
Submitted:
11 December 2023
Posted:
12 December 2023
You are already at the latest version
Abstract
The mechanistic influences of dopamine (DA) signaling and impact on motor function is nearly always interpreted from changes in nigrostriatal neuron terminals in striatum. This is a standard practice in studies of human Parkinson’s disease (PD) and aging, and related animal models of PD and aging-related parkinsonism. However, despite dozens of studies indicating an ambiguous relationship between changes in striatal DA signaling and motor phenotype, this perseverating focus on striatum continues. Although DA release in substantia nigra (SN) was first reported almost 50 years ago. assessment of nigral DA signaling changes in relation to motor function is rarely considered. Whereas DA signaling has been well-characterized in striatum at all 5 steps of neurotransmission (biosynthesis and turnover, storage, release, reuptake, and post-synaptic binding) in the nigrostriatal pathway, the depth of such interrogations in the SN, outside of cell counts, is sparse. However, there is sufficient evidence that these steps in DA neurotransmission in the SN are operational and regulated autonomously from striatum, and are present in human PD and aging, and related animal models. To complete our understanding of how nigrostriatal DA signaling affects motor function, it is past time to include interrogation of nigral DA signaling. This brief review highlights evidence that changes in nigral DA signaling at each step in DA neurotransmission are autonomous from those in striatum and changes in the SN alone can influence locomotor function. Accordingly, for full characterization of how nigrostriatal DA signaling affects locomotor activity, interrogation of DA signaling in SN is essential.
Keywords:
Substantia nigra
; dopamine
; tyrosine hydroxylase
; dopamine receptor
; striatum
; reuptake
; phosphorylation
; nigrostriatal
; Parkinson’s disease
; aging
Introduction:
Ever since dopamine (DA) and norepinephrine (NE) neuronal pathways were identified and functionally characterized in vivo [1,2,3,4,5,6,7], the depth and breadth of studies of how both neurotransmitters affect cognitive and motor behavior has been immense. The viability and function of the neuronal pathways that produce these neurotransmitters, nigrostriatal and ceruleo-cortical respectively, are significantly decreased in Parkinson’s disease (PD). As such, the 5 components of neurotransmission (biosynthesis, storage, release, reuptake, and post-synaptic function) have been studied for respective contributions to deficits in DA or NE signaling in PD. The range of approaches used to interrogate these pathways include defining PD-related genes and physiological regulation of catecholamine genes [8,9,10,11], expression of catecholamine-regulating enzymes and transporters [12,13,14,15,16,17,18,19,20], post-translational modification of biosynthesis enzymes [21,22,23,24,25,26,27,28], neuron electrophysiological properties [29,30,31,32,33,34], release and uptake [14,35,36,37,38,39,40], pre- and post-synaptic receptor function [22,30,34,41,42,43,44,45], basal ganglia circuit function [46,47,48,49,50,51,52], and growth factor signaling [53,54,55,56,57,58,59,60,61,62,63,64]. Clearly the investment of resources in these multiple areas of research is for the ultimate goals of understanding PD etiology, the consequences of DA or NE loss that arise from PD on motor and cognitive skills, and to identify a sound mechanistic rationale for effective treatments to delay or arrest disease progression. Notably, the vast majority of studies that focus on the relationship between motor function and DA signaling have evaluated one or more of the 5 components of neurotransmission in the striatum, the terminal field region of the nigrostriatal pathway. It is important to keep in mind that at the time of PD diagnosis, the striatal regions already show ~70-80% loss of DA-regulating proteins or aspects of DA signaling (such as DA release). This non-linear relationship brings up 2 yet to be resolved questions; why is motor impairment not detected prior to 80% loss, and second, why does the severity of motor impairment continue to worsen when loss in striatum reaches near 100% 4-5 years after diagnosis [20].
Insights of how striatal DA signaling affects locomotor function have reached a plateau
In the context of PD, DA is, by far, the most studied of the catecholamines, with NE running a distant second. Since 1962, there have been ~29,000 publications associated with DA and PD vs. ~1700 associated with NE and PD. The evidence for deficient nigrostriatal DA signaling as the primary cause of motor symptoms of PD is strong. Yet there still remains a critical unresolved issue that hampers progress; a continuous perseverating focus to attribute deficient DA signaling in the striatum as the sole culprit for motor impairment. This focus is undoubtedly driven by the longstanding working model of basal ganglia circuit dysfunction that arises from the loss of striatal DA due to the progressive loss of nigrostriatal neurons. It is argued that this striato-centric focus has generated a plateau in our understanding of exactly how any of the 5 steps of neurotransmission with deficient DA-regulating function in striatum actually impair motor function. [For definition purposes, the relation of nigrostriatal DA signaling to motor impairment will focus upon bradykinesia/hypokinesia, which is among 4 cardinal signs of PD which also include rigidity and postural instability, and tremor at rest.] Indeed, there are clinically-based examples of where improvements in striatal DA signaling did not equate to alleviate motor impairment in PD patients [60,64,65]. More evidence of this lack of alignment between striatal DA levels and severity of motor impairment is seen at the later stages of PD. Although the severity of motor impairment continues to worsen 4 to 5- years after PD diagnosis, loss of striatal DA-regulating proteins or signaling has already reached near 100% [20,66,67,68,69]. There is a comparable amount of evidence for this misalignment between striatal DA levels and motor function status in preclinical studies of rat PD models [22,57,58,59,70,71,72,73,74,75,76]. Motor impairment may be also present with far less than 80%, if any, striatal DA loss [54,65,70,72], or to the converse, motor impairment may not be present even though striatal DA loss meets or exceeds 80% [22,73,74]. Motor impairment can also be alleviated without any increase or recovery of striatal DA or DA-regulating protein loss [57,59,73,74,75,76]
It is not the position of this review to assert that striatal DA signaling does not influence on motor function. The weight of evidence showing the influence of striatal DA signaling on basal ganglia circuits is too great to list here. However, the incongruities between the level of locomotor function and DA signaling in striatum can no longer be ignored if we are to solve which critical dopaminergic element(s) are to be targeted to maximize effective therapeutic strategies. This brief review will present evidence that challenges the central dogma that compromised DA signaling in striatum is the sole deficiency of DA that impairs locomotor function. The overwhelming evidence that nigrostriatal DA signaling does affect locomotor function has been obtained from our knowledge of PD and from studies that experimentally modulate components of DA neurotransmission (biosynthesis, DA receptor function, etc.). The key question is where in the nigrostriatal pathway does DA have the greatest influence on locomotor function; particularly regarding the mechanisms that drive the initiation of self-generated movement. Although the evidence that nigral DA signaling can influence motor function is sparse, it has nonetheless been in existence since the 1980s [77,78,79,80,81]. The paucity of studies evaluating the SN is likely due to a prevailing presumption that neurotransmitter functions at the axon terminal are the sole influence of behavioral outcomes. Thus, interrogation of nigral DA signaling has not been considered in experimental designs to define how components of nigrostriatal DA signaling affect locomotor activity. In this light, it is reasonable to presume that the numerous ambiguities between striatal DA regulation and motor function that have accumulated in the literature over the past several decades could have been resolved if assessment of nigral DA signaling was included in the study design.
Dissecting the impact of the 5 components of DA neurotransmission on locomotor function
As goes with the loss of nigrostriatal neurons in PD, the loss of DA-regulating proteins and processes involved in neurotransmission follows. Interference with the functions of any of these proteins or processes can also affect locomotor function in naïve (non-PD) animal models. Tyrosine hydroxylase (TH) is the rate-limiting step of DA biosynthesis, converting tyrosine to L-dihydroxyphenylalanine (L-DOPA). Inhibition of TH with alpha-methyl-p-tyrosine (AMPT) decreases DA inhibits locomotor activity [7,82,83,84,85,86]. In humans, inhibition of hyperkinetic movements, such as chorea, dystonia, or dyskinesia, can also be produced by AMPT [87,88]. At the DA storage step, a process controlled by vesicular monoamine transporter 2 (VMAT2) imports monoamines like DA into synaptic vesicles. This function is inhibited by reserpine, which also inhibits locomotor activity [89,90,91], as first identified by a parkinsonian symptom side effect produced in hypertension treatment [92]. VMAT2 is expressed in both striatum and SN [93,94], which confers the capacity for storing DA for eventual release in the entire nigrostriatal pathway.
Once DA is packaged in synaptic vesicles, it can be released by neuronal activity or by modulation of transporter function through stimulant action. At the extracellular level, DA release from the nigrostriatal pathway is the step that delivers tissue content, via vesicular delivery, to the synapse [95,96,97,98], wherein DA has 4 fates, binding to the pre- or post-synaptic DA receptors, reuptake into the neuron, or diffusion away from the release site [99]. Drugs that target DA receptors, the post-synaptic DA D1 receptor or pre- and post-synaptic DA D2 receptor, also influence locomotor activity, and are targets for pharmacotherapy in PD treatment [100]. An acute regimen of antipsychotics such as haloperidol or either DA D1 or D2 receptor antagonists reduce locomotor activity [101,102,103,104,105]. Conversely, DA D1 or D2 agonists increase locomotor activity in rodents and primates [106,107,108] and improve motor functions in late-stage human PD [109,110,111]. The release of DA can also be modulated by DA D2 autoreceptor function [112] in both striatum and SN [31,113]. Functionally, the regulation of DA release by neuronal activity is critical for initiation of locomotor activity [114,115,116,117,118]. Deficits in DA release, as shown in aging studies or over-expression of alpha-synuclein, are associated with decreased locomotor activity [119,120,121], whereas increased DA release, as induced by amphetamine or methamphetamine [122,123,124], increases locomotor activity [125,126,127].
The termination of DA signaling occurs by reduction of extracellular DA levels in the synapse, largely (though not exclusively [99) through reuptake by the dopamine transporter (DAT) [128,129,130], a process that occurs in SN as well as striatum [131,132,133]. DAT protein expression is considerably greater in the striatum [94], and this difference may explain why DA release and uptake dynamics differ between these two regions [131,132,133]. Through constant trafficking between cytosol and plasma membrane, DAT function is dynamically regulated, including aging and in PD [134,135,136]. The DAT, like the DA D2 receptor, also has considerable interaction with other components of DA neurotransmission, including DA D2 receptors [34,113] and has considerable influence on maintaining DA tissue levels, TH expression and phosphorylation selectively in the striatum, but not in SN [137,138]. There is also evidence of plasticity in DA uptake under conditions where DA and DAT levels are particularly low. In such cases, the NE transporter may also transport DA, with inherently low DA innervation or from severe loss of nigrostriatal neuron terminals [139,140,141].
Given the considerable influence of DAT on DA homeostasis, locomotor activity is strongly affected by DAT expression levels. DAT knockout mice show a hyperkinetic phenotype [142,143]. This hyperkinetic phenotype is not likely explained by the low DA uptake capacity in the striatum due to DAT knockout, as DA tissue content levels are severely reduced to a level that is comparable to nigrostriatal lesion (>90% loss) [137,138]. Systemic delivery of nomifensine, a DAT inhibitor, increases locomotor activity [144], consistent with the hyperkinetic phenotype of the knockout [142,143]. While presumably this effect would be considered to be due to elevated extracellular DA levels in striatum from interference with DA uptake, we recently reported infusion of nomifensine in striatum did not increase locomotor activity in aged rats, despite a striatum-specific increase in extracellular DA levels produced by nomifensine infusion therein [145].
Approaches and outcomes needed to discern role of striatal and nigral DA signaling
In summary, there is considerable evidence that the proteins and processes associated with the 5 steps of DA neurotransmission in the nigrostriatal pathway are operational in both striatum and SN. Modifications of these functions can alter DA signaling dynamics in either region, although there are notable differences in the functional dynamics between these regions at some of these steps, such as DAT expression and reuptake capacity [94,131,140]. The release of DA occurs in both striatum and SN with activation of nigrostriatal neurons [96,115,146,147,148], and is associated with self-directed movement in rodents [115,148]. Thus, with DA release occurring in striatum and SN, it would seem to be experimentally challenging to decipher the role of DA signaling in either region in locomotor function. However, with localized delivery of DA-modulating compounds into striatum or SN, it is plausible to target one or more of these steps in one region to modify and isolate DA signaling dynamics. Thus, interference with a step in neurotransmission in one region would influence extracellular DA levels (or interfere with receptor function) therein, which would address the fact that DA release occurs simultaneously in both regions from neuronal activity. The critical outcome needed from this approach would be whether this region-specific modulation affected DA signaling not only in the targeted region, but also did not affect DA signaling in the non-targeted region (ie. targeting SN would not influence DA dynamics in striatum). This approach is feasible, and therefore it is possible to parse out the relative contributions of DA signaling in striatum or SN and respective impact on locomotor function [44,54,84,118,145,149,150,151]. Most importantly, as the functional status of each step in DA neurotransmission is established in normal and disease states in either striatum or SN, it is possible to infer what the loss of such functions in disease states has on locomotor function, based upon the results obtained from region-specific modulation of DA signaling.
Autonomy of DA biosynthesis in SN and impact on motor function in aging and PD
An experimental approach that can modulate DA signaling by targeting one of the 5 steps of neurotransmission in a specific region of the nigrostriatal pathway represents a means to emulate specific mechanisms of DA signaling that exist in vivo in normal or disease (including aging) states. For example, if TH levels are reduced selectively in the SN in a disease or aging model, then targeting TH activity in that region in a naïve or control animal can be useful to determine if the loss of TH is contributing to deficient DA signaling and locomotor function [149]. The specific targeting of SN or striatum to modulate DA signaling by targeting one of the 5 steps of neurotransmission is a critical experimental approach because differences in DA regulation exist at multiple steps in normal (or naïve) rodents, PD models, and in models of aging-related parkinsonism. Moreover, because such differences have also been identified in human PD and aging, it is feasible to determine, by experimental modulation within the SN or striatum, if any specific change in DA signaling is driving locomotor impairment. Most of the evidence that has evaluated how DA signaling from SN or striatum affects locomotor function has been obtained at the DA biosynthesis step.
Differences in TH expression, TH phosphorylation, and DA tissue content exist between the SN and striatum under normal [84,138,153,154], PD- [18,20,22,57,58,66,67,68,69,72,156] or aging-related conditions [54,61,84,145,150,152,155,156,157], both in animal models and in human PD [20,66,67,68,69,156,157,158] and aging [156,157,158,159,160,161,162]. In naïve (young and without nigrostriatal lesion) rodents and across multiple rat strains, TH protein expression is 3- to 4-fold greater in the striatum [18,22,61,62,84,138,145,150,152]. This difference in TH expression between striatum and SN is matched by differences in DA tissue content, which is ~15- to 25-fold greater in striatum [18,22,84,138,145,152]. The greater disparity in DA tissue levels as compared to the differences in TH protein between these two regions is likely due to 3- to 10-fold greater ser31 TH phosphorylation in the striatum [22,84,138,145,152,153,154]. Increased ser31 TH phosphorylation can alone increase DA biosynthesis [163] and the level of ser31 phosphorylation is highly correlative to DA tissue levels when accounting for inherent TH protein levels across 4 DA regions in vivo [84,138,152,153,154]. These results collectively indicate that DA biosynthesis capacity differs between striatum and SN at the levels of TH protein, ser31 TH phosphorylation, and DA tissue.content. As such, the effect of nigrostriatal lesion (reflecting PD) and aging on these components of DA biosynthesis would presumably play a significant role on regulation of DA signaling and ultimately locomotor function.
Autonomy of post-synaptic DA signaling in SN and impact on motor function
The activation of the DA D1 receptor, expressed on striatonigral neurons, in the SN mediates GABA release [30,42,47]. This release of GABA decreases the inhibitory output of the basal ganglia; a process that facilitates the generation of movement. Both aging and PD can affect D1 receptor expression. In the middle to late-middle stage of the lifespan, there is a 30% decrease in expression of this receptor in the rat SN, and smaller decrease in striatum [150] (Figure 1). This decrease is associated with an aging-related decrease in locomotor activity. In human aging, DA D1 receptor expression also decreases proportionally with age [174]. This decrease may be associated with the onset of mild bradykinesia beginning in late-middle age in humans. Previous work by Trevitt and colleagues modulated D1 receptor function in SN and striatum to evaluate relative impact on locomotor function in rats. They showed nigral infusion of a DA D1 receptor antagonist was highly potent in reducing operant behavior and open-field activity [44]. Decreased locomotor activity is also produced by DA D1 receptor antagonists following systemic delivery [105]. Thus, it is plausible that the locomotor-modulating action of DA D1 receptor drugs, in animal models and humans alike, is driven by modulation of its post-synaptic functions in the SN [103,105,106,107,108,109,110]. Thus, applied to physiologically-based DA-meditated changes in locomotor activity, it is plausible that through local release of GABA in the SN, driven by activation of DA D1 receptors following local DA release in the SN [30,42,47], the disinhibition of basal ganglia output from the SN pars reticulata neurons occurs. It is feasible that this sequence of events, initiated by DA release in the SN, provides the signal to increase locomotor activity (Figure 3). This work also suggests that the first onset of aging-related decreases in locomotor activity in the lifespan may be driven by decreased DA D1 receptor expression in the SN. Prevention of aging-related deficits in motor function is associated with increased DA D1 receptor expression, exclusively in the SN [145].
In PD, the DA D1 receptor has recently been identified as a novel target to treat motor impairment in the later stages of the disease [109,110,111]. The status of DA D1 receptor expression or function is far less known than the DA D2 receptor [175]. Our work in the 6-OHDA model indicates that the DA D1 receptor is upregulated, specifically in the SN, as nigrostriatal neuron and DA loss increase therein [22]. We speculate this increase is a response by the striatonigral neurons to maintain DA signaling in the SN. Notably, D1 receptor expression does not change in the early stages of neuronal loss, when DA tissue levels are unaffected. Thus, if the D1 receptor is upregulated in the latter stages of PD, it stands to reason that a D1 receptor agonist could substitute for DA, given the reduction in DA levels at the latter stage of neuron loss. In contrast to the changes in SN, D1 receptor expression is unchanged in striatum, despite the severe loss of DA beginning early after nigrostriatal neuron lesion.
In summary, multiple lines of evidence from human PD and aging and related animal models indicate that DA signaling in the SN plays a significant role in locomotor activity levels. Changes at the biosynthesis, reuptake, and post-synaptic signaling steps in the SN occur autonomously from changes (if any) in the striatum, making a clear case that augmenting DA signaling in the SN alone could be achieved by several possible strategies to alleviate locomotor impairment. Moreover, targeting specific steps of DA neurotransmission that are affected in aging and PD can reveal which deficit (and where in the nigrostriatal pathway) is responsible for decreasing DA signaling to impair locomotor acitivity. As long as there is a means to modulate one or more steps in DA neurotransmission, such as inhibition of DA biosynthesis [84,149,150], or augmenting it by infusion of L-DOPA [39,176,177,178] it is possible to pinpoint the most critical losses responsible for locomotor impairment.
Upstream regulators of DA signaling: the role of GDNF signaling in SN
There has been a great need to find treatment for PD that is disease modifying, in addition to a therapeutic approach that can reduce the amount of L-DOPA needed to maintain mobility without debilitating side effects such as L-DOPA-induced dyskinesia. In the 1990s, glial cell line-derived neurotrophic factor (GDNF) emerged as a top candidate for treatment of motor impairment in PD based upon encouraging preclinical studies in rodent and non-human primates [56,57,58,59]. Notably, GDNF had the rather remarkable attribute of long-term impact on constituents of DA signaling (such as increased DA tissue content and ser31 TH phosphorylation), particularly in the SN, after a single delivery [56,57,58,61,62,179]. These long-term effects of GDNF were eventually revealed in clinical trials, wherein motor benefits to patients endured for up to a year following discontinuation [180,181], and motor benefits realized while receiving GDNF [55,182]. In preclinical rat PD models, this long-term effect of GDNF may be driven by increased expression of its receptor, GFR-α1, specifically in the SN [53,183]. Notably, GFR-α1 itself alleviates TH and DA loss after 6-OHDA lesion in the SN, but not striatum [183], and can increase TH and DA levels, again selectively in the SN, with increased locomotor activity, in aged rats [54].
More recent clinical trials with GDNF reported failure to reach primary end point for improvement in motor scores in GDNF recipients relative to placebo control groups [60,64], leading the field to reconsider its value for treating the motor impairments of PD [184]. It should be briefly noted that in the failed trials there was evidence of increased DA signaling in the putamen [60,64]; an outcome representing more evidence of the ambiguity between striatal DA signaling and locomotor function. Retrograde transport of GDNF from striatum to the SN has been a well-documented physiological event [62,185,186,187,188]. Given the impact of GDNF or GFR-α1 in the SN on DA signaling and strong association with improved locomotor activity, it is likely that the trophic action of GDNF depends upon there being sufficient GFR-α1 levels in both striatum (for retrograde transport), and in the SN wherein the stimulating effects on DA signaling can occur [53,54,58,61,189]. This has been recently identified as a potential major challenge, as GFR-α1 expression progressively decreases in DA neurons as neuronal loss proceeds [63]
Conclusions:
We have known for nearly 50 years that DA is released from the somatodendritic region of nigrostriatal neurons in the SN [189,190] and that the 5 steps of DA neurotransmission that comprise DA signaling in striatum are also present, functional, and targetable in the SN. Moreover, substantial evidence shows that DA signaling is autonomously regulated in SN from striatum. Thus, it cannot be assumed that changes in DA signaling in one compartment are also occurring in the other compartment. Therefore, under physiological conditions, despite that DA release occurs in both striatum and SN during neuron activation, modulation at specific steps of DA neurotransmission in one of these two regions can alter the magnitude of DA release capacity in only one region. Given the multiple examples of studies that have shown incongruity between components of striatal DA signaling and locomotor function, it stands to reason that changes in DA signaling in the SN in these studies could have been the culpable mechanism. Given the autonomy of DA regulation between striatum and SN, changes in nigral DA signaling alone theoretically could influence locomotor function, and the evidence for this continues to increase. Indeed, although there is a substantially lesser number of studies of interrogating nigral DA signaling, and an even fewer number of studies that also measure locomotor activity against it, there is congruity with the direction of change in nigral DA. modulation and locomotor activity in a number of studies [44,54,71,78,79,80,118,145,149,151]. These results are also consistent with studies reporting changes in basal ganglia output from the SN as a result of modulating DA signaling specifically in the SN [30,47,191,192,193,194]. These results are applicable in PD and aging, as, the autonomy of DA signaling and components of DA neurotransmission exist at multiple levels [19,20,35,36,66,67,68,69,152,155,156,164,165,178]. This has direct implications to identify whether the striatum or SN is the source of DA signaling deficits that drives locomotor impairment and its severity in both conditions [195].
Future Directions:
The loss of nigrostriatal neurons in PD has paved the way in our understanding how DA loss affects motor function, and in general, how changes in DA signaling components affect locomotor function. However, it is past time to consider that the continuing loss of DA signaling components remaining in the SN may well be driving the worsening locomotor impairment in the patient. However, a collective epiphany in recognizing the role of nigral DA signaling in locomotor function will expand our understanding of the mechanisms, including those upstream of DA (such as GDNF signaling) that contribute to locomotor impairment. For example, with evidence for DA compensation occurring in the SN to mitigate the severity of locomotor decline, the inherent mechanisms driving it may represent targets to maintain locomotor function when TH protein loss is too great. It will be also important to delve further into understanding what striatal DA signaling is doing for maintaining locomotor function. For example, tremor at rest is a cardinal sign in PD. However in aging-related parkinsonism the evidence for its presence is scarce; notably TH and DA loss are nowhere near the severity in PD. Therefore, DA deficits in striatum may reach a level of severity that promote this involuntary movement. Finally, it should be a priority to determine what compartment of the nigrostriatal pathway should be targeted to maximize the efficacy of potential treatments, such as GDNF, on locomotor recovery. The potential for increased nigral DA signaling as a mechanism for locomotor recovery should stand as priority comparable to the attention that the striatum has garnered.
References
- Glowinski, J.; Axelrod, J.; Iversen, L.L. Regional studies of catecholamines in the rat brain, I.V. Effects of drugs on the disposition and metabolism of H3-norepinephrine and H3-dopamine. J. Pharmaco.l Exp. Ther. 1966, 153, 30–41. [Google Scholar]
- Glowinski, J.; Iversen, L.L. Regional studies of catecholamines in the rat brain, I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J. Neurochem. 1966, 13, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J. Noradrenaline: Fate and Control of its biosynthesis. Science 1971, 173, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Thierry AM, Blanc G, Sobel A, Stinus L, Glowinski J. 1973. Dopaminergic terminals in the rat cortex. Science 182: 499-501. 4- Coyle JT, Axelrod J. 1971. Development of the uptake and storage of L [3H] norepinephrine in the rat brain. J Neurchem 18: 2061-2075 .
- Carlsson, A.; Dahlstroem, A.; Fuxe, K.; Lindqvist, M. HISTOCHEMICAL AND BIOCHEMICAL DETECTION OF MONOAMINE RELEASE FROM BRAIN NEURONS. Life Sci. 1965, 4, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Anden, N.E.; Carlsson, A.; Dahlstroem, A.; Fuxe, K.; Hillarp, N.A.; Larsson, K. DEMONSTRATION AND MAPPING OUT OF NIGRO-NEOSTRIATAL DOPAMINE NEURONS. Life Sci. 1964, 3, 523–530. [Google Scholar]
- Rech, R.H.; Borys, H.K.; Moore, K.E. Alterations in behavior and brain catecholamine levels in rats treated with alpha-methyltyrosine. J. Pharmacol. Exp. Ther. 1966, 153, 412–419, ISSN 0022-3565. [Google Scholar] [PubMed]
- Nagatsu, T.; Nakashima, A.; Ichinose, H.; Kobayashi, K. Human tyrosine hydroxylase in Parkinson’s disease and in related disorders. J. Neural Trans. 2019, 126, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Kumer, S.C.; Vrana, K.E. Intricate regulation of tyrosine hydroxylase activity and gene expression. J. Neurochem. 1996, 67, 443–462. [Google Scholar] [CrossRef]
- Reed, X.; Bandrés-Ciga, S.; Blauwendraat, C.; Cookson, M.R. The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol. Dis. 2019, 124, 230–239. [Google Scholar] [CrossRef]
- Nishioka, K.; Imai, Y.; Yoshino, H.; Li, Y.; Funayama, M.; Hattori, N. Clinical Manifestations and Molecular Backgrounds of Parkinson's Disease Regarding Genes Identified From Familial and Population Studies. Front. Neurol. 2022, 13, 764917. [Google Scholar] [CrossRef] [PubMed]
- Chotibut, T.; Davis, R.W.; Arnold, J.C.; Frenchek, Z.; Gurwara, S.; Bondada, V.; Geddes, J.W.; Salvatore, M.F. Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson's model. Mol. Neurobiol. 2014, 49, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Pickel, V.M.; Beckley, S.C.; Joh, T.H.; Reis, D.J. Ultrastructural immunocytochemical localization of tyrosine hydroxylase in the neostriatum. Brain Res. 1981, 225, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Chotibut, T.; Apple, D.M.; Jefferis, R.; Salvatore, M.F. Dopamine transporter loss in 6-OHDA Parkinson’s model is unmet by parallel reduction in dopamine uptake. PLoS ONE 2012, 7, e52322. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.; Xu, M.; Bohlen, J.K.; Meshul, C.K. Differential ultrastructural alterations in the Vglut2 glutamatergic input to the substantia nigra pars compacta/pars reticulata following nigrostriatal dopamine loss in a progressive mouse model of Parkinson’s disease. Eur. J. Neurosci. 2020, 53, 2061–2077. [Google Scholar] [CrossRef] [PubMed]
- Fiorenzato, E.; Antonini, A.; Bisiachhi, P.; Weis, L.; Biundo, R. Asymmetric Dopamine Transporter Loss Affects Cognitive and Motor Progression in Parkinson's Disease. Mov. Disord. 2021, 36, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, L.C.; Dore, V.; Villemagne, V.L.; Xu, S.; Finkelstein, D.; Barnham, K.J.; Rowe, C. Utilizing 18F-AV-133 VMAT2 PET Imaging to Monitor Progressive Nigrostriatal Degeneration in Parkinson Disease. Neurology 2023. [CrossRef]
- Salvatore, MF. ser31 tyrosine hydroxylase phosphorylation parallels differences in dopamine recovery in nigrostriatal pathway following 6-OHDA lesion. J. Neurochem. 2014, 129, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Dovero, S.; Prunier, C.; Ravenscroft, P.; Chalon, S.; Guilloteau, D.; Crossman, A.R.; Bioulac, B.; Brotchie, J.M.; Gross, C.E. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned Macaque model of Parkinson's disease. J. Neurosci. 2001, 21, 6853–6861. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A Role for alpha -Synuclein in the Regulation of Dopamine Biosynthesis. J. Neurosci. 2002, 22, 3090–3099, ISSN 0270-6474. [Google Scholar] [CrossRef] [PubMed]
- Kasanga, E.A.; Han, Y.; Shifflet, M.K.; Navarrete, W.; McManus, R.; Parry, C.; Barahona, A.; Nejtek, V.A.; Manfredsson, F.P.; Kordower, J.H.; Richardson, J.R.; Salvatore, M.F. Nigral-specific increase in ser31 phosphorylation compensates for tyrosine hydroxylase protein and nigrostriatal neuron loss: Implications for delaying parkinsonian signs. Exp. Neurol. 2023, 368, 114509. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Salvatore, M.F.; Maiolo, S.A.; Bobrovskaya, L. Tyrosine hydroxylase as a sentinel for central and peripheral tissue responses in Parkinson's progression: Evidence from clinical studies and neurotoxin models. Prog. Neurobiol. 2018, 165–167, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Shehadeh, J.; Double, K.I.; Murphy, K.E.; Bobrovskaya, L.; Reyes, L.; Dunkely, P.R.; Halliday, G.M.; Dickson, P.W. Expression of tyrosine hydroxylase isoforms and phosphorylation at serine 40 in the human nigrostriatal system in Parkinson's disease. Neurobiol. Dis. 2019, 130, 104524. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Mori, K.; Kaneko, Y.S.; Hayashi, N.; Nagatsu, T.; Ota, A. Phosphorylation of the N-terminal portion of tyrosine hydroxylase triggers proteasomal digestion of the enzyme. Biochem. Biophys. Res. Commun. 2011, 407, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Kolacheva, A.; Alekperova, L.; Pavlova, E.; Bannikova, A.; Ugrumov, M.V. Changes in tyrosine hydroxylase activity and dopamine synthesis in the nigrostriatal system of mice in an acute model of Parkinson’s disease as a manifestation of neurodegeneration and neuroplasticity. Brain Sci. 2022, 12, 779. [Google Scholar] [CrossRef] [PubMed]
- Haycock, J.W.; Haycock, D.A. Tyrosine hydroxylase in rat brain dopaminergic nerve terminals: Multiple-site phosphorylation in vivo and in synaptosomes. J. Biol. Chem. 1991, 266, 5650–5657, ISSN 0021-9258. [Google Scholar] [CrossRef]
- Morgenroth, V.H.; Hegstrand, L.R.; Roth, R.H.; Greengard, P. Evidence for involvement of protein kinase in the activation by adenosine 3':5'-monophosphate of brain tyrosine 3-monooxygenase. J. Biol. Chem. 1975, 250, 1946–1948, ISSN 0021-9258. [Google Scholar] [CrossRef]
- Willard, A.M.; Islett, B.R.; Whalen, T.C.; Mastro, K.J.; Ki, C.S.; Mao, X.; Gittis, A.H. State transitions in the substantia nigra reticulata predict the onset of motor deficits in models of progressive depletion in mice. eLife 2019, 8, e42746. [Google Scholar] [CrossRef]
- Kliem, M.A.; Maidment, N.T.; Axkerson, L.C.; Chen, S.; Smith, Y.; Wichmann, T. Activation of nigral and pallidal dopamine D1-like receptors modulates basal ganglia outflow in monkeys. J. Neurophysiol. 2007, 98, 489–1500. [Google Scholar] [CrossRef]
- Dagra, A.; Miller, D.R.; Lin, M.; Gopinath, A.; Shaerzadeh, F.; Harris, S.; Sorrentino, Z.A.; Stoier, J.F.; Velasco, S.; Azar, J.; Alonge, A.R.; Lebowitz, J.J.; Ulm, B.; Bu, M.; Hansen, C.A.; Urs, N.; Giasson, B.I.; Khoshbouei, H. α-Synuclein-induced dysregulation of neuronal activity contributes to murine dopamine neuron vulnerability. npj Parkinsons Dis. 2021, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Matschke, L.A.; Komadowski, M.A.; Stohr, A.; Lee, B.; Henrick, M.T.; Griesbach, M.; Rinne, S.; Geibl, F.F.; Chiu, W.H.; Koprich, J.B.; Brotchie, J.M.; Kiper, A.K.; Dolga, A.M.; Oertel, W.H.; Decher, N. Enhanced firing of locus coeruleus neurons and SK channel dysfunction are conserved in distinct models of prodromal Parkinson's disease. Sci. Rep. 2022, 12, 3180. [Google Scholar] [CrossRef] [PubMed]
- Ellens, D.J.; Leventhal, D.K. Electrophysiology of Basal Ganglia and Cortex in Models of Parkinson Disease. J. Parkinson’s Dis. 2013, 3, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Mckie, P.M.; Shaerzadeh, F.; Gamble-George, J.; Miller, D.R.; Martyniuk, C.J.; Khoshbouei, H. In Parkinson's patient-derived dopamine neurons, the triplication of α-synuclein locus induces distinctive firing pattern by impeding D2 receptor autoinhibition. Acta Neuropathologica Commun. 2021, 9, 107. [Google Scholar] [CrossRef] [PubMed]
- Matuskey, D.; Tinaz, S.; Wilcox, K.C.; Naganawa, M.; Toyonaga, T.; Dias, M.; Henry, S.; Pittman, B.; Ropchan, J.; Nabulsi, N.; Suridjan, I.; Comley, R.A.; Huang, Y.; Finnema, S.J.; Carson, R.E. Synaptic Changes in Parkinson Disease Assessed with in vivo Imaging. Ann. Neurol. 2020, 87, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Saari, L.; Kivinen, K.; Gardberg, M.; Joutsa, J.; Noponen, T.; Kaasinen, V. Dopamine transporter imaging does not predict the number of nigral neurons in Parkinson disease. Neurology 2017, 88, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Creed, R.B.; Menallel, L.; Casey, B.; Dave, K.D.; Janssens, H.B.; Veinbergs, I.; van der Hart, M.; Rassoulpour, A.; Goldberg, M.S. Basal and Evoked Neurotransmitter Levels in Parkin, DJ-1, PINK1 and LRRK2 Knockout Rat Striatum. Neuroscience 2019, 409, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Chotibut, T.; Fields, V.; Salvatore, M.F. Norepinephrine transporter inhibition with desipramine exacerbates L-DOPA-induced dyskinesia: role for synaptic dopamine regulation in denervated nigrostriatal terminals. Mol. Pharmacol. 2014, 86, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Sarre, S.; Vandeneede, D.; Ebinger, G.; Michotte, Y. Biotransformation of L-DOPA to dopamine in the substantia nigra of freely moving rats: effect of dopamine receptor agonists and antagonists. J. Neurochem. 1990, 70, 1730–1739. [Google Scholar] [CrossRef]
- Perez, X.A.; Parameswaran, N.; Huang, L.Z.; O’Leary, K.T.; Wuik, M. Pre-synaptic dopaminergic compensation after moderate nigrostriatal damage in non-human primates. J. Neurochem. 2008, 105, 1861–1872. [Google Scholar] [CrossRef]
- Mela, F.; Marti, M.; Bido, S.; Cenci, M.A.; Morari, M. In Vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to l-DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol. Dis. 2012, 45, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Kliem, M.A.; Pare, J.F.; Khan, Z.U.; Wichmann, T.; Smith, Y. Ultrastructural localization and function of dopamine D1-like receptors in the substantia nigra pars reticulata and the internal segment of the globus pallidus of parkinsonian monkeys. Eur. J. Neurosci. 2010, 31, 836–851. [Google Scholar] [CrossRef] [PubMed]
- Mailman, R.B.; Yang, Y.; Huang, X. D.1.; not, D.2.; dopamine receptor activation dramatically improves MPTP-induced parkinsonism unresponsive to levodopa. Eur. J. Pharmacol. 2021, 892, 173760. [Google Scholar] [CrossRef]
- Trevitt, J.T.; Carlson, B.B.; Nowend, K.; Salamone, J.D. Substantia nigra pars reticulate is a highly potent site of action for the behavioral effects of the D1 antagonist SCH23390 in rat. Psychopharmacol. 2001, 156, 32–41. [Google Scholar]
- Tang, P.; Knight, W.C.; Li, H.; Guo, Y.; Perlmutter, J.S.; Benzinger, T.L.S.; Morris, J.C.; Xu, J. Dopamine D1 + D3 receptor density may correlate with parkinson disease clinical features. Ann. Clin. Transl. Neurol. 2021, 8, 224–237. [Google Scholar]
- Roedter, A.; Winkler, C.; Samil, M.; Walter, G.; Brandis, A.; Nikkhah., *!!! REPLACE !!!*. Comparison of unilateral and bilateral intrastriatal 6-hydroxydopamine-induced axon terminal lesions: Evidence for interhemispheric functional coupling of the two nigrostriatal pathways. J. Comp. Neurol. 2001, 432, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Radnikow, G. Misgeld U Dopamine D1 receptors facilitate GABAA synaptic currents in the rat substantia nigra pars reticulata, J. Neurosci. 1998, 18, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Dorval, A.D.; Grill, W.M. Deep brain stimulation of the subthalamic nucleus reestablishes neuronal information transmission in the 6-OHDA rat model of parkinsonism. J. Neurophysiol. 2014, 111, 1949–1959. [Google Scholar] [CrossRef]
- DeLong, M.R.; Wichmann, T. Basal Ganglia Circuits as Targets for Neuromodulation in Parkinson Disease. JAMA Neurol. 2015, 72, 1354–1360. [Google Scholar] [CrossRef]
- McGregor, M.M.; Nelson, A.B. Circuit mechanisms of Parkinson’s disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef]
- Calabresi, P.; Picconi, B.; Tozzi, A.; Ghiglieri, V.; Di Filippo, M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci. 2014, 17, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Foffani, G.; Dehay, B.; Bezard, E.; Obeso, J.A. Motor and non-motor circuit disturbances in early Parkinson disease: which happens first? Nat. Rev. Neurosci. 2022, 23, 115–128. [Google Scholar] [CrossRef]
- Zaman, V.; Boger, H.A.; Granholm, A.C.; Rohrer, B.; Moore, A.; Buhusi, M.; Gerhardt, G.A.; Hoffer, B.J.; Middaugh, L.D. The nigrostriatal dopamine system of aging GFRalpha-1 heterozygous mice: neurochemistry morphology behaviorEur, J. Neurosci. 2008, 28, 1557–1568. [Google Scholar]
- Pruett, B.S.; Salvatore, M.F. Nigral GFRα1 infusion in aged rats increases locomotor activity, nigral tyrosine hydroxylase, and dopamine content in synchronicity. Mol. Neurobiol. 2013, 47, 988–999. [Google Scholar] [CrossRef]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O'Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef]
- Grondin, R.; Cass, W.A.; Zhang, Z.; Stanford, J.A.; Gash, D.M.; Gerhardt, G.A. Glial Cell Line -Derived Neurotrophic Factor Increases Stimulus-Evoked Dopamine Release and Motor Speed in Aged Rhesus Monkeys. J. Neurosci. 2003, 23, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Gash, D.M.; Zhang, Z.; Ovadia, A.; Cass, W.A.; Yi, A.; Simmerman, L.; Russell, D.; Martin, D.; Lapchak, P.A.; Collins, F.; Hoffer, B.J. Gerhardt GA Functional recovery in parkinsonian monkeys treated with, G. D.N.F. Nature 1996, 380, 252–255. [Google Scholar] [CrossRef]
- Gerhardt, G.A.; Cass, W.A.; Huettl, P.; Brock, S.; Zhang, Z.; Gash, D.M. GDNF improves dopamine function in the substantia nigra but not the putamen of unilateral MPTP-lesioned rhesus monkeys. Brain Res. 1999, 817, 163–171. [Google Scholar] [CrossRef]
- Hoffer, B.J.; Hoffman, A.F.; Bowenkamp, K.E.; Huettl, P.; Hudson, J.; Martin, D.; Lin, L.F.; Gerhardt, G.A. Glial cell line-derived neurotrophic factor reverses toxin-induced injury to midbrain dopaminergic neurons in vivo. Neurosci. Lett. 1994, 182, 107–111. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; Brodsky, M.A.; Burchiel, K.; Kelly, P.; Dalvi, A.; Scott, B.; Stacy, M.; Turner, D.; Wooten, G.F.; Elias, W.J.; Laws, E.R.; Dhawan, V.; Stoessl, A.J.; Matcham, J.; Coffey, R.J.; Traub, M. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson's disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Salvatore, M.F.; Zhang, J.L.; Large, D.M.; Wilson, P.E.; Gash, C.R.; Thomas, T.C.; Haycock, J.W.; Bing, G.; Stanford, J.A.; Gash, D.M.; Gerhardt, G.A. Striatal GDNF administration increases tyrosine hydroxylase phosphorylation in rat striatum and substantia nigra. J. Neurochem. 2004, 90, 245–254. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Gerhardt, G.A.; Dayton, R.D.; Klein, R.L.; Stanford, J.A. Bilateral effects of unilateral GDNF administration on dopamine- and GABA-regulating proteins in the rat nigrostriatal system. Exp. Neurol. 2009, 219, 197–207. [Google Scholar] [CrossRef]
- Kasanga, E.A.; Han, Y.; Navarrete, W.; McManus, R.; Shifflet, M.K.; Parry, C.; Barahona, A.; Manfredsson, F.P.; Nejtek, V.A.; Richardson, J.R.; Salvatore, M.F. Differential expression of RET and GDNF family receptor, GFR-α1, between striatum and substantia nigra following nigrostriatal lesion: A case for diminished GDNF-signaling. Exp. Neurol. 2023, 366, 114435. [Google Scholar] [CrossRef]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson's disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef]
- Kordower, J.H.; Goetz, C.G.; Chu, Y.; Halliday, G.M.; Nicholson, D.A.; Musial, T.F.; Marmion, D.J.; Stoessl, A.J.; Freeman, T.B.; Olanow, C.W. Robust graft survival and normalized dopaminergic innervation do not obligate recovery in a Parkinson disease patient. Ann. Neurol. 2017, 81, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Shima, A.; Kambe, D.; Nishida, A.; et al. Motor progression and nigrostriatal neurodegeneration in Parkinson’s disease. Ann. Neurol. 2022, 92, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.K.; Tian, L.; Flores, H.; Su, Y.; Tabbal, S.D.; Loftin, S.K.; Moerlin, S.M.; Perlmutter, J.S. Validation of nigrostriatal positron emission tomography measures: critical limits. Ann. Neurol. 2013, 73, 390–396. [Google Scholar] [CrossRef]
- Perlmuttter, J.S.; Norris, S.A. Neuroimaging biomarkers for Parkinson’s disease: fact and fantasy. Ann. Neurol. 2014, 76, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Schröter, N.; Rijntjes, M.; Urbach, H.; Weiller, C.; Treppner, M.; Kellner, E.; Jost, W.H.; Sajonz, B.E.A.; Reisert, M.; Hosp, J.A.; Rau, A. Disentangling nigral and putaminal contribution to motor impairment and levodopa response in Parkinson’s disease. npj Parkinson’s Dis. 2022, 8, 132. [Google Scholar] [CrossRef]
- Pérez-Taboada, I.; Alberquilla, S.; Martin, E.D.; Anand, R.; Vietti-Michelina, S.; Tebeka, N.N.; Cantley, J.; Cragg, S.J.; Moratalla, R.; Vallejo, M. Diabetes Causes Dysfunctional Dopamine Neurotransmission Favoring Nigrostriatal Degeneration in Mice. Mov. Disord. 2020, 35, 1636–1648. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.D.; De Silva, S.; et al. Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol. Dis. 2014, 70, 190–203. [Google Scholar] [CrossRef]
- Blesa, J.; Pifl, C.; Sanchez-Gonzalez, M.A.; Juri, C.; Garcia-Cabezas, M.A.; Adanez, R.; Iglesias, E.; et al. The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: A PET, histological, and biochemical study. Neurobiol. Dis. 2012, 48, 79–91. [Google Scholar] [CrossRef]
- Petzinger, G.M.; Walsh, J.P.; Akopian, G.; Hogg, E.; Abernathy, A.; Arevalo, P.; Turnquist, P.; Vuckovic, M.; Fisher, B.E.; Togasaki, D.M.; Jakowec, M.W. Effects of treadmill exercise on dopaminergic transmission in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse model of basal ganglia injury. J. Neurosci. 2007, 27, 5291–5300. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, S.J.; Gross, N.B.; Fricks, A.N.; Casiano, B.D.; Nguyen, T.B.; Marshall, J.F. Running wheel exercise enhances recovery from nigrostriatal dopamine injury without inducing neuroprotection. Neuroscience 2007, 144, 1141–1151. [Google Scholar] [CrossRef]
- Churchill, M.J.; Pflibsen, L.; Sconce, M.D.; Moore, C.; Kim, K.; Meshul, C.K. Exercise in an animal model of Parkinson’s disease: Motor recovery but not restoration of the nigrostriatal pathway, Neuroscience. 2017, 359, 224–247.
- Robertson, G.S.; Robertson, H.A. Evidence that L-DOPA-induced rotational behavior is dependent on both striatal and nigral mechanisms. J. Neurosci. 1999, 9, 3326–3331. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.S.; Robertson, H.A. Evidence that the substantia nigra is a site of action for, L.-D.O.P.A. Neurosci. Lett. 1988, 89, 204–208. [Google Scholar] [CrossRef]
- Jackson, E.A.; Kelly, P.H. Role of nigral dopamine in amphetamine-induced locomotor activity. Brain Res. 1983, 278, 366–369. [Google Scholar] [CrossRef]
- Bradbury, A.J.; Costall, B.; Kelly, M.E.; Naylor, R.J.; Smith, J.A. Biochemical correlates of motor changes caused by the manipulation of dopamine function in the substantia nigra of the mouse. Neuropharmacology 1985, 24, 1155–1161. [Google Scholar] [CrossRef]
- Jackson, E.A.; Kelly, P.H. Effects of intranigral injections of dopamine agonists and antagonists, glycine, muscimol and N- methyl-D,L-aspartate on locomotor activity. Brain Res. Bull. 1984, 13, 309–317. [Google Scholar] [CrossRef]
- Ahlenius, S.; Anden, N.E.; Engel, J. Restoration of locomotor activity in mice by low L-DOPA doses after suppression by alpha-methyltyrosine but not by reserpine. Brain Res. 1973, 62, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C.; Jenner, P.; Marsden, C.D. The relative importance of dopamine and noradrenaline receptor stimulation for the restoration of motor activity in reserpine or alpha-methyl-p-tyrosine pre-treated mice. Pharmacol. Biochem. Behav. 1976, 4, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; Pruett, B.S. Dichotomy of Tyrosine Hydroxylase and Dopamine Regulation between Somatodendritic and Terminal Field Areas of Nigrostriatal and Mesoaccumbens Pathways. PLoS ONE 2012, 7, e29867. [Google Scholar] [CrossRef] [PubMed]
- Leng, A.; Mura, A.; Hengerer, B.; Feldon, J.; Ferger, B. Effects of blocking the dopamine biosynthesis and of neurotoxic dopamine depletion with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) on voluntary wheel running in mice. Behav. Brain Res. 2005, 154, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.A.; Marsh, S.T.; Hutchings, J.E.; Castañeda, E. Amphetamine-evoked rotation requires newly synthesized dopamine at 14 days but not 1 day after intranigral 6-OHDA and is consistently dissociated from sensorimotor behavior. Behav. Brain Res. 2009, 200, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Ankenman, R.; Salvatore, M.F. Low dose alpha-methyl-para-tyrosine (AMPT) in the treatment of dystonia and dyskinesia. J. Neuropsychiatry Clin. Neurosci. 2007, 19, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Bloemen, O.J.N.; de Koning, M.B.; Boot, E.; Booij, J.; van Amelsvoort, T.A. Challenge and Therapeutic Studies Using Alpha-Methyl-para-Tyrosine (AMPT) in Neuropsychiatric Disorders: A Review. Central Nervous System agents in Medicinal Chemistry. 2008, 8, 249–256. [Google Scholar] [CrossRef]
- Rubinstein, M.; Gershanik, O.; Stefano, F.J. Different roles of D-1 and D-2 dopamine receptors involved in locomotor activity of supersensitive mice. Eur. J. Pharmacol. 1988, 148, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.C.; Meurer, Y.S.R.; Bioni, V.S.; Cunha, D.M.G.; Goncalves, N.; Lopes-Silva, L.B.; Becegato, M.; Soares, M.B.L.; Marinho, G.F.; Santos, J.R.; Silva, R.H. Female Rats Are Resistant to Cognitive, Motor and Dopaminergic Deficits in the Reserpine-Induced Progressive Model of Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 757714. [Google Scholar] [CrossRef]
- Duty, S.; Jenner, P. Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef]
- May, R.H.; Voegele, G.E. Parkinsonian reactions following chlorpromazine and reserpine; similar reactions in the same patients. AMA Arch. Neurol. Psychiatry 1956, 75, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Nirenberg, M.J.; Chan, J.; Liu, Y.; Edwards, R.H.; Pickel, V.M. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J. Neurosci. 1996, 16, 4135–4145. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.M.; Salvatore, M.F.; Pruett, B.S.; Guerin, G.F.; Goeders, N.E. Biphasic dopamine regulation in mesoaccumbens pathway in response to non-contingent binge and escalating methamphetamine regimens in the Wistar rat. Psychopharmacology 2011, 215, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Nissbrandt, H.; Sundström, E.; Jonsson, G.; Hjorth, S.; Carolsson, A. Synthesis and Release of Dopamine in Rat Brain: Comparison Between Substantia Nigra Pars Compacta, Pars Reticulata, and Striatum. J. Neurochem. 1989, 52, 1170–1182. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, M.J.; Abercrombie, E.D. Biochemistry of Somatodendritic Dopamine Release in Substantia Nigra: An In Vivo Comparison with Striatal Dopamine Release. J. Neurochem. 1995, 65, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.; Westerink, B.H.C. Characterization and Pharmacological Responsiveness of Dopamine Release Recorded by Microdialysis in the Substantia Nigra of Conscious Rats. J Neurochem. 1991, 57, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.G.; Forbes, B.; Cheung, P.Y.; Martini, A.; Burrell, M.H.; Freestone, P.S.; Lipski, J. Action potential and calcium dependence of tonic somatodendritic dopamine release in the Substantia Nigra pars compacta. J. Neurochem. 2018, 148, 462–479. [Google Scholar] [CrossRef]
- Cragg, S.J.; Rice, M.E. Dancing past the DAT at a DA synapse. Trends Neurosci. 2004, 27, 270–277. [Google Scholar] [CrossRef]
- Kaasinen, V.; Vahlberg, T.; Stoessl, J.A.; Strafella, A.P.; Antonini, A. Dopamine receptors in Parkinson’s disease: A meta-analysis of imaging studies. Mov. Disord. 2021, 36, 1781–1791. [Google Scholar] [CrossRef]
- Biswas, B.; Carlsson, A. Potentiation by Neuroleptic Agents of the Inhibitory Action of Intraperitoneally Administered GABA on the Locomotor Activity of Mice. Pharmacol. Biochem. Behav. 1978, 6, 651–654. [Google Scholar] [CrossRef]
- Hillegaart, V.; Ahlenius, S. Effects of raclopride on exploratory locomotor activity, treadmill locomotion, conditioned avoidance behaviour and catalepsy in rats: behavioural profile comparisons between raclopride, haloperidol and preclamol. Pharmacol. Toxicol. 1987, 60, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Löschmann, P.A.; Smith, L.A.; Lange, K.W.; Jaehnig, P.; Jenner, P.; Marsden, C.D. Motor activity following the administration of selective D-1 and D-2 dopaminergic drugs to normal common marmosets. Psychopharmacology 1991, 105, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Ericson, H.; Radesäter, A.C.; Servin, E.; Magnusson, O.; Mohringe, B. Effects of intermittent and continuous subchronic administration of raclopride on motor activity, dopamine turnover and receptor occupancy in the rat. Pharmacol. Toxicol. 1996, 79, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.C.; Beninger, R.J. The D1 dopamine receptor antagonist, SCH 23390 reduces locomotor activity and rearing in rat. Pharmacol Biochem Behav 1985, 22, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.W.; Caramona, G.N. Effects of dopamine agonists and antagonists on locomotor activity in male and female rats. Pharmacol. Biochem. Behav. 2002, 72, 857–863. [Google Scholar] [CrossRef]
- Svensson, K.A.; Heinz, B.A.; Schaus, J.M.; Beck, J.P.; Hao, J.; Krushinski, J.H.; et al. An Allosteric Potentiator of the Dopamine D1 Receptor Increases Locomotor Activity in Human D1 Knock-In Mice without Causing Stereotypy or Tachyphylaxis. 2017, 360, 117–128.
- Mailman, R.B.; Yang, Y.; Huang, X. D.1.; not, D.2.; dopamine receptor activation dramatically improves MPTP-induced parkinsonism unresponsive to levodopa. Eur. J. Pharmacol. 2021, 892, 173760. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, S.H.; Hauser, R.A.; Pahwa, R.; Gray, D.; Duvvuri, S. Dopamine agonists in Parkinson’s disease: Impact of D1-like or D2-like dopamine receptor subtype selectivity and avenues for future treatment. Clin. Parkinsonism and Rel Disord. 2023, 9, 100212. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, S.; Liu, W.; Duvvuri, S.; Thayer, K.; Gray, D.L. Evaluation of D1/D5 partial agonist PF-06412562 in Parkinson’s disease following oral administration. Neurodegener. Dis. 2018, 18, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Xuemei, H.; Lewis, M.M.; Van Scoy, L.J.; De Jesus, S.; Eslinger, P.J.; Arnold, A.C.; Miller, A.J.; et al. The D1/D5 Dopamine Partial Agonist PF-06412562 in Advanced-Stage Parkinson’s Disease: A Feasibility Study. J. Parkinson’s Dis. 2020, 10, 1515–1527. [Google Scholar]
- Pothos, E.N.; Przedborski, S.; Davila, V.; Schmitz, Y.; Sulzer, D. D2-Like Dopamine Autoreceptor Activation Reduces Quantal Size in PC12 Cells. J. Neurosci. 1998, 18, 5575–5585. [Google Scholar] [CrossRef]
- Cragg, S.J.; Greenfield, S.A. Differential Autoreceptor Control of Somatodendritic and Axon Terminal Dopamine Release in Substantia Nigra, Ventral Tegmental Area, and Striatum. J. Neurosci. 1997, 17, 5738–5746. [Google Scholar] [CrossRef] [PubMed]
- Schultz, W.; Ruffieux, A.; Aebischer, P. The activity of pars compacta neurons of the monkey substantia nigra in relation to motor activation. Exp. Brain Res. 1983, 51, 377–387. [Google Scholar] [CrossRef]
- da Silva, J.A.; Tecuapetla, F.; Paixão, V.; Costa, R.M. Dopamine neuron activity before action Initiation gates and invigorates future movements. Nature 2018, 554, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Coddington, L.T.; Dudman, J.T. Learning from Action: Reconsidering Movement Sinaling in Midbrain Dopamine Neuron Activity. Neuron 2019, 104, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; da Silva, J.A.; Costa, R.M. What, If, and When to Move: Basal Ganglia Circuits and Self-Paced Action Initiation. Ann. Rev. Neurosci. 2019, 42, 459–483. [Google Scholar] [CrossRef] [PubMed]
- Bergquist, F.; Shahabi, H.N.; Nissbrandt, H. Somatodendritic dopamine release in rat substantia nigra influences motor performance on the accelerating rod. Brain Res. 2003, 973, 81–91. [Google Scholar] [CrossRef]
- Hebert, M.A.; Gerhardt, G.A. Normal and drug-induced locomotor behavior in aging: comparison to evoked DA release and tissue content in Fischer 344 rats. Brain Res. 1998, 797, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Yurek, D.M.; Hipkens, S.B.; Hebert, M.A.; Gash, D.M.; Gerhardt, G.A. Age-related decline in striatal dopamine release and motoric function in Brown Norway/Fischer 344 hybrid rats. Brain Res. 1998, 791, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, M.N.; Genc, O.; Bobela, W.; Mohanna, S.; Ardah, M.T.; El-Agnaf, O.M.; Cantoni, M.; Bensadoun, J.C.; Schneggenburger, R.; Knott, G.W.; Aebischer, P.; Schneider, B.L. Nigrostriatal overabundance of α-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathologica 2012, 123, 653–669. [Google Scholar] [CrossRef]
- Goodwin, J.S.; Larson, G.A.; Swant, J.; Sen, N.; Javitch, J.A.; Zahniser, N.R.; De Felice, L.J.; Khoshbouei, H. Amphetamine and methamphetamine differentially affect dopamine transporters in vitro and in vivo. J. Biol. Chem. 2009, 284, 2978–2989. [Google Scholar] [CrossRef]
- Kahlig, K.M.; Binda, F.; Khoshbouei, H.; Blakely, R.D.; McMahon, D.G.; Javitch, J.A.; Galli, A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc. Natl. Acad. Sci. 2005, 102, 3495–3500. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef] [PubMed]
- Rivière, G.J.; Byrnes, K.A.; Gentry, W.B.; Owens, S.M. Spontaneous Locomotor Activity and Pharmacokinetics of Intravenous Methamphetamine and Its Metabolite Amphetamine in the Rat. 1999, 291, 1220–1226.
- Laruelle, M.; Abi-Dargham, A.; van Dyck, C.H.; Rosenblatt, W.; Zea-Ponce, Y.; Zoghbi, S.S.; Baldwin, R.M.; Charney, D.S.; Hoffer, P.B.; Kung, H.F.; Innis, R.B. SPECT Imaging of Striatal Dopamine Release after Amphetamine Challenge. J. Nuc Med. 1995, 36, 1182–1190. [Google Scholar]
- Hall, D.A.; Stanis, J.J.; Avila, H.M.; Gulley, J.M. A comparison of amphetamine- and methamphetamine-induced locomotor activity in rats: evidence for qualitative differences in behavior. Psychopharmacology 2008, 195, 469–478. [Google Scholar] [CrossRef]
- Ciliax, B.J.; Heilman, C.; Demchyshyn, L.L.; Pristupa, Z.B.; Ince, E.; Hersch, S.M.; Niznik, H.B.; Levey, A.I. The dopamine transporter: immunochemical characterization and localization in brain. 1995, 15, 1714–1723.
- Nirenberg, M.J.; Vaughan, R.A.; Uhl, G.R.; Kuhar, M.J.; Pickel, V.M. The Dopamine Transporter Is Localized to Dendritic and Axonal Plasma Membranes of Nigrostriatal Dopaminergic Neurons. J. Neurosci. 1996, 16, 436–437. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.A.; Foster, J.D. Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol. Sci. 2013, 34, P489–P496. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.F.; Lupica, C.R.; Gerhardt, G.A. Dopamine transporter activity in the substantia nigra and striatum assessed by high-speed chronoamperometric recordings in brain slices. J. Pharmacol. Exp. Ther. 1998, 287, 487–496. [Google Scholar] [PubMed]
- Ford, C.P.; Gantz, S.C.; Phillips, P.E.M.; Williams, J.T. Control of extracellular dopamine at dendrite and axon terminals. J. Neurosci. 2010, 30, 6975–6983. [Google Scholar] [CrossRef] [PubMed]
- Cragg, S.J.; Rice, M.E.; Greenfield, S.A. Heterogeneity of Electrically Evoked Dopamine Release and Reuptake in Substantia Nigra, Ventral Tegmental Area, and Striatum. J. Neurophysiol. 1997, 77, 863–873. [Google Scholar] [CrossRef]
- Ma, S.Y.; Ciliax, B.J.; Stebbins, G.; Jaffar, S.; Joyvce, J.N.; Cochran, E.J.; Kordower, J.H.; Mash, D.C.; Levey, A.I.; Mufson, E.J. Dopamine transporter-immunoreactive neurons decrease with age in the human substantia nigra. J. Comp. Neurol. 1999, 409, 25–37. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Apparsundaram, S.; Gerhardt, G.A. Decreased plasma membrane expression of striatal dopamine transporter in aging. Neurobiol. Aging 2003, 24, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Bu, M.; Farrer, M.J.; Khoshbouei, H. Dynamic control of the dopamine transporter in neurotransmission and homeostasis. npj Parkinson’s Dis. 2021, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Gainetdinov, R.R.; Jaber, M.; Giros, B.; Wightman, R.M.; Caron, M.G. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc. Natl. Acad. Sci. USA 1998, 95, 4029–4034. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; Calipari, E.S.; Jones, S.R. Regulation of tyrosine hydroxylase expression and phosphorylation in dopamine transporter-deficient mice. ACS Chem. Neurosci. 2016, 7, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Morón, J.A.; Brockiington, A.; Wise Rocha, B.A.; Hope, B.T. Dopamine Uptake through the Norepinephrine Transporter in Brain Regions with Low Levels of the Dopamine Transporter: Evidence from Knock-Out Mouse Lines. J. Neurosci. 2002, 22, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Chotibut, T.; Apple, S.M.; Jefferis, R.; Salvatore, M.F. Dopamine Transporter Loss in 6-OHDA Parkinson’s Model Is Unmet by Parallel Reduction in Dopamine Uptake. PLoS ONE 2012, 7, e52322. [Google Scholar] [CrossRef]
- Chotibut, T.; Fields, V.; Salvatore, M.F. Norepinephrine transporter inhibition with desipramine exacerbates L-DOPA-induced dyskinesia: role for synaptic dopamine regulation in denervated nigrostriatal terminals. Mol. Pharmacol. 2014, 86, 675–685. [Google Scholar] [CrossRef]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Spielewoy, C.; Roubert, C.; Hamon, M.; Nosten-Bertrand, M.; Betancur, C.; Giros, B. Behavioural disturbances associated with hyperdopaminergia in dopamine-transporter knockout mice. Behav. Pharmacol. 2000, 11, 279–290. [Google Scholar] [CrossRef]
- Stanford, J.A.; Vorontsova, E.; Surgener, S.P.; Gerhardt, G.A.; Fowler, S.C. Aged Fischer 344 rats exhibit altered locomotion in the absence of decreased locomotor activity: exacerbation by nomifensine. Neurosci. Lett. 2002, 333, 195–198. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Kasanga, E.A.; Kelly, D.P.; Venable, K.E.; McInnis, T.R.; et al. Modulation of nigral dopamine signaling mitigates parkinsonian signs of aging: evidence from intervention with caloric restriction or inhibition of dopamine uptake. GeroScience 2023, 45, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Damsma, G.; Fibiger, H. Characterization of dopamine release in the substantia nigra by in vivo microdialysis in freely moving rats. J. Neurosci. 1991, 11, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Nieoullon, A.; Cheramy, A.; Glowinski, J. Nigral and striatal dopamine release under sensory stimuli. Nature 1977, 269, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Coddington, L.T.; Dudman, J.T. The timing of action determines reward prediction signals in identified midbrain dopamine neurons. Nat. Neurosci. 2018, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; McInnnis, T.R.; Cantu, M.A.; Apple, D.M.; Pruett, B.S. Tyrosine Hydroxylase Inhibition in Substantia Nigra Decreases Movement Frequency. Mol. Neurobiol. 2019, 56, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; Terrebonne, J.; Cantu, M.A.; McInnis, T.; Venable, K.; Kelley, P.; Kasanga, E.A.; Latimer, B.; Owens, C.L.; et al. Dissociation of striatal dopamine and tyrosine hydroxylase expression from aging-related motor decline: evidence from calorie restriction intervention. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 73, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.R.; Nissbrandt, H.; Bergquist, F. Partial depletion of dopamine in substantia nigra impairs motor performance without altering striatal dopamine neurotransmission. Eur. J. Neurosci. 2006, 24, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; Pruett, B.S.; Spann, S.L.; Dempsey, C. Aging Reveals a Role for Nigral Tyrosine Hydroxylase ser31 Phosphorylation in Locomotor Activity Generation. PLoS ONE 2009, 4, e8466. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; Garcia-Espana, A.; Goldstein, M.; Deutch, A.Y.; Haycock, J.W. Stoichiometry of tyrosine hydroxylase phosphorylation in the nigrostriatal and mesolimbic systems in vivo: effects of acute haloperidol and related compounds. J. Neurochem. 2000; 75, 225–232. [Google Scholar]
- Salvatore, M.F.; Pruett, B.S.; Dempsey, C.; Fields, V. Comprehensive profiling of dopamine regulation in substantia nigra and ventral tegmental area. J Vis Exp. 2012, 66, 4171. [Google Scholar] [CrossRef]
- Emborg, M.E.; Ma, S.Y.; Mufson, E.J.; Levey, A.I.; Taylor, M.D.; Brown, W.D.; Holden, J.E.; Kordower, J.H. Age-related declines in nigral neuronal function correlate with motor impairments in rhesus monkeys. J. Comp. Neurol. 1998, 401, 253–265. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson's disease: Substantia nigra regional selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef]
- Chu, Y.; Hirst, W.D.; Federoff, H.J.; Harms, A.S.; Stoessl, A.J.; Kordower, J.H. Nigrostriatal tau pathology in parkinsonism and Parkinson’s disease. 2023; awad388. [Google Scholar] [CrossRef]
- Ross, G.W.; Petrovich, H.; Abbott, R.D.; Nelson, J.; Markesbery, W.; Davis, D.; Hardman, J.; Launer, L.; Masaki, K.; Tanner, C.M.; White, L.R. Parkinsonian signs substantia nigra neuron density in descendent elders without, P.D. Ann. Neurol. 2004, 56, 532–529. [Google Scholar] [CrossRef]
- Buchman, A.S.; Shulman, J.M.; Nag, S.; Leurgans, S.E.; Arnold, S.E.; Morris, M.C.; Schneider, J.A.; Bennett, D.A. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann. Neurol. 2012, 71, 258–266. [Google Scholar] [CrossRef]
- Wolf, M.E.; LeWitt, P.A.; Bannon, M.J.; Dragovic, L.J.; Kapatos, G. Effect of aging on tyrosine hydroxylase protein content and the relative number of dopamine nerve terminals in human caudate. J. Neurochem. 1991, 56, 1191–1200. [Google Scholar] [CrossRef]
- Kish, S.J.; Shannak, K.; Rajput, A.; Deck, J.H.N.; Hornykiewicz, O. Aging produces a specific pattern of striatal dopamine loss: Implications for the etiology of idiopathic Parkinson's disease. J Neurochem 1992, 58, 642–648. [Google Scholar] [CrossRef]
- Haycock, J.W.; Becker, L.; Ang, L.; Yoshiaki, F.; Hornykiewicz, O.; Kish, S.J. Marked disparity between age-related changes in dopamine and other presynaptic dopaminergic markers in human striatum. J. Neurochem. 2003, 87, 574–585. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Waymire, J.C.; Haycock, J.W. Depolarization-stimulated catecholamine biosynthesis: involvement of protein kinases and tyrosine hydroxylase phosphorylation sites in situ. J. Neurochem. 2001, 79, 349–360. [Google Scholar] [CrossRef]
- Gerhardt, G.A.; Cass, W.A.; Yi, A.; Zhang, Z.; Gash, D.M. Changes in somatodendritic but not terminal dopamine regulation in aged rhesus monkeys. J. Neurochem. 2002, 80, 168–177. [Google Scholar] [CrossRef]
- Irwin, I.; DeLanney, L.E.; McNeill, T.; Chan, P.; Forno, L.S.; et al. Aging and the nigrostriatal dopamine system: a non-human primate study. Neurodegeneration 1994, 3, 251–265. [Google Scholar]
- Siddiqi, Z.; Kemper, T.L.; Killiany, R. Age-related Neuronal Loss from the Substantia Nigra-Pars Compacta and Ventral Tegmental Area of the Rhesus Monkey. J. Neuropath Exp. Neurol. 1999, 58, 959–971. [Google Scholar] [CrossRef]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurological Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Parkinson’s disease. Lancet 1990, 335, 948–952. [Google Scholar] [CrossRef]
- Collier, T.J.; Lipton, J.; Daley, B.F.; Palfi, S.; Chu, Y.; Sortwell, C.; Bakay, R.A.; Sladek, J.R.; Kordower, J.H. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: Diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol. Dis. 2007, 26, 56–65. [Google Scholar] [CrossRef]
- Pifl, C.; Hornykiewicz, O. Dopamine turnover is upregulated in the caudate/putamen of asymptomatic MPTP-treated rhesus monkeys. Neurochem. Int. 2006, 49, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, M.J. Do compensatory processes underlie the preclinical phase of neurodegenerative disease? Insights from an animal model of parkinsonism. Neurobiol. Dis. 1997, 4, 247–253. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Dileone, M.; Lopez-Gonzalez del Rey, N.; Hernandez, L.F.; Obeso, J.A. Compensatory mechanisms in Parkinson's disease: Circuits adaptations and role in disease modification. Exp. Neurol. 2017, 298, 148–161. [Google Scholar] [CrossRef]
- Sarre, S.; Yuan, H.; Jonkers, N.; Van Hemelrijck, A.; Ebinger, G.; Michotte, Y. In vivo characterization of somatodendritic dopamine release in the substantia nigra of 6-hydroxydopamine-lesioned rats. J. Neurochem. 2004, 90, 29–39. [Google Scholar] [CrossRef]
- 174. Suhara, T.; Fukuda, H.; Inoue, O.; Itoh, T.; Suzuki, K.; Yamasaki, T.; et al. Age-related changes in human D1 dopamine receptors measured by positron emission tomography. Psychopharmacology. 1991, 103, 41–45. [Google Scholar] [CrossRef]
- Kaasinen, V.; Vahlberg, T.; Stoessl, J.A.; Strafella, A.P.; Antonini, A. Dopamine receptors in Parkinson’s disease: A meta-analysis of imaging studies. Mov. Disord. 2021, 36, 1781–1791. [Google Scholar] [CrossRef]
- Shui, H.A.; Peng, Y.I.; Wu, R.M.; Tsai, Y.F. Evaluation of l-DOPA biotransformation during repeated l-DOPA infusion into the striatum in freely-moving young and old rats. Dev. Brain Res. 2000, 121, 123–131. [Google Scholar] [CrossRef]
- Orosz, D.; Bennett, J.P. Simultaneous microdialysis in striatum and substantia nigra suggests that the nigra is a major site of action of l-dihydroxyphenylalanine in the “Hemiparkinsonian” rat. Exp. Neurol. 1992, 115, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Sarre, S.; Herregodts, P.; Deleu, D.; Devrieze, A.; De Klippel, N.; Ebinger, G.; Michotte, Y. Biotransformation of L-DOPA in striatum and substantia nigra of rats with a unilateral, nigrostriatal lesion: a microdialysis study. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1992, 346, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Grondin, R.; Zhang, Z.; Yi, A.; Cass, W.A.; Maswood, N.; Andersen, A.H.; Elsberry, D.D.; Klein, M.C.; Gerhardt, G.A.; Gash, D.M. Chronic, controlled GDNF infusion promotes structural and functional recovery in advanced parkinsonian monkeys. Brain 2002, 125, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Slevin, J.T.; Gash, D.M.; Smith, C.D.; Gerhardt, G.A.; Kryscio, R.; Chebrolu, H.; Walton, A.; Wagner, R.; Young, A.B. Unilateral intraputamenal glial cell line-derived neurotrophic factor in patients with Parkinson disease: response to 1 year of treatment and 1 year of withdrawal. J. Neurosurg. 2012, 106, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.K.; Pavese, N.; Javed, S.; Hotton, G.R.; Brooks, D.J.; Gill, S.S. Benefits of putaminal GDNF infusion in Parkinson disease are maintained after GDNF cessation. Neurology 2013, 81, 13. [Google Scholar] [CrossRef] [PubMed]
- Slevin, J.T.; Gerhardt, G.A.; Smith, C.D.; Gash, D.M.; Kryscio, R.; Young, B. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputamenal infusion of glial cell line-derived neurotrophic factor. J. Neurosurg. 2005, 102, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Kasanga, E.A.; Owens, C.L.; Cantu, M.A.; Richard, A.D.; Davis, R.W.; McDivitt, L.M.; Blancher, B.; Pruett, B.S.; Tan, C.; Gajewski, A.; Manfredsson, F.P.; Nejtek, V.A.; Salvatore, M.F. GFR-α1 Expression in Substantia Nigra Increases Bilaterally Following Unilateral Striatal GDNF in Aged Rats and Attenuates Nigral Tyrosine Hydroxylase Loss Following 6-OHDA Nigrostriatal Lesion. ACS Chem. Neurosci. 2019, 10, 4237–4249. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Bjorklund, A.; Gash, D.M.; Whone, A.; Van Laar, A.; Kordower, J.H.; Bankiewicz, K.; Kieburtz, K.; Saarma, M.; Booms, S.; Huttunen, H.; et al. GDNF and Parkinson’s Disease: Where Next? A Summary from a Recent Workshop. J. Parkinsons Dis. 2020, 10, 875–891. [Google Scholar] [CrossRef] [PubMed]
- Tomac, A.; Widenfalk, J.; Lin, L.F. Retrograde axonal transport of glial cell line-derived neurotrophic factor in the adult nigrostriatal system suggests a trophic role in the adult. Proc. Natl. Acad. Sci. USA 1995, 92, 8274–8278. [Google Scholar] [CrossRef]
- Ibáñez, C.F.; Andressoo, J.O. Biology of GDNF and its receptors- relevance for disorders of the central nervous system. Neurobiol. Dis. 2017, 97, 80–89. [Google Scholar] [CrossRef]
- Leitner, M.L.; Molliver, D.C.; Osborne, P.A.; Vejsada, R.; Golden, J.P.; Lampe, P.A.; Kato, A.C.; Milbrandt, J.; Johnson, E.M. Analysis of the retrograde transport of glial cell line-derived neurotrophic factor (GDNF), Neurturin, and Persephin suggests that in vivo signaling for the GDNF family is GFRα coreceptor-specific. J. Neurosci. 1999, 19, 9322–9331. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; Ai, Y.; Fischer, B.; Zhang, A.M.; Grondin, R.C.; Zhang, Z.; Gerhardt, G.A.; Gash, D.M. Point source concentration of GDNF may explain failure of phase II clinical trial. Exp. Neurol. 2006, 202, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Geffen, L.B.; Jessell, T.M.; Cuello, A.C.; Iversen, L.L. Release of dopamine from dendrites in rat substantia nigra. Nature 1976, 260, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Cheramy, A.; Leviel, V.; Glowinski, J. Dendritic release of dopamine in the substantia nigra. Nature 1981, 289, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Waszczak, B.L.; Walters, J.R. Dopamine modulation of the effects of gamma-aminobutyric acid on substantia nigra pars reticulata neurons. Science 1983, 220, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Ruffieux, A.; Schultz, W. Dopaminergic activation of reticulate neurons in the substantia nigra. Nature 1980, 285, 240–241. [Google Scholar] [CrossRef]
- atuszewich, L.; Yamamoto, B.K. Modulation of GABA release by dopamine in the substantia nigra. Synapse 1999, 32, 29–36. [Google Scholar] [CrossRef]
- Lahiri, A.K.; Bevan, M.D. Dopaminergic transmission rapidly and persistently enhances excitability of D1 receptor-expressing striatal projection neurons. Neuron 2020, 106, 288–290. [Google Scholar] [CrossRef]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s disease: different sides of the same coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef]
Figure 1.
Molecular changes in components of DA signaling in SN are autonomous from those in striatum during aging. Unlike PD, loss of TH protein and tissue DA in striatum is substantially less in aging, with maximum loss ~50% being the most ever reported. Conversely in the SN, there are several aging-related changes occurring at the biosynthesis and receptor levels. Loss of the DA D1 receptor occurs in the middle to middle-late stages of the lifespan and is associated with the onset of locomotor decline [150]. Loss of TH protein occurs in the SN toward the latter (aged) part of the lifespan, with a decrease in DA tissue content [152]. Notably, there is also a decrease in site-specific TH phosphorylation at ser31 that occurs only in the SN. The magnitude of nigral TH protein and tissue DA loss in aging [152] is comparable to TH and DA loss at the onset of locomotor impairment in PD [19], suggesting that DA tissue loss arising from decreased ser31 TH phosphorylation and TH protein are mechanisms of hypokinesia seen in aging and in early PD.
Figure 1.
Molecular changes in components of DA signaling in SN are autonomous from those in striatum during aging. Unlike PD, loss of TH protein and tissue DA in striatum is substantially less in aging, with maximum loss ~50% being the most ever reported. Conversely in the SN, there are several aging-related changes occurring at the biosynthesis and receptor levels. Loss of the DA D1 receptor occurs in the middle to middle-late stages of the lifespan and is associated with the onset of locomotor decline [150]. Loss of TH protein occurs in the SN toward the latter (aged) part of the lifespan, with a decrease in DA tissue content [152]. Notably, there is also a decrease in site-specific TH phosphorylation at ser31 that occurs only in the SN. The magnitude of nigral TH protein and tissue DA loss in aging [152] is comparable to TH and DA loss at the onset of locomotor impairment in PD [19], suggesting that DA tissue loss arising from decreased ser31 TH phosphorylation and TH protein are mechanisms of hypokinesia seen in aging and in early PD.
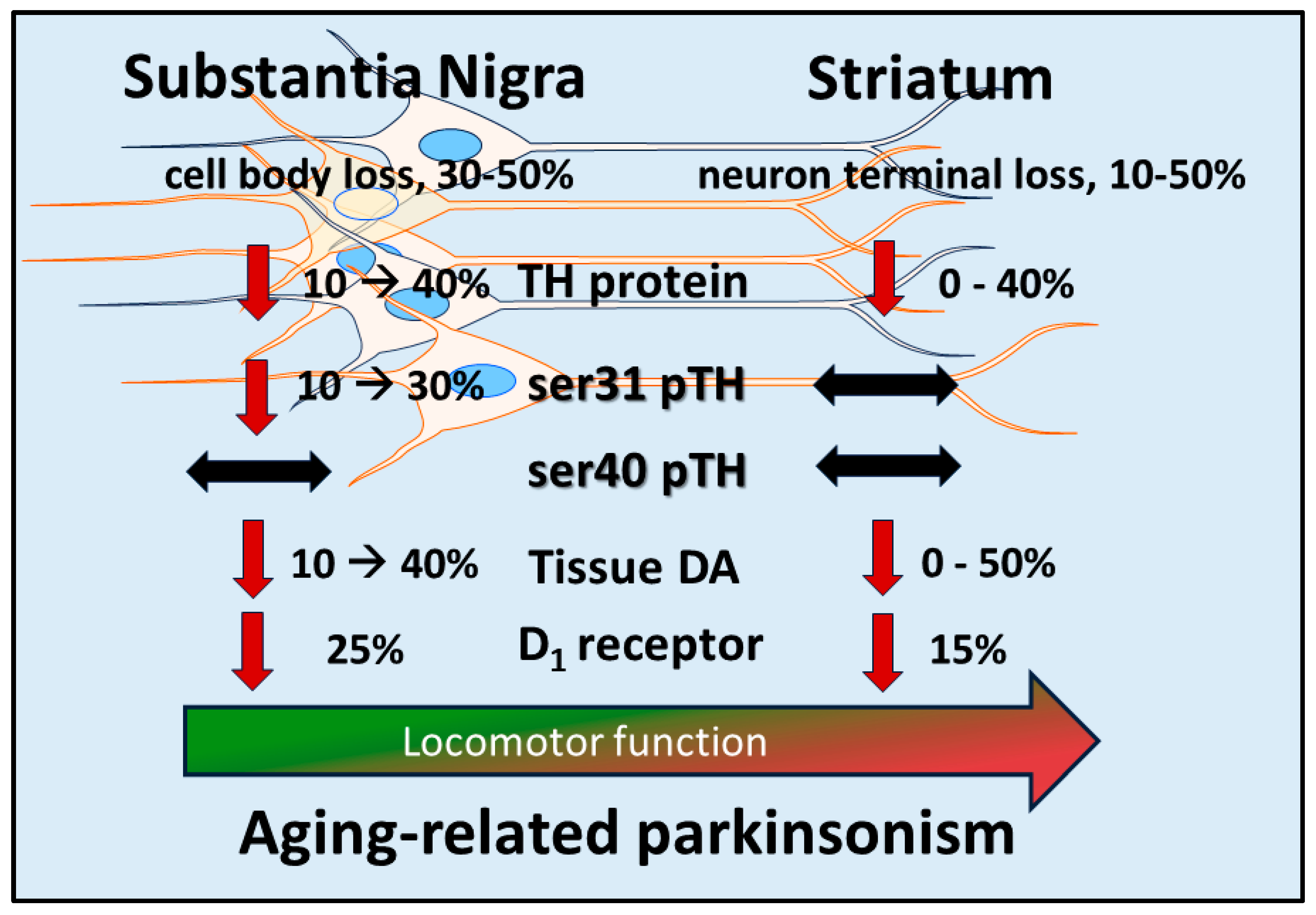
Figure 2.
Dichotomous molecular changes in DA signaling components in SN and striatum in response to neuronal loss. Induction of nigrostriatal neuron loss by 6-OHDA produces a progressive loss of neurons over 4 weeks. Loss of TH protein in SN is less than the magnitude of loss in the striatum at the earlier time points post-lesion, and tissue DA loss is substantially and consistently less in the SN than in striatum. In response to TH loss, there is a site-specific increase in TH phosphorylation at ser31 only in the SN; in striatum, there is a progressive decrease. This increase in ser31 in the SN offsets the progressive loss of TH therein to keep DA loss at a lesser level than TH. As DA tissue loss increases in the SN, the DA D1 receptor increases expression at the latter stages of neuron loss. The increase in both ser31 TH phosphorylation and DA D1 receptor in the SN are compensatory mechanisms to delay the onset of locomotor impairment and alleviate its severity.
Figure 2.
Dichotomous molecular changes in DA signaling components in SN and striatum in response to neuronal loss. Induction of nigrostriatal neuron loss by 6-OHDA produces a progressive loss of neurons over 4 weeks. Loss of TH protein in SN is less than the magnitude of loss in the striatum at the earlier time points post-lesion, and tissue DA loss is substantially and consistently less in the SN than in striatum. In response to TH loss, there is a site-specific increase in TH phosphorylation at ser31 only in the SN; in striatum, there is a progressive decrease. This increase in ser31 in the SN offsets the progressive loss of TH therein to keep DA loss at a lesser level than TH. As DA tissue loss increases in the SN, the DA D1 receptor increases expression at the latter stages of neuron loss. The increase in both ser31 TH phosphorylation and DA D1 receptor in the SN are compensatory mechanisms to delay the onset of locomotor impairment and alleviate its severity.
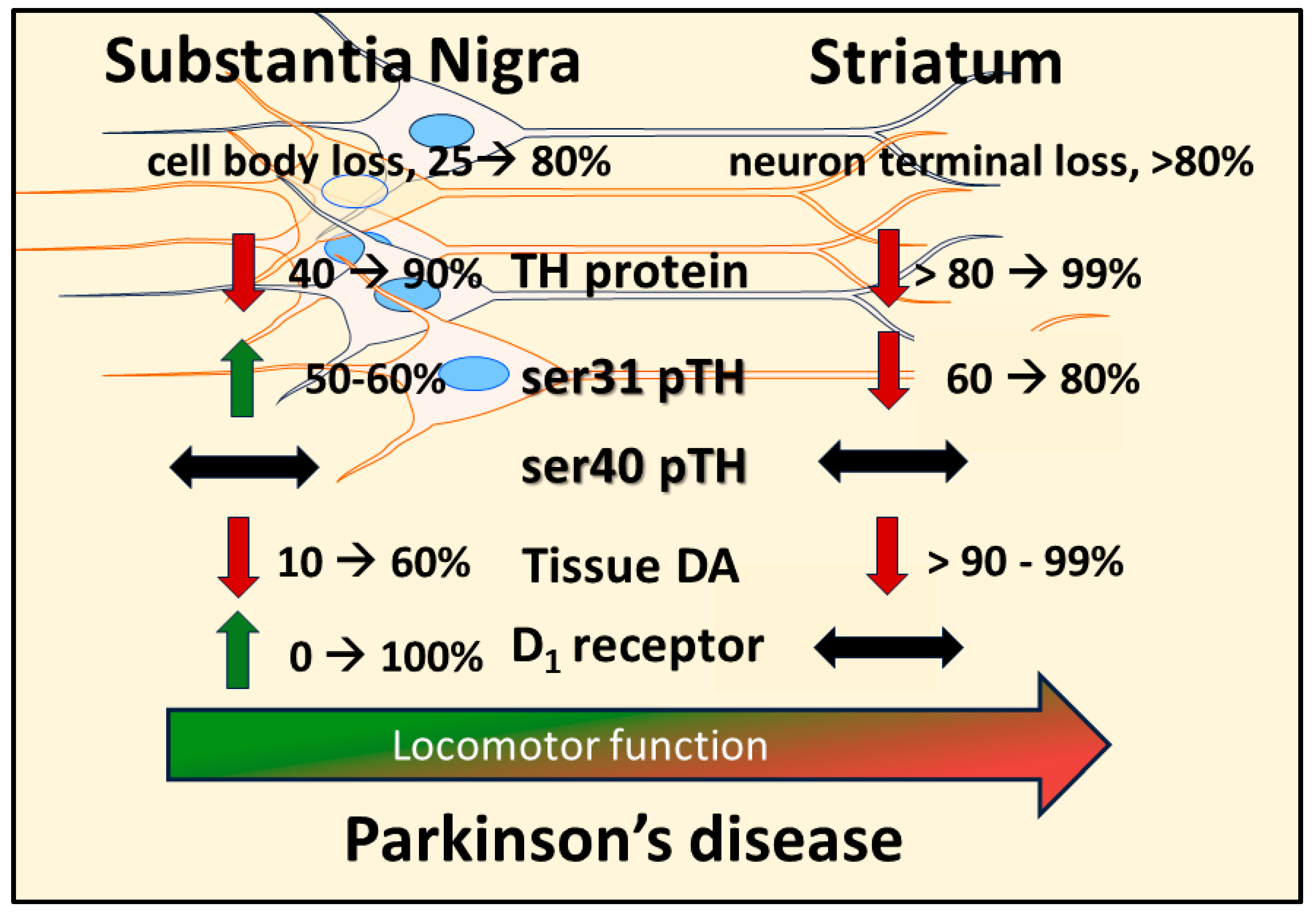
Figure 3.
Compensatory response in substantia nigra to maintain DA signaling. A. Early stage of nigrostriatal neuron loss. During the loss of nigrostriatal neurons, TH protein loss in SN precedes neuron loss. To maintain DA tissue levels in response to TH protein loss, TH phosphorylation at ser31 increases, offsetting the loss of DA that would otherwise occur (as seen in striatum) [22]. The local release of DA in the SN is sufficient to maintain GABA release from striatonigral terminals which is needed to mitigate tonic GABA release from the SNr efferents to promote locomotor activity. B. Late stage of nigrostriatal neuron loss. Although ser31 TH phosphorylation is still increased, the progressive loss of TH protein is sufficient to diminish DA tissue levels, although DA loss is still less than TH protein loss [22]. The decrease in DA tissue content is expected to diminish release. In response, the post-synaptic DA D1 receptor is upregulated on post-synaptic striatonigral terminals to increase responsivity against diminished synaptic DA levels. The overall plasticity of increased DA biosynthesis and DA D1 receptor expression in the SN is hypothesized to mitigate the severity of bradykinesia/hypokinesia arising from TH and neuron loss.
Figure 3.
Compensatory response in substantia nigra to maintain DA signaling. A. Early stage of nigrostriatal neuron loss. During the loss of nigrostriatal neurons, TH protein loss in SN precedes neuron loss. To maintain DA tissue levels in response to TH protein loss, TH phosphorylation at ser31 increases, offsetting the loss of DA that would otherwise occur (as seen in striatum) [22]. The local release of DA in the SN is sufficient to maintain GABA release from striatonigral terminals which is needed to mitigate tonic GABA release from the SNr efferents to promote locomotor activity. B. Late stage of nigrostriatal neuron loss. Although ser31 TH phosphorylation is still increased, the progressive loss of TH protein is sufficient to diminish DA tissue levels, although DA loss is still less than TH protein loss [22]. The decrease in DA tissue content is expected to diminish release. In response, the post-synaptic DA D1 receptor is upregulated on post-synaptic striatonigral terminals to increase responsivity against diminished synaptic DA levels. The overall plasticity of increased DA biosynthesis and DA D1 receptor expression in the SN is hypothesized to mitigate the severity of bradykinesia/hypokinesia arising from TH and neuron loss.
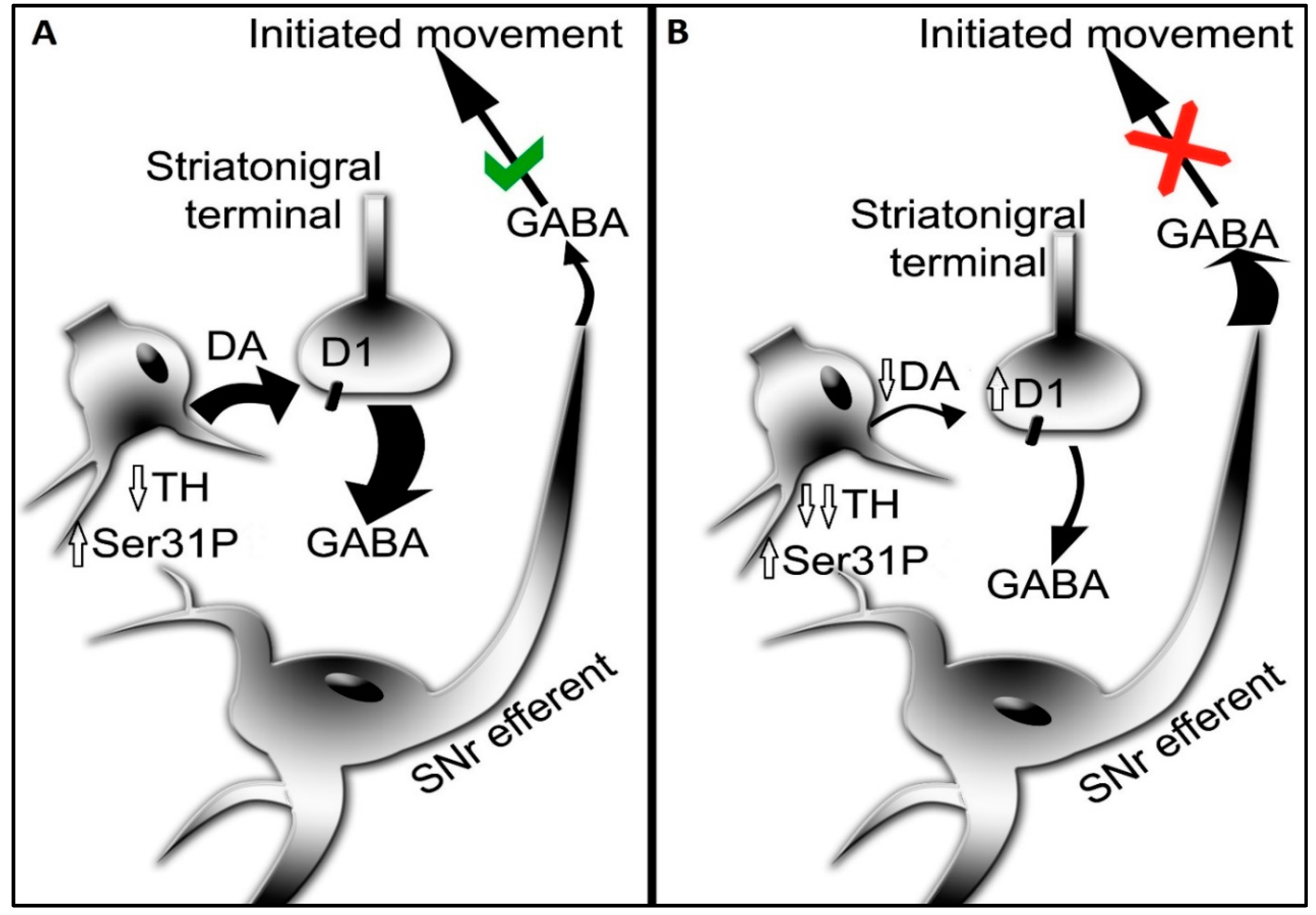
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



