
Submitted:
26 December 2023
Posted:
27 December 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
it has been just over three years since SARS-CoV-2 viral infection was first known as causing severe acute and highly transmissible respiratory syndrome coronavirus. During one of the worst global pandemics of the last century, caused by the COVID-19 disease, under the persistent immune pressure exerted by the newest vaccines, the SARS-CoV-2 virus is fastly mutating and becoming less severe, but more contagious. On the other hand, some studies [1,2,3] indicate that the elderly COVID-19 patients or those who have reported a virulent episode of this infection, experience some long-lasting neurological complications, including encephalopathy, encephalitis, cerebrovascular disease, Parkinson’s disease (PD), Alzheimer’s disease (AD) and prion disease (PrD). So, there are several recent reports of patients [4,5] developing PrD following the SARS-CoV-2 infection, but, to the best of our knowledge, this is the first case reported in Italy on an association of the PRNP E200K somatic mutation with gCJD. The rising global prevalence neurodegenerative complications following COVID-19 disease adds urgency to the study of this potential relationship.
Keywords:
genetic CJD
; mutation E200K
; COVID-19
; prion
1. Introduction
In 2020, after China, Italy was the first European country to be overwhelmed, at a never seen speed, by the COVID-19 pandemic, in the first period directly leading to a wide spectrum of clinical manifestations, which could vary from asymptomatic and mild flu-like symptoms to acute respiratory tract disorder, often fatal, or causing severe multysistemic complications. Since the end of 2020, while a massive vaccination campaign has begun worldwide, so decreasing morbidity and mortality, long-term pulmonary, cardiological, neurological and/or multi-organ complications have also been reported in COVID-19 infection, especially depending on age, pre-existing comorbidities and vaccination status. In particular, exacerbation of preexisting neurological disturbances was over all observed, suggesting that the SARS-CoV-2 neurotropism may generate a likely acceleration of neurodegenerative diseases like Parkinson’s disease (PD), Alzheimer’s disease (AD), Creutzfeld-Jakob disease (CJD). [1,2]
CJD is a fatal neurodegenerative disease caused by transmissible agents called prions which replicates in the CNS leading to characteristic neuropathological findings including spongiosis, glyosis, neuron loss and the deposition of the pathological prion protein (PrPSc). Humans transmissible spongiform encephalopathies (TSEs) include sporadic, genetic, iatrogenic and infectious forms. Surveillance of human TSEs has been inaugurated in Italy in 1993, and the sporadic Creutzfeldt–Jakob disease (sCJD) is nowadays considered the major human prion disease as well as in the Italy and worldwide. The identification and mandatory reporting of CJD in Italy has allowed to improve our understanding on the CJD pathogenesis, the variable CJD clinical phenotypes, the possible geographic clusters. [3]
The aim of this study is to describe the rare case of the COVID-19 interplay in a genetic CJD associated with E200K mutation and to provide an ongoing insight into the disease pathogenesis of an even greater global health challenge than acute infection.
2. Materials and Methods
A 51-year-old woman with a history of arterial hypertension and uterine fibromatosis presented to the emergency room (ER) with rapidly progressive cognitive deterioration and functional decline, as she had become unable to maintain the upright position and to walk in few weeks. Her relatives have reported that, approximately two months before, she was diagnosed with COVID-19 through a positive reverse transcriptase polymerase chain reaction (RT-PCR) on nasopharyngeal swabs. Despite a mild course of COVID-19, in the first days after healing she developed cognitive and motor slowing, paucity of speech with paraphasic errors and hypophony, unsteady gait with ataxia, insomnia, vertical gaze palsy and blurred vision.
On ER admission, she was alerted, but quite mute, except for occasional whispered monosyllables. She was unresponsive to verbal stimulation, whereas painful stimuli evoked normal flexion of upper limbs. Bilateral direct and indirect light reflexes were normal, but she had not blink reflex in response to threat, suggesting cortical blindness. There was a right limb hyperreflexia in absence of Hoffmann and Babinski signs. In few days after the admission, she was bedridden, presented extensor plantar responses, muscle rigidity and abnormal muscle tone, staring gaze, myoclonus and palmomental and glabellar reflexes, and required enteral nutrition via percutaneous endoscopic gastrostomy (PEG) given the rapidly deterioration clinical condition.
Comprehensive blood tests, including onconeural antibodies and neural surface antigens antibodies, urine sample, cerebrospinal fluid (CSF) analysis and total-body computed tomography were collected to rule out differential diagnoses, including vascular, infectious, toxic-metabolic, autoimmune and systemic diseases, malignancies and paraneoplastic antibody-mediated encephalopathies. Her diagnostic assessment also included a brain magnetic resonance imaging and an electroencephalography.
3. Results
Blood and CSF screening and microbiological tests resulted all negative. She was treated with thiamine replacement without any clinical improvement. After the electroencephalography, that have revealed diffuse pseudo-periodic sharp-waves complexes (Figure 1), CSF real-time quaking-induced conversion (RT-QuIC) resulted positive and PRNP sequencing on lymphocyte DNA revealed the E200K mutation with homozygosity for methionine (MET) at codon 129, thus confirming the diagnosis of Creutzfeldt-Jakob disease.
A magnetic resonance imaging (MRI) of the brain revealed restricted diffusion with corresponding FLAIR in caudate nucleus and putamen, bilaterally, and cortical ribboning in the right occipito-parietal cortex (Figure 2).
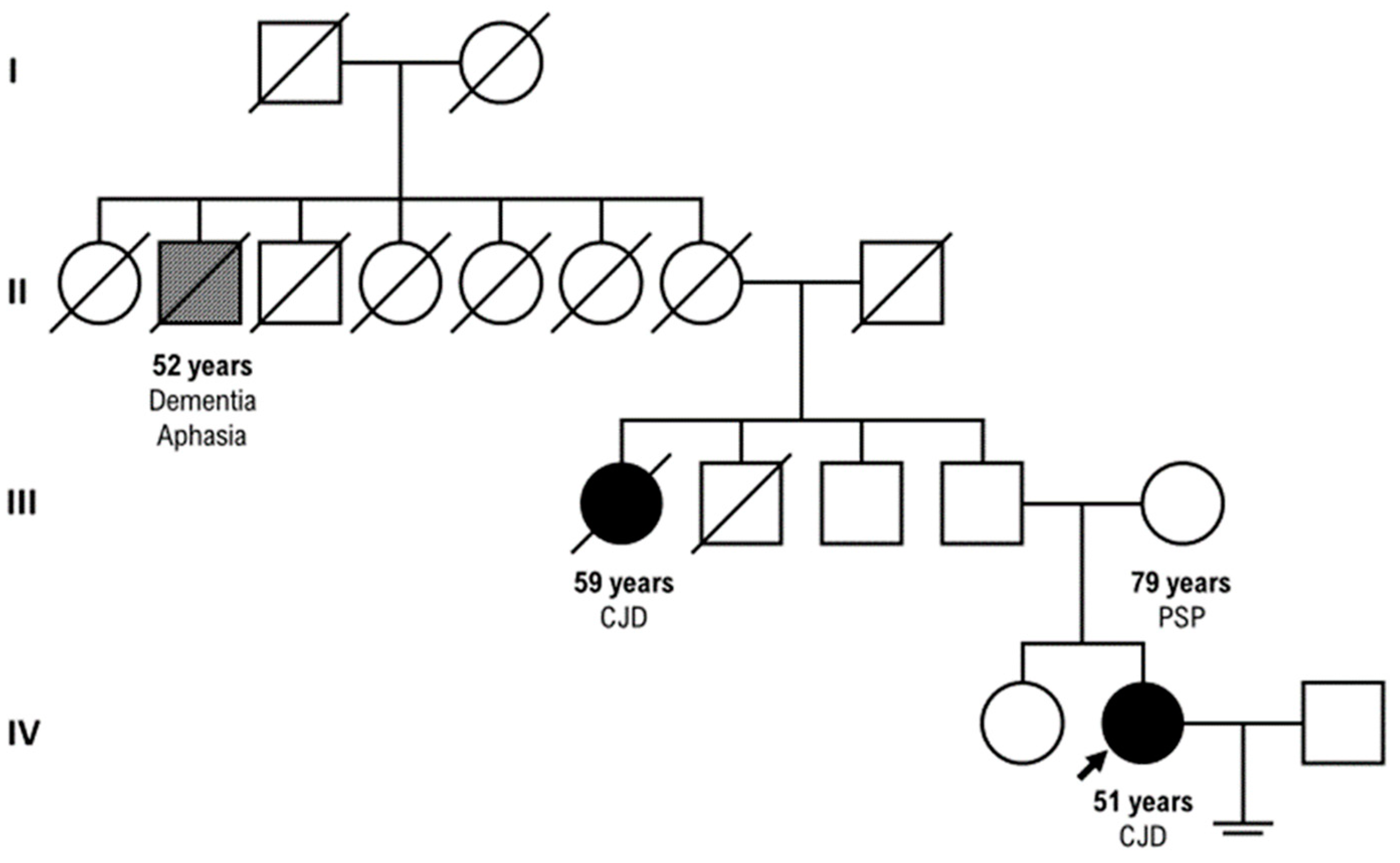
Our patient had a positive family history for CJD: her parental aunt who died at the age of 59 years with CJD (clinical course of three ys) and her paternal great-grand-uncle who died at the age of 52 years with dementia and aphasia, but PRNP sequencing unluckily was not tested in these relatives. (Figure 3) Our proband progressed up to mutism, akinetic and fully dependent state, and unfortunately died two months after the discharge.
4. Discussion
Before the COVID-19 outbreak, the principal cause of rapidly progressive dementia was certainly prion disease, excluding infectious, metabolic, vascular, neoplastic, and autoimmune disorders. A growing number of studies demonstrated that patients surviving COVID-19 can show neuroinflammatory activation of microglia and astrocytes, that might favor the fast development of neurodegenerative diseases, such as CJD. [6]
We know that CJD includes three forms: sporadic, inherited and acquired, comprising iatrogenic CJD (iCJD), variant CJD (vCJD) and Kuru. CJD is over all most likely diagnosed in countries with established large-scale surveillance systems, such as in Italy. [5]
In order to recognize early atypical/typical cases of prion disease, the application of genetic tests, such as real-time quaking induced conversion (RT-QuIC), has represented a turning point for the early diagnosis of CJD; on the other hand, it has allowed to identify unexpectedly PRNP mutations in patients apparently presenting with a sCJD, on the contrary, such as in our case, presenting with a genetic form of CJD. The most common until now described variants are E200K, P102L, D178N, 6-OPRI, 5-OPRI, A117V, and P105L, [7]
besides others additional variants that have been recently reported and classified as likely highly penetrant. [8] Interestingly, despite the classical genotype-phenotype correlation, converging toward the semplified series of symptoms (confusion, dementia, mutism, akinetic state), an unpredictable spectrum of onset and clinical presentation have been documented not only within the subtypes, but also within the same CJD family, even between monozygotic twins. It seems to suggest that an environmental, iatrogenic or behavioral or stressfull event might overall represent an effective trigger even for a genetic form of prion disease. [6,7]
About 10–15% of human prion diseases or transmissible spongiform encephalopathies (TSE) are associated to PRNP mutation, even if with variable penetrance and neuropathologic findings. [9]
In addition, E200K is certainly described highly prevalent in European countries. [10], especially in Poland, Slovakia, Greek and Italy, beyond in Chile, and in Jewish families of Libyans and Tunisians. [9]
A striking example of this phenotypic variability is our family, in which we could unfortunately detect PRNP sequencing only in our proband, who rapidly progressed and died three months after disease onset, differently from her parental aunt. Recent studies have reported that the K200 mutation of the PRNP gene, identified especially in the frontal cortex and hippocampus of sporadic CJD patients, has manifested high penetrance, as shown in our family. [10]
In particular, there is also evidence that the mutation at the codon 129 can be linked to different clinical phenotypes depending on whether the mutation co-segregates with methionine (FFI) or valine (genetic CJD), and mutation carriers can also develop different clinical phenotypes [9,10].
It is now becoming widely accepted ([9] that neurological manifestations, known as “neuro-COVID”, were reported to occur in 36% of COVID-19 patients and are likely favored by its neurotrophic and neuroinvasive features [10], inflammatory cytokine release and immunopathology [11].
It is an intriguing question to define the interesting associations between SARS-CoV2 infection and prion neurobiology.
Human prion diseases (PrD) include an expanding spectrum of progressive, and fatal neurodegenerative disorders caused by the accumulation and aggregation of a misfolded isoform (PrPSc) from the native cellular prion protein (PrPc), associated with systemic inflammation. Previous studies described some cases developing CJD during or following SARS-CoV-2 viral invasion, supporting a significant relationship between host immune-responses to SARS-CoV-2 and an exacerbation involving a systemic inflammation, a progressive acceleration of prion-like protein spread [12,13].
Furthermore, the SARS-CoV-2 ‘S1’ spike proteins contain some “prion-like” regions, amyloid peptide-binding and other domains that support the formation of pathogenic plaques in the CNS [14,15].
Recently, after the pre-mortem diagnostic revolution represented by the disease-specific real-time quaking induced conversion assay, which detects prion seeds in cerebrospinal fluid or brain tissue, the introduction of targeted sequencing of PRNP has allowed to diagnose the presence of protein-altering variants in PRNP. [16]
Our proband presented with the classical symptoms historically associated with genetic Creutzfeld-Jakob disease: early cognitive symptoms, such as memory decline, dementia; also ataxia, myoclonus, pyramidal and extrapyramidal signs, behavioral change, psychiatric symptoms such as hallucinations, delusions, and depression. [17]
She too presented the typical MRI findings described in a precedent study [18] about a singular cluster of high incidence of CJD occurring among Libyan Jews living in Israel and caused by familial transmission of the E200K mutation: four patients (mean age 60 years; one woman and three men). Their brain MRI showed gray matter atrophy, FLAIR or DWI hyperintensities in the basal ganglia and thalamus. Notably, their death occurred within 1 year of the MR studies, while our proband died earlier in three months.
Despite the phenotypic variability that often delays CJD diagnosis, clinicians should encourage the relatives to undertake a genetic counseling and testing, because in the era of genetically targeted therapies, [19,20] it might result useful for the stratification of patients in future preventive treatment trials for prion disease, to develop novel therapeutic strategies. On the other hand, considering the prolonged incubation time of prion diseases, our study do not answer the question whether SARS-CoV-2 might be able to induce protein misfolding in a clinically relevant way, but we cannot exclude that Long COVID may involve the induction of spontaneous prion emergence, especially in the elderly already at risk for neurodegenerative disease. [21] One hypothesis is that a retrograde transmission of the virus from the olfactory epithelium to the brainstem could cause a neurological damage. [22] Beyond this possibility, it is still unclear whether or not angiotensin converting enzyme-2 (ACE2) is the main route of entry of SARS-CoV-2 into neuronal, especially because ACE2 expression in the CNS is debated. [23]
Interestingly, a very recent German study shows incidence patterns of SARS-CoV-2 and SARS-CoV-2 immunization rates in relation to the incidence of a neurodegenerative/protein misfolding disease over time, and concludes that the SARS-CoV-2 pandemic and the vaccination programme against SARS-CoV-2 did not affect sCJD incidence or disease characteristics of sCJD in Germany, even if they cannot exclude that single disease courses may have been influenced by SARS-CoV-2 infections. [24]
5. Conclusions
Our case report confirms the importance of PRNP sequencing combined with deep phenotyping to address the genetic counseling in the CJD diseases, despite its rarity, that can unfortunately further isolate the affected families.
Further studies will be necessary to understand how the Sars-cov2 infection can be a trigger for CJD onset, particularly for the genetic forms, identifying the route of SARS-CoV-2 invasion into the brain in addition to determining the sequence of infection in different cell types in the central nervous system, so the temporal relationship between SARS-CoV-2 and prion disease onset.
Author Contributions
E.C. designed the study. L.P and A.Pet. contributed to the acquisition of data. A.P. and L.P. drafted the manuscript. E.C. and A.P. critically revised the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. The collection and scientific dissemination of data related to COVID-19 epidemics by every public health body, such as San Camillo Hospital, was authorized on February 27th, 2020, by the Italian Presidency of the Council of Ministers (Gazzetta Ufficiale Della Repubblica Italiana, Serie Generale, Parte Prima, 2020, Feb 28. Available from: https://www.gazzettaufficiale.it/eli/gu/2020/02/28/50/sg/pdf. ).
Informed Consent Statement
missing consent due to early death of the patient.
Data Availability Statement
Upon reasonable request, data presented in this study will be provided by emailing the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Farheen S.; Agrawal S.; Zubair S.; Agrawal A.; Jamal F.; Altaf I.; et al.. Patho-physiology of aging and immune-senescence: possible correlates with comorbidity and mortality in middle-aged and old COVID-19 patients. Front. Aging. 2021, 2, 748591. [CrossRef]
- Fu Y. W.; Xu H. S.; Liu S. J.. COVID-19 and neurodegenerative diseases. Eur. Rev. Med. Pharmacol Sci. 2022, 26, 4535–4544. [CrossRef]
- Zhao Y; Jaber VR; Lukiw WJ. SARS-CoV-2, long COVID, prion disease and neurodegeneration. Front Neurosci. 2022;16:1002770. [CrossRef]
- McGrath A; Pai H; Clack A. Rapid progression of probable Creutzfeldt-Jakob disease with concomitant COVID-19 infection. BMJ Case Rep. 2023 1;16(11):e254402. [CrossRef]
- Ciolac D.; Racila R.; Duarte C.; Vasilieva M.; Manea D.; Gorincioi N. et al.. Clinical and radiological deterioration in a case of Creutzfeldt-Jakob Disease following SARS-CoV-2 Infection: hints to accelerated age-dependent neurodegeneration. Biomedicines. 2021, 9, 1730. 10. [CrossRef]
- Koc HC, Xiao J, Liu W, Li Y, Chen G. Long COVID and its Management. Int J Biol Sci. 2022, 11;18(12):4768-4780. [CrossRef]
- Minikel E.V., Vallabh S.M., Lek M.et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016; 8: 322ra9. [CrossRef]
- Richards S., Aziz N., Bale S. et al.Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405-424. [CrossRef]
- Ladogana A; Gabor G; Kovacs, Chapter 13 - Genetic Creutzfeldt–Jakob disease,Editor(s): Maurizio Pocchiari, Jean Manson,Handbook of Clinical Neurology,Elsevier,Volume 153,2018,Pages 219-242,ISSN 0072-9752,ISBN 9780444639455.
- Won SY; Kim YC; Jeong BH; Elevated E200K Somatic Mutation of the Prion Protein Gene (PRNP) in the Brain Tissues of Patients with Sporadic Creutzfeldt-Jakob Disease (CJD). Int J Mol Sci. 2023, 2;24(19):14831. [CrossRef]
- Watson N; Brandel JP; Green A; Hermann P; Ladogana A;Lindsay T; Mackenzie J; Pocchiari M; Smith C; Zerr I; Pal S. The importance of ongoing international surveillance for Creutzfeldt-Jakob disease. Nat Rev Neurol. 2021;17(6):362-379. [CrossRef]
- Wu Z; McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020 7;323(13):1239-1242. [CrossRef]
- Guedes BF. NeuroCOVID-19: a critical review. Arq Neuropsiquiatr. 2022;80(5 Suppl 1):281-289. [CrossRef]
- Frontera JA; Thorpe LE; Simon NM; de Havenon A; Yaghi S; Sabadia SB; Yang D; Lewis A; Melmed K; Balcer LJ; Wisniewski T; Galetta SL. Post-acute sequelae of COVID-19 symptom phenotypes and therapeutic strategies: A prospective, observational study. PLoS One. 2022 29;17(9):e0275274. [CrossRef]
- Pogue A. I.; Lukiw W. J.. microRNA-146a-5p, neurotropic viral infection and prion disease (PrD). Int. J. Mol. Sci. 2021, 22, 9198. [CrossRef]
- Young M. J.; O'Hare M.; Matiello M.; Schmahmann J. D.. Creutzfeldt-Jakob disease in a man with COVID-19: SARS-CoV-2-accelerated neurodegeneration? Brain Behav. Immun. 2020, 89, 601–603. [CrossRef]
- Lukiw WJ; Jaber VR; Pogue AI; Zhao Y. SARS-CoV-2 Invasion and Pathological Links to Prion Disease. Biomolecules. 2022 7;12(9):1253. [CrossRef]
- Fulbright RK, Kingsley PB, Guo X, Hoffmann C, Kahana E, Chapman JC, Prohovnik I. The imaging appearance of Creutzfeldt-Jakob disease caused by the E200K mutation. Magn Reson Imaging. 2006;24(9):1121-9. [CrossRef]
- Tetz G.; Tetz V. Prion-like domains in spike protein of SARS-CoV-2 differ across its variants and enable changes in affinity to ACE2. Microorganisms. 2022, 10, 280. [CrossRef]
- Goldman JS; Vallabh SM. Genetic counseling for prion disease: Updates and best practices, Genetics in Medicine, Volume 24, Issue 10, 2022, Pages 1993-2003, ISSN 1098-3600. [CrossRef]
- Takada L.T.; Kim M.O., Cleveland R.W. et al. Genetic prion disease: experience of a rapidly progressive dementia center in the United States and a review of the literature Am J Med Genet B Neuropsychiatr Genet, 2017, 174 (1), pp. 36-69. [CrossRef]
- Mahalakshmi A.M., Ray B., Tuladhar S., Bhat A., Paneyala S., Patteswari D., Sakharkar M.K., Hamdan H., Ojcius D.M., Bolla S.R., et al. Does COVID-19 contribute to development of neurological disease? Immun. Inflamm. Dis. 2021;9:48–58. [CrossRef]
- Samuel J. Ahmad, Chaim M. Feigen, Juan P. Vazquez, Andrew J. Kobets, David J. Altschul. Neurological Sequelae of COVID-19. J. Integr. Neurosci. 2022, 21(3), 77. [CrossRef]
- Hermann P, Böhnke J, Bunck T, Goebel S, Jaeger VK, Karch A, Zerr I. Effect of SARS-CoV-2 incidence and immunisation rates on sporadic Creutzfeldt-Jakob disease incidence. Neuroepidemiology. 2023 Dec 12.
Figure 1.
Electroencephalography showing the fairly typical repetitive pattern in CJD.
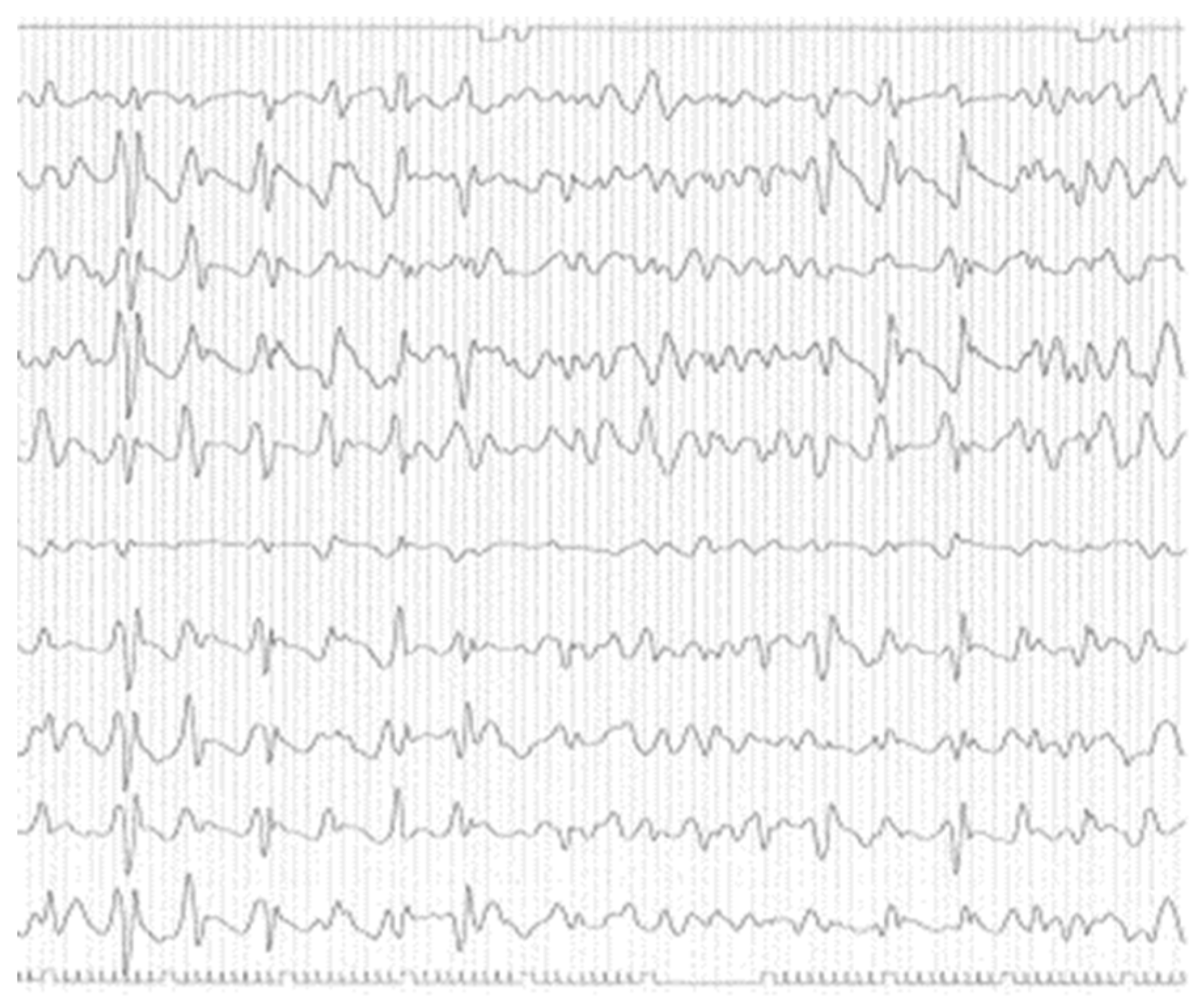
Figure 2.
Diffusion-weighted imaging abnormalities reported tipically in CJD in the cortical and basal ganglia.
Figure 2.
Diffusion-weighted imaging abnormalities reported tipically in CJD in the cortical and basal ganglia.
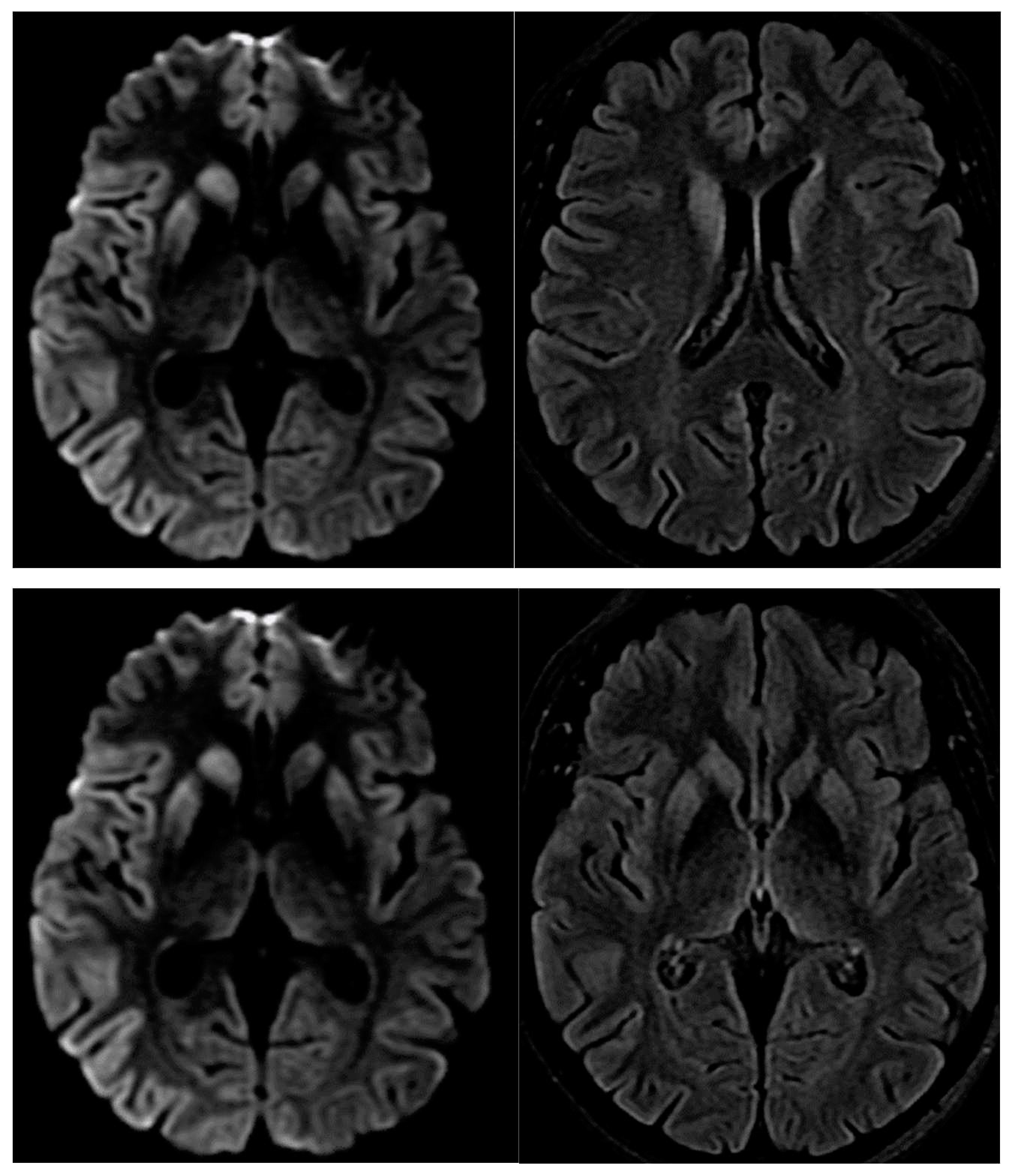
Figure 3.
Our three-generation family tree.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



