Submitted:
06 October 2023
Posted:
06 October 2023
You are already at the latest version
Abstract
Several genetic tools have been developed for Clostridium acetobutylicum utilizing 5-fluorouracil (5FU) or 5-fluorocytosine (5FC) resistance as a selection method. A method based on the integration, by single crossing-over, of a suicide plasmid (pCat-upp) followed by selection for the second crossing-over using a counter-selectable marker (the upp gene and 5FU resistance) was recently developed for genome editing in C. acetobutylicum. This method allows the modification of the genome without any marker or scar left in a strain of C. acetobutylicum that is ∆upp. Unfortunately, 5FU has strong mutagenic properties inducing mutations in the strain’s genome. After numerous applications of the pCat-upp/5FU system for genome modification in C. acetobutylicum, the bacteria became completely resistant to 5FU in the presence of the upp gene, resulting in failure in the selection for the second crossing-over. It was found that the potential repressor of the pyrimidine operon, PyrR, was mutated at position A115, leading to the 5FU resistance of the strain. We developed two plasmids, one overexpressing the native pyrR gene and a suicide plasmid carrying a non-mutated and optimized pyrR gene (pyrR*) and upp, which allowed us to restore the 5FU sensitivity of the strain. We also improved the pCat-upp/5FU system by reducing the concentration of 5FU from 1mM to 5 µM using a define synthetic medium.
Keywords:
Clostridium acetobutylicum
; genome edition
; 5FU
; PyrR
1. Introduction
In recent years, solventogenic Clostridia have garnered significant attention in the post-genomic era, primarily owing to the comprehensive sequencing and annotation of their genomes [1,2]. This wealth of genomic information has provided valuable insights into the metabolism of these industrially important strains, thereby catalyzing new approaches to genetic analysis, functional genomics, and metabolic engineering for the development of industrial strains geared towards biofuel and bulk chemical production.
To facilitate these endeavors, various reverse genetic tools have been devised for solventogenic Clostridia. These tools include markerless gene inactivation systems, employing methods such as homologous recombination with non-replicative [3,4,5] and replicative plasmids [6,7,8,9], as well as the insertion of group II introns [10,11]. For all homologous recombination-based methods involving two crossing-over, the use of a counterselection technique is imperative. This may involve employing CRISPR-Cas9 [12,13] or a counterselectable marker, which have been constructed using the codon-optimized mazF toxin gene from Escherichia coli (under the control of a lactose-inducible promoter) [7], the pyrE [8] gene (encoding an orotate phosphoribosyltransferase, leading to 5-fluoroorotate (5-FOA) toxicity), the upp gene (encoding an uracil phosphoribosyltransferase, leading to 5-fluorouracil (5-FU) toxicity) [5,9], or the codA gene [4] (encoding a cytosine deaminase that converts 5-Fluorocytosine to 5-FU, which is further transformed into a toxic compound by the product of the upp gene).
It is worth noting that while strategies relying on 5FC/5FU selection are highly effective, they should be employed cautiously. 5FU is a well-known anticancer drug recognized for its mutagenic properties in human cancers [14]. These mutagenic attributes have been demonstrated in various organisms, including Caenorhabditis elegans, E. coli, or Mycobacterium tuberculosis [15,16,17].
In our study, we will demonstrate that a mutation impairs the 5FU counterselection method. Upon identification of this issue, we endeavored to enhance the 5FU counterselection method for genome editing in C. acetobutylicum. This improvement involved the development of a corrective and preventive suicide plasmid, inspired by Foulquier et al. [5], featuring the introduction of a synthetic codon-optimized pyrR gene, referred to as pyrR*. Additionally, we created a corrective replicative plasmid, building upon the work of Raynaud et al. [18], which incorporated the native pyrR gene. These two plasmids effectively circumvented the unanticipated 5FU resistance observed in C. acetobutylicum, substantially improving our team's previously described method by also reducing the concentration of 5FU required by a factor of 200 (from 1 mM to 5 µM) using a synthetic define medium.
2. Materials and Methods
2.1. Bacterial strains, plasmids and oligonucleotides
2.2. Growth Conditions
E. coli strains were grown in Luria-Bertani (LB) medium. C. acetobutylicum strains were maintained as spores in synthetic medium (SM) at - 20°C as previously described or, for non-sporulating strains, directly in degassed and sterile serum bottles at - 80°C [21,22,23]. Spores were activated by heat shock at 80°C for 15 min. Strains were grown under anaerobic conditions at 37 °C in Clostridial Growth Medium (CGM) supplemented each time with 30 gL-1 of glucose [24] or in CGM supplemented with 20 gL-1 MES hydrate (Sigma Aldrich) , synthetic medium (SM) or in SM supplemented with 20 gL-1 MES hydrate or in Reinforced Clostridial Medium (RCM) (Millipore). The pH of CGM was adjusted at 6.0 or 5.2 with hydrochloric acid. The pH of RCM was adjusted at 5.8 with hydrochloric acid. The SM used for C. acetobutylicum growth contained per liter of deionized water: Glucose, 30 g; KH2PO4, 0.50 g; K2HPO4, 0.50 g; MgSO4.7H2O, 0.22 g; acetic acid, 2.3 mL; FeSO4.7H2O 10 mg; para amino benzoic acid, 8 mg; biotin, 0.08 mg, nickel (II) chloride, 3 mg; zinc chloride, 60 mg; nitriloacetic acid, 0.2 g. The pH of the medium was adjusted to 6.0 with ammonia. For solid media preparation, 1.5 % agar was added to liquid media. The media were supplemented as needed with the appropriate antibiotic at the following concentrations: for C. acetobutylicum, erythromycin (Ery) at 40 µg/mL, clarithromycin (Clari) at 40 µg/mL, and thiamphenicol (Tm) at 10 µg/mL; for E. coli, carbenicillin (Cb) at 100 µg/mL and chloramphenicol (Cm) at 30 µg/mL. Stocks of 5-Fluorouracil (5FU) and uracil (Sigma Aldrich) were prepared at 0.1 M in dimethyl sulfoxide (DMSO) and stored at - 20°C.
2.3. DNA manipulation
Genomic DNA was extracted from C. acetobutylicum strains using GenEluteTM Bacterial Genomic DNA Kits (Sigma-Aldrich). Plasmids DNA were extracted from E. coli using NucleoSpin® Plasmid or NucleoBond® Xtra Midi kits (Macherey-Nagel). Phusion DNA Polymerase (New England Biolabs (NEB)) was used to generate PCR products according to the supplier's standard protocols. OneTaq® 2X Master Mix with Standard Buffer (NEB) was used to screen colonies by PCR according to the supplier's standard protocols. Restriction enzymes, antartic phosphatase, T4 DNA ligase (NEB) were used according to the manufacturer's instructions. DNA fragments were purified from agarose gel using ZymocleanTM Large Fragment DNA Recovery Kit (Zymo Research). DNA PCR fragments were purified using NucleoSpin® Gel and PCR Clean-up (Macherey-Nagel). Plasmid DNA and DNA PCR fragments were sequenced using sanger method (Eurofins Genomics).
2.4. Design of pyrR*
The nucleotide sequence of pyrR gene (CA_C2113) was codon optimized to create a synthetic pyrR gene, named pyrR*, with low identity to the wildtype pyrR, in which some codons have been replaced by other codons with closed frequency in C. acetobutylicum using https://gcua.schoedl.de/. The synthetic gene was then synthesized (Geneart, Thermofisher Scientific).

Table 3.
Nucleotide sequence of wild-type pyrR gene and pyrR*gene.
| Gene name | Nucleotide sequence |
| wild-type pyrR synthetic pyrR (pyrR*) |
ATGAATTTAAAAGCAAAGATTTTAGATGATAAGGCTATGCAAAGGACTTTGACCAGAATAGCACATGAAATTATAGAAAAGAATAAAGGTATAGATGATATAGTACTAGTAGGAATAAAGAGAAGAGGAGTTCCAATAGCCGATAGAATAGCGGATATAATTGAAGAAATAGAAGGAAGTAAGGTTAAGCTAGGAAAAGTAGATATAACCTTATATAGAGACGATTTGTCTACGGTAAGTTCTCAACCAATAGTAAAAGATGAGGAAGTATATGAAGATGTAAAGGATAAGGTAGTAATACTTGTTGATGACGTTTTATATACAGGAAGAACATGCAGAGCAGCCATAGAAGCTATTATGCATAGAGGAAGACCAAAGATGATACAGCTTGCAGTTTTGATAGATAGGGGACATAGAGAACTTCCTATAAGGGCAGATTATGTTGGAAAAAATGTACCTACATCAAAAAGTGAATTGATATCGGTAAATGTTAAAGGAATAGATGAAGAGGATTCAGTAAACATTTATGAGTTGTAG ATGAATCTTAAAGCTAAGATTCTTGATGATAAGGCAATGCAAAGGACACTAACCAGAATAGCTCATGAAATAATAGAAAAGAATAAAGGAATAGATGATATAGTTTTGGTTGGAATAAAGAGAAGAGGAGTACCTATAGCGGATAGAATAGCCGATATAATAGAAGAAATAGAAGGATCAAAGGTAAAGTTGGGAAAAGTTGATATAACCCTTTATAGAGACGATCTATCAACCGTTTCAAGTCAACCTATAGTTAAAGATGAGGAAGTTTATGAAGATGTTAAGGATAAGGTTGTTATATTAGTTGATGACGTACTTTATACTGGAAGAACTTGCAGAGCTGCGATAGAAGCAATAATGCATAGAGGAAGACCTAAGATGATACAGTTAGCTGTACTAATAGATAGGGGACATAGAGAACTACCAATAAGGGCTGATTATGTAGGAAAAAATGTTCCAACTAGTAAATCAGAATTGATATCCGTAAATGTAAAAGGAATAGATGAAGAGGATAGTGTTAACATATATGAGCTATAG |
2.5. Construction of pCat-upp-pyrRmut
This plasmid was constructed, based on the pCat-upp described by Foulquier et al. [5] by introducing the pyrRmut gene containing the mutation g.344C>T encoding the PyrR A115V protein found in our mutant strain. The mutant pyrR gene was PCR amplified from total DNA from CAB1060 as template using the PSC 75 and PSC 76 primers containing BamHI restriction sites. The PCR fragment and the pCat-upp were digested by BamHI. The plasmid was dephosphorylated with antarctic phosphatase and the PCR fragment and the plasmid were purified. The PCR fragment was cloned by ligation into the plasmid to obtain pCat-upp-pyrRmut.
2.6. Construction of pCat-upp-pyrR*
This plasmid was constructed from the pCat-upp described by Foulquier et al. [5] by introducing the synthetic pyrR* gene under the control of the thiolase promoter. The entire pCat-upp plasmid was amplified with PSC 51 and PSC 52 for linearization. The pyrR* gene was PCR amplified primers using synthetic pyrR gene (from Geneart) as template. A first PCR was performed with PSC 61 and PSC 62 primers to amplify the pyrR gene with its native RBS and downstream homology arms. The first PCR fragment was then purified. A second PCR was performed on the first PCR product with PSC 72 and PSC 62 to introduce upstream homology arm. The final PCR fragment of pyrR* was cloned into linearized pCat-upp plasmid by recombination using the GeneArtTM Seamless Cloning and Assembly Kit (Thermofisher Scientific).
2.7. Construction of pCat-upp-pyrR*- Δldh
This plasmid was constructed based on the pCat-upp-pyrR* (this study) and the pCat-upp-Δldh described by Soucaille et al. in the patent WO 2016/042160 A1 [20]. The pCat-upp-pyrR* plasmid was linearized by digestion with BamHI restriction enzyme and dephosphorylated with antartic phosphatase. Then the pCat-upp-Δldh plasmid was digested with BamHI and the fragment containing the ldh homology arms was purified from agarose gel. The two fragments were then ligated using T4 DNA Ligase.
2.8. Construction of pCat-upp-pyrR*-Δldh::sadh-hydG
This plasmid was constructed based on the pCat-upp-pyrR*-Δldh plasmid (this study) by introducing an operon composed of sadh and hydG genes (GenBank: AF157307.2), with their own RBS, under the control of the ldh promoter. The pCat-upp-Δldh was digested by StuI, dephosphorylated and purified. sadh and hydG were amplified from a synthesized sadh_hydG genes (Genart, Thermofisher) as template. A first PCR was performed with PSC 104 and PSC 105 to amplify sadh_hydG genes and introduce the ldh promoter region upstream of sadh and the ldh terminator downstream of hydG. After purification, the first PCR product was then amplified with PSC 106 and PSC 107 to introduce upstream and downstream homology arms to recombine with the pCat-upp-pyrR*-Δldh plasmid. The final PCR fragment was cloned into the pCat-upp-pyrR*-Δldh by recombination using the GeneArtTM Seamless Cloning and Assembly Kit (Thermofisher Scientific).
2.9. Construction of pSOS95-pyrR
This plasmid was constructed based on the pSOS95 plasmid described by Raynaud et al. [18] by introducing the native pyrR gene under the control of the thiolase promoter. The pSOS95 plasmid was digested with BamHI and SfoI and purified from agarose gel. The pyrR gene was amplified using total DNA from the strain Δcac1502 as template. The PSC 58 and PSC 46 used for this amplification introduced the BamHI restriction site upstream of the pyrR gene RBS and the SfoI restriction site downstream of the pyrR gene. The PCR fragment was digested with BamHI and SfoI, purified. And cloned in the pSOS95 plasmid by ligation using T4 DNA Ligase.
2.10. Transformation protocol
Transformation of C. acetobutylicum was performed by electroporation according to the following protocol. From a culture of C. acetobutylicum in CGM at A620 between 1 and 2, a new serum bottle with 50 ml of CGM was inoculated at A620 of 0.1. When the culture reached A620 between 0.6 and 0.8, the culture has been placed on ice for 30 minutes and transferred under an anaerobic chamber (Jacomex) where all the following manipulations were performed. The cells were then harvested by centrifugation at 7000g for 15 minutes and washed in 10 mL of ice-cold electroporation buffer (EB) composed of 270 mM sucrose, 10 mM MES hydrate at pH 6.0. Then the pellet was resuspended in 500 µL of EB and cells were transferred into a sterile electrotransformation vessel (0.40 cm electrode gap x 1.00 cm) with 5-100 µg plasmid DNA. A 1.8 kV discharge was applied to the suspension from a 25 µF capacitor and a 400 Ω resistance in parallel using the Gene Pulser (Bio-Rad Laboratories, Richmond, CA). Cells were transferred directly to 10 mL of warm CGM and incubated for 6 hours at 37°C before plating on RCM supplemented with the required antibiotics.
2.11. Microbiological enumeration on solid media
C. acetobutylicum was cultivated in CGM until reaching an A620 of 0.55. Subsequently, the culture was transferred to an anaerobic chamber, and 100 µL of various dilutions (10-1 to 10-6) of the culture were plated onto CGM MES or SM MES agar supplemented with the necessary antibiotics, ranging from 0 µM to 200 µM for 5FU and from 0 µM to 50 µM for uracil. Following incubation at 37 °C for a period of 1 to 4 days, the resulting colonies were counted.
2.12. 5FU selection protocol
C. acetobutylicum was cultivated in CGM until reaching an A620 of 0.55. The spreading protocol was the same as described in subsection 2.11. After isolation, 50 colonies were picked and plated on a fresh plate with the same concentration of 5FU. The plates were incubated at 37 °C from 1 to 2 days. The colonies were then picked and patched onto plates with and without thiamphenicol to determine the percentage of double crossing-over. Colonies showing a double crossover phenotype were screened by PCR to verify that genome editing occurred.
2.13. Locus verification in C.acetobutylicum after metabolic engineering
After the genome edition of C. acetobuylicum, the different loci were checked by PCR amplification. In order to check for the insertion of point mutations, the genome was amplified by PCR and the PCR fragment obtained was sent for sequencing.
| Primers name | Function |
| PSC 39 – PSC 40 | pyrR gene sequencing |
| PSB 384 – PSB 385 | ldh locus verification |
2.14. Analytical procedures
Culture growth was monitored by measuring optical density over time using a spectrophotometer at A620. Glucose, acetate, butyrate, acetone, isopropanol, ethanol and butanol concentrations were measured using High Performance Liquid Chromatography (HPLC) analysis (Agilent 1200 series, Massy, France). The separations were performed on a Biorad Aminex HPX-87H column (300 mm x 7.8 mm), and detection was achieved using either a refractive index measurement or a UV absorbance measurement (210 nm). The operating conditions were as follows: temperature, 14 °C; mobile phase, H2SO4 (0.5 mM); and flow rate, 0.5 ml/min.
3. Results
3.1. Identification of the 5FU resistance of the strain
The strain CAB1060, as detailed by Nguyen et al. [19], was developed through the utilization of the upp/5FU counterselection method. This genome editing method, initially described by Croux et al. [8] and originally employing a replicative plasmid, was subsequently adapted into a suicide plasmid format, as outlined by Foulquier et al. [5].
After several genome modifications and the use of 5FU as a counter selection marker, the strain became no longer sensitive to 5FU even at a concentration of 1 mM and its entire genome was sequenced. Many random mutations were found including one that particularly caught our attention, the mutation g.344C>T located in the pyrR gene that introduced a A115V mutation in the PyrR protein. PyrR is a potential repressor of the pyrimidine operon and it has been shown in other organisms that mutations in the pyrR gene or a complete deletion of the pyrR gene can lead to 5FU resistance [16,17,25]. The hypothesis put forward was that the mutated protein no longer performed its regulatory function and the pyrimidine operon was overexpressed. This could result in overproduction of UMP, which protect bacteria from the toxic effects of 5FUMP. Based on these data, we hypothesized that the observed mutation could be responsible of 5FU resistance in the strain.
3.2. Evaluation of the 5FU sensitivity of C. acetobutylicum strain (non mutated in the pyrR gene)
The viability of the C. acetobutylicum ∆cac1502 strain was evaluated both on rich medium (CGM MES) and on synthetic medium (SM MES) in the presence of various concentrations of 5FU (Table 4). In the absence of 5FU, no significant differences could be observed between the two media. On the otherhand, the 5FU sensitivity of the strain was much higher when spread on SM MES (Table 4) compared to rich media.
According to Singh et al., exogenous uracil protects the bacteria from the toxicity of 5FU. This was validated in Mycobacterium tuberculosis in which the supplementation of uracil at 15.6 µM protected bacteria up to 25 µM 5FU [17]. On the basis of the literature, we have assumed that the yeast extract added to the rich medium contains between 25 µM and 50 µM of uracil [26]. Therefore, we tested the protective effect of uracil against 5FU in C. acetobutylicum by addition of uracil to the synthetic medium containing 5 µM of 5FU (Table 5).
As expected, we observed a protective effect of uracil but at very low concentration (5 µM) which is 5 to 10-fold lower than the expected concentration due to the addition of yeast extract. Based on these results, and in order to minimize the concentrations of 5FU used, all the following experiments were carried out in synthetic medium.
3.3. Construction of a C. acetobutylicum strain with pyrRmut and 5FU resistance validation
First, we tried to reproduce the occurrence of the mutation g.344C>T in pyrR gene obtained in the 5FU resistant CAB1060 strain. To test for the occurrence of mutations, we plated 2 x 108 cells of C. acetobutylicum strain Δcac1502 on SM MES agar plates with high concentrations of 5FU (25-50 µM). The occurrence of mutations in the pyrR and in upp genes were analyzed. No mutation in the upp was observed, but many mutations appeared at different positions in the pyrR gene (Table 6). However, the original mutation found in the pyrR gene in the 5FU resistant CAB1060 strain has not been obtained. To ensure that the mutation g.344C>T was responsible for the 5FU resistance of the CAB1060 strain, it was decided to introduce it into the C. acetobutylicum strain Δcac1502ΔuppΔcac3535 using a pCat-upp-pyrRmut. After the transformation with pCat-upp-pyrRmut and the 5FU selection, the insertion of g.344C>T mutation in pyrR gene was checked by sequencing. In the C. acetobutylicum strain Δcac1502ΔuppΔcac3535pyrRmut obtained, the pCat upp-Δldh was then integrated at the ldh locus. In a strain with a WT pyrR gene, the upp gene contained in the suicide plasmid should be sufficient to get a sensitivity to 5FU of the strain. But, as shown in Table 7, in the strain mutated in the pyrR gene, most of the cells were resistant to high concentrations of 5FU. This result showed that the single g.344C>T mutation in pyrR gene was sufficient to obtain a strain resistant to 5FU and confirmed our hypothesis concerning the 5FU resistance of the CAB1060 strain.
3.4. Restoration of the 5FU sensitivity in a strain mutated in pyrR gene
3.4.1. Restoration of 5FU sensitivity to 5FU resistant strains by overexpressing pyrR gene on a replicative plasmid
This first method consists of using a replicative plasmid overexpressing a native non-mutated version of pyrR, to overcome the problems of 5FU selection when a strain mutated in the pyrR gene has already integrated a pCat-upp. Viability of C. acetobutylicum strain Δcac1502ΔuppΔcac3535 pyrRmut with the pCat-upp-Δldh integrated at the ldh locus and the pSOS95-pyrR replicative plasmid was tested in the presence of erythromycin for the maintenance of the replicative plasmid overexpressing pyrR and/or thiamphenicol for the maintenance of the pCAT-upp-Δldh suicide vector. Whereas, previously, in the presence of Tm, most of the cells were resistant to 5FU (Table 7), by just overexpressing a WT pyrR gene in the same strain, we restore its sensitivity to 5FU even at low concentrations (Table 8).
Indeed, by overexpressing the pyrR gene, a selection with 5FU at a concentration of 5µM is sufficient to allow a high frequency (> 85 %) of double crossing-over in a pyrR mutant strain with an integrated pCat-upp in the genome. This frequency was even higher than 95 % at 10 µM of 5FU (Table 9). These results showed that it was possible to reverse 5FU resistance of a strain mutated in the pyrR gene with a pCat-upp integrated into its genome. The use of a replicative plasmid overexpressing a native pyrR gene allowed the excision of the suicide vector at very low 5FU concentrations.
3.4.2. Restoration of 5FU sensitivity to 5FU resistant strains by overexpressing a synthetic pyrR* gene on a suicide vector
The second method to overcome the problems of 5FU selection when a strain is mutated in the pyrR gene consists of using a codon optimised (for low identity to the wild type gene) version of the pyrR gene (pyrR*) directly on the suicide plasmid.
Viability of the C. acetobutylicum strain Δcac1502ΔuppΔcac3535 pyrRmut with a pCat-upp-pyrR*-Δldh integrated at the ldh locus was tested in the presence or absence of Tm for the maintenance of the suicide vector and no difference in viability was observed (Table 10).
Whereas previously, in the strain with the pCat-upp-Δldh, most of the cells were resistant to 5FU in the presence of Tm (Table 7), with the same suicide plasmid but containing the pyrR*, no colony were obtained (Table 10). These results validated the functionality of the synthetic pyrR* gene and showed that a single copy of pyrR* was sufficient to restore the sensitivity to 5FU of a resistant strain.
Indeed, by overexpressing the pyrR* gene directly on the suicide vector, a selection with 5FU at a concentration of 5 µM is sufficient to allow a high frequency (> 90 %) of double crossing-over in a pyrR mutant strain. This frequency was even higher than 98 % with 10 µM of 5FU (Table 11). Thus, overexpressing a single copy of pyrR* was sufficient to restore the sensitivity to 5FU of a resistant pyrRmut strain.
3.5. Preventive use of pyrR*
After demonstrating the efficiency of overexpressing pyrR curatively in a 5FU resistant strain, we wondered if we could use the same method preventively in a strain “non-mutated” for the pyrR gene to avoid the development of 5FU resistance. However, we first wanted to check that the overexpression of pyrR did not result in a too high 5FU sensitivity. Viability of the C. acetobutylicum strain Δcac1502ΔuppΔcac3535 with a pCat-upp-pyrR*-Δldh integrated at the ldh locus plasmid was assessed at low 5FU concentrations (5 and 10 µM) and the results showed that it was not affected (Table 12). In parallel, the frequency of double crossing-over, evaluated though the sensitivity to Tm, was shown to reach 100 % (Table 12). These results confirmed that overexpressing pyrR can be a method used both curatively and preventively.
3.6. Insertion of sadh and hydG from C. beijerinckii at ldh locus
After validating the new C. acetobutylicum genome editing method using pCat-upp-pyrR*, we wanted to test this tool to both delete and replace genes in a single step. The goal was to use the pCat-upp-pyrR*-Δldh::sadh-hydG suicide plasmid to delete the ldh gene and replace it with an operon, to produce isopropanol in the C. acetobutylicum strain Δcac1502ΔuppΔcac3535pyrRmut. This operon is composed of sadh and hydG genes from C. beijerinckii (Figure 1) which encoded for a primary-secondary alcohol dehydrogenase (SADH) [27] and a putative electron transfer protein (HydG) [28] from Clostridium beijerinckii NRRL B593, respectively. In the Figure 2 the different stages involved in the integration of the “isopropanol operon” at the ldh locus are described. After integration of the suicide vector at the ldh locus and 5FU selection at 5 µM, Tm sensitive colonies were selected and screened by PCR using external primers. Primers were designed outside the homology arms, to discriminate between WT revertants and mutants with the desired genotype (Δcac1502ΔuppΔcac3535 pyrRmut Δldh::sadh hydG) (Figure 3). After picking and patching colonies, a second time on 5FU, we obtained 92 % of viable colonies using 5FU at 5 µM. After selecting on Tm, all viable clones were shown to be sensitive to 5FU, i.e. 100 % excision of pCat-upp-pyrR* was achieved (Table 13). Both WT revertants and mutants with the desired genotype were obtained. In the figure 3, an example of two mutant clones (11 & 12) and one WT revertant clone (clone 15) is shown. The pCat-upp-pyrR* tool can therefore be used to delete genes and replace them with others in a single step.
3.7. Culture of C. acetobutylicum on synthethic medium for isopopanol production
Figure 4.
a Growth profile of the C. acetobutylicum strain ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG and C. acetobutylicum strain ∆cac1502∆upp∆cac3535 on SM b Monitoring glucose consumption and solvents production over time of C. acetobutylicum strain ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG c Monitoring glucose consumption and solvents production over time of C. acetobutylicum strain ∆cac1502∆upp∆cac3535 d Molar yields of solvents production. All the measurements shown are mean average (n=2). Errors bars represent the standard deviation.
Figure 4.
a Growth profile of the C. acetobutylicum strain ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG and C. acetobutylicum strain ∆cac1502∆upp∆cac3535 on SM b Monitoring glucose consumption and solvents production over time of C. acetobutylicum strain ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG c Monitoring glucose consumption and solvents production over time of C. acetobutylicum strain ∆cac1502∆upp∆cac3535 d Molar yields of solvents production. All the measurements shown are mean average (n=2). Errors bars represent the standard deviation.
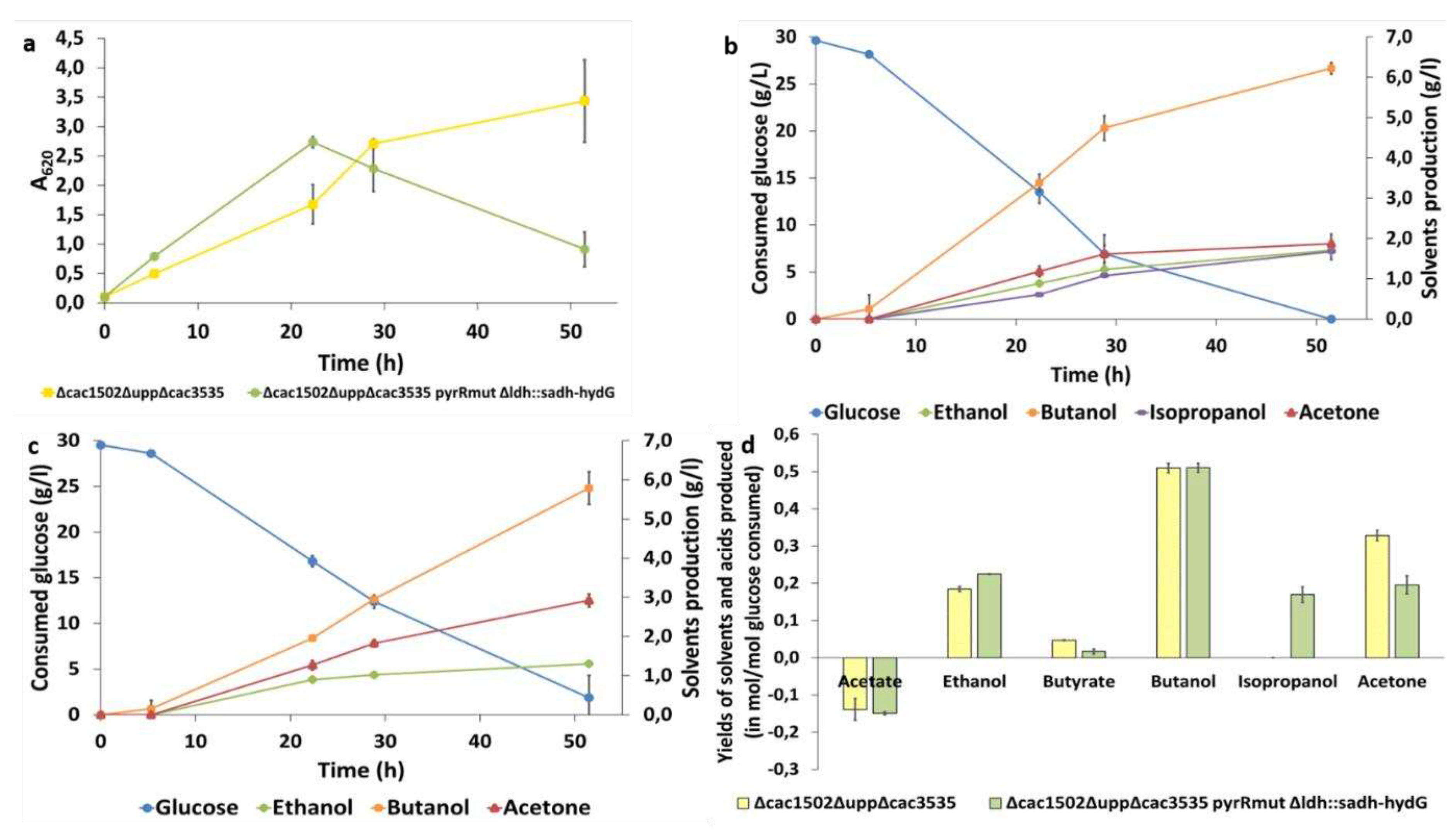
To validate the functionality of the “isopropanol operon”, the C. acetobutylicum strain ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG and the control strain, without isopropanol production pathway, were then cultured in synthetic media in serum bottles at an initial pH of 6.0. After 52 hours of culture, solvents and acids productions and final product yields were evaluated. The strain ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG had a growth rate of 0.15 h-1 during the first 25 h of cultures and reached an A620 maximum of 2.5 and then entered the lysis phase after 25 h of culture (Figure 4 a). The strain Δcac1502ΔuppΔcac3535, on the other hand, had a growth rate of 0.13 h-1 during the first 25 h of cultures, reached an A620 maximum of 2.9 and had not entered the lysis phase after 50 h of culture (Figure 4 a). Concerning isopropanol, its production was only detected in the C. acetobutylicum strain ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG . As expected, final molar yields showed that the isopropanol production observed is associated to a decreased acetone production in comparison to the control strain (acetone production is 1.7-fold-time higher in the control strain) (Figure 4 d). The isopropanol/acetone molar ratio was 0.87 in ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG strain. This strain also had higher acetate consumption, lower butyrate production and slightly higher ethanol production (Figure 4 d). The insertion of the “isopropanol operon” with the new pCat-upp-pyrR*/5FU system has been confirmed by the production of isopropanol in the strain ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG.
4. Discussion
In the present study, we have shown that overexposure of C. acetobutyicum to 5FU can make it resistant to this drug. Spontaneous mutations are induced in the bacterial chromosome, notably in the pyrR gene. As described in other publications, the pyrR gene encodes for PyrR, the repressor of the pyrimidine operon [16,17,25]. The presence of a mutation in this protein can lead to the cessation of its function, resulting in an overproduction of UMP. This overproduction of UMP can protect against the harmful effects of 5FUMP, a molecule that is toxic to bacteria. We observed the appearance of a mutation in the PyrR protein of C. acetobutylicum that completely avoids selection of the double crossing-over step when using the pCatupp/5FU system. The principle of the upp/5FU system was based on the use of a strain in which the upp gene has been deleted and the use of 5FU as a counter selection agent. The upp gene encoding for an uracil phosphoribosyl transferase (UPRTase) that could convert uracil to UMP and 5FU to 5FUMP. 5FUMP is a molecule that prevents cells from producing intermediates needed for DNA synthesis, thereby causing cell death (Figure 5) [17].
When the A115V PyrR mutation was discovered in C. acetobutylicum, we compared it with other mutations in homologous proteins already described in the literature. A conserved protein sequence required in PRPP binding can be found in many species. Ghode and Singh described the G125V and R126C mutations in M. tuberculosis as being in this conserved zone [16,17]. According to Ghode, a mutation in this region could block the production of 5FUMP. As A115V PyrR mutation is situated close to this site, it could be one of the reasons why our strain is resistant to 5FU (Figure 6). In addition, pyrR encodes the regulatory protein of the pyrimidine operon. According to Ghode and Fields [16,29], mutations in PyrR of M. tuberculosis or M. smegmatis, or when PyrR is completely deleted in B. subtilis the protein no longer performs its regulatory function and the pyrimidine operon is overexpressed [25]. This results in overproduction of UMP, which protects the bacteria from the toxic effects of 5FUMP.
To overcome this problem, we had to revise the protocol previously described by our team. First, we realized that the composition of the medium plays an important role in the resistance of the strain to 5FU. It is preferable to use a synthetic medium that is not supplemented with uracil. In fact, the yeast extract present in CGM brings uracil into the medium and thus protects against the toxic effect of 5FUMP. This hypothesis was tested by adding low concentrations of uracil to the synthetic medium. Bacterial growth was no longer affected by the presence of 5FU in the medium. After optimizing the medium for 5FU selection, we constructed two plasmids to restore the sensitivity of the strain to 5FU. The first plasmid is a replicative plasmid overexpressing a native version of pyrR. It is used to overcome the problems of 5FU selection when a strain mutated in the pyrR gene has already integrated a pCat-upp. The second plasmid is a pCat-upp containing a codon optimised version of the pyrR gene, called pyrR*. The 5FU selection problem for genome editing is directly bypassed by this method. With both of these strategies, the concentration of 5FU could be reduced from 1 mM to 5 µM, thus minimizing the risk of spontaneous mutation. Both the use of pyrR* and the use of the SM medium will be beneficial for all the counterselection methods involving upp and 5FU, as well as those utilizing codA and 5FC [4] .
Once the new protocol was established, we demonstrated that it was possible to both delete and insert genes of interest in C. acetobutylicum ∆cac1502∆upp∆cac3535 pyrRmut strain in a single step using the pCat-upp-pyrR*/5FU system. An isopropanol production pathway from C. beijerinckii was inserted at the ldh of C. acetobutylicum ∆cac1502∆upp∆cac3535 pyrRmut∆ldh::sadh-hydG strain using this technique. We decided to insert the sadh and hydG genes from C. beijerinckii NRRL 593 following a publication by Dusséaux et al. [27]. SADH is an NADPH-dependent primary-secondary alcohol dehydrogenase that catalyzes acetone reduction and HydG is a putative electron transfer protein [28,32]. hydG was introduced into the C. acetobutylicum ∆cac1502∆upp∆cac3535 pyrRmut strain genome at the same time as sadh, since these two genes are located in the same operon in C. beijerinkcii NRRL 593. It was assumed that the HydG activity would have a positive effect on the SADH activity, allowing the strain to obtain a better isopropanol production [28]. The final production of our C. acetobutylium ∆cac1502∆upp∆cac3535 pyrRmut ∆ldh::sadh-hydG strain is lower than the one obtained by Dusséaux (up to 4.7 g.L-1 of isopropanol produced in a culture of 30 h) with a lower molar ratio of isopropanol/acetone [27]. This can be explained by the fact that in this strain both gene were overexpressed in a multi copy replicative plasmid and under the control of the ptb promoter, which is a stronger promoter than the ldh promoter [33]. The strain had also been grown in pH-regulated batch cultures, whereas we grew our strain in serum bottles without pH regulation. Better results would probably be obtained if the “isopropanol operon” was introduced at the ptb buk locus and if the pH of the culture was controlled.
Author Contributions
Conceptualization, C.F. and P.S.; methodology, E.B.; validation, C.F. and P.S.; resources, P.S.; data curation, C.F.; writing—original draft preparation, E.B and C.F.; writing—review and editing, C.F. and P.S.; supervision, P.S and C.F.; project administration, P.S.; funding acquisition, P.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by ANR-19-COBI-0009 as part of the EraCoBioTech project, SynConsor4Butanol grant agreement number 722361.
Data Availability Statement
Not applicable.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nölling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, et al. Genome Sequence and Comparative Analysis of the Solvent-Producing Bacterium Clostridium acetobutylicum. J Bacteriol. 2001;183: 4823–4838. [CrossRef]
- Poehlein A, Solano JDM, Flitsch SK, Krabben P, Winzer K, Reid SJ, et al. Microbial solvent formation revisited by comparative genome analysis. Biotechnol Biofuels. 2017;10: 58. [CrossRef]
- Green EM, Boynton ZL, Harris LM, Rudolph FB, Papoutsakis ET, Bennett GN. Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiology. 1996;142: 2079–2086. [CrossRef]
- Huang C-N, Liebl W, Ehrenreich A. Restriction-deficient mutants and marker-less genomic modification for metabolic engineering of the solvent producer Clostridium saccharobutylicum. Biotechnol Biofuels. 2018;11: 264. [CrossRef]
- Foulquier C, Huang C-N, Nguyen N-P-T, Thiel A, Wilding-Steel T, Soula J, et al. An efficient method for markerless mutant generation by allelic exchange in Clostridium acetobutylicum and Clostridium saccharobutylicum using suicide vectors. Biotechnology for Biofuels. 2019;12: 31. [CrossRef]
- Harris LM, Welker NE, Papoutsakis ET. Northern, Morphological, and Fermentation Analysis of spo0A Inactivation and Overexpression in Clostridium acetobutylicum ATCC 824. J Bacteriol. 2002;184: 3586–3597. [CrossRef]
- Al-Hinai MA, Fast AG, Papoutsakis ET. Novel System for Efficient Isolation of Clostridium Double-Crossover Allelic Exchange Mutants Enabling Markerless Chromosomal Gene Deletions and DNA Integration. Appl Environ Microbiol. 2012;78: 8112–8121. [CrossRef]
- Heap JT, Ehsaan M, Cooksley CM, Ng Y-K, Cartman ST, Winzer K, et al. Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Research. 2012;40: e59–e59. [CrossRef]
- Croux C, Nguyen N-P-T, Lee J, Raynaud C, Saint-Prix F, Gonzalez-Pajuelo M, et al. Construction of a restriction-less, marker-less mutant useful for functional genomic and metabolic engineering of the biofuel producer Clostridium acetobutylicum. Biotechnol Biofuels. 2016;9: 23. [CrossRef]
- Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. The ClosTron: A universal gene knock-out system for the genus Clostridium. Journal of Microbiological Methods. 2007;70: 452–464. [CrossRef]
- Shao L, Hu S, Yang Y, Gu Y, Chen J, Yang Y, et al. Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res. 2007;17: 963–965. [CrossRef]
- Li Q, Chen J, Minton NP, Zhang Y, Wen Z, Liu J, et al. CRISPR-based genome editing and expression control systems in Clostridium acetobutylicum and Clostridium beijerinckii. Biotechnology Journal. 2016;11: 961–972. [CrossRef]
- Wilding-Steele T, Ramette Q, Jacottin P, Soucaille P. Improved CRISPR/Cas9 Tools for the Rapid Metabolic Engineering of Clostridium acetobutylicum. IJMS. 2021;22: 3704. [CrossRef]
- Christensen S, Van der Roest B, Besselink N, Janssen R, Boymans S, Martens JWM, et al. 5-Fluorouracil treatment induces characteristic T>G mutations in human cancer. Nat Commun. 2019;10: 4571. [CrossRef]
- Rosener B, Sayin S, Oluoch PO, García González AP, Mori H, Walhout AJ, et al. Evolved bacterial resistance against fluoropyrimidines can lower chemotherapy impact in the Caenorhabditis elegans host. eLife. 2020;9: e59831. [CrossRef]
- Ghode P, Ramachandran S, Bifani P, Sivaraman J. Structure and mapping of spontaneous mutational sites of PyrR from Mycobacterium tuberculosis. Biochemical and Biophysical Research Communications. 2016;471: 409–415. [CrossRef]
- Singh V, Brecik M, Mukherjee R, Evans JC, Svetlíková Z, Blaško J, et al. The Complex Mechanism of Antimycobacterial Action of 5-Fluorouracil. Chemistry & Biology. 2015;22: 63–75. [CrossRef]
- Raynaud C, Sarçabal P, Meynial-Salles I, Croux C, Soucaille P. Molecular characterization of the 1,3-propanediol (1,3-PD) operon of Clostridium butyricum. Proc Natl Acad Sci USA. 2003;100: 5010–5015. [CrossRef]
- Nguyen N-P-T, Raynaud C, Meynial-Salles I, Soucaille P. Reviving the Weizmann process for commercial n-butanol production. Nat Commun. 2018;9: 3682. [CrossRef]
- Soucaille P, Nguyen N-P-T, Percheron B, Croux C, Meynial-Salles I. Clostridium acetobutylicum strains unable to produce hydrogen and useful for the continuous production of chemicals and fuels. Institut National des Sciences Appliquées; WO 2016/42160 A1, 2016. pp. 33–34.
- Monot F, Martin J-R, Petitdemange H, Gay R. Acetone and Butanol Production by Clostridium acetobutylicum in a Synthetic Medium. Appl Environ Microbiol. 1982;44: 1318–1324. [CrossRef]
- Peguin S, Goma G, Delorme P, Soucaille P. Metabolic flexibility of CIostridiumacetobutylicumin response to methyl viologen addition. Applied Microbial and Cell Physiology. 1994;42: 611–616.
- Vasconcelos I, Girbal L, Soucaille P. Regulation of Carbon and Electron Flow in Clostridium acetobutylicum Grown in Chemostat Culture at Neutral pH on Mixtures of Glucose and Glycerolt. Journal of Bacteriology. 1994;176: 1443–1450.
- Wiesenborn DP, Rudolph FB, Papoutsakis ET. Thiolase from Clostridium acetobutylicum ATCC 824 and Its Role in the Synthesis of Acids and Solvents. Appl Environ Microbiol. 1988;54: 2717–2722. [CrossRef]
- Cho H-S, Ghim S-Y. Characterization of a PyrR-deficient Mutant of Bacillus subtilis by a Proteomic Approach.
- Vieira E, Brandão T, Ferreira IMPLVO. Evaluation of Brewer’s Spent Yeast To Produce Flavor Enhancer Nucleotides: Influence of Serial Repitching. J Agric Food Chem. 2013;61: 8724–8729. [CrossRef]
- Dusséaux S, Croux C, Soucaille P, Meynial-Salles I. Metabolic engineering of Clostridium acetobutylicum ATCC 824 for the high-yield production of a biofuel composed of an isopropanol/butanol/ethanol mixture. Metabolic Engineering. 2013;18: 1–8. [CrossRef]
- Jang Y-S, Malaviya A, Lee J, Im JA, Lee SY, Lee J, et al. Metabolic engineering of Clostridium acetobutylicum for the enhanced production of isopropanol-butanol-ethanol fuel mixture. Biotechnol Progress. 2013;29: 1083–1088. [CrossRef]
- Fields CJ, Switzer RL. Regulation of pyr gene expression in Mycobacterium smegmatis by PyrR-dependent translational repression. J Bacteriol. 2007;189: 6236–6245. [CrossRef]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research. 2022;50: D439–D444. [CrossRef]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596: 583–589. [CrossRef]
- Ismaiel AA, Zhu CX, Colby GD, Chen JS. Purification and characterization of a primary-secondary alcohol dehydrogenase from two strains of Clostridium beijerinckii. J Bacteriol. 1993;175: 5097–5105. [CrossRef]
- Girbal L, Mortier-Barrière I, Raynaud F, Rouanet C, Croux C, Soucaille P. Development of a Sensitive Gene Expression Reporter System and an Inducible Promoter-Repressor System for Clostridium acetobutylicum. Applied and Environmental Microbiology. 2003;69: 4985–4988. [CrossRef]
Figure 1.
Suicide plasmid for ldh replacement by sadh and hydG from C.beijerinckii
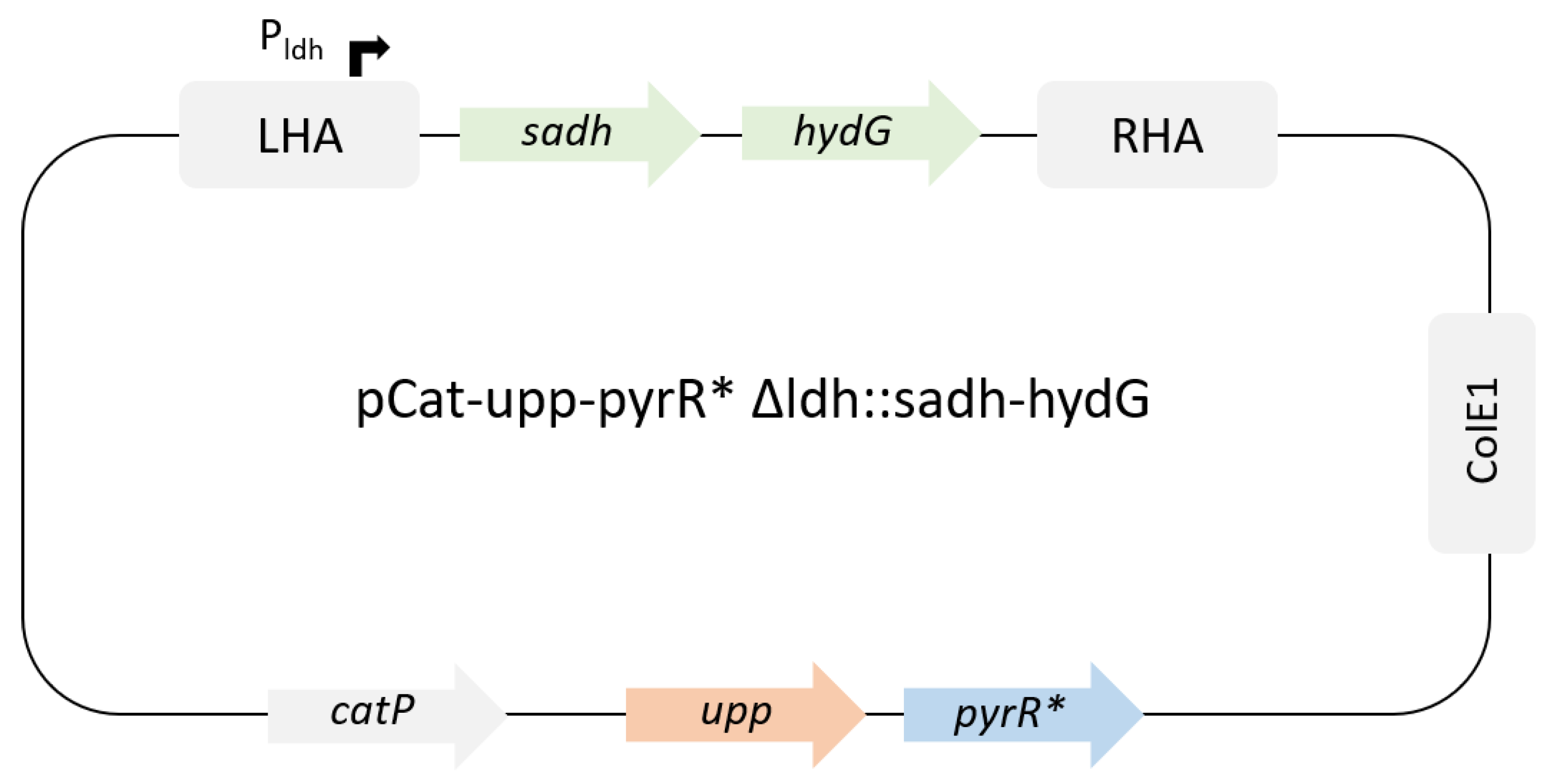
Figure 2.
Diagram representing the replacement of ldh by sadh and hydG from C. beijerinckii by allelic ex-change in Δcac1502ΔuppΔcac3535 pyrRmut. LHA: left homology arm; RHA: right homology arm. a 5’ integration of the suicide plasmid. The integrants are selected on thiamphenicol. b Double crossing-over induced by 5FU that cause the excision of the suicide plasmid.
Figure 2.
Diagram representing the replacement of ldh by sadh and hydG from C. beijerinckii by allelic ex-change in Δcac1502ΔuppΔcac3535 pyrRmut. LHA: left homology arm; RHA: right homology arm. a 5’ integration of the suicide plasmid. The integrants are selected on thiamphenicol. b Double crossing-over induced by 5FU that cause the excision of the suicide plasmid.
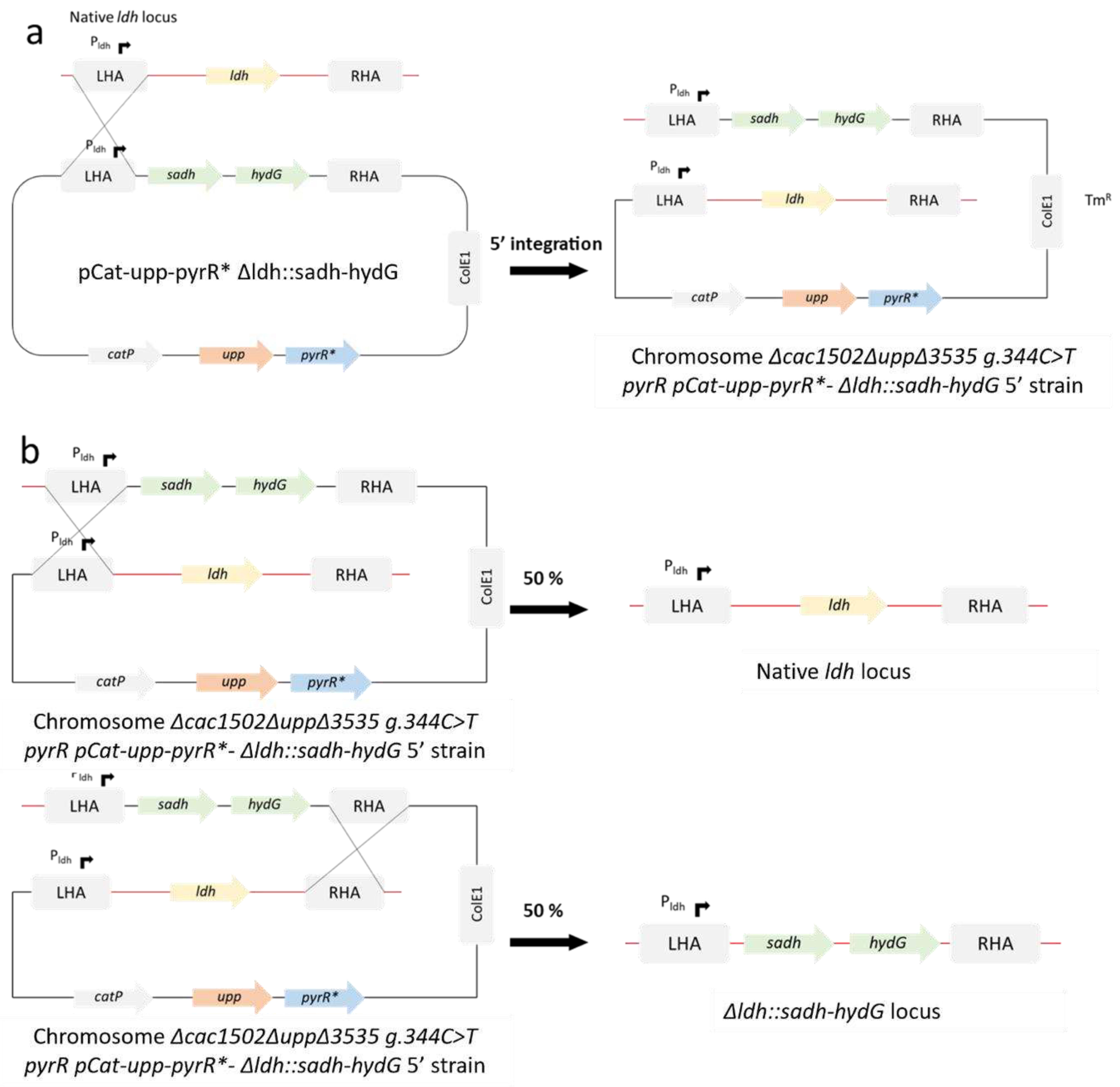
Figure 3.
Figure 3. a Screening of Δldh::sadh hydG mutant. The colonies were screened using PSB 384 and PSB 385 primers. Ladder: 1kb DNA ladder provided by New England Biolabs. b Schematic representation of Δldh::sadh hydG locus and native ldh locus.
Figure 3.
Figure 3. a Screening of Δldh::sadh hydG mutant. The colonies were screened using PSB 384 and PSB 385 primers. Ladder: 1kb DNA ladder provided by New England Biolabs. b Schematic representation of Δldh::sadh hydG locus and native ldh locus.

Figure 5.
Metabolism of 5FU. PRPP, phosphoribosyl pyrophosphatase; PPi, pyrophosphatse; UMP, uridine monophosphate; 5FU, 5-fluorouracile; 5FUMP, 5-fluorouridine monophosphate.
Figure 5.
Metabolism of 5FU. PRPP, phosphoribosyl pyrophosphatase; PPi, pyrophosphatse; UMP, uridine monophosphate; 5FU, 5-fluorouracile; 5FUMP, 5-fluorouridine monophosphate.
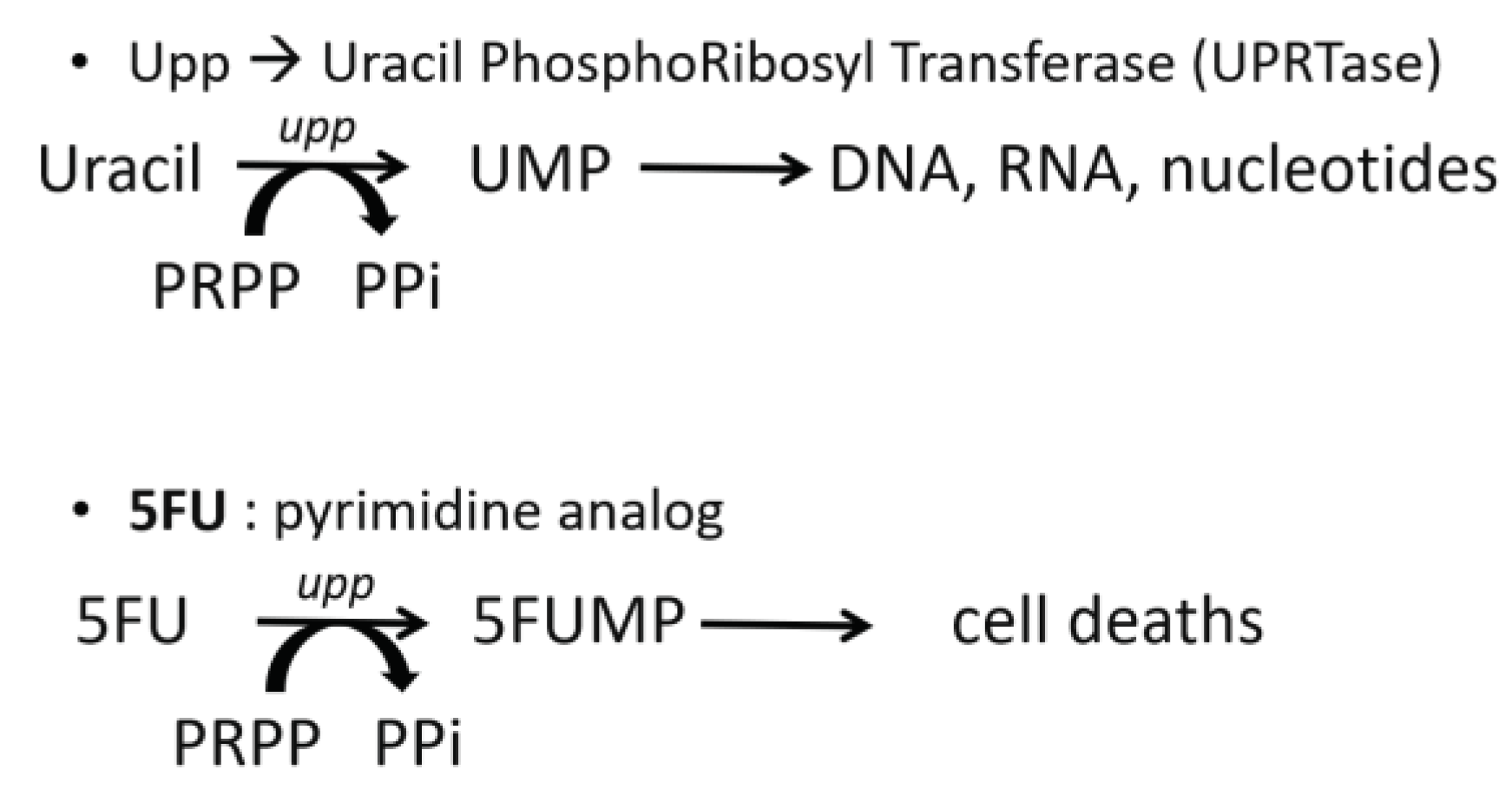
Figure 6.
a Multiple sequences alignment of PyrR. b Cartoon diagram of PyrR protein of C.acetobutylicum predicted by Alpha-fold [30,31]. The blue boxes show the amino acids implied in PRPP binding. The green boxes highlight G125 and R126 sites described by Ghod and Singh [16,17]. The pink boxes represent the A115 position, where C.acetobutylicum was mutated.
Figure 6.
a Multiple sequences alignment of PyrR. b Cartoon diagram of PyrR protein of C.acetobutylicum predicted by Alpha-fold [30,31]. The blue boxes show the amino acids implied in PRPP binding. The green boxes highlight G125 and R126 sites described by Ghod and Singh [16,17]. The pink boxes represent the A115 position, where C.acetobutylicum was mutated.
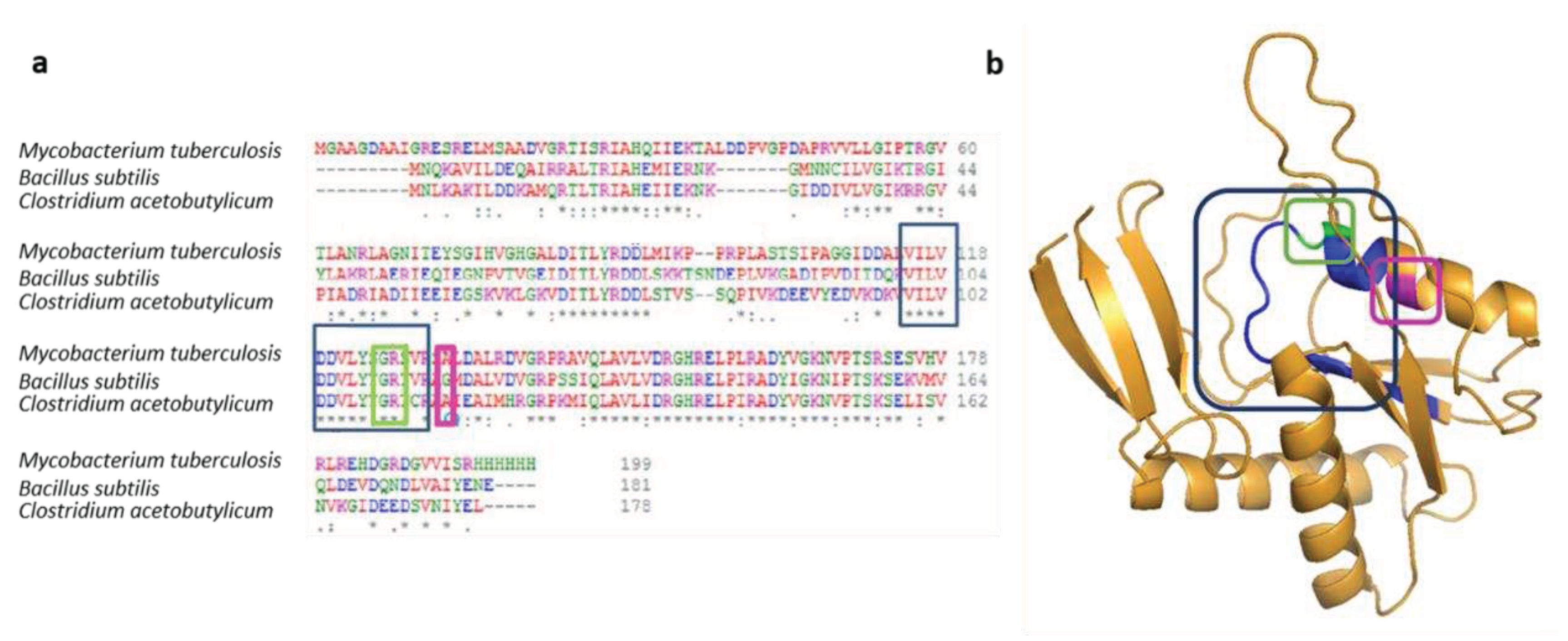
Table 1.
Bacterial strains and plasmids used in this study.
| Strain or plasmid | Relevant characteristics | Source or reference |
| Bacterial strains E. coli TOP10 |
Invitrogen |
|
|
C. acetobutylicum CAB1060 Δcac1502 Δcac1502ΔuppΔcac3535 Δcac1502ΔuppΔcac3535 pyrRmut Δcac1502ΔuppΔcac3535 pyrRmut Δldh::sadh hydG |
ΔCAC1502ΔuppΔptbΔbukΔctfABΔldhAΔrexA ΔthlA::atoB Δhbd::hbd1 ΔCA_C1502 ΔCA_C1502 ΔCA_C2879 ΔCA_3535 ΔCA_C1502 ΔCA_C2879 ΔCA_3535 CA_C2113 g.344C>T ΔCA_C1502 ΔCA_C2879 ΔCA_3535 CA_C2113 g.344C>T ΔCA_C0227:: CIBE_3470 HydG (Accession: P25981.3) |
[19] [9] [9] This study This study |
| Plasmid pCat-upp pCat-upp- pyrRmut pCat-upp-Δldh pCat-upp-pyrR* pCat-upp-pyrR*- Δldh pCat-upp-pyrR*-∆ldh::sadh-hydG pSOS95 pSOS95-pyrR |
CmR, upp, colE1 origin CmR, upp, pyrR edition cassette for C. acetobutylicum CmR, upp, ldh deletion cassette for C. acetobutylicum CmR upp pyrR* CmR, upp pyrR*, ldh deletion cassette for C. acetobutylicum CmR, upp pyrR*, ldh substitution cassette for sadh hydG for C. acetobutylicum ApR,MLSR , acetone operon, repL gene, colE1 origin ApR, MLSR, pyrR, repL gene, colE1 origin |
[5] This study [20] This study This study This study [18] This study |
Table 2.
Oligonucleotides used for PCR amplification.
| Primer name | 5’-3’ Oligonucleotide Sequence |
| PSC 39 PSC 40 PSC 46 PSC 51 PSC 52 PSC 58 PSC 61 PSC 62 PSC 72 PSC 75 PSC 76 PSC 104 PSC 105 PSC 106 PSC 107 PSB 384 PSB 385 |
GCATGCTCTTGTAGGTGATCCTT TGTTTACTGAATCCTCTTCATCTATTCC AAAAAAGGCGCCCTACAACTCATAAATGTTTACTGAATCCTC CAGAGTATTTAAGCAAAAACATCGTAGAAAT TTATTTTGTACCGAATAATCTATCTCCAGC AAAAAAGGATCCTTATACTGGAGGTGAGTGTATGAATTTAAAAG CCATGGTTATACTGGAGGTGAGTGTATGAATCTTAAAGCTAAGATTCTTGATGATAAGGC AAACACCGTATTTCTACGATGTTTTTGCTTAAATACTCTGCCATGGCTATAGCTCATATATGTTAACACTATCCTCTTC TCTTGGAGATGCTGGAGATAGATTATTCGGTACAAAATAACCATGGTTATACTGGAGGTGAGTG TTAATAGGATCCGAACCCATCAAATAAGAGTGCATATGG TATTAAGGATCCAGTCCTGCCCAACC AAATATAAATGAGCACGTTAATCATTTAACATAGATAATTAAATAGTAAAAGGAGGAACATATTTTATGAAAGGTTTTGC GGCAAAAGTTTTATAAACATGGGTACTGGTTATATTATATTATTTATGACTTTATTATTTATCACCTCTGCAACCACAGC TAGAGAAATTTTTAAAGATTTCTAAAGGCCTTTAACTTCATGTGAAAAGTTTGTTAAAATATAAATGAGCACGTTAATCATTTAA TCCACCCTTGGAGTTTAGGTCTTTTACCAGGCCTGAATACCCATGTTTATAGGGCAAAAGTTTTATAAACATGGGTACT GGGAAAGGTTTTAAGAGCGGCG CAACAATTGTCTCCGGTTTCAAGGG |
Table 4.
Bactericidal effect of 5FU on cac1502 on CGM MES and SM MES
| 5FU concentration (µM) | CGM MES (UFC/mL) | SM MES (UFC/mL) |
| 0 | 2.06 ± 0.41E7 | 1.75 + 0.25E7 |
| 5 | 1.65 ± 0.34E7 | 0 |
| 25 | 3.02 ± 0.52 E6 | 0 |
| 50 | 1.45 ± 0.28E6 | 0 |
| 100 | 0 | 0 |
| 200 | 0 | 0 |
Table 5.
Protective effect of uracil against 5FU in C. acetobutylicum ∆cac1502 strain. 0.1 ml of a 10-1 dilution of a CGM culture were spread on the different SM MES plates.
Table 5.
Protective effect of uracil against 5FU in C. acetobutylicum ∆cac1502 strain. 0.1 ml of a 10-1 dilution of a CGM culture were spread on the different SM MES plates.
| Uracil concentration (µM) | Number of colonies (5 µM 5FU) |
| 0 | 0 |
| 5 | Layer |
| 12.5 | Layer |
| 25 | Layer |
| 50 | Layer |
Table 6.
Spontaneous mutations founded in PyrR after 5FU exposition
| 5FU concentration (µM) | Amino acid change | Nucleotide change |
| 25 | R124G | g.370A>G |
| 25 | A47D | g.140C>A |
| 50 | R136X | T addition in aa 132 |
| 50 | P45L | g.134C>T |
| 50 | V85X | G deletion in aa 85 |
| 50 | E23K | g.67G>T |
Table 7.
Mutated pyrR strain viability on 5FU while maintaining a pCat-upp
| pCat-upp-Δldh |
|||
| 5FU concentration (µM) | 0 | 25 | 50 |
| SM MES + Tm (UFC/mL) | 9.52E7 | 9.14E7 | 8.46E7 |
Table 8.
Viability of Δcac1502ΔuppΔcac3535pyrRmut strain with a pCat-upp plasmid and a replicative pSOS95-pyrR plasmid
Table 8.
Viability of Δcac1502ΔuppΔcac3535pyrRmut strain with a pCat-upp plasmid and a replicative pSOS95-pyrR plasmid
| pCat-upp-Δldh + pSOS95-pyrR | ||||
| 5FU concentration (µM) | 0 | 5 | 10 | 25 |
| SM MES + Ery (UFC/mL) | 1.20E7 | 3.80E4 | 2.84E4 | 2.10E4 |
| SM MES + Ery + Tm (UFC/mL) | 6.28E6 | 0 | 0 | 0 |
Table 9.
Viability and frequency of double crossing-over of Δcac1502ΔuppΔcac3535pyrRmut strain with a pCat-upp plasmid and a replicative pSOS95-pyrR plasmid after 5FU selection
Table 9.
Viability and frequency of double crossing-over of Δcac1502ΔuppΔcac3535pyrRmut strain with a pCat-upp plasmid and a replicative pSOS95-pyrR plasmid after 5FU selection
| pCat-upp-Δldh + pSOS95-pyrR | |||
| 5FU concentration (µM) | 5 | 10 | 25 |
| Picked colonies viability (%) | 86 | 90 | 84 |
| Picked colonies Tm sensitivity (%) | 86 | 96 | 98 |
Table 10.
Viability of Δcac1502ΔuppΔcac3535pyrRmut strain with pCat-upp-pyrR*-Δldh plasmids
| pCat-upp-pyrR*-Δldh |
|||
| 5FU concentration (µM) | 0 | 25 | 50 |
| SM MES (UFC/mL) | 8.80E7 | 4.24E4 | 3.88E4 |
| SM MES + Tm (UFC/mL) | 7.60E7 | 0 | 0 |
Table 11.
Viability and frequency of double crossing-over of Δcac1502ΔuppΔcac3535pyrRmut strain with pCat-upp and a pCat-upp-pyrR* plasmids after 5 FU selection
Table 11.
Viability and frequency of double crossing-over of Δcac1502ΔuppΔcac3535pyrRmut strain with pCat-upp and a pCat-upp-pyrR* plasmids after 5 FU selection
| pCat-upp-Δldh |
pCat-upp-pyrR*-Δldh |
|||||||
| 5FU concentration (µM) | 5 | 10 | 25 | 50 | 5 | 10 | 25 | 50 |
| Picked colonies viability (%) | 100 | 98 | 98 | 76 | 88 | 96 | 60 | 52 |
| Picked colonies Thiamphenicol sensitivity (%) | 0 | 6 | 75 | 89 | 90 | 98 | 100 | 100 |
Table 12.
Viability and frequency of double crossing-over of Δcac1502ΔuppΔcac3535 strain with the pCat-upp-pyrR*/ 5 FU counter selection system
Table 12.
Viability and frequency of double crossing-over of Δcac1502ΔuppΔcac3535 strain with the pCat-upp-pyrR*/ 5 FU counter selection system
| pCat-upp-pyrR*-Δldh | ||
| 5FU concentration (µM) | 5 | 10 |
| Picked colonies viability (%) | 98 | 100 |
| Picked colonies Thiamphenicol sensitivity (%) | 100 | 100 |
Table 13.
Viability and frequency of double crossing-over of Δcac1502ΔuppΔcac3535pyrRmut strain with pCat--pyrR*-Δldh::sadh-hydG after 5FU selection.
Table 13.
Viability and frequency of double crossing-over of Δcac1502ΔuppΔcac3535pyrRmut strain with pCat--pyrR*-Δldh::sadh-hydG after 5FU selection.
| pCat-pyrR*-Δldh::sadh-hydG | |
| 5FU concentration (µM) | 5 |
| Picked colonies viability (%) | 92 |
| Picked colonies Thiamphenicol sensitivity (%) | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



