
Submitted:
23 August 2023
Posted:
24 August 2023
You are already at the latest version
Abstract
Artemia (brine shrimp), holds significant value as a live feed for larval fish and crustaceans, owing to their distinctive dietary requirements. However, it is vital to acknowledge that Artemia also carries potential risk as a vector of infection. We conducted a metagenomic analysis to explore the virome present in Artemia cysts collected from inland salt lakes across four distinct regions in Kazakhstan. The study identified the presence of dsDNA phages and RNA virus sequences, with a predominant representation from the Reoviridae, Nodaviridae, Dicistroviridae, Picornaviridae, Astroviridae, Tombusviridae, and Solemoviridae families. In general, this study has significantly enhanced our understanding of the virome of Artemia cysts in the saline lakes of Kazakhstan. It has also furnished crucial insights for the identification, surveillance, and management of viral infections that impact brine shrimp and fish species populations.
Keywords:
Artemia (brine shrimp)
; artemia cysts
; metagenomics analysis
; viral diversity
; saline lakes
1. Introduction
Viruses are extensively dispersed and abundant entities on Earth, particularly in both marine and freshwater environments [1,2,3]. The utilization of a conventional culture-based technique in the investigation of environmental viruses frequently leads to an underestimation of their prevalence and ecological importance [4]. However, the emergence of metagenomic analysis provided a reliable and efficient method for identifying genetic material from individual or pooled samples within a significantly shortened period [5,6]. Also, this methodology enables the discernment of known and novel viral species, thereby enabling the exploration of ecological associations between microorganisms and their respective natural environments [7,8]. Through metagenomics, we obtain deeper insights into the variety, dynamics, and ecological functions of viruses, enriching our knowledge of their influence on ecosystems.
In recent years, due to the significant impact of viruses on microbial communities and ecosystem functions, people have become increasingly interested in investigating the diversity of viruses in salt lakes [9,10,11]. Salt lakes are fascinating ecosystems characterized by high salt concentrations and unique physical and chemical properties [12]. These saline systems are distributed around the world, with prominent examples including the Great Salt Lake in Utah [13], the Dead Sea in the Middle East[14], and the Aral Sea in Central Asia [15]. Hypersaline viral communities across different salinity environments exhibit a remarkable degree of genetic consistency, indicating the formation of a cohesive community [10]. Moreover, viral populations exhibit similarities to the viruses found in hypersaline ponds and lakes that possess diverse salinity levels [9,16]. The awareness of the variety of viruses present in saline water environments contributes to our understanding of the intricate associations between viruses and their hosts, thereby unveiling the ecological dynamics that influence these exceptional habitats.
Artemia, often known as brine shrimp, is a small crustacean that lives in global saline water environments, and this species is of significant importance in upholding ecosystem equilibrium and productivity [17], while also providing sustenance for a wide array of aquatic organisms that rely on these habitats for survival [18,19,20]. Artemia, despite its widespread use as a live feed in diverse applications, presents a potential concern as a vector for transmitting infections into rearing systems [21,22]. Research has demonstrated that artemia has the likely to serve as a vector for parasites, bacteria, and viruses, thereby enabling the dissemination of these diseases over the food chain [23,24]. It is essential to understand the role of artemia as a possible medium for the spread of pathogens in order to establish efficient management methods and reduce the hazards associated with its utilization in aquaculture and other interconnected systems.
The primary objective of this study is to evaluate the viral diversity present in Artemia populations inhabiting salt lake ecosystems in four distinct locations in Kazakhstan. This will be achieved through the utilization of metagenomic analysis. The employed methodology facilitates a thorough investigation of viral populations present in salt lakes located in both the northern and southern areas. The findings of this study contribute to the advancement of knowledge regarding viral dynamics in saltwater lake ecosystems.
2. Materials and Methods
2.1. Sampling Sites and Collection
The Artemia cysts analyzed in this study were sourced from the Fisheries Research and Production Centre in Almaty and were collected from several salt lake ecosystems in four regions of Kazakhstan, namely North Kazakhstan, Pavlodar, Almaty, and Kyzylorda. Figure 1 and Supplementary Table S1 depict the sampling sites and their respective position coordinates, wherein the labeled numbers correspond to the names of the lakes in each region. The sampling sites in the North Kazakhstan region included Pasynki Lake (1), Smirnovka Lake (2), Zhamantuz Lake (3), and Kalibek Lake (4). The Pavlodar region includes several salt lakes, namely Kyzyltuz Lake (5), Ashchitakyr Lake (6), TuzKala Lake (7), and Kazy Lake (8). The Almaty region encompassed two distinct geographical locations, namely Ray Lakes (9) and Tuzkol Lake (10). Similarly, the Kyzylorda region featured Tushchybas Bay (11) and Qashqansu Bay (12) in the Aral Sea as prominent spots within its territory.
Sampling was conducted in the Pavlodar and Kyzylorda regions during the month of August 2021, but in the North Kazakhstan and Almaty areas, sampling happened in August 2022. In order to ensure proper preservation of the samples, they were promptly placed in a sealed icebox after collection and thereafter maintained in a biomedical freezer set at a temperature of -80 °C upon their arrival at the laboratory.
2.2. Viral Nucleic Acid Extraction and Sequencing
For viral nucleic acid extraction, approximately 10 mg of Artemia cysts were collected from each of the 12 saline lakes. These samples were then grouped based on their individual regions, resulting in the creation of four composite pools based on the sampling region. The pooled samples were homogenized using a TissueLyser (Qiagen) in conjunction with lysis buffer (Buffer RLT Plus, Qiagen) and 5 mm stainless steel beads, achieving comprehensive mixing through vigorous vortexing. Following this, the homogenized samples were centrifuged at a force of 10,000 times the acceleration due to gravity using a benchtop microcentrifuge for a duration of 5 min at a temperature of 4°C in order to remove any remnants of cyst debris. The centrifugation stage was performed twice in order to provide optimal separation. In addition, a 0.45 μm filter (Merck Millipore, MA, USA) was employed to exclude particles of eukaryotic and bacterial cell sizes from every pooled sample. The DNase kits (Turbo DNase from Thermo Fisher, Baseline-ZERO from Epicentre, and Benzonase from Novagen) and RNase kits (Promega, Madison, WI, USA) were utilized to enzymatically degrade unprotected nucleic acid present in the filtrates that were enriched with viral particles [25,26]. The residual viral RNA and DNA were subsequently isolated using the QIAamp viral RNA Minikit (Qiagen) in accordance with the protocol provided by the manufacturer.
Four pooled samples were produced, each containing virus sequences of both DNA and RNA, subjected to reverse transcription (RT) and double-stranded DNA construction utilizing QIASeq Stranded RNA Library Kit (Qiagen, Germany) along with a random hexamer primer at a concentration of 100 pmol. Subsequently, four libraries were generated via the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, USA) and sequenced on the Miseq Illumina platform. The obtained raw data were submitted to the NCBI SRA database with the accession number PRJNA1000065.
2.3. Metagenome Data Analysis
The data processing was performed on a Linux machine located locally, utilizing the DIAMOND + MEGAN analytic combination as described in reference [27]. Initially, the raw metagenomic data obtained from each pool underwent preprocessing using Fastp software (version 0.20.0) with the following parameters: “-n 0 -l 30 -5 -r -W 5 -cut_mean_quality 20” [28]. This preprocessing step aimed to exclude low-quality sequencing tails and adapters. This stage involved determining the overall quantity of clean reads acquired from the four pools. To ensure contamination control, the sequencing reads were aligned to the scaffold-level genome of Artemia sp. Kazakhstan ARC1039 (Assembly accession: ASM2916890v1) using BWA-mem v. 0.7 tools [29], and afterward removed from the dataset.
Subsequently, the quality-controlled reads were de novo assembled applying MEGAHIT v1.2.9, with “-k-min 21 -k-max 141 -k-step 20” [30]. In order to taxonomically annotate the assembled contigs, a comparison was made against the NCBI’s nr database (ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz) using DIAMOND blastx v. 0.9.24. The E-value cutoff used was <10-5 [31]. Subsequently, the reads were taxonomically classified, and the results were visualized using the default setting in MEGAN (MEtaGenome Analyzer, version 6.24.23, Tübingen, Germany) [27,32]. The MEGAN outputs, which were received in the form of summarised reads, were utilized to present a comprehensive overview of the taxonomic classification of the sequencing reads (Table S2). Exclusion criteria were applied to the reads originating from cellular organisms, including bacteria, archaea, and eukaryotes. The primary objective of the study was to specifically target and acquire reads resembling those of viruses.
To eliminate false positive virus hits, further analysis of virus-like reads was performed using DIAMOND Blastx (E-value <10-10) and compared with the NCBI virus reference sequence (RefSeq) database (https://ftp.ncbi.nih.gov/refseq/release/viral/) to identify sequences with higher similarity to viral sequences compared to non-viral sequences. The obtained results were further analyzed using MEGAN (v. 6.24.23) software, employing the lowest common ancestor (LCA) parameter. A minimum score cutoff of 100 and an e-value threshold of 1.0E-10 were applied during the analysis.
In order to attain a full classification of virus-like sequences within our dataset, an extensive collection of detailed information on various virus groupings was undertaken. These categories include double-strand DNA (dsDNA), single-strand positive-sense RNA (ssRNA(+)), and double-strand RNA (dsRNA). In addition, we gathered data from two key sources regarding well-documented virus hosts, including bacteria, invertebrates, vertebrates, and plants. The first material cited in this study was obtained from the ICTV website, which may be accessed at http://talk.ictvonline.org (Davison, 2017) [33]. The second source utilized in this study was the ViralZone database, a comprehensive repository of genetic and taxonomic data pertaining to viral diseases. The database may be accessed at the following URL: http://viralzone.expasy.org [34].
2.4. Phylogenetic Analysis
The phylogenetic analyses in our work were carried out with predicted amino acid sequences, as well as the nearest viral relatives identified using top BLASTx matches, and representative individuals from the corresponding viral species. The sequence alignment was performed using Clustal W [35], utilizing the default values. The aligned sequences underwent trimming in order to correspond to specific nucleotide orders of the viral sequences that were retrieved. The maximum-likelihood method was employed to construct a phylogenetic tree in MEGA-X (v. 10.2.6) [36]. The analysis incorporated 500 bootstrap resamples to provide statistical support. The Geneious Prime software (version 2023.2.1) was utilized to anticipate potential open reading frames (ORFs)[37].
3. Results
3.1. Overview of Sequencing Outcomes
The present investigation involved the gathering of twelve batches of Artemia cysts derived from saline lakes situated in the northern and southern regions of Kazakhstan (Figure 1). The Illumina MiSeq platform was utilized to generate a substantial number of raw paired-end reads (8,108,513 in total) from the four pooled cyst samples. After starting the Fastp filtering method, a significant fraction of the raw reads, ranging from 91.8% to 93.3% (7,486,652 reads), were successfully retained. After aligning the reads with the genome of Artemia parthenogenetica, the average number of reads in each pool was found to be 1.85 million (95% CI: 1.22–2.48 million reads), as shown in Table 1. All raw sequences obtained during this experiment have been stored in the NCBI Sequence Read Archive (www.ncbi.nlm.nih.gov/sra) under the accession codes SRR25455639, SRR25455640, SRR25455638, and SRR25455641.
3.2. Contig Analysis
The utilization of MEGAHIT resulted in the generation of a total of 39,863 contigs with diverse lengths. The contig selection process for each individual region is summarised in Table 1. The examination of the North Kazakhstan region yielded the discovery of a grand total of 18,182 contigs. The contigs had an average length of 487 base pairs (bp), with a maximum contig length of 9,836 bp. A comprehensive set of 5,526 contigs was identified in the samples obtained from the Pavlodar region, exhibiting varying lengths spanning from 468 bp to 9,067 bp. The cyst specimens collected from the areas of Almaty and Kyzylorda produced a combined total of 5,205 contigs, with lengths ranging from 507 bp to 9,717 bp, and 10,950 contigs, with lengths ranging from 473 bp to 11,780 bp, respectively.
Following the assembly of contigs, the homology-based
identification step was conducted by running BLASTx tools in Diamond and
applying an e-value threshold of less than 10-5. And contigs
originating from cellular organisms (eukaryotes, bacteria, and archaea) were
omitted from the obtained results. Furthermore, contigs lacking significant
similarity to any known amino acid sequences were also excluded. Subsequently,
a total of 168 reads (containing phages) were identified among the remaining
contigs. These putative viral contigs accounted for a proportion that varied
from 0.88% to 1.77% of the total contigs found in each pool, as shown in
Supplementary Figure S1. To enhance the
accuracy of the dataset, two measures were implemented. Initially, we
eliminated virus contigs that were identified as false positives and those
exhibiting limited levels of amino acid similarity to established viral
sequences. Furthermore, we retained contigs that were classified as potential
viral entities while excluding those that exhibited similarities to established
RNA or DNA viruses. Subsequently, the contigs that were kept passed additional
analysis. Consequently, a sum of 105 contigs was identified as the best match
with viral reads. The percentage of reads associated with viruses in each pool
ranged from 0.17% to 0.43% (Table 1).
3.3. Composition of the Artemia Viral Community
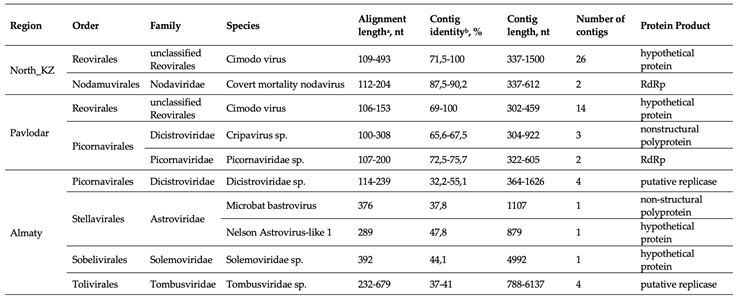
The identification of viral sequences in our study was mostly based on homology, which resulted in a restricted range of DNA and RNA virus groups. This was determined by the examination of sequence contigs. The contigs demonstrating a significant level of similarity to the viral genome were methodically categorized into seven viral families, comprising a total of nine distinct species. In addition, our research has effectively identified a collective of seven phage families, comprising a set of 25 unique species (Table 2, Tables S3 and S4). Furthermore, in addition to the aforementioned findings, there have been identifications of viruses that have yet to be classified.
The classification of these viral families was established by the analysis of their unique patterns of host infection. There are seven families of double-stranded (ds) DNA viruses that have been identified, most of which host bacteria, and one of these families remains unclassified. In addition, a total of six families were formed, including single-stranded (ss) RNA viruses, with a particular focus on positive-strand viruses. It is important to state that two of these viral families have been shown to have associations with vertebrate hosts, whereas another two were recognized for their ability to infect plants. Furthermore, a specific family demonstrated a unique capacity to infect invertebrates, while the other family consisted of a virus capable of infecting both vertebrates and invertebrates. A distinct viral family is identified within the taxonomy of viruses, which is defined by its association with a non-enveloped double-stranded (ds) RNA virus. The virus exhibits a distinct capacity to infect a diverse range of hosts, including both vertebrates and invertebrates. Additionally, the presence of unclassified RNA and DNA viruses was identified (Figure 2a).
A significant proportion of the viral sequences were linked to the dsRNA viruses within the Reoviridae family, making up 38.1% of the overall reads of virus-like sequences. The second largest virus group is dsDNA viruses, especially phages classified as Caudoviricetes, which consist of six virus families and unclassified Caudovirales. These accounted for 36.2% of the overall viral reads. Furthermore, an analysis of the viral sequence reads revealed that 17.1% of them were associated with ssRNA viruses. These ssRNA viruses belonged to several families, including Dicistroviridae (6.67%), Tombusviridae (3.81%), Astroviridae (1.9%), Picornaviridae (1.9%), Nodaviridae (1.9%), and Solemoviridae (0.95%). The viral reads that accounted for the remaining 8.6% were classified as unclassified viruses, as depicted in Figure 2b.
3.4. Distribution of Viral Families in Lakes of Different Regions
Through careful analysis of the data obtained, we found that some patterns of viral diversity of Artemia cysts among different regions emerge in Table 2 and Figure 2a. The Artemia cysts from the Almaty region are notable for their exceptional viral variety, which encompasses four distinct viral species from different families: Dicistroviridae, Tombusviridae, Astroviridae, and Solemoviridae. In materials from the Pavlodar region, which is known for its hypersaline ecosystems, there have been observations of four distinct viral species belonging to three viral families: Reoviridae, Picornaviridae, and Dicistroviridae. The subject of our inquiry now turns to the saline lakes found in the North Kazakhstan region, where two viral species can be identified belonging to the viral families Reoviridae and Nodaviridae. On the other hand, the Kyzylorda region is notable for its high occurrence of contigs that are classified within bacteriophage families. It is noteworthy that, other than an unidentified DNA virus, no other viral contigs were identified in this study. After doing a thorough comparative examination of viral diversity in different places, a significant observation becomes apparent. Although viral species and families exhibit diverse compositions, the overall amount of diversity is very low.
3.5. Characteristics of Selected Viruses
After characterizing the host properties, the dataset followed a process of excluding plant viral reads (Tombusviridae, Solemoviridae), bacteriophages, previously unidentified virus reads, and those that provide too few sequence reads (Astroviridae). This facilitated a shift in our attention towards a comprehensive examination of specific viruses associated with both vertebrate and invertebrate hosts, which are classified under the viral families Reoviridae, Nodaviridae, Dicistroviridae, and Picornaviridae. As part of this study, we performed an analysis and comparison of the partial genome sequences of these viruses with their closest relatives using phylogenetic analysis.
- Reoviridae
The viral family Reoviridae is categorized within the taxonomic order Reovirales. The viruses under consideration exhibit a non-enveloped, segmented double-stranded RNA genome and have the ability to infect a diverse array of hosts, encompassing mammals, birds, reptiles, fish, insects, and plants [38]. It is worth noting that the Reoviridae family has been found to be highly abundant in the pooled Artemia cysts collected from the North Kazakhstan and Pavlodar regions. Close relatives of unclassified Reoviridae, specifically the Cimodo virus (CMDV), were discovered during our investigation. CMDV was isolated from mosquitoes and consists of a total of 12 genomic segments [39]. The virus that has been recognized is classified as Artemia reo-like virus. A total of 40 contigs belonging to the Artemia reo-like virus were identified from the pooled samples, originating from two distinct regions. It has been observed that a distinct resemblance exists between the Artemia reo-like virus and the CMDV. The amino acid identities between them display a range from 71.5% to 100% in the North Kazakhstan region and 65.6% to 100% in the Pavlodar region, highlighting the genetic similarity between them.
To further investigate the genetic links between the Artemia reo-like virus and closely related reference sequences, comprehensive phylogenetic research was undertaken. The study involved creating a phylogenetic tree based on the sequence of a particular segment of the Artemia reo-like virus genome. The partial contig sequence (length 1500 nt) was selected because of its high amino acid identity and covering over 80% of the hypothetical protein segment 6 of CMDV. The result showed that there is a closer relatedness between the Artemia reo-like virus and CMDV (Accession No. KF880765.1) (Figure 3). Our data suggested that Artemia reo-like virus belonged to the unclassified Reoviridae family based on the result of a homology similarity and phylogenetic analysis.
- Nodaviridae
The family Nodaviridae is comprised of small, non-enveloped RNA viruses with positive-sense, single-stranded genomes. This family can be further divided into two separate genera known as alphanodavirus and betanodavirus. These viral species demonstrate a remarkable ability to infect a wide range of hosts, including insects, fish, and crustaceans [40].
In the course of our inquiry, we encountered a couple of viral sequences belonging to the Nodaviridae family, which we have designated as an Artemia noda-like virus, in Artemia cysts from the North Kazakhstan region. During the BLASTx analysis, an identity level of amino acid similarity, specifically 90%, was observed between the contig in query and the sequence of the Covert Mortality Nodavirus (CMNV). The analyzed sequences had similarities to the RNA-dependent RNA polymerase gene (RdRp) of CMNV isolated from Larimichthys, with the contigs covering approximately 11%–20% of the Open Reading Frame 1 (3132nt) of CMNV (Accession No. MW625911.1).
Following the previous evaluation of similarities, a thorough examination of the phylogenetic relationships was conducted (Figure 4). This investigation employed the contig sequence of the Artemia noda-like virus, focusing on the longest length, along with other viruses, particularly those resembling the CMNV. Based on phylogenetic research utilizing the partial genome sequence of Artemia noda-like virus, it was observed that the genome of this virus exhibited clustering with other viruses belonging to the CMNV in unclassified Nodaviridae.
- Dicistroviridae
The Dicistroviridae family comprises a group of small, non-enveloped RNA viruses with single-stranded genetic material that demonstrates a host range that includes insects and crustaceans. The family can be classified into two genera, namely Cripavirus and Aparavirus. The viruses belonging to the Dicistroviridae family exhibit tiny icosahedral geometries and feature positive-sense RNA genomes that are typically 8–10 kilobases in size [41,42].
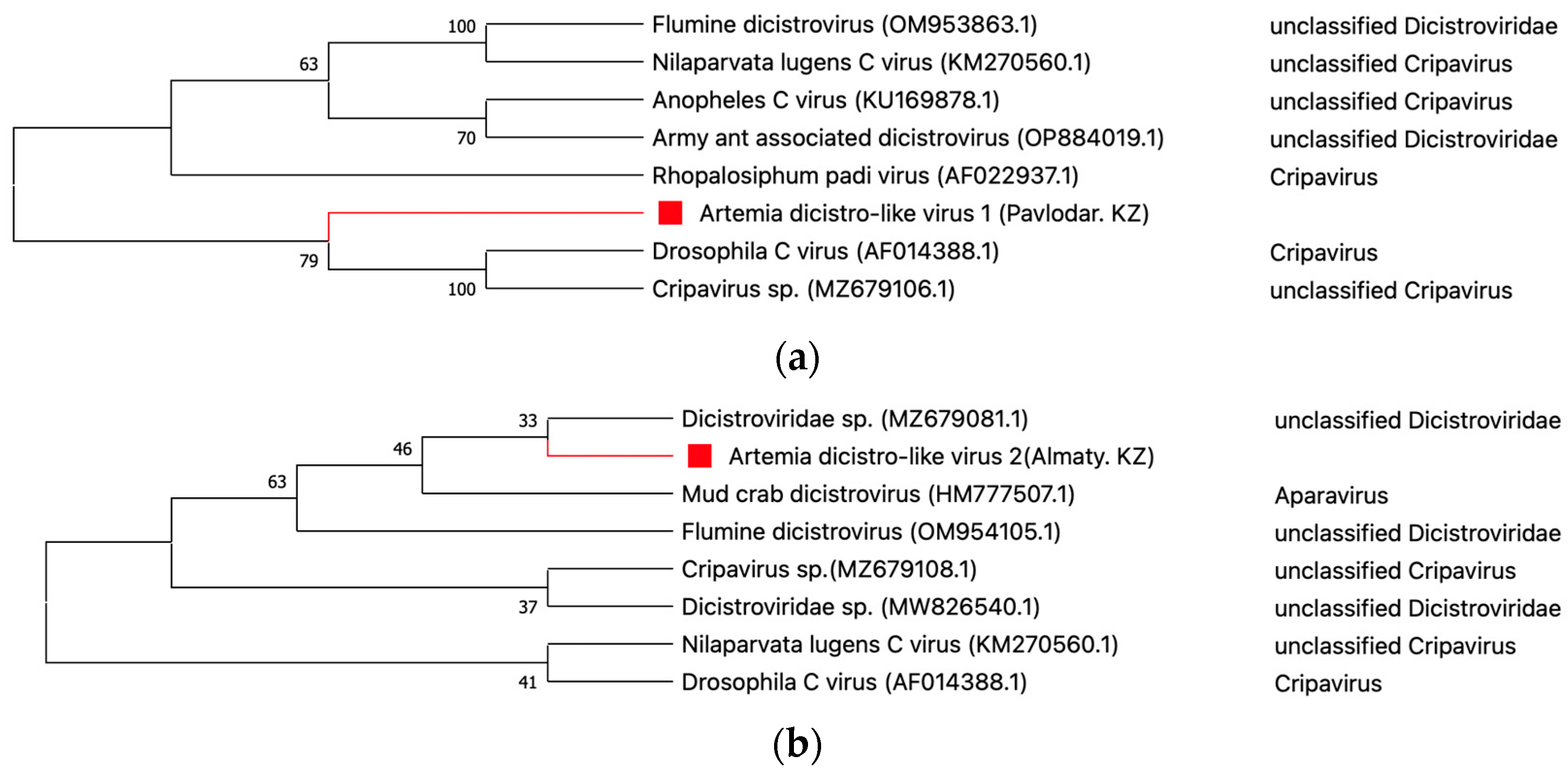
The current study has detected sequences of Artemia dicistro-like viruses in both the Pavlodar and Almaty regions. Within the pooled sample of Artemia originating from the Pavlodar region, a limited number of sequences (Artemia dicistro-like virus 1) displayed the highest resemblance to the Cripavirus genus. These sequences demonstrated an amino acid identity ranging from 65% to 68% for nonstructural polyprotein genes of Cripavirus sp. The Artemia dicistro-like virus 1, which has close phylogenetic clustering with members of the Cripavirus genus (Figure 4a), has been discovered to occupy approximately 17% of the ORF1 (5343 nt) of the drosophila C virus, which has a length of 9264 bp (Accession No. AF014388.1). On the other hand, the genomic sequence of Artemia dicistro-like virus 2 was detected in the Almaty region, exhibiting the highest resemblance to Dicistroviridae sp. The amino acid sequence similarity between the two entities varied from 32% to 55%. In phylogenetic analysis, the queried sequence was subjected to clustering with the Dicistroviridae sp. (Accession No. MZ679081.1) (Figure 4b).
Figure 5.
Phylogenetic relationships of Artemia dicistro-like virus 1 (a) and Artemia dicistro-like virus 2 (b). The nonstructural polyprotein genes of Artemia dicistro-like virus 1 (Pavlodar. KZ) and Artemia dicistro-like virus 2 (Almaty. KZ) were aligned with representative selected sequences from phylogenetically related viruses using a maximum likelihood approach to create the phylogenetic tree. The numbers indicated on the branches are bootstrap support values. Notably, a red square clearly highlights the virus that was found in this particular investigation.
Figure 5.
Phylogenetic relationships of Artemia dicistro-like virus 1 (a) and Artemia dicistro-like virus 2 (b). The nonstructural polyprotein genes of Artemia dicistro-like virus 1 (Pavlodar. KZ) and Artemia dicistro-like virus 2 (Almaty. KZ) were aligned with representative selected sequences from phylogenetically related viruses using a maximum likelihood approach to create the phylogenetic tree. The numbers indicated on the branches are bootstrap support values. Notably, a red square clearly highlights the virus that was found in this particular investigation.
- Picornaviridae
The family Picornaviridae comprises viruses that exhibit small, non-enveloped particles that possess a linear single-stranded RNA (+) genome with a size ranging from 7.1 to 8.9 kilobases (kb). The present genome exhibits polyadenylation and is comprised of a single ORF that encodes a polyprotein. These viruses exhibit significant diversity and possess a broad spectrum of hosts, infecting not just human beings but also several vertebrate and invertebrate species [43,44].
Artemia picorna-like viral sequences, closely resembling an unidentified virus from the Picornaviridae family, were identified in the Pavlodar region. Two contigs were generated by assembling the acquired sequences of Picornaviridae-like viruses. The initial contig exhibited a 76% amino acid identity, covering 54% of the partial polyprotein gene of Picornaviridae sp. (Accession No. ON162097.1). The second contig exhibited a 73% amino acid identity in the incomplete RdRp gene, spanning a length of 605 base pairs, which corresponds to 54% of the RdRp gene of Picornaviridae sp. (Accession No. ON161882.1). In order to further our comprehension of the evolutionary correlation, we performed a phylogenetic analysis utilizing the most extensive contig sequence (Figure 6). Consequently, the Artemia picorna-like virus exhibited clustering with the unclassified Picornaviridae Clinch picorna-like virus (Accession No. MT341476.1).
4. Discussion
Metagenomic research, coupled with homology-based data processing, has provided evidence for all three categories of viruses, encompassing both single-stranded and double-stranded RNA/DNA viruses. The viral groups under consideration consist of a combined total of 15 viral families and 35 viral species. These groups can infect a wide range of hosts, including vertebrates, invertebrates, plants, and microbes. The present study highlights the dynamic diversity observed mostly in dsDNA viruses, with a specific emphasis on the abundance of bacteriophages. The seven viral families in this specific category, together representing 36.2% of viral families, underscore their basic importance within the viral ecology. The prevalence of phages was not a surprise, since earlier studies have demonstrated the predominance of phage families in comparison to other viruses in the saltwater ecosystem [9,10,16,45]. However, it is crucial to acknowledge that bacteriophages did not constitute the primary focus of our investigation.
- Reoviridae
The findings of our analysis have shed light on the dominant viral group, the Reoviridae family, which accounts for 38% of the overall viral readings. As previously mentioned, this particular family is classified under a category of dsRNA viruses renowned for their capacity to infect a diverse array of hosts [38]. Significantly, our research has observed a plausible occurrence of cross-species transmission involving the Artemia reo-like virus detected in pooled Artemia cysts originating from the northern region of Kazakhstan. Significantly, the present virus has a considerable degree of genetic resemblance to the hypothetical protein of the Cimodo virus, which belongs to the Reoviridae family and was originally discovered in mosquitoes inhabiting an African rainforest. CMDV is purportedly responsible for the classification of a new genus in the Spinareovirinae subfamily [39]. The veracity of this association was additionally corroborated through the use of phylogenetic research, as depicted in Figure 3. Multiple study articles have provided evidence for the presence of diverse reo-like viruses, such as the Hubei reo-like virus, the French Guiana reovirus, and the Shenzhen reo-like virus [46,47,48]. A recent study documented the discovery of the diaphorina citri cimodo-like virus within the Asian citrus psyllid insect [49]. Moreover, the cimodo-like virus in brine shrimp has been detected in some countries’ brine shrimp cysts, including Russia, China, Iran, and the United States [50].
Moreover, the earlier result implies a plausible ecological interdependence between brine shrimp and mosquitoes, maybe facilitated by an unknown intermediary species. The complex interaction of viruses linked to aquatic animals highlights their ability to move across various species within aquatic ecosystems. According to these observations, additional extensive investigation is imperative to ascertain the potential transmission of these viruses to fish and other animals reliant on brine shrimp or inhabiting similar environments. It is imperative to comprehend the potential effects of these interactions and transmission pathways in order to get insight into the broader implications for aquatic ecosystems and their inhabited species.
- Nodaviridae
The present study not only emphasizes the widespread occurrence of the Reoviridae family but also reveals the intriguing existence of the Nodaviridae family inside the brine shrimp population. In the context of our research, we have identified a viral strain classified under the Nodaviridae family, specifically referred to as the Artemia noda-like virus, present within the Artemia population residing in North Kazakhstan. By employing phylogenetic analysis and homology-based identification techniques, we have identified sequences present in the Artemia noda-like virus that exhibit similarities with the covert mortality nodavirus (CMNV), an unclassified virus belonging to the Nodaviridae family. Previous studies have demonstrated that CMNV possesses the ability to cause several types of pathology to the tissues and organs of prominent farmed crustaceans [51,52]. Moreover, CMNV has exhibited its capacity to surpass species boundaries and infect a wide array of cultivated and wild fish species, obtained from various habitats including prawn ponds or drainage channels that have been impacted by CMNV [53,54]. This statement emphasizes the inherent capacity of CMNV to surpass taxonomic divisions, therefore highlighting its ability to infect both vertebrate and invertebrate taxa.
The identification of the Artemia noda-like virus in the northern region of Kazakhstan prompts inquiries regarding species interactions and the potential involvement of Artemia in the transmission of the virus. The discernible correlation between brine shrimp and other aquatic organisms underscores the necessity for comprehensive investigation in order to reveal the implications for aquatic ecosystems and various taxa in the North Kazakhstan region. Furthermore, an examination of CMNV-like sequences within the Artemia population has the potential to provide valuable knowledge regarding the virus’s ability to adapt and its effects on both invertebrates and vertebrates.
- Dicistroviridae
The identification of Dicistroviridae genome sequences among Artemia cysts originating from the Pavlodar and Almaty regions serves as a noteworthy illustration of their capacity to thrive in various ecological habitats. It is postulated that the occurrence of an unclassified Cripavirus within artemia cysts found in the salt lakes of the Pavlodar region could potentially contribute to the partial impact of these viruses on the indigenous invertebrate fauna in the indicated region. In contrast, the Artemia dicistro-like virus 2 found in the Almaty region exhibits lower levels of amino acid similarity when compared to the unclassified Dicistroviridae. Based on published research studies, the presence of the Dicistroviridae family has been observed in aquatic invertebrates inhabiting saltwater habitats. Two examples of viruses include the Crustacea picorna-like virus N14, which was found in the South China Sea, and the Bivalvia picorna-like virus D23, which was discovered in the East China Sea [55]. Nevertheless, the coverage levels of the genomes belonging to the Dicistroviridae family exhibited comparatively low values in both the regional pooled samples. Further investigation is necessary to ascertain the classification of these viruses within the Dicistroviridae family.
- Picornaviridae
The identification of members belonging to the Picornaviridae family within the Pavlodar region contributes to the advancement of our knowledge regarding the diversity of viruses. The family Picornaviridae is considered one of the most prominent and substantial collections of ssRNA viruses [43,44]. Significantly, there has been significant growth in the Picornavirales order in recent years. This expansion can be attributed to the identification of previously unidentified picornaviruses using advanced sequencing techniques, such as next-generation sequencing, across a wide range of animals, including vertebrates, arthropods, algae, humans, insects, and plants [55,56]. The investigation conducted by our research examined the detection of an unidentified virus belonging to the Picornaviridae family in the samples collected from the Pavlodar region. Although, the virus has a restricted number of contigs, posing a significant challenge in accurately identifying the true viral species.
Moreover, the prevalence of plant viruses, vertebrate viruses, and unidentified viruses is seen in almost all pooled samples. This statement highlights the critical necessity for additional investigative studies aimed at understanding the complex viral dynamics present among populations of Artemia. Nevertheless, it is important to acknowledge that certain crustacean host viruses that have been identified as detrimental to aquacultures, such as the white spot virus, yellow head virus, viral nervous necrosis (VNN), and Lymphocystis disease virus (LCDV), were not identified in the samples examined throughout the period of this investigation [22,57,58].
In addition, it is vital to point out the viral diversity that has been discovered within the examined populations of Artemia cysts. The limited diversity observed in this study may be influenced by various factors, including specific sampling locations, salinity degrees, timing of sampling, temperature conditions, and sample size [9,10,16,45]. As previously stated, the collecting sites for Artemia cysts in the North Kazakhstan and Pavlodar areas are located in the northern part of Kazakhstan, whereas the Almaty and Kyzylorda regions are located in the southern part of the country (Figure 1). It is important to note that the temperatures and seasonal circumstances in these regions also exhibit variations based on their respective geographical locations. The availability of sample collection locations in the southern region is comparatively limited in comparison to the northern region. Future studies should take into account these considerations, such as collecting larger sample sizes and minimizing variations in sample site factors, in order to conduct a full assessment of viral diversity in these ecosystems.
5. Conclusions
Overall, this pioneering research offers novel and significant perspectives on the artemia virus communities residing in saline aquatic environments across four distinct regions of Kazakhstan. Furthermore, these investigations serve as a foundation for further research endeavors aimed at exploring the evolutionary dynamics of viruses within their natural reservoir hosts. Additionally, they enable the anticipation of the potential impact of viral diseases on fish and crustacean aquaculture, thereby facilitating the advancement of sustainable production in this field.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Information about the sampling location in the four regions (sampling site number, sampling site, sampling location, sampling dates); Table S2: Overview of Sequencing Reads Classification at the Domain Level (DIAMOND Blastx and MEGAN-LCA) Figure S1: Proportion of the classification level of sequencing reads for different regions; Table S3: Summary of the total count of viral readings obtained from 15 distinct viral families and unclassified viruses; Table S4: Overview of contigs of Bacteriophage species identified using BLASTx in Artemia cysts collected from four regions
Author Contributions
Conceptualization, A.K and K.K.; methodology, T.S., K.K; software, M.K.; validation, T.S., Ye.K. and Zh.M.; formal analysis, Ye.K.; investigation, T.S., M.K., K.K.; resources, Ye.K., Zh.M., S.A.; data curation, Ye.K.; writing—original draft preparation, M.K.; writing—review and editing, A.K and K.K.; visualization, M.K.; supervision, A.K.; project administration, Zh.M.; funding acquisition, K.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Ecology and Natural Resources of the Republic of Kazakhstan (Grant No. BR10264205).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The obtained raw data were submitted to the NCBI Sequence Read Archive (SRA) database with the BioProject accession number PRJNA1000065.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Fuhrman, J.A. Marine Viruses and Their Biogeochemical and Ecological Effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Rohwer, F. Viral Metagenomics. Nat Rev Microbiol 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Elbehery, A.H.A.; Deng, L. Insights into the Global Freshwater Virome. Front Microbiol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Bergh, Ø.; BØrsheim, K.Y.; Bratbak, G.; Heldal, M. High Abundance of Viruses Found in Aquatic Environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Bassi, C.; Guerriero, P.; Pierantoni, M.; Callegari, E.; Sabbioni, S. Novel Virus Identification through Metagenomics: A Systematic Review. Life 2022, 12, 2048. [Google Scholar] [CrossRef] [PubMed]
- Varghese, F.S.; van Rij, R.P. Insect Virus Discovery by Metagenomic and Cell Culture-Based Approaches. Methods in Molecular Biology 2018, 1746, 197–213. [Google Scholar] [CrossRef]
- Rosario, K.; Breitbart, M. Exploring the Viral World through Metagenomics. Curr Opin Virol 2011, 1, 289–297. [Google Scholar] [CrossRef]
- Hall, R.J.; Wang, J.; Todd, A.K.; Bissielo, A.B.; Yen, S.; Strydom, H.; Moore, N.E.; Ren, X.; Huang, Q.S.; Carter, P.E.; et al. Evaluation of Rapid and Simple Techniques for the Enrichment of Viruses Prior to Metagenomic Virus Discovery. J Virol Methods 2014, 195, 194–204. [Google Scholar] [CrossRef]
- Bhattarai, B.; Bhattacharjee, A.S.; Coutinho, F.H.; Goel, R.K. Viruses and Their Interactions With Bacteria and Archaea of Hypersaline Great Salt Lake. Front Microbiol 2021, 12. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Ravet, V.; Colombet, J.; Bettarel, Y.; Auguet, J.-C.; Bouvier, T.; Lucas-Staat, S.; Vellet, A.; Prangishvili, D.; et al. Analysis of Metagenomic Data Reveals Common Features of Halophilic Viral Communities across Continents. Environ Microbiol 2016, 18, 889–903. [Google Scholar] [CrossRef]
- Oren, A. Microbial Life at High Salt Concentrations: Phylogenetic and Metabolic Diversity. Saline Syst 2008, 4, 2. [Google Scholar] [CrossRef]
- Waiser, M.J.; Robarts, R.D. Saline Inland Waters. In Encyclopedia of Inland Waters; Elsevier, 2009; pp. 634–644. [Google Scholar] [CrossRef]
- Baxter, B.K. Great Salt Lake Microbiology: A Historical Perspective. International Microbiology 2018, 21, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Bodaker, I.; Sharon, I.; Feingersch, R.; Rosenberg, M. Archaeal Diversity in the Dead Sea : Microbial Survival under Increasingly Harsh Conditions. Natural Resources and Environmental Issues 2009, 15. [Google Scholar]
- Izhitskiy, A.S.; Zavialov, P.O.; Sapozhnikov, P. V.; Kirillin, G.B.; Grossart, H.P.; Kalinina, O.Y.; Zalota, A.K.; Goncharenko, I. V.; Kurbaniyazov, A.K. Present State of the Aral Sea: Diverging Physical and Biological Characteristics of the Residual Basins. Sci Rep 2016, 6, 23906. [Google Scholar] [CrossRef]
- Motlagh, A.M.; Bhattacharjee, A.S.; Coutinho, F.H.; Dutilh, B.E.; Casjens, S.R.; Goel, R.K. Insights of Phage-Host Interaction in Hypersaline Ecosystem through Metagenomics Analyses. Front Microbiol 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Van Stappen, G.; Sui, L.; Hoa, V.N.; Tamtin, M.; Nyonje, B.; Medeiros Rocha, R.; Sorgeloos, P.; Gajardo, G. Review on Integrated Production of the Brine Shrimp Artemia in Solar Salt Ponds. Rev Aquac 2020, 12, 1054–1071. [Google Scholar] [CrossRef]
- Leger, P.; Bengtson, D. a; Simpson, K.L.; Sorgeloos, P. The Use and Nutritional Value of Artemia as a Food Source. Oceanogr.Mar.Biol.Ann.Rev. 1986, 24. [Google Scholar]
- Conceição, L.E.C.; Yúfera, M.; Makridis, P.; Morais, S.; Dinis, M.T. Live Feeds for Early Stages of Fish Rearing. Aquac Res 2010, 41, 613–640. [Google Scholar] [CrossRef]
- Lavens, P.; Sorgeloos, P. The History, Present Status and Prospects of the Availability of Artemia Cysts for Aquaculture. Aquaculture 2000, 181, 397–403. [Google Scholar] [CrossRef]
- Sivakumar, V.K.; Sarathi, M.; Venkatesan, C.; Sivaraj, A.; Hameed, A.S.S. Experimental Exposure of Artemia to Hepatopancreatic Parvo-like Virus and Subsequent Transmission to Post-Larvae of Penaeus Monodon. J Invertebr Pathol 2009, 102, 191–195. [Google Scholar] [CrossRef]
- Valverde, E.J.; Labella, A.M.; Borrego, J.J.; Castro, D. Artemia Spp., a Susceptible Host and Vector for Lymphocystis Disease Virus. Viruses 2019, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Lavens, P.; Sorgeloos, P. Manual on the Production and Use of Live Food for Aquaculture. 1996; Vol. 361. [Google Scholar]
- Avila-Villa, L.A.; Martínez-Porchas, M.; Gollas-Galván, T.; López-Elías, J.A.; Mercado, L.; Murguia-López, Á.; Mendoza-Cano, F.; Hernández-López, J. Evaluation of Different Microalgae Species and Artemia (Artemia Franciscana) as Possible Vectors of Necrotizing Hepatopancreatitis Bacteria. Aquaculture 2011, 318, 273–276. [Google Scholar] [CrossRef]
- Victoria, J.G.; Kapoor, A.; Li, L.; Blinkova, O.; Slikas, B.; Wang, C.; Naeem, A.; Zaidi, S.; Delwart, E. Metagenomic Analyses of Viruses in Stool Samples from Children with Acute Flaccid Paralysis. J Virol 2009, 83, 4642–4651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Pesavento, P.A.; Delwart, E. Faecal Virome of Cats in an Animal Shelter. J Gen Virol 2014, 95, 2553–2564. [Google Scholar] [CrossRef]
- Bağcı, C.; Patz, S.; Huson, D.H. DIAMOND+MEGAN: Fast and Easy Taxonomic and Functional Analysis of Short and Long Microbiome Sequences. Curr Protoc 2021, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H. [Heng Li - Compares BWA to Other Long Read Aligners like CUSHAW2] Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv preprint arXiv 2013. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Gautam, A.; Felderhoff, H.; Bağci, C.; Huson, D.H. Using AnnoTree to Get More Assignments, Faster, in DIAMOND+MEGAN Microbiome Analysis. mSystems 2022, 7. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus Taxonomy: The Database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Hulo, C.; de Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A Knowledge Resource to Understand Virus Diversity. Nucleic Acids Res 2011, 39, D576–D582. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Attoui, H.; Mertens, P.P.C.; Becnel, J.; Belaganahalli, S.; Bergoin, M.; Brussaard, C.P.; Chappell, J.D.; Ciarlet, M.; del Vas, M.; TSD, V.T. Ninth Report of the International Committee on Taxonomy of Viruses. Family: Reoviridae 2011, 541–603. [Google Scholar]
- Hermanns, K.; Zirkel, F.; Kurth, A.; Drosten, C.; Junglen, S. Cimodo Virus Belongs to a Novel Lineage of Reoviruses Isolated from African Mosquitoes. Journal of General Virology 2014, 95, 905–909. [Google Scholar] [CrossRef]
- Sahul Hameed, A.S.; Ninawe, A.S.; Nakai, T.; Chi, S.C.; Johnson, K.L. ICTV Virus Taxonomy Profile: Nodaviridae. Journal of General Virology 2019, 100, 3–4. [Google Scholar] [CrossRef]
- Warsaba, R.; Sadasivan, J.; Jan, E. Dicistrovirus-Host Molecular Interactions. Curr Issues Mol Biol 2020, 34, 83–112. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E. ICTV Virus Taxonomy Profile: Dicistroviridae. Journal of General Virology 2017, 98, 355–356. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. Journal of General Virology 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Ryu, W.-S. Picornavirus. In Molecular Virology of Human Pathogenic Viruses; 2017; pp. 153–164. [Google Scholar] [CrossRef]
- Gong, Z.; Liang, Y.; Wang, M.; Jiang, Y.; Yang, Q.; Xia, J.; Zhou, X.; You, S.; Gao, C.; Wang, J.; et al. Viral Diversity and Its Relationship With Environmental Factors at the Surface and Deep Sea of Prydz Bay, Antarctica. Front Microbiol 2018, 9. [Google Scholar] [CrossRef]
- Ribeiro, G.d.O.; Monteiro, F.J.C.; Rego, M.O. da S.; Ribeiro, E.S.D.; Castro, D.F. de; Caseiro, M.M.; Souza Marinho, R. dos S.; Komninakis, S.V.; Witkin, S.S.; Deng, X.; et al. Detection of RNA-Dependent RNA Polymerase of Hubei Reo-Like Virus 7 by Next-Generation Sequencing in Aedes Aegypti and Culex Quinquefasciatus Mosquitoes from Brazil. Viruses 2019, 11, 147. [Google Scholar] [CrossRef]
- Truchado, D.A.; Llanos-Garrido, A.; Oropesa-Olmedo, D.A.; Cerrada, B.; Cea, P.; Moens, M.A.J.; Gomez-Lucia, E.; Doménech, A.; Milá, B.; Pérez-Tris, J.; et al. Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery. Microorganisms 2020, 8, 1869. [Google Scholar] [CrossRef]
- Guo, L.; Lu, X.; Liu, X.; Li, P.; Wu, J.; Xing, F.; Peng, H.; Xiao, X.; Shi, M.; Liu, Z.; et al. Metatranscriptomic Analysis Reveals the Virome and Viral Genomic Evolution of Medically Important Mites. J Virol 2021, 95. [Google Scholar] [CrossRef]
- Britt, K.; Stevens, K.; Gebben, S.; Levy, A.; Al Rwahnih, M.; Batuman, O. Partial Genome Sequence of a Novel Reo-Like Virus Detected in Asian Citrus Psyllid (Diaphorina Citri) Populations from Florida Citrus Groves. Microbiol Resour Announc 2021, 10. [Google Scholar] [CrossRef]
- Dong, X.; Li, C.; Wang, Y.; Hu, T.; Zhang, F.; Meng, F.; Han, X.; Wang, G.; Qin, J.; Nauwynck, H.; et al. Diversity and Connectedness of Brine Shrimp Viruses in Global Hypersaline Ecosystems. 2023. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, Q.; Liu, S.; Yang, H.; Liu, S.; Zhu, L.; Yang, B.; Jin, J.; Ding, L.; Wang, X.; et al. A New Nodavirus Is Associated with Covert Mortality Disease of Shrimp. Journal of General Virology 2014, 95, 2700–2709. [Google Scholar] [CrossRef]
- Liu, S.; Wang, X.; Xu, T.; Li, X.; Du, L.; Zhang, Q. Vectors and Reservoir Hosts of Covert Mortality Nodavirus (CMNV) in Shrimp Ponds. J Invertebr Pathol 2018, 154, 29–36. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, T.; Wan, X.; Liu, S.; Wang, X.; Li, X.; Dong, X.; Yang, B.; Huang, J. Prevalence and Distribution of Covert Mortality Nodavirus (CMNV) in Cultured Crustacean. Virus Res 2017, 233, 113–119. [Google Scholar] [CrossRef]
- Liu, S.; Xia, J.; Tian, Y.; Yao, L.; Xu, T.; Li, X.; Li, X.; Wang, W.; Kong, J.; Zhang, Q. Investigation of Pathogenic Mechanism of Covert Mortality Nodavirus Infection in Penaeus Vannamei. Front Microbiol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Sanfaçon, H.; Wellink, J.; Le Gall, O.; Karasev, A.; van der Vlugt, R.; Wetzel, T. Secoviridae: A Proposed Family of Plant Viruses within the Order Picornavirales That Combines the Families Sequiviridae and Comoviridae, the Unassigned Genera Cheravirus and Sadwavirus, and the Proposed Genus Torradovirus. Arch Virol 2009, 154, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; He, Y.; Chen, X.; Kalim, U.; Wang, Y.; Yang, S.; Qi, H.; Cheng, H.; Lu, X.; Wang, X.; et al. Viral Metagenomics Reveals Diverse Viruses in the Feces Samples of Raccoon Dogs. Front Vet Sci 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Salgado, L.; Olveira, J.G.; Dopazo, C.P.; Bandín, I. Role of Rotifer ( Brachionus Plicatilis ) and Artemia ( Artemia Salina ) Nauplii in the Horizontal Transmission of a Natural Nervous Necrosis Virus (NNV) Reassortant Strain to Senegalese Sole ( Solea Senegalensis ) Larvae. Veterinary Quarterly 2020, 40, 205–214. [Google Scholar] [CrossRef]
- Walker, P.J.; Mohan, C. V. Viral Disease Emergence in Shrimp Aquaculture: Origins, Impact and the Effectiveness of Health Management Strategies. Rev Aquac 2009, 1, 125–154. [Google Scholar] [CrossRef]
Figure 1.
Locations of artemia cysts collection spots in four regions of Kazakhstan. The red dots in the map represent the sampling locations allocated to each region, and the marked numbers represent the names of the corresponding lakes. Source: Wikipedia.
Figure 1.
Locations of artemia cysts collection spots in four regions of Kazakhstan. The red dots in the map represent the sampling locations allocated to each region, and the marked numbers represent the names of the corresponding lakes. Source: Wikipedia.
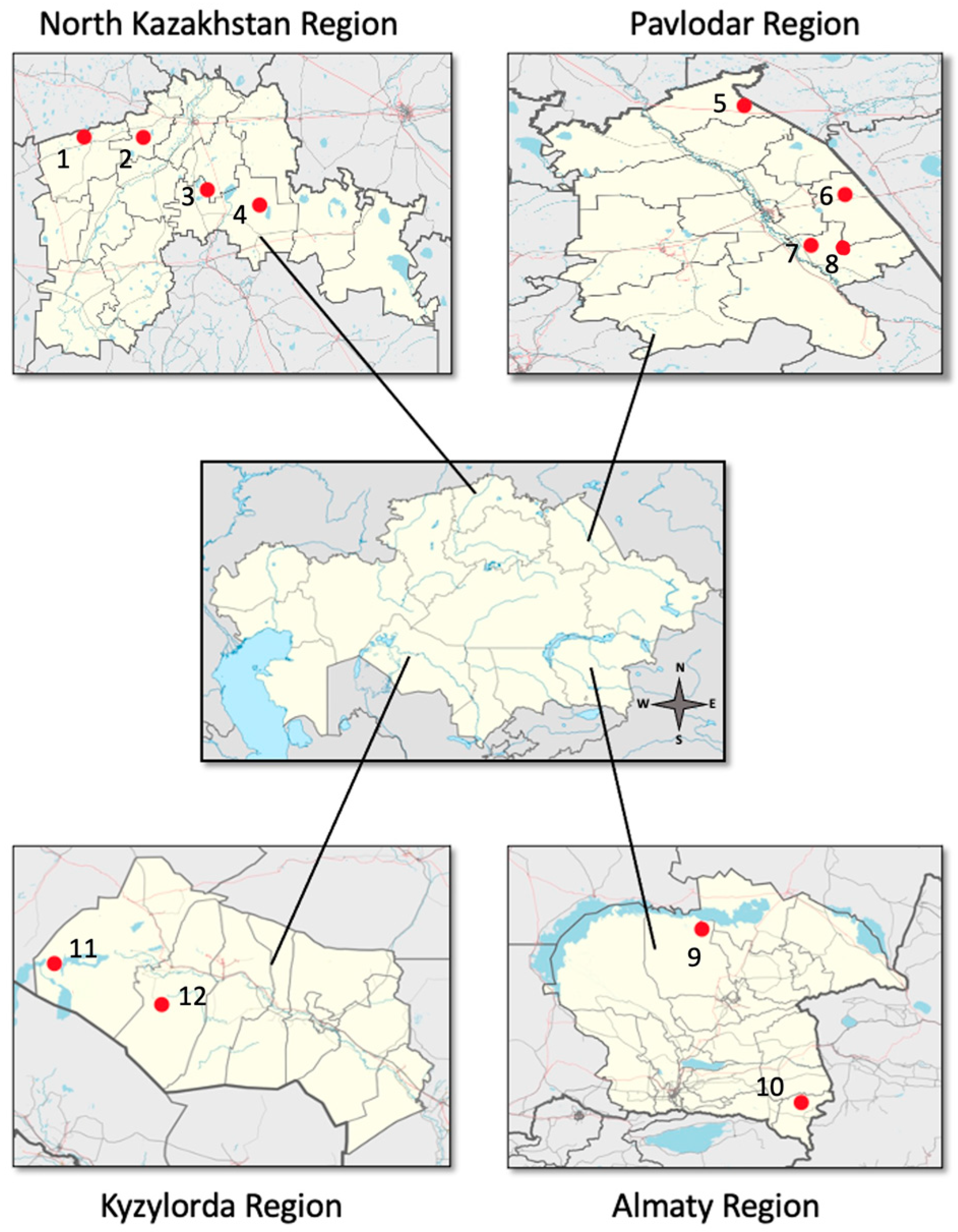
Figure 2.
Distribution of virus sequence reads within a specific virus family. (a) The distribution of several virus families among taxonomic classes. The y-axis of the graph depicts the number of individual viruses belonging to each viral family, while the x-axis displays the taxonomic categorization and corresponding viral family. (b) Proportion of virus sequences in different virus families within the classification.
Figure 2.
Distribution of virus sequence reads within a specific virus family. (a) The distribution of several virus families among taxonomic classes. The y-axis of the graph depicts the number of individual viruses belonging to each viral family, while the x-axis displays the taxonomic categorization and corresponding viral family. (b) Proportion of virus sequences in different virus families within the classification.
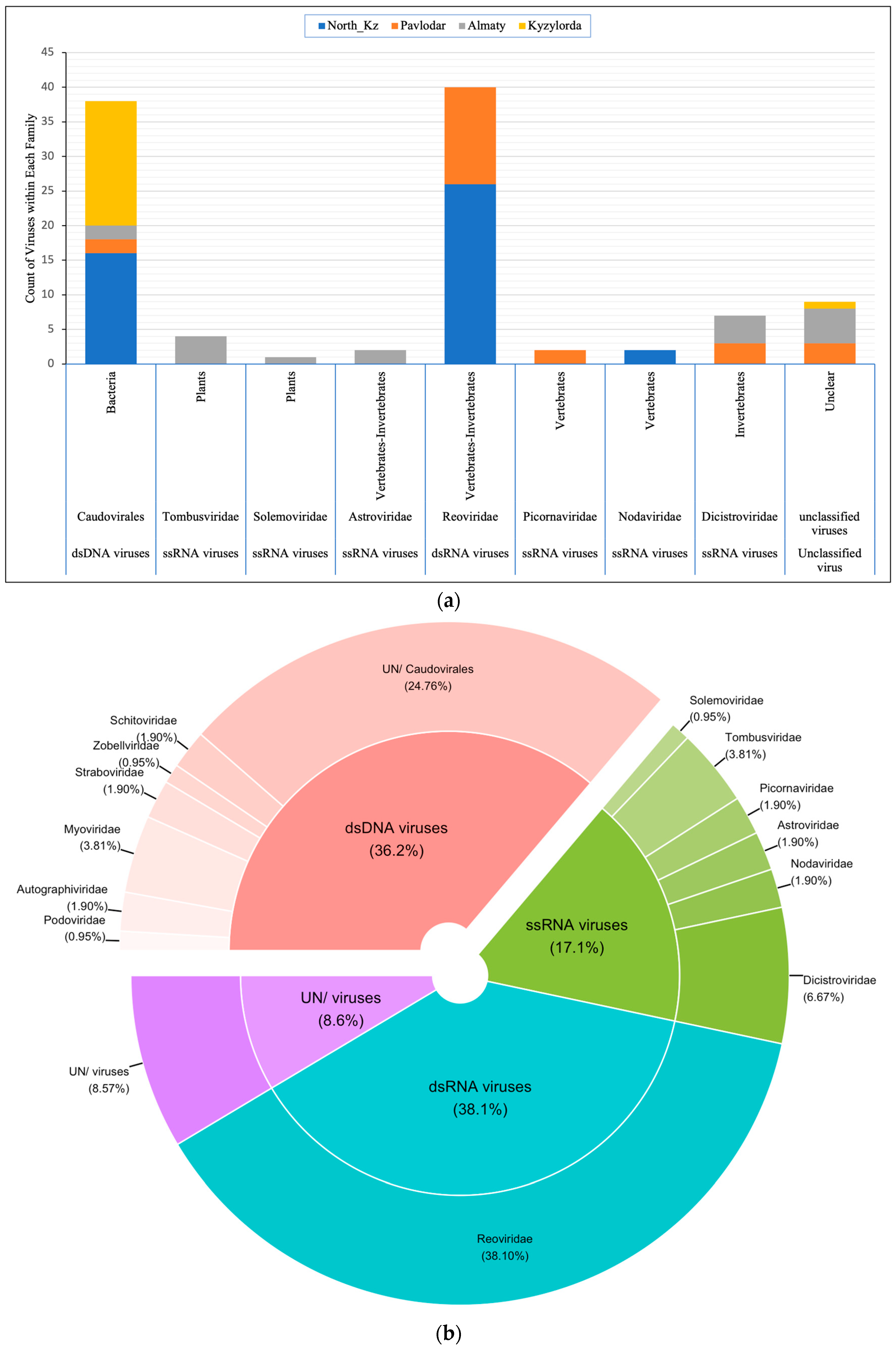
Figure 3.
Phylogenetic relationships of the Artemia reo-like virus. A phylogenetic tree was constructed using a maximum likelihood method, utilizing the alignment of the conserved portion of the partial hypothetical protein of Artemia reo-like virus (segment 6) and a set of representative sequences from phylogenetically related viruses. The numerical values assigned to the branches of the tree diagram represent bootstrap support levels. The virus found in this specific investigation is prominently indicated using a red square.
Figure 3.
Phylogenetic relationships of the Artemia reo-like virus. A phylogenetic tree was constructed using a maximum likelihood method, utilizing the alignment of the conserved portion of the partial hypothetical protein of Artemia reo-like virus (segment 6) and a set of representative sequences from phylogenetically related viruses. The numerical values assigned to the branches of the tree diagram represent bootstrap support levels. The virus found in this specific investigation is prominently indicated using a red square.
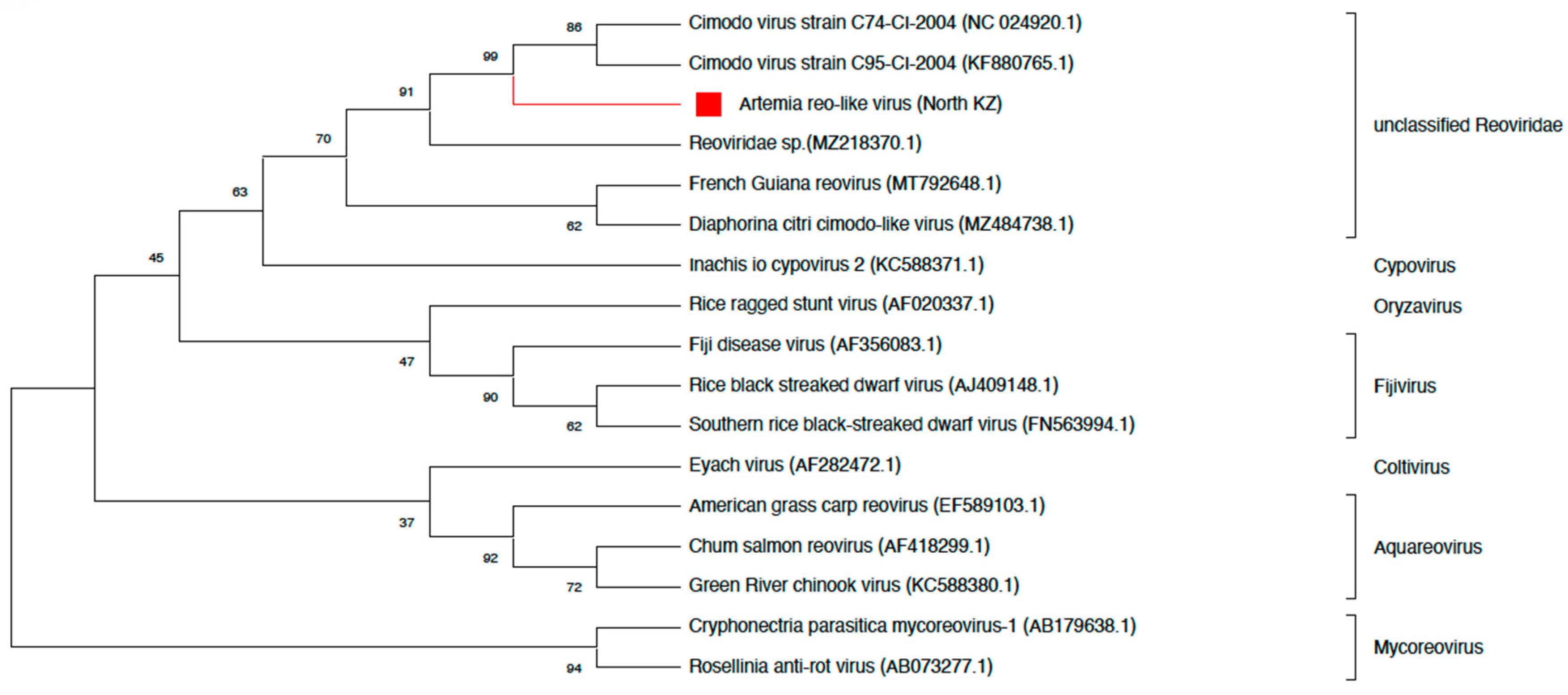
Figure 4.
Phylogenetic relationships of Artemia noda-like virus. The construction of the phylogenetic tree utilized a maximum likelihood methodology, employing the alignment of the RdRp partial genome of the Artemia noda-like virus alongside a carefully chosen set of representative sequences from phylogenetically similar viruses. The numerical values assigned to the branches of the tree diagram represent bootstrap support levels. The virus found in this specific investigation is prominently indicated using a red square.
Figure 4.
Phylogenetic relationships of Artemia noda-like virus. The construction of the phylogenetic tree utilized a maximum likelihood methodology, employing the alignment of the RdRp partial genome of the Artemia noda-like virus alongside a carefully chosen set of representative sequences from phylogenetically similar viruses. The numerical values assigned to the branches of the tree diagram represent bootstrap support levels. The virus found in this specific investigation is prominently indicated using a red square.

Figure 6.
Phylogenetic relationships of Artemia picorna-like virus. The construction of the phylogenetic tree involved the implementation of a maximum likelihood method. This method utilized the alignment of the incomplete RdRp gene of the Artemia picorna-like virus with a set of carefully chosen representative sequences from viruses that are phylogenetically related. The numerical numbers depicted on the branches signify bootstrap support values. Significantly, the virus found in this specific study is prominently emphasized within a red square.
Figure 6.
Phylogenetic relationships of Artemia picorna-like virus. The construction of the phylogenetic tree involved the implementation of a maximum likelihood method. This method utilized the alignment of the incomplete RdRp gene of the Artemia picorna-like virus with a set of carefully chosen representative sequences from viruses that are phylogenetically related. The numerical numbers depicted on the branches signify bootstrap support values. Significantly, the virus found in this specific study is prominently emphasized within a red square.


Table 1.
Summary of read and contig counting of pooled cysts of Artemia from four regions of Kazakhstan in each data processing step.
Table 1.
Summary of read and contig counting of pooled cysts of Artemia from four regions of Kazakhstan in each data processing step.
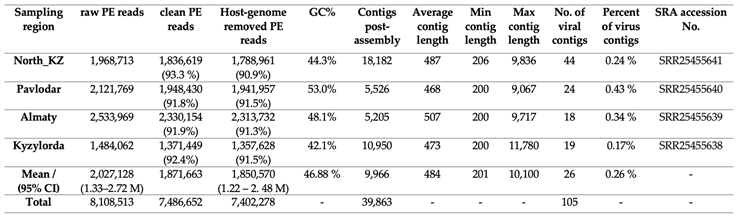 |
Table 2.
Contig count for viral species, families, and orders with a range of alignment length, alignment identity percentage, and contig length.
Table 2.
Contig count for viral species, families, and orders with a range of alignment length, alignment identity percentage, and contig length.
 |
a Contigs sequences over 100 nt in length were characterized. b Determination of identity levels using BLASTx with virus reference strains from NCBI RefSeq database (E-value <1 × 10-10).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



