
Submitted:
27 July 2023
Posted:
28 July 2023
You are already at the latest version
Abstract
2,3,5,6-Tetramethyl-1,4-diisocyanobenzene (1), 1,4-diisocyanobenzene (2), and 1,4-dicyanobenzene (3) were co-crystallized with 1,3,5-triiodotrifluorobenzene (1,3,5-FIB) to give three cocrystals, 1·1,3,5-FIB, 2·2(1,3,5-FIB), and 3·2(1,3,5-FIB), which were studied by XRD. Common feature of the three structures is the presence of I···Cisocyanide or I···Nnitrile HaBs occurred between an iodine -hole and the isocyanide C- (or the nitrile N-) atom. The diisocyanide and dinitrile cocrystals, 2·2(1,3,5-FIB) and 3·2(1,3,5-FIB), are isostructural thus providing a basis for accurate comparison of the two types of noncovalent linkages of CN/NC groups in the composition of structurally similar entities and in one crystal environment. The bonding situation was studied by a set of theoretical methods. Diisocyanides are more nucleophilic than the dinitrile and they exhibit stronger binding to 1,3,5-FIB. In all structures, the HaBs are mostly determined by the electrostatic interactions, but the dispersion and induction components also provide a noticeable contribution and make the HaBs attractive. Charge transfer has a small contribution (<5%) to the HaB and it is higher for the diisocyanide than for the dinitrile systems. At the same time, diisocyanide and dinitrile structures exhibit typical electron-donor and -acceptor properties in relation to the HaB donor.
Keywords:
halogen bond
; isocyanides
; nitriles
; QTAIM
; SAPT
; NBO analysis
1. Introduction
Halogen bonding (abbreviated as HaB) is currently among the most actively studied forces in the palette of noncovalent interactions.[1,2,3,4,5,6,7,8] This interest is determined by impressive applications of HaB in various fields of science. Recently published reviews focus on general aspects of HaB[9,10,11,12,13] and also more specific topics such as HaB-based crystal engineering,[14,15,16,17,18] sensoring,[19,20] molecular[20,21] and anion[11,22] recognition, noncovalent catalysis,[11,20,23,24,25,26,27] synthetic organometallic and coordination chemistry,[28] polymer chemistry,[29] and drug design.[30,31,32,33] It is noteworthy that HaB is involved in human physiology,[34,35] in particular in the functioning of thyroid hormones.[34]
Great diversity of the studied HaB systems, in many respects, is associated with a significant variety of accepting centers, which act as the nucleophilic components of Hal···Nu contacts. In the overwhelming majority of instances, HaB acceptors are represented by electronegative heteroatoms bearing a lone pair (abbreviated as LP). Owing to a lower electronegativity of C-atom compared to N, O, S, halogens etc., noncovalent interactions including LP of a carbon site are rare. They have been only identified in some cocrystals of iodoperfluoroarenes with aryl isocyanides[36,37,38] or highly nucleophilic persistent carbenes.[39]
In view of our interest in crystal engineering of noncovalent systems with C- or N-nucleophilic coformers, we attempted to compare HaBs, which include the acceptor centers of different nature in the composition of structurally similar entities and in one crystal environment. For this study we addressed the isomeric isocyanide and nitrile species and the goal was to establish how the C≡N/N≡C isomerism affects the HaB (Figure 1).
As HaB acceptors we chose homoditopic 2,3,5,6-tetramethyl-1,4-diisocyanobenzene (1), 1,4-diisocyanobenzene (2), 1,4-dicyanobenzene (3) and, as HaB donor, potentially trifunctional 1,3,5-triiodotrifluorobenzene (1,3,5-FIB) (Figure 2). We found that cocrystallization of diisocyanides 1 and 2 and also dinitrile 3 with 1,3,5-FIB afford cocrystals 1·1,3,5-FIB, 2·2(1,3,5-FIB), and 3·2(1,3,5-FIB), exhibiting halogen-bonded supramolecular architecture. These cocrystals were studied by single-crystal X-ray diffraction (XRD) followed by theoretical calculations to closely interrogate the HaB systems. Our findings—uncovering common features and differences in the two types of noncovalent linkages of isomeric C≡N/N≡C groups (Figure 1)—are detailed in the following sections.
2. Results
2.1. Crystal growth and structural motifs of the XRD structures
Diisocyanides 1 and 2 and dinitrile 3 were co-crystallized with 1,3,5-FIB (1:1 molar ratio between the components) on slow evaporation of their solutions at 20–23 °C. This procedure gave three cocrystals, 1·1,3,5-FIB, 2·2(1,3,5-FIB), and 3·2(1,3,5-FIB), which were studied by XRD. The homogeneity of the samples was confirmed in powder diffraction X-ray experiments (the Supporting
Materials).
The structures exhibit different molar ratios between the coformers, namely 1:2 for 2·2(1,3,5-FIB) and 3·2(1,3,5-FIB), and 1:1 for 1·1,3,5-FIB; for TG characterization see the Supporting Materials. In all structures, 1,3,5-FIB acts as a trifunctional 120°-orienting HaB donor, whereas the diisocyanides (1 and 2) and the dinitrile (3) function as homoditopic 180°-orienting HaB acceptors. In the structures of 1·1,3,5-FIB and 2·2(1,3,5-FIB), we observed several types of HaBs, namely I···C, σ-(I)-hole···iodine electron-belt, and I···F. In 3·2(1,3,5-FIB), I···N and σ-(I)-hole···iodine electron-belt contacts.
Although the crystal structures exhibit different supramolecular architectures, their common feature is the presence of I···Cisocyanide or I···Nnitrile HaBs occurred between an iodine σ-hole and the isocyanide C- (or the nitrile N-) atom (Table 1). These HaBs are characterized by rather short interatomic distances. For the diisocyanide cocrystals, the I···N distances are comparable with those previously observed in cocrystals of iodo(perfluoro)arenes with CNMes[36] or with 1 and 2.[38] For the dinitrile cocrystal, the I···N distance is similar to those found in C6F5I·NCMes (3.092(4) Å, EBIHEF)[40] and (2,3,5,6-tetramethyl-1,4-dicyanobenzene)·1,4-I2C6F4 (3.061(3) Å, HUMLOQ).[41] In addition, weak σ-(I)-hole···iodine electron-belt HaB was observed in the structures of 2·2(1,3,5-FIB) and 3·2(1,3,5-FIB) and σ-(I)-hole···fluorine, I···F HaB in 1·1,3,5-FIB (Table 1).
The diisocyanide and dinitrile cocrystals, 2·2(1,3,5-FIB) and 3·2(1,3,5-FIB), are isostructural, they exhibit similar cell parameters, and display the same supramolecular organization. In these structures, the I-atoms of the I···C contact (in 2·2(1,3,5-FIB)) or the I···N contact (in 3·2(1,3,5-FIB)) function as HaB acceptor site and are involved in two additional I···I HaBs (Figure 3). In these two cases, two relatively short contacts, which are different in 2·2(1,3,5-FIB) and 3·2(1,3,5-FIB), namely I···C and I···N, exist in a very similar environment; this provide a basis for an accurate comparison between the isocyanide and the isomeric nitrile groups. For this comparison, we performed appropriate quantum chemical calculations detailed in section 2.2.
In the structure of 1·1,3,5-FIB, a rather strong HaB is formed apparently because of an enhanced σ-hole acceptor ability of the isocyano C-atom in 1 due to the combined effect of four electron-donating methyl groups (Figure 4); tri(poly)-center HaBs were not observed.
We measured FTIR-ATR spectra for pure 1–3 and their cocrystals (see the Supporting Materials for details). The isocyanide-based cocrystals 1·1,3,5-FIB and 2·2(1,3,5-FIB) demonstrate high-frequency shift of ν(NC) band (18 and 13 cm–1) relatively to 1 and 2, while the difference between the dinitrile-involving structures, 3·2(1,3,5-FIB) and 3, is small (1 cm–1). This trend in experimental ν(NC) band shifts is in a good agreement with theoretical data (Table S4). Small changes in ν(NC) frequency for dinitrile-based system may indicate a weakening of the charge transfer (CT) for dinitrile as compared to isocyanides; this correlates well with the calculation of the CT by NBO method (section 2.2.4).
The high-freqiency shift induced by the HaB, resembles the situation with C-isocyanide/N-nitrile coordination to Lewis acidic centers. Siginficant ν(NC) increase (150 cm–1) was observed for the H3B·CNMe associate and this blue shift was rationalized by appropriate increase of CN force constant upon coordination.[42] High-frequency shift of ν(NC) band in IR and Raman spectra was observed for the homoleptic copper(I) [Cu(CNMe)4]+ and [Cu(NCMe)4]+ complexes as compared to the corresponding uncomplexed isocyanide and nitrile species.[43] This shift is greater for the isocyanide ligand (40–60 cm–1) and smaller for the nitrile (~15 cm–1).[43]
2.2. Theoretical calculations
The nature of C–I···C/N contacts occurred between the diisocyanides or the dinitrile and 1,3,5-FIB were studied by a set of computational methods including Molecular electrostatic potential (MEP), Quantum theory of atoms in molecules (QTAIM), Independent gradient model (IGMH), Electron localization function (ELF), Natural Bond Orbital (NBO), The domain based local pair-natural orbital coupled-cluster (DLPNO-CCSD(T)), and Symmetry Adapted Perturbation Theory (SAPT). We also performed a comparative analysis of HaB in the cocrystals to verify common features and differences of these noncovalent interactions.
To study HaB in our systems, geometry optimization was carried out for all bimolecular fragments, [(1–3)‧1,3,5-FIB]. The optimized structures are shown in Figure 5, and the relevant main geometric parameters are collected in Table S3. In all structures, the geometry optimization led to a reduction of N≡C (isocyanide) or С≡N (nitrile), and С–I distances (Table S3) and a slight decrease (by ~6°) of ∠(C–I···C/N). The lengths of the I···C/N HaBs increase by 0.03, 0.08, and 0.10 Å, respectively, for the two diisocyanide structures and one dinitrile structure. In general, the obtained optimized structures are consistent with the experimental XRD data.
2.2.1. Nucleophilicity and molecular electrostatic potential.
We determined the global (NNu) and local (NNuloc) nucleophilicity indexes and found that NNu decreases in the following order: 1 (2.35 eV) > 2 (1.80 eV) > 3 (1.40 eV). This order correlates well with Mayr’s experimental nucleophilicity N+ indexes.[44] At the same time, the analysis of local nucleophilicity showed that the NNuloc of the C-atom of the isocyanide group in 1 is approximately equal to the NNuloc of the C-atom in 2. This observation suggests that the interaction energy of HaB should be approximately the same for 1 and 2. The NNuloc at the N-atom of the nitrile group in 3 is slightly lower than that at any one of the C-atoms of the isocyanide groups (Table 2); this is in agreement with Vs,min MEP potentials of the HaB acceptors (Figure 6).
2.2.2. QTAIM-IGMH.
The QTAIM[45,46] analysis for all contact types revealed the presence of bond critical points (BCPs) and a bond path through the BCP. This bond path indicates the accumulation of the maximum electron density between the interacting nuclear attractors. The most important topological parameters at BCPs (namely, electron density (ρb), Laplacian (∇2ρb), and total energy density (Hb = Vb + Gb), potential and kinetic energy densities (Vb and Gb), and second eigenvalues of the Hessian matrix (λ2) are listed in Table 3. The molecular graphs for [(1–3)‧1,3,5-FIB] are given in Figure 7. Low values of ρb (0.015–0.019 a.u.), positive values of ∇2ρb (0.047–0.050 a.u.), and virtually zero values of Hb are typical for noncovalent interactions and their consideration confirms the presence of the closed-shell HaB. The ρb values for the HaB involving the nitrile are slightly lower than those found for the isocyanides; this indicates a weakening of the interaction energy in the nitrile system. The negative value of λ2 ranges from –0.012 to –0.010 a.u. showing that these interactions are attractive. Furthermore, the computed value of the electron localization function (ELF) at the BCP is slightly higher for both (diisocyanide)·1,3,5-FIB systems (Table 3) probably because of a higher contribution of the covalent component in the HaB (for detailed ELF consideration see section 2.2.3).
The IGMH[47,48,49,50,51] isosurface (Figure 7a, b) was also calculated for all types of contacts. This isosurface is represented by a drop-shaped green surface located between the HaB-donating I- and the HaB-accepting C- or N-atoms. Analysis of the calculated intrinsic bond strength index (IBSI,[52] the local stretching force constant) indices, similar to QTAIM, favors the weakening of HaB between 1,3,5-FIB and the dinitrile than that for diisocyanides. In all adducts, no auxiliary interactions were found and it means that the HaB is the main attractive interaction in the studied systems.
2.2.3. Electron localization function.
To understand the reason why do the isocyanide systems are characterized by a larger contribution of the covalent component, we additionally performed an ELF analysis[53] for [(1–3)‧1,3,5-FIB]. This analysis allows us to measure the excess kinetic energy caused by Pauli repulsion and also to visualize the localization of electrons. The values of the ELF function at high electron localization (the localization can be interpreted either as LPs, or chemical bonds) should exhibit a value close to 1, while for low electron localization the value of the ELF function tends to 0.[54]
The topological analysis of the gradient field of the ELF function is also very useful because this analysis leads to the division of the molecular space into non-overlapping basins of attractors.[55] These basins can be classified as basic (C(X), concentrated on atoms) and valence (V(X) or V(X,Y), concentrated between atoms). The basins could have a synaptic order indicating the number of basins linked together. Figure 8 shows that in all cases, the disynaptic basins between the I-atom of the HaB donor and the C- or N-atoms of the HaB acceptors were not observed; this fact additionally favors the noncovalent nature of the HaB.
Hence, the main attention was paid to the electron population and volume of the monosynaptic V(C) or V(N) and the dinosynaptic V(C,I) basins. The monosynaptic basins can be attributed to LPs on the C- or N- atoms, while the disynaptic basin V(C,I) is related to the localization of the electron density of the C–I bond; these basins are directly involved in the HaBs. The population of the V(C,I) basin for [(1 and 2)‧1,3,5-FIB], involving in the HaB, is higher (by 0.08 e) than the basins that are not involved in the HaB (Table 4).
The population of the monosynaptic basin V(C) is reduced by 0.01 e compared to the unbound С-atoms of the isocyanides. We also found that the interaction of the isocyanides with 1,3,5-FIB leads to a decrease in the local volume of the basin V(C) for LP(C) from 280 to 100 Å3 for [1‧1,3,5-FIB], and from 440 to 170 Å3 for [2‧1,3, 5-FIB]. Remarkably, the basin V(C) volume for LP(C) is also decreased from 280 to 100 Å3 for [1‧1,3,5-FIB] and 440 to 170 Å3 for [2‧1,3,5-FIB]. The analysis of the ELF basin for [3‧1,3,5-FIB] also shows an increase in the population of the V(C,I) basin, but the population of the monosynaptic V(N) basin remains unchanged. All these features are coherent with the weakening of the charge transfer (CT) for the dinitrile system in comparison with the diisocyanide systems.
The ELF data clearly indicate that the covalent component is caused by the outflow of electrons as a result of CT from the diisocyanides (or the dinitrile) to 1,3,5-FIB. However, the HaB with the dinitrile, in contrast to those for both diisocyanides, is characterized by a very weak CT.
2.2.4. Natural Bond Orbital approach.
Since the examination of the ELF results revealed the presence of CT, we performed an NBO analysis to characterize orbital interactions between the HaB donors and acceptors; NBO data are collected in Table 5. We found that two most important donor-acceptor interactions are associated with CT and they are responsible for stabilizing HaB by the HaB acceptors.
The first type is related to the donor-acceptor interaction with LP(C/N) to σ*-orbital (I–C) of 1,3,5-FIB (Figure 9). The second order perturbation energies E(2) for the transitions LP(C/N)→σ*(I–C) follow the order [1‧1,3,5-FIB] (9.6) ~ [2‧1,3,5-FIB] (10.2) > [3‧1,3,5-FIB] (5.2 kcal/mol). The second interaction type is associated with the LP(I)⟶σ*/π*(C–N)isocyanide and σ*/π*(N–C)nitrile bond orbitals. On the whole, the E(2) values also follow the general trend of the first type interaction, but are characterized by smaller E(2) values (Table 5). To study the directions of the charge flow, we analyzed the occupancy of the σ*(I–C) orbital of 1,3,5-FIB and also the total population of the orbitals associated with the (C–N)isocyanide or (N–C)nitrile bonds (Table 5). As can be inferred from consideration of the data gathered in Table 5, the population of the σ*(I–C) site increases by 50 me.
This increase is associated with the CT from the LP(C or N) orbitals of the diisocyanides (or the dinitrile) to the σ*(I–C) orbital of 1,3,5-FIB. At the same time, the population of the orbitals of the HaB acceptors and σ*(N≡C) and σ*(C≡N) bonds is increased by 16 me. This increase favors the presence of a reverse CT from LP(I)⟶σ*/π*(C≡N)isocyanide and σ*/π*(N≡C)nitrile bond orbitals. In these cases, the first effect prevails over the second one and it leads to an increase (by 0.01 Å) of the I–C bond length (Table S3). This observation indicates that for the isocyanides, the direct CT is 3-fold higher than the reverse CT due to the more pronounced σ-donor properties of the diisocyanides compared to the dinitrile. The direct CT decreases in a series [1‧1,3,5-FIB] ~ [2‧1,3,5-FIB] > [3‧1,3,5-FIB]. The latter trend is consistent with the weakening of the nucleophilicity in the order 1 ~ 2 > 3 (section 2.2.1; calculated indices are given in Table 2). Thus, the diisocyanides and the dinitrile exhibit typical electron-donor and π-acceptor properties with respect to the HaB donor.
2.2.5. Energy.
The interaction (EintSM) and binding (EbSM) energy data – obtained using the supramolecular approach and calculated at the DLPNO-CCSD(T))[56,57] level – are collected in Table 6. In general, EintSM values do not exceed –4 kcal/mol; this energy corresponds to weak noncovalent interactions. The values of EbSM are comparable to those of EintSM. This is due to the fact that the deformation energy of the HaB acceptors has a positive sign, while the deformation energy of the HaB donor is negative. Thus, the resultant energy responsible for the deformation of the monomers in the dimer geometry becomes low.
Finally, to closely interrogate the physical nature of the HaBs, we performed the decomposition of the interaction energy by the SAPT[58,59,60,61,62] method into electrostatic (Eelec), induction (Eind), dispersion (Edis), and exchange (Eexch) components (Figure 10). In general, the total interaction energies SAPT(0) (EintSAPT) correlate with the corresponding energies of the supramolecular interaction (Table 6), although the energies calculated using the DLPNO-CCSD(T) method are by 18% lower in absolute values than those calculated at the SAPT(0) level.
According to the obtained SAPT data (Table 6), HaBs are characterized by the predominance of electrostatic energy, which comprise approx. 60% of Σ(Eelec, Eind, and Edis). The dispersive attractive energy provides a significant contribution (~30%), while the repulsive Pauli energy has a 70% fraction. It has been proved[63,64] that the high directionality of HaB is determined by electrostatic and exchange-repulsion interactions and therefore we carefully analyzed the Eexch energy. Ongoing from [1‧1,3,5-FIB] (66%) to [2‧1,3,5-FIB] (71%), the contribution of the repulsion Pauli energy increases, and then it decreases in [3‧1,3,5-FIB] (43%). This trend is in a good agreement with the weakening of orbital interactions on the transition from the diisocyanide to the dinitrile systems. The highest value of Eexch for [2‧1,3,5-FIB] is probably associated with an increase in the basin volume LP(C) for [2‧1,3,5-FIB] than that for [1‧1,3,5-FIB]. As a result of the increase in the LP(C) volume, the Eexch energy also increases (Table 4). The inductive effect also makes some contribution to the HaB but it does not exceed 15%. In particular, the induction term includes both polarization and CT. However, SAPT method allows the estimate a contribution of CT in the induction energy.[65] An analysis of the obtained Ect data shows that the contribution of CT to the HaB interaction energy is relatively small (<30% and 6% of the induction energy for the diisocyanides and the dinitrile, respectively).
3. Discussion
In this work, we obtained cocrystals of two diisocyanides, 1 and 2, and one dinitrile, 3, with HaB-donating 1,3,5-FIB. A characteristic feature of all three structures is the occurrence of C···I or N···I HaB, correspondingly. Two cocrystals, 2·2(1,3,5-FIB) and 3·2(1,3,5-FIB), are isostructural, which facilitates the comparison of HaBs with two different acceptors (the terminal C- or N-atoms) under similar crystal environment. Results of our theoretical studies allowed us to identify common features and to verify differences in these two types of interactions (Table 7).
In summary, the examination of the obtained results revealed that the diisocyanides are more nucleophilic than the dinitrile and they exhibit stronger binding to 1,3,5-FIB. The HaBs in all structures are mostly determined by the electrostatic interactions, which is not, however, sufficient to compensate the repulsion Pauli energy. Considering all these, we led to the conclusion that the dispersion and induction components also provide a noticeable contribution to the HaB and make this interaction attractive. Charge transfer has a small contribution (<5%) to the HaB, but it is higher for the diisocyanide than for the dinitrile systems. At the same time, diisocyanide and dinitrile structures exhibit typical electron-donor and π-acceptor properties in relation to the HaB donor.
4. Materials and Methods
4.1. Materials and Instruments
Solvents, compounds 2 and 3, and 1,3,5-FIB were obtained from commercial sources and used as received. Diisocyanide 1 was prepared from commercial durene accordingly to the known four-step procedure.[38] FTIR-ATR spectra were obtained on Schimadzu IRAffinity-1 instrument. The TG studies were performed on a NETZSCH TG 209 F1 Libra thermoanalyzer; MnO2 powder was used as a standard. The initial weights of the samples were in the range 1.2–3.9 mg. The experiments were run in an open aluminum crucible in a stream of argon at a heating rate of 10 °/min. The final temperature was 450 °C. Processing of the thermal data was performed with Proteus analysis software.[66] The samples were examined by XRD phase analysis using Bruker “D8 DISCOVER” high resolution diffractometer with monochromated CuKα long focus source. Background correction and full-profile analyzes of powder diffraction patterns were carried out using the diffractometer software (TOPAS, 4.2, Bruker).
4.2. Cocrystal growth
1,3,5-FIB (82 mg, 0.16 mmol) and any one of 1–3 (0.16 mmol) were mixed in 1:1 molar ratio in hexane/methylene chloride mixture (2 mL; 1:1 v/v), and thus obtained solution was left for 2 d at 20–23 °C in a vial with poorly closed lid. During this time, the solvent was completely evaporated and released crystals of 1·1,3,5-FIB, 2·2(1,3,5-FIB), and 3·2(1,3,5-FIB) were mechanically separated and studied by XRD.
4.3. XRD studies
XRD experiments were performed using SuperNova, Single source at offset/far, HyPix3000 (for 1·1,3,5-FIB and 2·2(1,3,5-FIB)) and XtaLAB Synergy, Single source at home/near, HyPix (for 3·2(1,3,5-FIB)) diffractometers with monochromated CuKα radiation. All crystals were kept at 100 K during data collection. The structures were solved using ShelXT[67] structure solution program and refined by means using ShelXL[67] incorporated in Olex2[68] program package. Empirical absorption correction was accounted using spherical harmonics implemented in SCALE3 ABSPACK scaling algorithm (CrysAlisPro, 1.171.41.122a, Rigaku Oxford Diffraction). The structures can be obtained free of charge via CCDC database (CCDC numbers 2280076, 2280077, and 2281032).
4.4. Computational details
Full geometry optimization of the model clusters was carried out at the DFT level of theory using the PBE0[69,70] functional with the atom-pairwise dispersion correction with the Becke–Johnson damping scheme (D3BJ).[71,72] The ORCA package (version 5.0.3) was used for the calculation.[73,74] Zero-order regular approximation (ZORA)[75] was employed to account the relativistic effects. The optimization calculations based on the X-ray geometries were performed at the PBE0-D3BJ level with the ZORA-def2-TZVP(–f)[75] (for H, C, N, and F) and the SARC-ZORA-TZVP (for I) basis sets.[76] The Hessian matrix was calculated analytically for the optimized structures to prove the location of correct minima (no imaginary frequencies). Combination of the “resolution of identity” and the “chain of spheres exchange” algorithms (RIJCOSX)[77] in conjunction with the auxiliary basis sets SARC/J were used.[78] The SCF calculations were tightly converged “TightSCF”. This level of theory was used the QTAIM, ELF, IGMH, MEP, CDF, NBO and ETS-NOCV analyses. The natural bond orbital analysis was performed using the NBO 7.0 program.[79] The DLPNO-CCSD(T) method was applied for calculate single-point energy with the ZORA-def2-TZVP(–f) (for H, C, N, and F) and the SARC-ZORA-TZVP (for I) basis. The approach DLPNO-CCSD(T) used the level of accuracy “TightPNO”.
The QTAIM, ELF, IGMH, and MEP calculations were carried out using the Multiwfn 3.8 software[80,81,82] and results were visualized using the VMD program.[83] The SAPT calculations at the SAPT0 level were performed with the recommended basis sets aug-cc-pVTZ (for H, C, N, and F) and aug-cc-pVTZ-PP (for I) for the bimolecular clusters using the Psi4 package (version 1.7.1).[84] The CDF and ETS-NOCV analyses were carried out according to the methodology described in refs.[85,86,87] using the Multiwfn software.
The interaction and binding energies (Eint and Eb) were calculated for bimolecular clusters as
Eint(A···B) = E(AB) – E{A} – E{B}
Eb(A···B) = E(AB) – E(A) – E(B),
where A and B are molecules, E(AB), E(A), and E(B) are total energies of the corresponding optimized structures, E{A} and E{B} are total energies of A and B in the geometry of the optimized AB.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Crystal data for 1·1,3,5-FIB, 2·2(1,3,5-FIB), and 3·2(1,3,5-FIB); Table S2: Selected bond lengths and angles for 1·1,3,5-FIB, 2·2(1,3,5-FIB), and 3·2(1,3,5-FIB); Table S3: Main geometric parameters of the optimized structures; Table S4: Selected bands in the FTIR-ATR spectra; Table S5: TG data; Figures S1–S7: FTIR ATR spectra; Figures S8–S10: PDXRD data; Figures S11–S13: TG curves; Cartesian coordinates for the studied cocrystals.
Author Contributions
Conceptualization, A.S., E.K., V.K.; methodology, A.S., A.M., and E.K.; investigation, A.S, E.K., M.K., and A.M.; resources, A.S., E.K.; writing—original draft preparation, A.S., E.K., and N.B.; writing—review and editing, V.K.; visualization, E.K. and N.B.; supervision, V.K.; funding acquisition, A.S. All authors have read and agreed to the published version of the manuscript.
Funding
support from the Russian Science Foundation (project 22-23-00307) is greatly appreciated.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and supporting materials. Also CIFs are openly available in www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgments
The article is in commemoration of the 300th anniversary of St. Petersburg State University’s founding. Physicochemical studies were performed at the Center for X-ray Diffraction Studies and Center for Chemical Analysis and Materials Research, Center for Thermogravimetric and Calorimetric Research, Center for Magnetic Resonance while theoretical calculations were performed at Computational Center, liquid nitrogen for research was obtained from Cryogenic Department (all belonging to Saint Petersburg State University).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cornaton, Y.; Djukic, J.-P. Noncovalent Interactions in Organometallic Chemistry: From Cohesion to Reactivity, a New Chapter. Acc. Chem. Res. 2021, 54, 3828–3840. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. On the Importance of σ–Hole Interactions in Crystal Structures. Crystals 2021, 11, 1205. [Google Scholar] [CrossRef]
- Shukla, R.; Chopra, D. Chalcogen and pnictogen bonds: insights and relevance. Curr. Sci. 2021, 120, 1848–1853. [Google Scholar] [CrossRef]
- Miller, D.K.; Chernyshov, I.Y.; Torubaev, Y.V.; Rosokha, S.V. From weak to strong interactions: structural and electron topology analysis of the continuum from the supramolecular chalcogen bonding to covalent bonds. Phys. Chem. Chem. Phys. 2022, 8251–8259. [Google Scholar] [CrossRef] [PubMed]
- Resnati, G.; Metrangolo, P. Celebrating 150 years from Mendeleev: The Periodic Table of Chemical Interactions. Coordination Chemistry Reviews 2020, 420, 213409. [Google Scholar] [CrossRef]
- Brammer, L. Halogen bonding, chalcogen bonding, pnictogen bonding, tetrel bonding: origins, current status and discussion. Faraday Discussions 2017, 203, 485–507. [Google Scholar] [CrossRef]
- Brammer, L.; Peuronen, A.; Roseveare, T.M. Halogen bonds, chalcogen bonds, pnictogen bonds, tetrel bonds and other [sigma]-hole interactions: a snapshot of current progress. Acta Crystallographica Section C 2023, 79, 204–216. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chemical Reviews 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Li, B.; Zang, S.-Q.; Wang, L.-Y.; Mak, T.C.W. Halogen bonding: A powerful, emerging tool for constructing high-dimensional metal-containing supramolecular networks. Coordination Chemistry Reviews 2016, 308, 1–21. [Google Scholar] [CrossRef]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew Chem Int Ed Engl 2018, 57, 6004–6016. [Google Scholar] [CrossRef] [PubMed]
- Non-covalent Interactions in the Synthesis and Design of New Compounds; Abel M. Maharramov, K.T.M., Maximilian N. Kopylovich, Armando J. L. Pombeiro, Ed.; Wiley: 2016.
- Saha, B.K.; Veluthaparambath, R.V.P.; Krishna G., V. Halogen⋯Halogen Interactions: Nature, Directionality and Applications. Chemistry – An Asian Journal 2023, 18, e202300067. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Accounts of Chemical Research 2014, 47, 2514–2524. [Google Scholar] [CrossRef]
- Xin, D.; Matti, T.; Matti, H. Halogen Bonding in Crystal Engineering. In Recent Advances in Crystallography, Jason, B.B., Ed.; IntechOpen: Rijeka, 2012; p. Ch. 7. [Google Scholar]
- Teyssandier, J.; Mali, K.S.; De Feyter, S. Halogen Bonding in Two-Dimensional Crystal Engineering. ChemistryOpen 2020, 9, 225–241. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Bokach, N.A.; Yu. Kukushkin, V.; Frontera, A. Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering. Chemistry – A European Journal 2022, 28, e202103173. [Google Scholar] [CrossRef] [PubMed]
- Hein, R.; Beer, P.D. Halogen bonding and chalcogen bonding mediated sensing. Chemical Science 2022, 13, 7098–7125. [Google Scholar] [CrossRef]
- Zheng, J.; Suwardi, A.; Wong, C.J.E.; Loh, X.J.; Li, Z. Halogen bonding regulated functional nanomaterials. Nanoscale Advances 2021, 3, 6342–6357. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chemical Reviews 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Pancholi, J.; Beer, P.D. Halogen bonding motifs for anion recognition. Coordination Chemistry Reviews 2020, 416, 213281. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; da Silva, M.F.C.G.; Pombeiro, A.J. Noncovalent Interactions in Catalysis; Royal Society of Chemistry: 2019.
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent interactions in metal complex catalysis. Coordination Chemistry Reviews 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Breugst, M.; von der Heiden, D.; Schmauck, J. Novel Noncovalent Interactions in Catalysis: A Focus on Halogen, Chalcogen, and Anion-π Bonding. Synthesis 2017, 49, 3224–3236. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angewandte Chemie International Edition 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chemistry – A European Journal 2016, 22, 14434–14450. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Non-covalent interactions in the synthesis of coordination compounds: Recent advances. Coordination Chemistry Reviews 2017, 345, 54–72. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; Meyer, F. Halogen bonding in polymer science: from crystal engineering to functional supramolecular polymers and materials. Polymer Chemistry 2015, 6, 3559–3580. [Google Scholar] [CrossRef]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking Back, Looking Forward at Halogen Bonding in Drug Discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef]
- Ho, P.S. Halogen bonding in medicinal chemistry: from observation to prediction. Future Medicinal Chemistry 2017, 9, 637–640. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Pavan, B.; Ferretti, V. Can pharmaceutical co-crystals provide an opportunity to modify the biological properties of drugs? Drug Discov Today 2017, 22, 1134–1138. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Xu, Z.; Li, H.; Liu, H.; Zhu, W. Halogen bonding for rational drug design and new drug discovery. Expert Opin Drug Discov 2012, 7, 375–383. [Google Scholar] [CrossRef]
- Bayse, C.A. Halogen bonding from the bonding perspective with considerations for mechanisms of thyroid hormone activation and inhibition. New Journal of Chemistry 2018, 42, 10623–10632. [Google Scholar] [CrossRef] [PubMed]
- Marsan, E.S.; Bayse, C.A. A Halogen Bonding Perspective on Iodothyronine Deiodinase Activity. Molecules 2020, 25, 1328. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Novikov, A.S.; Boyarskiy, V.P.; Kukushkin, V.Y. The halogen bond with isocyano carbon reduces isocyanide odor. Nature Communications 2020, 11, 2921. [Google Scholar] [CrossRef] [PubMed]
- Mikherdov, A.S.; Popov, R.A.; Smirnov, A.S.; Eliseeva, A.A.; Novikov, A.S.; Boyarskiy, V.P.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y.; Bokach, N.A. Isocyanide and Cyanide Entities Form Isostructural Halogen Bond-Based Supramolecular Networks Featuring Five-Center Tetrafurcated Halogen···C/N Bonding. Crystal Growth & Design 2022, 22, 6079–6087. [Google Scholar]
- Smirnov, A.S.; Mikherdov, A.S.; Rozhkov, A.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y.; Bokach, N.A. Halogen Bond-Involving Supramolecular Assembly Utilizing Carbon as a Nucleophilic Partner of I⋅⋅⋅C Non-covalent Interaction. Chemistry – An Asian Journal 2023, 18, e202300037. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Kline, M.; Calabrese, J.C.; Davidson, F. SYNTHESIS OF A REVERSE YLIDE FROM A NUCLEOPHILIC CARBENE. Journal of the American Chemical Society 1991, 113, 9704–9705. [Google Scholar] [CrossRef]
- Wasilewska, A.; Gdaniec, M.; Polonski, T. CCDC 618388: Experimental Crystal Structure Determination. 2006. [CrossRef]
- Britton, D.; Gleason, W.B. Dicyanodurene-p-tetrafluorodiiodobenzene (1/1). Acta Crystallographica Section E 2002, 58, o1375–o1377. [Google Scholar] [CrossRef]
- Watari, F. Vibrational frequencies, assignments and normal-coordinate analysis for the methyl isocyanide-borane complex. Inorganic Chemistry 1982, 21, 1442–1446. [Google Scholar] [CrossRef]
- Bell, A.; Walton, R.A.; Edwards, D.A.; Poulter, M.A. Cationic copper(I) isocyanide complexes, [Cu(CNR)4]+ (R = CH3, C(CH3)3 and 2,6-(CH3)2C6H3): Preparations, spectroscopic properties and reactions with neutral ligands. A comparison of the vibrational spectra of [Cu(CNCH3)4]+, [Cu(NCCH3)4]+ and [Cu(NCCD3)4]+. Inorganica Chimica Acta 1985, 104, 171–178. [Google Scholar] [CrossRef]
- Tumanov, V.V.; Tishkov, A.A.; Mayr, H. Nucleophilicity Parameters for Alkyl and Aryl Isocyanides. Angewandte Chemie International Edition 2007, 46, 3563–3566. [Google Scholar] [CrossRef]
- Bader, R.F. A quantum theory of molecular structure and its applications. Chemical Reviews 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. Journal of Chemical Sciences 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Physical Chemistry Chemical Physics 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef]
- Zhu, S.; Asim Khan, M.; Wang, F.; Bano, Z.; Xia, M. Rapid removal of toxic metals Cu2+ and Pb2+ by amino trimethylene phosphonic acid intercalated layered double hydroxide: A combined experimental and DFT study. Chemical Engineering Journal 2020, 392, 123711. [Google Scholar] [CrossRef]
- Huang, W.; Lin, R.; Zhao, X.; Li, Q.; Huang, Y.; Ye, G. How does a weak interaction change from a reactive complex to a saddle point in a reaction? Computational and Theoretical Chemistry 2020, 1173, 112640. [Google Scholar] [CrossRef]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef]
- Klein, J.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. New Way for Probing Bond Strength. The Journal of Physical Chemistry A 2020, 124, 1850–1860. [Google Scholar] [CrossRef]
- Silvi, B.; Gillespie, R.J.; Gatti, C. Electron density analysis. In Comprehensive Inorganic Chemistry II, Reedijk, J., Poeppelmeier, K., Eds.; 2013; Volume 9, pp. 187-226.
- Fuster, F.; Sevin, A.; Silvi, B. Topological Analysis of the Electron Localization Function (ELF) Applied to the Electrophilic Aromatic Substitution. The Journal of Physical Chemistry A 2000, 104, 852–858. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. The Journal of Chemical Physics 2013, 139, 134101. [Google Scholar] [CrossRef] [PubMed]
- Altun, A.; Neese, F.; Bistoni, G. Local energy decomposition analysis of hydrogen-bonded dimers within a domain-based pair natural orbital coupled cluster study. Beilstein Journal of Organic Chemistry 2018, 14, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Parrish, R.M.; Sherrill, C.D.; Turney, J.M.; Schaefer, H.F. , 3rd. Large-scale symmetry-adapted perturbation theory computations via density fitting and Laplace transformation techniques: investigating the fundamental forces of DNA-intercalator interactions. J Chem Phys 2011, 135, 174107. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Jaeger, H.M.; Carrell, E.J.; Tschumper, G.S.; Sherrill, C.D. Accurate Interaction Energies for Problematic Dispersion-Bound Complexes: Homogeneous Dimers of NCCN, P2, and PCCP. Journal of Chemical Theory and Computation 2011, 7, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Hayes, I.; Stone, A. An intermolecular perturbation theory for the region of moderate overlap. Molecular Physics 1984, 53, 83–105. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation theory approach to intermolecular potential energy surfaces of van der Waals complexes. Chemical Reviews 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. WIREs Computational Molecular Science 2020, 10, e1452. [Google Scholar] [CrossRef]
- Grant Hill, J.; Legon, A.C. On the directionality and non-linearity of halogen and hydrogen bonds. Physical Chemistry Chemical Physics 2015, 17, 858–867. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Sensitivity of pnicogen, chalcogen, halogen and H-bonds to angular distortions. Chemical Physics Letters 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Stone, A.J.; Misquitta, A.J. Charge-transfer in Symmetry-Adapted Perturbation Theory. Chemical Physics Letters 2009, 473, 201–205. [Google Scholar] [CrossRef]
- NETZSCH Proteus Software v.6.1, Netzsch-Gerätebau Bayern, Germany, 2013.
- Sheldrick, G. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Physical review letters 1996, 77, 3865. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. The Journal of chemical physics 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. Journal of Computational Chemistry 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. The Journal of Chemical Physics 2010, 132, 154104. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Computational Molecular Science 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Computational Molecular Science 2022, 12, e1606. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Physical Chemistry Chemical Physics 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Rolfes, J.D.; Neese, F.; Pantazis, D.A. All-electron scalar relativistic basis sets for the elements Rb–Xe. Journal of Computational Chemistry 2020, 41, 1842–1849. [Google Scholar] [CrossRef]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. Journal of computational chemistry 2003, 24, 1740–1747. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Actinides. Journal of Chemical Theory and Computation 2011, 7, 677–684. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO7, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018.
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. Journal of Computational Chemistry 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. Journal of Molecular Graphics and Modelling 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. Journal of Computational Chemistry 2022, 43, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L. Psi4: an open-source ab initio electronic structure program. Wiley Interdisciplinary Reviews: Computational Molecular Science 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Nunzi, F.; Cesario, D.; Belpassi, L.; Tarantelli, F.; Roncaratti, L.F.; Falcinelli, S.; Cappelletti, D.; Pirani, F. Insight into the halogen-bond nature of noble gas-chlorine systems by molecular beam scattering experiments, ab initio calculations and charge displacement analysis. Physical Chemistry Chemical Physics 2019, 21, 7330–7340. [Google Scholar] [CrossRef]
- Belpassi, L.; Infante, I.; Tarantelli, F.; Visscher, L. The Chemical Bond between Au(I) and the Noble Gases. Comparative Study of NgAuF and NgAu+ (Ng = Ar, Kr, Xe) by Density Functional and Coupled Cluster Methods. Journal of the American Chemical Society 2008, 130, 1048–1060. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. Intermolecular interaction characteristics of the all-carboatomic ring, cyclo[18]carbon: Focusing on molecular adsorption and stacking. Carbon 2021, 171, 514–523. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Organic & Biomolecular Chemistry 2011, 9, 7168–7175. [Google Scholar]
- Chattaraj, P.K.; Duley, S.; Domingo, L.R. Understanding local electrophilicity/nucleophilicity activation through a single reactivity difference index. Organic & Biomolecular Chemistry 2012, 10, 2855–2861. [Google Scholar]
Figure 1.
Schematic representation of HaB in the isomeric isocyanide and nitrile systems.
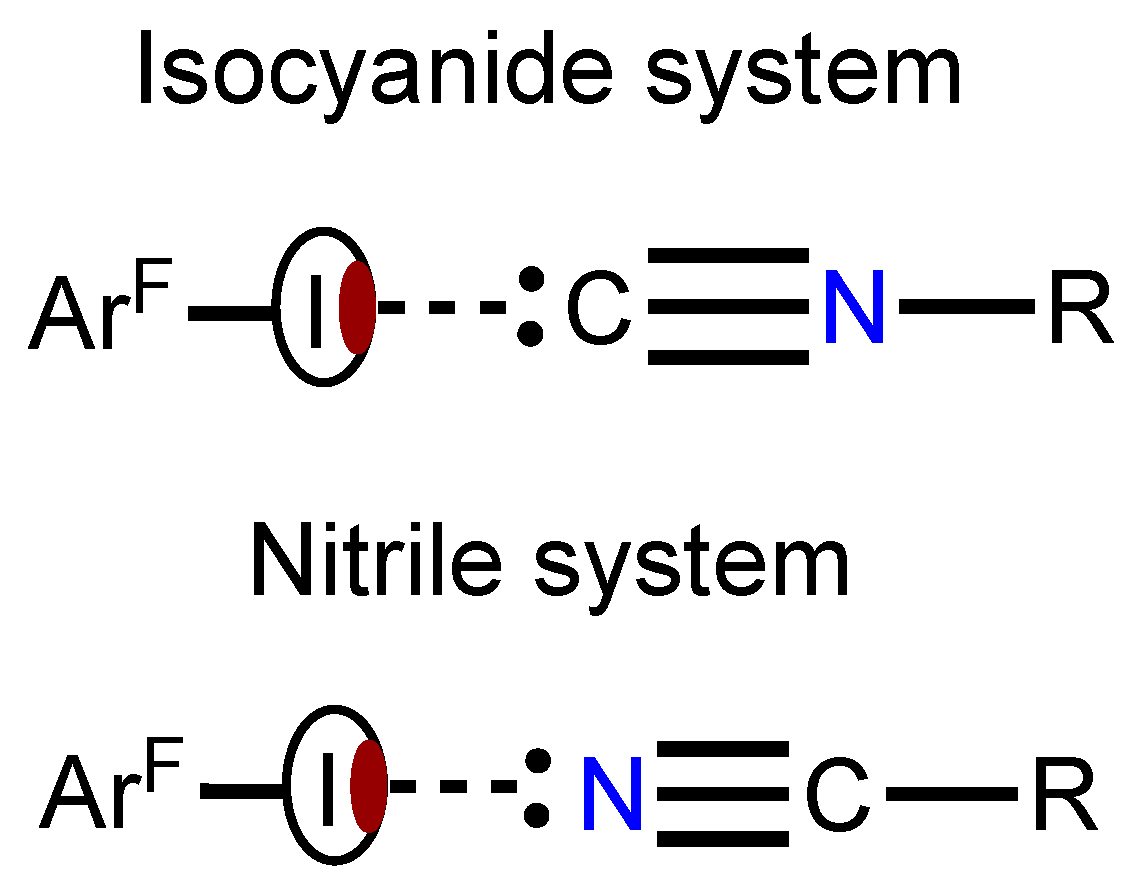
Figure 2.
Coformers of the HaB-based cocrystals and their numbering.
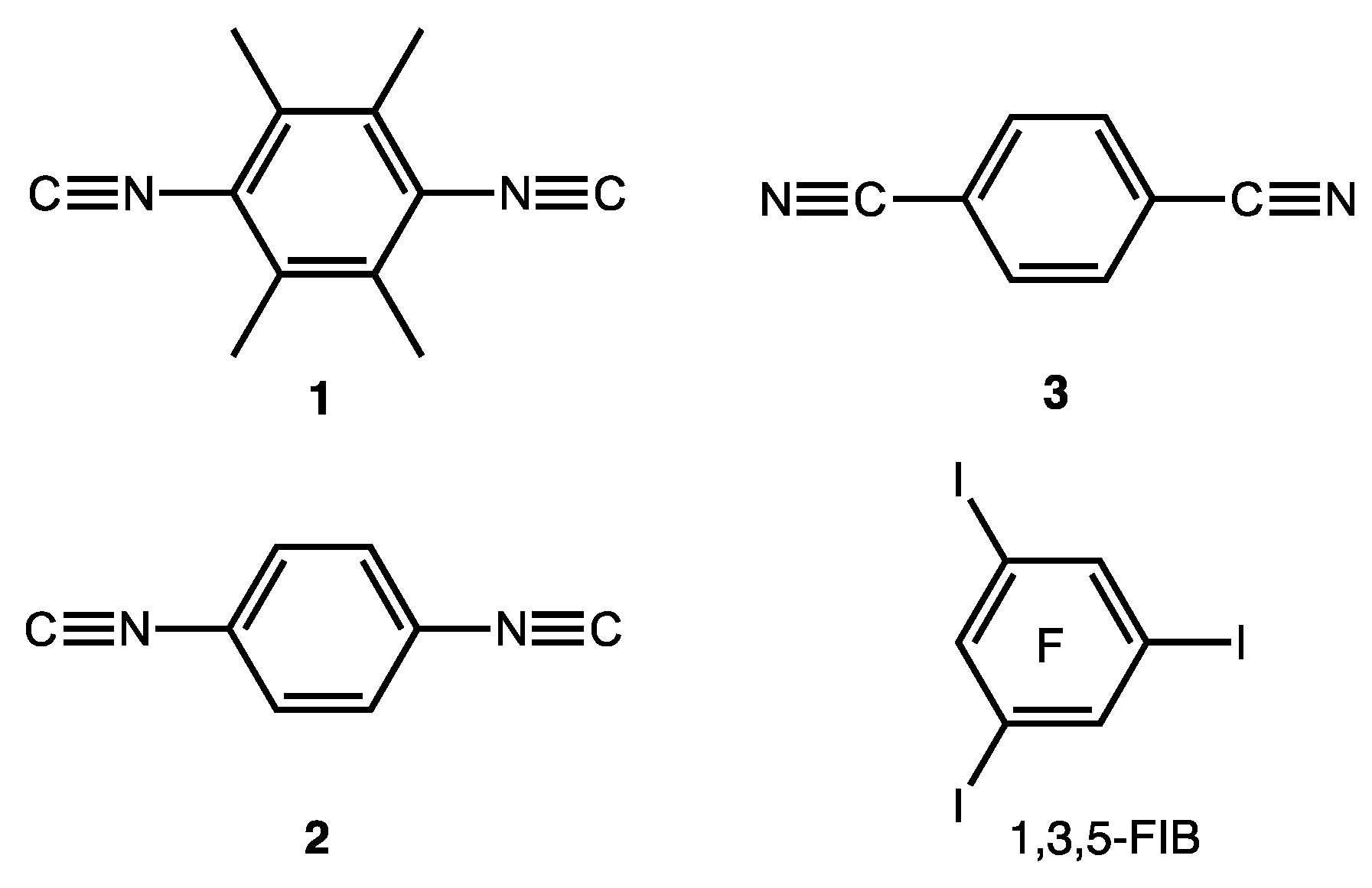
Figure 3.
Fragments of the molecular structures of 2·2(1,3,5-FIB) (left panel) and 3·2(1,3,5-FIB) (right panel); HaB is given by dotted lines.
Figure 3.
Fragments of the molecular structures of 2·2(1,3,5-FIB) (left panel) and 3·2(1,3,5-FIB) (right panel); HaB is given by dotted lines.
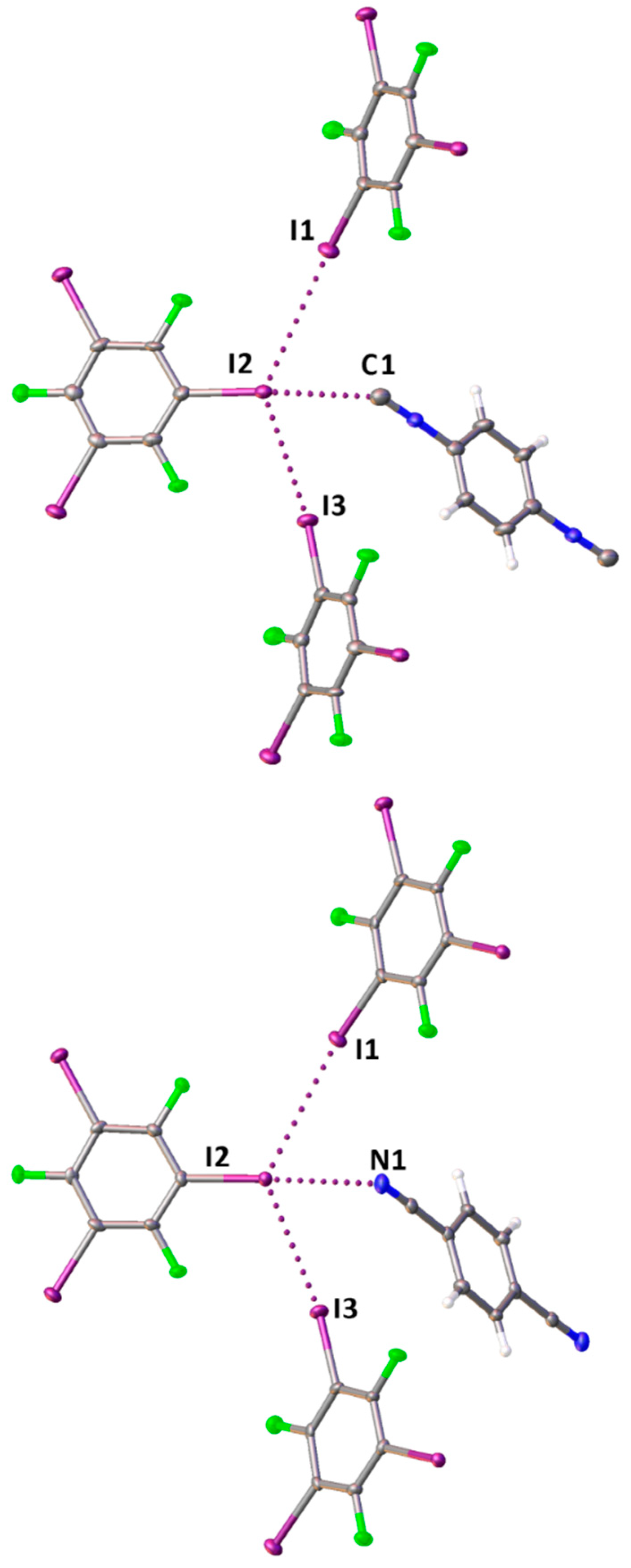
Figure 4.
A fragment of the molecular structure of 1·1,3,5-FIB displaying three two-center HaBs.
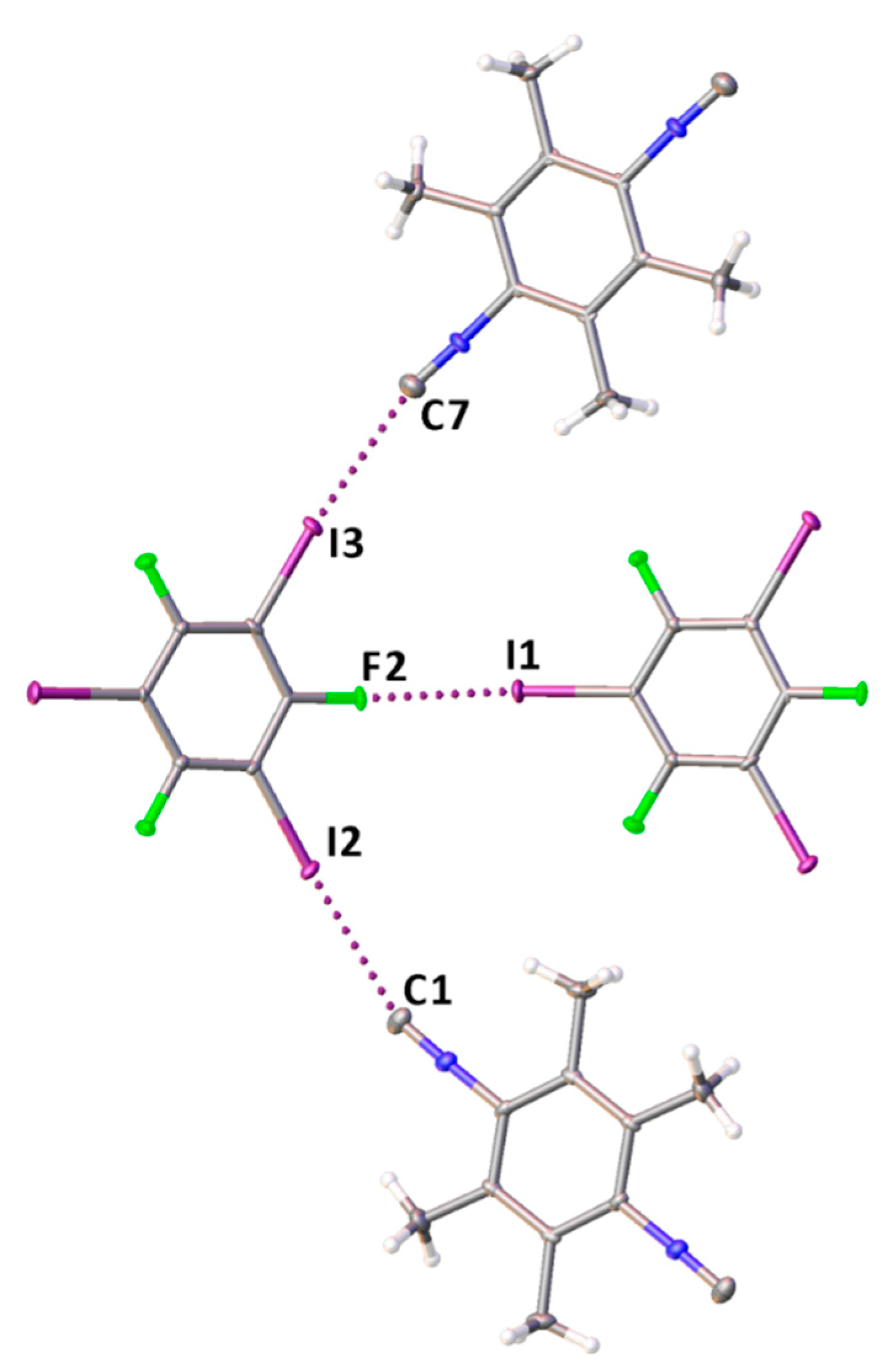
Figure 5.
Optimized structures of (a) [1‧1,3,5-FIB], (b) [2‧1,3,5-FIB], and (c) [3‧1,3,5-FIB].
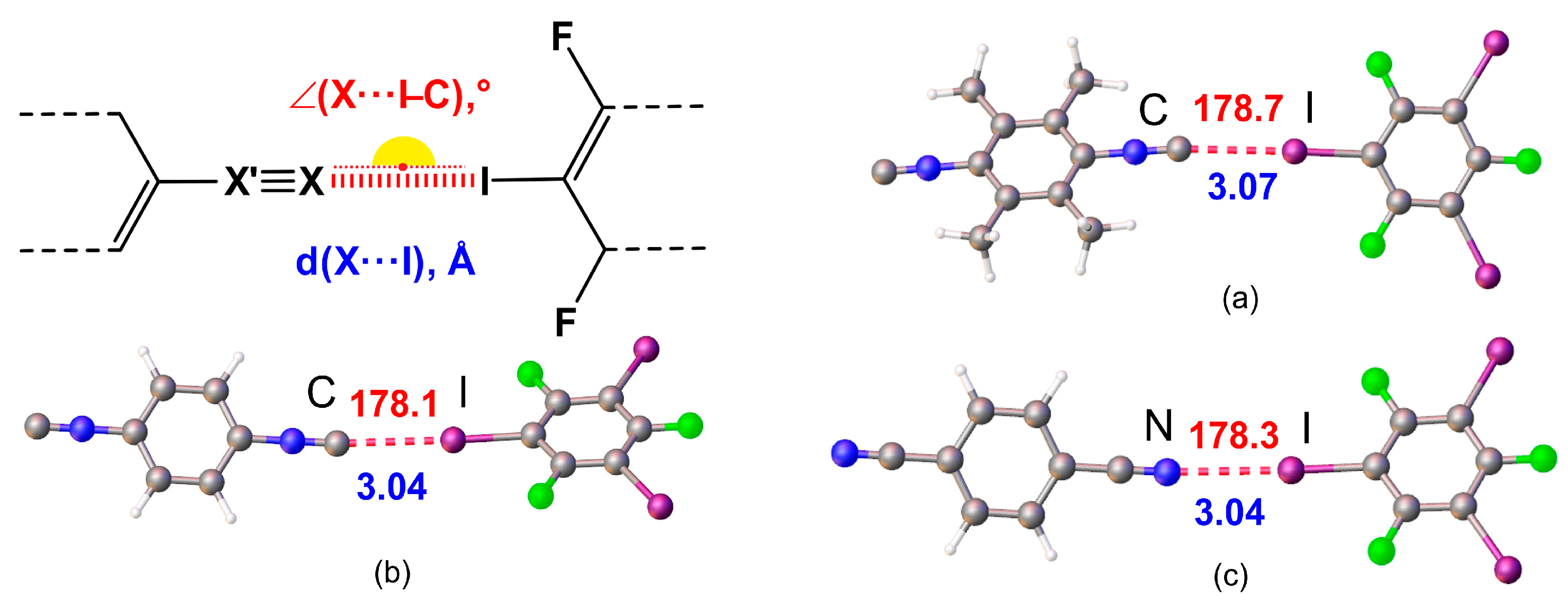
Figure 6.
MEP distribution in (a) 1, (b) 2, and (c) 3 calculated for the optimized structures (Vs,min MEP values in kcal/mol).
Figure 6.
MEP distribution in (a) 1, (b) 2, and (c) 3 calculated for the optimized structures (Vs,min MEP values in kcal/mol).
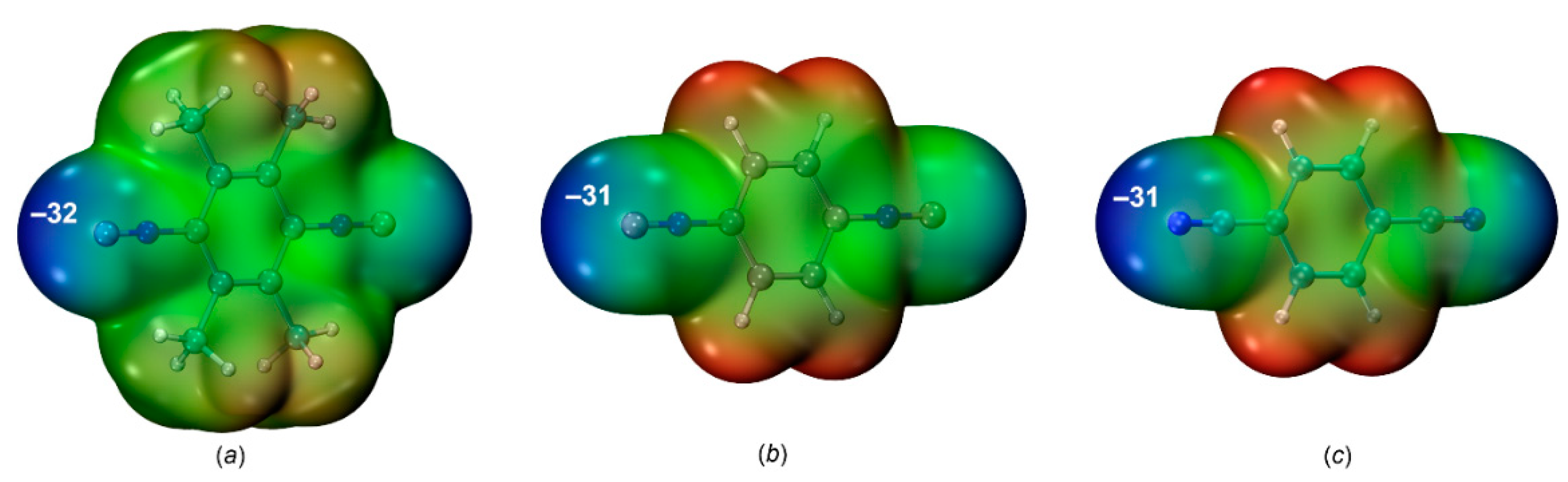
Figure 7.
The molecular graph (BCPs in red and bond paths as orange lines) and δginter isosurface for the structure of (a) [2‧1,3,5-FIB] and (b) [3‧1,3,5-FIB] (δginter = 0.01 a.u. and blue-cyan-green-yellow-red color scale −0.05 < sign(λ2)ρ(r) < 0.05); Contour line diagram of the Laplacian distribution, ∇2ρ(r), zero flux surfaces and bond paths in the C–I–C and C–I–N plane (bond critical points are shown by red dots) for the systems of [2‧1,3,5-FIB] (c) and [3‧1,3,5-FIB] (d).
Figure 7.
The molecular graph (BCPs in red and bond paths as orange lines) and δginter isosurface for the structure of (a) [2‧1,3,5-FIB] and (b) [3‧1,3,5-FIB] (δginter = 0.01 a.u. and blue-cyan-green-yellow-red color scale −0.05 < sign(λ2)ρ(r) < 0.05); Contour line diagram of the Laplacian distribution, ∇2ρ(r), zero flux surfaces and bond paths in the C–I–C and C–I–N plane (bond critical points are shown by red dots) for the systems of [2‧1,3,5-FIB] (c) and [3‧1,3,5-FIB] (d).
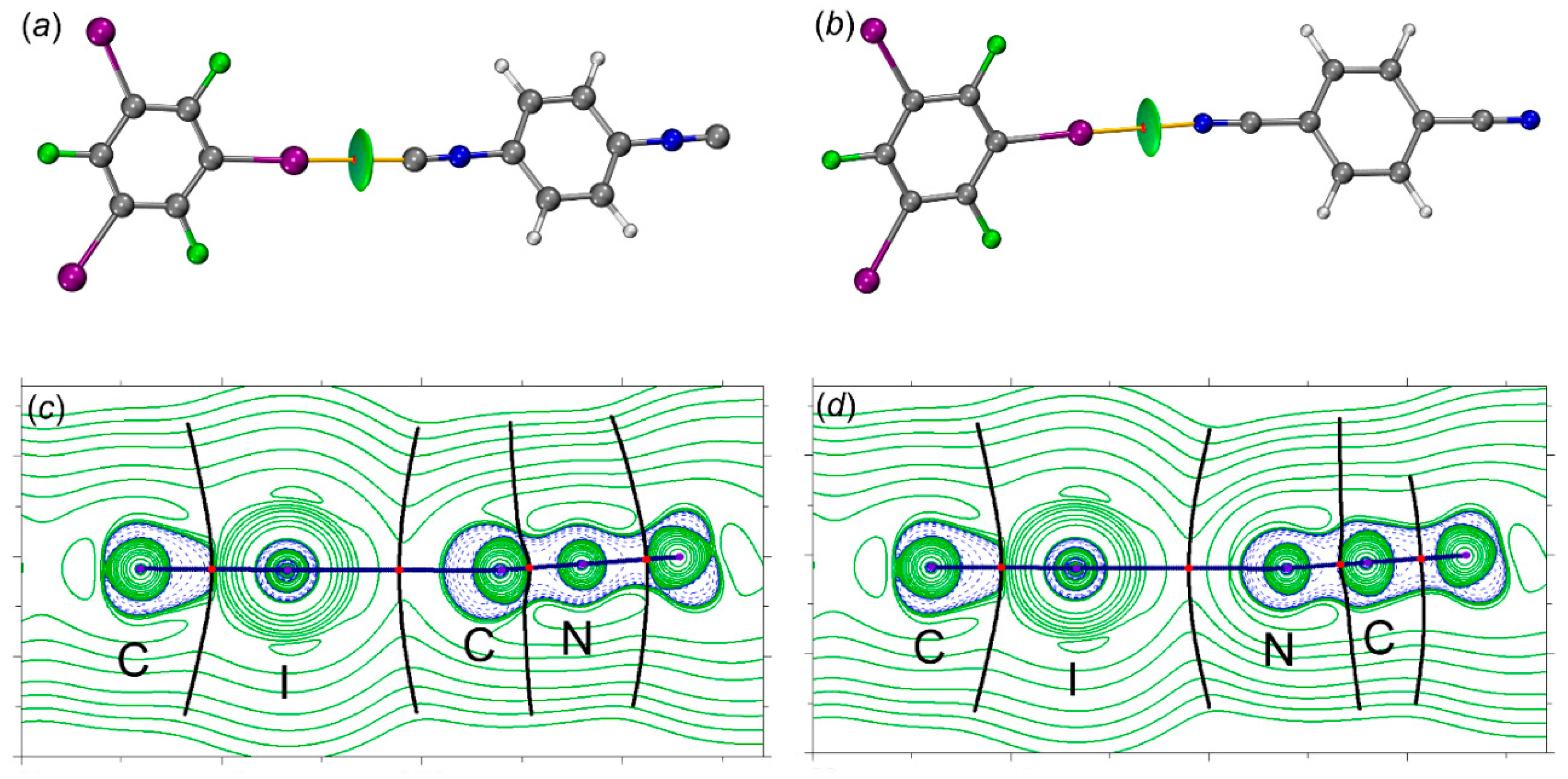
Figure 8.
ELF localization domains for the structures of (a) [2‧1,3,5-FIB] and (b) [3‧1,3,5-FIB]. Monosynaptic basins are shown in green, disynaptic basins are shown in red. *Values of the basin V(C or N) that are not involved in the HaB.
Figure 8.
ELF localization domains for the structures of (a) [2‧1,3,5-FIB] and (b) [3‧1,3,5-FIB]. Monosynaptic basins are shown in green, disynaptic basins are shown in red. *Values of the basin V(C or N) that are not involved in the HaB.
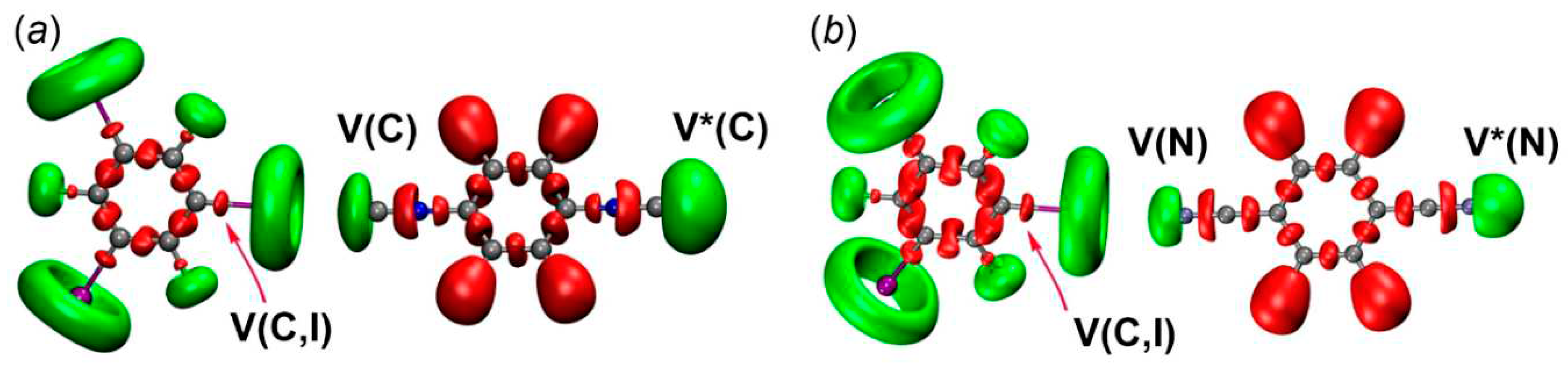
Figure 9.
Natural bond orbitals corresponding to the (a) LP(C) → σ*(I−C) transitions; (b) LP(I) → σ*(C−N); (c) LP(I) → π*(C=N) in [2‧1,3,5-FIB].
Figure 9.
Natural bond orbitals corresponding to the (a) LP(C) → σ*(I−C) transitions; (b) LP(I) → σ*(C−N); (c) LP(I) → π*(C=N) in [2‧1,3,5-FIB].
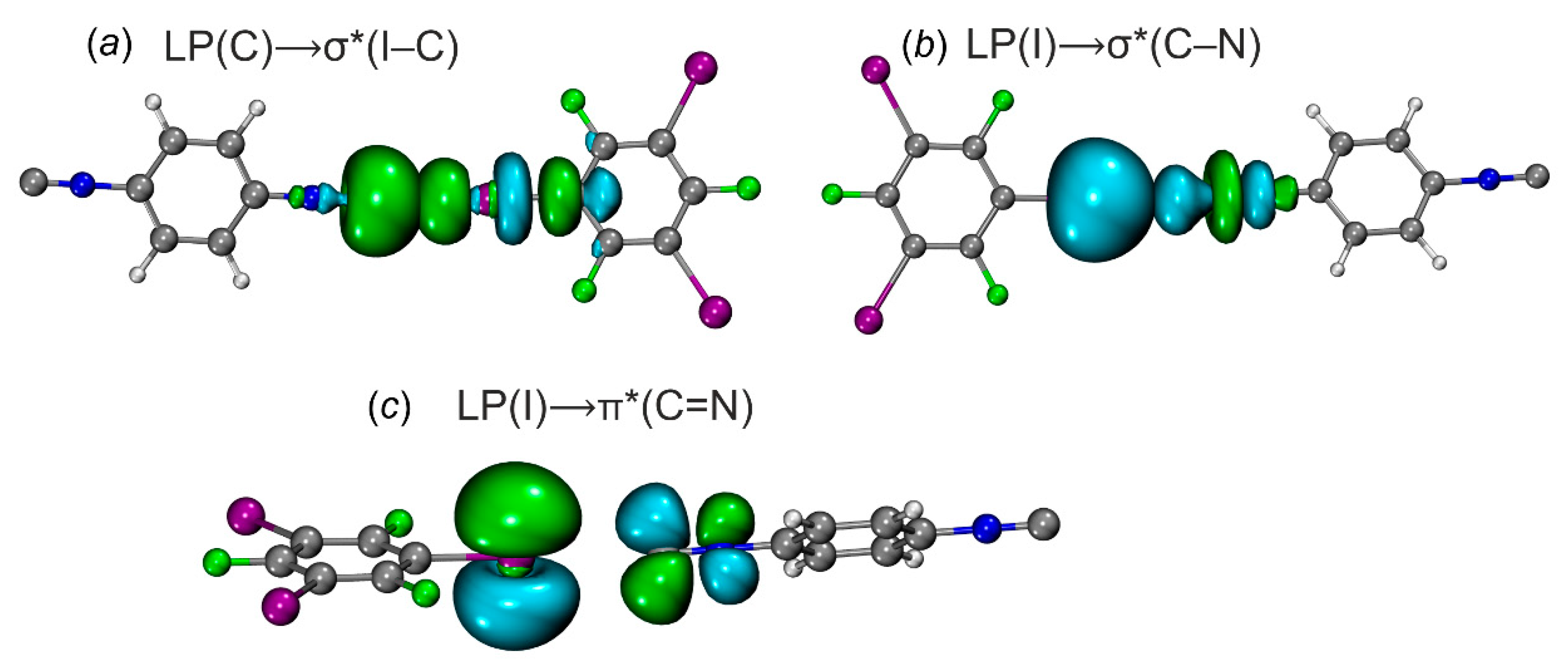
Figure 10.
Contribution to the sum of the attractive contributions to the interaction energy (%). Energies are in kcal/mol.
Figure 10.
Contribution to the sum of the attractive contributions to the interaction energy (%). Energies are in kcal/mol.
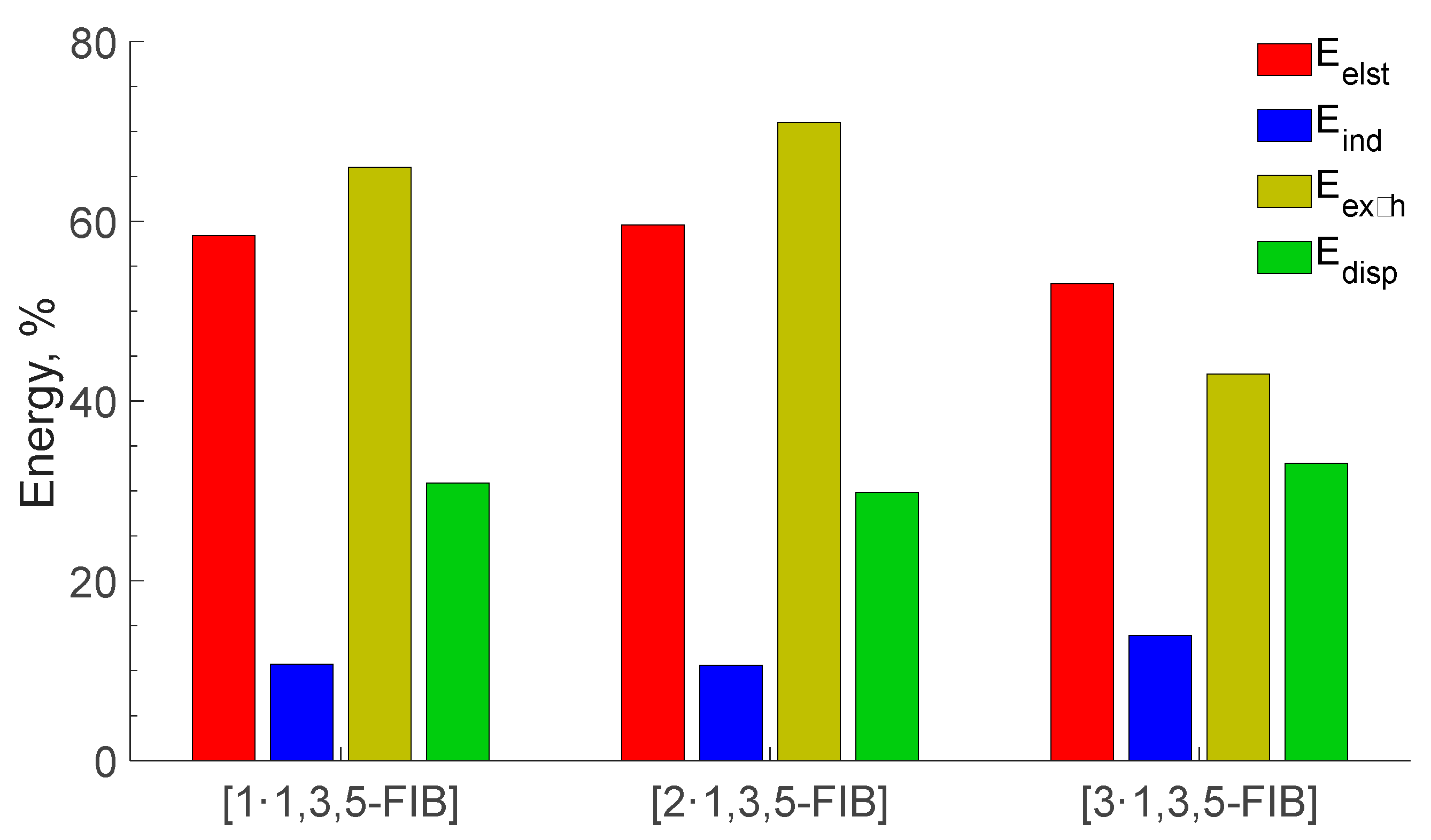

Table 1.
Geometrical parameters of HaB.
| Cocrystal | Contact I···Nu (Nu = C, I, or F) |
Interatomic distance, Å | Nc1 | Angle C–I···Nu, ° |
Angle I···Nu–Y, ° |
|---|---|---|---|---|---|
| 1·1,3,5-FIB | I2···C1 | 3.084(5) | 0.84 | 173.22(16) | 153.4(4) |
| I3···C7 | 3.035(5) | 0.82 | 173.38(16) | 159.5(4) | |
| I1···F2 | 2.992(3) | 0.87 | 171.54(13) | 136.9(3) | |
| 2·2(1,3,5-FIB) | I2···C1 | 2.957(6) | 0.80 | 172.0(2) | 138.4(5) |
| I1···I2 | 3.9561(5) | 1.00 | 163.51(14) | 116.60(15) | |
| I3···I2 | 3.8022(4) | 0.96 | 172.52(14) | 110.56(14) | |
| 3·2(1,3,5-FIB) | I2···N1 | 2.935(5) | 0.80 | 173.79(19) | 135.9(5) |
| I1···I2 | 3.9625(6) | 1.00 | 163.79(14) | 116.95(18) | |
| I3···I2 | 3.8122(5) | 0.96 | 171.32(14) | 111.63(17) |
1 Nc is the normalized contact which is here the ratio between the observed I···C, I···N, I···F, and I···I distances and the sum of van der Waals radii of involved atoms.
Table 2.
Global and local nucleophilicity of HaB acceptors.
| 1 | 2 | 3 | |
|---|---|---|---|
| NNu, eV | 2.35 | 1.80 | 1.40 |
| NNuloc (C or N), e*eV | 0.33 | 0.30 | 0.23 |
Table 3.
Electron density (ρb), its Laplacian (∇2ρb), potential, kinetic, and total energy densities (Vb, Gb, and Hb), second eigenvalue of the Hessian matrix (λ2), values (in a.u.), ELF at BCPs, IBSI, and δgpair.
Table 3.
Electron density (ρb), its Laplacian (∇2ρb), potential, kinetic, and total energy densities (Vb, Gb, and Hb), second eigenvalue of the Hessian matrix (λ2), values (in a.u.), ELF at BCPs, IBSI, and δgpair.
| Contact | Clusters | ρb | ∇2ρb | Vb | Gb | Hb | λ2 | ELF | IBSI | δgpair |
|---|---|---|---|---|---|---|---|---|---|---|
| I···C | [1‧1,3,5-FIB] | 0.0177 | 0.0467 | –0.0101 | 0.0109 | 0.0008 | –0.0115 | 0.09 | 0.032 | 0.027 |
| I···C | [2‧1,3,5-FIB] | 0.0186 | 0.0494 | –0.0109 | 0.0116 | 0.0007 | –0.0123 | 0.09 | 0.034 | 0.028 |
| I···N | [3‧1,3,5-FIB] | 0.0150 | 0.0505 | –0.0094 | 0.0110 | 0.0016 | –0.0103 | 0.05 | 0.025 | 0.024 |
Table 4.
Values of the basin electron population V(C or N), V(C,I), (in e), and electron volume VELF for basin V(C or N) (in Å3).
Table 4.
Values of the basin electron population V(C or N), V(C,I), (in e), and electron volume VELF for basin V(C or N) (in Å3).
| Clusters | V(C or N), e [VELF, Ǻ3] | V(C,I), e |
|---|---|---|
| [1‧1,3,5-FIB] | 2.62 [100]; 2.63* [380] | 1.75; 1.67*; 1.67* |
| [2‧1,3,5-FIB] | 2.60 [170]; 2.61* [610] | 1.76, 1.68*; 1.68* |
| [3‧1,3,5-FIB] | 3.31 [90]; 3.31* [460] | 1.74, 1.67*; 1.67* |
*Values of the basin V(C or N) and V(C,I) electron population that are not involved in the HaB.
Table 5.
Second order NBO perturbation energies (E(2), in kcal/mol), change of the occupancy of the σ*(I–C) and σ*/π*(C≡N) or σ*/π*(N≡C) NBO on the occurrence of HaB (Δocc, in me).
Table 5.
Second order NBO perturbation energies (E(2), in kcal/mol), change of the occupancy of the σ*(I–C) and σ*/π*(C≡N) or σ*/π*(N≡C) NBO on the occurrence of HaB (Δocc, in me).
| Clusters | Transition | E(2) | Δocc |
|---|---|---|---|
| [1‧1,3,5-FIB] | LP(C)⟶σ*(I–C) | 9.6 | 44 |
| LP(I)⟶σ*/π*(C≡N) | 2.1 | 15 | |
| [2‧1,3,5-FIB] | LP(C)⟶σ*(I–C) | 10.2 | 46 |
| LP(I)⟶σ*/π*(C≡N) | 2.3 | 16 | |
| [3‧1,3,5-FIB] | LP(N)⟶σ*(I–C) | 5.2 | 16 |
| LP(I)⟶σ*/π*(N≡C) | 1.8 | 12 |
Table 6.
Calculated values of the interaction and binding energies and their decomposition (in kcal/mol).
Table 6.
Calculated values of the interaction and binding energies and their decomposition (in kcal/mol).
| Cluster | Eelst | Eind (Ect) | Eexch | Edisp | EintSAPT* | EintSM | EbSM |
|---|---|---|---|---|---|---|---|
| [1‧1,3,5-FIB] | –8.7 | –1.6 (0.5) | 9.8 | –4.6 | –5.1 | –4.2 | –4.1 |
| [2‧1,3,5-FIB] | –9.0 | –1.6 (0.5) | 10.6 | –4.5 | –4.6 | –3.8 | –3.8 |
| [3‧1,3,5-FIB] | –6.1 | –1.6 (0.1) | 6.4 | –3.8 | –5.0 | –3.7 | –3.7 |
* Eint (SAPT) = Eelst + Eind + Edis + Eexch + δHF.
Table 7.
Comparison of the diisocyanides and dinitriles functioning as HaB acceptors.
| Methods (Descriptors) | HaB I···Ciso vs I···Nnitr | Comments |
|---|---|---|
| Global Nucleophilicity (NNu) | NNiso > NNnitr | Diisocyanides are more nucleophilic than the dinitrile |
| Local Nucleophilicity (NNuloc) | NNuloc(iso) > NNuloc(nitr) | |
| MEP (Vs,min) | Vs,miniso ~ Vs,minnitr | Electrostatic potentials are nearly the same for both systems |
| QTAIM (ρb) | ρbiso > ρbnitr | The electron density values at the BCP of the HaB are slightly higher for the diisocyanide system; this indicates a stronger binding of the diisocyanides to 1,3,5-FIB |
| ELF (V(C,I)) | V(C,I)iso > V(C,I)nitr | Charge transfer effect is higher for the isocyanide systems |
| NBO (Δocc (σ*(I–C)) | Δocc iso > Δocc nitr | |
| DLPNO-CCSD(T) (EbSM) | Ebiso > Ebnitr | Bond energy between the coformers is larger for the diisocyanide structures |
| SAPT (Eexch) | Eexchiso > Eexchnitr | Repulsive Pauli energy for the I···C/N HaBs is higher for the isocyanides. This is in agreement with increased orbital interactions for the diisocyanide systems |
| SAPT (Eelst) | ΔEelstiso ~ ΔEelstnitrl | SAPT results indicate the dominance of Coulomb interactions in the HaB of both systems; they contribute approximately 60% to the sum of negative energy components. The dispersion contribution is no more than 30% |
| SAPT (Edisp) | ΔEdispiso ~ ΔEdispnitrl |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



