
Submitted:
01 December 2023
Posted:
01 December 2023
You are already at the latest version
Abstract
Sarcomas are a heterogenous group of tumours that commonly carry poor prognosis with limited therapeutic options. Adolescents and young adults (AYA) with sarcoma are a unique and under-studied patient population that have only achieved modest survival gains compared to other groups. We present our institutional experience of AYA with sarcoma who underwent comprehensive molecular profiling (CMP) using either large-panel targeted DNA sequencing or whole genome and transcriptome sequencing and evaluated the feasibility and clinical impact of this approach. Genomic variants detected were determined to be clinically relevant and actionable following the Molecular Tumour Board evaluation. Clinicians provided feedback regarding the utility of testing three months after reporting.
Twenty-five patients who were recruited for CMP are included in this analysis. The median time from consent to final molecular report was 45 days (interquartile range 37-57). Potentially actionable variants were detected for 14 patients (56%) and new treatment recommendations were identified for 12 patients (48%). Pathogenic germline variants were identified in three patients (12%), and one patient had a change of diagnosis. Implementation for CMP for AYA with sarcoma is clinically valuable, feasible and should be increasingly integrated into routine clinical practice as technologies and turnaround times continue to improve.
Keywords:
Adolescents and young adults (AYA) cancer
; molecular profiling
; precision oncology
; genomics
; whole genome sequencing
; next-generation sequencing
; sarcoma
; diagnostic biomarkers
1. Introduction
Comprehensive molecular profiling (CMP) using next generation sequencing (NGS) is one of the cornerstones of precision oncology and can significantly impact clinical decision-making. Sarcomas are a complex group of tumours with heterogenous biology that commonly carry poor prognosis with limited therapeutic options. The use of genomic profiling in sarcoma has the potential to improve patient outcomes through its capacity to improve our understanding of the tumour biology, to provide pathological evaluation for an accurate diagnosis, reveal targeted therapeutic options, and assist in accurate prognostication [1,2]. Historically, CMP was not considered a routinely feasible option given barriers of access and expense, however, in the modern era of precision oncology these technologies are rapidly becoming more accessible.
Adolescents and young adults (AYA) with sarcoma have distinct biological and clinical features, and as a patient population have been underrepresented in clinical trials [3]. The prognosis for AYA with sarcoma is typically inferior to their younger counterparts and although underlying host and tumour factors likely contribute, this is poorly understood [4,5]. Moreover, onset of cancers associated with genetic predisposition syndromes frequently occur during AYA years and underscores the imperative to invest in molecular profiling initiatives for this age group [6]. CMP is hypothesized to enhance clinical care for AYA patients with sarcoma through the identification of actionable genomic biomarkers and/or germline predisposition, however, clinical evidence to support its use is lacking [7].
The aim of this study was to describe the frequency of actionable variants, feasibility and clinical impact of prospective CMP for AYA sarcoma patients at an adult tertiary referral sarcoma service.
2. Materials and Methods
This is an analysis of AYA patients with sarcoma whose tumour specimens underwent CMP via recruitment to the Victorian Comprehensive Cancer Centre (VCCC) PRECISION study. All included patients were treated and recruited at the Peter MacCallum Cancer Centre, Australia’s leading tertiary referral sarcoma service between 1 July 2019 and 31 July 2023. The institution is a predominantly adult service, although patients as young as 15 years are seen. Patients were eligible for inclusion in this study if they were aged between 15 and 39 years, which is consistent with major North American and European working group definitions of the AYA age range [3]. Other inclusion criteria included a histological diagnosis of sarcoma which was incurable but with a life expectancy of at least six months, and an ECOG performance status of 0 or 1. The study was approved by the institutional Human Research Ethics Committee. All participating patients provided written informed consent.
Eligible participants underwent either targeted sequencing (TS) or whole genome and transcriptome sequencing (WGTS) of their tumour tissue. WGTS was performed according to our previously described methodology [8]. TS was performed using either the TruSight Oncology 500 Assay (Illumina), or an in-house developed tumour-normal comprehensive targeted DNA panel test [9,10]. Both TS assays are designed to detect selected fusions. Choice of TS or WGTS was based on clinician discretion, however, WGTS was favoured as the test of choice if a newly obtained biopsy was possible. A newly obtained biopsy was performed where feasible, otherwise archival tumour specimens were used (for TS only). A matched germline sample (peripheral blood) was additionally sequenced for all patients who underwent WGTS, as well as in a subset of patients who underwent TS.
Detected genomic variants were classified into tiers by the level of evidence based on clinical significance according to the AMP / ASCO / CAP Guidelines 11. Clinically relevant driver alterations were further assessed for actionability by the Molecular Tumour Board. The final report, including results of molecular analysis and potential clinical implications, was then issued to the participant’s treating clinician. The molecular reports were then reviewed in conjunction with the clinical data by investigators of this study to assess for clinical significance and actionability of identified variants. “New potentially actionable variants” were defined as previously unidentified variants leading to a change in diagnosis and/or with therapeutic implications, meaning they could predict response or resistance to systemic therapy as per the OncoKB classification system (www.oncokb.org) of levels of actionability [12]. Unless these criteria were met, other variants that contributed diagnostic information, such as genomic rearrangements, or other biological information such as oncogenic drivers, were noted but not classified as ‘new potentially actionable variants’.
Prospective collection of clinical data was captured using questionnaires completed by treating clinicians at enrolment including: patient demographics, diagnostic and treatment information for sarcoma, known germline mutations, details of relevant previous molecular testing, ECOG performance status and availability of an appropriate archival specimen. Clinicians were invited to provide feedback regarding the utility and impact of molecular profiling three months after the report was issued. Retrospective review of the electronic medical record was conducted to extract additional clinical data including details of diagnosis, previous treatments and outcomes to further determine the clinical impact of CMP.
3. Results
3.1. Patient Characteristics
Between 1 July 2019 and 31 July 2023, 25 AYA patients with a histological diagnosis of sarcoma were recruited. Survival and disease status data were collected up to 4 September 2023, censored at death for a median follow-up of 3.2 years (IQR 1.3-6.3). Thirteen males and 12 females were enrolled. The median age was 26.6 years (IQR 22.5 - 31.8) at diagnosis, and 28.7 years (24.8 - 34.0) at enrolment for CMP. Twenty patients had soft tissue sarcomas (80%) with bone tumours comprising the remainder (20%). The majority of patients had received prior treatment with surgery (n=20, 80%), radiation therapy (n=14, 56%) and at least one line of systemic therapy (n=15, 60%) prior to CMP (Table 1).
At the time of sequencing, almost all AYA patients had metastatic disease (n=24, 96%) and of these 17 (68%) had primary progressive disease or were experiencing first recurrence. Three AYA patients were newly diagnosed at the time of recruitment to the study, two with a poor prognosis (cases 9 and 17) and one with two lesions of uncertain relationship (case 24). The remaining five cases were recruited to the study at disease recurrence or later.
Five AYA patients had CMP performed previously (TS n=2, RNA sequencing panel n= 2, circulating tumour DNA assay n=1). Those patients previously tested with TS or ctDNA went on to undergo WGTS. Two AYA patients had known germline aberrations prior to sequencing (RB1 and TP53).
At the time of sequencing, almost all AYA patients had metastatic disease (n=24, 96%) and of these 17 (68%) had primary progressive disease or were experiencing first recurrence. Three AYA patients were newly diagnosed at the time of recruitment to the study, two with a poor prognosis (cases 9 and 17) and one with two lesions of uncertain relationship (case 24). The remaining five cases were recruited to the study at disease recurrence or later.
Five AYA patients had CMP performed previously (TS n=2, RNA sequencing panel n= 2, circulating tumour DNA assay n=1). Those patients previously tested with TS or ctDNA went on to undergo WGTS. Two AYA patients had known germline aberrations prior to sequencing (RB1 and TP53).
3.2. Details and Feasibility of Molecular Testing
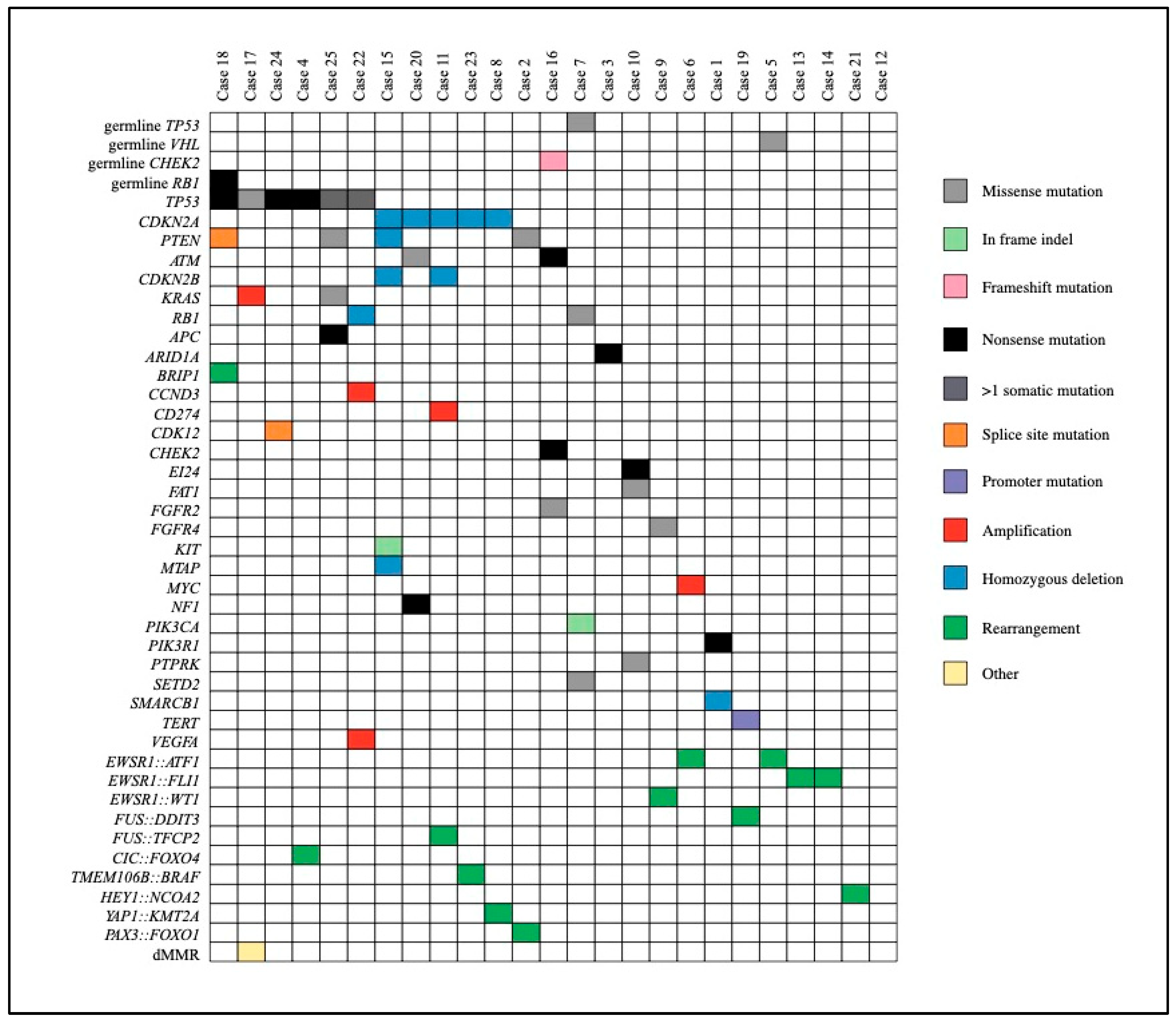
Fifteen AYA patients underwent CMP with WGTS and 10 with TS. A new biopsy was required in half of the cases (n=12, 48%). There were no patients who experienced a complication as a result of biopsy. There were no cases of CMP assay failure. The median time from consent to final molecular report was 45 days (IQR 37-57). Pathogenic variants predicted to drive tumour progression identified with CMP are described in Figure 1. As detailed in Table 2, more than half (n=14, 56%) of the AYA patients who underwent CMP had a new potentially actionable variant identified. Among the 14 patients with new actionable variants, 12 had at least one new treatment recommendation and/or clinical trial option identified as a result of CMP, six of whom had more than one identified option. All treatment suggestions were based on Tier IIC or lower level of evidence.
Three patients commenced new treatments as a result of their molecular profiling, two of whom did so via enrolment on a clinical trial (Case 9: TRK inhibitor; Case 18: Combination PARP inhibitor and anti-PD1 antibody). The third case (Case 23) had a favourable initial response to treatment with cobimetinib (by compassionate access) after a BRAF rearrangement was identified in a primary pancreatic spindle cell sarcoma, however, had progressive disease at 3.8 months and died approximately 5 months after treatment commenced [13]. The remaining nine AYA who had new treatments identified did not commence treatment for the following reasons: clinical circumstances precluded eligibility to enroll on a clinical trial (n=2); death due to unexpected severe medical complication (n=2); unable to access recommended treatment (n=1); treating clinician’s discretion (n=1); patient preference (n=1); and systemic therapy not indicated because able to achieve remission with standard therapies (n=2).
Two AYA patients had germline variants detected as part of molecular profiling. Case 5 had a germline VHL missense variant of uncertain significance identified and case 16 had a pathogenic germline CHEK2 frameshift deletion. Both patients remain in complete remission at follow up. Case 24 had a TP53 deletion detected on molecular profiling with TS, which in combination with a strong family history led to subsequent germline testing confirmatory for Li-Fraumeni syndrome.
One patient had their histological diagnosis revised as a consequence of molecular profiling. On a background of multiply recurrent desmoid fibromatosis and newly progressive metastatic disease, a further biopsy was performed which was sent for both RNA sequencing fusion panel as well as WGTS. Both tests identified a YAP1::KMT2A fusion supportive of a diagnosis of a rare subtype of sclerosing epithelioid fibrosarcoma (MUC4 negative on immunohistochemistry) [14]. WGTS also led to refinement of diagnosis through identification of gene fusions and/or fusion partners following initial testing with FISH break-apart probes in eight cases (32%). Details of gene fusion and fusion partners are shown in Figure 1.
Figure 1.
Driver Variants Identified Through Comprehensive Molecular Profiling. legend –Plot demonstrating driver variants and germline mutations identified across the study cohort.
Figure 1.
Driver Variants Identified Through Comprehensive Molecular Profiling. legend –Plot demonstrating driver variants and germline mutations identified across the study cohort.
3.3. Clinician Feedback
Three months after the molecular report was issued, treating clinicians provided feedback regarding their perception of the clinical impact of the CMP testing results. Clinical feedback analysis was provided and evaluable for 22 patients. In two-thirds of these cases (n=14, 64%) the treating clinician found that molecular profiling provided useful information aside from deciding current therapy. The most frequent reasons for this included: identification of future treatment options (n=8), clarifying the diagnosis (n=7), avoiding treatments (n=3), and clarifying or refining prognosis (n=2). Of the 11 cases where a potentially actionable variant was not identified, clinician feedback was available for 10 cases, three of whom found testing useful despite not finding an actionable variant.
4. Discussion
In this series we demonstrate the clinical utility of CMP among a cohort of AYA patients with sarcoma, most of whom had advanced and poor prognosis disease. At least one newly identified potentially actionable variant was identified in more than half the cohort (n=14, 56%), with the majority of these discoveries translating to new therapeutic options (n=12, 48%).
Our results are consistent with other reports, however frequencies of identification of variants with therapeutic potential vary widely. To our knowledge there is only one other published study focusing on CMP exclusively for AYA sarcoma patients. In their multisite European cohort of 48 patients, a very high frequency of actionable variants with therapeutic recommendations (81%) was identified by a combination of whole exome sequencing, methylation profiling and RNA sequencing [7]. More consistent with our study, studies of older adults using a variety of NGS platforms reported finding biomarkers with therapeutic potential in 36-56% [1,15,16,17]. A large study of 7494 sarcoma patients of all ages reported a lower overall rate of 31% and did not identify specific differences for the younger cohort (≤ 30 years), although AYA patients were not separated as a subgroup [18]. The variation in results is likely multifactorial, reflective of the heterogenous patient and tumour group, differences in testing platforms and assays, selection bias and the challenges of interpretation and classification of actionable variants that lend themselves to guiding therapeutic choices [19]. Within this study, of those patients who were tested using WGTS, 11/15 (73%) had actionable mutations, compared with only 3/10 (30%) of those tested using TS. Whilst this observation is noteworthy, it remains unclear which AYA subgroups should preferentially use WGTS. It should be noted that most TS panel tests do not include the ability to detect rarer fusions which are enriched in AYA sarcomas.
Implementation of CMP in our cohort was highly feasible. Repeat biopsies when required and testing occurred with universal success and without complication. The median turnaround time of six weeks from enrolment to reporting is shorter than other reports [7], although in the clinical context of poor prognosis disease with limited options for effective treatments, six weeks is significant and argues for implementing CMP earlier in the clinical journey. Of the 12 patients with new treatment options identified with CMP, only three were able to start this therapy. This is likely linked to the turnaround time, barriers to drug accessibility, and that testing occurred relatively late in the patient’s journey [20].
The majority of patients included in this study had testing when standard treatments were no longer effective, with only three patients having CMP at diagnosis. Performing upfront CMP at diagnosis of a metastatic or poor prognostic sarcoma, especially when the planned standard of care treatment does not depend on the results, would allow for advanced planning of personalised second-line and salvage therapies. However, whilst our experience advocates for earlier CMP implementation, consensus is lacking regarding the best time to perform molecular profiling. A relative lack of targeted therapeutic options at least in part contribute to this, as well as the understanding that changes in tumour biology such as clonal diversity and various mechanisms of treatment resistance occur over time [21,22]. Other factors contributing to reluctance to test earlier may include clinical concerns around risks of repeated biopsy, cost of testing and lack of consensus around which patients would benefit most as well as which testing platforms to utilise.
We add to a body of evidence demonstrating clinical value of CMP beyond the ability to identify new treatment options [23,24]. Of major importance to AYA patients, CMP led to identification of pathogenic germline variants in 12% of our cohort, which is similar to previous reports in this population [25,26], although this is notably less than the finding of 55% in a large multisite cohort of 1,162 adult sarcoma patients [27]. Our department now advocates for referral to a familial cancer centre to offer germline testing for all patients under 40 years with sarcoma, as well as for older patients with a suggestive personal or family history. Whilst only one patient had a formal change in diagnosis, CMP allowed diagnoses to be refined in several cases which was still deemed useful for clinicians. For example, identification of the exact oncogenic fusion driver in a tumour explained specific patterns of treatment resistance and poor prognosis.
A strength of our study is the inclusion of clinician perspectives regarding the clinical value of CMP for their AYA patients. The majority of clinicians found CMP to be useful aside from deciding about current treatment. The fact that several clinicians found profiling to be useful even following negative tests due to perceived benefits of clarifying diagnosis and avoiding future treatments underscores the clinical value of CMP as tool to improve care for AYA sarcoma patients.
Despite their unique biological and clinical care needs, AYA patients tend to fall in a gap between traditional adult and paediatric approaches. In the adult oncology setting where there is a much higher volume of patients, use of CMP tends to be more judicious and typically reserved for those with advanced disease with a high pre-test probability of identifying a molecular target. By contrast, in the paediatric setting in Australia and internationally, precision oncology efforts are now moving to offer CMP to all children with a new cancer diagnosis, irrespective of disease extent or prognosis and are expanding programs to include platforms such as in vitro drug testing, patient-derived xenograft models and phosphoproteomics in order to improve personalised treatment recommendations [28,29]. Differences in approach are reflective of known differences in biology between paediatric and adult sarcomas, however sarcoma subtypes experienced by AYA patients can span this divide and would greatly benefit from an individualized approach [30].
This study is limited by its small sample size, single-site design and potential selection bias from treating clinicians. Another important limitation of this, and other studies in this field, is the absence of patient-reported outcomes, which is especially important in AYA research [31]. The patient’s perspective of the experience of the process and outcomes of CMP could considerably contribute to assessments of clinical value and utility. High levels of satisfaction and perceived benefit have been reported by parents and adolescent patients enrolled in precision oncology programs in the paediatric setting [32].
There is an urgent need to enable access to clinical trials for AYA patients, especially those with a molecular focus in order to improve overall care and outcomes for this vulnerable group. The results of current adult and paediatric clinical trials are eagerly awaited to inform whether CMP in sarcomas can identify new promising therapeutic targets, whether this can be integrated into clinical practice and whether this translates in improved patient outcomes, although dedicated research specifically for the AYA group is needed [29,33]. Nevertheless, we identified that implementation of CMP for AYA with sarcoma is clinically valuable, feasible and should be increasingly integrated into routine clinical practice as technologies, cost and turnaround times continue to improve.
Author Contributions
SL and JL conceived and supervised the study. SL and ECA assessed variants as ‘potentially actionable’. ECA extracted and analysed data and wrote the manuscript. WZ, SN, LV, CM, JHAV and SMG analysed primary data. JD, LO, AH, SMG, WZ, SN, LV, CM, HX, SBF & JHAV provided manuscript review.
Funding
Whole genome and transcriptome sequencing and targeted panel sequencing were available through enrolment in the VCCC PRECISION program funded by the Victorian Comprehensive Cancer Centre (VCCC).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Human Research and Ethics Committee of the Peter MacCallum Cancer Centre (HREC/48455/PMCC-2018, approved 6 March 2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author and after approval from the VCCC PRECISION study investigators. The data are not publicly available due to ethics limitations.
Acknowledgments
We would like to acknowledge Kym Pham Stewart, Oliver Hofmann and the University of Melbourne Centre for Cancer Research Clinical Genomics Platform for performing whole genome/transcriptome sequencing analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lucchesi C, Khalifa E, Laizet Y, Soubeyran I, Mathoulin-Pelissier S, Chomienne C, et al. Targetable Alterations in Adult Patients With Soft-Tissue Sarcomas: Insights for Personalized Therapy. JAMA Oncol. 2018, 4, 1398–1404. [CrossRef] [PubMed]
- Lopes-Brás R, Lopez-Presa D, Esperança-Martins M, Melo-Alvim C, Gallego L, Costa L, et al. Genomic Profiling of Sarcomas: A Promising Weapon in the Therapeutic Arsenal. Int J Mol Sci. 2022, 23, 14227. [CrossRef] [PubMed]
- Ferrari A, Stark D, Peccatori FA, Fern L, Laurence V, Gaspar N, et al. Adolescents and young adults (AYA) with cancer: a position paper from the AYA Working Group of the European Society for Medical Oncology (ESMO) and the European Society for Paediatric Oncology (SIOPE). ESMO Open. 2021, 6, 100096. [CrossRef]
- Tricoli JV, Blair DG, Anders CK, Bleyer WA, Boardman LA, Khan J, et al. Biologic and clinical characteristics of adolescent and young adult cancers: Acute lymphoblastic leukemia, colorectal cancer, breast cancer, melanoma, and sarcoma. Cancer. 2016, 122, 1017–1028. [CrossRef] [PubMed]
- Wolfson JA, Kenzik KM, Foxworthy B, Salsman JM, Donahue K, Nelson M, et al. Understanding Causes of Inferior Outcomes in Adolescents and Young Adults With Cancer. J Natl Compr Canc Netw. 2023, 21, 881–888. [CrossRef]
- Sender L, Zabokrtsky KB. Adolescent and young adult patients with cancer: a milieu of unique features. Nat Rev Clin Oncol. 2015, 12, 465–480. [CrossRef] [PubMed]
- Morfouace M, Horak P, Kreutzfeldt S, Stevovic A, de Rojas T, Denisova E, et al. Comprehensive molecular profiling of sarcomas in adolescent and young adult patients: Results of the EORTC SPECTA-AYA international proof-of-concept study. Eur J Cancer. 2023, 178, 216–226. [CrossRef]
- Dall G, Vandenberg CJ, Nesic K, Ratnayake G, Zhu W, Vissers JHA, et al. Targeting homologous recombination deficiency in uterine leiomyosarcoma. J Exp Clin Cancer Res. 2023, 42, 112. [CrossRef] [PubMed]
- O’Haire S, Degeling K, Franchini F, Tran B, Luen SJ, Gaff C, et al. Comparing Survival Outcomes for Advanced Cancer Patients Who Received Complex Genomic Profiling Using a Synthetic Control Arm. Target Oncol. 2022, 17, 539–548. [CrossRef]
- Cameron D, Schröder J, Penington J, Do H, Molania R, Dobrovic A, et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017, 27, 2050–2060. [CrossRef]
- Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017, 19, 4–23. [CrossRef]
- Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017, 1–16. [CrossRef]
- Mitchell C, Malalasekera V, Gill AJ, Vissers JHA, Luen SJ, Grimmond SM, et al. Primary pancreatic spindle cell sarcoma with a TMEM106B::BRAF gene fusion treated with MEK inhibition. Pathology. 2023, 55, 127–129. [CrossRef]
- Kao YC, Lee JC, Zhang L, Sung YS, Swanson D, Hsieh TH, et al. Recurrent YAP1 and KMT2A Gene Rearrangements in a Subset of MUC4-negative Sclerosing Epithelioid Fibrosarcoma. Am J Surg Pathol. 2020, 44, 368–377. [CrossRef]
- Schipper LJ, Monkhorst KA-O, Samsom KA-O, Bosch LJW, Snaebjornsson P, van Boven H, et al. Clinical Impact of Prospective Whole Genome Sequencing in Sarcoma Patients. Cancers (Basel). 2022, 14, 436. [CrossRef] [PubMed]
- Cote GM, He J, Choy E. Next-Generation Sequencing for Patients with Sarcoma: A Single Center Experience. Oncologist. 2018, 23, 234–242. [CrossRef]
- Groisberg R, Hong DS, Holla V, Janku F, Piha-Paul S, Ravi V, et al. Clinical genomic profiling to identify actionable alterations for investigational therapies in patients with diverse sarcomas. Oncotarget. 2017, 8, 39254–39267. [CrossRef]
- Gounder MM, Agaram NP, Trabucco SE, Robinson V, Ferraro RA, Millis SZ, et al. Clinical genomic profiling in the management of patients with soft tissue and bone sarcoma. Nature Commun. 2022, 13, 3406. [CrossRef] [PubMed]
- Malone ER, Oliva M, Sabatini PJB, Stockley TL, Siu LL. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [CrossRef]
- Lewin J, Siu LL. Cancer genomics: the challenge of drug accessibility. Curr Opin Oncol. 2015, 27, 250–257. [CrossRef]
- Thapa B, Ahmed G, Szabo A, Kamgar M, Kilari D, Mehdi M, et al. Comprehensive genomic profiling: Does timing matter? Front Oncol. 2023, 13, 1025367. [CrossRef] [PubMed]
- Colomer R, Mondejar R, Romero-Laorden N, Alfranca A, Sanchez-Madrid F, Quintela-Fandino M. When should we order a next generation sequencing test in a patient with cancer? eClinicalMedicine. 2020, 25. [CrossRef]
- Villani A, Davidson S, Kanwar N, Lo WW, Li Y, Cohen-Gogo S, et al. The clinical utility of integrative genomics in childhood cancer extends beyond targetable mutations. Nat Cancer. 2023, 4, 203–221. [CrossRef]
- Jacobs C, Lapeire L. Translating Molecular Profiling of Soft Tissue Sarcomas into Daily Clinical Practice. Diagnostics (Basel). 2021, 11, 512. [CrossRef]
- Carvalho N, Santiago K, Maia J, Costa F, Formiga M, Soares D, et al. Prevalence and clinical implications of germline pathogenic variants in cancer predisposing genes in young patients across sarcoma subtypes. J Med Genet 2023. [CrossRef]
- Toss A, Quarello P, Mascarin M, Banna GA-O, Zecca M, Cinieri S, et al. Cancer Predisposition Genes in Adolescents and Young Adults (AYAs): a Review Paper from the Italian AYA Working Group. Curr Oncol Rep. 2022, 24, 843–860. [CrossRef]
- Ballinger ML, Goode DL, Ray-Coquard I, James PA, Mitchell G, Niedermayr E, et al. Monogenic and polygenic determinants of sarcoma risk: an international genetic study. Lancet Oncol. 2016, 17, 1261–1271. [CrossRef]
- Wong M, Mayoh C, Lau LMS, Khuong-Quang D-A, Pinese M, Kumar A, et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med. 2020, 26, 1742–1753. [CrossRef]
- Parsons DW JK, Patton DR, Winter CL, Coffey B, Williams PM, Roy-Chowdhuri S, Tsongalis GJ, Routbort M, Ramirez NC, Saguilig L, Piao J, Alonzo TA, Berg SL, Fox E, Hawkins DS, Abrams JS, Mooney M, Takebe N, Tricoli JV, Seibel NL; NCI-COG Pediatric MATCH Team. . Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol. 2022, 40, 2224–2234. [CrossRef]
- Reed DR, Naghavi A, Binitie O. Sarcoma as a Model for Adolescent and Young Adult Care. J Oncol Pract. 2019, 15, 239–247. [CrossRef]
- Roth ME, Parsons SK, Ganz PA, Wagner LI, Hinds PS, Alexander S, et al. Inclusion of a core patient-reported outcomes battery in adolescent and young adult cancer clinical trials. J Natl Cancer Inst. 2023, 115, 21–28. [CrossRef] [PubMed]
- Wakefield CE, Hetherington K, Robertson EG, Donoghoe MW, Hunter JD, Vetsch J, et al. Hopes, concerns, satisfaction and regret in a precision medicine trial for childhood cancer: a mixed-methods study of parent and patient perspectives. Br J Cancer. 2023. [CrossRef]
- Italiano A DD, Soubeyran I, Bellera C, Espérou H, Delmas C, Mercier N, Albert S, Poignie L, Boland A, Bourdon A, Geneste D, Cavaille Q, Laizet Y, Khalifa E, Auzanneau C, Squiban B, Truffaux N, Olaso R, Gerber Z, Wallet C, Bénard A, Blay JY, Laurent-Puig P, Deleuze JF, Lucchesi C, Mathoulin-Pelissier S; MULTISARC study group. Molecular profiling of advanced soft-tissue sarcomas: the MULTISARC randomized trial. BMC Cancer. 2021, 21, 1180. [CrossRef]

Table 1.
Patient and Tumour Characteristics.
| Case | Age (y) at Diagnosis | Sex | Histological Diagnosis | Primary Tumour Site | Disease Extent at Diagnosis | Lines of Systemic Treatment | Other Prior Treatment | Sequencing Platform |
| 1 | 22.9 | M | Alveolar rhabdomyosarcoma | Head & neck | Localised | 1 | Surgery, RT | WGTS |
| 2 | 26.6 | M | Alveolar rhabdomyosarcoma | Head & neck | Metastatic | 1 | RT | TS |
| 3 | 34.7 | F | Angiosarcoma | Thorax | Localised | 1 | Surgery, RT | TS |
| 4 | 27.5 | M | CIC-rearranged sarcoma | Extremity | Metastatic | 1 | RT | WGTS |
| 5 | 22.6 | M | Clear cell sarcoma of soft tissue | Abdomen | Localised | 0 | Surgery | WGTS |
| 6 | 20.3 | M | Clear cell sarcoma of soft tissue | Extremity | Localised | 0 | Surgery, RT | WGTS |
| 7 | 29.4 | M | Dedifferentiated chondrosarcoma | Pelvis | Localised | 1 | Surgery | TS |
| 8 | 19.6 | F | Desmoid fibromatosis, recurrent | Extremity | Localised | 1 | Surgery, RT | WGTS |
| 9 | 21.5 | M | Desmoplastic small round cell tumour | Abdomen | Metastatic | 1 | WGTS | |
| 10 | 23.0 | F | EBV-associated smooth muscle tumour | Abdomen | Metastatic | 1 | Surgery, RT | WGTS |
| 11 | 34.4 | F | Epithelioid sarcoma | Extremity | Localised | 2 | Surgery, RT | TS |
| 12 | 31.3 | F | Epithelioid sarcoma | Pelvis | Metastatic | 0 | Surgery, RT | TS |
| 13 | 15.7 | F | Ewing sarcoma | Thorax | Localised | 2 | Surgery, RT | TS |
| 14 | 31.8 | M | Ewing sarcoma | Head & neck | Metastatic | 3 | Surgery, RT | TS |
| 15 | 24.4 | F | Gastrointestinal Stromal Tumour | Abdomen | Metastatic | 1 | Surgery | WGTS |
| 16 | 21.7 | M | Hepatic sarcoma, NOS | Abdomen | Localised | 0 | Surgery | WGTS |
| 17 | 31.8 | F | Intimal Sarcoma of Pulmonary Artery | Thorax | Localised | 0 | Surgery | WGTS |
| 18 | 23.7 | M | Leiomyosarcoma - radiation induced | Head & neck | Localised | 0 | Surgery | WGTS |
| 19 | 34.7 | M | Malignant peripheral nerve sheath tumour | Head & neck | Localised | 0 | Surgery, RT | TS |
| 20 | 27.6 | F | Mesenchymal chondrosarcoma | Pelvis | Metastatic | 1 | RT | WGTS |
| 21 | 32.5 | F | Myxoid liposarcoma | Extremity | Localised | 0 | Surgery, RT | WGTS |
| 22 | 20.9 | F | Osteosarcoma | Extremity | Metastatic | 1 | Surgery | WGTS |
| 23 | 22.5 | F | Primary pancreatic sarcoma, NOS | Abdomen | Localised | 0 | Surgery | WGTS |
| 24 | 33.9 | M | Spindle cell sarcoma, NOS | Thorax | Metastatic | 0 | Surgery | TS |
| 25 | 28.2 | M | Teratoma with sarcomatous transformation, NOS | Thorax | Localised | 1 | Surgery | TS |
M – Male F – Female NOS – Not Otherwise Specified RT – Radiation Therapy WGTS – Whole Genome and Transcriptome Sequencing TS – Targeted Sequencing.
Table 2.
Newly identified potentially actionable variants and treatment recommendations.
| Case | Histological Diagnosis | Sequencing Platform | New Potentially Actionable Variant(s) | AMP/ASCO Tiers for clinical significancea | Proposed Treatment Optionsb | Treatment Started |
| 1 | Alveolar rhabdomyosarcoma | WGTS |
CD274 amplification CDKN2A homozygous deletion |
III III |
Anti PD-1/PD-L1 antibody | No |
| 5 | Clear cell sarcoma of soft tissue | WGTS | Germline VHL missense variant | N/A | ||
| 6 | Clear cell sarcoma of soft tissue | WGTS | MYC amplification | IIC | RNA Polymerase I inhibitor | No |
| 7 | Dedifferentiated chondrosarcoma | TS | PIK3CA in-frame deletion | IIC | PI3-kinase inhibitor | No |
| 8 | Desmoid fibromatosis, recurrent | WGTS |
YAP1::KMT2A fusion CDKN2A homozygous deletion SBS Mutation Signature 3 |
IIC IIC N/A |
TEAD inhibitor CDK4/6 inhibitor + checkpoint blockade PARP inhibitor + checkpoint blockade |
No |
| 9 | Desmoplastic small round cell tumour | WGTS |
FGFR4 missense variant High NTRK3 expressionc |
IIC N/A |
Pan TRK inhibitor | Yes |
| 15 | Gastrointestinal Stromal Tumour | WGTS | CDKN2A | IIC III |
CDK4/6 inhibitor + checkpoint blockade | No |
| 16 | Hepatic sarcoma, NOS | WGTS | Germline CHEK2 frameshift deletion ATM inactivating variant FGFR2 missense variant |
N/A III III |
PARP inhibitor + checkpoint blockade FGFR inhibitor |
No |
| 17 | Intimal Sarcoma of Pulmonary Artery | WGTS | High tumour mutational burden High microsatellite instability |
N/A N/A |
Anti PD-1/PD-L1 antibody | No |
| 18 | Leiomyosarcoma – radiation induced | WGTS | BRIP1 rearrangement | IIC | PARP inhibitor + checkpoint blockade | Yes |
| 19 | Malignant peripheral nerve sheath tumour | TS |
NF1 deletion ATM substitution CDKN2A deletion |
IIC IIC IID |
MEK + mTOR inhibition PARP inhibitor + checkpoint blockade CDK4/6 inhibitor + checkpoint blockade |
No |
| 21 | Myxoid liposarcoma | WGTS | TERT promoter variant | IID | None | |
| 23 | Primary pancreatic sarcoma, NOS | WGTS | TMEM106B::BRAF fusion | III | RAF dimer inhibitor + MEK inhibitor | Yes |
| 24 | Spindle cell sarcoma, NOS | TS |
TP53 deletiond CDK12 substitution |
IIC IID |
CDK4/6 inhibitor + checkpoint blockade | No |
WGTS – Whole Genome and Transcriptome Sequencing PD-L1 – Programmed Death-Ligand 1 TS – Targeted DNA Sequencing NOS – Not Otherwise Specified N/A – Not applicable. a High tumour mutational burden and microsatellite instability are considered actionable but are not included in the tiering system. b Proposed treatment options were based on clinical trials available at the time of Molecular Tumour Board meeting. c Identified on whole transcriptome sequencing. d Confirmed to be germline on subsequent testing.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



