
Submitted:
01 September 2023
Posted:
04 September 2023
You are already at the latest version
Abstract
With the rapid increase in diabetes worldwide, the number of patients with diabetic kidney disease (DKD), a complication of diabetes mellitus, is also on the rise. DKD is a major cause of chronic kidney disease progressing to end-stage renal failure; however, current medical treatments for DKD do not achieve satisfactory outcomes. Molecular hydrogen (H2) is an antioxidant that selectively reduces hydroxyl radicals, a reactive oxygen species with a very potent oxidative capacity. H2 was recently shown to exert not only antioxidant, but also anti-inflammatory, cell lethality-regulating, and signal transduction-regulating effects, and is now being applied clinically. Many factors contribute to the onset and progression of DKD, with mitochondrial dysfunction, oxidative stress, and inflammation being strongly implicated. Recent preclinical and clinical trials reported that substances with antioxidant properties may slow the progression of DKD. Therefore, we conducted a literature review on animal models and human clinical trials in which H2 showed efficacy against various renal diseases. This literature review and our previous findings collectively suggest that H2 exerts therapeutic effects in patients with DKD by improving mitochondrial function. Future large-scale clinical studies are needed to confirm these findings.
Keywords:
molecular hydrogen
; diabetic kidney disease
; mitochondrial dysfunction
; oxidative stress
; in-flammation
; reactive oxygen species
; medical application
1. Introduction
Kidney diseases include a wide variety of conditions, including glomerulonephritis and pyelonephritis caused by kidney inflammation and infection, as well as nephrosclerosis, diabetic kidney disease (DKD), and polycystic kidney disease caused by hypertension, atherosclerosis, and diabetes mellitus. DKD is one of the complications of diabetes mellitus and is also a major cause of chronic kidney disease (CKD) progressing to end-stage renal disease (ESRD) [1,2].
The International Diabetes Federation estimated that the global prevalence of diabetes mellitus would reach 10.5% (536.6 million individuals) in 2021 and 12.2% (783.2 million individuals) by 2045 [3]. With the rapid increase in diabetes mellitus worldwide, the number of patients with DKD also continues to rise, particularly in developed countries [1,2,3]. DKD is a risk factor not only for progression to ESRD, but also death from cardiovascular complications [4]. Approximately 30-40% of patients with diabetes mellitus develop DKD, making it a global public health and health economic issue [1,2,3].
Molecular hydrogen (H2) is a flammable, colorless, and odorless gaseous molecule. It functions as an antioxidant that directly reduces hydroxyl radicals (·OH) and peroxynitrite (ONOO−), which are reactive oxygen species (ROS) and reactive nitrogen species (RNS), respectively, with very potent oxidative capacities [5]. In addition, H2 exerts a number of indirect effects, including antioxidant, anti-inflammatory, and cell lethality-regulating effects, through the regulation of gene expression, in addition to its effects through nuclear factor erythroid-related factor 2 (Nrf-2) and intracellular signal transduction [6,7,8,9]. The total number of publications on the biological effects of H2 currently exceeds 2,000. H2 not only crosses the blood-brain barrier easily, but also biological membranes to reach mitochondria and protect cells from ROS- and RNS-induced cell damage [10]. Although the target molecules of H2 remain unknown, oxidized porphyrins were recently shown to catalyze the reaction of H2 with ·OH, thereby reducing oxidative stress [11].
Many factors are involved in the development and progression of DKD, among which mitochondrial dysfunction, oxidative stress, hyperglycemia, and inflammation have been strongly implicated [12,13,14,15]. Recent preclinical and clinical studies reported that new therapeutic agents that reduce oxidative stress may slow the progression of DKD [16,17,18,19,20]. Other studies demonstrated the efficacy of H2 in various animal renal disease models [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43]. Furthermore, H2 gas inhalation therapy or therapy with H2-enriched dialysis solution was shown to attenuate oxidative stress in dialysis patients [44,45,46,47]. However, to the best of our knowledge, the potential therapeutic effect of H2 on DKD has not been reported. This review is based on a literature review and our previous findings, and we propose that H2 exerts therapeutic effects in patients with DKD by improving mitochondrial function.
2. H2 Regulates Oxidative Stress
2.1. History and Progress of Research on Medical Applications of H2
In 1975, Dole et al. reported that H2 gas exerted anti-tumor effects. They showed that the inhalation of an 8 atm mixture of 2.5% oxygen and 97.5% H2 significantly regressed squamous cell carcinoma in mice [48]. In 1994, Abraini et al. demonstrated that the inhalation of a mixture of 49% H2, 50% helium, and 1% oxygen effectively prevented decompression sickness in divers [49]. In 2001, Gharib et al. showed the anti-inflammatory effects of hyperbaric H2 gas in a mouse model of chronic hepatitis induced by schistosomiasis infection [50]. Furthermore, in 2005, Yanagihara et al. found that saturated concentrations of neutral H2 water produced by water electrolysis reduced chemical oxidant-induced liver injury in rats [51].
In 2007, Ohsawa et al. reported the utility of H2 as a therapeutic antioxidant by selectively reducing ·OH and ONOO−, which have very potent oxidative capacities [5]. Medical applications of H2 have since attracted worldwide attention and marked advances have been achieved in this field. In 2023, oxidized porphyrins were shown to catalyze the reaction of H2 with ·OH [11]. A mechanism was proposed in which the potent oxidative capacity of ·OH is reduced in the presence of H2 and porphyrins, which activates Nrf-2 as a hormesis-like effect and induces the antioxidant enzyme heme oxygenase-1 (HO-1) [52].
2.2. ROS Production and Scavenging Systems
Oxygen is essential for the production of energy by breathing organisms and is used by the mitochondria of cells to generate adenosine triphosphate (ATP). However, 1-2% of consumed oxygen becomes ROS, which has potent oxidative properties [8,10,53,54]. ROS in humans is mainly superoxide, hydrogen peroxide, ·OH, and singlet oxygen [8,10,53,54]. Superoxide is formed when electrons leaking from the mitochondrial respiratory chain combine with oxygen. Superoxide is also produced by xanthine oxidase, which uses oxygen and xanthine as substrates, or the arachidonic acid cascade in vascular endothelial cells. Superoxide is a relatively potent ROS but is degraded to hydrogen peroxide by superoxide dismutase (SOD) [8,10,53,54]. Hydrogen peroxide is further decomposed into water and oxygen by catalase (CAT) and glutathione peroxidase (GPX). [8,10,53,54]. On the other hand, singlet oxygen is generated when oxygen reacts with pigments in the body that act as sensitizers when the organism is exposed to ultraviolet radiation (Figure 1).
The human body is equipped with antioxidant enzymes, such as SOD, CAT, and GPX, as described above, as a defense mechanism to suppress the formation of ROS. However, the function of antioxidant enzymes and the body’s defense capabilities against ROS decline with age [10,54]. Furthermore, when ROS are produced in large amounts due to mental and physical stress, excessive exercise, smoking, and exposure to ultraviolet light and irradiation, the balance between ROS production and scavenging systems is disrupted, and ROS that exceed the protective capacity of antioxidant enzymes appear [10,54]. The disruption of the balance between oxidation and anti-oxidation results in superoxide and hydrogen peroxide catalyzed by iron and copper ions producing ·OH, which has a very potent oxidative capacity [55]. ·OH is also generated in other biological reactions and when water, a biological substance, is exposed to irradiation. ·OH is present in the body for only one millionth of a second; however, during that time, it exhibits an oxidizing power 100-fold stronger than that of superoxide [56]. On the other hand, nitric oxide (NO·) reacts with superoxide to produce ONOO−, which is extremely oxidizing [8,53]. When ·OH and ONOO− are produced, they react with nucleic acids, lipids, and proteins in biological membranes and tissues, causing oxidative damage and even oxidizing DNA. Antioxidant enzymes cannot scavenge ·OH and ONOO−, while H2 selectively reduces ·OH and ONOO−, converting them to water (Figure 1) [8,10,53,54].
2.3. “.Beneficial” and “Detrimental” Effects of ROS
ROS have both “detrimental” and “beneficial” effects on organisms. Superoxide and hydrogen peroxide are cytotoxic at high concentrations, but function as second messengers in signal transduction mechanisms and as regulators of immune cell metabolism, activation, cell proliferation, cell differentiation, and apoptosis at low concentrations [10,54]. Hydrogen peroxide at high concentrations is converted by antioxidant enzymes to hypochlorous acid, which protects the body from bacterial attack. NO· is important for intracellular signal transduction and vasodilation and is used clinically as a medical gas. On the other hand, a difference between eustress and distress has been reported. Large amounts of ROS cause oxidative damage, whereas small amounts of ROS activate Nrf-2 and induce HO-1, exerting antioxidant effects [9,57]. Small amounts of ROS also induce the expression of the tumor suppressor gene, p53, which plays an important role in the prevention of cancer [58]. Furthermore, small amounts of ROS are crucial for maintaining homeostasis in organisms [59].
Oxidative stress caused by ROS causes mutations in normal cells and promotes their transformation into cancer cells. Therefore, a method to remove ROS and reduce oxidative stress by antioxidants was considered promising for cancer prevention and treatment. A large clinical trial on vitamin E supplementation was conducted [60,61]. However, contrary to expectations, the incidence of prostate cancer was significantly higher in patients treated with vitamin E [60,61]. Studies using mouse models of cancer also reported the cancer-promoting effects of N-acetylcysteine and vitamin E supplementation [62]. The mechanism by which these antioxidants promote cancer growth was elucidated by DeNicola et al. and Schafer et al [63,64]. Therefore, the effects of antioxidants on cancer are two-sided, either inhibiting or promoting carcinogenesis, depending on the conditions. H2 has not yet been shown to exert carcinogenic effects, whereas its carcinogenic inhibitory effects have been demonstrated [65].
3. Mitochondrial Involvement in Renal Disease
3.1. Mitochondrial Structure and Function
Mitochondria are intracellular organelles that produce more than 90% of intracellular energy and generate ATP by oxidative phosphorylation (OXPHOS) under aerobic conditions [8,10,53,54]. Mitochondria comprise an outer membrane, inner membrane, intermembrane lumen between the outer and inner membranes, a matrix surrounded by an inner membrane, and a crista lumen surrounded by a recessed crista membrane [66]. Mitochondria have their own genome, mitochondrial DNA (mtDNA), which is distinct from the nuclear genome (nDNA) [66]. This is attributed to the intracellular parasitism of the aerobic bacterium, Proteobacteria, into archaea approximately 2 billion years ago, which transformed them into mitochondria [67]. Therefore, the structure of mtDNA is more similar to the bacterial genome structure than to the eukaryotic nuclear genome. Mitochondrial respiratory chain complexes, complexes I-V, assemble in inner membrane cristae for efficient ATP production [66,68,69]. Many mitochondrial ROS (mtROS) are generated during this ATP production process, mainly from complexes I and III, which are normally removed by the antioxidant system [66,68,69]. Therefore, mitochondria maintain efficient energy production by the electron transfer system of the inner membrane respiratory chain complex and OXPHOS by ATP synthase.
3.2. Role of ROS in Renal Disease
Mitochondrial disease is caused by abnormalities in various genes involved in mitochondrial function and structural maintenance, including ATP synthesis, the transport of amino acids, lipids, and proteins, and the removal of oxidative stress within mitochondria [68,69]. These genetic abnormalities may result from both mtDNA aberrations and genetic mutations in nDNA [70]. When mitochondrial function is impaired because of these genetic abnormalities, organs with high energy demands become dysfunctional and exhibit a wide range of symptoms [68,71]. Mitochondrial diseases are called mitochondrial encephalomyopathies or mitochondrial myopathies depending on the location of the disorder [72].
Abnormal mitochondrial function is induced by various factors other than genetic abnormalities that decrease ATP production and increase mtROS, and cell lethality is caused by apoptotic signals, such as cytochrome c released from within damaged mitochondria [69,71]. This type of mitochondrial dysfunction has been observed in many metabolic and neurodegenerative diseases [73]. Among them, the kidney, which has the highest volume per tissue weight and oxygen consumption among organs after the heart and brain, respectively, relies upon mitochondrial ATP production for active mass transport energy metabolism in the tubular epithelium [69,74]. Furthermore, since podocytes, the glomerular vascular endothelium, and mesangial cells, which perform glomerular filtration functions, are active in mitochondrial metabolism, the kidney is subject to the failure of energy metabolism due to ischemia, hypoxia, and mitochondrial dysfunction from toxic substances [75,76,77,78,79].
With increases in the number of patients with diabetes mellitus, DKD has become the leading disease among ESRD [1]. Diabetes mellitus is considered to be the primary cause of the recent increase in the number of patients on dialysis [1,2,3,4]. It decreases the effects of insulin in cells, resulting in chronic hyperglycemia and dyslipidemia [80,81]. Therefore, abnormalities in signaling mechanisms, such as nutrient-sensitive AMP-activated protein kinase (AMPK) and mechanistic target of rapamycin complex 1, and mitochondrial dysfunction are observed [80,81]. In addition, oxidative stress is increased in diabetes mellitus due to the intracellular influx of excess glucose, which promotes the mitochondrial production of excess mtROS, activates the polyol metabolic pathway, activates protein kinase C, and leads to the accumulation of advanced glycation end products [82,83,84]. Oxidative stress also promotes the development of DKD.
However, physiological levels of ROS are essential for the regulation of intracellular signaling mechanisms and cellular homeostasis [10,54]. Mitochondrial membrane potential and respiratory regulation rates are reduced in diabetes mellitus and, conversely, ROS production is decreased, which inhibits OXPHOS and ATP production [80,85]. Continued reductions in OXPHOS and ATP production in mitochondria increase oxidative stress outside mitochondria due to the uncoupling of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and endothelial-type NO· synthase, releasing cytokines involved in inflammation and fibrosis and inducing cellular damage [86]. ROS derived from the cytoplasm are considered to play a more important role than mtROS in this case. The administration of AMPK activators to animal models of diabetes mellitus was previously shown to improve mitochondrial function and induce the production of mtROS, thereby decreasing albuminuria and attenuating DKD [80,86,87]. As part of the mitochondrion-targeted DKD therapeutic strategy, the utility of a treatment that improves mitochondrial function and induces physiological levels of ROS production has been proposed [86,87].
4. Effects of H2 on Various Renal Diseases
We herein investigated the literature for the preventive and therapeutic effects of H2 in animal renal disease models and human renal diseases [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47]. The following is a summary of studies that reported the effects of H2 on ischemia-reperfusion (I/R) injury, transplantation, CKD, drug-induced kidney injury, kidney stones, renal fibrosis, sepsis-related acute kidney injury (AKI), peritoneal dialysis (PD), and hemodialysis (HD) (Table 1).
4.1. Effects on Animal Disease Models
4.1.1. I/R Injury
Renal I/R injury is an important cause of AKI, a factor in the development of CKD. Shingu et al. investigated the protective effects of H2-rich saline solution (HRS) on renal I/R injury in rats. HRS improved mitochondrial morphology and significantly reduced blood urea nitrogen (BUN), creatinine (Cr), and 8-hydroxydeoxyguanosine (8-OHdG) [21].
Similarly, in a rat renal I/R injury model, Wang et al. found that HRS significantly suppressed BUN, Cr, malondialdehyde (MDA), 8-OHdG, tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, and myeloperoxidase (MPO), and significantly increased tissue SOD and CAT activities [22].
Li et al. also examined the effects of HRS on a rat model of renal I/R injury and showed that HRS significantly reduced renal tissue stromal congestion, edema, and hemorrhage, as well as BUN, Cr, B-cell/CLL lymphoma 2 (Bcl-2), caspase-3, -8, and -9, IL-6, and TNF-α, while Bcl-2-associated x (Bax) was significantly increased [23]. Furthermore, they reported that the protective effects of HRS may be attributed to its anti-apoptotic and anti-inflammatory effects.
Chen et al. investigated the protective effects of HRS in an I/R-induced AKI mouse model and showed that it significantly reduced renal tissue fibrosis, BUN, and Cr and increased the Klotho levels of anti-aging genes [24]. Moreover, they demonstrated that HRS increased damage-regulated autophagy modulator (Beclin-1) and microtubule-associated protein light chain 3-II (LC3-II) [24]. They suggested that HRS exerted its protective effects through the maintenance of Klotho expression and activation of autophagy in the kidney.
Xu et al. investigated the effects of HRS on a rat model of renal I/R injury and reported that HRS significantly decreased BUN, Cr, MDA, and 8-OHdG and increased HO-1 gene expression and SOD activity [25]. They suggested that HRS ameliorated renal I/R injury in rats by reducing oxidative stress and increasing HO-1 gene expression.
4.1.2. Transplantation
ROS is involved in the development of interstitial fibrosis and tubular atrophy in chronic allograft nephropathy (CAN). Cardinal et al. investigated the effects of drinking H2-rich water (HRW) in a rat allogeneic renal transplantation model and found that HRW improved graft function by decreasing BUN, Cr, and urinary protein, slowed CAN progression, decreased MDA, TNF-α, and IL-6, and further prolonged overall survival [26]. HRW also decreased the activation of inflammatory signaling pathways, such as mitogen-activated protein kinase (MAPK), suggesting its effectiveness at preventing CAN and prolonging renal allograft survival [26].
I/R injury is unavoidable in renal transplantation and affects both short- and long-term allograft survival rates. Abe et al. examined the inhibitory effects of H2-rich University of Wisconsin (HRUW) solution on I/R injury in a rat renal allograft model and demonstrated that it reduced MDA and 8-OHdG in renal allografts and decreased the numbers of tubular terminal transferase dUTP nick-end labeling (TUNEL)-stained cells and ED-1-positive cells in renal tubules [27]. It also reduced Cr and urinary protein, thereby improving renal function and prolonging recipient survival, which suggested that HRUW alleviated tubular damage and, in turn, reduced the development of interstitial fibrosis [27].
On the other hand, AKI has a significant impact on the survival of liver transplant recipients. Du et al. investigated the protective effects of HRS on AKI after orthotropic liver transplantation in rats and showed that HRW reduced histological damage and decreased BUN, Cr, MDA, and SOD [28]. At the same time, HRS significantly ameliorated apoptosis by suppressing caspase-3 and cytochrome c expression. Furthermore, the expression of Beclin-1 and LC3-II was up-regulated [28]. Chloroquine, an autophagy inhibitor, counteracted the protective effects of HRS [28]. These findings suggested that HRS prevents AKI by reducing apoptosis and activating autophagy.
4.1.3. CKD
The Dahl salt-sensitive (SS) rat is a CKD model animal that develops elevated blood pressure and kidney damage with aging. Zhu et al. investigated the effects of H2-dissolved electrolyte water (EW) on ischemia-induced cardiorenal injury in the Dahl SS rat [29]. Rats were fed EW or filtered water (FW), after which they underwent unilateral renal I/R. The control group receiving FW showed significant increases in MCP-1, methylglyoxal, and BUN [29]. In a histological examination of the kidneys and heart, significant increases in nitrotyrosine staining were detected in control rats [29]. However, these findings were significantly improved in EW-treated rats, suggesting the potential of EW to prevent CKD [29].
Zhu et al. also examined the effects of EW and FW on age-related cardiorenal injury in Dahl SS rats [30]. Albuminuria and cardiac remodeling increased in the FW group. Histologically, significant age-related changes were observed in the kidney and heart; however, these changes were significantly reduced and MDA and nitrotyrosine decreased in the EW group [30].
Xin et al. investigated the protective effects of HRW on renal damage in spontaneously hypertensive rats (SHR). HRW significantly reduced BUN and Cr, decreased ROS production, increased SOD, GPX, and CAT activities, and inhibited NADPH oxidase activity in SHR [31]. It also suppressed the expression of TNF-α, IL-6, and IL-1β. Moreover, HRW exerted ameliorative effects on mitochondrial morphology and function, including the suppression of mtROS production and mitochondrial swelling, and increased ATP production [31].
4.1.4. Drug-Induced Renal Injury
Cisplatin is an anticancer drug that is widely used in the treatment of a broad range of tumors; however, its application is limited by oxidative stress-induced nephrotoxicity. Nakashima-Kamimura et al. reported that when mice inhaled H2 gas or drank HRW, H2 reduced renal injury without impairing the anti-tumor activity of cisplatin [32]. In other words, H2 gas or HRW improved cisplatin-induced mortality and weight loss, ameliorated renal histological damage, and restored Cr and BUN [32].
Li et al. investigated the efficacy of HRW in a rat model of iron nitrilotriacetate-induced renal injury [33]. HRW decreased Cr, BUN, MDA, ONOO− production, and NADPH oxidase activity and increased CAT activity. HRW ameliorated mitochondrial dysfunction and oxidative stress, including kidney mitochondrial swelling, decreased ATP production, and increased mtROS production [33]. HRW also suppressed inflammation as indicated by the decreased expression of NF-κB, IL-6, and monocyte chemotactic protein-1 (MCP-1) in the kidney [33]. Furthermore, HRW suppressed vascular endothelial growth factor (VEGF) expression and signal transducer and activator of transcription 3 (STAT3) phosphorylation, thereby reducing the incidence of renal cell carcinoma and inhibiting tumor growth [33].
Oxidative stress induced by cyclosporin A is a major cause of chronic kidney injury. Lu et al. examined the mitigating effects of HRW on cyclosporine A-induced renal injury in rats and found that it decreased ROS production, MDA, and Kelch-like ECH-associated protein 1 (Keap1) and increased the expression of Nrf-2 and HO-1 [34]. They suggested that the effects of HRW involved the amelioration of oxidative stress through the activation of the Keap1/Nrf-2 signaling pathway.
4.1.5. Renal Stones
Peng et al. evaluated the protective effects of H2 gas against glyoxylate-induced renal calcium oxalate (CaOx) crystal deposition in mice and reported that it decreased MDA and 8-OHdG levels and increased SOD, GSH, and CAT activities [35]. They also showed that H2 gas reduced MCP-1 and increased IL-10 expression, indicating that H2 gas exerted protective effects against renal stone disease by reducing renal crystallization, renal oxidative damage, and inflammation [35].
4.1.6. Renal Fibrosis
Xu et al. examined the efficacy of HRS in a model of renal fibrosis induced by unilateral ureteral obstruction (UUO) in rats and showed that it significantly improved the renal injury score, apoptosis index, stromal fibrosis, and macrophage infiltration in renal tissue [36]. Additionally, HRS reduced MDA levels and increased SOD activity.
Furthermore, Xing et al. investigated the efficacy of HRW in a mouse model of renal fibrosis caused by UUO and showed that it suppressed Cr, BUN, and renal fibrosis [37]. They also examined the inhibitory effects of HRW on renal epithelial-mesenchymal transition (EMT) induced by transforming growth factor-β1 (TGF-β1) using human renal proximal tubular epithelial cells and showed that HRW abolished EMT and restored decreases in the expression of sirtuin-1 (Sirt1) [37]. Sirtinol, the inhibitor of Sirt1, abolished the inhibitory effects of HRW on EMT, indicating that HRW ameliorated renal injury and fibrosis by regulating Sirt1 [37].
Congenital obstructive nephropathy is commonly implicated in the pathophysiology of CKD, and the release of ROS contributes to the exacerbation of renal fibrosis. Mizutani et al. evaluated the efficacy of HRW in a rat model of UUO-induced renal injury [38]. HRW suppressed tubulointerstitial injury and reduced the area of interstitial fibrosis and frequency of TGF-β1-positive cells [38]. In addition, HRW restored decreases in Klotho mRNA expression.
4.1.7. Sepsis-Related AKI
Liu et al. investigated the combined effects of early infusion resuscitation and H2 gas on AKI occurring during septic shock in rats induced by lipopolysaccharide, and showed that the combination of both reduced BUN and Cr [39]. It also decreased MDA and reduced renal TNF-α and IL-6 levels more than infusion resuscitation alone [39]. These findings indicated that early infusion resuscitation combined with H2 gas exerted stronger protective effects against AKI.
Yao et al. examined the protective effects of the aerosol inhalation of HRS in a model of sepsis-related AKI in mice induced by cecum ligation and puncture [40]. AKI occurred during the early stage of sepsis, as evidenced by increases in BUN and Cr, renal fibrosis, and renal tubular epithelial cell apoptosis, and was accompanied by macrophage infiltration and the generation of inflammatory cytokines (IL-6 and TNF-α) [40]. In contrast, HRS aerosol inhalation increased the mRNA levels of anti-inflammatory cytokines (IL-4 and IL-13) and enhanced the generation of anti-inflammatory cytokines (IL-10 and TGF-β) in renal tissues, suggesting the utility of HRS aerosol inhalation for renal protection and the attenuation of inflammation in septic AKI [40].
4.1.8. Others
Guo et al. investigated the efficacy of HRS in a rat model of severe burn-induced early AKI and reported that HRW improved renal function (BUN and Cr) and attenuated tubular apoptosis [41]. Furthermore, the mechanisms underlying the AKI-ameliorating effect of HRW involved the inhibition of oxidative stress-induced apoptosis and inflammation, and these effects appeared to be mediated through the regulation of MAPK and nuclear factor (NF)-κB signaling pathways [41].
Shi et al. examined the protective effects of HRS on AKI and the underlying mechanisms in sodium taurocholate-induced acute pancreatitis in rats [42]. The findings obtained showed that HRS prevented the progression of the inflammatory cascade and alleviated oxidative damage in the kidney by inhibiting NF-κB activation and removing ROS [42].
Furthermore, Guan et al. analyzed the protective effects of H2 gas on renal damage caused by chronic intermittent hypoxia (CIH) in rats in terms of oxidative stress, autophagy, and ER stress [43]. H2 gas also improved renal function in rats with CIH, and alleviated histological damage, oxidative stress, and apoptosis. They also found that H2 gas ameliorated CIH-induced renal injury by suppressing oxidative stress-dependent MAPK activation, thereby reducing ER stress, and activating autophagy [43].
4.2. Effects on Human Diseases
4.2.1. PD
Oxidative stress derived from glucose degradation products is responsible for peritoneal degradation in patients with PD. Terawaki et al. investigated the effects of a H2-enriched dialysate (HED) on peritoneal oxidative stress in 6 patients with PD [44]. Based on the findings showing that the percentage of reduced albumin was higher and the percentage of oxidized albumin was lower in the effluent and serum of PD patients treated with HED than with the standard dialysate, HED appeared to reduce peritoneal and systemic oxidative stress [44].
4.2.2. HD
Nakayama et al. developed a dialysis system using a dialysate dissolved in H2 gas and investigated the efficacy of HED in 21 patients with HD. HED significantly reduced systolic blood pressure before and after dialysis [45]. Moreover, it significantly decreased MCP-1 and MPO, suggesting its potential to control uremia by attenuating inflammation [45].
Terawaki et al. investigated the effects of HED on oxidative stress in eight HD patients in a crossover study using a standard dialysate (SD) and HED and showed that HED significantly reduced the mean percentage of oxidized albumin in serum at the exit of the dialysis system more than SD [46].
Sokawa et al. examined the effects of H2 gas on oxidative stress and inflammatory responses in six HD patients [47]. The inhalation of H2 gas three times a week for two weeks did not affect the biological antioxidant potential (BAP). However, it significantly reduced diacron-reactive oxygen metabolites (d-ROMs) and C-reactive protein (CRP), and these effects persisted for two weeks after the discontinuation of H2 gas inhalation, indicating that the inhalation of H2 gas attenuated oxidative stress and inflammatory responses in HD patients [47].
5. Mechanism of Action of H2 on Renal Disease
The preventive and therapeutic effects of H2 on renal diseases have been reported in many studies in which H2 improved histological injuries and reduced serum BUN and Cr and urinary protein. On the other hand, based on a literature review, the mechanism of action of H2 on renal diseases may be mainly categorized as the improvement of mitochondrial function, antioxidant and anti-inflammatory effects, and the regulation of cell lethality and intracellular signal transduction. These mechanisms are not independent of each other, they interact to form the complex mechanism of H2 (Figure 2).
5.1. Improvement of Mitochondrial Function
Transmission electron microscopy is a commonly used method to examine morphological changes in mitochondria. The effects of H2 on mitochondrial morphology showed that H2 ameliorated mitochondrial swelling [21,31]. It also inhibited mtROS production, enhanced ATP production, and decreased NADPH oxidase activity [31,33], suggesting that its mechanisms of action involve ameliorative effects on mitochondrial morphology and function.
5.2. Antioxidant Effects
Using fluorescent reagents, H2 was shown to reduce the fluorescence intensity of ROS or RNS in renal tissue [33,34]. H2 also decreased MDA, a marker of lipid oxidation, and 8-OHdG, a marker of DNA oxidation [21,22,25,26,27,28,30,33,34,35,36,39]. The activities of antioxidant enzymes in renal tissue, such as SOD, CAT, and GPX, are used as markers to assess antioxidant activity; H2 increased the activities of these antioxidant enzymes [22,25,28,31,33,35,36]. Furthermore, oxidized albumin and d-ROMs are used as oxidative markers in human clinical studies and reduced albumin and BAP as antioxidant markers; H2 decreased oxidized albumin and d-ROMs and increased reduced albumin in PD or HD patients [44,46,47]. On the other hand, ONOO− modifies tyrosine residues exposed on the protein surface to produce nitrotyrosine. This nitrotyrosine has attracted attention as a nitrosative stress marker in various inflammatory diseases; H2 reduced ONOO− and inhibited nitrotyrosine production, thereby ameliorating oxidative stress, nitrosative stress, and inflammation [29,30]. Therefore, experimental findings indicate that H2 ameliorates renal injury through its antioxidant properties.
5.3. Anti-Inflammatory Effects
IL-1β, IL-6, and TNF-α are inflammatory cytokines, while IL-4, IL-10, IL-13, and TGF-β are anti-inflammatory cytokines. In experiments to examine the mRNA expression or protein levels of these cytokines in animal models, H2 decreased the former, while simultaneously increasing the latter [22,23,26,31,33,35,39,40]. In addition, MCP-1 and MPO have been used as markers of macrophage infiltration and inflammatory responses. In animal models of renal disease and HD patients, H2 decreased MCP-1 and MPO levels [33,35,45] and reduced CRP in human HD patients [47]. These findings suggest that H2 exerts protective effects against renal injury through its anti-inflammatory properties.
5.4. Regulation of Cell Lethality
Bcl-2 is a protein that promotes apoptosis, while Bax is a protein that inhibits apoptosis. The protease family, which is involved in apoptosis, includes caspase-3, -8, and -9. In addition, the TUNEL staining method is used to examine DAN fragmentation due to apoptosis. In a renal disease model, H2 not only suppressed Bcl-2 gene expression and increased Bax gene expression, but also suppressed the expression of caspase-3, -8, and -9 [23,28]. H2 also reduced TUNEL-positive cells in renal tubules [27]. On the other hand, Beclin-1 and LC3-II have been identified as regulators and markers of autophagy; H2 not only exerted ameliorative effects on renal injury, but also increased the expression of Beclin-1 and LC3-II [24,28]. However, chloroquine, an autophagy inhibitor, nullified the effects of H2 [28]. These findings indicate that H2 regulates cell lethality by inhibiting apoptosis and activating autophagy.
5.5. Regulatory Effects of Signal Transduction
Previous findings demonstrated that the mechanisms underlying the protective effects of H2 on renal diseases involved a decrease in Keap1 levels and an increase in Nrf-2 and HO-1 gene expression [25,34]. Therefore, the attenuation of oxidative stress and enhanced biological defense functions through the activation of the Keap1/Nrf-2 signaling pathway are involved in the effects of H2. Moreover, the suppression of signaling pathways, such as MAPK and NF-κB, were shown to play a role in the antioxidative and anti-inflammatory effects of H2 [26,33,41,43]. Furthermore, the inhibition of signaling pathways for STAT3 phosphorylation and VEGF expression as well as the activation of that for Sirt1 expression contributed to the effects of H2 [33,37,41]. Since sirtinol, a Sirt1 inhibitor, nullified the effects of H2, Sirt1 may be involved in the protective effects of H2 on renal disease [37]. Therefore, H2 exerts its ameliorative effects on renal injury through the activation or suppression of various signaling pathways.
6. Therapeutic Potential of H2 for DKD
6.1. Development Status of DKD Therapeutics
Bardoxolone methyl (BM) has attracted the most attention as a potential treatment for DKD. It is a novel synthetic triterpenoid with an oleic acid skeleton [16]. BM was initially developed as an anticancer agent [88]. Phase I clinical trials on cancer patients showed an increase in the estimated glomerular filtration rate (eGFR); therefore, BM was converted into a DKD treatment [88]. The main mechanism by which BM improves renal function is the activation of the Keap1/Nrf-2 pathway [16]. After a phase II open-label and double-blind placebo-controlled trial, a phase III double-blind placebo-controlled trial (AYAME study) in Japan was conducted to prove the efficacy and safety of BM [89]. However, although this clinical trial showed an improvement in eGFR, it failed to demonstrate that BM inhibited the development of ESRD, and, thus, further research on BM was discontinued [90]. Other substances that have been reported to exhibit similar mechanisms to BM include curcumin, isothiocyanate, cinnamic aldehyde, resveratrol, and α-lipoic acid [91,92,93,94,95]. These substances are in preclinical or early clinical trials and are expected to be developed as activators of the Keap1/Nrf-2 pathway in the future. In addition, sodium-glucose contransporter-2 (SGLT2) inhibitors exert their effects on oxidative stress and inflammation, while glucagon-like peptide 1 (GLP-1) receptor agonists act on the AMPK-mTOR-autophagy-ROS-signaling axis, and both were shown to be effective in animal models of DKD [17,18]. These SGLT2 inhibitors and GLP-1 receptor agonists are expected to be useful as therapeutic agents targeting oxidative stress.
On the other hand, DKD therapeutics that target the improvement of mitochondrial function have been developed. One highly mitochondrion-directed antioxidant is coenzyme-Q (MitoQ), which was found to be effective in animal models of I/R injury and DKD [19,96]. However, MitoQ has a number of limitations, such as becoming an oxidant itself, particularly at high doses, which reduces its efficacy [81,84,97]. Elamipretide (SS-31), which acts on cardiolipins in the mitochondrial inner membrane, was also effective in I/R injury and DKD models, and various clinical trials on SS-31 in DKD patients are ongoing [98]. Mitochonic acid 5 (MA-5), a novel synthetic indole compound characterized by its ability to increase intracellular ATP and decrease mtROS, has also attracted interest [99]. MA-5 was effective against I/R nephropathy and cisplatin-induced nephropathy [20]. It also improved cardiac and renal mitochondrial respiratory function and prolonged the lifespan of mice with mitochondrial disease, and clinical trials are ongoing in patients with mitochondrial disease [20,100].
6.2. Therapeutic Potential of H2 in the Etiology of DKD
Inflammation is both a cause and consequence of the onset and progression of DKD. Inflammation is triggered by inflammatory cytokines released by the innate immunity. Pathogens such as viruses and bacteria, substances produced when the body is damaged, and irritants in the environment serve as inflammation-inducing signals [101,102]. These external signals cause mitochondrial dysfunction and induce the excessive production of ROS [103,104,105]. Excessive mtROS production in mitochondria results in the release of oxidized mtDNA into the cytoplasm, which, in turn, leads to the formation of the nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome [103,104,105]. The NLRP3 inflammasome then activates caspase-1, which induces the release of mature inflammatory cytokines from immune cells, such as macrophages and neutrophils, resulting in inflammation (Figure 3) [103,104,105].
Advanced proteinuria is a progressive risk factor for the formation of interstitial lesions in DKD. In tubulointerstitial lesions associated with advanced proteinuria, tubular damage is induced by the excessive reabsorption of free fatty acids, a mechanism that involves the activation of the NLRP3 inflammasome through mitochondrial damage [106]. In addition, mineralocorticoid receptor (MR) activation is closely involved in renal inflammation and fibrosis, and MR activation has been shown to induce the production of mtROS [107]. Furthermore, the activation of caspase-1 in glomerular epithelial cells may be important for the formation of glomerulosclerotic lesions in DKD [108]. On the other hand, many studies that investigated the efficacy of H2 in inflammatory disease models suggested that the inhibition of mtROS production by H2 is involved in the mechanism by which H2 suppresses acute and chronic inflammation [109,110,111,112,113,114]. Therefore, we proposed a possible mechanism for the efficacy of H2 against inflammatory disease models that involves H2 reducing ∙OH and suppressing oxidative damage to mtDNA, which, in turn, inhibits a series of signaling pathways from activation of the NLRP3 inflammasome to the release of inflammatory cytokines [115]. H2 may ameliorate the formation of stromal lesions in patients with DKD by suppressing NLRP3 inflammasome activation and ameliorating chronic inflammation and fibrosis in the kidney (Figure 3).
Diabetic peripheral neuropathy (DNP) is another serious diabetic complication similar to DKD. Jiao et al. investigated the efficacy of HRS against DNP in a streptozotocin-induced diabetic rat model and showed that it significantly suppressed the behavioral, biochemical, and molecular biological effects of DNP in rats [116]. They also reported that 5-hydroxydecanoate, a selective inhibitor of the mitochondrial ATP-sensitive K+ (mitoKATP) channel, partially attenuated the therapeutic effects of HRS [116]. These findings indicate that the mechanism underlying the efficacy of HRS against DNP involves a protective effect on mitochondria through the activation of the mitoKATP pathway. Furthermore, we recently reported that the mechanism by which H2 is effective in animal models of various diseases, and human chronic inflammatory diseases, such as the “sequelae” of coronavirus infection 2019 (COVID-19) called post-COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), may involve the improvement of mitochondrial function [117,118]. These findings suggest that the therapeutic effects of H2 in patients with DKD involve the improvement of mitochondrial function (Figure 3).
6.3. Prospects for H2 as a Therapeutic Substance for DKD
The therapeutic effects of H2 have been observed in a wide range of diseases, and its efficacy has been reported in more than 130 clinical papers. Since no side effects of H2 were observed in these studies, H2 is a medical gas with excellent efficacy and safety [10,54,119]. In addition, H2 is a convenient gaseous molecule that may be inhaled directly as a gas or dissolved in water or saline solution for drinking or intravenous administration [10,54,119]. H2 also has excellent pharmacokinetic and intracellular kinetic characteristics [10,54,120]. Mitochondrial dysfunction, oxidative stress, inflammation, and cell lethality are closely related to the onset and progression of DKD; therefore, H2 may be effective against DKD. The mechanisms by which H2 exhibits efficacy in various animal renal disease models and human dialysis patients, as well as our previous findings, provide evidence for the therapeutic potential of H2 for DKD [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,115,117,118]. Large-scale clinical trials are needed to demonstrate this potential.
On the other hand, several limitations have been recognized in the study of medical applications of H2. A recent study reported that oxidized porphyrins function as target molecules of H2 and catalyze the reaction of H2 with ·OH [11]. However, the target molecules of H2 are still in the early stages, having only been partially elucidated [11]. Furthermore, information on dosages and usages for individual diseases, including optimal H2 concentrations, daily dosages, and durations of intake, remain unclear. Moreover, while the improvement of mitochondrial function by H2 may exert therapeutic effects on DKD, other mechanisms of H2 may be involved. In addition, the majority of DKD models used in animal studies exhibit minor clinical symptoms, which diverge from the clinical symptoms of human DKD. Therefore, further studies on the optimal dosage and usage of H2 for individual diseases, the mechanisms of action of H2, including its target molecules, and the development of animal models of DKD are warranted.
7. Conclusions
H2 has demonstrated efficacy in various animal models of renal disease and in dialysis patients, and the mechanisms of action of H2 include mitochondrial improvement, antioxidant and anti-inflammatory effects, and the regulation of cell lethality and intracellular signaling. H2 may ameliorate the pathogenesis of DKD because mitochondrial dysfunction, oxidative stress, inflammation, cell lethality, and intracellular signaling are also involved in the pathogenesis and progression of DKD. Our recent mechanistic studies on the efficacy of H2 against human chronic inflammatory diseases, such as post-COVID-19 and ME/CFS, suggest the therapeutic potential of H2 in patients with DKD. Future large-scale clinical trials are needed to confirm the effects of H2 on DKD.
Author Contributions
The following statements should be used: conceptualization, S-i.H.; methodology, S-i.H.; investigation, S-i.H.; data curation, S-i.H.; writing—original draft preparation, S-i.H.; writing—review and editing, Y.I., B.S., Y.T., and F.S.; supervision, Y.T. and F.S. All authors have read and agree to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical consent and approval were waived for this study because it is not a research study.
Data Availability Statement
No research data were collected.
Acknowledgments
The authors would like to thank Ms. Yoko Satoh of MiZ Company Limited for her advice throughout the submission of this review.
Conflicts of Interest
S.H., Y.I., B.S., and F.S. are employees of MiZ Company Limited. Y.T. declares that this research was conducted in the absence of any commercial or financial relationships that may be construed as a potential conflict of interest.
References
- Koye, D.N.; Magliano, D.J.; Nelson, R.G.; Pavkov, M.E. The global epidemiology of diabetes and kidney disease. Adv. Chronic Kidney Dis. 2018, 25, 121–132. [CrossRef]
- Bonner, R.; Albajrami, O.; Hudspeth, J.; Upadhyay, J. Diabetic kidney disease. Prim. Care 2020, 47, 645 –649.
- IDF Diabetes Atlas 2021. Available online: https://diabetesatlas.org/atlas/tenth-edition/ (accessed on 19 June 2023).
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Naicker, S.; Plattner, B.; Saran, R.; Wang A.Y.M.; Yang, C.W. Chronic kidney disease: global dimension and perspectives. Lancet 2013, 382, 260–272. [CrossRef]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katura, K.I.; Katayama, Y.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [CrossRef]
- Nogueira, J.E.; Passaglia, P.; Mota, C.M.D.; Santos, B.M., Batalhão, M.E.; Carnio, E.C.; Branco, L.G.S. Molecular hydrogen reduces acute exercise-induced inflammatory and oxidative stress status. Free Radic. Biol. Med. 2018, 129, 186–193. [CrossRef]
- Wang, D.; Wang, L.; Zhang, Y.; Zhao, Y.; Chen, G. Hydrogen gas inhibits lung cancer progression through targeting SMC3. Biomed. Pharmacother. 2018, 104, 788–797. [CrossRef]
- Hirano, S.i.; Ichikawa, Y.; Sato, B.; Takefuji, Y.; Satoh, F. Molecular hydrogen as a potential clinically applicable radioprotective agent. Int. J. Mol. Sci. 2021, 22, 4566. [CrossRef]
- Kawamura, T.; Wakabayashi, N.; Shigemura, N.; Huang, C.S.; Masutani, K.; Tanaka, Y, Nota, K, Peng, X.; Takahashi, T.; Billiar, T.R.; et al. Hydrogen gas reduces hyperoxic lung injury via the Nrf2 pathway in vivo. Am. J. Physiol. Lung. Cell Mol. Physiol. 2013, 304, L646–656. [CrossRef]
- Ohta, S. Molecular hydrogen as a novel antioxidant: Overview of the advantages of hydrogen for medical applications. Methods Enzymol. 2015, 555, 289–317.
- Jin, Z.; Zhao, P.; Gong, W.; Ding, W.; He, Q. Fe-porphyrin: a redox-related biosensor of hydrogen molecule. Nano Research 2023, 16, 2020–2025. [CrossRef]
- Wei, P. Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta. 2019, 496, 108–116. [CrossRef]
- Su, S.; Ma, Z.; Wu, H.; Xu, Z.; Yi, H. Oxidative stress as a culprit in diabetic kidney disease. Life Sci. 2023, 322, 121661. [CrossRef]
- Tanase, D.M.; Gosav E.M.; Anton, M.I.; Floria, M.; Isac, P.N.S.; Hurjui, L.L.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Oxidative stress and NRF2/Keap1/ARE pathway in Diabetic kidney disease (DKD): new perspective. Biomolecules 2022, 12, 1227.
- Galvan, D.L.; Mise, K.; Danesh, F.R. Mitochondrial regulation of diabetic kidney disease. Front. Med. 2021, 8, 745279. [CrossRef]
- Kanda, H.; Yamawaki, K. Bardoxolone methyl: drug development for diabetic kidney disease. Clin. Exp. Nephrol. 2020, 24, 857–864. [CrossRef]
- Liu, H.; Sridhar, V.S.; Boulet, J.; Dharia, A.; Khan, A.; Lawier, P.R.; Cherney, D.Z.I. Cardiorenal protection with Sglt2 inhibitors in patients with diabetes mellitus: from biomarkers to clinical outcomes in heart failure and diabetic kidney disease. Metab. Clin. Exp. 2022, 126, 154918. [CrossRef]
- Yang, S.; Lin, C.; Zhuo, X.; Wang, J.; Rao, S.; Xu, W.; Cheng, Y.; Yang, L. Glucagon-like peptide-1 alleviates diabetic kidney disease through activation of autophagy by regulating Amp-activated protein kinase-mammalian target of rapamycin pathway. Am. J. Phys. Endocrinol. Metab. 2020, 319, E1019–E1030. [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [CrossRef]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M. Hashizume, O.; Nagatoshi, S.; Matsuo, A.; Sato, T.; Kudo, T.; Matsuhashi, T.; et al. Mitochonic acid 5 binds mitochondria and ameliorates renal tubular and cardiac myocyte damage. J. Am. Soc. Nephrol. 2016, 27, 1925–1932.
- Shingu, C.; Koga, H.; Hagiwara, S.; Matsumoto, S.; Goto, K.; Yokoi, I.; Noguchi, T. Hydrogen-rich saline solution attenuates renal ischemia-reperfusion injury. J. Anesth. 2010, 24, 569–574. [CrossRef]
- Wang, F.; Yu, G.; Liu, S.Y.; Li, J.B.; Wang, J.F.; Bo, L.L.; Qian, L.R.; Sun, X.J.; Deng, X.M. Hydrogen-rich saline protects against renal ischemia/reperfusion injury in rats. J. Surg. Res. 2011, 167, e339–344. [CrossRef]
- Li, J.; Hong, Z.; Liu, H.; Zhou, J.; Cui, L.; Yuan, S.; Chu, X.; Yu, P. Hydrogen-rich saline promotes the recovery of renal function after ischemia/reperfusion injury in rats via anti-apoptosis and anti-inflammation. Front. Pharmacol. 2016, 7, 106. [CrossRef]
- Chen, J.; Zhang, H.; Hu, J.; Gu, Y.; Shen, Z.; Xu, L.; Jia, X.; Zhang, X.; Ding, X. Hydrogen-rich saline alleviates kidney fibrosis following AKI and retains Klotho expression. Front. Pharmacol. 2017, 8, 499. [CrossRef]
- Xu, X.; He, X.; Liu, J.; Qin, J.; Ye, J.; Fan, M. Protective effects of hydrogen-rich saline against renal ischemia-reperfusion injury by increased expression of heme oxygenase-1 in aged rats. Int. J. Clin. Exp. Pathol. 2019, 12, 1488–1496.
- Cardinal, J.S.; Zhan, J.; Wang, Y.; Sugimoto, R.; Tsung, A.; McCurry, K.R.; Billiar, T.R.; Nakao, A. Oral hydrogen water prevents chronic allograft nephropathy in rats. Kidney Int. 2010, 77, 101–109. [CrossRef]
- Abe, T.; Li, X.K.; Yazawa, K.; Hatayama, N.; Xie, L.; Sato, B.; Kakuta, Y.; Tsutahara K.; Okumi, M.; Tsuda, H.; et al. Hydrogen-rich University of Wisconsin solution attenuates renal cold ischemia-reperfusion injury. Transplantation 2012, 94, 14–21. [CrossRef]
- Du, H.; Sheng, M.; Wu, L.; Zhang, Y.; Shi, D.; Weng, Y.; Xu, R.; Yu, W. Hydrogen-rich saline attenuates acute kidney injury after liver transplantation via activating p53-mediated autophagy. Transplantation 2016, 100, 563–570. [CrossRef]
- Zhu, W.J.; Nakayama, M.; Mori, T.; Nakayama, K.; Katoh, J.; Murata, Y.; Sato, T.; Kabayama, S.; Ito, S. Intake of water with high levels of dissolved hydrogen (H2) suppresses ischemia-induced cardio-renal injury in Dahl salt-sensitive rats. Nephrol. Dial. Transplant. 2011, 26, 2112–2118. [CrossRef]
- Zhu, W.J.; Nakayama, M.; Mori, T.; Hao, K.; Terawaki, H.; Katoh, J.; Kabayama, S.; Ito. S. Amelioration of cardio-renal injury with aging in dahl salt-sensitive rats by H2-enriched electrolyzed water. Med. Gas. Res. 2013, 3, 26. [CrossRef]
- Xin, H.G.; Zhang, B.B.; Wu, Z.Q.; Hang, X.F.; Xu, W.S.; Ni, W.; Zhang, R.Q.; Miao, X.H. Consumption of hydrogen-rich water alleviates renal injury in spontaneous hypertensive rats. Mol. Cell Biochem. 2014, 392, 117–124. [CrossRef]
- Nakashima-Kamimura, N.; Mori, T.; Ohsawa, I.; Asoh, S.; Ohta. S. Molecular hydrogen alleviates nephrotoxicity induced by an anti-cancer drug cisplatin without compromising anti-tumor activity in mice. Cancer Chemother. Pharmacol. 2009, 64, 753–761. [CrossRef]
- Li, F.Y.; Zhu, S.X.; Wang, Z.P.; Wang, H.; Zhao, Y.; Chen, G.P. Consumption of hydrogen-rich water protects against ferric nitrilotriacetate-induced nephrotoxicity and early tumor promotional events in rats. Food Chem. Toxicol. 2013, 61, 248–254. [CrossRef]
- Lu, Y.; Li, C.F.; Ping, N.N.; Sun, Y.Y.; Wang, Z.; Zhao, G.X.; Yuan, S.H.; Zibrila, A.I.; Soong, L.; Liu, J.J. Hydrogen-rich water alleviates cyclosporine A-induced nephrotoxicity via the Keap1/Nrf2 signaling pathway. J. Biochem. Mol. Toxicol. 2020, 34, e22467.
- Peng, Z.; Chen, W.; Wang, L.; Ye, Z.; Gao, S.; Sun, X.; Guo, Z. Inhalation of hydrogen gas ameliorates glyoxylate-induced calcium oxalate deposition and renal oxidative stress in mice. Int. J. Clin. Exp. Pathol. 2015, 8, 2680–2689.
- Xu, B.; Zhang, Y.B.; Li, Z.Z.; Yang, M.W.; Wang, S.; Jiang, D.P. Hydrogen-rich saline ameliorates renal injury induced by unilateral ureteral obstruction in rats. Int. Immunopharmacol. 2013, 17, 447–452. [CrossRef]
- Xing, Z.; Pan, W.; Zhang, J.; Xu, X.; Zhang, X.; He, X.; Fan, M. Hydrogen rich water attenuates renal injury and fibrosis by regulation transforming growth factor-β induced Sirt1. Biol. Pharm. Bull. 2017, 40, 610–615. [CrossRef]
- Mizutani, A.; Endo, A.; A.; Saito, M.; Hara, T.; Nakagawa, M.; Sakuraya, K.; Murano, Y.; Nishizaki, N.; Hirano, D.; Fujinaga,S.; et al. Hydrogen-rich water reduced oxidative stress and renal fibrosis in rats with unilateral ureteral obstruction. Pediatr. Res. 2022, 91, 1695–1702.
- Liu, W.; Dong, X.S.; Sun, Y.Q.; Liu, Z. A novel fluid resuscitation protocol: provide more protection on acute kidney injury during septic shock in rats. Int. J. Clin. Exp. Med. 2014,15, 919–926.
- Yao, W.; Guo, A.; Han, X.; Wu, S.; Chen, C.; Luo, C.; Li, H.; Li, S.; Hei, Z. Aerosol inhalation of a hydrogen-rich solution restored septic renal function. Aging (Albany NY) 2019, 11, 12097–12113. [CrossRef]
- Guo, S.X.; Fang, Q.; You, C.G.; Jin, Y.Y.; Wang, X.G.; Hu, X.L.; Han, C.M. Effects of hydrogen-rich saline on early acute kidney injury in severely burned rats by suppressing oxidative stress induced apoptosis and inflammation. J. Transl. Med. 2015, 13, 183. [CrossRef]
- Shi, Q.; Liao, K.S.; Zhao, K.L.; Wang, W.X.; Zuo, T.; Deng, W.H.; Chen, C.; Yu, J.; Guo, W.Y.; He, X.B.; et al. Hydrogen-rich saline attenuates acute renal injury in sodium taurocholate-induced severe acute pancreatitis by inhibiting ROS and NF-κB pathway. Mediators Inflamm. 2015, 2015, 685043.
- Guan, P.; Sun, Z.M.; Luo, L.F.; Zhou, J.; Yang, S.; Zhao, Y.S.; Yu, F.Y.; An, J.R.; Wang, N.; Ji, E.S. Hydrogen protects against chronic intermittent hypoxia induced renal dysfunction by promoting autophagy and alleviating apoptosis. Life Sci. 2019, 225, 46–54. [CrossRef]
- Terawaki, H.; Hayashi, Y.; Zhu, W.J.; Matsuyama, Y.; Terada, T.; Kabayama, S.; Watanabe, T.; Era, S.; Sato, B.; Nakayama, M. Transperitoneal administration of dissolved hydrogen for peritoneal dialysis patients: a novel approach to suppress oxidative stress in the peritoneal cavity. Med. Gas Res. 2013, 3, 14. [CrossRef]
- Nakayama, M.; Nakano, H.; Hamada, H.; Itami, N.; Nakazawa, R.; Ito, S. A novel bioactive haemodialysis system using dissolved dihydrogen (H2) produced by water electrolysis: a clinical trial. Nephrol. Dial. Transplant. 2010, 25, 3026–3033. [CrossRef]
- Terawaki, H.; Zhu, W.J.; Matsuyama, Y.; Terada, T.; Takahashi, Y.; Sakurai, K.; Kabayama, S.; Miyazaki, M.; Itami, N.; Nakazawa, R. Effect of a hydrogen (H2)-enriched solution on the albumin redox of hemodialysis patients. Hemodial. Int. 2014, 18, 459–466. [CrossRef]
- Sokawa, S.; Matsuura, A.; Suga, Y.; Sokawa, Y.; Kojima, T.; Nakamura, H. Reduction of oxidative stress and CRP levels in hemodialysis patients by hydrogen gas inhalation. J. Jpn. Ass. Dial. Physicians 2021, 54, 433–439. [CrossRef]
- Dole, M.; Wilson, F.R.; Fife, W.P. Hyperbaric hydrogen therapy: A possible treatment for cancer. Science 1975, 190, 152–154. [CrossRef]
- Abraini, J.H.; Gardette-Chauffour, M.C.; Martinez, E.; Rostain, J.C.; Lemaire, C. Psychophysiological reactions in humans during an open sea dive to 500 m with a hydrogen-helium-oxygen mixture. J. Appl. Physiol. 1994, 76, 1113–1118. [CrossRef]
- Gharib, B.; Hanna, S.; Abdallahi, O.M.; Lepidi, H.; Gardette, B.; De Reggi, M.; Anti-inflammatory properties of molecular hydrogen: investigation on parasite-induced liver inflammation. C. R. Acad. Sci. III 2001, 324, 719–724. [CrossRef]
- Yanagihara, T.; Arai, K.; Miyamae, K.; Sato, B.; Shudo, T.; Yamada, M.; Aoyama, M. Electrolyzed hydrogen-saturated water for drinking use elicits an antioxidative effect: a feeding test with rats. Biosci. Biotechnol. Biochem. 2005, 69, 1985–1987. [CrossRef]
- Ohta, S. Molecular hydrogen may activate the transcription factor Nrf2 to alleviate oxidative stress through the hydrogen-targeted porphyrin. Aging Pathobiol. Ther. 2023, 5, 25–32. [CrossRef]
- Hirano, S.i.; Yamamoto, H.; Ichikawa, Y.; Sato, B.; Takefuji, Y. Molecular hydrogen as a novel antitumor agent: possible mechanisms underlying gene expression. Int. J. Mol. Sci. 2021, 22, 8724. [CrossRef]
- Ohta S. Molecular hydrogen as a preventive and therapeutic medical gas: Initiation, development and potential of hydrogen medicine. Pharmacol. Ther. 2014,144, 1–11. [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Biologically relevant metal ion-dependent hydroxyl radical generation. FEBS Lett. 1992, 307, 108–112. [CrossRef]
- Setsukinai, K.I.; Urano, Y.; Kakinuma, K.; Majima, H.J.; Nagano, T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem, 2003, 278, 3170–3175. [CrossRef]
- Li, S.; Takahara, T.; Que, W.; Fujino, M.; Guo, W.Z.; Hirano, S.i.; Ye, L.P.; Li, X.K. Hydrogen-rich water protects liver injury in nonalcoholic steatohepatitis though HO-1 enhancement via IL-10 and Sirt 1 signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G450–463.
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [CrossRef]
- Ohsawa, I. Biological responses to hydrogen molecule and its preventive effects on inflammatory disease. Curr. Pharm. Des. 2021, 27, 659–666. [CrossRef]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Growley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M. et al. Vitamin E and the risk of prostate cancer. The selenium and vitamin E cancer prevention trial (Select). J. Am. Med. Assoc. 2011, 306, 1549–1556.
- Chandel, N.S.; Tuveson D.A, The promise and perils of antioxidants for cancer patients. N Engl J Med, 2014, 371, 177–178. [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [CrossRef]
- DeNicola, G.M; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis, Nature, 2011, 475, 106–109. [CrossRef]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature, 2009, 461, 109–113. [CrossRef]
- Kawai, D.; Takaki, A.; Nakatsuka, A.; Wada, J.; Tamaki, N.; Yasunaka, T.; Koike, K.; Tsuzaki, R.; Matsumoto, K.; Miyake, Y. et al. Hydrogen-rich water prevents progression of nonalcoholic steatohepatitis and accompanying hepatocarcinogenesis in mice. Hepatology. 2012, 56, 912–921. [CrossRef]
- Zerbes, R.M.; van der Klei, I.J.; Veenhuis, M.; Pfanner, M.; van der Laan, M.; Bohnert, M. Mitofilin complexes: conserved organizers of mitochondrial membrane architecture. Bio. Chem. 2012, 393, 1247.
- Roger, A.G.; Muñoz-Gómez, S.A.; Kamikawa R. The origin and diversification of mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S, Hirano, M.; Koga, Y.; McFarlamd, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080.
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267. [CrossRef]
- Schapira, A.H. Mitochondrial disorders. Curr. Opin. Neurol. 2000, 13, 527–532.
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet, 2018, 391, 2560. [CrossRef]
- Guerrero-Molina, M.P.; Morales–Conejo, M.; Delmiro, A.; Morán, M.; Domínguez-González, C.; Arranz-Canales, E.; Ramos-González, A.; Arenas, J.; Martín, M.A.; de la Aleja, J.G. High-dose oral glutamine supplementation reduces elevated glutamate levels in cerebrospinal fluid in patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome. Eur. J. Neurol. 2023, 30, 538–547.
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236. [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondria energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646.
- Abe, Y.; Sakairi, T.; Kajiyama, H. Shrivastav, S.; Beeson, C. Kopp, J.B. Bioenergetic characterization of mouse podocytes. Am. J. Physiol. Cell Physiol. 2010, 299, C464–476. [CrossRef]
- Ozawa, S.; Ueda, S.; Imamura, H.; Mori, K.; Asanuma, K.; Yanagita, M.; Nakagawa, T. Glycolysis, but not mitochondria, responsible for intracellular ATP distribution in cortical area of podocytes. Sci. Rep. 2015, 5, 18575. [CrossRef]
- Imasawa, T.; Rossignol, R. Podocytes energy metabolism and glomerular disease. Int. J. Biochem. Cell Biol. 2013, 45, 2109–2118. [CrossRef]
- Fink, B.D.; Herlein, J.A.; O'Malley, Y.; Sivitz, W.I. Endothelial cell and platelet bioenergetics: effect of glucose and nutrient composition. PLoS One 2012, 7, e39430. [CrossRef]
- Czajka, A.; Malik, A.N. Hyperglycemia induced damage to mitochondria respiration in renal mesangial and tubular cells: Implication for diabetic nephropathy. Redox. Biol. 2016, 10, 100–107.
- Dugan, L.L.; You, Y.H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.E.: Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Invest. 2013, 123, 4888–4899. [CrossRef]
- Forbes, J.M.; Thorburn, D.R.; Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [CrossRef]
- Brownlee, M. The Pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005, 54, 1615–1625.
- Sagoo, M.K.; Gnudi, L. Diabetic nephropathy: Is there a role for oxidative stress? Free Radic. Biol. Med. 2018, 116, 50–63.
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [CrossRef]
- Sharma, K. Mitochondrial hormesis and diabetic complications. Diabetes 2015, 64, 663–672. [CrossRef]
- Coughlan, M.T.; Sharma, K. Challenging the dogma of mitochondrial reactive oxygen species overproduction in diabetic kidney disease. Kidney Int. 2016, 90, 272–279. [CrossRef]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [CrossRef]
- Nangaku, M.; Takama, H.; Ichikawa, K.; Mukai, K.; Kojima, M.; Suzuki, Y.; Watada, H.; Wada, T.; Ueki, K.; Narita, I.; et al. Randomized, double-blind, placebo-controlled phase 3 study of bardoxolone methyl in patients with diabetic kidney disease: design and baseline characteristics of Ayame study. Nephrol. Dial. Transplant. 2023, 38, 1204–1216. [CrossRef]
- A phase III double-blind placebo-controlled trial of bardoxolone methyl (AYAME trial) in Japan. News releases from Kyowa Kirin Co. Ltd. Available online: https://www.kyowakirin.co.jp/pressroom/news_releases/2023/20230510_01.html (accessed on 19 July 2023).
- Huang, J.; Huang, K.; Lan, T.; Xie, X.; Shen, X.; Liu, P.; Huang, H. Curcumin ameliorates diabetic nephropathy by inhibiting the activation of the SphK1-S1P signaling pathway. Mol. Cell. Endocrinol. 2013, 365, 231–240. [CrossRef]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 Beta/Fyn/Nrf2 signaling pathway. J. Nutr. Biochem. 2015, 26, 596–606. [CrossRef]
- El-Bassossy, H.M.; Fahmy, A.; Badawy, D. Cinnamaldehyde Protects from the Hypertension Associated with Diabetes. Food Chem. Toxicol. 2011, 49, 3007–3012. [CrossRef]
- Sattarinezhad, A.; Roozbeh, J.; Shirazi Yeganeh, B.; Omrani, G.R.; Shams, M. Resveratrol reduces albuminuria in diabetic nephropathy: A randomized double-blind placebo-controlled clinical trial. Diabetes Metab. 2019, 45, 53–59. [CrossRef]
- Jiang, Z.; Tan, Z.; Meng, Z.; Li, X. Curative effects of valsartan alone or combined with alpha-lipoic acid on inflammatory cytokines and renal function in early-stage diabetic kidney disease. J. Coll. Phys. Surg. Pak. 2019, 29, 1009–1011. [CrossRef]
- Sun, J.; Zhu, H.; Wang, X.; Cao, Q.; Li, Z.; Hung, H. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J. Endocrinol. 2019, 240, 445-465. [CrossRef]
- Szeto, H.H. Pharmacologic approaches to improve mitochondrial function in AKI and CKD. J. Am. Soc. Nephrol. 2017, 28, 2856–2865. [CrossRef]
- Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; Ge, M.; Santos, J.V.; Hernandez, Y.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Invest. 2019, 129, 3387–3400. [CrossRef]
- Matsuhashi, T.; Sato, T.; Kanno, S.I.; Suzuki, T.; Matsuo, A.; Oba, Y.; Kikusato, M.; Ogasawara, E.; Kudo, T.; Suzuki, K.; et al. Mitochonic acid 5 (MA-5) facilitates ATP synthase oligomerization and cell survival in various mitochondrial disease. EBioMedicine 2017, 20, 27–38. [CrossRef]
- Clinical trial of MA-5, a treatment for mitochondrial disease. Press releases from Tohoku University. Available online: https://www.tohoku.ac.jp/japanese/2021/12/press20211206-02-ma5.html (accessed on 24 July 2023).
- Mason, D.R.; Beck, P.L.; Muruve, D.A. Nucleotide-binding oligomerization domain-like receptors and inflammasomes in the pathogenesis of non-microbial inflammation and disease. J. Innate. Immun. 2012, 4, 16–30. [CrossRef]
- Wallet, S.M.; Puri, V.; Gibson, F.C. Linkage of infection to adverse systemic complication: Periodontal disease, toll-like receptors, and other pattern recognition systems. Vaccines 2018, 6, 21.
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21.
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non -transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [CrossRef]
- Nishi, Y.; Satoh, M.; Nagasu, H.; Kadoya, H.; Ihoriya, C.; Kidokoro, K.; Sasaki, T.; Kashihara, N. Selective estrogen receptor modulation attenuates proteinuria-induced renal tubular damage by modulating mitochondrial oxidative status. Kidney Int. 2013, 83, 662–673. [CrossRef]
- Kadoya, H.; Satoh, M.; Sasaki, T.; Taniguchi, S.; Takahashi, M.; Kashihara, N. Aldosterone is a critical danger signal for inflammasome activation in development of renal fibrosis in mice. FASEB J. 2015, 29, 3899–3910. [CrossRef]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Kohli, S.; Nazir, S.; Ghosh, S.; Ranjan, S.; Wang, H.; Madhusudhan, T.; Nawroth, P.P.; Isermann, B. Caspase-1, but not caspase-3, promotes diabetic nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2270–2275. [CrossRef]
- Ren, J.D.; Wu, X.B.; Jiang, R.; Hao, D.P.; Liu, Y. Molecular hydrogen inhibits lipopolysaccharide-triggered NLRP3 inflammasome activation in macrophages by targeting the mitochondrial reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 50–55. [CrossRef]
- Yang, L.; Guo, Y.; Fan, X.; Chen, Y.; Yang, B.; Liu, K.X.; Zhou, J. Amelioration of coagulation disorders and inflammation by hydrogen-rich solution reduces intestinal ischemia/reperfusion injury in rats through NF-kB/NLRP3 pathway. Mediat. Inflamm. 2020, 2020, 4359305.
- Zou, R.; Wang, M.H.; Chen, Y.; Fan, X.; Yang, B.; Du, J.; Wang, X.B.; Liu, K.X.; Zhou, J. Hydrogen-rich saline attenuates acute lung injury induced by limb ischemia/reperfusion via down-regulating chemerin and NLRP3 in rats. Shock 2018, 52, 134–141. [CrossRef]
- Chen, H.; Zhou, C.; Xie, K.; Meng, X.; Wang, Y.; Yu, Y. Hydrogen-rich saline alleviated the hyperpathia and microglia activation via autophagy mediated inflammasome inactivation in neuropathic pain rats. Neuroscience 2019, 421, 17–30. [CrossRef]
- Shao, A.;Wu, H.; Hong, Y.; Tu, S.; Sun, X.;Wu, Q.; Zhao, Q.; Zhang, J.; Sheng, J. Hydrogen-rich saline attenuated subarachnoid hemorrhage-induced early brain injury in rats by suppressing inflammatory response: Possible involvement of NF-kB pathway and NLRP3 inflammasome. Mol. Neurobiol. 2016, 53, 3462–3476.
- Zhuang, K.; Zuo, Y.C.; Scherchan, P.; Wang, J.K.; Yan, X.X.; Liu, F. Hydrogen inhalation attenuates oxidative stress related endothelial cells injury after subarachnoid hemorrhage in rats. Front. Neurosci. 2020, 13, 1441. [CrossRef]
- Hirano, S.i.; Ichikawa, Y.; Sato, B.; Yamamoto, H.; Takefuji, Y.; Satoh, F. Potential therapeutic applications of hydrogen in chronic inflammatory diseases: possible inhibiting role on mitochondrial stress. Int. J. Mol. Sci. 2021, 22, 2549. [CrossRef]
- Jiao, Y.; Yu, Y.; Li, B.; Gu, X.; Xie, K.; Wang, G.; Yu, Y. Protective effects of hydrogen-rich saline against experimental diabetic peripheral neuropathy via activation of the mitochondrial ATP-sensitive potassium channel channels in rats. Mol. Med. Rep. 2020, 21, 282–290. [CrossRef]
- Hirano, S.i.; Ichikawa, Y.; Sato, B.; Takefuji, Y.; Satoh, F. Molecular hydrogen as a medical gas for the treatment of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): possible efficacy based on a literature review. Front. Neurosci. 2022, 841310.
- Hirano, S.i.; Ichikawa, Y.; Sato, B.; Takefuji, Y.; Satoh, F. Successful treatment of myalgic encephalomyelitis/chronic fatigue syndrome using the hydrogen gas: a case report in four patients. Med Gas Res, 2024, 14, in press.
- Hirano, S.i.; Ichikawa, Y.; Sato, B.; Satoh, F.; Takefuji, Y. Hydrogen is promising for medical applications. Clean. Technol. 2020, 2, 529–541. [CrossRef]
- Liu, C.; Kurokawa, R.; Fujino, M.; Hirano, S.i.; Sato, B.; Li, X.K. Estimation of the hydrogen concentration in rat tissue using an airtight tube following the administration of hydrogen via various routes. Sci. Rep. 2014, 4, 5485. [CrossRef]
Figure 1.
ROS production and scavenging systems. Antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase, cannot scavenge ∙OH and ONOO–, which are potent oxidants. In contrast, H2 selectively scavenges ∙OH and ONOO–, converting them to water. ROS: reactive oxygen species; ∙OH: hydroxyl radicals; ONOO–: peroxynitrite.
Figure 1.
ROS production and scavenging systems. Antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase, cannot scavenge ∙OH and ONOO–, which are potent oxidants. In contrast, H2 selectively scavenges ∙OH and ONOO–, converting them to water. ROS: reactive oxygen species; ∙OH: hydroxyl radicals; ONOO–: peroxynitrite.
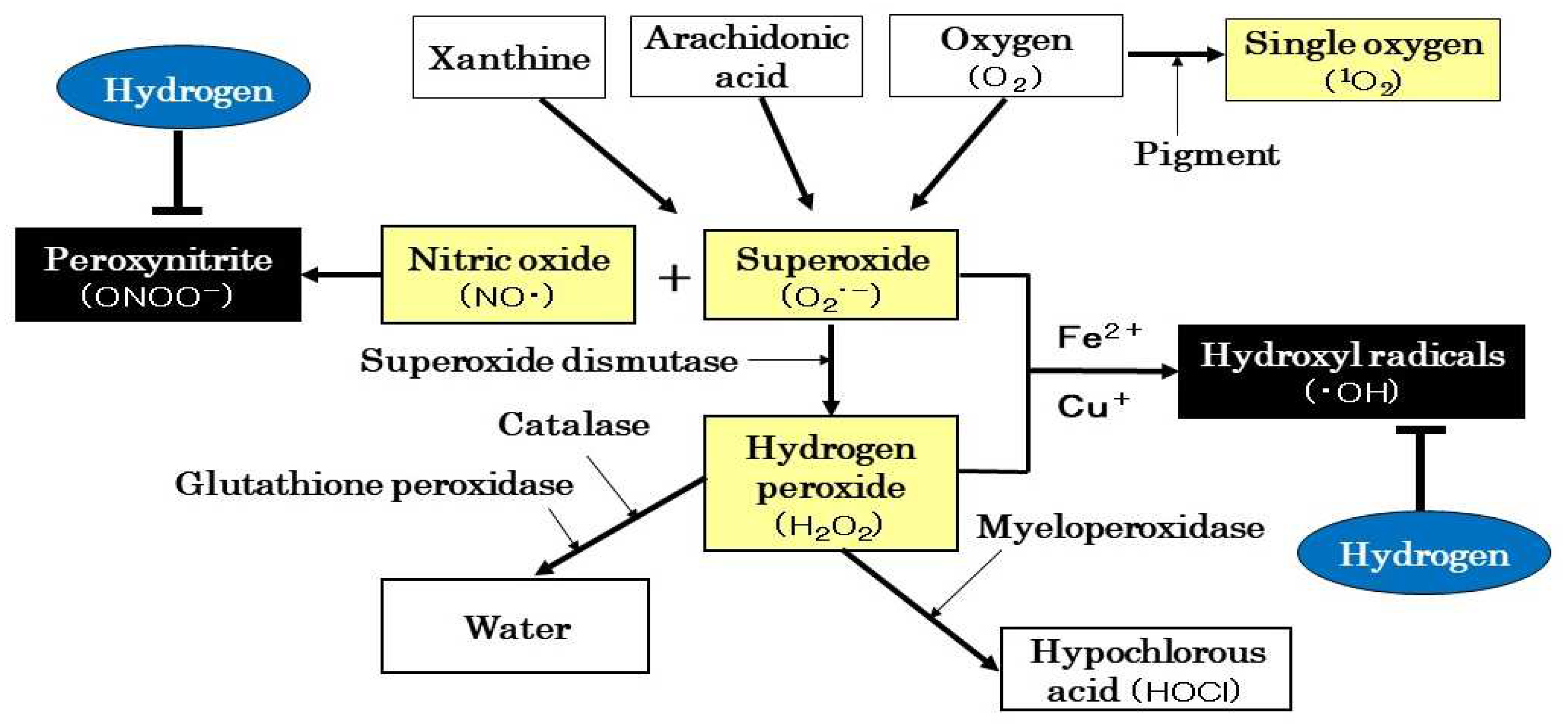
Figure 2.
Possible mechanisms of action of H2 in renal disease. Its effects are mainly categorized as the improvement of mitochondrial function, antioxidant and anti-inflammatory effects, and the regulation of cell lethality and intracellular signal transduction. Bax: Bcl-2-associated x; Beclin-1: damage-regulated autophagy modulator; Bcl-2: B-cell/CLL lymphoma 2; CAT: catalase; CRP: C-reactive protein; d-ROMs: diacron-reactive oxygen metabolites; GPX: glutathione peroxidase; H2: molecular hydrogen; HO-1: heme oxygenase-1; 8-OHdG: 8-hydroxydeoxyguanosine; IL: interleukin; Keap1: Kelch-like ECH-associated protein 1; LC3- II: microtubule-associated protein light chain 3-II; MPO: myeloperoxidase; MAPK: mitogen-activated protein kinase; MCP-1: monocyte chemotactic protein-1; MDA: malondialdehyde; NADPH: nicotinamide adenine dinucleotide phosphate; NF-κB: nuclear factor-κB; Nrf-2: nuclear factor erythroid-related factor 2; ROS: reactive oxygen species; SOD: superoxide dismutase; STAT3: signal transducer and activator of transcription 3; Sirt1: sirtuin-1; TNF-α: tumor necrosis factor-α; TUNEL: tubular terminal transferase dUTP nick-end labeling; TGF-β: transforming growth factor-β1; VEGF: vascular endothelial growth factor; ↑: increase; ↓: decrease.
Figure 2.
Possible mechanisms of action of H2 in renal disease. Its effects are mainly categorized as the improvement of mitochondrial function, antioxidant and anti-inflammatory effects, and the regulation of cell lethality and intracellular signal transduction. Bax: Bcl-2-associated x; Beclin-1: damage-regulated autophagy modulator; Bcl-2: B-cell/CLL lymphoma 2; CAT: catalase; CRP: C-reactive protein; d-ROMs: diacron-reactive oxygen metabolites; GPX: glutathione peroxidase; H2: molecular hydrogen; HO-1: heme oxygenase-1; 8-OHdG: 8-hydroxydeoxyguanosine; IL: interleukin; Keap1: Kelch-like ECH-associated protein 1; LC3- II: microtubule-associated protein light chain 3-II; MPO: myeloperoxidase; MAPK: mitogen-activated protein kinase; MCP-1: monocyte chemotactic protein-1; MDA: malondialdehyde; NADPH: nicotinamide adenine dinucleotide phosphate; NF-κB: nuclear factor-κB; Nrf-2: nuclear factor erythroid-related factor 2; ROS: reactive oxygen species; SOD: superoxide dismutase; STAT3: signal transducer and activator of transcription 3; Sirt1: sirtuin-1; TNF-α: tumor necrosis factor-α; TUNEL: tubular terminal transferase dUTP nick-end labeling; TGF-β: transforming growth factor-β1; VEGF: vascular endothelial growth factor; ↑: increase; ↓: decrease.
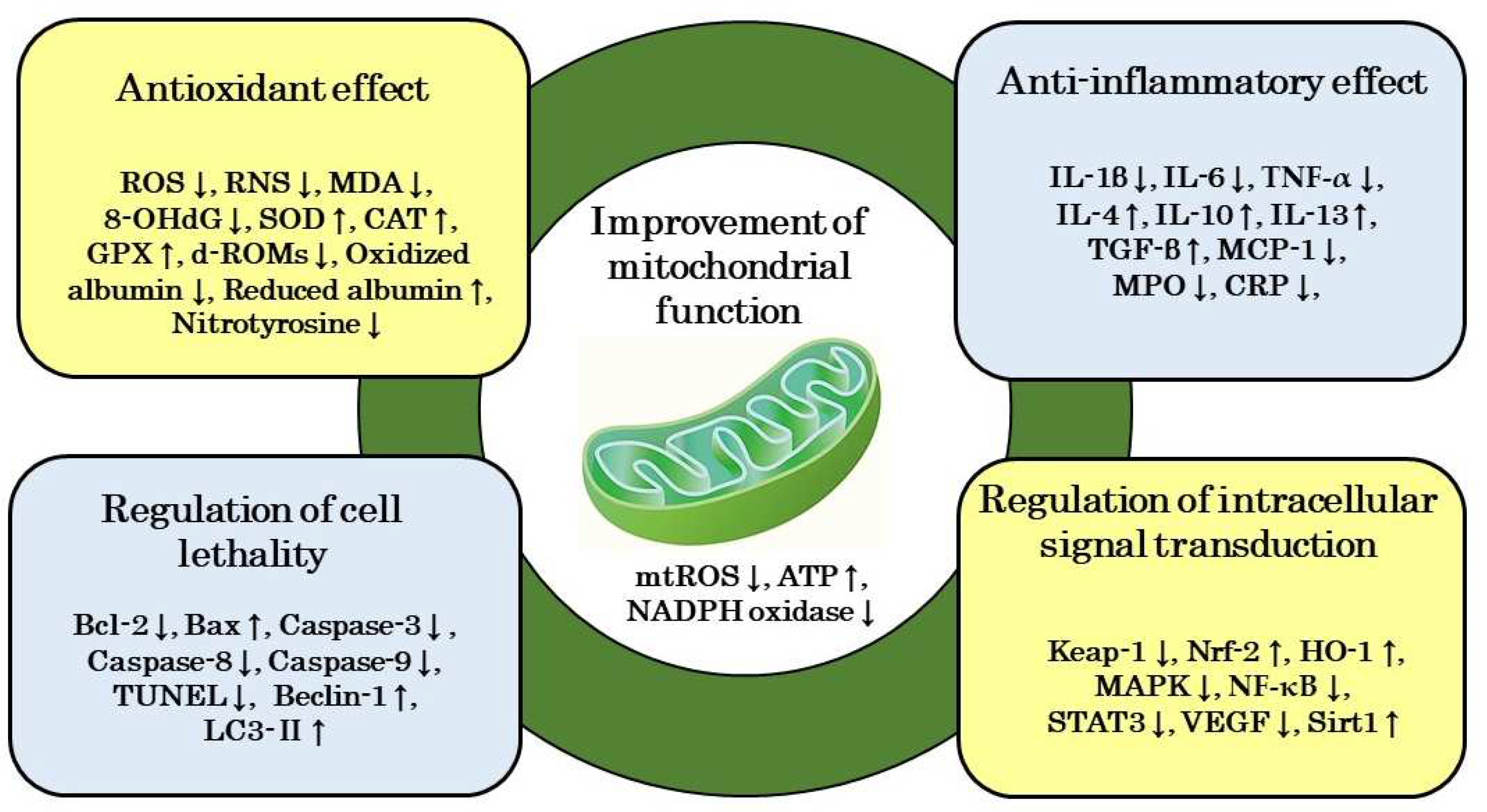
Figure 3.
A possible mechanism by which H2 ameliorates mitochondrial dysfunction in patients with DKD. H2 ameliorates mitochondrial dysfunction by scavenging ·OH and blocks the cascade from NLRP3 activation to the release of inflammatory cytokines, which attenuates chronic inflammation and fibrosis in the kidney. ASC: apoptosis-associated speck-like protein containing a caspase recruitment domain; ATP: adenosine triphosphate; DKD: diabetic kidney disease; FAO: fatty acid oxidation; mtDNA: mitochondrial DNA; mtROS: mitochondrial reactive oxygen species; NLRP3: nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3; ∙OH: hydroxyl radicals; OXPHOS: oxidative phosphorylation; TCA: tricarboxylic acid cycle.
Figure 3.
A possible mechanism by which H2 ameliorates mitochondrial dysfunction in patients with DKD. H2 ameliorates mitochondrial dysfunction by scavenging ·OH and blocks the cascade from NLRP3 activation to the release of inflammatory cytokines, which attenuates chronic inflammation and fibrosis in the kidney. ASC: apoptosis-associated speck-like protein containing a caspase recruitment domain; ATP: adenosine triphosphate; DKD: diabetic kidney disease; FAO: fatty acid oxidation; mtDNA: mitochondrial DNA; mtROS: mitochondrial reactive oxygen species; NLRP3: nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3; ∙OH: hydroxyl radicals; OXPHOS: oxidative phosphorylation; TCA: tricarboxylic acid cycle.

Table 1.
Summary of effects of molecular hydrogen (H2) in animal renal disease models and human renal diseases
Table 1.
Summary of effects of molecular hydrogen (H2) in animal renal disease models and human renal diseases
| Species | Type of H2 | Effects of H2 | Ref. | |
| Diseases | Changes in biomarkers | |||
| Rats | HRS | AKI | Swelling of Mt.↓, BUN↓, Cr↓, 8-OHdG↓ | [21] |
| Rats | HRS | I/R injury | BUN↓, Cr↓, MDA↓, 8-OHdG↓, TNF-α↓, IL-1β↓, IL-6↓, MPO↓, SOD↑, CAT↑ | [22] |
| Rats | HRS | I/R injury | Tissue injury↓, BUN↓, Cr↓, Bcl-2↓, Caspase-3, -8, and -9↓, IL-6↓, TNF-α↓, Bax↑ | [23] |
| Mice | HRS | AKI | Tissue injury↓, BUN↓, Cr↓, Klotho↑, Beclin-1↑, LC3- II↑ | [24] |
| Rats | HRS | I/R injury | BUN↓, Cr↓, MDA↓, 8-OHdG↓, HO-1↑, SOD↑ | [25] |
| Rats | HRW | Renal Transplantation | Overall survival↑, BUN↓, Cr↓, Urinary protein↓, MDA↓, TNF-α↓, IL-6↓, MAPK↓ | [26] |
| Rats | HRUW | Renal Transplantation | Overall survival↑, MDA↓, 8-OHdG↓, TUNEL-stained cells↓, ED-1-positive cells↓, Cr↓, Urinary protein↓ | [27] |
| Rats | HRW | AKI | BUN↓, Cr↓, MDA↓, SOD↑, Caspase-3↓, Cytochrome C↓, Beclin-1↑, LC3- II↑ | [28] |
| Rats | EW | CKD | MCP-1↓, Methylglyoxal↓, BUN↓, Nitrotyrosine staining↓ | [29] |
| Rats | EW | CKD | Age-related histological changes↓, Albuminuria↓, Cardiac remodeling↓, MDA↓, Nitrotyrosine staining↓ | [30] |
| Rats | HRW | CKD | BUN↓, Cr↓, ROS↓, SOD↑, GPX↑, CAT↑, NADPH oxidase↓, TNF-α↓, IL-6↓, IL-1β↓ | [31] |
| Mice | HRW/H2 gas | Cisplatin-induced injury | Histological injury ↓, BUN↓, Cr↓ | [32] |
| Rats | HRW | Fe-NTA-induced injury | Cr↓, BUN↓, MDA↓, ONOO−↓, NADPH oxidase↓, CAT↑, mtROS↓, NF-κB↓, IL-6↓, MCP-1↓, VEGF↓, STAT3↓ | [33] |
| Rats | HRW | Cyclosporin A-induced injury | ROS↓, MDA↓, Keap1↓, Nrf-2↑, HO-1↑ | [34] |
| Mice | H2 gas | Renal stones | MDA↓, 8-OHdG↓, SOD↑, GSH↑, CAT↑, MCP-1↓, IL-10↑ | [35] |
| Rats | HRS | Renal fibrosis | Injury score↓, Apoptosis index↓, Stromal fibrosis↓, MDA↓, SOD↑ | [36] |
| Mice | HRW | Renal fibrosis | Cr↓, BUN↓, Fibrosis↓, EMT↓, Sirt1↑ | [37] |
| Rats | HRW | Renal fibrosis | Fibrosis↓, TGF-β1-positive cells↓, Klotho↑ | [38] |
| Rats | H2 gas | Sepsis-related AKI | BUN↓, Cr↓, MDA↓, TNF-α↓, IL-6↓ | [39] |
| Mice | HRS | Sepsis-related AKI | IL-4 ↑, IL-13↑, IL-10↑, TGF-β↑ | [40] |
| Rats | HRS | Burn-induced AKI | BUN↓, Cr↓, Tubular apoptosis↓, Inflammation↓, MAPK↓, NF-κB↓ | [41] |
| Rats | HRS | AKI | NF-κB↓, ROS↓ | [42] |
| Rats | H2 gas | Hypoxia-induced injury | Renal function↑, Histological damage↓, Oxidative stress↓, Apoptosis↓, MAPK↓ | [43] |
| Humans | HED | PD | Reduced albumin↑, Oxidized albumin↓ | [44] |
| Humans | HED | HD | SBP↓, MCP-1↓, MPO↓ | [45] |
| Humans | HED | HD | Oxidized albumin↓ | [46] |
| Humans | H2 gas | HD | d-ROMs↓, CRP↓ | [47] |
AKI: acute kidney disease; Bax: Bcl-2-associated x; Beclin-1: damage-regulated autophagy modulator; Bcl-2: B-cell/CLL lymphoma 2; BUN: blood urea nitrogen; CAT: catalase; Cr: creatinine; CRP: C-reactive protein; CKD: chronic kidney disease; ↓: decrease; d-ROMs: diacron-reactive oxygen metabolites; EMT: epithelial-mesenchymal transition; GPX: glutathione peroxidase; H2: molecular hydrogen; HRS: hydrogen-rich saline; HRW: hydrogen-rich water; HRUW: H2-rich University of Wisconsin; HED: H2-enriched dialysate; HD: hemodialysis; HO-1: heme oxygenase-1; 8-OHdG: 8-hydroxydeoxyguanosine; Fe-NTA: iron nitrilotriacetate; ↑: increase; IL: interleukin; Keap1: Kelch-like ECH-associated protein 1; LC3-II: microtubule-associated protein light chain 3-II; Mt.: mitochondria; MPO: myeloperoxidase; mtROS: mitochondrial ROS; MAPK: mitogen-activated protein kinase; MCP-1: monocyte chemotactic protein-1; MDA: malondialdehyde; NADPH: nicotinamide adenine dinucleotide phosphate; NF-κB: nuclear factor-κB; Nrf-2: nuclear factor erythroid-related factor 2; PD: peritoneal dialysis; ONOO−: peroxynitrite; Ref.: reference; ROS: reactive oxygen species; SOD: superoxide dismutase; STAT3: signal transducer and activator of transcription 3; Sirt1: sirtuin-1; SBP: systolic blood pressure; TNF-α: tumor necrosis factor-α; TUNEL: tubular terminal transferase dUTP nick-end labeling; TGF-β: transforming growth factor-β1; VEGF: vascular endothelial growth factor
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



