
Submitted:
25 September 2023
Posted:
26 September 2023
You are already at the latest version
Abstract
Gliomas, the most prevalent and lethal form of brain cancer, are known to exhibit metabolic alterations that facilitate tumor growth, invasion, and resistance to therapies. Peroxisomes, essential organelles responsible for fatty acid oxidation and reactive oxygen species (ROS) homeostasis, rely on the receptor PEX5 for the import of metabolic enzymes into their matrix. However, the prognostic significance of PEX5 for glioma patients remains unclear. We developed a robust prognosis model based on PEX5-dependent signature. This signature not only serves as a robust prognosis model capable of accurately predicting outcomes for glioma patients but also effectively distinguishes several clinicopathological features, including the grade, isocitrate dehydrogenase (IDH) mutation, and 1p19q codeletion status. Furthermore, we developed a nomogram that integrates the prognostic model with other clinicopathological factors, demonstrating highly accurate performance in estimating patient survival. Patients classified into the high-risk group based on our prognostic model exhibit an immunosuppressive microenvironment. Finally, we validated that the peroxisomal localization of the signature genes depends on PEX5 and demonstrated that PEX5 is required for cell growth, migration and invasion of glioma cells. These findings identify the PEX5-dependent signature as a promising prognostic tool for gliomas.
Keywords:
Glioma
; peroxisome
; PEX5
; prognosis
; immune microenvironment
1. Introduction
Gliomas are the most common and deadly type of cancer in the central nervous system in adults, accounting for 80% of all malignant brain tumors [1]. Glioblastoma (GBM), an extremely aggressive form of glioma, makes up 50% of newly diagnosed glioma cases and has a median overall survival (OS) of less than two years [2]. Despite advances in standard treatments like surgery, radiotherapy, and chemotherapy, therapeutic resistance remains a significant challenge due to the highly infiltrative nature of malignancy [3]. To address this, previous studies have identified various molecular markers, such as mutations in isocitrate dehydrogenase 1 and 2 genes (IDH1 and IDH2, collectively referred to as IDH) and co-deletion of 1p/19q, to aid in molecular pathological diagnosis, treatment options, and prognostic assessment [4]. However, the prognosis for glioma patients remains poor, necessitating the development of an improved prognostic model for the disease.
Peroxisomes are crucial single membrane-bound organelles present in all eukaryotic cells. Their importance for mammalian physiology is underscored by the existence of severe inherited human diseases resulting from the complete or partial loss of peroxisomal functions [5]. Peroxisomes are involved in essential cellular metabolism, most notably FA oxidation as well as the synthesis and breakdown of ROS [5,6]. Additionally, peroxisomes participate in various other processes such as glyoxylate detoxification, the synthesis and breakdown of ether phospholipids, and amino acid metabolism [5,6].
To enable these metabolic pathways, peroxisomes contain a variety of metabolic enzymes in the matrix. The import of matrix proteins is typically facilitated by peroxisomal targeting signals (PTSs), such as PTS1, which comprises a non-cleaved, C-terminal tripeptide, SKL, or its conserved variants [7], or PTS2, which consists of a nonapeptide sequence, (R/K)(L/V/I/Q)XX(L/V/I/H/Q)(L/S/G/A/K)X(H/Q)(L/A/F), located near the N terminus of the cargo protein [8]. PEX5 functions as the receptor for recognition and import of PTS1-containing proteins [9,10], while PEX7 is the receptor for PTS2 proteins. Remarkably, PEX5 acts as a co-receptor for the import of PTS2 proteins by binding to the PEX7-cargo complexes [11,12]. Additionally, membrane proteins harbor targeting signals (mPTS), recognized by the receptor PEX19 [13].
One of the hallmarks of glioma progression is metabolic reprogramming, which allows tumor cells to adapt and survive in a hostile microenvironment [14]. In gliomas, alterations in metabolic pathways, such as fatty acid, lipid, and redox metabolism, are not only a consequence of genetic and epigenetic changes, but they also contribute to tumor growth, invasion, and therapy resistance [14,15]. Accumulating evidence indicates that peroxisomes actively participate in cancer, a disease characterized by abnormal metabolism [16]. A significant proportion of gliomas carry mutations in IDH1 and IDH2 genes [17]. It is worth mentioning that IDH1 is partially localized within peroxisomes [18]. Several studies have linked the expression of peroxisomal genes and proteins to the malignancy grade of gliomas [19,20,21]. However, the prognostic significance of peroxisomes for glioma patients remains unclear.
In our study, we focused on the receptor PEX5, as it plays a crucial role in the matrix import of major peroxisomal enzymes. We developed a PEX5-dependent prognosis model and evaluated its sensitivity, specificity, and accuracy in predicting the survival of glioma patients. Furthermore, we knocked out the PEX5 gene and assessed the essentiality of PEX5 in glioma cell growth, migration, and invasion.
2. Materials and Methods
2.1. Data acquisition and processing
We retrieved expression data and clinical information for glioma patients from multiple public databases. Specifically, we obtained the TCGA RNA-seq datasets from the UCSC Xena website, which consisted of 529 Lower Grade Glioma (LGG) samples and 173 Glioblastoma (GBM) samples. Additionally, we downloaded two RNA-seq datasets, CGGA_693 (693 samples) and CGGA_325 (325 samples), from the Chinese Glioma Genome Atlas. We also acquired the GSE16011 microarray dataset (284 samples) from the Gene Expression Omnibus (GEO) database.
For the RNA-seq cohorts, we obtained both the FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) values and raw count data for expression analysis. To ensure the quality of the prognosis model analysis, we filtered out patient data with missing survival time or status information. After applying this criterion, we obtained a total of 691, 657, 313, and 131 glioma patient data for the TCGA, CGGA_693, CGGA_325, and GSE16011 cohorts, respectively. The detailed clinicopathological characteristics of glioma patients were summarized in Tables S1–S4.
2.2. Construction of the PEX5-based prognostic gene signature
Identification of PEX5-dependent peroxisomal metabolic enzymes was carried out by searching the Peroxisome database (http://www.peroxisomedb.org/home.jsp) and selecting those with PTS1 or PTS2 signals. The resulting genes were then subjected to least absolute shrinkage and selection operator (LASSO) regression analysis using the glmnet R package. Multivariate Cox regression analysis was subsequently performed with the survival R package to establish an optimal PEX5-based risk signature. A prognostic risk score was generated for each patient with the following formula: risk score = expression level of gene1 x j1 + expression level of gene2 x j2 + .... + expression level of genex x jx, where j represents the coefficient. We used TCGA data as the training cohort, while CGGA_693, CGGA_325, and GSE16011 data were used as validation cohorts.
2.3. Survival analysis
The glioma patients were categorized into low- and high-risk groups based on their median risk score. A risk plot was generated using an in-house "risk_plot" R function to illustrate the distribution of survival status of the patients and the expression of the risk signature genes in the different risk groups. To evaluate the sensitivity and specificity of the risk signatures in predicting outcomes, we established time-dependent receiver operating characteristic (ROC) curves using the R package "survivalROC". For the ROC analysis, a higher area under the curve (AUC) indicates better accuracy. The AUC values range from 0.5 to 1.0, where 0.5 represents random chance, and 1.0 represents perfect discrimination. Generally, an AUC value greater than 0.7 is considered a reasonable estimate [22]. To compare the OS between different groups of glioma patients, we conducted Kaplan-Meier survival analyses using the R "survival" and "survminer" packages.
2.4. Independent prognostic role of the risk signature
To determine whether the PEX5-based risk score and other clinicopathological factors were independent prognostic factors for glioma patients, univariate and multivariate Cox regression analyses were conducted using the "survival" R package. These clinicopathological factors included age, gender, tumor grade, IDH mutation status, 1p19q codeletion status, and MGMT promoter methylation status. Results were presented using forest plots generated by the "forestplot" R package.
2.5. Construction of the Nomogram
To develop a prognostic nomogram for glioma patients, the "rms" R package was utilized. The nomogram included the PEX5-based risk score and the aforementioned clinicopathological factors, and was used to predict survival probabilities at 1, 3, and 5 years. The accuracy of the nomogram was evaluated through calibration curves and decision curve analysis (DCA) for the one-, three-, and five-year survival rates of glioma patients. DCA is a method for evaluating the clinical benefit of alternative models [23]. In the context of nomograms, DCA evaluates net benefits at various threshold probabilities. To provide a meaningful comparison, two reference curves are typically plotted: the treat-all-patients scheme, representing the highest clinical costs, and the treat-none scheme, indicating no clinical benefit.
2.6. Differential expression gene (DEG) and functional enrichment Analysis
Raw count data from the three RNA-seq cohorts (TCGA, CGGA_693, and CGGA_325) was obtained, and genes with expression detected in at least half of the samples were selected for DEG analysis using the "limma" R package between the low- and high- risk group. Statistically significant DEGs were defined as those with an adjusted p value (adj. p) < 0.05 and a |fold change (FC)| >= 2, and were visualized in volcano plots. To determine gene function enrichment, the human c5.go.bp gene set was downloaded from the MSigDB database. Genes were pre-ranked based on differential expression (log2FC), and a GSEA analysis of the c5.go.bp gene set was conducted using the GSEA() function in the "clusterProfiler" R package.
2.7. Plasmid construction
The PEX5 sgRNA sequence (sgPEX5-1: 5’ ttcgtgcggcagattggcga 3’; sgPEX5-2: 5’ acgagccaagtcagctatag 3’) was subcloned into the lentiCRISPRv2 vector. The C-terminal tripeptide, SKL, was in frame cloned into the GFP tag at the C terminus to generate the GFP-PTS1 construct. The nonapeptide sequence, RLQSIKDHL, was in frame cloned into the GFP tag at the N terminus to generate the PTS2-GFP construct. The coding regions for ACOX1, HMGCL, PIPOX, GSTK1, or CAT were cloned into the pEGFP-C1 construct with an in-frame N-terminal GFP tag.
2.8. Cell culture and PEX5 knockout cell establishment
293T and U251 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Biological Industries, 01-052-1A), supplemented with 10% fetal bovine serum (FBS) (Biological Industries, 04-001-1A), 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco, 15070063). The cells were incubated in a humidified 5% CO2 incubator at 37°C.
To establish PEX5 knockout cells, the PEX5 KO targeting vector was co-transfected with psPAX2 (Addgene, 12,260) and pMD2.G (Addgene, 12,259) into 293T cells. The virus in the culture media was filtered through a 0.45 μm filter and used to infect U251 cells. Forty-eight hours after infection, cells were selected with puromycin (1 μg/ml). The surviving cell population was digested and single cells were plated into 96-well plates. The single-cell populations were then expanded and subjected to PEX5 knockout detection by DNA sequencing and western blot.
2.9. Western blot
Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktails (Roche, 05892970001). Equal amounts of proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes of 0.45 µm pore size. The membranes were then blocked with 5% skim milk, followed by incubation with α-Actin (Sigma, A1978) or α-PEX5 (Cell Signaling Technology, 83020S) antibodies overnight at 4°C. Subsequently, the membranes were incubated with dye 800-conjugated secondary antibodies and the fluorescence signals were captured using a Licor Odyssey-CLx machine.
2.10. Immunofluorescence
Cells grown on cover slides were transfected with GFP fusion constructs using a homemade polycation PEI reagent. After 24 hours, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. They were then blocked with 1% BSA and incubated with the primary α-GFP antibody (Proteintech, 50430-2-A) overnight at 4°C. Following a washing step with PBS, the cells were incubated with fluorescent dye-conjugated secondary antibodies and stained with 1μg/ml DAPI to label the nuclei. The subcellular localization of proteins was visualized using an Olympus FV3000 laser scanning confocal microscope.
2.11. CCK-8 assays
To assess cell proliferation, cells were plated into 96-well plates at a density of 1500 cells/well. At the indicated days, cell viability was measured using a Cell Counting Kit-8 (CCK-8) assay. The absorbance was measured at 450 nm using a microplate reader (Multiskan FC, Thermo Fisher Scientific).
2.12. Colony formation assay
Cells were seeded in 6-well plates (200 cells/well) and maintained in growth median for 14 days. Cell colonies were stained with 0.1% crystal violet solution. The colonies were photographed and the colony number was calculated.
2.13. Cell cycle
Cells were digested with 0.25% trypsin into single cells and washed with ice-cold PBS. The cells were fixed with 70% ice-cold ethanol for 2 hours and then washed with PBS before being resuspended in PI staining buffer (Beyotime, C1052). After a 30-minute incubation at room temperature in the dark, the cells were subjected to flow cytometry analysis using a BD flow cytometer. The cell cycle distribution was analyzed using FlowJo software.
2.14. Wound-healing assay
Cells were plated in 6-well plates and allowed to reach approximately 90% confluence. A sterile 10 µl pipette tip was used to create a scratch wound, and the cells were washed twice with PBS buffer. The cells were then incubated in growth media, and the wound area was imaged at the indicated times using an inverted fluorescence microscope. The wound healing rate was calculated as follows: (wound width at 0h – wound width at the indicated time) / wound width at 0h.
2.15. Transwell
Cells were cultured in DMEM supplemented with 1% FBS in the upper chamber of a polycarbonate filter membrane with an 8.0 µm pore size (Corning-Costar, Cambridge, MA, USA). The lower chamber was filled with DMEM supplemented with 10% FBS. After 36 hours of incubation, non-invading cells on the upper membranes were carefully removed by cotton swabs. The migrated cells on the lower membranes were fixed with 4% polyoxymethylene and stained with a 0.1% crystal violet solution. Five randomly selected fields for each transwell membrane were photographed, and the numbers of migrated cells were recorded.
2.16. Statistical analysis
Experimental data were analyzed at least three times. Statistical analysis was performed using GraphPad Prism software or R packages. Data are presented as the mean ± SD. The statistical tests, including Student’s t test, Wilcox test, and log-rank test, were indicated in the respective figure legends. A p-value of <0.05 was considered statistically significant, and the significance levels were denoted as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
3. Results
3.1. Construction of a prognostic gene signature in the TCGA training cohort
Human peroxisomal metabolic pathways play a critical role in various metabolic pathways, including fatty acid (FA) oxidation, glyoxylate metabolism, ether phospholipid biosynthesis, amino acid metabolism, antioxidant system, and proteases. Using the peroxisome database (http://www.peroxisomeDB.org), we identified 45 human peroxisomal enzymes involved in these pathways, 35 of which contained the predicted PTS1 or PTS2 signals (Figure 1A). We investigated whether the PTS1 or PTS2-containing metabolic enzymes, whose peroxisomal localization depends on PEX5, could be used to establish a prognostic model for glioma.
To develop a prognostic model for glioma, we used the TCGA glioma dataset as the training cohort and performed the least absolute shrinkage and selection operator (LASSO) Cox regression analysis on the 35 genes (Figure 1B-1C). This resulted in a signature consisting of 5 genes (ACOX1, HSD17B4, ACOT4, CROT, ECH1), 2 genes (HMGCL, PIPOX), 3 genes (GSTK1, PRDX5, CAT), and the LONP2 gene, involved in FA oxidation, amino acid metabolism, antioxidant system, and proteases, respectively (Figure 1A, 1D).
Among these genes, ACOX1, HSD17B4, ECH1, PRDX5, and LONP2 were identified as protective factors for glioma survival, as indicated by hazard ratios (HRs) less than 1 (Figure 1D). Conversely, ACOT4, CROT, HMGCL, PIPOX, GSTK1, and CAT were identified as risk factors, with HRs greater than 1 (Figure 1D).
3.2. Evaluation of the prognosis model
To assess the prognostic value of our PEX5-dependent model, we computed a risk score for each patient based on the mRNA expression levels of the 11 signature genes and LASSO Cox regression coefficients. Using the risk scores, we employed ROC curve analysis and Kaplan-Meier analysis to evaluate the sensitivity, specificity, and survival prediction of the model. In addition to the TCGA training cohort, we validated the model using two RNA-seq datasets (CGGA_693 and CGGA_325) and a microarray glioma dataset (GSE16011).
Based on the median risk score, we divided patients in each cohort into low- and high-risk groups. Figure 2A depicts the distribution of risk scores and survival times for each patient, along with the expression levels of the signature genes. Our analysis demonstrated that the PEX5-dependent model had excellent sensitivity and specificity for predicting 1-, 3-, and 5-year OS in all four cohorts, with area under curve (AUC) values of 0.87, 0.87, and 0.79 for the TCGA cohort, and 0.72, 0.71, 0.71 for the CGGA_693 cohort, 0.69, 0.77, 0.83 for the CGGA_325 cohort, and 0.79, 0.87, 0.81 for the GSE16011 cohort, respectively (Figure 2B). Furthermore, KM analysis showed that patients in the high-risk group had worse outcomes than those in the low-risk group across all cohorts (Figure 2C). These results suggest that our model has strong predictive power.
Different clinicopathological features have been shown to influence the survival outcomes of glioma patients. For instance, patients with a higher World Health Organization (WHO) grade or IDH wild-type status are typically associated with a poor prognosis [24,25]. We examined the four cohorts containing the patient grade information and observed significantly higher risk scores among the GBM population (WHO grade IV) (Figure S1A). Additionally, the three RNA-seq cohorts included patient information on IDH and 1p19q codeletion status, and our analysis showed that patients with wild-type IDH or 1p19q non-codeletion status had significantly higher risk scores in these cohorts (Figure S1B-S1C). These findings highlight the prognostic value of the PEX5-dependent signature and demonstrate its ability to differentiate between different clinicopathological features in glioma patients.
3.3. Construction and validation of the nomogram
We examined the independence of PEX5-dependent risk score from other known prognostic factors, such as age, gender, WHO grade, IDH mutation status, 1p19q codeletion status, and MGMT promoter methylation status. Using univariate Cox analysis, we found that the PEX5-dependent risk score, along with other prognostic factors, was significantly associated with OS in the TCGA training cohort (Figure 3A). Multivariate Cox regression analysis revealed that the PEX5-dependent risk score, along with age, grade, and 1p19q codeletion status, significantly correlated with OS (Figure 3B). These findings suggest that the risk score is an independent prognostic variable for glioma patients in the TCGA training cohort. To validate our findings, we extended our analysis to two independent cohorts (CGGA-325 and CGGA_693), for which clinicopathological parameters were available. Consistently, our results indicated that the PEX5-based risk score was an independent predictor of OS across these cohorts (Figure S2A-S2B, S2D-S2E).
We developed nomograms to predict 1-, 3-, and 5-year OS in glioma patients using the TCGA cohort. The nomograms incorporated seven independent risk factors, including the PEX5 dependent risk score, age, gender, WHO grade, IDH mutation, 1p19q codeletion, and MGMT promoter methylation statuses (Figure 3C). In the model, each risk factor was assigned points based on its contribution to OS (Figure 3C). Calibration curves indicated a good match between the actual and predicted OS at the 1-, 3-, and 5-year intervals (Figure 3D).
The clinical benefits of the nomogram were compared to those of IDH mutation or WHO grade. DCA curves demonstrated that the nomogram exhibited superior predictive capabilities for OS compared to IDH mutation or WHO grade. This superiority was observed across various threshold probabilities in both the training cohort (TCGA) and the validation cohorts (CGGA_693 and CGGA_325). Furthermore, the nomogram consistently yielded higher net benefits when compared to both the treat-all-patients (All) scheme and the treat-none (None) scheme (Figure 3E).
We constructed the nomogram model for the CGGA-693 and CGGA-325 cohorts by integrating six risk factors, including risk score, age, gender, WHO grade, IDH mutation, and 1p19q codeletion statuses (Figure S2C, S2F). Since the majority of patients in these cohorts lacked information for the MGMT promoter methylation status, we omitted it from the nomogram construction. Consistently, these models demonstrated accurate prediction of patient survival for 1-, 3-, and 5-year OS in both the CGGA-693 and CGGA-325 cohorts (Figure 3D-3E).
3.4. High-risk group had an immunosuppressive feature
Gliomas exhibit a distinct tumor microenvironment, largely due to the presence of the blood-brain barrier [26]. During glioma progression, the infiltrating macrophages, Tregs and MDSCs have been shown to have the protumor and immunosuppressive effects [26,27]. To examine the potential role of these immune cells in the prognosis value of PEX5-dependent model, we computed the abundance of the three types of immune cells in each glioma patient through ssGSEA [28]. Our analysis revealed that the abundance of all the three-cell population was significantly upregulated in the high-risk group, and positively correlated with the risk score in the three examined cohorts (TCGA, CGGA_693, and CGGA_325) (Figure 4A-4B).
We divided the glioma samples into different immune subtypes (C1-C6 subtypes) and found that the high-risk group had significantly more C4 and less C5 immune subtypes than the low-risk group in all three cohorts (Figure 4C). It is worth noting that the prognosis of the C4 immune subtype in tumors is worse than that of the C5 immune subtype [29]. These results suggest that the enhanced activity of the macrophages, Tregs and MDSCs may contribute to the immunosuppressive microenvironment and worse prognosis in the high-risk group.
We analyzed the negative immune regulators, such as cancer-immunity cycle inhibitors [30] and immune checkpoints [31]. Through differential gene expression analysis through the limma package, we found that 567 and 611 genes were downregulated and upregulated, respectively in the high-risk group across all the three RNA-seq cohorts (Figure S3A-S3B). The Venn analysis showed that 10 cancer-immunity cycle inhibitors and 13 checkpoint genes were among the upregulated gene list (Figure 4D). Remarkably, five genes (PDCD1LG2, CD48, IDO1, LAIR1, PDCD1) were found to be present in both the cancer-immunity cycle inhibitors and checkpoint genes categories. Overall, we identified 18 upregulated negative regulators (PDCD1LG2, CD48, IDO1, LAIR1, MICB, ICAM1, PDCD1, VEGFA, IL10, CCL2, TNFSF14, CD80, CD44, ICOS, CD70, NRP1, CD28, TNFRSF9) in the high-risk group. Notably, the expression of each of these negative regulators was positively correlated with the risk score (Figure 4E).
To demonstrate the immunotherapy sensitivity differentiated by the model, we used Tumor Immune Dysfunction and Exclusion (TIDE) [32]. Our analysis showed that the high-risk group had higher TIDE scores than the low-risk group (Figure 4F), suggesting that the high-risk group is more likely to resist immunotherapy than the low-risk group.
3.5. The peroxisomal localization of the signature genes depends on PEX5.
To investigate the role of the import receptor PEX5 in determining the subcellular localization of signature genes, we designed a specific sgRNA to target the PEX5 gene in glioma U251 cells. We successfully generated the PEX5 knockout (KO, sgPEX5-1) cells, as confirmed by DNA sequencing and western blot analysis (Figure 5A-5B). As expected, PEX5 KO resulted in the defective import of PTS1- and PTS2-containing proteins, as demonstrated by PTS1- and PTS2-reporter constructs (Figure 5C).
Subsequently, we transfected U251 cells with constructs expressing each of the five signature genes: ACOX1, HMGCL, PIPOX, GSTK1, and CAT. We observed their peroxisomal localizations (Figure 5C). However, the peroxisomal localizations were found to be disrupted in PEX5 KO cells (Figure 5C). Furthermore, we performed immunostaining with a CAT antibody and observed that the endogenous CAT proteins lost their typical peroxisomal localization in the absence of PEX5 (Figure 5D). These findings collectively underscore the pivotal role of PEX5 in governing the peroxisomal localization of the signature genes.
3.6. PEX5 knockout inhibits the growth, migration, and invasion of glioma cells
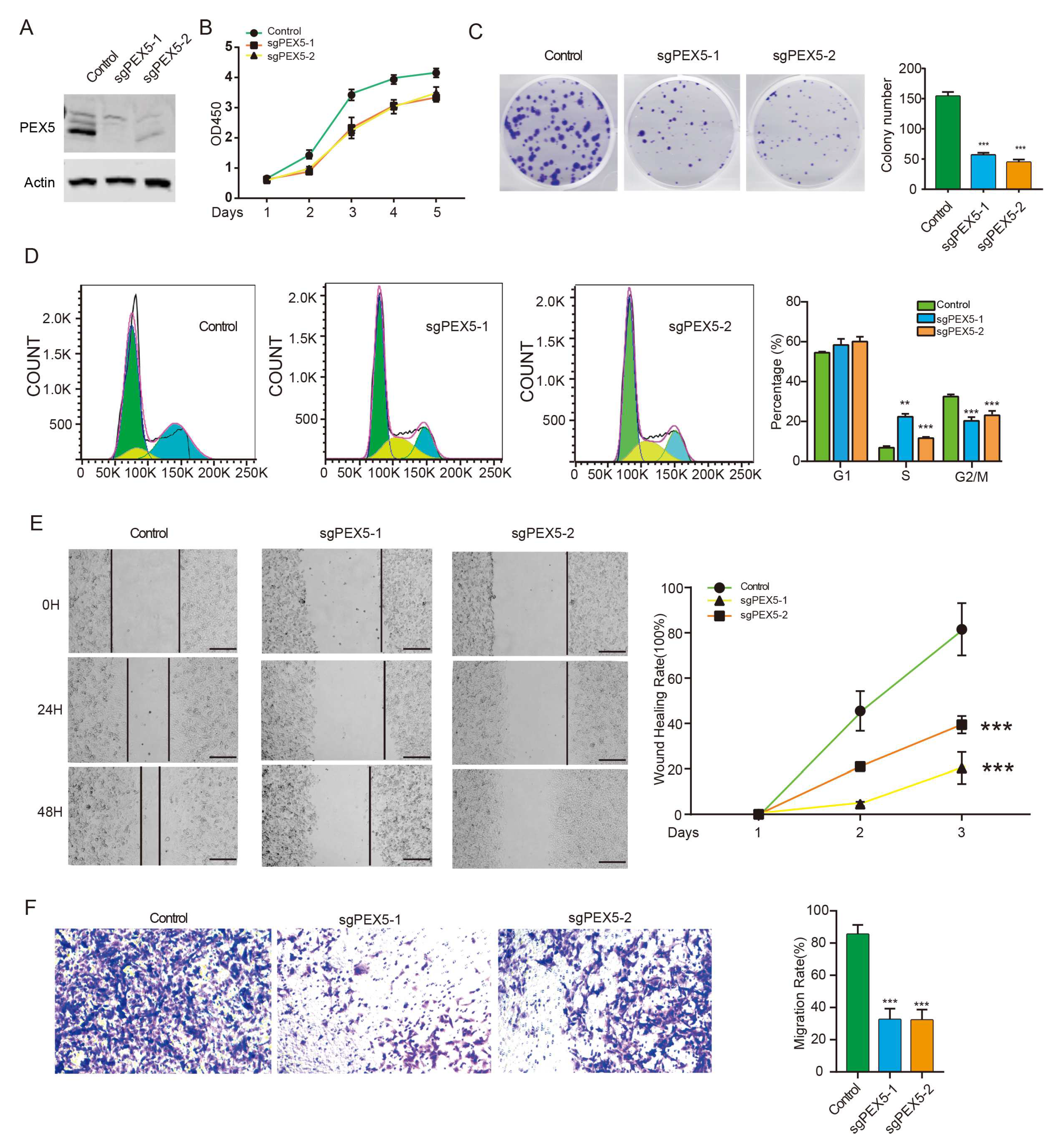
As the accurate peroxisomal localization of signature genes is pivotal in regulating their metabolic activity, we investigated the role of our prognosis model by assessing the cellular activity of glioma cells with PEX5 KO. To exclude the off-target effect, we developed an alternative PEX5 sgRNA and subsequently established the PEX5 KO (sgPEX5-2) cells, as validated by western blot analysis (Figure 6A). The CCK8 and colony formation assays revealed that PEX5 KO severely suppressed the cell viability and anchorage independent growth (Figure 6B-6C). Cell cycle analysis showed an increase and decrease in the percentage of cells in S and G2/M phases, respectively, while the percentage of cells in G1 phase was unchanged (Figure 6D). These findings suggested that PEX5 KO inhibits cell growth by arresting the cell cycle from S to G2M phase. Furthermore, our wound-healing and trans-well assays demonstrated a remarkable impairment of migration and invasion in PEX5 KO cells (Figure 6E-6F). In summary, these findings underscore the pivotal role of PEX5 in glioma cell growth, migration, and invasion, further corroborating the significance of our PEX5-dependent prognosis model.
4. Discussion
Within our prognostic model, several genes are involved in FA oxidation (FAO) and redox metabolism, alterations of which have been observed during glioma progression and are associated with malignancy [14]. It is worth noting that these metabolic processes also occur in mitochondria, albeit with different enzymes and reactions involved [33]. Take the FAO for example, two types of FAO occur in mammalian cells: α-oxidation and β-oxidation. α-Oxidation is a unique process that takes place in peroxisomes and is responsible for removing a single carbon from certain lipids at the carboxy terminus, enabling their subsequent β-oxidation [34]. Very-long-chain fatty acids (VLCFAs), which exceed 22 carbons in length, cannot be metabolized in the mitochondria and must undergo catabolism in peroxisomes [35]. While most of the research on FAO regards β-oxidation within the mitochondria in glioma cells [36,37], the study of peroxisomal FAO is very limited. Our PEX5-dependent prognosis model takes advantage of the unique peroxisomal metabolism and highlights the pivotal role of PEX5-dependent peroxisomal metabolism in glioma development.
Gliomas present a complex and unique immune microenvironment, largely due to the presence of the blood-brain barrier [26]. Within this microenvironment, various immune cell populations coexist, including macrophages, resident microglia, MDSCs, T cells, natural killer (NK) cells, and a small number of B cells [27]. Studies have shown that macrophages, Tregs, and MDSCs exert protumor and immunosuppressive effects during the progression of gliomas [26,27]. In gliomas, there is a noticeable shift in the polarization of tumor-associated macrophages (TAMs) from an M1 phenotype to an M2 phenotype, resulting in the suppression of the local immune response [38,39]. Recent research has linked TAMs to the promotion of tumor cell proliferation and the creation of an immunosuppressive environment through the recruitment of Tregs and MDSCs [40,41]. Furthermore, MDSCs contribute to the maintenance of an immunosuppressive environment by inhibiting various effector cells and promoting T-regulatory cell function [41].
Our findings indicate that patients in the high-risk group exhibit an immunosuppressive microenvironment characterized by the upregulation of macrophages, Tregs, and MDSCs, as well as the presence of certain negative immune regulators. Moreover, these patients demonstrate higher TIDE scores, indicating reduced sensitivity to immunotherapy. These findings offer insights into the potential mechanisms underlying the unfavorable prognosis associated with high-risk gliomas. Currently, immunotherapy has shown limited success in the treatment of glioma patients [27], and a major challenge lies in identifying the patients most likely to respond to this treatment. Therefore, our results provide valuable guidance for the development of personalized immunotherapy approaches for glioma patients.
Peroxisomes play a crucial role in compartmentalizing immune response pathways, serving as signaling platforms for initiating immune signaling cascades [42,43,44]. Additionally, peroxisomes act as immune metabolic hubs, influencing cellular metabolites such as ROS and unsaturated fatty acids. This functionality enables them to regulate immune cell development and activation, and modulate inflammatory pathways across different immune cells and tissues [45,46,47,48]. Impairment of macrophage activity has been observed in cells deficient in peroxisome biogenesis genes, such as PEX5 [45,47], or the antioxidant enzyme CAT [45]. These findings, in conjunction with our results, suggest that metabolic alterations may contribute to the establishment of an immunosuppressive microenvironment in high-risk gliomas. However, further research is necessary to identify the specific metabolic enzymes and products involved in this process.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Distribution of risk scores in gliomas with different clinicopathological features; Figure S2: Establishment of the nomogram using the CGGA_693 or CGGA_325 cohort ; Figure S3: Differential expressed genes between the low and high-risk groups; Table S1-S4: The clinicopathological characteristics of glioma patients in the TCGA (Table S1), CGGA_693 (Table S2), CGGA_325 (Table S3), and GSE16011 (Table S4) cohorts were indicated.
Author Contributions
Conceptualization, X.Q., B.W., and W.W.; methodology, X.Q., and B.W.; software, X.Q., and B.W.; validation, X.Q., B.W., X.L., and Y.S.; formal analysis, X.Q., B.W., X.L., and Y.S.; investigation, X.Q., and B.W.; resources, X.Q., X.L., and Y.S.; data curation, X.L., and Y.S.; writing—original draft preparation, X.Q., B.W., W.W.; writing—review and editing, W.W.; visualization, X.Q., B.W.; supervision, W.W.; project administration, W.W.; funding acquisition, W.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Nos. 32070756 and 81903016).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J Clin Oncol 2017, 35, 2402–2409. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Rodriguez-Camacho, A.; Flores-Vazquez, J.G.; Moscardini-Martelli, J.; Torres-Rios, J.A.; Olmos-Guzman, A.; Ortiz-Arce, C.S.; Cid-Sanchez, D.R.; Perez, S.R.; Macias-Gonzalez, M.D.S.; Hernandez-Sanchez, L.C.; et al. Glioblastoma Treatment: State-of-the-Art and Future Perspectives. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol 2019, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Aitchison, J.D. Peroxisomes take shape. Nat Rev Mol Cell Biol 2013, 14, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Keller, G.A.; Hosken, N.; Wilkinson, J.; Subramani, S. A conserved tripeptide sorts proteins to peroxisomes. J Cell Biol 1989, 108, 1657–1664. [Google Scholar] [CrossRef]
- Swinkels, B.W.; Gould, S.J.; Bodnar, A.G.; Rachubinski, R.A.; Subramani, S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J 1991, 10, 3255–3262. [Google Scholar] [CrossRef]
- Gatto, G.J., Jr.; Geisbrecht, B.V.; Gould, S.J.; Berg, J.M. Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5. Nat Struct Biol 2000, 7, 1091–1095. [Google Scholar] [CrossRef]
- Wang, W.; Subramani, S. Role of PEX5 ubiquitination in maintaining peroxisome dynamics and homeostasis. Cell Cycle 2017, 16, 2037–2045. [Google Scholar] [CrossRef]
- Braverman, N.; Dodt, G.; Gould, S.J.; Valle, D. An isoform of pex5p, the human PTS1 receptor, is required for the import of PTS2 proteins into peroxisomes. Hum Mol Genet 1998, 7, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Harano, T.; Honsho, M.; Ghaedi, K.; Mukai, S.; Tanaka, A.; Kawai, A.; Shimizu, N.; Fujiki, Y. The mammalian peroxin Pex5pL, the longer isoform of the mobile peroxisome targeting signal (PTS) type 1 transporter, translocates the Pex7p.PTS2 protein complex into peroxisomes via its initial docking site, Pex14p. J Biol Chem 2000, 275, 21703–21714. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. PEX19 is a predominantly cytosolic chaperone and import receptor for class 1 peroxisomal membrane proteins. J Cell Biol 2004, 164, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas - an emerging landscape of actionable co-dependency targets. Nat Rev Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. Cancer metabolism as a central driving force of glioma pathogenesis. Brain Tumor Pathol 2016, 33, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.S.; Di Pietro, E.; Jangal, M.; Goncalves, C.; Witcher, M.; Braverman, N.E.; Del Rincon, S.V. Peroxisomes and cancer: The role of a metabolic specialist in a disease of aberrant metabolism. Biochim Biophys Acta Rev Cancer 2018, 1870, 103–121. [Google Scholar] [CrossRef]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 2013, 13, 345. [Google Scholar] [CrossRef]
- Geisbrecht, B.V.; Gould, S.J. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem 1999, 274, 30527–30533. [Google Scholar] [CrossRef]
- Benedetti, E.; Galzio, R.; Laurenti, G.; D'Angelo, B.; Melchiorre, E.; Cifone, M.G.; Fanelli, F.; Muzi, P.; Coletti, G.; Alecci, M.; et al. Lipid metabolism impairment in human gliomas: expression of peroxisomal proteins in human gliomas at different grades of malignancy. Int J Immunopathol Pharmacol 2010, 23, 235–246. [Google Scholar] [CrossRef]
- Ruokun, C.; Yake, X.; Fengdong, Y.; Xinting, W.; Laijun, S.; Xianzhi, L. Lentivirus-mediated silencing of HSDL2 suppresses cell proliferation in human gliomas. Tumour Biol 2016, 37, 15065–15077. [Google Scholar] [CrossRef]
- Laurenti, G.; Benedetti, E.; D'Angelo, B.; Cristiano, L.; Cinque, B.; Raysi, S.; Alecci, M.; Ceru, M.P.; Cifone, M.G.; Galzio, R.; et al. Hypoxia induces peroxisome proliferator-activated receptor alpha (PPARalpha) and lipid metabolism peroxisomal enzymes in human glioblastoma cells. J Cell Biochem 2011, 112, 3891–3901. [Google Scholar] [CrossRef] [PubMed]
- Linden, A. Measuring diagnostic and predictive accuracy in disease management: an introduction to receiver operating characteristic (ROC) analysis. J Eval Clin Pract 2006, 12, 132–139. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Saville, B.R.; Lewis, R.J. Decision curve analysis. JAMA 2015, 313, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Noorbakhsh, A.; Hirshman, B.R.; Zhou, T.; Tang, J.A.; Chang, D.C.; Carter, B.S.; Chen, C.C. Survival trends of grade I, II, and III astrocytoma patients and associated clinical practice patterns between 1999 and 2010: A SEER-based analysis. Neurooncol Pract 2016, 3, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, L.; Zhang, H.; Zhang, Y.; Ju, H.; Wang, X.; Ren, H.; Zhu, X.; Dong, Y. The immunosuppressive microenvironment and immunotherapy in human glioblastoma. Front Immunol 2022, 13, 1003651. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Schmidt, M.H.H.; Schumann, U. The Glioma Immune Landscape: A Double-Edged Sword for Treatment Regimens. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol 2017, 47, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Li, K.; Zhang, W.; Wan, C.; Zhang, J.; Jiang, P.; Liu, X.S. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.A.; Wanders, R.J. Alpha-oxidation. Biochim Biophys Acta 2006, 1763, 1403–1412. [Google Scholar] [CrossRef]
- Sassa, T.; Kihara, A. Metabolism of very long-chain Fatty acids: genes and pathophysiology. Biomol Ther (Seoul) 2014, 22, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Miska, J.; Chandel, N.S. Targeting fatty acid metabolism in glioblastoma. J Clin Invest 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528 e520. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep 2016, 17, 2445–2459. [Google Scholar] [CrossRef]
- Vidyarthi, A.; Agnihotri, T.; Khan, N.; Singh, S.; Tewari, M.K.; Radotra, B.D.; Chatterjee, D.; Agrewala, J.N. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol Immunother 2019, 68, 1995–2004. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin Cancer Res 2019, 25, 4202–4210. [Google Scholar] [CrossRef]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of Type I and III Interferon Response by Mitochondrial and Peroxisomal MAVS and Inhibition by Hepatitis C Virus. PLoS Pathog 2015, 11, e1005264. [Google Scholar] [CrossRef]
- Choi, Y.B.; Choi, Y.; Harhaj, E.W. Peroxisomes support human herpesvirus 8 latency by stabilizing the viral oncogenic protein vFLIP via the MAVS-TRAF complex. PLoS Pathog 2018, 14, e1007058. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.R.; Marques, M.; Ribeiro, D. Peroxisomes and Innate Immunity: Antiviral Response and Beyond. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, F.; Sheshachalam, A.; Braverman, N.E.; Rachubinski, R.A.; Simmonds, A.J. Peroxisome-Mediated Metabolism Is Required for Immune Response to Microbial Infection. Immunity 2018, 48, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Facciotti, F.; Ramanjaneyulu, G.S.; Lepore, M.; Sansano, S.; Cavallari, M.; Kistowska, M.; Forss-Petter, S.; Ni, G.; Colone, A.; Singhal, A.; et al. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat Immunol 2012, 13, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Srinu, T.; Karnati, S.; Garikapati, V.; Linke, M.; Kamalyan, L.; Mali, S.R.; Sudan, K.; Kollas, A.; Schmid, T.; et al. A New Immunomodulatory Role for Peroxisomes in Macrophages Activated by the TLR4 Ligand Lipopolysaccharide. J Immunol 2017, 198, 2414–2425. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bulow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in Immune Response and Inflammation. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
Figure 1.
Identifying prognostic genes for developing a PEX5-dependent risk model. The six metabolic pathways in peroxisomes were depicted. The proteins with predicted PTS1 or PTS2 signals were denoted with red and blue fonts, respectively. The genes enclosed within the box represent the 11 signature genes selected by the LASSO model, as illustrated in Panel B and C below. The 35 PTS1- or PTS2- containing genes in panel (A) were subjected to the LASSO coefficient analysis, using the TCGA data set. Selection of the optimal parameter (lambda), as indicated by the first black dotted line, in the LASSO model in panel (B). Forest plot of the 11 signature genes selected by the LASSO model.
Figure 1.
Identifying prognostic genes for developing a PEX5-dependent risk model. The six metabolic pathways in peroxisomes were depicted. The proteins with predicted PTS1 or PTS2 signals were denoted with red and blue fonts, respectively. The genes enclosed within the box represent the 11 signature genes selected by the LASSO model, as illustrated in Panel B and C below. The 35 PTS1- or PTS2- containing genes in panel (A) were subjected to the LASSO coefficient analysis, using the TCGA data set. Selection of the optimal parameter (lambda), as indicated by the first black dotted line, in the LASSO model in panel (B). Forest plot of the 11 signature genes selected by the LASSO model.
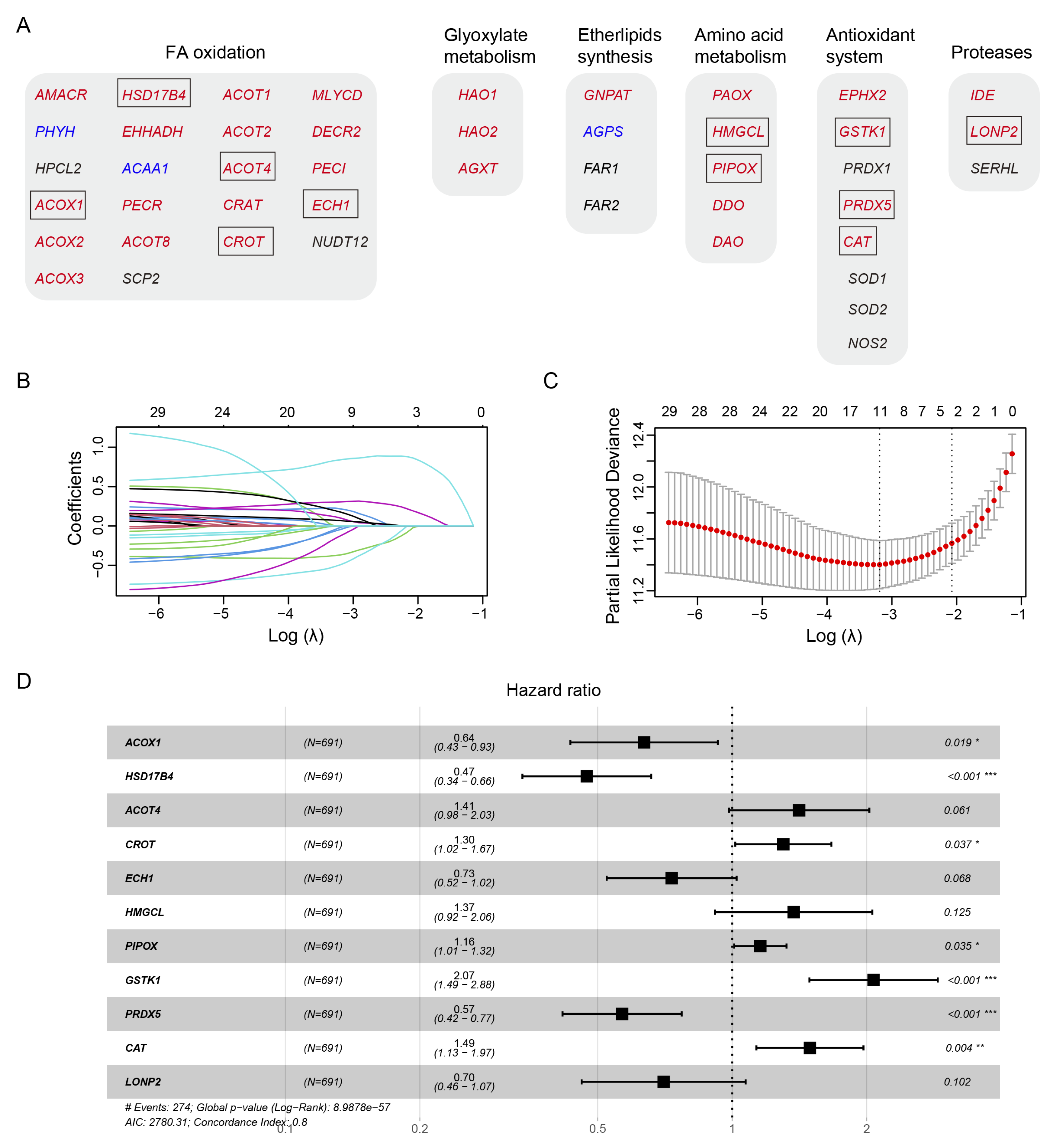
Figure 2.
Assessment of the prognostic prediction ability of the 11-gene signature. (A) Distributions of risk scores, OS, and survival status, and heatmaps of gene expression profiles of the signature genes in the indicated cohorts. The dotted line indicates patients were median-dichotomized into the low-risk group and high-risk group. (B) ROC curves of the 11-gene signature in the training TCGA dataset, and the three validation datasets: CGGA_693; CGGA_325; GSE16011. (C) KM curve of the prognosis signature in the indicated four cohorts (log-rank test).
Figure 2.
Assessment of the prognostic prediction ability of the 11-gene signature. (A) Distributions of risk scores, OS, and survival status, and heatmaps of gene expression profiles of the signature genes in the indicated cohorts. The dotted line indicates patients were median-dichotomized into the low-risk group and high-risk group. (B) ROC curves of the 11-gene signature in the training TCGA dataset, and the three validation datasets: CGGA_693; CGGA_325; GSE16011. (C) KM curve of the prognosis signature in the indicated four cohorts (log-rank test).
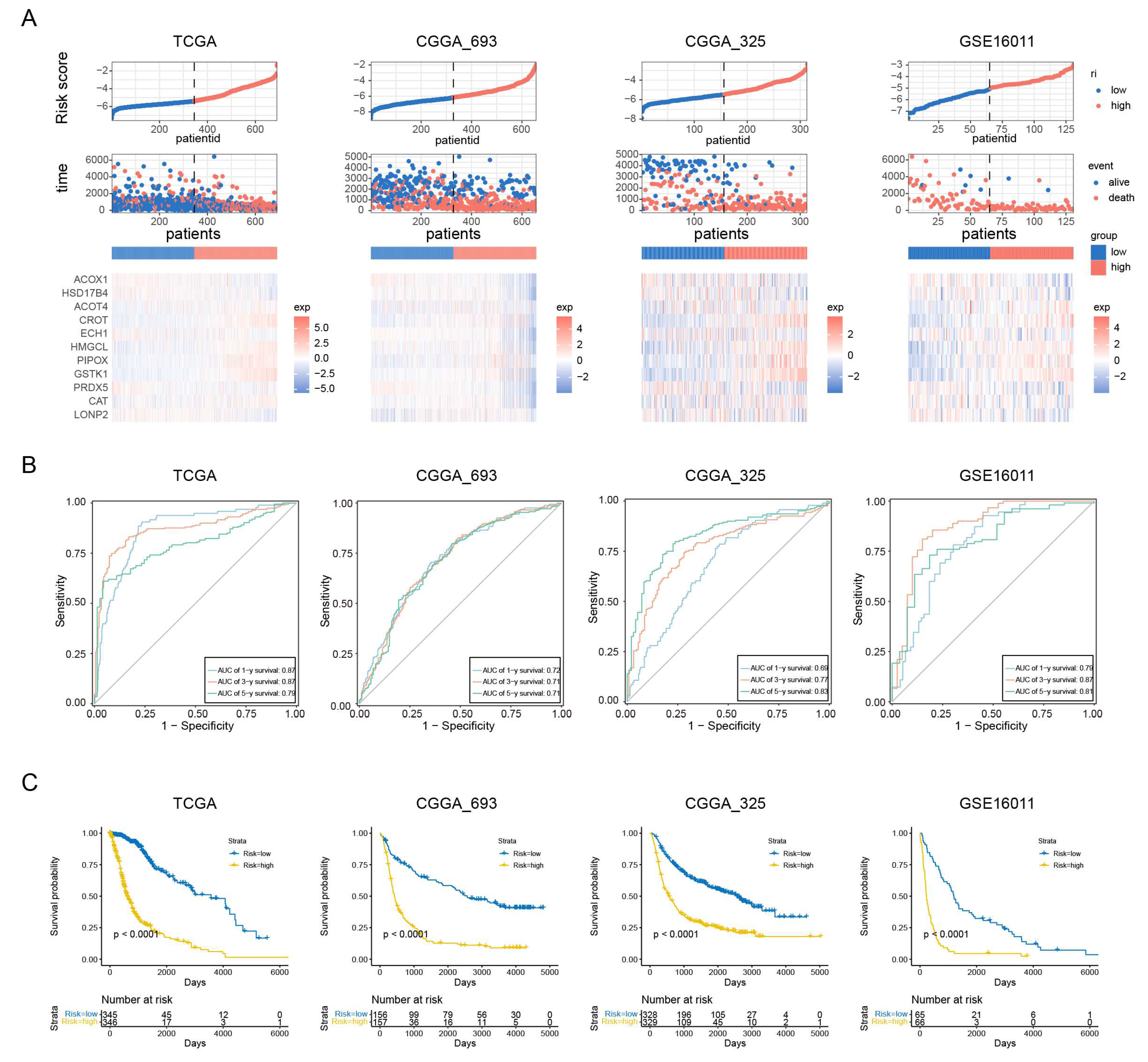
Figure 3.
Establishment and assessment of the nomogram. (A, B) Forest plot of the univariate (A) and multivariate (B) Cox regression analyses of the indicated parameters in the TCGA cohort. IDH, IDH mutation status; grade, WHO grade; 1p19q, 1p19q codeletion status; Meth, MGMT promoter methylation status. The X-axis indicated the hazard ratio values. A hazard ratio of 1 signified no difference in survival risk. (C) The nomogram plot was constructed based on the risk score, IDH, age, gender, grade, and meth. The points of the factors indicate their corresponding contribution to the survival probability. The total points of each patient provided the estimated 3-year and 5-year survival times. (D) Calibration plot of the nomogram based on the TCGA, CGGA_693, and CGGA_325 datasets. (E) DCA of the nomogram for the 1-, 3-and 5-year OS in the TCGA, CGGA_693 and CGGA_325 datasets.
Figure 3.
Establishment and assessment of the nomogram. (A, B) Forest plot of the univariate (A) and multivariate (B) Cox regression analyses of the indicated parameters in the TCGA cohort. IDH, IDH mutation status; grade, WHO grade; 1p19q, 1p19q codeletion status; Meth, MGMT promoter methylation status. The X-axis indicated the hazard ratio values. A hazard ratio of 1 signified no difference in survival risk. (C) The nomogram plot was constructed based on the risk score, IDH, age, gender, grade, and meth. The points of the factors indicate their corresponding contribution to the survival probability. The total points of each patient provided the estimated 3-year and 5-year survival times. (D) Calibration plot of the nomogram based on the TCGA, CGGA_693, and CGGA_325 datasets. (E) DCA of the nomogram for the 1-, 3-and 5-year OS in the TCGA, CGGA_693 and CGGA_325 datasets.
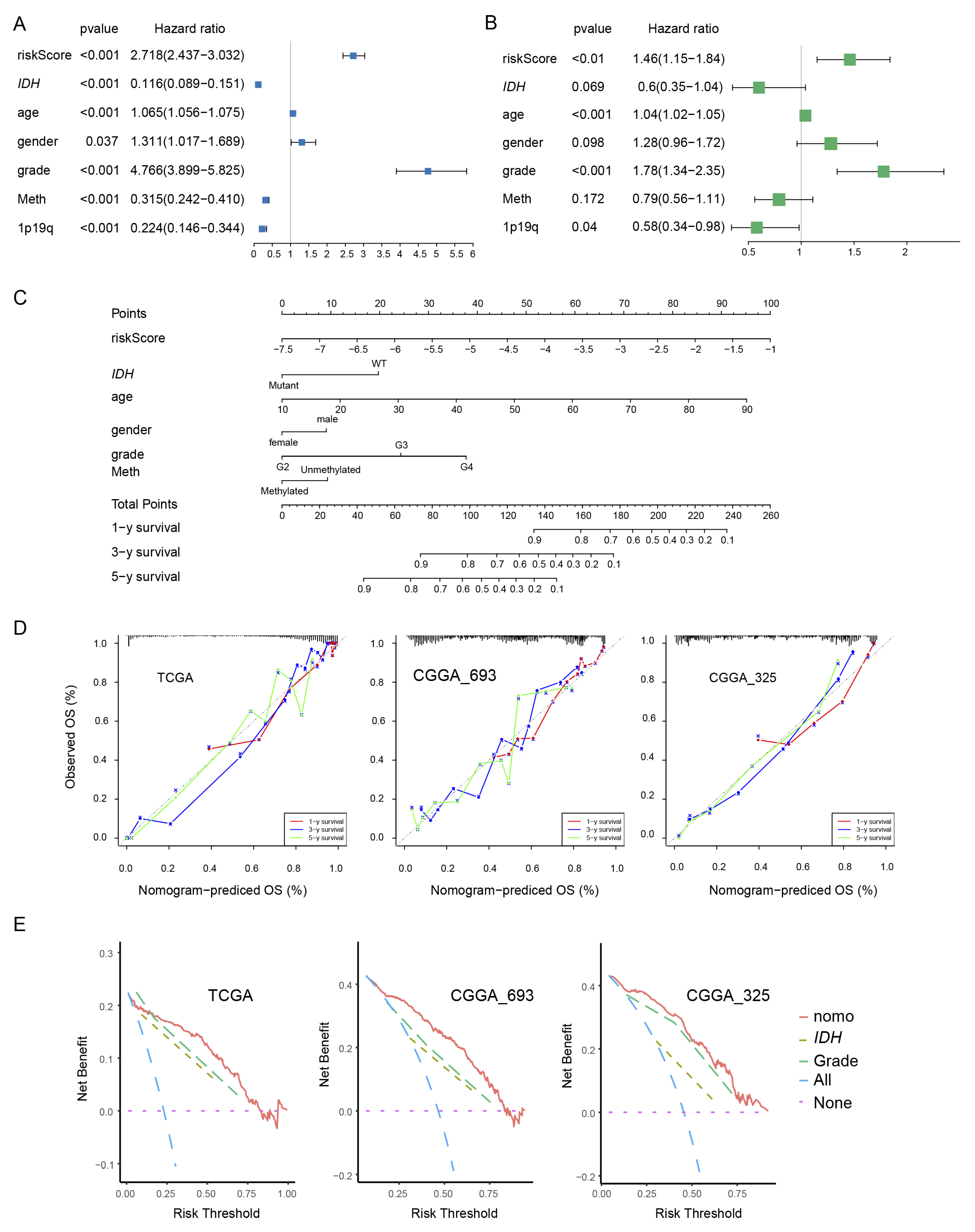
Figure 4.
Comparisons of immunosuppressive features between the low- and high-risk gliomas. The abundance of macrophages, MDSCs, and Tregs is compared between the low- and high-risk groups in the three cohorts. ***, p < 0.001 (Wilcoxon test). (A) The correlation of the risk score with the abundance of macrophages, MDSCs, and Tregs is demonstrated in the three cohorts. Pearson correlation was used to calculate the correlation coefficient (R) and p-values. (B) The distribution of immune subtypes is compared between the low- and high-risk groups in the three cohorts. (C) Intersection analysis of commonly upregulated genes in the high-risk group among three cohorts with genes that inhibit the cancer-immunity cycle (TIP_down) or immune checkpoints. Heatmaps depict the expression levels of these overlapping genes between the low- and high-risk groups in the three cohorts. Genes that belong to both the cancer-immunity cycle inhibitors and checkpoint genes categories are denoted by the "#" label." (D) Correlation matrix visualization showing the relationship between the risk score and the overlapping genes from panel (A). Pearson correlation coefficient (R) values were indicated by the color scale. *, p < 0.05; **, p < 0.01; ***, p < 0.001. (E) Comparison of the TIDE scores between the low- and high-risk groups in the three cohorts. *, p < 0.05; ***, p < 0.001 (Wilcoxon test).
Figure 4.
Comparisons of immunosuppressive features between the low- and high-risk gliomas. The abundance of macrophages, MDSCs, and Tregs is compared between the low- and high-risk groups in the three cohorts. ***, p < 0.001 (Wilcoxon test). (A) The correlation of the risk score with the abundance of macrophages, MDSCs, and Tregs is demonstrated in the three cohorts. Pearson correlation was used to calculate the correlation coefficient (R) and p-values. (B) The distribution of immune subtypes is compared between the low- and high-risk groups in the three cohorts. (C) Intersection analysis of commonly upregulated genes in the high-risk group among three cohorts with genes that inhibit the cancer-immunity cycle (TIP_down) or immune checkpoints. Heatmaps depict the expression levels of these overlapping genes between the low- and high-risk groups in the three cohorts. Genes that belong to both the cancer-immunity cycle inhibitors and checkpoint genes categories are denoted by the "#" label." (D) Correlation matrix visualization showing the relationship between the risk score and the overlapping genes from panel (A). Pearson correlation coefficient (R) values were indicated by the color scale. *, p < 0.05; **, p < 0.01; ***, p < 0.001. (E) Comparison of the TIDE scores between the low- and high-risk groups in the three cohorts. *, p < 0.05; ***, p < 0.001 (Wilcoxon test).
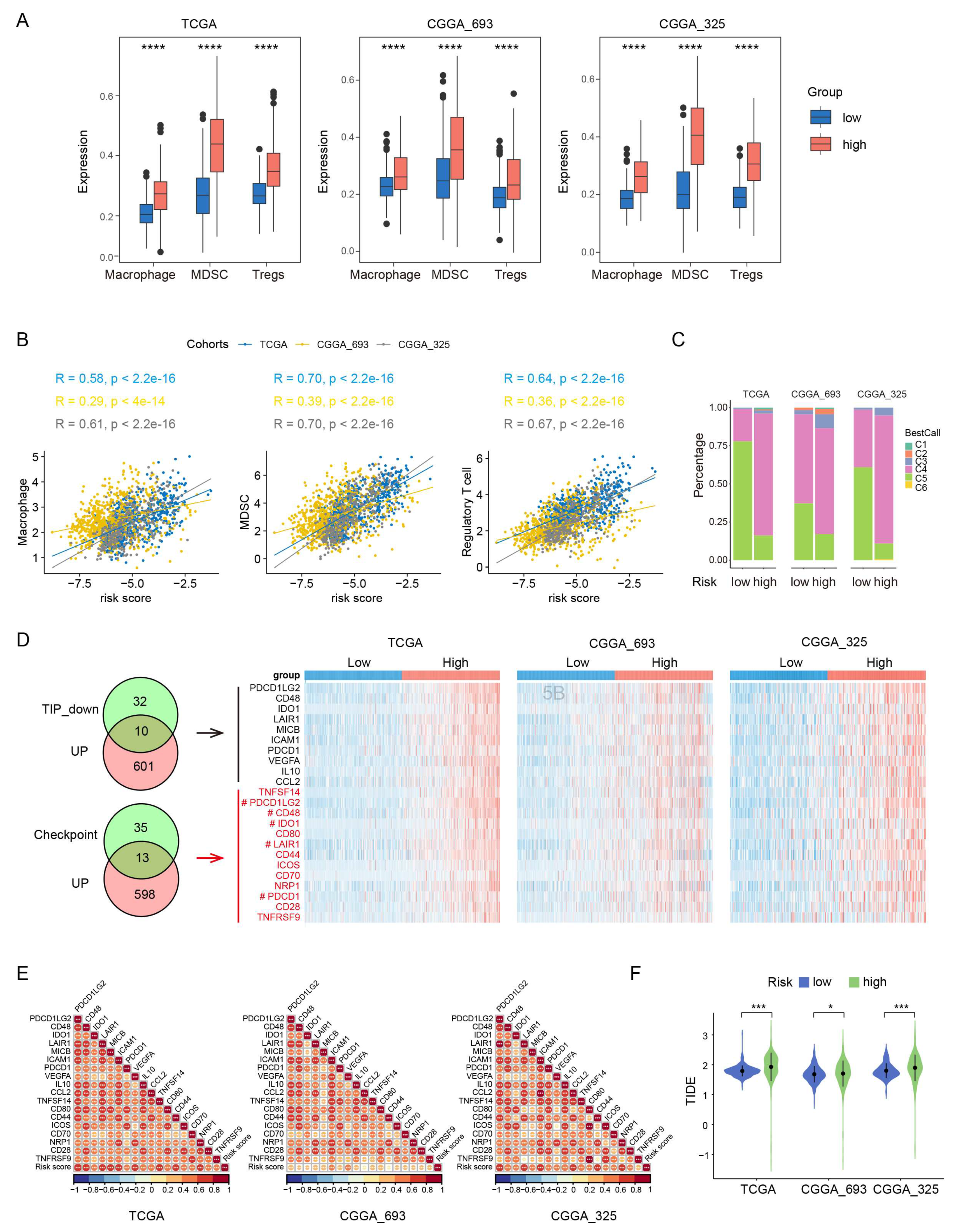
Figure 5.
PEX5 KO inhibits the peroxisomal localization of the signature genes. (A) The genomic DNA of U251(sgPEX5-1) cells was extracted, and the regions surrounding the PEX5 sgRNAs were amplified and sequenced. The deletion region by the sgRNAs is indicated. (B) PEX5 detection in the U251 (sgPEX5-1) cells by western blot. (C) The control and sgPEX5-1 U251 cells were transfected with the GFP-PTS1 or GFP-PTS2 reporter constructs, or the indicated constructs. The cells were stained with GFP antibodies to enhance the fluorescent signal. Scale bars, 10 µm. (D) The control and sgPEX5-1 U251 cells were co-stained with PMP70 and CAT antibodies. Scale bars, 10 µm.
Figure 5.
PEX5 KO inhibits the peroxisomal localization of the signature genes. (A) The genomic DNA of U251(sgPEX5-1) cells was extracted, and the regions surrounding the PEX5 sgRNAs were amplified and sequenced. The deletion region by the sgRNAs is indicated. (B) PEX5 detection in the U251 (sgPEX5-1) cells by western blot. (C) The control and sgPEX5-1 U251 cells were transfected with the GFP-PTS1 or GFP-PTS2 reporter constructs, or the indicated constructs. The cells were stained with GFP antibodies to enhance the fluorescent signal. Scale bars, 10 µm. (D) The control and sgPEX5-1 U251 cells were co-stained with PMP70 and CAT antibodies. Scale bars, 10 µm.

Figure 6.
PEX5 KO inhibits proliferation, migration, and invasion of U251 cells. (A) detection of PEX5 in the Control, sgPEX5-1, and sgPEX5-2 U251 cells. U251 cells (control, sgPEX5-1, sgPEX5-2) were plated, and subjected to the CCK8 (B), colony formation (C), flowcytometry (D), migration (E), and transwell (F) assays. Data are presented as the mean ± SD and representative of three independent experiments. **, p < 0.01; ***, p < 0.001 (unpaired two-tailed Student’s t test).
Figure 6.
PEX5 KO inhibits proliferation, migration, and invasion of U251 cells. (A) detection of PEX5 in the Control, sgPEX5-1, and sgPEX5-2 U251 cells. U251 cells (control, sgPEX5-1, sgPEX5-2) were plated, and subjected to the CCK8 (B), colony formation (C), flowcytometry (D), migration (E), and transwell (F) assays. Data are presented as the mean ± SD and representative of three independent experiments. **, p < 0.01; ***, p < 0.001 (unpaired two-tailed Student’s t test).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



