Submitted:
13 September 2023
Posted:
14 September 2023
Read the latest preprint version here
Abstract
Phylogenomics has enriched our understanding of the Tree of Life. Non-vertical modes of evolution—such as hybridization/introgression and horizontal gene transfer—deviate from a strictly bifurcating tree model, mirroring a network-like or reticulate structure. Here, we present an overview of a phylogenomic workflow for inferring organismal histories, calibrating those histories to evolutionary time, and detecting reticulate evolution. Mitigating analytical sources of error facilitates accurate reconstructions of evolutionary history and, in turn, characterization of non-vertical modes of evolution. Workflows and methods discussed herein may aid in the rigorous inference of organismal histories in geologic time and reticulation, providing a clearer understanding of the evolutionary process.
Keywords:
phylogenetics
; hybridization
; introgression
; horizontal gene transfer
; lateral gene transfer
; phylogenetic incongruence
; gene-tree-species-tree discordance
Introduction
Phylogenomics—phylogenetic analysis using genome-scale data—has been used to infer the evolutionary history of diverse lineages across the Tree of Life, including animals, fungi, plants, bacteria, archaea, and viruses (Dunn et al. 2008; Misof et al. 2014; Wickett et al. 2014; Worobey et al. 2016; Simion et al. 2017; Parks et al. 2018; Shen et al. 2018; One Thousand Plant Transcriptomes Initiative 2019; Zhu et al. 2019; Coleman et al. 2021; Galindo et al. 2021; Li et al. 2021; Tahon et al. 2021). These studies have resolved numerous phylogenetic controversies, deepening our understanding of life's history (Capella-Gutiérrez et al. 2012; King and Rokas 2017; Williams et al. 2019; Pipes et al. 2021; Steenwyk et al. 2023b). Phylogenomics has also proven useful for delineating lineage relationships at various taxonomic scales, ranging from species to higher-order relationships (Díaz-Tapia et al. 2017; Muñoz-Gómez et al. 2017; Mateo-Estrada et al. 2019; Bringloe et al. 2021; Steenwyk et al. 2022a; Sierra-Patev et al. 2023), and provides the necessary framework for comparative evolutionary genomic studies, such as determining gene duplication and loss events or studying phenotypic innovation (Zhang et al. 2014a; Steenwyk et al. 2019a; Fernández and Gabaldón 2020; Shen et al. 2020; Phillips et al. 2021; Li et al. 2022a; Opulente et al. 2023).
However, errors may be introduced during analytical steps, such as orthology and site-wise homology inference, resulting in inaccurate reconstructed evolutionary histories (Martín-Durán et al. 2017; Ashkenazy et al. 2019; Emms and Kelly 2019; Steenwyk et al. 2023b). Similarly, the inferred timing of ancestor-to-descendent divergence events can be erroneous due to misspecifying molecular clock models, fossil calibrations, or other factors (Ho and Duchêne 2014; Tao et al. 2020; Carruthers and Scotland 2021). Careful consideration of these analytical sources of error during experimental design is crucial for improving the accuracy of phylogenomic inference (Steenwyk et al. 2023b).
Incongruence between the evolutionary histories of single loci and organisms (locus-tree-species-tree incongruence or discordance) can also arise from biological factors (Steenwyk et al. 2023b). These factors include reticulate evolutionary processes like hybridization/introgression, the interbreeding between distinct lineages, which can disrupt inferences of both the timing and pattern of historical divergences (Rieseberg et al. 2007; Racimo et al. 2015; Barley et al. 2018; Gonçalves et al. 2018; Gonçalves and Gonçalves 2019; Steenwyk et al. 2020b; Li et al. 2022b; Suvorov et al. 2022; Tiley et al. 2023). Hybridization/introgression has been documented in plants, algae, fungi, and a variety of animals among other lineages (Rieseberg et al. 2007; Neafsey et al. 2010; Stukenbrock 2016; Edelman et al. 2019; Edger et al. 2019; Sousa et al. 2019; Mixão and Gabaldón 2020; Steenwyk et al. 2020b; Bringloe et al. 2021; Wang et al. 2022). Among humans, loci originating from admixture events between early humans and Neanderthals have been associated with adaptation, phenotypic variation, and disease risk, including for severe COVID-19 (Sankararaman et al. 2016; Simonti et al. 2016; Dannemann and Kelso 2017; Dannemann et al. 2017; Zeberg and Pääbo 2020). Hybridization can also result in allopolyploid wherein the genome of the hybrid organism encodes (nearly) the entire genome of both parents. Allopolyploidy has been observed in numerous plants, fungi, and a few vertebrates (Ozkan et al. 2001; Session et al. 2016; Edger et al. 2019; Steenwyk et al. 2020b; Chen et al. 2022; Session and Rokhsar 2023). Genome evolution in allopolyploids can be rapid, marked by pronounced loss of genetic material (Ozkan et al. 2001), or be relatively stable (Steenwyk et al. 2020b). In either case, introgression/hybridization results in novel combinations of genes and genetic backgrounds that can result in distinct phenotypic profiles (Steenwyk et al. 2020b; Bautista et al. 2021).
Another mode of reticulate evolution, horizontal gene transfer—the transfer of genetic material without sexual reproduction—also causes locus-tree-species-tree incongruence and has been documented in diverse organisms, especially among prokaryotes and archaea (Galtier 2007; Yue et al. 2012; Van Etten and Bhattacharya 2020; Arnold et al. 2022; Gonçalves and Gonçalves 2022; Gophna and Altman-Price 2022; Li et al. 2022b; Steenwyk et al. 2023a). Horizontal gene transfer can be advantageous, endowing recipient organisms with potentially novel functionality (Gonçalves and Gonçalves 2019; Kominek et al. 2019; Li et al. 2022b). In certain cases, complex patterns of horizontal gene transfer or lateral acquisition of entire gene clusters can occur, resulting in new metabolic capabilities such as alcohol fermentation and the biosynthesis of thiamine and siderophores (Gonçalves et al. 2018; Gonçalves and Gonçalves 2019; Kominek et al. 2019). Horizontally acquired genes can also facilitate adaptation to extreme environments. For example, ice-binding proteins originating from bacteria are thought to contribute to algal adaptation to Arctic environments (Dorrell et al. 2023) and mercuric reductase, an enzyme responsible for converting mercury to a less toxic form, was transferred from bacteria to extremophilic algae commonly isolated from environments with a high mercury concentration (Schönknecht et al. 2013). Among protists, approximately 1% of gene repertoires are estimated to have been horizontally acquired (Van Etten and Bhattacharya 2020). These observations emphasize the significance of horizontal gene transfer as a major evolutionary mode.
From data acquisition to divergence time estimation, this review outlines notable steps for phylogenomic inference, discusses potential analytical sources of error, and explores methodologies for detecting reticulate evolution. By rigorously considering sources of error, researchers can disentangle analytical and biological factors contributing to locus-tree-species-tree incongruence, thereby enhancing the accuracy of phylogenomic inference, and facilitating the characterization of reticulate evolutionary processes.
A Workflow for Robust Phylogenomic Inference
Data Acquisition and Preparation
The first step of phylogenomic tree inference involves acquiring high-quality genomic/transcriptomic data from the target taxa (Figure 1A) (Cheon et al. 2020; Kapli et al. 2020). The abundance of available genomic/transcriptomic resources, accessible through online repositories like the National Center for Biotechnology Information (NCBI), enables researchers to address worthwhile questions without generating new data. Nevertheless, expanded taxon sampling through novel ‘omic resources can improve phylogenomic inference, help address contentious issues, and provide a valuable resource to the community (Pollock et al. 2002; Dunn et al. 2008; Wiens and Tiu 2012; Shen et al. 2018; Blaxter et al. 2022; Steenwyk et al. 2023b). To improve taxon sampling, researchers can apply a variety of methods to the taxa of interest, including targeted sequencing, reduced representation sequencing, transcriptomics, or whole-genome sequencing (Dunn et al. 2008; Peterson et al. 2012; One Thousand Plant Transcriptomes Initiative 2019; Hale et al. 2020). Applying standard methods for sequence data processing and quality control, such as removing low-quality data and contaminated sequences, is crucial (Zhou and Rokas 2014). Numerous tools exist for removing low-quality sequencing data, such as Trimmomatic and fastp (Bolger et al. 2014; Chen et al. 2018). Contaminated sequences can be purged from datasets using dinucleotide odds ratios or coverage-versus-lengths plots (Schmieder and Edwards 2011; Douglass et al. 2019).
For the sake of simplicity, the following sections of this review focus on phylogenomic inference using alignments of protein-coding sequences. However, it is important to note that different data sources require tailored approaches to facilitate the subsequent steps. For instance, when working with genomic data, an additional step of gene boundary predictions is necessary (Stanke et al. 2006; Brůna et al. 2021). Similarly, SNP-based phylogenomics, which is also suitable for detecting hybridization/introgression, or synteny data, an emerging phylogenomic marker of collinearity between two or more genomes (Rokas and Holland 2000; Bringloe et al. 2021; Parey et al. 2023; Schultz et al. 2023; Steenwyk and King 2023), requires unique considerations not covered here.
Gene Orthology Determination
Genes that arise from speciation events in a shared ancestor are termed orthologous genes and serve as the foundation for phylogenomic analysis (Gabaldón and Koonin 2013). Relationships among orthologous genes can be described as one of three categories: one-to-one, one-to-many, and many-to-many (Fernández et al. 2019). Considering two genomes, one-to-one orthologs are encoded in each genome once; one-to-many orthologs are encoded in one genome once and the other multiple times; and many-to-many orthologs refer to a gene with multiple copies in each genome. Although methods have been developed to infer organismal histories using one-to-many and many-to-many orthologs (Zhang et al. 2020; Smith et al. 2022), most phylogenomic analyses rely on one-to-one orthologous genes and will be the focus of the present article. Moreover, single-copy orthologs are often the substrate of many downstream molecular evolution analyses such as measures of selection, relative evolutionary rates, and gene-gene coevolution (Chikina et al. 2016; Kowalczyk et al. 2019; Steenwyk et al. 2021, 2022d; Álvarez-Carretero et al. 2023).
Inferring orthology can be accomplished through two main approaches: global and targeted inference. A commonly utilized framework for global orthology inference involves all-to-all sequence similarity calculations followed by clustering (Figure 1B). Several tools implement this approach, including OrthoFinder, OrthoMCL, or MMseqs2 (Li et al. 2003; Emms and Kelly 2015, 2019; Steinegger and Söding 2018). To perform all-to-all sequence similarity calculations, software like BLAST, DIAMOND, or MMseqs2 can be employed (Camacho et al. 2009; Steinegger and Söding 2018; Buchfink et al. 2023). Sequence similarity scores can be influenced by biases arising from differences in sequence lengths. Correcting this analytical error can enhance the accuracy of orthology inference (Emms and Kelly 2015). Subsequently, graph-based clustering methods, such as the Markov Cluster Process (Van Dongen 2008), are applied to categorize genes into distinct orthologous groups of genes. During Markov clustering, the inflation value will impact the granularity of orthologous groups of genes (Brohée and Van Helden 2006). Relaxed inflation values may combine orthologous groups of genes whereas stringent inflation values may oversplit them. Oversplitting can also stem from the inability to detect remote homology among orthologs encoded in distantly related species or rapidly evolving taxa (Weisman et al. 2020). There is no golden rule for an appropriate inflation value; the best value will likely be dataset-dependent. The resulting clusters of orthologous genes can serve as proxies for gene families.
In phylogenomic inference, single-copy gene families, orthologous genes encoded in each taxon once, are preferred as they (presumably) have not experienced duplication or loss (Li et al. 2017). Organismal histories are often inferred using hundreds to thousands of loci to avoid inadequate sampling of gene loci, a known source of error while inferring organismal histories (Rokas et al. 2003; Steenwyk et al. 2023b). However, as the number of taxa and evolutionary distances between them increases, the number of single-copy orthologous genes tends to decrease (Emms and Kelly 2018). For instance, in a dataset comprising 42 plants with complex evolutionary histories involving dynamic gene gain and loss events (Clark and Donoghue 2018), no single-copy orthologous genes were identified (Emms and Kelly 2018). In such cases, no gene families are fit for standard phylogenomic analyses.
To overcome this challenge, tree decomposition algorithms that partition multi-copy gene families into subgroups of single-copy orthologous genes and pruning algorithms that remove species-specific paralogs (i.e., duplicated genes observed in a single species) can be useful (Kocot et al. 2013; Willson et al. 2022). The software OrthoSNAP combines tree splitting and pruning to identify single-copy orthologs nested within larger gene families (Steenwyk et al. 2022c). This process is analogous to snapping branches in a tree; thus, the resulting subgroups of single-copy orthologs are termed SNAP-OGs (splitting and pruning). Single-copy gene families and SNAP-OGs are statistically indistinguishable across diverse measures of information content (Steenwyk et al. 2022c), indicating their suitability for phylogenomic analyses.
While global orthology inference offers valuable insights, it often demands significant computational resources. An alternative approach involves the identification of pre-determined single-copy genes (Figure 1B). One method is the BUSCO pipeline (Benchmarking Universal Single-Copy Ortholog), which identifies near-universally single-copy orthologous genes within a genome or transcriptome (Waterhouse et al. 2018). BUSCO uses predefined near-universally single-copy orthologs from OrthoDB (Kriventseva et al. 2019). This analysis also serves as a useful quality control measure for assessing assembly gene content completeness (Waterhouse et al. 2018). When researchers prefer to use a custom set of predefined single-copy orthologous genes, orthofisher can be used (Steenwyk and Rokas 2021). Additionally, various sequence similarity search tools such as HMMER, BLAST, and DIAMOND can be employed to support the identification of single-copy orthologous genes (Camacho et al. 2009; Eddy 2011; Buchfink et al. 2023). If the putative orthologs identified using this approach are multi-copy, the same splitting and pruning procedure in OrthoSNAP can identify SNAP-OGs (Steenwyk et al. 2022c). If the dataset contains a small number of genes, single-locus phylogenies can also be visually inspected, and paralogous sequences can be manually removed.
Multiple Sequence Alignment and Trimming
Once a curated set of phylogenomic markers has been obtained, the next step is multiple sequence alignment (Figure 1C), which aims to determine the site-wise homology across a group of sequences. Progressive alignment is a widely used strategy for multiple sequence alignment and involves an iterative pairwise alignment approach (Feng and Doolittle 1987). Over time, advancements have incorporated additional biological information to improve multiple sequence alignment. For instance, PRANK utilizes a phylogeny-aware approach, while 3DCoffee incorporates protein structure information (O’Sullivan et al. 2004; Löytynoja and Goldman 2005).
Several databases, such as BAliBASE and PREFAB (Thompson et al. 1999; Edgar 2004), have been developed to evaluate the effectiveness of these approaches. By using gold standard alignments from such databases or alignments generated by simulation software like INDELible (Fletcher and Yang 2009), the accuracy of multiple sequence alignment algorithms can be assessed using metrics like sum-of-pairs (pairwise alignment accuracy) and column scores (column-wise alignment accuracy) (Thompson et al. 1999; Steenwyk et al. 2021). Additionally, secondary structure predictions derived from the aligned sequences can be compared to known secondary structures to evaluate alignment accuracy (Sievers and Higgins 2020). Despite the establishment of standards, variable outcomes among benchmarking studies indicate that there is currently no universally superior algorithm for multiple sequence alignment (Raghava et al. 2003; Wang et al. 2018b; Sievers and Higgins 2020). Generating study-specific simulated alignments based on expected phylogenetic diversity can help determine which alignment strategy performs well for a given dataset.
Multiple sequence alignments are commonly subjected to trimming, which involves the removal of specific sites or blocks of sites within alignments. Traditionally, sites are trimmed because they are highly variable, which is thought to stem from erroneously inferred site homology, or are devoid of phylogenetic informativeness due to saturation by multiple substitutions (Talavera and Castresana 2007; Criscuolo and Gribaldo 2010). However, comparing multiple sequence alignment trimming algorithms has revealed that these methods often lead to poorer phylogenetic inference for single-gene analyses (Tan et al. 2015). This finding suggests that current trimming methods may inadvertently remove phylogenetically informative sites. In contrast, the software ClipKIT uses an alternative approach where informative sites (e.g., parsimony informative sites) are retained and all others removed, outperforming other algorithms under diverse evolutionary scenarios (Steenwyk et al. 2020a). A secondary advantage of trimming multiple sequence alignments is that shorter alignments require fewer computational resources, an important consideration for environmentally conscious computing practices (Kumar 2022; Steenwyk et al. 2023b). To address other types of errors in multiple sequence alignments, tools such as TAPER, Divviers, or MACSE can be employed (Ranwez et al. 2018; Ali et al. 2019; Zhang et al. 2021).
Model Selection
In molecular phylogenetics, the identification of an optimal model of sequence evolution occurs after alignment trimming and before tree inference. Although substitution models may, at times, not have a large impact on phylogenetic inference, model misspecification is a well-appreciated source of error (Shen et al. 2018; Abadi et al. 2019; Betancur-R. et al. 2019; Cao et al. 2022; Steenwyk et al. 2023b). Accordingly, substitution models have been a major research focus. Two main categories of substitution models exist: site homogeneous and site heterogeneous. Site homogeneous models employ the same parameters of character frequencies and substitution rates across the entire alignment (Galtier and Gouy 1995). Site heterogeneous mixture models allow for different character frequencies at each site (Lartillot et al. 2007; Si Quang et al. 2008). Site homogeneous models may be more prone to model misspecification, which can be overcome by generating alignment-specific time-reversible and time-non-reversible substitution models (Rodríguez-Ezpeleta et al. 2007; Minh et al. 2021). Site heterogeneous models have a larger parameter space and are robust to over-parameterization but require substantial computational resources (Baños et al. 2022). To accelerate computation, site heterogeneous models can be approximated using posterior mean site frequency profiles (Wang et al. 2018a).
Software tools that perform model testing are often integrated into phylogenetic tree inference software. For example, ModelFinder is included in the IQ-TREE toolkit (Kalyaanamoorthy et al. 2017; Minh et al. 2020b). These tools employ statistical frameworks—such as likelihood ratio testing and calculations of information criterion—to facilitate assessing model fit (Darriba et al. 2012, 2020; Kalyaanamoorthy et al. 2017).
Gene Alignment Concatenation or Coalescence-based Methods for Inferring Organismal History
There are two commonly used approaches for organismal tree inference from genome-scale datasets: gene alignment concatenation (or simply concatenation) and coalescence (Figure 1D) (Rokas et al. 2003; Liu et al. 2009a; Steenwyk et al. 2023b). Each employs different theoretical and statistical frameworks. Concatenation primarily utilizes maximum likelihood or Bayesian statistics. Coalescence relies on the multi-species coalescent model, which accounts for discordance between single loci and species-trees stemming from processes like incomplete lineage sorting.
The concatenation approach combines the aligned and trimmed orthologous genes into a single matrix. SequenceMatrix, FASconCAT-G, and PhyKIT can concatenate multiple sequence alignments (Vaidya et al. 2011; Kück and Longo 2014; Steenwyk et al. 2021). The resulting supermatrix can then be analyzed using various software that employs maximum likelihood statistical frameworks, such as RAxML-NG and IQ-TREE (Kozlov et al. 2019; Minh et al. 2020b), or Bayesian frameworks, such as MrBayes, BEAST, PhyloBayes, and RevBayes (Huelsenbeck and Ronquist 2001; Ronquist et al. 2012; Höhna et al. 2016; Bouckaert et al. 2019; Lartillot 2020). The RevBayes software allows users to customize how parameters, priors, and data are directly connected in the model, either in the Rev language or in the R programming language (Höhna et al. 2017; Charpentier and Wright 2022). In concatenation approaches, a single substitution model can be applied to the entire alignment, or the supermatrix can be partitioned, with different substitution models applied to each partition (Kainer and Lanfear 2015).
Comparative studies have systematically evaluated the performance of tools implementing different flavors of heuristics for maximum likelihood calculations and tree rearrangement methods, revealing variation in their performance (Zhou et al. 2018). This observation suggests that software choice is an important consideration. More broadly, software choice is encompassed in a newly described source of error: treatment errors, the negative impact of software choice, or data treatment upon stochastic and systematic errors (Steenwyk et al. 2023b).
In coalescent-based methods, two different approaches—one-step and two-step methods—are commonly employed for phylogenomic inference. In one-step approaches, single-locus phylogenies are estimated simultaneously with the species tree. This methodology is implemented in software such as BEST, StarBeast, and bpp (Liu et al. 2008; Yang and Rannala 2010; Douglas et al. 2022). Similarly, SVDquartets derives species trees from aligned, unlinked single nucleotide polymorphism data using the coalescent framework (Chifman and Kubatko 2014). In two-step approaches, as implemented in STAR and ASTRAL (Liu et al. 2009b; Zhang et al. 2018), involve first inferring individual single-locus phylogenies and then constructing a summary tree from the collection of single-locus phylogenies. One-step approaches are computationally expensive and may be difficult to apply to large phylogenomic datasets. Two-step approaches may be susceptible to errors resulting from inaccurate single-locus tree inference (Degnan and Rosenberg 2009). Collapsing poorly supported branches before applying summary tree methods can help mitigate the impact of uncertainty in single-locus phylogenies (Steenwyk et al. 2023b).
Although both concatenation and coalescence methods are widely used in species tree inference, there is currently no definitive guidance on when to prefer one method. However, it has been consistently observed that concatenation and coalescence can yield different topologies and varying levels of gene-wise support (Gatesy et al. 2017; Shen et al. 2018, 2021; Steenwyk et al. 2019b, 2023b; Li et al. 2020). The performance of these methods may vary depending on factors such as the data characteristics—the number and length of sampled loci, completeness of taxon sampling, the extent of reticulate evolution or incomplete lineage sorting among taxa, and the evolutionary diversity of the taxa, for example. A comprehensive study is needed to elucidate evolutionary scenarios where one method outperforms the other.
Examining Bipartition Support
Assessing the confidence in the resulting phylogenetic tree is crucial for identifying unstable bipartitions and gaining insights into lineages with reticulate evolutionary histories (Steenwyk et al. 2023b). Various methods have been developed to evaluate the support for different branches in a phylogeny and identify poorly supported bipartitions.
Bootstrapping is one of the earliest and most widely used methods for evaluating confidence in phylogenetic tree inference (Figure 1E). This statistical procedure involves resampling sites from an alignment with replacement to create multiple replicates of the original data (Felsenstein 1985). Each replicate is then used to infer a phylogenetic tree, and the frequency of observing a particular branch among the bootstrap replicates quantifies the support for that clade. Bootstrapping is typically performed with many replicates, such as 100 or more, to obtain robust support values. However, bootstrapping can be computationally intensive and time-consuming.
As phylogenomic datasets have become larger, alternatives have been introduced to expedite computation. For example, RAxML implements a rapid bootstrap approach (Stamatakis et al. 2008) and IQ-TREE employs an ultrafast bootstrap approximation method (Hoang et al. 2018). The "bag of little bootstraps" can also reduce the computational burden of long alignments by combining resampling with subsampling (Sharma and Kumar 2021). The gradual "transfer" distance method has also been proposed for bootstrap inference in phylogenies with hundreds to thousands of taxa (Lemoine et al. 2018).
Other methods to evaluate confidence use branching patterns observed among single-gene phylogenies. These include single-locus or -site support frequencies (also known as concordance factors) and quartet mapping (Figure 1E), as implemented in IQ-TREE, ASTRAL, and PhyKIT (Zhang et al. 2018; Minh et al. 2020b; Steenwyk et al. 2021). Calculations of internode certainty, an entropy-based measure of branch support, can help identify when alternative topologies are well supported among a collection of single-locus or bootstrap trees (Salichos and Rokas 2013; Salichos et al. 2014; Kobert et al. 2016; Zhou et al. 2020).
Phylogenomic subsampling is another approach for exploring tree space and identifying unstable bipartitions (Figure 1E) (Edwards 2016; Steenwyk et al. 2023b). In phylogenomic subsampling, subsamples of loci in a full phylogenomic data matrix are used to re-infer organismal histories. Bipartitions incongruent between the full and subsampled matrices warrant further investigation and can be considered unstable. Subsampling is typically done by selecting, for example, half of the loci in a full data matrix with the desirable feature associated with phylogenetic signal. These features—such as alignment length, average bipartition support, treeness divided by relative composition variability, and the number of parsimony and variable sites, among others (Phillips and Penny 2003; Shen et al. 2016, 2018; Steenwyk et al. 2019b, 2020a, 2021, 2022b; Mongiardino Koch 2021; McCarthy et al. 2023; Redmond et al. 2023)—aim to capture the information content of a locus.
Time-calibration of inferred phylogenetic divergences
Divergence times among branches in a phylogenomic analysis can be estimated by using fossils, mutation rates, or other temporal evidence to calibrate a molecular clock model (Ho and Phillips 2009; Dos Reis et al. 2016, 2018; Tiley et al. 2020). This procedure converts the relative divergences of molecular substitution rates to absolute time, often in units of thousands or millions of years ago. The resulting time-calibrated phylogenies, which are often referred to as ‘timetrees’ or ‘chronograms,’ differ from uncalibrated phylogenies (‘phylograms’) in that the former is directly comparable to other types of time-scaled data. Timetrees can be used to investigate causal eco-evolutionary dynamics relative to a broad array of independent evidence; for example, past changes in global temperature versus rates of lineage divergence (Oliveros et al. 2019; Schubert et al. 2019; S. Meseguer and Condamine 2020; Feijó et al. 2022); co-diversification among taxa (Sabrina Pankey et al. 2022; Nelsen et al. 2023); and rates of speciation in related clades (Harvey et al. 2020; Upham et al. 2021).
Approaches to estimating divergence times can be divided into node dating, tip dating, and fossil-free dating. Node dating places temporal constraints (i.e., calibrations) on a bifurcating internal node of a phylogeny, whereas tip dating places calibrations on terminal taxa that existed at some time in the past (Ho and Phillips 2009; Heath et al. 2014). The ages of serially sampled taxa—usually fossils or viruses and other microbes (Stadler and Yang 2013)—are the most reliable data for calibrating divergence times in phylogenomic datasets. Fossils and their associated ages (typically a confidence interval for the age of rock formations above and below a fossil discovery) can calibrate divergence times at either nodes or tips, typically using a probability distribution to incorporate age uncertainty (Ho and Phillips 2009; Stadler and Yang 2013). A fossil's phylogenetic position relative to living members of a given clade must be inferred or assumed based on other data for that fossil to serve as a time calibration (Parham et al. 2012). Viruses and other microbes evolve rapidly enough that samples collected in the last few decades offer valuable tip calibrations, analogous to the role of fossils in longer-lived mammals or plants (Volz et al. 2013; Andréoletti et al. 2022). The resulting ‘phylodynamic’ analyses help expose the population-dynamic processes that generate the phylogenetic patterns inferred from phylogenomic datasets. Phylodynamics is a promising integration of phylo-centric fields, enabling advances in cell biology, epidemiology, and macroevolution (Stadler et al. 2021; Andréoletti et al. 2022).
Clock models are used to extrapolate species divergence times from calibrated nodes. Strict clock models assume a fixed mutation rate in all branches, which is often violated when comparing more distant relatives (e.g., the 2% per million years rate used for bird mitochondrial genes; (Ho 2007)). However, strict clocks may lack biological realism. To address this, the strict clock assumption can be relaxed, such as in autocorrelated clock models wherein closely related branches have similar mutation rates or, in uncorrelated models, each branch is given an independent rate (Drummond et al. 2006; Lepage et al. 2007; Steenwyk and Rokas 2023). Relaxed clocks allow greater flexibility for handling the inherent molecular-rate variation among lineages, and thus are in wide use today for all types of time-calibration approaches, including fossil dating and multi-species coalescent approaches (Dos Reis et al. 2016, 2018; Flouri et al. 2022). These latter approaches enable the simultaneous estimation of species divergence times and ancestral population sizes using phylogenomic data and can be quite accurate when mutation rates are known from pedigrees (Tiley et al. 2020).
What if no fossils or other serial samples are available for a particular taxon? Two main options exist to calibrate divergences: use a fixed, strict clock model to project estimates back from tips or secondary calibrations. Secondary calibrations use previously estimated (from primary fossil or rate calibrations) divergence times of a sister taxon or outgroup to calibrate an internal or root node in the clade of interest (Shaul and Graur 2002). However, caution is required to avoid specifying overly precise secondary calibrations (Schenk 2016).
Choosing which software to use for divergence-time estimation involves a trade-off between available compute resources and the desired level of biological realism. At one extreme, the most realistic models (e.g., BPP and StarBEAST (Flouri et al. 2018; Douglas et al. 2022)) will perform Bayesian inference to estimate multi-species coalescent parameters across thousands of gene genealogies, considering multiple rate priors, and integrating across both phylogenetic and temporal uncertainty to yield a posterior distribution of time-scaled trees. However, these ‘full methods’ do not scale to large numbers of taxa or distant relatives (Tiley et al. 2020; Jiao et al. 2021).
At the other extreme, concatenated sequence data are used step-wise to first estimate the phylogenetic tree topology in units of substitutions/site, and then calibrations are applied in a second step of divergence-time estimation. Step-wise methods most commonly use maximum-likelihood (e.g., r8s, treePL, RelTime; (Sanderson 2003; Smith and O’Meara 2012; Tao et al. 2020)), but can also be implemented using Bayesian inference in programs like BEAST or MrBayes, which often requires fixing the tree topology. Midway between these extremes is the use of concatenated sequence data to perform simultaneous estimation of topology and divergence times, generally as implemented in a Bayesian framework (e.g., BEAST, MCMCtree, MrBayes, PhyloBayes, RevBayes). This latter approach has been implemented in large datasets (e.g., 800 taxa by 40,000 sites; (Upham et al. 2019)), and continues to be aided by GPU-based computing libraries (Ayres et al. 2019). Strategies for setting priors can impact divergence time estimation and are an important consideration (Barba-Montoya et al. 2017).
During divergence time estimation, a range of dates are typically plausible under the experimental conditions. Thus, divergence times are depicted as a confidence interval. Divergence times can also be inferred using a bootstrapping approach for intractably large datasets. Overall, the choices of node, tip, or fossil-free dating; strict or relaxed clocks; and concatenation or multi-species coalescent methods depend upon the question of interest, available molecular and morphological data, and prevalence of locus-tree-species-tree incongruence.
Detecting Reticulate Evolution
Reticulate evolutionary processes, such as hybridization/introgression and horizontal gene transfer, result in loci with evolutionary histories that differ from organismal history (Dobzhansky 1982; Abbott et al. 2013; Steenwyk et al. 2023b). There is a spectrum of outcomes for hybridization ranging from adaptive changes due to ecological selection, or compromised viability or fertility due to hybrid incompatibilities (Racimo et al. 2015; Moran et al. 2021). Similarly, horizontal gene transfer endows recipient organisms with novel genetic material and can be adaptive or not (Schönknecht et al. 2013; Gonçalves and Gonçalves 2019; Arnold et al. 2022; Gophna and Altman-Price 2022; Li et al. 2022b; Dorrell et al. 2023). The impact of horizontal gene transfer is well appreciated among prokaryotes and archaea but also occurs in the eukaryotes (Shen et al. 2018; Gonçalves and Gonçalves 2019; Lartillot 2020; Arnold et al. 2022; Gophna and Altman-Price 2022; Li et al. 2022b).
Hybridization/introgression
Hybridization/introgression can be adaptive or not, either expanding the ecological repertoire of organisms as exemplified by sunflower adaptation to novel environments or leading to the reabsorption of incipient species (Mallet 2005, 2008; Racimo et al. 2015; Buck et al. 2023). Hybrid progeny can have improved growth and reproductive success or conversely be sterile (Zanewich et al. 2018; Qiao et al. 2019; Allen et al. 2020; Adavoudi and Pilot 2021). Hybridization has been observed in microbial pathogens and thus may contribute to higher or lower organismal virulence (Lin et al. 2009; Depotter et al. 2016; Mixão and Gabaldón 2020). Hybridization can also result in neutral or negative fitness outcomes (Allen et al. 2020; Adavoudi and Pilot 2021).
The identification of hybridization/introgression events can be accomplished by utilizing phylogenetic and comparative genomic methodologies (Scannell et al. 2006; Marcet-Houben and Gabaldón 2015; Ortiz-Merino et al. 2017; Mixão and Gabaldón 2020; Steenwyk et al. 2020b, 2023a). Among phylogenetic approaches, it is crucial to discriminate between incongruences among single-locus phylogenies stemming from hybridization instead of incomplete lineage sorting, the random sorting of ancestral alleles that can, at times, result in locus-tree-species-tree incongruence (Yu et al. 2013). To make this distinction, two nearly equally supported topologies among multiple single-locus phylogenies indicate hybridization, especially if hybridization was a recent event. Conversely, in the case of incomplete lineage sorting, the alternative topology is expected to be observed less frequently, especially among more recent divergences (Steenwyk et al. 2019b). The expected degree of incongruence stemming from incomplete lineage sorting can be modeled using the multispecies coalescent model. Deviations from that model may indicate a hybridization event (Degnan and Rosenberg 2009). Calculating internode certainty can also help quantify the degree of support for the two most prevalent topologies (Salichos and Rokas 2013; Kobert et al. 2016).
Many methods have been developed to detect hybridization events more directly using phylogenetic trees (Hibbins and Hahn 2022). One pioneering approach is the utilization of the D-statistic, also known as the ABBA-BABA test, which employs biallelic site patterns within a phylogenetic framework (Figure 2) (Green et al. 2010). Specifically, the ABBA-BABA test examines asymmetric support between ABBA and BABA patterns at biallelic sites, which suggests an introgression/hybridization event; in contrast, equal proportions of ABBA and BABA site patterns would suggest no introgression/hybridization. Leveraging genome-scale data, the ABBA-BABA test accurately quantifies introgression across a wide parameter space (Zheng and Janke 2018).
Discerning the direction of introgression poses a greater challenge (Martin et al. 2015). To address this, the Dp-statistic, an extension of the ABBA-BABA test, incorporates patterns observed across all variable sites, thus enabling the determination of gene flow directionality (Hamlin et al. 2020). In the context of a symmetric five-taxon phylogeny, the DFOIL statistic, another extension of the ABBA-BABA test built on the partitioned D-statistic (Eaton and Ree 2013), explicitly examines all potential introgression events, facilitating precise and sensitive detection of the extent and direction of introgression (Pease and Hahn 2015). Several other methodologies, including the F4-ratio, fd, the f3-statistic, and algorithms implemented in the HyDe software, have been developed to identify introgression events (Green et al. 2010; Reich et al. 2012; Martin et al. 2015; Blischak et al. 2018; Jacobs et al. 2018). Note, these methods examine rates of tree incongruences and are distinct from F-statistics, such as Fst, which are related to population genetic analysis.
In the context of allopolyploid hybrids, where the genome of the hybrid organism contains (nearly) the complete genetic complement of both parental genomes and, therefore, two or more copies of most genes, supplementary methods play a crucial role in detecting and unraveling the evolution of a hybrid genome. Ancient allopolyploid events can be identified by a burst of gene duplications (Chain et al. 2011; Marcet-Houben and Gabaldón 2015; Session et al. 2016), but would benefit from other lines of evidence such as synteny information. In allopolyploids, it may be helpful to determine the parent of origin for each gene in a hybrid genome. This can be accomplished by evaluating phylogenetic distances between the parental species and the gene copies encoded in the hybrid (Steenwyk et al. 2023a). Additionally, Ks plots, which visualize sequence divergence between loci in the hybrid taxon and their closest homolog in a parental species, prove valuable in assigning parent-of-origin for loci within a hybrid genome (Ortiz-Merino et al. 2017; Steenwyk et al. 2020b).
Horizontal gene transfer
Horizontal gene transfer, or lateral gene transfer, is the acquisition of genetic material from other organisms without sexual reproduction (Mallet 2005). Horizontal gene transfer is considered a major mode of evolution in prokaryotes and archaea wherein the mechanism of transfer—conjugation, transduction, and transformation—is better understood (Galtier 2007; Arnold et al. 2022; Gonçalves and Gonçalves 2022; Gophna and Altman-Price 2022). In recent years, horizontal gene transfer in eukaryotes is better appreciated (Coelho et al. 2013; Gonçalves et al. 2018; Husnik and McCutcheon 2018; Shen et al. 2018; Zhou et al. 2018; Gonçalves and Gonçalves 2019; Van Etten and Bhattacharya 2020; Irwin et al. 2021). One hypothesized transfer mechanism among microeukaryotes serves as the foundation for the “you are what you eat” hypothesis, which posits that organisms that phagocytose prey may be more susceptible to rare and accidental nucleic acid integration from prey DNA (Sibbald et al. 2020). In support of this hypothesis, recent analyses found that carnivorous mammal genomes contain a greater prevalence of DNA-based transposable elements than do herbivores or omnivores, suggestive of origins via horizontal gene transfer from ingested prey or their viruses (Osmanski et al. 2023).
Horizontal gene transfer is also well appreciated in the context of mitochondrial and plastid organellogenesis. These organelles arose from the ancient assimilation and degradation of endosymbiotic bacteria in eukaryotic cells, during which many endosymbiont genes were lost or transferred to the host nucleus (Cenci et al. 2018; Ponce-Toledo et al. 2018; Sibbald and Archibald 2020). Charting endosymbiotic transfers has aided in estimating the order and mechanisms of higher-order plastid acquisitions and revealed that many plastid-related genes have highly chimeric origins (Stiller et al. 2014; Dorrell et al. 2017; Strassert et al. 2021). These observations may support scenarios of ancient serial plastid replacements before permanent plastid integration (Minge et al. 2010; Ponce-Toledo et al. 2018; Morozov and Galachyants 2019). Transferred genes have also provided evidence of relic plastids, illuminating patterns of plastid gain and loss (Cenci et al. 2018; Gawryluk et al. 2019). Some barriers to horizontal gene transfer are also known, such as multicellularity and incompatible genetic codes (Yue et al. 2012; Shen et al. 2018).
The methods employed for detecting horizontally acquired loci vary in precision and accuracy. Early methods relied on identifying deviations in gene sequence characteristics. In the case of very recent prokaryote-to-eukaryote horizontal gene transfer, detection could be achieved by observing genes that deviate in guanine-cytosine content, intron content, gene order, and codon usage across the host genome (Friedman and Ely 2012; Zhang et al. 2014b; Jaramillo et al. 2015; Gonçalves and Gonçalves 2022). In the phylogenomic era, these methods are often employed to support the identification of horizontal gene transfer events rather than serving as primary detection tools.
Another approach is to calculate the alien index—a score that compares the similarity between sequences within the target group and sequences from outgroup taxa (Gladyshev et al. 2008; Alexander et al. 2016)—of all genes in a host genome. Loci exhibiting alien indices indicative of potential horizontal gene transfer are then selected for further investigation through phylogenetic inference, the gold standard approach for horizontal gene transfer detection. Several software tools have been developed to calculate alien indices or similar metrics for assessing horizontal gene transfer. Examples include AvP, HGTector, and HGTphyloDetect (Zhu et al. 2014; Koutsovoulos et al. 2022; Yuan et al. 2023).
Phylogenetic trees that suggest horizontal gene transfer events are characterized by the confident placement of one or a few sequences within an unexpected taxonomic group (Figure 3). For instance, in the case of prokaryote-to-eukaryote horizontal gene transfer, sequences in a eukaryotic genome may be nested deep within a prokaryotic lineage (Coelho et al. 2013; Gonçalves et al. 2018; Husnik and McCutcheon 2018; Shen et al. 2018; Zhou et al. 2018; Gonçalves and Gonçalves 2019; Kominek et al. 2019; Van Etten and Bhattacharya 2020; Irwin et al. 2021; Li et al. 2022b). The evidence for horizontal gene transfer can be strengthened using topology tests, such as the Kishino-Hasegawa and Shimodaira-Hasegawa tests (Kishino and Hasegawa 1989; Shimodaira and Hasegawa 1999). These tests compare the likelihood of a phylogeny constrained to reflect a vertical evolutionary scenario (the null hypothesis) with the observed topology, reflecting the occurrence of horizontal gene transfer (the alternative hypothesis) (Gonçalves et al. 2018; Shen et al. 2018).
Although putatively transferred genes should be investigated using molecular phylogenetics, accurate phylogenomic species tree inference may not be crucial since it is widely accepted that the taxa involved are of distinct lineages; for example, fungi and bacteria are well known to comprise different clades. However, when examining horizontal gene transfer among more closely related taxa, robust species tree inference becomes essential (Ropars et al. 2015). The potential for shared ancestral variation or asymmetric gene loss patterns among closely related organisms increases the risk of erroneously inferring horizontal gene transfer events (i.e., type I error / false positive). Horizontally transferred loci may also complicate accurate inference of organismal histories; thus, purging phylogenomic data matrices from potentially horizontally acquired loci may be warranted. Lastly, to rule out putatively horizontally transferred genes stemming from contamination, this analysis is often restricted to well-assembled contigs, devoid of contamination signatures (Shen et al. 2018; Li et al. 2022b).
Phylogenetic Networks
Hybridization/introgression and horizontal gene transfer challenge the strictly bifurcating tree model. Phylogenetic networks can help explore organismal histories in the presence of these factors (Huson 1998; Lutteropp et al. 2022; Steenwyk et al. 2023b), and to large extent can be viewed as complementary to strictly bifurcating phylogenetic analyses (Blair and Ané 2020). Phylogenetic networks can be broadly categorized into reticulate networks, which capture reticulate evolutionary processes (Huson et al. 2005), and splits networks, which represent all splits in a collection of single-locus phylogenies (Huson 1998). Additionally, visualizing the density of phylogenetic trees can reveal common branching patterns across these collections, which can help identify regions of the tree that require further investigation. DensiTree is a tool commonly employed to visualize the density of phylogenetic tree topologies in a posterior distribution of trees (Bouckaert 2010). Gene-support frequencies (or quartet frequencies) and gene-wise phylogenetic signals offer additional insights to scrutinize and assess alternative branching patterns (Shen et al. 2017, 2021; Sayyari and Mirarab 2018; Steenwyk et al. 2019b; Minh et al. 2020a). The methods discussed herein facilitate rigorous phylogenomic inference and detection of reticulate evolution in organismal histories.
Conclusion
Knowledge of organismal history underpins many evolutionary studies. This article outlines the many branching choices that investigators must make when performing phylogenomic inference, and some of the known pathways toward assembling robust methodological pipelines. The resulting analyses of genome-wide evolutionary signatures can, in turn, be used to enrich our understanding of life's history and the evolutionary process. Ameliorating analytical factors driving apparent incongruence will facilitate more accurate inference of organismal histories. True histories of reticulate evolution will deepen our understanding of the Tree of Life, adding a dimension of ‘webiness.’ Future computational improvements, reduced cost of high-quality genome sequencing, and algorithmic advances culminating in more chromosome-scale genome assemblies will pave the way for increasing larger datasets and greater elucidation of the Tree of Life. Such advances will also bring new challenges and opportunities. To encourage the continued investigation of biodiversity via phylogenomics, we hope this article provides helpful guidance for scientific investigation and pedagogy alike.
Funding
J.L.S. is a Howard Hughes Medical Institute Awardee of the Life Sciences Research Foundation. N.S.U. was supported by Arizona State University start-up funds. H.V. received support from the Australian Biological Resources Study (4-G046WSD) and the Australian Research Council (DP200101613).
Acknowledgments
We thank Drs. Xing-Xing Shen and Yuanning Li for helpful discussion over the years. J.L.S. thanks Dr. Antonis Rokas, who taught him to embrace phylogenomic incongruence. Chat-GPT was used to edit portions of the manuscript; AI-generated text was further edited.
Competing Interests
J.L.S. is a scientific advisor for WittGen Biotechnologies. J.L.S. is an advisor for ForensisGroup Inc.
References
- Abadi, S.; Azouri, D.; Pupko, T.; Mayrose, I. Model selection may not be a mandatory step for phylogeny reconstruction. Nat Commun 2019, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.; Albach, D.; Ansell, S.; Arntzen, J. W.; Baird, S. J. E.; Bierne, N.; Boughman, J.; Brelsford, A.; Buerkle, C. A.; Buggs, R.; Butlin, R. K.; Dieckmann, U.; Eroukhmanoff, F.; Grill, A.; Cahan, S. H.; Hermansen, J. S.; Hewitt, G.; Hudson, A. G.; Jiggins, C.; Jones, J.; Keller, B.; Marczewski, T.; Mallet, J.; Martinez-Rodriguez, P.; Möst, M.; Mullen, S.; Nichols, R.; Nolte, A. W.; Parisod, C.; Pfennig, K.; Rice, A. M.; Ritchie, M. G.; Seifert, B.; Smadja, C. M.; Stelkens, R.; Szymura, J. M.; Väinölä, R.; Wolf, J. B. W.; Zinner, D. Hybridization and speciation. J. Evol. Biol 2013, 26, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Adavoudi, R.; Pilot, M. Consequences of Hybridization in Mammals: A Systematic Review. Genes 2021, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W. G.; Wisecaver, J. H.; Rokas, A.; Hittinger, C. T. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 4116–4121. [Google Scholar] [CrossRef] [PubMed]
- Ali, R. H.; Bogusz, M.; Whelan, S. Identifying Clusters of High Confidence Homologies in Multiple Sequence Alignments. Molecular Biology and Evolution 2019, 36, 2340–2351. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.; Ryan, H.; Davis, B. W.; King, C.; Frantz, L.; Irving-Pease, E.; Barnett, R.; Linderholm, A.; Loog, L.; Haile, J.; Lebrasseur, O.; White, M.; Kitchener, A. C.; Murphy, W. J.; Larson, G. A mitochondrial genetic divergence proxy predicts the reproductive compatibility of mammalian hybrids. Proc. R. Soc. B 2020, 287, 20200690. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Carretero, S.; Kapli, P.; Yang, Z. Beginner’s Guide on the Use of PAML to Detect Positive Selection. Molecular Biology and Evolution 2023, 40. [Google Scholar] [CrossRef]
- Andréoletti, J.; Zwaans, A.; Warnock, R. C. M.; Aguirre-Fernández, G.; Barido-Sottani, J.; Gupta, A.; Stadler, T.; Manceau, M. The Occurrence Birth–Death Process for Combined-Evidence Analysis in Macroevolution and Epidemiology. Systematic Biology 2022, 71, 1440–1452. [Google Scholar] [CrossRef]
- Arnold, B. J.; Huang, I.-T.; Hanage, W. P. Horizontal gene transfer and adaptive evolution in bacteria. Nat Rev Microbiol 2022, 20, 206–218. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Sela, I.; Karin, E. Levy; Landan, G.; Pupko, T. Multiple Sequence Alignment Averaging Improves Phylogeny Reconstruction. Systematic Biology 2019, 68, 117–130. [Google Scholar] [CrossRef]
- Ayres, D. L.; Cummings, M. P.; Baele, G.; Darling, A. E.; Lewis, P. O.; Swofford, D. L.; Huelsenbeck, J. P.; Lemey, P.; Rambaut, A.; Suchard, M. A. BEAGLE 3: Improved Performance, Scaling, and Usability for a High-Performance Computing Library for Statistical Phylogenetics. Systematic Biology 2019, 68, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Ba#xF1os, H.; Susko, E.; Roger, A. J. Is Over-parameterization a Problem for Profile Mixture Models? Evolutionary Biology 2022. [Google Scholar]
- Barba-Montoya, J.; Reis, M. Dos; Yang, Z. Comparison of different strategies for using fossil calibrations to generate the time prior in Bayesian molecular clock dating. Molecular Phylogenetics and Evolution 2017, 114, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Barley, A. J.; Brown, J. M.; Thomson, R. C. Impact of Model Violations on the Inference of Species Boundaries Under the Multispecies Coalescent. Systematic Biology 2018, 67, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Bautista, C.; Marsit, S.; Landry, C. R. Interspecific hybrids show a reduced adaptive potential under DNA damaging conditions. Evol Appl 2021, 14, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Betancur-R., R.; Arcila, D.; Vari, R. P.; Hughes, L. C.; Oliveira, C.; Sabaj, M. H.; Ortí, G. Phylogenomic incongruence, hypothesis testing, and taxonomic sampling. Evolution 2019, 73, 329–345. [Google Scholar] [CrossRef]
- Blair, C.; Ané, C. Phylogenetic Trees and Networks Can Serve as Powerful and Complementary Approaches for Analysis of Genomic Data. Systematic Biology 2020, 69, 593–601. [Google Scholar] [CrossRef]
- Blaxter, M.; Archibald, J. M.; Childers, A. K.; Coddington, J. A.; Crandall, K. A.; Palma, F. Di; Durbin, R.; Edwards, S. V.; Graves, J. A. M.; Hackett, K. J.; Hall, N.; Jarvis, E. D.; Johnson, R. N.; Karlsson, E. K.; Kress, W. J.; Kuraku, S.; Lawniczak, M. K. N.; Lindblad-Toh, K.; Lopez, J. V.; Moran, N. A.; Robinson, G. E.; Ryder, O. A.; Shapiro, B.; Soltis, P. S.; Warnow, T.; Zhang, G.; Lewin, H. A. Why sequence all eukaryotes? Proc. Natl. Acad. Sci. U.S.A 2022, 119, e2115636118. [Google Scholar] [CrossRef]
- Blischak, P. D.; Chifman, J.; Wolfe, A. D.; Kubatko, L. S. HyDe: a Python package for genome-scale hybridization detection. Systematic Biology 2018, 67, 821–829. [Google Scholar] [CrossRef]
- Bolger, A. M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bouckaert, R. R. DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 2010, 26, 1372–1373. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T. G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; Maio, N. De; Matschiner, M.; Mendes, F. K.; Müller, N. F.; Ogilvie, H. A.; Plessis, L. du; Popinga, A.; Rambaut, A.; Rasmussen, D.; Siveroni, I.; Suchard, M. A.; Wu, C.-H.; Xie, D.; Zhang, C.; Stadler, T.; Drummond, A. J. BEAST 2. 5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol 2019, 15, e1006650. [Google Scholar] [PubMed]
- Bringloe, T. T.; Zaparenkov, D.; Starko, S.; Grant, W. S.; Vieira, C.; Kawai, H.; Hanyuda, T.; Filbee-Dexter, K.; Klimova, A.; Klochkova, T. A.; Krause-Jensen, D.; Olesen, B.; Verbruggen, H. Whole-genome sequencing reveals forgotten lineages and recurrent hybridizations within the kelp genus Alaria (Phaeophyceae). J. Phycol 2021, 57, 1721–1738. [Google Scholar] [CrossRef] [PubMed]
- Brohée, S.; Helden, J. Van. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 2006, 7, 488. [Google Scholar] [CrossRef]
- Brůna, T.; Hoff, K. J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics and Bioinformatics 2021, 3. [Google Scholar] [CrossRef]
- Buchfink, B.; Ashkenazy, H.; Reuter, K.; Kennedy, J. A.; Drost, H.-G. Sensitive clustering of protein sequences at tree-of-life scale using DIAMOND DeepClust. Genomics 2023. [Google Scholar]
- Buck, R.; Vecchyo, D. Ortega-Del; Gehring, C.; Michelson, R.; Flores-Rentería, D.; Klein, B.; Whipple, A. V.; Flores-Rentería, L. Sequential hybridization may have facilitated ecological transitions in the Southwestern pinyon pine syngameon. New Phytologist 2023, 237, 2435–2449. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T. L. BLAST+: architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef]
- Cao, Z.; Li, M.; Ogilvie, H. A.; Nakhleh, L. The Impact of Model Misspecification on Phylogenetic Network Inference. Evolutionary Biology 2022. [Google Scholar]
- Capella-Gutiérrez, S.; Marcet-Houben, M.; Gabaldón, T. Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi. BMC Biol 2012, 10, 47. [Google Scholar] [CrossRef]
- Carruthers, T.; Scotland, R. W. Uncertainty in Divergence Time Estimation. Systematic Biology 2021, 70, 855–861. [Google Scholar] [CrossRef]
- Cenci, U.; Sibbald, S. J.; Curtis, B. A.; Kamikawa, R.; Eme, L.; Moog, D.; Henrissat, B.; Maréchal, E.; Chabi, M.; Djemiel, C.; Roger, A. J.; Kim, E.; Archibald, J. M. Nuclear genome sequence of the plastid-lacking cryptomonad Goniomonas avonlea provides insights into the evolution of secondary plastids. BMC Biol 2018, 16, 137. [Google Scholar] [CrossRef]
- Chain, F. J.; Dushoff, J.; Evans, B. J. The odds of duplicate gene persistence after polyploidization. BMC Genomics 2011, 12, 599. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, C. P.; Wright, A. M. Revticulate: An R framework for interaction with RevBayes. Methods Ecol Evol 2022, 13, 1177–1184. [Google Scholar] [CrossRef]
- Chen, L.; Xu, J.; Sun, X.; Xu, P. Research advances and future perspectives of genomics and genetic improvement in allotetraploid common carp. Reviews in Aquaculture 2022, 14, 957–978. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.; Zhang, J.; Park, C. Is Phylotranscriptomics as Reliable as Phylogenomics? Molecular Biology and Evolution 2020, 37, 3672–3683. [Google Scholar] [CrossRef]
- Chifman, J.; Kubatko, L. Quartet Inference from SNP Data Under the Coalescent Model. Bioinformatics 2014, 30, 3317–3324. [Google Scholar] [CrossRef]
- Chikina, M.; Robinson, J. D.; Clark, N. L. Hundreds of Genes Experienced Convergent Shifts in Selective Pressure in Marine Mammals. Mol Biol Evol 2016, 33, 2182–2192. [Google Scholar] [CrossRef]
- Clark, J. W.; Donoghue, P. C. J. Whole-Genome Duplication and Plant Macroevolution. Trends in Plant Science 2018, 23, 933–945. [Google Scholar] [CrossRef]
- Coelho, M. A.; Gonçalves, C.; Sampaio, J. P.; Gonçalves, P. Extensive Intra-Kingdom Horizontal Gene Transfer Converging on a Fungal Fructose Transporter Gene. PLoS Genet 2013, 9, e1003587. [Google Scholar] [CrossRef] [PubMed]
- Coleman, G. A.; Davín, A. A.; Mahendrarajah, T. A.; Szánthó, L. L.; Spang, A.; Hugenholtz, P.; Szöllősi, G. J.; Williams, T. A. A rooted phylogeny resolves early bacterial evolution. Science 2021, 372. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol 2010, 10, 210. [Google Scholar] [CrossRef]
- Dannemann, M.; Kelso, J. The Contribution of Neanderthals to Phenotypic Variation in Modern Humans. The American Journal of Human Genetics 2017, 101, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Dannemann, M.; Prüfer, K.; Kelso, J. Functional implications of Neandertal introgression in modern humans. Genome Biol 2017, 18, 61. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A. M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Molecular Biology and Evolution 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G. L.; Doallo, R.; Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 2012, 9, 772–772. [Google Scholar] [CrossRef]
- Degnan, J. H.; Rosenberg, N. A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends in Ecology & Evolution 2009, 24, 332–340. [Google Scholar]
- Depotter, J. R.; Seidl, M. F.; Wood, T. A.; Thomma, B. P. Interspecific hybridization impacts host range and pathogenicity of filamentous microbes. Current Opinion in Microbiology 2016, 32, 7–13. [Google Scholar] [CrossRef]
- Díaz-Tapia, P.; Maggs, C. A.; West, J. A.; Verbruggen, H. Analysis of chloroplast genomes and a supermatrix inform reclassification of the Rhodomelaceae (Rhodophyta). J. Phycol 2017, 53, 920–937. [Google Scholar] [CrossRef]
- Dobzhansky, T. Genetics and the Origin of Species; Columbia university press, 1982. [Google Scholar]
- Dorrell, R. G.; Gile, G.; McCallum, G.; Méheust, R.; Bapteste, E. P.; Klinger, C. M.; Brillet-Guéguen, L.; Freeman, K. D.; Richter, D. J.; Bowler, C. Chimeric origins of ochrophytes and haptophytes revealed through an ancient plastid proteome. eLife 2017, 6, e23717. [Google Scholar] [CrossRef] [PubMed]
- Dorrell, R. G.; Kuo, A.; Füssy, Z.; Richardson, E. H.; Salamov, A.; Zarevski, N.; Freyria, N. J.; Ibarbalz, F. M.; Jenkins, J.; Karlusich, J. J. Pierella; Steindorff, A. Stecca; Edgar, R. E.; Handley, L.; Lail, K.; Lipzen, A.; Lombard, V.; McFarlane, J.; Nef, C.; Vanclová, A. M. Novák; Peng, Y.; Plott, C.; Potvin, M.; Vieira, F. R. J.; Barry, K.; Vargas, C. De; Henrissat, B.; Pelletier, E.; Schmutz, J.; Wincker, P.; Dacks, J. B.; Bowler, C.; Grigoriev, I. V.; Lovejoy, C. Convergent evolution and horizontal gene transfer in Arctic Ocean microalgae. Life Sci. Alliance 2023, 6, e202201833. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, M.; Donoghue, P. C. J.; Yang, Z. Bayesian molecular clock dating of species divergences in the genomics era. Nat Rev Genet 2016, 17, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, M.; Gunnell, G. F.; Barba-Montoya, J.; Wilkins, A.; Yang, Z.; Yoder, A. D. Using Phylogenomic Data to Explore the Effects of Relaxed Clocks and Calibration Strategies on Divergence Time Estimation: Primates as a Test Case. Systematic Biology 2018, 67, 594–615. [Google Scholar] [CrossRef]
- Douglas, J.; Jiménez-Silva, C. L.; Bouckaert, R. StarBeast3: Adaptive Parallelized Bayesian Inference under the Multispecies Coalescent. Systematic Biology 2022, 71, 901–916. [Google Scholar] [CrossRef]
- Douglass, A. P.; O’Brien, C. E.; Offei, B.; Coughlan, A. Y.; Ortiz-Merino, R. A.; Butler, G.; Byrne, K. P.; Wolfe, K. H. Coverage-Versus-Length Plots, a Simple Quality Control Step for de Novo Yeast Genome Sequence Assemblies. G3 GenesGenomesGenetics 2019, 9, 879–887. [Google Scholar] [CrossRef]
- Drummond, A. J.; Ho, S. Y. W.; Phillips, M. J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol 2006, 4, e88. [Google Scholar] [CrossRef]
- Dunn, C. W.; Hejnol, A.; Matus, D. Q.; Pang, K.; Browne, W. E.; Smith, S. A.; Seaver, E.; Rouse, G. W.; Obst, M.; Edgecombe, G. D.; Sørensen, M. V.; Haddock, S. H. D.; Schmidt-Rhaesa, A.; Okusu, A.; Kristensen, R. M.; Wheeler, W. C.; Martindale, M. Q.; Giribet, G. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 2008, 452, 745–749. [Google Scholar] [CrossRef]
- Eaton, D. A. R.; Ree, R. H. Inferring Phylogeny and Introgression using RADseq Data: An Example from Flowering Plants (Pedicularis: Orobanchaceae). Systematic Biology 2013, 62, 689–706. [Google Scholar] [CrossRef]
- Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput Biol 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Edelman, N. B.; Frandsen, P. B.; Miyagi, M.; Clavijo, B.; Davey, J.; Dikow, R. B.; García-Accinelli, G.; Belleghem, S. M. Van; Patterson, N.; Neafsey, D. E.; Challis, R.; Kumar, S.; Moreira, G. R. P.; Salazar, C.; Chouteau, M.; Counterman, B. A.; Papa, R.; Blaxter, M.; Reed, R. D.; Dasmahapatra, K. K.; Kronforst, M.; Joron, M.; Jiggins, C. D.; McMillan, W. O.; Palma, F. Di; Blumberg, A. J.; Wakeley, J.; Jaffe, D.; Mallet, J. Genomic architecture and introgression shape a butterfly radiation. Science 2019, 366, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Edger, P. P.; Poorten, T. J.; VanBuren, R.; Hardigan, M. A.; Colle, M.; McKain, M. R.; Smith, R. D.; Teresi, S. J.; Nelson, A. D. L.; Wai, C. M.; Alger, E. I.; Bird, K. A.; Yocca, A. E.; Pumplin, N.; Ou, S.; Ben-Zvi, G.; Brodt, A.; Baruch, K.; Swale, T.; Shiue, L.; Acharya, C. B.; Cole, G. S.; Mower, J. P.; Childs, K. L.; Jiang, N.; Lyons, E.; Freeling, M.; Puzey, J. R.; Knapp, S. J. Origin and evolution of the octoploid strawberry genome. Nat Genet 2019, 51, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S. V. Phylogenomic subsampling: a brief review. Zool Scr 2016, 45, 63–74. [Google Scholar] [CrossRef]
- Emms, D. M.; Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 2019, 20, 238. [Google Scholar] [CrossRef]
- Emms, D. M.; Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 2015, 16, 157. [Google Scholar] [CrossRef]
- Emms, D. M.; Kelly, S. STAG: Species Tree Inference from All Genes. Evolutionary Biology 2018. [Google Scholar]
- Feijó, A.; Ge, D.; Wen, Z.; Cheng, J.; Xia, L.; Patterson, B. D.; Yang, Q. Mammalian diversification bursts and biotic turnovers are synchronous with Cenozoic geoclimatic events in Asia. Proc. Natl. Acad. Sci. U.S.A 2022, 119, e2207845119. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: an approach using the bootstrap. evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Feng, D.-F.; Doolittle, R. F. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol 1987, 25, 351–360. [Google Scholar] [CrossRef]
- Fernández, R.; Gabaldón, T. Gene gain and loss across the metazoan tree of life. Nat Ecol Evol 2020, 4, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Fernández, R.; Gabaldón, T.; Dessimoz, C. Orthology: definitions, inference, and impact on species phylogeny inference. 2019. [Google Scholar] [CrossRef]
- Fletcher, W.; Yang, Z. INDELible: A Flexible Simulator of Biological Sequence Evolution. Molecular Biology and Evolution 2009, 26, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Flouri, T.; Huang, J.; Jiao, X.; Kapli, P.; Rannala, B.; Yang, Z. Bayesian Phylogenetic Inference using Relaxed-clocks and the Multispecies Coalescent. Molecular Biology and Evolution 2022, 39. [Google Scholar] [CrossRef] [PubMed]
- Flouri, T.; Jiao, X.; Rannala, B.; Yang, Z. Species Tree Inference with BPP Using Genomic Sequences and the Multispecies Coalescent. Molecular Biology and Evolution 2018, 35, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.; Ely, B. Codon Usage Methods for Horizontal Gene Transfer Detection Generate an Abundance of False Positive and False Negative Results. Curr Microbiol 2012, 65, 639–642. [Google Scholar] [CrossRef]
- Gabaldón, T.; Koonin, E. V. Functional and evolutionary implications of gene orthology. Nat Rev Genet 2013, 14, 360–366. [Google Scholar] [CrossRef]
- Galindo, L. J.; López-García, P.; Torruella, G.; Karpov, S.; Moreira, D. Phylogenomics of a new fungal phylum reveals multiple waves of reductive evolution across Holomycota. Nat Commun 2021, 12, 4973. [Google Scholar] [CrossRef]
- Galtier, N. A Model of Horizontal Gene Transfer and the Bacterial Phylogeny Problem. Systematic Biology 2007, 56, 633–642. [Google Scholar] [CrossRef]
- Galtier, N.; Gouy, M. Inferring phylogenies from DNA sequences of unequal base compositions. Proceedings of the National Academy of Sciences 1995, 92, 11317–11321. [Google Scholar] [CrossRef]
- Gatesy, J.; Meredith, R. W.; Janecka, J. E.; Simmons, M. P.; Murphy, W. J.; Springer, M. S. Resolution of a concatenation/coalescence kerfuffle: partitioned coalescence support and a robust family-level tree for Mammalia. Cladistics 2017, 33, 295–332. [Google Scholar] [CrossRef]
- Gawryluk, R. M. R.; Tikhonenkov, D. V.; Hehenberger, E.; Husnik, F.; Mylnikov, A. P.; Keeling, P. J. Non-photosynthetic predators are sister to red algae. Nature 2019, 572, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, E. A.; Meselson, M.; Arkhipova, I. R. Massive Horizontal Gene Transfer in Bdelloid Rotifers. Science 2008, 320, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.; Gonçalves, P. Multilayered horizontal operon transfers from bacteria reconstruct a thiamine salvage pathway in yeasts. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 22219–22228. [Google Scholar] [CrossRef]
- Gonçalves, C.; Wisecaver, J. H.; Kominek, J.; Oom, M. S.; Leandro, M. J.; Shen, X.-X.; Opulente, D. A.; Zhou, X.; Peris, D.; Kurtzman, C. P.; Hittinger, C. T.; Rokas, A.; Gonçalves, P. Evidence for loss and reacquisition of alcoholic fermentation in a fructophilic yeast lineage. eLife 2018, 7, e33034. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, P.; Gonçalves, C. Horizontal gene transfer in yeasts. Current Opinion in Genetics & Development 2022, 76, 101950. [Google Scholar]
- Gophna, U.; Altman-Price, N. Horizontal Gene Transfer in Archaea—From Mechanisms to Genome Evolution. Annu. Rev. Microbiol 2022, 76, 481–502. [Google Scholar] [CrossRef] [PubMed]
- Green, R. E.; Krause, J.; Briggs, A. W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M. H.-Y. A draft sequence of the Neandertal genome. science 2010, 328, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Hale, H.; Gardner, E. M.; Viruel, J.; Pokorny, L.; Johnson, M. G. Strategies for reducing per-sample costs in target capture sequencing for phylogenomics and population genomics in plants. Appl Plant Sci 2020, 8. [Google Scholar] [CrossRef]
- Hamlin, J. A. P.; Hibbins, M. S.; Moyle, L. C. Assessing biological factors affecting postspeciation introgression. Evolution Letters 2020, 4, 137–154. [Google Scholar] [CrossRef]
- Harvey, M. G.; Bravo, G. A.; Claramunt, S.; Cuervo, A. M.; Derryberry, G. E.; Battilana, J.; Seeholzer, G. F.; McKay, J. S.; O’Meara, B. C.; Faircloth, B. C.; Edwards, S. V.; Pérez-Emán, J.; Moyle, R. G.; Sheldon, F. H.; Aleixo, A.; Smith, B. T.; Chesser, R. T.; Silveira, L. F.; Cracraft, J.; Brumfield, R. T.; Derryberry, E. P. The evolution of a tropical biodiversity hotspot. Science 2020, 370, 1343–1348. [Google Scholar] [CrossRef]
- Heath, T. A.; Huelsenbeck, J. P.; Stadler, T. The fossilized birth–death process for coherent calibration of divergence-time estimates. Proc. Natl. Acad. Sci. U.S.A. 2014, 111. [Google Scholar] [CrossRef] [PubMed]
- Hibbins, M. S.; Hahn, M. W. Phylogenomic approaches to detecting and characterizing introgression. Genetics 2022, 220. [Google Scholar] [CrossRef] [PubMed]
- Ho, S. Y. M. Calibrating molecular estimates of substitution rates and divergence times in birds. J Avian Biology 2007, 38, 409–414. [Google Scholar] [CrossRef]
- Ho, S. Y. W.; Duchêne, S. Molecular-clock methods for estimating evolutionary rates and timescales. Mol Ecol 2014, 23, 5947–5965. [Google Scholar] [CrossRef] [PubMed]
- Ho, S. Y. W.; Phillips, M. J. Accounting for Calibration Uncertainty in Phylogenetic Estimation of Evolutionary Divergence Times. Systematic Biology 2009, 58, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D. T.; Chernomor, O.; Haeseler, A. von; Minh, B. Q.; Vinh, L. S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Molecular Biology and Evolution 2018, 35, 518–522. [Google Scholar] [CrossRef]
- H#xF6hna, S.; Landis, M. J.; Heath, T. A. Phylogenetic Inference Using RevBayes. Current Protocols in Bioinformatics 2017, 57. [Google Scholar]
- Höhna, S.; Landis, M. J.; Heath, T. A.; Boussau, B.; Lartillot, N.; Moore, B. R.; Huelsenbeck, J. P.; Ronquist, F. RevBayes: Bayesian Phylogenetic Inference Using Graphical Models and an Interactive Model-Specification Language. Syst Biol 2016, 65, 726–736. [Google Scholar] [CrossRef]
- Huelsenbeck, J. P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Husnik, F.; McCutcheon, J. P. Functional horizontal gene transfer from bacteria to eukaryotes. Nature Reviews Microbiology 2018, 16, 67–79. [Google Scholar] [CrossRef]
- Huson, D. H. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Huson, D. H.; Kl#xF6pper, T.; Lockhart, P. J.; Steel, M. A. Reconstruction of Reticulate Networks from Gene Trees. Pp. 233–249 in S. Miyano, J. Mesirov, S. Kasif, S. Istrail, P. A. Pevzner, and M. Waterman, eds. Research in Computational Molecular Biology. Springer Berlin Heidelberg, Berlin, Heidelberg. 2005. [Google Scholar]
- Irwin, N. A. T.; Pittis, A. A.; Richards, T. A.; Keeling, P. J. Systematic evaluation of horizontal gene transfer between eukaryotes and viruses. Nat Microbiol 2021, 7, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.; Carruthers, M.; Eckmann, R.; Yohannes, E.; Adams, C. E.; Behrmann-Godel, J.; Elmer, K. R. Rapid niche expansion by selection on functional genomic variation after ecosystem recovery. Nat Ecol Evol 2018, 3, 77–86. [Google Scholar] [CrossRef]
- Jaramillo, V. D. A.; Sukno, S. A.; Thon, M. R. Identification of horizontally transferred genes in the genus Colletotrichum reveals a steady tempo of bacterial to fungal gene transfer. BMC Genomics 2015, 16, 2. [Google Scholar] [CrossRef]
- Jiao, X.; Flouri, T.; Yang, Z. Multispecies coalescent and its applications to infer species phylogenies and cross-species gene flow. National Science Review 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Kainer, D.; Lanfear, R. The Effects of Partitioning on Phylogenetic Inference. Molecular Biology and Evolution 2015, 32, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B. Q.; Wong, T. K. F.; Haeseler, A. Von; Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Kapli, P.; Yang, Z.; Telford, M. J. Phylogenetic tree building in the genomic age. Nat Rev Genet 2020, 21, 428–444. [Google Scholar] [CrossRef]
- King, N.; Rokas, A. Embracing Uncertainty in Reconstructing Early Animal Evolution. Current Biology 2017, 27, R1081–R1088. [Google Scholar] [CrossRef]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol 1989, 29, 170–179. [Google Scholar] [CrossRef]
- Kobert, K.; Salichos, L.; Rokas, A.; Stamatakis, A. Computing the Internode Certainty and Related Measures from Partial Gene Trees. Mol Biol Evol 2016, 33, 1606–1617. [Google Scholar] [CrossRef] [PubMed]
- Kocot, K. M.; Citarella, M. R.; Moroz, L. L.; Halanych, K. M. PhyloTreePruner: A Phylogenetic Tree-Based Approach for selection of Orthologous sequences for phylogenomics. Evol Bioinform Online 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Kominek, J.; Doering, D. T.; Opulente, D. A.; Shen, X.-X.; Zhou, X.; DeVirgilio, J.; Hulfachor, A. B.; Groenewald, M.; Mcgee, M. A.; Karlen, S. D.; Kurtzman, C. P.; Rokas, A.; Hittinger, C. T. Eukaryotic Acquisition of a Bacterial Operon. Cell 2019, 176, 1356–1366. [Google Scholar] [CrossRef] [PubMed]
- Koutsovoulos, G. D.; Noriot, S. Granjeon; Bailly-Bechet, M.; Danchin, E. G. J.; Rancurel, C. AvP: A software package for automatic phylogenetic detection of candidate horizontal gene transfers. PLoS Comput Biol 2022, 18, e1010686. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Meyer, W. K.; Partha, R.; Mao, W.; Clark, N. L.; Chikina, M. RERconverge: an R package for associating evolutionary rates with convergent traits. Bioinformatics 2019, 35, 4815–4817. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A. M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Kriventseva, E. V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F. A.; Zdobnov, E. M. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research 2019, 47, D807–D811. [Google Scholar] [CrossRef]
- Kück, P.; Longo, G. C. FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front Zool 2014, 11, 81. [Google Scholar] [CrossRef]
- Kumar, S. Embracing Green Computing in Molecular Phylogenetics. Molecular Biology and Evolution 2022, 39. [Google Scholar] [CrossRef]
- Lartillot, N. PhyloBayes: Bayesian Phylogenetics Using Site-heterogeneous Models. P. 1.5:1--1.5:16 in C. Scornavacca, F. Delsuc, and N. Galtier, eds. Phylogenetics in the Genomic Era. No commercial publisher | Authors open access book. 2020. [Google Scholar]
- Lartillot, N.; Brinkmann, H.; Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol 2007, 7, S4. [Google Scholar] [CrossRef]
- Lemoine, F.; Entfellner, J.-B. Domelevo; Wilkinson, E.; Correia, D.; Felipe, M. Dávila; Oliveira, T. De; Gascuel, O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452–456. [Google Scholar] [CrossRef]
- Lepage, T.; Bryant, D.; Philippe, H.; Lartillot, N. A General Comparison of Relaxed Molecular Clock Models. Molecular Biology and Evolution 2007, 24, 2669–2680. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C. J.; Roos, D. S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; David, K. T.; Shen, X.-X.; Steenwyk, J. L.; Halanych, K. M.; Rokas, A. Feature frequency profile-based phylogenies are inaccurate. Proc. Natl. Acad. Sci. U.S.A 2020, 117, 31580–31581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Steenwyk, J. L.; LaBella, A. L.; Harrison, M.-C.; Groenewald, M.; Zhou, X.; Shen, X.-X.; Zhao, T.; Hittinger, C. T.; Rokas, A. Contrasting modes of macro and microsynteny evolution in a eukaryotic subphylum. Current Biology 2022a, S0960982222016700. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Z.; Liu, C.; Shi, Z.; Pang, L.; Chen, C.; Chen, Y.; Pan, R.; Zhou, W.; Chen, X.; Rokas, A.; Huang, J.; Shen, X.-X. HGT is widespread in insects and contributes to male courtship in lepidopterans. Cell 2022b, 185, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Steenwyk, J. L.; Chang, Y.; Wang, Y.; James, T. Y.; Stajich, J. E.; Spatafora, J. W.; Groenewald, M.; Dunn, C. W.; Hittinger, C. T.; Shen, X.-X.; Rokas, A. A genome-scale phylogeny of the kingdom Fungi. Current Biology 2021, 31, 1653–1665. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Torre, A. R. De La; Sterck, L.; Cánovas, F. M.; Avila, C.; Merino, I.; Cabezas, J. A.; Cervera, M. T.; Ingvarsson, P. K.; Peer, Y. Van De. Single-Copy Genes as Molecular Markers for Phylogenomic Studies in Seed Plants. Genome Biology and Evolution 2017, 9, 1130–1147. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Patel, S.; Litvintseva, A. P.; Floyd, A.; Mitchell, T. G.; Heitman, J. Diploids in the Cryptococcus neoformans Serotype A Population Homozygous for the α Mating Type Originate via Unisexual Mating. PLoS Pathog 2009, 5, e1000283. [Google Scholar] [CrossRef]
- Liu, L.; Pearl, D. K.; Brumfield, R. T.; Edwards, S. V. Estimating species trees using multiple-allele DNA sequence data. Evolution 2008, 62, 2080–2091. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, L.; Kubatko, L.; Pearl, D. K.; Edwards, S. V. Coalescent methods for estimating phylogenetic trees. Molecular Phylogenetics and Evolution 2009a, 53, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, L.; Pearl, D. K.; Edwards, S. V. Estimating Species Phylogenies Using Coalescence Times among Sequences. Systematic Biology 2009b, 58, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Löytynoja, A.; Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proceedings of the National Academy of Sciences 2005, 102, 10557–10562. [Google Scholar] [CrossRef] [PubMed]
- Lutteropp, S.; Scornavacca, C.; Kozlov, A. M.; Morel, B.; Stamatakis, A. NetRAX: accurate and fast maximum likelihood phylogenetic network inference. Bioinformatics 2022, 38, 3725–3733. [Google Scholar] [CrossRef]
- Mallet, J. Hybridization as an invasion of the genome. Trends in Ecology & Evolution 2005, 20, 229–237. [Google Scholar]
- Mallet, J. Hybridization, ecological races and the nature of species: empirical evidence for the ease of speciation. Phil. Trans. R. Soc. B 2008, 363, 2971–2986. [Google Scholar] [CrossRef]
- Marcet-Houben, M.; Gabaldón, T. Beyond the Whole-Genome Duplication: Phylogenetic Evidence for an Ancient Interspecies Hybridization in the Baker’s Yeast Lineage. PLoS Biol 2015, 13, e1002220. [Google Scholar] [CrossRef]
- Martin, S. H.; Davey, J. W.; Jiggins, C. D. Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Molecular biology and evolution 2015, 32, 244–257. [Google Scholar] [CrossRef]
- Martín-Durán, J. M.; Ryan, J. F.; Vellutini, B. C.; Pang, K.; Hejnol, A. Increased taxon sampling reveals thousands of hidden orthologs in flatworms. Genome Res 2017, 27, 1263–1272. [Google Scholar] [CrossRef]
- Mateo-Estrada, V.; Graña-Miraglia, L.; López-Leal, G.; Castillo-Ramírez, S. Phylogenomics Reveals Clear Cases of Misclassification and Genus-Wide Phylogenetic Markers for Acinetobacter. Genome Biology and Evolution 2019, 11, 2531–2541. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C. G. P.; Mulhair, P. O.; Siu-Ting, K.; Creevey, C. J.; O’Connell, M. J. Improving Orthologous Signal and Model Fit in Datasets Addressing the Root of the Animal Phylogeny. Molecular Biology and Evolution 2023, 40. [Google Scholar] [CrossRef] [PubMed]
- Minge, M. A.; Shalchian-Tabrizi, K.; Tørresen, O. K.; Takishita, K.; Probert, I.; Inagaki, Y.; Klaveness, D.; Jakobsen, K. S. A phylogenetic mosaic plastid proteome and unusual plastid-targeting signals in the green-colored dinoflagellate Lepidodinium chlorophorum. BMC Evol Biol 2010, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Minh, B. Q.; Dang, C. C.; Vinh, L. S.; Lanfear, R. QMaker: Fast and Accurate Method to Estimate Empirical Models of Protein Evolution. Systematic Biology 2021, 70, 1046–1060. [Google Scholar] [CrossRef]
- Minh, B. Q.; Hahn, M. W.; Lanfear, R. New Methods to Calculate Concordance Factors for Phylogenomic Datasets. Molecular Biology and Evolution 2020a, 37, 2727–2733. [Google Scholar] [CrossRef]
- Minh, B. Q.; Schmidt, H. A.; Chernomor, O.; Schrempf, D.; Woodhams, M. D.; Haeseler, A. von; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Molecular Biology and Evolution 2020b, 37, 1530–1534. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R. S.; Donath, A.; Mayer, C.; Frandsen, P. B.; Ware, J.; Flouri, T.; Beutel, R. G.; Niehuis, O.; Petersen, M.; Izquierdo-Carrasco, F.; Wappler, T.; Rust, J.; Aberer, A. J.; Aspöck, U.; Aspöck, H.; Bartel, D.; Blanke, A.; Berger, S.; Böhm, A.; Buckley, T. R.; Calcott, B.; Chen, J.; Friedrich, F.; Fukui, M.; Fujita, M.; Greve, C.; Grobe, P.; Gu, S.; Huang, Y.; Jermiin, L. S.; Kawahara, A. Y.; Krogmann, L.; Kubiak, M.; Lanfear, R.; Letsch, H.; Li, Y.; Li, Z.; Li, J.; Lu, H.; Machida, R.; Mashimo, Y.; Kapli, P.; McKenna, D. D.; Meng, G.; Nakagaki, Y.; Navarrete-Heredia, J. L.; Ott, M.; Ou, Y.; Pass, G.; Podsiadlowski, L.; Pohl, H.; Reumont, B. M. Von; Schütte, K.; Sekiya, K.; Shimizu, S.; Slipinski, A.; Stamatakis, A.; Song, W.; Su, X.; Szucsich, N. U.; Tan, M.; Tan, X.; Tang, M.; Tang, J.; Timelthaler, G.; Tomizuka, S.; Trautwein, M.; Tong, X.; Uchifune, T.; Walzl, M. G.; Wiegmann, B. M.; Wilbrandt, J.; Wipfler, B.; Wong, T. K. F.; Wu, Q.; Wu, G.; Xie, Y.; Yang, S.; Yang, Q.; Yeates, D. K.; Yoshizawa, K.; Zhang, Q.; Zhang, R.; Zhang, W.; Zhang, Y.; Zhao, J.; Zhou, C.; Zhou, L.; Ziesmann, T.; Zou, S.; Li, Y.; Xu, X.; Zhang, Y.; Yang, H.; Wang, J.; Wang, J.; Kjer, K. M.; Zhou, X. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef]
- Mixão, V.; Gabaldón, T. Genomic evidence for a hybrid origin of the yeast opportunistic pathogen Candida albicans. BMC Biol 2020, 18, 48. [Google Scholar] [CrossRef]
- Mongiardino Koch, N. Phylogenomic Subsampling and the Search for Phylogenetically Reliable Loci. Molecular Biology and Evolution 2021, 38, 4025–4038. [Google Scholar] [CrossRef]
- Moran, B. M.; Payne, C.; Langdon, Q.; Powell, D. L.; Brandvain, Y.; Schumer, M. The genomic consequences of hybridization. eLife 2021, 10, e69016. [Google Scholar] [CrossRef]
- Morozov, A. A.; Galachyants, Y. P. Diatom genes originating from red and green algae: Implications for the secondary endosymbiosis models. Marine Genomics 2019, 45, 72–78. [Google Scholar] [CrossRef]
- Muñoz-Gómez, S. A.; Mejía-Franco, F. G.; Durnin, K.; Colp, M.; Grisdale, C. J.; Archibald, J. M.; Slamovits, C. H. The New Red Algal Subphylum Proteorhodophytina Comprises the Largest and Most Divergent Plastid Genomes Known. Current Biology 2017, 27, 1677–1684. [Google Scholar] [CrossRef]
- Neafsey, D. E.; Barker, B. M.; Sharpton, T. J.; Stajich, J. E.; Park, D. J.; Whiston, E.; Hung, C.-Y.; McMahan, C.; White, J.; Sykes, S.; Heiman, D.; Young, S.; Zeng, Q.; Abouelleil, A.; Aftuck, L.; Bessette, D.; Brown, A.; FitzGerald, M.; Lui, A.; Macdonald, J. P.; Priest, M.; Orbach, M. J.; Galgiani, J. N.; Kirkland, T. N.; Cole, G. T.; Birren, B. W.; Henn, M. R.; Taylor, J. W.; Rounsley, S. D. Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res 2010, 20, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Nelsen, M. P.; Moreau, C. S.; Boyce, C. Kevin; Ree, R. H. Macroecological diversification of ants is linked to angiosperm evolution. Evolution Letters 2023, 7, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, C. H.; Field, D. J.; Ksepka, D. T.; Barker, F. K.; Aleixo, A.; Andersen, M. J.; Alström, P.; Benz, B. W.; Braun, E. L.; Braun, M. J.; Bravo, G. A.; Brumfield, R. T.; Chesser, R. T.; Claramunt, S.; Cracraft, J.; Cuervo, A. M.; Derryberry, E. P.; Glenn, T. C.; Harvey, M. G.; Hosner, P. A.; Joseph, L.; Kimball, R. T.; Mack, A. L.; Miskelly, C. M.; Peterson, A. T.; Robbins, M. B.; Sheldon, F. H.; Silveira, L. F.; Smith, B. T.; White, N. D.; Moyle, R. G.; Faircloth, B. C. Earth history and the passerine superradiation. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 7916–7925. [Google Scholar] [CrossRef] [PubMed]
- One Thousand Plant Transcriptomes Initiative. 2019. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685.
- Opulente, D. A.; LaBella, A. L.; Harrison, M.-C.; Wolters, J. F.; Liu, C.; Li, Y.; Kominek, J.; Steenwyk, J. L.; Stoneman, H. R.; VanDenAvond, J.; Miller, C. R.; Langdon, Q. K.; Silva, M.; Gon#xE7alves, C.; Ubbelohde, E. J.; Li, Y.; Buh, K. V.; Jarzyna, M.; Haase, M. A. B.; Rosa, C. A.; #x10Cadež, N.; Libkind, D.; DeVirgilio, J. H.; Hulfachor, A. B.; Kurtzman, C. P.; Sampaio, J. P.; Gon#xE7alves, P.; Zhou, X.; Shen, X.-X.; Groenewald, M.; Rokas, A.; Hittinger, C. T. 2023. Genomic and ecological factors shaping specialism and generalism across an entire subphylum. Evolutionary Biology.
- Ortiz-Merino, R. A.; Kuanyshev, N.; Braun-Galleani, S.; Byrne, K. P.; Porro, D.; Branduardi, P.; Wolfe, K. H. Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. PLoS Biol 2017, 15, e2002128. [Google Scholar] [CrossRef]
- Osmanski, A. B.; Paulat, N. S.; Korstian, J.; Grimshaw, J. R.; Halsey, M.; Sullivan, K. A. M.; Moreno-Santillán, D. D.; Crookshanks, C.; Roberts, J.; Garcia, C.; Johnson, M. G.; Densmore, L. D.; Stevens, R. D.; Consortium†, Zoonomia; Rosen, J.; Storer, J. M.; Hubley, R.; Smit, A. F. A.; Dávalos, L. M.; Karlsson, E. K.; Lindblad-Toh, K.; Ray, D. A.; Andrews, G.; Armstrong, J. C.; Bianchi, M.; Birren, B. W.; Bredemeyer, K. R.; Breit, A. M.; Christmas, M. J.; Clawson, H.; Damas, J.; Palma, F. Di; Diekhans, M.; Dong, M. X.; Eizirik, E.; Fan, K.; Fanter, C.; Foley, N. M.; Forsberg-Nilsson, K.; Garcia, C. J.; Gatesy, J.; Gazal, S.; Genereux, D. P.; Goodman, L.; Grimshaw, J.; Halsey, M. K.; Harris, A. J.; Hickey, G.; Hiller, M.; Hindle, A. G.; Hubley, R. M.; Hughes, G. M.; Johnson, J.; Juan, D.; Kaplow, I. M.; Karlsson, E. K.; Keough, K. C.; Kirilenko, B.; Koepfli, K.-P.; Korstian, J. M.; Kowalczyk, A.; Kozyrev, S. V.; Lawler, A. J.; Lawless, C.; Lehmann, T.; Levesque, D. L.; Lewin, H. A.; Li, X.; Lind, A.; Lindblad-Toh, K.; Mackay-Smith, A.; Marinescu, V. D.; Marques-Bonet, T.; Mason, V. C.; Meadows, J. R. S.; Meyer, W. K.; Moore, J. E.; Moreira, L. R.; Moreno-Santillan, D. D.; Morrill, K. M.; Muntané, G.; Murphy, W. J.; Navarro, A.; Nweeia, M.; Ortmann, S.; Osmanski, A.; Paten, B.; Paulat, N. S.; Pfenning, A. R.; Phan, B. N.; Pollard, K. S.; Pratt, H. E.; Ray, D. A.; Reilly, S. K.; Rosen, J. R.; Ruf, I.; Ryan, L.; Ryder, O. A.; Sabeti, P. C.; Schäffer, D. E.; Serres, A.; Shapiro, B.; Smit, A. F. A.; Springer, M.; Srinivasan, C.; Steiner, C.; Storer, J. M.; Sullivan, K. A. M.; Sullivan, P. F.; Sundström, E.; Supple, M. A.; Swofford, R.; Talbot, J.-E.; Teeling, E.; Turner-Maier, J.; Valenzuela, A.; Wagner, F.; Wallerman, O.; Wang, C.; Wang, J.; Weng, Z.; Wilder, A. P.; Wirthlin, M. E.; Xue, J. R.; Zhang, X. Insights into mammalian TE diversity through the curation of 248 genome assemblies. Science 2023, 380, eabn1430. [Google Scholar] [CrossRef]
- O’Sullivan, O.; Suhre, K.; Abergel, C.; Higgins, D. G.; Notredame, C. 3DCoffee: combining protein sequences and structures within multiple sequence alignments. Journal of molecular biology 2004, 340, 385–395. [Google Scholar] [CrossRef]
- Ozkan, H.; Levy, A. A.; Feldman, M. Allopolyploidy-Induced Rapid Genome Evolution in the Wheat ( Aegilops – Triticum ) Group. Plant Cell 2001, 13, 1735–1747. [Google Scholar] [PubMed]
- Parey, E.; Louis, A.; Montfort, J.; Bouchez, O.; Roques, C.; Iampietro, C.; Lluch, J.; Castinel, A.; Donnadieu, C.; Desvignes, T.; Bucao, C. Floi; Jouanno, E.; Wen, M.; Mejri, S.; Dirks, R.; Jansen, H.; Henkel, C.; Chen, W.-J.; Zahm, M.; Cabau, C.; Klopp, C.; Thompson, A. W.; Robinson-Rechavi, M.; Braasch, I.; Lecointre, G.; Bobe, J.; Postlethwait, J. H.; Berthelot, C.; Crollius, H. R.; Guiguen, Y. Genome structures resolve the early diversification of teleost fishes. Science 2023, 379, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Parham, J. F.; Donoghue, P. C. J.; Bell, C. J.; Calway, T. D.; Head, J. J.; Holroyd, P. A.; Inoue, J. G.; Irmis, R. B.; Joyce, W. G.; Ksepka, D. T.; Patané, J. S. L.; Smith, N. D.; Tarver, J. E.; Tuinen, M. Van; Yang, Z.; Angielczyk, K. D.; Greenwood, J. M.; Hipsley, C. A.; Jacobs, L.; Makovicky, P. J.; Müller, J.; Smith, K. T.; Theodor, J. M.; Warnock, R. C. M.; Benton, M. J. Best Practices for Justifying Fossil Calibrations. Systematic Biology 2012, 61, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Parks, D. H.; Chuvochina, M.; Waite, D. W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Pease, J. B.; Hahn, M. W. Detection and Polarization of Introgression in a Five-Taxon Phylogeny. Systematic Biology 2015, 64, 651–662. [Google Scholar] [CrossRef]
- Peterson, B. K.; Weber, J. N.; Kay, E. H.; Fisher, H. S.; Hoekstra, H. E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Phillips, M. A.; Steenwyk, J. L.; Shen, X.-X.; Rokas, A. Examination of Gene Loss in the DNA Mismatch Repair Pathway and Its Mutational Consequences in a Fungal Phylum. Genome Biology and Evolution 2021, 13. [Google Scholar] [CrossRef]
- Phillips, M. J.; Penny, D. The root of the mammalian tree inferred from whole mitochondrial genomes. Molecular Phylogenetics and Evolution 2003, 28, 171–185. [Google Scholar] [CrossRef]
- Pipes, L.; Wang, H.; Huelsenbeck, J. P.; Nielsen, R. Assessing Uncertainty in the Rooting of the SARS-CoV-2 Phylogeny. Molecular Biology and Evolution 2021, 38, 1537–1543. [Google Scholar] [CrossRef]
- Pollock, D. D.; Zwickl, D. J.; McGuire, J. A.; Hillis, D. M. Increased Taxon Sampling Is Advantageous for Phylogenetic Inference. Systematic Biology 2002, 51, 664–671. [Google Scholar] [CrossRef]
- Ponce-Toledo, R. I.; Moreira, D.; López-García, P.; Deschamps, P. Secondary Plastids of Euglenids and Chlorarachniophytes Function with a Mix of Genes of Red and Green Algal Ancestry. Molecular Biology and Evolution 2018, 35, 2198–2204. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Liu, W.; Zhang, Y.; Zhang, Y.; Li, Q. Q. Genetic admixture accelerates invasion via provisioning rapid adaptive evolution. Mol Ecol 2019, 28, 4012–4027. [Google Scholar] [CrossRef]
- Racimo, F.; Sankararaman, S.; Nielsen, R.; Huerta-Sánchez, E. Evidence for archaic adaptive introgression in humans. Nature Reviews Genetics 2015, 16, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Raghava, G.; Searle, S. M.; Audley, P. C.; Barber, J. D.; Barton, G. J. OXBench: A benchmark for evaluation of protein multiple sequence alignment accuracy. BMC Bioinformatics 2003, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Ranwez, V.; Douzery, E. J. P.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the Alignment of Coding Sequences Accounting for Frameshifts and Stop Codons. Molecular Biology and Evolution 2018, 35, 2582–2584. [Google Scholar] [CrossRef]
- Redmond, A. K.; Casey, D.; Gundappa, M. K.; Macqueen, D. J.; McLysaght, A. Independent rediploidization masks shared whole genome duplication in the sturgeon-paddlefish ancestor. Nat Commun 2023, 14, 2879. [Google Scholar] [CrossRef]
- Reich, D.; Patterson, N.; Campbell, D.; Tandon, A.; Mazieres, S.; Ray, N.; Parra, M. V.; Rojas, W.; Duque, C.; Mesa, N. Reconstructing native American population history. Nature 2012, 488, 370–374. [Google Scholar] [CrossRef]
- Rieseberg, L. H.; Kim, S.-C.; Randell, R. A.; Whitney, K. D.; Gross, B. L.; Lexer, C.; Clay, K. Hybridization and the colonization of novel habitats by annual sunflowers. Genetica 2007, 129, 149–165. [Google Scholar] [CrossRef]
- Rodríguez-Ezpeleta, N.; Brinkmann, H.; Roure, B.; Lartillot, N.; Lang, B. F.; Philippe, H. Detecting and overcoming systematic errors in genome-scale phylogenies. Systematic biology 2007, 56, 389–399. [Google Scholar] [CrossRef]
- Rokas, A.; Holland, P. W. H. Rare genomic changes as a tool for phylogenetics. Trends in Ecology & Evolution 2000, 15, 454–459. [Google Scholar]
- Rokas, A.; Williams, B. L.; King, N.; Carroll, S. B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Mark, P. Van Der; Ayres, D. L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M. A.; Huelsenbeck, J. P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Systematic Biology 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Ropars, J.; Vega, R. C. Rodríguez de la; López-Villavicencio, M.; Gouzy, J.; Sallet, E.; Dumas, É; Lacoste, S.; Debuchy, R.; Dupont, J.; Branca, A.; Giraud, T. Adaptive Horizontal Gene Transfers between Multiple Cheese-Associated Fungi. Current Biology 2015, 25, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- S. Meseguer, A.; Condamine, F. L. Ancient tropical extinctions at high latitudes contributed to the latitudinal diversity gradient*. Evolution 2020, 74, 1966–1987. [Google Scholar] [CrossRef]
- Sabrina Pankey, M.; Plachetzki, D. C.; Macartney, K. J.; Gastaldi, M.; Slattery, M.; Gochfeld, D. J.; Lesser, M. P. Cophylogeny and convergence shape holobiont evolution in sponge–microbe symbioses. Nat Ecol Evol 2022, 6, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Salichos, L.; Rokas, A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 2013, 497, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Salichos, L.; Stamatakis, A.; Rokas, A. Novel Information Theory-Based Measures for Quantifying Incongruence among Phylogenetic Trees. Molecular Biology and Evolution 2014, 31, 1261–1271. [Google Scholar] [CrossRef]
- Sanderson, M. J. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef]
- Sankararaman, S.; Mallick, S.; Patterson, N.; Reich, D. The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Current Biology 2016, 26, 1241–1247. [Google Scholar] [CrossRef]
- Sayyari, E.; Mirarab, S. Testing for Polytomies in Phylogenetic Species Trees Using Quartet Frequencies. Genes 2018, 9, 132. [Google Scholar] [CrossRef]
- Scannell, D. R.; Byrne, K. P.; Gordon, J. L.; Wong, S.; Wolfe, K. H. Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature 2006, 440, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Schenk, J. J. Consequences of Secondary Calibrations on Divergence Time Estimates. PLoS ONE 2016, 11, e0148228. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Fast Identification and Removal of Sequence Contamination from Genomic and Metagenomic Datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef] [PubMed]
- Schönknecht, G.; Chen, W.-H.; Ternes, C. M.; Barbier, G. G.; Shrestha, R. P.; Stanke, M.; Bräutigam, A.; Baker, B. J.; Banfield, J. F.; Garavito, R. M.; Carr, K.; Wilkerson, C.; Rensing, S. A.; Gagneul, D.; Dickenson, N. E.; Oesterhelt, C.; Lercher, M. J.; Weber, A. P. M. Gene Transfer from Bacteria and Archaea Facilitated Evolution of an Extremophilic Eukaryote. Science 2013, 339, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Marcussen, T.; Meseguer, A. S.; Fjellheim, S. The grass subfamily Pooideae: Cretaceous–Palaeocene origin and climate-driven Cenozoic diversification. Global Ecol Biogeogr 2019, geb.12923. [Google Scholar] [CrossRef]
- Schultz, D. T.; Haddock, S. H. D.; Bredeson, J. V.; Green, R. E.; Simakov, O.; Rokhsar, D. S. Ancient gene linkages support ctenophores as sister to other animals. Nature 2023. [Google Scholar] [CrossRef]
- Session, A. M.; Rokhsar, D. S. Transposon signatures of allopolyploid genome evolution. Nat Commun 2023, 14, 3180. [Google Scholar] [CrossRef]
- Session, A. M.; Uno, Y.; Kwon, T.; Chapman, J. A.; Toyoda, A.; Takahashi, S.; Fukui, A.; Hikosaka, A.; Suzuki, A.; Kondo, M.; Heeringen, S. J. Van; Quigley, I.; Heinz, S.; Ogino, H.; Ochi, H.; Hellsten, U.; Lyons, J. B.; Simakov, O.; Putnam, N.; Stites, J.; Kuroki, Y.; Tanaka, T.; Michiue, T.; Watanabe, M.; Bogdanovic, O.; Lister, R.; Georgiou, G.; Paranjpe, S. S.; Kruijsbergen, I. Van; Shu, S.; Carlson, J.; Kinoshita, T.; Ohta, Y.; Mawaribuchi, S.; Jenkins, J.; Grimwood, J.; Schmutz, J.; Mitros, T.; Mozaffari, S. V.; Suzuki, Y.; Haramoto, Y.; Yamamoto, T. S.; Takagi, C.; Heald, R.; Miller, K.; Haudenschild, C.; Kitzman, J.; Nakayama, T.; Izutsu, Y.; Robert, J.; Fortriede, J.; Burns, K.; Lotay, V.; Karimi, K.; Yasuoka, Y.; Dichmann, D. S.; Flajnik, M. F.; Houston, D. W.; Shendure, J.; DuPasquier, L.; Vize, P. D.; Zorn, A. M.; Ito, M.; Marcotte, E. M.; Wallingford, J. B.; Ito, Y.; Asashima, M.; Ueno, N.; Matsuda, Y.; Veenstra, G. J. C.; Fujiyama, A.; Harland, R. M.; Taira, M.; Rokhsar, D. S. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 2016, 538, 336–343. [Google Scholar] [CrossRef]
- Sharma, S.; Kumar, S. Fast and accurate bootstrap confidence limits on genome-scale phylogenies using little bootstraps. Nat Comput Sci 2021, 1, 573–577. [Google Scholar] [CrossRef]
- Shaul, S.; Graur, D. Playing chicken ( Gallus gallus ): methodological inconsistencies of molecular divergence date estimates due to secondary calibration points. Gene 2002, 300, 59–61. [Google Scholar] [CrossRef]
- Shen, X.-X.; Hittinger, C. T.; Rokas, A. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat Ecol Evol 2017, 1, 0126. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Opulente, D. A.; Kominek, J.; Zhou, X.; Steenwyk, J. L.; Buh, K. V.; Haase, M. A. B.; Wisecaver, J. H.; Wang, M.; Doering, D. T.; Boudouris, J. T.; Schneider, R. M.; Langdon, Q. K.; Ohkuma, M.; Endoh, R.; Takashima, M.; Manabe, R.; Čadež, N.; Libkind, D.; Rosa, C. A.; DeVirgilio, J.; Hulfachor, A. B.; Groenewald, M.; Kurtzman, C. P.; Hittinger, C. T.; Rokas, A. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 2018, 175, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Salichos, L.; Rokas, A. A Genome-Scale Investigation of How Sequence, Function, and Tree-Based Gene Properties Influence Phylogenetic Inference. Genome Biol Evol 2016, 8, 2565–2580. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Steenwyk, J. L.; LaBella, A. L.; Opulente, D. A.; Zhou, X.; Kominek, J.; Li, Y.; Groenewald, M.; Hittinger, C. T.; Rokas, A. Genome-scale phylogeny and contrasting modes of genome evolution in the fungal phylum Ascomycota. Sci. Adv 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Steenwyk, J. L.; Rokas, A. Dissecting Incongruence between Concatenation- and Quartet-Based Approaches in Phylogenomic Data. Systematic Biology 2021, 70, 997–1014. [Google Scholar] [CrossRef]
- Shimodaira, H.; Hasegawa, M. Multiple Comparisons of Log-Likelihoods with Applications to Phylogenetic Inference. Molecular Biology and Evolution 1999, 16, 1114–1116. [Google Scholar] [CrossRef]
- Si Quang, L.; Gascuel, O.; Lartillot, N. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics 2008, 24, 2317–2323. [Google Scholar] [CrossRef]
- Sibbald, S. J.; Archibald, J. M. Genomic Insights into Plastid Evolution. Genome Biology and Evolution 2020, 12, 978–990. [Google Scholar] [CrossRef]
- Sibbald, S. J.; Eme, L.; Archibald, J. M.; Roger, A. J. Lateral Gene Transfer Mechanisms and Pan-genomes in Eukaryotes. Trends in Parasitology 2020, 36, 927–941. [Google Scholar] [CrossRef]
- Sierra-Patev, S.; Min, B.; Naranjo-Ortiz, M.; Looney, B.; Konkel, Z.; Slot, J. C.; Sakamoto, Y.; Steenwyk, J. L.; Rokas, A.; Carro, J.; Camarero, S.; Ferreira, P.; Molpeceres, G.; Ruiz-Dueñas, F. J.; Serrano, A.; Henrissat, B.; Drula, E.; Hughes, K. W.; Mata, J. L.; Ishikawa, N. K.; Vargas-Isla, R.; Ushijima, S.; Smith, C. A.; Donoghue, J.; Ahrendt, S.; Andreopoulos, W.; He, G.; LaButti, K.; Lipzen, A.; Ng, V.; Riley, R.; Sandor, L.; Barry, K.; Martínez, A. T.; Xiao, Y.; Gibbons, J. G.; Terashima, K.; Grigoriev, I. V.; Hibbett, D. A global phylogenomic analysis of the shiitake genus Lentinula. Proc. Natl. Acad. Sci. U.S.A 2023, 120, e2214076120. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D. G. QuanTest2: benchmarking multiple sequence alignments using secondary structure prediction. Bioinformatics 2020, 36, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Simion, P.; Philippe, H.; Baurain, D.; Jager, M.; Richter, D. J.; Franco, A. Di; Roure, B.; Satoh, N.; Quéinnec, É; Ereskovsky, A.; Lapébie, P.; Corre, E.; Delsuc, F.; King, N.; Wörheide, G.; Manuel, M. A Large and Consistent Phylogenomic Dataset Supports Sponges as the Sister Group to All Other Animals. Current Biology 2017, 27, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Simonti, C. N.; Vernot, B.; Bastarache, L.; Bottinger, E.; Carrell, D. S.; Chisholm, R. L.; Crosslin, D. R.; Hebbring, S. J.; Jarvik, G. P.; Kullo, I. J.; Li, R.; Pathak, J.; Ritchie, M. D.; Roden, D. M.; Verma, S. S.; Tromp, G.; Prato, J. D.; Bush, W. S.; Akey, J. M.; Denny, J. C.; Capra, J. A. The phenotypic legacy of admixture between modern humans and Neandertals. Science 2016, 351, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Smith, M. L.; Vanderpool, D.; Hahn, M. W. Using all Gene Families Vastly Expands Data Available for Phylogenomic Inference. Molecular Biology and Evolution 2022, 39. [Google Scholar] [CrossRef]
- Smith, S. A.; O’Meara, B. C. treePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 2012, 28, 2689–2690. [Google Scholar] [CrossRef]
- Sousa, F.; Neiva, J.; Martins, N.; Jacinto, R.; Anderson, L.; Raimondi, P. T.; Serrão, E. A.; Pearson, G. A. Increased evolutionary rates and conserved transcriptional response following allopolyploidization in brown algae: GENOME EVOLUTION IN ALLOPOLYPLOID ALGAE. Evolution 2019, 73, 59–72. [Google Scholar] [CrossRef]
- Stadler, T.; Pybus, O. G.; Stumpf, M. P. H. Phylodynamics for cell biologists. Science 2021, 371, eaah6266. [Google Scholar] [CrossRef]
- Stadler, T.; Yang, Z. Dating Phylogenies with Sequentially Sampled Tips. Systematic Biology 2013, 62, 674–688. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Systematic Biology 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Steenwyk, J.; King, N. From Genes to Genomes: Opportunities and Challenges for Synteny-based Phylogenies. Preprints; 2023. [Google Scholar] [CrossRef]
- Steenwyk, J. L.; Balamurugan, C.; Raja, H. A.; Gon#xE7alves, C.; Li, N.; Martin, F.; Berman, J.; Oberlies, N. H.; Gibbons, J. G.; Goldman, G. H.; Geiser, D. M.; Hibbett, D. S.; Rokas, A. Phylogenomics reveals extensive misidentification of fungal strains from the genus Aspergillus. Evolutionary Biology 2022a. [Google Scholar]
- Steenwyk, J. L.; Buida, T. J.; Gonçalves, C.; Goltz, D. C.; Morales, G.; Mead, M. E.; LaBella, A. L.; Chavez, C. M.; Schmitz, J. E.; Hadjifrangiskou, M.; Li, Y.; Rokas, A. BioKIT: a versatile toolkit for processing and analyzing diverse types of sequence data. Genetics 2022b, 221. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J. L.; Buida, T. J.; Labella, A. L.; Li, Y.; Shen, X.-X.; Rokas, A. PhyKIT: a broadly applicable UNIX shell toolkit for processing and analyzing phylogenomic data. Bioinformatics 2021, 37, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J. L.; Buida, T. J.; Li, Y.; Shen, X.-X.; Rokas, A. ClipKIT: A multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biol 2020a, 18, e3001007. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J. L.; Goltz, D. C.; Buida, T. J.; Li, Y.; Shen, X.-X.; Rokas, A. OrthoSNAP: A tree splitting and pruning algorithm for retrieving single-copy orthologs from gene family trees. PLoS Biol 2022c, 20, e3001827. [Google Scholar] [CrossRef]
- Steenwyk, J. L.; Knowles, S. L.; Bastos, R.; Balamurugan, C.; Rinker, D.; Mead, M. E.; Roberts, C. D.; Raja, H. A.; Li, Y.; Colabardini, A. C.; Castro, P. A.; Reis, T. F.; Canovas, D.; Sanchez, R. L.; Lagrou, K.; Torrado, E.; Rodrigues, F.; Oberlies, N. H.; Zhou, X.; Goldman, G.; Rokas, A. Evolutionary origin, population diversity, and diagnostics for a cryptic hybrid pathogen. Evolutionary Biology 2023a. [Google Scholar]
- Steenwyk, J. L.; Li, Y.; Zhou, X.; Shen, X.-X.; Rokas, A. Incongruence in the phylogenomics era. Nature Reviews Genetics 2023b. [Google Scholar] [CrossRef]
- Steenwyk, J. L.; Lind, A. L.; Ries, L. N. A.; Reis, T. F. dos; Silva, L. P.; Almeida, F.; Bastos, R. W.; Silva, T. F. de C. Fraga da; Bonato, V. L. D.; Pessoni, A. M.; Rodrigues, F.; Raja, H. A.; Knowles, S. L.; Oberlies, N. H.; Lagrou, K.; Goldman, G. H.; Rokas, A. Pathogenic Allodiploid Hybrids of Aspergillus Fungi. Current Biology 2020b, 30, 2495–2507. [Google Scholar] [CrossRef]
- Steenwyk, J. L.; Opulente, D. A.; Kominek, J.; Shen, X.-X.; Zhou, X.; Labella, A. L.; Bradley, N. P.; Eichman, B. F.; Čadež, N.; Libkind, D.; DeVirgilio, J.; Hulfachor, A. B.; Kurtzman, C. P.; Hittinger, C. T.; Rokas, A. Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. PLoS Biol 2019a, 17, e3000255. [Google Scholar] [CrossRef]
- Steenwyk, J. L.; Phillips, M. A.; Yang, F.; Date, S. S.; Graham, T. R.; Berman, J.; Hittinger, C. T.; Rokas, A. An orthologous gene coevolution network provides insight into eukaryotic cellular and genomic structure and function. Sci. Adv 2022d, 8, eabn0105. [Google Scholar] [CrossRef]
- Steenwyk, J. L.; Rokas, A. orthofisher: a broadly applicable tool for automated gene identification and retrieval. G3 GenesGenomesGenetics 2021, 11, jkab250. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J. L.; Rokas, A. The dawn of relaxed phylogenetics. PLoS Biol 2023, 21, e3001998. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J. L.; Shen, X.-X.; Lind, A. L.; Goldman, G. H.; Rokas, A. A Robust Phylogenomic Time Tree for Biotechnologically and Medically Important Fungi in the Genera Aspergillus and Penicillium. mBio 2019b, 10, e00925–19. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nat Commun 2018, 9, 2542. [Google Scholar] [CrossRef]
- Stiller, J. W.; Schreiber, J.; Yue, J.; Guo, H.; Ding, Q.; Huang, J. The evolution of photosynthesis in chromist algae through serial endosymbioses. Nat Commun 2014, 5, 5764. [Google Scholar] [CrossRef] [PubMed]
- Strassert, J. F. H.; Irisarri, I.; Williams, T. A.; Burki, F. A molecular timescale for eukaryote evolution with implications for the origin of red algal-derived plastids. Nat Commun 2021, 12, 1879. [Google Scholar] [CrossRef]
- Stukenbrock, E. H. The Role of Hybridization in the Evolution and Emergence of New Fungal Plant Pathogens. Phytopathology® 2016, 106, 104–112. [Google Scholar] [CrossRef]
- Suvorov, A.; Kim, B. Y.; Wang, J.; Armstrong, E. E.; Peede, D.; D’Agostino, E. R. R.; Price, D. K.; Waddell, P. J.; Lang, M.; Courtier-Orgogozo, V.; David, J. R.; Petrov, D.; Matute, D. R.; Schrider, D. R.; Comeault, A. A. Widespread introgression across a phylogeny of 155 Drosophila genomes. Current Biology 2022, 32, 111–123. [Google Scholar] [CrossRef]
- Tahon, G.; Geesink, P.; Ettema, T. J. G. Expanding Archaeal Diversity and Phylogeny: Past, Present, and Future. Annu. Rev. Microbiol 2021, 75, 359–381. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Systematic Biology 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Tan, G.; Muffato, M.; Ledergerber, C.; Herrero, J.; Goldman, N.; Gil, M.; Dessimoz, C. Current Methods for Automated Filtering of Multiple Sequence Alignments Frequently Worsen Single-Gene Phylogenetic Inference. Syst Biol 2015, 64, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Tamura, K.; Kumar, S. Efficient Methods for Dating Evolutionary Divergences. Pp. 197–219 in S. Y. W. Ho, ed. The Molecular Evolutionary Clock. Springer International Publishing, Cham. 2020. [Google Scholar]
- Thompson, J. D.; Plewniak, F.; Poch, O. BAliBASE: a benchmark alignment database for the evaluation of multiple alignment programs. Bioinformatics (Oxford, England) 1999, 15, 87–88. [Google Scholar] [CrossRef]
- Tiley, G. P.; Flouri, T.; Jiao, X.; Poelstra, J. W.; Xu, B.; Zhu, T.; Rannala, B.; Yoder, A. D.; Yang, Z. Estimation of species divergence times in presence of cross-species gene flow. Systematic Biology 2023, 72, 820–836. [Google Scholar] [CrossRef] [PubMed]
- Tiley, G. P.; Poelstra, J. W.; Reis, M. Dos; Yang, Z.; Yoder, A. D. Molecular Clocks without Rocks: New Solutions for Old Problems. Trends in Genetics 2020, 36, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Upham, N. S.; Esselstyn, J. A.; Jetz, W. Inferring the mammal tree: Species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLoS Biol 2019, 17, e3000494. [Google Scholar] [CrossRef] [PubMed]
- Upham, N. S.; Esselstyn, J. A.; Jetz, W. Molecules and fossils tell distinct yet complementary stories of mammal diversification. Current Biology 2021, 31, 4195–4206. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D. J.; Meier, R. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Van Dongen, S. Graph Clustering Via a Discrete Uncoupling Process. SIAM J. Matrix Anal. & Appl 2008, 30, 121–141. [Google Scholar]
- Van Etten, J.; Bhattacharya, D. Horizontal Gene Transfer in Eukaryotes: Not if, but How Much? Trends in Genetics 2020, 36, 915–925. [Google Scholar] [CrossRef]
- Volz, E. M.; Koelle, K.; Bedford, T. Viral Phylodynamics. PLoS Comput Biol 2013, 9, e1002947. [Google Scholar] [CrossRef]
- Wang, H.-C.; Minh, B. Q.; Susko, E.; Roger, A. J. Modeling Site Heterogeneity with Posterior Mean Site Frequency Profiles Accelerates Accurate Phylogenomic Estimation. Systematic Biology 2018a, 67, 216–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-S.; Murray, G. G. R.; Mann, D.; Groves, P.; Vershinina, A. O.; Supple, M. A.; Kapp, J. D.; Corbett-Detig, R.; Crump, S. E.; Stirling, I.; Laidre, K. L.; Kunz, M.; Dalén, L.; Green, R. E.; Shapiro, B. A polar bear paleogenome reveals extensive ancient gene flow from polar bears into brown bears. Nat Ecol Evol 2022, 6, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, H.; Cai, Y. A benchmark study of sequence alignment methods for protein clustering. BMC Bioinformatics 2018b, 19, 529. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R. M.; Seppey, M.; Simão, F. A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E. V.; Zdobnov, E. M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Molecular Biology and Evolution 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Weisman, C. M.; Murray, A. W.; Eddy, S. R. Many, but not all, lineage-specific genes can be explained by homology detection failure. PLoS Biol 2020, 18, e3000862. [Google Scholar] [CrossRef]
- Wickett, N. J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M. S.; Burleigh, J. G.; Gitzendanner, M. A.; Ruhfel, B. R.; Wafula, E.; Der, J. P.; Graham, S. W.; Mathews, S.; Melkonian, M.; Soltis, D. E.; Soltis, P. S.; Miles, N. W.; Rothfels, C. J.; Pokorny, L.; Shaw, A. J.; DeGironimo, L.; Stevenson, D. W.; Surek, B.; Villarreal, J. C.; Roure, B.; Philippe, H.; dePamphilis, C. W.; Chen, T.; Deyholos, M. K.; Baucom, R. S.; Kutchan, T. M.; Augustin, M. M.; Wang, J.; Zhang, Y.; Tian, Z.; Yan, Z.; Wu, X.; Sun, X.; Wong, G. K.-S.; Leebens-Mack, J. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. U.S.A. 2014, 111. [Google Scholar] [CrossRef]
- Wiens, J. J.; Tiu, J. Highly Incomplete Taxa Can Rescue Phylogenetic Analyses from the Negative Impacts of Limited Taxon Sampling. PLoS ONE 2012, 7, e42925. [Google Scholar] [CrossRef]
- Williams, T. A.; Cox, C. J.; Foster, P. G.; Szöllősi, G. J.; Embley, T. M. Phylogenomics provides robust support for a two-domains tree of life. Nat Ecol Evol 2019, 4, 138–147. [Google Scholar] [CrossRef]
- Willson, J.; Roddur, M. S.; Liu, B.; Zaharias, P.; Warnow, T. DISCO: Species Tree Inference using Multicopy Gene Family Tree Decomposition. Systematic Biology 2022, 71, 610–629. [Google Scholar] [CrossRef]
- Worobey, M.; Watts, T. D.; McKay, R. A.; Suchard, M. A.; Granade, T.; Teuwen, D. E.; Koblin, B. A.; Heneine, W.; Lemey, P.; Jaffe, H. W. 1970s and ‘Patient 0’ HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 2016, 539, 98–101. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Bayesian species delimitation using multilocus sequence data. Proceedings of the National Academy of Sciences 2010, 107, 9264–9269. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Barnett, R. M.; Nakhleh, L. Parsimonious Inference of Hybridization in the Presence of Incomplete Lineage Sorting. Systematic Biology 2013, 62, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Lu, H.; Li, F.; Nielsen, J.; Kerkhoven, E. J. HGTphyloDetect: facilitating the identification and phylogenetic analysis of horizontal gene transfer. Briefings in Bioinformatics 2023, 24, bbad035. [Google Scholar] [CrossRef]
- Yue, J.; Hu, X.; Sun, H.; Yang, Y.; Huang, J. Widespread impact of horizontal gene transfer on plant colonization of land. Nat Commun 2012, 3, 1152. [Google Scholar] [CrossRef] [PubMed]
- Zanewich, K. P.; Pearce, D. W.; Rood, S. B. Heterosis in poplar involves phenotypic stability: cottonwood hybrids outperform their parental species at suboptimal temperatures. Tree Physiology 2018, 38, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Zeberg, H.; Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020, 587, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 2018, 19, 153. [Google Scholar] [CrossRef]
- Zhang, C.; Scornavacca, C.; Molloy, E. K.; Mirarab, S. ASTRAL-Pro: Quartet-Based Species-Tree Inference despite Paralogy. Molecular Biology and Evolution 2020, 37, 3292–3307. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Y.; Braun, E. L.; Mirarab, S. TAPER: Pinpointing errors in multiple sequence alignments despite varying rates of evolution. Methods Ecol Evol 2021, 12, 2145–2158. [Google Scholar] [CrossRef]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D. M.; Lee, C.; Storz, J. F.; Antunes, A.; Greenwold, M. J.; Meredith, R. W.; Ödeen, A.; Cui, J.; Zhou, Q.; Xu, L.; Pan, H.; Wang, Z.; Jin, L.; Zhang, P.; Hu, H.; Yang, W.; Hu, J.; Xiao, J.; Yang, Z.; Liu, Y.; Xie, Q.; Yu, H.; Lian, J.; Wen, P.; Zhang, F.; Li, H.; Zeng, Y.; Xiong, Z.; Liu, S.; Zhou, L.; Huang, Z.; An, N.; Wang, J.; Zheng, Q.; Xiong, Y.; Wang, G.; Wang, B.; Wang, J.; Fan, Y.; Fonseca, R. R. Da; Alfaro-Núñez, A.; Schubert, M.; Orlando, L.; Mourier, T.; Howard, J. T.; Ganapathy, G.; Pfenning, A.; Whitney, O.; Rivas, M. V.; Hara, E.; Smith, J.; Farré, M.; Narayan, J.; Slavov, G.; Romanov, M. N.; Borges, R.; Machado, J. P.; Khan, I.; Springer, M. S.; Gatesy, J.; Hoffmann, F. G.; Opazo, J. C.; Håstad, O.; Sawyer, R. H.; Kim, H.; Kim, K.-W.; Kim, H. J.; Cho, S.; Li, N.; Huang, Y.; Bruford, M. W.; Zhan, X.; Dixon, A.; Bertelsen, M. F.; Derryberry, E.; Warren, W.; Wilson, R. K.; Li, S.; Ray, D. A.; Green, R. E.; O’Brien, S. J.; Griffin, D.; Johnson, W. E.; Haussler, D.; Ryder, O. A.; Willerslev, E.; Graves, G. R.; Alström, P.; Fjeldså, J.; Mindell, D. P.; Edwards, S. V.; Braun, E. L.; Rahbek, C.; Burt, D. W.; Houde, P.; Zhang, Y.; Yang, H.; Wang, J.; Consortium, Avian Genome; Jarvis, E. D.; Gilbert, M. T. P.; Wang, J.; Ye, C.; Liang, S.; Yan, Z.; Zepeda, M. L.; Campos, P. F.; Velazquez, A. M. V.; Samaniego, J. A.; Avila-Arcos, M.; Martin, M. D.; Barnett, R.; Ribeiro, A. M.; Mello, C. V.; Lovell, P. V.; Almeida, D.; Maldonado, E.; Pereira, J.; Sunagar, K.; Philip, S.; Dominguez-Bello, M. G.; Bunce, M.; Lambert, D.; Brumfield, R. T.; Sheldon, F. H.; Holmes, E. C.; Gardner, P. P.; Steeves, T. E.; Stadler, P. F.; Burge, S. W.; Lyons, E.; Smith, J.; McCarthy, F.; Pitel, F.; Rhoads, D.; Froman, D. P. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014a, 346, 1311–1320. [Google Scholar] [CrossRef]
- Zhang, R.; Ou, H.-Y.; Gao, F.; Luo, H. Identification of Horizontally-transferred Genomic Islands and Genome Segmentation Points by Using the GC Profile Method. CG 2014b, 15, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Janke, A. Gene flow analysis method, the D-statistic, is robust in a wide parameter space. BMC bioinformatics 2018, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lutteropp, S.; Czech, L.; Stamatakis, A.; Looz, M. V.; Rokas, A. Quartet-Based Computations of Internode Certainty Provide Robust Measures of Phylogenetic Incongruence. Systematic Biology 2020, 69, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Rokas, A. Prevention, diagnosis and treatment of high-throughput sequencing data pathologies. Mol Ecol 2014, 23, 1679–1700. [Google Scholar] [CrossRef]
- Zhou, X.; Shen, X.-X.; Hittinger, C. T.; Rokas, A. Evaluating Fast Maximum Likelihood-Based Phylogenetic Programs Using Empirical Phylogenomic Data Sets. Molecular Biology and Evolution 2018, 35, 486–503. [Google Scholar] [CrossRef]
- Zhu, Q.; Kosoy, M.; Dittmar, K. HGTector: an automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genomics 2014, 15, 717. [Google Scholar] [CrossRef]
- Zhu, Q.; Mai, U.; Pfeiffer, W.; Janssen, S.; Asnicar, F.; Sanders, J. G.; Belda-Ferre, P.; Al-Ghalith, G. A.; Kopylova, E.; McDonald, D.; Kosciolek, T.; Yin, J. B.; Huang, S.; Salam, N.; Jiao, J.-Y.; Wu, Z.; Xu, Z. Z.; Cantrell, K.; Yang, Y.; Sayyari, E.; Rabiee, M.; Morton, J. T.; Podell, S.; Knights, D.; Li, W.-J.; Huttenhower, C.; Segata, N.; Smarr, L.; Mirarab, S.; Knight, R. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat Commun 2019, 10, 5477. [Google Scholar] [CrossRef]
Figure 1.
A workflow for phylogenomic inference. (A) The first step in a phylogenomic workflow is data acquisition and preparation. This often entails identifying gene boundaries in genome assemblies or assembling transcripts in transcriptomes. (B) The next step is to identify orthologs using (left) de novo approaches—for example, all-by-all sequence similarity calculations followed by graph-based clustering—or (right) from predetermined sets of orthologs. (C) Orthologous groups of genes suitable for phylogenomics (i.e., one-to-one orthologs and SNAP-OGs) are subsequently aligned and trimmed. (D) The resulting multiple sequence alignments can be (left) concatenated into a supermatrix or (right) collections of single-locus phylogenies can be used in a coalescence-based approach. (E) Support for the inferred phylogeny can be evaluated using multiple approaches, such as bootstrap statistics, gene support frequencies / concordance factors, and phylogenomic subsampling. (F) Divergence time estimates can be inferred using node dating, tip dating, or fossil-free analyses. Branch lengths represent substitutions per site in the phylogeny on the left and time on the right. Grey boxes in the timetree represent confidence intervals. Silhouette images were obtained from PhyloPic (https://www.phylopic.org/); credit goes to their respective contributors.
Figure 1.
A workflow for phylogenomic inference. (A) The first step in a phylogenomic workflow is data acquisition and preparation. This often entails identifying gene boundaries in genome assemblies or assembling transcripts in transcriptomes. (B) The next step is to identify orthologs using (left) de novo approaches—for example, all-by-all sequence similarity calculations followed by graph-based clustering—or (right) from predetermined sets of orthologs. (C) Orthologous groups of genes suitable for phylogenomics (i.e., one-to-one orthologs and SNAP-OGs) are subsequently aligned and trimmed. (D) The resulting multiple sequence alignments can be (left) concatenated into a supermatrix or (right) collections of single-locus phylogenies can be used in a coalescence-based approach. (E) Support for the inferred phylogeny can be evaluated using multiple approaches, such as bootstrap statistics, gene support frequencies / concordance factors, and phylogenomic subsampling. (F) Divergence time estimates can be inferred using node dating, tip dating, or fossil-free analyses. Branch lengths represent substitutions per site in the phylogeny on the left and time on the right. Grey boxes in the timetree represent confidence intervals. Silhouette images were obtained from PhyloPic (https://www.phylopic.org/); credit goes to their respective contributors.
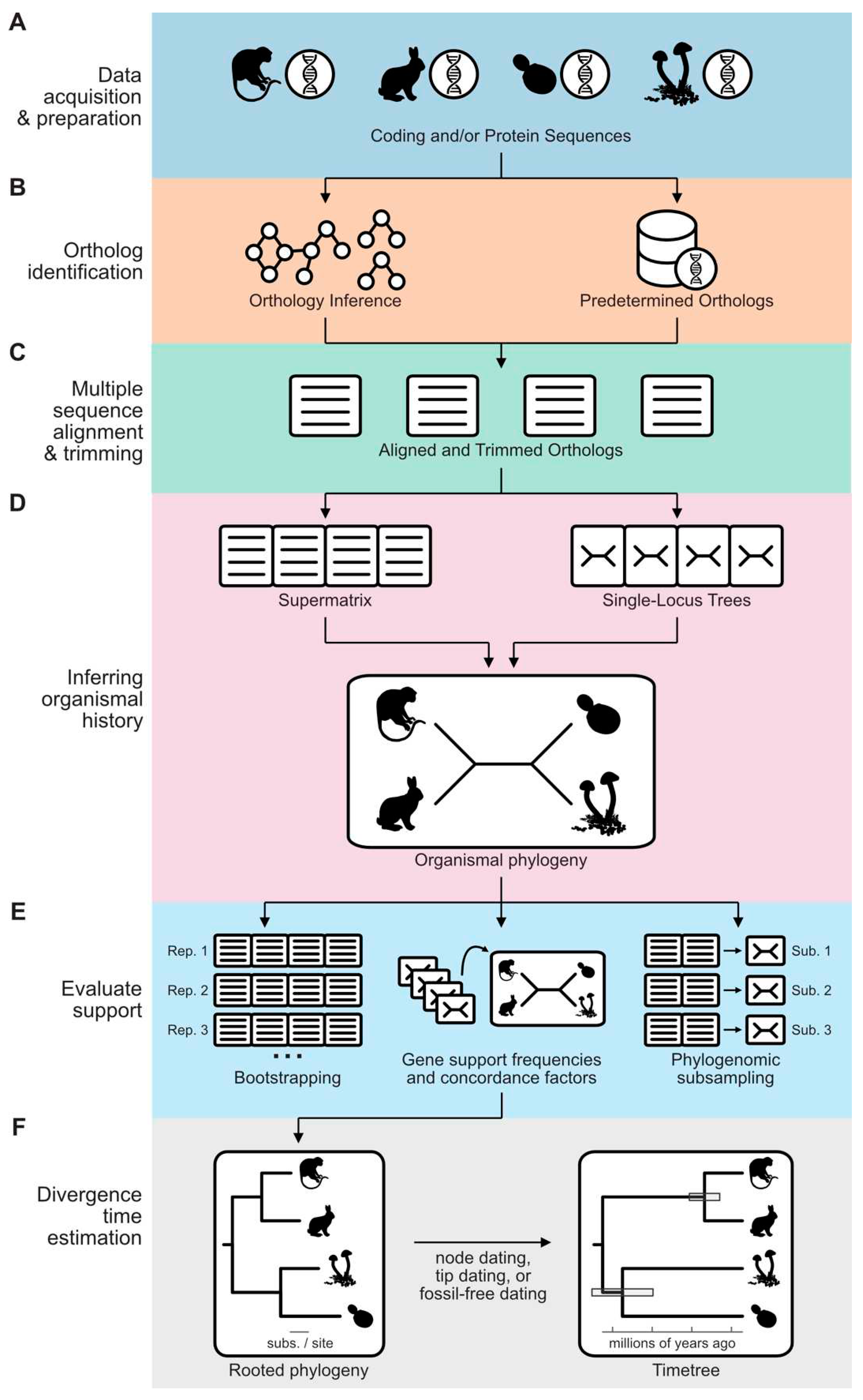
Figure 2.
The ABBA-BABA test for detecting introgression/hybridization. To detect introgression/hybridization in the (A) four-taxon case (represented as T1 through T4 where T4 is the outgroup taxa), the D-statistic or ABBA-BABA test can be used. (B) The orange dot represents a mutation from the ancestral allele ‘A’ (blue) to a derived allele ‘B’ (orange). The BBAA pattern, which is not directly accounted for in the ABBA-BABA test, is a biallelic site that follows the organismal phylogeny. Asymmetric patterns of ABBA and BABA biallelic site patterns suggest the occurrence of an introgression/hybridization event. The ABBA pattern can arise from (C) incomplete lineage sorting or (D) introgression/hybridization, whereas the (E) BABA pattern can arise from incomplete lineage sorting; thus, unequal frequencies of ABBA and BABA patterns are suggestive of introgression/hybridization.
Figure 2.
The ABBA-BABA test for detecting introgression/hybridization. To detect introgression/hybridization in the (A) four-taxon case (represented as T1 through T4 where T4 is the outgroup taxa), the D-statistic or ABBA-BABA test can be used. (B) The orange dot represents a mutation from the ancestral allele ‘A’ (blue) to a derived allele ‘B’ (orange). The BBAA pattern, which is not directly accounted for in the ABBA-BABA test, is a biallelic site that follows the organismal phylogeny. Asymmetric patterns of ABBA and BABA biallelic site patterns suggest the occurrence of an introgression/hybridization event. The ABBA pattern can arise from (C) incomplete lineage sorting or (D) introgression/hybridization, whereas the (E) BABA pattern can arise from incomplete lineage sorting; thus, unequal frequencies of ABBA and BABA patterns are suggestive of introgression/hybridization.

Figure 3.
Phylogenetic signatures of a horizontal gene transfer event. To detect horizontal gene transfer, (A) organismal histories are compared to (B) single-locus phylogenies. Horizontal gene transfer is suggested when sequences are placed within an unexpected taxonomic group in single-locus phylogenies. Here, an example of prokaryote-to-eukaryote transfer is depicted wherein an organism from the fungal kingdom (orange) is monophyletic with prokaryotic sequences (blue). The horizontal gene transfer event is depicted as a grey arrow. Silhouette images were obtained from PhyloPic (https://www.phylopic.org/); credit goes to their respective contributors.
Figure 3.
Phylogenetic signatures of a horizontal gene transfer event. To detect horizontal gene transfer, (A) organismal histories are compared to (B) single-locus phylogenies. Horizontal gene transfer is suggested when sequences are placed within an unexpected taxonomic group in single-locus phylogenies. Here, an example of prokaryote-to-eukaryote transfer is depicted wherein an organism from the fungal kingdom (orange) is monophyletic with prokaryotic sequences (blue). The horizontal gene transfer event is depicted as a grey arrow. Silhouette images were obtained from PhyloPic (https://www.phylopic.org/); credit goes to their respective contributors.
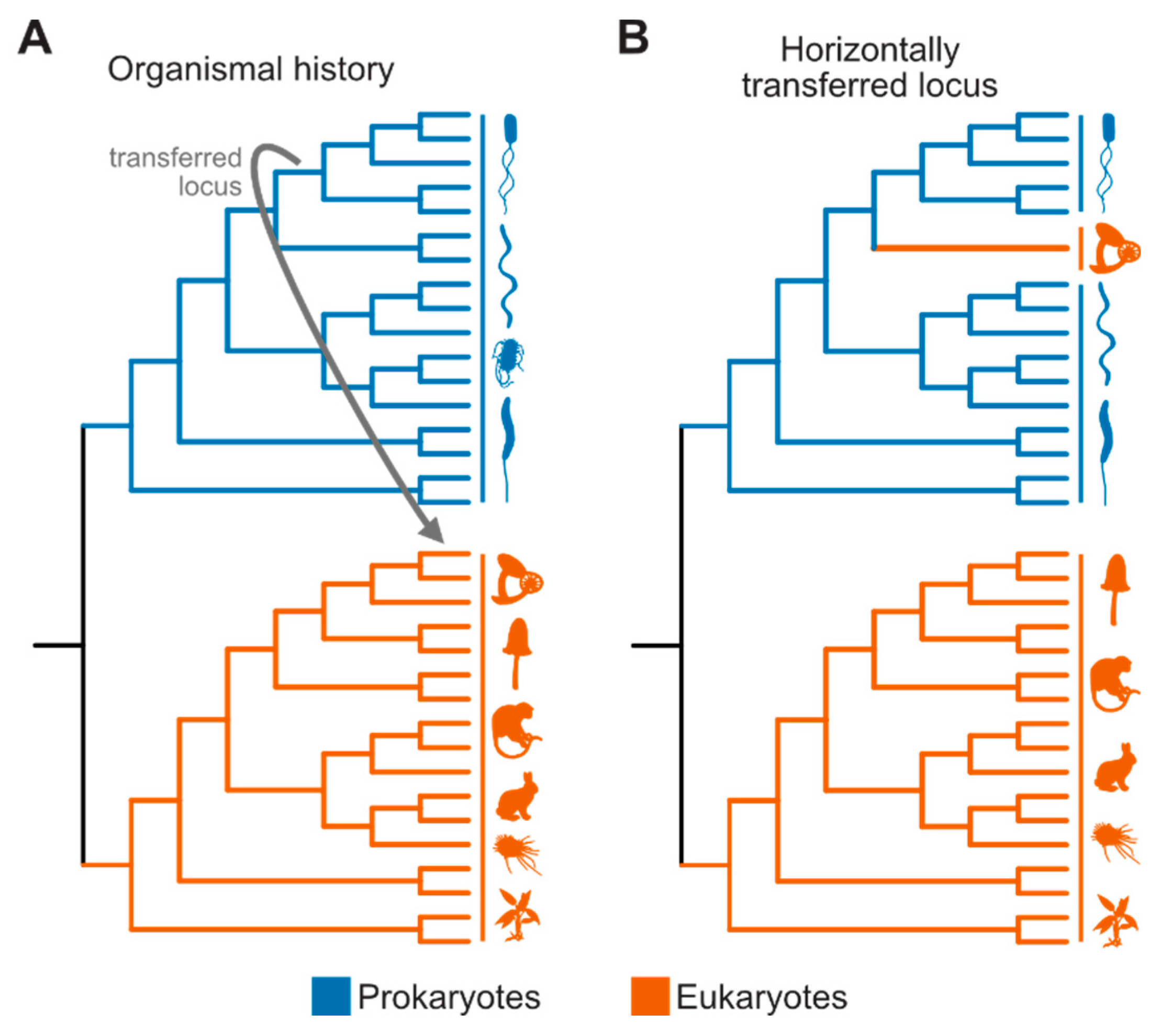
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



