
Submitted:
24 December 2023
Posted:
26 December 2023
You are already at the latest version
Abstract
Background: One of the most astounding discoveries of recent times is the recognition that cancer embodies a transition from a higher level of metazoan cell organization to a more foundational pre-metazoic state. This shift is steered by genes housed within the ancestral genome compartment, pervasive across all metazoan genomes, encompassing humans, and governed by a pre-metazoic gene regulatory mechanism (aGRN). This work aims to highlight the emerging field of evolutionary cancer cell biology (ECCB), which points to the deep homology between cancer and protist life cycles tracing back to the common ancestor of amoebozoans, metazoans, and fungi (AMF). The ECCB analysis reveals the essence of the non-gametogenic germline of the AMF ancestor, which serves as a blueprint for all metazoan germlines and stem cell lineages and controls the life cycle of cancer. Every germ and stem cell lineage of humans and metazoans traces its lineage back to this Urgermline, transmitting crucial processes such as asymmetric cell cycling, differentiation, stemness, and phenomena like GST and SGT (aka EMT and MET) to their subsequent evolutionary descendants. Oxygen-sensitive germline and stem cells suffer DNA-DSB due to stress and oxygen ranges reminiscent of ancestral hyperoxia, leading to cell senescence. Cells that can overcome senescence can proliferate as DSCD cells with defective symmetric cell division, paving the way for malignancy and PGCC cancers. Conclusions: Understanding cancer from its evolutionary origins may help break some of the logjams in cancer prevention and open up new therapeutic pathways.
Keywords:
cancer
; senescence
; asymmetric and symmetric division
; loss of function
; DNA-damage repair
; reprogramming
; malignancy
1. Introduction
In recent years, the emerging field of evolutionary cancer cell biology (ECCB) has made significant advances, leading to the realization that understanding cancer and carcinogenesis requires consideration of their evolutionary history [1]. Evolutionary life cycle biology has illuminated homologies between the germ and soma life cycles developed by the common AMF ancestor, including its non-gametogenic (NG) Urgermline, and the homologous life cycles of cancer and protists (Table 1). In this context, the NG germline of cancer emerges as a germline with ancestral imprints [2]. Evolutionary cancer cell biology views cancer as a disease caused by the reactivation of an ancient unicellular genome module (premetazoan genome) conserved in the genomes of humans and metazoans [3].
Cancer cells possesse a hybrid genome comprising both constitutive premetazoan genes and more recent multicellular genes. The transition from multicellular to unicellular cell state occurs by genome reprogramming under the control of an ancient gene regulatory network (aGRN). Urgermline genes and their developmental programs not only play a role in oncogenesis and tumorigenesis but also regulate asymmetric cell cycling and asymmetric cell division in non-cancerous and cancerous cells, including stemness and differentiation [4].
DSCD cells in humans and metazoans are defective pre-cancerous cells originating from normal stem cells. This aberrant phenotype arises from severe DNA damage and oxidative stress, needing repair. These cells proliferate through defective symmetric cell divisions (DSCD) and persist within specific niches for repair. Given that metazoans lack effective repair mechanisms for severe genomic damage, human DSCD cells employ an ancient unicellular repair pathway involving cell and nuclear fusion, resulting in hyperpolyploid giant nuclei capable of repairing and remodeling pre-metazoan genomes as evolved by the common AMF ancestor. The multinuclear genome repair syncytya are known as MGRS.
In humans and metazoans, this inadequate ancestral repair mechanisms acts as a genome reprogramming program that governs and regulates the DSCD genome through the gene regulatory network (aGRN) evolved by the common AMF ancestor. However, this ancient genome repair program does not regenerate normal human stem cells. Instead, it generates stem cells imprinted with a unicellular pattern that contributes to the development of cancer (primary CSCs). The regulatory aGRN network reprograms the DSCD genome into a premetazoan-like genome responsible for PGCC cancers, featuring unicellular germ and soma life cycles. Repair and reprogramming by hyperpolyploid MGRS structures are unknown in humans and do not occur in the polyploidy of hepatocytes, cardiomyocytes and mesenchymal stromal cells. Repair and reprogramming via MGRS structures are unknown in humans and do not occur in the endopolyploidy of hepatocytes, cardiomyocytes and mesenchymal stromal cells [5].
Polyploidy As described by Liu et al. 2010, there are various degrees of endopolyploidy: around 80 % of cardiomyocites are tetraploid, carrying two diploid nuclei (2x2N). Approximately 8 % are mononucleated with a diploid 2N or a tetraploid 4N nucleus, and 10 % are octoploid (4N×2). The remaining cells have even an higher ploidy, up to 32N [6].
Researchers propose that the increase in ploidy observed in hepatocytes and cardiomyocytes may be a specific response to intense mitotic stimuli and genotoxicity. As a consequence, cells lack certain parts of the cell division machinery, and adopt an abortive cell cycle mode known as "mitotic slippage," also referred to as mitotic arrest, mitotic block, or mitotic exit [7]. Endopolyploidization occurs through endocycles in individual cells exposed to genotoxic agents like excessive oxygen, irradiation, or chemotherapeutics, which are unable to undergo cell and nuclear fusion. In contrast, hyperpolyploidization through homotypic cell fusion (MGRS) represents an ancient genome repair and rebuilding pathway evolved by the common AMF ancestor. It remains inactive in healthy cells of metazoans, mammals, and humans, including their stem cells. However, when reactivated to repair severely damaged DSCD genomes, it leads to the genome reprogramming towards malignancy. Both forms of polyploidization are encompassed under the term PGCCs in tumors.
Numerous cancer cells and stem cells in aneuploid and tetraploid stages lack the capacity for repair, eventually undergoing apoptosis. The ability to repair genotoxic damage is only realized as cells progress from endopolyploidy to hyperpolyploidy. MGRS structures highlight that lower levels of endopolyploidy (8-16N), observed during the initial phase of MGRS development, yield non-viable daughter nuclei. However, in the subsequent phase, the accumulation of multiple genome copies (>64N) within the giant hyperpolyploid nucleus facilitates the repair and restoration of genomic integrity. This process leads to the creation of viable spores (buds) and the emergence of new generations of CSCs [8].
As accurately defined by Storchova and Pellman twenty years ago, endopolyploidy, is controlled by a specific cellular program involving the acquisition of multiple centromers, aberrant mitosis, genomic instability, apoptosis and/or uncontrolled proliferation“[9]. The researchers also proposed that such a program might enable cardiomyocytes to adapt to different conditions, like hypertrophic growth, aiding in avoiding potentially harmful effects of cell divisions in the working myocardium that must maintain high blood pressure throughout life. We now know that endopolyploidy is also a genome repair mechanism and not merely a modified form of quiescence. It indicates DNA double-strand break damages and the need for damaged cells to undergo repair to regain function.
PGCC cancers are cancer types that employ the uncommon, ancient polyploid or hyperpolyploid genome repair and remodeling pathway. The terms PGCC (Polyploid Giant Cancer Cells) and MGRS (Multinucleated Giant Repair Structures) are synonymous with giant cell structures capable of repairing severe DNA damage in stressed germline cells and stem cell lineages, as well as remodeling or reprogramming nonfunctional genomes. MGRSs, in particular, are multinucleated giant cell structures utilized by premetazoan cell systems to repair dysfunctional germline cells that lack stemness and differentiation potential. In the context of human precancerous DSCD cells, which require repair, the ancient MGRS pathway is employed for precancerous to cancerous cell reprogramming and carcinogenic transformation and the generation of primary CSCs. PGCCs present in cancers are MGRS-like polyploid giant cells capable of repairing cancer germ cells and stem cells damaged by stress. PGCCs aneuploid and tetraploid can also be induced through irradiation and chemotherapeutics.
Genome reprogramming drive human DSCDs into the non-gametogenic germ and soma life cycle of cancer. From an evolutionary perspective, changes in environmental conditions inside or outside the niche initiate DSCD cell repair, which is conducted under the auspices of the only available unicellular repair program and its aGRN network. Consequently, the DSCD cell and its offsprings fall under the definitive control of the aGRN. In this manner, cancer adheres to systemic rules and programs that trace back more than 1000 My in the past (Figure 1). Considering the logic and rationality of adaptationist evolution, as well as the retention and selection of evolutionary traits, cancer is regarded as an anomaly and, ultimately, an evolutionary disaster.
Currently, efforts are underway to identify precancerous cell phenotypes that trigger oncogenesis in all evolutionarily controlled PGCC cancers, regardless of their tissue or origin, as well as to uncover the fundamental principles governing transformation. Evidence indicates that premalignant DSCD cells are dysfunctional cells lacking stemness and differentiation capacity. Despite their origin as stem cells, precancerous DSCD cells cannot give rise to either non-cancerous or cancerous stem cells. They survive and proliferate through defective symmetric cell cycling and defective symmetric cell division. A distinction can be made between early proliferating and older, dormant DSCDs that remain quiescent in the niche for years or undergo slow cycling.
These evolutionary insights have sparked severe controversies regarding the origin of cancer. The central controversy revolves around whether carcinogenesis is solely initiated by genetic alterations, mutations, and driver genes, as taught by the mutational theory. Evolutionary cancer cell biology posits that hyperpolyploidy-related cancers (PGCC cancers) result not from mutations but rather from inappropriate use of old repair mechanisms employed by DSCD cells during the repair process. The molecular processes involved in the reprogramming and subordination of DSCDs to the ancient gene regulatory network aGRN are currently unknown. Somatic mutations are considered secondary in this context.
Mutationist researchers argue that cancer results from an accumulation of genetic alterations and mutations, asserting that the non-mutational origin of cancer, stemming from precancerous DSCD cells through DNA damage repair and reprogramming via hyperpolyploid PGCC-like structures, is overstated. They contend that evolutionary PGCC cancers may be a minor phenomenon (Anna Dart, personal communication).
However, this perspective is flawed. Firstly, driver mutations identified in tumor cells only provide indirect evidence of ongoing carcinogenesis. Secondly, driver mutations and gain-of-function mutations have been identified in a maximum of 40% of tumors [10]. and not in experiments directly inducing transformations. Thirdly, the repair of damaged non-malignant cells through the formation of polyploid giant cell structures and genome reconstruction in single-cell systems is a common occurrence. The primary argument against the anti-evolutionary critique lies in the assertion that most cancers cannot progress to the polyploid and damaged state. However, the opposite is true. Colorectal cancer statistics indicate that over 90% of undifferentiated colorectal cancers exhibit PGCCs [11]. PGCCs are present in approximately 37 % of all human tumors [12].
2. Premetazoan ancestry; symmetric and asymmetric cell phenotypes, and DSCD cells
The non-gametogenic (NG) Urgermline evolved over a thousand million years ago as an oxygen-sensitive hypoxic cell line. As ambient oxygen levels increased, the oxygen-sensitive Urgermline, with its limited DNA damage repair ability, evolved a somatic sister cell line resistant to oxygen and an interactive germ and soma cell system (G+S) capable of maintaining genome integrity. From this point in evolution, these two cell lines have distinct roles: the Urgermline primarily produced stem cells, while the somatic cell line’s function was to safeguard genomic integrity under all environmental conditions. This division of labor persisted throughout the life cycles of protists and cancer.
Under normoxic conditions, both cell lines proliferate through ACD phenotypes capable of asymmetric cell division (ACD). Of the asymmetric progeny, one daughter cell (d1 cell) was self-renewing, while the other daughter cell (d2 cell) entered either a stem cell state, like the germline, or an undifferentiated resting state, like the somatic cell line [1,2]. When the optimal normoxic conditions changed slightly but did not reach hyperoxia, both cell lines transitioned to symmetric cell division phenotypes, resulting in identical daughter cells. This represented a case of phenotypic plasticity without persistent DNA damage and loss of function. When optimal living conditions were restored, both symmetric cycling phenotypes reverted to the ACD phenotype, and the germline once again produced stem cells.
Hyperoxic conditions damaged the DNA of the germline, causing the ACD phenotype to shift into a defective DSCD cell type, devoid of stemness and differentiation potential. These cells remained viable and continued to proliferate through defective symmetric cell cycling but were unable to revert to the normal ACD phenotype. In contrast, somatic cycling phenotypes remained fully functional under hyperoxia conditions and wer able to return to the asymmetric cell cycle.
Hyperoxia, with oxygen levels exceeding 6.0% O2, altered the germline genome and forced germline cells into an irreparable DSCD cell state characterized by defective DNA replication, spindle defects, multinucleation, faulty and premature cell division caused by immature nuclei [1,2,3,4]. These DNA defects cannot be repaired by common DNA repair mechanisms such as homologous recombination (HR). The DSCD cell represented an HR-deficient germline cell that had lost its functionality and differentiation capacity. When cells exposed to hyperoxia returned to normoxia, somatic cells with an intact genome could transform into functional germ cells through soma-to-germ cell transition, generating efficient new germline clones with stemness and ACD potential.
To repair the damaged DSCD genome, the common ancestor of amoebozoans, metazoans and fungi developed repair mechanisms involving cell and nuclear fusion [1,4,13,14]. DSCD cells became fusible, forming multinucleated syncytia. Individual syncytia nuclei fused into hyperpolyploid giant nuclei capable of excising defective DNA regions and restoring genome integrity. Such multinucleated genome repair structures (MGRSs) existed in protists and were known as PGCCs in metazoan cancers.
In protists, the DSCD repair system comprised a multi-step process with four distinct stages. Stage I involved DSCD proliferation or, if sufficient growth resources were not available, a quiescent phase in the niche. Stage II saw cell fusion; DSCD cells became fusible and fused to form multinucleated syncytia. Stage III involved nuclear fusion; individual MGRS nuclei fused into giant hyperpolyploid nuclei capable of repairing DNA damage and reconstructing the functional genome. Stage IV was characterized by cellularization and the formation of spores (buds), restarting the G+S life cycle and generating germline stem cells.
DSCD cells have a long evolutionary history and arose whenever the germline was exposed to hyperoxia with oxygen levels above 6.0%. Defective DSCD phenotype displayed irreparable DNA defects and loss of function, driving self-renewal, stemness, and differentiation. Their repair through cell and nuclear fusion is a characteristic of protists and cancer, absent in healthy humans and mammals.
3. Precancerous DSCD-like cells in humans
DSCD-like phenotypes have also been identified in humans and mammals, manifesting in various tissues and emerging as prime candidates for oncogenic transformation. These cell types are referred to as "very small embryonic-like cells" (VSELs), based on specific pluripotency markers. Researchers have suggested that VSEL cells have a connection with pluripotent stem cells and extragonadal germ cells, which give rise to extragonadal germ cell tumors. According to Barati et al. (2021), VSEL cells may originate from the proximal epiblast, with researchers believing that "a group of pluripotent primordial germ cells resides in a dormant state in other tissues" [15,16]. These cells can be found in various locations, including the bone marrow, brain, pancreas, thymus, intestinal epithelium, as well as the surface epithelium of the ovaries and testes in both humans and rodents [17,18].
Alternatively, it has been proposed that VSELs are a type of migratory stem cell released from the bone marrow niche into the bloodstream under stressful conditions [19]. VSELs have also been isolated from human embryonic stem cell cultures and are presumed to be defective DSCD-like cells that arise in tissues and cultures when oxygen concentrations exceed 6.0% O2. Hence, there are compelling reasons to categorize them within the broader DSCD family.
In our perspective, cells within the DSCD family represent the precancerous cell-of-origin for PGGC cancers. The DSCD family can be defined as disrupted tissue-resident counterparts of adult stem or progenitor cells. It is increasingly apparent that oncogenic transformation is a multistep process analogous to the DSCD repair process observed in protists.
As we unravel how non-gametogenic protist NG germlines respond to stressors and oxygen levels exceeding 6.0%, resulting in irreversible DNA damage and loss of function, as well as understanding the limited repair mechanisms available to them, rooted in unicellular imprints, the evolutionary underpinnings of oncogenic transformation in PGCC cancers have become clearer.
4. Malignant transformation to PGCC cancers
4.1. The point of no return: ACD disruption and DSCD hyperproliferation
Tumor researchers have demonstrated that asymmetric cell division and differentiation in Drosophila may function as a tumor suppressor mechanism, whereas the disruption of ACD can drive cells towards abnormal proliferation and genomic instability. As early as 2014, Gomez-Lopez et al. described how the loss of function in key ACD regulators could potentially be an initiating factor in oncogenesis [20]. According to their findings, ACD disruption results in "hyperproliferative phenotypes in situ, characterized by more symmetric divisions and the generation of misspecified progeny that fail to exit the cell cycle for differentiation, instead continuously proliferating." The loss of ACD cell fate, coupled with increased proliferation and evasion of cell cycle control, was associated with the loss of polarity, spindle control, and mitotic errors - early events in cancer development that align with evolutionary events of ancestral origin observed in protists.
In a 2018 study focused on the normal cellular response to replication stress (RSR), McGrail et al. discovered that extensive DNA damage typically leads to senescence and apoptosis. However, defects in RSR can also result in hyperproliferation, ultimately leading to early tumorigenesis [21]. RSR defects in progenitor cells can elevate cancer risk and place non-malignant cells in a cancer stem cell (CSC)-like state [22]. DNA damage, RSR defects, and hyperproliferation together contribute to genomic instability, serving as key features of early cancer development.
VSELs and EGCs not only express pluripotency markers but also embryonal/ fetal markers. Changes in environmental conditions within the precancerous niche or exiting the niche could induce cells of the DSCD family to continue the symmetric cell cycle, with their offspring entering a fusible cell state.
4.2. Temporary quiescence in precancerous DSCDs and the role of the Igf2/H19 complex
When precancerous DSCD cells reach a point of irreversibility, they face two options: hyperproliferative symmetric proliferation or entry into a temporary quiescent state within the niche, awaiting repair. Disrupted VSEL and EGC phenotypes typify tissue-resident cells, most of which remaining dormant within tissues and niches. The Igf2/H19 complex exhibits antagonistic effects, capable of both promoting and suppressing growth. Downregulation of the growth factor Igf2 leads cellular dormancy, while the upregulation of the tumor suppressor gene H19 stimulates growth and proliferation [23].
Igf2 (insulin-like growth factor 2) plays a pivotal role in regulating cell proliferation, growth, migration, differentiation and survival. Sequence analysis revealed a significant homology with pro-insulin, leading to their classification as IGF1 and IGF2 [24]. The growth-promoting and beneficial functions of IGF2 during embryonic and fetal development, as well as placental growth, are well documented. Bergman et al. noted that one of the first syndromes associating IGF2 expression with a growth disorder was Beckwith-Wiedemann syndrome [25]. This syndrome is particularly interesting noteworthy as it links abnormal growth with subsequent tumour development (as occurring in DSCDs). While overexpression of IGF2 is frequently observed in Wilms’ tumor and was long thought to promote tumors, it was thought that IGF2 can induce apoptosis and necrosis in Wilms' tumor cells.
According to Shoshani et al. [5], endopolyploidization, distinct from hyperpolyploidization via MGRS, potentially counteracts malignancy under environmental stress and respond to stress by increasing H19 expression. Excessive stress leads to the formation of diploid transformed diploid cells and unstable endopolyploid cells, fostering malignancy. Conversely, some cells may evade malignant transformation by undergoing polyploidization, associated with the repression of H19 expression. The researchers suspect that disorders that occur before polyploidization could lead to stable, non-tumorigenic endopolyploid cells. In their experiments, the authors found that diploid cell tumorigenicity could be reduced through artificial tetraploidization, likely due to the concurrent reduction of H19. Therefore, endopolyploidization, particularly tetraploidization coupled with H19 suppression, might exert a cancer-preventative effect by maintaining a noncancerous state. However, it remains unclear which molecular pathways associated with endopolyploidization and tetraploidy protect against transformation and tumorigenesis.
As Gamaev et al. noted, the oncofetal long noncoding RNA H19 is postnatally repressed in most tissues but re-expressed in many cancers, though its role in carcinogenesis remains contentious [26]. Expression levels of H19 and Igf2 varied in nontumor liver tissues and were notably downregulated in most matched tumors. In the nontumor liver tissue of several aged females, H19 was primarily expressed in hepatocytes, coinciding with hepatocyte proliferation.
When considering the involvement of the Igf2/H19 complex in the fate of stem cells and DSCD cells, a rather intricate scenario emerges. It's evident that the regression of Igf2 triggers a temporary quiescent state, while increased H19 levels could prompt these cells to resume growth and proliferation. Reinstating Igf2 expression terminates the transient quiescence, fostering abnormal growth, proliferation, and contributing to tumorigenesis.
Conversely, heightened H19 expression might drive normal stem cells towards DSCD transformation, leading to malignancy, while regression of H19 would result in non-tumorigenic endopolyploid cells, notably tetraploid cells. Consequently, the Igf2/H19 complex functions as a promoter, contributor, or accomplice in malignancy.
4.3 Cell and nuclear fusion; hyperpolyploidization
Cell fusion through homologous cells, as well as the formation of MGRS/PGCC giant cell structures, occur in both protists and cancer. In humans, precancerous DSCD cells exploit the genes of the ancient MGRS repair mechanism, but in irradiated or chemotherapeutically treated cell populations, genome reconstitution can also happen through tetraploidization without cell and nuclear fusion.
There is limited information available on the oncogenic transformation and fusion ability of late VSELs/EGCs cells, but more extensive data exist regarding the homotypic and heterotypic fusion potential of cancer germline cells. Homotypic cell fusions have been observed in glioblastoma cancer cells after irradiation, but they occur more frequently in cultures, indicating that irradiation cannot eliminate the ability of germline-like cells and stem cells to transform into fusible phenotypes [27,28,29,30]. Kaur et al. demonstrated that, after irradiation, mononucleated radioresistant cells enter the G2/M phase [27]. Similar to protist DSCD cells, non-proliferative, radioresistant cells are highly motile and fuse homotypically, thereby facilitating the repair of severe DNA damage. Consequently, homotypic cell fusion is a response of cancer radio-resistant cells to overcome extrinsic stress. It plays a role in the initiation of cancer and is crucial for the formation of cancer stem cells [31]. Homotypic cell fusion is inherited from the Urgermline and serves to repair severe DNA damage and restore genetic integrity. In protists, it contributes to the generation of new germline clones with ACD and differentiation potential, as well as new stem cells with increased virulence [1,2,3,4,13,14].
Heterotypic cell fusions have also been observed in the generation of tumor CSCs, resulting from the fusion of stem and differentiated cells [32,33]. Wei et al. [34] reported spontaneous heterotypic cell fusions in lung cancer and mouse CSCs, whose progeny exhibited a reduced proliferation rate and stemness. Dittmar et al. [35] also observed stem cell-like features in hybrid cells derived from experimentally fused epithelial breast cells and breast cancer cells.
4.4 Reprogramming of human DSCDs
As described earlier, homotypic fusion is part of an ancient mechanism to restore genomic integrity. It evolved in the Urgermline and is inherited in all metazoans, also exploited by cancer cell systems. However, while the MRGS of protists successfully reconstructs and remodels their genome through this unicellular repair mechanism via MRGS structures, the same mechanism is dysfunctional when used for the defective multicellular DSCD cells, serving for cancerous reprogramming and genome remodeling.
There is limited information available about homotypic cell fusion and DSCD genome remodeling during carcinogenesis. Most data refer to the induction of cancerous PGCCs after irradiation and chemotherapeutic stress, particularly in cancer recurrence and metastases. In general, the number of PGCCs increases in advanced tumor stages and is considered an indicator of poor prognosis.
According to recent work by Liu et al.[36], PGCCs, mostly occurring in hypoxic regions, acquire stemness qualities, which are even more profound in their multiple progeny. In 2016, Niu et al.[37], had the right intuition when they hypothesized that the progeny of precancerous PGCCs acquire a new genome. According to the researchers, the giant cell could be the cell in which genomic changes occur. This viewpoint reinforces our perspective.
Recent evolutionary cancer genome theory reveals the deep homology between MGRS and PGCC structures [1]. The non-mutational process of genome reprogramming that occurs in the hyperpolyploid nuclei of precancerous human MGRS cells or the tetraploid nuclei of several PGCCs is deeply homologous to the ancient genome repair process evolved by the common AMF ancestor. In protists, ancient MGRS and their hyperpolyploid nuclei restore genome integrity and generate spores and buds that form new germline clones with ACD capability and stemness potential, whereas precancerous MGRSs in humans induce reactivation of the ancient AMF gene module with genome hybridization, oncogenesis, and the generation of CSCs. Therefore, it remains highly questionable whether PGCCs can be generally considered as a "genomically modified embryo," as suggested by the aforementioned investigators [36,37].
5. Benign tumors and carcinogenesis
Benign tumors consist of mixed cell phenotypes of different origin interrupted on the way of oncogenic transformation by genes of tissular development. Genome changing and oncogenic transformation is still pending. Nonetheless, Valet and Narbonne [38] consider 2022 benign tumor cells as dysfunctional non-cancerous cell types capable to complete oncogenic transformation and form malign tumors Benign tumors result by stem cell deregulation [39,40], however the dysregulation occurred at different stages of stem cell development and under the influence of different stimuli (niche stimuli) and growth factors [41,42,43,44,45,46].
5.1. Niche signaling and formation of benign tumors
Stem cells in the niche are in a quiescent or low symmetric cycling cell state until some of their progeny leave the niche, are not exposed to stress, and differentiate [43,47]. In essence, stem cells are “primed” for asymmetric cell cycling (ACD phenotype) and differentiation. Normally, the niche environment maintains the undifferentiated, undamaged phenotype by preventing differentiation, but when Notch signals were blocked, all cells in the niche were rapidly stimulated to differentiate [48,49,50]. According to Valet and Narbonne [38], two types of niche signals lead to stem cell deregulation and benign tumor formation: (i) early constitutive niche signals that lead to undifferentiated tumors, and (ii) later defective homeostatic signals that lead to differentiated tumors. The authors believe that "undifferentiated benign tumors can arise when a mutation constitutively activates niche signaling“, and shows that “mutations in genes encoding several highly conserved proteins prevent homeostatic germ stem cell regulation in C. elegans. These include loss-of-function in DAF-18/PTEN, PAR-4/LKB1 and AAK-1/AMPK, and a gain-of-function in LET-60/Ras. However, how these genes work together to maintain germline homeostasis is not yet fully understood“.
5.2. How benign tumor cells become malignant
According to Valet and Narbone [38], benign tumors “acquires changes that disrupt homeostatic stem cell regulation by their differentiated progeny and prevent proper stem cell differentiation, respectively stem cell quiescence“.. The authors propose that mutations affecting terminal differentiation promote the development of benign or abnormally differentiated tumors. These mutations can lead to increased stem cell proliferation, hastening their progression toward cancer.
Researchers perceive the transformation of a normal stem cell into a cancer stem cell as a series of genetic and epigenetic changes resulting in the acquisition of multiple cancer hallmarks [51]. It is assumed that this transformation typically occurs over several years, involving multiple intermediate stages displaying progressive deregulation, often referred to as "pre-cancerous" stem cells [52]. These significant proliferative changes may be responsible for the formation of benign tumors.
Evolutionary cancer cell biology ECCB hypothesizes that, in benign tumors, one or more cells experience irreversible DNA damage and enter a DSCD-like cell state under the influence of environmental stressors. Through subsequent symmetric cell divisions, DNA repair, genome reprogramming, and polyploidization, with or without cell fusion, these cells break out of the benign state and initiate the pathway for cancer stem cell formation. Polyploidization, hyperpolyploidization, and PGCCs structures are common features in undifferentiated malignant tumors.
6. Can early pluripotent stem cells undergo irreversible DNA-damage and DSCD pathway?
There is limited data indicating the occurrence of irreversible DNA damage, DSCDs, and reprogramming by PGCC-like structures arising from early-stage pluripotent and embryonic stem cells. Most researchers, instead, assume that weakened adult stem cells are reprogrammed for carcinogenesis.
An exception to this is the rather extravagant hypothesis proposed by Jinsong Liu's research group [53], which suggests the existence of a pre-embryogenic program disrupted by PGCC processes. According to this perspective, "PGCCs represent a stress-induced mechanism that enables the tolerance of extensive genomic errors and the creation of a new biological system [54,55]. Within this framework, PGCCs are thought to have uniform genomic consequences, as they establish an “altered genome systems ready for cancer's macroevolutionary selection, followed by the activation of molecular pathways to facilitate subclonal expansion for microevolution". This concept entails a "PGCC life cycle" without the activation of a differentiation program.
Jinsong Liu,, the mentor of this research group, theorizes that the maturation of the blastocyst and its implantation disrupt the embryo's "germ cell life cycle" and hinder the differentiation of cancer stem cells. In his view, as humans age, certain structures formed in the unimplanted blastocyst through the dedifferentiation of senescent somatic cells and nuclear reprogramming would give rise to a "giant cell-like cycle" and a "neoplastic life code." These "blastomere-like giant cancer cells" would be the PGCCs that awaken the endogenously-suppressed embryonic program to new life [56].
However, this histopathological interpretation contradicts molecular embryological findings and disregards the evolutionary history of oncogenesis. PGCCs are not a stress-induced mechanism that creates a new biological system; instead, they represent an ancient genome reconstruction mechanism that repairs genomically damaged CSCs and non-gametogenic cancer germline cells [4,13,14]. These cells fuse to form a homotypic syncytium with a greatly hyperpolyploid nucleus, which reconstructs genome integrity and gives rise to new cancer germline clones. These clones regain asymmetric cell cycling and stemness potential and differentiate into naive CSCs.
The present paper scrutinizes whether, and to what extent, the embryological literature corroborates Jinsong Liu's assumptions. Unfortunately, no data could be found to support these assertions. Conversely, Ribeiro Reily Rocha et al. [57] have noted that our understanding of the molecular processes associated with tissue degeneration and premature aging in older patients with deficits in DNA damage repair and responses remains "unclear." There is substantial evidence to suggest that such abnormalities are linked to stem cell defects, but the underlying mechanisms are not yet fully understood. In the following, we briefly review the characteristics of pluripotent stem cells and whether irreversible DNA damage of DSCD pattern can occur in these cells.
6.1. Naive and primed pluripotency
During early embryonic development, a distinction is made between naïve and primed pluripotency. The naïve state of pluripotency occurs before the implantation of the blastocyst, while the primed state of pluripotency is observed afterward. To clarify, the naïve state of pluripotency, as referred to by Liu [56], corresponds to the pluripotency of the inner cell mass whereas the primed state pertains to the pluripotency of the epiblast. Genomic integrity is crucial for both the ICM and epiblast. It is essential for embryonic and fetal development, and it is appropriately safeguarded [58].
Mouse embryonic stem cells (mESCs) derived from the early 3.5-day-old ICM can be maintained in culture. These cells are capable of retaining the naïve pluripotent state but can also transition to a primed pluripotent state, eventually differentiating into ESCs of lower potency [59,60,61,62,63,64,65]. It has become evident that blastocyst pluripotency gives rise to pluripotent ESCs, and following implantation, epiblast cells develop into somatic cells and primordial germ cells PGCs, which are the precursors of sperm and oocytes. Differentiation of somatic cells and PGCs has also been observed in cultures. This underscores the fact that the transition from totipotency to naive pluripotency and further to primed pluripotency is orchestrated and regulated by the epigenome and epigenomic reprogramming [66]. As of now, there is no evidence to suggest that, under the influence of stressors, pluripotent stem cells or embryonic stem cells can irreversibly transition to a damaged DSCD state, fusible cells, or MGRS/PGCC-like phenotypes.
As highlighted by Frosina several years ago [58], maintaining genomic stability in pluripotent PSCs and embryonic ESCs is crucial, as "genetic alterations in these progenitor cells compromise the genomic stability and functionality of entire cell lineages." In mice, mutation rates are lower in mise embryonic stem cells than in adult somatic cells due to robust molecular mechanisms and effective DNA damage repair, which counteract persistent DNA defects and loss of function.
6.2. Molecular Mechanisms Preventing DNA Damage
6.2.1. S-Phase Extension
As pointed out by Choi et al. [67], ESCs and their progenitors exhibit unique cell cycle traits characterized by a short G1 phase and an extended S phase. For instance, mouse mESCs have a cell division time of approximately 12 hours, featuring an unusually brief S-phase lasting only 3 hours. In asynchronous cell cultures, a greater number of cells spend a prolonged period in the S phase. The short G1 phase, the absence of a G1/S checkpoint, and the extended S phase play crucial roles in maintaining pluripotency. PSCs exposed to stress do not linger in the G1 phase but progress smoothly into the S-phase [68,69,70,71,72]. Conversely, the extended S phase facilitates error-free DNA double-strand break (DSB) repair. Key homologous recombination (HR) proteins such as RAD51, RAD52, and RAD54 enhance “efficient replication fork progression by preventing replication fork collapse and repairing DNA breaks during prolonged S phases" [73,74]. In essence, cell cycle regulatory mechanisms empower ESCs to tolerate the effects of DNA damage.
The prolonged S phase also favors epigenetic regulation and the preservation of genome stability, possibly contributing to the stabilization of the pluripotent state. There is evidence that proteins directly involved in DNA replication play a stabilizing role in pluripotency and the PSC state [67]. According to Dalton and Coverdell, the duration of the G1 phase governs pluripotency. An extended G1 phase hampers entry into the S phase, disrupts the pluripotent state, and induces differentiation [75]. ESCs maintain the integrity of their genome, avoid functional losses, and do not change into defective DSCD-like cell types.
6.2.2. Accumulation of HR Factors
Numerous researchers believe that the constitutive expression and accumulation of HR factors during the extended S phase are essential for preventing DNA breaks and maintaining genome integrity [67]. It is evident that impaired HR repair could destabilize the pluripotent state, while the accumulation of HR factors ensures an effective DNA damage response (DDR) and error-free HR repair. In mESCs, the accumulation of HR factors is approximately 6-fold higher than in somatic cells, resulting in a much lower mutation rate compared to somatic cells and enhanced genomic stability. It can be argued that increased HR protein activity secures both pluripotency and genomic integrity, enabling self-renewal and differentiation [74,76].
In the absence of HR factors, increased DNA damage leads to a loss of genomic integrity and apoptosis [77,78,79]. When HR factors are depleted, ESCs are arrested at the S/M checkpoint, resulting in cell death as a dramatic consequence. Abundant HR factors facilitate DNA damage repair [73]. During differentiation processes, HR factors diminish continuously. As differentiation initiates, mESC cells transition from the HR-mediated repair pathway to non-homologous end joining (NHEJ) [80]. This shift to NHEJ and the reduction in HR-related factors result in inefficient DNA repair, making mESCs incapable of protecting replication forks, ultimately leading to a high frequency of DNA breaks and cell death.
6.3. Less Protected: The FA-DNA Repair Pathway
Fanconi anemia (FA) is a rare inherited disorder characterized by gene defects in DNA repair that predispose individuals to various hematological cancers, sporadic cancers, and solid tumors [81]. Gene mutations result in (i) the defective FA phenotype, (ii) genome instability, and (iii) additional mutations in somatic cells. Studies have established a connection between Fanconi anemia and cancer, primarily due to gene defects disrupting the FA DNA-repair pathway.
The relatively low mutation frequency observed in pluripotent stem cell states is, in part, attributed to error-free homologous recombination [82]. The error-free HR repair pathway requires the Fanconi anemia DNA repair pathway. DNA repair-effector complexes that form in response to endogenous DNA lesions under the influence of metabolic stressors cause loss-of-function mutations in FA genes. These mutations lead to hereditary disorders characterized by bone marrow failure, developmental defects, and an elevated cancer risk [83].
Human induced pluripotent stem cells (iPSCs) also rely on the FA DNA repair pathway. Upon loss of the FA pathway, induced iPSCs retain pluripotency but undergo significant G2 cell cycle arrest and apoptosis. FA-deficient ESCs can maintain their phenotype but exhibit limited self-renewal. Chlon et al.'s work [82] suggests that the HR machinery engages in double-strand break (DSB) repair in FA-deficient G2 arrested cells, indicating that HR-mediated repair stalls in these cells beyond RAD51 recruitment. iPSCs employ both FA and HR-mediated repair during the G2 phase of the cell cycle, and the inability of FA-deficient iPSCs to repair this damage likely results in G2 arrest [82]. In FA-deficient iPSCs, unrepaired damage coincides with profound G2 arrest and apoptosis.
Researchers have identified 16 to 22 FA genes responsible for maintaining genomic stability. Mutations in any of these genes lead to defects in DNA damage repair responses, resulting in FA disease [84,85]. The FA pathways play a pivotal role in DNA repair and cancer suppression, both in inherited and sporadic cancers.
Fanconi anemia patients face a significantly increased risk of developing acute myeloid leukemia, along with bone marrow failure. Additionally, they are at risk for head and neck squamous cell carcinoma, particularly in areas of rapid cell reproduction, such as the oral cavity, vulva, esophagus, gastrointestinal tract, and anus. The incidence of these cancers is 500-700 times higher compared to the general population.
The FA-DNA repair pathway is not exclusive to PSCs or ESCs and is not triggered by specific loss-of-function in embryonic PSCs. It is conserved across vertebrates, invertebrates, plants, and yeast, based on shared biochemical and physiological functions, including activation of downstream DNA repair pathways such as nucleotide excision repair, translesion synthesis, and homologous recombination [86].
7. Adult stem cells, DNA-damage and PGCC cancers
In the past decade, it was widely assumed that oncogenic transformation in adult stem cells or progenitor cells must result from a gradual accumulation of acquired mutations over time. These mutations would age these cells, compromise their genomic stability, induce epigenetic alterations, and ultimately lead to cellular dysfunction [87,88,89,90,91]. This often resulted in permanent or transient cell cycle arrest, also known as senescence, and sometimes led to programmed cell death (altruistic suicide.)
However, from an evolutionary perspective, a substantial part of accumulated data now supports the notion that oncogenic transformation, at least in the context of PGCC cancers, is a process involving genome reprogramming. This reprogramming is orchestrated by the conserved ancient gene regulatory network aGRN, which essentially controls the cancer life cycle by reshaping the multicellular genome of DSCD cells [1,3,4].
7.1. Long-term quiescence and stem cell aging in the niche favour the transition into the DSCD phenotype
In the classical stem cell biology stem cells entstehen aus dem ACD phenotype of the non-gametogenic NG germline. Germline cells with ACD, stemness and differentiation potential performed asymmetric celldivisions and give to non-identical germ cells: one daughter cell (d1) is committed for self-renewing the germline and the other (d2) is programmed for a stem cell fate and cell differentiation [1,2]. In cancer research, it is often confusion between NG germline (germline clones) and stem cell lineages. Dependent of environmental conditions, d2 cells can become quiescent in the niche, and quiescent cells exiting the niche can reenter mitotic cell cycles or commit for differentiation. Dependant of environmental cues, non-commtited can proliferate asymmetrically (ACD phenotype) or accumulate symmetrically (SCD phenotype).
In higher metazoan organisms and humans, the quiescence inside the niche maintain stemness and differentiationg potential. Stem cell niches typically maintain a hypoxic environment, which serves to protect adult stem cells from stressors like elevated oxygen levels and oxidative stress. Non-proliferating cell in the niche or quiescent cells perform very slow cell mitotically correct cell cycles. They are not exposed to deletorious factors such as excess oxygen causing severe DNADSB damage. Mäßige quescence prevent cell aging and maintains the stemness and differentiation potential in the quiescent niche.
The prolonged state of quiescence or a lifelong persistence of stem and progenitor germline cells in the, makes them vulnerable to changing neech conditions or external environment. As a result, DNA damage accumulation, disruptions in the cell cycle, decline in function such as loss of stemnes were observed [92,93,94]. Degenerative processes in deepening stem cells quiescence have been connected to the age dependent decline in regenerative potential of tissue [95].
Quiescence deepening flead to delayed reactivation of [96]. The duration of quiescence affects cell viability and proliferation potential. Es wird erwartet, dass a better understanding of the molecular processes that regulate the maintenance of proliferation potential and the mechanisms mediating reactivation of proliferation after quiescence will contribute to comprehension of stem cell aging [95]. Even Hanahan and Weinberg hypothesized in 2011 [97]. that failure of the programs that negatively regulate cell division upon withdrawal of growth signals would lead to uncontrolled proliferation, that is a hallmark of DSCD cells. According to Fujimaki et al. there is a parallel between quiescence, cellular senescence and aging [98]. This recent finding and our own results suggest that ROS accumulation favors deeper quiescence
Long-term quiescence has detrimental consequences. They often bypass DNA damage checkpoints and cell cycle repair pathways, and their DNA damage response and repair tends to be modest at best [99]. When these cells re-enter the G1 phase, they primarily rely on non-homologous end-joining (NHEJ) processes to repair long-standing DNA damage. Unfortunately, NHEJ is less effective compared to the homologous recombination repair mechanisms typically active during the S phase. Conversely, short-term quiescence can help prevent DNA damage, as it involves lower metabolic activity, offering protection against the acquisition of DNA damage [93].
7.2. DSCDs hallmarks: defective symmetric cell cycling, aggressive proliferation and readiness for reprogramming
As reported by Mas-Bargues et al. and other researchers, cellular senescence represents a state of permanent or irreversible cell cycle arrest triggered by adverse stress stimuli, such as hyperoxia [100]. While senescent cells remain viable and metabolically active, they are generally unresponsive to mitogenic or oncogenic signals and lack specific adult stem cell functions [101]. Notably, human stem cells cultured under atmospheric oxygen conditions exhibit an increase in senescence markers compared to cells cultured under lower oxygen tension conditions [102,103,104]. This underscores the influence of environmental oxygen levels on the maintenance, survival, and proliferation of stem cells. Cultures maintained at lower oxygen levels delay senescence, inhibit the expression of senescence-related genes like p21 and p16, and prevent cell cycle arrest [100].
Milanovic et al. [105] observed that cells released from temporary senescence reenter the cell cycle with a "strongly enhanced clonogenic growth potential" compared to nearly identical populations that had experienced similar exposure to chemotherapy but had never undergone senescence. This research demonstrated that such cells, previously senescent, possess a significantly higher potential for initiating tumors. Cellular senescence, a response to severe cellular stressors, results in the generation of post-senescent cells with a considerably detrimental potential, marked by a much more aggressive growth phenotype.
The author of this article holds the view that the post-senescent cells described by Mas-Bargues et al. and Milanovic et al. [100,105] exhibit all the essential features of DSCDs. Unfortunately, both articles do not provide information on the transition of post-senescent cell stages into the cancer life cycle. The assertions made by Milanovic et al., namely, that proliferating post-senescent cells serve as precursors of cancer cells and cancer stem cells, and that senescence-associated stem cells act as precursors of tumors, align with the core messages of this work.
8. Germ stem cells (GSCs) for DSCD conversion
It would be expected that human and mammalian germline cells, which are largely related to the non-gametogenic Urgermline and not as well protected against DNA damage as pluripotent PSCs and embryonic ESCs, could be also dysregulated into DSCDs, similar to stressed adult stem cells, as described above.
8.1. Differentiation of primordial germ cells and germline stem cells
Primordial germ cells (PGCs) originate from the 6.5- to 7.0-day-old post-implantation epiblast, which is programmed for germ cell differentiation s rather than somatic cells [91]. The 6.5- to 7.0-day-old epiblast carries the genetic and epigenetic information necessary for germline specification. According to researchers, PGC specification involves three key events: (i) repression of somatic programming, (ii) regaining of pluripotency, and (iii) genome-wide epigenetic reprogramming [106,107]. Primordial PGCs, which are precursors of various maturation stages of germ cell lineages, undergo a process of proliferation and migration from the yolk sac through the hindgut toward the genital ridge, where subsequent sex determination occurs [108]. However, they can also migrate further along the midline of the body, which explains the emergence and topography of extragonadal germ cell tumors (GCTs).
Cheng et al. [108] refer to germline stem cells as "the unique cell type that produces more stem cells via self-renewal or different progenitor cells of germline development and finally differentiates into specialized cells, spermatozoa, and ova for producing offspring." In mammals, GSCs mainly include (1) primordial germ cells from embryos as embryonic pluripotent stem cells, (2) induced PGC-like cells from embryonic stem cells or induced pluripotent stem cells (iPSCs), as well as (3) spermatogonial stem cells and (4) female germline stem cells.
PGCs are the founder cells of the gonadal germline and the source of totipotency, contributing to the formation of new organisms. PGC-like cells can be generated in vitro from embryonic stem cells or induced pluripotent stem cells. In vivo, the germline produces germ stem cells (GSCs) through long-term mitotic self-renewal.
8.2. Gonadal and extra-gonadal germ cell tumors (GCTs)
Human germ cell tumors (GCTs) are a group of neoplasms originating from germ cells that contain both immature and mature elements. They primarily arise in gonads; however, 1–5% of all germ cell tumors occur extragonadally along the midline of the body, where migrating primordial germ cells are located during embryogenesis. It is believed that extragonadal germ cell tumors GCT tumors arise from PGCs that have been misplaced to aberrant ectopic sites [109,110] Nevertheless, the majority of GCTs are benign.
Several studies have demonstrated the presence of PGCs outside of the gonads long after gonadal differentiation. Interestingly, these extragonadal PGCs do not lose their functionality and can differentiate and mature into oocytes [111,112,113]. They are not irreparably dysfunctional DSCD cells but rather represent intermediary stages shortly before transitioning. On the other hand, some extragonadal germ cells display malignant potential, with researchers believing that "they might have undergone malignant transformation during embryonic development" [112]. Some investigators have proposed that extragonadal germ cell tumors originate from specific progenitor cells referred to as "carcinoma in situ" or "germ cell neoplasia in situ," which represent transformed PGCs or gonocytes [114].
The author of this work believes there is no contradiction in these different statements because, during their journey through oncogenic transformation, extragonadal germ cells progress through a series of intermediate cell states. This progression ranges from nearly intact extragonadal cells that have not yet lost their function to dysregulated precursors such as "carcinoma in situ" or "germ cell neoplasia in situ," that have irreversibly lost their cellular fate and correspond to the evolutionary DSCD cells described by the author here. It is evident that extragonadal germ cell tumors do not result from gene mutations but rather from the reprogramming of their cells-of-origin in the target niches. Dysregulation of human PGCs in terms of migration, colonization, and differentiation contributes to significant diseases such as germ cell tumors and ovarian cancer [115].
As stated by Cheng et al. [108], the relatively rare extragonadal GCTs "arise in various human organs and tissues, such as the brain, head/neck, lung, thymus, heart/mediastinum, sacrococcygeal region, abdomen, retroperitoneum, vagina, and placenta, all of which are also sites of germ cell tumors." It is widely accepted that extragonadal GCT cells originating from the mismigration of human PGCs fail to undergo apoptosis.
8.3. Germ cell tumors, dysregulated DSCD pathway and epigenetic controlled plasticity
According to Oosterhuis et al., germ cell tumors are seldom caused by somatic driver mutations but rather by exogenously induced developmental defects in their cells of origin [110], These researchers suggest that GCTs result from the „reprogramming of their cells of origin due to failures in cell-intrinsic mechanisms and niche environmental factors that control latent cell developmental potency“. The cell of origin is sensitive to DNA damage, can override its control mechanisms, and thus be reprogrammed into GCTs. This precisely represents the evolutionary pathway through which DSCDs evolve.
Lobo et al. also argue for non-mutagenic evolution [107]. More than 95% of testicular neoplasms originate from germ cells arrested in their differentiation. The researchers believe that errors in the regulation of the developmental potential of ESCs and early PGCs can lead to extragonadal tumors early in life. In contrast, deregulation of the developmental potential of later germline cells leads to a variety of tumors preferentially localized in the gonads mainly after childhood.
De Felici et al. also believe that aberrant PGCs arise without genetic manipulation [109], and consider that GCT tumors arise (1) either from germ cells that remain incompletely determined in the germline and undergo a dysregulated cell cycle, (2) from germ cells on the path to somatic differentiation, or (3) from PGCs that do not receive differentiation signals, such as extragonadal PGCs, but remain viable and are prone to oncogenic transformation. Such altered PGCs are largely eliminated from the niche due to the predominant expression of cell death factors. Nonetheless, some of them may also survive for extended periods without differentiation, increasing the risk of cancer.
According to Müller et al., germ cell tumors develop early during the specification, migration, or colonization of primordial germ cells in the genital ridge [116]. It is widely believed that the precursor of GCTs arises during early germ cell development in the fetus until the arrival of late PGCs in the gonads. Since driver mutations could not be identified, the authors propose that the epigenetic machinery also plays a crucial role. They suggest that SOX2 and SOX17 determine either an embryonic stem cell-like fate (embryonal carcinoma) or a PGC-like cell fate (seminoma), and that factors secreted by the microenvironment, such as several inhibiting molecules, dictate the fate decision of „germ cell neoplasia in situ“. This indicates the vital role of the microenvironment in GCT plasticity and the transformation of PGCs into germ cell neoplasia in situ [110,111].
In contrast to the investigators above, the author of this work fundamentally distinguishes between epigenetically controlled cell plasticity, allowing for phenotype reversibility, such as the reversible conversion from asymmetric to symmetric cycling phenotypes, and the irreversibility of DNA-damaging processes leading to DSCD cells. These processes pave the way into ontogenesis. When severe stress conditions, such as hyperoxia, stress the asymmetric cycling ACD phenotype, a transition into a defective DSCD phenotype with aberrant symmetric cell cycling, loss of stemness, and differentiation occurs, and this process is irreversible.
Conclusions
Cancer cell biology can be traced back to its origins in premetazoan cell biology. This connection becomes particularly apparent in PGCC (polyploid giant cancer cell) cancers, where an ancient premetazoan gene regulatory network, known as aGRN, plays a pivotal role in controlling their development and evolution. PGCC cancers exhibit highly unusual structures, featuring endopolyploid, aneuploid and tetraploid cells as well as hyperpolyploid giant cells with characteristics reminiscent of premetazoan organisms.
Some cancer researchers, who may lack a deep understanding of premetazoan cell biology, weakly argue that the principles of evolution do not universally apply to all cancers. They often attribute the occurrence of tumors to numerous somatic mutations that occur after the initial carcinogenic event, overlooking the fact that PGCCs are prevalent in certain types of cancer, such as glioblastomas and colorectal carcinomas, where they are found in over 90% of cases. Consequently, the mutagenic theory, which suggests that mutations are solely responsible for malignant transformation, is increasingly being challenged. This challenge is supported by a growing body of evidence in the field of cell biology [105,107,109,110,111,115,116].
Both in the premetazoan era and later, stress and the stress response played pivotal evolutionary role. Under stress influence, the common AMF ancestor of amoebozoans, metazoans and fungi evolved a second somatic cell line over 1 billion years ago. It established a dual cell system comprising oxygen-sensitive germ cells and oxygen-resistant somatic cells. Recent hypotheses suggest a profound evolutionary connection between the emergence of new cell types and the ancestral response to stress. Stress' role in evolution can also be traced back to the development of basic somatic and germ cell types in multicellular organisms [117,118]. Stress governs the reversible conversion of asymmetric to symmetric cycling phenotypes, along with epigenetically reversible transitions from germ to somatic cells (GST, MET) and from somatic to germ cells (SGT, EMT).
Nonetheless, stress has the potential to trigger irreversible DNA damage, affecting both premetazoan cell systems and healthy human individuals, resulting in the formation of a defective cell type. This particular cell type, marked by severe and irreversible DNA damage, does not undergo apoptosis and can either persist through defective symmetric proliferation (DSCD) or enter a temporary quiescent state while awaiting repair. Remarkably, both premetazoans and precancerous cell systems in humans utilize the same DNA repair mechanism, governed by the ancient gene regulatory network (aGRN). In premetazoans, this hyperpolyploid repair pathway serves to restore genome integrity by mending DNA damage. However, in humans, the aGRN regulatory network orchestrates a different outcome, reprogramming DSCD cells towards the path of carcinogenesis.
Human DSCDs are derived from ACD phenotypes capable of stemness and differentiation (Figure 2). All these phenotypes have the potential to serve as the cell-of-origin for PGCC cancers. Nevertheless, the ACD types less likely to give rise to these cancers include pluripotent and embryonic stem cells, which possess stronger defense mechanisms against DNA damage, thanks to more robust prevention and repair mechanisms and reduced exposure to stress conditions.
Human DSCD cells have close affiliations with the VSEL and EGC phenotypes, collectively constituting the broader DSCD family. All members of the human DSCD family exhibit characteristics of loss of function, which include a decrease in stemness and a reduced capacity for differentiation, akin to DSCD cells found in protists and in the common AMF ancestor. Controlled by the ancient gene regulatory network, most human DSCD cells undergo a similar process of DNA DSB damage repair involving cell and nuclear fusion, resulting in the formation of multinucleated genome repair syncytia with hyperpolyploid giant nuclei (MGRS). In humans, the ancient GRN via MGRS repair reprograms the DSCD genome, reactivating old unicellular genes from the conserved AMF genome. This multi-stage MGRS process, which consists of a first phase of endopoliploidization leading to defective daughter nuclei and a second phase of hyperpolyploidization in which the daughter nuclei fuse to form a giant nucleus, plays a crucial role in genome remodeling and ultimately in reprogramming to malignant transformation.
The hybrid genome of non-gametogenic cancer germline is fully under the control of the ancestral aGRN regulatory network, while the somatic cell lines of cancers germ and soma life cycle are not. As a consequence, dysregulated somatic processes result in numerous DNA mutations, clearly distinguishing cancer cells and CSCs from normal somatic human cells and HSCs.
In summary, the current data suggest that the malignant tansformation leading to PGCC cancers is a non-mutational reprogramming process rooted in ancestral imprinting and transition to a lower level of systemic cell organization.The cell-of-origin for PGCC cancers is a dysfunctional DSCD in need of repair, but the available repair mechanisms are less adequate and derive from ancient AMF common ancestror and premetazoan organisms. They repair the DNADSB damage of DSCDs while reprogramming their genome for malignancy (Figure 2). Controlled by the ancient aGRN network, this process involves uploading old unicellular genes and downloading multicellular genes. The reprogramming process may manifest many years later, affecting quiescent DSCD cells within their niches. PGCC-like MGRS structures give rise to viable spores and buds, initiating the germ and soma (G+S) life cycle of cancer and generating primary, naive CSCs.
Aknowledgements:
This work, along with all my previous works from the last 10 years, would not have been possible without the unwavering support and understanding of my family. They have relieved me of the daily life burdens and provided me with the peace necessary for scientific work and reflection. I want to extend special thanks to my wife, the German microbiologist Dr. Eugenia R. Niculescu, who has consistently supported my scientific endeavors and offered valuable professional insights. I would also like to express my gratitude to Mr. Gregor Jaruga, who has been responsible for designing illustrations for my publications for many years, consistently delivering excellent graphic work.
Abbreviations
ACD phenotype, cell dividing by asymmetric cell division; AMF, amoebozoan, metazoan and fungi; DSCD, damaged cell capable of symmetric cell division; EGC, extragonadal germ cell; aGRN, ancestral gene regulatory network; G+S, germ and soma lifde cycle; GCT, germ cell tumor; MGRS, multinucleated genome repair syncytia; NG, non-gametogenic; PGC, primordial germ cell; PGCC, polyploid giant cancer cell; PSC, pluripotent stem cell; SCD, symmetric cell division; SGT, soma-to-germ transition; EMT, epithelial-mesenchymal transition; VSEL, very small embryonic like cell.
Appendix A
Comments and responses to the preprint V1
1. The first comment claimed that "cancer is not merely a mutational or genetic disease, but rather a non-mutational genome-altering condition," which appears subjective and inconsistent with current studies highlighting numerous cancers carrying proto-oncogene mutations such as p53, Myc, as well as driver genes and hub genes. How does the author address this apparent contorversy?
Second, the author need to talk more about DNA modifications, which are not necessarily correlate with DNA mutation. Especially the presented concept “defective symmetric cell divisions” is more reasonable because the disruption of the ACD balance, which probably due to the systematic/network changes, not solely the DNA damage accumulation.
Author’s response:
There are several approaches that require clarification. Firstly, the evolutionary perspective of cancer cell biology (ECCB) interprets cancer as the transition of multicellular cells afflicted with significant DNA double-strand break (DSB) defects, to a lower level of cell organization. 10.20944/preprints202309.2156.v1; 10.20944/preprints202311.0383.v1.
According to the ECCB, adult stem cells exposed to an oxygen content exceeding 6.0% O2 (pre-metazoic hyperoxia) experience DNA double-strand breaks (DNA DSB). The damaged cells irreversibly lose their capacity for asymmetric cell differentiation, stemness and differentiation potential. In other words, normal oxygen levels in tissue disrupt the balance between asymmetric (ACD) and symmetric cell division (SCD) irreversibly. Unlike senescent stem cells of aging, oxygen-damaged stem cells escape apoptosis or bypass it. They either persist in the normoxic niche awaiting repair or proliferate outside the niche through defective symmetric cell division (DSCD), characterized by spindle defects, binucleation with mature and immature nuclei, premature cell division and await repair. The DSCD phenotyp is an ancestral phenotyp that initially appeared in the common AMF ancestor under hyperoxic stress conditions.
Homologous recombination (HR) and non-homologous end joining (NHEJ) associated with the DSCD cell cycle are insufficient to repair DNA damage. Conversely, multiple HR and NHEJ cycles linked to endopolyploidization processes, as observed in the 8C-16C cysts of protists, are necessary to reverse DNA DSB damage. However, severe DNA DSB damage impedes the reversal through multiple HR and NHEJ cycles. The resulting 8-16 daughter nuclei are non-viable and must fuse to form a hyperpolyploid giant nucleus with repair capacity (as observed in MGRS and PGCC giant nuclei). Such giant nuclei can excise damaged genome material and restore a functional genome. Unfortunately, MGRSs/PGCCs follow a pre-metazoan pattern of repair and the DSCD cell undergoes reprogramming into a transformed cell, fostering malignancy and a cancer life cycle with pre-metazoan imprinting. The reprogrammed multicellular DSCD cell s shifts to a low level of cell organization.
Secondly, genes of the two levels of organization within the human genome are related and deeply homologous to the genes of the lower cell organization system evolved by the common AMF ancestor. However, they are not identical due to further evolution in late premetazaoans, early metazoans and transitional forms. Changes from “healthy“ human genes to their homologous of lower organization cell sytems like cancer were formerly perceived as "mutations" by molecular and genetic researchers when the evolutionary aspects of cancer were less understood.
In the ECCB view, however, such mutations are more akin to a return to a more ancestral gene state. This applies to onco-genes, proto-oncogenes, as well as genes like p53, Myc, etc., from healthy individuals and tumors. Genes from pre-metazoans are retained in the genome of metazoans and remain partially dormant. The transition from the high-level proto-oncogene to the lower-level gene state in cancer should not be viewed as an acquired gene mutation via DNA changes, but rather as the utilization of evolutionary gene variants.
see: Proto-oncogenes of P. brassicae are specifically activated during the multinucleate cell division stage
see: Proto-Oncogenes, Oncogenes and Orthologous Tumor Protein
2. The second comment claimed that “there are compelling reasons to categorize VSELs within the broader DSCD family”. However, it seems that the VSELs are the stem cells that can undergo asymmetric cell divisions based on the current research.
Author’s response:
The DSCD cell family comprises former stem cells that are exposed to metabolic or ancestral hyperoxic stress due to the oxygen content in the tissue and bloodstream. They react with severe DNA double-strand break damage (DNA DSB), loss of function, loss of stemness and asymmetric cell division (ACD) as well as loss of differentiation potential. Unlike aging stem cells, DSCD cells can survive for extended periods within the niche or proliferate, but only through defective symmetric cell division, characterized by binucleation, the presence of mature and immature nuclei, and abnormal mitotic spindles. Their offspring can fuse to MGRS/PGCC structures capable of reprogramming and generating mononucleated spores (buds) that contribute to cancer stem cell (CSC) generation.
The ECCB believes that DSCD cells can persist for extended periods in suitable human and metazoan niches and considers many of the long-lived quiescent ASCs vulnerable when leaving the niche and migrating into tissues and the bloodstream. According to the ECCB, this includes both extragonadal germ cells and very small embryonic-like (VSEL) cells, along with many other stem cells exiting the quiescence niche, and transitioning to a migratory state. All these cells have a normal phenotype and a defective phenotype with severe DNA double-strand break damage and a heightened cancer risk.
Their progeny, experiencing severe double-strand break damage due to tissue oxygen levels resembling ancestral hyperoxia, are identified as DSCDs. VSELs are found in a dormant state within quiescence niches in the bone marrow, brain, pancreas, thymus, intestinal epithelium, as well as a type of migratory stem cell released from the bone marrow into the bloodstream under stressful conditions [17,18,19].
In the ECCB's opinion, the term VSEL is an overarching classification. VSELs exist as a non-damaged stem cell phenotype but also as a transformed, damaged DSCD phenotype. The normal VSEL/SC phenotype is capable of both asymmetric and symmetric cell division, while the VSEL/DSCD phenotype is not. It loses function and ACD potential completely and, like all other irreparable DSCD cells, requires repair through hyperpolyploid mechanisms.
In 2016, Bhartiya et al. demonstrated that VSELs serve as a backup pool for adult stem cells and are mobilized under stress conditions. However, an imbalance in VSEL function may result in uncontrolled proliferation and cancer (https://doi.org/10.1093/humupd/dmw030). They propose that “cancer could possibly occur by aberrant expansion of VSELs that are pluripotent and majorly uncommitted cells with global unmethylated chromatin. However, the concept of global demethylation resulting in cancer is not well accepted, as major heterogeneity in marker expression cannot be explained.” This work supports the DSCD concept. The authors stated that "most likely the function of VSELs can be altered by not yet well understood changes in the niche that lead to uncontrolled proliferation" and referred to the cells with altered function as "transformed VSELs".
References
- Niculescu, V.F. The evolutionary cancer genome theory and ist reasoning. Genet Med Open. 2023, 1, 100809. [Google Scholar] [CrossRef]
- Niculescu, V.F. Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies. Genes Dis. 2022, 9, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. Cancer exploits a late premetazoan gene module conserved in the human genome. Genes Dis. 2023, 10, 1136–1138. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. Attempts to restore loss of function in damaged ACD cells open the way to non-mutational oncogenesis. Genes Dis. 2024, 11, 101109. [Google Scholar] [CrossRef]
- Shoshani, O.; Massalha, H.; Shani, N.; Kagan, S.; Ravid, O.; Madar, S.; Trakhtenbrot, L.; Leshkowitz, D.; Rechavi, G.; Zipori, D. Polyploidization of Murine Mesenchymal Cells Is Associated with Suppression of the Long Noncoding RNA H19 and Reduced Tumorigenicity. Cancer Res 2012, 72, 6403–6413. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yue, S.; Chen, X.; Kubin, T.; Braun, T. Regulation of Cardiomyocyte Polyploidy and Multinucleation by CyclinG1. Circ. Res. 2010, 106, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.A; Rieder, C.L. Mitotic Checkpoint Slippage in Humans Occurs via Cyclin B Destruction in the Presence of an Active Checkpoint. Curr Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, D; Gosh, S.K. Cellular Events of Multinucleated Giant Cells Formation During the Encystation of Entamoeba invadens. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef]
- Tessema, M.; Rossi, M.R.; Picchi, M.A.; Yingling, C.M.; Lin, Y.; Ramalingam, S.S.; Belinsky, S.A. Common cancer-driver mutations and their association with abnormally methylated genes in lung adenocarcinoma from never-smokers. Lung Cancer 2018, 123, 99–106. [Google Scholar] [CrossRef]
- White-Gilbertson, S.; Voelkel-Johnson, C. Giants and monsters: Unexpected characters in the story of cancer recurrence. Adv. Cancer Res. 2020, 148, 201–232. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. The evolutionary cancer gene network theory versus embryogenic hypotheses. Med Oncol. 2023, 40, 114. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. On the role of polyploid giant cells in oncogenesis and tumorigenesis. Med Oncol. 2023, 40, 232. [Google Scholar] [CrossRef] [PubMed]
- Barati, M.; Akhondi, M.; Mousavi, N.S.; Haghparast, N.; Ghodsi, A.; Baharvand, H.; Ebrahimi, M.; Hassani, S.-N. Pluripotent Stem Cells: Cancer Study, Therapy, and Vaccination. Stem Cell Rev. Rep. 2021, 17, 1975–1992. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-M.; Liu, R.; Klich, I.; Wu, W.; Ratajczak, J.; Kucia, M.; Ratajczak, M.Z. Molecular signature of adult bone marrow-purified very small embryonic-like stem cells supports their developmental epiblast/germ line origin. Leukemia 2010, 24, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M. Z.; Ratajczak, J.; Kucia, M. Very Small Embryonic-Like Stem Cells (VSELs). Circ. Res. 2019, 124, 208–210. [Google Scholar] [CrossRef]
- Zuba-Surma, E.K.; Wojakowski, W.; Ratajczak, M.Z.; Dawn, B. Very Small Embryonic-Like Stem Cells: Biology and Therapeutic Potential for Heart Repair. Antioxid. Redox Signal 2011, 15, 1821–1834. [Google Scholar] [CrossRef]
- Ratajczak, M, Z.; Bujko, K.; Mack, A.; Kucia, M.; Ratajczak, J. Cancer from the perspective of stem cells and misappropriated tissue regeneration mechanisms. Leukemia 2018, 32, 2519–2526. [Google Scholar] [CrossRef]
- Gómez-López, S.; Lerner, R.G.; Petritsch, C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell. Mol. Life Sci. 2014, 71, 575–597. [Google Scholar] [CrossRef]
- McGrail, D.J.; Lin, C.C.J.; Dai, H.; Mo, W.; Li, Y.; Stephan, C.; Davies, P.; Lu, Z.; Mills, G.B.; Lee, J.S.; Lin, S.Y. Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype. Cell Rep. 2018, 23, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Peters, J. The role of genomic imprinting in biology and disease: an expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Rinderknecht, E; Humbe, l.R.E. The amino acid sequence of human insulin like growth factor I and its structural homology with pro-insulin. J. Biol. Chem. 1978; 253, 2769–2776. [Google Scholar] [CrossRef]
- Bergman, D.; Halje, M.; Nordin, M.; Engström, W. Insulin-Like Growth Factor 2 in Development and Disease: A Mini-Review. Gerontology 2013, 59, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Gamaev, L.; Mizrahi, L.; Friehmann, T.; Rosenberg, N.; Pappo, O.; Olam, D.; Zeira, E.; Halpern, K.B.; Caruso, S.; Zucman-Rossi, J.; et al. The pro-oncogenic effect of the lncRNA H19 in the development of chronic inflammation-mediated hepatocellular carcinoma. Oncogene 2020, 40, 127–139. [Google Scholar] [CrossRef]
- Kaur, E.; Rajendra, J.; Jadhav, S.; Shridhar, E.; Goda, J.S.; Moiyadi, A.; Dutt, S. Radiation-induced homotypic cell fusions of innately resistant glioblastoma cells mediate their sustained survival and recurrence. Carcinogenesis 2015, 36, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Tanner, M.; Köninki, K.; Isola, J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011, 13, R46. [Google Scholar] [CrossRef] [PubMed]
- Malpica, A.; Deavers, M.T.; Lu, K.; Bodurka, D.C.; Atkinson, E.N.; Gershenson, D.M.; Silva, E.G. Grading Ovarian Serous Carcinoma Using a Two-Tier System. Am. J. Surg. Pathol. 2004, 28, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, H.; Yang, X.; Fan, L.; Tian, S.; Niu, R.; Yan, M.; Zheng, M.; Zhang, S. Cell Fusion-Related Proteins and Signaling Pathways, and Their Roles in the Development and Progression of Cancer. Front. Cell Dev. Biol. 2022, 9, 809668. [Google Scholar] [CrossRef]
- Wang, H.-F.; Xiang, W.; Xue, B.-Z.; Wang, Y.-H.; Yi, D.-Y.; Jiang, X.-B.; Zhao, H.-Y.; Fu, P. Cell fusion in cancer hallmarks: Current research status and future indications (Review). Oncol. Lett. 2021, 22, 530. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Nagler, C.; Schwitalla, S.; Reith, G.; Niggemann, B.; Zänker, K.S. Recurrence cancer stem cells-made by cell fusion? Med. Hypotheses 2009, 73, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.-J.; Nickoloff, J.A.; Chen, W.-H.; Liu, H.-Y.; Lo, W.-C.; Chang, Y.-T.; Yang, P.-C.; Wu, C.-W.; Williams, D.F.; Gelovani, J.G.; et al. FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget 2014, 5, 9514–9529. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Schwitalla, S.; Seidel, J.; Haverkampf, S.; Reith, G.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zänker, K.S. Characterization of hybrid cells derived from spontaneous fusion events between breast epithelial cells exhibiting stem-like characteristics and breast cancer cells. Clin. Exp. Metastasis 2011, 28, 75–90. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Y.; Wu, M.; Liu, J.; Wu, H.; Xu, C.; Chen, L. Hypoxia-induced polypoid giant cancer cells in glioma promote the transformation of tumor-associated macrophages to a tumor-supportive phenotype. CNS Neurosci. Ther. 2022, 28, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Zhang, J.; Zhang, N.; Mercado-Uribe, I.; Tao, F.; Han, Z.; Pathak, S.; Multani, A.S.; Kuang, J.; Yao, J.; et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis 2016, 5, e281. [Google Scholar] [CrossRef]
- Valet, M.; Narbonne, P. Formation of benign tumos by stem cell deregulation. PLoS Genet. 2022, 18, e1010434. [Google Scholar] [CrossRef] [PubMed]
- Humphries, A.; Cereser, B.; Gay, L.J.; Miller, D.S.J.; Das, B.; Gutteridge, A.; Elia, G.; Nye, E.; Jeffery, R.; Poulsom, R.; et al. Lineage tracing reveals multipotent stem cells maintain human adenomas and the pattern of clonal expansion in tumor evolution. Proc. Natl. Acad. Sci. USA 2013, 110, E2490–E2499. [Google Scholar] [CrossRef]
- Jones, S.; Chen, W.-D.; Parmigiani, G.; Diehl, F.; Beerenwinkel, N.; Antal, T.; Traulsen, A.; Nowak, M.A.; Siegel, C.; Velculescu, V.E.; et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 4283–4288. [Google Scholar] [CrossRef]
- Qin, H.; Bao, D.; Tong, X.; Hu, Q.; Sun, G.; Huang, X. The role of stem cells in benign tumors. Tumor Biol. 2016, 37, 15349–15357. [Google Scholar] [CrossRef]
- Jiang, H.; Tian, A.; Jiang, J. Intestinal stem cell response to injury: lessons from Drosophila. Cell. Mol. Life Sci. 2016, 73, 3337–3349. [Google Scholar] [CrossRef]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef]
- Morrison, S.J.; Spradling, A.C. Stem Cells and Niches: Mechanisms That Promote Stem Cell Maintenance throughout Life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Narbonne, P.; Roy, R. Regulation of germline stem cell proliferation downstream of nutrient sensing. Cell Div. 2006, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.; Gururaja-Rao, S.; Banerjee, U. Nutritional regulation of stem and progenitor cells in Drosophila. Development 2013, 140, 4647–4656. [Google Scholar] [CrossRef]
- Lopez-Garcia, C.; Klein, A.M.; Simons, B.D.; Winton, D.J. Intestinal Stem Cell Replacement Follows a Pattern of Neutral Drift. Science 2010, 330, 822–825. [Google Scholar] [CrossRef]
- Kimble, J.; White, J. On the control of germ cell development in Caenorhabditis elegans. Dev. Biol. 1981, 81, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.; Kimble, J. glp-1 Is required in the germ line for regulation of the decision between mitosis and meiosis in C. elegans. Cell 1987, 51, 589–599. [Google Scholar] [CrossRef]
- Henderson, S.T.; Gao, D.; Lambie, E.J.; Kimble, J. lag-2 may encode a signaling ligand for the GLP-1 and LIN-12 receptors of C. elegans. Development 1994, 120, 2913–2924. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Fischer, M.; Mitrou, S.; Hübner, K. Proliferative Activity of Undifferentiated Cells (Blast Cells) in Preleukaemia. Acta Haematol. 1976, 55, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, Y.; Zhang, X.; Sood, A.K.; Liu, J. Spatiotemporal view of malignant histogenesis and macroevolution via formation of polyploid giant cancer cells. Oncogene 2023, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H. Genomic chaos: rethinking genetics, evolution, and molecular medicine. Academic Press, 2019.
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2022, 81, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. The “life code”: a theory that unifies the human life cycle and the origin of human tumors. Semin. Cancer Biol. 2019, 60, 680–397. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Lerner, L.K.; Okamoto, O.K.; Marchetto, M.C.; Menck, C.F.M. The role of DNA repair in the pluripotency and differentiation of human stem cells. Mutat. Res. Mol. Mech. Mutagen. 2013, 752, 25–35. [Google Scholar] [CrossRef]
- Frosina, G. The Bright and the Dark Sides of DNA Repair in Stem Cells. J. Biomed. Biotechnol. 2010, 2010, 845396. [Google Scholar] [CrossRef] [PubMed]
- Devika, A.S.; Montebaur, A.; Saravanan, S.; Bhushan, R.; Koch, F.; Sudheer, S. Human ES Cell Culture Conditions Fail to Preserve the Mouse Epiblast State. Stem Cells Int. 2021, 2021, 8818356. [Google Scholar] [CrossRef]
- Brons, I.G.; Smithers, L.E.; Trotter, M.W.; Rugg-Gunn, P.; Sun, B.; Chuva de Sousa Lopes, S.M.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A.; Vallier, L. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191–195. [Google Scholar] [CrossRef]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D.G. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 2007, 448, 196–199. [Google Scholar] [CrossRef]
- Ying, Q.-L.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519–523. [Google Scholar] [CrossRef]
- Guo, G.; Yang, J.; Nichols, J.; Hall, J.S.; Eyres, I.; Mansfield, W.; Smith, A. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development 2009, 136, 1063–1069. [Google Scholar] [CrossRef]
- Tosic, J.; Kim, G.-J.; Pavlovic, M.; Schröder, C.M.; Mersiowsky, S.-L.; Barg, M.; Hofherr, A.; Probst, S.; Köttgen, M.; Hein, L.; et al. Eomes and Brachyury control pluripotency exit and germ-layer segregation by changing the chromatin state. Nature Cell Biology 2019, 21, 1518–1531. [Google Scholar] [CrossRef]
- Li, Y.; Tanaka, T.S. Self-Renewal, Pluripotency and Tumorigenesis in Pluripotent Stem Cells Revisited. 2014. [CrossRef]
- Reik, W.; Surani, M.A. Germline and Pluripotent Stem Cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a019422. [Google Scholar] [CrossRef]
- Choi, E.-H.; Yoon, S.; Koh, Y.E.; Seo, Y.-J.; Kim, K.P. Maintenance of genome integrity and active homologous recombination in embryonic stem cells. Exp. Mol. Med. 2020, 52, 1220–1229. [Google Scholar] [CrossRef]
- Ahuja, A.K.; Jodkowska, K.; Teloni, F.; Bizard, A.H.; Zellweger, R. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 2016, 15, 10660. [Google Scholar] [CrossRef] [PubMed]
- Pauklin, S.; Madrigal, P.; Bertero, A.; Vallier, L. Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes Dev. 2016, 30, 421–433. [Google Scholar] [CrossRef]
- Liu, L.; Michowski, W.; Inuzuka, H.; Shimizu, K.; Nihira, N.T.; Chick, J.M.; Li, N.; Geng, Y.; Meng, A.Y.; Ordureau, A.; et al. G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nat. Cell Biol. 2017, 19, 177–188. [Google Scholar] [CrossRef]
- Boward, B.; Wu, T.; Dalton, S. Concise Review: Control of Cell Fate Through Cell Cycle and Pluripotency Networks. Stem Cells 2016, 34, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Suvorova, E.S.; Francia, M.; Striepen, B.; White, M.W. A Novel Bipartite Centrosome Coordinates the Apicomplexan Cell Cycle. PLoS Biol. 2015, 13, e1002093. [Google Scholar] [CrossRef]
- Choi, E.-H.; Yoon, S.; Kim, K.P. Combined Ectopic Expression of Homologous Recombination Factors Promotes Embryonic Stem Cell Differentiation. Mol. Ther. 2018, 26, 1154–1165. [Google Scholar] [CrossRef]
- Choi, E.-H.; Yoon, S.; Park, K.-S.; Kim, K.P. The Homologous Recombination Machinery Orchestrates Post-replication DNA Repair During Self-renewal of Mouse Embryonic Stem Cells. Sci. Rep. 2017, 7, 11610. [Google Scholar] [CrossRef]
- Dalton, S.; Coverdell, P. Linking the cell cycle to cell fate decisions. Trends Cell Biol. 2015, 25, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Tichy, E.D.; Stambrook, P.J. DNA repair in murine embryonic stem cells and differentiated cells. Exp. Cell Res. 2008, 314, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Dannenmann, B.; Lehle, S.; Hildebrand, D.G.; Kübler, A.; Grondona, P.; Schmid, V.; Holzer, K.; Fröschl, M.; Essmann, F.; Rothfuss, O.; et al. High Glutathione and Glutathione Peroxidase-2 Levels Mediate Cell-Type-Specific DNA Damage Protection in Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, O.; Knobloch, L.; Fornsaglio, J.; Varum, S.; Easley, C.; Schatten, G. DNA Damage Responses in Human Induced Pluripotent Stem Cells and Embryonic Stem Cells. PLoS ONE 2010, 5, e13410. [Google Scholar] [CrossRef] [PubMed]
- Serrano, L.; Liang, L.; Chang, Y.; Deng, L.; Maulion, C.; Nguyen, S.; Tischfield, J.A. Homologous Recombination Conserves DNA Sequence Integrity Throughout the Cell Cycle in Embryonic Stem Cells. Stem Cells Dev. 2011, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Stambrook, P.J.; Tichy, E.D. Preservation of genomic integrity in mouse embryonic stem cells. Adv. Exp. Med. Biol. 2010, 695, 59–75. [Google Scholar]
- Chen, H.; Zhang, S.; Wu, Z. Fanconi anemia pathway defects in inherited and sporadic cancers. Transl. Paediatr. 2014; 3, 300–304. [Google Scholar] [CrossRef]
- Chlon, T.M.; Ruiz-Torres, S.; Maag, L.; Mayhew, C.N.; Wikenheiser-Brokamp, K.A.; Davies, S.M.; Mehta, P.; Myers, K.C.; Wells, J.M.; Wells, S.I. Overcoming Pluripotent Stem Cell Dependence on the Repair of Endogenous DNA Damage. Stem Cell Rep. 2016, 6, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 668, 4–10. [Google Scholar] [CrossRef]
- Akkari, Y.M.N.; Bateman, R.L.; Reifsteck, C.A.; Olson, S.B.; Grompe, M. DNA Replication Is Required To Elicit Cellular Responses to Psoralen-Induced DNA Interstrand Cross-Links. Mol. Cell. Biol. 2000, 20, 8283–8289. [Google Scholar] [CrossRef]
- Dan, C.; Pei, H.; Zhang, B.; Zheng, X.; Ran, D.; Du, C. Fanconi anemia pathway and its relationship with cancer. Genome Instab. Dis. 2021, 2, 175–183. [Google Scholar] [CrossRef]
- Dong, H.; Nebert, D.W.; Bruford, E.A.; Thompson, D.C.; Joenje, H.; Vasiliou, V. Update of the human and mouse Fanconi anemia genes. Hum. Genom. 2015, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Pérez, L.M.; de Lucas, B.; Gálvez, B.G. Unhealthy Stem Cells: When Health Conditions Upset Stem Cell Properties. Cell. Physiol. Biochem. 2018, 46, 1999–2016. [Google Scholar] [CrossRef] [PubMed]
- Burkhalter, M.D.; Rudolph, K.L.; Sperka, T. Genome instability of ageing stem cells—Induction and defence mechanisms. Ageing Res. Rev. 2015, 23, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.R.; de Haan, G. Epigenetic perturbations in aging stem cells. Mamm Genome 2016, 27, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Suomalainen, A.; Hamalainen, R.H. Stem cells, mitochondria and aging. Biochim. Biophys. Acta 2015, 1847, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.; Thuret, S. The systemic milieu as a mediator of dietary influence on stem cell function during ageing. Ageing Res. Rev. 2015, 19, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, T.; Takubo, K. Hypoxia regulates the hematopoietic stem cell niche. Pflugers Arch 2015. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H. Changes in Regenerative Capacity through Lifespan. Int. J. Mol. Sci. 2015, 16, 25392–25432. [Google Scholar] [CrossRef]
- Mandal, P.K.; Blanpain, C.; Rossi, D.J. DNA damage response in adult stemcells: pathways and consequences. Nat. Rev. Mol. Cell Biol. 2011, 12, 198–202. [Google Scholar] [CrossRef]
- Olmedo, M.; Mata-Cabana, A.; Rodríguez-Palero, M.J.; García-Sánchez, S.; Fernández-Yañez, A.; Merrow, M.; Artal-Sanz, M. Prolonged quiescence delays somatic stem cell-like divisions in Caenorhabditis elegans and is controlled by insulin signaling. Aging Cell 2020, 19, e13085. [Google Scholar] [CrossRef]
- Kwon, J.S.; Everetts, N.J.; Wang, X.; Wang, W.; Della Croce, K.; Xing, J.; Yao, G. Controlling Depth of Cellular Quiescence by an Rb-E2F Network Switch. Cell Rep. 2017, 20, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, K.; Li, R.; Chen, H.; Della Croce, K.; Zhang, H.H.; Xing, J.; Bai, F.; Yao, G. Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc. Natl. Acad. Sci. USA 2019, 116, 22624–22634. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Wagers, A.J.; Manz, M.G.; Prohaska, S.S.; Scherer, D.C.; Beilhack, G.F.; Shizuru, J.A.; Weissman, I.L. Biology of Hematopoietic Stem Cells and Progenitors: Implications for Clinical Application. Annu. Rev. Immunol. 2003, 21, 759–806. [Google Scholar] [CrossRef] [PubMed]
- Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Inglés, M.; Gimeno-Mallench, L.; El Alami, M.; Viña-Almunia, J.; Gambini, J.; Viña, J.; Borrás, C. Relevance of Oxygen Concentration in Stem Cell Culture for Regenerative Medicine. Int. J. Mol. Sci. 2019, 20, 1195. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.K. Cellular senescence and aging. Oral Dis. 2016, 22, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Ko, Y.J.; Lee, M.W.; Park, H.J.; Park, Y.J.; Kim, D.-I.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Effect of low oxygen tension on the biological characteristics of human bone marrow mesenchymal stem cells. Cell Stress Chaperones 2016, 21, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-C.; Chen, Y.-J.; Yew, T.-L.; Chen, L.-L.; Wang, J.-Y.; Chiu, C.-H.; Hung, S.-C. Hypoxia inhibits senescence and maintains mesenchymal stem cell properties through down-regulation of E2A-p21 by HIF-TWIST. Blood 2011, 117, 459–469. [Google Scholar] [CrossRef]
- Vono, R.; Garcia, E.J.; Spinetti, G.; Madeddu, P. Oxidative Stress in Mesenchymal Stem Cell Senescence: Regulation by Coding and Noncoding RNAs. Antioxidants Redox Signal. 2018, 29, 864–879. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Yao, R.; Luo, H.; Shuai, L. Germline specification from pluripotent stem cells. Stem Cell Res. Ther. 2022, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Gillis, A.J.M.; Jerónimo, C.; Henrique, R.; Looijenga, L.H.J. Human Germ Cell Tumors are Developmental Cancers: Impact of Epigenetics on Pathobiology and Clinic. Int. J. Mol. Sci. 2019, 20, 258. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Shang, D.; Zhou, R. Germline stem cells in humans. Signal Transduct. Target. Ther. 2022, 7, 345. [Google Scholar] [CrossRef] [PubMed]
- De Felici, M.; Klinger, F.G.; Campolo, F.; Balistreri, C.R.; Barchi, M.; Dolci, S. To Be or Not to Be a Germ Cell: The Extragonadal Germ Cell Tumor Paradigm. Int. J. Mol. Sci. 2021, 22, 5982. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Teilum, G. Classification of endodermal sinus tumour (mesoblatoma vitellinum) and so-called “embryonal carcinoma” of the ovary. Acta Pathol. Microbiol. Scand. 1965, 64, 407–429. [Google Scholar] [CrossRef]
- Wrobel, K.H.; Suss, F. Identification and temporospatial distribution of bovine primordial germ cells prior to gonadal sexual differentiation. Anat. Embryol. 1998, 197, 451–467. [Google Scholar] [CrossRef] [PubMed]
- Rich, I. Primordial germ cells are capable of producing cells of the hematopoietic system in vitro. Blood 1995, 86, 463–472. [Google Scholar] [CrossRef]
- Berney, D.M.; Looijenga, L.H.J.; Idrees, M.; Oosterhuis, J.W.; Rajpert-De Meyts, E.; Ulbright, T.M.; Skakkebaek, N.E. Germ cell neoplasiain situ(GCNIS): Evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology 2016, 69, 7–10. [Google Scholar] [CrossRef]
- Siegel, R. L.; Miller, K. D.; Fuchs, H. E.; Jemal, A. Cancer statistics. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.R.; Skowron, M.A.; Albers, P.; Nettersheim, D. Molecular and epigenetic pathogenesis of germ cell tumors. Asian J. Urol. 2021, 8, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Erkenbrack, E.M.; Love, A.C. Stress-Induced Evolutionary Innovation: A Mechanism for the Origin of Cell Types. BioEssays 2019, 41, e1800188. [Google Scholar] [CrossRef] [PubMed]
- Nedelcu, A.M.; Michod, R.E. Stress Responses Co-Opted for Specialized Cell Types During the Early Evolution of Multicellularity. BioEssays 2020, 42, 2000029. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic universal tree updated from Forterre (2015) after Woese et al (1990). Amorpha is the last assemblage descended from the common ancestor LECA (Eukaria domain). It contains the three sister clades Amoebozoa, Metazoa, and Fungi. https://doi.org/10.3389/fmicb.2015.00717; (CC BY 2015).
Figure 1.
Schematic universal tree updated from Forterre (2015) after Woese et al (1990). Amorpha is the last assemblage descended from the common ancestor LECA (Eukaria domain). It contains the three sister clades Amoebozoa, Metazoa, and Fungi. https://doi.org/10.3389/fmicb.2015.00717; (CC BY 2015).
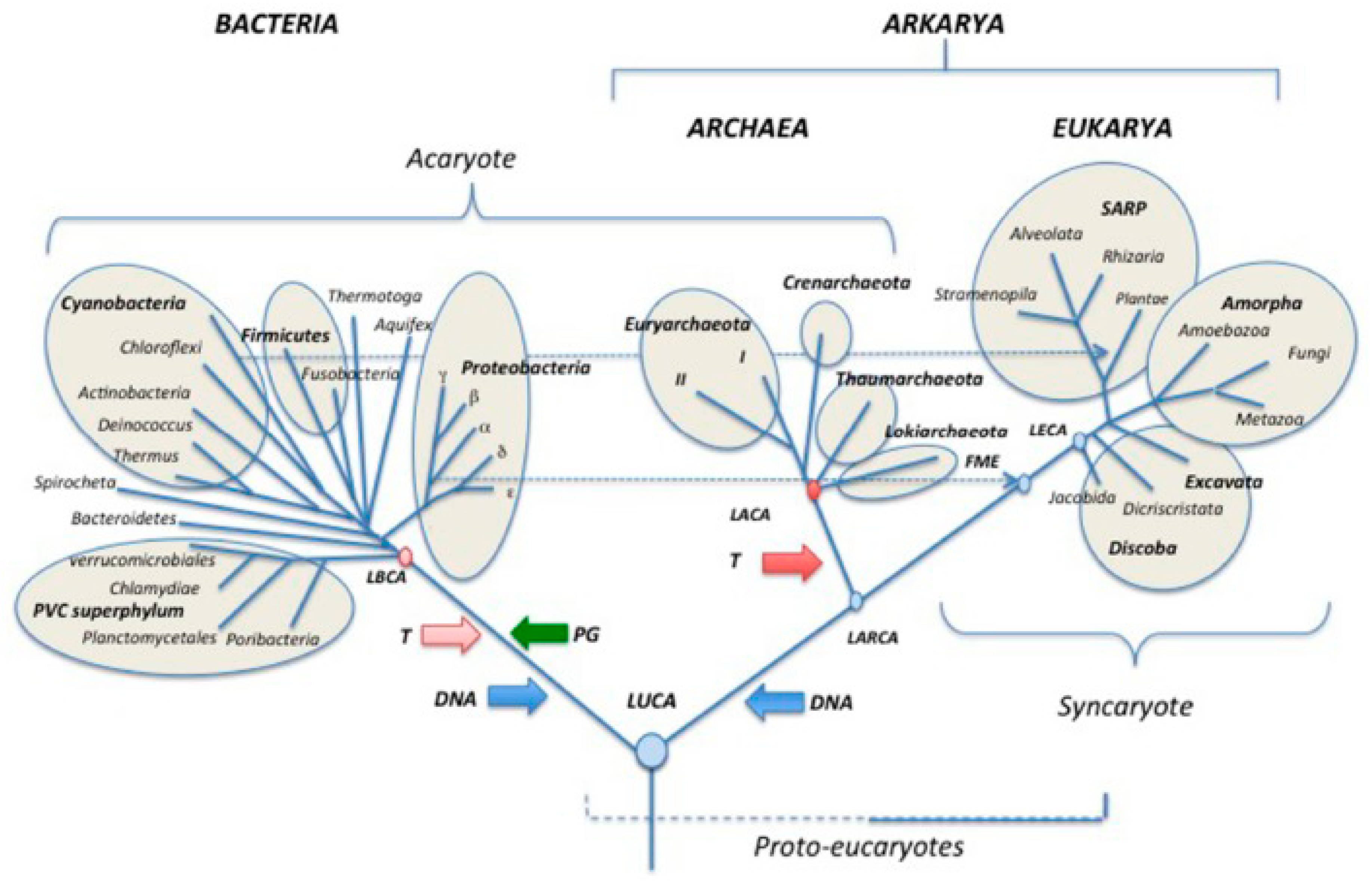
Figure 2.
The road to cancer. Upper hemisphere: all ACD-phenotypes including pluripotent PSCs, embryonic ESCs, adult stem cells ASCs, germ stem cells GSCs, and transition forms between pluripotency and multipotency are oxygen-sensitive cells (green). Under stress conditions such as oxygen excess and germline hyperoxia above 6.0% O2, they suffer severe and irreparable DNA DSB damage leading to loss of stemness, differentiation ability, and ACD potential (orange). They survive and convert into a DSCD cell type capable of defective symmetric cell cycling and symmetric cell division (SCD). DSCD cells are nonapoptotic and survive in appropriate niches (blue). Lower hemisphere: altered environmental conditions in or outside the niche induce DSCD cells to undergo a multi-stage repair process of ancient premetazoic imprinting: (i) DSCD proliferate through defective symmetric cell cycles and give rise to fusible daughter cells (red, I); (ii) the DSCD progeny form multinucleated genome repair syncytia (MGRS) by cell fusion (red, II); Likewise, benign tumors can arise from DSCD cells (iii) Within of MGRS, single nuclei fuse to form giant hyperpolyploid capable of DNA DSB repair (red III); unfortunately, the ancient premetazoic mechanisms cannot repair the defective DSCD genome to a normal human genome and reconstruct the genome according to premetazoan blueprints (red iv). At the end of the oncogenic transformation, the giant MGRS structure form spores (buds) with stemness function and ACD potential. They undergo the G+S life cycle rules and the a GRN control. The non gametogenic NG cancer germline produces primary CSCs (black). Bottom: the graphical staging of oncogenesis.
Figure 2.
The road to cancer. Upper hemisphere: all ACD-phenotypes including pluripotent PSCs, embryonic ESCs, adult stem cells ASCs, germ stem cells GSCs, and transition forms between pluripotency and multipotency are oxygen-sensitive cells (green). Under stress conditions such as oxygen excess and germline hyperoxia above 6.0% O2, they suffer severe and irreparable DNA DSB damage leading to loss of stemness, differentiation ability, and ACD potential (orange). They survive and convert into a DSCD cell type capable of defective symmetric cell cycling and symmetric cell division (SCD). DSCD cells are nonapoptotic and survive in appropriate niches (blue). Lower hemisphere: altered environmental conditions in or outside the niche induce DSCD cells to undergo a multi-stage repair process of ancient premetazoic imprinting: (i) DSCD proliferate through defective symmetric cell cycles and give rise to fusible daughter cells (red, I); (ii) the DSCD progeny form multinucleated genome repair syncytia (MGRS) by cell fusion (red, II); Likewise, benign tumors can arise from DSCD cells (iii) Within of MGRS, single nuclei fuse to form giant hyperpolyploid capable of DNA DSB repair (red III); unfortunately, the ancient premetazoic mechanisms cannot repair the defective DSCD genome to a normal human genome and reconstruct the genome according to premetazoan blueprints (red iv). At the end of the oncogenic transformation, the giant MGRS structure form spores (buds) with stemness function and ACD potential. They undergo the G+S life cycle rules and the a GRN control. The non gametogenic NG cancer germline produces primary CSCs (black). Bottom: the graphical staging of oncogenesis.
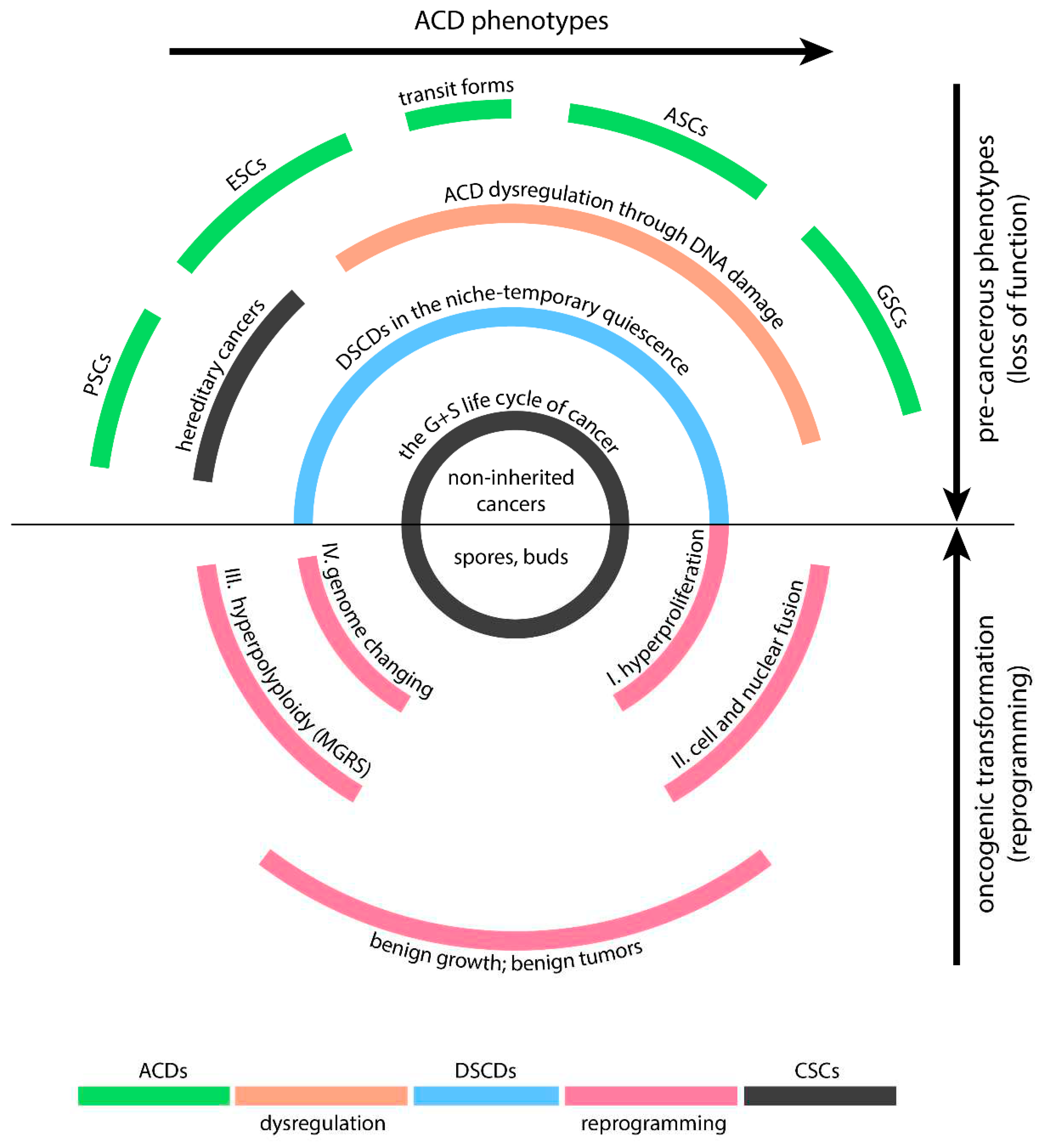

Table 1.
The life cycles of amoebozoans and cancer. Homologies and differences.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



