Submitted:
22 September 2023
Posted:
25 September 2023
Read the latest preprint version here
Abstract
The p53 tumor suppressor protein is an activator of transcription. Diverse stress factors lead to various sets of posttranslational modifications of p53 what results in different sets of upregulated genes. We noticed that actinomycin D and nutlin-3a (A+N) synergize in inducing activating phosphorylations of p53 and upregulation of selected p53-target genes. Here we found that one of these genes is DUSP13, which codes for poorly-studied, dual-specificity phosphatase having at least two isoforms, one expressed in testis and the other in skeletal muscles. We found that in cancer cells exposed to A+N, DUSP13 is expressed from an alternative promoter in intron 3, what results in expression of isoform named TMDP-L1. The luciferase reporter tests demonstrated that this promoter is activated by both endogenous and ectopically expressed p53. We showed for the first time that mRNA expressed from this promoter actually produces the protein, which can be detected by Western blotting in all examined cancer cell lines with wild-type p53 exposed to A+N. In some cell lines it is also induced by clinically relevant camptothecin. This isoform, fused with green fluorescent protein localizes in perinuclear region of cells. Thus, TMDP-L1 isoform may be an important element of p53-regulated stress response system.
Keywords:
p53
; MDM2
; DUSP13
; alternative promoter
; dual-specificity phosphatase
1. Introduction
The p53 tumor suppressor protein impacts on functioning of a cell by acting as an activator of at least hundreds of genes. The early-identified p53-regulated genes coded for proteins promoting apoptosis, cell cycle arrest and DNA repair. However, as many more genes directly activated by p53 have been discovered, it was realized that p53 regulates wide spectrum of biological functions including metabolism, aging, angiogenesis, immunity, and more [1,2,3]. The p53 binds as a tetramer to its response element (RE) RRRCWWGYYYRRRCWWGYYY (R–A or G, Y–C or T, W–A or T) consisting of two decameric half sites divided further into four pentameric quarter sites (RRRCW or WGYYY). Thus, each p53 monomer binds to each quarter site of the p53 RE. This arrangement allows for some flexibility in the selection of p53 binding sites, which sometime can deviate from the consensus sequence [4].
Recently, we published the transcriptomic data showing changes in gene expression in A549 lung cancer cell line exposed to actinomycin D and nutlin-3a (henceforth abbreviated to A+N) [5]. Earlier, we noticed that these two substances synergize in activation of p53 and in activation of some p53-regulated genes [6,7]. The mechanism of synergy is not known. Actinomycin D inhibits RNA polymerase I, what induces nucleolar stress and release of some ribosomal proteins out of nucleolus. Some of these proteins bind to MDM2, which is the negative regulator of p53. MDM2, bound by ribosomal proteins, does not destabilize p53, which accumulates and activates its target genes [8]. However, probably it is only a part of the mechanism, by which actinomycin D activates p53 because we noticed, that this drug also promotes p53 phosphorylation by an undefined kinase [9]. Phosphorylation of p53 by various kinases on different amino acids is crucial for stimulation of p53 as a transcription regulator [10]. MDM2 inhibits p53 by promoting its degradation but also by concealing its transcription-activating domain [11,12]. Nutlin-3a is a molecule, which binds to MDM2 and fills its p53 interaction pocket, what prevents p53-MDM2 interaction, what in turn results in p53 stabilization and activation of p53-target genes [13]. We hypothesize, that when actinomycin D and nutlin-3a are applied together they synergize because actinomycin D activates p53-phosphorylating kinases and nutlin-3a prevents MDM2 from covering p53, what gives the kinases easy contact with p53 and promotes its strong phosphorylation. By employing this p53 activation mode (A+N) and a cell line (A549) never used before in high-throughput searches for p53-regulated genes, we were able to identify new candidate p53-target genes [5].
One of the genes strongly activated by A+N is DUSP13. We selected it for detailed study not only because of its activation by A+N (55-fold by RNA-Seq), but also because its expression, as suggested by the location of mapped sequencing reads, starts from an alternative promoter in intron 3 [5]. We hypothesized that p53 activated by A+N, induces the expression of DUSP13 from this alternative promoter.
Initially DUSP13 was cloned as a gene coding for a protein abundantly expressed in testis [14]. The regulation of DUSP13 is very complicated and unusual so we considered it interesting to learn how p53 contributes to this regulation. DUSP13 codes for two isoforms of proteins, which function as dual-specificity phosphatases removing phosphate groups from serines/threonines as well as from tyrosines. DUSP13 codes these isoforms utilizing the alternative open reading frames (ORF) what is extremely rare in eukaryotes. Exons 1-3 of DUSP13 code for muscle-restricted dual specificity phosphatase (MDSP), whereas exons 7-9 encode testis and skeletal muscle specific dual specificity phosphatase (TMDP). The RT-PCR analysis of RNA from skeletal muscles also revealed the existence of mRNAs able to code another isoforms, related to TMDP named TMDP-L1 and TMDP-L2, however, the Western blotting analysis of proteins extracted from muscles did not reveal the presence of these proteins [15]. These results generate several questions, e.g., are these mRNA forms translated, was the employed Western blotting method sensitive enough to detect their presence, and if expressed, what is their physiological function?
Our RNA-Seq data [5] revealed that activation of p53 by A+N in lung cancer cells may induce the expression of a DUSP13 isoform related to TMDP. This is unexpected observation, because so far the expression of DUSP13 was detected only in skeletal muscle and testis. Our results suggested that the p53 has the potential to activate expression of DUSP13 in other tissues. Interestingly, TMDP was found to inactivate stress-activated kinases named MAPKs [16] what suggests that p53 may modulate the stress response of a cell by activation of this DUSP13 isoform.
2. Materials and Methods
2.1. Cell Culture and Treatment
A549 (lung adenocarcinoma, American Type Culture Collection-ATCC, Manassas, VA, USA), NCI-H292 (mucoepidermoid pulmonary carcinoma, ATCC) and U-2 OS (osteosarcoma, ATCC) cells were grown as previously described [6]. NCI-H460 (lung cancer, ATCC) were cultured in RPMI-1640 supplemented with 2 mM L-glutamine, 4.5 g/L glucose and 1 mM sodium pyruvate, and 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA). A375 melanoma cell line (ATCC) was cultured in high-glucose DMEM supplemented with 10% FBS and AGS gastric adenocarcinoma cell line (ATCC) was cultured in McCoy’s 5A medium with 10% FBS. All culture media were supplemented with 1% penicillin–streptomycin (Sigma-Aldrich, St. Louis, MO, USA). The cells were grown at 37°C/5% CO2.
The stock solutions of chemicals were prepared in DMSO: actinomycin D (10 µM; Sigma-Aldrich, St. Louis, MO, USA), camptothecin (10 mM; Calbiochem-Merck, Darmstadt, Germany), nutlin-3a (10 mM; Selleck Chemicals LLC, Houston, TX, USA), idasanutlin (10 mM; MedChemExpress, Monmouth Junction, NJ, USA) and RG7112 (10 mM; MedChemExpress). Stock solutions were diluted in culture medium to the following concentrations: 5 nM actinomycin D, 5 µM nutlin-3a, 5 µM camptothecin, 5 µM idasanutlin and 5 µM RG7112. Control cells were mock-treated with medium containing DMSO.
The generation of p53-deficient A549 and U-2 OS cells using CRISPR/Cas9 technology was described earlier [5].
2.2. Semi-Quantitative Real-Time PCR
Total RNA samples were isolated from cells using the RNeasy mini kit (Qiagen, Hilden, Germany). The cDNA was synthesized with MuLV reverse transcriptase and random hexamers (Applied Biosystems, Foster City, CA, USA). Measurements of mRNA levels were performed using Real-Time 2x PCR Master Mix SYBR (A&A Biotechnology, Gdynia, Poland). We used the following primers to measure the expression of DUSP13 by RT-PCR: 5′-GAT ACA TCC GAG CTG CCC TC-3′ and 5′-GCC TCT ACC AGC GTC ATG TT-3′. The primers for internal control ACTB were as follows: 5′-GCA AGC AGG AGT ATG ACG AG-3′ and 5′-CAA ATA AAG CCA TGC CAA TC-3′. Amplifications were performed on a CFX96 Real-Time System (Bio-Rad, Hercules, CA, USA). In each PCR run, cDNA samples were amplified in triplicate. Relative quantitation of mRNA was carried out using the ΔΔCT method with ACTB as a reference. Mean and standard deviation were calculated from three or four biological replicates.
2.3. Western Blotting
The preparation of the whole-cell lysates using IP buffer, supplemented with protease and phosphatase inhibitors as well as preparation of the concentrated conditioned medium were described previously [7]. Aliquots of lysates (35–50 µg) were separated by SDS-PAGE on 8% or 13% gels and electro-transferred onto PVDF membranes. Before incubation with primary antibody, the membranes were incubated for 1 h at room temperature in blocking solution (5% skim milk in PBS with 0.1% Tween-20). The anti-phospho-Ser37 p53 and anti-phospho-Ser392 p53 antibodies were from Cell Signaling Technology (Danvers, MA, USA). Anti-p53 (DO-1), and loading control anti-HSC70 (B-6) antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-DUSP13 antibody (rabbit polyclonal) was from Proteintech (Rosemont, IL, USA). The HRP-conjugated anti-GFP (green fluorescent protein) antibody (B-2) was from Santa Cruz Biotechnology. All incubations with primary antibodies were performed overnight at 4oC in blocking solution. HRP-conjugated secondary antibodies (anti-mouse, anti-rabbit) were detected by chemiluminescence (SuperSignal West Pico or SuperSignal West Femto chemiluminescent substrate, Thermo Fisher Scientific, Waltham, MA, USA).
2.4. Molecular Cloning, Site-Directed Mutagenesis and Luciferase Reporter Assay
The alternative promoter region of DUSP13 was cloned into the pGL3-Basic reporter vector, which encodes firefly luciferase (Promega, Madison, WI, USA). The sequences of primers used to amplify the promoter are: 5′-TTTT ACG CGT CCA CCT CTG CTT CCT CTA CA-3′ and 5′-TTTT CTC GAG CCA GCT CTG GAA GAG AGA TGA-3′. The primers were designed to contain the restriction sites for MluI and XhoI (underlined). Amplified DNA was ligated into the respective sites of pGL3-Basic plasmid. PCR was performed with PfuPlus! DNA polymerase mix (EURx, Gdańsk, Poland) to ensure high fidelity DNA amplification. The inserted DNA was sequenced to ensure that the clones contained no mutations.
The mutations of CWWG (W-A or T) sequence in the putative p53 response element from DUSP13 promoter were created using GeneArt Site-Directed Mutagenesis PLUS kit (Life Technologies, Carlsbad, CA, USA) using following oligonucleotides: DUSP13 forward (5′-GGTGACTGGCCTGGGGCGTCTTGGGAGCTGGAAC-3′), DUSP13 complementary reverse (5′-GTTCCAGCTCCCAAGACGCCCCAGGCCAGTCACC-3′), (the sites of mutation are underlined).
The luciferase reporter assay was performed as described recently [7]. In short, U-2 OS cells were co-transfected using FuGENE 6 (Promega, Madison, WI, USA) with a combination of reporter vector, encoding firefly luciferase under the control of tested promoter (wild type or mutant), and expression vector pC53-SN3, encoding wild-type p53 or pC53-SCX3 encoding Val143Ala p53 mutant (a gift from Dr. Bert Vogelstein and Dr. Kenneth W. Kinzler from Johns Hopkins University, Baltimore, MD, USA [17]). As a negative control, the p53 plasmid was replaced by empty vector. The transfection mixture also contained pRL-TK vector, encoding Renilla sp. luciferase under the control of herpes simplex virus thymidine kinase (HSV-TK) promoter (internal control). The next day, the cells were washed with culture medium and incubated with fresh medium for an additional 24 h. The cells were lysed with PLB buffer from the Dual Luciferase Reporter Assay system (Promega, Madison, WI, USA) and the activities of the luciferases were measured. Firefly luciferase activity was normalized against Renilla sp. luciferase activity what produced normalized firefly luciferase activity (NFLA). The NFLA in control cells was set as 1 and the activity in experimental cells was expressed as its fold-change. Each transfection was performed in triplicate in three independent experiments.
To test how the endogenous p53 impacts on the activity of the cloned DUSP13 promoter, the cells were transfected with the DUSP13 reporter vector and the abovementioned pRL-TK control vector. After 24 h, the medium with the transfection mixture (Fugene 6 + DNA) was removed and the cells were exposed either to control medium or the medium with A+N to activate the endogenous p53. After 24 hours the activities of both luciferases were measured and normalized firefly luciferase activity was calculated as described in the previous paragraph.
To generate DUSP13-EGFP chimeric cDNA, we first isolated RNA from A549 cells exposed to A+N and then we generated cDNA as described above. We employed a triple-ligation method to clone cDNA of the DUSP isoform taking advantage of the ClaI restriction site naturally occurring in the middle of its cDNA sequence. The 5′ part of cDNA was amplified with the following primers: 5′-TTTT AAG CTT ACA GAG CTC ATC TCT CTT CC-3′ and 5′-CAG ACC TCA TCG ATA TGG TTC-3′. This PCR product, containing the ClaI siste (bold), was digested with HinDIII and ClaI restriction enzymes. The HinDIII was created by PCR primer (underlined). The 3′ part of cDNA was amplified with the following primers: 5′-AAC CAT ATC GAT GAG GTC TGG-3′, and 5′-AGG GTC AGG GAT CCT GGC T-3′. This PCR product was digested with ClaI and BamHI (underlined). These products were ligated into HinDIII and BamHI sites of pcDNA3.1(+) plasmid. The whole insert was sequenced to ensure that the clone without mutations was selected. In this way we generated expression vector coding for unmodified isoform of DUSP13. Subsequently we amplified cDNA of the DUSP13 isoform from this plasmid using the following primers: 5′-TTTTGCTAGCCCTGCCATGGGGCTCTGCCAC-3′ and 5′-TTTTAAGCTTTCCGAACCGCCCCGTCTCCCG-3′. In the first primer the NheI restriction site was created (underlined) and the location of start codon is marked in bold. In the second primer the HinDIII site was created (underlined) and the last codon of DUSP13 isoform (Phe, in reverse) is marked by bold font. The PCR product was ligated into NheI and HinDIII sites of pcDNA3.1(+) plasmid containing EGFP cDNA cloned into BamHI and EcoRI sites. In this way we ligated DUSP13 cDNA in frame with EGFP cDNA. The last PCR step was needed to remove the stop codon of DUSP13 isoform. The correctness of the constructed chimeric gene was confirmed by sequencing. To find out if the fusion protein is expressed, we transfected the plasmids coding for chimeric protein or for only EGFP to U-2 OS cells using FuGene 6. The next day, cells were harvested and the lysates were prepared. The expression of proteins was examined by Western blotting as described above. In order to observe the cellular localization of the fusion protein DUSP13-EGFP (or control EGFP), the U-2 OS cells were seeded on chambered coverglasses and on the next day, the cells were transfected with the plasmids using FuGENE 6. The localization of proteins in living cells was observed starting 24 hours post-transfection with Zeiss confocal microscope.
3. Results
The results of high-throughput sequencing of mRNA molecules (RNA-Seq) can easily reveal the alternative splicing and the use of alternative promoters of genes. In our recently-published RNA-Seq results [5], we noticed that the transcription of DUSP13 gene in A549 cells exposed to A+N starts from the alternative promoter located in intron 3 (Figure 1A). Based on the pattern of exon splicing (Figure 1A), we conclude that the mRNA in the cells exposed to A+N codes for the isoform of DUSP13 called TMDP-L1, which is related to TMDP isoform, but shows longer ORF.
The location of p53 binding sites in DNA can be detected using sequencing of DNA isolated from chromatin immunoprecipitated with anti-p53 antibody (ChIP-Seq). The results of these investigations performed on various cells growing in control conditions or exposed to various stress factors were published in numerous papers. They can be viewed by the ChIP-Atlas platform [18]. Interestingly, we noticed two p53 ChIP-Seq peaks within intron 3 of DUSP13, however, these peaks were detected only in cells ectopically expressing engineered p53 molecules (p53EE/RR) with strong cooperative binding of p53 monomers (Figure 1B). Wild-type p53 did not bind to this locus [19] neither did the endogenous p53 in MCF-7 cells exposed as indicated on Figure 1B. Thus, apparently in order to bind to this DNA sequence, p53 must be properly modified what enables it to take the conformation similar to the one of p53EE/RR mutant [19]. Is it possible that this p53 modification is brought about by the treatment of cells with A+N?
In order to find out if the fragment of intron 3 can function as a promoter activated by p53, we cloned it into the reporter vector. The cloned fragment encompasses the region of the two p53 ChIP-Seq peaks as marked on Figure 1B (red line). Moreover, we noticed that there is a sequence closely matching the consensus site of p53 response element (Figure 2A). The DUSP13 sequence shows only two mismatches, what makes this sequence highly probable p53 response element (Figure 2A). We mutated its four critical sequence positions as shown on Figure 2A. The reporter assays performed on U-2 OS cell line demonstrated that wild-type p53 stimulated the wild-type promoter approximately 10-fold (Figure 2B). The activation of the promoter by the mutant p53 was significantly weaker. However, the mutant promoter still can be activated by wild-type p53, hence the p53-response element must be located elsewhere in the cloned region.
In order to find out if this promoter can be activated by the endogenous p53, we transfected into U-2 OS cells the reporter vector coding firefly luciferase controlled by the cloned wild-type DUSP13 promoter. As an internal control, we transfected a reporter vector with the gene for the luciferase from Renilla sp. transcriptionally controlled by HSV-TK promoter to control for transfection efficiency and stress induced by A+N. Twenty four hours after transfection, control cells were mock-treated, whereas the experimental cells were exposed to A+N for 24 hours. Subsequently, the cells were harvested and the activities of both luciferases were measured. The firefly luciferase activity was divided by Renilla luciferase activity producing the normalized firefly luciferase activity (NFLA). In cells exposed to A+N the value of NFLA increased more than 50-fold indicating that exposure to A+N stimulates the activity of the cloned promoter, probably by activating the endogenous p53 (Figure 2C). To test the involvement of p53, we performed this experiment on p53-deficient U-2 OS cells and their controls prepared using CRISPR/Cas9 technology as described by us earlier [5]. To show that these cells are p53-deficient we exposed them to A+N or camptothecin (a precursor of anticancer drugs–topotecan and irinotecan) and we examined the expression of total p53 or its form with phosphorylated Ser37 - the amino acid located in transcription activating domain, a fragment, which is a target of CRISPR/Cas9-generated DNA break. The Ser37 is phosphorylated on activated p53. The p53-deficient cells express low amount of total p53 and do not express p53 with the activating phosphorylation on Ser37 (Figure 2D). In control cells for knockdown, the exposure to A+N activates DUSP13 promoter more than 30-fold, whereas in p53-deficient cells only five-fold (Figure 2E). Thus, the activity of p53 is required to strongly activate the alternative promoter of DUSP13 following exposure to A+N.
To find out if the activation of endogenous DUSP13 gene is controlled by p53, we performed experiment with p53-deficient A549 cells prepared by CRISPR/Cas9 technology as described previously [5]. The p53-deficient cells and their controls were exposed to A+N or to another strong activator of p53–camptothecin. After exposing cells to these compounds either RNA samples were isolated for RT-PCR or protein lysates were prepared for Western blotting. Both analyses gave concordant results, namely that A+N induces stronger activation of DUSP13 than camptothecin and that wild-type p53 is indispensable for activation of DUSP13 by either compound (Figure 3A,B). This Western blot also shows that the protein coded by the alternative mRNA of DUSP13 is actually produced in cells exposed to A+N (or camptothecin) and can be detected by the antibody employed in our experiments (Figure 3B).
Subsequently, using Western blotting, we examined expression of DUSP13 in various cancer cell lines exposed to actinomycin D, nutlin-3a, A+N and camptothecin. We selected three lung cancer cell lines (A549, NCI-H460, NCI-H292), osteosarcoma cell line (U-2 OS), melanoma cell line (A375) and gastric adenocarcinoma cell line (AGS). All cell lines had wild-type gene coding for p53 protein. The cells were exposed to the compounds for 48 hours (Figure 4). What is clearly visible is very strong synergy between actinomycin D and nutlin-3a in activation of DUSP13 gene in all cell lines tested and weaker activation of the gene by camptothecin. DUSP13 protein is barely detectable in cells exposed to actinomycin D or nutlin-3a acting alone, whereas the accumulation of DUSP13 in cells exposed to both compounds is very strong. The exception is NCI-H292 cell line showing relatively high expression of DUSP13 protein even in cells exposed to nutlin-3a acting alone. Thus, in some cells, this gene can by activated without the need of double treatment. After long exposure of the signals on the blot, another protein is detected by the employed antibody in A549 cells treated with A+N. This protein form has a molecular mass of approximately 20 kDa compared with the approximately 30 kDa of the major form (Figure 4A). We suspect that it may be DUSP13 translated from an AUG codon downstream the major translation start site (see below).
To find out if other antagonists of p53-MDM2 interaction have similar impact on DUSP13 expression as nutlin-3a we exposed cells to idasanutlin or RG7112 (reviewed by Kocik et al., [20]) either alone or in combination with actinomycin D. As is demonstrated on Figure 5 these two compounds also synergize with actinomycin D in activation of DUSP13. Moreover, at the concentration used in the experiment (5 µM) they induce DUSP13 also when acting alone (Figure 5).
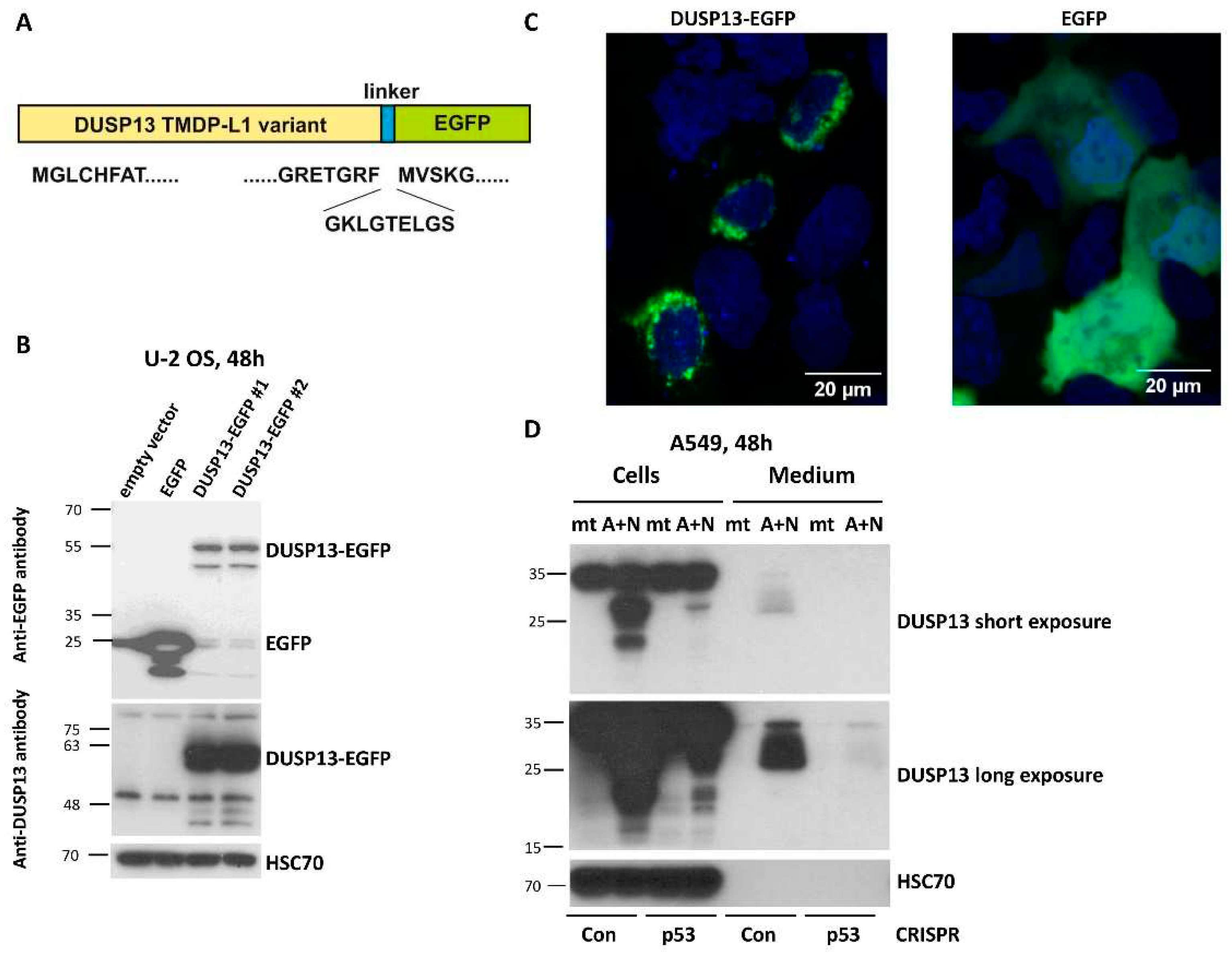
To gain insight into biological function of TMDP-L1 isoform of DUSP13, we amplified its cDNA sequence from A549 cells exposed to A+N and cloned it into an expression vector in front of the coding sequence of the green fluorescent protein EGFP (Figure 6A). This chimeric cDNA codes for the chimeric protein with EGFP attached to the C-terminal fragment of TMDP-L1. We wanted to keep the native form of the N-terminus of DUSP13, because it may contain a signaling sequence guiding the protein to its destination within a cell or extracellular space. Subsequently, we transfected the vector with the chimeric gene to U-2 OS cells and we observed the molecular weight of the fusion protein by Western blotting (Figure 6B) and its localization by confocal microscopy of living cells (no fixation) (Figure 6C). According to the Western blotting (Figure 6B) the size of the DUSP13-EGFP fusion protein (55 kDa) is about 30 kDa higher when compared to the size of EGFP alone (25 kDa), what is in agreement with the expected size of this version of DUSP13 (32 kDa). The fusion protein was detected by antibody directed against EGFP as well as by antibody directed against DUSP13 used in this work (Figure 6B). The microscopic observations demonstrated that the DUSP13-EGFP fusion protein is localized in cytoplasmic region around the nucleus, what contrasts with the localization pattern of EGFP, which localizes in cytoplasm and nucleus without any aggregation in perinuclear region (Figure 6C).
The N-terminus of TMDP-L1 of DUSP13 contains a sequence, which was predicted with high probability as a signal peptide MGLCHFATLALILLVLLEALAQAD [21]. This peptide targets a protein to the secretory pathway encompassing endoplasmic reticulum, Golgi apparatus, trans Golgi network, secretory vesicles and plasma membrane. To find out if this protein is secreted, we analyzed both–the cell lysates and conditioned media from control mock-treated cells and cells exposed to A+N for 48 hours. We performed the experiment on the aforementioned p53-deficient A549 cells and their controls. The proteins from cell lysates and concentrated media were separated by electrophoresis, transferred to membranes and probed with anti-DUSP13 antibody. In the lysates the antibody detected two protein forms (approximately 20 kDa and 30 kDa) but only in p53-proficient cells exposed to A+N (Figure 6D). In the medium, the smear of protein with the size of about 30 kDa and higher was detected, but again, only in case of p53-proficient A+N-treated cells. In the medium, the 20 kDa protein was not visible even after overexposure of detection film (Figure 6D). If this shorter protein is translated from an alternative, downstream start codon, then it lacks the signal peptide for secretion. Moreover, the lack of 20 kDa protein in the medium suggests that DUSP13 does not passively spill to extracellular space–in this case both forms would be detected. In conclusion, a fraction of the endogenous DUSP13 protein from A549 cells exposed to A+N is secreted to the extracellular space.
4. Discussion
The expression of DUSP13 dual specificity phosphatase is best-studied in mice. Exons 1-3 code for the open reading frame of MDSP isoform expressed in diaphragm and skeletal muscles, whereas exons 7-9 encode open reading frame of TMDP isoform expressed in testis. The transcript for MDSP also contains exons 4, 5, 7, 8, 9, which encode long 3’-UTR [15]. Genes encoding alternative ORFs producing two distinct proteins are rare in higher eukaryotes. The isoform, which we found expressed in A549 cells exposed to A+N is similar to TMDP. This transcript was found for the first time in muscles and was named TMDP-L1, but the encoded protein was not detected by Western blotting [15]. We found that the expression of TMDP-L1 is induced in A549 cells exposed to A+N. In these conditions, A549 cells produce a protein with the size expected for TMDP-L1 (approximately 30 kDa), which can be detected by the antibody employed in our experiments. The DUSP13 mRNA and protein can also be upregulated by another substance strongly activating p53–camptothecin, although in this case the upregulation is significantly weaker. Activation of DUSP13 measured at mRNA or protein level is strongly attenuated in p53-deficient cells what indicates that p53 plays role in this process, apparently in direct fashion because both ectopically expressed and endogenous p53 is able to activate the cloned alternative promoter of DUSP13. However, there must be something special in the interaction between p53 and the alternative promoter of DUSP13 driving the expression of TMDP-L1. First, the data published by Schlereth et al. [19] demonstrated that this DNA fragment is not bound by ectopically expressed wild-type p53 but only by a mutant version of this protein (Figure 1B), which promotes cooperative binding between p53 monomers. Thus, apparently in this region there is no classic p53 response element and p53 is able to bind this region only when the protein is modified or associates with other proteins, in a way which promotes reciprocal binding of p53 monomers in their tetrameric conformation. Second, the mutation of a plausible p53 response element identified in silico by Tebaldi et al. [4] in intron 3 of DUSP13 did not destroy the ability of the cloned promoter containing this mutant sequence to respond to p53. Thus, this cloned promoter contains other, less obvious p53 binding site. Third, in all examined cell lines actinomycin D and nutlin-3a strongly synergized in stimulation of the expression of DUSP13 protein. Actinomycin D or nutlin-3a acting alone in spite of activation of p53, did not stimulate the expression of DUSP13, the exception being NCI-H292 cells, where this isoform is induced by nutlin-3a (Figure 4B). Thus, “regular” activation of p53 is not able to stimulate the expression of this protein. Thus, to activate the expression of this isoform of DUSP13, p53 requires a special set of modifications or interactions with other proteins. These conditions are provided by the exposure to A+N. Interestingly, a recent review by Fischer et al. [22] provides a summary of transcriptomic studies searching for p53-regulated genes. In these analyses, DUSP13 was identified as p53 target only in 12 out of 57 studies. This confirms that this gene is regulated by p53 only in specific conditions.
DUSP13 is not a well-studied protein. As of this writing “DUSP13” phrase in PubMed returns only 18 papers (as of 01 September 2023). The gene was cloned by Nakamura et al. [14]. The crystal structure of TMDP isoform was determined [23]. Interestingly, TMDP variant when overexpressed with DUSP4 in A549 cell line attenuates TGFβ1-induced migration and drug resistance [24]. Recently, DUSP13 was found to belong to three-gene signature that can accurately distinguish COVID-19 patients from healthy controls [25]. This finding indicates that DUSP13 may participate in some immune-related activity. Another recently published data indicate that DUSP13 is a protein, which can be detected in blood plasma and may be associated with the increased risk of atrial fibrillation [26]. The expression of DUSP13 was reduced after epithelial-mesenchymal transition of ovarian cancer cell line triggered by TGFβ1 [27]. The oncology research focusing on DUSP13 is very limited [24,27,28,29,30]. In some studies DUSP13 was not a major subject but appeared as a hit in high-throughput analyses. In the latest of these reports, the authors found that a regulatory axis involving lncRNA PVT1, miR-378c and DUSP13 is involved in microvascular invasion in hepatocellular carcinoma. Moreover, based on various types of data analyses, the authors suggested that DUSP13 may be involved in lipid metabolism, glycosyl compound metabolic process and xenobiotic metabolic process [30].
The physiological role of TMDP-L1 isoform upregulated by p53 is not known. The TMDP expressed in testis inhibits stress-activated MAPK kinases and suppresses AP-1-dependent gene expression [16]. Whether a similar role is played by TMDP-L1 variant induced by A+N in p53-dependent manner remains to be determined. Our data clearly indicate that this isoform of DUSP13 is a part of the p53-regulated stress-response system, what may prompt other researchers to better study this form of DUSP13. Our data also show that DUSP13 expression is not limited to testis or muscles but can be found in cancer cells with wild-type p53 exposed to the clinically relevant substances (e.g. camptothecin), experimental drug combination (A+N) or novel antagonists of MDM2-p53 interaction (idasanutlin and RG7112). This line of research may be continued because there is an indispensable tool–the antibody, which can detect TMDP-L1 expression, what is important because, in our experience, there is scarcity of commercially available and usable antibodies recognizing poorly studied proteins.
5. Conclusions
In A549 lung cancer cell line, the treatment with actinomycin D and nutlin-3a (A+N) induces expression of TMDP-L1 variant of DUSP13 dual-specificity phosphatase starting from the alternative promoter located in intron 3. Cloned alternative promoter can be activated by both ectopically expressed and endogenous p53 protein. In cells with knocked-down expression of p53, the activation of DUSP13 in response to either A+N or camptothecin is attenuated. The protein of the expected size can be detected by Western blotting in various cancer cell lines with wild-type p53 exposed to A+N. DUSP13 protein can also be found in culture medium of exposed cells. DUSP13 TMDP-L1 variant with the attached green fluorescent protein localizes in perinuclear region of cells. Thus, the expression of DUSP13 is not limited, as observed earlier, to skeletal muscles or testis, but can also be induced by strongly activated p53 in cancer cells, what indicates that it can be important element of the p53-dependent stress-response system.
Author Contributions
Conceptualization, M.R.; Methodology, M.R., M.K. and B.Ł.-S.; Validation, A.G.-K. and A.B.; Formal Analysis, B. Ł.-S. and M.K.; Investigation, M.R., M.K., B.Ł.-S., A.G.-K. and A.B.; Writing–Original Draft Preparation, M.R.; Writing–Review & Editing, M.K. and B.Ł.-S.; Visualization, M.R., M.K. and B.Ł.-S.; Supervision, M.R.; Project Administration, M.R.; Funding Acquisition, M.R., M.K.
Funding
This research was funded by National Science Center (NCN), Poland, grant numbers: 2013/11/B/NZ5/03190 and 2019/35/O/NZ5/02600 to MR and grand number DEC-2018/02/X/NZ5/00234 to M.K.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequencing files (fastq) used to generate Figure 1A are available at Sequence Read Archive under the accession number PRJNA757776. Other, processed sequencing data are available from the corresponding author upon request.
Acknowledgments
The technical assistance of Ms. Patrycja Jakubowska is highly appreciated.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levine, A.J. The many faces of p53: Something for everyone. J. Mol. Cell Biol. 2019, 11, 524–530. [Google Scholar] [CrossRef]
- Shen, J.; Wang, Q.; Mao, Y.; Gao, W.; Duan, S. Targeting the p53 signaling pathway in cancers: Molecular mechanisms and clinical studies. MedComm (2020) 2023, 4, e288. [Google Scholar] [CrossRef]
- Łasut-Szyszka, B.; Rusin, M. The Wheel of p53 Helps to Drive the Immune System. Int. J. Mol. Sci. 2023, 24, 7645. [Google Scholar] [CrossRef]
- Tebaldi, T.; Zaccara, S.; Alessandrini, F.; Bisio, A.; Ciribilli, Y.; Inga, A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genom. 2015, 16, 464. [Google Scholar] [CrossRef] [PubMed]
- Łasut-Szyszka, B.; Małachowska, B.; Gdowicz-Kłosok, A.; Krześniak, M.; Głowala-Kosińska, M.; Zajkowicz, A.; Rusin, M. Transcriptome Analysis of Cells Exposed to Actinomycin D and Nutlin-3a Reveals New Candidate p53-Target Genes and Indicates That CHIR-98014 Is an Important Inhibitor of p53 Activity. Int. J. Mol. Sci. 2021, 22, 11072. [Google Scholar] [CrossRef]
- Zajkowicz, A.; Gdowicz-Kłosok, A.; Krześniak, M.; Ścieglińska, D.; Rusin, M. Actinomycin D and nutlin-3a synergistically promote phosphorylation of p53 on serine 46 in cancer cell lines of different origin. Cell Signal. 2015, 27, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Krześniak, M.; Zajkowicz, A.; Gdowicz-Kłosok, A.; Głowala-Kosińska, M.; Łasut-Szyszka, B.; Rusin, M. Synergistic activation of p53 by actinomycin D and nutlin-3a is associated with the upregulation of crucial regulators and effectors of innate immunity. Cell Signal. 2020, 69, 109552. [Google Scholar] [CrossRef]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef]
- Krześniak, M.; Zajkowicz, A.; Matuszczyk, I.; Rusin, M. Rapamycin prevents strong phosphorylation of p53 on serine 46 and attenuates activation of the p53 pathway in A549 lung cancer cells exposed to actinomycin D. Mech. Ageing Dev. 2014, 139, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef]
- Thut, C.J.; Goodrich, J.A.; Tjian, R. Repression of p53-mediated transcription by MDM2: A dual mechanism. Genes Dev. 1997, 11, 1974–1986. [Google Scholar] [CrossRef]
- Momand, J.; Wu, H.H.; Dasgupta, G. MDM2--master regulator of the p53 tumor suppressor protein. Gene 2000, 242, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Shima, H.; Watanabe, M.; Haneji, T.; Kikuchi, K. Molecular cloning and characterization of a novel dual-specificity protein phosphatase possibly involved in spermatogenesis. Biochem. J. 1999, 344, 819–825. [Google Scholar] [CrossRef]
- Chen, H.H.; Luche, R.; Wei, B.; Tonks, N.K. Characterization of two distinct dual specificity phosphatases encoded in alternative open reading frames of a single gene located on human chromosome 10q22.2. J. Biol. Chem. 2004, 279, 41404–41413. [Google Scholar] [CrossRef]
- Katagiri, C.; Masuda, K.; Nomura, M.; Tanoue, K.; Fujita, S.; Yamashita, Y.; Katakura, R.; Shiiba, K.; Nomura, E.; Sato, M.; Tanuma, N.; Shima, H. DUSP13B/TMDP inhibits stress-activated MAPKs and suppresses AP-1-dependent gene expression. Mol. Cell. Biochem. 2011, 352, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Markowitz, S.; Fearon, E.R.; Willson, J.K.; Vogelstein, B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science 1990, 249, 912–915. [Google Scholar] [CrossRef]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef]
- Schlereth, K.; Beinoraviciute-Kellner, R.; Zeitlinger, M.K.; Bretz, A.C.; Sauer, M.; Charles, J.P.; Vogiatzi, F.; Leich, E.; Samans, B.; Eilers, M.; et al. DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2010, 38, 356–368. [Google Scholar] [CrossRef]
- Kocik, J.; Machula, M.; Wisniewska, A.; Surmiak, E.; Holak, T.A.; Skalniak, L. Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists. Cancers (Basel) 2019, 11, 1014. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Schwarz, R.; Riege, K.; DeCaprio, J.A.; Hoffmann, S. TargetGeneReg 2.0: A comprehensive web-atlas for p53, p63, and cell cycle-dependent gene regulation. NAR Cancer 2022, 4, zcac009. [Google Scholar] [CrossRef]
- Kim, S.J.; Jeong, D.G.; Yoon, T.S.; Son, J.H.; Cho, S.K.; Ryu, S.E.; Kim, J.H. Crystal structure of human TMDP, a testis-specific dual specificity protein phosphatase: Implications for substrate specificity. Proteins 2007, 66, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Guler, S.; Altunok, T.H.; Sarioglu, A.; Zik, B.; Asmaz, D.; Kayapunar, N.; Sonmez, O.; Tepedelen, B.E.; Yalcin, A. Overexpression of dual-specificity phosphatases 4 and 13 attenuates transforming growth factor β1-induced migration and drug resistance in A549 cells in vitro. Biochem. Biophys. Res. Commun. 2022, 606, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lai, G.; Liu, H.; Deng, J.; Li, K.; Xie, B. A Novel 3-Gene Signature for Identifying COVID-19 Patients Based on Bioinformatics and Machine Learning. Genes (Basel) 2022, 13, 1602. [Google Scholar] [CrossRef]
- Ning, Z.; Huang, Y.; Lu, H.; Zhou, Y.; Tu, T.; Ouyang, F.; Liu, Y.; Liu, Q. Novel Drug Targets for Atrial Fibrillation Identified through Mendelian Randomization Analysis of the Blood Proteome. Cardiovasc. Drugs Ther. 2023. ahead of print. [Google Scholar] [CrossRef]
- Güler, S.; Yalçın, A. Expression of dual-specificity phosphatases in TGFß1-induced EMT in SKOV3 cells. Turk. J. Med. Sci. 2023, 53, 640–646. [Google Scholar] [CrossRef]
- Fridley, B.L.; Armasu, S.M.; Cicek, M.S.; Larson, M.C.; Wang, C.; Winham, S.J.; Kalli, K.R.; Koestler, D.C.; Rider, D.N.; Shridhar, V.; et al. Methylation of leukocyte DNA and ovarian cancer: Relationships with disease status and outcome. BMC Med. Genom. 2014, 7, 21. [Google Scholar] [CrossRef]
- Au, C.H.; Ho, D.N.; Ip, B.B.K.; Wan, T.S.K.; Ng, M.H.L.; Chiu, E.K.W.; Chan, T.L.; Ma, E.S.K. Rapid detection of chromosomal translocation and precise breakpoint characterization in acute myeloid leukemia by nanopore long-read sequencing. Cancer Genet. 2019, 239, 22–25. [Google Scholar] [CrossRef]
- Su, R.; Zhang, H.; Zhang, L.; Khan, A.R.; Zhang, X.; Wang, R.; Shao, C.; Wei, X.; Xu, X. Systemic analysis identifying PVT1/DUSP13 axis for microvascular invasion in hepatocellular carcinoma. Cancer Med. 2023, 12, 8937–8955. [Google Scholar] [CrossRef]
Figure 1.
Treatment with A+N activates an alternative promoter in intron 3 of DUSP13 gene. A. Genome browser (IGV) views of RNA-Seq reads mapped to DUSP13 gene in control mock-treated (mt) A549 cells and in cells exposed to A+N for 30h. The raw sequencing data are deposited in the Sequence Read Archive under accession number PRJNA757776. B. Genome browser (IGV) views of p53 binding peaks in 5′ part of DUSP13 gene. Using ChIP-Atlas tool [18] we imported publicly available coverage tracks from five ChIP-Seq experiments aimed at finding p53 binding sites in: MCF-7 cell line exposed to ionizing radiation (IR) and Nutlin (sample ID SRX2924018), MCF-7 cells treated with Nutlin (sample ID SRX2060922), SAOS-2 cell line ectopically expressing wild-type p53 (sample ID SRX016980), SAOS-2 cells ectopically expressing pair of engineered p53 molecules with strong cooperative binding of p53 monomers (sample ID ERX181467) and SW480 cell line exposed to TNFα (Sample ID SRX4235879). The red, thick horizontal line marks the location of the cloned promoter.
Figure 1.
Treatment with A+N activates an alternative promoter in intron 3 of DUSP13 gene. A. Genome browser (IGV) views of RNA-Seq reads mapped to DUSP13 gene in control mock-treated (mt) A549 cells and in cells exposed to A+N for 30h. The raw sequencing data are deposited in the Sequence Read Archive under accession number PRJNA757776. B. Genome browser (IGV) views of p53 binding peaks in 5′ part of DUSP13 gene. Using ChIP-Atlas tool [18] we imported publicly available coverage tracks from five ChIP-Seq experiments aimed at finding p53 binding sites in: MCF-7 cell line exposed to ionizing radiation (IR) and Nutlin (sample ID SRX2924018), MCF-7 cells treated with Nutlin (sample ID SRX2060922), SAOS-2 cell line ectopically expressing wild-type p53 (sample ID SRX016980), SAOS-2 cells ectopically expressing pair of engineered p53 molecules with strong cooperative binding of p53 monomers (sample ID ERX181467) and SW480 cell line exposed to TNFα (Sample ID SRX4235879). The red, thick horizontal line marks the location of the cloned promoter.
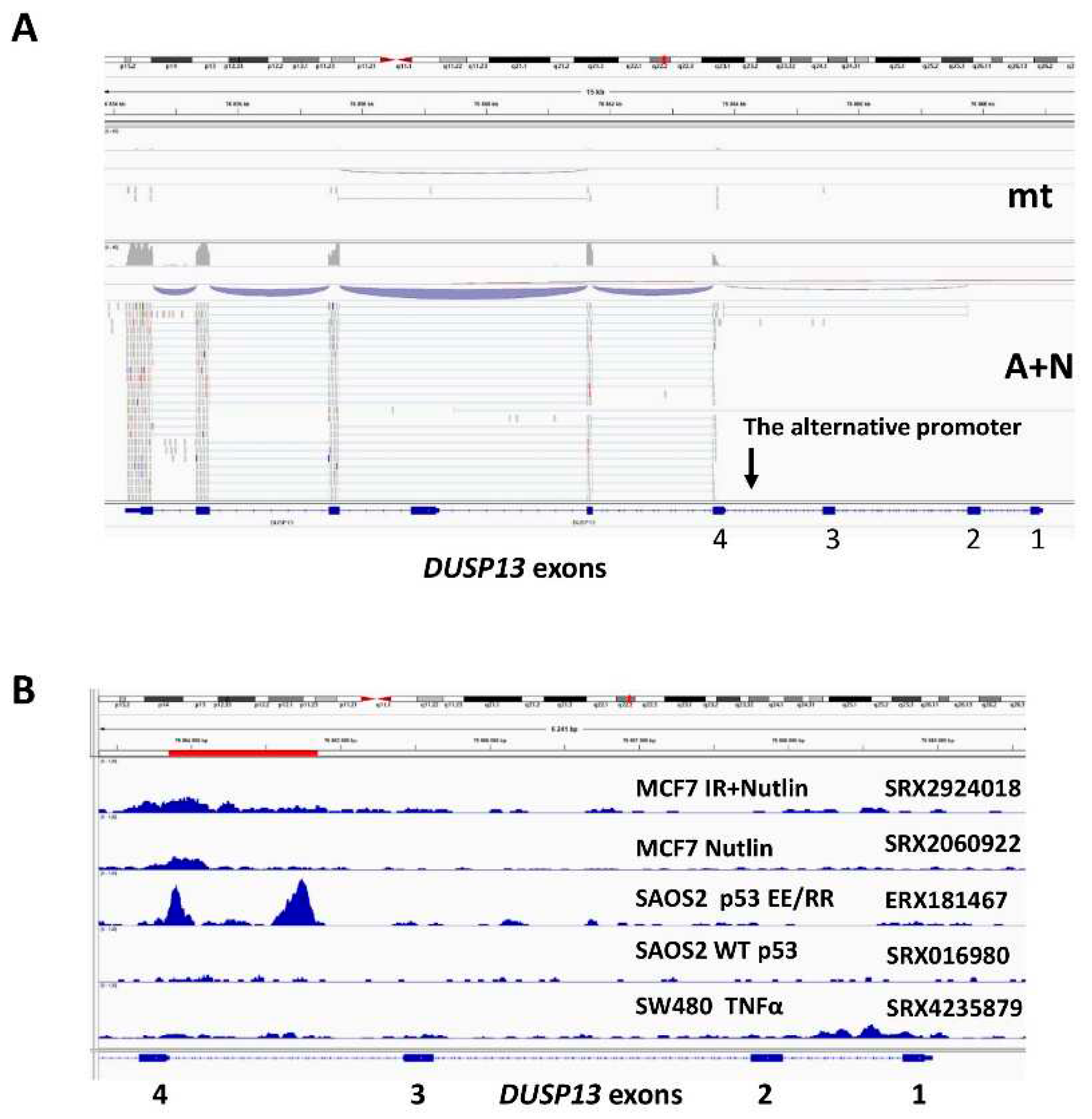
Figure 2.
The p53 protein activates the alternative promoter of DUSP13. A. The sequence of putative p53-response element within the alternative DUSP13 promoter is shown together with the consensus sequence of the response element and the mutations we generated. B. The relative values of NFLA in U-2 OS cells transfected with (bars from left to right): plasmid with wild-type DUSP13 promoter and empty expression vector, plasmid with wild-type DUSP13 promoter and wild-type p53 expression vector, plasmid with wild-type DUSP13 promoter and mutant p53 expression vector, plasmid with mutant DUSP13 promoter and empty expression vector, plasmid with mutant DUSP13 promoter and wild-type p53 expression vector, plasmid with mutant DUSP13 promoter and mutant p53 expression vector. The means and standard deviations from three biological repeats performed in triplicate are shown. C. The relative value of NFLA in U-2 OS cells transfected with plasmid with wild-type DUSP13 promoter growing in control conditions (mock-treatment, mt) or exposed to A+N. The results represent means and standard deviations from three biological replicates, p value was calculated by paired t test, ** p≤0.01. The calculation was performed using GraphPad Prism version 9.5.1 (2023) for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com. D. The expression of total p53, p53 with phosphorylated Ser37 and HSC70 as a loading control in U-2 OS cells with p53 expression knocked-down by CRISPR/Cas 9 (CRISPR-p53) and in control cells for knockdown (CRISPR-Con) growing in control conditions (mock-treatment, mt) or exposed to A+N or camptothecin (CPT). The expression of indicated proteins was determined by Western blotting. E. The relative values of NFLA in U-2 OS cells transfected with plasmid with wild-type DUSP13 promoter. The transfection was performed in p53-deficient cells (CRISPR-p53) and their controls (CRISPR-Con) exposed to A+N for 24 h (A+N) or mock-treated (mt). The results represent means and standard deviations from three biological replicates, p value was calculated by multiple t test, *** p≤0.001. The calculation was performed using GraphPad Prism version 9.5.1 (2023) for Windows.
Figure 2.
The p53 protein activates the alternative promoter of DUSP13. A. The sequence of putative p53-response element within the alternative DUSP13 promoter is shown together with the consensus sequence of the response element and the mutations we generated. B. The relative values of NFLA in U-2 OS cells transfected with (bars from left to right): plasmid with wild-type DUSP13 promoter and empty expression vector, plasmid with wild-type DUSP13 promoter and wild-type p53 expression vector, plasmid with wild-type DUSP13 promoter and mutant p53 expression vector, plasmid with mutant DUSP13 promoter and empty expression vector, plasmid with mutant DUSP13 promoter and wild-type p53 expression vector, plasmid with mutant DUSP13 promoter and mutant p53 expression vector. The means and standard deviations from three biological repeats performed in triplicate are shown. C. The relative value of NFLA in U-2 OS cells transfected with plasmid with wild-type DUSP13 promoter growing in control conditions (mock-treatment, mt) or exposed to A+N. The results represent means and standard deviations from three biological replicates, p value was calculated by paired t test, ** p≤0.01. The calculation was performed using GraphPad Prism version 9.5.1 (2023) for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com. D. The expression of total p53, p53 with phosphorylated Ser37 and HSC70 as a loading control in U-2 OS cells with p53 expression knocked-down by CRISPR/Cas 9 (CRISPR-p53) and in control cells for knockdown (CRISPR-Con) growing in control conditions (mock-treatment, mt) or exposed to A+N or camptothecin (CPT). The expression of indicated proteins was determined by Western blotting. E. The relative values of NFLA in U-2 OS cells transfected with plasmid with wild-type DUSP13 promoter. The transfection was performed in p53-deficient cells (CRISPR-p53) and their controls (CRISPR-Con) exposed to A+N for 24 h (A+N) or mock-treated (mt). The results represent means and standard deviations from three biological replicates, p value was calculated by multiple t test, *** p≤0.001. The calculation was performed using GraphPad Prism version 9.5.1 (2023) for Windows.
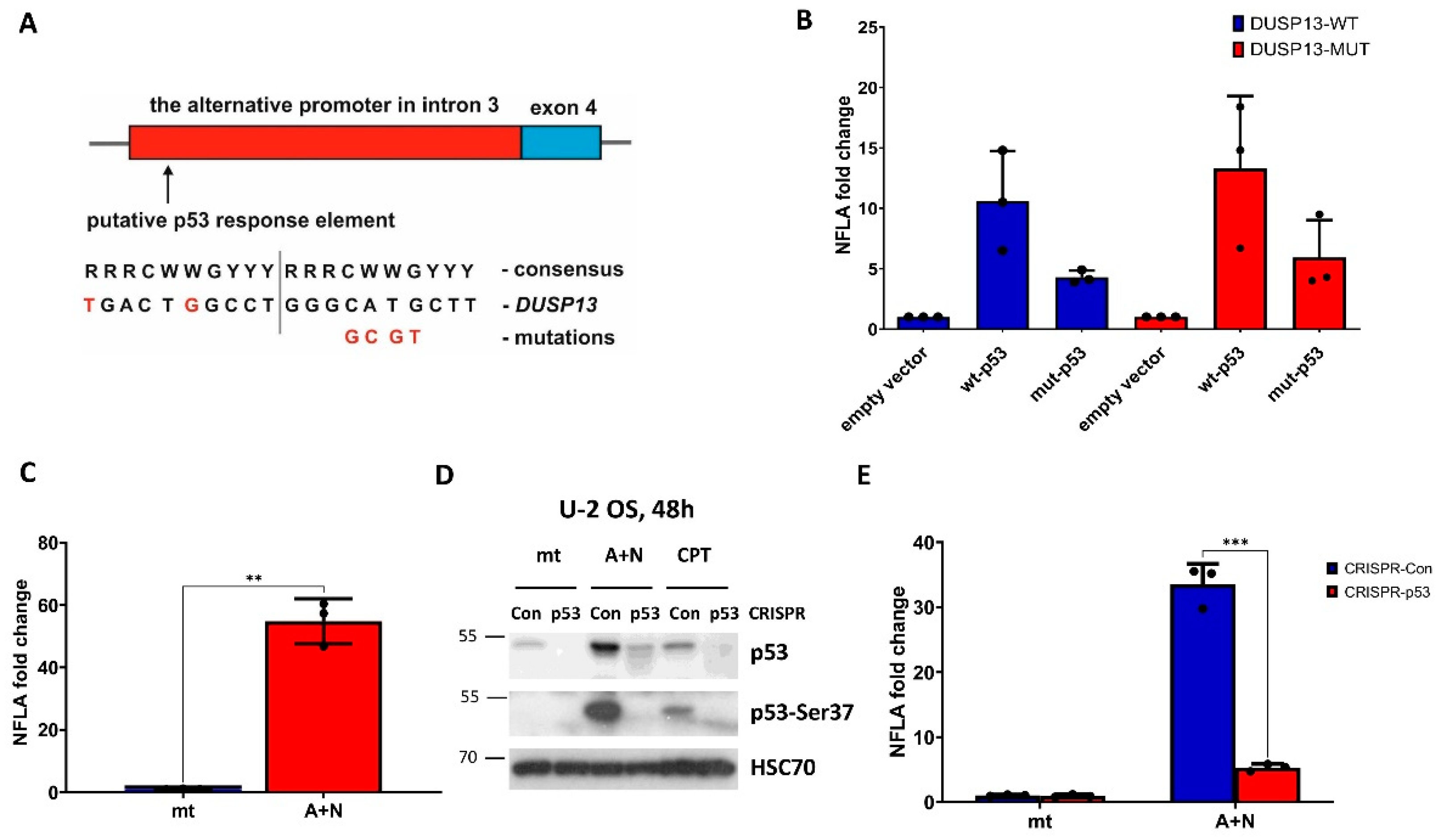
Figure 3.
Expression of DUSP13 measured at mRNA and protein level is attenuated in p53-deficient cells. A. The expression of DUSP13 mRNA measured by semi-quantitative RT-PCR in mock-treated control cells (mt) or the cells exposed to A+N or camptothecin (CPT) for 30 h. The p53 knock-down was performed by CRISPR/Cas9 technology. The results represent means and standard deviations from three biological replicates, p values were calculated by multiple t test, ** p≤0.01, *** p≤0.001. The calculations were performed using GraphPad Prism version 9.5.1 (2023) for Windows. B. The expression of p53, its form with phosphorylated Ser-37 and DUSP13 in p53-deficient cells (CRISPR-p53) and their controls (CRISPR-Con) prepared as described in A and exposed to A+N or camptothecin (CPT) for 48 hours or mock-treated (mt). The expression of indicated proteins was determined by Western blotting.
Figure 3.
Expression of DUSP13 measured at mRNA and protein level is attenuated in p53-deficient cells. A. The expression of DUSP13 mRNA measured by semi-quantitative RT-PCR in mock-treated control cells (mt) or the cells exposed to A+N or camptothecin (CPT) for 30 h. The p53 knock-down was performed by CRISPR/Cas9 technology. The results represent means and standard deviations from three biological replicates, p values were calculated by multiple t test, ** p≤0.01, *** p≤0.001. The calculations were performed using GraphPad Prism version 9.5.1 (2023) for Windows. B. The expression of p53, its form with phosphorylated Ser-37 and DUSP13 in p53-deficient cells (CRISPR-p53) and their controls (CRISPR-Con) prepared as described in A and exposed to A+N or camptothecin (CPT) for 48 hours or mock-treated (mt). The expression of indicated proteins was determined by Western blotting.
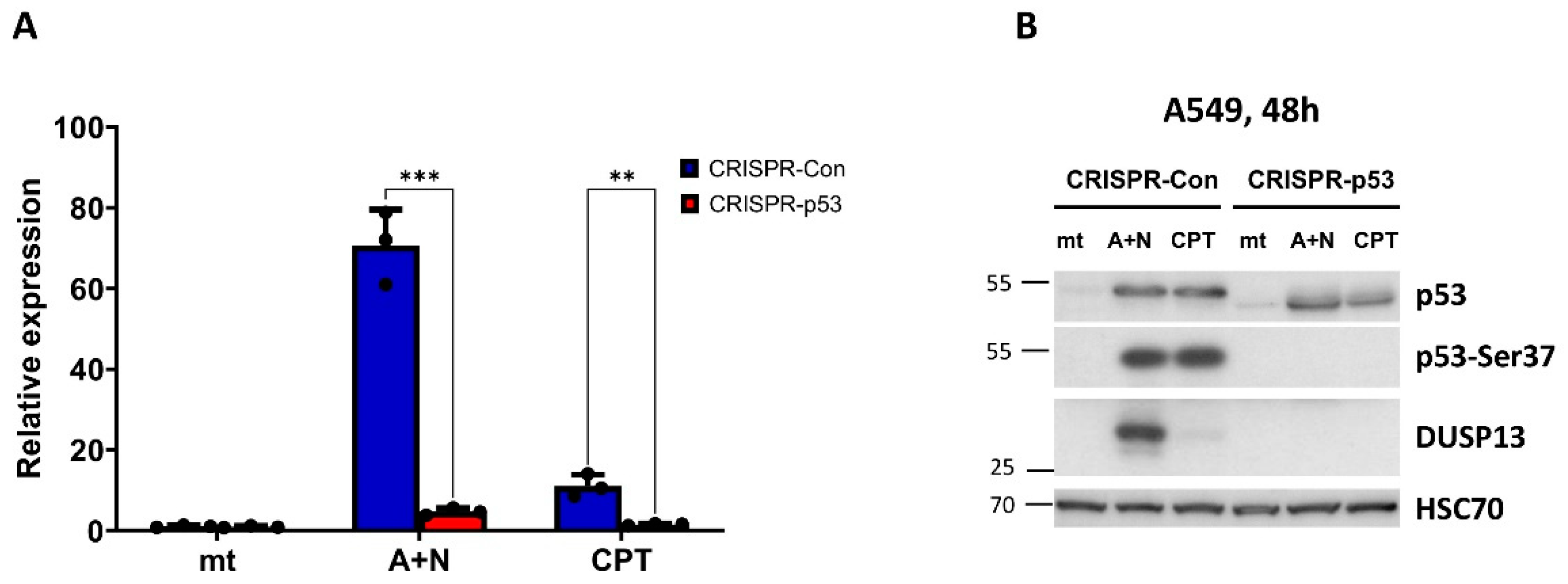
Figure 4.
The expression of DUSP13 is induced by A+N in all examined cell lines with wild-type p53 gene. A-E. Western blots showing expression of indicated proteins in selected cell lines with wild-type p53 mock-treated (mt) or incubated for 48 h with actinomycin D (ActD), nutlin-3a (Nut), both substances (A+N) or camptothecin (CPT).
Figure 4.
The expression of DUSP13 is induced by A+N in all examined cell lines with wild-type p53 gene. A-E. Western blots showing expression of indicated proteins in selected cell lines with wild-type p53 mock-treated (mt) or incubated for 48 h with actinomycin D (ActD), nutlin-3a (Nut), both substances (A+N) or camptothecin (CPT).
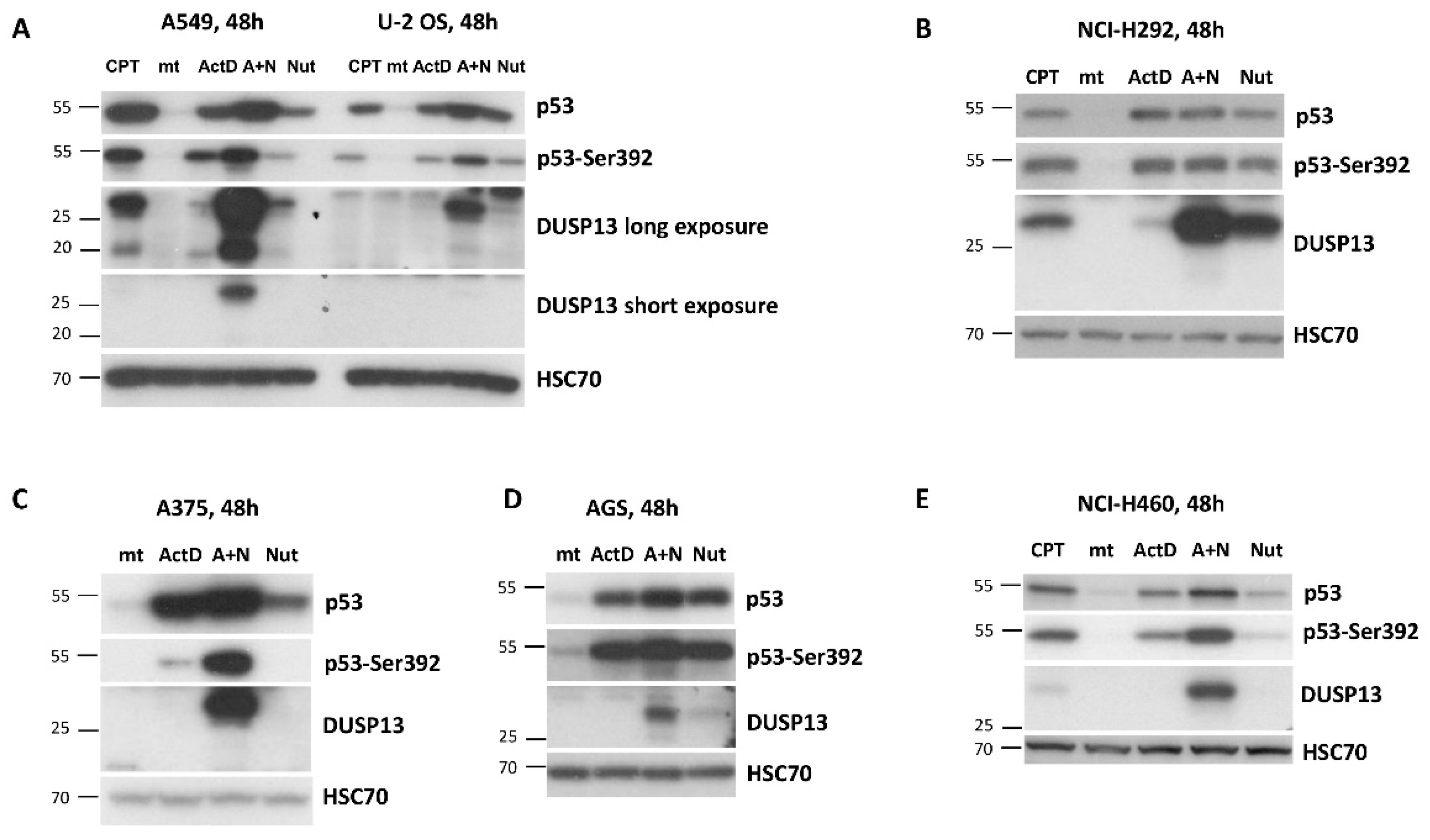
Figure 5.
The other antagonists of MDM2-p53 interaction, idasanutlin and RG7112, also synergize with actinomycin D in inducing expression of DUSP13. A549 cells were exposed as shown to various combinations of actinomycin D and the antagonists of MDM2-p53 interaction: nutlin-3a, idasanutlin and RG7112. The expression of indicated proteins was determined by Western blotting.
Figure 5.
The other antagonists of MDM2-p53 interaction, idasanutlin and RG7112, also synergize with actinomycin D in inducing expression of DUSP13. A549 cells were exposed as shown to various combinations of actinomycin D and the antagonists of MDM2-p53 interaction: nutlin-3a, idasanutlin and RG7112. The expression of indicated proteins was determined by Western blotting.
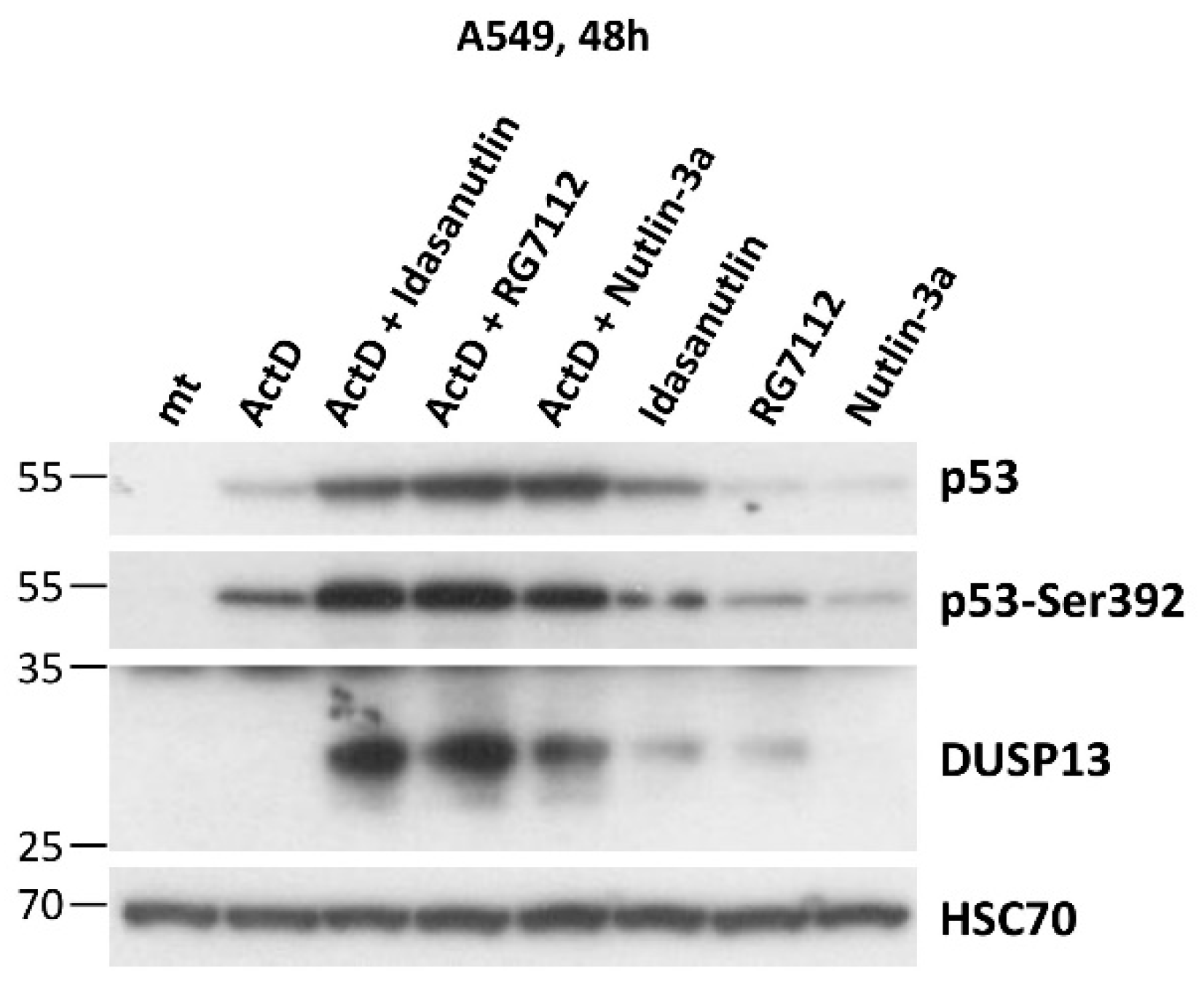
Figure 6.
The DUSP13 isoform with the attached enhanced green fluorescent protein localizes to the perinuclear region of U-2 OS cells. A. The structure of the fusion protein expressed from the engineered plasmid. The variant of DUSP13 is fused by the plasmid-coded linker to the enhanced green fluorescent protein (EGFP). The amino acid sequences of the DUSP13 N- and C- ends, the linker and the start of EGFP are shown. B. The Western blot showing the expression of EGFP and DUSP13-EGFP fusion protein expressed from two clones of the plasmid. The positions of the molecular weight markers (kDa) are placed on the left. The expected size of the unmodified DUSP13 variant is 32 kDa. Two separate blots were probed either with anti-EGFP antibody or with anti-DUSP13 antibody. C. The localization of the proteins expressed from the plasmids transfected to U-2 OS cells. The living cells were observed using Zeiss confocal microscope 24 h after the start of transfection. The nuclei were stained using Hoechst 33342. D. The expression of DUSP13 in cells and in concentrated medium of cells growing in control condition (mock-treatment, mt) or exposed to A+N for 48 h. We used p53 knockdown cells (CRISPR-p53) and the controls for knockdown with wild-type p53 (CRISPR-Con). The expression of indicated proteins was determined by Western blotting. Both panels show the same blot with short (upper) and long (bottom) exposure times.
Figure 6.
The DUSP13 isoform with the attached enhanced green fluorescent protein localizes to the perinuclear region of U-2 OS cells. A. The structure of the fusion protein expressed from the engineered plasmid. The variant of DUSP13 is fused by the plasmid-coded linker to the enhanced green fluorescent protein (EGFP). The amino acid sequences of the DUSP13 N- and C- ends, the linker and the start of EGFP are shown. B. The Western blot showing the expression of EGFP and DUSP13-EGFP fusion protein expressed from two clones of the plasmid. The positions of the molecular weight markers (kDa) are placed on the left. The expected size of the unmodified DUSP13 variant is 32 kDa. Two separate blots were probed either with anti-EGFP antibody or with anti-DUSP13 antibody. C. The localization of the proteins expressed from the plasmids transfected to U-2 OS cells. The living cells were observed using Zeiss confocal microscope 24 h after the start of transfection. The nuclei were stained using Hoechst 33342. D. The expression of DUSP13 in cells and in concentrated medium of cells growing in control condition (mock-treatment, mt) or exposed to A+N for 48 h. We used p53 knockdown cells (CRISPR-p53) and the controls for knockdown with wild-type p53 (CRISPR-Con). The expression of indicated proteins was determined by Western blotting. Both panels show the same blot with short (upper) and long (bottom) exposure times.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



