Submitted:
13 November 2023
Posted:
13 November 2023
You are already at the latest version
Abstract
This study investigates novel ferrocene-based electrochemical sensors for metal cation detection through the design, synthesis and characterisation of ferrocene derivatives. Specifically, the re-search determines the redox potentials of ferrocene versus decamethylferrocene to provide in-sight into the redox potential variations between them. The investigation also examines how electrochemical oxidation of the ferrocene moiety can modulate host affinity for transition metals cations via effects such as electrostatic interactions and changes to coordination chemistry. Metal ion coordination to receptors containing functional groups like imine and quinoline is explored to elucidate selectivity mechanisms. These findings advance the fundamental understanding of ferrocene electrochemistry and host-guest interactions, supporting the development of improved cation sensors with optimised recognition properties, sensitivity, and selectivity. Overall, the work lays the necessary groundwork for applications in analytical chemistry and sensor technologies through customised ferrocene-derived materials.
Keywords:
ferrocene
; electrochemical sensors
; metal cation
; host-guest interaction
1. Introduction
The landscape of molecular sensing and recognition has been redefined by the versa-tile synthetic chemistry of ferrocene (Fc), an organometallic compound that has emerged as a cornerstone in developing electrochemical molecular receptors for various cationic species [1,2,3,4,5]. The fascination with Fc in this context stems from its well-established synthetic routes and the accessibility of its redox couple, which confers it with exceptional utility for constructing molecular receptors. These receptors often in-corporate Fc functionalities strategically, serving as signalling or reporter groups. The redox responses of these functionalities undergo perturbations upon binding guest molecules. Alternatively, Fc can serve as a structural component, allowing for precise control over the topology of the guest binding site. Remarkably, Fc-based receptors, especially those assuming dual roles, exhibit a diverse range of functionalities that transcend the capabilities of purely organic architectures [6].
The design paradigm for redox-active receptors commonly employs spacer groups that covalently link the host (metal ion binding moieties) to the Fc unit. This strategic arrangement capitalises on the coexistence of the redox-active centre and the receptor functionality within the same molecular entity. This framework enables Fc-based molecular receptors to engage in concurrent or sequential processes of electron transfer and guest binding (guest = metal cations), with their mutual influence being particularly evident between alkali and alkaline earth metal ions and crown ether or similar oxygen-containing ligands. However, recent investigations have revealed that when hosts containing amine functionalities are involved (e.g., cyclen, cyclam), the interplay between electron transfer and molecular recognition takes on a more intricate nature [7,8,9,10].
Fc-based molecular receptors can be designed to bind anionic, cationic or neutral guest molecules [11,12]. Nevertheless, this review article explores Fc-based sensors and molecular receptors tailored for cationic species, shedding light on their intricate designs, redox mechanisms, and sensing responses. We examine the foundational reasons underlying the suitability of Fc as a molecular scaffold for this task, considering factors such as the effect of solvents and substituents on Fc redox potential. We discuss the interactions between Fc and crown ethers as opposed to amine-containing hosts and illustrate these concepts using concrete sensor examples, outlining the resultant changes in electrochemical potentials. By navigating through relevant literature and dissecting prominent case studies, we aim to provide an overview of the advancements in electrochemical Fc-based sensing platforms. Through this exploration, we unveil the intricate mechanisms governing their operation and emphasise their pivotal role in shaping the modern landscape of molecular sciences.
2. Why Ferrocene?
Ferrocene stands out because of its unique structure: a central iron atom neatly sandwiched between two cyclopentadienyl rings. This structural arrangement leads to some fascinating electrochemical properties that have attracted the curiosity of many researchers. The redox potential of Fc, as discussed below, is known to vary based on its proximity to the host. This observation, though noteworthy, isn’t an isolated phenomenon. When diving deeper into the electrochemical behaviour of Fc, it becomes apparent that several other factors also affect the mentioned redox potential. These factors encompass the nature of the solvent, the type of supporting electrolyte used, and the specific substituents connected to the Fc molecule. The ability of Fc to exhibit changes in its electrochemical responses under these varied conditions underscores its versatility and has cemented its place as a subject of intense study in electrochemistry.
In the extensive body of research surrounding Fc, one aspect has been given less prominence than it deserves: the role of the solvent in modulating Fc redox potential. While ferrocene is often put on a pedestal as an internal reference redox scale or system (IRRS), it is crucial to take a step back and consider the entirety of the system, including the solvent. Neglecting or underestimating the influence of the solvent could lead to a biased or incomplete picture of how ferrocene behaves in different experimental setups. These interactions involve intricate electrostatic interplays between the solvent or electrolyte and specific components of the ferrocene molecule. Such interactions encompass the iron centre and the cyclopentadienyl ring components [13]. Research by Yang et al. has shed light on this phenomenon through a comprehensive analysis of the molecular electrostatic potential (MEP) of Fc and its oxidised ferrocenium ion (Fc+ or [FeIII(η5-C5H5)2]+) counterpart [14]. The study elucidated the distribution of electrostatic charges, revealing remarkable insights into the solvation behaviour of Fc. The MEP analysis uncovered negative electrostatic potentials concentrated atop the cyclopentadienyl rings alongside a distinctive planetary-like charge distribution encircling the central iron atom. This distribution significantly fosters electrostatic interactions between Fc and positively charged molecules or ions, including those with electron-acceptor functionalities. Notably, the positively charged regions on the sides of the cyclopentadienyl rings facilitate interactions with electron-donating components from the surrounding solvent or electrolyte.
Interestingly, in the case of Fc+, the MEP analysis distinctly presents positive electrostatic potentials. This configuration favours electrostatic interactions between Fc+ and negatively charged ions or molecules characterised by electron-donor groups. The discernible MEP differences between Fc and its Fc+ counterpart assume a pivotal role, elucidating the observed variances in mass transport properties within viscous solvents, such as ionic liquids [15,16,17,18,19]. This intricate understanding of solvation effects and their consequential impact on Fc and Fc+ behaviour in distinct environments holds significant promise for advancing our comprehension of their behaviour in diverse contexts. One of which is the demystification of Fc as the optimal IRRS. An IRRS is a reversible or nearly reversible redox system that provides a known and stable, solvent-independent reference point in non-aqueous solvents where reliable reference electrodes are difficult to establish or stabilise [19,20,21]. The IRRS is generally used with a quasi-reference electrode [22,23,24,25,26,27].
The history of establishing an IRRS that remains unaffected by the solvent for comparing redox potentials in non-aqueous systems is extensive. This chronicle commenced with the introduction of the Rb|Rb+ or Rb(Hg)|Rb+ redox couples and subsequently advanced with the proposition of employing organometallic redox pairs [28]. To streamline the comparative process and enhance simplicity, IUPAC advocated for the adoption of specific IRRS in non-aqueous contexts, namely, Fc/Fc+ (also referred to as Fc0/+) and bis(biphenyl)chromium(0)|bis(biphenyl)chromium(I), BCr/BCr+ [28]. These selections were made arbitrarily from various published redox systems [28]. It is noteworthy to observe how an initial arbitrary selection of redox systems, including Fc, has permeated the present scientific landscape, leading to the enduring perception of Fc as an incontrovertible IRRS. This underscores the intricate interplay between historical conventions, pragmatic considerations, and the trajectory of scientific understanding that collectively shape the enduring reputation of Fc.
A range of essential attributes characterises an effective IRRS, several initially outlined by Gritzner and Kuta in the IUPAC recommendation [28]. Additionally, a revised set of properties has been introduced by our research group [29]. This updated framework expands and refines the criteria that define a reliable IRRS, reflecting advancements in our understanding and methodological approaches. The evolution of these properties not only demonstrates the maturation of the field but also underscores the commitment to enhancing the accuracy and applicability of IRRS in diverse electrochemical settings.
Decamethylferrocene (DmFc or [FeII(η5-C5(CH3)5)2]) exhibits notably diminished solvent-solute interactions in organic solvent systems, a contrast that surpasses at least one order of magnitude in comparison to ferrocene. This divergence arises due to the methyl-substituent groups within the cyclopentadienyl rings of DmFc, which effectively impede both specific and non-specific interactions. This obstruction originates from the hindered accessibility of organic solvent and supporting electrolyte molecules to the metal centre and the cyclopentadienyl ring [13,30,31,32]. The veracity of this observation is further corroborated by the analysis of X-ray diffraction patterns of DmFc, which substantiates that the inter-ring methyl groups within DmFc adhere to Van der Waals distances [33]. Consequently, owing to its intrinsic characteristics, DmFc proves to be more appropriate than Fc as an IRRS within organic solvents, where it undergoes a reversible one-electron oxidative transfer process, leading to the formation of decamethylferrocenium (DmFc+ or [FeIII(η5-C5(CH3)5)2]+), which is reduced back to DmFc when the potential scanning direction is reverted. The DmFc0/+ mid-point potential, Em, value associated with this electron transfer is lower than that of Fc due to the electron-donating influence imparted by the methyl groups (inductive effect). These methyl groups direct electron density towards the metal ion, facilitating the extraction of an electron by the electrode. Table 1 shows the difference in the Em values between Fc0/+ and DmFc0/+, which fluctuates from 0.614 ± 0.005 V in dichloromethane/0.1 M [Bu4N][TFAB] ([TFAB] = [B(C6F5)4]–) to 0.413 ± 0.005 V in tetrahydrofuran/0.1 M [Bu4N][BF4], representing a difference of 0.201 V due to solvent and supporting electrolyte effects. Furthermore, a change of 0.152 V can be observed by transitioning from tetrahydrofuran to 2,2,2-trifluoroethanol as the organic solvent while maintaining constant the nature and concentration (0.1 M [Bu4N][ClO4]) of the supporting electrolyte. To mitigate uncertainties when comparing Em in Table 1, all potential values cited are referenced against the DmFc0/+ potential scale. Consequently, as the values in this table signify the disparity in Em, the chances exist for the absolute Em values of the Fc0/+ and DmFc0/+ IRRS to exhibit a more significant variation than that presented here.
When considering the impact of substituents on the cyclopentadienyl rings, the solvent’s role becomes even more pronounced. Substituents modulate the electron density around the iron centre by their electron-donating or withdrawing nature. This change in electron density, in turn, can influence the solvation shell and the nature of solvent interactions with ferrocene and its derivatives (Table 2). For instance, an electron-donating substituent might increase the negative charge density on the cyclopentadienyl ring. In a polar solvent, which already affects the redox potential of Fc through the stabilisation of charged species, the increase in negative charge might result in an even more pronounced shift of its Em to more negative potential values. However, the effect might be attenuated or manifest differently in a non-polar solvent. Conversely, electron-withdrawing substituents could exacerbate or mitigate solvent effects, contingent on the specific properties of the solvent used and the nature of the substituent. The interplay between solvent effects and substituent-induced electronic modulation offers a dynamic continuum, with each factor potentially amplifying or modulating the other, resulting in a rich variability of electrochemical behaviour.
The influence of substituents on the Em of Fc is an exciting aspect of molecular electrochemistry that warrants in-depth investigation. It is well-established that the electron density around the central iron atom can be modulated by introducing various substituents on the surrounding ligands. Depending on the electronic nature of the substituent, such modulation can either stabilise or destabilise the oxidised form of the complex, and its effects rest predominantly upon the electronic principles of induction. When an electron-donating group is appended to the cyclopentadienyl ring of Fc, it can engage in inductive donation. This introduces electron density to the π-system of the cyclopentadienyl ring, which in turn delocalises to the central iron atom. Such an enhancement of electron density around the metal cation predisposes it to a greater likelihood of electron loss, making oxidation more favourable, thus shifting the Em to more negative potential values respect to that of unmodified Fc (Table 2).
In contrast, electron-withdrawing groups operate on induction and resonance withdrawal principles. These groups extract electron density from the cyclopentadienyl ring. Additionally, certain withdrawing groups can engage in resonance structures that pull electron density away from the cyclopentadienyl ring and into the substituent. As a result of this electron density diminishment, the central iron cation becomes less prone to lose an electron, translating to a shifting of the Em to more positive potential values respect to that of unmodified Fc (Table 2).
Table 2 shows the Em values of different substituted Fc derivatives versus Fc0/+, which is equal to -0.570 V vs. Fc0/+ IRRS for DmFc in dichloromethane/0.1 M [Bu4N][ClO4] and 0.640 V vs. Fc0/+ for 1,1′-bis(trifluoromethyl)ferrocene in the same solvent/supporting electrolyte system, representing a difference of 1.21 V. Furthermore, a change of 0.141 V can be observed for 1,1′,3,3′-tetra(t-butyl)ferrocene by transitioning from acetonitrile (0.1 M [Bu4N][ClO4]) to toluene containing 0.5 M [Hex4N][ClO4] as the supporting electrolyte.
The Em of Fc is also affected by the distance between the ferrocenyl moiety and substituents that either donate or withdraw electrons [10,37,38]. When analysing a scenario where an electron-withdrawing group is attached to the Fc unit (e.g., Fc-COOH), it becomes evident that increasing methylene groups progressively diminishes the electron-withdrawing effect on Fc—specifically, introducing a single methylene group between the Fc and the carboxylic group results in an Em shift of 0.245 V towards more negative potential values (Table 2). The magnitude of this shift reduces upon subsequent additions of methylene groups, with further negative shifts of 0.016 and 0.025 V observed following the incorporation of a second and third methylene unit, respectively. In this new scenario (e.g., Fc-(CH2)3-COOH), the inductive donating property of methylene towards the cyclopentadienyl rings of Fc begins to dominate the resultant Em of Fc.
The resultant molecule often exhibits compromised stability when the Fc unit is directly linked with electron-donating groups such as amines or hydroxyls. This is exemplified by Fc-NH2, whose stability depends on various factors such as temperature, pH, and the presence of oxidising agents (e.g., oxygen from air). Consequently, research on these molecule families typically commences with one methylene unit, as seen in Fc-CH2-OH and Fc-CH2-NH2 (Table 2). Interestingly, the observed Em shift magnitude towards negative values closely aligns with those exhibited by Fc connected to carboxylic groups bearing variable counts of methylene groups as a bridge. As an illustration, the negative Em shifts of 0.041, 0.032, and 0.040 V are documented upon increasing the number of methylene groups from Fc-CH2-COOH, Fc-CH2-NH2, and Fc-CH2-OH to Fc-(CH2)3-COOH, Fc-(CH2)3-NH2, and Fc-(CH2)3-OH, respectively.
From these observations, it is possible to deduct the pivotal role of methylene group quantity in shaping the overall reactivity and behaviour of ferrocene-based compounds. Expanding the methylene chain effectively serves as a strategic buffer, isolating the electron-withdrawing effects and safeguarding the inherent chemical stability of Fc. Conversely, the judicious placement of electron-donating entities proximal to the ferrocene unit presents opportunities to modulate its Em and alter its oxidation pathway (see below).
Furthermore, it is essential to consider the spatial orientation and steric hindrance offered by substituents. While electronic effects often dominate, steric factors can influence the approach and interaction of molecules at the electrode surface during redox processes, potentially altering observed redox potentials. Thus, while the electronic properties of substituents provide a foundational understanding of their effects on redox potential, the holistic picture must also account for three-dimensional spatial factors.

Table 2.
Redox potentials of substituted Fc in different organic solvents containing different supporting electrolytes.
Table 2.
Redox potentials of substituted Fc in different organic solvents containing different supporting electrolytes.
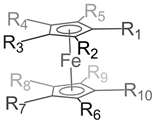 |
Solvent | Electrolyte |
Em vs. Fc0/+ (V) |
Ref |
|---|---|---|---|---|
| R1-10: H | DCM | 0.1 M [Bu4N][ClO4] | 0.000 | [2] |
| R1-10: CH3 | DCM | 0.1 M [Bu4N][ClO4] | -0.570 | [2] |
| R1-5: CH3; R6-10: H | DCM | 0.1 M [Bu4N][ClO4] | -0.270 | [2] |
| R1,10: CH3; R2-9: H | CH3CN | 0.1 M [Bu4N][ClO4] | -0.113 | [39] |
| CH3CN | 0.1 M [Bu4N][PF6] | -0.096 | [40] | |
| MeOH | 0.1 M [Bu4N][ClO4] | -0.104 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | -0.075 | [39] | |
| R2-9: CH3; R1,10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.406 | [40] |
| R1,3,7,10: t-Bu; R2,4,5,6,8,9: H | CH3CN | 0.1 M [Bu4N][ClO4] | -0.238 | [39] |
| CH3CN | 0.1 M [Bu4N][PF6] | -0.233 | [40] | |
| MeOH | 0.1 M [Bu4N][ClO4] | -0.229 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | -0.097 | [39] | |
| R1: n-Bu; R2-10: H | CH3CN | 0.1 M [Bu4N][ClO4] | -0.062 | [39] |
| MeOH | 0.1 M [Bu4N][ClO4] | -0.055 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | -0.073 | [39] | |
| R1-10: CH2Ph | DCM | 0.1 M [Bu4N][ClO4] | -0.070 | [2] |
| R1,10: CF3; R2-9: H | DCM | 0.1 M [Bu4N][ClO4] | 0.640 | [2] |
| R1: CH=CH2; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.022 | [41] |
| R1: CH2OH; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.029 | [41] |
| CH3CN | 0.1 M [Bu4N][ClO4] | -0.012 | [39] | |
| CH3CN | 0.1 M [Bu4N][PF6] | 0.016 | [38] | |
| MeOH | 0.1 M [Bu4N][ClO4] | 0.005 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | -0.044 | [39] | |
| R1: (CH2)2OH; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.046 | [38] |
| R1: (CH2)3OH; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.052 | [38] |
| R1: (CH2)4OH; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.054 | [38] |
| R1: CH(CH3)OH; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | -0.008 | [41] |
| R1,10: CH(CH3)OH; R2-9: H | CH3CN | 0.1 M [Et4N][ClO4] | -0.013 | [41] |
| R1: CH2CONH2; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.003 | [38] |
| R1: (CH2)2CONH2; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.027 | [38] |
| R1: (CH2)3CONH2; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.049 | [38] |
| R1: COOH; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.239 | [41] |
| CH3CN | 0.1 M [Bu4N][ClO4] | 0.234 | [39] | |
| CH3CN | 0.1 M [Li][ClO4] | 0.239 | [37] | |
| MeOH | 0.1 M [Bu4N][ClO4] | 0.233 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | 0.157 | [39] | |
| R1: CH2COOH; R2-10: H | CH3CN | 0.1 M [Li][ClO4] | -0.006 | [37] |
| R1: (CH2)2COOH; R2-10: H | CH3CN | 0.1 M [Li][ClO4] | -0.022 | [37] |
| R1: (CH2)3COOH; R2-10: H | CH3CN | 0.1 M [Li][ClO4] | -0.047 | [37] |
| R1: COOCH3; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.243 | [41] |
| CH3CN | 0.1 M [Bu4N][ClO4] | 0.237 | [39] | |
| MeOH | 0.1 M [Bu4N][ClO4] | 0.263 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | 0.214 | [39] | |
| R1,10: COOCH3; R2-9: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.470 | [41] |
| R1: COCH3; R2-10: H | CH3CN | 0.1 M [Bu4N][ClO4] | 0.244 | [39] |
| MeOH | 0.1 M [Bu4N][ClO4] | 0.271 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | 0.191 | [39] | |
| R1,10: COCH3; R2-9: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.482 | [41] |
| R1: COPh; R2-10: H | CH3CN | 0.1 M [Bu4N][ClO4] | 0.250 | [39] |
| MeOH | 0.1 M [Bu4N][ClO4] | 0.272 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | 0.214 | [39] | |
| R1: CONH2; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | 0.183 | [38] |
| R1: CHO; R2-10: H | CH3CN | 0.1 M [Bu4N][ClO4] | 0.285 | [39] |
| MeOH | 0.1 M [Bu4N][ClO4] | 0.304 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | 0.259 | [39] | |
| R1: CH2NH2; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.014 | [38] |
| R1: (CH2)2NH2; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.037 | [38] |
| R1: (CH2)3NH2; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.046 | [38] |
| R1: (CH2)4NH2; R2-10: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.060 | [38] |
| R1: CH2N(CH3)2; R2-10: H | CH3CN | 0.1 M [Bu4N][ClO4] | -0.004 | [39] |
| MeOH | 0.1 M [Bu4N][ClO4] | 0.046 | [39] | |
| Toluene | 0.5 M [Hex4N][ClO4] | 0.009 | [39] | |
| CH3CN | 0.1 M [Bu4N][PF6] | -0.023 | [42] | |
| DCM:CH3CN 1:4 | 0.1 M [Bu4N][PF6] | 0.003 | [7] | |
| R1,10: CH2N(CH3)2; R2-9: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.017 | [42] |
| R1,10: (CH2)2N(CH3)2; R2-9: H | CH3CN | 0.1 M [Bu4N][PF6] | -0.077 | [42] |
| R1,10: CH2N(CH2Ph)2; R2-9: H | DCM | 0.2 M [Bu4N][PF6] | -0.001 | [9] |
| R1,10: C(CH3)=N(CH2)5CH3; R2-9: H | DCM:CH3CN 1:1 | 0.1 M [Bu4N][PF6] | 0.211 | [43] |
R1,10: 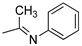 ; R2-9: H ; R2-9: H |
DCM:CH3CN 1:1 | 0.1 M [Bu4N][PF6] | 0.289 | [43] |
R1,10: 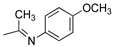 ; R2-9: H ; R2-9: H |
DCM:CH3CN 1:1 | 0.1 M [Bu4N][PF6] | 0.245 | [43] |
R1,10: 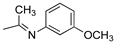 ; R2-9: H ; R2-9: H |
DCM:CH3CN 1:1 | 0.1 M [Bu4N][PF6] | 0.261 | [43] |
R1,10: 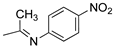 ; R2-9: H ; R2-9: H |
DCM:CH3CN 1:1 | 0.1 M [Bu4N][PF6] | 0.435 | [43] |
R1,10: 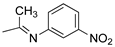 ; R2-9: H ; R2-9: H |
DCM:CH3CN 1:1 | 0.1 M [Bu4N][PF6] | 0.390 | [43] |
| R1,10: SH; R2-9: H | DCM | 0.1 M [Bu4N][ClO4] | 0.200 | [2] |
| R1: S(CH2)2OH; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.010 | [41] |
| R1: SCH2CH(CH3)COOH; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.039 | [41] |
| R1: CH(CH3)SPh; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.020 | [41] |
| R1: CH(Ph)SPh; R2-10: H | CH3CN | 0.1 M [Et4N][ClO4] | 0.043 | [41] |
R1: 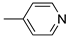 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.180 | [44] |
R1,10: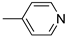 ; R2-9: H ; R2-9: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.350 | [44] |
R1: 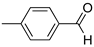 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.170 | [44] |
R1,10: 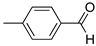 ; R2-9: H ; R2-9: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.180 | [44] |
R1: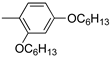 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.020 | [44] |
R1: 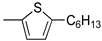 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.090 | [44] |
R1: 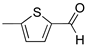 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.130 | [44] |
R1: 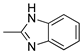 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.075 | [45] |
R1: 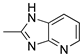 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.085 | [45] |
R1,10: 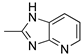 ; R2-9: H ; R2-9: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.205 | [45] |
R1,10: 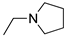 ; R2-9: H ; R2-9: H |
DCM | 0.2 M [Bu4N][PF6] | 0.002 | [9] |
R1,10: 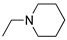 ; R2-9: H ; R2-9: H |
DCM | 0.2 M [Bu4N][PF6] | -0.001 | [9] |
R1,10: 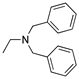 ; R2-9: H ; R2-9: H |
DCM | 0.2 M [Bu4N][PF6] | -0.001 | [9] |
R1: 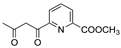 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][ClO4] | 0.366 | [45] |
R1: 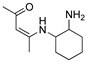 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [Bu4N][PF6] | 0.100 | [46] |
R1: 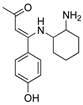 ; R2-10: H ; R2-10: H |
CH3CN | 0.1 M [BuN][PF6] | 0.120 | [46] |
3. Ferrocene-Based Electrochemical Sensors Using Oxygen-Containing Host Molecules
Electrochemical molecular recognition refers to the integrated chemical processes by which a redox-responsive receptor molecule recognises and electrochemically senses a guest species [47]. A suitable recognition requires a potential change larger than the experimental error of the electrochemical technique used. For example, the mid-point potential change (∆Em) should be larger than ±0.005 V when cyclic voltammetry is used. However, this change could be larger than ±0.001 V for potentiometric titrations [12].
Oxygen-containing ferrocene derivatives are vital for alkali and alkaline earth metal ions sensing [48,49]. The mechanism of molecular recognition can be described using a squares scheme where Fc-H, Fc+-H, G, Fc-HG, and Fc+-HG represent the ferrocene host, ferrocenium host, guest, ferrocene host-guest complex, and ferrocenium host-guest complex, respectively (Scheme 1), were Em1 and Em2 are the mid-point potentials for the Fc-H/Fc+-H and Fc-HG/Fc+-HG redox processes and k1 and k2 are the host-guest binding constant for Fc-H and Fc+-H, respectively [6,47]. The vertical reactions in Scheme 1 represent the guest binding at the host site, which can shift the Em of Fc pendant group connected to the host. The horizontal reactions represent the electrochemical electron transfer, where Em1 ≠ Em2 for a suitable electrochemical molecular recognition once the guest ion binds to the host. The larger the shift, the better the electrochemical molecular recognition.
Crown ethers have gained particular interest as hosts within the family of macrocycles. This is due to their unique interwoven structures composed of oxygen and carbon atoms, making them well-suited for binding a wide range of metal ions [50,51,52]. The first group of cation receptors known for their redox activity that researchers explored consisted of ferrocene crown ether conjugates 1-4 [53,54,55,56]. These Fc-based electrochemical sensors were developed for alkali cations such as sodium, potassium, caesium, and lithium, where the electron-rich crown ether acts as the host centre. Inserting a cation to the crown ether of 1–3 significantly results in a substantial Em shift of Fc to more positive potential values. For example, cyclic voltammograms of 0.2 mM pentaoxa[13]ferrocenophane 1 recorded in CH2Cl2/0.1 M [Bu4N][PF6] showed a positive Em shift of 0.17 V after addition of 1 mM NaClO4 [56]. This effect was explained by electrostatic repulsion forces between ferrocenium and the guest Na+.
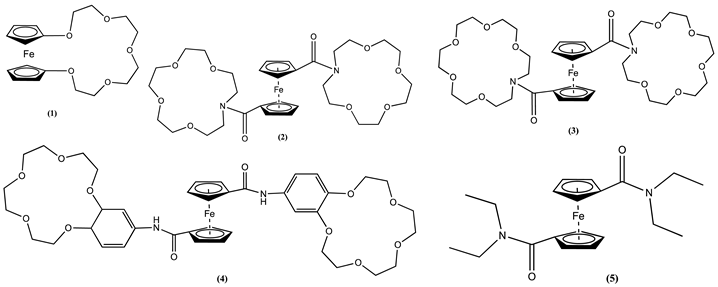
In the case of molecule 2, the Em of Fc collected in CH3CN containing 0.2 M [Bu4N][BF4] as the supporting electrolyte showed a significant positive shift of 0.040, 0.020 and 0.070 V after one equivalent of Na+, K+ and Li+ ions were added, respectively. Similar shifts of 0.035, 0.020, and 0.075 V were observed for molecule 3 after adding one equivalent of the respective ions [53]. Nevertheless, the Em of 4 remained unchanged in the presence of Na+, K+ and Li+ ions, which was argued as related to the considerable distance between Fc and host [55]. Notably, the ability to sense alkali cations is not exclusive to the crown ether host. For example, ferrocene amide 5 has a carbonyl oxygen donor, which acts as a host site for Li+, inducing a positive Em shift of 0.390 V compared to free 5 [53,57].
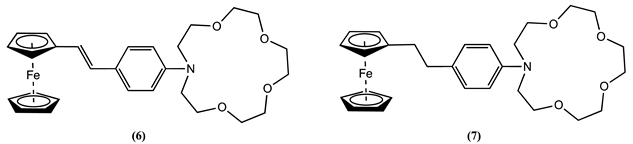
Additional investigations into Fc-crown ethers have shown that electronic conjugation can convey a redox reaction when cations bind, even when the binding unit is apart from the Fc reporter group. This can be further explained using molecules 6 and 7 [58]. In molecule 6, an aza-crown is connected to Fc through an E-stilbene-like linkage. Cyclic voltammetry of 6 in CH3CN containing 0.2 M [Bu4N][BF4] as the supporting electrolyte showed a positive Em shift of 0.120 V when Mg2+ was added. Conversely, for molecule 7, no discernible cation-induced Em shift was noticed, which was rationalised as a consequence of the lack of a channel for electronic communication between the host and Fc due to its saturated linker.
Receptor 8, where an aryl aza-crown is directly bonded to Fc, shows a positive Em shift of 0.040 V for K+, 0.090 V for Na+ and 0.110 V for Mg2+ in acetonitrile containing 0.2 M [Bu4N][BF4] as the supporting electrolyte [57]. It was hypothesised that the magnitude of the Em shift of Fc could be related to the charge/radius ratio of cation guests.
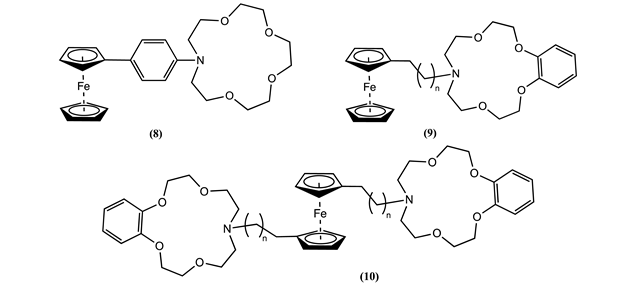
Two series of ferrocene-linked benzoaza[15]crown-5 host molecules 9 and 10 were synthesized with varying alkyl spacer lengths (n = 0, 1, and 3). Cyclic and square wave voltammetry were employed to investigate the electrochemical behaviour of these ligands in the presence of proton and various metal cations [59]. Significant positive Em shifts were observed upon cation binding, supporting Coulombic origin of the through-space interactions. The ΔEm varied linearly with the inverse of the distance between Fc and the bound cation, in agreement with Coulomb’s law. For the optimal condition (n = 0), 9 ΔEm of 0.163, 0.068, and 0.040 V and 10 ΔEm of 0.402, 0.292, and 0.159 V for H+, Mg2+ and Ba2+ were observed [59].
Fc connected to cryptands is also well-studied as a molecular sensor for cations [60]. Cyclic voltammetry studies showed a positive Em shift in the presence of alkaline earth and lanthanide cations [61,62]. Molecule 11 showed the most significant Em positive shift of 0.295 V for Be2+ and 0.254 V for Dy3+. These studies have established a broad correlation between the change in redox potential and the charge density of the cationic species.
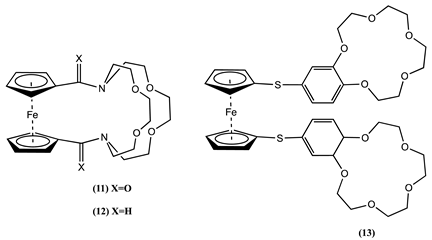
When alkyl amines replaced the amide groups in 11 as in 12, a more significant Em shift was observed after cations binding [63]. For example, the Ca2+ binding to 11 and 12 showed a positive Em shift of 0.155 and 0.275 V, respectively. Also, molecule 12 has an exceptional selectivity for Ag+, displaying a positive ∆Em of 0.282 V after Ag+ binding. It was hypothesised that the flexibility of the alkyl linker greatly influences the coupling between Fc and the cryptand host site.
Interestingly, molecule 13, which has the Fc connected to two crown ether ligands via sulphur, showed a 0.070 V positive ∆Em for Na+ binding and a 0.060 V negative ∆Em for K+ ion [48]. The source of this anomalous behaviour is due to conformational changes upon K+ binding. 13C-NMR and FAB-MS confirmed that the larger size of K+ causes the receptor molecule to form a 1:1 sandwich complex, whereas the smaller size of the Na+ ion produces a 1:2 host:guest complex. The 1:2 complex may have the two crown ether units in a trans-like configuration with respect to the Fc unit. However, the 1:1 sandwich confirmation forces the two crown ether units to the same side of the Fc, forming a cis-like configuration, increasing the Fc instability, and justifying the negative ∆Em observed.
Molecules 14, 15 and 16 are examples of Fc linked to varying sizes of cryptands via electron donor oxygen atoms [64]. Upon careful analysis of cyclic voltammetry data for these molecules and their respective alkali and alkaline earth metal complexes in CH3CN, two trends can be observed: a) Larger the size of the cryptands (14 < 15 < 16), the smaller the positive ∆Em observed; b) the largest positive ∆Em was observed for metal ions whose sizes are complementary to the cryptand size. For molecule 14, the most significant positive ∆Em of 0.380, 0.360 and 0.305 V were obtained for Ca2+, Sr2+, and Ba2+, respectively.
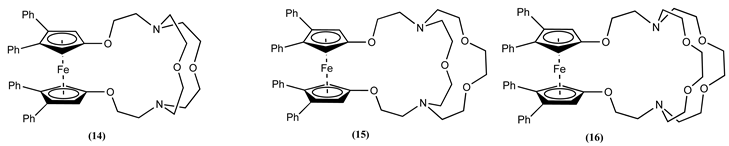
After complexation with Na+, 14, 15 and 16 showed a positive ∆Em of 0.215, 0.180 and 0.080 V, respectively. This trend supports the theory that the most considerable anodic shift was produced by metal ions whose sizes complement the size of the cryptand connected to the Fc unit.
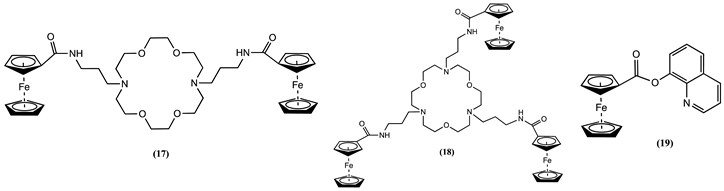
Host units containing di- or tri-aza crown ethers featuring multiple Fc units (17 and 18) have been synthesised [65]. Cyclic voltammetry experiments carried out in CH3CN/0.2 M [Bu4N][BF4] showed that the progressive addition of two equivalent K+ to solutions of 17 or 18 showed a positive ∆Em of 0.050 and 0.085 V, respectively.
Receptor 19 was synthesised by connecting Fc to an 8-hydroxyquinoline unit [66]. Cyclic voltammogram of 19 in CH3CN/0.15 M [Bu4N][ClO4] shows a chemically reversible Fc0/+ one-electron process. However, 19 exhibits a negative ∆Em of 0.149 V after adding one equivalent of Hg2+. This shift was attributed to Hg2+, which enhances the electron density at the Fc centre of 19. Based on fluorescence data, it was hypothesised that the Hg2+ ion binds to the oxygen atoms of the ester group, increasing the intramolecular charge transfer, a phenomenon linked to the influence of heavy atoms [67].
4. Ferrocene-Based Electrochemical Sensors Using Nitrogen-Containing Host Molecules
The interaction between Fc and amines has been the subject of extensive research due to its potential wide application range, including the design of metal ion receptors and sensors [8,9,10,63,68,69,70,71,72]. However, despite numerous studies, the details of this interaction and the oxidation mechanism of ferrocene-bearing amine compounds have remained elusive until recently, where the oxidation mechanism of N,N-dimethylaminomethylferrocene (20, Fc-CH2-N(CH3)2), which can be considered as the parent ligand for many Fc receptors presented in this review, was evaluated and the oxidation product identified (Scheme 2) [7].
At fast scan rates (n), the oxidation process was found to be a simple reversible one-electron oxidation, where the Fc moiety is oxidised to form the ferrocenium cation 20+. This cation is then further oxidised at more positive potentials to form 202+, triggering a reaction sequence that leads to the formation of various intermediate species. Interestingly, the presence of the amine moiety was found to enhance the reactivity of the ferrocenium cation formed on oxidation.
At slow n, additional steps were observed in the oxidation mechanism. These steps included the direct oxidation of the ferrocenium cation and the formation of a terminal methylene radical 20a2+. This intermediate 20a2+ is formed via two distinct routes: i) through direct oxidation of 20+ when the potential scan is extended to more positive values (as observed at high scan rates), and ii) through an intramolecular electron transfer from a nitrogen atom to the iron(III) centre (k1 =0.05 s-1), effectively regenerating the Fc unit and producing a radical cation designated as 20′+, which can then be further oxidised to form 202+ and subsequently deprotonation to yield 20a2+. A following-up disproportionation reaction regenerates 20+ and forms the iminium derivative 20b2+. Water molecules, either existing as contaminants in the organic solvent or introduced during the extraction of the product, may react with 20b2+ to produce the secondary amine 20c2+ along with formaldehyde as a side product [7]. Although without product identification, a similar mechanism was postulated for 21-26 and related molecules, arriving after oxidation to the corresponding iminium cation [8,9,10].
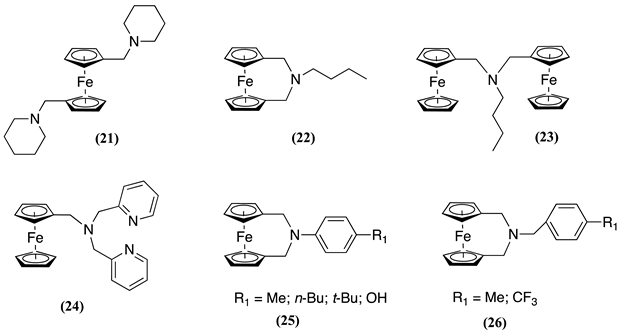
The consequence of this finding is that the electrochemical oxidation of the Fc moiety could potentially modify the host affinity for transition metal ions through three distinct mechanisms: a) the development of electrostatic repulsion due to the close spatial proximity of the oxidised Fe(III) centre (as part of the ferrocenium ion) to the binding site; b) the induction of a positive charge on a host nitrogen atom by substitution of a lone electron pair, which diminishes the host ability to complement the target ion; and c) the interaction of the iminium species with water or other nucleophilic molecules leading to the opening of host ring, diminishing the inherent preorganisation of the amine-containing hosts [7]. Consequently, the square scheme described above for oxygen-containing hosts to describe molecular recognition processes may not extend to nitrogen-containing host systems and caution must be taken when conducting such analyses [47,49].
The Fc bridged cyclam 27 was evaluated by cyclic voltammetry in CH3CN/ [Bu4N][PF6] showed a significant positive ∆Em of 0.360 V for Co2+, 0.380 V for Ni2+, 0.410 V for Cu2+, and 0.470 V for Zn2+ [73]. The significant positive Em shift in the presence of metal ions could be a through-space electrostatic interaction, which causes a substantial perturbation of the Em of Fc. Molecule 27 was also used as a selective sensor for Cu2+, which showed an ∆Em of 0.210 V in the presence of Cu2+ in 70:30 1,4-dioxane:0.1 M KNO3 aqueous solution (pH 5.0). However, the Em was not affected by the presence Ni2+, Zn2+, Cd2+, Hg2+ and Pb2+ ions [74].
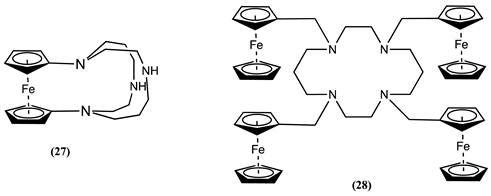
The electrochemical behaviour of 28 in 1:1 DCM:MeOH/[Bu4N][PF6] was studied in the absence and presence of Cu2+ [75]. The addition of Cu2+ resulted in a positive Em shift of 0.094 V. However, no Em changes were observed when 28 was in contact with Ni2+, which was hypothesised as related to the slow complexation kinetics.
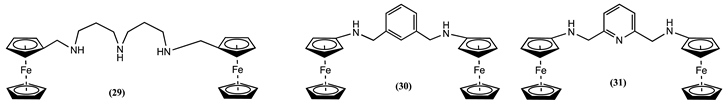
Acyclic receptor molecules 29-31 were synthesised, and their interaction with transition metals was evaluated [76]. It was observed that the added metal ions may directly coordinate with host molecules or protonate them by acting as an acid. For example, 29 co-ordinates with Ni2+ and Cu2+, generating a positive Em shift of 0.058 and 0.098 V, respectively. Instead, the addition of Zn2+ undergoes a protonation pathway of the receptor, causing a positive Em shift of 0.162 V. In contrast, 30 exclusively follows the protonation pathway due to the large bite angle of the host group. Adding Ni2+, Cu2+, and Ca2+ to 30 showed a positive Em shift of 0.175, 0.169 and 0.160 V, respectively. Meanwhile, the pyridine nitrogen atom in 31 participates in the coordination of metal ions. Its interaction with Ni2+, Cu2+ and Zn2+ resulted in a positive Em shift of 0.115, 0.105 and 0.075 V, respectively.
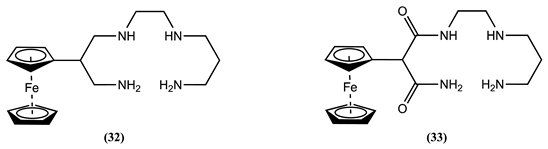
The electrochemical sensor 32 forms a square-planar complex with Ni2+, showing a slight positive Em shift of 0.025 V in an aqueous 0.1 M NaClO4 solution [77]. However, due to the presence of two electron-deficient amide nitrogen atoms in 33, the dioxotetraamino host shows a poor tendency towards complexation [77]. This behaviour changes when Ni2+ is complexed under basic conditions, where nitrogen deprotonations result in a double negative charge on the ring system of 33, forming a stable four-coordinated system that generates a negative Em shift of 0.042 V [11,77].
Recently, we synthesised cyclen and cyclam macrocycles bearing ferrocene pendants and investigated their ability to work as electrochemical sensors for transitional metal cations [7]. The cyclic voltammograms 34 in 1:4 CH2Cl2:CH3CN/0.1 M [Bu4N][PF6] showed upon addition of Ni2+, Cd2+, Zn2+, Cu2+ and Co2+ ions a positive Em shift of 0.054, 0.057, 0.078, 0.100, and 0.120 V, respectively. Similarly, when Zn2+ co-ordinates with molecule 35, a positive Em shift of 0.098 V was observed.
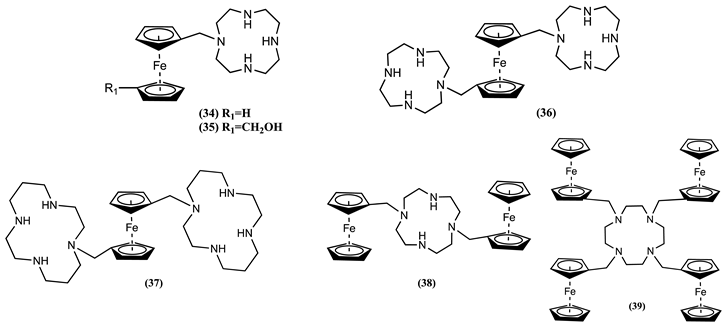
Addition of Ni2+, Cd2+, Zn2+, Cu2+ and Co2+ ions to a solution of 36 in 1:4 CH2Cl2:CH3CN/0.1 M [Bu4N][PF6] generates a positive Em shift of 0.148, 0.160, 0.203, 0.240, and 0.280 V, respectively. Similarly, cyclic voltammograms of 37 showed a more significant response in the presence of the mentioned ions, generating a positive Em shift of 0.184 V for Ni2+, 0.179 V for Cd2+, 0.183 V for Zn2+, 0.239 V for Cu2+ and 0.259 V for Co2+.
The cyclic voltammetric behaviour of 38, where two Fc units are linked via the cyclen host group, was studied in detail before and after complexation with 3d and 4d transition metal cations. The complexation reaction between 38 and Ni2+, Cd2+, Zn2+, Cu2+ and Co2+ ions resulted in a positive Em shift of 0.064, 0.060, 0.088, 0.111, and 0.132 V, respectively. Similarly, 39, where four Fc units are attached to the cyclen host group, was also investigated. Adding Cd2+, Zn2+, Cu2+ and Co2+ to 39 resulted in a positive ∆Em of 0.053, 0.047, 0.078, and 0.022 V, respectively. However, no response was observed after the addition of Ni2+ ions.
Based on electrochemical data, the charge density of transitional metal ions can be correlated with the ∆Em observed, except for Ni2+. The magnitude of the Em shift follows the order 36 ≈ 37 >> 34 ≈ 35 ≈ 38 > 39 and depends on the number of macrocycles linked to Fc. Nonetheless, no significant change in Em can be obtained by increasing the number of Fc groups attached to a guest macrocycle [7].
The redox behaviour of 40-44 was studied in CH3CN/0.1 M [Bu4N][ClO4] [45]. Receptor 40 showed a positive Em shift of 0.250 V after adding Pb2+, Zn2+, or Hg2+ ions. However, 41 shows a maximum response after adding one equivalent of Pb2+, presenting a positive Em shift of 0.150 V. Similarly, 41 showed a positive ∆Em of 0.090 and 0.120 V for Zn2+ and Hg2+ ions. This shift was very specific, and the cyclic voltammogram of 41 was not affected by the addition of large quantities of Li+, Na+, K+, Ca2+, Mg2+, Ni2+ and Cd2+ ions. Likewise, 42 showed a positive Em shift of 0.120 V in the presence of Pb2+. Consequently, the positive Em shift for the monosubstituted Fc after complexing with Pb2+ is 42 < 41 < 40.
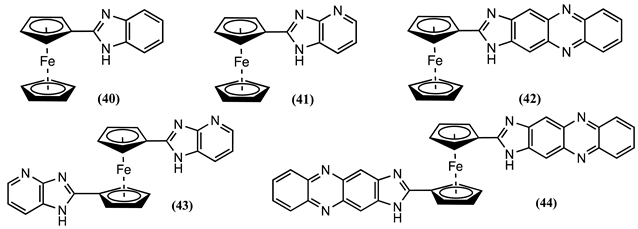
Receptors 43 and 44 also display selectivity for Pb2+ and Zn2+. Molecule 43 showed a positive Em shift of 0.180 and 0.190 V upon adding one equivalent of Pb2+ and Zn2+, respectively. A comparable trend was also found for 44, which undergoes a positive ∆Em of 0.110 and 0.170 V for the same cations. The ∆Em observed for Zn2+ starkly contrasts observations made with the related molecules 41 and 42, which is in agreement with previous works, where the complexation of Zn2+ ions to 1,1′-disubstituted ferrocene resulted in a large positive Em shift with respect to 1-monosubstituted ferrocene [7,78]. The receptor 45, which differs from 40 by a methyl group in the benzimidazole ring, has been electrochemically studied in CH3CN/0.1 M [Bu4N][ClO4] [79]. It displayed specific sensing capability for Sn2+ ions, showing a positive Em shift of 0.230 V after adding one equivalent of Sn2+ ions. Similarly, the ability of 46 and 47 to sense different metal ions was assessed in 1:1 CH3CN:DCM/0.01M [Bu4N][PF6] [80]. In the presence of Hg2+ and Pb2+, 46 showed a positive Em shift of 0.050 and 0.060 V, whereas 47 showed an ∆Em of 0.150 V for Hg2+ and 0.040 V for Pb2+ after complexation. Notably, the presence of a 3-pyridyl unit in the sensors, regardless of location, significantly enhanced their ability to detect Hg2+ selectively and sensitively. It is worth noting that the 3-pyridyl unit did not directly coordinate with metal ions despite its influence on the improved performance.
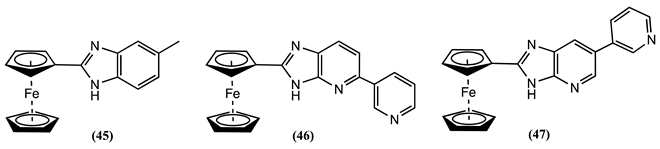
Sensors 48-50 have been synthesised by connecting a Fc unit to a pyridyl moiety using a β-diketone bridge [81]. The complexing properties of these sensors towards Zn2+, Hg2+, Co2+, Cu2+, Mn2+, Cd2+, and Ni2+ ions were investigated in ethanol/0.1 M LiClO4. The largest positive Em shifts were obtained after adding Cu2+ and Cd2+ to a solution of 48, showing ∆Em = 0.072 and 0.067 V, respectively. Meanwhile, positive Em shifts of 0.102 V for Cu2+ and 0.109 V for Mn2+ were reported for 49.
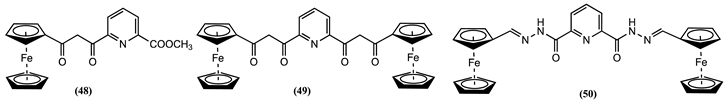
Similarly, 50 results in a positive Em shift of 0.053 V for Hg2+ and 0.054 V for Mn2+ ions. Yet, the receptors 49 and 50 exhibit significantly lower redox potential shifts when treated with Zn2+, Hg2+, Co2+, Cd2+ and Ni2+ ions.
Meanwhile, naphthalimide was connected to a ferrocenyl-chalcone group using a triazole linker, and the electrochemical properties of the resulting molecule 51 to sense Cu2+ ion in CH3CN/0.01 M [Bu4N][ClO4] was evaluated [82]. A 0.5 mM 51 solution showed a positive ∆Em of 0.020 V upon adding 30 µM Cu2+ ion.
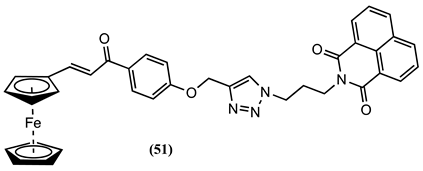
Two receptors, 52 and 53, which incorporate Fc and rhodamine containing triazole, were synthesised, and their electrochemical properties to sense metal cations in CH3CN/0.1 M [Bu4N][ClO4] explored [83]. The Em of these receptors was not affected by the presence of Na+, Mg2+, K+, Ca2+, Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Pb2+, Cd2+, and Tl+ ions. However, introducing one equivalent of Hg2+ led to a positive Em shift of 0.200 and 0.250 V for 52 and 53, respectively. NMR studies of the 52-Hg2+ complex indicated that Hg2+ ions coordinate with the nitrogen atoms of the triazole ring, the imine, the oxygen atoms linked directly to the methylene, and the amide carbonyl group [83]. A similar molecule, 54, shows a negative ∆Em of 0.050 V upon adding one equivalent of Hg2+. HR-MS and FT-IR studies suggest that the spirolactam ring undergoes an opening process when Hg2+ binds to 54. Consequently, it is believed that the pyridine nitrogen atom and two amide oxygen atoms of 54 potentially form coordination bonds with Hg2+, creating a chelating complex in conjunction with a solvent molecule [83].
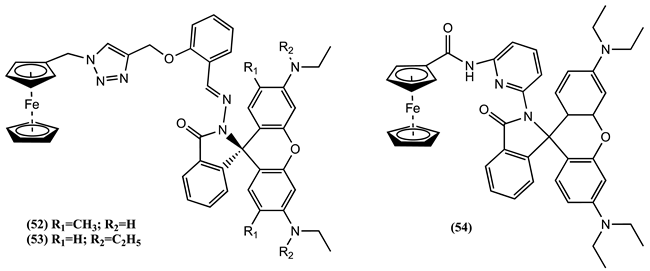
The strained asymmetric sensor 55 was synthesised and electrochemically evaluated in DCM/0.1 M [Bu4N][ClO4] [84]. The cyclic voltammograms revealed two closely spaced, reversible oxidation peaks at 0.510 and 0.740 V vs. SCE. The initial reversible oxidation process was assigned to the oxidation of the single-substituted Fc unit, and the second one to the oxidation of the 1,1′-disubstituted Fc unit. While the addition of Ca2+ ions did not cause any Em changes, a noticeable change was observed upon introducing one equivalent of Mg2+ ions, where a positive Em shift of 0.340 V was observed in the second peak. Meanwhile, a small positive ∆Em of 0.030 V was reported for the first peak. This sensor was able to distinguish between Mg2+ and Ca2+ ions without being affected by the presence of excess amount of Li+, Na+, and K+ ions [84].
Molecule 56 was studied in 4:1 DCM:CH3CN/0.1 M [Bu4N][PF6] [85]. The cyclic voltammogram displayed two electrochemically reversible one-electron peaks at Em = -0.060 and 0.460 V vs. Fc0/+. Adding increasing quantities of Zn2+ into a solution of 56 led to the partial disappearance of the peak at -0.060 V and the emergence of a new peak at 0.020 V. However, there were no noticeable Em changes in the second peak of the receptor. Unsymmetrical related molecules 57-59 were evaluated with respect to a range of cations, including Li+, Na+, K+, Ca2+, Mg2+, Ni2+, Cd2+, Zn2+, Pb2+, Hg2+, and Cu2+ in 4:1 or 3:2 CH3CN:DCM/ 0.1 M [Bu4N][PF6 [86]. Each free receptor displayed two reversible one-electron redox processes. Compound 57 shows these processes at 0.050 and 0.250 V vs. Fc0/+, 58 at 0.050 and 0.310 V, and 59 at 0.030 and 0.240 V. In all three cases, the initial oxidation process pertains to the Fc unit with a single substitution and the second oxidation wave is associated with the Fc unit bearing two substitutions.
Adding Pb2+ ions to 57 generates a positive Em shift of 0.050 V for the first process and a negative Em shift of 0.150 V for the second. A similar situation was observed after adding Zn2+, with a positive Em shift of 0.060 V for the first process and a negative ∆Em of 0.140 V for the second. In the case of 58, the second process presented a positive Em shift of 0.050 V for Cd2+, 0.130 V for Ni2+, 0.150 V for Pb2+, and 0.140 V for Zn2+. Nevertheless, adding Pb2+ ions to 59 generates a positive Em shift of 0.040 V for the first process and a negative ∆Em of 0.160 V for the second. Similarly, adding Zn2+ causes a positive Em shift of 0.010 V for the first process and a negative ∆Em of 0.200 V for the second one [86]. As a result of this experiment, the addition of Pb2+ and Zn2+ to 57 and 59 resulted in a concurrent positive shift of the lower Em and a negative shift of the higher Em. This causes a single wave to appear in the cyclic voltammograms of the final complex due to the overlapping of the two oxidation processes part of the receptors. Nevertheless, the most noteworthy change occurs in the higher oxidation process when Ni2+, Cd2+, Zn2+, and Pb2+ are added to probe 58. This process experiences a reduction in intensity until it completely disappears when one equivalent of metal ion is present. Concurrently, the lower oxidation process remains unchanged. This observation implies that the triazole group plays a minor role in interacting with metal cations during complexation. As a result, the primary binding process is likely concentrated in the imine arm of the various receptors, with particular emphasis on receptor 58, which incorporates an additional quinoline ring [86].
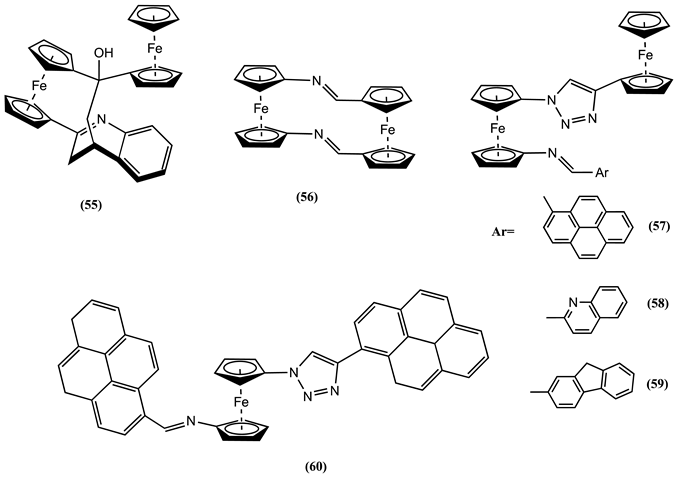
A related molecule 60, containing one Fc unit, was studied in 4:1 CH3CN:DCM/0.1 M [Bu4N][PF6] [87]. Free 60 showed a one-electron reversible process at 0.195 V vs Fc0/+ IRRS. A new reversible process at 0.045 and 0.200 V more positive potential values was observed after adding Zn2+ and Pb2+, respectively. Notably, the cyclic voltammetric behaviour of 60 is not affected by adding other ions, such as Li+, Na+, K+, Mg2+, Ca2+, Ni2+, Cu2+, Cd2+, and Hg2+.
The receptor 61 was studied in CH3CN containing 0.1 M [Bu4N][PF6] as the supporting electrolyte. Meanwhile, receptor 62 was evaluated in 9:1 CH2Cl2:DMF/0.1 M [Bu4N][PF6] [88]. Each free receptor displayed a characteristic reversible one-electron redox process, with Em= 0.575 V for 61 and Em = 0.470 V vs. DmFc0/+ IRRS for 62. The Em of 61 shifted in the positive direction by 0.333, 0.282, 0.224, 0.274, and 0.361 V after adding Zn2+, Pb2+, Ni2+, Cd2+, and Hg2+, respectively. In contrast, 62 shows a positive ∆Em = 0.260 V in the presence of Hg2+. Based on NMR and theoretical studies, it was postulated that the coordination of metal ions to receptor 61 occurs via imine and quinoline nitrogens. Similarly, the ferrocene appended phenolic hydroxyl Schiff base 63 was studied in MeOH/0.1 M [Bu4N][ClO4] [89]. The addition of Al3+, Cr3+, or Fe3+ to a solution of 63 resulted in a positive shift in the Em of 0.028, 0.044, and 0.015 V, respectively.
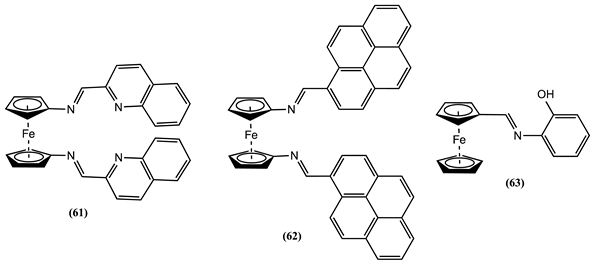
Cyclic voltammetric experiments were performed using a 3:7 CH3CN:H2O/0.1 M [Bu4N][ClO4] solution of 64 and 65. Their ability to detect the presence of Li+, Na+, K+, Ca2+, Mg2+, Cr3+, Zn2+, Fe3+, Ni2+, Co2+, Cu2+, Cd2+, Hg2+, and Pb2+ ions was evaluated [90]. Both receptors 64 and 65 displayed a reversible one-electron oxidation process, in which Em are not perturbed by the presence of the mentioned ions, except for Hg2+, where the addition of one equivalent of this ion generates a positive Em shift of 0.076 and 0.043 V for 64 and 65, respectively. No Hg2+ ion sensing ability improvement is obtained with 66 or 67. Receptor 66 can be seen as the dimeric version of 64, and its Em shifts by 0.078 V in the positive direction after the addition of Hg2+ [91]. However, the Em of receptor 67 shifts by 0.033 V when exposed to Hg2+ ions [92]. Based on 1H-NMR titration analysis, it was inferred that the Hg2+ binds to the triazole ring nitrogen atom and the oxygen atom in the OCH2 bridging group [91].
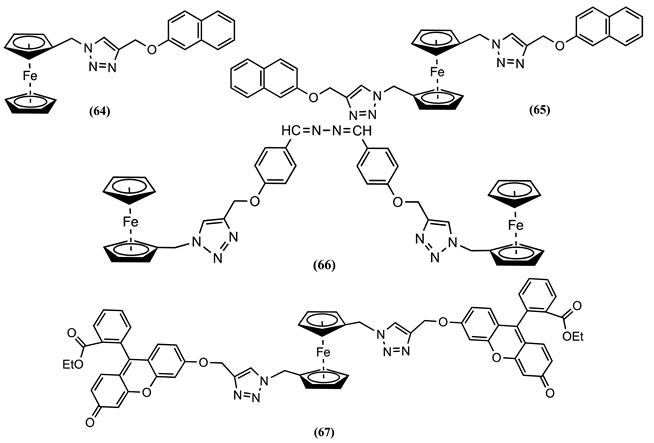
Receptors 68 and 69 containing monosubstituted Fc pendants and 70 and 71 containing disubstituted Fc pendants have been synthesised and electrochemically studied in CH3CN containing 0.1 M [Bu4N][PF6] as the supporting electrolyte [93]. The addition of Ni2+, Hg2+, and Pb2+ to 68 resulted in the appearance of a new oxidation process at a more positive ∆Em of 0.053, 0.025, and 0.014 V, respectively. In contrast, 69 showed a positive Em shift of 0.072 V upon adding an Hg2+ ion. A similar response was observed with the related receptor 70, which showed a reversible two-electron process at Em = 0.544 V vs. DmFc0/+ IRRS for the terminal Fc groups and a reversible one-electron process at Em = 0.953 V vs. DmFc0/+ related to the disubstituted central Fc group [93]. The addition of Zn2+, Hg2+, and Pb2+ to 70 produced a negligible Em shift for the terminal Fc groups but an Em shift of 0.032, 0.076, and 0.022 V, respectively, for the central Fc group. In the case of 71, only the Hg2+ ion produced a significant response, showing a positive Em shift of 0.035 V after adding one equivalent of the mentioned ion.
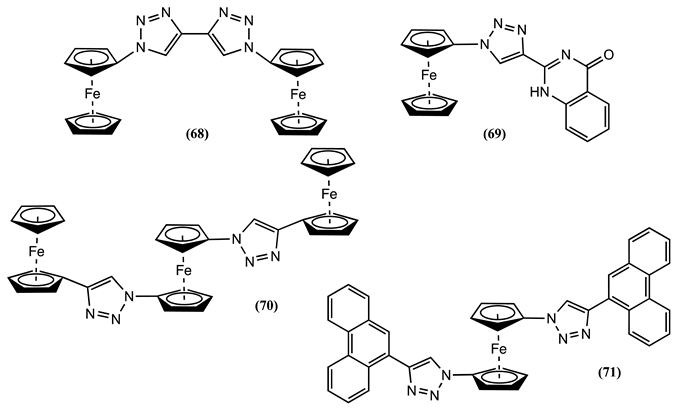
Cyclic voltammetry of 72 in CH3CN containing 0.01M [Bu4N][ClO4] as the supporting electrolyte showed a one-electron reversible process, which Em shifter positively by 0.065 V after the addition of Fe3+ ions to the solution [94]. Meanwhile, Ferrocene quinazolines 73 was studied in 1:1 ethanol:H2O/0.1M [Bu4N][ClO4] [95]. The introduction of Hg2+ to this solution caused the appearance of a new irreversible process at 0.136 V more positive to the Em of 73. Similarly, adding Pb2+ to a solution of 73 resulted in a negative Em shift of 0.025 V [95].
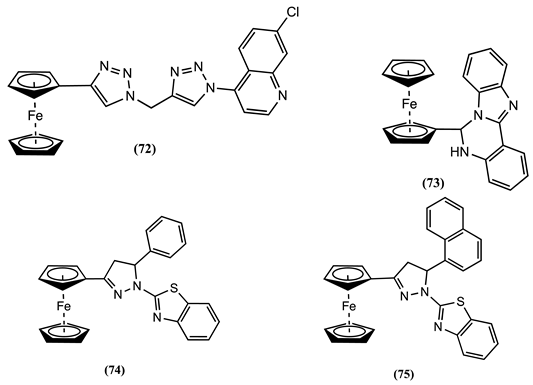
The interaction of 74 and 75 with various metal ions was systematically studied in acetonitrile using cyclic voltammetry and differential pulse voltammetry [96]. The addition of Hg2+ ion to this solution produces the appearance of a new process at 0.078 and 0.100 V more positive potentials with the consequent disappearance of the process related to the free 74 and 75 receptors, respectively. Conversely, when one equivalent of Cu2+ ions was introduced to the receptor solutions, a single process was observed, showing a positive Em shift of 0.042 V for receptor 74 and 0.048 V for receptor 75. The 1H NMR analysis of the 74-Hg2+ complex showed that the ion interacts with the nitrogen atoms in 1H-pyrazoline and 2-benzothiazole. This finding is supported by theoretical computations conducted using density functional theory [96].
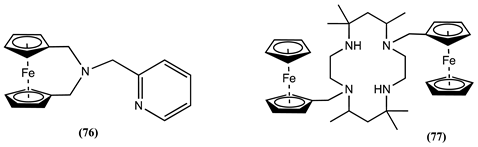
Sensor 76 was studied in a CH3OH/H2O solution containing 0.1M KNO3 as the supporting electrolyte [97]. The addition of 0.5 equivalents of Cu2+ to this solution resulted in the appearance of two reversible one-electron redox processes. The first one was related to the redox process of the free 76, and the second, appearing at 0.125 V more positive potentials, was assigned to the 76-Cu2+ complex. When one equivalent of Cu2+ was introduced, the redox process of the free 76 disappeared. Interestingly, the Em of 76 is not affected by the presence of other metal ions, such as Mg2+, Ni2+, Zn2+, Pb2+, and Cd2+.
Receptor 77 was investigated in a 7:3 dioxane:water containing [Bu4N][ClO4] as the supporting electrolyte, revealing its pH-dependent behaviour, shifting Em towards more positive potentials as the pH decreases from 12 to 5 [98]. Additionally, the presence of Mn2+, Co2+, Ni2+, Cu2+, and Zn2+ shifts the Em of 77 in the positive potential direction by 0.046 V for Cu2+, 0.020 V for Co2+, 0.015 V for Mn2+, 0.013 V for Ni2+, and 0.009 V for Zn2+ [98].
5. Conclusions
Significant advances have been made in developing Fc receptors capable of selectively sensing metal cations. This review presented a detailed discussion of how oxygen and nitrogen-containing macromolecular receptors featuring Fc moiety can initially form bonds and then exhibit shifts in redox potentials in response to positively charged guest molecules. This interaction occurs through electrostatic perturbation across space or linked conjugated bonds connecting the Fc unit to the metal cation host binding unit. The general factors that affect the magnitude of the redox potential shift upon binding of the cations to the guest site depend on i) the complementarity between the ferrocene-containing host and guest cation, encompassing the thermodynamics and kinetics of their binding interactions; ii) the charge-to-size ratio of the metal cation; iii) the closeness of the binding site to the redox active Fc; iv) the type of chemical bond that connects the host to the Fc pendant; and e) the nature of the solvent used during the experiment. This review also examined the complexity involved in the interplay between electron transfer and molecular recognition where hosts with amine functionalities such as cyclen and cyclam are utilised.
So far, most cation recognition has been conducted in organic solvents. This presents a significant challenge when attempting to use these ferrocene-based molecular receptors in aqueous solvents, a requirement for ion recognition in biological systems. To add to the complexity, competing redox active molecules against ferrocene probes might also influence the sensor’s specificity. These challenges could open an excellent opportunity to enhance the ability of receptors to selectively bind cation guests, particularly in intricate aqueous environments and integrate them into electrochemical device platforms.
Author Contributions
Conceptualization, A.K.V.M. and A.A.J.T.; methodology, A.K.V.M. and A.A.J.T.; software, A.K.V.M. and A.A.J.T.; validation, A.K.V.M. and A.A.J.T.; formal analysis, A.K.V.M. and A.A.J.T.; investigation, A.K.V.M. and A.A.J.T.; resources, A.K.V.M. and A.A.J.T.; data curation, A.K.V.M. and A.A.J.T.; writing—original draft preparation, A.K.V.M. and A.A.J.T.; writing—review and editing, A.K.V.M. and A.A.J.T.; visualization, A.K.V.M. and A.A.J.T.; supervision, A.K.V.M. and A.A.J.T.; project administration, A.K.V.M. and A.A.J.T.; funding acquisition, A.K.V.M. and A.A.J.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lehn, J.M. Supramolecular Chemistry; Wiley-VCH: New York, USA, 1995; pp. I–X. [Google Scholar]
- Zanello, P. Inorganic Electrochemistry: Theory, Practice and Application; The Royal Society of Chemistry: Cambridge (UK), 2003. [Google Scholar]
- Molina, P.; Tárraga, A.; Caballero, A. Ferrocene-Based Small Molecules for Multichannel Molecular Recognition of Cations and Anions. European Journal of Inorganic Chemistry 2008, 2008, 3401–3417. [Google Scholar] [CrossRef]
- Togni, A.; Hayashi, T. , (Eds.) Ferrocenes: homogeneolls catalysis, organic synthesis, materials science. VCH: New York, USA, 1995.
- Casado, C.M.; Alonso, B.; García-Armada, M.P. 7.02—Ferrocenes and Other Sandwich Complexes of Iron. In Comprehensive Organometallic Chemistry IV, Parkin, G., Meyer, K., O’hare, D., Eds.; Elsevier: Oxford, 2022; pp. 3–45. [Google Scholar]
- Stepnicka, P. , (Ed.) Ferrocenes: Ligands, Materials and Biomolecules. John Wiley & Sons, Ltd.: Chichester, UK, 2008.
- Torriero, A.A.J.; Zeng, Z.; Mruthunjaya, A.K.V.; Bond, A.M. Electrochemical Properties of Cyclen and Cyclam Macrocycles Bearing Ferrocenyl Pendants and Their Transition Metal Complexes. Journal of Electroanalytical Chemistry 2023, 945, 117687. [Google Scholar] [CrossRef]
- Milaeva, E.R.; Tyurin, V.Y.; Shpakovsky, D.B.; Moiseeva, A.A.; Gracheva, Y.A.; Antonenko, T.A.; Maduar, V.V.; Osolodkin, D.I.; Palyulin, V.A.; Shevtsova, E.F. Redox-active metal complexes with 2,2′-dipicolylamine containing ferrocenyl moiety: Synthesis, electrochemical behavior and biological activity. Journal of Organometallic Chemistry 2017, 839, 60–70. [Google Scholar] [CrossRef]
- Dwadnia, N.; Allouch, F.; Pirio, N.; Roger, J.; Cattey, H.; Fournier, S.; Penouilh, M.-J.; Devillers, C.H.; Lucas, D.; Naoufal, D.; et al. Aminomethyl-Substituted Ferrocenes and Derivatives: Straightforward Synthetic Routes, Structural Characterization, and Electrochemical Analysis. Organometallics 2013, 32, 5784–5797. [Google Scholar] [CrossRef]
- Osakada, K.; Sakano, T.; Horie, M.; Suzaki, Y. Functionalized ferrocenes: Unique properties based on electronic communication between amino group of the ligand and Fe center. Coordination Chemistry Reviews 2006, 250, 1012–1022. [Google Scholar] [CrossRef]
- Fabbrizzi, L. The ferrocenium/ferrocene couple: a versatile redox switch. ChemTexts 2020, 6, 22. [Google Scholar] [CrossRef]
- Bayly, S.R.; Beer, P.D.; Chen, G.Z. Ferrocene Sensors. In Ferrocenes: Ligands, Materials and Biomolecules Stepnicka, P., Ed.; John Wiley & Sons: Chichester, England, 2008; pp. 281–318. [Google Scholar]
- Noviandri, I.; Brown, K.N.; Fleming, D.S.; Gulyas, P.T.; Lay, P.A.; Masters, A.F.; Phillips, L. The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple for Studying Solvent Effects on the Thermodynamics of Electron Transfer. J. Phys. Chem. B 1999, 103, 6713–6722. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, L. Theoretical investigations of ferrocene/ferrocenium solvation in imidazolium-based room-temperature ionic liquids. PCCP Phys. Chem. Chem. Phys. 2013, 15, 2669–2683. [Google Scholar] [CrossRef]
- Torriero, A.A.J. Characterization of decamethylferrocene and ferrocene in ionic liquids: argon and vacuum effect on their electrochemical properties. Electrochimica Acta 2014, 137, 235–244. [Google Scholar] [CrossRef]
- Torriero, A.A.J.; Sunarso, J.; Forsyth, M.; Pozo-Gonzalo, C. Assessment of permethylated transition-metal sandwich complexes as internal reference redox systems in ionic liquids. PCCP Phys. Chem. Chem. Phys. 2013, 15, 2547–2553. [Google Scholar] [CrossRef]
- Torriero, A.A.J.; Howlett, P.C. Ionic liquid effects on the redox potential of ferrocene. Electrochemistry Communications 2012, 16, 84–87. [Google Scholar] [CrossRef]
- Torriero, A.A.; Sunarso, J.; Howlett, P.C. Critical evaluation of reference systems for voltammetric measurements in ionic liquids. Electrochimica Acta 2012, 82, 60–68. [Google Scholar] [CrossRef]
- Torriero, A.A.J. Reference systems for voltammetric measurements in ionic liquids. In Electrochemistry in Ionic Liquids. Volume 1: Fundamentals, Torriero, A.A.J., Ed.; Springer: Switzerland, 2015; Volume 1. [Google Scholar]
- Torriero, A.A.J.; Sunarso, J.; Howlett, P.C. Critical Evaluation of Reference Systems for Voltammetric Measurements in Ionic Liquids. Electrochimica Acta 2012, 82, 60–68. [Google Scholar] [CrossRef]
- Torriero, A.A.J. On Choosing Ferrocene as an Internal Reference Redox Scale for Voltammetric Measurements: A Cautionary Tale. Medicinal & Analytical Chemistry International Journal 2019, 3, 000151. [Google Scholar] [CrossRef]
- Inzelt, G.; Lewenstam, A.; Scholz, F. Handbook of reference electrodes; Springer: Berlin, 2013. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical methods: fundamentals and applications, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2001. [Google Scholar]
- Izutzu, K. Electrochemistry in nonaqueous solutions; Wiley-VCH: NY, USA, 2002. [Google Scholar]
- Smith, T.J.; Stevenson, K.J. Reference electrodes. In Handbook of electrochemistry, Zoski, C.G., Ed.; Elsevier: Amsterdam, 2007; pp. 73–110. [Google Scholar]
- Torriero, A.A.J.; Bond, A.M. Critical Evaluation of Electrochemistry in Ionic Liquids. In Electroanalytical Chemistry Research Trends, Hayashi, K., Ed.; Nova Science Publishers, Inc.: New York, 2009. [Google Scholar]
- Torriero, A.A.J. Understanding the Differences between a Quasi-Reference Electrode and a Reference Electrode. Medicinal & Analytical Chemistry International Journal 2019, 3, 000144. [Google Scholar]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potentials in nonaqueous solvents. Pure and Applied Chemistry 1983, 56, 461–466. [Google Scholar] [CrossRef]
- Torriero, A.A.J.; Feldberg, S.; Zhang, J.; Simonov, A.; Bond, A. On choosing a reference redox system for electrochemical measurements: a cautionary tale. Journal of Solid State Electrochemistry 2013, 17, 3021–3026. [Google Scholar] [CrossRef]
- Aranzaes, J.R.; Daniel, M.-C.; Astruc, D. Metallocenes as references for the determination of redox potentials by cyclic voltammetry—Permethylated iron and cobalt sandwich complexes, inhibition by polyamine dendrimers, and the role of hydroxy—containing ferrocenes. Canadian Journal of Chemistry 2006, 84, 288–299. [Google Scholar] [CrossRef]
- Ruiz, J.; Astruc, D. Permethylated electron-reservoir sandwich complexes as references for the determination of redox potentials. Suggestion of a new redox scale. Comptes Rendus de l’Academie des Sciences, Serie IIc: Chimie 1998, 1, 21–27. [Google Scholar] [CrossRef]
- Matsumoto, M.; Swaddle, T.W. The Decamethylferrocene(+/0) Electrode Reaction in Organic Solvents at Variable Pressure and Temperature. Inorganic Chemistry 2004, 43, 2724–2735. [Google Scholar] [CrossRef]
- Freyberg, D.P.; Robbins, J.L.; Raymond, K.N.; Smart, J.C. Crystal and molecular structures of decamethylmanganocene and decamethylferrocene. Static Jahn-Teller distortion in a metallocene. Journal of the American Chemical Society 1979, 101, 892–897. [Google Scholar] [CrossRef]
- Barriere, F.; Geiger, W.E. Use of Weakly Coordinating Anions to Develop an Integrated Approach to the Tuning of Delta E1/2 Values by Medium Effects. J. Am. Chem. Soc. 2006, 128, 3980–3989. [Google Scholar] [CrossRef]
- Bennett, M.A.; Bhargava, S.K.; Bond, A.M.; Burgar, I.M.; Guo, S.X.; Kar, G.; Priver, S.H.; Wagler, J.; Willis, A.C.; Torriero, A.A.J. Synthesis, X-ray structure and electrochemical oxidation of palladium(II) complexes of ferrocenyldiphenylphosphine. Dalton Transactions 2010, 39, 9079–9090. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Torriero, A.A.J.; Belousoff, M.J.; Bond, A.M.; Spiccia, L. Synthesis, X-ray Structure of Ferrocene Bearing bis(Zn-cyclen) Complexes and the Selective Electrochemical Sensing of TpT. Chemistry - A European Journal 2009, 15, 10988–10996. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.F.; Neuse, E.W.; Thomas, H.G. Electrochemical characterization of some ferrocenylcarboxylic acids. Transition Metal Chemistry 1987, 12, 301–306. [Google Scholar] [CrossRef]
- Nonjola, P.T.N.; Siegert, U.; Swarts, J.C. Synthesis, Electrochemistry and Cytotoxicity of Ferrocene-Containing Amides, Amines and Amino-Hydrochlorides. Journal of Inorganic and Organometallic Polymers and Materials 2015, 25, 376–385. [Google Scholar] [CrossRef]
- Zhong, Z.H.; Matsumura-Inoue, T.; Ichimura, A. Solvent effect on the redox potential of ferrocene derivatives using an ultramicroelectrode. Analytical Sciences 1992, 8, 877–879. [Google Scholar] [CrossRef]
- Paul, A.; Borrelli, R.; Bouyanfif, H.; Gottis, S.; Sauvage, F. Tunable Redox Potential, Optical Properties, and Enhanced Stability of Modified Ferrocene-Based Complexes. ACS Omega 2019, 4, 14780–14789. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.; Sochaj, K. Cyclic voltammetry of some ferrocenophanes in acetonitrile. Electrochimica Acta 1991, 36, 689–694. [Google Scholar] [CrossRef]
- Plenio, H.; Yang, J.; Diodone, R.; Heinze, J. Redox-Switched Bonding of Protons to Ferrocenophanes, Ferrocene Cryptands, and Simple Ferrocene Amines. Correlation of X-ray Structural Data and Cyclic Voltammetry Derived Redox Potentials. Inorganic Chemistry 1994, 33, 4098–4104. [Google Scholar] [CrossRef]
- Lee, T.-Y.; Chiang, P.-R.; Tsai, M.-C.; Lin, C.-Y.; Huang, J.-H. From diacetylferrocene to 1,1′-ferrocenyldiimines: Substituent effects on synthesis, molecular structure, electrochemical behavior and optical absorption property. Journal of Molecular Structure 2009, 935, 102–109. [Google Scholar] [CrossRef]
- Manfredi, N.; Decavoli, C.; Boldrini, C.L.; Coluccini, C.; Abbotto, A. Ferrocene Derivatives Functionalized with Donor/Acceptor (Hetero)Aromatic Substituents: Tuning of Redox Properties. Energies 2020, 13. [Google Scholar] [CrossRef]
- Zapata, F.; Caballero, A.; Espinosa, A.; TaÌrraga, A.; Molina, P. Imidazole-Annelated Ferrocene Derivatives as Highly Selective and Sensitive Multichannel Chemical Probes for Pb(II) Cations. The Journal of Organic Chemistry 2009, 74, 4787–4796. [Google Scholar] [CrossRef] [PubMed]
- Celedón, S.; Hamon, P.; Artigas, V.; Fuentealba, M.; Kahlal, S.; Carrillo, D.; Saillard, J.-Y.; Hamon, J.-R.; Manzur, C. Ferrocene functionalized enantiomerically pure Schiff bases and their Zn(ii) and Pd(ii) complexes: a spectroscopic, crystallographic, electrochemical and computational investigation. New Journal of Chemistry 2022, 46, 3948–3960. [Google Scholar] [CrossRef]
- Paul D., Beer; Gale, P.A.; Chen, G.Z. Electrochemical molecular recognition: pathways between complexation and signalling. Journal of the Chemical Society, Dalton Transactions 1999, 1897–1910. [Google Scholar] [CrossRef]
- Beer, P.D.; Danks, J.P.; Hesek, D.; McAleer, J.F. A potassium-selective sulfide-linked redox-active ferrocene ionophore that exhibits extraordinary electrochemical recognition behaviour. Journal of the Chemical Society, Chemical Communications, 1993; 1735–1737. [Google Scholar] [CrossRef]
- Beer, P.D. Transition Metal and Organic Redox-Active Macrocycles Designed to Electrochemically Recognize Charged and Neutral Guest Species. In Advances in Inorganic Chemistry; Sykes, A.G., Ed.; Academic Press, 1992; Volume 39, pp. 79–157. [Google Scholar]
- Pedersen, C.J. Cyclic polyethers and their complexes with metal salts. Journal of the American Chemical Society 1967, 89, 7017–7036. [Google Scholar] [CrossRef]
- Lindoy, L.F. The Chemistry of Macrocyclic Ligand Complexes; Cambridge University Press: Cambridge, 1989. [Google Scholar]
- Dobler, M. Macrocyclic chemistry: aspects of organic and inorganic supramolecular chemistry by B. Dietrich, P. Viout and J.-M. Lehn. Acta Crystallographica Section B 1993, 49, 1074–1074. [Google Scholar] [CrossRef]
- Beer, P.D.; Keefe, A.D.; Sikanyika, H.; Blackburn, C.; McAleer, J.F. Metallocene bis(aza-crown ether) ligands and related compounds. Their syntheses, co-ordination chemistry, and electrochemical properties. Journal of the Chemical Society, Dalton Transactions 1990, 3289–3294. [Google Scholar] [CrossRef]
- Saji, T.; Kinoshita, I. Electrochemical ion transport with ferrocene functionalized crown ether. Journal of the Chemical Society, Chemical Communications 1986, 716–717. [Google Scholar] [CrossRef]
- Beer, P.D.; Sikanyika, H.; Slawin, A.M.Z.; Williams, D.J. The synthesis, coordination and electrochemical studies of metallocene bis(crown ether) receptor molecules. Single-crystal x-ray structure of a ferrocene bis(crown ether) potassium complex. Polyhedron 1989, 8, 879–886. [Google Scholar] [CrossRef]
- Saji, T. ELECTROCHEMICALLY SWITCHED CATION BINDING IN PENTAOXA [13] FERROCENOPHANE. Chemistry Letters 1986, 15, 275–276. [Google Scholar] [CrossRef]
- Beer, P.D.; Sikanyika, H.; Blackburn, C.; McAleer, J.F.; Drew, M.G.B. Redox responsive crown ethers containing a direct link between the ferrocene redox-active centre and benzo crown ether. Crystal structure of a ferrocene benzo-15-crown-5 sodium complex. Journal of Organometallic Chemistry 1988, 356, C19–C22. [Google Scholar] [CrossRef]
- Beer, P.D.; Blackburn, C.; McAleer, J.F.; Sikanyika, H. Redox-responsive crown ethers containing a conjugated link between the ferrocene moiety and a benzo crown ether. Inorganic Chemistry 1990, 29, 378–381. [Google Scholar] [CrossRef]
- Jin, S.; Wang, D.; Jin, X.; Chen, G.Z. Intramolecular Electrostatics: Coulomb’s Law at Sub-Nanometers. ChemPhysChem 2004, 5, 1623–1629. [Google Scholar] [CrossRef]
- Hall, C.D. Macrocycles and Cryptands Containing the Ferrocene Unit. In Ferrocenes: homogeneolls catalysis, organic synthesis, materials science, Togni, A., Hayashi, T., Eds.; VCH: New York, USA, 1995; pp. 279–316. [Google Scholar]
- Hall, C.D.; Sharpe, N.W.; Danks, I.P.; Sang, Y.P. Cyclic voltammetry studies on the complexation of metal cations by cryptands containing the ferrocene unit. Journal of the Chemical Society, Chemical Communications 1989, 0, 419–421. [Google Scholar] [CrossRef]
- Dennis Hall, C.; Chu, S.Y.F. Cyclic voltammetry of cryptands and cryptates containing the ferrocene unit. Journal of Organometallic Chemistry 1995, 498, 221–228. [Google Scholar] [CrossRef]
- Medina, J.C.; Goodnow, T.T.; Rojas, M.T.; Atwood, J.L.; Lynn, B.C.; Kaifer, A.E.; Gokel, G.W. Ferrocenyl iron as a donor group for complexed silver in ferrocenyldimethyl[2.2]cryptand: a redox-switched receptor effective in water. J. Am. Chem. Soc. 1992, 114, 10583–10595. [Google Scholar] [CrossRef]
- Plenio, H.; Aberle, C. Oxaferrocene Cryptands as Efficient Molecular Switches for Alkali and Alkaline Earth Metal Ions. Organometallics 1997, 16, 5950–5957. [Google Scholar] [CrossRef]
- Beer, P.D.; Crowe, D.B.; Ogden, M.I.; Drew, M.G.B.; Main, B. Ammonium redox-responsive receptors containing multiple ferrocene and quinone redox-active centres attached to di- and tri-aza crown ether macrocycles. Journal of the Chemical Society, Dalton Transactions, 1993; 2107–2116. [Google Scholar] [CrossRef]
- Shi, L.; Song, W.; Li, Y.; Li, D.-W.; Swanick, K.N.; Ding, Z.; Long, Y.-T. A multi-channel sensor based on 8-hydroxyquinoline ferrocenoate for probing Hg(II) ion. Talanta 2011, 84, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-H.; Chen, F.-R.; Zhou, Y.-F.; Wang, J.-N.; Zhang, H.; Xu, J.-G. Enhanced fluorescence sensing of hydroxylated organotins by a boronic acid-linked Schiff base. Chemical Communications 2009, 4179–4181. [Google Scholar] [CrossRef]
- Zanello, P.; Cinquantini, A.; Fontani, M.; Giardiello, M.; Giorgi, G.; Landis, C.R.; Kimmich, B.F.M. Redox behavior of boronato-functionalized 1,1′-bis(diphenylphosphino)ferrocenes. Journal of Organometallic Chemistry 2001, 637-639, 800–804. [Google Scholar] [CrossRef]
- El Ghachtouli, S.; Cadiou, C.; Déchamps-Olivier, I.; Chuburu, F.; Aplincourt, M.; Turcry, V.; Le Baccon, M.; Handel, H. Spectroscopy and Redox Behaviour of Dicopper(II) and Dinickel(II) Complexes of Bis(cyclen) and Bis(cyclam) Ligands. European Journal of Inorganic Chemistry 2005, 2005, 2658–2668. [Google Scholar] [CrossRef]
- Mruthunjaya, A.K.V.; Torriero, A.A.J. Mechanistic Aspects of the Electrochemical Oxidation of Aliphatic Amines and Aniline Derivatives. Molecules 2023, 28, 471. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Belousoff, M.J.; Spiccia, L.; Bond, A.M.; Torriero, A.A.J. Macrocycles Bearing Ferrocenyl Pendants and their Electrochemical Properties upon Binding to Divalent Transition Metal Cations. ChemPlusChem 2018, 83, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Torriero, A.A.J.; Bond, A.M.; Spiccia, L. Fluorescent and Electrochemical Sensing of Polyphosphate Nucleotides by Ferrocene Functionalised with Two Zn(II)-(TACN)(pyrene) Complexes. Chemistry - A European Journal 2010, 16, 9154–9163. [Google Scholar] [CrossRef]
- Plenio, H.; Aberle, C. Synthesis of a ferrocene bridged cyclam: a new redox-active macrocycle and the structure of a nickel(II) complex with strongly coupled metal centers. Chemical Communications 1998, 2697–2698. [Google Scholar] [CrossRef]
- Plenio, H.; Aberle, C.; Al Shihadeh, Y.; Lloris, J.M.; Martínez-Máñez, R.; Pardo, T.; Soto, J. Ferrocene–Cyclam: A Redox-Active Macrocycle for the Complexation of Transition Metal Ions and a Study on the Influence of the Relative Permittivity on the Coulombic Interaction between Metal Cations. Chemistry – A European Journal 2001, 7, 2848–2861. [Google Scholar] [CrossRef] [PubMed]
- Tendero, M.J.L.; Benito, A.; Cano, J.; Lloris, J.M.; Martínez-Máñez, R.; Soto, J.; Edwards, A.J.; Raithby, P.R.; Rennie, M.A. Host molecules containing electroactive cavities obtained by the molecular assembly of redox-active ligands and metal ions. Journal of the Chemical Society, Chemical Communications, 1995; 1643–1644. [Google Scholar] [CrossRef]
- D. Beer, P.; K. Smith, D. Tunable bis(ferrocenyl) receptors for the solution-phase electrochemical sensing of transition-metal cations. Journal of the Chemical Society, Dalton Transactions 1998, 417–424. [Google Scholar] [CrossRef]
- De Santis, G.; Fabbrizzi, L.; Licchelli, M.; Pallavicini, P.; Perotti, A. A redox-switchable ligand for which the binding ability is enhanced by oxidation of its ferrocene unit. Journal of the Chemical Society, Dalton Transactions, 1992; 3283–3284. [Google Scholar] [CrossRef]
- Zapata, F.; Caballero, A.; Espinosa, A.; Tárraga, A.; Molina, P. A Simple but Effective Ferrocene Derivative as a Redox, Colorimetric, and Fluorescent Receptor for Highly Selective Recognition of Zn2+ Ions. Organic Letters 2007, 9, 2385–2388. [Google Scholar] [CrossRef]
- Nedunchezhian, K.; Nallathambi, S. Mono- and di-ferrocene conjugated 5-methyl benzimidazole based multi-channel receptors for cations/anions with their antimicrobial and anticancer studies. New Journal of Chemistry 2023, 47, 4656–4666. [Google Scholar] [CrossRef]
- Zhang, B.; Suo, Q.; Li, Q.; Hu, J.; Zhu, Y.; Gao, Y.; Wang, Y. Multiresponsive chemosensors based on ferrocenylimidazo[4,5-b]pyridines: Solvent-dependent selective dual sensing of Hg2+ and Pb2+. Tetrahedron 2022, 120, 132878. [Google Scholar] [CrossRef]
- Tian, H.-j.; Tang, R.-r.; Li, S.-f.; Luo, Y.-m. Synthesis, characterization and electrochemical recognition of metal ions of three new ferrocenyl derivatives containing pyridyl moiety. Journal of Central South University 2013, 20, 3379–3384. [Google Scholar] [CrossRef]
- Kaur, S.; Shalini, *!!! REPLACE !!!*; Ahmad Shiekh, B.; Kumar, V.; Kaur, I. Triazole-tethered naphthalimide-ferrocenyl-chalcone based voltammetric and potentiometric sensors for selective electrochemical quantification of Copper(II) ions. Journal of Electroanalytical Chemistry 2022, 905, 115966. [Google Scholar] [CrossRef]
- Arivazhagan, C.; Borthakur, R.; Ghosh, S. Ferrocene and Triazole-Appended Rhodamine Based Multisignaling Sensors for Hg2+ and Their Application in Live Cell Imaging. Organometallics 2015, 34, 1147–1155. [Google Scholar] [CrossRef]
- Lopez, J.L.; Tárraga, A.; Espinosa, A.; Velasco, M.D.; Molina, P.; Lloveras, V.; Vidal-Gancedo, J.; Rovira, C.; Veciana, J.; Evans, D.J.; et al. A New Multifunctional Ferrocenyl-Substituted Ferrocenophane Derivative: Optical and Electronic Properties and Selective Recognition of Mg2+ Ions. Chemistry – A European Journal 2004, 10, 1815–1826. [Google Scholar] [CrossRef]
- Otón, F.; Ratera, I.; Espinosa, A.; Wurtz, K.; Parella, T.; Tárraga, A.; Veciana, J.; Molina, P. Selective Metal-Cation Recognition by [2.2]Ferrocenophanes: The Cases of Zinc- and Lithium-Sensing. Chemistry – A European Journal 2010, 16, 1532–1542. [Google Scholar] [CrossRef]
- Otón, F.; González, M.d.C.; Espinosa, A.; Tárraga, A.; Molina, P. Synthesis, Structural Characterization, and Sensing Properties of Clickable Unsymmetrical 1,1′-Disubstituted Ferrocene–Triazole Derivatives. Organometallics 2012, 31, 2085–2096. [Google Scholar] [CrossRef]
- González, M.a.d.C.; Otón, F.; Orenes, R.A.; Espinosa, A.; Tárraga, A.; Molina, P. Ferrocene–Triazole–Pyrene Triads as Multichannel Heteroditopic Recognition Receptors for Anions, Cations and Ion Pairs. Organometallics 2014, 33, 2837–2852. [Google Scholar] [CrossRef]
- Sola, A.; Otón, F.; Espinosa, A.; Tárraga, A.; Molina, P. Aldimines generated from aza-Wittig reaction between bis(iminophosphoranes) derived from 1,1′-diazidoferrocene and aromatic or heteroaromatic aldehydes: electrochemical and optical behaviour towards metal cations. Dalton Transactions 2011, 40, 12548–12559. [Google Scholar] [CrossRef]
- Guo, L.; Yan, L.; Xie, R.; Han, L.; Zhu, N. Multi-channel sensing of trivalent metal ions using a simple ferrocenyl Schiff base probe with AIE property. Journal of Molecular Structure 2024, 1295, 136629. [Google Scholar] [CrossRef]
- Bhatta, S.R.; Bheemireddy, V.; Vijaykumar, G.; Thakur, A. Triazole appended mono and 1,1′ di-substituted ferrocene-naphthalene conjugates: Highly selective and sensitive multi-responsive probes for Hg(II). Sensors and Actuators B: Chemical 2017, 240, 640–650. [Google Scholar] [CrossRef]
- Bhatta, S.R.; Mondal, B.; Lima, S.; Thakur, A. Metal-coordination driven intramolecular twisting: a turn-on fluorescent-redox probe for Hg2+ ions through the interaction of ferrocene nonbonding orbitals and dibenzylidenehydrazine. Dalton Transactions 2019, 48, 8209–8220. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, S.R.; Pal, A.; Sarangi, U.K.; Thakur, A. Ferrocene appended fluorescein-based ratiomeric fluorescence and electrochemical chemosensor for Fe3+ and Hg2+ ions in aqueous media: Application in real samples analysis. Inorganica Chimica Acta 2019, 498, 119097. [Google Scholar] [CrossRef]
- Romero, T.; Orenes, R.A.; Tárraga, A.; Molina, P. Preparation, Structural Characterization, Electrochemistry, and Sensing Properties toward Anions and Cations of Ferrocene-Triazole Derivatives. Organometallics 2013, 32, 5740–5753. [Google Scholar] [CrossRef]
- Kamal, A.; Kumar, S.; Kumar, V.; Mahajan, R.K. Selective sensing ability of ferrocene appended quinoline-triazole derivative toward Fe (III) ions. Sensors and Actuators B: Chemical 2015, 221, 370–378. [Google Scholar] [CrossRef]
- Pandey, R.; Gupta, R.K.; Shahid, M.; Maiti, B.; Misra, A.; Pandey, D.S. Synthesis and Characterization of Electroactive Ferrocene Derivatives: Ferrocenylimidazoquinazoline as a Multichannel Chemosensor Selectively for Hg2+ and Pb2+ Ions in an Aqueous Environment. Inorganic Chemistry 2012, 51, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Kiran Kumar, C.; Trivedi, R.; Giribabu, L.; Niveditha, S.; Bhanuprakash, K.; Sridhar, B. Ferrocenyl pyrazoline based multichannel receptors for a simple and highly selective recognition of Hg2+ and Cu2+ ions. Journal of Organometallic Chemistry 2015, 780, 20–29. [Google Scholar] [CrossRef]
- Chen, L.; Cui, X.; Cheng, H.; Chen, X.; Song, M.; Tang, M.; Wei, D.; Wu, Y. Syntheses, structures of N-(substituted)-2-aza-[3]-ferrocenophanes and their application as redox sensor for Cu2+ ion. Applied Organometallic Chemistry 2012, 26, 449–454. [Google Scholar] [CrossRef]
- Krishnapriya, K.R.; Sampath, N.; Ponnuswamy, M.N.; Kandaswamy, M. Synthesis and electrochemical sensing behaviour of a new ferrocene functionalized tet ‘a’ macrocyclic receptor towards transition metal ions. Applied Organometallic Chemistry 2007, 21, 311–317. [Google Scholar] [CrossRef]
Scheme 1.
Square scheme showing coupled electron transfer and guest transfer to and from a ferrocene host molecule.
Scheme 1.
Square scheme showing coupled electron transfer and guest transfer to and from a ferrocene host molecule.
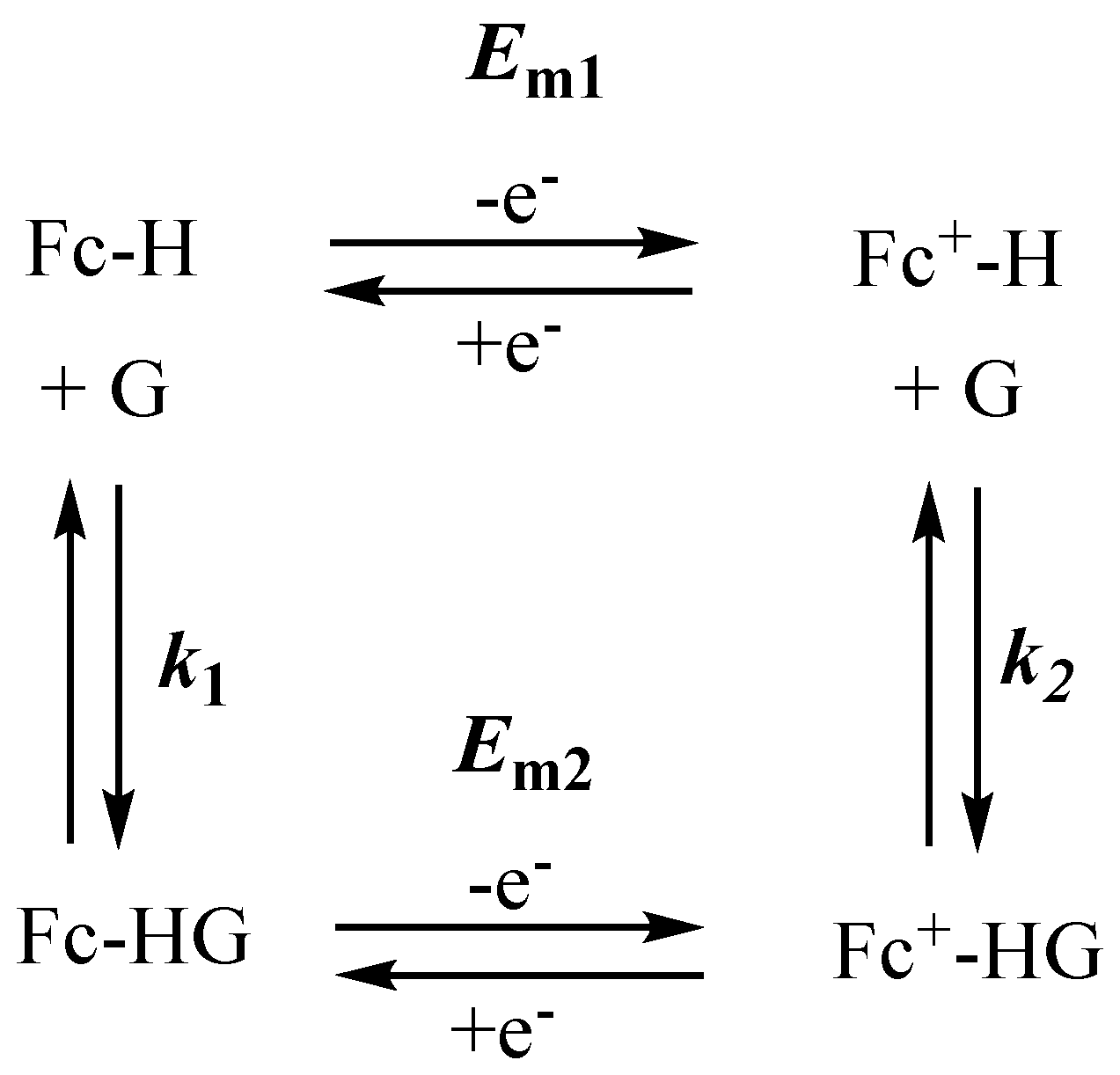
Scheme 2.
Schematic representation of the mechanism postulated for the oxidation of 20.
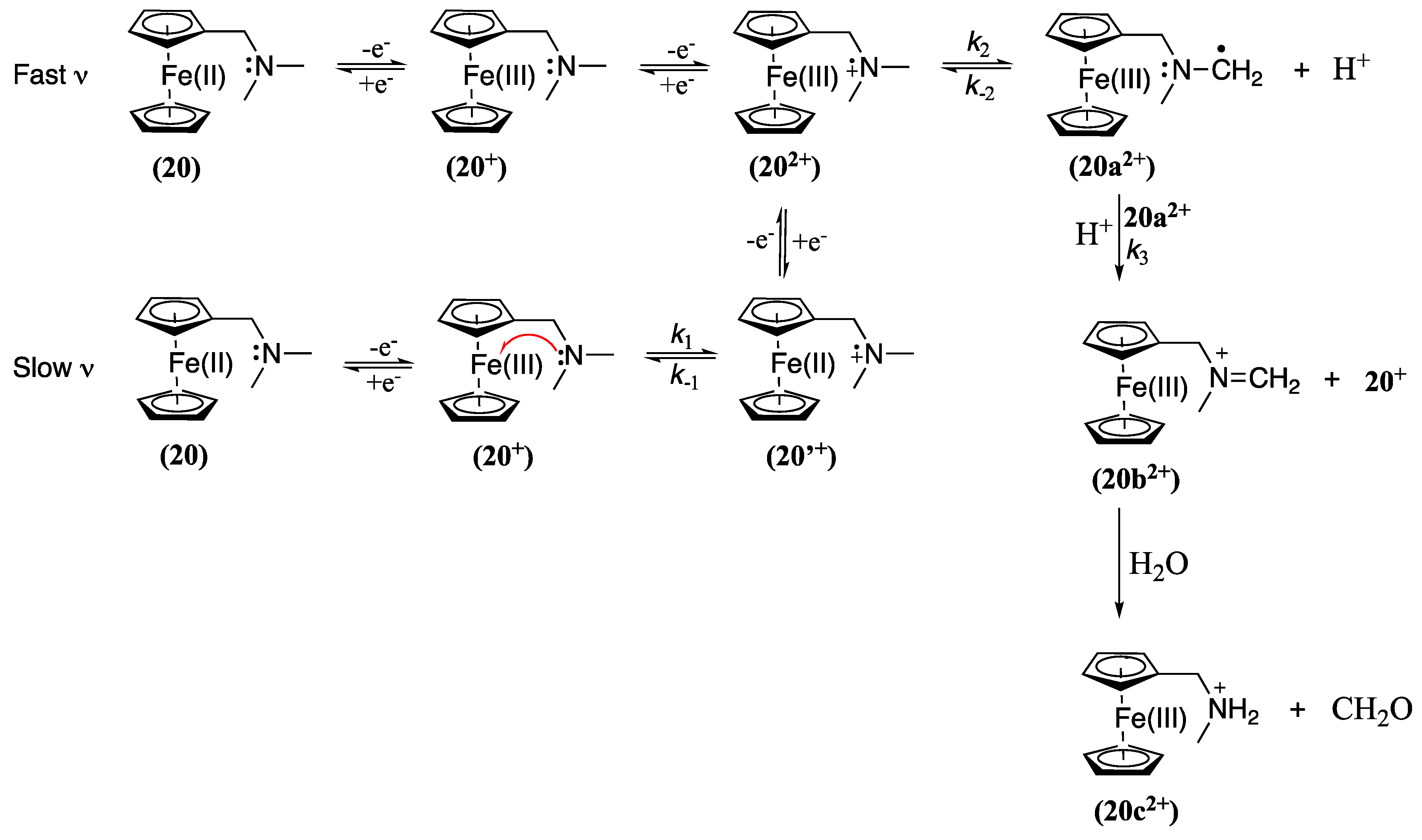
Table 1.
Redox potentials of Fc vs. DmFc0/+ IRRS in different organic solvents containing different supporting electrolytes.
Table 1.
Redox potentials of Fc vs. DmFc0/+ IRRS in different organic solvents containing different supporting electrolytes.
| Solvent | Electrolyte |
Em of Fc0/+ vs. DmFc0/+ (V) |
Ref. |
|---|---|---|---|
| 1,2-dibromoethane | 0.1 M [Bu4N][ClO4] | 0.475±0.007 | [13] |
| 1,2-dichloroethane | 0.1 M [Bu4N][ClO4] | 0.532±0.001 | [13] |
| 1,2-dichlorobenzene | 0.1 M [Bu4N][ClO4] | 0.535±0.001 | [13] |
| 2-propanol | 0.1 M [Bu4N][CF3SO3] | 0.455±0.003 | [13] |
| 2,2,2-trifluoroethanol | 0.1 M [Bu4N][ClO4] | 0.575±0.004 | [13] |
| Acetone | 0.1 M [Bu4N]Cl | 0.451±0.005 | [34] |
| 0.1 M [Bu4N][ClO4] | 0.479±0.004 | [13] | |
| 0.1 M [Bu4N][PF6] | 0.487±0.005 | [34] | |
| 0.1 M [Bu4N][TFAB] | 0.504±0.005 | [34] | |
| Acetonitrile (CH3CN) | 0.1 M [Bu4N]Cl | 0.501±0.005 | [34] |
| 0.1 M [Bu4N][ClO4] | 0.505±0.002 | [13] | |
| 0.1 M [Bu4N][PF6] | 0.509±0.003 | [35] | |
| 0.1 M [Bu4N][TFAB] | 0.517±0.005 | [34] | |
| Acetonitrile:dichloromethane (80:20) | 0.1 M [Bu4N][PF6] | 0.512±0.003 | [36] |
| Aniline | 0.1 M [Bu4N][ClO4] | 0.527±0.004 | [13] |
| Anisole | 0.1 M [Bu4N][PF6] | 0.518±0.005 | [34] |
| 0.1 M [Bu4N][TFAB] | 0.607±0.005 | [34] | |
| Benzonitrile | 0.1 M [Bu4N]Cl | 0.524±0.005 | [34] |
| 0.1 M [Bu4N][ClO4] | 0.523±0.001 | [13] | |
| 0.1 M [Bu4N][PF6] | 0.530±0.005 | [34] | |
| 0.1 M [Bu4N][TFAB] | 0.543±0.005 | [34] | |
| Benzyl alcohol | 0.1 M [Bu4N][ClO4] | 0.508±0.003 | [13] |
| Bromobenzene | 0.1 M [Bu4N][ClO4] | 0.489±0.005 | [13] |
| Chlorobenzene | 0.1 M [Bu4N][ClO4] | 0.497±0.001 | [13] |
| Chloroform | 0.1 M [Bu4N][ClO4] | 0.483±0.001 | [13] |
| Dichloromethane (DCM) | 0.1 M [Bu4N]Cl | 0.534±0.005 | [34] |
| 0.1 M [Bu4N][ClO4] | 0.532±0.002 | [13] | |
| 0.1 M [Bu4N][ClO4] | 0.570±0.002 | [2] | |
| 0.1 M [Bu4N][PF6] | 0.548±0.003 | [35] | |
| 0.1 M [Et4N][BF4] | 0.541±0.003 | [17] | |
| 0.1 M [Bu4N][TFAB] | 0.614±0.005 | [34] | |
| 0.1 M [C4mPyr][FAP] | 0.589±0.003 | [17] | |
| 0.1 M [C2mim][FAP] | 0.590±0.003 | [17] | |
| 0.1 M [C2mim][B(CN)4] | 0.588±0.003 | [17] | |
| 0.1 M [C4mim][NTf2] | 0.570±0.003 | [17] | |
| 0.1 M [C4mPyr][NTf2] | 0.568±0.003 | [17] | |
| 0.1 M [C2mim][FSI] | 0.569±0.003 | [17] | |
| 0.1 M [C3mim][FSI] | 0.568±0.003 | [17] | |
| 0.1 M [C4mPyr][N(CN)2] | 0.564±0.003 | [17] | |
| 0.1 M [C4mim][PF6] | 0.556±0.003 | [17] | |
| 0.1 M [C4mim][BF4] | 0.557±0.003 | [17] | |
| 0.1 M [C4mim][CF3SO3] | 0.556±0.003 | [17] | |
| Diethyl ether | 0.1 M [Bu4N][BArF24] | 0.550±0.005 | [34] |
| 0.1 M Na[BArF24] | 0.583±0.005 | [34] | |
| Dimethyl sulfoxide | 0.1 M [Bu4N][PF6] | 0.486±0.005 | [34] |
| 0.1 M [Bu4N][TFAB] | 0.493±0.005 | [34] | |
| 0.1 M [Bu4N][ClO4] | 0.468±0.001 | [13] | |
| Ethanol | 0.1 M [Bu4N][ClO4] | 0.473±0.005 | [13] |
| Formamide | 0.1 M [Bu4N][ClO4] | 0.510±0.003 | [13] |
| Methanol (MeOH) | 0.1 M [Bu4N][ClO4] | 0.497±0.002 | [13] |
| Nitrobenzene | 0.1 M [Bu4N][ClO4] | 0.514±0.002 | [13] |
| Nitromethane | 0.1 M [Bu4N]Cl | 0.505±0.005 | [34] |
| 0.1 M [Bu4N][ClO4] | 0.516±0.004 | [13] | |
| 0.1 M [Bu4N][PF6] | 0.510±0.005 | [34] | |
| 0.1 M [Bu4N][TFAB] | 0.516±0.005 | [34] | |
| N-methylformamide | 0.1 M [Bu4N][ClO4] | 0.510±0.002 | [13] |
| N,N-dimethylformamide (DMF) | 0.1 M [Bu4N]Cl | 0.475±0.005 | [34] |
| 0.1 M [Bu4N][ClO4] | 0.458±0.003 | [13] | |
| 0.1 M [Bu4N][PF6] | 0.478±0.005 | [34] | |
| 0.1 M [Bu4N][TFAB] | 0.493±0.005 | [34] | |
| N,N-dimethylacetamide | 0.1 M [Bu4N][ClO4] | 0.455±0.008 | [13] |
| Propylene carbonate | 0.1 M [Bu4N][ClO4] | 0.495±0.002 | [13] |
| Pyridine | 0.1 M [Bu4N][ClO4] | 0.517±0.004 | [13] |
| Tetrahydrofuran | 0.1 M [Bu4N][BF4] | 0.413±0.005 | [34] |
| 0.1 M [Bu4N][CF3SO3] | 0.438±0.005 | [34] | |
| 0.1 M [Bu4N][ClO4] | 0.423±0.005 | [34] | |
| 0.427±0.002 | [13] | ||
| 0.1 M [Bu4N][PF6] | 0.446±0.005 | [34] | |
| 0.1 M [Bu4N][BPh4] | 0.485±0.005 | [34] | |
| 0.1 M Na[BArF24] | 0.502±0.005 | [34] | |
| 0.1 M [Bu4N][TFAB] | 0.484±0.005 | [34] | |
| 0.1 M [Bu4N][BArF24] | 0.521±0.005 | [34] | |
| Toluene | 0.1 M [Bu4N][BF4] a | 0.430±0.005 | [34] |
Abbreviations: [Bu4N] = tetrabutylammonium; [PF6] = hexafluorophosphate; [ClO4] = perchlorate; [BF4] = tetrafluoroborate; [CF3SO3] = trifluoromethanesulfonate; [TFAB] = tetrakis(pentafluorophenyl)borate; [Et4N] = tetraethylammonium; [C4mPyr] = 1-butyl-1-methylpyrrolidinium; [C2mim] = 1-ethyl-3-methylimidazolium; [C4mim] = 1-butyl-3-methylimidazolium; [FAP] = tris(pentafluoroethyl)trifluorophosphate; [B(CN)4] = tetracyanoborate; [NTf2] = Bis(trifluoromethanesulfonyl)amide; [FSI] = bis(fluorosulfonyl)imide; [N(CN)2] = dicyanamide; [BArF24] = bis(trifluoromethyl)phenyl)borate; [BPh4] = Tetraphenylborate. a the toluene:[Bu4N][BF4] electrolyte is of the 3:1 stoichiometry.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



