
Submitted:
24 October 2023
Posted:
25 October 2023
You are already at the latest version
Abstract
Inflammation involves the activation of innate immune cells and is believed to play an important role in the development and progression of both infectious and non-infectious diseases such as neurodegeneration, rheumatoid arthritis or cancer. Inflammation in the brain is marked by the upregulation of the translocator protein (TSPO) in microglia. Moreover, high TSPO levels are found, for example in macrophages during rheumatoid arthritis and in malignant tumor cells compared to its relatively low physiological expression. The same applies for cycloxygenase-2 (COX-2), which is constitutively expressed in the kidney, brain, thymus and gastrointestinal tract, but induced in microglia, macrophages and synoviocytes during inflammation. This puts TSPO and COX-2 in the spotlight as important targets for the diagnosis of inflammation. Imaging modalities, such as positron emission tomography and single-photon emission tomography, can be used to localize inflammatory processes and to track their progression over time. They could also enable to monitor the efficacy of therapy and predict its outcome. This review focuses on the current development of PET and SPECT tracers not only for the detection of neuroinflammation, but also for emerging diagnostic measures in infectious and other non-infectious diseases like rheumatic arthritis, cancer, cardiac inflammation and in lung diseases.
Keywords:
Tracer development
; neuroinflammation
; pneumonia
; myocarditis
; translocator protein (TSPO)
; cyclooxygenase-2 (COX-2)
; positron emission tomography (PET)
; single-photon emission tomography (SPECT)
1. The Translocator protein (TSPO)
1.1. Introduction
The translocator protein (TSPO) 18 kDa was originally described as peripheral benzodiazepine receptor (PBR), a secondary binding site for diazepam, in order to differentiate it from the central benzodiazepine receptor (CBR), which constitutes a part of the GABAA receptor complex. The discovery of PBR in the central nervous system (CNS) (located in the ependymal and glial cells) proved that these receptors are not only be found in the periphery [1]. TSPO is a highly hydrophobic protein with five transmembrane helical domains located in the outer membrane of mitochondria [2,3,4,5]. It is best known for its role in cholesterol transport from the outer mitochondrial membrane to the inner membrane, where cholesterol is metabolized to pregnenolone, a product necessary for steroidogenesis. Consequently, TSPO is found in high concentrations in steroid-producing organs, the adrenal cortex, in testis and ovary, but also in heart and kidney [3,4]. It is important to note that TSPO is expressed at very low levels in resting microglia in the healthy brain [4].
The emerging interest in TSPO as a PET-imaging target is attributed to its pronounced upregulation during inflammation. In neuroinflammation, for instance, pro-inflammatory activation of the microglia upon acute injury or during progressive neurodegeneration has been observed and for a while now has been believed to contribute to dementia as a cause and a downstream effect [6,7,8,9]. The pathological changes seen in neurodegenerative diseases (NDD), such as abnormal accumulation of protein aggregates as found in Alzheimer’s disease (AD), dementia with Lewy bodies (DLB), frontotemporal dementia (FTD) or Parkinson syndrome have been linked to microglial and astroglial activation into proinflammatory states [9,10,11,12]. Immunohistochemical (IHC) staining together with PET-imaging have further confirmed the increased expression of TPSO in a variety of animal models with neuroinflammatory conditions as found in NDD and this colocalizes to reactive astroglia and activated microglia [7,9].
Despite limitations such as: high lipophilicity [13], result-confounding radiometabolites [14], and most importantly, intersubject variability, which is a consequence of the TSPO rs6971 single nucleotide polymorphism (SNP) [7,15] associated with existing TSPO PET-tracers [8], there are still ongoing efforts to develop novel PET tracers with optimal lipophilicity as well as diminished susceptibility to the rs6971 SNP [7]. Fortunately, the 4 domains needed for interactions of ligands with TSPO have already been discovered and they include one hydrogen-bond donor group and three major lipophilic regions [4]. Based on this motif several small-molecule-TSPO radiotracers have been synthesized. Moreover, PET studies revealed high TSPO binding in various dementias, pointing at the great potential of using TSPO as a biomarker for exploring the contribution of neuroinflammation in the pathogenesis of dementia and monitoring the progression of the [9] neurodegenerative disease. [9]. Interestingly, recent evidence suggests that increased neuronal activity is also accompanied by increased TPSO expression, which could provide insight into some disease processes [16]. Consequently, the detection of neuroinflammation through TSPO detection may not be cell type specific and may also involve non-inflammatory neuronal activity [17].
1.2. First generation of TSPO radiotracer
[11. C](R)PK11195
The first nonbenzodiazepine-type ligand discovered and most studied PET TSPO tracer worldwide (Figure 1) is a carbon-11 labeled isoquinoline carboxamide derivative identified in the early 1980s as a TSPO antagonist [18,19]. In vitro binding assays using [3H]PK11195 showed a Kd of 1.4 nM in rat brain tissue, and Kd ranging from 4.3-6.6 nM in human brain tissue [20].
Despite its high lipophilicity (log P 3.4) [13], it still remains the most widely used TSPO-radiotracer due to its apparent lack of susceptibility to the rs6971 SNP [21,22]. Another important shortcoming of the tracer is that it can only be radiolabeled with the short-lived carbon-11 (20 minutes half-life), which is only available in centers with an on-site cyclotron and fundamentally limits its usage in routine clinical practice. High plasma protein binding and non-specific binding, both consequences of its high lipophilicity, result in both poor brain-uptake and poor signal-to-noise ratio as observed in PET studies in healthy controls [4,6,23]. Moreover, its low BPND (non-displaceable binding potential) and low receptor affinity were also attributed to its high lipophilicity, which may also facilitate unspecific binding of the tracer in the periphery [23]. This was confirmed by comparative blocking experiments carried out in the same healthy controls. These studies revealed that [11C](R)PK11195 has a low BPND of 0.8, which is roughly corresponds to a low signal to noise ratio [9], which means that it does not have the sensitivity needed to detect mild neuroinflammation, and therefore [11C](R)PK11195 cannot be used for early diagnosis and the tracking of subtle changes in disease therapy.
So far, clinical studies carried out with [11C](R)PK11195 provided mixed results. Some initial studies reported high brain TSPO PET signal compared to controls in patients with AD, Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and those at risk of Huntington’s disease [10,24,25,26,27,28,29,30,31]. In early PD patients, the tracer detected activated microglia in the nigrostriatal brain regions. Additionally, it showed a difference in the distribution of microglia in PD and Lewy body dementia (LBD), which supported the conclusion that these diseases proceed differently based on nigrostriatal and cortical distribution of the activated microglia [11,32].
Figure 1.
Structures of [11C](R)PK11195) and second generation TSPO tracers.
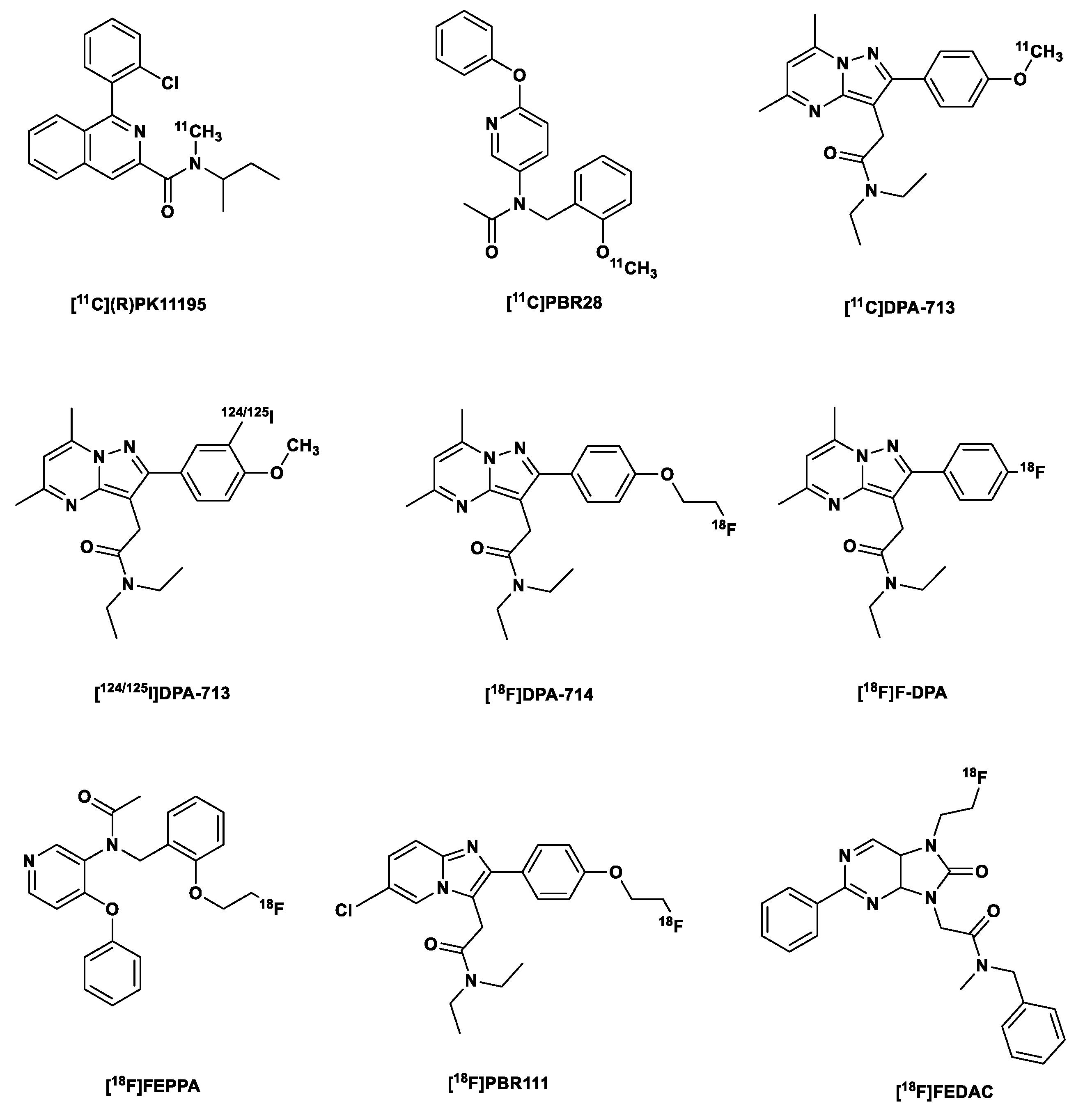
1.3. Second generation TSPO tracers
The high lipophilicity of [11C](R)PK11195 and the related limitations mentioned already led to the development of a second generation of TSPO tracers, which include but are not limited to the following: the phenoxyarylacetamide derivatives ([11C]PBR28, [18F]FEPPA, [11C]DAA1106), pyrazolopyrimidines [11C]DPA-713, [123I]/[124I]/[125I]DPA-713, [18F]DPA-714 (PBR099), and [18F]F-DPA (Figure 1) [33] most of these have been studied in humans, especially in mild cognitive impairment (MCI) and AD patients, and in patients with Parkinson syndrome [6,10,11].
Reduced lipophilicity accompanied by improved signal-to-noise ratio was observed with this generation of TSPO tracers which facilitated the discovery of the human rs6971 SNP. The rs6971 SNP results in a non-conservative substitution at the 147th amino acid of TSPO, where alanine is exchanged for threonine (A147T). Codominant expression of this genetic trait results in three genotypes: low (LAB), mixed (MAB) and high affinity binders (HAB) as was discovered with [11C]PBR28 and [11C]DPA-713. HAB subjects are homozygous (A/A) for wild-type (WT) TSPO, while the MAB and LAB are heterozygous (A/T) and homozygous (T/T) respectively for the A147T TSPO [7,15]. The tracers bound with low affinity in approximately 5-25% of human donors (LAB), around 50-65% in the HAB, and about 30% in MAB. In the MAB group, some ligands showed a two-site binding affinity while others displayed a Ki between that of the HAB and LAB [7,34,35,36].
For [11C]PBR28, in which this feature was first discovered, the difference between HAB and LAB was up to 40-fold. The polymorphism had such a huge effect on the binding of [11C]PBR28 that subjects who were homozygous LAB showed no detectable brain signals from the tracer [9]. The SNP polymorphism might not have been detected with [11C](R)PK11195 due to its low specific binding in the brain, however, the sensitivity to SNP rs6971was observed in peripheral organs with much higher expressions of TSPO such as the heart and the lung [21,22].
The discovery of the rs6971 SNP in the TSPO gene in the second generation TSPO led to a change in the design of clinical experimentation, with studies performed based on genotype tests, or with the exclusion of LAB which comprise ~10% of the population [6,7,9,14]. These new paradigms led to more consistent findings in studies of neuroinflammation in AD patients, based on increased tracer retention than with the first-generation tracer [6]. Moreover, in comparison to controls there were consistent higher TSPO PET brain signals in patients with AD, ALS, MCI, PD and multiple sclerosis (MS) [6,7,37]. Full-blockade (receptor/transporter) studies conducted in monkeys in another study showed that the evaluated second generation TSPO radiotracers have a higher specific binding than [11C](R)PK11195. For example, [11C]PBR28 showed a specific binding 80-fold higher than that of [11C](R)PK11195 in vivo in monkey brain [21]. Further studies with [11C]PBR28 and other TSPO tracers in healthy subjects based on baseline and blocking studies revealed the BPND of the TSPO tracers in HAB. [11C]DPA-713 showed the best BPND of 7.3, followed by the third generation TSPO tracer, [11C]ER176 with BPND 4.2, and [11C](R)PK11195 with BPND 0.75, which was 1.5 times less than that of [11C]PBR28 with a BPND of 1.2 [9,14,22].
Compared to [11C](R)PK11195, [11C]DPA-713 not only provided the highest signal to background ratio in HAB, but also showed an accumulation of radiometabolites in the brain, which was consistent with increased distribution volume (VT). Although brain radiometabolites usually are detrimental to the quantification of PET images, in this case, their effect was diluted due to the high specific binding of [11C]DPA-713 in HAB and MAB, and further mollified by the relatively low concentration of the brain radiometabolites (compared to the general brain uptake). The effects of the brain radiometabolites, however, cannot be ignored in the case of LAB, for whom this could pose significant impediment [22], since there are barely any specific signals in the first place.
In a PET study using the Herpes Simplex encephalitis virus-1 (HSV-1) rat model, the fluoroethylated analog of [11C]DPA-713, that is [18F]DPA-714 showed lower non-specific binding than [11C](R)PK11195, but slightly lower specific binding than its predecessor [11C]DPA-713 in the infected rat brain. To determine specific binding, the average tissue/plasma ratio of the rat model pretreated with non-radioactive PK11195 was subtracted from the tissue/plasma ratio of the control and rat models, for each rat separately. Nevertheless, its brain uptake was lower than that of [11C]DPA-713 and [11C](R)PK11195 in both, healthy and infected brain tissues, but with comparable specific uptake to [11C](R)PK11195. Despite its relatively low performance, [18F]DPA-714 was found to be an agonist at the TSPO receptor, which might take advantage of the high affinity state of TSPO resulting in improved binding of the tracer to TSPO in chronically activated microglia cells during progressive neurodegeneration [12].
[18F]F-DPA like [18F]DPA-714 is another interesting fluorine-18 labeled pyrazolopyrimidine tracer but unlike the latter, the fluorine atom is bound to a Csp2 aromatic carbon. The tracer was discovered as early as 2001 by Selleri et al. Its affinity to TSPO (1.7 nM) and selectivity over CBR (> 1 μM) in a competition binding experiment was comparable to that of its fluoroethylated analog [18F]DPA-714 which were 0.91 nM and > 1 μM respectively [38].
Until recently, the radiosynthesis of [18F]F-DPA has been fraught with difficulties, which resulted to low radiochemical yields (RCY) and low molar activity (Am). Radiosynthesis via [18F]fluoride resulted in <3% RCY probably owing to lack of ring activator. Keller et al. [38] were able to improve RCY via electrophilic radiofluorination via carrier-added [18F]F2 but improved RCY (15%) was accompanied by a low Am (7.8 GBq/μmol). Wang et al. [39] were able to obtain the tracer in higher RCY (45%) using via [18F]fluoride by using an optimized spirocyclic iodonium ylide (SCIDY) precursor. Am was also higher (96 GBq/μmol) compared that obtained by Keller et al [38]. Automation of synthesis [40] further improved RCY to 15.6% (non-decay corrected), with >5-fold increased Am. Further optimization of synthesis conditions using the same precursor resulted in up to almost 20% RCY [41]. Although, [18F]DPA-714 obtained with low Am was still demonstrated the ability to distinguish between healthy and AD transgenic mice [41], it might not be able to detect subtle changes in certain cases.
[18F]F-DPA displayed saturable specific binding to TSPO in rodent models of neuroinflammation (AD and cerebral ischemia). Around 1.2 – 1.6-fold higher brain uptake of the tracer was seen in AD mice compared to age-matched controls. It also demonstrated high uptake into the ischemic brain hemisphere in rats with focal cerebral ischemia. Uptake was diminished (by 80%) by co-administration with PK11195 (3 mg/kg), which indicated specific in vivo uptake and low non-specific binding. Specific binding in autoradiography in vitro was also determined by co-incubation with PK11195. In a simplified reference tissue model BPND was determined to be 2.5-fold higher than that of [11C]PK11195 [39]. Compared to [18F]DPA-714, [18F]F-DPA displayed higher in vivo stability in the plasma (2.5-fold) at 90 min post injection (p.i). At the same time point, the brain content of the unchanged [18F]F-DPA was 1.7-fold higher compared to [18F]DPA-714 in healthy rats [38]. This was expected since the aryl-18F bond present in [18F]F-DPA is stronger than the Csp3-18F bond in [18F]DPA-714.
López-Picón et al. [42] demonstrated in comparative PET studies that [18F]F-DPA might be a better tracer than [18F]DPA-714 and [11C]PBR28 in detecting low levels of inflammation. Compared to the other two tracers, it displayed higher brain uptake and washout as well as higher transgenic-to-wild-type standardized uptake value ratios (SUVR) to the SUVR obtained from the cerebellum.
1.4. Third generation TSPO tracers
Even though genotypic stratification proved helpful in clinical studies with second generation tracers, there was still the need to develop PET TSPO tracers, which do not discriminate between the genotypes, i.e. between wild type and A147T mutations.
With this in mind, Zanotti-Fregonara et al. developed [11C]ER176, a quinazoline analog of [11C](R)PK11195, (Figure 2) with very diminished sensitivity to the human SNP rs6971 [5,43], and a BPND ratio in HAB and LAB of 1.3:1. This affinity ratio was even much higher for [11C]PBR28 at 55:1 [43]. This means that LAB do not need to be further excluded in clinical studies with this tracer, however, due to a slight sensitivity to the genotype, the VT values have to be corrected for genotype a posteriori [14]. Another unique advantage of [11C]ER176 over the other TSPO PET tracers apart from having the second highest BPND of 4.2 after [11C]DPA-713, was the absence of brain radiometabolites, which resulted in more time-stable values of VT in both HABs and LABs after the blockade of TSPO receptors by XBD173 [14]. So far, clinical studies have been carried out only in healthy subjects, hence there is still a need to perform these studies in patients with neuroinflammation [7].
In addition to the already mentioned properties, [11C]ER176 showed a higher affinity (3.1 times) in rat kidney mitochondrial membranes. Moreover, it exhibits less lipophilicity (1.3 times) in comparison to its direct analog [11C](R)PK11195 [5], which resulted in higher unbound fractions in the plasma, improved brain uptake and PET signal (in the monkey brain), and higher specific binding confirmed by up to 80% signal blockade after the administration of cold (R)PK11195 [14]. There is an ongoing clinical study, which aims at assessing the usefulness of [11C]ER176 for the accurate quantification of microglial activation in patients with AD [44].
To make up for the short half-life of the carbon-11 labeled [11C]ER176, its fluorine-18 labeled analog, [18F]BIBD-239 was developed by Chen et al [45]. [18F]BIBD-239 (IC50 5.24 nM) showed slightly improved affinity for TSPO compared to [11C]ER176 (IC50 5.94 nM). [18F]BIBD-239 also showed similar binding modes and sites to Ala147-TSPO andThr147-TSPO in theoretical simulation, a characteristic, which suggests low sensitivity to the rs6971 polymorphism. It also showed nearly 2 %ID/g in an ICR mouse model, but was able to clearly detect focal cerebral ischemia in SD rats with mild focal ischemia in PET studies in vivo. In vitro autoradiography experiments with the ischemic rat brain correlated with the PET studies. Clear tumor PET images with [18F]BIBD-239 were also obtained in a GL261 mouse model. Additionally, at 60 min p.i. no metabolites of [18F]BIBD-239 were found in the brain even though the biotransformation of the tracer was fast in the periphery as indicated by low plasma content of the tracer at 60 min p.i..
The [18F]fluorine-labeled TSPO tracer flutriciclamide [18F]GE-180, a tricyclic indole derivative discovered by Wadsworth et al., also belongs to this tracer generation (Figure 2). The S-enantiomer displayed superior properties in comparison to the D-enantiomers in terms of binding affinity and pharmacokinetics, with 4.4-fold affinity to the target and a faster clearance from the striatum (a low expressing region), and a relatively high percentage of the parent tracer (94 %) in the brain at 60 min p.i. [46]. Flutriciclamide also showed better imaging characteristics in compared to [11C](R)PK11195 and [18F]fluorine-labeled DPA-713, that is [18F]DPA-714, in preclinical studies [47], but failed to show the extent of microglia/macrophage activation as accurately as [11C]DPA-713, which also afforded earlier detection of inflammation [48].
In healthy subjects, however, [18F]GE-180 showed poor imaging properties with a very low brain uptake, an almost flat time activity curve, and due to high uptake in blood vessels kinetic modeling was difficult [49,50,51]. In comparison to [11C]PBR28, it showed a 20-fold lower VT, in spite of its higher plasma concentration of [18F]GE-180 owing to its low brain uptake [50]. Zanotti-Fregonara et al. pointed out that this was at odds with the high lesion-to-background ratio observed in brain tumors and MS lesion sites, which suggests that the disruption of the BBB, which is a hallmark in these pathologies, likely facilitated the brain entry of both the tracer and its radiometabolites. This was supported by gadolinium uptake especially into MS brain tissue. This means that the signals observed were nonspecific [52]. Albert et al. countered that the high uptake of the tracer in MS lesions, glioma and in other neurological diseases correlates with disease severity and outweighs its underperformance in healthy subjects, for whom the tracer was not developed [53]. A prior study carried out by Sridharan et al. had shown that the TSPO ligand XBD173 competitively displaced [18F]GE-180 in brain tissue of MS patients, hence the uptake into MS lesions might be specific after all [53,54].
Another important criticism of the tracer was referring to the proposed in vivo insensitivity to TSPO polymorphism, which was surprisingly owing to a 15:1 affinity ratio in vitro between HAB and LAB, which was less than what was displayed by [11C]DPA-713. It was argued by Zanotti-Fregonara et al. that the poor image quality of the PET, a consequence of low brain uptake, made such a glaring difference undetectable in vivo [34]. In response, Albert et al. contended that lack of allelic discrimination in vivo should be a sought-after quality for a TSPO tracer but not the reverse. However, further experiments need to be carried out in order to generally understand allelic dependence on affinity of TSPO tracers [53]. Also, further experiments will be carried out on brain tumor patients in order to spatially compare [18F]GE-180-PET with histopathological analysis of tissue samples from stereotactic biopsies [53].
A study by López-Picón et al. determined the longitudinal relationship between the deposition of Aβ and neuroinflammation in the APP23 AD mouse model with both the TSPO-tracer and the Aβ tracer [11C]PIB. It was observed that there was a clear age-dependent increase of [11C]PIB with increased β-amyloid aggregation in the frontal cortex (FC), parietotemporal cortex (PTC), thalamus and hippocampus [43]. Like [11C]PIB, [18F]GE-180 convincingly correlated neuroinflammation and Aβ deposition in the PVC and thalamus, but [18F]GE-180 displayed minimal binding in areas of early amyloid deposition (FC and hippocampus). Moreover, the binding of [18F]GE-180 reached a plateau earlier in the pathogenesis of the AD in contrast to [11C]PIB. This is an indication that [18F]GE-180 might only be a useful tracer for early detection of pathological neuroinflammation in AD, but not useful for the long-term tracking of disease progression [55].
A more recent longitudinal PET study (supported by autoradiography) carried out by Holzgreve et al. [56] with [18F]GE-180 in an orthotopic syngeneic GL261 glioblastoma (GBM) mouse model demonstrated a continuous uptake of the tracer over time, which overlapped with contrast-enhancement in CT and tissue-established observations. Therefore, it is concluded that the tracer might prove useful for the imaging of brain tumors.
An analog of [18F]GE-180 was recently developed by Qiao, et al. [2], by exchanging one of the ethyl substituents on the amide nitrogen with a butyl substituent. Varying the substituents on this functional group is believed to influence the HAB/LAB ratio. Like its predecessor [18F]GE-180, the S-enantiomer based on a rat heart TSPO assay showed a higher binding affinity (1.04 nM) than the R-enantiomer (21-fold lesser).
[18F](S)GE-387 (Figure 2) showed little loss in affinity to A147T TSPO when evaluated for sensitivity to TSPO polymorphism in an assay using human embryonic kidney cell lines, with a LAB/HAB ratio of 1.3, that is comparable to [11C](R)PK11195 (1.2). A further in vivo PET analysis in healthy rats showed a modest distribution of the racemic mixture of both enantiomers of the tracer in the brain, hence, there is a possibility that in pathology there might be a higher uptake of the tracer. Further, biological evaluation is still being carried on both enantiomers of the tracer [2].
The efficacy of [18F](S)GE-387 as a useful TPSO tracer was additionally evaluated by Ramakrishnan et al. [57] in a lipopolysaccharide (LPS)-induced (10 µg/4 µL) neuroinflammation rat model corroborated the claims by Qiao, et al. [2]. They observed that the tracer can discern inflamed from healthy brain regions three days after the injection of LPS in their brains. Likewise, they confirmed low sensitivity of the tracer to the TPSO rs6971 polymorphism in genotyped human brain tissue.
Other recently reported third generation TPSO tracers include [18F]CB251 and [18F]BS224. The former is a chemically-modified version of the second generation tracer [18F]PBR111. [18F]PBR111 exhibited specific binding to TSPO, which facilitates both in vivo visualization and quantification of neuroinflammation [58,59]. Unfortunately, it is sensitive to the TPSO polymorphism [59]. [18F]CB251 on the other hand, in addition to showing high cellular uptake in modified cells and LPS-induced neuroinflammation mice showed comparably low sensitivity to the rs6971 polymorphism and based on PET/MRI studies sensitivity to changes in the severity of neuroinflammation [60].
[18F]BS224 is a direct analog of the [18F]CB251 (Figure 2), with the [18F]fluoroethyl group exchanged for a Csp2[18F]fluorine atom, that is, directly bound to the benzene ring. This was done in a bid to reduce in vivo defluorination observed in the fluoroethylated [18F]CB251. In vitro competitive inhibition assays with membrane proteins showed that [18F]BS224 has low sensitivity to the rs6971 polymorphism with an almost equal affinity to HAB (IC50 0.59 nM) and LAB (IC50 0.45 nM): LAB/HAB ratio 0.76. Moreover, the LAB/HAB ratio of [18F]BS224 was comparable to that of [11C]PK11195 (0.83), which is believed to be insensitive to the polymoprphism. Although the fluoroethylated [18F]CB251 showed an overall higher affinity to both LAB and HAB, the LAB/HAB ratio was nonetheless higher (1.14), making it in regard to the former two the worst in this regard. Moreover, [18F]BS224 in rats models of LPS-induced inflammatory and ischemic stroke clearly labeled inflammatory lesions with a high BPND = 1.43 ± 0.17 and 1.57 ± 0.37, respectively [61].
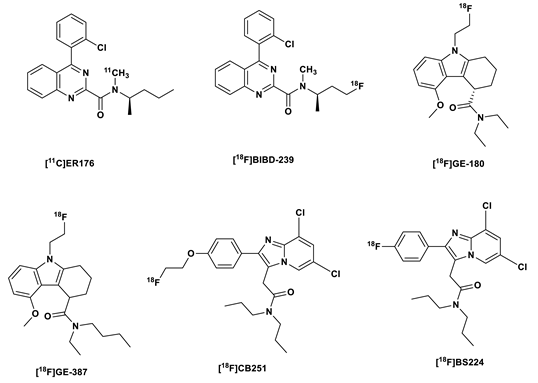
There has been marked improvement in the susceptibility of the TSPO tracers to SNP from the second to the more recent generation of tracers, however, this is still remains a factor that has to be considered in the further development of TSPO tracers. Therefore, efforts are still being put forward to better understand the difference in the binding requirements to both wild type and the A147T variant [62,63,64] by carrying out high throughput screening of ligands [65], identification of other binding sites in the TSPO protein [66], understanding in vivo kinetics and physiological modulators of binding, and brain penetration of radiometabolites, especially, the effects of these radiometabolites on the PET signals in LAB [14,67].
2. The Cyclooxygenase-2 (COX-2) enzyme
2.1. Introduction
Cyclooxygenase-2 is one of the two COX isozymes also known as prostaglandin-endoperoxide synthase (PTGS) [68], which is induced in response to inflammation and pain in the body [69,70,71]. The other isoform of the enzyme, COX-1, is predominantly expressed constitutively and plays a role in the maintenance of organ homeostasis including gastric cytoprotection, and maintenance of renal function [72,73,74,75]. Recent findings suggested that COX-2 is also constitutively expressed in the kidney where it plays a role in modulation of vascular tone in addition to regulation of hydric balance [75]. While constitutive COX-2 mRNA expression can be detected in the gut and kidney, high COX-2 mRNA levels are found in the normal (rat) brain [76]; in which it was also discovered that strength and endurance training increases the expression of COX-2, which is involved in neuronal plasticity and learning [77].
Recently, however, Shrestha et al. observed a lack of displaceable binding of [11C]MC1 (IC50 COX-2 3 nM; selectivity over COX-1 > 3000-fold [78]) to COX-2 in monkey brain at baseline, whereas specific binding was observed in the ovary [74], which corroborated previous results obtained by Min-Jeong et al. [79]. These findings suggested that COX-2 might not be expressed in a healthy human brain at all, which makes it a distinguished target for the evaluation of inflammatory processes in the brain.
COX-2 is present intracellularly on the luminal side of the smooth endoplasmic reticulum, Golgi apparatus and the nuclear membrane [80,81,82], with its active site located on the membrane bound portion of the enzyme. This is a constraint in terms of drug delivery, since only highly lipophilic ligands could transverse both the cell and the organelle membranes. Even so, very lipophilic ligands are known to bind non-specifically, and suffer from slower clearance, hence longer waiting times are needed for the ligands (radiotracers) to be cleared from non-target tissues in order to obtain better target-to-background ratios [83,84].
As already discussed for the TSPO 18 kDa protein, COX-2 expression is upregulated during pathology, both in acute and chronic inflammatory conditions. However, neuroinflammatory responses to pathological stimuli differs: the upregulation of COX-2 occurs quicker but transient, with a return to baseline in a matter of hours [74]; in contrast, TSPO upregulation could take several days and may last for weeks [85]. Moreover, the location of COX-2 after induction is largely restricted to neurons and brain endothelial cells [70,86] whereas TSPO is minimally upregulated in neurons, but highly in microglia and astroglia. This transient upregulation of COX-2 expression in neuroinflammatory responses might allow the usage of COX-2 tracers for the measurement of acute as well as chronic inflammation, conversely TSPO tracers will be useful to image chronic inflammatory conditions as could be found in NDDs [74].
Moreover, many premalignant neoplasms are marked by the overexpression of COX-2 [81], with elevated upregulation in a variety of cancers such as breast, colorectal and gastric cancers [72,81,83]. It also plays a role as a biomarker and effector enzyme in neural damage both after brain trauma, and in the ageing brain and associated pathological conditions [81], for example, in NDDs like PD [87] and AD.
The molecular mechanisms underlying COX-2 expression in certain types of cancers and inflammation have been widely investigated. However, there still remains a controversy regarding the exact role of COX-2 in NDDs and carcinogenesis, which is further aggravated by the discrepancies between in the anti-cancer effects of some COX-2 inhibitors in vitro and their lack of therapeutic efficacy in vivo. With an effective tracer for in vivo imaging it will be possible to understand the elaborate role of COX-2 in the pathogenesis of various diseases, and to confirm or disprove hypotheses regarding its contribution and regulation in different pathologies [72,81,83].
So far, several COX-2 imaging agents have been synthesized both for PET and SPECT imaging, with only [11C]MC1 making it so far to clinical trials [75]. The fact that the evaluation of COX-2 expression is only possible via elaborate ex vivo analyses makes the development of tracers for this enzyme quite challenging, for instance, COX-2 mRNA and protein are unstable in vitro and undergo quick degradation [72].
In this review, some interesting carbon-11 and fluorine-18 labeled COX-2 PET tracers together with some Iodine-125 & 123 and Tc-99m-labeled SPECT tracers are discussed.
2.1. PET COX-2 radiotracers
2.1.1. [11C]Carbon labeled COX-2 tracers
Earlier in the development of [11C]carbon-labeled COX-2 tracers, mostly FDA approved COX inhibitors were used, even though among these celecoxib is the only COX-2 inhibitor still approved for use in clinical practice. Rofecoxib was withdrawn from the market due to increased cardiovascular risks after long-term use of high doses [88]. The latter of course does not constitute a problem since radiotracers are used short term and at sub-therapeutic concentrations with insignificant pharmacological effects [72]. These COX-inhibitors were radiolabeled to enable a smooth transition into clinical practice and to facilitate the translation of data obtained via PET studies into data of clinical relevance.
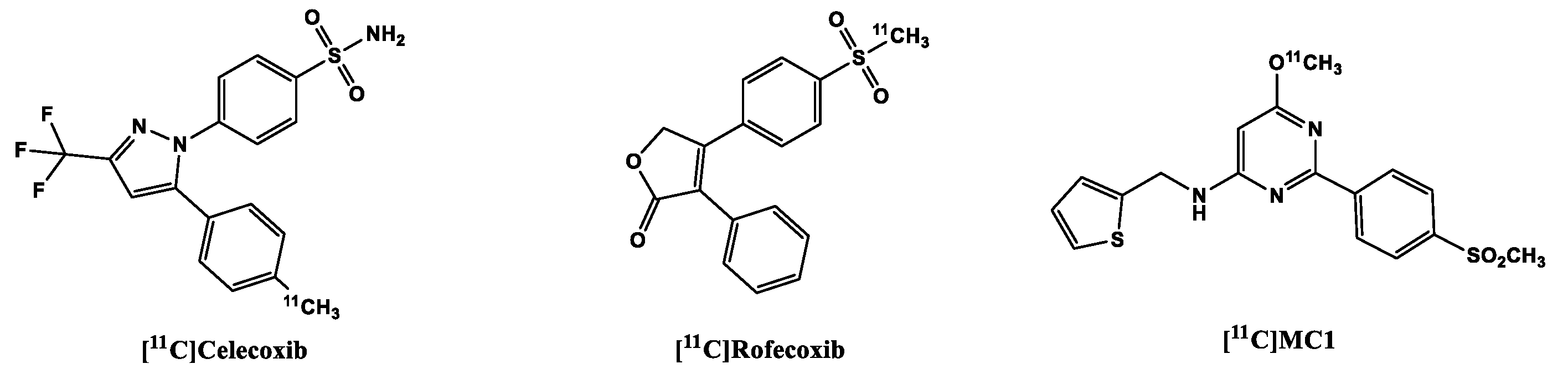
Celecoxib (Figure 3) is a COX-2 inhibitor currently still applied in clinical practice [89]) and its analogs were deemed lead structures for the development of COX-2radiotracers. In an experiment to evaluate drug transport mechanisms in biliary excretion, carbon-11 labeled celecoxib (Ki COX-2 40 nM; selectivity over COX-1 425-fold) [90] was found to have low in vivo stability. It was quickly metabolized to carboxylic acid and hydroxymethyl-containing products [91], and due to the presence of the sulfonamide functional group present in the molecule there was high blood retention of the tracer. It is believed that although the binding site of carbonic anhydrase (CA) and COX-2 have similar shapes, the presence of zinc in the catalytic site of the former increases its affinity to the sulfonamide derivatives, due to perhaps the strong bond between the zinc-containing catalytic site and the primary amine in the sulfonamide moiety [92]. Other derivatives of Celecoxib were disqualified as in vivo imaging of COX-2 due to high unspecific binding as, for example, seen in tracers developed by Fujisaki, et al. [93] and suboptimal affinity to the target as seen in the tracers developed by Gao et al., probably due to the compromise of the sulfonamide group, a functional group necessary for affinity to the target [94].
[11C]carbon-labeled rofecoxib (Figure 3) with Ki COX-2 18-44.6 nM and Ki COX-1 50000 nM performed slightly better than the celecoxib tracers, but not in all experiments: rofecoxib showed specific binding in healthy rat brains, but unfortunately the same result could not be replicated in inflammation models, a failure partly blamed on the inadequacy of the models, especially the sterile inflammation model [68,95].
Unlike the other already-mentioned COX-2 tracers, [11C]MC1 (Figure 4) contains a 6-membered heterocyclic pyrimidine ring in the centre of the molecule instead of a 5-membered ring and a sulfone moiety instead of the CA-susceptible sulfonamide group. It showed good brain uptake in a monkey brain with radioactivity peaking in brain at 2.9 SUV at 2 min post injection (p.i.) that quickly reduced to 1.16 SUV at 40 min p.i. [78]. Subsequent examination of the tracer in LPS-treated rhesus macaques (LPS injected in their brains), two patients with rheumatoid arthritis as well as two healthy participants revealed that [11C]MC1 can be used to evaluate inflammation both in the brain and in the periphery [74]. This makes it the first PET radioligand that was able to successfully image and quantify COX-2 expression/regulation in vivo. Recruitment for clinical trials with this ligand for PET Imaging of cyclooxygenases in dementia, rheumatoid arthritis and myositis are ongoing [96].
Figure 3.
Structure of 11C-labeled COX-2 tracers.
2.1.2. [18F]Fluorine-labeled COX-2 tracers
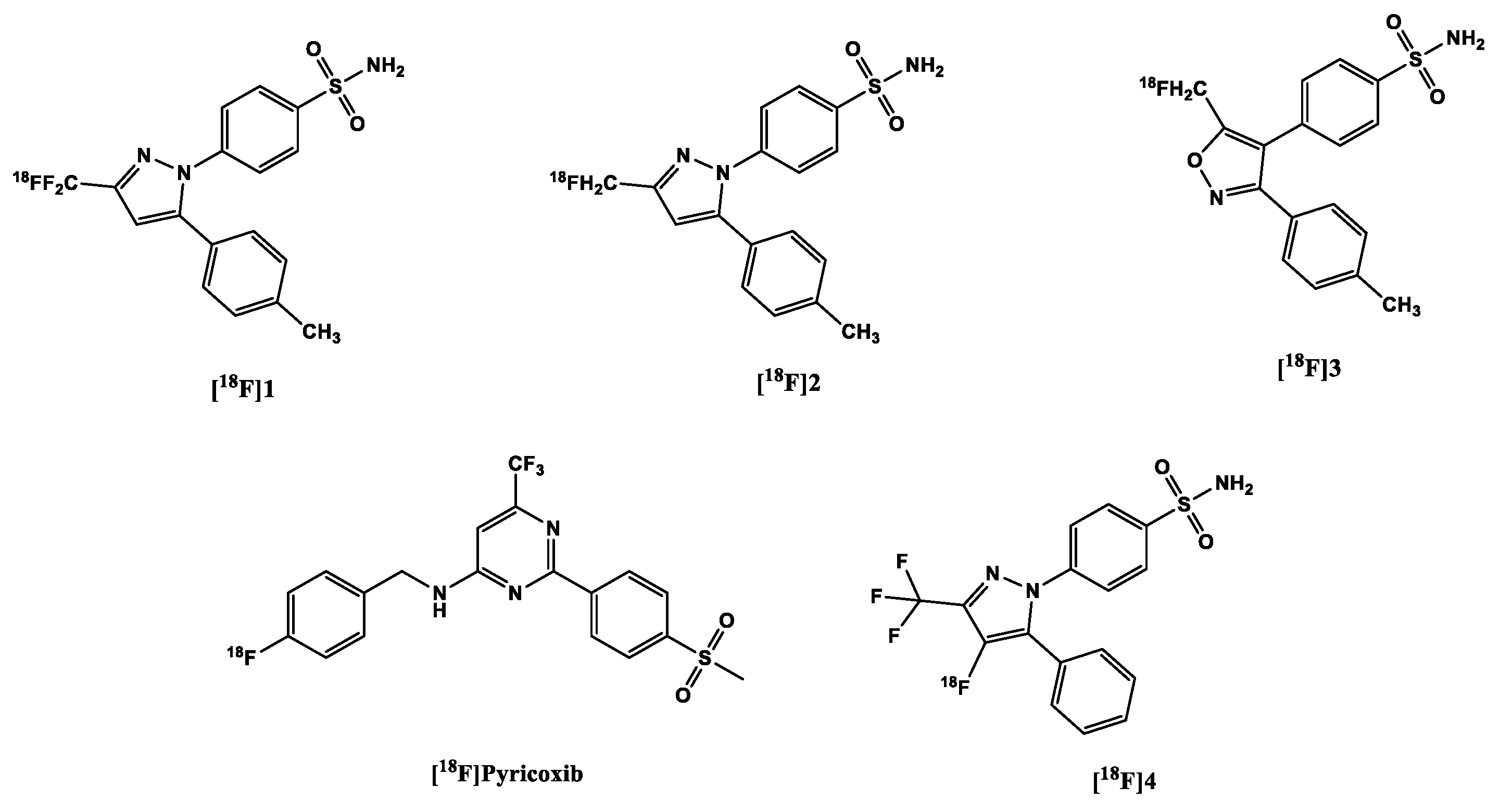
As with the [11C]carbon-labeled COX-2 tracers, the aim was to label COX-2 inhibitors already used in clinical practice, without structural alteration. Hence, celecoxib was radiofluorinated to obtain a [18F]trifluoromethyl analog [18F]1 (Figure 4) via nucleophilic substitution reaction (SNAr) using a bromodifluoromethyl precursor by Prabhakaran et al. [90]. PET scans in rats and baboons showed a high in vivo instability of the tracer partly due to high defluorination confirmed by the high bone uptake compared to the heart and brain in rats. Although defluorination was slower in baboons, there remained only 17% of the parent tracer in the plasma at 60 min p.i.. Although the rate of biotransformation of the tracer might be reduced in humans owing to the slower metabolic rate in compared to both mammals [97], this nevertheless impedes further development perspectives of this tracer for in vivo imaging.
To counter in vivo defluorination, Uddin et al. [98] developed another celecoxib analog [18F]2, for which a Ki of 160 nM to COX-2 and a Ki of 80 nM to COX-1 was reported in free enzyme assays and a Ki of 80 nM to COX-2 in whole cell assays. In contrast, this analog bore a [18F]fluoromethyl group instead of a [18F]trifluoromethyl group [98]. The specificity of the tracer was confirmed in the following experiments:
- i)
- a PET study comparing the uptake of the tracer in both inflamed (carrageenan-treated rat paws) and non-inflamed tissues (non-treated). It showed 1.53-fold increase in the former over the non-treated paws [98];
- ii)
- pre-dosing with celecoxib (10mg/kg), which significantly decreased tracer uptake in the inflamed rat paw (there was just only 1.7x times decrease in uptake [98];
- iii)
- experiments in COX-2 null mice further confirmed the specificity of the tracer; there was no increased tracer uptake in the inflamed carrageenan-treated paws of these mice compared to controls, i.e. non-inflamed carrageenan-treated paws of the same COX-2 knock-out mice with a ratio of 1.08. This contrasts significantly with the uptake in the inflamed paws of wild-type mice versus the control paws (1.48) [98];
- iv)
- results obtained using nude mice with both COX-2 positive 1483 HNSCC tumor and COX-2 negative HCT116 tumor suggest that the difference in the uptake in both tumors correlates to the difference in their expression of COX-2 (3-fold higher in the COX-2 positive tumor). The blocking of COX-2 active site in the former prevents the binding of the tracer (with tumor to muscle uptake ratio 1.01-fold), in comparison to control (2.94-fold) [98].
Importantly, it was observed that [18F]2 underwent minimal defluorination in vivo [98]. Compared to the carbon in the fluoromethyl moiety in [18F]2, the geminal carbon in [18F]1 bears a higher positive partial charge albeit having a carbon-fluorine bond stronger than that in [18F]2 [99]. The electron deficiency of position C3 in the pyrazole moiety to which the fluoro-substituted methyl moieties are bonded [100] further increases the positive partial charge on the geminal carbon and vice versa. This makes both the geminal carbon and the carbon in position C3 more susceptible to nucleophilic attacks [101] and eventual defluorination in vivo.
The valdecoxib analog [18F]3 (Figure 4) also underwent rapid defluorination in mice in vivo although it was labeled to obtain [18F]fluoromethyl group as [18F]2 (Figure 4) but for the replacement of the pyrazole ring with an isoxazole ring [102]. The weak N-O bond in the isoxazole ring might make the tracer more susceptible to in vivo metabolism [103]. In this study, however, the blood-pool retention of [18F]2 was not reported, although it also bears a sulfonamide moiety as well with a sub-nanomolar affinity to CA [92]. Further studies with the tracer are still needed to determine its efficacy as an in vivo imaging agent for detection of COX-2.
Based on the lead structure ([18F]pyricoxib) (Figure 4), which is based on a diarylpyrimidine backbone (reminiscent of the already mentioned [11C]MC1) originally reported by Swarbrick et al. [104], Tietz et al. recently developed a new class of potent and selective radiofluorinated COX-2 inhibitors.
They discovered that by altering the benzyl group substituent results (in position 4) resulted in varying degrees of affinity to COX-2: sterically bulky substituents (phenyl and tert-butyl) in this position gave quite low affinity (˃ 10 000 nM) to the enzyme compared to celecoxib (40 nM). Although still modestly sterically bulky but strongly electron-withdrawing the nitro-substituent at this position fared better (86 nM). The less bulky electronegative halo-substituents F (7 nM) and Cl (6 nM) outperformed the abovementioned substituent [84,105]. The more sterically bulky Br (48 nM) performed poorly compared to the other two but outperformed the nitro, phenyl and tert-butyl functional groups. The fact that a methyl substituent (5 nM) and a methoxy substituent (7 nM) outperformed or performed as well as the best halo-substituents is an indication that steric bulk plays a major role in the affinity of the ligands to COX-2, although it could also be seen that the successful halo-substituents are also capable of donating a lone pair of electrons in their resonance forms. Notwithstanding, all the ligands showed more selectivity over COX-1 (˃ 100 µM) than the sulfonamide and pyrimidine-bearing celecoxib (15 µM). Moreover, it was found out that affinity to COX-2 is compromised when the methylsulfone group was exchanged for the sulfonamide group. The corresponding sulfonamide counterparts of select methylsulfone compounds were less potent than the latter, the 4-fluoro-sulfonamide of [18F]pyricoxib showed 5.6x less potency [105].
Further studies were conducted on the lead compound ([18F]pyricoxib) by the same group [72]. The tracer showed a high uptake and retention in human colon adenocarcinoma HCA-7 and human colorectal carcinoma HCT-116. There was, however, also sufficient uptake and retention of the tracer in COX-2 negative HCT-116 cells indicative of a good passive diffusion owing to its high lipophilicity (log P 3.37). In any case, the uptake in COX-2 negative cells was significantly lower than in COX-2 positive HCA-7 cells. Results from IHC analysis confirmed a high expression of COX-2 in both HCA-7 and HCT-116 tumors, with higher detection signals in the former. A CD68 (a marker for tumor-associated macrophages) staining showed that the COX-2 positive staining was not due to infiltration of COX-2 expressing macrophages in inflammatory responses to cancer cell inoculation and tumor growth.
Uptake of [18F]pyricoxib was diminished in a concentration dependent manner in response to pretreatment of the HCA-7 cells with some COX-2 inhibitors, which serves as proof that cell retention of the tracer was specific. There was, however, still some non-specific intracellular binding, reasons for which were not further analyzed.
In vivo PET imaging studies in HCA-7 and HCT116 tumor-bearing NIH-III nude mice corroborated the COX-2 mediated tracer uptake and retention in HCA-7 tumors. Pre-administration of 2 mg of celecoxib to each of the mice used in the experiment resulted in a 16% decrease in tumor uptake of the tracer 1-hour p.i.. Selective COX-2 mediated uptake of the tracer in HCA-7 tumors was shown in a biodistribution study carried out on celecoxib-treated and control HCA-7 tumors-bearing NIH-III mice, which showed a 50% blocking effect by celecoxib. Nonetheless, there was a high uptake in the muscle, which the authors believed was caused by interaction of the tracer with a non-COX target. However, the tracer showed sufficient in vivo metabolic stability in mice, with up to 60% of the parent tracer still present at 120 min p.i..
More recently, Lebedev et al. labeled a metabolically stable celecoxib analog 4 (Figure 4) (IC50 COX-2 1.7 nM) via an automated electrochemical radiosynthesis route, with the F-18 attached directly on the pyrazole ring, with the intention of improving metabolic stability and specificity [81]. The tracer showed a direct correlation with increasing LPS concentrations in whole cell assays [98], with a corresponding decrease in uptake after blockage of the active sites by celecoxib (32 µg/mL). Ex vivo biodistribution study confirmed the uptake of the tracer in the brain (2.2 ± 0.7 %ID/g) at 60 min p.i., with the bone uptake below that of the blood and the brain, and almost the same as the muscle uptake. Dynamic PET/CT scans conducted in healthy mice showed good pharmacokinetics with background reducing within an hour of p.i. without any apparent defluorination as was confirmed by lack of skull/vertebrae retention of the tracer. The results also demonstrated a lack of retention of the tracer in the blood even though the tracer contains the sulfonamide group, which appeared to be the undoing of previously developed tracers.
The major drawback presently is the low decay-corrected yield of 2% (isolated RCY) and specific activity of 3 Ci/mmol. The authors are still working on improving the above in order to allow for translation to clinical practice [81].
Figure 4.
Structure of 18F-labeled COX-2 tracers.
2.2. SPECT COX-2 radiotracers
2.2.1. [123,125. I]Iodine-labeled COX-2 tracers
As mentioned earlier, the location of the COX-2 target calls for a tracer with a high lipophilic profile in order to allow the tracer to passively transverse both the cellular and organelle membranes [80,81,82]. Of course, there is the disadvantage that highly lipophilic tracers may bind indiscriminately to tissues in vivo and for this reason requires longer waiting times in order to allow for sufficient clearance of the tracers from non-target sites for a reasonably high imaging contrast to be achieved [84]. Longer waiting times of up to 4 hours require longer-lived isotopes (of which fluorine-18 falls slightly short, much less carbon-11), which would allow for longer imaging experiments, in order to yield better imaging contrast [84]. Moreover, COX-2 inhibitors inhibit COX-2 in a time-dependent manner hence the longer-lived the radioisotope the better, in order to ensure that binding equilibrium is achieved before the complete decay of the radioisotope [106]. Although some of tracers already mentioned in this review may provide acceptable detection results with shorter-lived radioisotopes, it is still however preferable to have other options.
To this end the gamma emitters 123 or 125I and 99mTc gamma emitters find useful application. Due to their comparably longer half-lives of 13.2 hours, 59.4 days, and 6.0 hours respectively, these radionuclides are easier to handle relative to their positron emitting counterparts. The resolution of SPECT images is nonetheless considerably lower in comparison to PET [72]. In addition, the steric bulk of iodine (a size comparable to that of a phenyl group) makes it one of the lesser used halogens in small molecules, which is in sharp contrast [74,79] to the relatively small size of fluorine an isosteric analog of hydrogen [75,80]. Also the steric effects caused by the presence of iodine in small molecules limits its inclusion in binding pocket of the COX-2 active site [84].
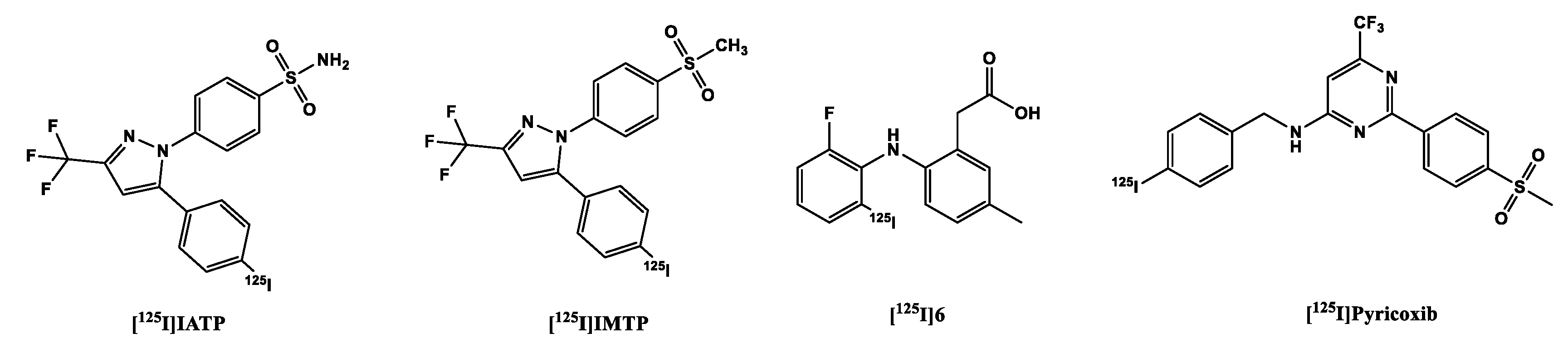
Kabalka et al. developed a 123I analog of celecoxib: the methyl group was replaced with [123I]5 (Figure 5), to retain affinity (IC50 COX-2 8.2 nM) and specificity, since halogens are better tolerated in position C4 [107]. Further biological evaluation was carried out by Schuller et al. in nicotine-derived nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) treated hamsters, which resulted in the induction of adenocarcinomas in their pancreas and lungs. Biodistribution experiments performed on these hamsters showed higher uptake of the tracer in the liver, pancreas, and a slightly higher uptake in the lung. Additional whole-body imaging with the 123I-labeled tracer showed increased uptake in the pancreas in one of the hamsters, and the liver of two of the hamsters, which correlated with the IHC staining of the organs. The third hamster showed neither accumulation in the already -mentioned organs nor overexpression of COX-2 via IHC. However, the results showed the distribution of the tracer correlates with the overexpression of COX-2, and that the uptake of the tracer might be mediated by COX-2. These results corroborated the resulted obtained from the in vitro evaluation of the tracer, in which it was observed that there was higher uptake in the NNK pretreated cells than the non-treated controls. This uptake was diminished after pre-incubation with celecoxib, a proof that uptake was due to presence of COX-2 in the cells [72,108].
Kuge, et al. in 2006 provided additional information on a compound developed by Kabalka et al. (125I-IATP) (Figure 5), and a methylsulfone derivative (125I-IMTP). Both tracers showed good binding to the COX-2 enzyme (125I-IMTP 5.2 µM and 125I-IATP 8.2 µM) [106] and selectivity over COX-1 (> 100 µM), resulting in approximately 19 and 12 fold-selectivity. However, Uddin, et al [109] found the affinity to COX-1 to be much higher > 4 µM, which means that the radioligands lack the required selectivity and hence cannot be used for the in vivo imaging of COX-2.
As usual, the sulfonamide derivative showed a higher retention in the blood than the methylsulfone derivative at all time points. The brain concentration of both tracers in healthy rats was quite low at 10 min p.i. 0.25 and 0.23 %ID/g for [125I]IMTP and [125I]IATP respectively; at 180 min 40% of the methylsulfone derivative remained in the brain while the brain concentration of the other remained almost unchanged at the same time point. The stability of the tracers was shown by their low uptake in the thyroid and the stomach [106].
In addition to the celecoxib derivatives, Kuge et al. [110] developed a lumiracoxib analog 6 ((Figure 6)), with the chlorine substituent in the lumiracoxib molecule substituted for iodine. This led to decreased affinity to COX-2 from 0.8 µM for lumiracoxib to 2.5 µM in the same assay, which might be due to the relative larger size of the iodine substituent, which was too big for the COX-2 binding pocket perhaps as pointed out by Tietz, et al. [84]. Notwithstanding, it also showed tracer uptake in LPS/IFN-g-stimulated macrophages in comparison to the non-stimulated ones. In biodistribution experiments, it showed no in vivo deiodination, suggested by the low uptake in the thyroid and stomach. Due to the presence of a carboxylic acid group in the molecule, which decreased the lipophilicity of the tracer and suggests possible ionization in vivo, there was very low uptake of the tracer in the brain 0.04% ID/g at 10 min.
More recently, Tietz, et al. reported a tracer [125I]pyricoxib, a radioiodinated derivative of the [18F]fluorine-labeled pyricoxib, in the hope that it will retain the same affinity to COX-2 and selectivity over COX-1 as tracer [18F]pyricoxib. This however was not the case as [125I]pyricoxib displayed ~12x less inhibitory potency than [18F]pyricoxib, and ~5x less than the ligand with a bromine atom in the 4-benzyl position. The steric bulk of iodine was believed to have played a role in this loss of potency, although it showed a better IC50 than the other already mentioned radio-iodinated COX-2 inhibitors, with improved selectivity over COX-1 as well. The group plan to radioiodinate the ligand with 124I in order to use the tracer for PET imaging as well as SPECT imaging [84].
Figure 5.
Structure of Radioiodinated COX-2 tracers.
2.3.2. 99. mTc-labeled COX-2 tracers
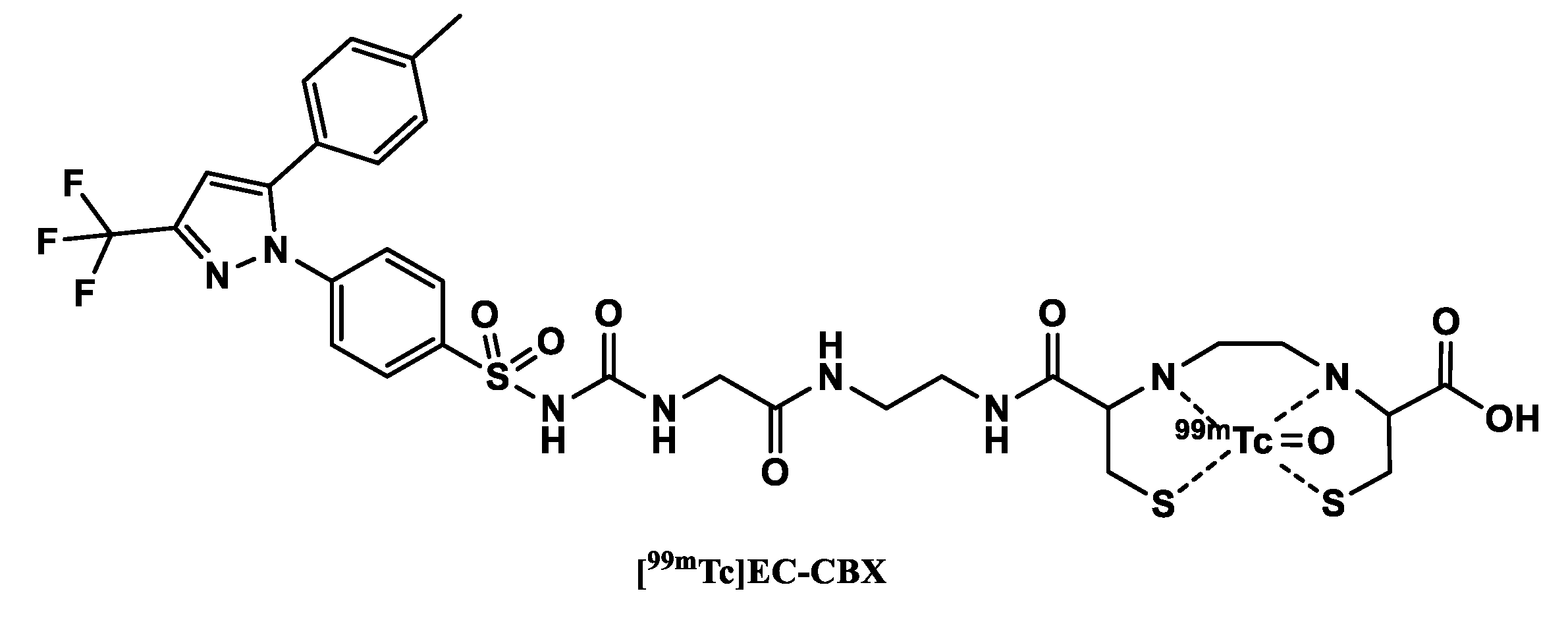
In 2004, Yang et al. [111] reported a 99mTc-labeled celecoxib (Figure 6) with the sulfonamide group modified by reacting it with ethyl 2-isocyanatoacetate to obtain an intermediate (ester form of CBX). In a subsequent aminolysis reaction with ethylenediamine, EA-CBX was synthesized. These modifications not only enabled the attachment of the chelator but also allowed sufficient distance between the pharmacologically active part of the tracer and the chelator. In the final step, EC–CBX was synthesized in a condensation reaction of EA-CBX with L,L-ethylenedicysteine (EC). The precursor EC–CBX was then labeled via [99mTc]pertechnetate.
An in vitro cellular tracer uptake assay in three cancer cell lines surprisingly showed a significant increase in tracer uptake after pretreatment with celecoxib. The authors suggested that this might be due to the induction of COX-2 expression by celecoxib. A more plausible explanation was suggested by Tietz et al. [72] who believe that the increased uptake was due to inhibition of P-glycoprotein by celecoxib in parallel to blocking COX-2 mediated uptake of the tracer.
Biodistribution experiments in breast tumor bearing rats showed that the uptake of the tracer was higher in tumors in comparison to the administration of only the 99mTc-labeled chelator ([99mTc]EC). The tracer additionally displayed favorable tumor/muscle ratios as a function of time (tumor/muscle ratio of 4.3 at 30 min & 9.7 at 4h). The tumor/blood ratios were less than one due to high blood retention of the tracer. Planar scintigraphy was also performed in tumor inoculated nude mice, rats, and rabbits. Planar images confirmed that the visualization of the tumors could be done from 30 min-4 h p.i. in the animal models.
Based on their results, the authors believe that the tracer could be used for the assessment of COX-2 expression of cancer. The EC–CBX may also be chelated with other isotopes as (68Ga, 61Cu) for PET imaging and 188Re for internal radionuclide therapy [111]. Since the sulfonamide functional group is believed to be essential for binding to the COX-2 enzyme, attaching a chelating agent directly to this group might diminish the affinity of the tracer to the target [112].
Figure 6.
Structure of 99mTc-labeled COX-2 tracers.
Some years later, Farouk et al. [113] presented another 99mTc-labeled celecoxib. Celecoxib in this case was labeled without a chelator, but in optimized conditions and, notably, at room temperature. The structure of the final complex formed, however, was not revealed by the authors.
Biodistribution studies carried out in healthy mice showed a gradual decrease in the blood uptake of the tracer with up to 47 % of the tracer concentration at 15 min p.i. remaining at 4 h p.i. The kidneys, however, showed high uptake, an indication of renal excretion to the urine. Biodistribution experiments carried out in sterile inflamed mice showed nearly the same biodistribution of the tracer as in healthy mice. More importantly, the inflamed muscle showed higher tracer uptake 1.7x, 2.1x and 2.5x at 15 min, 1h and 4h respectively than the non-inflamed tissue. The authors suspected that this higher uptake might be due to increased vascularization of the inflamed muscle, which facilitated a higher supply of the tracer to the area of interest. No additional blocking experiments were performed to determine the specificity of the uptake.
Chadha, et al. [114] carried out further experiments with the same tracer as Farouk et al. [113], which they radiolabeled using a different approach. They discovered a higher uptake of the tracer in the intestines of the 1,2-dimethylhydrazine (DMH) treated rats in comparison to healthy control rats. A significant uptake (2x, 2.9x and 2,4x higher at 0.5h, 2h and 4 h p.i. respectively) was also observed in the tumor site relative to the site adjacent to the tumor at all time points. Planar SPECT images at 4 h p.i. showed significant uptake of the tracer in the tumor site confirmed with histopathological studies, which suggest that the tracer has better selectivity towards neoplastic tissue in the colon of the DMH treated rats in comparison to control rats.
In conclusion, considerable progress has been made so far in the development of COX-2 tracers that can be used for the in vivo imaging of the COX-2 enzymes considering the obstacles met at the beginning, which included low metabolic stability, high blood retention of the sulfonamide analogs due to off-target binding to CA, and off-target binding to p-glycoprotein, which is a nuisance as it decreases the tumor-muscle ratio, and suitability of biological models for the in vivo evaluation of the prospective tracers.
Although this is only a minor problem, there are still concerns about selectivity over COX-1. Recently, the methylsulfone tracers [11C]MC1 [74,78], [18F]pyricoxib [72,105], [125I]pyricoxib [84], and the celecoxib analog [18F]6 [81] were developed and they showed minimal off-target binding to CA. Nevertheless, there is still a need for an improved and validated biological model, which will serve as gold standard for the evaluation of new COX-2 tracers and enable cross over between studies.
3. PET and SPECT imaging of TSPO and COX-2 in diseases
3.1. Introduction
Inflammation is significantly involved in a wide variety of diseases, which are not limited only to neurodegenerative pathologies. As a matter of fact, chronic (sub-clinical) inflammation is believed to be the most significant cause of death in the world, with not less than 50% associated with inflammation-related diseases like cancer, stroke, ischemic heart disease, chronic kidney disease, diabetes mellitus, non-alcoholic fatty liver disease, and the already mentioned NDD [115,116].
3.2. Pulmonary inflammation
The CNS communicates with other organs such as the skin [117], intestines, kidneys, and lungs [118,119,120,121]. As such, the activities of the microbiomes in these organs could in turn affect brain activities especially in relation to disease development [118,120,121]. The lungs, for instance, communicate directly with the external environment and, therefore, it is not surprising that emerging evidence show that CNS diseases are related to air pollution, which increases the risk of AD, PD, autism, cognitive dysfunction in the elderly and correlates with higher incidences of stroke. Albeit not directly, air pollution may activate microglia in due course and induce or accelerate inflammation in the brain through immune, endocrine or other mechanisms [121].
Moreover, researchers at the Institute for Neuroimmunology and Multiple Sclerosis Research at University of Göttingen established that changes in the lung microbiome plays a role in the susceptibility of the brain to autoimmune inflammation such as multiple sclerosis [120]. The neuropathology of NDD such as AD and PD are also linked to pulmonary bacteria [118]. Moreover, clinical studies showed that chronic obstructive pulmonary disease (COPD) is linked to cerebrovascular disease by an increment in white matter lesions, and predisposes to ischemic as well as hemorrhagic stroke [121,122]. Chronic lung pathologies or prolonged exposure to air pollutants could facilitate brain inflammation. Recent experimental evidence suggests that there are several lung-to-brain pathways involved in signaling to the brain during acute lung inflammation induced by the intratracheal injection of LPS in mice, which include circulating cytokines and immune cells [123].
Biomarkers of inflammation such as TSPO are upregulated in activated macrophages in COPD, asthma, as well as malaria-associated acute respiratory distress syndrome (MA-ARDS). Although specific studies investigating the use of TSPO and COX-2 imaging in lung inflammation are limited, it is conceivable that these imaging modalities could be explored in the context of pulmonary inflammatory conditions, such as pneumonia, bronchitis, COPD, and other respiratory disorders characterized by inflammation. PET based TPSO radiotracers could aid the visualization and the study of lung inflammation in order to better understand the mechanism of lung inflammation and lung-to-brain communication [121].
Goggi et al [124], was able to longitudinally image TSPO in the lungs of rodent models of MA-ARDS using the second generation TSPO tracer [18F]FEPPA (Figure 1). [18F]FEPPA was able to track changes in pulmonary accretion of interstitial inflammatory macrophages and MHC-II-positive alveolar macrophages in Plasmodium berghei ANKA-infected mice in correlation with parasitemia.
In a study by Chen et al [125], using another second generation TSPO tracer ([11C]PBR28) (Figure 1), the authors demonstrated that [11C]PBR28 can better distinguish macrophage-dominant inflammation from neutrophilic-dominant than [18F]FDG in mouse models of lung inflammation. Moreover, they showed that [11C]PBR28 can quantify M2-polarized TSPO-expressing-macrophages in the lungs. There was around 3-fold increase in this macrophage phenotype 49 days post Sendai virus (SeV) infection compared to the monocyte-deficient mouse model with attenuated chronic inflammatory response also on day 49 post infection. In addition, minimal recruitment of macrophages was observed in WT mice on the third day post infection. Still, no change in [11C]PBR28 uptake was observed 24h after the intranasal instillation of endotoxin (LPS) in WT mice. This was well in line with the fact [126] that an M1 macrophage-driven inflammatory response predominates with an abundance of neutrophils at this stage of the infection. Finally, they were able to confirm the recruitment of macrophage (TSPO) to mouse lungs using immunostaining [125]. This was in agreement with a similar experiment using human lung sections [125]. In human lung sections, the intensity of TSPO staining was significantly higher in CD68+ cells compared to neutrophils. These experiments revealed that [11C]PBR28 uptake not only depends on macrophage recruitment but may also depend on the macrophage phenotype.
Contrary to the study by Chen et al. [125], a previous study by Hatori et al. [127] was able to demonstrate that the second generation tracer [18F]FEDAC (Figure 1) can detect TSPO expressed in macrophages and neutrophils in lungs of mice instilled with LPS . Considering the duration of the experiment, it is apparent that only M1-polarized macrophages and neutrophils predominated, hence, were the main source of signals. There was a significant difference in the uptake of [18F]FEDAC in this mouse model of LPS-induced lung inflammation compared to the uptake of [11C](R)-PK11195 24h after LPS instillation [127]. Western blot assay confirmed an increased TSPO expression over time (2, 6 and 24 h) after LPS instillation compared to controls. IHC analysis further substantiated that TSPO was expressed by activated neutrophils and macrophages after pulmonary LPS instillation, with time-dependent recruitment of macrophages over a span of 24 h. However, it remains to be determined whether [18F]FEDAC may also label M2-polarized macrophages like [11C]PBR28 in cases of chronic inflammation or whether its efficacy is only limited to acute pulmonary inflammation.
The reported study by Chen et al. [125] corroborated a study by Jones et al. [128] conducted nearly twenty years prior to Chen and colleagues. In their study in rabbits, Jones et al. monitored the kinetics of lung macrophages after particle challenge to the lung using the first generation TSPO tracer [11C]R-PK11195. Overall, this study also showed that TSPO tracers can be applied in the assessment of effects of airborne pollutants.
Using [11C]DPA-713, Shah et al. [129] sought to unearth the mechanism of organ-level pathophysiology that ensue during Ebola virus infections. Longitudinal PET imaging together with various disease biomarkers and IHC indicated that, compared to baseline values, there is sustained loss of TSPO in lungs and spleen owing to the local depletion of monocytes, macrophages and dendritic cells. This is accompanied by a reactive haematopoietic activation in the bone marrow. Similar approach using TSPO PET ligands could help to noninvasively assess the pathogenesis and progression of other systemic inflammatory and infectious diseases in real time.
The incorporation of longer-lived iodine radioisotopes in DPA-713 has further facilitated the study of other infectious pulmonary diseases like tuberculosis (TB) as well as SARS-CoV-2 infection. Ordonez, et al. [130] used the iodine-125 (t1/2 59.5 days) radiolabeled [125I]DPA-713 to monitor the potency of a novel bedaquiline-containing TB drug regimen. This regimen is planned for patients with multidrug-resistant (MDR) tuberculosis [130]. TB remains still an important cause of human death amongst curable diseases and its complete elimination by 2050 is the target of the World Health Organisation (WHO). TB-associated inflammation is characterized by the presence of activated macrophages, which TSPO tracers like [125I]DPA-713 could image and consequently furnish invaluable real-time information about the outcome of TB treatment and the possibility of relapses. Pulmonary SPECT with [125I]DPA-713 established significant correlation with the bactericidal potency of TB treatments both standard and bedaquiline-containing compared to [18F]FDG PET in a C3HeB/FeJ TB model. Moreover, a time-dependent decrease in the cytokine levels interferon-ɣ (IFN-ɣ) and tumor necrosis factor-α (TNF-α) was detected with tuberculosis treatments, which correlated with [125I]DPA-713 uptake. Being a low-energy gamma emitter, iodine-125 was exchanged with the higher energy emitting iodine-124, which has better tissue penetration. [124I]DPA-713 PET showed up to 4-fold higher uptake in the infected tuberculosis lesions than uninfected controls [130]. Ruiz-Bedoya et al. [131] also used [124I]DPA-713 to evaluate the immune response in a hamster model of SARS-CoV-2 infection. PET/CT scans established that the tracer was trapped by activated macrophage cells in pulmonary lesions. Additionally, there were significant gendered differences observed for [124I]DPA-713 PET/CT in the hamsters: female hamsters displayed higher PET activity compared to the males. These findings were confirmed via optical imaging, immunofluorescence and flow cytometry.
3.3. Rheumatoid arthritis (RA)
RA a heterogenic immunopathology characterized by chronic synovial inflammation (resulting oftentimes in structural joint damage) can also be diagnosed by non-invasive PET tracers of inflammation[132,133]. By this route, quantitative measurements of changes in perfusion can be conducted in order to monitor and predict the efficacy of disease-modifying anti-rheumatic drugs. This is necessary since some RA patients fail to respond to therapy [134,135]. Histopathological analysis of synovial tissue biopsies, the classical method to evaluate treatment efficacy and immunopathological features of RA, is invasive [136]. The samples are acquired via ultrasound-guided biopsies or arthroscopy. Moreover, this classical method is delimited by the number of joints that can be examined [132,136].
The first generation TSPO tracer [11C]R-PK11195 (Figure 1) was able to image upregulated TSPO in activated macrophages in RA. Van der Laken et al. [138], were able to quantitatively visualize (significantly) synovitis in severely inflamed joints and joints with mild/moderate signs of inflammation. Interestingly, [11C]R-PK11195 was also able to visualize subclinical disease activity in the uninflamed knee contralateral to the inflamed knee. PBR staining of the sublining of synovial tissue correlated significantly with the uptake of [11C]R-PK11195 in the joints, as well as CD68 staining of macrophages. However, macrophage-specificity of the CD68 staining was later disproved by Narayan et al. [137]. Gent et al. [138] also corroborated the usefulness of [11C]R-PK11195 for the detection of subclinical RA. Using the same tracer, they demonstrated that [11C]R-PK11195 can visualize subclinical RA in patients who expressed the anti-citrullinated protein antibodies (ACPAs). A two-year follow-up confirmed the previous discovery. Gent et al. further demonstrated that [11C]R-PK11195 PET scans, can predict flares in RA patients [139], even better than MRI scans [140]. These were patients without clinical arthritis during or after treatment. Nevertheless, being a relatively lipophilic tracer (ClogP 4.6), its background uptake in periarticular tissues was relatively high and, thus, hindered the visualization of more subtle synovitis.
Narayan et al. [137] subsequently also showed its uptake in the tibiofemoral joint of RA patients with a significantly higher signal than in healthy controls using the comparatively less lipophilic tracer [11C]PBR28 (ClogP 3.4). These results were confirmed by both, synovial tissue autoradiography (with [3H]PBR28) and IHC staining. Interestingly, immunofluorescence showed that in addition to the macrophages there are other TSPO expressing cells in the RA pannus such as CD4+-T lymphocytes and fibroblast-like synoviocytes (FLS) (activated stromal cells). In mRNA and [3H]PBR28 studies in vitro, the highest expression of TSPO was seen in activated FLS cells, nonactivated M0 macrophages, and activated M2 reparative macrophages. Low expression of TSPO was observed in M1 macrophages, monocytes, unstimulated FLS cells and, with the overall lowest expression in activated and nonactivated CD4+ T cells. This remarkable contribution of activated FLS cells to TSPO PET signals from the RA pannus would be beneficial in the determination of responses to FLS cells-targeted therapies.
Bruijnen et al. [136] studied the relatively less lipophilic O-[18F]fluoroethylated DPA-TSPO tracer [18F]DPA-714 (Figure 2) (ClogP 3.3) and its O-[11C]methylated analog [11C]DPA-713 (ClogP 3.1) in RA patients. [18F]DPA-714 PET showed only a marginal improvement over [11C]R-PK11195. Even with a slightly lower background uptake of [18F]DPA-714 (Figure 2), the target-to-background (T/B) ratio of both tracers was not significantly different. The mean SUV values of PET-positive joints did not differ between the two tracers. A combination of lower background uptake of [11C]DPA-713 and higher absolute uptake in the inflamed joints resulted in up to a two-fold higher T/B ratio than that of the two other tracers. The outperformance of [18F]DPA-714 by [11C]DPA-713 may be related to the lower lipophilicity of the latter. Both DPA tracers showed no susceptibility to SNP polymorphism, unlike in brain imaging, which is indicative of the absence of the effect TSPO polymorphism in RA imaging.
Interestingly, it has also been reported that the COX-2 can serve as a biomarker for RA PET imaging. Shrestha et al. [74], using the PET tracer [11C]MC1(Figure 3), demonstrated that there was significantly higher uptake of the tracer in inflamed joints consistent with symptoms of the patients with RA. A PET scan with the third generation TSPO tracer [11C]ER176 (Figure 2) was also carried out, which showed similar results as [11C]MC1. The specificity of [11C]MC1 uptake was determined in another scan after a per os administration of 400 mg of celecoxib. Celecoxib showed no effect on the uptake of the [11C]ER176, but decreased the uptake of [11C]MC1 in the joints with a more consistent blocking of brain uptake of the tracer.
3.4. Cardiac pathology
TSPO upregulation has been observed in cardiac diseases like myocarditis [143], large vessel vasculitis [141], arrhythmia [142], atherosclerosis [143] and cardiac hypertrophy [144]. In cardiovascular diseases such as myocardial infarction (MI) [145] and artrial arrhythmia [142], TSPO could play the role of more than a diagnostic biomarker, as it can also serve as a therapeutic target. For this reason, the quantification of TSPO expression by TSPO tracers might provide invaluable information, which would help to individualize patient treatment.
Mou et al. [146] employed [18F]F-DPA (Figure 1) from the DPA family (Figure 3) to evaluate cardiac inflammation in rats post MI. [18F]F-DPA displayed high stable cardiac uptake with fast clearance from nearby organs such as the lungs, which enabled improved heart imaging. There was higher normalized SUV ratio (NSR) of [18F]F-DPA to [13N]NH3 in the infarct and peri-infarct regions compared to remote regions. Blocking studied with PK11195 confirmed the specificity of the tracer for TSPO as also did H&E staining. NSR evaluation using [13N]NH3 as reference was absolutely necessary since TSPO is also highly expressed in healthy cardiomyocytes and, therefore, may confound the results of cardiac inflammation. Moreover, the uptake in of [18F]F-DPA was lower in the infarct region compared to the remote region, which negatively affected visualization. Overall, higher uptake in the remote compared to the in infarct region suggests that cardiac TSPO imaging is a combination of mitochondrial dysfunction in dying cardiomyocytes and activated inflammatory cells [147].
Using a polar map of [99mTc]sestamibi as a reference, Thackeray et al. [147,148] analyzed high signals of [18F]flutriciclamide ([18F]GE180) (Figure 2) in infarcted regions in mice. [18F]GE180 was able to image infiltrating CD68+ macrophages in the acutely infarcted regions one-week post MI. It was also able to autonomously predict contractile dysfunction eight weeks post MI, during which TSPO signals were associated with mitochondrial dysfunction in failing but non-infarcted cardiomyocytes remote to the infarcted regions. They also assessed the influence of MI on both cardiac and brain inflammation using [18F]GE180 (Figure 2). Neuroinflammation is believed to correspond to cardiac dysfunctions in the early phase of post-infarct myocardial inflammation and in the late phase of chronic heart failure. This effect could attributed to increased proinflammatory cytokines, diminished cerebral blood flow as well as increasing levels of angiotensin II [148]. Moreover, elevated cerebral tumor necrosis factor-α and activated microglia were observed in mice with congestive heart failure, which was accompanied by cognitive impairment [149].
4. Conclusions
In this review, we highlighted several interesting TSPO and COX-2 PET and SPECT tracers. The background of the development of some of the tracers were provided in order to highlight some of the pitfalls that should be avoided in the development of future tracers. Apart from the biomarkers of inflammation mentioned in this review, there exist other targets, which may be more suitable for the investigation to specific pathologies. Nevertheless, TSPO and COX-2 are the most studied biomarkers of inflammation, which explains the high number of tracers already developed for these targets. Some of these tracers can be labeled with both radioisotopes for PET and SPECT imaging. Radiolabeling with SPECT radioisotopes enables to monitor pharmacokinetics of tracers with slow clearance from the organs of interest. This facilitates preclinical assessments since imaging can be performed hours post administration. In particular, the positron-emitting iodine-124 is useful in this regard. Additionally, there is also a potential to translate the use of such labeled tracers to clinical settings.
Inflammatory responses should be actively halted when their detrimental effects outweigh their benefits in order to avoid unnecessary bystander damage to tissues. Fortunately, some of the mentioned TSPO tracers such as [11C]R-PK11195 (in RA cases) are even able to detect inflammation before clinical presentation of symptoms. Importantly, the development of tracers to track inflammation may facilitate the development of drugs for the treatment of both, infectious and non-infectious diseases like cancer or neuroinflammation associated with neurodegeneration and autoimmune disorders.
Funding
BY, CC and CR are supported by the Flexifunds initiative “Lung-brain axis in health and disease” by the Research Campus Mid-Hessen (FCMH); CR and CC are supported by EU Joint Programme – Neurodegenerative Disease Research (JPND): SOLID JPND2021-650-233; Federal Ministry of Education and Research: 01ED2207; CC is supported by ERA-NET NEURON project MINERVA, DLR 100582004; CR is supported by the German Research Foundation, RU 1397/8-1, and the APC was partially funded by Philipps-University of Marburg.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapère, J.-J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.-R.; et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Fisher, E.; McMurray, L.; Milicevic Sephton, S.; Hird, M.; Kuzhuppilly-Ramakrishnan, N.; Williamson, D.J.; Zhou, X.; Werry, E.; Kassiou, M.; et al. Radiosynthesis of (R,S)-18 FGE387: A Potential PET Radiotracer for Imaging Translocator Protein 18 kDa (TSPO) with Low Binding Sensitivity to the Human Gene Polymorphism rs6971. ChemMedChem 2019, 14, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Damont, A.; Hinnen, F.; Kuhnast, B.; Schöllhorn-Peyronneau, M.-A.; James, M.; Luus, C.; Tavitian, B.; Kassiou, M.; Dollé, F. Radiosynthesis of [ 18 F]DPA-714, a selective radioligand for imaging the translocator protein (18 kDa) with PET. J. Label. Compd. Radiopharm. 2008, 51, 286–292. [Google Scholar] [CrossRef]
- Largeau, B.; Dupont, A.-C.; Guilloteau, D.; Santiago-Ribeiro, M.-J.; Arlicot, N. TSPO PET Imaging: From Microglial Activation to Peripheral Sterile Inflammatory Diseases? Contrast Media Mol. Imaging 2017, 2017, 6592139. [Google Scholar] [CrossRef] [PubMed]
- Zanotti-Fregonara, P.; Zhang, Y.; Jenko, K.J.; Gladding, R.L.; Zoghbi, S.S.; Fujita, M.; Sbardella, G.; Castellano, S.; Taliani, S.; Martini, C.; et al. Synthesis and evaluation of translocator 18 kDa protein (TSPO) positron emission tomography (PET) radioligands with low binding sensitivity to human single nucleotide polymorphism rs6971. ACS Chem. Neurosci. 2014, 5, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Valkimadi, P.-E.; Pagano, G.; Cousins, O.; Dervenoulas, G.; Politis, M. Applications of amyloid, tau, and neuroinflammation PET imaging to Alzheimer’s disease and mild cognitive impairment. Hum. Brain Mapp. 2019, 40, 5424–5442. [Google Scholar] [CrossRef] [PubMed]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.; Vugts, D.J.; Windhorst, A.D.; Mach, R.H. PET Imaging of Microglial Activation-Beyond Targeting TSPO. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Henter, I.D.; Innis, R.B. Imaging Translocator Protein as a Biomarker of Neuroinflammation in Dementia. Adv. Pharmacol. 2018, 82, 163–185. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A. TSPO imaging in parkinsonian disorders. Clin. Transl. Imaging 2016, 4, 183–190. [Google Scholar] [CrossRef]
- Belloli, S.; Morari, M.; Murtaj, V.; Valtorta, S.; Moresco, R.M.; Gilardi, M.C. Translation Imaging in Parkinson’s Disease: Focus on Neuroinflammation. Front. Aging Neurosci. 2020, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Doorduin, J.; Klein, H.C.; Dierckx, R.A.; James, M.; Kassiou, M.; Vries, E.F.J. de. 11C-DPA-713 and 18F-DPA-714 as new PET tracers for TSPO: a comparison with 11C-(R)-PK11195 in a rat model of herpes encephalitis. Mol. Imaging Biol. 2009, 11, 386–398. [Google Scholar] [CrossRef] [PubMed]
- James, M.L.; Fulton, R.R.; Henderson, D.J.; Eberl, S.; Meikle, S.R.; Thomson, S.; Allan, R.D.; Dolle, F.; Fulham, M.J.; Kassiou, M. Synthesis and in vivo evaluation of a novel peripheral benzodiazepine receptor PET radioligand. Bioorg. Med. Chem. 2005, 13, 6188–6194. [Google Scholar] [CrossRef]
- Fujita, M.; Kobayashi, M.; Ikawa, M.; Gunn, R.N.; Rabiner, E.A.; Owen, D.R.; Zoghbi, S.S.; Haskali, M.B.; Telu, S.; Pike, V.W.; et al. Comparison of four 11C-labeled PET ligands to quantify translocator protein 18 kDa (TSPO) in human brain: (R)-PK11195, PBR28, DPA-713, and ER176-based on recent publications that measured specific-to-non-displaceable ratios. EJNMMI Res. 2017, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Rodichkin, A.N.; McGlothan, J.L.; La Acanda De Rocha, A.M.; Azzam, D.J. Imaging neuroinflammation with TSPO: A new perspective on the cellular sources and subcellular localization. Pharmacol. Ther. 2022, 234, 108048. [Google Scholar] [CrossRef] [PubMed]
- Notter, T.; Schalbetter, S.M.; Clifton, N.E.; Mattei, D.; Richetto, J.; Thomas, K.; Meyer, U.; Hall, J. Neuronal activity increases translocator protein (TSPO) levels. Mol. Psychiatry 2021, 26, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Camsonne, R.; Crouzel, C.; Comar, D.; Mazière, M.; Prenant, C.; Sastre, J.; Moulin, M.; Syrota, A. Synthesis of N-(11C) methyl, N-(methyl-1 propyl), (chloro-2 phenyl)-1 isoquinoleine carboxamide-3 (PK 11195): A new ligand for peripheral benzodiazepine receptors. J. Label Compd. Radiopharm. 1984, 21, 985–991. [Google Scholar] [CrossRef]
- Yuan, J.; Yao, J.-Q.; Fang, X.-X.; Dai, W.; Wang, Y.-H.; Zhang, L.-M.; Li, Y.-F. Involvement of regulation of the excitation:inhibition functional balance in the mPFC in the antidepressant-anxiolytic effect of YL-IPA08, a novel TSPO ligand. Metab. Brain Dis. 2022, 37, 2305–2314. [Google Scholar] [CrossRef]
- Jučaite, A.; Cselényi, Z.; Arvidsson, A.; Ahlberg, G.; Julin, P.; Varnäs, K.; Stenkrona, P.; Andersson, J.; Halldin, C.; Farde, L. Kinetic analysis and test-retest variability of the radioligand 11C(R)-PK11195 binding to TSPO in the human brain - a PET study in control subjects. EJNMMI Res. 2012, 2, 15. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Fujita, M.; Fujimura, Y.; Kimura, N.; Jenko, K.J.; Kannan, P.; Hong, J.; Morse, C.L.; Zoghbi, S.S.; Gladding, R.L.; et al. Comparison of (11)C-(R)-PK 11195 and (11)CPBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. NeuroImage 2010, 49, 2924–2932. [Google Scholar] [CrossRef]
- Kobayashi, M.; Jiang, T.; Telu, S.; Zoghbi, S.S.; Gunn, R.N.; Rabiner, E.A.; Owen, D.R.; Guo, Q.; Pike, V.W.; Innis, R.B.; et al. 11C-DPA-713 has much greater specific binding to translocator protein 18 kDa (TSPO) in human brain than 11C-(R)-PK11195. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2018, 38, 393–403. [Google Scholar] [CrossRef]
- Ching, A.S.C.; Kuhnast, B.; Damont, A.; Roeda, D.; Tavitian, B.; Dollé, F. Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Into Imaging 2012, 3, 111–119. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. The Lancet, 358(9280), 461-467. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.-L.; et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PloS One 2012, 7, e52941. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with 11C(R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Gulyás, B.; Tóth, M.; Schain, M.; Airaksinen, A.; Vas, A.; Kostulas, K.; Lindström, P.; Hillert, J.; Halldin, C. Evolution of microglial activation in ischaemic core and peri-infarct regions after stroke: a PET study with the TSPO molecular imaging biomarker ((11))Cvinpocetine. J. Neurol. Sci. 2012, 320, 110–117. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, F.; Kosaka, J.; Ota, M.; Higuchi, M.; Ito, H.; Fujimura, Y.; Nozaki, S.; Takahashi, S.; Mizukami, K.; Asada, T.; et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with ¹¹CDAA1106. Psychiatry Res. 2012, 203, 67–74. [Google Scholar] [CrossRef]
- Iannaccone, S.; Cerami, C.; Alessio, M.; Garibotto, V.; Panzacchi, A.; Olivieri, S.; Gelsomino, G.; Moresco, R.M.; Perani, D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Park. Relat. Disord. 2013, 19, 47–52. [Google Scholar] [CrossRef]
- Dollé, F.; Luus, C.; Reynolds, A.; Kassiou, M. Radiolabelled molecules for imaging the translocator protein (18 kDa) using positron emission tomography. Curr. Med. Chem. 2009, 16, 2899–2923. [Google Scholar] [CrossRef]
- Owen, D.R.J.; Gunn, R.N.; Rabiner, E.A.; Bennacef, I.; Fujita, M.; Kreisl, W.C.; Innis, R.B.; Pike, V.W.; Reynolds, R.; Matthews, P.M.; et al. Mixed-Affinity Binding in Humans with 18-kDa Translocator Protein Ligands. Journal of Nuclear Medicine, 52(1), 24-32. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2011, 52, 24–32. [Google Scholar] [CrossRef]
- Owen, D.R.; Howell, O.W.; Tang, S.-P.; Wells, L.A.; Bennacef Idriss; Bergstrom, M.; Gunn, R.N.; Rabiner, E.A.; Wilkins, M.R.; Reynolds, R.; et al. Two Binding Sites for [3H]PBR28 in Human Brain: Implications for TSPO PET Imaging of Neuroinflammation. Journal of cerebral blood flow and metabolism official journal of the International Society of Cerebral Blood Flow and Metabolism. Available online: https://journals.sagepub.com/doi/10.1038/jcbfm.2010.63?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed (accessed on 14 October 2020).
- Chauveau, F.; van Camp, N.; Dollé, F.; Kuhnast, B.; Hinnen, F.; Damont, A.; Boutin, H.; James, M.; Kassiou, M.; Tavitian, B. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2009, 50, 468–476. [Google Scholar] [CrossRef]
- Terada, T.; Yokokura, M.; Yoshikawa, E.; Futatsubashi, M.; Kono, S.; Konishi, T.; Miyajima, H.; Hashizume, T.; Ouchi, Y. Extrastriatal spreading of microglial activation in Parkinson’s disease: a positron emission tomography study. Ann. Nucl. Med. 2016, 30, 579–587. [Google Scholar] [CrossRef]
- Keller, T.; Krzyczmonik, A.; Forsback, S.; Picón, F.R.L.; Kirjavainen, A.K.; Takkinen, J.; Rajander, J.; Cacheux, F.; Damont, A.; Dollé, F.; et al. Radiosynthesis and Preclinical Evaluation of 18FF-DPA, A Novel Pyrazolo1,5apyrimidine Acetamide TSPO Radioligand, in Healthy Sprague Dawley Rats. Mol. Imaging Biol. 2017, 19, 736–745. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, R.; Fujinaga, M.; Yang, J.; Zhang, Y.; Hatori, A.; Kumata, K.; Yang, J.; Vasdev, N.; Du, Y.; et al. A Facile Radiolabeling of 18FFDPA via Spirocyclic Iodonium Ylides: Preliminary PET Imaging Studies in Preclinical Models of Neuroinflammation. J. Med. Chem. 2017, 60, 5222–5227. [Google Scholar] [CrossRef]
- Wang, L.; Yao, S.; Tang, R.; Zhu, H.; Zhang, L.; Gong, J.; Chen, Q.; Collier, T.L.; Xu, H.; Liang, S.H. A concisely automated synthesis of TSPO radiotracer 18 FFDPA based on spirocyclic iodonium ylide method and validation for human use. J. Label. Comp. Radiopharm. 2020, 63, 119–128. [Google Scholar] [CrossRef]
- Keller, T.; López-Picón, F.R.; Krzyczmonik, A.; Forsback, S.; Takkinen, J.S.; Rajander, J.; Teperi, S.; Dollé, F.; Rinne, J.O.; Haaparanta-Solin, M.; et al. Comparison of high and low molar activity TSPO tracer 18FF-DPA in a mouse model of Alzheimer’s disease. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2020, 40, 1012–1020. [Google Scholar] [CrossRef]
- López-Picón, F.R.; Keller, T.; Bocancea, D.; Helin, J.S.; Krzyczmonik, A.; Helin, S.; Damont, A.; Dollé, F.; Rinne, J.O.; Haaparanta-Solin, M.; et al. Direct Comparison of 18FF-DPA with 18FDPA-714 and 11CPBR28 for Neuroinflammation Imaging in the same Alzheimer’s Disease Model Mice and Healthy Controls. Mol. Imaging Biol. 2022, 24, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Lohith, T.G.; Shrestha, S.; Telu, S.; Zoghbi, S.S.; Castellano, S.; Taliani, S.; Da Settimo, F.; Fujita, M.; Pike, V.W.; et al. 11C-ER176, a Radioligand for 18-kDa Translocator Protein, Has Adequate Sensitivity to Robustly Image All Three Affinity Genotypes in Human Brain. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 320–325. [Google Scholar] [CrossRef]
- Columbia University. Imaging Inflammation in Alzheimer’s Disease With 11C-ER176: NCT03744312, AAAR6570. Available online: https://clinicaltrials.gov/ct2/show/NCT03744312 (accessed on 3 November 2022).
- Chen, H.; Jiang, Z.; Cheng, X.; Zheng, W.; Sun, Y.; Yu, Z.; Yang, T.; Zhang, L.; Yan, J.; Liu, Y.; et al. 18FBIBD-239: 18F-Labeled ER176, a Positron Emission Tomography Tracer Specific for the Translocator Protein. Mol. Pharm. 2022, 19, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, H.; Jones, P.A.; Chau, W.-F.; Durrant, C.; Fouladi, N.; Passmore, J.; O’Shea, D.; Wynn, D.; Morisson-Iveson, V.; Ewan, A.; et al. ¹⁸FGE-180: a novel fluorine-18 labelled PET tracer for imaging Translocator protein 18 kDa (TSPO). Bioorganic Med. Chem. Lett. 2012, 22, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Hanani, R.; Hibbs, D.; Damont, A.; Da Pozzo, E.; Selleri, S.; Dollé, F.; Martini, C.; Kassiou, M. Pyrazolo1,5-apyrimidine acetamides: 4-Phenyl alkyl ether derivatives as potent ligands for the 18 kDa translocator protein (TSPO). Bioorganic Med. Chem. Lett. 2010, 20, 5799–5802. [Google Scholar] [CrossRef] [PubMed]
- Chaney, A.; Cropper, H.C.; Johnson, E.M.; Lechtenberg, K.J.; Peterson, T.C.; Stevens, M.Y.; Buckwalter, M.S.; James, M.L. 11C-DPA-713 Versus 18F-GE-180: A Preclinical Comparison of Translocator Protein 18 kDa PET Tracers to Visualize Acute and Chronic Neuroinflammation in a Mouse Model of Ischemic Stroke. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2019, 60, 122–128. [Google Scholar] [CrossRef]
- Feeney, C.; Scott, G.; Raffel, J.; Roberts, S.; Coello, C.; Jolly, A.; Searle, G.; Goldstone, A.P.; Brooks, D.J.; Nicholas, R.S.; et al. Kinetic analysis of the translocator protein positron emission tomography ligand 18FGE-180 in the human brain. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2201–2210. [Google Scholar] [CrossRef]
- Zanotti-Fregonara, P.; Pascual, B.; Rizzo, G.; Yu, M.; Pal, N.; Beers, D.; Carter, R.; Appel, S.H.; Atassi, N.; Masdeu, J.C. Head-to-Head Comparison of 11C-PBR28 and 18F-GE180 for Quantification of the Translocator Protein in the Human Brain. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1260–1266. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Jenko, K.J.; Hines, C.S.; Lyoo, C.H.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Hyde, T.; Kleinman, J.E.; Pike, V.W.; et al. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zanotti-Fregonara, P.; Veronese, M.; Pascual, B.; Rostomily, R.C.; Turkheimer, F.; Masdeu, J.C. The validity of 18F-GE180 as a TSPO imaging agent. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1205–1207. [Google Scholar] [CrossRef]
- Albert, N.L.; Unterrainer, M.; Brendel, M.; Kaiser, L.; Zweckstetter, M.; Cumming, P.; Bartenstein, P. In response to: The validity of 18F-GE180 as a TSPO imaging agent. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Lepelletier, F.-X.; Trigg, W.; Banister, S.; Reekie, T.; Kassiou, M.; Gerhard, A.; Hinz, R.; Boutin, H. Comparative Evaluation of Three TSPO PET Radiotracers in a LPS-Induced Model of Mild Neuroinflammation in Rats. Mol. Imaging Biol. 2017, 19, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Francisco, R. López-Picón; Anniina Snellman; Olli Eskola; Semi Helin; Olof Solin; Merja Haaparanta-Solin; Juha O. Rinne. Neuroinflammation Appears Early on PET Imaging and Then Plateaus in a Mouse Model of Alzheimer Disease. J. Nucl. Med. 2018, 59, 509–515. [Google Scholar] [CrossRef]
- Holzgreve, A.; Pötter, D.; Brendel, M.; Orth, M.; Weidner, L.; Gold, L.; Kirchner, M.A.; Bartos, L.M.; Unterrainer, L.M.; Unterrainer, M.; et al. Longitudinal 18FGE-180 PET Imaging Facilitates In Vivo Monitoring of TSPO Expression in the GL261 Glioblastoma Mouse Model. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Ramakrishnan, N.K.; Hird, M.; Thompson, S.; Williamson, D.J.; Qiao, L.; Owen, D.R.; Brooks, A.F.; Scott, P.J.H.; Bacallado, S.; O’Brien, J.T.; et al. Preclinical evaluation of (S)-18FGE387, a novel 18-kDa translocator protein (TSPO) PET radioligand with low binding sensitivity to human polymorphism rs6971. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 125–136. [Google Scholar] [CrossRef]
- Hobson, B.A.; Rowland, D.J.; Sisó, S.; Guignet, M.A.; Harmany, Z.T.; Bandara, S.B.; Saito, N.; Harvey, D.J.; Bruun, D.A.; Garbow, J.R.; et al. TSPO PET Using 18FPBR111 Reveals Persistent Neuroinflammation Following Acute Diisopropylfluorophosphate Intoxication in the Rat. Toxicol. Sci. 2019, 170, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Ottoy, J.; Picker, L. de; Verhaeghe, J.; Deleye, S.; wyffels, L.; Kosten, L.; Sabbe, B.; Coppens, V.; Timmers, M.; van Nueten, L.; et al. 18F-PBR111 PET Imaging in Healthy Controls and Schizophrenia: Test–Retest Reproducibility and Quantification of Neuroinflammation. J. Nucl. Med. 2018, 59, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, H.; Bae, S.-H.; Lee, S.-Y.; Kim, Y.-H.; Na, J.; Lee, C.-H.; Lee, M.S.; Ko, G.B.; Kim, K.Y.; et al. 18FCB251 PET/MR imaging probe targeting translocator protein (TSPO) independent of its Polymorphism in a Neuroinflammation Model. Theranostics 2020, 10, 9315–9331. [Google Scholar] [CrossRef]
- Lee, S.H.; Denora, N.; Laquintana, V.; Mangiatordi, G.F.; Lopedota, A.; Lopalco, A.; Cutrignelli, A.; Franco, M.; Delre, P.; Song, I.H.; et al. Radiosynthesis and characterization of [18F]BS224: a next-generation TSPO PET ligand insensitive to the rs6971 polymorphism. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 110–124. [Google Scholar] [CrossRef]
- Li, F.; Liu, J.; Zheng, Y.; Garavito, R.M.; Ferguson-Miller, S. Protein structure. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–558. [Google Scholar] [CrossRef]
- Jaremko, M.; Jaremko, Ł.; Giller, K.; Becker, S.; Zweckstetter, M. Structural Integrity of the A147T Polymorph of Mammalian TSPO. Chembiochem 2015, 16, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, M.; Jaremko, Ł.; Giller, K.; Becker, S.; Zweckstetter, M. Backbone and side-chain resonance assignment of the A147T polymorph of mouse TSPO in complex with a high-affinity radioligand. Biomol. NMR Assign. 2016, 10, 79–83. [Google Scholar] [CrossRef]
- Berroterán-Infante, N.; Tadić, M.; Hacker, M.; Wadsak, W.; Mitterhauser, M. Binding Affinity of Some Endogenous and Synthetic TSPO Ligands Regarding the rs6971 Polymorphism. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Midzak, A.S.; Akula, N.; Rone, M.B.; Papadopoulos, V. Computational modeling and biological validation of novel non-steroidal ligands for the cholesterol recognition/interaction amino acid consensus (CRAC) motif of the mitochondrial translocator protein (TSPO). Pharmacol. Res. 2015, 99, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Stathis, M.; Coughlin, J.M.; Pomper, M.; Slusher, B.S. The Low-Affinity Binding of Second Generation Radiotracers Targeting TSPO is Associated with a Unique Allosteric Binding Site. J. Neuroimmune Pharmacol. 2018, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- ENZYME - 1.14.99.1 Prostaglandin-endoperoxide synthase. Available online: https://enzyme.expasy.org/EC/1.14.99.1 (accessed on 1 January 2021).
- Yamagata, K.; Matsumura, K.; Inoue, W.; Shiraki, T.; Suzuki, K.; Yasuda, S.; Sugiura, H.; Cao, C.; Watanabe, Y.; Kobayashi, S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J. Neurosci. 2001, 21, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Rummel, C.; Sachot, C.; Poole, S.; Luheshi, G.N. Circulating interleukin-6 induces fever through a STAT3-linked activation of COX-2 in the brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1316–R1326. [Google Scholar] [CrossRef] [PubMed]
- Steiner, A.A.; Hunter, J.C.; Phipps, S.M.; Nucci, T.B.; Oliveira, D.L.; Roberts, J.L.; Scheck, A.C.; Simmons, D.L.; Romanovsky, A.A. Cyclooxygenase-1 or -2--which one mediates lipopolysaccharide-induced hypothermia? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R485–R494. [Google Scholar] [CrossRef]
- Tietz, O.; Wuest, M.; Marshall, A.; Glubrecht, D.; Hamann, I.; Wang, M.; Bergman, C.; Way, J.D.; Wuest, F. PET imaging of cyclooxygenase-2 (COX-2) in a pre-clinical colorectal cancer model. EJNMMI Res. 2016, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Le Fur, G.; Perrier, M.L.; Vaucher, N.; Imbault, F.; Flamier, A.; Benavides, J.; Uzan, A.; Renault, C.; Dubroeucq, M.C.; Guérémy, C. Peripheral benzodiazepine binding sites: Effect of PK 11195, 1-(2-chlorophenyl)-n-methyl-n-(1-methylpropyl)-3-isoquinolinecarboxamide. Life Sciences, 32(16), 1839-1847. Life Sci. 1983, 32, 1839–1847. [Google Scholar] [CrossRef]
- Shrestha, S.; Kim, M.-J.; Eldridge, M.; Lehmann, M.L.; Frankland, M.; Liow, J.-S.; Yu, Z.-X.; Cortes-Salva, M.; Telu, S.; Henter, I.D.; et al. PET measurement of cyclooxygenase-2 using a novel radioligand: upregulation in primate neuroinflammation and first-in-human study. J. Neuroinflammation 2020, 17, 140. [Google Scholar] [CrossRef]
- Goetz Moro, M.; Vargas Sanchez, P.K.; Lupepsa, A.C.; Baller, E.M.; Nobre Franco, G.C. Biología de la ciclooxigenasa en la función renal – Revisión de la literatura. Rev. Colomb. Nefrol. 2017, 4, 27. [Google Scholar] [CrossRef]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. USA 1994, 91, 12013–12017. [Google Scholar] [CrossRef] [PubMed]
- Krüger, K.; Bredehöft, J.; Mooren, F.C.; Rummel, C. Different effects of strength and endurance exercise training on COX-2 and mPGES expression in mouse brain are independent of peripheral inflammation. J. Appl. Physiol. (1985) 2016, 121, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Michelle Cortes; Prachi Singh; Cheryl Morse; Saurav Shrestha; Kimberly Jenko; Aneta Kowalski; Sami Zoghbi; Masahiro Fujita; Robert Innis; Victor Pike. Synthesis of PET radioligands as potential probes for imaging COX-2 in neuroinflammation. J. Nucl. Med. 2015, 56, 1092.
- Min-Jeong Kim; Stal Shrestha; Mark Eldridge; Michelle Cortes; Prachi Singh; Jeih-San Liow; Robert Gladding; Sami Zoghbi; Masahiro Fujita; Victor Pike; et al. Novel PET radioligands show that, in rhesus monkeys, COX-1 is constitutively expressed and COX-2 is induced by inflammation. J. Nucl. Med. 2017, 58, 203.
- Sejdiu, B.I.; Tieleman, D.P. COX-1 – lipid interactions: arachidonic acid, cholesterol, and phospholipid binding to the membrane binding domain of COX-1, 2020.
- Lebedev, A.; Jiao, J.; Lee, J.; Yang, F.; Allison, N.; Herschman, H.; Sadeghi, S. Radiochemistry on electrodes: Synthesis of an 18F-labelled and in vivo stable COX-2 inhibitor. PloS One 2017, 12, e0176606. [Google Scholar] [CrossRef]
- Patel, R.; Attur, M.G.; Dave, M.; Abramson, S.B.; Amin, A.R. Regulation of Cytosolic COX-2 and Prostaglandin E2 Production by Nitric Oxide in Activated Murine Macrophages. J. Immunol. 1999, 162, 4191–4197. [Google Scholar] [CrossRef] [PubMed]
- Tietz, O.; Marshall, A.; Wuest, M.; Wang, M.; Wuest, F. Radiotracers for molecular imaging of cyclooxygenase-2 (COX-2) enzyme. Curr. Med. Chem. 2013, 20, 4350–4369. [Google Scholar] [CrossRef]
- Tietz, O.; Dzandzi, J.; Bhardwaj, A.; Valliant, J.F.; Wuest, F. Design and synthesis of (125)IPyricoxib: A novel (125)I-labeled cyclooxygenase-2 (COX-2) inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 1516–1520. [Google Scholar] [CrossRef]
- Martín, A.; Boisgard, R.; Thézé, B.; van Camp, N.; Kuhnast, B.; Damont, A.; Kassiou, M.; Dollé, F.; Tavitian, B. Evaluation of the PBR/TSPO radioligand (18)FDPA-714 in a rat model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2010, 30, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Matsumura, K.; Yamagata, K.; Watanabe, Y. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1 beta: a possible site of prostaglandin synthesis responsible for fever. Brain Res. 1996, 733, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- New Scientist. Up to 140,000 heart attacks linked to Vioxx. Available online: https://www.newscientist.com/article/dn6918-up-to-140000-heart-attacks-linked-to-vioxx/?ignored=irrelevant (accessed on 3 January 2021).
- fda/cder. label.
- Prabhakaran, J.; Underwood, M.D.; Parsey, R.V.; Arango, V.; Majo, V.J.; Simpson, N.R.; van Heertum, R.; Mann, J.J.; Kumar, J.S.D. Synthesis and in vivo evaluation of 18F-4-5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-ylbenzenesulfonamide as a PET imaging probe for COX-2 expression. Bioorg. Med. Chem. 2007, 15, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Takashima-Hirano, M.; Takashima, T.; Katayama, Y.; Wada, Y.; Sugiyama, Y.; Watanabe, Y.; Doi, H.; Suzuki, M. Efficient sequential synthesis of PET Probes of the COX-2 inhibitor 11Ccelecoxib and its major metabolite 11CSC-62807 and in vivo PET evaluation. Bioorg. Med. Chem. 2011, 19, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Casini, A.; Heine, A.; Kuhn, D.; Supuran, C.T.; Scozzafava, A.; Klebe, G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. J. Med. Chem. 2004, 47, 550–557. [Google Scholar] [CrossRef]
- Fujisaki, Y.; Kawamura, K.; Wang, W.-F.; Ishiwata, K.; Yamamoto, F.; Kuwano, T.; Ono, M.; Maeda, M. Radiosynthesis and in vivo evaluation of 11C-labeled 1,5-diarylpyrazole derivatives for mapping cyclooxygenases. Ann. Nucl. Med. 2005, 19, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, M.; Miller, K.D.; Hutchins, G.D.; Zheng, Q.-H. Synthesis of carbon-11 labeled celecoxib derivatives as new candidate PET radioligands for imaging of inflammation. Appl. Radiat. Isot. 2009, 67, 2019–2024. [Google Scholar] [CrossRef]
- Vries, E.F.J. de; Doorduin, J.; Dierckx, R.A.; van Waarde, A. Evaluation of (11)Crofecoxib as PET tracer for cyclooxygenase 2 overexpression in rat models of inflammation. Nucl. Med. Biol. 2008, 35, 35–42. [Google Scholar] [CrossRef]
- Search results for 11C-MC1 - Clinical Trials Registry - ICH GCP. Available online: https://ichgcp.net/clinical-trials-registry/research/list?intr=11C-MC1 (accessed on 8 January 2021).
- Barrio, J.R.; Satyamurthy, N.; Huang, S.C.; Keen, R.E.; Nissenson, C.H.; Hoffman, J.M.; Ackermann, R.F.; Bahn, M.M.; Mazziotta, J.C.; Phelps, M.E. 3-(2’-18Ffluoroethyl)spiperone: in vivo biochemical and kinetic characterization in rodents, nonhuman primates, and humans. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 1989, 9, 830–839. [Google Scholar] [CrossRef]
- Uddin, M.J.; Crews, B.C.; Ghebreselasie, K.; Huda, I.; Kingsley, P.J.; Ansari, M.S.; Tantawy, M.N.; Reese, J.; Marnett, L.J. Fluorinated COX-2 inhibitors as agents in PET imaging of inflammation and cancer. Cancer Prev. Res. (Phila) 2011, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Lemal, D.M. Perspective on fluorocarbon chemistry. J. Org. Chem. 2004, 69, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Secrieru, A.; O’Neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2019, 25. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kumadaki, I. Reactions of aromatic trifluoromethyl compounds with nucleophilic reagents. Acc. Chem. Res. 1978, 11, 197–204. [Google Scholar] [CrossRef]
- Toyokuni, T.; Kumar, J.S.D.; Walsh, J.C.; Shapiro, A.; Talley, J.J.; Phelps, M.E.; Herschman, H.R.; Barrio, J.R.; Satyamurthy, N. Synthesis of 4-(5-18Ffluoromethyl-3-phenylisoxazol-4-yl)benzenesulfonamide, a new 18Ffluorinated analogue of valdecoxib, as a potential radiotracer for imaging cyclooxygenase-2 with positron emission tomography. Bioorganic Med. Chem. Lett. 2005, 15, 4699–4702. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Qi, J.; Ren, X.; Zhang, J.; Li, H.; Xiao, H.; Wang, R.; Liu, Z.; Meng, L.; Ma, N.; et al. Developing Isoxazole as a Native Photo-Cross-Linker for Photoaffinity Labeling and Chemoproteomics. Angew. Chem. Int. Ed Engl. 2022, 61, e202209947. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, M.E.; Beswick, P.J.; Gleave, R.J.; Green, R.H.; Bingham, S.; Bountra, C.; Carter, M.C.; Chambers, L.J.; Chessell, I.P.; Clayton, N.M.; et al. Identification of 4-4-(methylsulfonyl)phenyl-6-(trifluoromethyl)-2-pyrimidinyl amines and ethers as potent and selective cyclooxygenase-2 inhibitors. Bioorganic Med. Chem. Lett. 2009, 19, 4504–4508. [Google Scholar] [CrossRef] [PubMed]
- Tietz, O.; Sharma, S.K.; Kaur, J.; Way, J.; Marshall, A.; Wuest, M.; Wuest, F. Synthesis of three 18F-labelled cyclooxygenase-2 (COX-2) inhibitors based on a pyrimidine scaffold. Org. Biomol. Chem. 2013, 11, 8052–8064. [Google Scholar] [CrossRef]
- Kuge, Y.; Katada, Y.; Shimonaka, S.; Temma, T.; Kimura, H.; Kiyono, Y.; Yokota, C.; Minematsu, K.; Seki, K.-i.; Tamaki, N.; et al. Synthesis and evaluation of radioiodinated cyclooxygenase-2 inhibitors as potential SPECT tracers for cyclooxygenase-2 expression. Nucl. Med. Biol. 2006, 33, 21–27. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Mereddy, A.R.; Schuller, H.M. Synthesis of an iodine-123-labeled celecoxib analogue: a potential spect agent. J. Label. Compd. Radiopharm. 2005, 48, 295–300. [Google Scholar] [CrossRef]
- Schuller, H.M.; Kabalka, G.; Smith, G.; Mereddy, A.; Akula, M.; Cekanova, M. Detection of overexpressed COX-2 in precancerous lesions of hamster pancreas and lungs by molecular imaging: implications for early diagnosis and prevention. ChemMedChem 2006, 1, 603–610. [Google Scholar] [CrossRef]
- Uddin, M.J.; Crews, B.C.; Ghebreselasie, K.; Tantawy, M.N.; Marnett, L.J. I-Celecoxib Analogues as SPECT Tracers of Cyclooxygenase-2 in Inflammation. ACS Med. Chem. Lett. 2011, 2, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Kuge, Y.; Obokata, N.; Kimura, H.; Katada, Y.; Temma, T.; Sugimoto, Y.; Aita, K.; Seki, K.-i.; Tamaki, N.; Saji, H. Synthesis and evaluation of a radioiodinated lumiracoxib derivative for the imaging of cyclooxygenase-2 expression. Nucl. Med. Biol. 2009, 36, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.J.; Bryant, J.; Chang, J.Y.; Mendez, R.; Oh, C.-S.; Yu, D.-F.; Ito, M.; Azhdarinia, A.; Kohanim, S.; Edmund Kim, E.; et al. Assessment of cyclooxygense-2 expression with 99mTc-labeled celebrex. Anticancer. Drugs 2004, 15, 255–263. [Google Scholar] [CrossRef]
- Méric, J.-B.; Rottey, S.; Olaussen, K.; Soria, J.-C.; Khayat, D.; Rixe, O.; Spano, J.-P. Cyclooxygenase-2 as a target for anticancer drug development. Crit. Rev. Oncol. Hematol. 2006, 59, 51–64. [Google Scholar] [CrossRef]
- Farouk, N.; El-Tawoosy, M.; Ayoub, S.; El-Bayoumy, A.S. Optimization of the reaction conditions for the preparation of 99mTc-celecoxib and its biological evaluation. J Radioanal Nucl Chem 2011, 290, 685–690. [Google Scholar] [CrossRef]
- Vijayta D Chadha, Pearl laird, Gowsia Jan and Anna Ara Khan. Radiosynthesis, Biodistribution and Scintigraphic Imaging of 99mtc-Celecoxib in Experimental Rat Model of Colon Carcinogenesis. Available online: https://austinpublishinggroup.com/nuclear-medicine-radiotherapy/fulltext/ajnmr-v2-id1010.php (accessed on 28 February 2021).
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-X.; Zhou, M.; Ma, H.-L.; Qiao, Y.-B.; Li, Q.-S. The Role of Chronic Inflammation in Various Diseases and Anti-inflammatory Therapies Containing Natural Products. ChemMedChem 2021, 16, 1576–1592. [Google Scholar] [CrossRef]
- Peters, E.; Del Rey, A.; Krüger, K.; Rummel, C. 2nd European Psychoneuroimmunology Network Autumn School: The Skin-Brain Axis and the Breaking of Barriers. Neuroimmunomodulation 2023, 30 Suppl 1, 3–7. [Google Scholar] [CrossRef]
- Bajinka, O.; Simbilyabo, L.; Tan, Y.; Jabang, J.; Saleem, S.A. Lung-brain axis. Crit. Rev. Microbiol. 2022, 48, 257–269. [Google Scholar] [CrossRef]
- Rummel, C.; Del, R.A.; Bähr, L.; Krüger, K.; Peters, E. 1st European Psychoneuroimmunology Network (EPN) Autumn School: Lung-Brain Axis in Health and Disease. Neuroimmunomodulation 2022, 29 Suppl 2. [CrossRef]
- Hosang, L.; Canals, R.C.; van der Flier, F.J.; Hollensteiner, J.; Daniel, R.; Flügel, A.; Odoardi, F. The lung microbiome regulates brain autoimmunity. Nature 2022, 603, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, W.; Lin, F.; Li, W.; Wang, P.; Liao, G.; Zhang, L. Functional Two-Way Crosstalk Between Brain and Lung: The Brain-Lung Axis. Cell. Mol. Neurobiol. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alzghool, O.M.; van Dongen, G.; van de Giessen, E.; Schoonmade, L.; Beaino, W. α-Synuclein Radiotracer Development and In Vivo Imaging: Recent Advancements and New Perspectives. Mov. Disord. 2022, 37, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.; Schäffer, J.; Herden, C.; Pflieger, F.J.; Reiche, S.; Körber, S.; Kitagawa, H.; Welter, J.; Michels, S.; Culmsee, C.; et al. n-3 Polyunsaturated Fatty Acids Modulate LPS-Induced ARDS and the Lung-Brain Axis of Communication in Wild-Type versus Fat-1 Mice Genetically Modified for Leukotriene B4 Receptor 1 or Chemerin Receptor 23 Knockout. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Goggi, J.L.; Claser, C.; Hartimath, S.V.; Hor, P.X.; Tan, P.W.; Ramasamy, B.; Abdul Rahman, H.; Cheng, P.; Chang, Z.W.; Nguee, S.Y.T.; et al. PET Imaging of Translocator Protein as a Marker of Malaria-Associated Lung Inflammation. Infect. Immun., 89. [CrossRef]
- Chen, D.L.; Agapov, E.; Wu, K.; Engle, J.T.; Solingapuram Sai, K.K.; Arentson, E.; Spayd, K.J.; Moreland, K.T.; Toth, K.; Byers, D.E.; et al. Selective Imaging of Lung Macrophages Using 11CPBR28-Based Positron Emission Tomography. Mol. Imaging Biol. 2021, 23, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Mandhair, H.; Smyth, E.; Dakin, S.G.; Kiriakidis, S.; Wells, L.; Owen, D.; Sabokbar, A.; Taylor, P. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ’M1’ human macrophages. PloS One 2017, 12, e0185767. [Google Scholar] [CrossRef] [PubMed]
- Hatori, A.; Yui, J.; Yamasaki, T.; Xie, L.; Kumata, K.; Fujinaga, M.; Yoshida, Y.; Ogawa, M.; Nengaki, N.; Kawamura, K.; et al. PET imaging of lung inflammation with 18FFEDAC, a radioligand for translocator protein (18 kDa). PloS One 2012, 7, e45065. [Google Scholar] [CrossRef]
- Jones, H.A.; Valind, S.O.; Clark, I.C.; Bolden, G.E.; Krausz, T.; Schofield, J.B.; Boobis, A.R.; Haslett, C. Kinetics of lung macrophages monitored in vivo following particulate challenge in rabbits. Toxicol. Appl. Pharmacol. 2002, 183, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Swati Shah; Sanhita Sinharay; Reema Patel; Jeffrey Solomon; Ji Hyun Lee; William Schreiber-Stainthorp; Falguni Basuli; Xiang Zhang; Katie R. Hagen; Rebecca Reeder; et al. PET imaging of TSPO expression in immune cells can assess organ-level pathophysiology in high-consequence viral infections. Proc. Natl. Acad. Sci. U. S. A. 2022, 119. [CrossRef]
- Ordonez, A.A.; Pokkali, S.; DeMarco, V.P.; Klunk, M.; Mease, R.C.; Foss, C.A.; Pomper, M.G.; Jain, S.K. Radioiodinated DPA-713 imaging correlates with bactericidal activity of tuberculosis treatments in mice. Antimicrob. Agents Chemother. 2015, 59, 642–649. [Google Scholar] [CrossRef]
- Ruiz-Bedoya, C.A.; Mota, F.; Ordonez, A.A.; Foss, C.A.; Singh, A.K.; Praharaj, M.; Mahmud, F.J.; Ghayoor, A.; Flavahan, K.; Jesus, P. de; et al. 124I-Iodo-DPA-713 Positron Emission Tomography in a Hamster Model of SARS-CoV-2 Infection. Mol. Imaging Biol. 2022, 24, 135–143. [Google Scholar] [CrossRef]
- van der Krogt, J.M.A.; van Binsbergen, W.H.; van der Laken, C.J.; Tas, S.W. Novel positron emission tomography tracers for imaging of rheumatoid arthritis. Autoimmun. Rev. 2021, 20, 102764. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, M.; van Steenbergen, H.W.; Jong, P.H.P. de; van der Helm-van Mil, A.H.M. Unraveling heterogeneity within ACPA-negative rheumatoid arthritis: the subgroup of patients with a strong clinical and serological response to initiation of DMARD treatment favor disease resolution. Arthritis Res. Ther. 2022, 24, 4. [Google Scholar] [CrossRef]
- Novella-Navarro, M.; Plasencia, C.; Tornero, C.; Navarro-Compán, V.; Cabrera-Alarcón, J.L.; Peiteado-López, D.; Nuño, L.; Monjo-Henry, I.; Franco-Gómez, K.; Villalba, A.; et al. Clinical predictors of multiple failure to biological therapy in patients with rheumatoid arthritis. Arthritis Res. Ther. 2020, 22, 284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, J.; Xie, D.; He, D.; Lu, A.; Liang, C. Toward Overcoming Treatment Failure in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 755844. [Google Scholar] [CrossRef]
- Bruijnen, S.T.G.; Verweij, N.J.F.; Gent, Y.Y.J.; Huisman, M.C.; Windhorst, A.D.; Kassiou, M.; van de Ven, P.M.; Lammertsma, A.A.; Hoekstra, O.S.; Voskuyl, A.E.; et al. Imaging disease activity of rheumatoid arthritis by macrophage targeting using second generation translocator protein positron emission tomography tracers. PloS One 2019, 14, e0222844. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Owen, D.R.; Mandhair, H.; Smyth, E.; Carlucci, F.; Saleem, A.; Gunn, R.N.; Rabiner, E.A.; Wells, L.; Dakin, S.G.; et al. Translocator Protein as an Imaging Marker of Macrophage and Stromal Activation in Rheumatoid Arthritis Pannus. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1125–1132. [Google Scholar] [CrossRef]
- Gent, Y.Y.J.; Voskuyl, A.E.; Kloet, R.W.; van Schaardenburg, D.; Hoekstra, O.S.; Dijkmans, B.A.C.; Lammertsma, A.A.; van der Laken, C.J. Macrophage positron emission tomography imaging as a biomarker for preclinical rheumatoid arthritis: findings of a prospective pilot study. Arthritis Rheum. 2012, 64, 62–66. [Google Scholar] [CrossRef]
- Gent, Y.Y.; Ahmadi, N.; Voskuyl, A.E.; Hoetjes, N.; van, K.C.; Britsemmer, K.; Turkstra, F.; Boers, M.; Hoekstra, O.S.; van, d.L.C. Detection of subclinical synovitis with macrophage targeting and positron emission tomography in patients with rheumatoid arthritis without clinical arthritis. J. Rheumatol. 2014, 41. [Google Scholar] [CrossRef] [PubMed]
- Gent, Y.Y.J.; Ter Wee, M.M.; Voskuyl, A.E.; den Uyl, D.; Ahmadi, N.; Dowling, C.; van Kuijk, C.; Hoekstra, O.S.; Boers, M.; Lems, W.F.; et al. Subclinical synovitis detected by macrophage PET, but not MRI, is related to short-term flare of clinical disease activity in early RA patients: an exploratory study. Arthritis Res. Ther. 2015, 17, 266. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, F.; Gaemperli, O.; Kinderlerer, A.R.; Lamare, F.; Shalhoub, J.; Davies, A.H.; Rimoldi, O.E.; Mason, J.C.; Camici, P.G. Imaging of vascular inflammation with 11C-PK11195 and positron emission tomography/computed tomography angiography. J. Am. Coll. Cardiol. 2010, 56, 653–661. [Google Scholar] [CrossRef]
- Li, J.; Xiao, J.; Liang, D.; Zhang, H.; Zhang, G.; Liu, Y.; Zhang, Y.; Liu, Y.; Yu, Z.; Yan, B.; et al. Inhibition of mitochondrial translocator protein prevents atrial fibrillation. Eur. J. Pharmacol. 2010, 632, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, S.; Silvola, J.M.U.; Kiugel, M.; Liljenbäck, H.; Savisto, N.; Li, X.-G.; Thiele, A.; Lehmann, L.; Heinrich, T.; Vollmer, S.; et al. 18-kDa translocator protein ligand 18F-FEMPA: Biodistribution and uptake into atherosclerotic plaques in mice. J. Nucl. Cardiol. 2017, 24, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Maulik, S.K.; Kumar, S. Oxidative stress and cardiac hypertrophy: a review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef]
- Xiao, J.; Liang, D.; Zhang, H.; Liu, Y.; Li, F.; Chen, Y.-H. 4’-Chlorodiazepam, a translocator protein (18 kDa) antagonist, improves cardiac functional recovery during postischemia reperfusion in rats. Exp. Biol. Med. (Maywood) 2010, 235, 478–486. [Google Scholar] [CrossRef]
- Mou, T.; Tian, J.; Tian, Y.; Yun, M.; Li, J.; Dong, W.; Lu, X.; Zhu, Z.; Mi, H.; Zhang, X.; et al. Automated synthesis and preliminary evaluation of 18FFDPA for cardiac inflammation imaging in rats after myocardial infarction. Sci. Rep. 2020, 10, 18685. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, J.T.; Bengel, F.M. Molecular Imaging of Myocardial Inflammation With Positron Emission Tomography Post-Ischemia: A Determinant of Subsequent Remodeling or Recovery. JACC Cardiovasc. Imaging 2018, 11, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, J.T.; Hupe, H.C.; Wang, Y.; Bankstahl, J.P.; Berding, G.; Ross, T.L.; Bauersachs, J.; Wollert, K.C.; Bengel, F.M. Myocardial Inflammation Predicts Remodeling and Neuroinflammation After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 263–275. [Google Scholar] [CrossRef]
- Meissner, A.; Visanji, N.P.; Momen, M.A.; Feng, R.; Francis, B.M.; Bolz, S.-S.; Hazrati, L.-N. Tumor Necrosis Factor-α Underlies Loss of Cortical Dendritic Spine Density in a Mouse Model of Congestive Heart Failure. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



