
Submitted:
09 February 2024
Posted:
09 February 2024
You are already at the latest version
Abstract
With the aim to develop novel scaffolds for sustained release of drugs, we initially developed an easy approach for the synthesis of α,ω-homobifunctional mercaptoacyl poly(alkyl oxide)s. This was based on the esterification of the terminal hydroxyl groups of poly(alkyl oxide)s with suitably S-4-methoxytrityl (Mmt) protected mercapto acids, followed by removal of the acid labile S-Mmt group. This method allowed the efficient synthesis of the title compounds in high yield and purity, which were further used in the development a thioether cross-linked liposome scaffold, by thia-Michael reaction of the terminal thiol groups with preformed nano-sized liposomes bearing maleimide groups on their surface. The reaction process was followed by 1H-NMR, using a Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion NMR experiment (1H-NMR CPMG), which allowed real-time monitoring and optimization of the reaction process. The thioether cross-linked liposomal scaffold that was synthesized was proven to preserve the nanosized characteristics of the initial liposomes and allowed sustained release of calcein (which was used as a chromophore), providing evidence for the efficient synthesis of a novel drug release scaffold consisted of nanoliposome building blocks.
Keywords:
S-4-methoxytityl mercapto acids
; α
; ω-homofunctional mercaptoacyl poly(alkyl oxide)s
; thia-Michael addition
; thioether cross-linked liposomes
1. Introduction
Poly(alkyl oxide)s (PAOs) are biomacromolecules with the general formula HO-(CH2-CHR-O)x-H. Poly(ethylene glycol) (PEG) 1 (Figure 1), where the R group of PAOs is H, also known as poly(ethylene oxide) (PEO) or poly(oxyethylene) (POE), depending on its molecular weight, is a synthetic linear polyether with wide range of biomedical applications, due to its excellent water solubility, biocompatibility, low toxicity, low immunogenicity and antigenicity. In addition, PEG is non-biodegradable, yet it is readily excretable after administration into living organisms, exhibiting excellent pharmacokinetic and biodistribution behavior. All these properties make PEG particularly attractive and an ideal linker to graft organic molecules and biomolecules. In addition, PEG-conjugated drugs have been approved by the U.S. Food and Drug Administration (FDA) for safe use in humans [1,2,3,4].
Besides PEG, other types of PAOs, such as pluronics 2 (Figure 1) also represent an important class of biomacromolecules. They are composed of poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol), also abbreviated as “triblock PEO-PPO-PEO copolymers”, where PPO represents poly(propylene oxide) chains. The PEO block is considered hydrophilic and water soluble, while the PPO block is hydrophobic and water insoluble. The combination of these hydrophilic/lipophilic chains, introduce special properties to these polymers. Thus, in aqueous media, these block copolymers self-assemble into micelles, with a hydrophobic PPO center core being formed and a hydrophilic PEO outer shell that interfaces with water. Since these micelles are amphiphilic, they are able to entrap lipophilic molecules in the central hydrophobic core area. Consequently, pluronic micelles are also effectively used as drug carriers [5,6,7].
Both PEG and pluronic have been widely functionalized on their hydroxyl groups with a variety of groups or molecules, increasing their applicability. In fact, hetero- and homo-bifunctional PEG derivatives are very useful, acting as cross-linking agents or spacers between two chemical entities [8]. Especially the homobifunctional dithiol poly(ethylene glycol) (PEG) derivatives (Figure 2), which represent synthetic polyethers containing two thiol functionalities of the same type, are of high interest. For example, they are used in simple and rapid cross linking reactions between homodithiol functionalized poly(ethylene glycols) and drugs, peptides or proteins, based on the properties of thiol group to act as a nucleophile, mainly by the thiol-maleimide method, where the thiol group is used as a Michael donor [9,10,11], or in thiol-ene click chemistry [12,13].
Homofunctional dithiol PEGs of type 4, where thiol containing building blocks have been introduced into the hydroxyl end groups of the PEG molecules (Figure 2) have been used as alternatives to dithiol PEGs of type 3 (where the hydroxyl end groups have been replaced by thiol groups) [14,15,16,17]. Such derivatives (4) can be synthesized more easily and have the advantage of structural diversity.
Despite the high interest, the commercial availability of homobifunctional PEGs derived from 1 (and the more complicated 2), is extremely low and the cost is very high. By searching the commercial availability of PEG-dithiols, this is limited to the replacement of the hydroxyl end groups by the thiol group (type 3), while the commercial availability of homobifunctional poly(alkyl oxide)s of type 4 are limited to 3-mercaptopropionate derivatives, with pentaerythritol tetrakis(3-mercaptopropionate) being the most representative.
Among several methods that have been reported for the synthesis of homobifunctional poly(alkyl oxide)s of type 4, the simplest one is based on the direct esterification of both end sides of PEG chain with mercapto acids, by the reaction of hydroxyl end groups with mercapto acids in presence of p-toluenesulfonic acid (or sulfuric acid) as catalyst. In this method, a high excess of the mercapto acid (10-20 fold excess) in respect to the hydroxyl groups is used, and the reaction is performed in toluene under high temperature (reflux conditions) for 24 h and the azeotropic mixture is periodically collected. Moreover, high amounts of dithiothreitol (DTT), in parallel with a flow of nitrogen, is needed to prevent disulfide oxidation [18,19,20,21,22,23,24,25,26]. In another method, which has been used for the synthesis of pluronic di-mercaptopropionate, a three-step procedure was used. Initially, dithiodipropionyl chloride was prepared by reacting 3,3-dithiodipropionic acid and high excess of thionyl chloride to afford dithiodipropionyl chloride which was further reacted with pluronic. The product was further reduced by DTT to afford pluronic di-mercaptopropionate [27,28]. In a third reported method, the transesterification of methyl 3-mercaptopropionate with tetraethylene glycol and poly(ethylene glycol)s catalyzed by Candida antarctica Lipase B without the use of solvent, was studied [29]. However, all these methods have significant drawbacks.
By a synthetic point of view, although the third method can be considered as “green”, the use of an heterogenous catalyst is not practical for laboratory use, while the two other reported methods require drastic reaction conditions such as high temperatures, long reaction times, high excess of catalysts and mercapto acid, as well as the need of high amounts of DTT in parallel with the use of nitrogen flow during the reaction. Therefore, the introduction of new methods to overcome these limitations, for the synthesis of homobifunctional PEGs of type 4, is a challenge. Thus, in the first part of this work, we considered the synthesis of α,ω-bis-mercaptoacyl poly(alkyl oxide)s of type 4, by a simple and efficient method.
In the next part of this work, the synthesized products were used in the development of a drug release scaffold composed by thioether cross-linked nanoliposomes. Liposomes are mainly composed by phospholipids and cholesterol, thus, between the various nanoparticle types, they are usually preferred as drug carriers because they are structurally versatile, highly biocompatible and non-toxic. Thus, nanoliposomes become more and more useful in drug product development as drug delivery systems [30,31]. Liposome pegylation, which refers to the coating of the vesicle surface with polyethylene glycol molecules, can efficiently reduce the rapid clearance of liposomes by macrophages and prolong their circulation time in the blood [32,33]. Thus, in this work, we considered the synthesis of cross-linked nanoliposomes, by the reaction of nanoliposomes bearing maleimide groups on their surface with α,ω-homofunctional PEG-dithiols of type 4. To this end, we efficiently synthesized a liposomal scaffold, which was proven to maintain the nanosized characteristics of the initial liposome forms. We also proved that the synthesized scaffold enables sustained release of a chromophore (calcein), thus proving its potential use as a novel drug release scaffold consisted of nanoliposome building blocks.
2. Results and Discussion
2.1. Rationale
In order to synthesize innovating cross-linked nano-sized liposomes, we considered the use of homo-functional poly(alkyl oxide)s as cross-linking agent. Searching the literature, we found the preparation of liposome-crosslinked hybrids by the crosslinking reaction of end functionalized 4-arm PEG-thiols and maleimide functionalized liposomes, through a thia-Michael type addition. By this method, liposome-crosslinked hybrid hydrogels were formed, as the result of extensive cross-linking between the end functionalized 4-arm PEG-dithiols and maleimide functionalized liposomes [34]. The 4-arm PEG-dithiols were synthesized by the reaction of a 4-arm PEG and a mercapto acid in toluene under refluxing conditions (at about 155 oC) in presence of p-toluenesulfonic acid as catalyst. DTT and a flow of nitrogen were used to avoid oxidation, while the released water during the esterification reaction was collected by using a Dean Stark trap. The synthesized product should be stored under nitrogen at -20 °C (to avoid any oxidation).
Inspired by this work, we designed a novel cross-linked liposomal scaffold for slow release of drugs, where the initial nano-sized characteristics of liposomes would be preserved after the cross-linking reaction. To this end, we considered the replacement of 4-arm PEG-dithiols with α,ω-bis-mercapto poly(alkyl oxide)s, as a structural modification which would reduce the extensive cross-linking, and allow the development of nano-sized scaffolds (instead of liposome-crosslinked hybrid hydrogels).
In this approach, we considered the use of high-molecular-weight end-type PEG-dithiols, such as α,ω-bis-mercaptoacyl poly(alkyl oxide)s, as molecular tools of particular interest. However, the synthetic approaches that have been developed so far, for the synthesis of α,ω-bis-mercapto poly(alkyl oxide)s are not attractive and need to be replaced by simple and more efficient methods, avoiding high temperatures, sophisticated equipment, oxidation problems and by-products formation. Thus, a novel method for the synthesis of α,ω-bis-mercaptoacyl poly(alkyl oxide)s was initially developed, which was further used in the synthesis the proposed nano-sized scaffold.
2.2. Synthesis of functionalized α,ω-bis-mercaptoacyl poly(alkyl oxide)s
2.2.1. Selection and Synthesis of S-Mmt mercapto acids
For the synthesis of α,ω-bis-mercaptoacyl poly(alkyl oxide)s we considered the esterification of the hydroxy end groups of poly(alkyl oxide)s with suitably S-protected mercapto acids. Among the several protecting groups of thiols, the trityl-type protecting groups, such as trityl (Trt) and 4-methoxytrityl (Mmt), are very attractive. In fact, both groups are well-known for their acid lability, however, S-Trt-group requires treatment with concentrated trifluoroacetic acid (TFA) and is reversible, while the use of S-Mmt group has been proposed as the very acid-labile alternative of S-Trt protecting group, allowing simple and effective removal of the S-Mmt group even with 1-3% TFA in dichloromethane (DCM) and triethyl silane (TES) [35]. Thus, we previously reported methods for the synthesis of S-Mmt mercapto acids 6 as the very acid-labile alternative of S-Trt mercapto acids 5 [36].
Figure 3.
General structure of S-Trt mercapto acids 5 and S-Mmt mercapto acids 6. r=1, R΄=CH3; r=2-5, R΄=H.
Figure 3.
General structure of S-Trt mercapto acids 5 and S-Mmt mercapto acids 6. r=1, R΄=CH3; r=2-5, R΄=H.
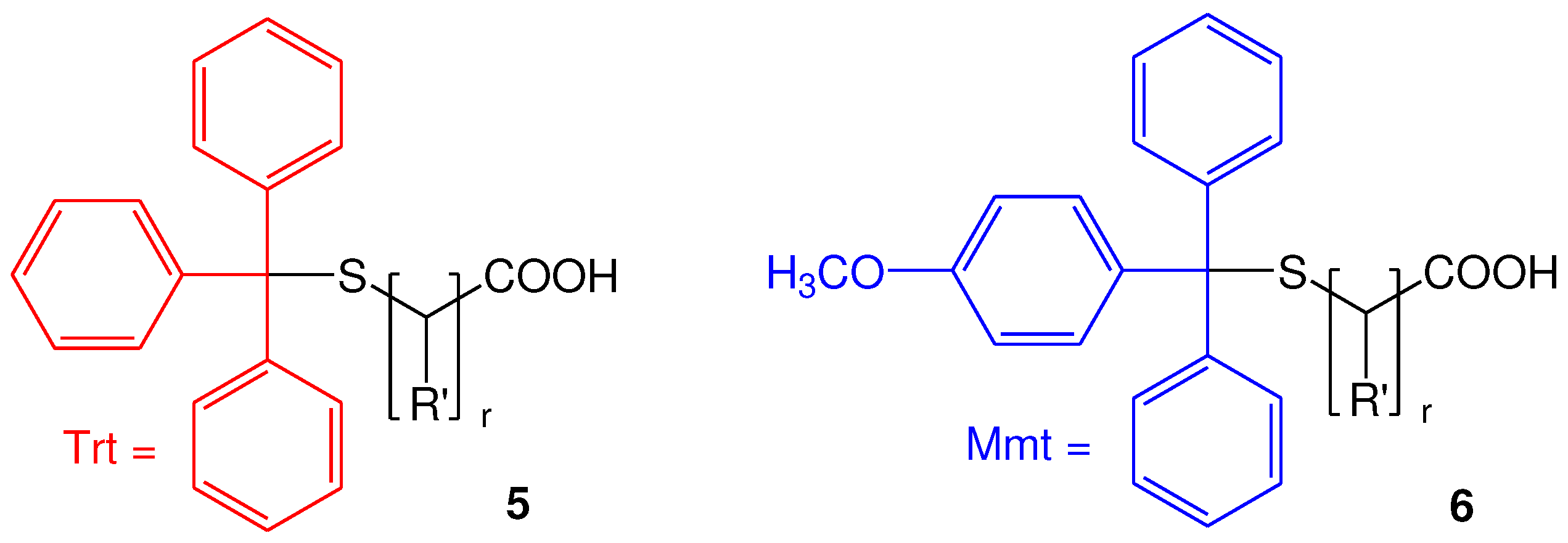
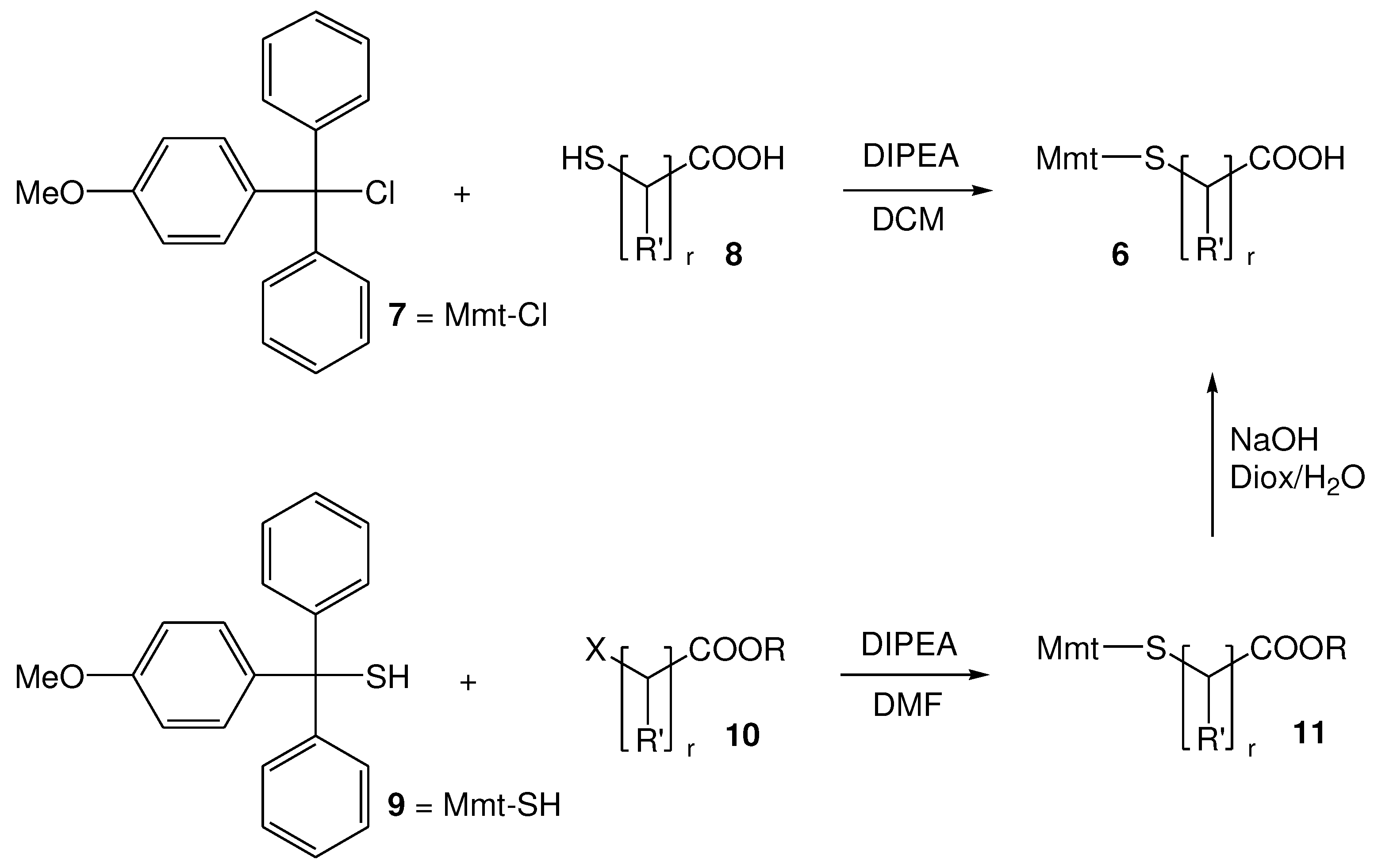
In fact, for the synthesis of 6, two methodologies have been proposed. In the first method, 4-methoxytrityl chloride (Mmt-Cl) 7 is reacted with commercially available mercapto acids 8, in presence of N, N΄-diisopropylethylamine (DIPEA), while in the second method, 4-methoxytrityl thiol (Mmt-SH) 9 is reacted with halo acids or esters 10, to initially form the S-Mmt-mercapto esters 11, which are further hydrolyzed, under basic conditions, to afford the S-Mmt-mercapto acids 6.
Scheme 1.
Methods that have been proposed for the synthesis of 6; r=1, R΄=CH3; r=2-5, R΄=H.
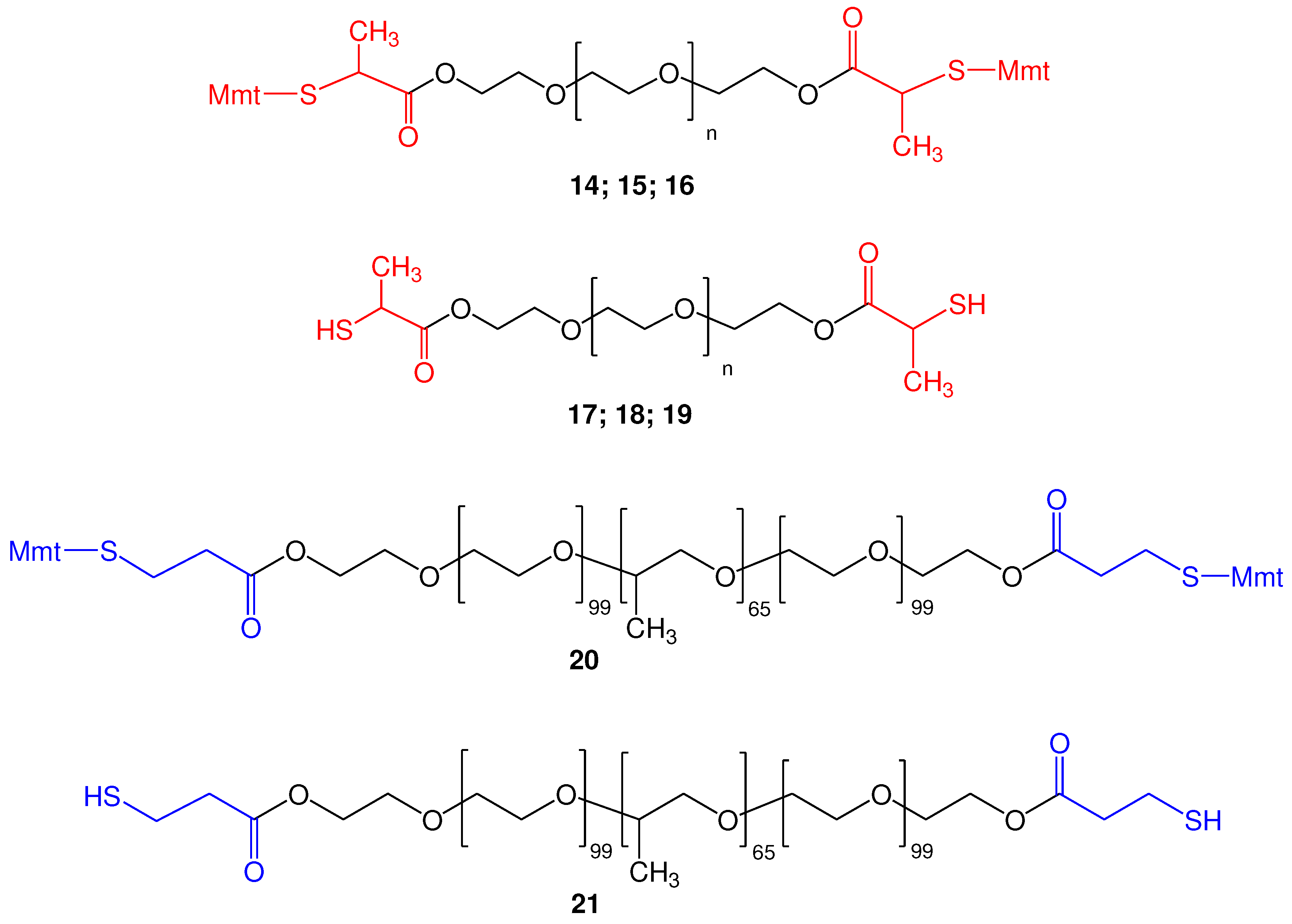
In this work, we considered the synthesis and further use of two S-Mmt-mercaptoacids: (a) S-Mmt-2-mercaptopropionic acid (6a; R΄=CH3; r=1) and (b) S-Mmt-3-mercaptopropionic acid (6b; R΄=H; r=2). Regarding the method of choice for their synthesis, considering that both mercapto acids are commercially available, the first method was chosen. For the synthesis, 2-mercaptopropionic acid (thiolactic acid) (8a; R΄=CH3; r=1) and 3-mercaptopropionic acid (8b; R΄=H; r=2) were reacted with Mmt-Cl 7 in presence of DIPEA, and 6a/b were isolated in high yield and purity, as confirmed by NMR analysis and hplc (Figures S1–S5). These (6a/b) were further used in the synthesis of α,ω-bis-2-mercaptopropionyl polyethylene glycols (abbreviated PEG-dithiols hereafter) and α,ω-bis-3-mercaptopropionyl poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) copolymers (abbreviated pluronic-dithiols hereafter).
2.2.2. Synthesis of di-S-Mmt-PEG-dithiols and PEG-dithiols
The proposed synthetic approach is shown in Scheme 2. In brief, we propose the reaction of PEG 1 with S-Mmt mercapto acids 6 by the Steglich esterification method. This requires the use of N, N΄-diisoproplylcarbodiimide (DΙC) and 4-(dimethylamino)pyridine (DMAP) as the carboxylic acid activating system (Scheme 2) [37,38], to initially afford the S-Mmt mercaptoacylated poly(ethylene glycol)s 12 (abbreviated di-S-Mmt-PEG-dithiols hereafter).
In this approach, the use S-Mmt protecting group during esterifiaction reaction has a double role, that is to: (a) prevent oxidation of the thiol groups of mercapto acids to the corresponding disulfides, (b) prevent the formation of thioesters and/or thiolactones (which would be formed due to inter molecular, and/or intra molecular nucleophilic attack of the free thiol groups to the activated carboxylic acid groups) and/or polymerization processes (due to the formation of active thioester/thiolactone intermediates along with the presence of free thiol groups) [39,40,41]. In addition, there are two obvious advantages after esterification reaction, that is to: (a) prevent disulfide formation, by initially obtaining the S-Mmt protected dithiols, ultimately increasing potential storage times, a common problem associated with thiol-containing therapeutics, and (b) allow an easy deprotection of the S-Mmt group (as previously explained). In fact, treatment of 12 with 1-3% TFA and TES in DCM, enables fast and irreversible cleavage of S-Mmt group. The use of TES is known to allow irreversible removal of S-Mmt group, by donating a hydride to the resulting Mmt cation, driving the equilibrium towards cleavage [42]. It is reasonable that, the presence of TES in the cleavage mixture, or during concentration of the cleavage mixture, may also form the corresponding sililated product, due to the formation of Si-S bonds. However, although thiosilanes are thermally stable, the Si-S bond is readily cleaved by many protic solvents, particularly those containing the O-H group [43]. Thus, in order to ensure complete cleavage of Si-S bonds of silylthioethers, further treatment with a suitable solvent, such as methanol (MeOH), is required, to enable silane alcoholysis and afford the desired PEG-dithiol 13.
Thus, by using this method, we were able to obtain the S-Mmt-PEG-dithiols 14 (PEG 10.000), 15 (PEG 4.000), and 16 (PEG 1.000), which upon treatment with TFA in DCM/TES (95:5) and further treatment with MeOH, afforded the corresponding deprotected PEG-dithiols 17 (PEG 10.000), 18 (PEG 4.000), and 19 (PEG 1.000) (Figure 4). All products (14-19) were obtained in high yield and purity, as confirmed by 1H and 13C-NMR analysis (Figures S6–S15). In addition, 1H-NMR analysis clearly indicated the quantitative esterification of both hydroxyl end groups of PEG with the applied S-Mmt mercapto acids and quantitative removal of S-Mmt protecting groups. ESI-MS analysis of 17, 18 and 19 was also indicative of the synthesized products (Figures S16–S18).
2.2.3. Synthesis of di-S-Mmt-pluronic-dithiols and pluronic-dithiols
Besides the linear poly(ethylene glycol) (PEG) polymer, our method was also applied in the esterification of the more complicated thermosensitive polyoxyethylene-polyoxypropylene triblock copolymer (PEO-PPO-PEO). For this, we used pluronic F127, which is composed of Poly(ethylene glycol)100-Poly(propylene glycol)65-Poly(ethylene glycol)100. This was acylated with S-Mmt-3-mercaptopropionic acid (6; R΄=H; r=2), which resulted in the synthesis of the di-S-Mmt-di-3-mercaptopropionyl pluronic 20 in high yield and purity. Cleavage of S-Mmt group with TFA in DCM/TES (95:5) and further treatment with MeOH (as previously explained), resulted to α,ω-di-3-mercaptopropionyl pluronic 21 in high yield and purity. 1H and 13C-NMR analysis was indicative of the synthesized products and proved the complete esterification of both hydroxyl end groups (Figures S19-S22). ESI-MS spectrum analysis was also performed and indicates the successful synthesis of 23 (Figure S23).
2.3. Thia-Michael reaction with pre-formed liposomes maleimides
2.3.1. General method
The general idea for the synthesis of nano-sized cross-linked liposomes is shown in Scheme 3. According to this approach, α,ω-bis-mercaptoacyl poly(alkyl oxides) (PAO-dithiols) react with preformed nano-sized liposomes 22 bearing maleimide groups on their surface, under thia-Michael addition conditions. It is obvious that the presence of a liposome dispersion in the reaction mixture enables, not only the linkage of one end group of PAO-dithiol with one maleimide group, as shown in the non-isolated intermediate 23, but also allows further reaction of the second available thiol group (of the initially formed 23) with a second available maleimide group. Obviously, the presence of maleimide groups on every liposomal particle, enables multi-directed thia-Michael reaction, which ensures the formation of a cross-linked liposomal scaffold consisted of thioether groups and poly(alkyl oxide) linkages between liposomes (24).
2.3.2. Monitoring of the reaction progress; Optimization
For this synthesis, we chose the high-molecular-weight di-2-mercaptopropionyl PEG10.000 17 (abbreviated PEG-dithiol hereafter), to be reacted with nano-sized SUV-type (Small Unilamellar Vesicles) liposomes, consisted of Phosphatidylcholine (PC), Cholesterol (Chol), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000-Mal) in a molar ratio of PC/Chol/DSPE-PEG2000-Mal 2:1:0.03 mol/mol (abbreviated PC-Lip-Mal hereafter), and the reaction progress of PEG-dithiol with the surface available maleimide groups was monitored, in real-time conditions, using a Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion NMR experiment (1H-NMR CPMG). This pulse sequence is known to eliminate the broad proton resonances of high molecular weight macromolecules (such as proteins, lipids, polysaccharides and other macromolecules), facilitating the observation of the signals of low molecular weight molecules [44,45,46]. By applying this method directly on preformed PC-Lip-Mal liposomes we were able to identify the maleimide groups. Figure 5 shows the 1D 1H-NMR CPMG spectrum of PC-Lip-Mal at a lipid concentration of 20 mg/ml (in D2O). The single sharp peak at 6.81 ppm corresponds to the protons of maleimide group that are present on the surface of liposomes. This was a clear indication that a Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion NMR experiment could be useful for monitoring the surface available maleimide protons, and therefore, the reaction progress of PEG-dithiol with maleimide groups on the surface of liposomes.
Another issue was to ensure the chemical stability over hydrolysis of maleimide groups when incubated in D2O at 27 oC (the conditions for the reaction of PEG-dithiol with liposome maleimides). As depicted in Figure 6, maleimide groups are not hydrolyzed for at least 3 h, indicating that liposome maleimides remain at their activated form and may react with PEG-dithiol for at least this time period.
Next, PEG-dithiol and PC-Lip-Mal were reacted in aqueous media (D2O) as the final step of our synthetic approach. For this reaction the lipid concentration was adjusted at 20 mg/ml, and liposomes were incubated at 27 oC (where our initial experiments/stability was performed) with a 2.5 molar excess of PEG-dithiol (in respect to maleimide groups). The reaction progress was monitored by the 1D 1H-NMR using the CPMG experiment. As depicted in Figure 7, the peak at 6.93 ppm, which corresponds to maleimide protons, decreases in time, and finally disappears within 2 h, indicating that all available maleimide groups have been reacted with the PEG-dithiol in this time period. In Figure 7, the marked in red area, which corresponds to the area where maleimide protons appear, has been expanded, and the peak which corresponds to maleimide protons has been indicated by a red arrow. It should be noted that CMPG method also suppressed the broad proton resonances of PEG-dithiol (which was added in the reaction mixture), allowing monitoring of the reaction progress by following the progressive decrease of the peak which corresponds to maleimide protons.
2.4. Effect of thioether cross-linking on liposome size
In order to investigate the effect of the molar excess of PEG dithiol and concentration of the reaction of the reaction mixture (i.e. liposome concentration), on the size of the liposomal system that is formed, we tested: (a) different excesses of PEG-dithiol in respect to maleimide groups (2.5, 5.0, 10.0 times molar excess), and (b) two liposome concentrations (of 20 and 40 mg/ml). In addition, two liposomal compositions were prepared; PC-Lips-Mal SUV liposomes consisted of PC/Chol/DSPE-PEG2000-Mal (2:1:0.03) mol/mol; and control liposomes (without maleimide groups) consisted of PC/Chol/DSPE-PEG2000-OMe (2:1:0.03) mol/mol. The physicochemical characteristics of the synthesized systems are summarized in Table 2 (for 20 mg/ml) and Table 3 (for 40 mg/ml) liposome concentrations.
As it is seen, increasing the excess of PEG-dithiol and lipid concentration, there is a significant effect on the hydrodynamic diameter (size) of PC-Lips-Mal, possibly because more extensive cross-linking between the PEG-dithiol and maleimide groups takes place. In brief, at 20 mg/ml lipid concentration, the size of the control liposomes (without maleimide) did not change during their reaction with PEG-dithiol (105.1 vs 101.8 nm), while the size of the corresponding PC-Lip-Mal increased about 30% (from 101.8 to 128.90 nm) for 2.5 molar excess of PEG-dithiol, while a 50% increase (from 101.8 to 153.10 nm) was seen for the highest tested PEG-dithiol excess (x10). A similar behavior was seen for the PC-Lip-Mal of 40 mg/ml, where the size of the control liposomes (without maleimide groups) remained unchanged during the incubation of liposomes with PEG-dithiol (89.38 vs 90.93 nm), while the PC-Lips-Mal showed about 40% increase for the 2.5 molar excess (from 90.93 to 138.30 nm) and about 50% increase for the highest tested excess (x10) (from 90.93 to 148 nm). Based on these findings, it can be safely said that the increase in size of PC-Lips-Mal, upon incubation with PEG-dithiol, is attributed to the reaction of PEG-dithiol with the surface-available maleimide groups of PC-Lip-Mal, which results in size increment.
The polydispersity index (PDI) is another important parameter for the homogeneity of a sample. Although the incubation of PEG-dithiol with control liposomes (without maleimide groups) had no effect on their size distribution, the reaction of PC-Lip-Mal with 2.5 molar excess (at 20 mg/ml) resulted in a PDI value of 0.199, slightly higher than the initial PDI value, indicative of an homogeneous sample. The corresponding value for the sample of 40 mg/ml was slightly higher (0.244), indicating a less homogenous sample. Higher excesses of PEG-dithiol (5.0 and 10.0 times relative to maleimide group) resulted in considerable increase of the polydispersity index (>0.25). The ζ-potential of all liposome structures (before and after the addition of PEG-dithiol) ranged between -2.20 to -2.65 mV.
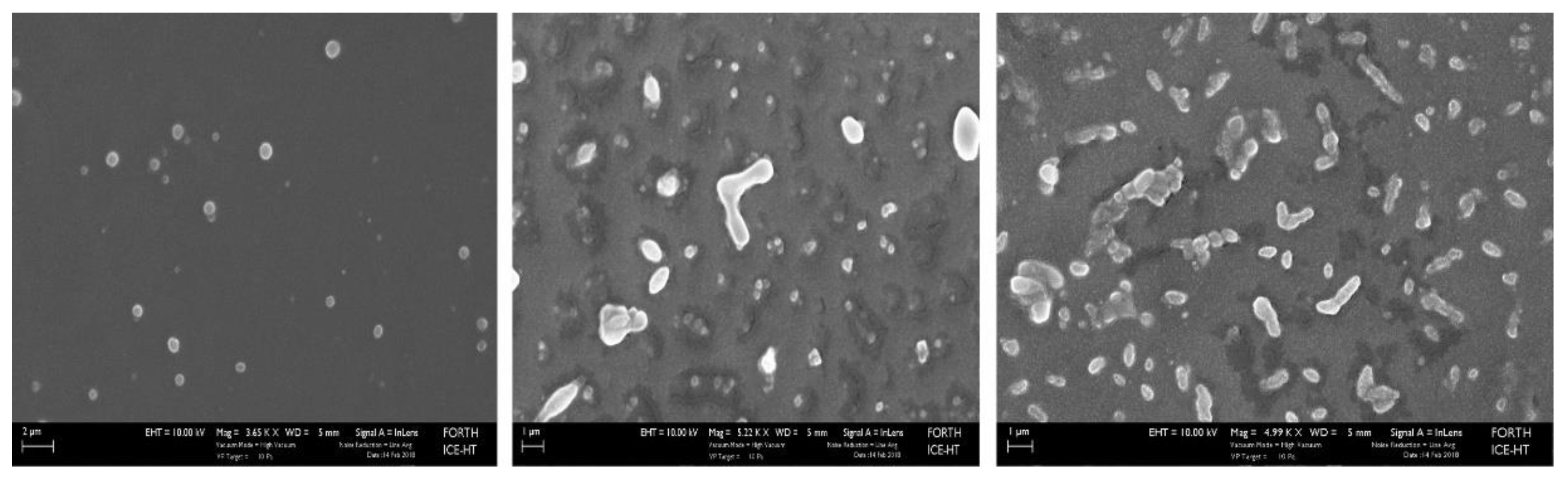
2.5. Scanning Electron Microscope (SEM) images
Representative SEM images of PC-Lip-Mal before reaction with PEG-dithiol (A) and after reaction with PEG-dithiol (B and C), verifies the cross-linking of liposomes, providing further evidence of the successful reaction of PEG-dithiols with the liposomal maleimide groups and the formation of nano-sized cross-linked liposomes.
Figure 8.
SEM images of PC-Lip-Mal (control liposomes) (A; scale bar: 2μm) and thioether cross-linked liposomes after the reaction of PC-Lip-Mal with PEG-dithiol (B, C; scale bar: 1 μm).
Figure 8.
SEM images of PC-Lip-Mal (control liposomes) (A; scale bar: 2μm) and thioether cross-linked liposomes after the reaction of PC-Lip-Mal with PEG-dithiol (B, C; scale bar: 1 μm).
2.6. Effect of thioether cross-linking on liposome scaffold viscosity
Further studies regarding the effect of the reaction of PEG-dithiol with liposome maleimide groups on the liposomes viscosity were performed. To this end, the reaction conditions were applied on Lips-Mal (PC/ Chol/ DSPE-PEG2000-Maleimide (2: 1: 0.03) mol/mol) as well as control liposomes (PC/ Chol/ DSPE-PEG2000-OMe (2: 1: 0.03) mol/mol). Two liposome concentrations were used (20 and 40 mg/ml), while the excess of PEG-dithiol in respect to maleimide groups was adjusted to 0.1, 2.5, 5.0 and 10.0 mg/ml (according to our previous investigations). A control liposome dispersion, with liposomes, was also used. The results of this experiment are shown in Figure 9.
As can be seen, the reaction of PEG-dithiol with Lips-Mal of 20 mg/ml resulted in a low, however considerable increase of liposome viscosity (Figure 9A), while a significantly higher increase of liposome dispersity was seen for the concentration of 40 mg/ml (Figure 9B). This was a clear effect of the reaction of PEG-dithiol with the surface available maleimide groups, providing further evidence for the successful development of a nano-sized (as described in section 2.4) thioether cross-linked liposome scaffold.
2.7. Evaluation of thioether cross-linked liposomes as drug eluting systems
In order to evaluate the potential of the synthesized cross-linked thioether type liposomes (abbreviated LIP-di-thioether-PEG hereafter) to be used as sustained release drug delivery systems, their integrity in buffer phosphate (PBS) at pH = 7.4 and Fetal Bovine Serum (FBS), was evaluated.
It is well know that liposome integrity is decreased in blood, due to liposome interaction with blood components, and their stability in blood can be increased by increasing their membrane integrity, by using more rigid lipids, the addition of cholesterol (Chol), and by coating liposome surface with PEG chains. Thus, in this set of experiments, PC lipid was replaced with hydrogenated PC (HPC) lipid, which is known to form more stable liposomes, while Chol and PEG were also used [47]. Furthermore, calcein (a chomophore) was encapsulated into liposomes, as a method to evaluate liposomes integrity and calcein release, when liposomes are incubated with PBS and FBS. The calcein leakage method is used as a prefered method to evaluate the integrity of liposome types, due to the fact that calcein is entrapped in the vesicles at a concentration where its fluoresence is quenched, and FI is increased as calcein leaks out of the vesicle (and is diluted in the liposome dispersion media) [48].
In this set of experiments, the following calcein encapsulated liposome compositions were prepared: (a) HPC/Chol/DSPE-PEG2000-OMe (2:1:0.03) mol/mol liposomes (control liposomes) and (b) HPC/Chol/DSPE-PEG2000-Mal liposomes (2:1:0.03) mol/mol (abbreviated HPC-Lip-Mal hereafter). The lipid concentration of control liposomes and HPC-Lip-Mal was set at 13.5 and 14.0 mg/ml (close to the optimal lipid concentration of the reaction of PEG-dithiol with PC-Lip-Mal). Next, calcein liposomes (control and HPC-Lip-Mal) were reacted with PEG-dithiol, under the optimimal conditions (2.5 molar excess, 2 h, 27 oC).
Liposome hydrodynamic diameter and polydispersity index (for both liposome compositions), before and after their reaction with PEG-dithiol, was measured (Table 4). As can be seen, the hydrodynamic diameter and polydispersity index of liposomes (control and HPC-Lip-Mal), before and after their reaction with PEG-dithiol, are consistent with our findings for PC-Lip-Mal liposomes. In brief, the size of HPC-Lip-Mal was increased from 100.3 nm to 145.7 nm (about 50% increment), whereas the size of control liposomes did not change (99.4 nm vs 100.8 nm).
The polydispersity index of the cross-linked liposomes was also increased [from 0.199 (before reaction) to 0.317 (after reaction)]. Regarding the PDI values, although the PDI value of HPC-Lip-Mal is higher (0.317) than the corresponding after reaction PDI value of PC-LIP-Mal (0.199), the liposome sample is still homogeneous (in the sense that no aggregates were present in the sample), as can be seen by the size distribution curves (supplementary file, Figure S24). The ζ-potential values ranged between -2.20 and -2.65 mV for all liposomes (similar values were seen for the PC-Lip-Mal reacted liposomes).
Regarding the integrity of liposomes, this was investigated by measuring the % latency of calcein encapsulating liposomes during their incubation in buffer phosphate (PBS) pH = 7.4 and fetal bovine serum (FBS) at 27 oC. The cross-linked liposomes, which were formed by the reaction of HPC-Lip-Mal with PEG-dithiol, and the corresponsing control liposomes (without maleimide groups/cross-linking) were used. The liposome concentrations that were studied, as determined by an initial optimization process, were fixed at 4 mg/ml, 2 mg/ml, 1 mg/ml, 0.5 mg/ml, 0.25 mg/ml. The results showed that, in presence of PBS, all liposome structures, control and cross-linked liposomes, were stable (up to 72 h) for all tested concentrations with % Latency values >95% (in all cases), indicating the high stability of the liposomal scaffolds (analytical graphs of liposome stability in PBS are shown in the supplementary file; Figure S25). On the contrary, in presence of FBS, both types of liposomes (control and cross-linked) showed an increase of the released calcein as lipid concentration was lowered (from 4 to 0.25 mg/ml), indicating the lower stability of the liposomal scaffold. It is noteworthy that calcein release was higher for control liposomes, than the corresponding crosslinked liposomes, at all time points (up to 72 hours) (Figure 10).
It is apparent that the cross-linked liposome scaffolds behaved differently than the corresponding control liposomes, by exhibiting higher stability (higher % latency) in presence of FBS, as a result of their cross-linking, after the reaction of the liposome surface with the PEG-dithiol, obviously resulting in a more rigid liposome scaffold. This effect becomes obvious at lower concentrations, where chemical modification of liposomes is more significantly involved in liposome stability, since fewer vesicles are incubated in the same amount of serum (and consequently of the serum components that are responsible for liposome membrane destabilization).
3. Materials and Methods
3.1. General Information
3.1.1. Synthesis of α,ω-bis-mercaptoacyl poly(alkyl oxide)s
Poly(ethylene glycol) (PEG) with average molecular weight 1.000 (950-1050) , 4.000 (3.500-4.500), 10.000 (9.000-12.500) and Poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (Pluronic F127) with an average MW 12.600 were purchased from Sigma-Aldrich (Darmstadt, Germany). In addition, 2-mercaptopropionic acid and 3-mercaptopropionic acid, as well as the reagents N,N′-diisopropylcarbodiimide (DIC), 4-(dimethylamino)pyridine (DMAP), trifluoroacetic acid (TFA) and triethylsilane (TES), were also purchased from Sigma-Aldrich (Darmstadt, Germany). 4-methoxytrityl chloride (Mmt-Cl) was gifted from CBL Patras S.A. All other chemicals were purchased from Sigma-Aldrich (Darmstadt, Germany). All solvents were of analytical or hplc grade and were purchased from Merck (Darmstadt, Germany).
Thin layer chromatography (TLC) was performed on silica gel 60 F254 plates (Merck, Darmstadt, Germany) and spot detection was carried out by UV light, and by charring with an aqueous solution of K2CO3/KMnO4. Flash column chromatography was performed on silica gel 230–400 mesh (Merck, Darmstadt, Germany). Hplc analysis was performed on a LiChrospher 100, RP-18, 250-4, 5μm column, using acetonitrile (AcCN) and water (H2O) in presence of 0.08% TFA as mobile phases. ESI-MS spectra were recorded on a Micromass Platform L.C. (Manchester, UK) at 30 eV. NMR spectra of the synthesized compounds were obtained at 600 MHz, on a Bruker Avance III HD spectrometer. NMR spectra of functionalized liposomes and real-time monitoring of the reaction of PEG-di-thiols with the functionalized liposomes were recorded at 27 oC on a Bruker Avance III High-Definition four-channel 700 MHz NMR spectrometer equipped with a cryogenically cooled 5 mm 1H/13C/15N/D Z-gradient probe (TCI). Chemical shifts (δ) were referenced to the corresponding solvent peaks and are reported in ppm.
3.1.2. Liposome preparations
Egg phosphatidylocholine (PC), Phosphatidylcholine, hydrogenated (HPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000-Mal) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000-OMe) were purchased from Lipoid, Germany. Cholesterol (Chol), Calcein, Triton X-100, Sephadex G-50, and Sepharose CL-4B were purchased from Sigma-Aldrich (Darmstadt, Germany). Fetal bovine serum (FBS) was obtained from Biosera (Nuaille, France). All solvents used were of analytical or hplc grade and were purchased from Merck (Darmstadt, Germany). Any other chemicals used were purchased from Sigma-Aldrich (Darmstadt, Germany).
For the liposome preparation a bath sonicator (Branson, Thermo Fisher Scientific, Waltham, MA, USA) or a microtip-probe high-intensity sonicator (Sonics and Materials, Leics, UK) was used. For liposome purification, size exclusion chromatography was performed on Sepharose CL-4B (Sigma-Aldrich, Darmstadt, Germany). A Shimatzu RF-1501 spectrofluorometer (Shimatzu, Kyoto, JP) was used for the measurement of the fluorescence intensity (FI) of calcein in samples (EX/EM 490 nm/525 nm; 5 nm slits).
3.1.3. Liposome physicochemical characterization
The mean hydrodynamic diameter and the polydispersity index (PDI) of the vesicles (dispersed in PBS at 0.2 mg/mL phospholipid concentration) was measured by dynamic light scattering (DLS) at 25 °C (173o angle) on a Malvern Nano-ZS (Malvern Instruments, Worcestershire, UK). Zeta potential (ζ-potential) values were measured also at 25 °C in the same samples by Doppler electrophoresis. Viscosity was recorded on a falling-ball viscometer (Anton Paar GmbH, Austria).
3.2. Synthetic Procedures
3.2.1. S-Mmt-mercaptopropionic acids (S-Mmt-2-mercaptopropionic acid 6a; S-Mmt-3-mercaptopropionic acid 6b)
4-methoxytrityl chloride (7; 2.5 g; 8.1 mmol) was dissolved in DCM (5.5 ml) and to this solution 2-mercaptopropionic acid (8a; 7.36 mmol; 0.64 ml) or 3-mercaptopropionic acid (8b; 7.36 mmol; 0.64 ml) was added and the reaction mixture was stirred at rt for about 30 min (the reaction progress was monitored by TLC). The reaction mixture was then concentrated on a rotary evaporator under reduced pressure and the oily product that was formed was diluted with ethyl acetate (EtOAc) and washed with water (x3). The organic layer was collected, and further dried over sodium sulfate and the filtrates were concentrated until the formation of an oily residue. To this, ether was added, and the resulting solution was placed at 4 oC, until the formation of white crystals. These were finally filtered and washed with ether (x3) to afford 6a and 6b as white solids: 6a (2.56 gr; yield: 92%); m.p. (153-155 oC); 6b (2.50 gr; yield 90%); m.p. 137-138 oC; 6a: 1H-NMR (MeOH-d4, 600 MHz) δ 7.46-7.40 (m, 4H, H-Ar), 7.35-7.31 (m, 2H, H-Ar); 7.31-7.25 (m, 4H, H-Ar); 7.25-7.19 (m, 2H, H-Ar), 6.87-6.82 (m, 2H, H-Ar), 3.79 (s, 3H, H-c), 2.92 (q, J = 7.3 Hz, 1H, H-b), 1.05 (d, J = 7.3 Hz, 3H, H-a); 13C-NMR (CDCl3, 150 MHz) δ 178.59 (C-e), 158.28, 144.48, 136.20, 130.87, 129.48, 127.96, 126.85, 113.21 (C-Ar), 67.88 (C-d), 55.22 (C-c), 42.43 (C-b), 18.58 (C-a); 6b: 1H-NMR (MeOH-d4, 600 MHz) δ 7.41-7.35 (m, 4H, H-Ar), 7.31-7.23 (m, 6H, H-Ar), 7.23-7.17 (m, 2H, H-Ar), 6.86-6.80 (m, 2H, H-Ar), 3.78 (s, 3H, H-c), 2.40 (t, J = 7.2 Hz, 2H, H-a), 2.20 (t, J = 7.2 Hz, 2H, H-b); 13C-NMR (CDCl3, 150 MHz) δ 176.87 (C-e), 158.12, 144.86, 136.63, 130.74, 129.45, 127.91, 126.66, 113.19 (C-Ar), 66.44 (C-d), 55.23 (C-c), 33.24 (C-b), 26.55 (C-a).
3.2.2. Di-S-Mmt-di-2-mercaptopropionyl PEG10.000 (14)
First, 6a (0.40 gr; 1.05 mmol) and DMAP (0.14 gr; 1.155 mmol) were placed into a round bottom flask and completely dissolved in DCM (4 ml). DIC (1.155 mmol; 0.18 ml) was then added and the resulting mixture was stirred for 5 min at room temperature. The activated 6a was then transferred to another flask where PEG10.000 (5 gr, 0.5 mmol) had been dissolved in DCM (8 ml), and the resulting reaction mixture was stirred overnight at room temperature. The reaction progress was monitored by TLC, where disappearance of the starting material (PEG), and formation of one product (14) was clear evidence of the reaction progress/completion. The reaction mixture was then filtered, and the filtrates were further extracted with DCM and 10% citric acid. The two layers were separated, and the organic layer was further washed with water (x2). Finally, the organic layer was dried over magnesium sulfate and the filtrates were concentrated under reduced pressure until the formation of an oily residue. To this, EtOAc (25 ml) was added, until the oil was dissolved, and then, diethyl ether (Et2O) was slowly added (25 ml) and the solution was allowed to stand at room temperature until the formation of white crystals began. Then, the solution was placed at 4 oC to allow complete crystallization. The white crystals that were formed were filtered and washed with a mixture of EtOAc:Et2O (1:1) (x1) and Et2O (x2) and then dried over P2O5 to afford 14 as white solid (4.96 gr; yield: 92.5%); m.p.: 55-57 oC; 1H-NMR (MeOH-d4, 600 MHz) δ 7.46-7.39 (m, 8H, H-Ar), 7.37-7.26 (m, 12H, H-Ar), 7.26-7.20 (m, 4H, H-Ar), 6.89-6.83 (m, 4H, H-Ar), 4.12-4.08 (m, 2H, H-c), 3.97-3.93 (m, 2H, H-c), 3.79 (s, 6H, H-g), 3.77-3.46 (H-d,e,f), 2.95 (q, J = 7.2 Hz, 2H, H-b), 1.12 (d, J = 7.3 Hz, 6H, H-a); 13C-NMR (CDCl3, 150 MHz) δ 173.54 (C-i), 158.15, 144.68, 144.56, 136.36, 130.88, 129.54, 129.48, 127.86, 126.69, 113.11 (C-Ar), 70.54, 68.74 (C-d,e,f), 67.67 (C-h), 64.04 (C-c), 55.20 (C-g), 42.33 (C-b), 18.67 (C-a).
3.2.3. Di-2-mercaptopropionyl PEG10.000 (17)
A mixture of 5% TFA in DCM/TES (95:5) (80 ml) was added into a round flask containing 14 (4.0 gr; 0.37 mmol) and the mixture was stirred at room temperature, where a gradual discoloration of the cleavage mixture was observed. Complete/irreversible cleavage was confirmed by adding a few drops of concentrated TFA, until no color reappeared. TLC was also indicative of a complete/irreversible cleavage of S-Mmt groups. Then, the cleavage mixture was concentrated under reduced pressure, and the resulting oily product was treated with an excess of MeOH and concentrated again (twice). To the oily product that was finally formed, Et2O (40 ml) was added, and the mixture was left at room temperature until precipitation began and then it was placed at 4 oC to allow complete precipitation. The solid was finally filtered and washed twice with Et2O and dried over P2O5 to afford 17 as white solid (3.26 gr; yield: 86%); m.p.: 57-58 oC; 1H-NMR (CDCl3, 600 MHz) δ 4.30-4.26 (m, 4H, H-c), 3.77.3.49 (H-b, d, e, f), 2.19 (d, 2H, J = 8.3 Hz, SH), 1.52 (d, J = 7.0Hz, 6H, H-a); 13C-NMR (CDCl3, 150 MHz) δ 173.69 (C-g), 70.54, 68.91 (C-d, e, f), 64.44 (C-c), 35.54 (C-b), 20.97 (C-a).
3.2.4. Di-S-Mmt-di-2-mercaptopropionyl PEG4.000 (15)
First, 6a (0.2 gr; 0.525 mmol) and DMAP (70.55 mg; 0.58 mmol) were completely dissolved in DCM (4 ml). DIC (0.58 mmol; 90.42 μl) was added and the mixture was stirred for 5 min at room temperature. The activated 6a was then added to another flask where PEG4.000 (1.0 gr, 0.25 mmol) had been dissolved in DCM (3 ml), and the resulting mixture was stirred overnight at room temperature. The reaction progress was monitored by TLC, where disappearance of the starting material (PEG), and formation of one product (15) was clear evidence of the reaction progress/completion. The reaction mixture was then filtered, and the filtrates were further extracted in DCM and 10% citric acid. The two layers were separated, and the organic layer was further washed with water (x2). Finally, the organic layer was dried over magnesium sulfate and the filtrates were concentrated under reduced pressure until the formation of an oily residue. This was further crystallized in boiling ethanol (EtOH) and the crystalline product was filtered at 4oC (and further recrystallized in EtOH). The crystals were collected and dried over P2O5 to afford 15 as white solid (1.05 gr; yield: 94%); m.p.: 47-48 oC; 1H-NMR (CDCl3, 600 MHz) δ 7.46-7.40 (m, 8H, H-Ar), 7.35-7.31 (m, 4H, H-Ar), 7.29-7.24 (m, 8H, H-Ar), 7.22-7.17 (m, 4H, H-Ar), 6.78-6.82 (m, 4H, H-Ar), 4.13-4.08 (m, 2H, H-c), 3.98-3.93 (m, 2H, H-c), 3.78 (s, 6H, H-g), 3.77-3.50 (H-d,e,f), 2.98 (q, J = 7.3 Hz, 2H, H-b), 1.16 (d, J = 7.3 Hz, 6H, H-a); 13C-NMR (CDCl3, 150 MHz) δ 173.55 (C-i), 158.16, 144.68, 144.56, 136.36, 130.88, 129.54, 129.48, 127.86, 126.69, 113.11 (C-Ar), 70.54, 68.74 (C-d,e,f), 67.67 (C-h), 64.04 (C-c), 55.20 (C-g), 42.33 (C-b), 18.67 (C-a).
3.2.5. Di-2-mercaptopropionyl PEG4.000 (18)
A mixture of 5% TFA in DCM/TES (95:5) (10 ml) was added into a round flask containing 15 (0.5 gr), and the mixture was stirred at room temperature, where a gradual discoloration of the cleavage mixture was observed. Complete/irreversible cleavage was confirmed by adding a few drops of concentrated TFA, until no color reappeared. TLC was also indicative of a complete/irreversible cleavage of S-Mmt groups. Then, the cleavage mixture was concentrated under reduced pressure, and the resulting oily product was treated with an excess of MeOH and concentrated again (twice). To the oily product that was formed, Et2O was added, and the mixture was left at room temperature until precipitation started and then it was placed at 4 oC to allow complete precipitation. The precipitate was finally filtered, washed with Et2O (x2) and further dried over P2O5 to afford 18 as white solid (0.41 gr; yield: 93%); m.p.: 50-51.5 oC; 1H-NMR (CDCl3 600 MHz) δ 4.29-4.26 (m, 4H, H-c), 3.77.3.49 (H-b, d, e, f), 2.19 (d, 2H, J = 8.3 Hz, SH), 1.51 (d, J = 7.0 Hz, 6H, H-a); 13C NMR (CDCl3, 150 MHz) δ 173.70 (C-g), 70.55, 68.91 (C-d, e, f), 64.44 (C-c), 35.54 (C-b), 20.97 (C-a).
3.2.6. Di-S-Mmt-di-2-mercaptopropionyl PEG1.000 (16)
First, 6a (1.6 gr; 4.2 mmol) and DMAP (0.56 gr; 4.6 mmol) were completely dissolved in DCM (4 ml). DIC (4.6 mmol; 0.72 ml) was added and the mixture was stirred for 5 min at room temperature. The activated 6a was then transferred to another flask where PEG1.000 (2.0 gr, 2.0 mmol) had been dissolved in DCM (3 ml), and the reaction mixture was stirred overnight at room temperature. The reaction progress was monitored by TLC, where disappearance of the starting material (PEG1.000), and formation of one product (16) was clear evidence of the reaction progress/completion. The reaction mixture was then filtered, and the filtrates were further extracted in DCM and 10% citric acid. The two layers were separated, and the organic layer was further washed with water (x2). Finally, the organic layer was dried over magnesium sulfate and the filtrates were concentrated under reduced pressure until the formation of 16 as an oily residue, which was directly used in the next step.
3.2.7. Di-2-mercaptopropionyl PEG1.000 (19)
A mixture of 5% TFA in DCM/TES (95:5) (80 ml) was poured into the round flask containing 16, and the mixture was stirred at room temperature, where a gradual discoloration of the cleavage mixture was observed. Complete/irreversible cleavage of Mmt groups was confirmed by adding a few drops of concentrated TFA, until no color reappeared. TLC analysis was also indicative of a complete/irreversible cleavage of S-Mmt groups. Then, the cleavage mixture was concentrated, and the resulting oily product that was formed was treated with an excess of MeOH and evaporated again (twice). The resulting oily product was washed several times with hexane (Hex) and finally dried over P2O5 to afford 19 as oil (2.1 gr; yield: 89.4% over two steps); 1H-NMR (CDCl3 600 MHz) δ 4.25-4.31 (m, 4H, H-c), 3.80.3.49 (H-b, d, e, f), 2.19 (d, 2H, J = 8.3 Hz, SH), 1.51 (d, 6H, J = 7.0 Hz, H-a); 13C NMR (CDCl3, 150 MHz) δ 173.70 (C-g), 70.55, 68.91 (C-d, e, f), 64.44 (C-c), 35.54 (C-b), 20.97 (C-a).
3.2.8. Di-S-Mmt-di-3-mercaptopropionyl Pluronic (20)
First, 6b (0.126 gr; 0.33 mmol) and DMAP (44.8 mg; 0.37 mmol) were completely dissolved in DCM (0.5 ml). DIC (0.37 mmol; 57.4 μl) was then added and the mixture was stirred for 5 min at room temperature. The activated 6b was then transferred to another flask where pluronic (2.0 gr; 0.159 mmol) had been dissolved in DCM (3 ml), and the reaction mixture was stirred overnight at room temperature. The reaction progress was monitored by TLC analysis, where disappearance of the starting material (pluronic), and formation of one product (20), was clear evidence of the reaction progress/completion. Then the reaction mixture was filtered, and the filtrates were extracted in DCM and 10% citric acid. The two layers were separated, and the organic layer was further washed with water (x2). The organic layer was dried over magnesium sulfate and filtered, and the filtrates were concentrated until the formation of an oily residue. To this, Et2O (40 ml) was added, and the white precipitate that was formed was filtered and further washed with Et2O (x3), and finally dried over P2O5 to afford 20 as a white solid (1.97 gr; yield: 93%); m.p.: 48.5-50 oC; 1H-NMR (CDCl3, 600 MHz) δ 7.40-7.35 (m, 8H, H-Ar), 7.30-7.22 (m, 12H, H-Ar), 7.20-7.16 (m, 4H, H-Ar), 6.81-6.77 (m, 4H, H-Ar), 4.19-4.14 (m, 4H, H-c), 3.77 (s, 6H, H-j), 3.75-3.58 (H-d, e, f), 3.57-3.48 (m, 130H, H-h), 3.41-3.33 (m, 65H, H-g), 2.43 (t, J =7.3 Hz, 4H, H-a), 2.27 (t, 4H, J =7.3 Hz, H-b), 1.14-1.09 (m, 195H, H-i); 13C-NMR (CDCl3, 150 MHz) δ 171.82 (C-l), 158.07, 144.94, 136.69, 130.74, 129.45, 127.87, 126.60, 113.15 (C-Ar), 75.50, 75.35, 75.30, 75.10, 73.34, 72.94, 72.89, 72.85, 72.80, 70.82, 70.54, 69.02, 68.49, 63.72 (C-PEO, C-PPO), 68.49 (C-k), 55.20 (C-j), 33.45 (C-b), 26.86 (C-a), 17.43, 17.30 (C-i).
3.2.9. Di-3-mercaptopropionyl Pluronic (21)
A mixture of 5% TFA in DCM/TES (95:5) (26.5 ml) was added into a round flask containing 20 and the mixture was stirred at room temperature, where a gradual discoloration of the cleavage mixture was observed. Complete/irreversible cleavage of Mmt groups was confirmed by adding a few drops of concentrated TFA, until no color reappeared. TLC was also indicative of a complete/irreversible cleavage of S-Mmt groups. Then, the cleavage mixture was concentrated under reduced pressure, and the resulting oily product was treated with an excess of MeOH and concentrated again (twice). To the oily product that was formed, Et2O (30 ml) was added, until the formation of a white solid, and the suspension was left to stand at room temperature, overnight, to allow complete precipitation. The precipitate was finally filtered, washed with Et2O (x3) and dried over P2O5 to afford 21 as white solid (1.85 gr; yield: 91% over two steps); m.p.: 51-52.5 oC; 1H-NMR (CDCl3, 600 MHz) δ 4.26-4.22 (m, 4H, H-c), 3.76-3.57 (H-d, e, f), 3.56-3.43 (m, 130H, H-h), 3.41-3.31 (m, H65, H-g), 2.78-2.72 (m, 4H, H-a), 2.68-2.62 (m, 4H, H-b), 1.66 (t, 2H, J = 8.3 Hz, SH), 1.16-1.05 (m, 195H, H-i); 13C-NMR (CDCl3, 150 MHz) δ 171.62 (C-j), 75.50, 75.34, 75.30, 75.10, 73.34, 72.94, 72.90, 72.85, 72.81, 72,56, 70.82, 70.54, 70.29, 69.02, 68.58, 68.49, 63.87, 61.69 (C-c, d, e, f, g, h), 33.99 (C-b), 32.99 (C-a), 17.43, 17.31 (C-i).
3.2.10. Liposome Preparation Procedure
Small unilamellar vesicle (SUV) -type liposomes, were prepared by the thin film hydration method and high-intensity sonication. In brief, the appropriate amounts of lipids (PC or HPC; Chol; DSPE-PEG2000-OMe or DPSE-PEG2000-Mal) were dissolved in a chloroform (CHCl3)/MeOH (2:1 v/v) mixture and the resulting mixture was evaporated under reduced pressure until the formation of a thin film lipid layer. The lipid film was treated with N2 gas and was subsequently connected to a vacuum pump for 12 h, in order to remove any traces of organic solvent. The lipid film was next hydrated with PBS buffer (pH 7.40) at room temperature -in case of PC containing liposomes-, or at 60 oC -for HPC containing liposomes-. For calcein encapsulating liposomes a 100 mM solution of calcein, prepared in the same buffer, was used for thin film hydration. After complete lipid hydration and formation of multilamellar vesicle (MLV) -type liposomes, the liposome dispersion was placed under the microtip of a probe sonicator for 10 min, or until the liposome dispersion was completely clear. The liposome dispersion was then allowed to stand for 1 h at a temperature above the lipid transition temperature to anneal any structural defects. The calcein encapsulating liposomes were purified from non-entrapped calcein by size-exclusion chromatography on a Sepharose 4B-CL column that was eluted with PBS pH 7.40. The Stewart assay was used for the measurement of the phospholipid content/concentration of liposomes.
3.2.11. Reaction of di-thioether-PEGs with pre-formed LIP-maleimides
Pre-formed liposomes-maleimide of the appropriate lipid concentration were incubated with PEG-di-thiols at 27 oC in PBS pH 7.40. The obtained cross-linked thioether liposome scaffolds were purified from the non-reacted PEG-di-thiol with size-exclusion chromatography on a Sepharose 4B-CL column eluted with PBS pH 7.40. The lipid concentration, before and after chromatographic purification, was measured by the Stewart assay.
3.2.12. Liposome Integrity Studies
To monitor the integrity of liposomes (control and after their reaction with PEG-di-thiol), calcein-encapsulating liposomes (4 mg/ml, 2 mg/ml, 1 mg/ml, 0.5 mg/ml, 0.25 mg/ml phospholipid) were prepared. Calcein was encapsulated in liposomes at 100 mM concentration (where calcein fluorescence is quenched), and liposomes were incubated at 37 °C in PBS as well as in FCS (80% v/v). Calcein latency (%) values were calculated at various time points, by taking samples from the incubated liposomes and measuring calcein FI. In brief, at each time point, 20 μl of vesicle dispersions were taken and diluted in 4 ml of PBS buffer. The FI was measured (EX 470 nm, EM 520 nm) before and after the addition of Triton X-100 at a final concentration of 1% v/v (that ensures complete liposome disruption and release of all encapsulated (and latent) dye. Latency (%) was calculated from the equation: Latency (%) = 100 x [(1.1x Fat) – Fbt]/ (1.1 x Fat), where Fbt and Fat are calcein fluorescence intensities before and after the addition of Triton X-100, respectively (for Fat multiplication by 1.1 was applied for correction due to dilution).
3.2.13. Scanning Electron Microscopy (SEM)
The vesicular morphology of the liposome preparations was recorded by means of SEM. SEM images of PC-Lip-Mal before their reaction with PEG-di-thiol and after their reaction with PEG-di-thiol were recorded. In brief, 100 ul of each sample were placed on clean specimen mounts which were dried overnight first in a fume hood and then under vacuum (2.25 Pa). Subsequently, the samples were sputtered with gold and observed in a JEOL JSM-35 operating at an acceleration voltage of 25 kV with 0° tilt (×500).
4. Conclusions
In this manuscript, we propose a new approach for the simple and efficient synthesis of homobifunctional mercaptoacylated poly(alkyl oxide)s, using Poly(ethylene glycol)s and the more complicated Poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (Pluronic), as examples. Our method is based on the esterification of suitably S-Mmt protected mercapto acids with the end group hydroxyls, by the use of DIC and DMAP as the carboxylic acid activating agents. The advantages of this approach are summarized below: (a) The use of commercially available or synthesized S-Mmt mercapto acids allows structural diversity of the added mercaptoacyl groups; (b) Previously reported methods make use of high excess of the mercapto acid (usually a x10 to x20 times excess), as well as relatively harsh reaction conditions and specially designed glassware (reflux conditions for 24 h and the azeotropic mixture is periodically collected by Dean-Stark trap). In our approach, an equimolar amount of the S-Mmt-mercapto acid in respect to the available hydroxyl groups is used and the reaction is performed at room temperature by the use of simple esterification reagents. In addition, no reducing agents and/or inert conditions are required, minimizing the necessary glassware to a single flask. Moreover, there are no unpleasant odors during the reaction or work up/purification procedures (usually encountered in presence of thiols); (c) Protection of the thiol-groups during the esterification reaction ensures that no byproducts are formed during the reaction process, while cleavage of the labile S-Mmt group can be achieved fast and irreversibly in mild acidic conditions; (d) The applied conditions allows quantitative functionalization of both α,ω-hydroxyls; (e) The S-Mmt-protected poly(alkyl oxide)s can be stored until the corresponding free thiols are needed, avoiding the formation of any oxidation by-products during long term storage. According to our knowledge, this is the first time that S-Mmt-marcapto acids were used for the synthesis of homobifunctional mercaptoacylated poly(alkyl oxide)s, while the applied methods allow an easy and efficient synthesis of the title compounds.
Further, we proved the pharmacological interest of the synthesized compounds, as cross-linking agents, by the synthesis of a novel biomacromolecular nano-sized liposome scaffold, containing cross-linked liposomes via thioether PEG linkages. This was achieved by the reaction of pre-formed liposomes bearing maleimide groups on their surface with PEG-dithiol. The reaction progress -and completion- was followed by using a 1H-NMR Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiment (1H-NMR CPMG). According to our knowledge, this is the first time that 1H-NMR CPMG was used for the real time monitoring of the reaction between surface available nano-sized particle functionalities and organic compounds of any type, including PEG-dithiol macromolecules.
The liposomal scaffold that was synthesized is the third novelty of this work. According to our knowledge, this is the first cross-linked liposomal scaffold that has been synthesized up to date which maintains the nano characteristics of the initial liposomes. This scaffold allowed the sustained release of a chromophore (calcein) providing evidence for the efficient synthesis of an innovating nano-sized liposome scaffold for sustained release of drugs.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1. 1H-NMR of S-Mmt-2-mercaptopropionic acid (thiolactic acid) in MeOH-d4; Figure S2. 13C-NMR of S-Mmt-2-mercaptopropionic acid (thiolactic acid) in CDCl3; Figure S3. 1H-NMR of S-Mmt-3-mercaptopropionic acid in MeOH-d4; Figure S4. 13C-NMR of S-Mmt-3-mercaptopropionic acid in CDCl3; Figure S5. Analytical hplc of S-Mmt-3-mercaptopropionic acid (A1, A2) and S-Mmt-2-mercaptopropionic acid (thiolactic acid) (B1, B2) at 214 and 265 nm; Column: LiChrospher 100, RP-18, 250-4, 5 μm; Mobile phase solvents: AcCN/water (0.08% TFA); Gradient conditions: 20% to 100% AcCN in 30 min; Figure S6. 1H-NMR of di-S-Mmt-di-2-mercaptopropionyl PEG10.000 in MeOH-d4; Figure S7. 13C-NMR of di-S-Mmt-di-2-mercaptopropionyl PEG10.000 in CDCl3; Figure S8. 1H-NMR of di-2-mercaptopropionyl PEG10.000 in CDCl3; Figure S9. 13C-NMR of di-2-mercaptopropionyl PEG10.000 in CDCl3; Figure S10. 1H-NMR of di-S-Mmt-2-mercaptopropionyl PEG4.000 in CDCl3; Figure S11. 13C-NMR of di-S-Mmt-2-mercaptopropionyl PEG4.000 in CDCl3; Figure S12. 1H-NMR of di-2-mercaptopropionyl PEG4.000 in CDCl3; Figure S13. 1H-NMR of di-2-mercaptopropionyl PEG4.000 in CDCl3; Figure S14. 1H-NMR of di-2-mercaptopropionyl PEG1.000 in CDCl3; Figure S15. 1H-NMR of di-2-mercaptopropionyl PEG1.000 in CDCl3; Figure S16. ESI-MS of di-2-mercaptopropionyl PEG10.000; (M+12H)/12; Figure S17. ESI-MS of di-2-mercaptopropionyl PEG4.000; (M+6H)/6; Figure S18. ESI-MS of di-2-mercaptopropionyl PEG1.000; (M+2H)/2, M+H; Figure S19. 1H-NMR of di-S-Mmt-di-3-mercaptopropionyl pluronic in CDCl3; Figure S20. 13C-NMR of di-S-Mmt-di-3-mercaptopropionyl pluronic in CDCl3; Figure S21. 1H-NMR of di-3-mercaptopropionyl pluronic in CDCl3; Figure S22. 13C-NMR of di-3-mercaptopropionyl pluronic in CDCl3; Figure S23. di-3-mercaptopropionyl Pluronic F127; M+14H/14, M+15H/15; Figure S24. Size distribution before (A) and after reaction (B) for control liposomes. Size distribution before (C) and after reaction (D) for Lips-Mal; Figure S25. % Latency of calcein by the incubation of liposomes in PBS pH=7.40 at 27 oC. Control liposomes (HPC-Lip; PBS-NO MAL) and thioether cross-linked liposomes (HPC-LIP-di-thioether-PEG; PBS-MAL) at different concentrations (4 mg/ml, 2 mg/ml, 1 mg/ml, 0.5 mg/ml, 0.25 mg/ml).
Author Contributions
Conceptualization, S.A. and S.M.; methodology, S.A., S.M.; investigation, G.K., S.K., S.M.; validation, G.K., S.K., S.M., P.K.; formal analysis, G.K., S.K., S.M., P.K.; supervision, S.M., S.A., data curation, G.K., S.K., S.M.; project administration, S.A., S.M.; funding acquisition, S.A.; writing - original draft, S.M.; writing - review and editing, S.M., S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors.
Acknowledgments
We acknowledge access to 700 MHz NMR through the project “INSPIRED-The National Research Infrastructures on Integrated Structural Biology, Drug Screening Efforts and Drug target functional characterization” (MIS 5002550), funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF, 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund) and EU FP7 REGPOT CT-2011-285950 – “SEE-DRUG” project for the purchase of UPAT's 700 MHz NMR; We also acknowledge Professor Georgios Spyroulias, Department of Pharmacy, School of Health Sciences, University of Patras, Rio Patras 26510, Greece, who successfully applied the Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence for the real-time monitoring of the reaction between PEG-dithiols and liposome maleimide groups; Part of this work is from the master’s thesis of G.K. and S.K., submitted to the Department of Pharmacy, University of Patras; The authors are grateful to CBL Patras, Patras Industrial Area, Block 1, 25018 Patras, Greece and Professor Emeritus Kleomenis Barlos, for kindly providing 4-methoxytrityl chloride.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Bré, L.P.; Zheng, Y.; Pêgo, A.P.; Wang, W. Taking tissue adhesives to the future: From traditional synthetic to new biomimetic approaches. Biomater. Sci. 2013, 1, 239-253. [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28-51. [CrossRef]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today (2005) 10, 1451–1458. [CrossRef]
- Alconcel, S.N.S.; Baas, A.S.; Maynard, H.D. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [CrossRef]
- Rahdar, A.; Kazemi, S.; Askari, F. Pluronic as nano-carier for drug delivery systems. Nanomed. Res. J. 2018, 3, 174-179. [CrossRef]
- Yu, J.; Qiu, H.; Yin, S.; Wang, H.; Li, Y. Polymeric Drug Delivery System Based on Pluronics for Cancer Treatment. Molecules 2012, 26, 3610. [CrossRef]
- Batrakova, E.V.; Kabanov, A.V. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J. Control. Release 2008, 130, 98-106. [CrossRef]
- Hutanu, D.; Frishberg, M.D.; Guo, L.; Darie, C.C. Recent Applications of Polyethylene Glycols (PEGs) and PEG Derivatives. Mod. Chem. Appl. 2014, 2, 1-6. [CrossRef]
- Harris, J.M.; Kozlowski, A. Poly(ethylene glycol) derivatives with proximal reactive groups. US Patent: US6437025B1.
- Ravasco, J.M.; Faustino, H.; Trindade, A.; Gois, P.M.P. Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chem. Eur. J. 2018, 25, 43-59. [CrossRef]
- Renault, K.; Fredy, J.W.; Renard, P.Y.; Sabot, C. Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjug. Chem. 2018, 29, 2497-2513. [CrossRef]
- Kharkar, P.M.; Rehmann, M.S.; Skeens, K.M.; Maverakis, E.; Kloxin, A.M. Thiol-ene Click Hydrogels for Therapeutic Delivery. ACS Biomater. Sci. Eng. 2016, 2, 165-179. [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry, Angewandte Chemie (International ed. in English) 2010, 49, 1540-1573. [CrossRef]
- Goessl, A.; Tirelli, N.; Hubbell, J.A. A Hydrogel System for Stimulus-Responsive, Oxygen-Sensitive In Situ Gelation. J. Biomater. Sci. Polym. Ed. 2004, 15, 895-904. [CrossRef]
- Buwalda, S.J.; Dijkstra, P.J.; Feijen, J. In Situ Forming Poly(Ethylene Glycol)-Poly(L-Lactide) Hydrogels via Michael Addition: Mechanical Properties, Degradation, and Protein Release. Macromol. Chem. Phys. 2012, 213, 766–775. [CrossRef]
- Hiemstra, C.; van der Aa, L.J.; Zhong, Z.; Dijkstra, P.J.; Feijen, J. Novel In Situ Forming, Degradable Dextran Hydrogels by Michael Addition Chemistry: Synthesis, Rheology, and Degradation. Macromolecules 2007, 40, 1165–1173. [CrossRef]
- Yoshimoto, K.; Hirase, T.; Nemoto, S.; Hatta, T.; Nagasaki, Y. Facile Construction of Sulfanyl-Terminated Poly(Ethylene Glycol)-Brushed Layer on a Gold Surface for Protein Immobilization by the Combined use of Sulfanyl-Ended Telechelic and Semitelechelic Poly(Ethylene Glycol)s. Langmuir 2008, 24, 9623-9629. [CrossRef]
- Nie, T.; Baldwin, A.; Yamaguchi, N.; Kiick, K.L. Production of Heparin-Functionalized Hydrogels for the Development of Responsive and Controlled Growth Factor Delivery Systems. J. Control. Release 2007, 122, 287-296. [CrossRef]
- Belair, D.G.; Miller, M.J.; Wang, S.; Darjatmoko, S.R.; Binder, B.Y.; Sheibani, N.; Murphy, W.L. Differential Regulation of Angiogenesis using Degradable VEGF-Binding Microspheres. Biomaterials 2016, 93, 7-37. [CrossRef]
- Yu, H.; Feng, Z.G.; Zhang, A.Y.; Sun, L.G.; Qian, L. Synthesis and Characterization of Three-Dimensional Crosslinked Networks Based on Self-Assembly of α-Cyclodextrins with Thiolated 4-arm PEG using a Three-Step Oxidation. Soft. Matter. 2006, 2, 343-349. [CrossRef]
- Du, Y.J.; Brash, J.L. Synthesis and Characterization of thiol-terminated Poly(Ethylene Oxide) for Chemisorption to Gold Surface. J. Appl. Polym. Sci. 2003, 90, 594-607. [CrossRef]
- Wan, J.K.S.; Depew, M.C. Some Mechanistic Insights in the Behaviour of Thiol Containing Antioxidant Polymers in Lignin Oxidation Processes. Res. Chem. Intermed. 1996, 22, 241–253. [CrossRef]
- Yang, T.; Long, H.; Malkoch, M.; Kristofer Gamstedt E., Berglund, L.; Hult, A. Characterization of Well-Defined Poly(Ethylene Glycol) Hydrogels Prepared by Thiol-Ene Chemistry. Polym. Chem. 2011, 49, 4044-4054. [CrossRef]
- Zhang, H.J.; Xin, Y.; Yan, Q.; Zhou, L.L.; Peng, L.; Yuan, J.Y. Facile and Efficient Fabrication of Photoresponsive Microgels via Thiol-Michael Addition. Macromol. Rapid Commun. 2012, 33, 1952-1957. [CrossRef]
- Zustiak, S.P.; Leach, J.B. Hydrolytically Degradable Poly(Ethylene Glycol) Hydrogel Scaffolds with Tunable Degradation and Mechanical Properties. Biomacromolecules 2010, 11, 1348-1357. [CrossRef]
- Tong, X.; Lee, S.; Bararpour, L.; Yang, F. Long-term Controlled Protein Release from Poly(ethylene glycol) Hydrogels by Modulating Mesh Size and Degradation. Macromol. Biosci. 2015, 15, 1679-1686. [CrossRef]
- Picheth, G.F.; da Silva, L.C.E.; Giglio, L.P.; Plivelic, T.S.; de Oliveira, M.G. S-nitrosothiol-terminated Pluronic F127: Influence of microstructure on nitric oxide release. J. Colloid Interface Sci. 2020, 576, 457–467. [CrossRef]
- Niu, G.; Zhang, H.; Song, L.; Cui, L.; Cao, H.; Zheng, Y.; Zhu, S.; Yang, Z.; Yang, H. Thiol/acrylate-modified PEO-PPO-PEO triblocks used as reactive and thermosensitive copolymers. Biomacromolecules 2008, 9, 2621–2628. [CrossRef]
- Mulay, P.; Shrikhande, G.; Puskas, J. Synthesis of Mono- and Dithiols of Tetraethylene Glycol and Poly(ethylene glycol)s via Enzyme Catalysis. Catalysts 2019, 9, 228. [CrossRef]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019, 164, 640-653. [CrossRef]
- Liu, P.; Chen, G.; Zhang, J. A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives. Molecules 2022, 27, 1372. [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [CrossRef]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: a review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319-3329. [CrossRef]
- Liang, Y.; Kiick, K.L. Liposome-cross-linked hybrid hydrogels for glutathione- triggered delivery of multiple cargo molecules. Biomacromolecules 2016, 17, 601-614. [CrossRef]
- Spears, R. J.; McMahon, C.; Chudasama, V. Cysteine protecting groups: applications in peptide and protein science. Chem. Soc. Rev. 2021, 50, 11098-11155. [CrossRef]
- Mourtas, S.; Gatos, D.; Kalaitzi, V.; Katakalou, C.; Barlos, K. S-4-Methoxytrityl mercapto acids: synthesis and application. Tetrahedron Lett. 2001, 42, 6965-6967. [CrossRef]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. 1978, 17, 522-524. [CrossRef]
- Tsakos, M.; Schaffert, E.S.; Clement, L.L.; Villadsen, N.L.; Poulsen, T.B. Ester coupling reactions – an enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 2015, 32, 605-632. [CrossRef]
- McCourt, R.O.; Scanlan, E.M. A Sequential Acyl Thiol-Ene and Thiolactonization Approach for the Synthesis of δ-Thiolactones. Org. Lett. 2019, 21, 3460-3464. [CrossRef]
- Espeel, P.; Du Prez, F.E.; One-pot multi-step reactions based on thiolactone chemistry: A powerful synthetic tool in polymer science. Eur. Polym. J. 2015, 62, 247-272. [CrossRef]
- Suzuki, M.; Makimura, K.; Matsuoka, S. Thiol-Mediated Controlled Ring-Opening Polymerization of Cysteine-Derived β-Thiolactone and Unique Features of Product Polythioester. Biomacromolecules 2016, 17, 1135−1141. [CrossRef]
- Pearson, D.A.; Blanchette, M.; Baker, M.L.; Guindon, C.A. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989, 30, 2739-2742. [CrossRef]
- Chung, M.-K.; Schla, M. A Catalytic Synthesis of Thiosilanes and Silthianes: Palladium Nanoparticle-Mediated Cross-Coupling of Silanes with Thio Phenyl and Thio Vinyl Ethers through Selective Carbon-Sulfur Bond Activation. J. Am. Chem. Soc. 2004, 126, 7386–7392. [CrossRef]
- Kleckner, I.R.; Foster, M.P. An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta. 2011, 1814, 942-968. [CrossRef]
- Palmer, A.G.; Kroenke, C.D.; Loria, J.P. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Meth. Enzymol. 2001, 339, 204–238. [CrossRef]
- Palmer, A.G.; Grey, M.J.; Wang, C. Solution nmr spin relaxation methods for characterizing chemical exchange in high-molecular-weight systems. Meth. Enzymol. 2005, 394, 430–465. [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297-315.
- Marazioti, A.; Papadia, K.; Kannavou, M.; Spella, M.; Basta, A.; de Lastic, A-L; Rodi, M.; Mouzaki, A.; Samiotaki, M.; Panayotou, G.; Stathopoulos, G.T.; Antimisiaris, S.G. Cellular vesicles: New insights in engineering methods, interaction with cells and potential for brain targeting. JPET 2019, 370, 772–785. [CrossRef]
Figure 1.
General structure of Poly(ethylene glycol) 1 and Pluronic 2.
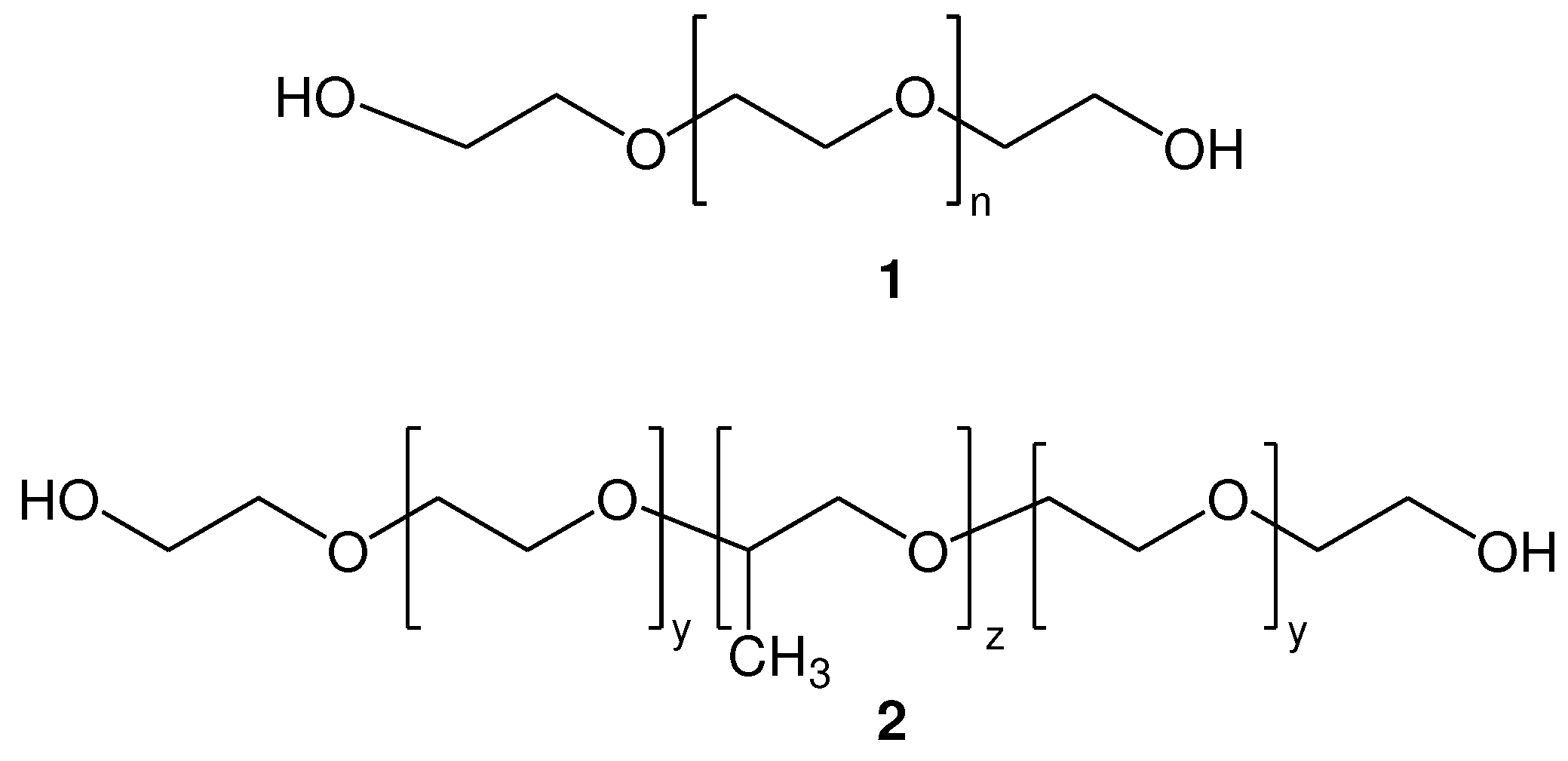
Figure 2.
General structure of dithiol poly(ethylene glycols) 3 and homobifunctional dithiol poly(ethylene glycols) 4.
Figure 2.
General structure of dithiol poly(ethylene glycols) 3 and homobifunctional dithiol poly(ethylene glycols) 4.
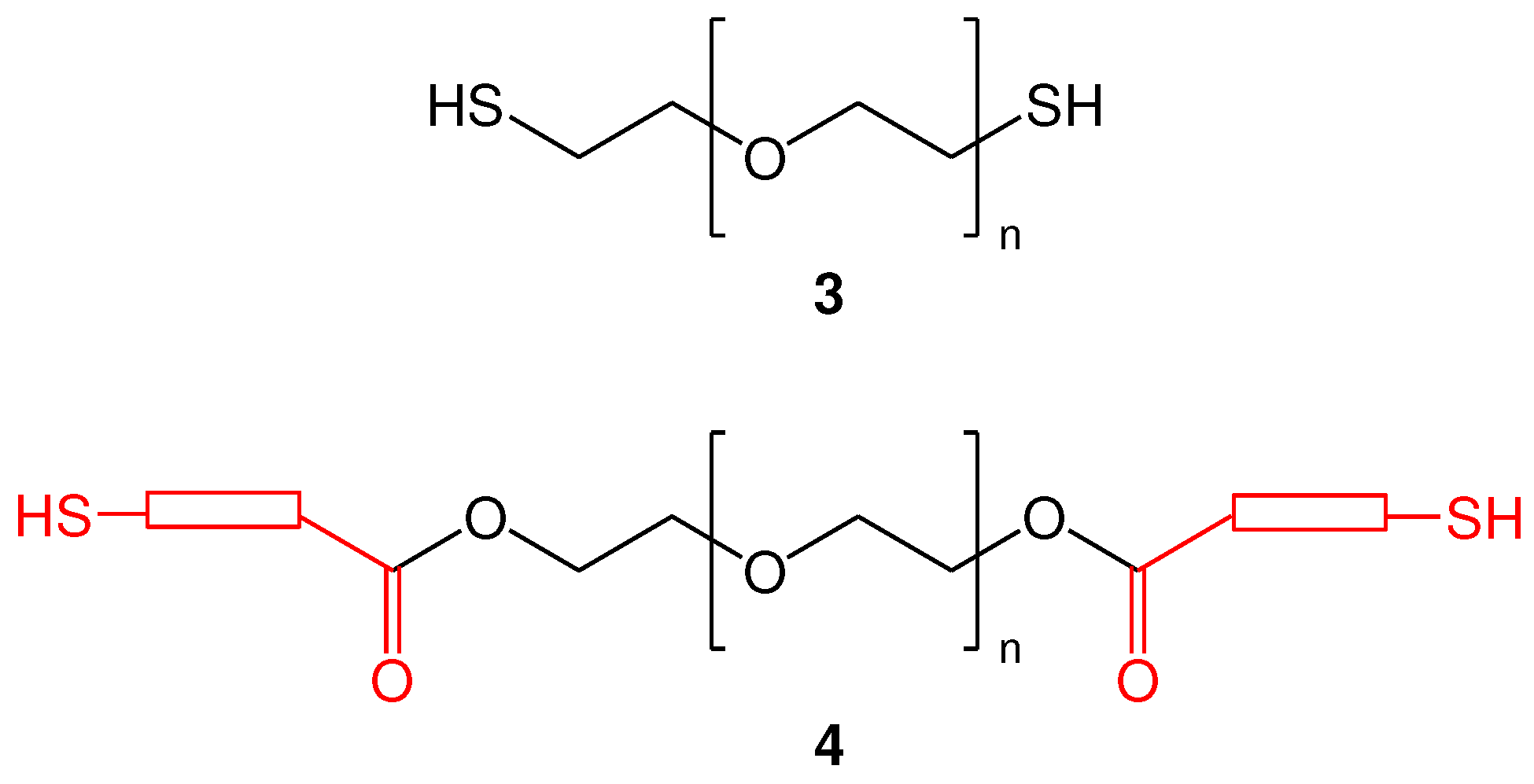
Scheme 2.
Method for the synthesis of homobifunctional di-S-Mmt-PEG-dithiols (12) and PEG-dithiols (13).
Scheme 2.
Method for the synthesis of homobifunctional di-S-Mmt-PEG-dithiols (12) and PEG-dithiols (13).
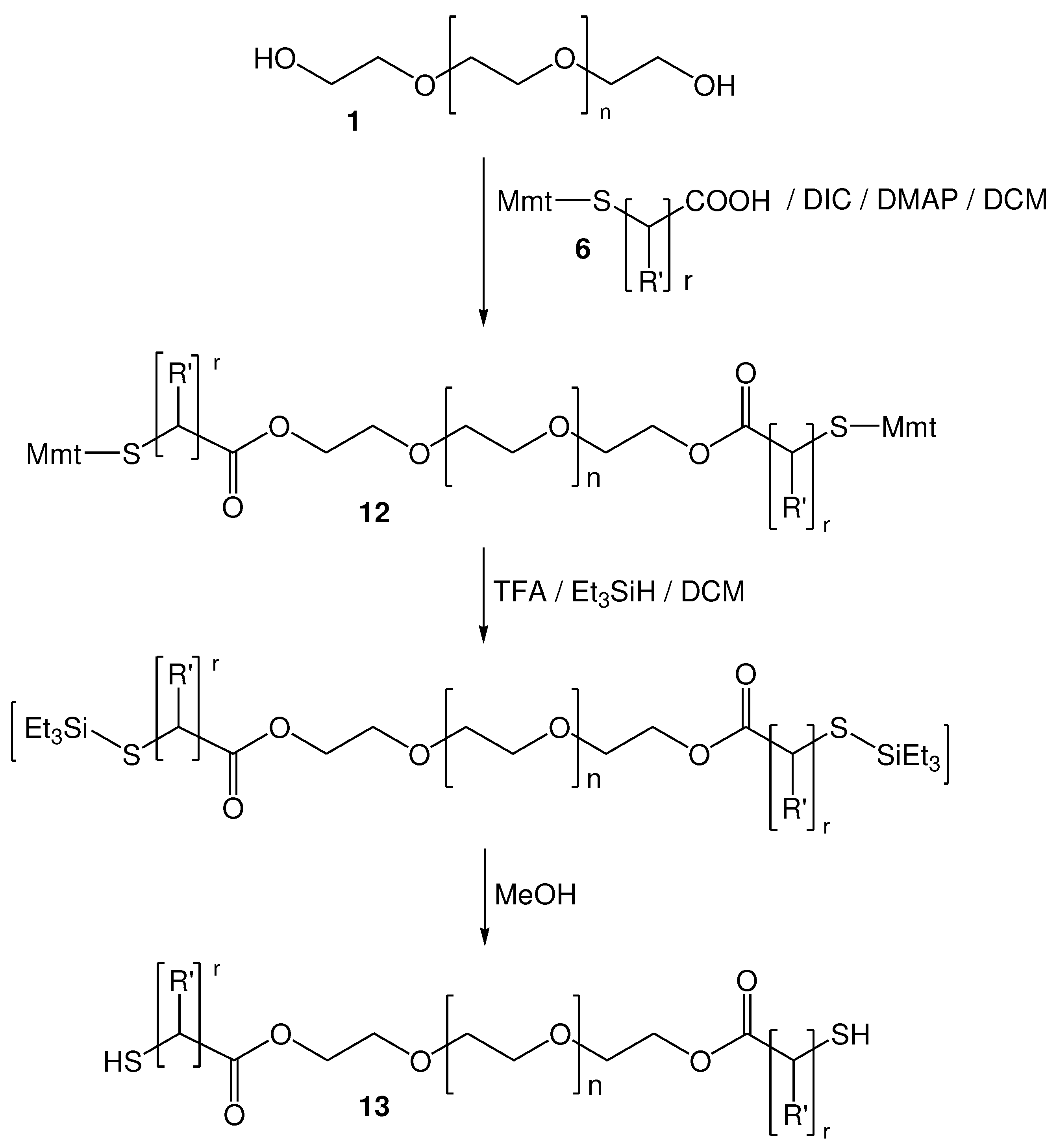
Figure 4.
di-S-Mmt-PEG-dithiols 14 (PEG 10.000); 15 (PEG 4.000); 16 (PEG 1.000), PEG-dithiols (17-19), di-S-Mmt-Pluronic-dithiol (20) and Pluronic-dithiol (21) that were synthesized.
Figure 4.
di-S-Mmt-PEG-dithiols 14 (PEG 10.000); 15 (PEG 4.000); 16 (PEG 1.000), PEG-dithiols (17-19), di-S-Mmt-Pluronic-dithiol (20) and Pluronic-dithiol (21) that were synthesized.
Scheme 3.
Thia-Michael reaction of α,ω-bis-mercaptoacyl poly(alkyl oxide) (PAO-dithiol) with nano-sized liposomes bearing maleimide groups on their surface, to afford a nano-sized cross-linked thioether type liposomal scaffold. In our case, di-2-mercaptopropionyl PEG10.000 17 and nano-sized SUV type liposomes were used.
Scheme 3.
Thia-Michael reaction of α,ω-bis-mercaptoacyl poly(alkyl oxide) (PAO-dithiol) with nano-sized liposomes bearing maleimide groups on their surface, to afford a nano-sized cross-linked thioether type liposomal scaffold. In our case, di-2-mercaptopropionyl PEG10.000 17 and nano-sized SUV type liposomes were used.
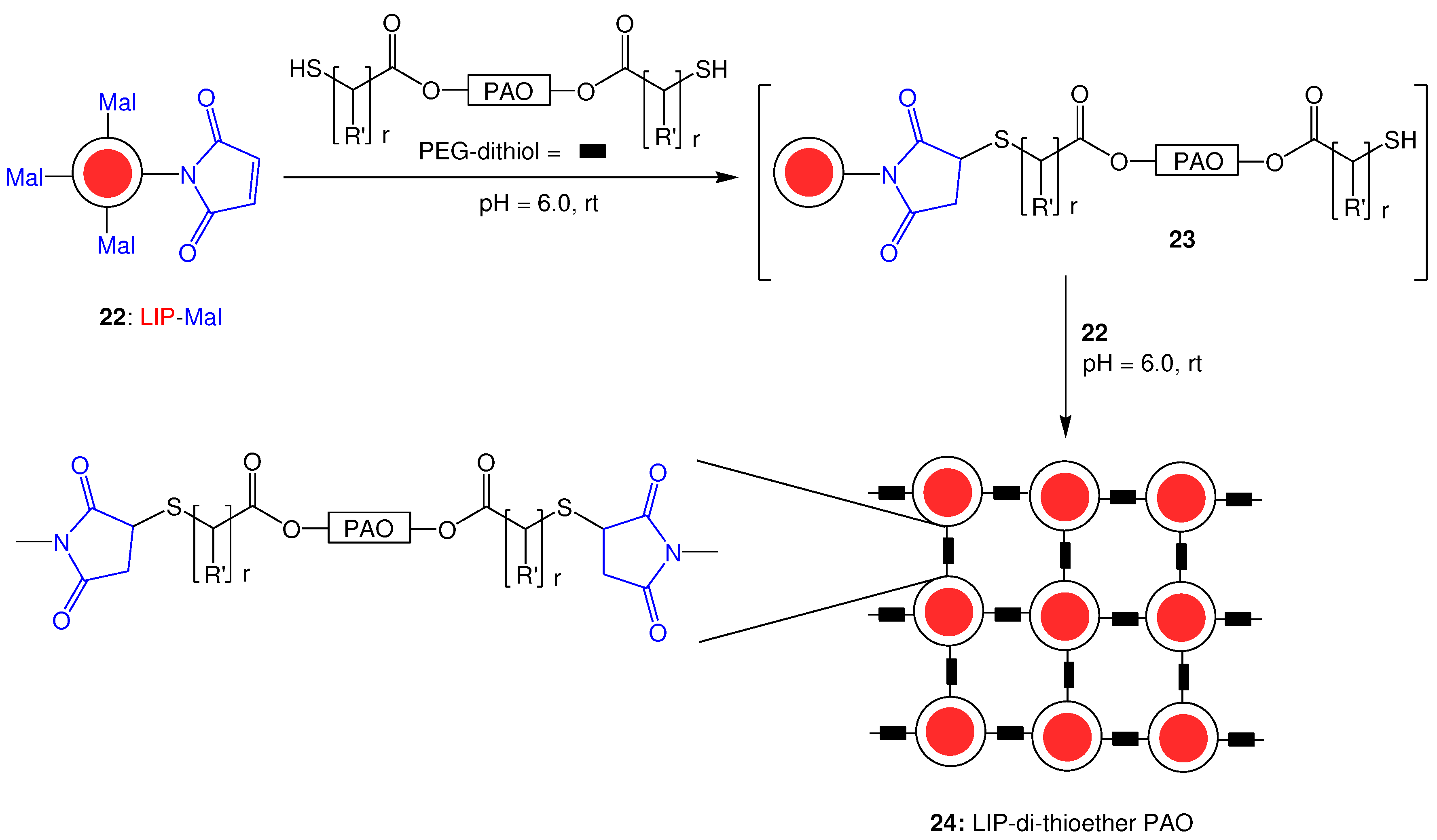
Figure 5.
1H-NMR spectrum of PC/Chol/DSPE-PEG2000-Mal (2:1:0.03) mol/mol liposomes in D2O using CPMG pulse sequence at 700 MHz, T=27 oC.
Figure 5.
1H-NMR spectrum of PC/Chol/DSPE-PEG2000-Mal (2:1:0.03) mol/mol liposomes in D2O using CPMG pulse sequence at 700 MHz, T=27 oC.
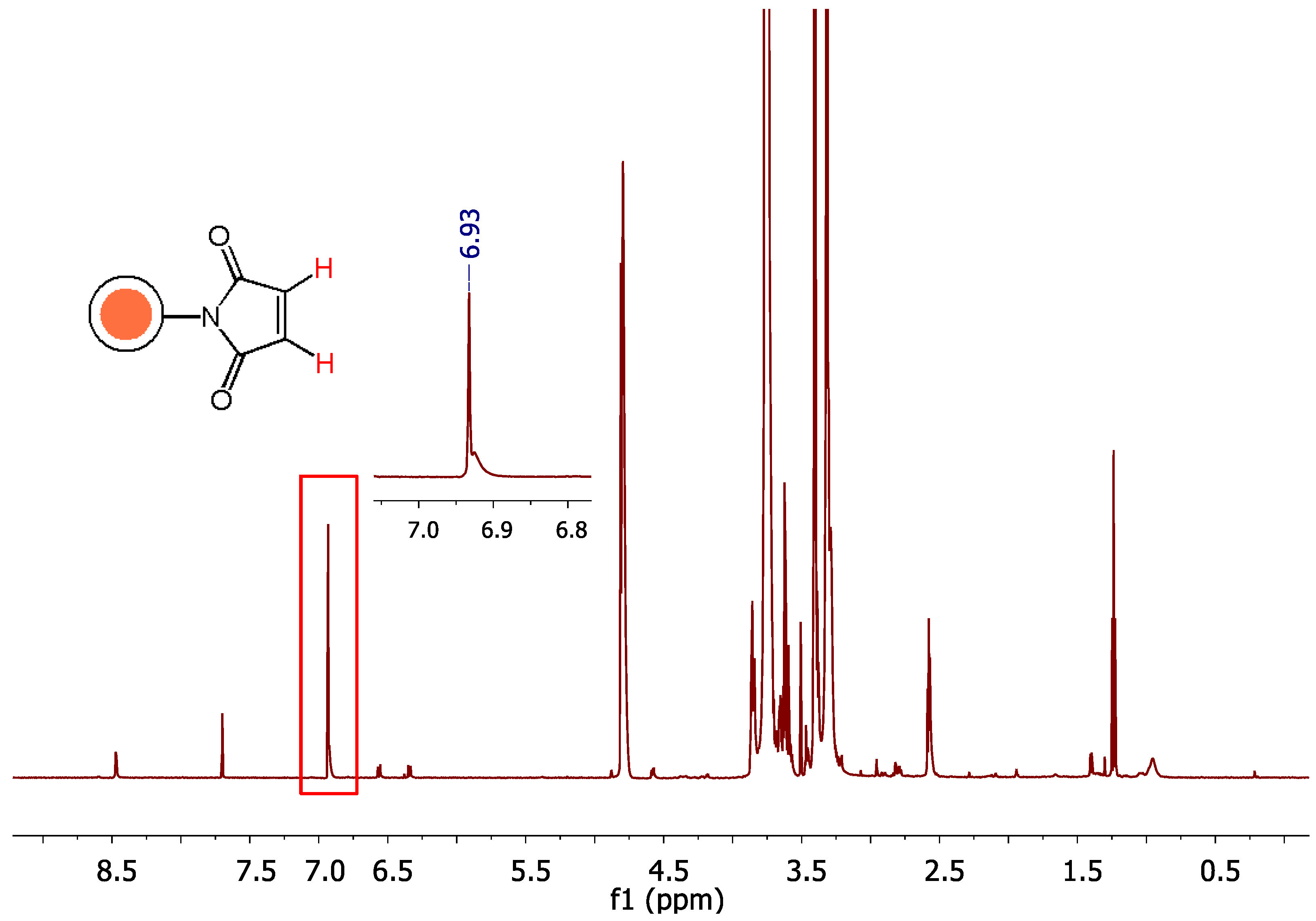
Figure 6.
1H-NMR analysis of PC/Chol/DSPE-PEG2000-Mal (2:1:0.03) mol/mol liposomes in D2O using CPMG pulse sequence at 700 MHz, T=27 oC, at t = 0, 45, 75, 180 min.
Figure 6.
1H-NMR analysis of PC/Chol/DSPE-PEG2000-Mal (2:1:0.03) mol/mol liposomes in D2O using CPMG pulse sequence at 700 MHz, T=27 oC, at t = 0, 45, 75, 180 min.
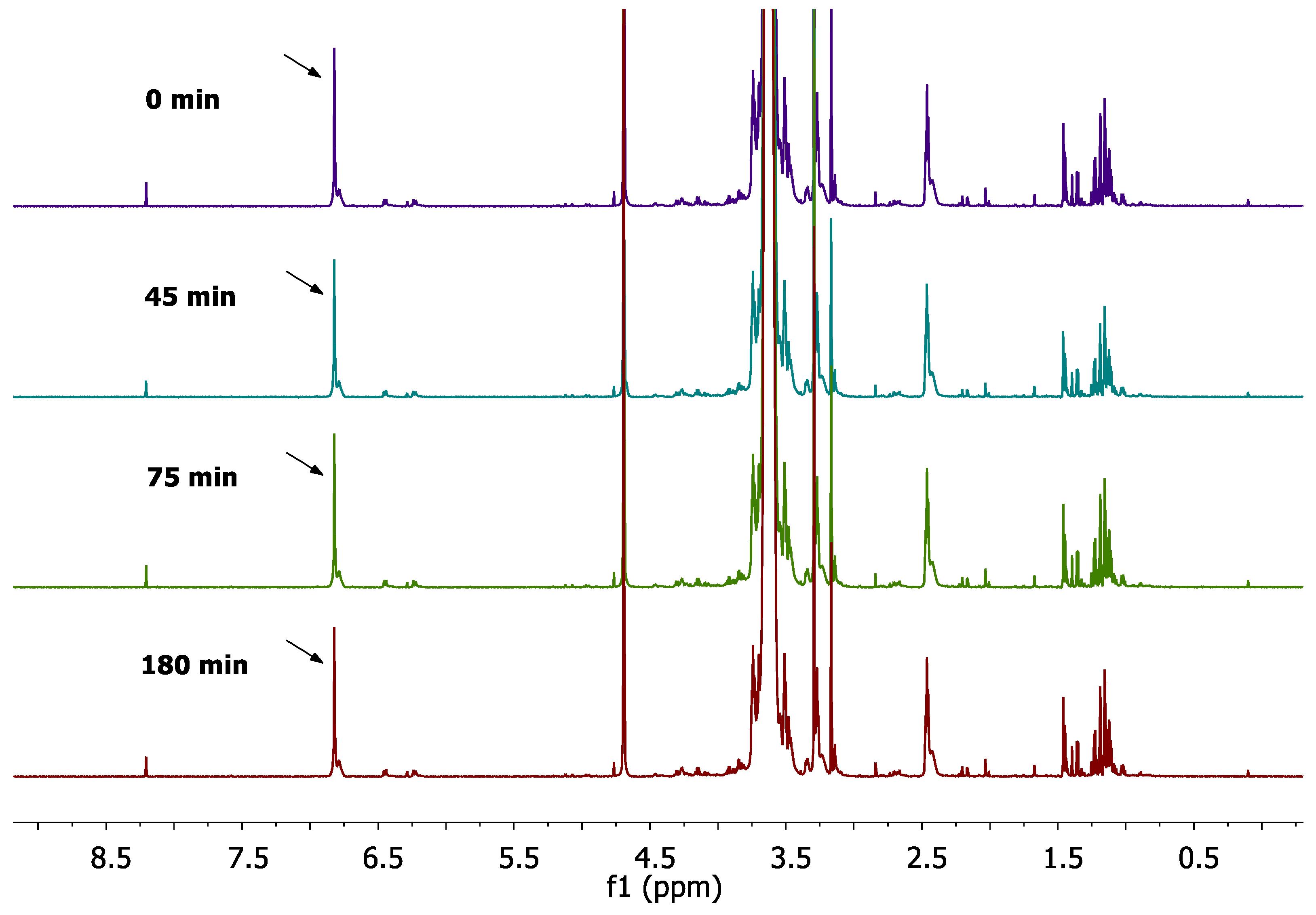
Figure 7.
Real-time monitoring of the reaction progress between pre-formed PC-Lip-Mal and PEG-dithiol in D2O using 1H-NMR CPMG pulse sequence at 700 MHz, T=27 oC.
Figure 7.
Real-time monitoring of the reaction progress between pre-formed PC-Lip-Mal and PEG-dithiol in D2O using 1H-NMR CPMG pulse sequence at 700 MHz, T=27 oC.
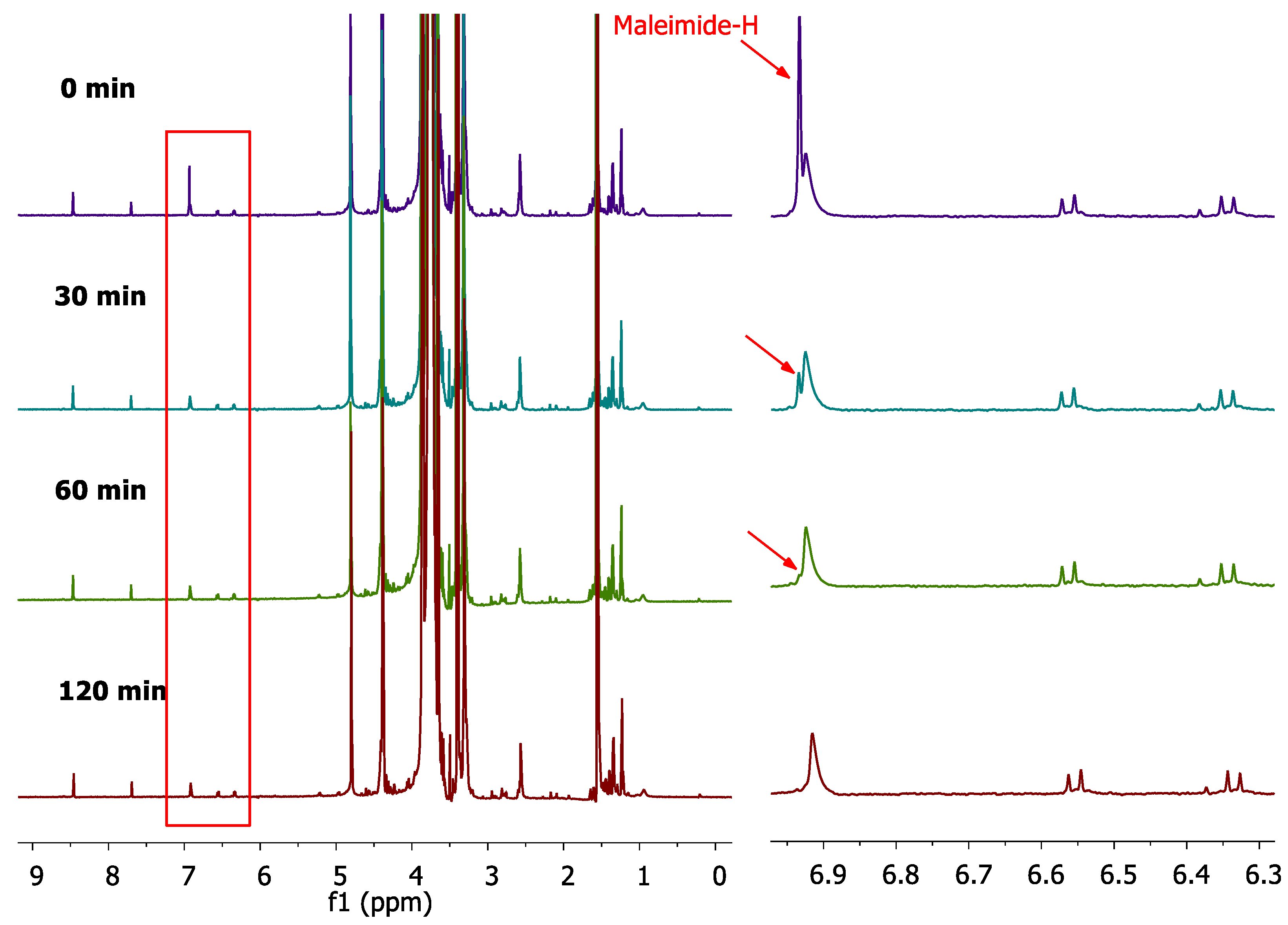
Figure 9.
Viscosity (mPa*S) of liposome scaffold by the reaction of PEG-dithiol with Lips-Mal, at two concentrations; 20 mg/ml (A) and 40 mg/ml (B). Asterisks show the significance of comparisons (*P≤ 0.05, **P≤ 0.01, ***P ≤0.001).
Figure 9.
Viscosity (mPa*S) of liposome scaffold by the reaction of PEG-dithiol with Lips-Mal, at two concentrations; 20 mg/ml (A) and 40 mg/ml (B). Asterisks show the significance of comparisons (*P≤ 0.05, **P≤ 0.01, ***P ≤0.001).
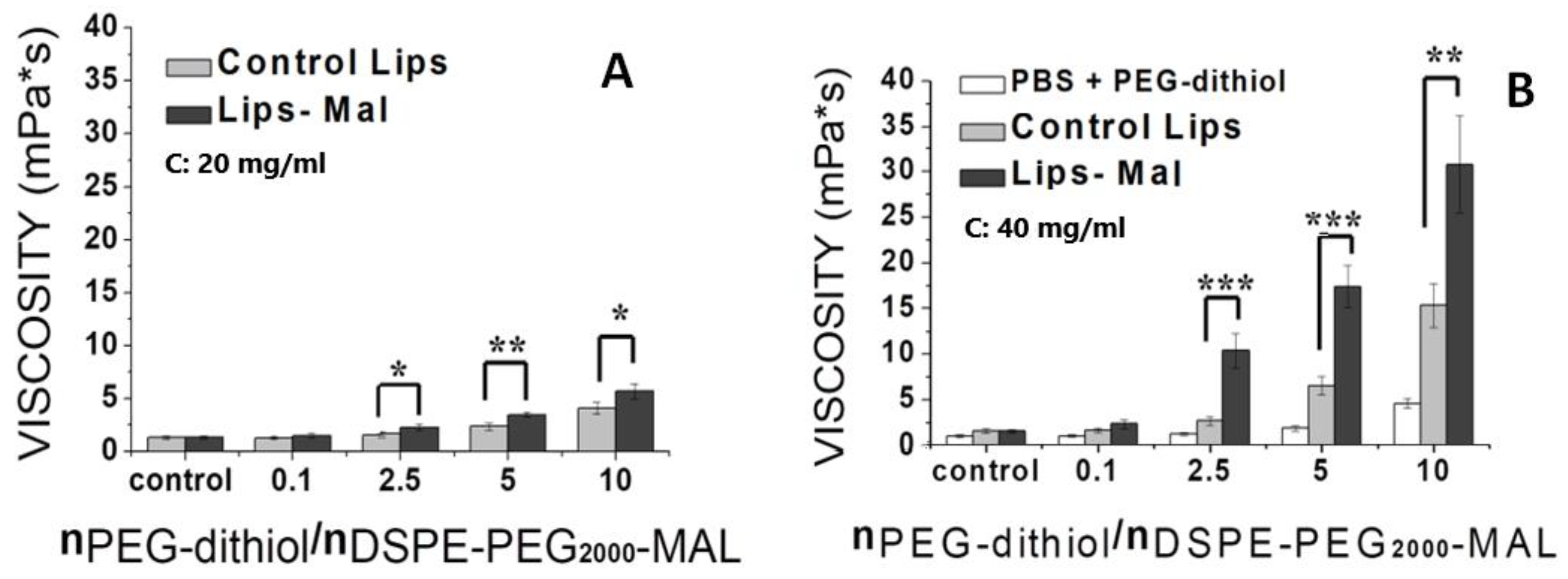
Figure 10.
% Latency of calcein by the incubation of control liposomes (HPC-Lip) and thioether cross-linked liposomes (HPC-LIP-di-thioether-PEG) at different concentrations (4 mg/ml, 2 mg/ml, 1 mg/ml, 0.5 mg/ml, 0.25 mg/ml) in FBS at 27 oC.
Figure 10.
% Latency of calcein by the incubation of control liposomes (HPC-Lip) and thioether cross-linked liposomes (HPC-LIP-di-thioether-PEG) at different concentrations (4 mg/ml, 2 mg/ml, 1 mg/ml, 0.5 mg/ml, 0.25 mg/ml) in FBS at 27 oC.
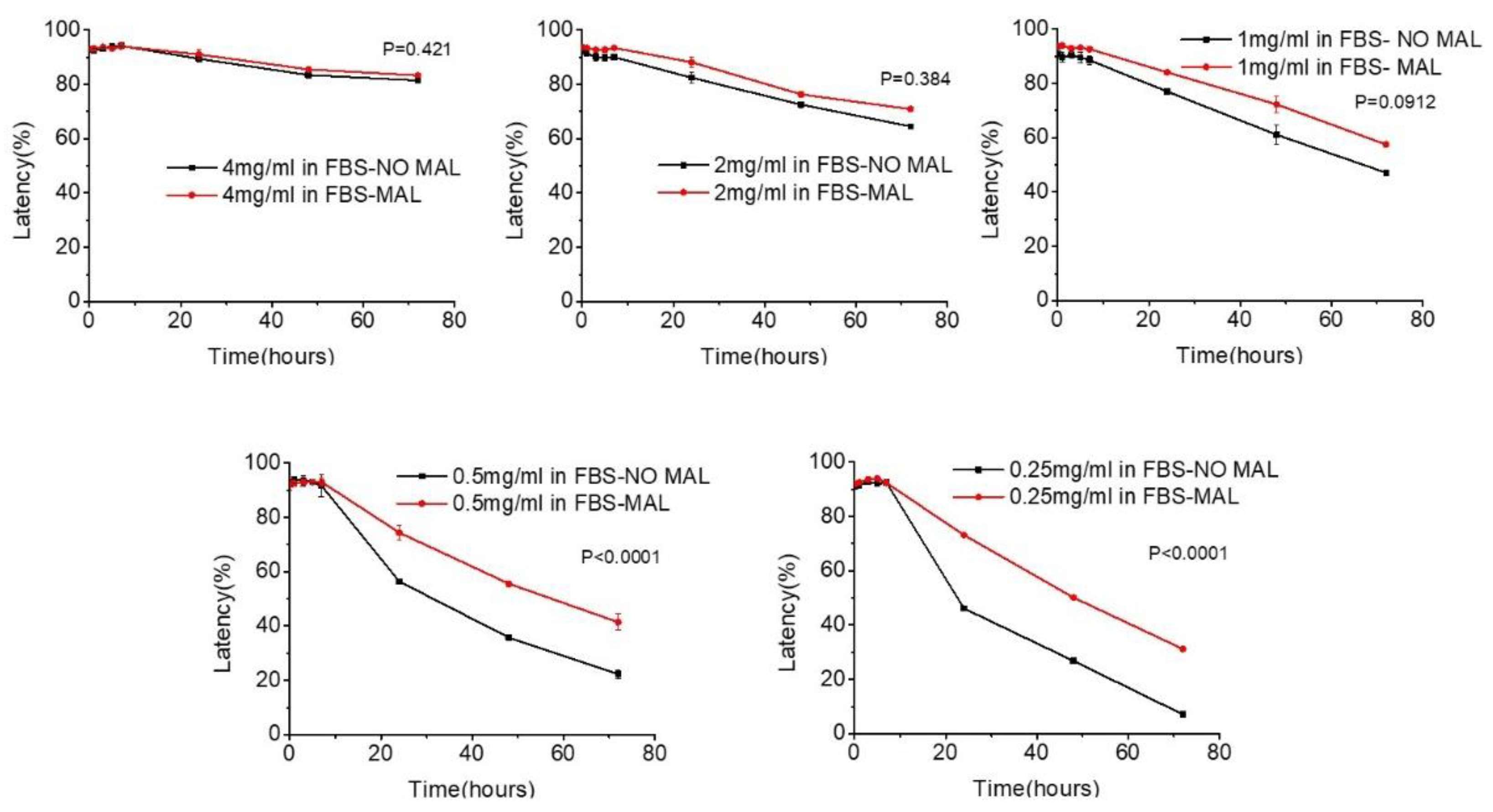

Table 2.
Mean hydrodynamic diameter (nm) and polydispersity index (PDI) for control liposomes (PC/Chol/DSPE-PEG2000-OMe (2: 1: 0.03 mol/mol)) and Lips-Mal (PC/Chol/DSPE-PEG2000-Mal (2: 1: 0.03 mol/mol)) before and after their reaction with PEG-dithiol, at several mol PEG-dithiol/mol maleimide ratios, and lipid concentration of 20 mg/ml.a.
Table 2.
Mean hydrodynamic diameter (nm) and polydispersity index (PDI) for control liposomes (PC/Chol/DSPE-PEG2000-OMe (2: 1: 0.03 mol/mol)) and Lips-Mal (PC/Chol/DSPE-PEG2000-Mal (2: 1: 0.03 mol/mol)) before and after their reaction with PEG-dithiol, at several mol PEG-dithiol/mol maleimide ratios, and lipid concentration of 20 mg/ml.a.
| Control Lips + PEG-dithiol | PC-Lip-Mal + PEG-dithiol | ||||
|---|---|---|---|---|---|
| Sample No b | mol PEG-dithiol/ mol Maleimide | Mean hydrodynamic diameter (nm) | PDI | Mean hydrodynamic diameter (nm) | PDI |
| 1 | - | 105.10 ± 2.07 | 0.158 | 101.80 ± 0.47 | 0.190 |
| 2 | 2.5 | 96.48 ± 0.74 | 0.199 | 128.90 ± 1.3 | 0.199 |
| 3 | 5 | 111.60 ± 0.95 | 0.231 | 121.30 ± 1.4 | 0.318 |
| 4 | 10 | 105.50 ± 3.7 | 0.260 | 153.10 ± 10.4 | 0.409 |
a The values reported are mean values from three measurements of three independent samples. b Sample 1 corresponds to liposomes (control and PC-Lip-Mal) at time zero (before the addition of PEG-dithiol); Samples 2-4 correspond to liposomes (control and PC-lip-Mal) treated with different excess of PEG-dithiol.
Table 3.
Mean hydrodynamic diameter (nm) and polydispersity index (PDI) for control liposomes (PC/ Chol/ DSPE-PEG2000-OMe (2: 1: 0.03 mol/mol)) and Lips-Mal (PC/ Chol/ DSPE-PEG2000-Maleimide (2: 1: 0.03 mol/mol)) before and after their reaction with PEG-dithiol, at several mol PEG-dithiol/mol maleimide ratios, and lipid concentration of 40 mg/ml. a.
Table 3.
Mean hydrodynamic diameter (nm) and polydispersity index (PDI) for control liposomes (PC/ Chol/ DSPE-PEG2000-OMe (2: 1: 0.03 mol/mol)) and Lips-Mal (PC/ Chol/ DSPE-PEG2000-Maleimide (2: 1: 0.03 mol/mol)) before and after their reaction with PEG-dithiol, at several mol PEG-dithiol/mol maleimide ratios, and lipid concentration of 40 mg/ml. a.
| Control Lips + PEG-dithiol | PC-Lip-Mal + PEG-dithiol | ||||
|---|---|---|---|---|---|
| Sample No b | mol PEG-dithiol/ mol Maleimide | Mean hydrodynamic diameter (nm) | PDI | Mean hydrodynamic diameter (nm) | PDI |
| 1 | - | 89.38 ± 0.85 | 0.154 | 90.93 ± 0.98 | 0.178 |
| 2 | 2.5 | 89.06 ± 0.79 | 0.169 | 138.30 ± 2.2 | 0.244 |
| 3 | 5 | 91.90 ± 1.6 | 0.246 | 140.20 ± 3.4 | 0.260 |
| 4 | 10 | 100.60 ± 1.4 | 0.261 | 148.00 ± 4.5 | 0.375 |
a The values reported are mean values from three measurements of three independent samples. b Sample 1 corresponds to liposomes (control and PC-Lip-Mal) at time zero (before the addition of PEG-dithiol); Samples 2-4 correspond to liposomes (control and PC-lip-Mal) treated with different excess of PEG-dithiol.
Table 4.
Mean hydrodynamic diameter (nm), polydispersity index (PDI) and ζ-potential (mV) values for control liposomes (HPC/Chol/DSPE-PEG2000-OMe (2: 1: 0.03) mol/mol) and HPC-Lip-Mal (HPC/Chol/DSPE-PEG2000-Maleimide (2: 1: 0.03) mol/mol) before and after their reaction with PEG-dithiol (2.5 molar excess) at lipid concentration of 13.5 and 14.0 mg/ml.
Table 4.
Mean hydrodynamic diameter (nm), polydispersity index (PDI) and ζ-potential (mV) values for control liposomes (HPC/Chol/DSPE-PEG2000-OMe (2: 1: 0.03) mol/mol) and HPC-Lip-Mal (HPC/Chol/DSPE-PEG2000-Maleimide (2: 1: 0.03) mol/mol) before and after their reaction with PEG-dithiol (2.5 molar excess) at lipid concentration of 13.5 and 14.0 mg/ml.
| Lipid Composition |
Mean hydrodynamic diameter (nm) |
PDI | ||
|---|---|---|---|---|
| Before Reaction |
After reaction |
Before reaction |
After Reaction |
|
| Control Lips | 99.4 ± 0.543 | 100.8 ± 0.757 | 0.180 | 0.192 |
| HPC-Lip-Mal | 100.3 ± 0.621 | 145.7 ± 2.875 | 0.199 | 0.317 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



