Submitted:
22 April 2024
Posted:
23 April 2024
You are already at the latest version
Abstract
Human papillomavirus (HPV) uses the cellular trafficking machinery to reach the cell nucleus and initiate a successful infection. The HPV capsid consists of two proteins: L1 (the major capsid protein) and L2 (minor capsid protein), and L2 is known to be responsible for trafficking the viral genome to the nucleus. From previous studies, we know that many cellular proteins interact with L2 and are involved in the trafficking process at different stages of the infectious entry process. To further investigate how HPV-16 achieves successful viral entry, we have performed a mass spectrometry analysis to identify novel interacting proteins that associate with incoming HPV-16 virions. We show that HPV-16 pseudovirions interact with a group of cellular cargo proteins (COPI), and here we confirm that this is mediated by the direct binding of L2, but not L1, to the COPB1 subunit. We also show that the ability of L2 to interact with COPB1 is conserved in both low- and high-risk HPV types. Furthermore, we also show that the interaction is essential for successful infection entry of multiple HPV types, thereby identifying a novel constituent of the infectious entry pathway of the HPV.
Keywords:
HPV
; virus entry
; COPI
1. Introduction
Human papillomavirus (HPV) is a small, non-enveloped DNA virus that infects skin or mucosal cells. The circular, double-stranded viral genome is approximately 8 kb in length. It is estimated that 20% of human cancers are caused by infections, and that approximately 12% of the total human cancer burden has a viral association [1]. HPV causes 5% of all human cancers and approximately 100% of all cervical cancers [2,3]. HPV is also responsible for a very high percentage of other anogenital cancers, and up to 70% of head-and-neck cancers, which includes those of the oral cavity, oropharynx, sinus, tonsil, and larynx [4,5]. Overall, over 600,000 new cancer cases every year are caused by high-risk HPV types, with types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 68 being defined by WHO as cancer-causing in the cervix [6]. In addition, HPV infection is the most common viral sexually transmitted infection worldwide [7,8,9].
HPV virions are composed of two structural proteins L1 and L2, both of which are required for a successful viral entry, L1 is required for cell attachment and L2 is required for successful viral genome trafficking to the cell nucleus [10,11,12]. From previous studies we know that L1 is the major capsid protein [13] and L2 is the minor capsid proteins [14,15], Previous research has shown that HPV can enter cells through clathrin-dependent or non-clathrin-dependent pathways [16,17]; it then travels to the early endosome, and from the early endosome to the late endosome [18]. During the acidification process the viral capsid begins a process of disassembly [19] and this allows a small portion of L2 to become exposed and span the endosomal membrance into the cytosol [20]. This exposure of L2 sequences allows the viral protein to recruit various cargo sorting components [21]. This in turn promotes endosomal tubulation and trafficking of the L2 DNA complex away from the late endosome [22,23], whilst the majority of L1 is left behind and is subsequently degraded within the lysozome [24,25]. During this process the L2/DNA complex is transported to the trans-Golgi network (TGN) [26,27], and stays there until mitosis [28,29], when the nuclear envelope breaks down and thereby allows access to nuclear components and subsequent nuclear retention [25,30]. Whilst a great deal is already known about the different cellular partners which facilitate this entry process and trafficking steps within the cell, such as SNX17 [31], CCT-3 [32], SNX27 and proteins of the retromer machinery [12,33], we were nonetheless interested in investigating other possible L2 interacting partners which may play a role in the infectious entry process. To do this we performed a proteomic analysis of L2 interacting proteins. Amongst these, we found that, we have found that COPB1, a component of the COP1 cargo protein complex is an interacting partner of L2 during HPV infectious entry; furthermore HPV requires. We have found that HPV requires COPI cargo proteins for a successful infection.
As from previous studies we know that there are two types of COP protein COPI and COPII [34,35,36], they are composed of five subunits COPI is composed of (α-COP), (β’-COP), (ε-COP (β-COP), (γ-COP), (δ-COP), (ζ-COP) and COPII is composed of Sar1, Sec23, Sec24, Sec13, and Sec31 [34,37], each subunit perform a specific function [38], COPII proteins transport newly synthesized proteins from ER endoplasmic reticulum to the TGN, [39,40,41] While COPI proteins transports recycling proteins from cis Golgi networks to the ER [42,43], Recently, it has also been shown that COPI protein is involved in trafficking of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [44,45] and Nairoviruses [46]. Basically both COP proteins are required for transportation of large molecules from one enclosed compartment to another, formation of membrane domains within the cells [47,48], being large transport carrier proteins, their connection with the cytoskeleton and interaction with the target organelles are the basic functions of COP proteins [49,50,51].
2. Materials and Methods
2.1. Mass Spectrometry Analysis
FLAG-HA-tagged HPV-16 L2 expression plasmid or, as a control, empty plasmid were transfected into HEK293 cells. After 48 hours, cell extracts were produced using mass spectrometry lysis buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 50 mM NaF, 1 mM EDTA, 0.5% NP-40), and they were then incubated for one hour at 4°C on a rotating wheel with EZview Red Anti-HA affinity gel beads (Sigma). The beads were washed using phosphate-buffered saline (PBS) after first being washed with wash buffer (25 mM HEPES [pH 7.0], 0.1% NP-40, and 150 mM NaCl), and then proteome analysis was performed as previously described [52]
In short, 50 ng of sequencing-grade trypsin in 20 mM triethanolamine bicarbonate (pH 8.5) was used for 12 hours at room temperature to elute proteins from the affinity beads. Following digestion, the beads’ supernatant was extracted, and they were then quickly incubated with another aliquot of 20 mM triethanolamine bicarbonate (pH 8.5). This aliquot was then extracted and combined with the first supernatant. Formic acid was added to 0.1% to halt the reactions. The resultant mixture was then desalted using C18 STAGE (Stop and Go Extraction) tips and lyophilized until it was completely dry. Nanobore columns were constructed using in-house constructed columns packed with 15 cm of 3-μm Ascentis RPA particles (Sigma) using a high-pressure column loader. After being desalted, the samples were reconstituted in 0.1% formic acid and put onto the column. A discontinuous gradient of 0% to 80% acetonitrile in 0.1% formic acid was used to create the column 60 minutes and poured straight into the Amazon ETD ion trap mass spectrometer’s opening (Bruker). During the liquid chromatography (LC) separation, eight data-dependent tandem mass spectrometry (MS/MS) scans were conducted in succession, after one complete scan (375 to 1,700 m/z). Using the Global Proteome Machine interfaced with X!Tandem, raw data files from the Amazon ETD instrument were converted to mgf files by a data analysis software package (Bruker). Then, the files were searched against the Ensembl human protein database and the NCBInr Viral database. The matches were filtered at a 1% false discovery rate.
2.2. Cell Culture and Transfections
The human embryonic kidney cells (HEK293 and HEK293TT-21), and human skin keratinocytes (HaCaT) were cultured in Dulbecco’s modified Eagle medium, supplemented with 10% foetal bovine serum, 100 ug/ml penicillin-streptomycin, and 300 mg/ml glutamine.
HEK293 and HEK293TT cells were transfected using the calcium phosphate precipitation technique (22). For siRNA experiments, HaCaT cells were transfected using Lipofectamine RNAiMAX (Invitrogen) with an ON-TARGETplus SMARTpool of siRNAs against COPB1 (Dharmacon): (5′-CCAAGAUUGCAUUGCGCUA), (5′-CAUAUAAGAAUUCGUGCAA), (5′-AUUAUUAAGGAGAGCGACA), and (5′-CCUCAUGACUUCGCAAAUA). In parallel, a scrambled siSTABLE nontargeting siRNA (Dharmacon) was employed as a control.
2.3. Antibodies
The following primary antibodies were employed in this study: Mouse anti-HPV-16 L1 ; Santa Cruz), Cat #sc-47699) 1:1000 dilutions, with rabbit anti-COPB1 (Abcam) Cat # ab205020]1:1500 dilutions, In Western blotting tests, the secondary antibodies horseradish peroxidase-conjugated swine anti-rabbit (Cat #PO399), and rabbit anti-mouse (Dako), Cat #DK25 (Dako) 1:5000 dilutions, as well as the anti-HA mouse (Santa Cruz, Cat # sc-7392), 1:2000 dilution), HA rabbit (Thermofisher, Cat #MA5-27915), 1:2000 dilution, were used.
2.4. Plasmids
The plasmids expressing FLAG-HA-tagged HPV-16 L2 have been previously described [22], as have the expression vectors for the HPV-16 L1 and HPV-16 L2 full-length GST fusion proteins [52]. DNA sequencing was used to authenticate the plasmids produced by the GENEART site-directed mutagenesis system (Invitrogen) for the production of shortened versions of GST-tagged HPV-16 L2, which were made using custom-designed oligonucleotides (Eurofins MWG). HPV-16 L1 and L2 were expressed in a codon-optimized bicistronic fashion using plasmid p16shell L2-3×FLAG-HA, in which L2 is tagged with FLAG and hemagglutinin (HA) tags [53]. The firefly luciferase gene-carrying pGL3 Luci construct was obtained from Promega. Genscript was used to create the murine Papilloma virus L2 protein construct. The plasmids expressing HPV-2, HPV-5, BPV-1, SfPV-1, Mus musculus papillomavirus type 1 (MmuPV-1), and Merkel Cell Polyoma Virus (MCPyV) capsid proteins were the gift of Dr Chris B. Buck; HPV-16 and HPV-31 capsid protein expression plasmids were the gift of Dr Michelle A. Ozbun; HPV-18 capsid protein expression plasmid was the gift of Dr Samuel K. Campos, the GST BPV-1 L2 construct was the gift of Dr Patricio I. Meneses, and the GST HPV-11 L2 construct was the gift of Dr Bob Garcea, all of which were sequenced for verification.
2.5. Fusion Protein Purification and Binding Assays
As previously reported [54], GST-tagged fusion proteins or GST alone were produced, purified, and immobilized on glutathione agarose (Sigma). After thorough washing, the GST bead-bound proteins were identified by western blotting with selected primary and secondary antibodies and by Ponceau staining. HaCat cell lysates were treated with GST-tagged fusion proteins for one hour at 4°C.
2.6. Infectivity Assays
In order to assess the infectivity in HaCaT cells, they were treated for 48 hours with either a control siRNA (siCTRL) or a siRNA specifically targeting COPB1. HPV-16 PsVs were added to these cells at concentrations of 150 vge/cell. Using a luciferase assay equipment (Promega), firefly luciferase activity was measured 48 hours later as an indicator of infection. To ensure that the luciferase assays used equivalent protein inputs, the total cell protein was measured. A one-tailed t test was used to assess the statistical significance of the decrease in luciferase activity upon si-COPB1-treated cells, relative to the luciferase readings for siCTRL-treated cells in each experiment. Additionally, the cellular lysates were utilised to validate COPB1’s knockdown effectiveness by western blot. 25*10^5 HEK-293 TT cells were seeded in 15 cm dishes (10 dishes) and incubated overnight at 37°C, the cells were overexpressed with (15ug/15cm plate) HPV p16 Shell (L1and L2 capsid proteins of HPV) with (30ug/ 15cm plate) Luci gene (reporter of capsid proteins), for 48 hours, then the cells were harvested and the pellet were transferred in to siliconized tubes, and 9.5M magnesium chloride were added the same as the volume of pellet, 10% Triton in PBS were added 1/10 volume of pellet, exonuclease and endonuclease enzymes were also added, then we vortexed it and incubate it for overnight at 37°C. then 5M NaCl were added, and the tube were freeze-thawed three times in dry ice, and then the supernatant were collected and we washed the pellet with 1ml HBS, two times, and mixed the washed supernatant with the lysates supernatant. Then a gradient of CSCL were prepared in ultracentrifuge tubes, and we added the lysate on the top and spin for overnight (16 hrs) at 20,000-RPM at 4C. then we got a band in the middle of the tube between heavy and light CSCL in the middle of the tube which contained the HPV PsVs. The band were extracted with a syringe and transferred in to filter containing falcon tubes and washed three times with HBS buffer, and then we runned a 2 ul 2ul of the PsVs in a SDS-PAGE and the gel were stained with Coomassie brilliant blue for 15 minutes and distained over-night with de-stained buffer, and L1 and L2 proteins were detected. And we measured the protein concentration with nanodrop.
Pseudovirions Quantification
LUCI DNA was extracted from PsVs by incubating 10 ul of each PsVs preparation with 5 ul of extraction buffer (20 mM Tris (pH 8.0), 20 mM DTT, 20 mM EDTA, 2.0% SDS, and 0.2% Proteinase K) at 50°C for 15 min. Then, 1.5 ul of the extract was run on an agarose gel to confirm extraction of reporter genome (Luci).
The extracted DNA was purified using a PCR purification kit and eluted by adding 30 ul of nuclease free water.
A standard curve was made for qPCR, as follows: standard stock 2*109 plasmid/µl was used. To make this, 1-2 µg of PGL3 plasmid was digested with BglII enzyme and run on an agarose gel. After excision and purification of the linear plasmid, it was quantified by nanodrop and the copy number (plasmid/ng) determined.
The qPCR was set up with different concentrations using a standard and DNA from the PsVs; we used single digested PGL3 plasmid with BglII as a positive control and water as negative control and every sample was analysed in duplicate.
Next, the VGE and MOI were calculated, based on the qPCR results and the cell numbers that were used; the calculations were done as below. 10: represents the amount of PsV preparation [in microliters] used for the genome extraction. The “ul of prep. used” is the amount of PsV per well used in the infection (0.2 in this case) and the “number of cells” refers to the cells present in the well when the infection is performed. Then, we obtained the mean value (second column) and calculated the copy number per microliter of genome purification (third column, in this case 5 in 500 dilution). Next, to obtain the VGE for each experiment we followed the formula: vge = copy number/ul (considering dilution factor)* ul of prep used / (number of cells *10).
2.7. Synthetic HPV Particle Production and Labeling
HEK293TT cells were used to create HPV-2,5,16,18,31, PMU, BPV, and PsVs with a luciferase reporter plasmid (pGL3 Luci), as previously reported [55]. Using solutions of bovine serum albumin (BSA) at various concentrations as standards, the purity and capsid protein content of the PsV preparations were assessed using SDS-PAGE and Coomassie brilliant blue staining. Using a standard curve of the published plasmid DNA, the encapsidated DNA was subjected to real-time PCR analysis, allowing the copy number of each PsVs preparation to be determined. The HPV-16 L1-only VLPs were produced by employing a PsV-like procedure.
2.8. Cell Viability Assay
A Trypan Blue exclusion assay was used to count the number of live cells. After being seeded in a 6-well plate, the HaCaT cells were treated with either a control siRNA (siCTRL) or siRNA that selectively targeted COPB1 for 48 hours. The cells were trypsinized, collected, and stained with 0.2% trypan blue after 48 hours. Using a typical hemocytometer, cells were counted directly under a microscope to measure the quantity of viable cells. A one-tailed t test was used to assess statistical significance after normalising the number of viable cells in siCOPB1-treated samples to the number of viable cells obtained for siCTRL-treated samples in each experiment. Western blotting of the cellular lysates was used to verify siCOPB1’s effectiveness.
3. Results
3.1. Mass Spectrometry Assay Identifies COPB1 Cargo Protein as an HPV-16 Interacting Protein
To begin to investigate which cellular proteins might be involved in HPV virus entry, we performed mass spectrometry assays. We transfected HEK293 cells with HPV capsid proteins, p16 shell L1/L2 proteins where L2 is HA and FLAG tagged, or with empty vector pCDNA (as a control). After overnight incubation, cells were lysed, the HPV p16 shell L1/L2 proteins where L2 protein is HA and FLAG tagged was immunoprecipitated with anti-HA-conjugated agarose beads and the co-precipitated proteins were analyzed by mass spectrometry. As can be seen in Figure 1A, from this analysis we found several interesting cargo proteins that coprecipitated in the samples containing HPV p16 shell L1/L2 proteins where L2 protein is HA and FLAG tagged, but were absent from the empty vector samples. These proteins are found in the cellular compartments where HPV is trafficked (endosome, TGN, and Nucleus), and include: Adaptor protein 2 (AP2) in the cell membrane, Adaptor protein 1 (AP1) in endosomes, Adaptor protein 3 (AP3) in the TGN, COPB1 protein between the TGN and ER, and SEC16A at the ER exit site. We were particularly interested in L1 and L2’s interaction with COPB1, as this has not previously been described. To validate this interaction, we performed western blots on the cell extracts used in the immunoprecipitation: Figure 1B shows a representative western blot, performed to confirm the binding of COPB1 with HPV-16 pseudovirion (L1/L2).
3.2. GST-L2 Fusion Protein Binds COPB1 In Vitro
Having found a number of new candidate interacting partners of HPV-16 L2, we first wanted to determine whether COPB1 proteins could associate with HPV-16 L2 in simple protein-protein interaction assays in vitro. We also wanted to ascertain whether these interactions were specific for HPV-16 L2 or whether they were conserved among other HPV types. In order to do this, HACAT cell lysates were incubated with GST alone, or GST-11 L2 (a low-risk virus), GST-16 L2 or GST-18 L2 (high-risk viruses) and GST MmuPV-L2 (murine papillomavirus) fusion proteins immobilised on Glutathione-agarose overnight at 4°C. After extensive washing, bound proteins were eluted and analysed by SDS-PAGE and western blot, probed with antibodies against COPB1 endogenous proteins. As can be seen in Figure 2, HPV-16 L2 interacts strongly with COPB1. Interestingly, similar results are also obtained with HPV-11 and HPV-18 L2 and MmuPV L2 proteins, indicating that these interactions are conserved across multiple Papillomavirus types.
3.3. HPV-16 and HPV-11 L2 Interact with COPB1 In Vivo
From the GST pulldown, we have shown that there is a strong binding between HPV-16 L2 and COPB1. Therefore, we further proceeded to confirm the interaction between HPV-16 and HPV-11 L2 and COPB1 by performing a co-immunoprecipitation. We transfected 5ug of each plasmid HA- and FLAG-tagged HPV-16 and HPV-11 L2 proteins were overexpressed overnight in a 10cm dish seeded with 500,000 HEK293 cells. Cell lysates were immunoprecipitated with anti-HA agarose beads; the bound proteins were analyzed by SDS-PAGE and western blot, probed for endogenous COPB1. The results in Figure 3 show that there is a strong interaction between HPV-16 L2 and COPB1, compared with control (empty vector).
3.4. HPV-16 L2, but Not L1, Binds to COPB1
We were further interested to know whether the COPB1 binding to L2 is specific, or whether it can also bind to L1, and furthermore whether the L2 interaction with COPB1 could be affected by the presence of L1. To examine this, we repeated the co-immunoprecipitation experiment: HA- and FLAG-tagged HPV-16 L2 and L1 proteins were overexpressed overnight in HEK293 cells. Cell lysates were immunoprecipitated with anti-HA agarose beads, then the bound proteins were analyzed by SDS-PAGE and western blot, probed for endogenous COPB1. The results in Figure 4 confirm that there is a strong interaction between HPV 16 L2 and COPB1 whilst L1 fails to interact with COPB1 and has no influence over the L2-COPB1 association.
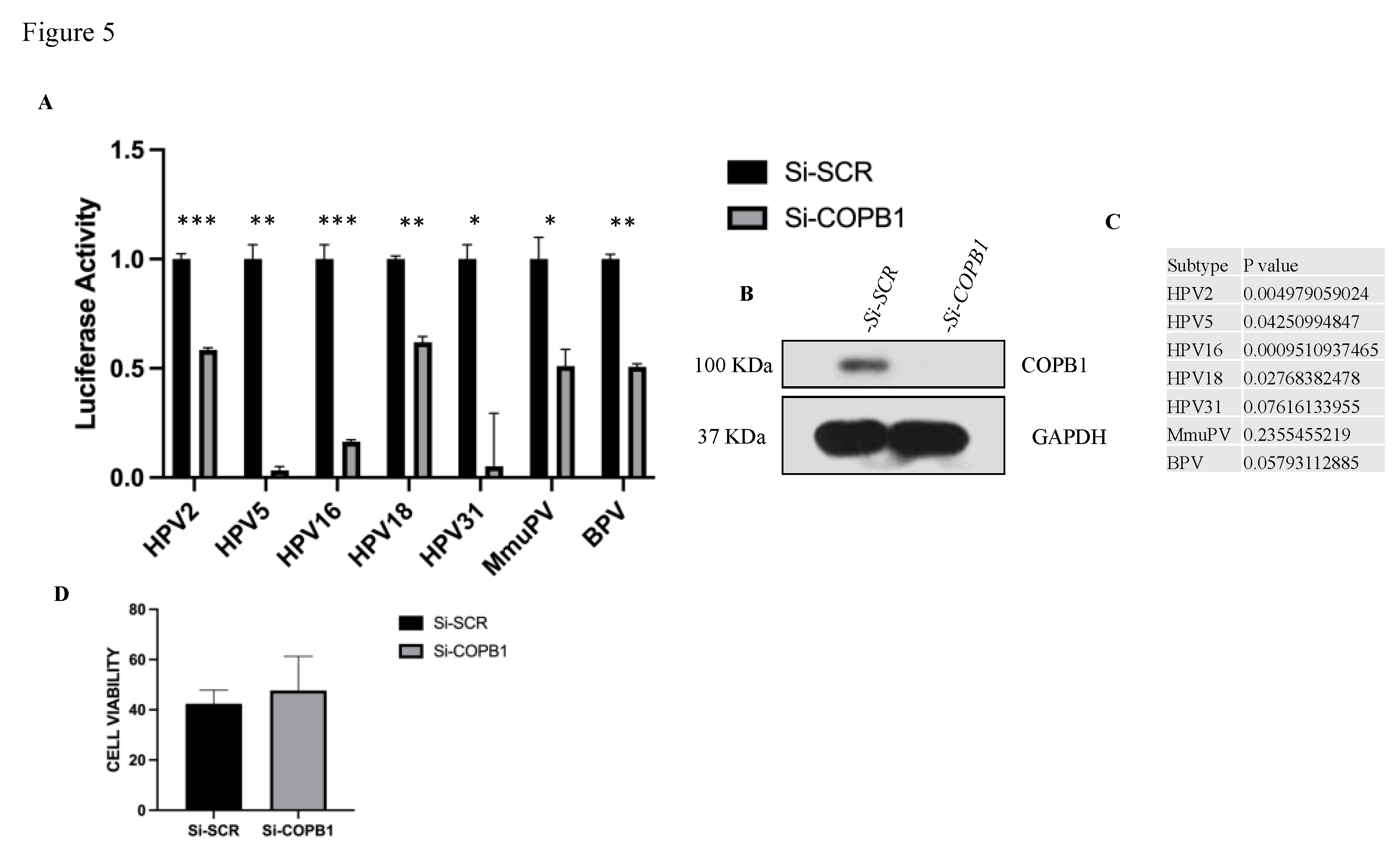
3.5. HPV Infection Requires COPB1
Since L2 is essential for HPV infection, we were interested to know how much the COPB1 interaction might contribute to a successful infection. To examine this, we transfected HACAT cells with siRNAs to knockdown COPB1 expression. After 48h, the cells were infected with 150 VGE of PsVs from HPV-2, -5, -16, -18, -31 as well as PsVs from bovine and murine papillomaviruses (BPV-1 and MmuPV, respectively). each having a Luciferase-expressing plasmid as a reporter genome; after a further 48h the cells were harvested, and Luciferase activity was measured by luminometer. The protein concentrations were also measured, and the luciferase activities normalized by protein concentration. The COPB1 knockdown levels were also confirmed by western blot. As shown by the data in Figure 5, knockdown of COPB1 results in a marked decrease in infectivity of all HPV types (more than 80% with HPV-16), compared with control, although it is interesting to note that there is wide variation among the different types as to the degree of loss of infectivity, with HPV5, 16 and 31 being particularly susceptible. Interestingly BPV and MmuPV infectivity is also reduced, indicating that the requirement for COPB1 is conserved across many different Papillomavirus types and we have also checked the cell viability upon COPB1 knockdown compare to control A Trypan Blue exclusion assay was used to count the number of live cells. After being seeded in a 6-well plate, the HaCaT cells were treated with either a control siRNA (siCTRL) or siRNA that selectively targeted COPB1 for 48 hours. The cells were trypsinized, collected, and stained with 0.2% trypan blue after 48 hours. Using a typical hemocytometer, cells were counted directly under a microscope to measure the quantity of viable cells. A one-tailed t test was used to assess statistical significance after normalizing the number of viable cells in siCOPB1-treated samples to the number of viable cells obtained for siCTRL-treated samples in each experiment. Western blotting of the cellular lysates was used to verify siCOPB1’s effectiveness.
4. Discussion
From previous studies we know that HPV entry is a multistep process, and that HPV uses the cellular machinery for entry and trafficking to the cell nucleus [56]. Using a proteomic screen we have found several interesting cargo proteins as potential interacting partners of HPV-16 L2. One of these cargo proteins was COPB1, which is a COPI protein subunit that is responsible for cargo binding and has the function of transporting cargo from TGN to ER [57]. After confirming the binding between COPBI and HPV-11, -16, and -18 L2 proteins, through GST pulldown and co-immunoprecipitation assays, we investigated whether COPI has any role in the infectious entry process of HPV. Upon knockdown of COPB1 we found that there is more than 80% reduction in HPV-16 PsVs infectivity. We also investigated the effect of COPB1 knockdown on the infectivity of both high-risk and low-risk HPV types, and found a similar requirement for COPB1 for infectivity of diverse HPV types. Finally we also analysed the effects on infectivity of other papillomaviruses - Bovine papillomavirus (BPV) and Murine papillomavirus (MmuPV) - and found that COPB1 knockdown reduces BPV and MmuPV infectivity, Interestingly, we also confirmed that MmuPV L2 can bind COPB1 in GST pulldown assays. As we can see in Figure 5 that some of HPVs are more sensitive upon COPB1 knockdown and some are less sensitive, which could be due to binding strength, as from GST pulldown in Figure 2, we can see that COPB1 interacts strong with HPV-16 L2 as compared to HPV-18 L2 and the same goes with infectivity (HPV-16 is more sensitive upon COPB1 knockdown as compared to HPV-18). Thus, from this study, we have understood that COPB1 is a common cargo from TGN towards cell Nucleus for many HPV types, as it is localized in specific parts of the cell that are traversed by most viruses en route to the cell nucleus through this path. This suggests that COPB1 could be a good target for blocking virus entry into the cell nucleus. Recently it has been also found that COPBI interacts with HPV-16 L2 through the R302/5A L2 [58], It would also be important to check the infectivity of newly synthesized viruses from nucleus upon COPII protein knockdown and to check their interaction with virus cargo proteins.
Author Contributions
Conceptualization, L.B.; Experimental methodology, L.B and O.B.; Experimental investigation, E.R and O.B.; writing—original draft preparation, E.R.; writing—review and editing, L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was unfunded.
Acknowledgments
We are extremely grateful to David Pim and Martina Bergant for experimental advice and to Miranda Thomas for revision of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schelhaas M, Shah B, Holzer M, Blattmann P, Kühling L, Day PM, Schiller JT, Helenius A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012;8[4]:e1002657. Epub 2012 Apr 19. [CrossRef] [PubMed]
- Marušič MB, Mencin N, Ličen M, Banks L, Grm HŠ. 2010. Modification of human papillomavirus minor capsid protein L2 by sumoylation. J Virol 84:11585–11589.
- Szymonowicz KA, Chen J. Biological and clinical aspects of HPV-related cancers. Cancer Biol Med. 2020 Nov 15;17[4]:864-878. Epub 2020 Dec 15. [CrossRef] [PubMed]
- Marur, Shanthi, Gypsyamber D’Souza, William H Westra, and Arlene A Forastiere. ‘HPV-Associated Head and Neck Cancer: A Virus-Related Cancer Epidemic’. The Lancet Oncology 11, no. 8 [August 2010]: 781–89. [CrossRef]
- Aksoy P, Gottschalk EY, Meneses PI. HPV entry into cells. Mutat Res Rev Mutat Res. 2017 Apr-Jun;772:13-22. [CrossRef] [PubMed]
- Chan CK, Aimagambetova G, Ukybassova T, Kongrtay K, Azizan A. Human Papillomavirus Infection and Cervical Cancer: Epidemiology, Screening, and Vaccination-Review of Current Perspectives. J Oncol. 2019 Oct 10;2019:3257939. [CrossRef] [PubMed]
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol. 2004 Mar 29;164[7]:1065-76. [CrossRef] [PubMed]
- Xie J, Zhang P, Crite M, Lindsay CV, DiMaio D. Retromer stabilizes transient membrane insertion of L2 capsid protein during retrograde entry of human papillomavirus. Sci Adv. 2021 Jun 30;7[27]:eabh4276. [CrossRef] [PubMed]
- Skoulakis, A., Fountas, S., Mantzana-Peteinelli, M. et al. Prevalence of human papillomavirus and subtype distribution in male partners of women with cervical intraepithelial neoplasia [CIN]: a systematic review. BMC Infect Dis 19, 192 [2019]. [CrossRef]
- Thomas M, Massimi P, Jenkins J, Banks L. 1995. HPV-18 E6 mediated inhibition of p53 DNA binding activity is independent of E6 induced degradation. Oncogene 10:261–268.
- Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med 119:445–462.
- Keiffer TR, Soorya S, Sapp MJ. Recent Advances in Our Understanding of the Infectious Entry Pathway of Human Papillomavirus Type 16. Microorganisms. 2021 Oct 1;9(10):2076. [CrossRef] [PubMed]
- Bazan SB, de Alencar Muniz Chaves A, Aires KA, Cianciarullo AM, Garcea RL, Ho PL. Expression and characterization of HPV-16 L1 capsid protein in Pichia pastoris. Arch Virol. 2009;154(10):1609-17. Epub 2009 Sep 10. [CrossRef] [PubMed]
- Darshan MS, Lucchi J, Harding E, Moroianu J. The l2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J Virol. 2004 Nov;78(22):12179-88. [CrossRef] [PubMed]
- Tsakogiannis D, Nikolaidis M, Zagouri F, Zografos E, Kottaridi C, Kyriakopoulou Z, Tzioga L, Markoulatos P, Amoutzias GD, Bletsa G. Mutation Profile of HPV16 L1 and L2 Genes in Different Geographic Areas. Viruses. 2022 Dec 31;15[1]:141. [CrossRef] [PubMed]
- Skoulakis, A., Fountas, S., Mantzana-Peteinelli, M. et al. Prevalence of human papillomavirus and subtype distribution in male partners of women with cervical intraepithelial neoplasia [CIN]: a systematic review. BMC Infect Dis 19, 192 [2019]. [CrossRef]
- Park SY, Guo X. Adaptor protein complexes and intracellular transport. Biosci Rep. 2014 Jul 29;34[4]:e00123. [CrossRef] [PubMed]
- Siddiqa A, Broniarczyk J, Banks L. ‘Papillomaviruses and Endocytic Trafficking’. International Journal of Molecular Sciences 19, no. 9 [September 2018]: 2619. [CrossRef]
- Morante AV, Baboolal DD, Simon X, Pan EC, Meneses PI. Human Papillomavirus Minor Capsid Protein L2 Mediates Intracellular Trafficking into and Passage beyond the Endoplasmic Reticulum. Microbiol Spectr. 2022 Jun 29;10[3]:e0150522. Epub 2022 May 24. [CrossRef] [PubMed]
- Calton CM, Bronnimann MP, Manson AR, Li S, Chapman JA, Suarez-Berumen M, Williamson TR, Molugu SK, Bernal RA, Campos SK. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog. 2017 May 2;13[5]: e1006200. [CrossRef] [PubMed]
- Broniarczyk J, Ring N, Massimi P, Giacca M, Banks L. HPV-16 virions can remain infectious for 2 weeks on senescent cells but require cell cycle re-activation to allow virus entry. Sci Rep. 2018 Jan 16;8[1]:811. [CrossRef] [PubMed]
- Graham FL, Van der Eb A. 1973. A new technique for the assay of infectivity of adenovirus 5 DNA. Virology 52:456–467.
- Siddiqa A, Broniarczyk J, Banks L. Papillomaviruses and Endocytic Trafficking. Int J Mol Sci. 2018 Sep 4;19[9]:2619. [CrossRef] [PubMed]
- DiGiuseppe S, Bienkowska-Haba M, Guion LGM, Keiffer TR, Sapp M. Human Papillomavirus Major Capsid Protein L1 Remains Associated with the Incoming Viral Genome throughout the Entry Process. J Virol. 2017 Jul 27;91[16]:e00537-17. [CrossRef] [PubMed]
- Broniarczyk J, Massimi P, Pim D, Bergant Marušič M, Myers MP, Garcea RL, Banks L. Phosphorylation of Human Papillomavirus Type 16 L2 Contributes to Efficient Virus Infectious Entry. J Virol. 2019 Jun 14;93[13]:e00128-19. [CrossRef] [PubMed]
- DiGiuseppe S, Keiffer TR, Bienkowska-Haba M, Luszczek W, Guion LG, Müller M, Sapp M. Topography of the Human Papillomavirus Minor Capsid Protein L2 during Vesicular Trafficking of Infectious Entry. J Virol. 2015 Oct;89[20]:10442-52. Epub 2015 Aug 5. [CrossRef] [PubMed]
- Broniarczyk J, Massimi P, Bergant M, Banks L. Human Papillomavirus Infectious Entry and Trafficking Is a Rapid Process. J Virol. 2015 Sep;89[17]:8727-32. Epub 2015 Jun 10. [CrossRef] [PubMed]
- Day PM, Thompson CD, Schowalter RM, Lowy DR, Schiller JT. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J Virol. 2013 Apr;87[7]:3862-70. Epub 2013 Jan 23. [CrossRef] [PubMed]
- Siddiqa A, Massimi P, Pim D, Broniarczyk J, Banks L. 2018. Human papillomavirus 16 infection induces VAP-dependent endosomal tubulation. J Virol 92:1–14.
- Day PM, Weisberg AS, Thompson CD, Hughes MM, Pang YY, Lowy DR, Schiller JT. Human Papillomavirus 16 Capsids Mediate Nuclear Entry during Infection. J Virol. 2019 Jul 17;93[15]:e00454-19. [CrossRef] [PubMed]
- Bergant M, Banks L. SNX17 facilitates infection with diverse papillomavirus types. J Virol. 2013 Jan;87[2]:1270-3. Epub 2012 Oct 31. [CrossRef] [PubMed]
- Bugnon Valdano M, Massimi P, Broniarczyk J, Pim D, Myers M, Gardiol D, Banks L. Human Papillomavirus infection requires the CCT Chaperonin Complex. J Virol. 2021 ;95[11]:e01943-20. Epub 2021 Mar 17. [CrossRef] [PubMed]
- Pim D, Broniarczyk J, Siddiqa A, Massimi P, Banks L. Human Papillomavirus 16 L2 Recruits both Retromer and Retriever Complexes during Retrograde Trafficking of the Viral Genome to the Cell Nucleus. J Virol. 2021 Jan 13;95[3]:e02068-20. [CrossRef] [PubMed]
- Duden, R. ER-to-Golgi transport: COP I and COP II function [Review]. Mol Membr Biol. 2003 Jul-Sep;20[3]:197-207. [CrossRef] [PubMed]
- McMahon HT, Mills IG. COP and clathrin-coated vesicle budding: different pathways, common approaches. Curr Opin Cell Biol. 2004 Aug;16[4]:379-91. [CrossRef] [PubMed]
- Beck R, Rawet M, Wieland FT, Cassel D. The COPI system: molecular mechanisms and function. FEBS Lett. 2009 Sep 3;583[17]:2701-9. Epub 2009 Jul 22. Erratum in: FEBS Lett. 2009 Nov 3;583[21]:3541. Ravet, M [corrected to Rawet, M]. [CrossRef] [PubMed]
- Matsuoka, K., Morimitsu, Y., Uchida, K., & Schekman, R. [1998]. Coat Assembly Directs v-SNARE Concentration into Synthetic COPII Vesicles. Molecular Cell, 2[5], 703-708. [CrossRef]
- Szul T, Sztul E. ‘COPII and COPI Traffic at the ER-Golgi Interface’. Physiology 26, no. 5 [October 2011]: 348–64. [CrossRef]
- Schoppe J, Schubert E, Apelbaum A, Yavavli E, Birkholz O, Stephanowitz H, Han Y, Perz A, Hofnagel O, Liu F, Piehler J, Raunser S, Ungermann C. Flexible open conformation of the AP-3 complex explains its role in cargo recruitment at the Golgi. J Biol Chem. 2021 Nov;297[5]:101334. Epub 2021 Oct 22. [CrossRef] [PubMed]
- 40. Golgi Apparatus. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26941/.
- Peotter J, Kasberg W, Pustova I, Audhya A. COPII-mediated trafficking at the ER/ERGIC interface. Traffic. 2019 Jul;20[7]:491-503. Epub 2019 May 30. [CrossRef] [PubMed]
- Gomez-Navarro N, and Miller EA. ‘COP-Coated Vesicles’. Current Biology 26, no. 2 [25 January 2016]: R54–57. [CrossRef]
- Sohn K, Orci L, Ravazzola M, Amherdt M, Bremser M, Lottspeich F, Fiedler K, Helms JB, Wieland FT. A major transmembrane protein of Golgi-derived COPI-coated vesicles involved in coatomer binding. J Cell Biol. 1996 Dec;135[5]:1239-48. [CrossRef] [PubMed]
- Pérez-Pulido AJ, Asencio-Cortés G, Brokate-Llanos AM, Brea-Calvo G, Rodríguez-Griñolo R, Garzón A, Muñoz MJ. Serial co-expression analysis of host factors from SARS-CoV viruses highly converges with former high-throughput screenings and proposes key regulators. Brief Bioinform. 2021 Mar 22;22(2):1038-1052. [CrossRef] [PubMed]
- Tiwari R, Mishra AR, Gupta A, Nayak D. Structural similarity-based prediction of host factors associated with SARS-CoV-2 infection and pathogenesis. J Biomol Struct Dyn. 2022 Aug;40(13):5868-5879. Epub 2021 Jan 28. [CrossRef] [PubMed]
- Fuller J, Álvarez-Rodríguez B, Todd EJAA, Mankouri J, Hewson R, Barr JN. Hazara Nairovirus Requires COPI Components in both Arf1-Dependent and Arf1-Independent Stages of Its Replication Cycle. J Virol. 2020 Aug 17;94(17):e00766-20. [CrossRef] [PubMed]
- Drake MT, Zhu Y, Kornfeld S. The assembly of AP-3 adaptor complex-containing clathrin-coated vesicles on synthetic liposomes. Mol Biol Cell. 2000 Nov;11[11]:3723-36. [CrossRef] [PubMed]
- Duden R. ER-to-Golgi transport: COP I and COP II function [Review]. Mol Membr Biol. 2003 Jul-Sep;20[3]:197-207. [CrossRef] [PubMed]
- Bonifacino, J., Lippincott-Schwartz, J. Coat proteins: shaping membrane transport. Nat Rev Mol Cell Biol 4, 409–414 [2003]. [CrossRef]
- van Vliet, C., Thomas, E. C., Merino-Trigo, A., Teasdale, R. D. & Gleeson, P. A. Intracellular sorting and transport of proteins. Progress in Biophysics and Molecular Biology 83, 1–45 (2003).
- Cui, L., Li, H., Xi, Y. et al. Vesicle trafficking and vesicle fusion: mechanisms, biological functions, and their implications for potential disease therapy. Mol Biomed 3, 29 (2022). [CrossRef]
- Darsow T, Burd CG, Emr SD. Acidic di-leucine motif essential for AP-3-dependent sorting and restriction of the functional specificity of the Vam3p vacuolar t-SNARE. J Cell Biol. 1998 Aug 24;142[4]:913-22. [CrossRef] [PubMed]
- Broniarczyk J, Massimi P, Bergant M, Banks L. Human Papillomavirus Infectious Entry and Trafficking Is a Rapid Process. J Virol. 2015 Sep;89[17]:8727-32. Epub 2015 Jun 10. [CrossRef] [PubMed]
- Bergant Marušič M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13:455–467.
- Zhang W, Kazakov T, Popa A, DiMaio D. 2014. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires γ-secretase activity. mBio 5:e01777-14.
- Siddiqa A, Broniarczyk J, Banks L. ‘Papillomaviruses and Endocytic Trafficking’. International Journal of Molecular Sciences 19, no. 9 [September 2018]: 2619. [CrossRef]
- Fuller J, Álvarez-Rodríguez B, Todd EJAA, Mankouri J, Hewson R, Barr JN. Hazara Nairovirus Requires COPI Components in both Arf1-Dependent and Arf1-Independent Stages of Its Replication Cycle. J Virol. 2020 Aug 17;94(17):e00766-20. [CrossRef] [PubMed]
- Harwood MC, Woo TT, Takeo Y, DiMaio D, Tsai B..HPV is a cargo for the COPI sorting complex during virus entry.Sci. Adv.9,eadc9830(2023). [CrossRef]
Figure 1.
(A) Mass spectrometry data showing different proteins from different parts of the cell that were precipitated from HPV-16 HA-L1/L2-expressing HEK293 cells: loge shows the base-10 log of the expectation that any particular protein assignment was made at random (E-value), the other two panels show, respectively, the number of unique peptides and the total number of peptides of each protein, precipitated from HPV-16 HA-L1/L2-expressing HEK293 cells, but not from control cells. (B) Western blot showing that COPB1 co-precipitates with pShell: HPV-16.
Figure 1.
(A) Mass spectrometry data showing different proteins from different parts of the cell that were precipitated from HPV-16 HA-L1/L2-expressing HEK293 cells: loge shows the base-10 log of the expectation that any particular protein assignment was made at random (E-value), the other two panels show, respectively, the number of unique peptides and the total number of peptides of each protein, precipitated from HPV-16 HA-L1/L2-expressing HEK293 cells, but not from control cells. (B) Western blot showing that COPB1 co-precipitates with pShell: HPV-16.
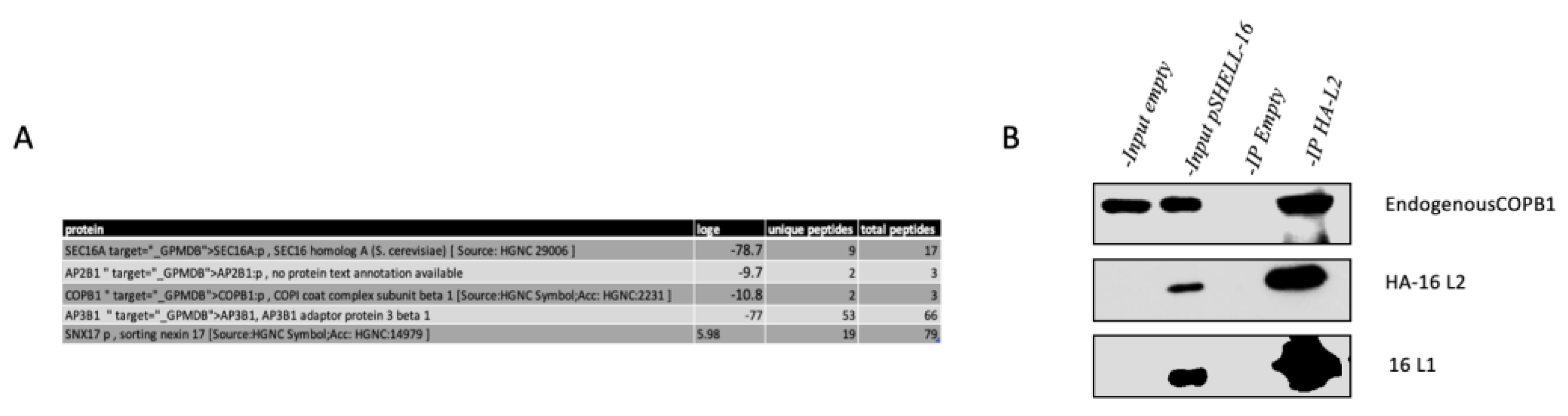
Figure 2.
GST pulldown assay using HaCaT cell lysate with GST alone, GST-HPV-16 L2, GST-HPV-11 L2, and GST-HPV-18 L2 and GST-MmuPV L2; the bound proteins were visualized by SDS-PAGE and western blot, probed for endogenous COPB1. The lower panel shows the GST-L2 proteins stained with Ponceau Red. Arrows indicate the binding of HPV-16 L2 with COPBI, compared with 5% of input. (B) shows Binding of GST-MmuPV L2 with COPB1.
Figure 2.
GST pulldown assay using HaCaT cell lysate with GST alone, GST-HPV-16 L2, GST-HPV-11 L2, and GST-HPV-18 L2 and GST-MmuPV L2; the bound proteins were visualized by SDS-PAGE and western blot, probed for endogenous COPB1. The lower panel shows the GST-L2 proteins stained with Ponceau Red. Arrows indicate the binding of HPV-16 L2 with COPBI, compared with 5% of input. (B) shows Binding of GST-MmuPV L2 with COPB1.
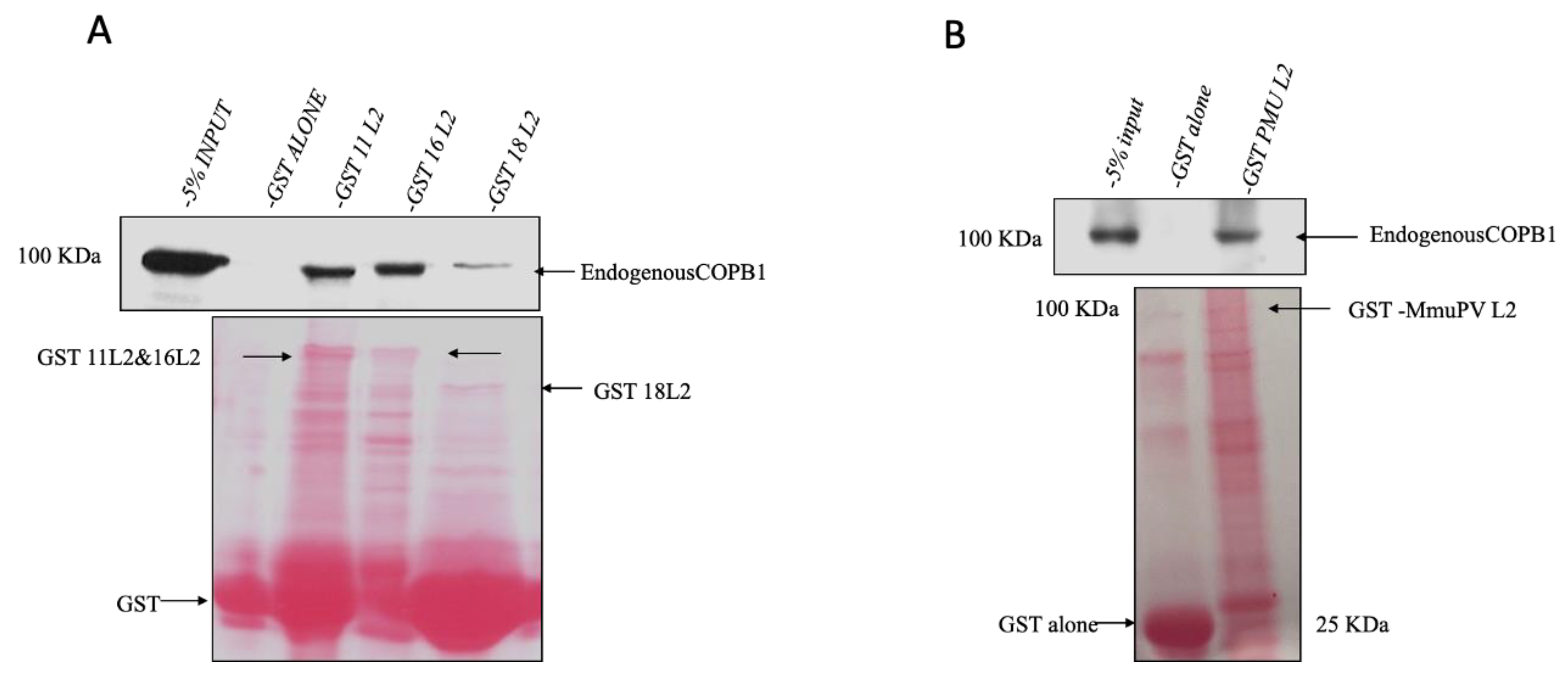
Figure 3.
(A) pCA:HPV-16 L2 and (B) pCA:HPV-11 L2 (HA and FLAG tagged) were overexpressed in HEK293 cells. Cell lysates were immunoprecipitated with anti-HA agarose beads and the bound proteins were analyzed by SDS-PAGE and western blot, probed for endogenous COPB1.
Figure 3.
(A) pCA:HPV-16 L2 and (B) pCA:HPV-11 L2 (HA and FLAG tagged) were overexpressed in HEK293 cells. Cell lysates were immunoprecipitated with anti-HA agarose beads and the bound proteins were analyzed by SDS-PAGE and western blot, probed for endogenous COPB1.
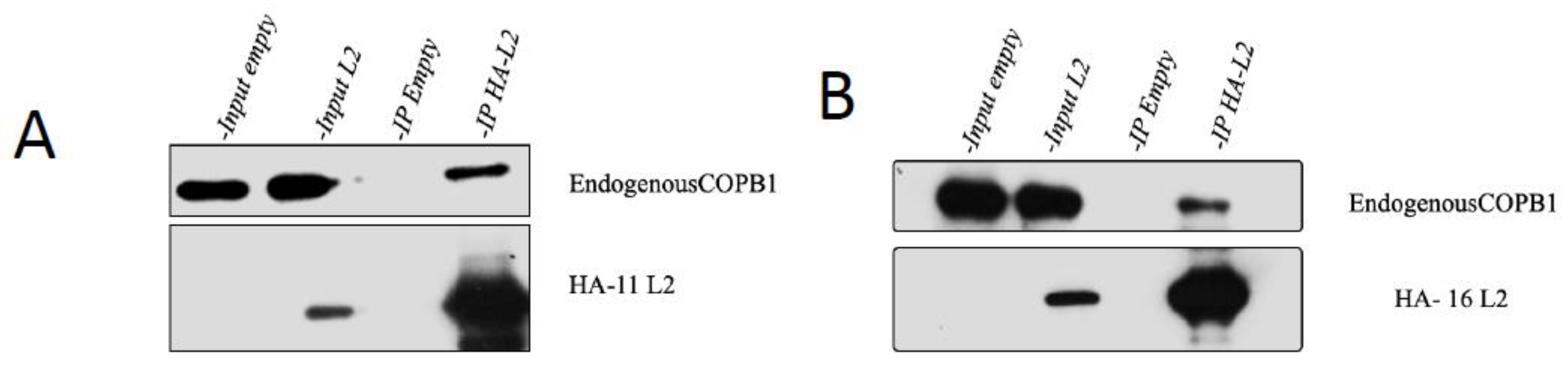
Figure 4.
HPV-16 L2, but not L1, binds to COPB1. pCA:HPV-16 L2 and pCA:HPV-16 L1 (HA and FLAG tagged) were overexpressed in HEK293 cells. Cell lysates were immunoprecipitated with anti-HA agarose beads. The bound proteins were analyzed by SDS-PAGE and western blot, probed for endogenous COPB1 (high exposure Blot is kept for detection of L2 protein in overexpressed L2 input and the bands overlapping in HPV-16 L1 lane are the antibody light chain).
Figure 4.
HPV-16 L2, but not L1, binds to COPB1. pCA:HPV-16 L2 and pCA:HPV-16 L1 (HA and FLAG tagged) were overexpressed in HEK293 cells. Cell lysates were immunoprecipitated with anti-HA agarose beads. The bound proteins were analyzed by SDS-PAGE and western blot, probed for endogenous COPB1 (high exposure Blot is kept for detection of L2 protein in overexpressed L2 input and the bands overlapping in HPV-16 L1 lane are the antibody light chain).
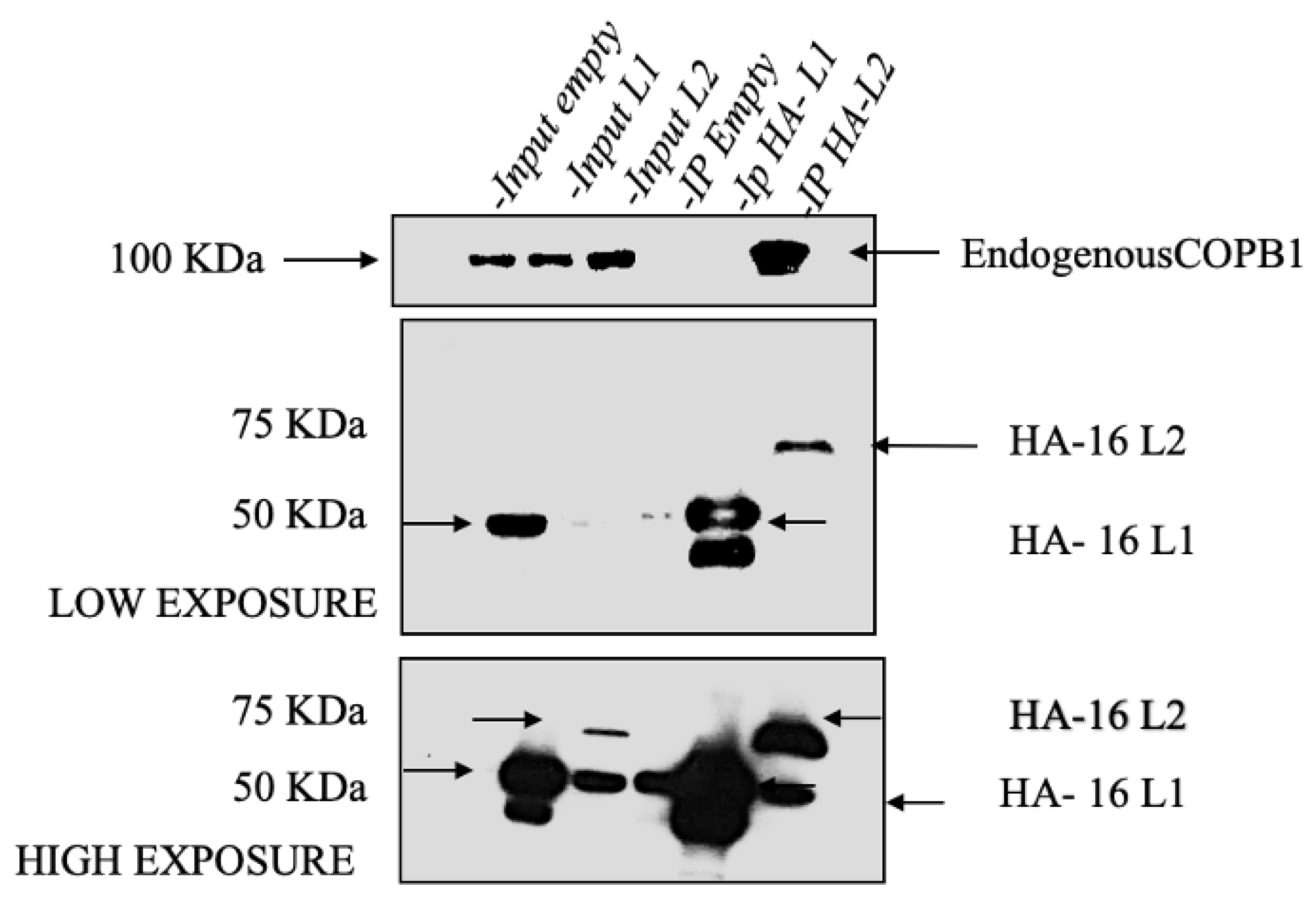
Figure 5.
HPV 16 requires COPB1 cargo protein for infectivity. (A) histogram shows the changes in levels of infectivity of HPV-2, HPV-5, HPV-16, HPV-18 and HPV-31 upon COPB1knockdown, as measured by luciferase activity. MmuPV and BPV-1 were also included as controls. (B) The knockdown of COPB1 was confirmed by western blot. (C) shows the P values of each HPV type’s infectivity, the stars indicate the significance: *** indicates a P value equal or less than 0.0005, ** when P value is equal or less than 0.005, and * when p value is less than 0.05. (D) shows the effects on cell viability upon transfection with si-SCR and si-COPB1.
Figure 5.
HPV 16 requires COPB1 cargo protein for infectivity. (A) histogram shows the changes in levels of infectivity of HPV-2, HPV-5, HPV-16, HPV-18 and HPV-31 upon COPB1knockdown, as measured by luciferase activity. MmuPV and BPV-1 were also included as controls. (B) The knockdown of COPB1 was confirmed by western blot. (C) shows the P values of each HPV type’s infectivity, the stars indicate the significance: *** indicates a P value equal or less than 0.0005, ** when P value is equal or less than 0.005, and * when p value is less than 0.05. (D) shows the effects on cell viability upon transfection with si-SCR and si-COPB1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



