, V.M.; Sorokina, E.M.; Moore, J.S.; Hunt, T.K.; Hauer-Jensen, M.; Velazquez, O.C.; Thom, S.R. Lactate stimulates vasculogenic stem cells via the thioredoxin system and engages an autocrine activation loop involving hypoxia-inducible factor 1. Mol Cell Biol 2008, 28, 6248–6261. [Google Scholar] [CrossRef]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; et al. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid Redox Signal, 1115. [Google Scholar] [CrossRef]
- Beckert, S.; Farrahi, F.; Aslam, R.S.; et al. Lactate stimulates endothelial cell migration. Wound Repair Regen. [CrossRef]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer research 2011, 71, 2550–2560. [Google Scholar] [CrossRef]
- Walenta, S.; Salameh, A.; Lyng, H.; Evensen, J.F.; Mitze, M.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in head and neck tumors with incidence of metastasis. The American journal of pathology 1997, 150, 409. [Google Scholar] [PubMed]
- Schwickert, G.; Walenta, S.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in human cervical cancer with incidence of metastasis. Cancer research 1995, 55, 4757–4759. [Google Scholar] [PubMed]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; Pestell, R.G. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Shegay, P.V.; Zabolotneva, A.A.; Shatova, O.P.; Shestopalov, A.V.; Kaprin, A.D. Evolutionary view on lactate-dependent mechanisms of maintaining cancer cell stemness and reprimitivization. Cancers 2022, 14, 4552. [Google Scholar] [CrossRef] [PubMed]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PloS one 2012, 7, e46571. [Google Scholar] [CrossRef] [PubMed]
- Samuvel, D.J.; Sundararaj, K.P.; Nareika, A.; Lopes-Virella, M.F.; Huang, Y. Lactate boosts TLR4 signaling and NF-κB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. The Journal of Immunology 2009, 182, 2476–2484. [Google Scholar] [CrossRef] [PubMed]
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-Secreted Lactic Acid Promotes IL-23/IL-17 Proinflammatory Pathway. J. Immunol 2008, 180, 7175–7183. [Google Scholar] [CrossRef] [PubMed]
- Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; Mueller-Klieser, W. Glycolytic metabolism and tumour response to fractionated irradiation. Radiotherapy and oncology 2010, 94, 102–109. [Google Scholar] [CrossRef]
- Stern, R. Hyaluronidases in cancer biology. InHyaluronan in Cancer Biology 2009 (pp. 207-220). Academic Press. Elsevier. Robert Stein Editor. First Edition Amsterdam, The Netherlands.
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Frontiers in oncology 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is lactate an oncometabolite? Evidence supporting a role for lactate in the regulation of transcriptional activity of cancer-related genes in MCF7 breast cancer cells. Frontiers in oncology 2020, 9, 1536. [Google Scholar] [CrossRef] [PubMed]
- Izzo, L.T.; Wellen, K.E. Histone lactylation links metabolism and gene regulation. Nature 2019, 574, 492–493. [Google Scholar] [CrossRef] [PubMed]
- Xin, Q.; Wang, H.; Li, Q.; Liu, S.; Qu, K.; Liu, C.; Zhang, J. Lactylation: a Passing Fad or the Future of Posttranslational Modification. Inflammation 2022, 45, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, L.; Gu, Y.; Cang, W.; Sun, P.; Xiang, Y. Lactate-Lactylation Hands between Metabolic Reprogramming and Immunosuppression. Int J Mol Sci 2022, 23, 11943. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The tortuous path of lactate shuttle discovery: From cinders and boards to the lab and ICU. Journal of sport and health science 2020, 9, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Hopp, A.K.; Grüter, P.; Hottiger, M.O. Regulation of glucose metabolism by NAD+ and ADP-ribosylation. Cells 2019, 8, 890. [Google Scholar] [CrossRef] [PubMed]
- Kocianova, E.; Piatrikova, V.; Golias, T. Revisiting the Warburg Effect with Focus on Lactate. Cancers 2022, 14, 6028. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Wan, J.; Cai, K.; Song, H.; Wang, Y.; Sun, W.; Hu, J. Understanding the contribution of lactate metabolism in cancer progress: a perspective from isomers. Cancers 2022, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Harman, E.M.; Curry, S.H.; Baumgartner, T.G.; Misbin, R.I. Treatment of lactic acidosis with dichloroacetate. New England Journal of Medicine 1983, 309, 390–396. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Lorenz, A.C.; Thomas, R.G.; Harman, E.M. Dichloroacetate in the treatment of lactic acidosis. Annals of internal medicine 1988, 108, 58–63. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Kerr, D.S.; Barnes, C.; Bunch, S.T.; Carney, P.R.; Fennell, E.M.; Felitsyn, N.M.; Gilmore, R.L.; Greer, M.; Henderson, G.N.; Hutson, A.D. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 2006, 117, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Irsigler, K.; Brändle, J.; Kaspar, L.; Kritz, H.; Lageder, H.; Regal, H. Treament of biguanide-induced lactic acidosis with dichloroacetate. 3 case histories. Arzneimittel-Forschung 1979, 29, 555–559. [Google Scholar] [PubMed]
- Stacpoole, P.W.; Wright, E.C.; Baumgartner, T.G.; Bersin, R.M.; Buchalter, S.; Curry, S.H.; Duncan, C.A.; Harman, E.M.; Henderson, G.N.; Jenkinson, S.; Lachin, J.M. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. New England Journal of Medicine 1992, 327, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Agbenyega, T.; Angus, B.J.; Bedu-Addo, G.; Ofori-Amanfo, G.; Henderson, G.; Szwandt, I.S.; O'BRIEN, R. , Stacpoole, P.W. Pharmacokinetics and pharmacodynamics of dichloroacetate in children with lactic acidosis due to severe malaria. QJM: An International Journal of Medicine 1995, 88, 341–349. [Google Scholar] [PubMed]
- Stacpoole, P.W.; Barnes, C.L.; Hurbanis, M.D.; Cannon, S.L.; Kerr, D.S. Treatment of congenital lactic acidosis with dichloroacetate. Archives of disease in childhood 1997, 77, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; Harry, G. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Plas, D.R.; Thompson, C.B. Cell metabolism in the regulation of programmed cell death. Trends in Endocrinology & Metabolism 2002, 13, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Agbenyega, T.; Planche, T.; Bedu-Addo, G.; Ansong, D.; Owusu-Ofori, A.; Bhattaram, V.A.; Nagaraja, N.V.; Shroads, A.L.; Henderson, G.N.; Hutson, A.D.; Derendorf, H. Population kinetics, efficacy, and safety of dichloroacetate for lactic acidosis due to severe malaria in children. The Journal of Clinical Pharmacology 2003, 43, 386–396. [Google Scholar] [CrossRef] [PubMed]
- 2005. Dichloroacetic Acid in Drinking-water Background document for development of WHO Guidelines for Drinking-water Quality. Downloaded December 26, 2023 from https://cdn.who.int/media/docs/default-source/wash-documents/wash-chemicals/dichloroaceticacid0505.pdf?sfvrsn=2246389b_4.
- Stacpoole, P.W. Clinical Physiology and Pharmacology of GSTZ1/MAAI. Biochemical Pharmacology 2023, 115818. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.H.; Lorenz, A.; Chu, P.I.; Limacher, M.; Stacpoole, P.W. Disposition and pharmacodynamics of dichloroacetate (DCA) and oxalate following oral DCA doses. Biopharm Drug Dispos 1991, 12, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Pharmacokinetics of Sodium Dichloroacetate. PhD dissertation. Gainesville, FL:University of Florida, 1987. Downloaded from https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Chu+P-I.+Pharmacokinetics+of+Sodium+Dichloroacetate.+PhD+dissertation.+Gainesville%2C+FL%3AUniversity+of+Florida%2C+1987.&btnG= Accessed February 2023.
- Evans, O.B. Dichloroacetate tissue concentration and its relationship to hypolactatemia and pyruvate dehydrogenase activity by dichloroacetate. Biochemical Pharmacology 1982, 31, 3124–3126. [Google Scholar] [CrossRef] [PubMed]
- Shroads, A.L.; Guo, X.; Dixit, V.; Liu, H.P.; James, M.O.; Stacpoole, P.W. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. Journal of Pharmacology and Experimental Therapeutics 2008, 324, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.C.; Smeltz, M.G.; Hu, Z.; Rowland-Faux, L.; Zhong, G.; Lorenzo, R.J.; Cisneros, K.V.; Stacpoole, P.W.; James, M.O. Regulation of dichloroacetate biotransformation in rat liver and extrahepatic tissues by GSTZ1 expression and chloride concentration. Biochem Pharmacol 2018, 152, 236–243. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- James, M.O.; Yan, Z.; Cornett, R.; Jayanti, V.M.; Henderson, G.N.; Davydova, N.; Katovich, M.J.; Pollock, B.; Stacpoole, P.W. Pharmacokinetics and metabolism of [14C] dichloroacetate in male sprague-dawley rats: Identification of glycine conjugates, including hippurate, as urinary metabolites of dichloroacetate. Drug Metabolism and Disposition 1998, 26, 1134–1143. [Google Scholar] [PubMed]
- Wells, P.G.; Moore, G.W.; Rabin, D.; Wilkinson, G.R.; Oates, J.A.; Stacpoole, P.W. Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia 1980, 19, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. The pharmacology of dichloroacetate. Metabolism-Clinical and Experimental 1989, 38, 1124–1144. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Moore, G.W.; Kornhauser, D.M. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N Engl J Med 1978, 298, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Leon, A.; Schultz, I.R.; Xu, G.; Bull, R.J. Pharmacokinetics and metabolism of dichloroacetate in the F344 rat after prior administration in drinking water. Toxicology and applied pharmacology 1997, 146, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Lukas, G.; Vyas, K.H.; Brindle, S.D.; Le Sher, A.R.; Wagner, W.E. Biological disposition of sodium dichloroacetate in animals and humans after intravenous administration. Journal of pharmaceutical sciences 1980, 69, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Maisenbacher, H.W.; Shroads, A.L.; Zhong, G.; Daigle, A.D.; Abdelmalak, M.M.; Samper, I.S.; Mincey, B.D.; James, M.O.; Stacpoole, P.W. Pharmacokinetics of oral dichloroacetate in dogs. Journal of biochemical and molecular toxicology 2013, 27, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.J.; Lane, J.R.; Turkel, C.C.; Capparelli, E.V.; Dziewanowska, Z.; Fox, A.W. Dichloroacetate: population pharmacokinetics with a pharmacodynamic sequential link model. The Journal of Clinical Pharmacology 2001, 41, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Henderson, G.N.; Yan, Z.; James, M.O. Clinical pharmacology and toxicology of dichloroacetate. Environmental health perspectives 1998, 106, 989. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. British journal of cancer 2008, 99, 989. [Google Scholar] [CrossRef]
- Stockwin, L.H.; Yu, S.X.; Borgel, S.; Hancock, C.; Wolfe, T.L.; Phillips, L.R.; Hollingshead, M.G.; Newton, D.L. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. International journal of cancer 2010, 127, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Constantin-Teodosiu, D. Regulation of muscle pyruvate dehydrogenase complex in insulin resistance: effects of exercise and dichloroacetate. Diabetes & metabolism journal 2013, 37, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M.; McCormack, J.G.; Rutter, G.A.; Burnett, P.; Edgell, N.J.; Moule, S.K.; Diggle, T.A. The hormonal regulation of pyruvate dehydrogenase complex. Advances in enzyme regulation 1996, 36, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Schoenmann, N.; Tannenbaum, N.; Hodgeman, R.M.; Raju, R.P. Regulating mitochondrial metabolism by targeting pyruvate dehydrogenase with dichloroacetate, a metabolic messenger. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1667. [Google Scholar] [CrossRef]
- Popov, K.M.; Zhao, Y.; Shimomura, Y.; Kuntz, M.J.; Harris, R.A. Branched-chain alpha-ketoacid dehydrogenase kinase. Molecular cloning, expression, and sequence similarity with histidine protein kinases. Journal of Biological Chemistry 1992, 267, 13127–13130. [Google Scholar] [CrossRef] [PubMed]
- Gudi, R.; Melissa, M.B.; Kedishvili, N.Y.; Zhao, Y.; Popov, K.M. Diversity of the pyruvate dehydrogenase kinase gene family in humans. Journal of Biological Chemistry 1995, 270, 28989–28994. [Google Scholar] [CrossRef] [PubMed]
- Rowles, J.; Scherer, S.W.; Xi, T.; Majer, M.; Nickle, D.C.; Rommens, J.M.; Popov, K.M.; Harris, R.A.; Riebow, N.L.; Xia, J.; Tsui, L.C. Cloning and characterization of PDK4 on 7q21. 3 encoding a fourth pyruvate dehydrogenase kinase isoenzyme in human. Journal of Biological Chemistry 1996, 271, 22376–22382. [Google Scholar] [CrossRef]
- Simon, M.C. Coming up for air: HIF-1 and mitochondrial oxygen consumption. Cell Metab 2006, 3, 150–151. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell metabolism 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.; Saramäki, A.; Malinen, M.; Rieck, M.; Väisänen, S.; Huotari, A.; Herzig, K.H.; Müller, R.; Carlberg, C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor β/δ. Journal of molecular biology 2007, 372, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Peters, J.M.; Harris, R.A. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor α. Biochemical and biophysical research communications 2001, 287, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer research 2011. [Google Scholar] [CrossRef]
- Yeaman, S.J.; Hutcheson, E.T.; Roche, T.E.; Pettit, F.H.; Brown, J.R.; Reed, L.J.; Watson, D.C.; Dixon, G.H. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry 1978, 17, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- Bowker-Kinley, M.M.; Davis, I.W.; Pengfei, W.U.; Harris, A.R.; Popov, MK. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochemical Journal 1998, 329, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.S.; Ramos, H.; Soares, J.; Saraiva, L. p53 and glucose metabolism: an orchestra to be directed in cancer therapy. Pharmacological research 2018, 131, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Li, J.; Chuang, J.L.; Chuang, D.T. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 2007, 15, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Morley, A. An evolutionary perspective on the Crabtree effect. Frontiers in molecular biosciences 2014, 1, 17. [Google Scholar] [CrossRef]
- Saunier, E.; Benelli, C.; Bortoli, S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. International journal of cancer 2016, 138, 809–817. [Google Scholar] [CrossRef]
- Holness, M.J.; and Sugden, M.C. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans 2003, 31, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, S.; Cooper, R.H.; Randle, P.J. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochemical Journal 1974, 141, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: new home for an orphan drug? . Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 2014, 1846, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, S.; Randle, P.J. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication). Biochem J 1973, 134, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Lorini, M.; Ciman, M. Hypoglycaemic action of diisopropylammonium salts in experimental diabetes. Biochemical pharmacology 1962, 11, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Albatany, M.; Li, A.; Meakin, S.; Bartha, R. Dichloroacetate induced intracellular acidification in glioblastoma: in vivo detection using AACID-CEST MRI at 9.4 Tesla. Journal of neuro-oncology 2018, 136, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Anemone, A.; Consolino, L.; Conti, L.; Reineri, F.; Cavallo, F.; Aime, S.; Longo, D.L. In vivo evaluation of tumour acidosis for assessing the early metabolic response and onset of resistance to dichloroacetate by using magnetic resonance pH imaging. International journal of oncology 2017, 51, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Albatany, M.; Li, A.; Meakin, S.; Bartha, R. In vivo detection of acute intracellular acidification in glioblastoma multiforme following a single dose of cariporide. International journal of clinical oncology 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, X.; Xiong, H.; Zhou, P.; Ni, Z.; Yang, T.; Zhang, Y.; Zeng, Y.; He, J.; Yang, F.; Zhang, N. Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells. Oncotarget 2017, 8, 51748. [Google Scholar] [CrossRef] [PubMed]
- Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Treading slowly through hypoxic waters: dichloroacetate to the rescue! J Physiol 2018, 596, 2957–2958. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor JA 3rd. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol Cancer Ther 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lee, W.D.; Leitner, B.P.; Zhu, W.; Fosam, A.; Li, Z.; Gaspar, R.C.; Halberstam, A.A.; Robles, B.; Rabinowitz, J.D.; Perry, R.J. Dichloroacetate as a novel pharmaceutical treatment for cancer-related fatigue in melanoma. American Journal of Physiology-Endocrinology and Metabolism 2023, 325, E363–E375. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razek, E.A.; Mahmoud, H.M.; Azouz, A.A. Management of ulcerative colitis by dichloroacetate: Impact on NFATC1/NLRP3/IL1B signaling based on bioinformatics analysis combined with in vivo experimental verification. Inflammopharmacology 2023, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. JNCI: Journal of the National Cancer Institute 2017, 109, djx071. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and cancer: an overview towards clinical applications. Oxidative medicine and cellular longevity 2019, 2019, 8201079. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Felts, J.M. Diisopropylammonium dichloroacetate (DIPA) and sodium dichloroacetate (DCA): Effect on glucose and fat metabolism in normal and diabetic tissue. Metabolism 1970, 19, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Harwood, H.J.; Varnado, C.E. Regulation of rat liver hydroxymethylglutaryl coenzyme A reductase by a new class of noncompetitive inhibitors. Effects of dichloroacetate and related carboxylic acids on enzyme activity. The Journal of clinical investigation 1983, 72, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.W.; Swift, L.L.; Rabinowitz, D.; Crofford, O.B.; Oates, J.A.; Stacpoole, P.W. Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Atherosclerosis 1979, 33, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Stakišaitis, D.; Kapočius, L.; Kilimaitė, E.; Gečys, D.; Šlekienė, L.; Balnytė, I.; Palubinskienė, J.; Lesauskaitė, V. Preclinical Study in Mouse Thymus and Thymocytes: Effects of Treatment with a Combination of Sodium Dichloroacetate and Sodium Valproate on Infectious Inflammation Pathways. Pharmaceutics 2023, 15, 2715. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; Ding, J. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Qu, J.; Li, P.; Sun, Z. Histone lactylation regulates cancer progression by reshaping the tumor microenvironment. Frontiers in Immunology 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Huang, D.; Jiang, Y.; Hou, J.; Tian, M.; Li, J.; Sun, L.; Zhang, Y.; Zhang, T.; Li, Z.; Li, Z. Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non-small cell lung cancer. Frontiers in oncology 2021, 11, 647559. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ye, Z.; Li, Z.; Jing, D.S.; Fan, G.X.; Liu, M.Q.; Zhuo, Q.F.; Ji, S.R.; Yu, X.J.; Xu, X.W.; Qin, Y. Lactate-induced protein lactylation: A bridge between epigenetics and metabolic reprogramming in cancer. Cell proliferation 2023, e13478. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Lv, Y.; Dai, X. Lactate, histone lactylation and cancer hallmarks. Expert Reviews in Molecular Medicine 2023, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Peng, Q.; Zheng, J.; Yang, Y.; Zhang, X.; Ma, A.; Qin, Y.; Qin, Z.; Zheng, X. The function and mechanism of lactate and lactylation in tumor metabolism and microenvironment. Genes & Diseases 2023, 10, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zou, X.; Yang, S.; Zhang, A.; Li, N.; Ma, Z. Identification of lactylation related model to predict prognostic, tumor infiltrating immunocytes and response of immunotherapy in gastric cancer. Frontiers in Immunology 2023, 14, 1149989. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Li, Y.; Li, S.; Liu, S.; Cheng, Y. Hypoxia-induced SHMT2 protein lactylation facilitates glycolysis and stemness of esophageal cancer cells. Molecular and Cellular Biochemistry 2024, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Zheng, Z.; Bian, C.; Chang, S.; Bao, J.; Yu, H.; Xin, Y.; Jiang, X. Functions and mechanisms of lactylation in carcinogenesis and immunosuppression. Frontiers in Immunology 2023, 14, 1253064. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ying, T.; Yuan, J.; Wang, Y.; Su, X.; Chen, S.; Zhao, Y.; Zhao, Y.; Sheng, J.; Teng, L.; Luo, C. BRAFV600E restructures cellular lactylation to promote anaplastic thyroid cancer proliferation. Endocrine-Related Cancer 2023, 30. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Zhao, X.; Liu, X. Hypoxia induced β-catenin lactylation promotes the cell proliferation and stemness of colorectal cancer through the wnt signaling pathway. Experimental Cell Research 2023, 422, 113439. [Google Scholar] [CrossRef] [PubMed]
- Rho, H.; Terry, A.R.; Chronis, C.; Hay, N. Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell metabolism 2023, 35, 1406–1423. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fan, W.; Li, N.; Ma, Y.; Yao, M.; Wang, G.; He, S.; Li, W.; Tan, J.; Lu, Q.; Hou, S. YY1 lactylation in microglia promotes angiogenesis through transcription activation-mediated upregulation of FGF2. Genome Biology 2023, 24, 87. [Google Scholar] [CrossRef]
- Wang, N.; Wang, W.; Wang, X.; Mang, G.; Chen, J.; Yan, X.; Tong, Z.; Yang, Q.; Wang, M.; Chen, L.; Sun, P. Histone lactylation boosts reparative gene activation post–myocardial infarction. Circulation Research 2022, 131, 893–908. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ji, Z.; Gong, Y.; Fan, L.; Xu, P.; Chen, X.; Miao, J.; Zhang, K.; Zhang, W.; Ma, P.; Zhao, H. Numb/Parkin-directed mitochondrial fitness governs cancer cell fate via metabolic regulation of histone lactylation. Cell reports 2023, 42. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Huang, H.; Li, M.; Chen, Y. Proteomic analysis identifies PFKP lactylation in SW480 colon cancer cells. Iscience 2024, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; He, T.; Meng, D.; Lv, W.; Ye, J.; Cheng, L.; Hu, J. BZW2 Modulates Lung Adenocarcinoma Progression through Glycolysis-Mediated IDH3G Lactylation Modification. Journal of Proteome Research 2023, 22, 3854–3865. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Mao, L.; Wang, J.; Zhang, X.; Wu, M.; Wen, Q.; Yu, S.C. Beyond metabolic waste: lysine lactylation and its potential roles in cancer progression and cell fate determination. Cellular Oncology 2023, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Varner, E.L.; Trefely, S.; Bartee, D.; von Krusenstiern, E.; Izzo, L.; Bekeova, C.; O'Connor, R.S.; Seifert, E.L.; Wellen, K.E.; Meier, J.L.; Snyder, N.W. Quantification of lactoyl-CoA (lactyl-CoA) by liquid chromatography mass spectrometry in mammalian cells and tissues. Open Biology 2020, 10, 200187. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Yruela, C.; Bæk, M.; Monda, F.; Olsen, C.A. Chiral posttranslational modification to lysine ε-amino groups. Accounts of Chemical Research 2022, 55, 1456–1466. [Google Scholar] [CrossRef]
- Patel, R.; Kumar, A.; Lokhande, K.B.; Swamy, K.V.; Sharma, N.K. Molecular Docking and Simulation Studies Predict Lactyl-CoA as the Substrate for P300 Directed Lactylation. This manuscript has been released as a Pre-Print at “bioRxiv” downloaded from https://chemrxiv.org/engage/api-gateway/chemrxiv/assets/orp/resource/item/60c74e8d4c8919380fad3a55/original/molecular-docking-and-simulation-studies-predict-lactyl-co-a-as-the-substrate-for-p300-directed-lactylation.pdf. Accessed 02/21/2024.
- Wu, H.; Huang, H.; Zhao, Y. Interplay between metabolic reprogramming and post-translational modifications: from glycolysis to lactylation. Frontiers in Immunology 2023, 14. [Google Scholar] [CrossRef]
- Shin, E.; Koo, J.S. Glucose metabolism and glucose transporters in breast cancer. Frontiers in cell and developmental biology 2021, 9, 728759. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chen, X.; Wang, J.; Liu, B. Glycolysis-related lncRNA TMEM105 upregulates LDHA to facilitate breast cancer liver metastasis via sponging miR-1208. Cell Death & Disease 2023, 14, 80. [Google Scholar] [CrossRef] [PubMed]
- Schreier, A.; Zappasodi, R.; Serganova, I.; Brown, K.A.; Demaria, S.; Andreopoulou, E. Facts and Perspectives: Implications of tumor glycolysis on immunotherapy response in triple negative breast cancer. Frontiers in Oncology 2023, 12, 1061789. [Google Scholar] [CrossRef] [PubMed]
- Godoy, A.; Ulloa, V.; Rodríguez, F.; Reinicke, K.; Yañez, A.J.; García, M.D.; Medina, R.A.; Carrasco, M.; Barberis, S.; Castro, T.; Martínez, F. Differential subcellular distribution of glucose transporters GLUT1–6 and GLUT9 in human cancer: ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. Journal of cellular physiology 2006, 207, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D'agostino, D.; Forlì, F. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell death & disease 2014, 5, e1336. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; Park, J.H. Inhibition of aerobic glycolysis represses Akt/mTOR/HIF-1α axis and restores tamoxifen sensitivity in antiestrogen-resistant breast cancer cells. PloS one 2015, 10, e0132285. [Google Scholar] [CrossRef] [PubMed]
- Littleflower, A.B.; Parambil, S.T.; Antony, G.R.; Subhadradevi, L. The determinants of metabolic discrepancies in aerobic glycolysis: Providing potential targets for breast cancer treatment. Biochimie 2024, 220(107) 121. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.L.; Eubank, W.B.; Mankoff, D.A. FDG PET, PET/CT, and breast cancer imaging. Radiographics 2007, 207, S215–S229. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Parish, C.R.; Board, P.G.; Blackburn, A.C. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast cancer research and treatment 2010, 120, 253–260. [Google Scholar] [CrossRef]
- Gang, B.P.; Dilda, P.J.; Hogg, P.J.; Blackburn, A.C. Dichloroacetate reverses the Warburg effect, inhibiting growth and sensitizing breast cancer cells towards apoptosis. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31-Apr 4; Chicago, IL. Philadelphia (PA): AACR; C.ancer Res 2012;72(8 Suppl):Abstract nr 3228.
- Harting, T.P.; Stubbendorff, M.; Hammer, S.C.; Schadzek, P.; Ngezahayo, A.; Escobar, H.M.; Nolte, I. Dichloroacetate affects proliferation but not apoptosis in canine mammary cell lines. PloS one 2017, 12, e0178744. [Google Scholar] [CrossRef] [PubMed]
- De Preter, G. , Neveu, M.-A., Danhier, P., Brisson, L., Payen, V.L., Porporato, P.E., … Gallez, B. (2016). Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. ( 7(3), 2910–2920. [CrossRef] [PubMed]
- Blackburn, A.C.; Rooke, M.; Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Board, P.G. Reversal of the glycolytic phenotype with dichloroacetate in a mouse mammary adenocarcinoma model. Pathology 2010, 42, S61–S62. [Google Scholar] [CrossRef]
- Wang, M.; Liao, C.; Hu, Y.; Pan, Q.; Jiang, J. Sensitization of breast cancer cells to paclitaxel by dichloroacetate through inhibiting autophagy. Biochemical and biophysical research communications 2017, 489, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.H. , Seo, S.-K., Park, Y., Kim, E.-K., Seong, M.-K., Kim, H.-A., … Park, I.-C. (). Dichloroacetate potentiates tamoxifen-induced cell death in breast cancer cells via downregulation of the epidermal growth factor receptor. Oncotarget 2016, 7, 59809–59819. [Google Scholar] [CrossRef]
- Haugrud, A.B.; Zhuang, Y.; Coppock, J.D.; Miskimins, W.K. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Breast cancer research and treatment 2014, 147, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Board, P.G.; Blackburn, A.C. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Molecular cancer 2011, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Lam, Y.M.; Leung, Y.C.; Hu, X.; Chen, X.; Cheung, E.; Tam, K.Y. Combined use of arginase and dichloroacetate exhibits anti-proliferative effects in triple negative breast cancer cells. J Pharm Pharmacol 2019, 71, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Robey, I.F.; Martin, N.K. Bicarbonate and dichloroacetate: evaluating pH altering therapies in a mouse model for metastatic breast cancer. BMC cancer 2011, 11, 235. [Google Scholar] [CrossRef]
- Hong, S.E.; Shin, K.S.; Lee, Y.H.; Seo, S.K.; Yun, S.M.; Choe, T.B.; Kim, H.A.; Kim, E.K.; Noh, W.C.; Kim, J.I.; Hwang, C.S. Inhibition of S6K1 enhances dichloroacetate-induced cell death. Journal of Cancer Research and Clinical Oncology 2015, 141. [Google Scholar] [CrossRef] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677. [Google Scholar] [CrossRef]
- Lefort, N.; Brown, A.; Lloyd, V.; Ouellette, R.; Touaibia, M.; Culf, A.S.; Cuperlovic-Culf, M. 1H NMR metabolomics analysis of the effect of dichloroacetate and allopurinol on breast cancers. Journal of pharmaceutical and biomedical analysis 2014, 93, 77–85. [Google Scholar] [CrossRef] [PubMed]
- 172 de Mey, S.; Dufait, I.; Jiang, H.; Corbet, C.; Wang, H.; Van De Gucht, M.; Kerkhove, L.; Law, K.L.; Vandenplas, H.; Gevaert, T.; Feron, O. Dichloroacetate radiosensitizes hypoxic breast cancer cells. International Journal of Molecular Sciences 2020, 21, 9367. [Google Scholar] [CrossRef]
- Gholami, L.; Attari, F.; Talkhabi, M.; Saadatpour, F. MDA-MB-231. Nova Biologica Reperta 2023 10(1):1-10Doi: 10.29252/nbr.10.1.1 Downloaded from https://scholar.google.com/scholar?cluster=12666777980647358402&hl=en&as_sdt=0,5&as_ylo=2023. Accessed January 2024. 2023. [Google Scholar]
- Cutruzzolà, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose metabolism in the progression of prostate cancer. Frontiers in physiology 2017, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: connecting the dots. Molecular cancer 2006, 5, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Salminen, E.; Hogg, A.; Binns, D.; Frydenberg, M.; Hicks, R. Investigations with FDG-PET scanning in prostate cancer show limited value for clinical practice. Acta Oncologica 2002, 41, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Akhurst, T.; Osman, I.; Nunez, R.; Macapinlac, H.; Siedlecki, K.; Verbel, D.; Schwartz, L.; Larson, S.M.; Scher, H.I. Fluorinated deoxyglucose positron emission tomography imaging in progressive metastatic prostate cancer. Urology 2002, 59, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yacoub, S.; Shiverick, K.T.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C.J. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. The Prostate 2008, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.H.; Withers, H.G.; Garbens, A.; Love, H.D.; Magnoni, L.; Hayward, S.W.; Moyes, C.D. Hypoxia and the metabolic phenotype of prostate cancer cells. Biochimica et Biophysica Acta (BBA)-Bioenergetics 2009, 1787, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Harting, T.; Stubbendorff, M.; Willenbrock, S.; Wagner, S.; Schadzek, P.; Ngezahayo, A.; Escobar, H.M.; Nolte, I. The effect of dichloroacetate in canine prostate adenocarcinomas and transitional cell carcinomas in vitro. International journal of oncology 2016, 49, 2341–2350. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Liang, H.; Guan, G. Dichloroacetate enhances the cytotoxic effect of Cisplatin via decreasing the level of FOXM1 in prostate cancer. Int J Clin Med 2016, 9, 11044–11050. [Google Scholar]
- Olszewski, U.; Poulsen, T.T.; Ulsperger, E.; Poulsen, H.S.; Geissler, K.; Hamilton, G. In vitro cytotoxicity of combinations of dichloroacetate with anticancer platinum compounds. Clinical pharmacology: advances and applications. [CrossRef]
- Chaudhary, A.K.; Bhat, T.A.; Kumar, S.; Kumar, A.; Kumar, R.; Underwood, W.; Koochekpour, S.; Shourideh, M.; Yadav, N.; Dhar, S.; Chandra, D. Mitochondrial dysfunction-mediated apoptosis resistance associates with defective heat shock protein response in African–American men with prostate cancer. British journal of cancer 2016, 114, 1090. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.W.; Kasai, M.; Yamamoto, S.; Fukuhara, H.; Karashima, T.; Kurabayashi, A.; Furihata, M.; Hanazaki, K.; Inoue, K.; Ogura, S.I. Metabolic shift towards oxidative phosphorylation reduces cell-density-induced cancer-stem-cell-like characteristics in prostate cancer in vitro. Biology Open 2023, 12, bio059615–10. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Xia, L.; Oyang, L.; Liang, J.; Tan, S.; Wu, N.; Yi, P.; Pan, Q.; Rao, S.; Han, Y.; Tang, Y. The POU2F1-ALDOA axis promotes the proliferation and chemoresistance of colon cancer cells by enhancing glycolysis and the pentose phosphate pathway activity. Oncogene 2022, 41, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.S.; Wang, J.Y.; Chung, F.Y.; Lee, S.C.; Huang, M.Y.; Kuo, C.W.; Yang, M.J.; Lin, S.R. Significance of the glycolytic pathway and glycolysis related-genes in tumorigenesis of human colorectal cancers. Oncology reports 2008, 19, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Madhok, B.M.; Yeluri, S.; Perry, S.L.; Hughes, T.A.; Jayne, D.G. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. British journal of cancer 2010, 102, 1746. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Hill, D.K.; Andrejeva, G.; Boult, J.K.; Troy, H.; Fong, A.L.; Orton, M.R.; Panek, R.; Parkes, H.G.; Jafar, M.; Koh, D.M. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. British journal of cancer 2014, 111, 375. [Google Scholar] [CrossRef] [PubMed]
- Delaney, L.M.; Ho, N.; Morrison, J.; Farias, N.R.; Mosser, D.D.; Coomber, B.L. Dichloroacetate affects proliferation but not survival of human colorectal cancer cells. Apoptosis 2015, 20, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cheng, X.; Pan, S.; Wang, L.; Dou, W.; Liu, J.; Shi, X. Dichloroacetate attenuates the stemness of colorectal cancer cells via trigerring ferroptosis through sequestering iron in lysosomes. Environmental toxicology 2021, 36, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Xie, G.; He, J.; Li, J.; Pan, F.; Liang, H. Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. BioMed Research International. 2011. [Google Scholar] [CrossRef]
- Liang, Y.; Hou, L.; Li, L.; Li, L.; Zhu, L.; Wang, Y.; Huang, X.; Hou, Y.; Zhu, D.; Zou, H.; Gu, Y. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene 2020, 39, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhu, D.; Zhu, L.; Hou, Y.; Hou, L.; Huang, X.; Li, L.; Wang, Y.; Li, L.; Zou, H.; Wu, T. Dichloroacetate overcomes oxaliplatin chemoresistance in colorectal cancer through the miR-543/PTEN/Akt/mTOR pathway. Journal of Cancer 2019, 10, 6037. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Andrews, D.; Blackburn, A.C. Long-term stabilization of stage 4 colon cancer using sodium dichloroacetate therapy. World journal of clinical cases 2016, 4, 336. [Google Scholar] [CrossRef]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; Bartek, J. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the V600EBRAF oncogene. Oncotarget 2013, 4, 584. [Google Scholar] [CrossRef]
- Falkenius, J.; Lundeberg, J.; Johansson, H.; Tuominen, R.; Frostvik-Stolt, M.; Hansson, J.; Brage, S.E. High expression of glycolytic and pigment proteins is associated with worse clinical outcome in stage III melanoma. Melanoma research 2013, 23, 452–460. [Google Scholar] [CrossRef]
- Koch, A.; Ebert, E.V.; Seitz, T.; Dietrich, P.; Berneburg, M.; Bosserhoff, A.; Hellerbrand, C. Characterization of glycolysis-related gene expression in malignant melanoma. Pathology-Research and Practice 2020, 216, 152752. [Google Scholar] [CrossRef] [PubMed]
- Franco-Molina, M.A.; Mendoza-Gamboa, E.; Sierra-Rivera, C.A.; Zapata-Benavides, P.; Hernandez, D.F.; Chávez-Reyes, A.; Rivera-Morales, L.G.; Tamez-Guerra, R.; Rodríguez-Padilla, C. In vitro and in vivo antitumoral activitiy of sodium dichloroacetate (DCA-Na) against murine melanoma. African Journal of Microbiology Research 2012, 6, 4782–4796. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 6, 37281. [Google Scholar] [CrossRef]
- Pópulo, H.; Caldas, R.; Lopes, J.M.; Pardal, J.; Máximo, V.; Soares, P. Overexpression of pyruvate dehydrogenase kinase supports dichloroacetate as a candidate for cutaneous melanoma therapy. Expert opinion on therapeutic targets 2015, 19, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, C.; Dahl, C.; Basse, A.L.; Ma, T.; Guldberg, P. Bioenergetic modulation with dichloroacetate reduces the growth of melanoma cells and potentiates their response to BRAF V600E inhibition. Journal of translational medicine 2014, 12, 247. [Google Scholar] [CrossRef]
- Zheng, M.F.; Shen, S.Y. DCA increases the antitumor effects of capecitabine in a mouse B16 melanoma allograft and a human non-small cell lung cancer A549 xenograft. Cancer chemotherapy and pharmacology 2013, 72, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Andrews, D.; Shainhouse, J.; Blackburn, A.C. Long-term stabilization of metastatic melanoma with sodium dichloroacetate. World journal of clinical oncology 2017, 8, 371. [Google Scholar] [CrossRef]
- Reuss, A.M.; Groos, D.; Buchfelder, M.; Savaskan, N. The acidic brain—glycolytic switch in the microenvironment of malignant glioma. International journal of molecular sciences 2021, 22, 5518. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. Journal of Biological Chemistry 2011, 286, 32843–32853. [Google Scholar] [CrossRef]
- Sanzey, M.; Abdul Rahim, S.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PloS one 2015, 10, e0123544. [Google Scholar] [CrossRef] [PubMed]
- Stanke, K.M.; Wilson, C.; Kidambi, S. High expression of glycolytic genes in clinical glioblastoma patients correlates with lower survival. Frontiers in Molecular Biosciences 2021, 8, 752404. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Wilson, E.B.; Short, S.; Melcher, A.A.; Biggs, M.; Diakos, C.I.; Howell, V.M. Glycolysis and fatty acid oxidation inhibition improves survival in glioblastoma. Frontiers in Oncology 2021, 11, 633210. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; Abdulkarim, B. Metabolic modulation of glioblastoma with dichloroacetate. Science translational medicine 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zhao, X.; Ren, W.; Wang, X.; Yu, K.F.; Li, D.; Zhang, X.; Zhang, Q. Antitumor activity of dichloroacetate on C6 glioma cell: in vitro and in vivo evaluation. OncoTargets and therapy. [CrossRef]
- Kumar, K. , Wigfield, S., Gee, H.E., Devlin, C.M., Singleton, D., Li, J.-L., … Ivan, M. (2013). Dichloroacetate Reverses the Hypoxic Adaptation to Bevacizumab and Enhances its Antitumor Effects in Mouse Xenografts. ( 91(6), 749–758. [CrossRef] [PubMed]
- Morfouace, M.; Lalier, L.; Oliver, L.; Cheray, M.; Pecqueur, C.; Cartron, P.F.; Vallette, F.M. Control of glioma cell death and differentiation by PKM2–Oct4 interaction. Cell death & disease 2014, 5, e1036. [Google Scholar] [CrossRef] [PubMed]
- Kolesnik, D.L.; Pyaskovskaya, O.N.; Boichuk, I.V.; Solyanik, G.I. Hypoxia enhances antitumor activity of dichloroacetate. Experimental oncology. 2: 4). [PubMed]
- Vella, S.; Conti, M.; Tasso, R.; Cancedda, R.; Pagano, A. Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. International journal of cancer 2012, 130, 1484–1493. [Google Scholar] [CrossRef]
- Sradhanjali, S.; Tripathy, D.; Rath, S.; Mittal, R.; Reddy, M.M. Overexpression of pyruvate dehydrogenase kinase 1 in retinoblastoma: A potential therapeutic opportunity for targeting vitreous seeds and hypoxic regions. PloS one 2017, 12, e0177744. [Google Scholar] [CrossRef]
- Park, J.M.; Recht, L.D.; Josan, S.; Merchant, M.; Jang, T.; Yen, Y.F.; Hurd, R.E.; Spielman, D.M.; Mayer, D. Metabolic response of glioma to dichloroacetate measured in vivo by hyperpolarized 13C magnetic resonance spectroscopic imaging. Neuro-oncology 2013, 15, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Fedorchuk, A.G.; Pyaskovskaya, O.N.; Gorbik, G.V.; Prokhorova, I.V.; Kolesnik, D.L.; Solyanik, G.I. Effectiveness of sodium dichloroacetate against glioma C6 depends on administration schedule and dosage. Exp Oncol. [PubMed]
- Wicks, R.T.; Azadi, J.; Mangraviti, A.; Zhang, I.; Hwang, L.; Joshi, A.; Bow, H.; Hutt-Cabezas, M.; Martin, K.L.; Rudek, M.A.; Zhao, M. Local delivery of cancer-cell glycolytic inhibitors in high-grade glioma. Neuro-oncology 2014, 17, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Korsakova, L.; Krasko, J.A.; Stankevicius, E. Metabolic-targeted combination therapy with dichloroacetate and metformin suppresses glioblastoma cell line growth in vitro and in vivo. in vivo 2021, 35, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of glioblastoma cells to irradiation by modulating the glucose metabolism. Molecular cancer therapeutics 2015, 14, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M. , Shen, H., McKelvey, K.J., Gee, H.E., & Hau, E. (2021). Targeting glucose metabolism of cancer cells with dichloroacetate to radiosensitize high-grade gliomas. International Journal of Molecular Sciences. [CrossRef]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 2016, 7, 56456. [Google Scholar] [CrossRef] [PubMed]
- Prokhorova, I.V.; Pyaskovskaya, O.N.; Kolesnik, D.L.; Solyanik, G.I. Influence of metformin, sodium dichloroacetate and their combination on the hematological and biochemical blood parameters of rats with gliomas C6. Experimental oncology. 2018. Т. 40, № 3. — С. 205-210. [PubMed]
- Kolesnik, D.L.; Pyaskovskaya, O.N.; Yurchenko, O.V.; Solyanik, G.I. Metformin enhances antitumor action of sodium dichloroacetate against glioma C6. Experimental Oncology 2019, 41, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yu, M.; Tsoli, M.; Chang, C.; Joshi, S.; Liu, J.; Ryall, S.; Chornenkyy, Y.; Siddaway, R.; Hawkins, C.; Ziegler, D.S. Targeting reduced mitochondrial DNA quantity as a therapeutic approach in pediatric high-grade gliomas. Neuro-oncology 2020, 22, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Y.; Zhang, L.; Zhao, C.; Wang, D. Phosphorylated form of pyruvate dehydrogenase α1 mediates tumor necrosis factor α-induced glioma cell migration. Oncology Letters 2021, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Duraj, T.; García-Romero, N.; Carrión-Navarro, J.; Madurga, R.; Ortiz de Mendivil, A.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg effect: Oxidative and glycolytic phenotypes coexist within the metabolic heterogeneity of glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Griguer, C.E.; Oliva, C.R.; Gillespie, G.Y. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. Journal of neuro-oncology. [CrossRef]
- Shibao, S.; Minami, N.; Koike, N.; Fukui, N.; Yoshida, K.; Saya, H.; Sampetrean, O. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro-oncology 2018, 20, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Boag, J.M.; Beesley, A.H.; Firth, M.J.; Freitas, J.R.; Ford, J.; Hoffmann, K.; Cummings, A.J.; de Klerk, N.H.; Kees, U.R. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia 2006, 20, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Hulleman, E.; Kazemier, K.M.; Holleman, A.; VanderWeele, D.J.; Rudin, C.M.; Broekhuis, M.J.; Evans, W.E.; Pieters, R.; Den Boer, M.L. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood 2009, 113, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Buentke, E.; Nordström, A.; Lin, H.; Björklund, A.C.; Laane, E.; Harada, M.; Lu, L.; Tegnebratt, T.; Stone-Elander, S.; Heyman, M.; Söderhäll, S.; Porwit, A.; Ostenson, C.G.; Shoshan, M.; Tamm, K.P.; Grandér, D. Glucocorticoid-induced cell death is mediated through reduced glucose metabolism in lymphoid leukemia cells. Blood Cancer, J 2011, 1, e31. [Google Scholar] [CrossRef] [PubMed]
- Samuels, A.L.; Heng, J.Y.; Beesley, A.H.; Kees, U.R. Bioenergetic modulation overcomes glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia. Br J Haematol 2014, 165, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.; McBrayer, S.K.; Rosen, S.T. Targeting the Warburg effect in hematological malignancies: from PET to therapy. Curr Opin Oncol 2009, 21, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Howman, R.A.; Neeson, P.J.; Berridge, M.V.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. Journal of leukocyte biology 2011, 89, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Xia, J.; Xu, H.; Frech, I.; Tricot, G.; Zhan, F. NEK2 promotes aerobic glycolysis in multiple myeloma through regulating splicing of pyruvate kinase. Journal of hematology & oncology 2017, 10, 1–1. [Google Scholar] [CrossRef] [PubMed]
- Gavriatopoulou, M.; Paschou, S.A.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Metabolic disorders in multiple myeloma. International Journal of Molecular Sciences 2021, 22, 11430. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, W.Y.; McGee, S.L.; Connor, T.; Mottram, B.; Wilkinson, A.; Whitehead, J.P.; Vuckovic, S.; Catley, L. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. British journal of cancer 2013, 108, 1624. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Sasano, T.; Arima, Y.; Kushima, S.; Tsujita, K.; Matsuoka, M.; Hata, H. A novel PDK1 inhibitor, JX06, inhibits glycolysis and induces apoptosis in multiple myeloma cells. Biochemical and Biophysical Research Communications 2022, 587, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S. , Kawano, Y., Yuki, H., Okuno, Y., Nosaka, K., Mitsuya, H., & Hata, H. (2013). PDK1 inhibition is a novel therapeutic target in multiple myeloma. ( 108(1), 170–178. [CrossRef] [PubMed]
- Tian, D.D.; Bennett, S.K.; Coupland, L.A.; Forwood, K.; Lwin, Y.; Pooryousef, N.; Tea, I.; Truong, T.T.; Neeman, T.; Crispin, P.; D’Rozario, J. GSTZ1 genotypes correlate with dichloroacetate pharmacokinetics and chronic side effects in multiple myeloma patients in a pilot phase 2 clinical trial. Pharmacology research & perspectives 2019, 7, e00526. [Google Scholar] [CrossRef] [PubMed]
- Voltan, R.; Rimondi, E.; Melloni, E.; Gilli, P.; Bertolasi, V.; Casciano, F.; Rigolin, G.M.; Zauli, G.; Secchiero, P. Metformin combined with sodium dichloroacetate promotes B leukemic cell death by suppressing anti-apoptotic protein Mcl-1. Oncotarget 2016, 7, 18965. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kant, S.; Singh, S.M. Antitumor and chemosensitizing action of dichloroacetate implicates modulation of tumor microenvironment: a role of reorganized glucose metabolism, cell survival regulation and macrophage differentiation. Toxicology and applied pharmacology 2013, 273, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Flavin, D.F. Non-Hodgkin's lymphoma reversal with dichloroacetate. Journal of oncology. 2010; Article ID 414726. [CrossRef]
- Strum, S.B.; Adalsteinsson, Ö.; Black, R.R.; Segal, D.; Peress, N.L.; Waldenfels, J. Case report: sodium dichloroacetate (DCA) inhibition of the “Warburg Effect” in a human cancer patient: complete response in non-Hodgkin’s lymphoma after disease progression with rituximab-CHOP. Journal of bioenergetics and biomembranes 2013, 45, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Abramek, J.; Bogucki, J.; Ziaja-Sołtys, M.; Stępniewski, A.; Bogucka-Kocka, A. Effect of sodium dichloroacetate on apoptotic gene expression in human leukemia cell lines. Pharmacological Reports 2019, 71, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Agnoletto, C.; Melloni, E.; Casciano, F.; Rigolin, G.M.; Rimondi, E.; Celeghini, C.; Brunelli, L.; Cuneo, A.; Secchiero, P.; Zauli, G. Sodium dichloroacetate exhibits anti-leukemic activity in B-chronic lymphocytic leukemia (B-CLL) and synergizes with the p53 activator Nutlin-3. Oncotarget 2014, 5, 4347. [Google Scholar] [CrossRef] [PubMed]
- Agnoletto, C.; Brunelli, L.; Melloni, E.; Pastorelli, R.; Casciano, F.; Rimondi, E.; Rigolin, G.M.; Cuneo, A.; Secchiero, P.; Zauli, G. The anti-leukemic activity of sodium dichloroacetate in p53mutated/null cells is mediated by a p53-independent ILF3/p21 pathway. Oncotarget 2015, 6, 2385. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Sadowska, M.; Carter-Cooper, B.; Bhatnagar, V.; van der Merwe, I.; Levis, M.J.; Sausville, E.A.; Lapidus, R.G. Perturbation of cellular oxidative state induced by dichloroacetate and arsenic trioxide for treatment of acute myeloid leukemia. Leukemia research 2015, 39, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Queckborner, S.; Turnbull, A.; Michie, C.O.; Williams, A.R.; Rye, T.; Gourley, C.; Langdon, S.P. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC cancer 2018, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, L.; Chai, W.; Zhao, T.; Jin, X.; Guo, X.; Han, L.; Yuan, C. Dichloroacetic acid upregulates apoptosis of ovarian cancer cells by regulating mitochondrial function. OncoTargets and therapy. 1729. [Google Scholar] [CrossRef]
- Saed, G.M.; Fletcher, N.M.; Jiang, Z.L.; Abu-Soud, H.M.; Diamond, M.P. Dichloroacetate induces apoptosis of epithelial ovarian cancer cells through a mechanism involving modulation of oxidative stress. Reproductive sciences 2011, 18, 1253–1261. [Google Scholar] [CrossRef]
- Štarha, P.; Trávníček, Z.; Vančo, J.; Dvořák, Z. Half-sandwich Ru (II) and Os (II) bathophenanthroline complexes containing a releasable dichloroacetato ligand. Molecules 2018, 23, 420. [Google Scholar] [CrossRef] [PubMed]
- Priego-Hernández, V.D.; Arizmendi-Izazaga, A.; Soto-Flores, D.G.; Santiago-Ramón, N.; Feria-Valadez, M.D.; Navarro-Tito, N.; Jiménez-Wences, H.; Martínez-Carrillo, D.N.; Salmerón-Bárcenas, E.G.; Leyva-Vázquez, M.A.; Illades-Aguiar, B. Expression of HIF-1α and Genes Involved in Glucose Metabolism Is Increased in Cervical Cancer and HPV-16-Positive Cell Lines. Pathogens 2022, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, B.S.; Yu, D.H.; Lu, Q.; Ma, J.; Qi, H.; Fang, C.; Chen, H.Z. Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. International journal of oncology 2011, 38, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Zhang, Y.Z.; Wang, Y.S.; Ma, X.X. Identification of novel cell glycolysis related gene signature predicting survival in patients with endometrial cancer. Cancer cell international 2019, 19, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.Y.; Huggins, G.S.; Debidda, M.; Munshi, N.C.; De Vivo, I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecologic oncology 2008, 109, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.H.; Zhou, C.; Bae-Jump, V. Dichloroacetate inhibits cell proliferation and induces apoptosis in human endometrial cancer cell lines. Cancer Research. 2016 Jul 15;76(14_Supplement):2993. [CrossRef]
- Li, X.B.; Gu, J.D.; Zhou, Q.H. Review of aerobic glycolysis and its key enzymes–new targets for lung cancer therapy. Thoracic cancer 2015, 6, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Smolle, E.; Leko, P.; Stacher-Priehse, E.; Brcic, L.; El-Heliebi, A.; Hofmann, L.; Quehenberger, F.; Hrzenjak, A.; Popper, H.H.; Olschewski, H.; Leithner, K. Distribution and prognostic significance of gluconeogenesis and glycolysis in lung cancer. Molecular oncology 2020, 14, 2853–2867. [Google Scholar] [CrossRef]
- Xu, J.Q.; Fu, Y.L.; Zhang, J.; Zhang, K.Y.; Ma, J.; Tang, J.Y.; Zhang, Z.W.; Zhou, Z.Y. Targeting glycolysis in non-small cell lung cancer: Promises and challenges. Frontiers in Pharmacology 2022, 13, 1037341. [Google Scholar] [CrossRef] [PubMed]
- Oylumlu, E.; yin Ng, Y. EVALUATION OF ANTIPROLIFERATIVE EFFECTS OF SODIUM DICHLOROACETATE ON LUNG ADENOCARCINOMA. Turkish Journal of Biochemistry/Turk Biyokimya Dergisi. 2019 Dec 3;44. Downloaded from https://openurl.ebsco.com/EPDB%3Agcd%3A11%3A2866491/detailv2?sid=ebsco%3Aplink%3Ascholar&id=ebsco%3Agcd%3A140410362&crl=c Accessed 20 december 2023.
- Allen, K.T.; Chin-Sinex, H.; DeLuca, T.; Pomerening, J.R.; Sherer, J.; Watkins III JB, Foley, J. ; Jesseph, J.M.; Mendonca, M.S. Dichloroacetate alters Warburg metabolism, inhibits cell growth, and increases the X-ray sensitivity of human A549 and H1299 NSC lung cancer cells. Free Radical Biology and Medicine 2015, 89, 263–273. [Google Scholar] [CrossRef]
- Lu, X.; Zhou, D.; Hou, B.; Liu, Q.X.; Chen, Q.; Deng, X.F.; Yu, Z.B.; Dai, J.G.; Zheng, H. Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer. Cancer management and research. 1231. [Google Scholar] [CrossRef]
- Sun, H. , Zhu, A., Zhou, X., & Wang, F. (2017). Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget, 2017; 8, 52642–52650. [Google Scholar] [CrossRef]
- Yang, Z.; Tam, K.Y. Anti-cancer synergy of dichloroacetate and EGFR tyrosine kinase inhibitors in NSCLC cell lines. European journal of pharmacology 2016, 789, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Al-Azawi, A.; Sulaiman, S.; Arafat, K.; Yasin, J.; Nemmar, A.; Attoub, S. Impact of sodium dichloroacetate alone and in combination therapies on lung tumor growth and metastasis. International journal of molecular sciences 2021, 22, 12553. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhou, Y.; Wu, P.; Ran, M.; Xu, N.; Shan, W.; Sha, O.; Tam, K.Y. Dichloroacetophenone biphenylsulfone ethers as anticancer pyruvate dehydrogenase kinase inhibitors in non-small cell lung cancer models. Chemico-Biological Interactions 2023, 378, 110467. [Google Scholar] [CrossRef] [PubMed]
- Fiebiger, W.; Olszewski, U.; Ulsperger, E.; Geissler, K.; Hamilton, G. In vitro cytotoxicity of novel platinum-based drugs and dichloroacetate against lung carcinoid cell lines. Clinical and Translational Oncology 2011, 13, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kolesnik, D.L.; Pyaskovskaya, O.N.; Boychuk, I.V.; Dasyukevich, O.I.; Melnikov, O.R.; Tarasov, A.S.; Solyanik, G.I. Effect of dichloroacetate on Lewis lung carcinoma growth and metastasis. Experimental oncology. 1: 2). [PubMed]
- Feng, M. , Wang, J., & Zhou, J. (2023). Unraveling the therapeutic mechanisms of dichloroacetic acid in lung cancer through integrated multi-omics approaches: metabolomics and transcriptomics. Frontiers in Genetics. [CrossRef]
- Penticuff, J.C. , Woolbright, B.L., Sielecki, T.M., Weir, S.J., and Taylor, J.A. MIF family proteins in genitourinary cancer: Tumorigenic roles and therapeutic potential. Nat. Rev. Urol. 2019, 16, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Nobre, C.C.; de Araújo, J.M.; Fernandes, T.A.; Cobucci, R.N.; Lanza, D.C.; Andrade, V.S.; Fernandes, J.V. Macrophage migration inhibitory factor (MIF): biological activities and relation with cancer. Pathology & Oncology Research. [CrossRef]
- Yasasever, V. , Camlica, H., Duranyildiz, D., Oguz, H., Tas, F., & Dalay, N. (2007). Macrophage migration inhibitory factor in cancer. Cancer investigation, 25(8), 715-719. [CrossRef] [PubMed]
- Zheng, X. , Wei, Y., Huang, T., Wei, X., Sun, S., Wang, T., & Zhao, Z. (2023). Role of macrophage migration inhibitory factor for stem cells. Chinese Journal of Tissue Engineering Research, 27(15), 2395. [CrossRef]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat Rev Dis Primers 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Morita, M.; Tanuma, N. A metabolic vulnerability of small-cell lung cancer. Oncotarget 2018, 9, 32278–32279. [Google Scholar] [CrossRef]
- Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer stem cells in small cell lung cancer cell line H446: higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS One 2016, 11, e0154576. [Google Scholar] [CrossRef]
- Grass, G.D.; Scott, K.E.; Fernandez, M.R.; Stewart, P.A.; Kang, Y.P.; Yang, C.; DeNicola, G.M.; Bannister, T.D.; Cubitt, C.L.; Haura, E.B.; Cleveland, J.L. Targeting Lactate Transport in Small-Cell Lung Cancer. International Journal of Radiation Oncology, Biology, Physics 2018, 102, e182. [Google Scholar] [CrossRef]
- Choi, J.E.; Sebastian, C.; Ferrer, C.M.; Lewis, C.A.; Sade-Feldman, M.; LaSalle, T.; Gonye, A.; Lopez, B.G.; Abdelmoula, W.M.; Regan, M.S.; Cetinbas, M. A unique subset of glycolytic tumour-propagating cells drives squamous cell carcinoma. Nature metabolism 2021, 3, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Somers, K.D.; Merrick, M.A.; Lopez, M.E.; Incognito, L.S.; Schechter, G.L.; Casey, G. Frequent p53 mutations in head and neck cancer. Cancer Research 1992, 52, 5997–6000. [Google Scholar] [PubMed]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; Freilino, M. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer discovery 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed]
- McBride, S.M.; Rothenberg, S.M.; Faquin, W.C.; Chan, A.W.; Clark, J.R.; Ellisen, L.W.; Wirth, L.J. Mutation frequency in 15 common cancer genes in high-risk head and neck squamous cell carcinoma. Head & neck 2014, 36, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Sok, J.C.; Coppelli, F.M.; Thomas, S.M.; Lango, M.N.; Xi, S.; Hunt, J.L.; Freilino, M.L.; Graner, M.W.; Wikstrand, C.J.; Bigner, D.D.; Gooding, W.E. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clinical Cancer Research 2006, 12, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.A.; Boyle, J.O.; Koch, W.M.; Goodman, S.N.; Hruban, R.H.; Eby, Y.J.; Couch, M.J.; Forastiere, A.A.; Sidransky, D. Association between cigarette smoking and mutation of the p53 gene in squamous-cell carcinoma of the head and neck. New England Journal of Medicine 1995, 332, 712–717. [Google Scholar] [CrossRef]
- Landor, S.K.; Mutvei, A.P.; Mamaeva, V.; Jin, S.; Busk, M.; Borra, R.; Grönroos, T.J.; Kronqvist, P.; Lendahl, U.; Sahlgren, C.M. Hypo-and hyperactivated Notch signaling induce a glycolytic switch through distinct mechanisms. Proceedings of the National Academy of Sciences 2011, 108, 18814–18819. [Google Scholar] [CrossRef]
- Slaninova, V.; Krafcikova, M.; Perez-Gomez, R.; Steffal, P.; Trantirek, L.; Bray, S.J.; Krejci, A. Notch stimulates growth by direct regulation of genes involved in the control of glycolysis and the tricarboxylic acid cycle. Open biology 2016, 6, 150155. [Google Scholar] [CrossRef] [PubMed]
- Bi, P.; Kuang, S. Notch signaling as a novel regulator of metabolism. Trends in Endocrinology & Metabolism 2015, 26, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Najy, A.J.; Huang, W.; Sethi, S.; Snyder, M.; Sakr, W.; Dyson, G.; Hüttemann, M.; Lee, I.; Ali-Fehmi, R.; Franceschi, S. HPV-associated differential regulation of tumor metabolism in oropharyngeal head and neck cancer. Oncotarget 2017, 8, 51530. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Agriesti, F.; Scrima, R.; Laurenzana, I.; Perrone, D.; Tataranni, T.; Mazzoccoli, C.; Muzio, L.L.; Capitanio, N.; Piccoli, C. Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment. Oncotarget 2015, 6, 1217. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Zhang, H.F.; Guo, Y.Z.; Wang, P.Y.; Liu, Z.S.; Gao, H.D.; Xie, W.L. Combination of Taxol® and dichloroacetate results in synergistically inhibitory effects on Taxol-resistant oral cancer cells under hypoxia. Molecular medicine reports 2015, 11, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Park, J.Y.; Kim, E.H.; Jang, H.J.; Kwon, M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Letters 2016, 371, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.F.; Mazurczak, M.; Dib, E.G.; Bleeker, J.S.; Geeraerts, L.H.; Tinguely, M.; Lohr, M.M.; McGraw, S.C.; Jensen, A.W.; Ellison, C.A.; Black, L.J. Phase II study of dichloroacetate, an inhibitor of pyruvate dehydrogenase, in combination with chemoradiotherapy for unresected, locally advanced head and neck squamous cell carcinoma. Investigational New Drugs 2022, 40, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Inanc, S.; Keles, D.; Eskiizmir, G.; Basbinar, Y.A.; OKTAY, G. Metformin And Dichloroacetate Combination Exert A Synergistic Effect On Cell Viability Of Oral Squamous Cell Carcinoma. ENT Updates 2019, 9, 68–73. [Google Scholar] [CrossRef]
- Morais, M.; Dias, F.; Teixeira, A.L.; Medeiros, R. MicroRNAs and altered metabolism of clear cell renal cell carcinoma: Potential role as aerobic glycolysis biomarkers. Biochimica et Biophysica Acta (BBA)-General Subjects 2017, 1861, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.; Liu, M.; Xu, Y.; Wu, G. Glycolysis-related genes serve as potential prognostic biomarkers in clear cell renal cell carcinoma. Oxidative medicine and cellular longevity. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Linehan, W.M.; Srinivasan, R. Aerobic glycolysis: a novel target in kidney cancer. Expert review of anticancer therapy 2013, 13, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Balan, M.; Sabarwal, A.; Choueiri, T.K.; Pal, S. Metabolic reprogramming in renal cancer: Events of a metabolic disease. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 2021, 1876, 188559. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Poma, J.; Trilla-Fuertes, L.; López-Vacas, R.; López-Camacho, E.; García-Fernández, E.; Pertejo, A.; Lumbreras-Herrera, M.I.; Zapater-Moros, A.; Díaz-Almirón, M.; Dittmann, A.; Fresno Vara, J.Á. Proteomics Characterization of Clear Cell Renal Cell Carcinoma. Journal of Clinical Medicine 2023, 12, 384. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Xavier, C.E.; Emaldi, M.; Mingo, J.; Øyjord, T.; Mælandsmo, G.M.; Fodstad, Ø.; Errarte, P.; Larrinaga, G.; Llarena, R.; López, J.I.; Pulido, R. The expression pattern of pyruvate dehydrogenase kinases predicts prognosis and correlates with immune exhaustion in clear cell renal cell carcinoma. Scientific Reports 2023, 13, 7339. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Dromparis, P.; Saleme, B.; Gurtu, V.; Watson, K.; Paulin, R.; Zervopoulos, S.; Stenson, T.; Sutendra, G.; Pink, D.B.; Carmine-Simmen, K. Metabolic modulation of clear-cell renal cell carcinoma with dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase. European urology 2016, 69, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Q.; Tang, H.Y.; Wang, C.D.; Sang, B.T.; Liu, X.; Yi, F.P.; Liu, G.L.; Wu, X.M. Influence of Dichloroacetate on Wilms’ Tumor in vitro. Annals of Clinical & Laboratory Science 2022, 52, 101–108. [Google Scholar]
- Kalay, S.; Dogan, A.; Turkan, A.; Demiroglu-Zergeroglu, A. Dicholoroacetate exerts anti-cancer activity on human renal cell carcinoma cells. Turkish Journal of Biochemistry 2017, 42, 577–585. [Google Scholar] [CrossRef]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose metabolism in pancreatic cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef] [PubMed]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; Wright, E.M. Functional expression of sodium-glucose transporters in cancer. Proceedings of the National Academy of Sciences 2015, 112, E4111–E4119. [Google Scholar] [CrossRef] [PubMed]
- Rajeshkumar, N.V. , Yabuuchi, S., Pai, S.G., De Oliveira, E., Kamphorst, J.J., Rabinowitz, J.D.,... & Dang, C.V. Treatment of pancreatic cancer patient–derived xenograft panel with metabolic inhibitors reveals efficacy of phenformin. Clinical Cancer Research, 2017; 23, 5639–5647. [Google Scholar] [CrossRef]
- Tavares-Valente, D.; Cannone, S.; Greco, M.R.; Carvalho, T.M.; Baltazar, F.; Queirós, O.; Agrimi, G.; Reshkin, S.J.; Cardone, R.A. Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell. Cancers 2023, 15, 3868. [Google Scholar] [CrossRef]
- Shimoni-Sebag, A.; Abramovich, I.; Agranovich, B.; Sirovsky, Y.; Stossel, C.; Atias, D.; Raitses-Gurevich, M.; Glick-Gorman, Y.; Margalit, O.; Regev, D.; Tal, R. The pentose-phosphate pathway induces pancreatic cancer radioresistance, a preclinical study with clinical validation. Cancer Research. 2023 Apr 4;83(7_Supplement):2411. [CrossRef]
- Feuerecker, B. , Biechl, P., Veltkamp, C., Saur, D., & Eisenreich, W. Metabolic response of pancreatic carcinoma cells under treatment with dichloroacetate. Metabolites, 2021, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, T. , Agriesti, F., Pacelli, C., Ruggieri, V., Laurenzana, I., Mazzoccoli, C.,... & Piccoli, C. (). Dichloroacetate affects mitochondrial function and stemness-associated properties in pancreatic cancer cell lines. Cells 2019, 8, 478. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Xiong, X.; Huang, G.; Liu, J.; Sheng, S.; Wang, H.; Qin, W. Dichloroacetate enhances adriamycin-induced hepatoma cell toxicity in vitro and in vivo by increasing reactive oxygen species levels. PLoS One 2014, 9, e92962. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.J.; Xie, P.; Dai, X.F.; Chen, L.X.; Sun, L.B.; Li, T.; He, W.H.; Xu, Z.Z.; Huang, G.; He, F.T.; Lian, J.Q. Dichloroacetate enhances the antitumor effect of pirarubicin via regulating the ROS-JNK signaling pathway in liver cancer cells. Cancer Drug Resistance 2020, 3, 947. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Lee, M.; Park, M.; Kim, S.Y.; Shim, M.S.; Lee, C.Y.; Choi, D.H.; Cho, Y. Metformin and dichloroacetate suppress proliferation of liver cancer cells by inhibiting mTOR complex 1. International Journal of Molecular Sciences 2021, 22, 10027. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Akazawa, T.; Aoki, M.; Kuze, B.; Mizuta, K.; Ito, Y.; Inoue, N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. International journal of cancer 2013, 133, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Rooke, M.; Coupland, L.A.; Truong, T.; Blackburn, A.C. Dichloroacetate is an effective treatment for sarcoma models in vitro and in vivo. Cancer & Metabolism. [CrossRef]
- Ishiguro, T.; Ishiguro, M.; Ishiguro, R.; Iwai, S. Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproliferative effect on malignant tumors. Oncology letters 2012, 3, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013, 32, 1638. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.M.; Baghdadi, H.; Ahmed, N.S.; Almaramhy, H.H.; Mahmoud, A.A.; El-Sawy, S.A.; Ayat, M.; Elshazley, M.; Abdel-Aziz, W.; Abdel-Latif, H.M.; Ibrahim, W. Dichloroacetate is an antimetabolite that antagonizes acetate and deprives cancer cells from its benefits: A novel evidence-based medical hypothesis. Medical Hypotheses 2019, 122, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Khyzhnyak, S.V.; Sorokina, L.V.; Stepanova, L.I.; Kaplia, A.A. Functional and dynamic state of inner mitochondrial membrane of sarcoma 37 in mice under administration of sodium dichloroacetate. The Ukrainian biochemical journal. 2014, 106–118. [Google Scholar] [CrossRef]
- Choi, Y.W.; Lim, I.K. Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer letters 2014, 346, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Hur, H.; Ham, I.H.; Yun, J.; Lee, J.Y.; Shim, W.; Kim, Y.B.; Lee, G.; Han, S.U.; Cho, Y.K. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Experimental cell research 2014, 321, 219–230. [Google Scholar] [CrossRef]
- Badr, M.M.; Qinna, N.A.; Qadan, F.; Matalka, K.Z. Dichloroacetate modulates cytokines toward T helper 1 function via induction of the interleukin-12–interferon-γ pathway. OncoTargets and Therapy, 2014, 7, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yuan, P.; Yu, W.; Lin, J.; Xu, A.; Xu, X.; Lou, J.; Yu, T.; Qian, C.; Liu, B.; Song, J. Mitochondria-targeting polymer micelle of dichloroacetate induced pyroptosis to enhance osteosarcoma immunotherapy. ACS nano 2022, 16, 10327–10340. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.K. , Yan, S., Lam, J.S.M., Feng, Y., Khan, M., Chen, C.,... & Ho, J.C.M. (2022). Disturbance of the Warburg effect by dichloroacetate and niclosamide suppresses the growth of different sub-types of malignant pleural mesothelioma in vitro and in vivo. Frontiers in Pharmacology. [CrossRef]
- Qin, H.; Zheng, G.; Li, Q.; Shen, L. Metabolic reprogramming induced by DCA enhances cisplatin sensitivity through increasing mitochondrial oxidative stress in cholangiocarcinoma. Frontiers in Pharmacology. 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Maimouni, S.; Zong, W.X. The pleiotropic effects of glutamine metabolism in cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Klose, K.; Packeiser, E.M.; Müller, P.; Granados-Soler, J.L.; Schille, J.T.; Goericke-Pesch, S.; Kietzmann, M.; Escobar, H.M.; Nolte, I. Metformin and sodium dichloroacetate effects on proliferation, apoptosis, and metabolic activity tested alone and in combination in a canine prostate and a bladder cancer cell line. PLoS One 2021, 16, e0257403. [Google Scholar] [CrossRef] [PubMed]
- Gurav, A.; Sivaprakasam, S.; Bhutia, Y.D.; Boettger, T.; Singh, N.; Ganapathy, V. Slc5a8, a Na+-coupled high-affinity transporter for short-chain fatty acids, is a conditional tumour suppressor in colon that protects against colitis and colon cancer under low-fibre dietary conditions. Biochem J 2015, 469, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Gopal, E.; Miyauchi, S.; Prasad, P.D. Biological functions of SLC5A8, a candidate tumour suppressor. Biochem Soc Trans. 2005, 33, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Di Bernardo, J. and Rhoden, K.J. Atlas of genetcs and cytogenetcs in oncology and haematology. 2009. Downloaded from https://atlasgeneticsoncology.org/gene/44089/slc5a8 Accessed 01/24/2024.
- Whitman, S.P.; Hackanson, B.; Liyanarachchi, S.; Liu, S.; Rush, L.J.; Maharry, K.; Margeson, D.; Davuluri, R.; Wen, J.; Witte, T.; Yu, L. DNA hypermethylation and epigenetic silencing of the tumor suppressor gene, SLC5A8, in acute myeloid leukemia with the MLL partial tandem duplication. Blood, The Journal of the American Society of Hematology 2008, 112, 2013–2016. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Myeroff, L.; Smiraglia, D.; Romero, M.F.; Pretlow, T.P.; Kasturi, L.; Lutterbaugh, J.; Rerko, R.M.; Casey, G.; Issa, J.P.; Willis, J. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proceedings of the National Academy of Sciences 2003, 100, 8412–8417. [Google Scholar] [CrossRef] [PubMed]
- Porra, V.; Ferraro-Peyret, C.; Durand, C.; Selmi-Ruby, S.; Giroud, H.; Berger-Dutrieux, N.; Decaussin, M.; Peix, J.L.; Bournaud, C.; Orgiazzi, J.; Borson-Chazot, F.; Dante, R.; Rousset, B. Silencing of the tumor suppressor gene SLC5A8 is associated with BRAF mutations in classical papillary thyroid carcinomas. J Clin Endocrinol Metab 2005, 90, 3028–3035. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Maunakea, A.; Jun, P.; Bollen, A.W.; Hodgson, J.G.; Goldenberg, D.D.; Weiss, W.A.; Costello, J.F. Shared epigenetic mechanisms in human and mouse gliomas inactivate expression of the growth suppressor SLC5A8. Cancer Res 2005, 65, 3617–3623. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Helm, J.F.; Zheng, W.; Ly, Q.P.; Hodul, P.J.; Centeno, B.A.; Malafa, M.P. Silencing of the candidate tumor suppressor gene solute carrier family 5 member 8 (SLC5A8) in human pancreatic cancer. Pancreas 2008, 36, e32–e39. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Toyota, M.; Akino, K.; Suzuki, H.; Kusano, M.; Satoh, A.; Mita, H.; Sasaki, Y.; Nojima, M.; Yanagihara, K.; Hinoda, Y.; Tokino, T.; Imai, K. Aberrant methylation and histone deacetylation associated with silencing of SLC5A8 in gastric cancer. Tumour Biol. [CrossRef]
- Li, B.; Li, X.; Ni, Z.; Zhang, Y.; Zeng, Y.; Yan, X.; Huang, Y.; He, J.; Lyu, X.; Wu, Y.; Wang, Y. Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells. Oncotarget 2016, 7, 59458. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.P.; Poff, A.M.; Koutnik, A.P.; D’Agostino, D.P. Complex I inhibition augments dichloroacetate cytotoxicity through enhancing oxidative stress in VM-M3 glioblastoma cells. PloS one 2017, 12, e0180061. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. Journal of neurochemistry 2002, 80, 780–787. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. Journal of Biological Chemistry 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; Chandel, N.S. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife, 2014; 3. [Google Scholar] [CrossRef]
- Algire, C.; Moiseeva, O.; Deschênes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prevention Research 2012, 5, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O'Neill, L.A. Metformin inhibits the production of reactive oxygen species from NADH: ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. Journal of Biological Chemistry 2015, 290, 20348–20359. [Google Scholar] [CrossRef]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proceedings of the National Academy of Sciences 2014, 111, E2501–E2509. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochemical Journal 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; De, B.B.; Gunsalus, I.C.; Hornberger CS, Jr. Crystalline alpha-lipoic acid; a catalytic agent associated with pyruvate dehydrogenase. Science. [CrossRef]
- Korotchkina, L.G.; Sidhu, S.; Patel, M.S. R-lipoic acid inhibits mammalian pyruvate dehydrogenase kinase. Free Radic Res, 1083. [Google Scholar] [CrossRef]
- Konrad, T.; Vicini, P.; Kusterer, K.; Höflich, A.; Assadkhani, A.; Böhles, H.J.; Sewell, A.; Tritschler, H.J.; Cobelli, C.; Usadel, K.H. alpha-Lipoic acid treatment decreases serum lactate and pyruvate concentrations and improves glucose effectiveness in lean and obese patients with type 2 diabetes. Diabetes care 1999, 22, 280–287. [Google Scholar] [CrossRef] [PubMed]
- 349. Mahban Rahimifard, Mona Navaei-Nigjeh, Maryam Baeeri, et al. Multiple protective mechanisms of alpha-lipoic acid in oxidation, apoptosis and inflammation against hydrogen peroxide induced toxicity in human lymphocytes. Molecular and Cellular Biochemistry. [CrossRef]
- Schwartz, L.; Abolhassani, M.; Guais, A.; Sanders, E.; Steyaert, J.M.; Campion, F.; Israël, M. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: preliminary results. Oncology reports 2010, 23, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Nickel, A.; Daniel, H. α-lipoic acid induces apoptosis in human colon cancer cells by increasing mitochondrial respiration with a concomitant O2−. -generation. Apoptosis 2005, 10, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Moungjaroen, J.; Nimmannit, U.; Callery, P.S.; Wang, L.; Azad, N.; Lipipun, V.; Chanvorachote, P.; Rojanasakul, Y. Reactive oxygen species mediate caspase activation and apoptosis induced by lipoic acid in human lung epithelial cancer cells through Bcl-2 down-regulation. Journal of Pharmacology and Experimental Therapeutics 2006, 319, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Simbula, G.; Columbano, A.; Ledda-Columbano, G.M.; Sanna, L.; Deidda, M.; Diana, A.; Pibiri, M. Increased ROS generation and p53 activation in α-lipoic acid-induced apoptosis of hepatoma cells. Apoptosis 2007, 12, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Feuerecker, B.; Pirsig, S.; Seidl, C.; Aichler, M.; Feuchtinger, A.; Bruchelt, G.; Senekowitsch-Schmidtke, R. Lipoic acid inhibits cell proliferation of tumor cells in vitro and in vivo. Cancer biology & therapy 2012, 13, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Pyaskovskaya, O.N.; Kolesnik, D.L.; Fedorchuk, A.G.; Prokhorova, I.V.; Solyanik, G.I. 2-Deoxy-D-glucose enhances dichloroacetate antitumor action against Lewis lung carcinoma. Exp Oncol 2016, 38, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhong, X.; Yuan, Y. Does baking soda function as a magic bullet for patients with cancer? A mini review. Integrative cancer therapies. 1534. [Google Scholar] [CrossRef]
- Ayyanathan, K. , Kesaraju, S., Dawson-Scully, K., & Weissbach, H. (2012). Combination of Sulindac and Dichloroacetate Kills Cancer Cells via Oxidative Damage. ( 7(7), e39949. [CrossRef]
- Dhar, S.; Lippard, S.J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proceedings of the National Academy of Sciences 2009, 106, 22199–22204. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.K.; Marrache, S.; Harn, D.A.; Dhar, S. Mito-DCA: a mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS chemical biology 2014, 9, 1178–1187. [Google Scholar] [CrossRef]
- Hanberry, B.S.; Berger, R.; Zastre, J.A. High-dose vitamin B1 reduces proliferation in cancer cell lines analogous to dichloroacetate. Cancer chemotherapy and pharmacology 2014, 73, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Ghosh, M.; Dutta, S.K. A potent tumoricidal co-drug ‘Bet-CA’-an ester derivative of betulinic acid and dichloroacetate selectively and synergistically kills cancer cells. Scientific reports 2015, 5, 7762. [Google Scholar] [CrossRef] [PubMed]
- Chen, H. , Liang, K., Hou, C. et al. Dichloroacetic acid and rapamycin synergistically inhibit tumor progression. J. Zhejiang Univ. Sci. B. [CrossRef]
- Bonner, M.Y.; Karlsson, I.; Rodolfo, M.; Arnold, R.S.; Vergani, E.; Arbiser, J.L. Honokiol bis-dichloroacetate (Honokiol DCA) demonstrates activity in vemurafenib-resistant melanoma in vivo. Oncotarget 2016, 7, 12857. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T. , Ishiguro, R.H., Ishiguro, M., Toki, A., & Terunuma, H. (2022). Synergistic anti-tumor effect of dichloroacetate and ivermectin. Cureus. [CrossRef]
- Marco-Brualla, J.; de Miguel, D.; Martínez-Lostao, L.; Anel, A. DR5 Up-Regulation Induced by Dichloroacetate Sensitizes Tumor Cells to Lipid Nanoparticles Decorated with TRAIL. Journal of Clinical Medicine 2023, 12, 608. [Google Scholar] [CrossRef] [PubMed]
- Mironiuk-Puchalska, E.; Karatsai, O.; Żuchowska, A.; Wróblewski, W.; Borys, F.; Lehka, L.; Rędowicz, M.J.; Koszytkowska-Stawińska, M. Development of 5-fluorouracil-dichloroacetate mutual prodrugs as anticancer agents. Bioorganic Chemistry 2023, 140, 106784. [Google Scholar] [CrossRef] [PubMed]
- Korga, A.; Ostrowska, M.; Iwan, M.; Herbet, M.; Dudka, J. Inhibition of glycolysis disrupts cellular antioxidant defense and sensitizes HepG2 cells to doxorubicin treatment. FEBS Open Bio 2019, 9, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor III, JA. The role of pyruvate dehydrogenase kinase-4 (PDK4) in bladder cancer and chemoresistance. Molecular Cancer Therapeutics 2018, 17, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Fekir, K.; Dubois-Pot-Schneider, H.; Desert, R.; Daniel, Y.; Glaise, D.; Rauch, C.; Morel, F.; Fromenty, B.; Musso, O.; Cabillic, F.; Corlu, A. Retrodifferentiation of human tumor hepatocytes to stem cells leads to metabolic reprogramming and chemoresistance. Cancer research 2019, 79, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Skeberdytė, A.; Sarapinienė, I.; Aleksander-Krasko, J.; Stankevičius, V.; Sužiedėlis, K.; Jarmalaitė, S. Dichloroacetate and salinomycin exert a synergistic cytotoxic effect in colorectal cancer cell lines. Scientific reports 2018, 8, 17744. [Google Scholar] [CrossRef]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol reduces cancer risk: a population-based cohort study. Medicine. [CrossRef]
- Wang, F.; Liu, H.; Wang, F.; Xu, R.; Wang, P.; Tang, F.; Zhang, X.; Zhu, Z.; Lv, H.; Han, T. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Molecular medicine reports 2018, 17, 5213–5221. [Google Scholar] [CrossRef]
- Al-Wadei, H.A.; Al-Wadei, M.H.; Schuller, H.M. Prevention of pancreatic cancer by the beta-blocker propranolol. Anti-cancer drugs 2009, 20, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Ma, Q.; Wang, L.; Hu, H.; Li, J.; Zhang, D.; Zhang, M. Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncology reports 2009, 22, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Sukhatme, V.; Meheus, L.; Rooman, I.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)—Propranolol as an anti-cancer agent. ecancermedicalscience. [CrossRef]
- Lucido, C.T.; Miskimins, W.K.; Vermeer, P.D. Propranolol promotes glucose dependence and synergizes with dichloroacetate for anti-cancer activity in HNSCC. Cancers 2018, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Guha, I.; Chatterjee, A.; Banerji, A. Anti-cancer potential of all-trans retinoic acid (ATRA): A Review. InProceedings of the Zoological Society 2013 Jun (Vol. 66, pp. 1–7). Springer-Verlag. [CrossRef]
- Hoffman, E.; Mielicki, W. All-trans retinoic acid (ATRA) in prevention and cancer therapy. Advances in Hygiene and Experimental Medicine. 2010 Jun 9;64. Downloaded from http://www.phmd.pl/fulltxt.php?ICID=911863. Accesed February 2023.
- Abildgaard, C.; Dahl, C.; Abdul-Al, A.; Christensen, A.; Guldberg, P. Inhibition of retinoic acid receptor β signaling confers glycolytic dependence and sensitization to dichloroacetate in melanoma cells. Oncotarget 2017, 8, 84210. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Chen, Q.; Jiang, F.; Yu, D.; Mao, Q.; Xia, W.; Shi, R.; Wang, J.; Xu, L. Diisopropylamine dichloroacetate enhances radiosensitization in esophageal squamous cell carcinoma by increasing mitochondria-derived reactive oxygen species levels. Oncotarget 2016, 7, 68170. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yu, J.; Ge, S.; Su, Y.; Fan, X. Novel insight into metabolic reprogrammming in cancer radioresistance: A promising therapeutic target in radiotherapy. International Journal of Biological Sciences 2023, 19, 811. [Google Scholar] [CrossRef] [PubMed]
- Toledo, G.F.; Nagamine, M.K.; Nowosh, V.; Machado, F.T.; Massoco, C.O.; Souza-Pinto, N.C.; Dagli, M.L. Antineoplastic effects of sodium dichloroacetate and omeprazole, alone or in combination, on canine oral mucosal melanoma cells. Frontiers in Veterinary Science. [CrossRef]
- Romero, Y.; Castillejos-López, M.; Romero-García, S.; Aguayo, A.S.; Herrera, I.; Garcia-Martin, M.O.; Torres-Espíndola, L.M.; Negrete-García, M.C.; Olvera, A.C.; Huerta-Cruz, J.C.; Velázquez-Cruz, R. Antitumor therapy under hypoxic microenvironment by the combination of 2-methoxyestradiol and sodium dichloroacetate on human non-small-cell lung cancer. Oxidative Medicine and Cellular Longevity. 2020. [Google Scholar] [CrossRef]
- Ma, W.; Zhao, X.; Wang, K.; Liu, J.; Huang, G. Dichloroacetic acid (DCA) synergizes with the SIRT2 inhibitor Sirtinol and AGK2 to enhance anti-tumor efficacy in non-small cell lung cancer. Cancer Biology & Therapy 2018, 19, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Dyrstad, S.E.; Lotsberg, M.L.; Tan, T.Z.; Pettersen, I.K.; Hjellbrekke, S.; Tusubira, D.; Engelsen, A.S.; Daubon, T.; Mourier, A.; Thiery, J.P.; Dahl, O. Blocking aerobic glycolysis by targeting pyruvate dehydrogenase kinase in combination with EGFR TKI and ionizing radiation increases therapeutic effect in non-small cell lung cancer cells. Cancers 2021, 13, 941. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, S.L.; Hu, X.; Tam, K.Y. Inhibition of pyruvate dehydrogenase kinase 1 enhances the anti-cancer effect of EGFR tyrosine kinase inhibitors in non-small cell lung cancer. European Journal of Pharmacology 2018, 838, 41–52. [Google Scholar] [CrossRef]
- Frisch, A.; Wang, Y.; Wang, Y.; Lontos, K.; Rivadeneira, D.; Delgoffe, G. 100 Redirecting glucose flux during in vitro expansion improves the in vivo performance of adoptive T cell therapies for cancer. Journal for ImmunoTherapy of Cancer. A: 1;9(Suppl 2). [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Karioti, A.; Antoniadi, G.; Antoniadis, N.; Liakopoulos, V.; Stefanidis, I. Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes. Journal of basic and clinical physiology and pharmacology 2013, 24, 271–276. [Google Scholar] [CrossRef]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential regulation of human Treg and Th17 cells by fatty acid synthesis and glycolysis. Frontiers in immunology 2019, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Calcutt, N.A.; Lopez, V.L.; Bautista, A.D.; Mizisin, L.M.; Torres, B.R.; Shroads, A.L.; Mizisin, A.P.; Stacpoole, P.W. Peripheral neuropathy in rats exposed to dichloroacetate. J Neuropathol Exp Neurol 2009, 68, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Engelstad, K. ; Wei Y et al (2006) Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 66:324–330. [CrossRef]
- Brandsma, D.; Dorlo, T.P.; Haanen, J.H.; Beijnen, J.H.; Boogerd, W. Severe encephalopathy and polyneuropathy induced by dichloroacetate. Journal of neurology 2010, 257, 2099–2100. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.M. Trial of dichloroacetate in MELAS: toxicity overshadows the assessment of potential benefit. Neurology 2006, 66, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Felitsyn, N.; Stacpoole, P.W.; Notterpek, L. Dichloroacetate causes reversible demyelination in vitro: potential mechanism for its neuropathic effect. Journal of neurochemistry 2007, 100, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Deck, M.; Van Hameren, G.; Campbell, G.; Bernard-Marissal, N.; Devaux, J.; Berthelot, J.; Lattard, A.; Médard, J.J.; Gautier, B.; Guelfi, S.; Abbou, S. Physiology of PNS axons relies on glycolytic metabolism in myelinating Schwann cells. Plos one 2022, 17, e0272097. [Google Scholar] [CrossRef] [PubMed]
- Babetto, E.; Wong, K.M.; Beirowski, B. A glycolytic shift in Schwann cells supports injured axons. Nature neuroscience 2020, 23, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Bouçanova, F.; Chrast, R. Metabolic interaction between Schwann cells and axons under physiological and disease conditions. Frontiers in cellular neuroscience 2020, 14, 148. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; Diaz, F. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Martyniuk, C.J.; James, M.O.; Calcutt, N.A. Dichloroacetate-induced peripheral neuropathy. International Review of neurobiology 2019, 145, 211–238. [Google Scholar] [CrossRef] [PubMed]
- Shroads, A.L.; Langaee, T.; Coats, B.S.; Kurtz, T.L.; Bullock, J.R.; Weithorn, D.; Gong, Y.; Wagner, D.A.; Ostrov, D.A.; Johnson, J.A.; Stacpoole, P.W. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. The Journal of Clinical Pharmacology 2012, 52, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Washington, J.T.; Quintyne, N.J. Dichloroacetate induces different rates of cell death in cancer and noncancer cell lines in vitro. Tumori Journal 2012, 98, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.J.; Sanchez, I.M.; Nelson, M.A.; Larson, J.L.; Lansing, A.J. Liver tumor induction in B6C3F1 mice by dichloroacetate and trichloroacetate. Toxicology 1990, 63, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.L.; Bull, R.J. Metabolism and lipoperoxidative activity of trichloroacetate and dichloroacetate in rats and mice. Toxicology and applied pharmacology 1992, 115, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Schwab, S.; Efferth, T. Fatal liver and bone marrow toxicity by combination treatment of dichloroacetate and artesunate in a glioblastoma multiforme patient: case report and review of the literature. Frontiers in Oncology 2016, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.M.; Bull, R.J. Early induction of reparative hyperplasia in the liver of B6C3F1 mice treated with dichloroacetate and trichloroacetate. Toxicology 1990, 64, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Guo, X. , Dixit, V., Liu, H., Shroads, A.L., Henderson, G.N., James, M.O. and Stacpoole, P.W., 2006. Inhibition and recovery of rat hepatic glutathione S-transferase zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug metabolism and disposition. [CrossRef]
- Hassoun, E.A.; Dey, S. Dichloroacetate-and trichloroacetate-induced phagocytic activation and production of oxidative stress in the hepatic tissues of mice after acute exposure. Journal of biochemical and molecular toxicology 2008, 22, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, E. , Cearfoss, J., Mamada, S., Al-Hassan, N., Brown, M., Heimberger, K. and Liu, M.C., 2014. The effects of mixtures of dichloroacetate and trichloroacetate on induction of oxidative stress in livers of mice after subchronic exposure. Journal of Toxicology and Environmental Health, Part, A.; 77. [CrossRef]
- James, M.O.; Stacpoole, P.W. Pharmacogenetic considerations with dichloroacetate dosing. Pharmacogenomics 2016, 17, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial involvement in cisplatin resistance. International journal of molecular sciences 2019, 20, 3384. [Google Scholar] [CrossRef]
- Bruzzese, F.; Rocco, M.; Castelli, S.; Di Gennaro, E.; Desideri, A.; Budillon, A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Molecular cancer therapeutics 2009, 8, 3075–3087. [Google Scholar] [CrossRef]
- Su, L. , Zhang, H., Yan, C., Chen, A., Meng, G., Wei, J., … Ding, Y. Superior anti-tumor efficacy of diisopropylamine dichloroacetate compared with dichloroacetate in a subcutaneous transplantation breast tumor model. Oncotarget, 2016; 7, 65721–65731. [Google Scholar] [CrossRef]
- She, W. , Liu, T., Li, H., Wang, Z., Guo, Z., Liu, Y., & Liu, Y. (2023). Reprogramming Energy Metabolism with Synthesized PDK Inhibitors Based on Dichloroacetate Derivatives and Targeted Delivery Systems for Enhanced Cancer Therapy. Journal of Medicinal Chemistry 2023, 66, 21–14683. [Google Scholar] [CrossRef] [PubMed]
- Trapella, C.; Voltan, R.; Melloni, E.; Tisato, V.; Celeghini, C.; Bianco, S.; Fantinati, A.; Salvadori, S.; Guerrini, R.; Secchiero, P.; Zauli, G. Design, synthesis, and biological characterization of novel mitochondria targeted dichloroacetate-loaded compounds with antileukemic activity. Journal of Medicinal Chemistry 2016, 59, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Fereidoonnezhad, M.; Tabaei, S.M.; Sakhteman, A.; Seradj, H.; Faghih, Z.; Faghih, Z.; Mojaddami, A.; Sadeghian, B.; Rezaei, Z. Design, synthesis, molecular docking, biological evaluations and QSAR studies of novel dichloroacetate analogues as anticancer agent. Journal of Molecular Structure 2020, 1221, 128689. [Google Scholar] [CrossRef]
- Yang, Y.; Shang, P.; Cheng, C.; Wang, D.; Yang, P.; Zhang, F.; Li, T.; Lu, A.; Zhao, Y. Novel N-phenyl dichloroacetamide derivatives as anticancer reagents: design, synthesis and biological evaluation. European journal of medicinal chemistry 2010, 45, 4300–4306. [Google Scholar] [CrossRef] [PubMed]
- Zajac, J.; Kostrhunova, H.; Novohradsky, V.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. Potentiation of mitochondrial dysfunction in tumor cells by conjugates of metabolic modulator dichloroacetate with a Pt (IV) derivative of oxaliplatin. Journal of Inorganic Biochemistry 2016, 156, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wu, X.Y.; Luo, O.Y.; Su, L.; Ding, Y.T.; Jiang, Y.; Yu, D.C. Diisopropylamine dichloroacetate alleviates liver fibrosis through inhibiting activation and proliferation of hepatic stellate cells. Int. J. Clin. Exp. Med 2019, 12, 3440–3448. [Google Scholar]
- Yamane, K.; Indalao, I.L.; Chida, J.; Yamamoto, Y.; Hanawa, M.; Kido, H. Diisopropylamine dichloroacetate, a novel pyruvate dehydrogenase kinase 4 inhibitor, as a potential therapeutic agent for metabolic disorders and multiorgan failure in severe influenza. PloS one 2014, 9, e98032. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhang, H.; Yan, C.; Chen, A.; Meng, G.; Wei, J.; Yu, D.; Ding, Y. Superior anti-tumor efficacy of diisopropylamine dichloroacetate compared with dichloroacetate in a subcutaneous transplantation breast tumor model. Oncotarget 2016, 7, 65721. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Marier, D.; Marsden, E.; Andrews, D.; Eliaz, I. A novel form of dichloroacetate therapy for patients with advanced cancer: a report of 3 cases. Altern Ther Health Med. 2014 Oct 1;20(Suppl 2):21-8. PMID 25362214.
- Khan, A.; Ghen, M. A 15 Year Evolution of Dichloroacetate-Based Metabolic Cancer Therapy: A Review with Case Reports. Medical Research Archives. [CrossRef]
- Ishiguro, T.; Ishiguro, R.; Ishiguro, M.; Iwai, S. Co-treatment of dichloroacetate, omeprazole and tamoxifen exhibited synergistically antiproliferative effect on malignant tumors: in vivo experiments and a case report. Hepato-gastroenterology 2012, 59, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Lemmo, W.; Tan, G. Prolonged Survival After Dichloroacetate Treatment of Non-Small-Cell Lung Carcinoma-Related Leptomeningeal Carcinomatosis. Journal of Medical Cases 2016, 7, 136–142. [Google Scholar] [CrossRef]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; et al. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Christofk, H.R.; Hosmer, W.; et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J Cancer Research and Clinical Oncology. 2014, 140, 443–52. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.F.; Mazurczak, M.; Dib, E.G.; Bleeker, J.S.; Geeraerts, L.H.; Tinguely, M.; Lohr, M.M.; McGraw, S.C.; Jensen, A.W.; Ellison, C.A.; Black, L.J.; Puumala, S.E.; Reed, V.J.; Miskimins, W.K.; Lee, J.H.; Spanos, W.C. Phase II study of dichloroacetate, an inhibitor of pyruvate dehydrogenase, in combination with chemoradiotherapy for unresected, locally advanced head and neck squamous cell carcinoma. Invest New Drugs 2022, 40, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Heshe, D.; Hoogestraat, S.; Brauckmann, C.; Karst, U.; Boos, J.; Lanvers-Kaminsky, C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer chemotherapy and pharmacology 2011, 67, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Shahrzad, S.; Lacombe, K.; Adamcic, U.; Minhas, K.; Coomber, B.L. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer letters 2010, 297, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.M.; Jajeh, J.; Guinan, P.; Rubenstein, M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer research 2009, 29, 4579–4588. [Google Scholar] [PubMed]
- Zwicker, F.; Kirsner, A.; Peschke, P.; Roeder, F.; Debus, J.; Huber, P.E.; Weber, K.J. Dichloroacetate induces tumor-specific radiosensitivity in vitro but attenuates radiation-induced tumor growth delay in vivo. Strahlentherapie und Onkologie 2013, 189, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Feuerecker, B.; Seidl, C.; Pirsig, S.; Bruchelt, G.; Senekowitsch-Schmidtke, R. DCA promotes progression of neuroblastoma tumors in nude mice. American journal of cancer research 2015, 5, 812. [Google Scholar] [PubMed]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nature reviews cancer 2020, 20, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Sounidaki, M.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. In human alloreactive CD4+ T-cells, dichloroacetate inhibits aerobic glycolysis, induces apoptosis and favors differentiation towards the regulatory T-cell subset instead of effector T-cell subsets. Molecular medicine reports 2016, 13, 3370–3376. [Google Scholar] [CrossRef] [PubMed]
- Rostamian, H.; Khakpoor-Kooshe, M.; Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Tavassolifar, M.J.; Pawelek, J.M.; Mirzaei, H.R.; Hadjati, J. Restricting Tumor Lactate Metabolism using Dichloroacetate Improves T Cell Functions. BMC Cancer, 06 Jan 2022, 22(1):39. [CrossRef]
- Schoonjans, C.A.; Joudiou, N.; Brusa, D.; Corbet, C.; Feron, O.; Gallez, B. Acidosis-induced metabolic reprogramming in tumor cells enhances the anti-proliferative activity of the PDK inhibitor dichloroacetate. Cancer letters 2020, 470, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Ferriero, R. , Iannuzzi, C., Manco, G., & Brunetti-Pierri, N. Differential inhibition of PDKs by phenylbutyrate and enhancement of pyruvate dehydrogenase complex activity by combination with dichloroacetate. Journal of inherited metabolic disease 2015, 38, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Škorja Milić, N. , Dolinar, K., Miš, K., Matkovič, U., Bizjak, M., Pavlin, M.,... & Pirkmajer, S. (2021). Suppression of pyruvate dehydrogenase kinase by dichloroacetate in cancer and skeletal muscle cells is isoform specific and partially independent of HIF-1α. International journal of molecular sciences. [CrossRef]
- Tiersma, J.F. , Evers, B., Bakker, B.M., Jalving, M., & de Jong, S. (2022). Pyruvate dehydrogenase kinase inhibition by dichloroacetate in melanoma cells unveils metabolic vulnerabilities. ( 23(7), 3745. [CrossRef] [PubMed]
- Gang, B.P. , Rooke, M., Sun, R.C., Banskota, S., Mishra, S., Dahlstrom, J.E., & Blackburn, A.C. (2023). Pyruvate dehydrogenase kinase expression profile is a biomarker for cancer sensitivity to dichloroacetate-mediated growth inhibition. bioRxiv. [CrossRef]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Hardy, M.; Topchyan, P.; Zander, R.; Volberding, P.; Cui, W.; Kalyanaraman, B. Potent inhibition of tumour cell proliferation and immunoregulatory function by mitochondria-targeted atovaquone. Scientific reports 2020, 10, 17872. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Segaran, A.; Lord, S. Targeting OXPHOS and the electron transport chain in cancer; Molecular and therapeutic implications. InSeminars in Cancer Biology, 86. [CrossRef]
- Gurtu, V.; Webster, L.; Kinnaird, A.; Boukouris, A.; White, C.; Nagendran, J.; Freed, D.H.; Wilkins, M.R.; Michelakis, E. Targeting Pyruvate Dehydrogenase Kinase With Dichloroacetate in Pulmonary Arterial Hypertension: A Variable Clinical Response Driven by Genetic Polymorphisms in Sirtuin-3 and Uncoupling Protein-2. Circulation. 2017 Nov 14;136(suppl_1):A17443-.
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell metabolism 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W. , & Dang, C.V. (). Multifaceted roles of glycolytic enzymes. Trends in biochemical sciences 2005, 30, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Parczyk, J. , Ruhnau, J., Pelz, C., Schilling, M., Wu, H., Piaskowski, N.N.,... & Klein, A. (2021). Dichloroacetate and PX-478 exhibit strong synergistic effects in a various number of cancer cell lines. BMC cancer. [CrossRef]
- Li, S.; Wu, L.; Feng, J.; Li, J.; Liu, T.; Zhang, R.; Xu, S.; Cheng, K.; Zhou, Y.; Zhou, S.; Kong, R. In vitro and in vivo study of epigallocatechin-3-gallate-induced apoptosis in aerobic glycolytic hepatocellular carcinoma cells involving inhibition of phosphofructokinase activity. Scientific reports 2016, 6, 28479. [Google Scholar] [CrossRef]
- Qiu, X.; Jiang, Z.; Luo, Y.; Tian, D.; Song, T.; Li, Q. PPP3CB Inhibits Cell Proliferation and the Warburg Effect in Bladder Cancer by Blocking PDHK1. Frontiers in Bioscience-Landmark 2024, 29, 48. [Google Scholar] [CrossRef] [PubMed]
- Zajac, J.; Kostrhunova, H.; Novohradsky, V.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. Potentiation of mitochondrial dysfunction in tumor cells by conjugates of metabolic modulator dichloroacetate with a Pt (IV) derivative of oxaliplatin. Journal of Inorganic Biochemistry 2016, 156, 89–97. [Google Scholar] [CrossRef]
- Sun, L. , Jiang, Y., Yan, X., Dai, X., Huang, C., Chen, L.,... & Lian, J. (2021). Dichloroacetate enhances the anti-tumor effect of sorafenib via modulating the ROS-JNK-Mcl-1 pathway in liver cancer cells. Experimental Cell Research, 1275. [Google Scholar] [CrossRef]
- Babu, E.; Ramachandran, S.; CoothanKandaswamy, V.; Elangovan, S.; Prasad, P.D.; Ganapathy, V.; Thangaraju, M. Role of SLC5A8, a plasma membrane transporter and a tumor suppressor, in the antitumor activity of dichloroacetate. Oncogene 2011, 30, 4026. [Google Scholar] [CrossRef] [PubMed]
- Kailavasan, M.; Rehman, I.; Reynolds, S.; Bucur, A.; Tozer, G.; Paley, M. NMR-based evaluation of the metabolic profile and response to dichloroacetate of human prostate cancer cells. NMR in biomedicine 2014, 27, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Crabb, D.W.; Harris, R.A. Mechanism responsible for the hypoglycemic actions of dichloroacetate and 2-chloropropionate. Archives of Biochemistry and Biophysics 1979, 198, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Kawata, Y.; Moritoh, Y.; Watanabe, M. Dichloroacetate, a pyruvate dehydrogenase kinase inhibitor, ameliorates type 2 diabetes via reduced gluconeogenesis. Heliyon 2022, 8, e08889. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E.; Nance, S. Selective reduction of fatty acid oxidation in colonocytes: correlation with ulcerative colitis. Lipids 1990, 25, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Hayek, A.; Woodside, W.F. Short-term influences of dichloroacetate on genetically hyperlipemic rats. Metabolism 1980, 29, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Nayak, M.K.; Dhanesha, N.; Doddapattar, P.; Rodriguez, O.; Sonkar, V.K.; Dayal, S.; Chauhan, A.K. Dichloroacetate, an inhibitor of pyruvate dehydrogenase kinases, inhibits platelet aggregation and arterial thrombosis. Blood Adv 2018, 2, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Min, B.K.; Oh, C.J.; Park, S.; Lee, J.M.; Go, Y.; Park, B.Y.; Kang, H.J.; Kim, D.W.; Kim, J.E.; Yoo, E.K.; Kim, H.E.; Kim, M.J.; Jeon, Y.H.; Kim, Y.H.; Lee, C.H.; Jeon, J.H.; Lee, I.K. Therapeutic effect of dichloroacetate against atherosclerosis via hepatic FGF21 induction mediated by acute AMPK activation. Exp Mol Med 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Horne, A.W.; Ahmad, S.F.; Carter, R.; Simitsidellis, I.; Greaves, E.; Hogg, C.; Morton, N.M.; Saunders, P.T.K. Repurposing dichloroacetate for the treatment of women with endometriosis. Proc Natl Acad Sci U S A 2019, 116, 25389–25391. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Bolatto, C.; Trías, E.; Gandelman, M.; Radi, R.; Barbeito, L.; Cassina, P. Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PloS one 2012, 7, e34776. [Google Scholar] [CrossRef] [PubMed]
- Shangraw, R.E.; Fisher, D.M. Pharmacokinetics and pharmacodynamics of dichloroacetate in patients with cirrhosis. Clinical Pharmacology & Therapeutics 1999, 66, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Shangraw, R.E.; Jahoor, F. Mechanism of dichloroacetate-induced hypolactatemia in humans with or without cirrhosis. Metabolism 2004, 53, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.C.; Solayman, M.H.; Lorenzo, R.J.; Langaee, T.; Stacpoole, P.W.; James, M.O. GSTZ1 expression and chloride concentrations modulate sensitivity of cancer cells to dichloroacetate. Biochimica et Biophysica Acta (BBA)-General Subjects 2016, 1860, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Terado, T.; Tambe, Y.; Mukaisho, K.I.; Kageyama, S.; Kawauchi, A.; Inoue, H. Cryptotanshinone, a novel PDK 4 inhibitor, suppresses bladder cancer cell invasiveness via the mTOR/β-catenin/N-cadherin axis. International journal of oncology 2021, 59, 1–1. [Google Scholar] [CrossRef] [PubMed]
- Tambe, Y.; Terado, T.; Kim, C.J.; Mukaisho, K.; Yoshida, S.; Sugihara, H.; Tanaka, H.; Chida, J.; Kido, H.; Yamaji, K.; et al. Antitumor activity of potent pyruvate dehydrogenase kinase 4 inhibitors from plants in pancreatic cancer. Mol Carcinog. 58:1726–1737. 2019. [CrossRef]
- 468 Sestito, S.; Bacci, A.; Chiarugi, S.; Runfola, M.; Gado, F.; Margheritis, E.; Gul, S.; Riveiro, M.E.; Vazquez, R.; Huguet, S.; Manera, C. Development of potent dual PDK1/AurA kinase inhibitors for cancer therapy: Lead-optimization, structural insights, and ADME-Tox profile. European Journal of Medicinal Chemistry 2021, 226, 113895. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.; Yadav, V.K.; Kuo, K.T.; Pikatan, N.W.; Lin, C.S.; Chien, M.H.; Lee, W.H.; Hsiao, M.; Chiu, S.C.; Yeh, C.T.; Tsai, J.T. PDK1 inhibitor BX795 improves cisplatin and radio-efficacy in oral squamous cell carcinoma by downregulating the PDK1/CD47/Akt-mediated glycolysis signaling pathway. International journal of molecular sciences 2021, 22, 11492. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, C.; De Franco, M.; Carbone, D.; Bassani, D.; Pavan, M.; Cascioferro, S.; Parrino, B.; Cirrincione, G.; Dall’Acqua, S.; Moro, S.; Gandin, V. 1, 2, 4-Amino-triazine derivatives as pyruvate dehydrogenase kinase inhibitors: Synthesis and pharmacological evaluation. European Journal of Medicinal Chemistry 2023, 249, 115134. [Google Scholar] [CrossRef]
- Mitchel, J.; Bajaj, P.; Patil, K.; Gunnarson, A.; Pourchet, E.; Kim, Y.N.; Skolnick, J.; Pai, S.B. Computational identification of stearic acid as a potential PDK1 inhibitor and in vitro validation of stearic acid as colon cancer therapeutic in combination with 5-fluorouracil. Cancer Informatics. 2021 Dec;20:11769351211065979. [CrossRef]
- Chen, Q.; Han, H.; Lin, F.; Yang, L.; Feng, L.; Lai, X.; Wen, Z.; Yang, M.; Wang, C.; Ma, Y.; Yin, T. Novel shikonin derivatives suppress cell proliferation, migration and induce apoptosis in human triple-negative breast cancer cells via regulating PDK1/PDHC axis. Life Sciences 2022, 310, 121077. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Cenciarelli, C.; Hasan, A. Potential of antibody–drug conjugates (ADCs) for cancer therapy. Cancer Cell International 2022, 22, 1–2. [Google Scholar] [CrossRef]
- Wei, G.; Sun, J.; Luan, W.; Hou, Z.; Wang, S.; Cui, S.; Cheng, M.; Liu, Y. Natural product albiziabioside A conjugated with pyruvate dehydrogenase kinase inhibitor dichloroacetate to induce apoptosis-ferroptosis-M2-TAMs polarization for combined cancer therapy. Journal of medicinal chemistry 2019, 62, 8760–8772. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The first evidence of DCA anti-tumor effects and its mechanisms. Plas and Thompson have shown that glycolytic metabolism increased resistance to apoptosis [72].
Figure 1.
The first evidence of DCA anti-tumor effects and its mechanisms. Plas and Thompson have shown that glycolytic metabolism increased resistance to apoptosis [72].
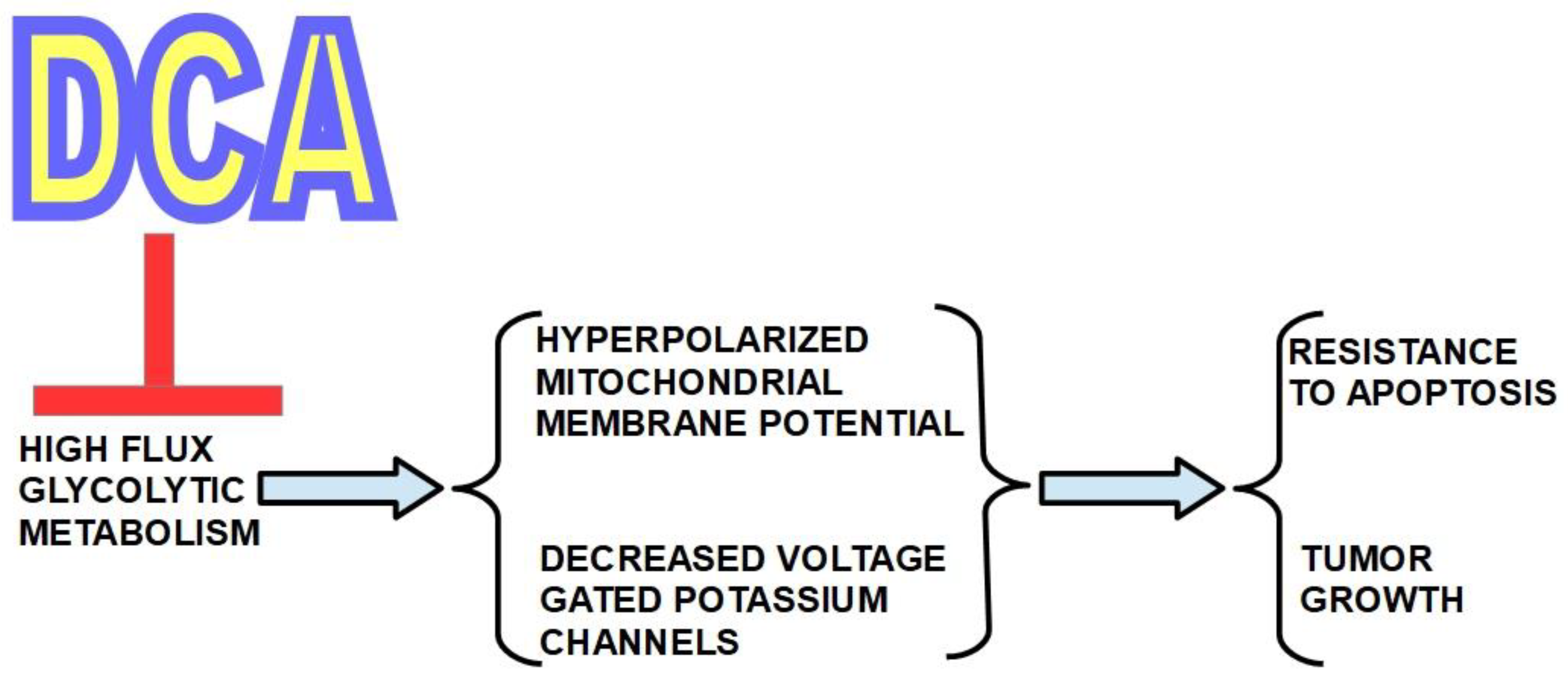
Figure 2.
DCA is sold over the counter and is produced by many laboratories. The quality of the product from these laboratories is not well established. These particulars explain many of the doubts about DCA’s real value as a therapeutic tool.
Figure 2.
DCA is sold over the counter and is produced by many laboratories. The quality of the product from these laboratories is not well established. These particulars explain many of the doubts about DCA’s real value as a therapeutic tool.

Figure 3.
Chemical structure of DCA, also known as dichloroethanoic acid. It is a halogenated carboxylic acid and a structural analog of pyruvate. Lower panel shows its metabolization to glyoxylate by the enzyme glutathione transferase zeta 1 (GSTZ1) also known as maleylacetoacetate isomerase (MAAI). GSTZ1 dechlorinates DCA and GSTZ1 genetic variations influence the metabolic rate of DCA processing [75]. Glyoxylate is oxidized to oxalate which is excreted by the urine. DCA has a high oral bioavailability [76].
Figure 3.
Chemical structure of DCA, also known as dichloroethanoic acid. It is a halogenated carboxylic acid and a structural analog of pyruvate. Lower panel shows its metabolization to glyoxylate by the enzyme glutathione transferase zeta 1 (GSTZ1) also known as maleylacetoacetate isomerase (MAAI). GSTZ1 dechlorinates DCA and GSTZ1 genetic variations influence the metabolic rate of DCA processing [75]. Glyoxylate is oxidized to oxalate which is excreted by the urine. DCA has a high oral bioavailability [76].
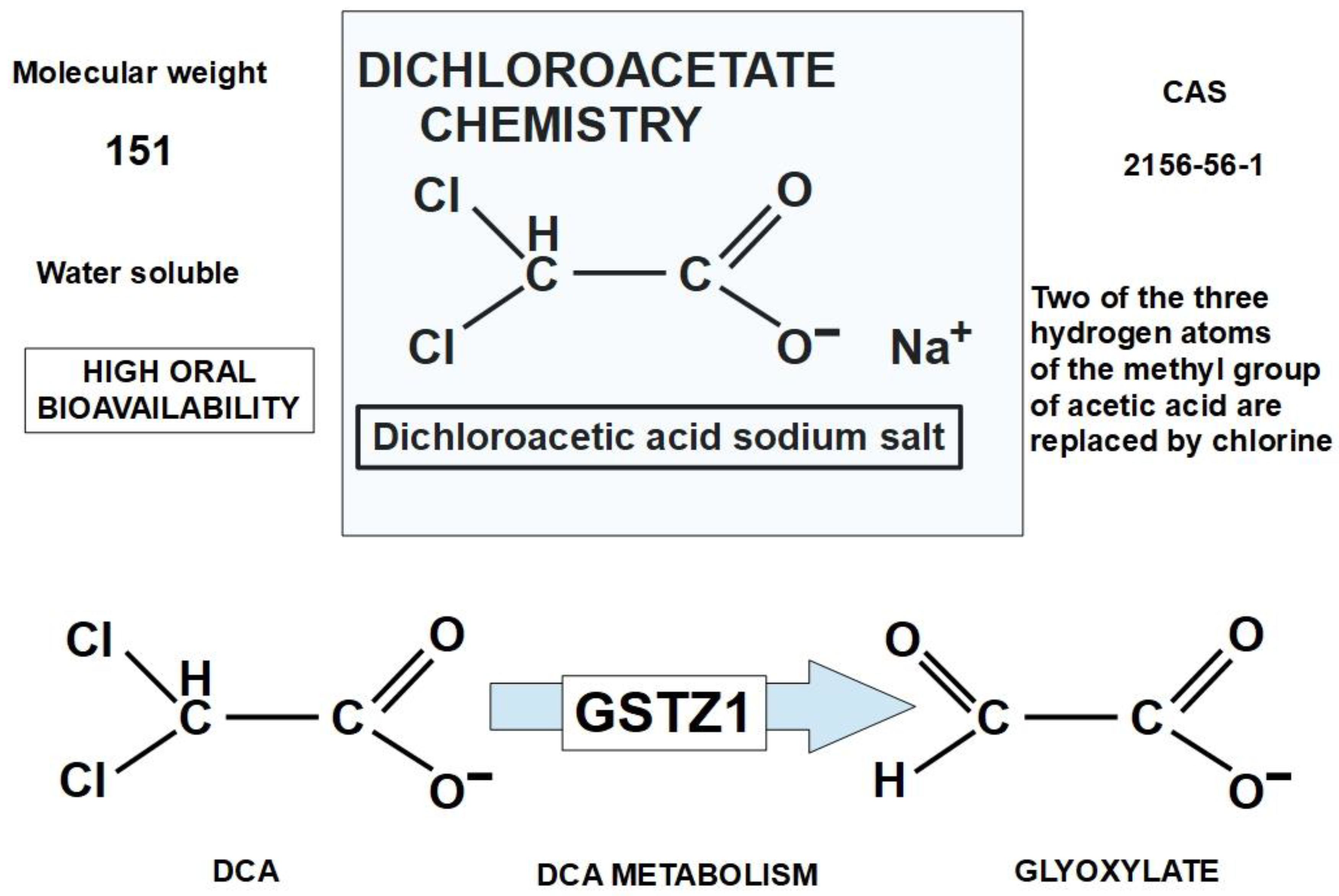
Figure 4.
The PDH enzymatic complex introduces pyruvate into the Krebs cycle after removing a molecule of CO2 and binding it to coenzyme A, forming acetyl-CoA.
Figure 4.
The PDH enzymatic complex introduces pyruvate into the Krebs cycle after removing a molecule of CO2 and binding it to coenzyme A, forming acetyl-CoA.
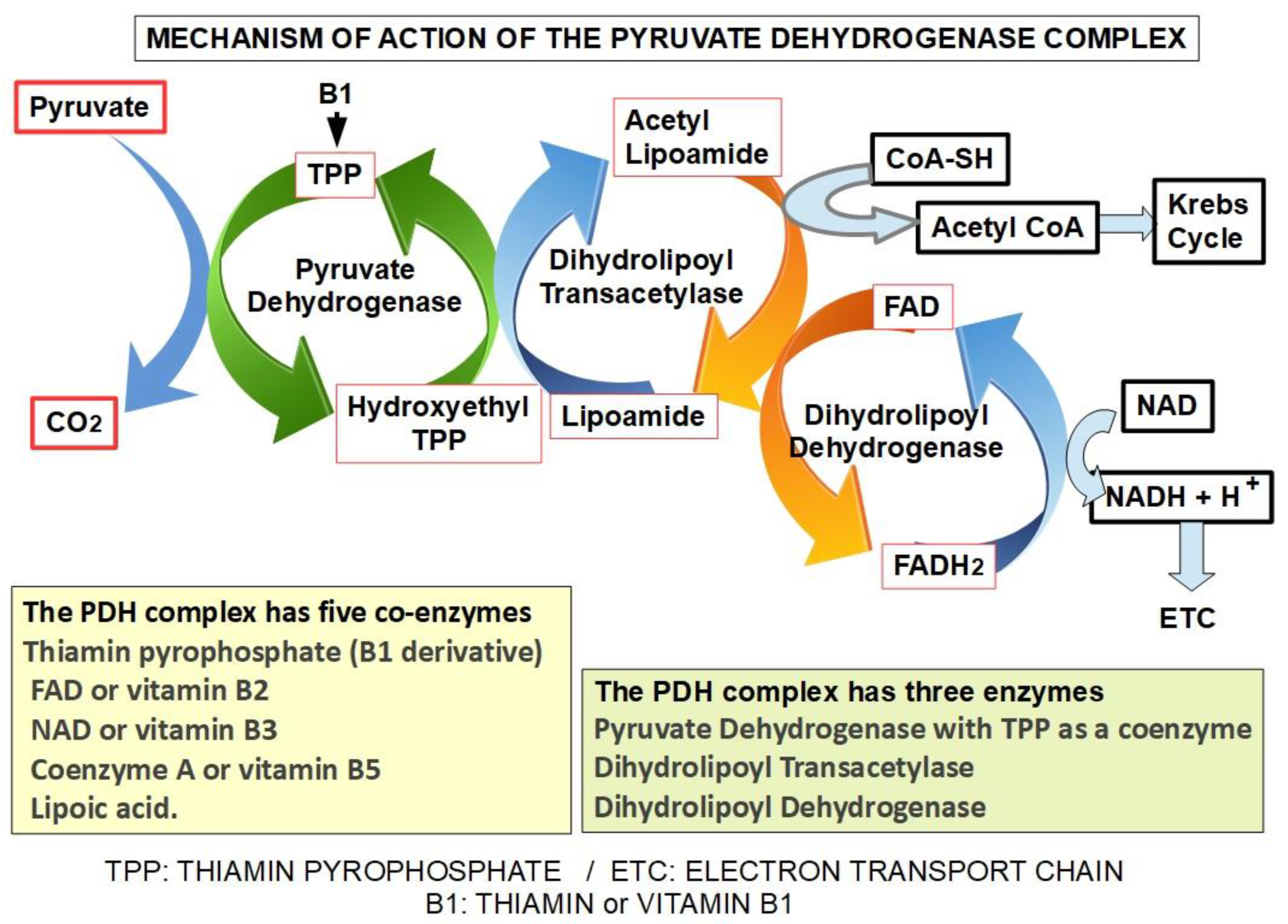
Figure 7.
Summary of DCA’s multiple pharmacological effects, many of which that stem from PDK inhibition. This includes effects on lipid and glucose metabolism, effects on cell stemness, angiogenesis and pH. The summary is based on references [119,120,121,122,123,124,125,126,127].
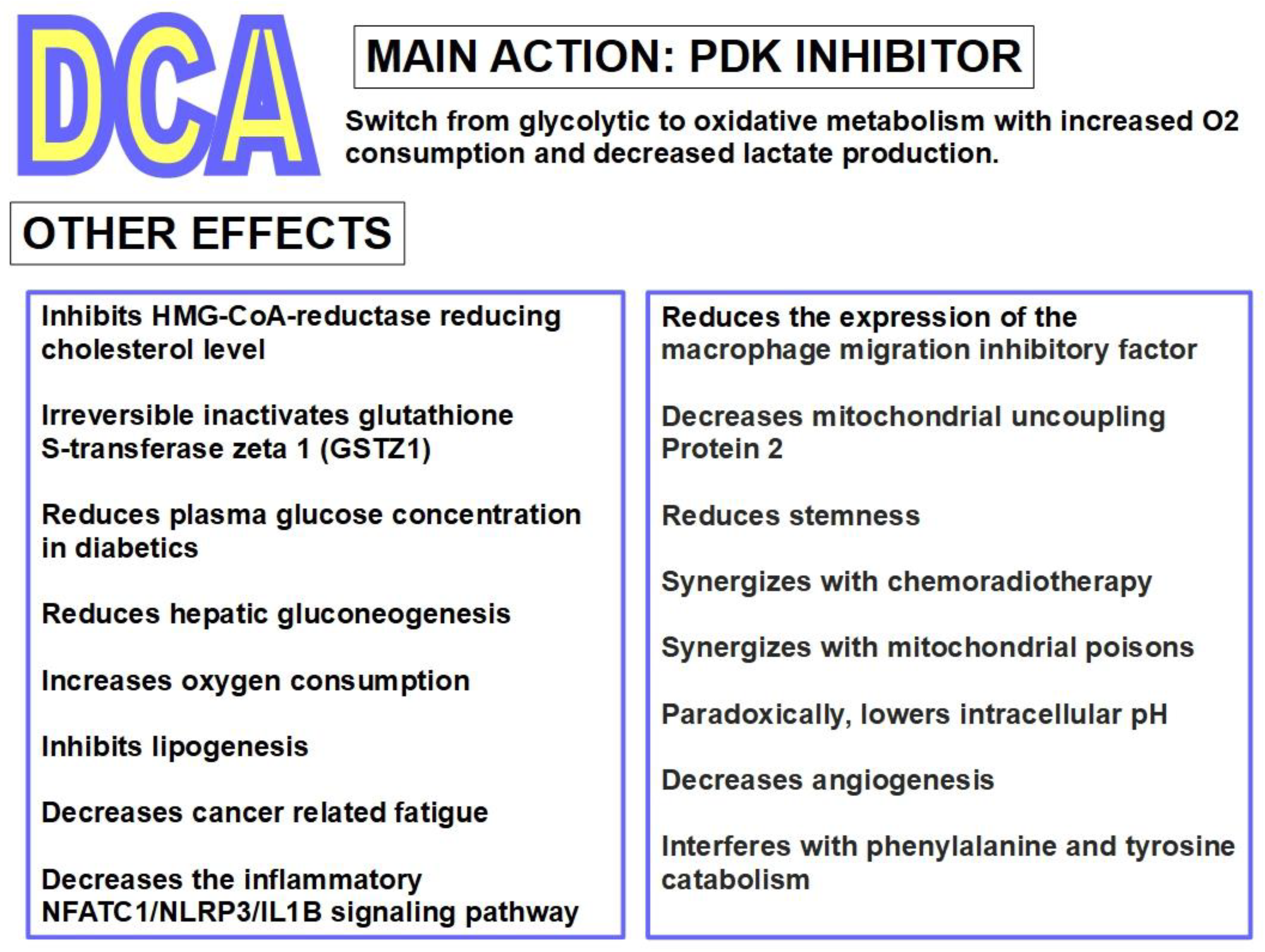
Figure 8.
Histone and protein lactylation in cancer and its prevention by DCA are facts supported by evidence [145]. However, there are still certain questions and unknowns. These are:.
Figure 8.
Histone and protein lactylation in cancer and its prevention by DCA are facts supported by evidence [145]. However, there are still certain questions and unknowns. These are:.
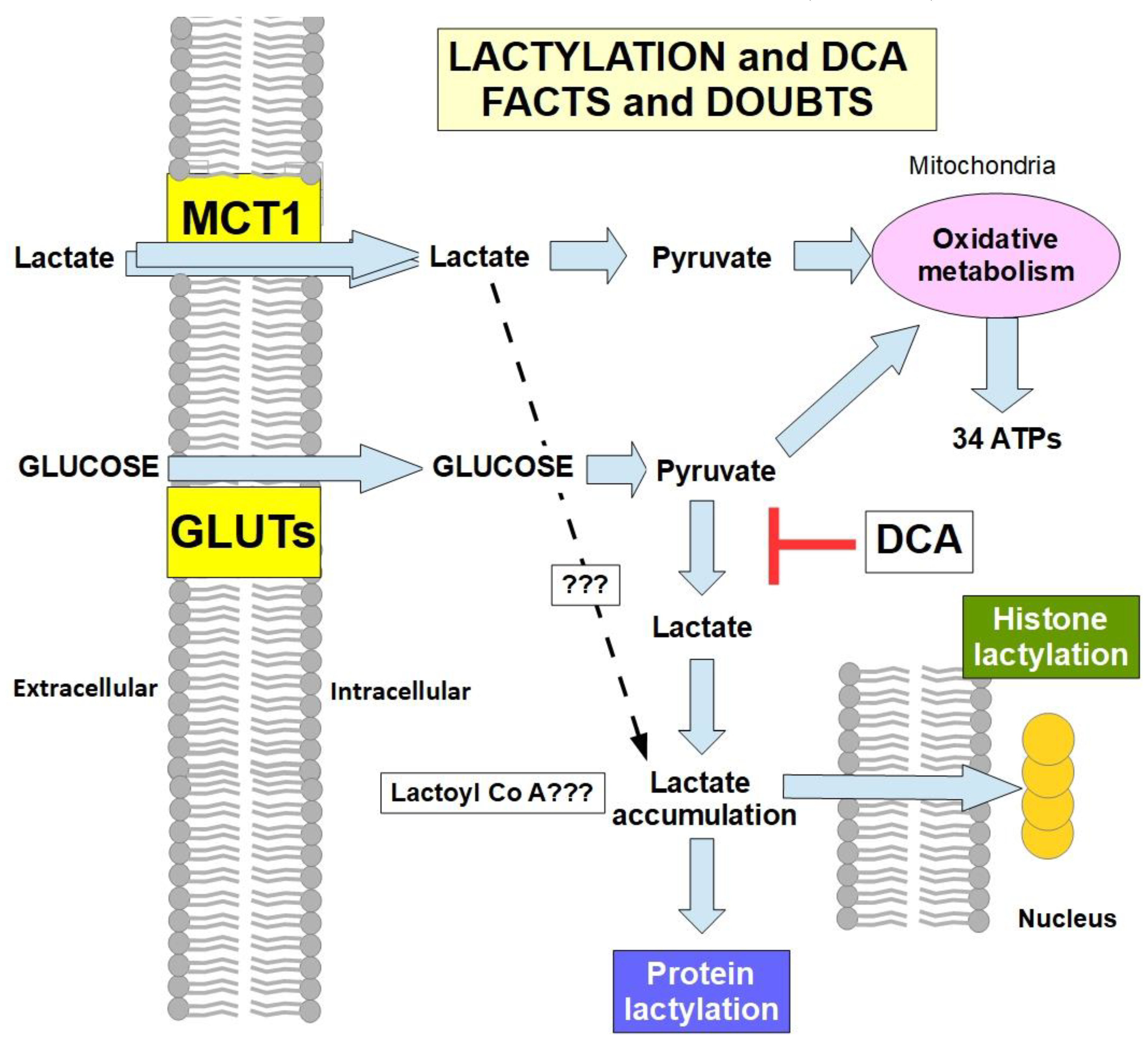
Figure 9.
DCA modifies glycolytic metabolism and decreases tumor growth in mice with NSCLC cell xenografts. DCA induced down-regulation of MIF seems to be an essential anti-tumor mechanism of this compound in this tumor type.
Figure 9.
DCA modifies glycolytic metabolism and decreases tumor growth in mice with NSCLC cell xenografts. DCA induced down-regulation of MIF seems to be an essential anti-tumor mechanism of this compound in this tumor type.
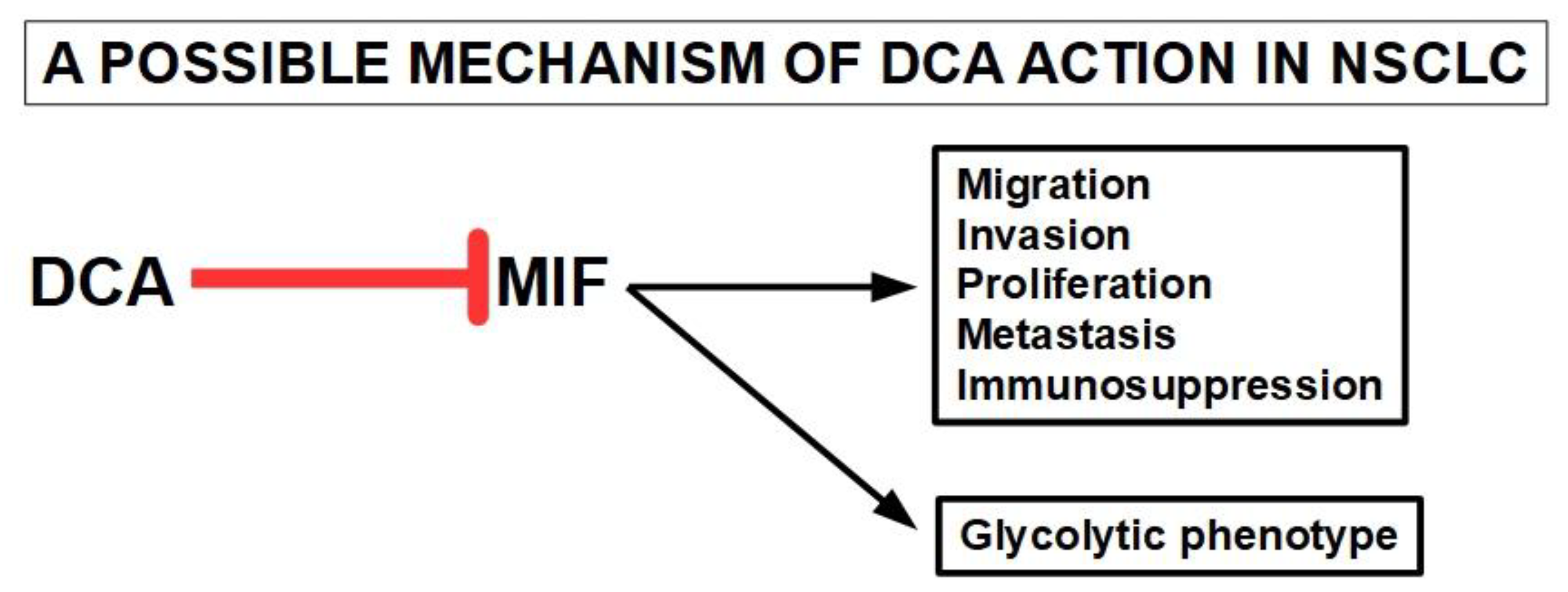
Figure 10.
Schematic diagram of pathways of activation of the most frequently found mutations in HNSCC and their pro-glycolytic pathways. Red boxes represent the average percentage of frequency of occurrence of these mutations. EGFR has the highest frequency and is the usual driver gene [284]. The tumor suppressor TP53 inactivating mutations deregulate its anti-glycolytic effects [285]. Notch signaling recruits hexokinase to the mitochondrial membrane promoting glycolysis. Inactivating mutations of Notch can also promote glycolysis by depressing oxidative phosphorylation [286,287,288].
Figure 10.
Schematic diagram of pathways of activation of the most frequently found mutations in HNSCC and their pro-glycolytic pathways. Red boxes represent the average percentage of frequency of occurrence of these mutations. EGFR has the highest frequency and is the usual driver gene [284]. The tumor suppressor TP53 inactivating mutations deregulate its anti-glycolytic effects [285]. Notch signaling recruits hexokinase to the mitochondrial membrane promoting glycolysis. Inactivating mutations of Notch can also promote glycolysis by depressing oxidative phosphorylation [286,287,288].
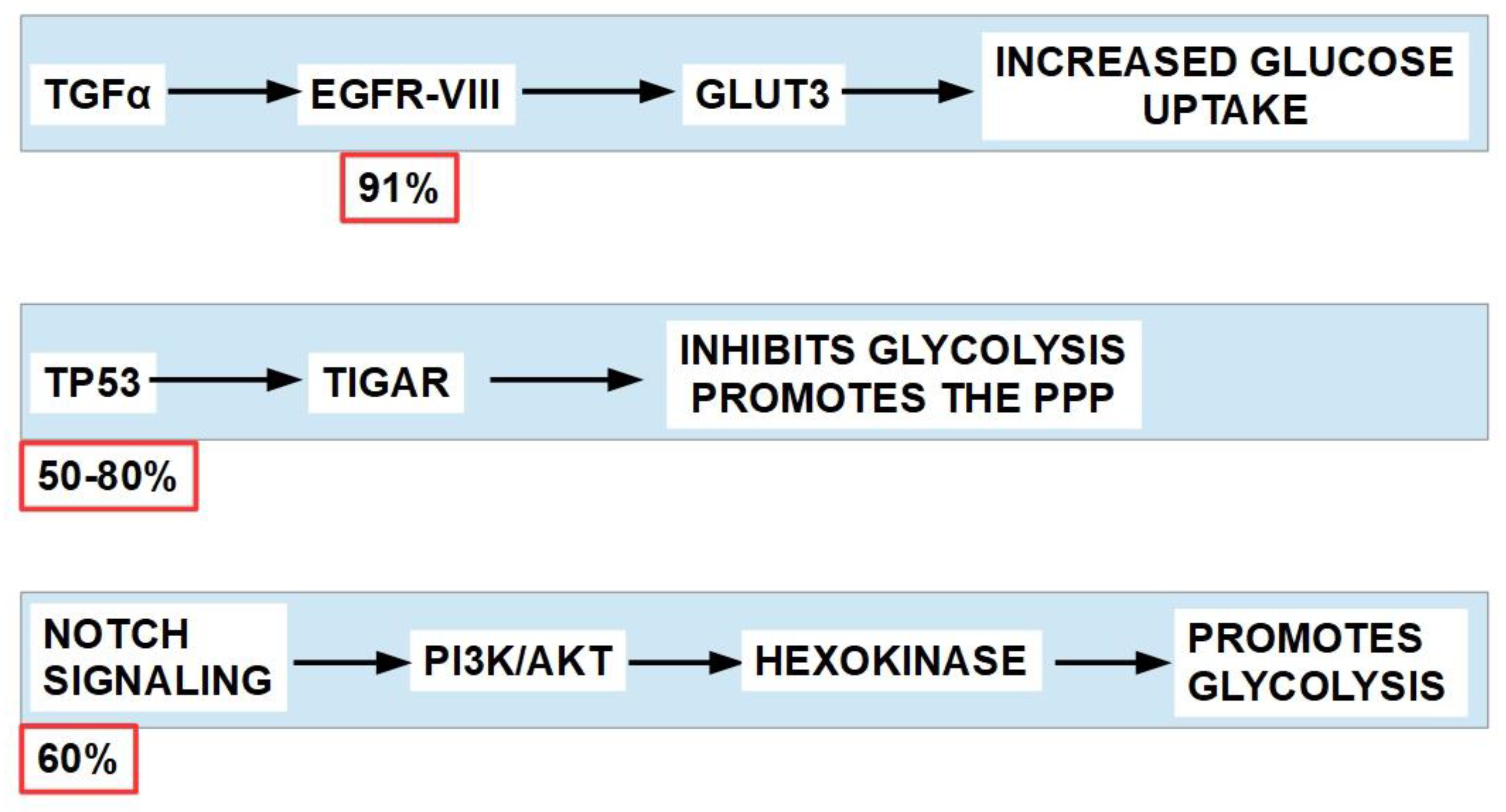
Figure 11.
Proposed structure of SMCT1 illustrating the compounds it can transport into the cell on the left side, and inhibitors of transport on the right.
Figure 11.
Proposed structure of SMCT1 illustrating the compounds it can transport into the cell on the left side, and inhibitors of transport on the right.
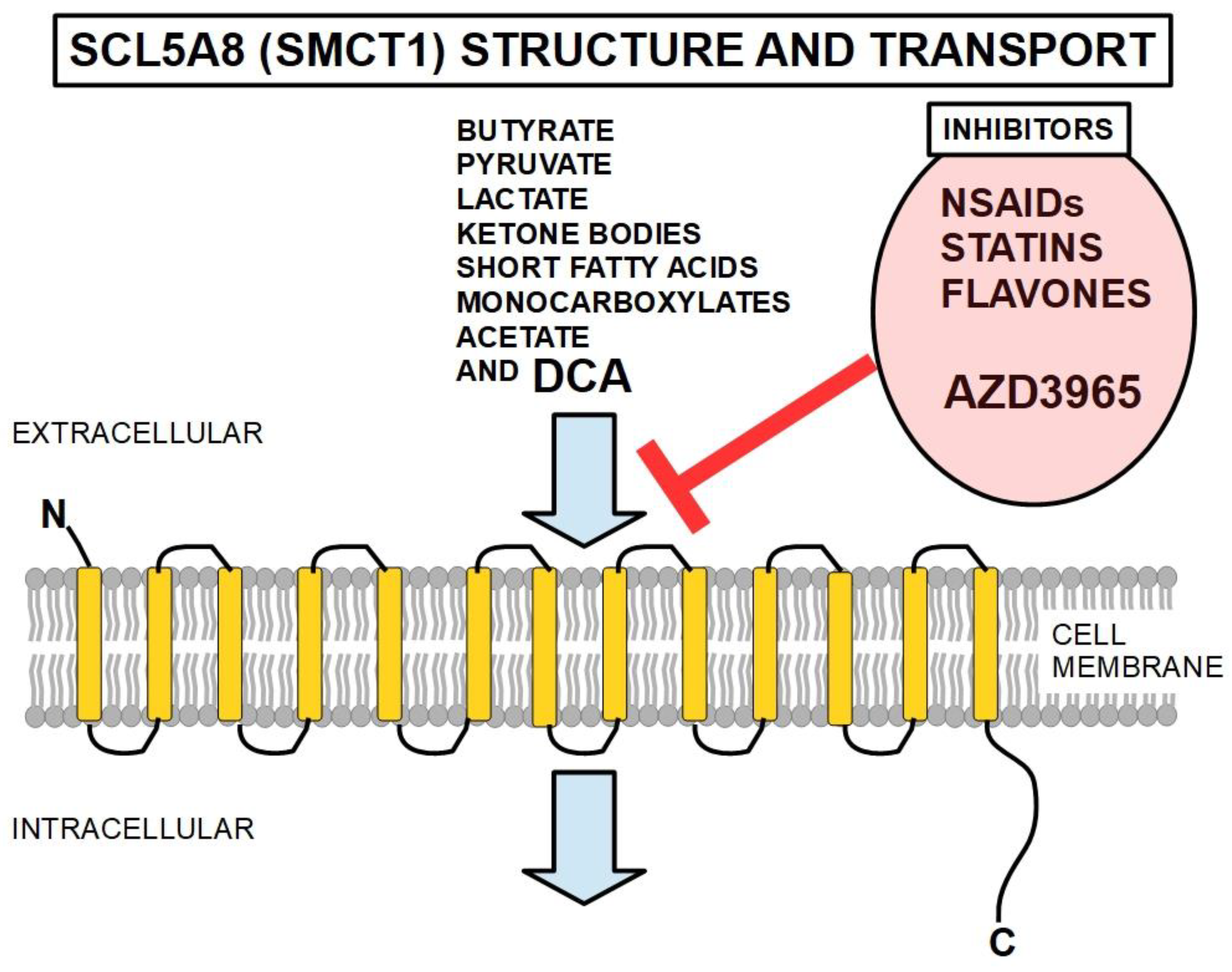
Figure 12.
Proposed mechanism by which inhibition of PDK and Complex I increase ROS and create an oxidative stress. Mitochondria release high rates of superoxide in the presence of the electron transport chain (ETC) inhibitors. The figure is based on references [339] and [340] but is controversial. According to Wheaton et al. [341] metformin does not increase the production of reactive oxygen species [342,343] while rotenone does. However, other research shows increased ROS production with metformin [344]. Therefore, the explanation for the synergy between DCA and metformin is still in question. According to other authors, metformin inhibits complex I through ubiquinone inhibition but stimulates ROS production through flavin in complex I [345].
Figure 12.
Proposed mechanism by which inhibition of PDK and Complex I increase ROS and create an oxidative stress. Mitochondria release high rates of superoxide in the presence of the electron transport chain (ETC) inhibitors. The figure is based on references [339] and [340] but is controversial. According to Wheaton et al. [341] metformin does not increase the production of reactive oxygen species [342,343] while rotenone does. However, other research shows increased ROS production with metformin [344]. Therefore, the explanation for the synergy between DCA and metformin is still in question. According to other authors, metformin inhibits complex I through ubiquinone inhibition but stimulates ROS production through flavin in complex I [345].
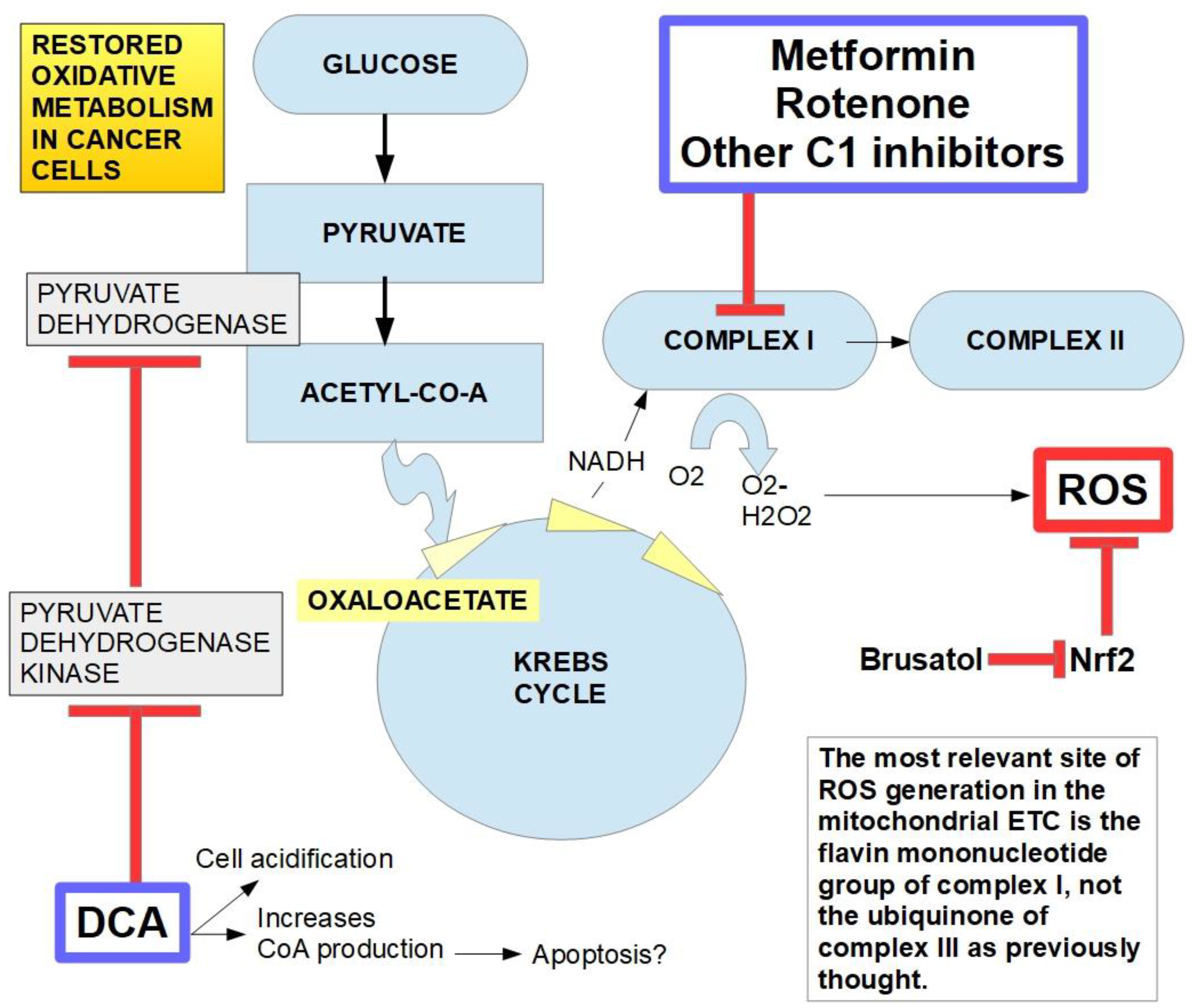
Figure 13.
Chemical formula of diisopropyl dichloroacetate.
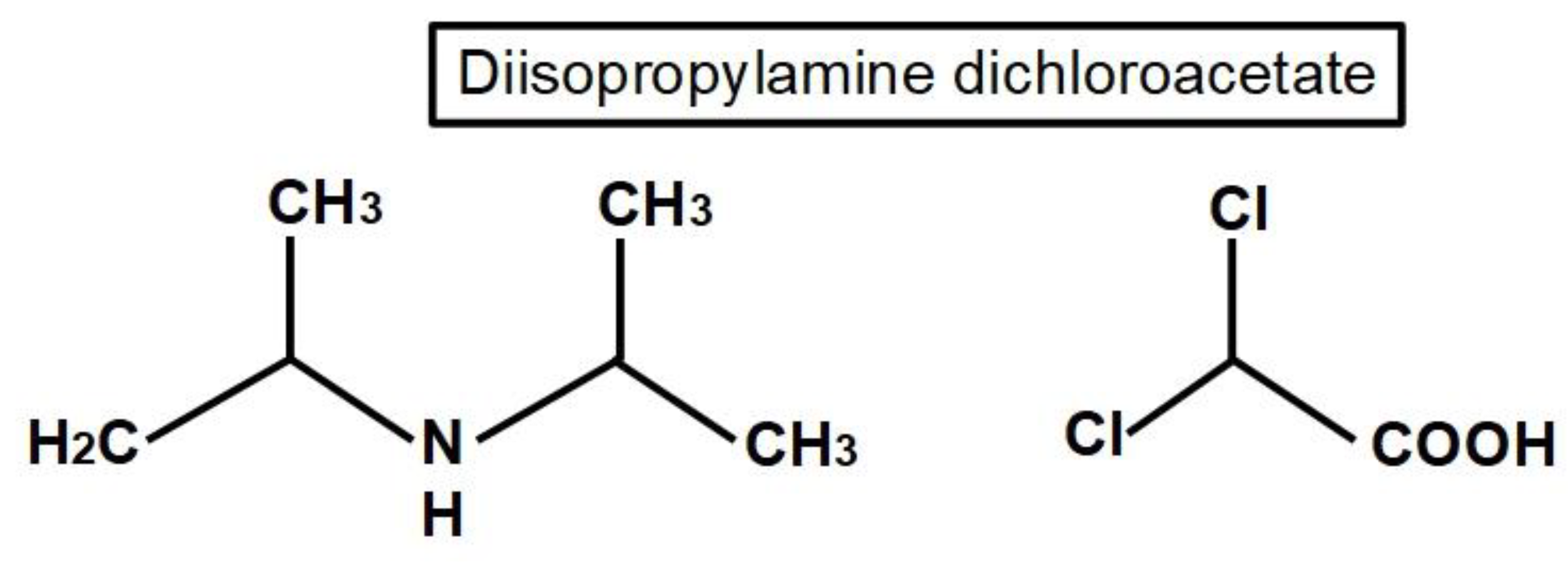
Figure 14.
Chemical structure of four different experimental DCA derivatives that have shown increased cytotoxic effects. The DCA moieties are circled.
Figure 14.
Chemical structure of four different experimental DCA derivatives that have shown increased cytotoxic effects. The DCA moieties are circled.
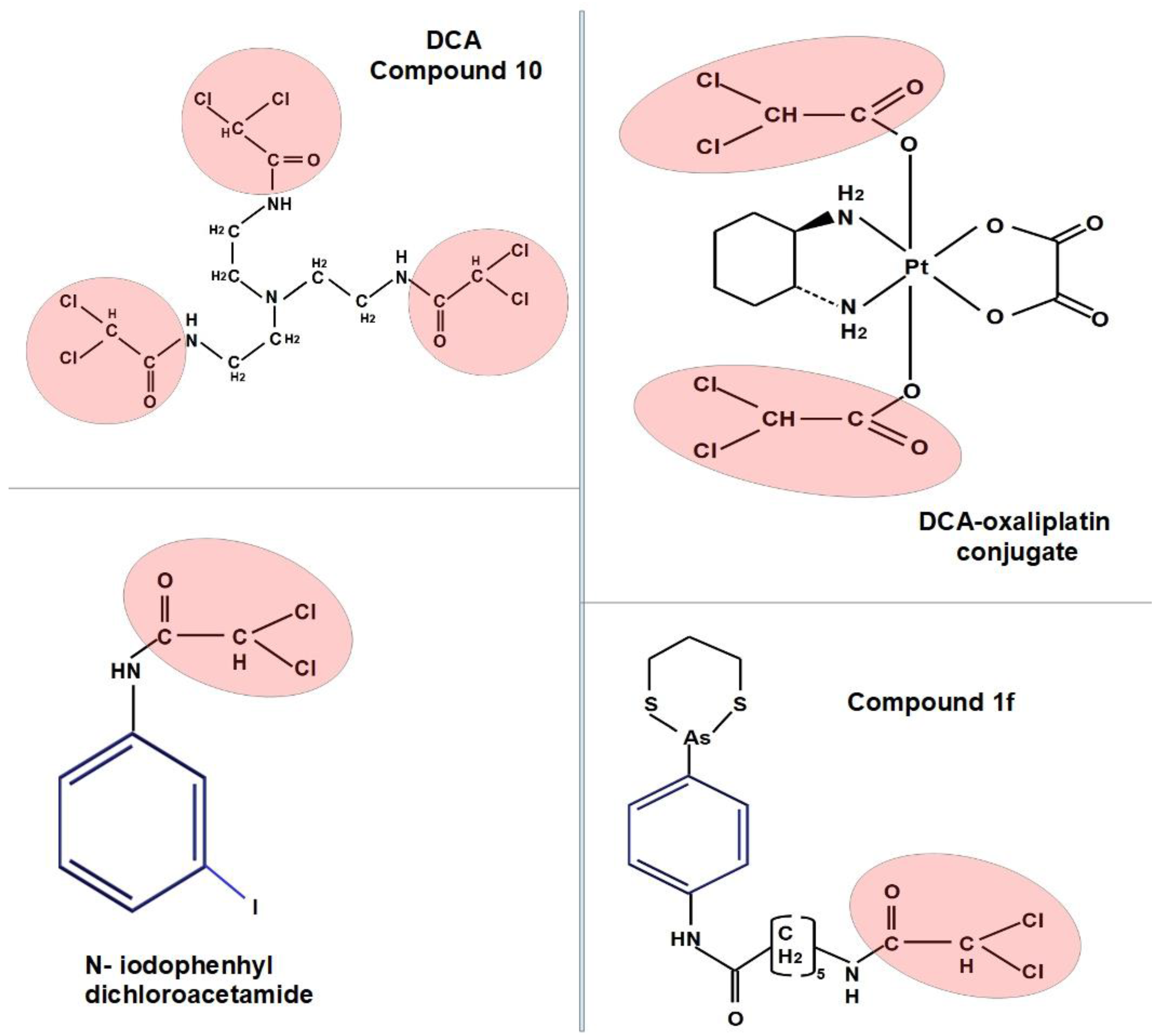
Figure 15.
The figure illustrates the maximum tolerable DCA concentration that can be reached safely in humans with administration of a high but safe dose (left line). The peak concentration can only be maintained for a short time. The few in vitro experiments that showed cytostatic effects were performed between 1 and 2 mM (center panel). Most in vitro experiments published in the literature were carried out with concentrations above 10 mM and up to 50 mM (right bar). These may have also shown cytotoxic effects.
Figure 15.
The figure illustrates the maximum tolerable DCA concentration that can be reached safely in humans with administration of a high but safe dose (left line). The peak concentration can only be maintained for a short time. The few in vitro experiments that showed cytostatic effects were performed between 1 and 2 mM (center panel). Most in vitro experiments published in the literature were carried out with concentrations above 10 mM and up to 50 mM (right bar). These may have also shown cytotoxic effects.
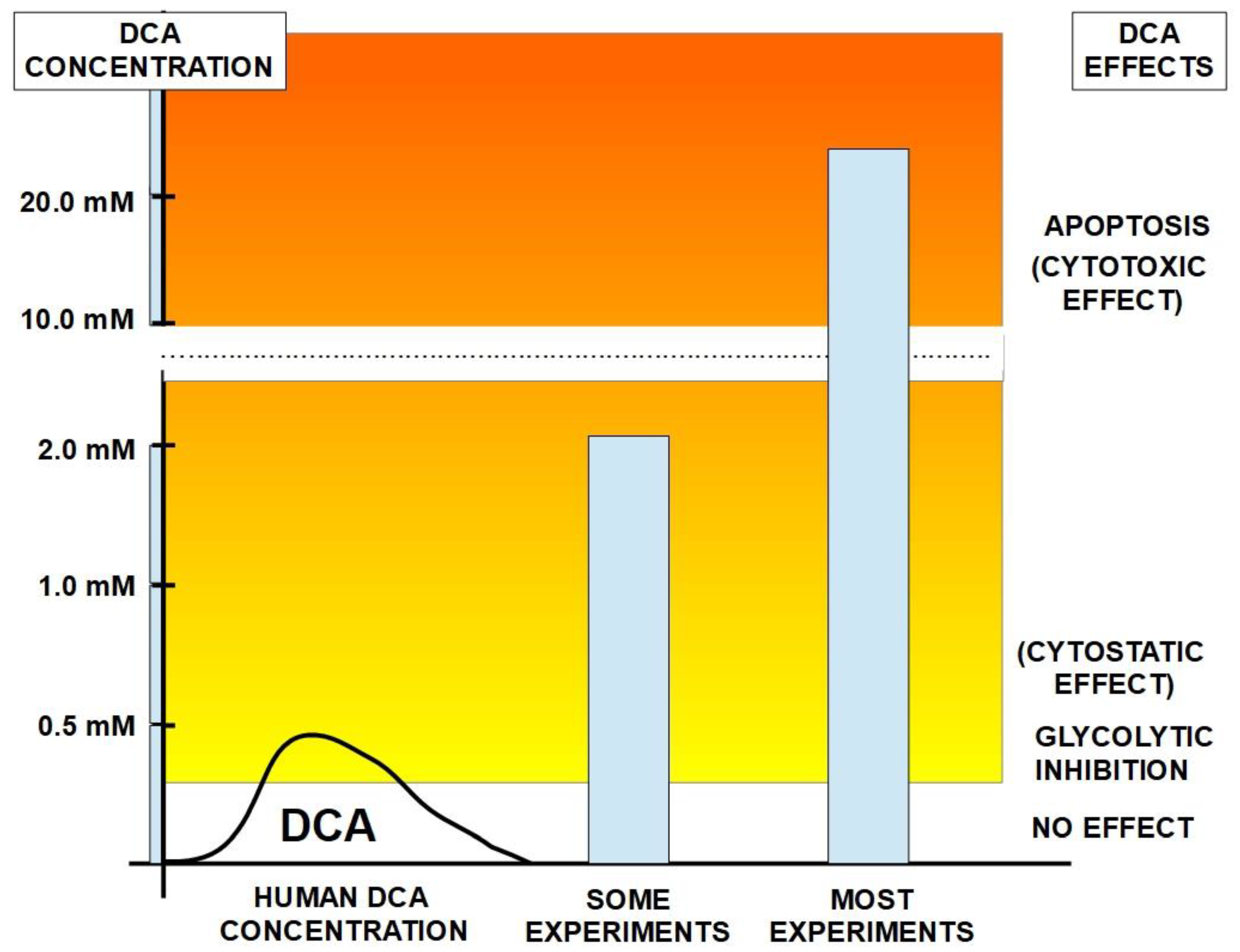

Table 1.
Glioblastoma – retinoblastoma.
| Reference | Findings |
|---|---|
| Michelakis ED et al, 2010 [209] | The authors tested whether DCA can reverse cancer-specific metabolic and mitochondrial remodeling in glioblastoma. Freshly isolated glioblastomas from 49 patients showed mitochondrial hyperpolarization, which was rapidly reversed by DCA. A separate experiment with five patients had glioblastoma treated with oral DCA for up to 15 months. Three patients showed evidence of tumor regression. DCA depolarized mitochondria, increased mitochondrial reactive oxygen species, and induced apoptosis in GBM cells, as well as in putative GBM stem cells. DCA therapy also inhibited the hypoxia-inducible factor–1α, promoted p53 activation, and suppressed angiogenesis both in vivo and in vitro. |
| Duan, Y.; et al. 2013 [210] | DCA inhibited cell proliferation, induced apoptosis, and arrested C6 glioma cells in S phase. In vivo antitumor testing indicated that DCA markedly inhibited the growth of glioma tumors in brain tumor-bearing rats and tumor-bearing nude mice. DCA significantly induced the ROS production and decreased the mitochondrial membrane potential in tumor tissues. Antiangiogenic effects were also found. |
| Kumar K et al. 2013 [211] | DCA synergistically enhanced the results of bevacizumab antiangiogenic treatment in a xenotransplant mouse model of glioblastoma. |
| Morfouace, M. et al. 2014 [212] | Oct4 is a major regulator of cell pluripotency. DCA increased the amount of Oct4-pyruvate kinase 2 complexes which inhibited Oct4- dependent gene expression, inducing differentiation of glioma stem cells. |
| Kolesnik DL et al. 2014 [213] | Hypoxia enhanced the cytotoxic effects of DCA in glioblastoma cells inducing necrosis of tumor cells. |
| Vella et al. 2012 [214] | DCA had anticancer effects on NB (neuroblastoma) tumor cells which was selectively directed to very malignant NB cells. More differentiated/less malignant NB cells were refractory to DCA treatment. |
| Sradhanjali S et al. 2017 [215] | DCA reduced retinoblastoma cell line and retinoblastoma explant growth and potentiated carboplatin inhibition of retinoblastoma cell growth. |
| Park, J.M.; et al. 2013 [216] | C6 glioma was transplanted into rat brains. Magnetic Resonance imaging in vivo showed that DCA modified PDH flux much more in gliomas compared to normal brains. |
| Fedorchuk, A.G.; et al. 2016 [217] | Experiments with rats with transplanted C6 glioma cells showed that different administration schedules of DCA could cause ambiguous effects: inhibition of tumor growth or stimulation. Prolonged daily administration showed the best antitumor effects. Under hypoxic conditions, anticancer efficacy of DCA was significantly increased. |
| Wicks, R.T.; et al. 2014 [218] | Local delivery (wafers) of DCA to experimental brain tumors in rats caused a significantly increased survival compared with controls and with oral administration of the drug. |
| Korsakova, et al. 2021 [219] | The authors found synergistic apoptotic effects of metformin and DCA in glioblastomas cells in vitro and in vivo with dose dependent cytotoxicity. Cytotoxic activity required DCA concentrations above 10 mM and metformin 5 mM. There were no effects with concentration of 5 mM DCA and 2.5 mM metformin. |
| Shen, H.; et al. 2015 [220] | DCA increased radiosensitivity in orthotopic glioblastoma-bearing mice and of high-grade gliomas. Also Reviewed in Cook, 2021 [221] |
| Jiang, W.; et al. 2016 [222] | DCA increased cytotoxic activity of phenformin on glioblastoma stem cells. |
| Prokorhova, I.V.; et al. 2018 [223] | Co-administration of metformin with DCA improved anemia and thrombocytopenia, produced by glioma C6 growth. |
| Kolesnik, D.L.; et al. 2019 [224] | It was found that metformin increased the cytotoxic activity of DCA against C6 glioma cells, in vitro and in vivo. |
| Shen, H.; et al. 2021 [225] | DCA associated with metformin and radiotherapy increased apoptosis in pediatric glioma cells in vitro and in vivo. A triple combination of DCA, metformin and radiation therapy had more potent effects. |
| Yang, Z.; et al. 2021 [226] | Phosphorylated PDH-A1 mediated tumor necrosis factor-α (TNF-α) -induced cell migration in glioma cells. DCA decreased phosphorylated PDH-A1 levels and reduced glioma cell migration and invasion. |
Table 2.
Other tumors.
| References | Organ/Cell | Findings |
|---|---|---|
| Ohashi T et al. 2013 [314] | Macrophages | DCA targets macrophages to suppress activation of the IL-23/IL-17 pathway and prevents the expression of arginase 1 (ARG1) induced by lactic acid. Lactic acid-pretreated macrophages inhibited CD8+T-cell proliferation, but CD8+T-cell proliferation was restored when macrophages were pretreated with lactic acid and DCA. Although DCA treatment alone did not suppress tumor growth, it increased antitumor immunotherapeutic activity. |
| Rooke M et al. 2014 [315] | Sarcoma models in vitro and in vivo. | Three types of cells (mouse fibrosarcoma S180, mouse osteosarcoma K7M2 and human fibrosarcoma HT1080-luc2) were tested with DCA with and without doxorubicin. DCA alone significantly decreased viability at a concentration of 5 mM. At 0.5 mM viability only decreased 15%. Importantly, there were no signs of apoptosis, therefore DCA inhibited proliferation but did not induce apoptosis. DCA had additive effects with low concentrations of doxorubicin. |
| Ishiguro T et al. 2012 [316] | Fibrosarcoma and colon cancer cells, normal human fibroblasts. | The combination of DCA and omeprazol exhibited more potent antitumor activity than DCA alone in tumor cells and did not affect the proliferation of normal cells. Caspase-dependent apoptosis through superoxide production was the suggested the mechanism of action. |
| Sutendra, G.; et al. 2013 [317] | NSCLC and 2335 mammary cells | In a rat xenotransplanted model, DCA reduced NSCLC and breast cancer tumor vascularity in vivo. DCA also inhibited HIF-1 α in tumor cell lines. |
| El Sayed SM et al. 2019 [318] | Cancer cells | The authors propose that DCA is an antagonist to acetate competing for enzymes, thus its therapeutic action would be caused by acetate deprivation. |
| Khyzhnyak, S.V. et al. 2014 [319] | Mouse sarcoma | Examination of sarcoma mitochondria from mice under DCA treatment showed they had a decrease in lactic acid levels and an increase in PDH activity with a decrease in the electron system transport activity and an increase in ROS. |
| Choi YW et al. 2014 [320] | HeLa cells | DCA and metformin acted synergistically enhancing cytotoxicity on cancer cells. |
| Xuan Y et al. 2014 [321] | Gastric cancer cells | DCA decreased resistance to 5-fluorouracil in highly hypoxic gastric cancer cells. |
| Badr MM et al. 2014 [322] | Fibrosarcoma | DCA showed immunomodulatory effects through the interleukin 12-interferon γ pathway. |
| Jin, J. et al. 2022 [323] | Osteosarcoma cell lines | DCA mitochondrial targeting micelles induced pyroptosis increasing the effects of immunotherapy. |
| Lam, S.K.; et al, 2022 [324] | Mesothelioma | The combination of DCA with niclosamide blocked growth and proliferation of different mesothelioma cells in vitro and in vivo. |
| Qin H et al. 2023 [325] | Cholangiocarcinoma | DCA increased sensitivity to cisplatin, changing glycolytic metabolism to an oxidative one and increasing ROS. Chloroquine further increased this sensitivity. |
Table 3.
[86].
Table 3.
[86].
| Dose in Humans | Concentration Range | Peak Concentration | |
|---|---|---|---|
| IV 10 mg/kg | 19.9 µg/ml | 24.7 µg/ml | 0.16 mM |
| IV 20 mg/kg | 57.3 µg/ml | 74.9 µg/ml | 0.49 mM |
| Oral 25 mg/kg | 41 µg/ml (0.27mM)(slow metabolizer) | ||
| Oral 25 mg/kg | 30 µg/ml (0.20 mM)(quick metabolizer) | ||
| Other sources | |||
| IV 25 mg/kg | 130 µg/ml (0.86 mM) | ||
| After 5 infusions | 163 µg/ml (1.08 mM) | ||
| DOSE IN RATS | |||
| 100 mg/Kg | 120 µg/ml | 164 µg/ml | 1.08 mM |
| DOSE IN DOGS | |||
| 100 mg/Kg | 447 µg/ml | 508 µg/ml | 3.36 mM |
Note: peak concentrations only last for a very short time because the half life of DCA in plasma is short. Therefore, average concentrations are far lower than peak concentrations.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



