
Submitted:
26 July 2023
Posted:
28 July 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The COVID-19 pandemic cost 7-8 million deaths worldwide, creating an unprecedented health and economic crisis. Affecting 700 million people globally, the magnitude of this pandemic is far from anything that humanity has encountered in recent times. A detailed investigation revealed that more than the SARS-CoV2 virus, the hyperactive immune system mediated injury as the real cause of mortality. Cytokine storm following viral infection leads to the surge of proinflammatory cytokines resulting in ARDS and lung injury. Anti-inflammatory intervention with anti-IL6 (anti-IL-6 receptor monoclonal antibodies (mAbs) (e.g., sarilumab, tocilizumab) and anti-IL-6 mAbs (i.e., siltuximab) and /or steroid-based approach lead to substantial protection and prevent death thereby implying the role of inflammation in COVID-19. In this short review, we summarized the dysregulated immune system in COVID-19 infection, investigating in detail the virus–host immune cross talks and presenting the possibilities of therapeutic intervention.
Keywords:
COVID-19 SARS-CoV-2
; Cytokine Storm
; PANoptosis
; Inflammation
; Host response
Running Title: COVID-19 Infection and Immune Dysregulation
Abbreviations:
- COVID-19 - Coronavirus infection disease 2019
- SARS-CoV-2 - Severe acute respiratory syndrome coronavirus 2
- MERS - Middle East Respiratory Syndrome
- S-protein - Spike protein
- ACE2 - Angiotensin-Converting Enzyme 2
- RBD - Receptor-Binding Domain
- ARDS - Acute respiratory distress syndrome
- PAMPs - Pathogen-Associated Molecular Patterns
- PRRs - Pattern Recognition Receptors
- DAMPs - Damage Associated Molecular Patterns
- TLR - Toll-like receptor
- NLR - Nucleotide-binding oligomerization domain-like receptors
- RLR - Retinoic Acid-Inducible Gene I Like Receptor
- ISG - IFN-Stimulated Gene
- TRAF - TNF-Receptor Associated Factor
- IKK - IĸB kinase
- TBK - TANK-binding kinase
- RAGE - Receptor for advanced glycation end-products
- RAAS - Renin-Angiotensin-Aldosterone System
- AT1 - Type 1 Angiotensin II Receptor
Introduction
Coronavirus infection disease 2019 (COVID-19) is an infectious disease caused by Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and has been affecting the world since 2019. As of January 2022, the outbreak of SARS-CoV-2 and its varied variants has spread to all the continents, with more than 600 million registered cases and over 6 million mortalities worldwide. Despite the observed low mortality rate of COVID-19 when compared with other SARS epidemics, its broadcasting has led to devastating effects on the healthcare system and the harmonization of national economics worldwide.
It's been just 2.2 years since the first case of COVID-19, but the response by both the medical personnel and the scientific community has been humongous, with a varied number of scientific research reports, meta-analyses, prospective cohorts, etc. However, in order to curb the pandemic, there have been numerous decisions taken in a hurry based on some anecdotal or poor evidence (usage of ibuprofen, hydroxychloroquine, ACE inhibitors, and lopinavir-ritonavir), inciting misperception among medical personnel and the general public 1–3. These challenges are potentially aggravated, subjecting to the gap in the knowledge in the understanding of COVID-19 pathophysiology and the complexity of treatment validation. However, we are still learning the dynamics of SARS-CoV-2 infection as a persuasive goal to understand the emergence of the various new variants that come with multiple COVID-19 phenotypes. Though all of the new variants (as of now) manifest with acute respiratory disorders, several non-respiratory subjects and post-COVID complications have also been reported. With the emerging points to consider, this clinical picture directs to the fact that SARS-CoV-2 dysregulates the host-immune response towards the infection, thereby fueling misleading research and biased hypotheses.
So as to implement optimal management strategies in order to improve the medical outcomes regarding COVID-19, similarities in pathophysiology, symptoms, autopsy reports, along with in-depth analysis of COVID-19-positive patients are deemed necessary. This paper aims to summarize the pathophysiology of COVID-19 and the related and unrelated disease mechanisms that underline several respiratory and non-respiratory manifestations. We also try to solve the question of whether the virus contributes towards the host's death or is it the complex array of cellular and humoral immune-inflammatory mechanisms that underlie the misfortune? Moreover, several systematic investigations and autopsy studies of SARS-CoV-2-associated deaths are still considered necessary to provide evidence for the epidemiological clusters and the organotropism of the virus and its variants.
Pathophysiology of COVID-19
Cellular mechanisms post SARS-CoV-2 entry into the host
As a phylogenetic member of the large coronavirus family, SARS-CoV-2 shares many similarities with SARS-CoV and Middle East Respiratory Syndrome (MERS), which were responsible for the two previous epidemics in the early 21st century. Alike SARS-CoV, the primary route of virus entry and transmission is expelled respiratory droplets that are absorbed by the mucous membranes. Both viruses exhibit similar coding domains (S1 and S2) of the Spike protein (S-protein), which facilitates its entry into the human host. The receptor-binding domain (RBD) in S1 interacts with the human host cell surface's angiotensin-converting enzyme 2 (ACE2), thus mediating SARS-CoV internalization 4. Several biophysical and structural evidence has stated the extensive affinity of ACE2 with SARS-CoV-2, with respect to that of its other members, thus hypothesizing the contagious nature of the virus 5. Viral entry also has been seen to be associated with TMPRSS2 protease activity, and it's the synergy between ACE2 and TMPRSS2 which leads to virus entry into the host cell membrane. Expression of both TMPRSS2 and ACE2 has been seen to occur largely in alveolar epithelial cells, with the former being extensively dispersed compared to ACE2 receptors, signifying that ACE2 receptors might play a rate-determining factor in the entry of the virus 6. However, ACE2 expression is found to be higher in adults when compared with children; thus infection rate is seen higher in adults 7,8. Other proteases like cathepsin B/L also play a vital role in contributing to viral entry in the absence of TMPRSS2 9. Extensive bioinformatics analyses have also deduced that furin, a type-I membrane-bound protease, also has been seen associated with several viral entries, including coronavirus 10. Furin’s action on the S-protein of SARS-CoV-2 influences its cellular entry by revealing the fusion domains and increasing the chances of pathogenesis, thus idealizing as an additional pathway for SARS-CoV-2 to enter 10–12. Furin has also been reported in several circulating cells like T cells, which work in a forward-feedback loop, further facilitating in contributing towards the replication of the virus and inducing cytokine storm in several patients 12.
Physiology of SARS-CoV-2 in the Human Host
Although SARS-CoV-2 is well known for targeting the lung epithelial cells, it has been seen in several extra pulmonary manifestations, including myocardial dysfunctions, thrombotic complications, hepatocellular injuries, neurologic illnesses, ocular and dermatological complications etc. 13. Considering the importance of the cellular surfaces proteins, like ACE2, TMPRSS2, cathepsins, and furin, in aiding SARS-CoV-2 entry into the human host, several transcriptomic gene analyses have been reported. In a Genotype-Tissue Expression Project data, ACE2 has been found to be expressed highly in the ileum and testis (>10 transcripts per million) and low in other organs like the heart, kidney, thyroid, and adipose tissues (> 5 transcripts per million) 14. Single-cell RNA-Seq data analysis by Zou et al. revealed that ACE2 expression was also highly expressed in cell types like alveolar type 2 epithelial cells, respiratory epithelial cells, myocardial cells, esophageal epithelial cells, etc., thereby forming physiological barriers against viral entry 15. However, ACE2, albeit serving as an entry point for SARS-CoV-2, increases the susceptibility of the human host to COVID-19 infection (as explained in Figure 1). Findings by Xu et al. also supplemented that ACE2 expressions in oral mucosal epithelial cells are highly notable, and SARS-CoV-2 infection is also susceptible in the oral cavity 16.
Neurological symptoms associated with COVID-19 patients also speculated the tendency of SARS-CoV-2 to invade the brain barriers and trigger neurological damage 17,18. In a prospective study conducted in New York, neurologic disorders conferred a high risk of in-hospital mortality 18. Further genome sequencing has verified the virus's existence in patients' cerebrospinal fluid. Possible explanations could be that the SARS-CoV-2 virus could have breached the olfactory mucosa lining the cribriform plate and must have transverse along the perineuronal space to access the cerebrospinal fluid in the subarachnoid space 19. Drainage of the virus-loaded CSF also might have triggered an immune response, thereby triggering COVID-19-affiliated encephalitis 20,21.
Gastroscopy examinations have shown that though viruses usually cannot endure the stomach’s acidic environment, there have been several shreds of evidence stating that the GI tract might be a potential transmission route of the SARS-CoV-2 virus 22. Several case studies have also proved the point by showing that COVID-19 patients present GI symptoms like nausea and diarrhea prior to the development of fever 23. As mentioned above, the expression of ACE2 and TMPRSS2 are high in absorptive enterocytes and the epithelial cells of the ileum; the GI symptoms perceived in COVID-19 patients may result from the virus’s invasion across the gut-epithelial barrier. Elevated levels of fecal calprotectin and IL-6, along with the clinical symptoms of diarrhea in COVID-19 patients, also suggest the acute inflammatory response in the GI tract 24,25.
Though lung tissues were one of the first and most critical organs affected by COVID-19, they expressed modest levels of ACE2. However, the presence of alveolar type 2 epithelial cells and their affiliated higher expression of ACE2 provided the platform for SARS-CoV-2 to replicate and upregulate cytokines like TGF-β1 and CTGF, ECM proteins and infection, further resulting in pulmonary fibrosis 26,27.
Clinical Manifestations of COVID-19
COVID-19 Pneumonia
The most common clinical manifestations constitute cough, fever, fatigue, muscle pain, anorexia, and loss of smell and taste 28. However, apart from these, other symptoms like sore throat, rhinorrhea, headache, GI symptoms also have been seen to precede the respiratory symptoms. Some patients who show severe symptoms like dyspnea (around day 5), often require hospitalization by day 7-8 and manifest hypoxemia and bilateral pneumonia. Out of the hospitalized patients, some deteriorate abruptly after the inception of dyspnea and succumb to respiratory failure 29,30. COVID pneumonia, with clinical symptoms like fever, cough, tachypnoea, higher tidal volume, and respiratory distress, in adults, has also been described as a potentiating reason for patients registering in hospitals with hypoxia 23,31. Though clinical management for hypoxia remains a contentious issue, several hypotheses have been proposed to explicate hypoxia, reduced diffusion capacity, oxygen receptor chemo sensitivity, and loss of hypoxic vasoconstrictive mechanism 32–35. In fact, an unusual phenomenon that was observed in the earlier months of COVID-19, which played a significant role in major mortalities, was silent hypoxemia, also known as happy hypoxia, characterized by the low partial pressure of arterial oxygen, yet low or mild respiratory discomfort 36,37. Moreover, altered mechanisms of the lungs, associated with alveolar collapse, pulmonary inflammation, fibrosis, and atelectasis, further impair lung functions causing lung tissue hypoxia.
ARDS and Lung Fibrosis
Acute respiratory distress syndrome (ARDS), an impairment of oxygenation, is one of the most severe complications of COVID-19. Though respiratory support is crucial and high-flow oxygen is often provided via invasive or non-invasive ventilation, ARDS is concomitant with sustained hospitalization and high mortality 28. Associated with high D-dimer concentrations and low compliance, intermittent prone positions help in improving gaseous exchange, reducing the ventilation/ lung-perfusion mismatch. However, this technique hasn't been proven clinically.
One of the initial mechanisms for ARDS is the cytokine storm, which uncontrollably stimulates an inflammatory response resulting from the enormous amounts of proinflammatory cytokines (IL-1β, IL-6, IL-12, IL-18, IL-33, IFN-α, IFN-γ, TNF-α and TGF-β etc) and chemokines (CCL2, CCL3, CCL5, CXCL8, CXCL9, CXCL10 etc) by the immune-effector cells in the viral infection 29,38–40. Characterized by the deterioration of the function of the lungs and respiratory failure, lung fibrosis is correlated with ARDS but with a poor prognosis. Driven by pro-fibrotic factors like TGF-β, secretion of the same promotes repair and perseverance of infection-mediated damage in COVID-19 injured lungs. However, severe damage causes excessive TGF- β signaling causing EMT and endothelial-to-mesenchymal transitions, thereby causing pulmonary fibrosis 41–45. Pulmonary fibrosis is a common complication of SARS and MERS. Several postmortems have revealed a high section of COVID-19 cases with fibrosis 46,47. The progression of ground-glass opacities in CT scans due to thick reticular pattern, irregular interface, and interstitial thickening are suggested to be early signs of COVID-19 pulmonary fibrosis 48.
Coagulopathy and endothelial damage
A stage of coagulopathy and endothelial damage also arises in severe COVID-19 cases, with frequent reports of microvascular thrombosis or disseminated intravascular coagulation. Several pieces of evidence also state the association of pulmonary failure with deranged coagulation. Hypercoagulability symptoms like high circulating D-dimer concentrations, elevated prothrombin time, increased fibrinogen, and thrombocytopenia are some of the common ones found in patients with severe COVID-19 29,49. Though the pathobiology of hypercoagulability in COVID-19 cases remains unclear, numerous autopsy reports have identified diffused alveolar damage, severe endothelial damage, and coagulopathic symptoms in the pulmonary microvasculature in patients with severe COVID-19, hence establishing the relation 50,51. Evidence of alveolar damage accompanied by thrombotic microangiopathy also supports the fact that endothelial injury stimulates pulmonary circulation thrombosis 52,53.
Endotheliitis, as referred by Varga et al. 54, can also be associated with refractory COVID-19-related ARDS with loss of hypoxic vasoconstriction, intra-alveolar fibrin, alveolar damage, edema, hemorrhage, and hypoperfused intrapulmonary blood flow 55. However, not just being restricted to the lungs, other vascular disorders, including stasis, disturbed endothelial barrier, endothelium localized inflammations, disturbed permeability control, and prothrombotic endothelial cell state, also act as proactive factors localized to the brain, heart, gut, liver, and kidney, thus revealing several other extrapulmonary manifestations 13,51,54,56,57.
The Immune System Response
Though the pathophysiology of SARS-CoV-2 isn't fully comprehensive, relative literature about SARS-CoV and MERS and recent literature have shown that SARS-CoV-2 has many defense mechanisms, making it challenging to eradicate. Similar to SARS-CoV, SARS-CoV-2 contains 4 structural proteins, envelope (E), membrane (M), nucleocapsid (N), and spike (S), and 14 open reading frames (ORFs) 58. Following the entry of SARS-CoV-2 into the cell by binding to the ACE2 on the epithelial cell surfaces, the viral genome is transferred into the cytoplasm and is translated into two polyproteins and structural proteins, after which they replicate. Post replication, the viral particles germinate into the endoplasmic reticulum-Golgi apparatus complex and fuse with the plasma membrane to release the virus 59.
Subsequent antigen production stimulates the human body's immune response (both cellular and humoral). Inflammatory responses are also triggered by Pathogen-Associated molecular patterns (PAMPs) in correlation with the cytosolic and endosomal pattern recognition receptors (PRRs) to limit infection and promote viral clearance 60. Armed with an array of PRRs that recognize PAMPs or DAMPs (Damage Associated Molecular Patterns), to stimulate inflammatory pathways and immune responses, the immune system, in accordance with the PRR-affiliated inflammasomes, Toll-like receptors (TLRs), Nucleotide-binding oligomerization domain-like receptors (NLRs) and retinoic acid-inducible gene I like receptors (RLRs), have been seen to stimulate signaling pathways against COVID-19 60. The infected lung and the alveolar epithelial cells trigger the innate immune response by secreting IL-8, which acts as a chemoattractant for T-lymphocytes and neutrophils. TLRs, in coordination with MyD88 and TRIF, possess a heterogeneous expression of the immune cell population 61. Several analyses have suggested that TLR2 recognizes the E protein of the virus and induces innate immune cell activation 62–64. Other TLRs like TLR1, TLR4, and TLR6 have been seen to show affinity towards the S protein of SARS-CoV-2, thereby contributing towards the release of proinflammatory cytokines 65. Moreover, X chromosomal TLR7 genetic anomalies have been observed in several young patients suffering from COVID-19, suggesting the pro-active role of TLR7 in the infection 66,67. The single-stranded RNA in the virus aggravates the immune response post-recognition by TLR7, which is expressed on dendritic cells and macrophages. TLR7, further stimulates several transcriptional factors, proinflammatory cytokines like IL-1, IL-6, MCP-1, TNF-α, and IFN-1, and putative signaling pathways like NF-ĸB, JAK/ STAT, sphingosine-1-phosphate (S1P), AP-1, interferon response factor 3 (IRF3) and IRF7 68; thus establishing the correlation of type I IFN with cytokine secretion in CD4+ T cells polarization 69. Activation of these pathways further endures other pathways like ERK/Ras, PI3/Akt/eNOS, and PLC/Ca2+ downstream pathways, which regulate the migration and trafficking of numerous immune cells, including NK cells, B and T lymphocytes, and dendritic cells 70. JAK1 TYK2 kinases phosphorylate STAT 1/2 and form a complex with IRF9 to commence transcription of IFN-stimulated genes (ISGs) 71. Following this, neutrophils get rapidly recruited to the infection sites and respond by their neutrophil extracellular traps and defensin secretion to kill the viruses 72. Early expressions of IFN-α, IFN-γ, CXCL10, CCL12, and ISG-encoded proteins were also noted in patients who recovered from SARS 71.
RNAs derived from the genomic intermediates of the virus can be detected by RLRs, namely LGP2, RIG-1, and MDA5. RIG-1 and MDA5 are key regulators and are associated with pathways like IFN and mitochondrial antiviral signaling (MAVS) signalosome, which further activate TNF-receptor associated factor (TRAF)-3, IĸB kinase (IKK) and TANK-binding kinase (TBK)-1 to mediate transcription of type I and III IFNs. Subsequent production and stimulation of IFNs activate downstream signaling via IFN receptors in an autocrine and paracrine manner to secrete vivid ISGs of different antiviral functions 73–76. However, silencing of the genes encoding MDA5 or LGP2 in human lung epithelial (Calu-3) cells cause a reduced type I IFN expression during COVID-19 infection 77. On the contrary, siRNA silencing for gene encoding for RIG-1 did not show any difference in Calu-3 cells, though this study remains quite contentious 78,79. Type I IFNs and other cytokines are also stimulated by NLRs in SARS-CoV-2 infections, with NLRP3 being one of the most characterized inflammasome sensors. Triggered in response to DAMPs and PAMPs, caspase-1 gets activated, thereby stimulating the release of IL-1β, IL-18, and cleavage of gasdermin D, thus leading to membrane ruptures 80,81. Increased levels of IL-1β and IL-18 have also been correlated with disease severity and mortality in patients suffering from SARS-CoV-2. PAMPs derived from ORF3a (viroporin), ORF8b, the viral RNA, and the E protein of SARS-CoV-2 have been demonstrated to stimulate NLRP3 inflammasome 82. The IFN pathways are also hindered by innate-signaling proteins like NSP13, NSP15, and ORF-9b. NSP13 interacts with TLE1, TLE3, and TLE5, and on the other hand, ORF-9c interferes with NLRX1, NDFIP2, and F2RL1, which are also involved in the NF-ĸB pathways 83. Further, NLRC1 has also been noted to contribute to COVID-19 response and cytokine release, and silencing the gene encoding for NLRC1 has been seen to reduce IFN-1β expression 77.
Active viral replication, however, opens the window for immune intervention, but prolonged replication later results in hyperproduction of type I IFN with an influx of macrophages and neutrophils. This heavy dependence upon type I IFN responses further culminates in controlling the replication and induces an effective adaptive immune response. In general, both the B and T cells play a vital role in viral clearance, wherein most patients try to develop a humoral response within the first 10-14 days of the infection. Poor humoral responses (developed by B cells) have been associated with ineffective COVID-19 virus clearance in some patients, suggesting its importance in SARS-CoV-2 neutralization 84. Since the polarization of B cell follows a linear process through pro B cell, pre B cell, immature B cell, and finally, a matured B cell, studies have shown the partial reliance of VDJ gene segments during the making of these virus-specific antibodies. Concerning SARS-CoV-2, antibody development towards IgG heavy chain IGHV3-23 and IGHV3-7 was identified, thereby helping us with antibody and vaccine development 85. Nevertheless, certain types of mature B cells, known as double-negative (DN) B cells, which are known to lack expressions of memory markers (CD27) and IgD, have also been shown to promote cytokine release and autoantibody progression, thus enhancing COVID-19 pathogenesis 86,87. Studies have reported that CD8+ T cell responses demonstrated better and more frequently with respect to CD4+ cells, thereby correlating with disease severity 88. Additionally, the virus-specific T cells from severe patients were associated with a higher frequency of polyfunctional CD4+ T cells (IL-2, IFN-γ, and TNF-α) and CD8+ T cells (IFN-γ and TNF-α) in comparison to moderate patients, thus postulating that strong T cell responses correlate with higher neutralizing antibodies 88. Furthermore, Th17 cells, neutrophils, and other granulocytes mediate the production of interleukins (IL-1, IL-6, IL-8, IL-17), MCP-1, C-CSF, CM-CSF, TNF-α, and PGE2, which further stimulate the production of monocytes, neutrophils, and other immune cells, thus trying to eradicate SARS-CoV-2 89–91.
Antibiotics also play a multifactorial role in viral clearance by binding to viral proteins expressed on the infected cell surface and recruiting NK cells to kill the infected cell by antibody-dependent cell cytotoxicity mechanism 92. Antibodies bind to the receptor-binding domain (RBD) of the S protein of SARS-CoV-2 and effectively block virus-ACE2 interaction, thereby neutralizing the virus 93–95. However, the neutralizing antibody responses have poor durability, putting patients at a high susceptibility to re-infection after a few years of infection 96,97. Typical production of IgM and IgG constitute the antibody profile against the virus. However, it has been reported that IgM disappears by the end of week 12, but IgG persists longer 98. Numerous studies have also suggested that SARS-CoV-2-based humoral immunity also does not last long. A steep decline of anti-SARS-CoV-2 antibodies has also been observed in asymptomatic COVID-19 patients, postulating their minimal protection against the viral infection 99–101.
Is that all?
As mentioned above, the magnitude and durability of COVID-19 infection correlate with the immune response. Several pieces of evidence have stated the defense mechanism of SARS-CoV-2, wherein it inhibits IFN1 by regulating IFN-β synthesis, which further results in managing or aggravating viral replication or induction of an adaptive immune response 68,102. In several circumstances, it has also been observed that the virus dampens the attack by virtue of immune dysregulation caused by the same infection, thus derailing into a cytokine storm and T-cell exhaustion, further weakening the overall body's immune response. This also partly clarifies the more extended incubation period (2-11 days) compared to influenza, which usually has an incubation period of 2-4 days 103. Indeed, the virus has also been seen to reduce antigen presentation on MHC I and II 104–106.
T cell death and exhaustion
Several autopsies and hospital reports have reported that patients with pneumonia abruptly succumb to respiratory failure and require mechanical ventilation 107. Lethal sepsis arising from bacterial community-acquired pneumonia and sudden deterioration after 7-8 days of the first symptom derived from the immune dysfunction hypothesis 108. Further analysis also stated that there was a depletion in CD4+ and CD8+ frequency cells, γδ-T cells, and features of lymphopenia with increased D-dimers and hepatic dysfunction were noted 109. There have been several hypotheses behind the cause of lymphopenia, with one being the inhibition of T cell circulation and apoptosis of memory CD8+CD44high T cells by cytokines like the class I IFN and TNF-α, thereby promoting retention in lymphoid organs 109–111. Necroptosis has also been associated with T cell death in which NSP12 of the SARS-CoV-2 interactome interacts with receptor-interacting serine/ threonine-protein kinase 1 (RIPK1) 112. Though studies have suggested retention of T cells as a primary component in the deterioration of immune response, other investigations also have demonstrated that surviving T cells get functionally exhausted and express high-level T-cell immunoglobulin mucin-3 (TIM3) and programmed cell death protein 1 (PD-1) in SARS-CoV-2 patients 113. Decreased productions of IFN-γ and IL-21 also support the exhaustion of CD8+ and CD4+ T cells 114,115. Other immune cells like cytotoxic lymphocytes and NK cells possess an upregulation of NKG2A (NK group 2 member A), an inhibitor of CD8+ T cells, and a downregulation of CD107a+, granzyme B+, IFN-γ+, and IL-2, consistent with the functional exhaustion of the immune cells 114,116.
Cytokine Storm
Release of proinflammatory cytokines, including IL-2, IL-6, IL-7, G-CSF, CXCL10, MCP-1, CCL2, macrophage inflammatory protein 1α (MIP-1 α), and TNF-α in severe COVID-19 patients, dictates the severity of the infection. Circulating cytokines like IL-6 and IL-1β were also found in large quantities in the patient's immune profile, thereby mediating Th17 cell differentiation, and promoting further IL-6 and IL-17 production, thus worsening the clinical status of the patient 29,117. IL-17, produced by Th17 cells, also plays a significant role in recruiting monocytes and neutrophils and activating other downstream cytokines and cascades like IL-1, IL-6, IL-8, IL-21, TNF-α, and MCP-1, thereby exacerbating the inflammation 16,118. IL-22 also has been seen to enhance the expression of mucins, fibrinogens, and anti-apoptotic proteins, thereby increasing the chances of life-threatening edema and ARDS 119. These cytokines further drive inflammation in the lungs and promote virus persistence by expressing Bcl-2 and Bcl-xL 120,121. This also results in an elevated D-dimer, CRP, procalcitonin, Cr, coagulopathy, and hyperferritinemia profile, which further turns into a typical macrophage activation syndrome 122–124. Consistent with the cytokine profile of the other coronaviruses, this cytokine storm leads to severe clinical phenotypes, namely ARDS, tissue hypoxia, and death 125. Ironically, the very immune system which usually acts to fight the infection potentially harms the host in doing so.
With the dysregulated secretion of inflammatory cytokines like TNF and IFN-γ, the combination of the same further characterizes a life-threatening condition mediated by inflammatory cell death (PANoptosis) 126. Dependent on PANoptosomes, caspases, inflammasome components, and the synergism of TNF and IFN-γ, PANoptosis also depends upon several signaling pathways and transcriptional factors like STAT1 and IRF1, which further activates caspase-8, Z DNA binding protein 1 (ZBP1), TGFβ activated kinase-1 (TAK1) and RIPK1 affiliated cell death 127–135. As a whole, PANoptosis works on a positive feedback loop wherein cytokine secretion causes PANoptosis (Figure 2), which further results in more cytokine secretion, thus culminating into a cytokine storm, thereby promoting inflammation and disease progression 82.
The cytokine release syndrome plays a vital and decisive role in COVID-19 patients. Cytokine production occurs via two pathways, either recognizes the virus by pattern-recognition receptors like TLR3, TLR7, TLR8, and TLR9 or through induction of DAMPS released from SARS-CoV-2 damaged cells 136,137. Upon lung or alveolar injury, the epithelial or endothelial or parenchymal cells release inflammatory mediators which mutually elicit multiple inflammatory cytokines and chemokines. Other than the inflammatory cytokines mentioned above, the type I and type III interferon responses and the IL 1-IL 6 axis further constitute pertinent biological signaling pathways 85,138–142. However, other controversies exist regarding IL-6, IL-8, and TNF signatures across varied acute conditions. Apart from the supporting evidence cited above, few studies have reported varying mRNA expressions of IL-1β and IL-6 in the autopsy reports of 4 individuals who succumbed to COVID-19 143. The same was, in fact, reproduced in a SARS-CoV-2-infected lung organoid using human iPSCs 144.
Cytokine responses have also been observed in other organs, given that ACE2 is robustly found in other organs and epithelial cells. Cytokine storms in COVID-19 infections have been seen to trigger violent assaults on the body, causing massive excitation of the immune system, triggering an uncontrolled systemic inflammation (Figure 2), leading to ARDS and multi-organ failure, and finally causing death 145. Encompassing a broad range of severity, several neurological disorders have been associated with cytokine storm, eg: immune effector cell-associated neurotoxicity syndrome, traumatic brain injury, encephalopathy, hemophagocytic lymphohistiocytosis, cytokine storm-associated encephalopathy (as proposed by Pensato et al.), etc. 146,147. Conditions from mild proteinuria to advanced acute kidney injury have been noted in the renal system due to cytokine storms and COVID-19 148. Altered liver chemistry with progressive cases like cirrhosis has also been reported with respect to cytokine storms 149,150. Several meta-analyses have also deduced that heart failure is a common complication in COVID-19 cases, with cytokine storm being the primary inducer. Other conditions like myocarditis, cardiac fibrosis, calcium dyshomeostasis, and cardiomyocyte pyroptosis, amongst a few, are some of the manifestations observed as a result of cytokine storm 151.
Endocrinal factors
Lifestyle comorbidities like hypertension, obesity and Diabetes have also been associated with COVID-19 complications 152. ACE2 is expressed in metabolic tissues, and organs like the thyroid, pancreas, adrenal, pituitary, ovaries, etc. infection in any of the organs leads to disruption in the endocrinal system. Evidence confirms the dysregulation of glycemic homeostasis as an outcome in COVID-19 patients suffering from diabetes 153. A retrospective study stated that patients with controlled glycemia showed less mortality in comparison to patients with high glycemic variability 153. Associated with hyperinflammation and enhanced oxidative stress, Diabetes, in addition to infection, further exacerbates endothelial or organ damage via TLR4 and RAGE (Receptor for advanced glycation end-products) 154,155. In addition to it, COVID-19 infection is also known to dysregulate the potassium axis, leading to hypokalemia, further affecting glucose homeostasis in diabetic patients 156. TMPRSS2 has also been correlated with androgenic hormones, which further describes the disparity in COVID-19 infection based on gender 9. Though more or less, SARS-CoV-2 infects both men and women equally, women's mortality was found to be twice as high as that of men 157. The renin-angiotensin-aldosterone system (RAAS) has also been reported to be highly active in COVID-19 patients 158. Angiotensin II promotes hyperinflammation by stimulating IL-6 in vascular and endothelial muscle cells via the type 1 angiotensin II receptor (AT1) 159.
Other Immune Cells
Mild peripheral neutrophilia has been documented in patients with COVID-19, wherein highly stimulated CD11β+, CD38+, HLA-DR+ (an MHC class II cell surface molecule), and myeloid-derived suppressor cells 160,161. Increased MDSCs have also been correlated with inflammation, hematopoiesis, and increased activation of peripheral immature neutrophil granulocytes, CD10- and CD16low 162,163. Neutrophilic infiltrations were also associated with SARS-CoV-2, where chemokine production and an activated Th17 response were reported, which likely contribute to the pathogenesis of the virus 164. Circulating monocytes and other leukocyte subsets were also seen to undergo fluctuations and trajectories. Dysregulation of monocytes also led to a decrease in CD16+ and a shift towards CD14+ monocytes, though this transfer resolves in later stages of the infection 85,105. An enhanced CD169 expression with elevated productions of IL-1 and IL-6 also was shown in both COVID-19 patients' lungs and mouse models of SARS-CoV-2 infection 55. Though monocytes haven't been reported to alter in COVID-19 substantially, the feedback loop between T cells and macrophages drives the limited alveolitis towards the development of persistent alveolar inflammation and further lung damage 165.
OMICRON: What new does it bring in?
With the recent breakthrough of the new SARS-CoV-2, Omicron, formerly designated as B.1.1.529, a variant of concern is known to have 32 mutations within the S- protein 166. Consisting of all the key mutations of the previous variants, including K417N, E48A, N501Y, and others, this variant can also change the sensitivity of the virus to neutralizing antibodies 167,168. Zhang et al. further showed that the newly made mutations did cause significant changes, concerning neutralization sensitivity, against numerous human sera samples pre-infected with the previous variants of COVID-19 169. The mutations also enhance the S-protein's affinity to ACE2 receptors, further contributing to its risk of transmissibility (3 mutations at cleavage site) and evasion in the immune system 170. However, a hypothesis by Dejnirattisai et al. states that changes found in RBD-62, which also happens to serve as an anchorage for ACE2, are more prone to mutational changes, including those to reduce ACE2 affinity, so as to evade antibody neutralization 171. Recent research has stated the mutation of NSP6 in the Omicron variant. NSP6 has been known to interfere with type I IFN pathways, by occluding the stimulation of tank-binding kinase, thus presenting a huge number of autophagosomes. In addition, NSP13, NSP15, and ORF-9b also hinder IFN pathways. Though not much research has been done in this field, we can speculate that mutations in NSP6 can lead to an appreciable amount of modifications, establishing a critical host-virus relationship, signifying immune escape, and SARS-CoV-2 pathogenicity.
Several investigations have been made in order to analyze the immune response toward this new variant. When compared with its older lineages, Omicron's neutralization has been found to be significantly low (explained via an antigenic map in Figure 3). However, Omicron's higher transmission rate and infectivity pose a major concern 172,173. Lung pathology reports of Omicron-infected hamsters suggested that neutralizing antibodies produced after Omicron infection demonstrated substantial reduction or negligible neutralization against its other variants 174. However, despite the broad neutralization escape against Omicron, 70-80% of the T cell responses were found to be cross-reactive 175. Peptide pool simulation results detected no differences in typical cytokine production in virtue of both the wildtype and Omicron variants of COVID-19, suggesting that T cell epitopes remain minimally affected, irrespective of the variant. In fact, booster vaccinations also led to the maintenance of variant-specific CD4+ and CD8+ T cell responses 176. Apart from T cell production, a decreased IL-2 and IL-2 plus other cytokine release was also noted 177.
Taking the world by storm, this new Omicron variant has a very high transmission and infection rate compared to the rest of the COVID-19 variants. It has also been seen to evade natural and vaccine-induced immunity 178; however, booster vaccinations (mechanism explained in Figure 4) have been seen to improve protection against Omicron. Compared to the last Delta variant, a thumping 91% decrease in mortality was noticed in a study associated with Omicron infections 179. Nevertheless, despite its mild severity, the sheer increase in Omicron cases has put the world on a high alert with the global economy at stake. In fact, the newer variants of Omicron, BA.1 and BA.2 have also replaced the previous Delta variants in their ability of immune escape and more transmissibility. A real-world study in Denmark showed that BA.2 is more infectious than BA.1, as it possessed more immune escape properties 180,181. The numerous mutations in the N-terminal and RBD region of the Omicron variants attribute to the immune escape properties 182. However, the latest lineages of Omicron, BA.4 and BA.5's neutralization capacities have reduced by 7 folds and 3 folds in unvaccinated and vaccinated individuals, respectively 183. NAb titers against these sub-variants have been observed to be lower than that of their initial ancestors, BA.1 and BA.2, suggesting their increased neutralization escape 184,185. In context to immune evasion, BA.4 and BA.5 have demonstrated escape from BA.1 and BA.2 induced immunity, about the reason for re-infections in several cases 184,186.
However, several analyses have shown that people subjected to Omicron variants had significantly low odds in terms of hospitalization or mortality when compared with the other variants 179,180,187–189. A high rate of asymptomatic cases also was observed, suggesting the milder symptoms of the variant, thereby confirming the lower pathogenicity of the same. In vitro studies have demonstrated that Omicron stimulated multinuclear syncytia in several cell lines when compared to other variants 190–192. Activation of signaling pathways like NF-ĸB seemed less efficient in the new variant, implying a slighter inflammatory response and extremely low mortality 193–195. Though some countries have initiated booster doses to control the spread of this variant, more research is required on its influence on cytokines, vaccine-based immune responses, post-COVID-19 complications, and mortality rates associated with Omicron. In fact, as per data of 18th December 2022, about 649 million cases have been reported globally, signifying the revival of new variants of SARS-CoV-2 in the world 196. Thus, further research is on demand whether Omicron stays as the last variant of its kind or is it going to script yet another deadly chapter in the history of a pandemic.
CONCLUSION
Mostly involving respiratory manifestations, the COVID-19 was also known to be involved with effecting other organs, followed by complications and dysregulation of host responses. Though the immune system plays the most vital role in fighting COVID-19, ironically it also turns out to be harmful, and eventually becomes the primary cause of COVID-19 affiliated death. Thus amidst the notion of acknowledging the reason behind the innumerable deaths due to COVID-19, it can be stated that the derangement of the immune system, post infection, is the culprit. Armed with this knowledge, therapeutic guidelines could be potentially updated, thereby allowing medical professionals to provide optimum care for patients with COVID-19.
DECLARATIONS
Author's contribution
OSS drafted the manuscript and made the images under the supervision of SK and RD. AN, RM and KP assisted in references and reading the draft for corrections and critical comments. KP assisted in clinical correlation. SK and RD overseen the entire manuscript.
Funding Statement
We express our gratitude to IMRG grant, from AIIMS Delhi received by SK to support this work.
Conflicts of Interests
No competing interests were disclosed.
Ethics approval
Not Applicable.
Availability of data and materials
Not Applicable.
References
- Osuchowski MF, Aletti F, Cavaillon JM, et al. SARS-CoV-2/COVID-19: Evolving Reality, Global Response, Knowledge Gaps, and Opportunities. Shock. 2020;54(4):416-437. https://doi.org/10.1097/SHK.0000000000001565. [CrossRef]
- Salian VS, Wright JA, Vedell PT, et al. COVID-19 Transmission, Current Treatment, and Future Therapeutic Strategies. Mol Pharm. 2021;18(3):754-771. https://doi.org/10.1021/acs.molpharmaceut.0c00608. [CrossRef]
- Rochwerg B, Parke R, Murthy S, et al. Misinformation During the Coronavirus Disease 2019 Outbreak: How Knowledge Emerges From Noise. Crit Care Explor. 2020;2(4):e0098. https://doi.org/10.1097/cce.0000000000000098. [CrossRef]
- Zhou P, Yang X Lou, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270-273. https://doi.org/10.1038/s41586-020-2012-7. [CrossRef]
- Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (80- ). 2020;367(6483):1260-1263. https://doi.org/10.1126/science.aax0902. [CrossRef]
- Sungnak W, Huang N, Bécavin C, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26(5):681-687. https://doi.org/10.1038/s41591-020-0868-6. [CrossRef]
- Rezaei N. COVID-19 affects healthy pediatricians more than pediatric patients. Infect Control Hosp Epidemiol. 2020;41(9):1106-1107. https://doi.org/10.1017/ice.2020.139. [CrossRef]
- Jia HP, Look DC, Shi L, et al. ACE2 Receptor Expression and Severe Acute Respiratory Syndrome Coronavirus Infection Depend on Differentiation of Human Airway Epithelia. J Virol. 2005;79(23):14614-14621. https://doi.org/10.1128/jvi.79.23.14614-14621.2005. [CrossRef]
- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271-280.e8. https://doi.org/10.1016/j.cell.2020.02.052. [CrossRef]
- Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176(February):104742. https://doi.org/10.1016/j.antiviral.2020.104742. [CrossRef]
- Cheng YW, Chao TL, Li CL, et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020;33(2):108254. https://doi.org/10.1016/j.celrep.2020.108254. [CrossRef]
- Abassi ZA, Skorecki K, Heyman SN, Kinaneh S, Armaly Z. Covid-19 infection and mortality: A physiologist's perspective enlightening clinical features and plausible interventional strategies. Am J Physiol - Lung Cell Mol Physiol. 2020;318(5):L1020-L1022. https://doi.org/10.1152/AJPLUNG.00097.2020. [CrossRef]
- Gupta A, Madhavan M V., Sehgal K, et al. Extrapulmonary manifestations of COVID-19. Nat Med. 2020;26(7):1017-1032. https://doi.org/10.1038/s41591-020-0968-3. [CrossRef]
- Lonsdale J, Thomas J, Salvatore M, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580-585. https://doi.org/10.1038/ng.2653. [CrossRef]
- Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020;14(2):185-192. https://doi.org/10.1007/s11684-020-0754-0 RESEARCH. [CrossRef]
- Xu H, Zhong L, Deng J, et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12(1):1-5. https://doi.org/10.1038/s41368-020-0074-x. [CrossRef]
- Conde Cardona G, Quintana Pájaro LD, Quintero Marzola ID, Ramos Villegas Y, Moscote Salazar LR. Neurotropism of SARS-CoV 2: Mechanisms and manifestations. J Neurol Sci. 2020;412:116824. https://doi.org/10.1016/j.jns.2020.116824. [CrossRef]
- Frontera JA, Sabadia S, Lalchan R, et al. A Prospective Study of Neurologic Disorders in Hospitalized Patients With COVID-19 in New York City. Neurology. 2021;96(4):e575-e586. https://doi.org/10.1212/WNL.0000000000010979. [CrossRef]
- Gänger S, Schindowski K. Tailoring formulations for intranasal nose-to-brain delivery: A review on architecture, physico-chemical characteristics and mucociliary clearance of the nasal olfactory mucosa. Pharmaceutics. 2018;10(3). https://doi.org/10.3390/pharmaceutics10030116. [CrossRef]
- Stevenson PG, Hawke S, Sloan DJ, Bangham CR. The immunogenicity of intracerebral virus infection depends on anatomical site. J Virol. 1997;71(1):145-151. https://doi.org/10.1128/jvi.71.1.145-151.1997. [CrossRef]
- Ye M, Ren Y, Lv T. Encephalitis as a clinical manifestation of COVID-19. Brain Behav Immun. 2020;88(April):945-946. https://doi.org/10.1016/j.bbi.2020.04.017. [CrossRef]
- Zhang H, Kang Z, Gong H, et al. Digestive system is a potential route of COVID-19: An analysis of single-cell coexpression pattern of key proteins in viral entry process. Gut. 2020;69(6):1010-1018. https://doi.org/10.1136/gutjnl-2020-320953. [CrossRef]
- Zhao X-Y, Xu X-X, Yin H-S, et al. Clinical Characteristics of Patients with 2019 Coronavirus disease in a non-Wuhan area of Hubei Province, China: a retrospective study. BMC Infect Dis. 2020;20(311). https://doi.org/10.21203/rs.3.rs-15734/v1. [CrossRef]
- Effenberger M, Grabherr F, Mayr L, et al. Faecal calprotectin indicates intestinal inflammation in COVID-19. Gut. 2020;69(8). [CrossRef]
- Lin L, Jiang X, Zhang Z, et al. Gastrointestinal symptoms of 95 cases with SARS-CoV-2 infection. Gut. 2020;69(6):997-1001. https://doi.org/10.1136/gutjnl-2020-321013. [CrossRef]
- Madissoon E, Wilbrey-Clark A, Miragaia RJ, et al. ScRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2019;21(1):1-16. https://doi.org/10.1186/s13059-019-1906-x. [CrossRef]
- Xu J, Xu X, Jiang L, Dua K, Hansbro PM, Liu G. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir Res. 2020;21(1):1-12. https://doi.org/10.1186/s12931-020-01445-6. [CrossRef]
- Mao L, Jin H, Wang M, et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):683-690. https://doi.org/10.1001/jamaneurol.2020.1127. [CrossRef]
- Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497-506. https://doi.org/10.1016/S0140-6736(20)30183-5. [CrossRef]
- Shi H, Han X, Jiang N, et al. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis. 2020;20(4):425-434. https://doi.org/10.1016/S1473-3099(20)30086-4. [CrossRef]
- Duffin J. Measuring the ventilatory response to hypoxia. J Physiol. 2007;584(1):285-293. https://doi.org/10.1113/jphysiol.2007.138883. [CrossRef]
- Brochard L, Slutsky A, Pesenti A. Mechanical ventilation to minimize progression of lung injury in acute respiratory failure. Am J Respir Crit Care Med. 2017;195(4):438-442. https://doi.org/10.1164/rccm.201605-1081CP. [CrossRef]
- Lang M, Som A, Mendoza DP, et al. Hypoxaemia related to COVID-19: vascular and perfusion abnormalities on dual-energy CT. Lancet Infect Dis. 2020;20(12):1365-1366. https://doi.org/10.1016/S1473-3099(20)30367-4. [CrossRef]
- Mo X, Jian W, Su Z, et al. Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur Respir J. 2020;55(6):2-5. https://doi.org/10.1183/13993003.01217-2020. [CrossRef]
- Tobin MJ, Laghi F, Jubran A. Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med. 2020;202(3):356-360. https://doi.org/10.1164/rccm.202006-2157CP. [CrossRef]
- Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN. The pathophysiology of happy hypoxemia in COVID-19. Respir Res. 2020;21:198. https://doi.org/10.1186/s12931-020-01462-5. [CrossRef]
- Siswanto, Gani M, Fauzi AR, et al. Possible silent hypoxemia in a COVID-19 patient: A case report. Ann Med Surg. 2020;60:583-586. https://doi.org/10.1016/j.amsu.2020.11.053. [CrossRef]
- Williams AE, Chambers RC. The mercurial nature of neutrophils: Still an enigma in ARDS? Am J Physiol - Lung Cell Mol Physiol. 2014;306(3). https://doi.org/10.1152/ajplung.00311.2013. [CrossRef]
- Cameron MJ, Bermejo-Martin JF, Danesh A, Muller MP, Kelvin DJ. Human immunopathogenesis of severe acute respiratory syndrome (SARS). Virus Res. 2008;133(1):13-19. https://doi.org/10.1016/j.virusres.2007.02.014. [CrossRef]
- Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39(5):529-539. https://doi.org/10.1007/s00281-017-0629-x. [CrossRef]
- Wilson MS, Wynn TA. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2(2):103-121. https://doi.org/10.1038/mi.2008.85. [CrossRef]
- Eapen MS, Lu W, Gaikwad AV, et al. Endothelial to mesenchymal transition: a precursor to post-COVID-19 interstitial pulmonary fibrosis and vascular obliteration? Eur Respir J. 2020;56(4):2-4. https://doi.org/10.1183/13993003.03167-2020. [CrossRef]
- Chen W. A potential treatment of COVID-19 with TGF-β blockade. Int J Biol Sci. 2020;16(11):1954-1955. https://doi.org/10.7150/ijbs.46891. [CrossRef]
- Gu J, Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol. 2007;170(4):1136-1147. https://doi.org/10.2353/ajpath.2007.061088. [CrossRef]
- Hui DS, Joynt GM, Wong KT, et al. Impact of severe acute respiratory syndrome (SARS) on pulmonary function, functional capacity and quality of life in a cohort of survivors. Thorax. 2005;60(5):401-409. https://doi.org/10.1136/thx.2004.030205. [CrossRef]
- Menezes RG, Rizwan T, Saad Ali S, et al. Postmortem findings in COVID-19 fatalities: A systematic review of current evidence. Leg Med. 2021;54(October 2020):102001. https://doi.org/10.1016/j.legalmed.2021.102001. [CrossRef]
- Edler C, Schröder AS, Aepfelbacher M, et al. Dying with SARS-CoV-2 infection-an autopsy study of the first consecutive 80 cases in Hamburg, Germany (International journal of legal medicine (2020) 134 4 (1275-1284)). Int J Legal Med. 2020;134(5):1977. https://doi.org/10.1007/s00414-020-02336-7. [CrossRef]
- Yu M, Liu Y, Xu D, Zhang R, Lan L, Xu H. Prediction of the development of pulmonary fibrosis using serial thin-section ct and clinical features in patients discharged after treatment for COVID-19 pneumonia. Korean J Radiol. 2020;21(6):746-755. https://doi.org/10.3348/kjr.2020.0215. [CrossRef]
- Leisman DE, Ronner L, Pinotti R, et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir Med. 2020;8(12):1233-1244. https://doi.org/10.1016/S2213-2600(20)30404-5. [CrossRef]
- Schaller T, Hirschbühl K, Burkhardt K, et al. Postmortem Examination of Patients with COVID-19. JAMA - J Am Med Assoc. 2020;323(24):2518-2520. https://doi.org/10.1001/jama.2020.8907. [CrossRef]
- Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. https://doi.org/10.1056/nejmoa2015432. [CrossRef]
- Buja LM, Wolf D, Zhao B, et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): Report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol. 2020;48:107233. https://doi.org/10.1016/j.carpath.2020.107233. [CrossRef]
- Sadegh Beigee F, Pourabdollah Toutkaboni M, Khalili N, et al. Diffuse alveolar damage and thrombotic microangiopathy are the main histopathological findings in lung tissue biopsy samples of COVID-19 patients. Pathol Res Pract. 2020;216(10):153228. https://doi.org/10.1016/j.prp.2020.153228. [CrossRef]
- Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417-1418. https://doi.org/10.1016/S0140-6736(20)30937-5. [CrossRef]
- Osuchowski MF, Winkler MS, Skirecki T, et al. The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir Med. 2021;9(6):622-642. https://doi.org/10.1016/S2213-2600(21)00218-6. [CrossRef]
- Su H, Yang M, Wan C, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98(1):219-227. https://doi.org/10.1016/j.kint.2020.04.003. [CrossRef]
- Kral AH, Lambdin BH, Wenger LD, Davidson PJ. Multiorgan and Renal Tropism of SARS-CoV-2. N Engl J Med. 2020;383(6):589-590. https://doi.org/10.1056/nejmc2015435. [CrossRef]
- Srinivasan S, Cui H, Gao Z, et al. Structural genomics of SARS-COV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses. 2020;12(4). https://doi.org/10.3390/v12040360. [CrossRef]
- Li X, Geng M, Peng Y, Meng L, Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal. 2020;10(2):102-108. https://doi.org/10.1016/j.jpha.2020.03.001. [CrossRef]
- Kanneganti TD. Intracellular innate immune receptors: Life inside the cell. Immunol Rev. 2020;297(1):5-12. https://doi.org/10.1111/imr.12912. [CrossRef]
- Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5(SEP):1-8. https://doi.org/10.3389/fimmu.2014.00461. [CrossRef]
- Zheng M, Karki R, Williams EP, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. 2021;22(7):829-838. https://doi.org/10.1038/s41590-021-00937-x. [CrossRef]
- Potapov I, Kanneganti TD, del Sol A. Fostering experimental and computational synergy to modulate hyperinflammation. Trends Immunol. 2022;43(1):4-7. https://doi.org/10.1016/j.it.2021.11.004. [CrossRef]
- Jung S, Potapov I, Chillara S, del Sol A. Leveraging systems biology for predicting modulators of inflammation in patients with COVID-19. Sci Adv. 2021;7(6):1-11. https://doi.org/10.1126/sciadv.abe5735. [CrossRef]
- Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92(10):2105-2113. https://doi.org/10.1002/jmv.25987. [CrossRef]
- Asano T, Boisson B, Onodi F, et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci Immunol. 2021;6(62). https://doi.org/10.1126/sciimmunol.abl4348. [CrossRef]
- Van Der Made CI, Simons A, Schuurs-Hoeijmakers J, et al. Presence of Genetic Variants among Young Men with Severe COVID-19. JAMA - J Am Med Assoc. 2020;324(7):663-673. https://doi.org/10.1001/jama.2020.13719. [CrossRef]
- Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific J Allergy Immunol. 2020;38(1):1-9. https://doi.org/10.12932/AP-200220-0772. [CrossRef]
- Marcken M de, Dhaliwal K, Danielsen AC, Gautron AS, Dominguez-Villar M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci Signal. 2019;12(605):1-19. https://doi.org/10.1126/scisignal.aaw1347. [CrossRef]
- Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11(6):403-415. https://doi.org/10.1038/nri2974. [CrossRef]
- De Wit E, Van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: Recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523-534. https://doi.org/10.1038/nrmicro.2016.81. [CrossRef]
- Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med. 2020;217(6):1-7. https://doi.org/10.1084/jem.20200652. [CrossRef]
- Wack A, Terczyńska-Dyla E, Hartmann R. Guarding the frontiers: The biology of type III interferons. Nat Immunol. 2015;16(8):802-809. https://doi.org/10.1038/ni.3212. [CrossRef]
- Loo YM, Gale M. Immune Signaling by RIG-I-like Receptors. Immunity. 2011;34(5):680-692. https://doi.org/10.1016/j.immuni.2011.05.003. [CrossRef]
- Horner SM, Liu HM, Park HS, Briley J, Gale M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011;108(35):14590-14595. https://doi.org/10.1073/pnas.1110133108. [CrossRef]
- Stark GR, Darnell JE. The JAK-STAT Pathway at Twenty. Immunity. 2012;36(4):503-514. https://doi.org/10.1016/j.immuni.2012.03.013. [CrossRef]
- Yin X, Riva L, Pu Y, et al. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Rep. 2021;34(2). https://doi.org/10.1016/j.celrep.2020.108628. [CrossRef]
- Rebendenne A, Chaves Valadão AL, Tauziet M, et al. SARS-CoV-2 Triggers an MDA-5-Dependent Interferon Response Which Is Unable To Control Replication in Lung Epithelial Cells. J Virol. 2021;95(8). https://doi.org/10.1128/jvi.02415-20. [CrossRef]
- Thorne LG, Reuschl A, Zuliani-Alvarez L, et al. SARS-CoV-2 sensing by RIG-I and MDA5 links epithelial infection to macrophage inflammation. EMBO J. 2021;40(15):1-17. https://doi.org/10.15252/embj.2021107826. [CrossRef]
- Christgen S, Kanneganti TD. Inflammasomes and the fine line between defense and disease. Curr Opin Immunol. 2020;62:39-44. https://doi.org/10.1016/j.coi.2019.11.007. [CrossRef]
- Kayagaki N, Kornfeld OS, Lee BL, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591(7848):131-136. https://doi.org/10.1038/s41586-021-03218-7. [CrossRef]
- Diamond MS, Kanneganti T-D. Innate immunity: the first line of defense against SARS-CoV-2. Nat Immunol. 2022;23(2):165-176. https://doi.org/10.1038/s41590-021-01091-0. [CrossRef]
- Anand U, Jakhmola S, Indari O, et al. Potential Therapeutic Targets and Vaccine Development for SARS-CoV-2/COVID-19 Pandemic Management: A Review on the Recent Update. Front Immunol. 2021;12(December 2019):1-27. https://doi.org/10.3389/fimmu.2021.658519. [CrossRef]
- Ye X, Xiao X, Li B, et al. Low Humoral Immune Response and Ineffective Clearance of SARS-Cov-2 in a COVID-19 Patient With CLL During a 69-Day Follow-Up. Front Oncol. 2020;10(July):1-7. https://doi.org/10.3389/fonc.2020.01272. [CrossRef]
- Wen W, Su W, Tang H, et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020;6(1). https://doi.org/10.1038/s41421-020-0168-9. [CrossRef]
- You X, Zhang R, Shao M, et al. Double Negative B Cell Is Associated With Renal Impairment in Systemic Lupus Erythematosus and Acts as a Marker for Nephritis Remission. Front Med. 2020;7(April):1-9. https://doi.org/10.3389/fmed.2020.00085. [CrossRef]
- Zhu L, Yin Z, Ju B, et al. Altered frequencies of memory B cells in new-onset systemic lupus erythematosus patients. Clin Rheumatol. 2018;37(1):205-212. https://doi.org/10.1007/s10067-017-3877-1. [CrossRef]
- Li CK, Wu H, Yan H, et al. T Cell Responses to Whole SARS Coronavirus in Humans. J Immunol. 2008;181(8):5490-5500. https://doi.org/10.4049/jimmunol.181.8.5490. [CrossRef]
- Casillo GM, Mansour AA, Raucci F, et al. Could IL-17 represent a new therapeutic target for the treatment and/or management of COVID-19-related respiratory syndrome? Pharmacol Res. 2020;156(April):104791. https://doi.org/10.1016/j.phrs.2020.104791. [CrossRef]
- Yazdanpanah F, Hamblin MR, Rezaei N. The immune system and COVID-19: Friend or foe? Life Sci. 2020;256(May):117900. https://doi.org/10.1016/j.lfs.2020.117900. [CrossRef]
- Bhattacharjee A, Saha M, Halder A, Debnath A, Mukherjee O. Therapeutics and Vaccines: Strengthening Our Fight Against the Global Pandemic COVID-19. Curr Microbiol. 2021;78(2):435-448. https://doi.org/10.1007/s00284-020-02310-x. [CrossRef]
- Klasse PJ. Neutralization of Virus Infectivity by Antibodies: Old Problems in New Perspectives. Adv Biol. 2014;2014:1-24. https://doi.org/10.1155/2014/157895. [CrossRef]
- Ju B, Zhang Q, Ge J, et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584(7819):115-119. https://doi.org/10.1038/s41586-020-2380-z. [CrossRef]
- To KKW, Tsang OTY, Leung WS, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect Dis. 2020;20(5):565-574. https://doi.org/10.1016/S1473-3099(20)30196-1. [CrossRef]
- Amanat F, Stadlbauer D, Strohmeier S, et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med. 2020;26(7):1033-1036. https://doi.org/10.1038/s41591-020-0913-5. [CrossRef]
- Mo H, Zeng G, Ren X, et al. Longitudinal profile of antibodies against SARS-coronavirus in SARS patients and their clinical significance. Respirology. 2006;11(1):49-53. https://doi.org/10.1111/j.1440-1843.2006.00783.x. [CrossRef]
- Wu LP, Wang NC, Chang YH, et al. Duration of antibody responses after severe acute respiratory syndrome. Emerg Infect Dis. 2007;13(10):1562-1564. https://doi.org/10.3201/eid1310.070576. [CrossRef]
- Dan JM, Mateus J, Kato Y, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science (80- ). 2021;371(6529). https://doi.org/10.1126/science.abf4063. [CrossRef]
- Mahal BA, Alshalalfa M, Kensler KH, et al. Rapid Decay of Anti–SARS-CoV-2 Antibodies in Persons with Mild Covid-19. N Engl J Med. 2020;383(11):1083-1085. https://doi.org/10.1056/nejmc2000069. [CrossRef]
- Seow J, Graham C, Merrick B, et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat Microbiol. 2020;5(12):1598-1607. https://doi.org/10.1038/s41564-020-00813-8. [CrossRef]
- Long QX, Tang XJ, Shi QL, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med. 2020;26(8):1200-1204. https://doi.org/10.1038/s41591-020-0965-6. [CrossRef]
- Infantino M, Damiani A, Li Gobbi F, et al. Serological assays for sars-cov-2 infectious disease: Benefits, limitations and perspectives. Isr Med Assoc J. 2020;22(4):203-210.
- Lessler J, Reich NG, Brookmeyer R, Perl TM, Nelson KE, Cummings DA. Incubation periods of acute respiratory viral infections: a systematic review. Lancet Infect Dis. 2009;9(5):291-300. https://doi.org/10.1016/S1473-3099(09)70069-6. [CrossRef]
- Zhang Y, Zhang J, Chen Y, et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through potently downregulating MHC-I. bioRxiv. Published online 2020. https://doi.org/10.1101/2020.05.24.111823. [CrossRef]
- Wilk AJ, Rustagi A, Zhao NQ, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. 2020;26(7):1070-1076. https://doi.org/10.1038/s41591-020-0944-y. [CrossRef]
- Giamarellos-Bourboulis EJ, Netea MG, Rovina N, et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020;27(6):992-1000.e3. https://doi.org/10.1016/j.chom.2020.04.009. [CrossRef]
- Arabi YM, Murthy S, Webb S. COVID-19: a novel coronavirus and a novel challenge for critical care. Intensive Care Med. 2020;46(5):833-836. https://doi.org/10.1007/s00134-020-05955-1. [CrossRef]
- Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. 2020;71(15):762-768. https://doi.org/10.1093/cid/ciaa248. [CrossRef]
- Dong P, Ju X, Yan Y, et al. γδ T Cells Provide Protective Function in Highly Pathogenic Avian H5N1 Influenza A Virus Infection. Front Immunol. 2018;9(December):1-11. https://doi.org/10.3389/fimmu.2018.02812. [CrossRef]
- Bahl K, Kim S-K, Calcagno C, et al. IFN-Induced Attrition of CD8 T Cells in the Presence or Absence of Cognate Antigen during the Early Stages of Viral Infections. J Immunol. 2006;176(7):4284-4295. https://doi.org/10.4049/jimmunol.176.7.4284. [CrossRef]
- Shiow LR, Rosen DB, Brdičková N, et al. CD69 acts downstream of interferon-α/β to inhibit S1P 1 and lymphocyte egress from lymphoid organs. Nature. 2006;440(7083):540-544. https://doi.org/10.1038/nature04606. [CrossRef]
- Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459-468. https://doi.org/10.1038/s41586-020-2286-9. [CrossRef]
- Diao B, Wang C, Tan Y, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol. 2020;11(May):1-7. https://doi.org/10.3389/fimmu.2020.00827. [CrossRef]
- Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020;17(5):533-535. https://doi.org/10.1038/s41423-020-0402-2. [CrossRef]
- Oja AE, Saris A, Ghandour CA, et al. Divergent SARS-CoV-2-specific T- and B-cell responses in severe but not mild COVID-19 patients. Eur J Immunol. 2020;50(12):1998-2012. https://doi.org/10.1002/eji.202048908. [CrossRef]
- Chen G, Wu D, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620-2629. https://doi.org/10.1172/JCI137244. [CrossRef]
- Wong CK, Lam CWK, Wu AKL, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136(1):95-103. https://doi.org/10.1111/j.1365-2249.2004.02415.x. [CrossRef]
- Rokni M, Ghasemi V, Tavakoli Z. Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: Comparison with SARS and MERS. Rev Med Virol. 2020;30(3). https://doi.org/10.1002/rmv.2107. [CrossRef]
- Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. 2020;53(3):368-370. https://doi.org/10.1016/j.jmii.2020.03.005. [CrossRef]
- Davis BK, Wen H, Ting JPY. The Inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707-735. https://doi.org/10.1146/annurev-immunol-031210-101405. [CrossRef]
- Hou W, Jin Y-H, Kang HS, Kim BS. Interleukin-6 (IL-6) and IL-17 Synergistically Promote Viral Persistence by Inhibiting Cellular Apoptosis and Cytotoxic T Cell Function. J Virol. 2014;88(15):8479-8489. https://doi.org/10.1128/jvi.00724-14. [CrossRef]
- Lin L, Lu L, Cao W, Li T. Hypothesis for potential pathogenesis of SARS-CoV-2 infection–a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect. 2020;9(1):727-732. https://doi.org/10.1080/22221751.2020.1746199. [CrossRef]
- Li X, Xu S, Yu M, et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol. 2020;146(1):110-118. https://doi.org/10.1016/j.jaci.2020.04.006. [CrossRef]
- Vita G, Syambani Z. Crucial laboratory parameters in COVID-19 diagnosis and prognosis: An updated meta-analysis. Med Clin. 2020;155(4):143-151. [CrossRef]
- Rokni M, Hamblin MR, Rezaei N. Cytokines and COVID-19: friends or foes? Hum Vaccines Immunother. 2020;16(10):2363-2365. https://doi.org/10.1080/21645515.2020.1799669. [CrossRef]
- Karki R, Kanneganti TD. The 'cytokine storm': molecular mechanisms and therapeutic prospects. Trends Immunol. 2021;42(8):681-705. https://doi.org/10.1016/j.it.2021.06.001. [CrossRef]
- Karki R, Sharma BR, Tuladhar S, et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell. 2021;184(1):149-168.e17. https://doi.org/10.1016/j.cell.2020.11.025. [CrossRef]
- Kuriakose T, Man SM, Subbarao Malireddi RK, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2). https://doi.org/10.1126/sciimmunol.aag2045. [CrossRef]
- Malireddi RKS, Gurung P, Kesavardhana S, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity–independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med. 2020;217(3):1-13. https://doi.org/10.1084/jem_20191644. [CrossRef]
- Malireddi RKS, Gurung P, Mavuluri J, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med. 2018;215(4):1023-1034. https://doi.org/10.1084/jem.20171922. [CrossRef]
- Gurung P, Anand PK, Malireddi RKS, et al. FADD and Caspase-8 Mediate Priming and Activation of the Canonical and Noncanonical Nlrp3 Inflammasomes. J Immunol. 2014;192(4):1835-1846. https://doi.org/10.4049/jimmunol.1302839. [CrossRef]
- Schwarzer R, Laurien L, Pasparakis M. New insights into the regulation of apoptosis, necroptosis, and pyroptosis by receptor interacting protein kinase 1 and caspase-8. Curr Opin Cell Biol. 2020;63:186-193. https://doi.org/10.1016/j.ceb.2020.02.004. [CrossRef]
- Fritsch M, Günther SD, Schwarzer R, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575(7784):683-687. https://doi.org/10.1038/s41586-019-1770-6. [CrossRef]
- Newton K, Wickliffe KE, Maltzman A, et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature. 2019;575(7784):679-682. https://doi.org/10.1038/s41586-019-1752-8. [CrossRef]
- Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti T-D. RIPK1 Distinctly Regulates Yersinia -Induced Inflammatory Cell Death, PANoptosis . ImmunoHorizons. 2020;4(12):789-796. https://doi.org/10.4049/immunohorizons.2000097. [CrossRef]
- Totura AL, Whitmore A, Agnihothram S, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. MBio. 2015;6(3):1-14. https://doi.org/10.1128/mBio.00638-15. [CrossRef]
- Onofrio L, Caraglia M, Facchini G, Margherita V, Placido S De, Buonerba C. Toll-like receptors and COVID-19: A two-faced story with an exciting ending. Futur Sci OA. 2020;6(8):10-13. https://doi.org/10.2144/fsoa-2020-0091. [CrossRef]
- Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355-362. https://doi.org/10.1038/s41577-020-0331-4. [CrossRef]
- Sinha P, Matthay MA, Calfee CS. Is a "cytokine Storm" Relevant to COVID-19? JAMA Intern Med. 2020;180(9):1152-1154. https://doi.org/10.1001/jamainternmed.2020.3313. [CrossRef]
- Chen X, Zhao B, Qu Y, et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated with Drastically Elevated Interleukin 6 Level in Critically Ill Patients with Coronavirus Disease 2019. Clin Infect Dis. 2020;71(8):1937-1942. https://doi.org/10.1093/cid/ciaa449. [CrossRef]
- Gao Y, Li T, Han M, et al. Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19. J Med Virol. 2020;92(7):791-796. https://doi.org/10.1002/jmv.25770. [CrossRef]
- Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46(5):846-848. https://doi.org/10.1007/s00134-020-05991-x. [CrossRef]
- Bösmüller H, Traxler S, Bitzer M, et al. The evolution of pulmonary pathology in fatal COVID-19 disease: an autopsy study with clinical correlation. Virchows Arch. 2020;477(3):349-357. https://doi.org/10.1007/s00428-020-02881-x. [CrossRef]
- Yang L, Han Y, Nilsson-Payant BE, et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell. 2020;27(1):125-136.e7. https://doi.org/10.1016/j.stem.2020.06.015. [CrossRef]
- Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420-422. https://doi.org/10.1016/S2213-2600(20)30076-X. [CrossRef]
- Pensato U, Muccioli L, Cani I, et al. Brain dysfunction in COVID-19 and CAR-T therapy: cytokine storm-associated encephalopathy. Ann Clin Transl Neurol. 2021;8(4):968-979. https://doi.org/10.1002/acn3.51348. [CrossRef]
- Kempuraj D, Selvakumar GP, Ahmed ME, et al. COVID-19, Mast Cells, Cytokine Storm, Psychological Stress, and Neuroinflammation. Neuroscientist. 2020;26(5-6):402-414. https://doi.org/10.1177/1073858420941476. [CrossRef]
- Ahmadian E, Hosseiniyan Khatibi SM, Razi Soofiyani S, et al. Covid-19 and kidney injury: Pathophysiology and molecular mechanisms. Rev Med Virol. 2021;31(3):1-13. https://doi.org/10.1002/rmv.2176. [CrossRef]
- Premkumar M, Kedarisetty CK. Cytokine storm of covid-19 and its impact on patients with and without chronic liver disease. J Clin Transl Hepatol. 2021;9(2):256-264. https://doi.org/10.14218/JCTH.2021.00055. [CrossRef]
- Cococcia S, Lenti MV, Santacroce G, Achilli G, de Andreis FB, Sabatino A Di. Liver-spleen axis dysfunction in COVID-19. World J Gastroenterol. 2021;27(35):5919-5931. https://doi.org/10.3748/wjg.v27.i35.5919. [CrossRef]
- Peng X, Wang Y, Xi X, et al. Promising Therapy for Heart Failure in Patients with Severe COVID-19: Calming the Cytokine Storm. Cardiovasc Drugs Ther. 2021;35(2):231-247. https://doi.org/10.1007/s10557-020-07120-8. [CrossRef]
- Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475-481. https://doi.org/10.1016/S2213-2600(20)30079-5. [CrossRef]
- Zhu L, She ZG, Cheng X, et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020;31(6):1068-1077.e3. https://doi.org/10.1016/j.cmet.2020.04.021. [CrossRef]
- Marshall RJ, Armart P, Hulme KD, et al. Glycemic variability in Diabetes increases the severity of influenza. MBio. 2020;11(2). https://doi.org/10.1128/mBio.02841-19. [CrossRef]
- Nielsen TB, Pantapalangkoor P, Yan J, et al. Diabetes exacerbates infection via hyperinflammation by signaling through TLR4 and RAGE. MBio. 2017;8(4). https://doi.org/10.1128/mBio.00818-17. [CrossRef]
- Pal R, Bhansali A. COVID-19, diabetes mellitus and ACE2: The conundrum. Diabetes Res Clin Pract. 2020;162(January). [CrossRef]
- Gebhard C, Regitz-Zagrosek V, Neuhauser HK, Morgan R, Klein SL. Impact of sex and gender on COVID-19 outcomes in Europe. Biol Sex Differ. 2020;11(1):1-13. https://doi.org/10.1186/s13293-020-00304-9. [CrossRef]
- Busse LW, Chow JH, McCurdy MT, Khanna AK. COVID-19 and the RAAS - A potential role for angiotensin II? Crit Care. 2020;24(1):1-4. https://doi.org/10.1186/s13054-020-02862-1. [CrossRef]
- Han Y, Runge MS, Brasier AR. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-κb transcription factors. Circ Res. 1999;84(6):695-703. https://doi.org/10.1161/01.RES.84.6.695. [CrossRef]
- Carissimo G, Xu W, Kwok I, et al. Whole blood immunophenotyping uncovers immature neutrophil-to-VD2 T-cell ratio as an early marker for severe COVID-19. Nat Commun. 2020;11(1):1-12. https://doi.org/10.1038/s41467-020-19080-6. [CrossRef]
- Bordoni V, Sacchi A, Cimini E, et al. An inflammatory profile correlates with decreased frequency of cytotoxic cells in coronavirus disease 2019. Clin Infect Dis. 2020;71(16):2272-2275. https://doi.org/10.1093/cid/ciaa577. [CrossRef]
- Silvin A, Chapuis N, Dunsmore G, et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182(6):1401-1418.e18. https://doi.org/10.1016/j.cell.2020.08.002. [CrossRef]
- Schulte-Schrepping J, Reusch N, Paclik D, et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182(6):1419-1440.e23. https://doi.org/10.1016/j.cell.2020.08.001. [CrossRef]
- Schaefer IM, Padera RF, Solomon IH, et al. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod Pathol. 2020;33(11):2104-2114. https://doi.org/10.1038/s41379-020-0595-z. [CrossRef]
- Grant RA, Morales-Nebreda L, Markov NS, et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature. 2021;590(7847):635-641. https://doi.org/10.1038/s41586-020-03148-w. [CrossRef]
- Karmakar S. SENDING THE WRONG MESSAGE. The Daily Guardian. 2022.
- Li Q, Nie J, Wu J, et al. SARS-CoV-2 501Y.V2 variants lack higher infectivity but do have immune escape. Cell. 2021;184(9):2362-2371.e9. https://doi.org/10.1016/j.cell.2021.02.042. [CrossRef]
- Garcia-Beltran WF, Lam EC, St. Denis K, et al. Erratum: Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity (Cell (2021) 184(9) (2372–2383.e9), (S0092867421002981), (10.1016/j.cell.2021.03.013)). Cell. 2021;184(9):2523. https://doi.org/10.1016/j.cell.2021.04.006. [CrossRef]
- Zhang L, Li Q, Liang Z, et al. The significant immune escape of pseudotyped SARS-CoV-2 variant Omicron. Emerg Microbes Infect. 2022;11(1):1-5. https://doi.org/10.1080/22221751.2021.2017757. [CrossRef]
- Ingraham NE, Ingbar DH. The omicron variant of SARS-CoV-2: Understanding the known and living with unknowns. Clin Transl Med. 2021;11(12):1-6. https://doi.org/10.1002/ctm2.685. [CrossRef]
- Dejnirattisai W, Huo J, Zhou D, Schreiber G, Stuart DI, Screaton GR. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell. 2022;185:1-18. https://doi.org/10.1016/j.cell.2021.12.046. [CrossRef]
- Sharma V, Rai H, Gautam DNS, Prajapati PK, Sharma R. Emerging evidence on Omicron (B.1.1.529) SARS-CoV-2 variant. J Med Virol. 2022;(January). https://doi.org/10.1002/jmv.27626. [CrossRef]
- Arora S, Grover V, Saluja P, et al. Literature Review of Omicron: A Grim Reality Amidst COVID-19. Microorganisms. 2022;10(2):1-7. https://doi.org/10.3390/microorganisms10020451. [CrossRef]
- Mohandas S, Yadav PD, Sapkal G, et al. Pathogenicity of SARS-CoV-2 Omicron in Syrian hamsters and its neutralization with different Variants of Concern. bioRxiv. Published online 2022:2022.01.19.477013. http://biorxiv.org/content/early/2022/01/20/2022.01.19.477013.abstract.
- Keeton R, Tincho MB, Ngomti A, et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature. Published online 2022:1-5. https://doi.org/10.1038/s41586-022-04460-3. [CrossRef]
- GeurtsvanKessel CH, Geers D, Schmitz KS, et al. Divergent SARS CoV-2 Omicron-specific T- and B-cell responses in COVID-19 vaccine recipients. Sci Immunol. Published online 2022:2021.12.27.21268416. https://doi.org/10.1126/sciimmunol.abo2202. [CrossRef]
- Jergovic M, Coplen CP, Uhrlaub JL, et al. Resilient T cell responses to B.1.1.529 (Omicron) SARS-CoV-2 variant. medRxiv. 2022;529:2022.01.16.22269361. https://www.medrxiv.org/content/10.1101/2022.01.16.22269361v1%0Ahttps://www.medrxiv.org/content/10.1101/2022.01.16.22269361v1.abstract.
- Singhal T. The Emergence of Omicron: Challenging Times Are Here Again! Indian J Pediatr. 2022;(0123456789). https://doi.org/10.1007/s12098-022-04077-4. [CrossRef]
- Lewnard J, Hong V, Patel M, Kahn R, Lipsitch M, Tartof SY. Clinical outcomes among patients infected with Omicron (B.1.1.529) SARS-CoV-2 variant in southern California. medRxiv. Published online 2022. https://doi.org/10.1101/2022.01.11.22269045. [CrossRef]
- Fonager J, Bennedbak M, Bager P, et al. Molecular epidemiology of the SARS-CoV-2 variant Omicron BA.2 sub-lineage in Denmark, 29 November 2021 to 2 January 2022. Eurosurveillance. 2022;27(10):1-7. https://doi.org/10.2807/1560-7917.ES.2022.27.10.2200181. [CrossRef]
- Huang J, Zeng G. Letter to the editor : Epidemiology of the SARS-CoV-2 variant Omicron BA . 2 – vigilance needed. Eurosurveillance. 2022;27(13):22-00254. https://doi.org/10.2807/1560-7917.ES.2022.27.13.2200254. [CrossRef]
- Fan Y, Li X, Zhang L, Wan S, Zhang L, Zhou F. SARS-CoV-2 Omicron variant: recent progress and future perspectives. Signal Transduct Target Ther. 2022;7(1):141. https://doi.org/10.1038/s41392-022-00997-x. [CrossRef]
- Shrestha LB, Foster C, Rawlinson W, Bull RA. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev Med Virol. 2022;e2381. https://doi.org/10.1002/rmv.2381. [CrossRef]
- Tuekprakhon A, Huo J, Nutalai R, et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell. Published online 2022:2422-2433. https://doi.org/10.1016/j.cell.2022.06.005. [CrossRef]
- Hachmann NP, Miller J, Collier AY, et al. Neutralization Escape by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4, and BA.5. N Engl J Med. 2022;387:86-88. https://doi.org/10.1056/nejmc2206576. [CrossRef]
- Xia S, Wang L, Zhu Y, Lu L. Origin , virological features , immune evasion and intervention of SARS-CoV-2 Omicron sublineages. Signal Transduct Target Ther. 2022;7:241. https://doi.org/10.1038/s41392-022-01105-9. [CrossRef]
- Houhamdi L, Gautret P, Hoang VT, Fournier PE, Colson P, Raoult D. Characteristics of the first 1119 SARS-CoV-2 Omicron variant cases, in Marseille, France, November−December 2021. J Med Virol. 2022;94(5):2290-2295. https://doi.org/10.1002/jmv.27613. [CrossRef]
- Ulloa AC, Buchan SA, Daneman N, Brown KA. Estimates of SARS-CoV-2 Omicron Variant Severity in Ontario, Canada. JAMA - J Am Med Assoc. 2022;327(13):1286-1288. https://doi.org/10.1001/jama.2022.2274. [CrossRef]
- Wolter N, Jassat W, Walaza S, et al. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: a data linkage study. Lancet. 2022;399(10323):437-446. https://doi.org/10.1016/S0140-6736(22)00017-4. [CrossRef]
- Rajah MM, Hubert M, Bishop E, et al. SARS-CoV-2 Alpha, Beta, and Delta variants display enhanced Spike-mediated syncytia formation. EMBO J. 2021;40(24):1-17. https://doi.org/10.15252/embj.2021108944. [CrossRef]
- Suzuki R, Yamasoba D, Kimura I, et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature. 2022;603(March). https://doi.org/10.1038/s41586-022-04462-1. [CrossRef]
- Meng B, Abdullahi A, Ferreira IATM, et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature. 2022;603(7902):706-714. https://doi.org/10.1038/s41586-022-04474-x. [CrossRef]
- Bojkova D, Widera M, Ciesek S, Wass MN, Michaelis M, Cinatl J. Reduced interferon antagonism but similar drug sensitivity in Omicron variant compared to Delta variant of SARS-CoV-2 isolates. Cell Res. 2022;32(3):319-321. https://doi.org/10.1038/s41422-022-00619-9. [CrossRef]
- Du X, Tang H, Gao L, et al. Omicron adopts a different strategy from Delta and other variants to adapt to host. Signal Transduct Target Ther. 2022;7(1):5-7. https://doi.org/10.1038/s41392-022-00903-5. [CrossRef]
- Shuai H, Chan JFW, Hu B, et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature. 2022;603(7902):693-699. https://doi.org/10.1038/s41586-022-04442-5. [CrossRef]
- Weekly epidemiological update on COVID-19 - 21 December 2022. Published 2022. Accessed December 21, 2022. https://www.who.int/publications/m/item/covid-19-weekly-epidemiological-update---21-december-2022.
Figure 1.
Schematic Representation of Pathogenesis of SARS-CoV-2 infection.
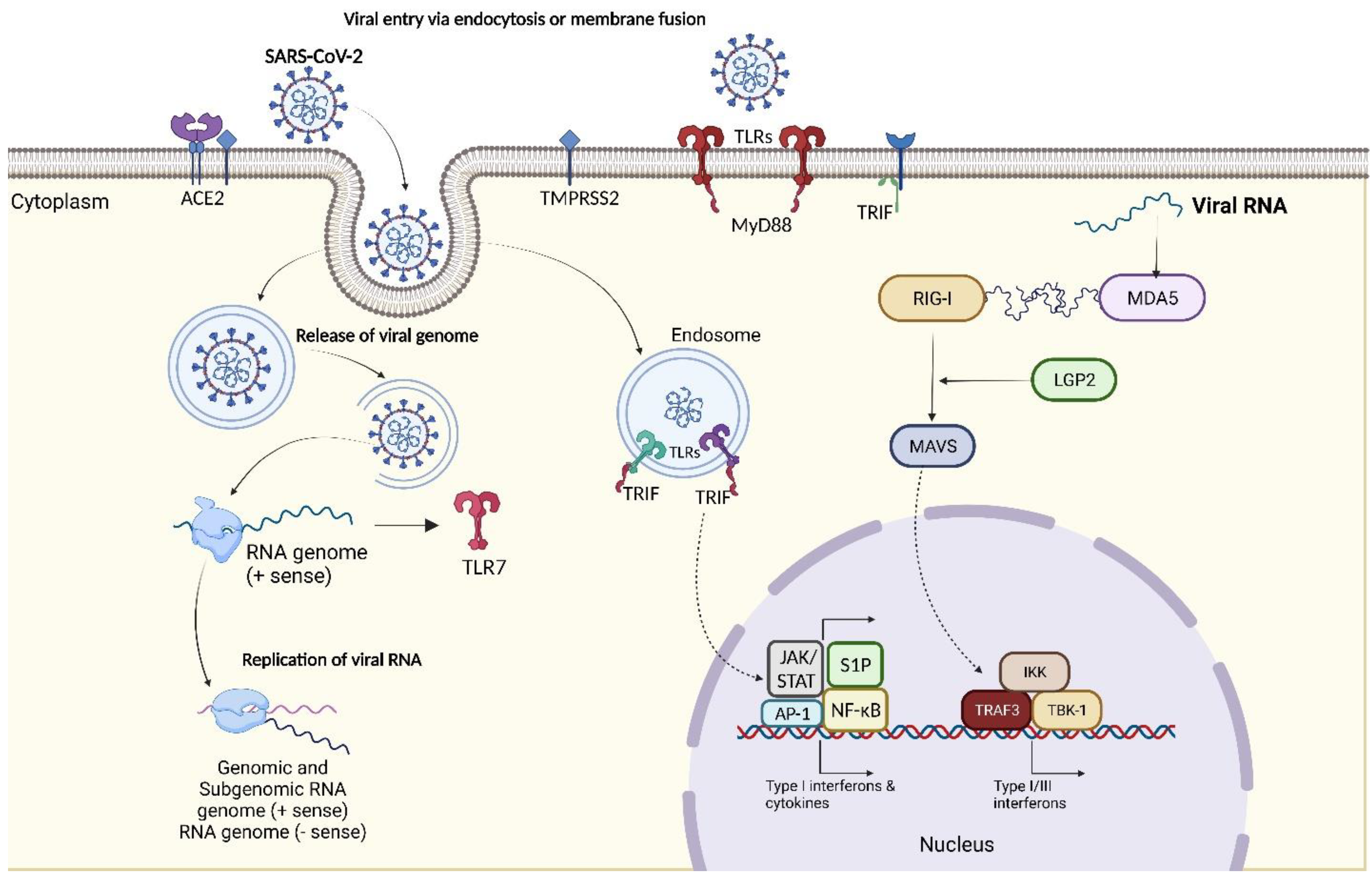
Figure 2.
Clinical Manifestations of COVID-19.
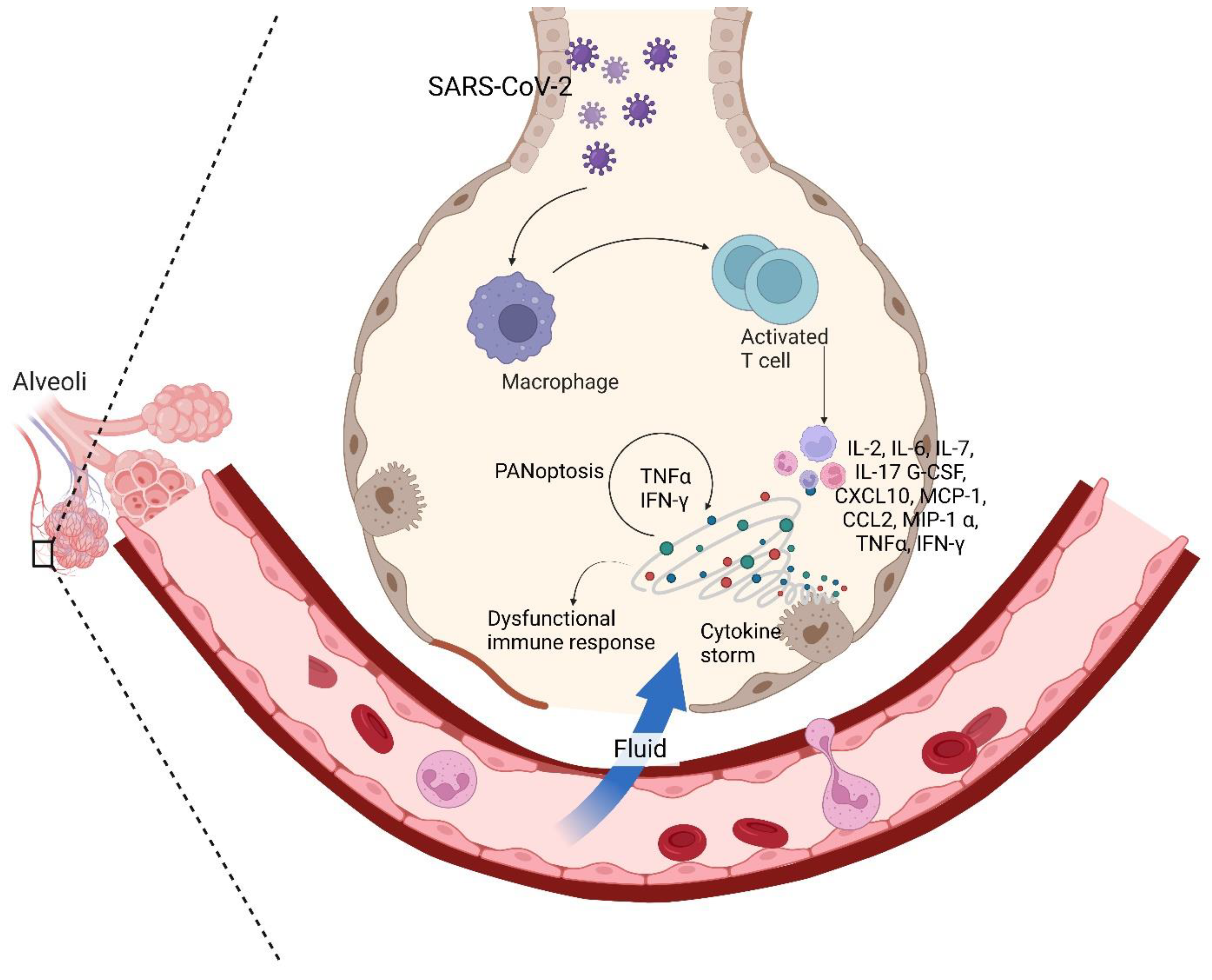
Figure 3.
Antigenic Map of SARS-CoV-2 variants.
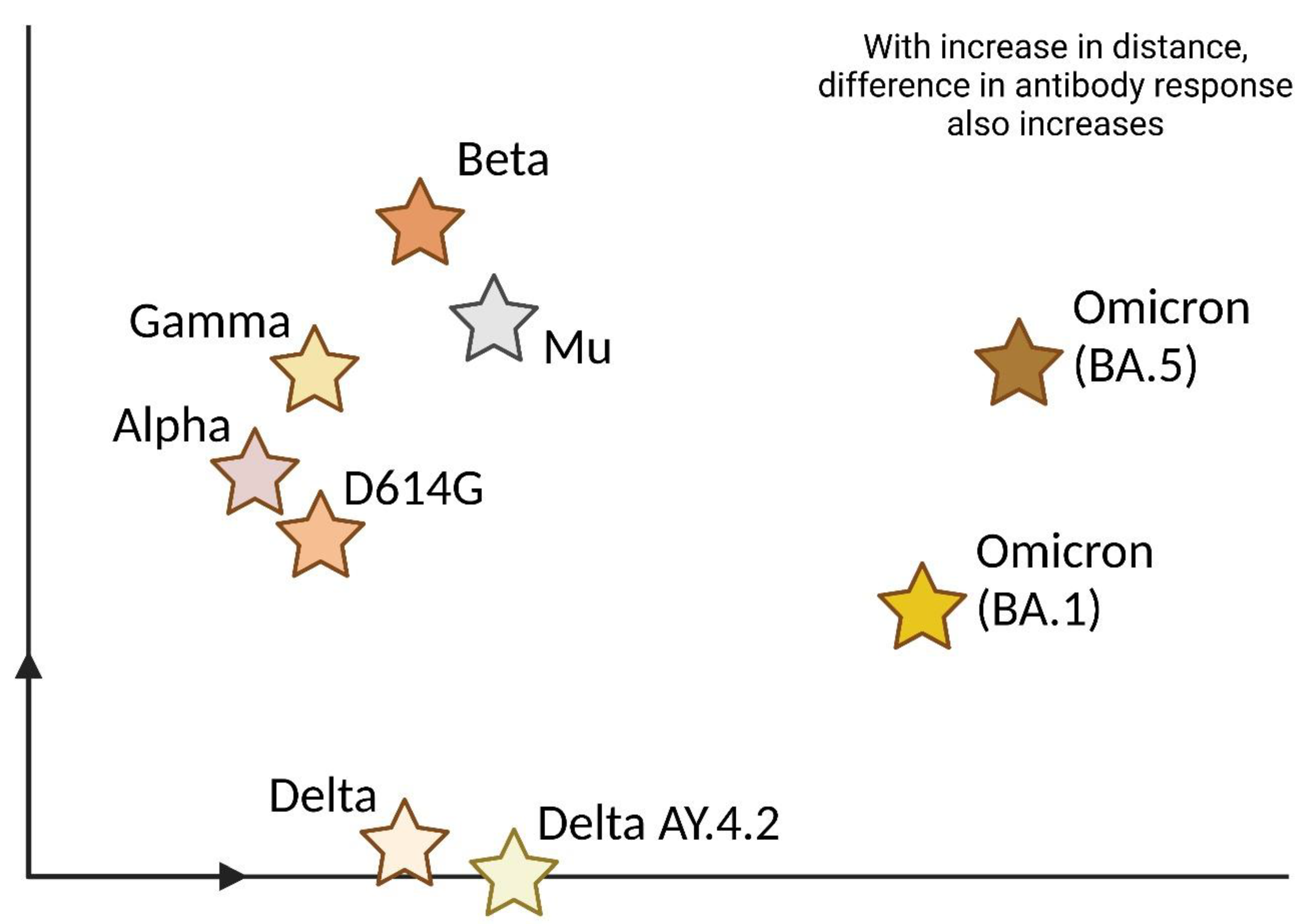
Figure 4.
Scheme of the mechanism of action of mRNA vaccines.
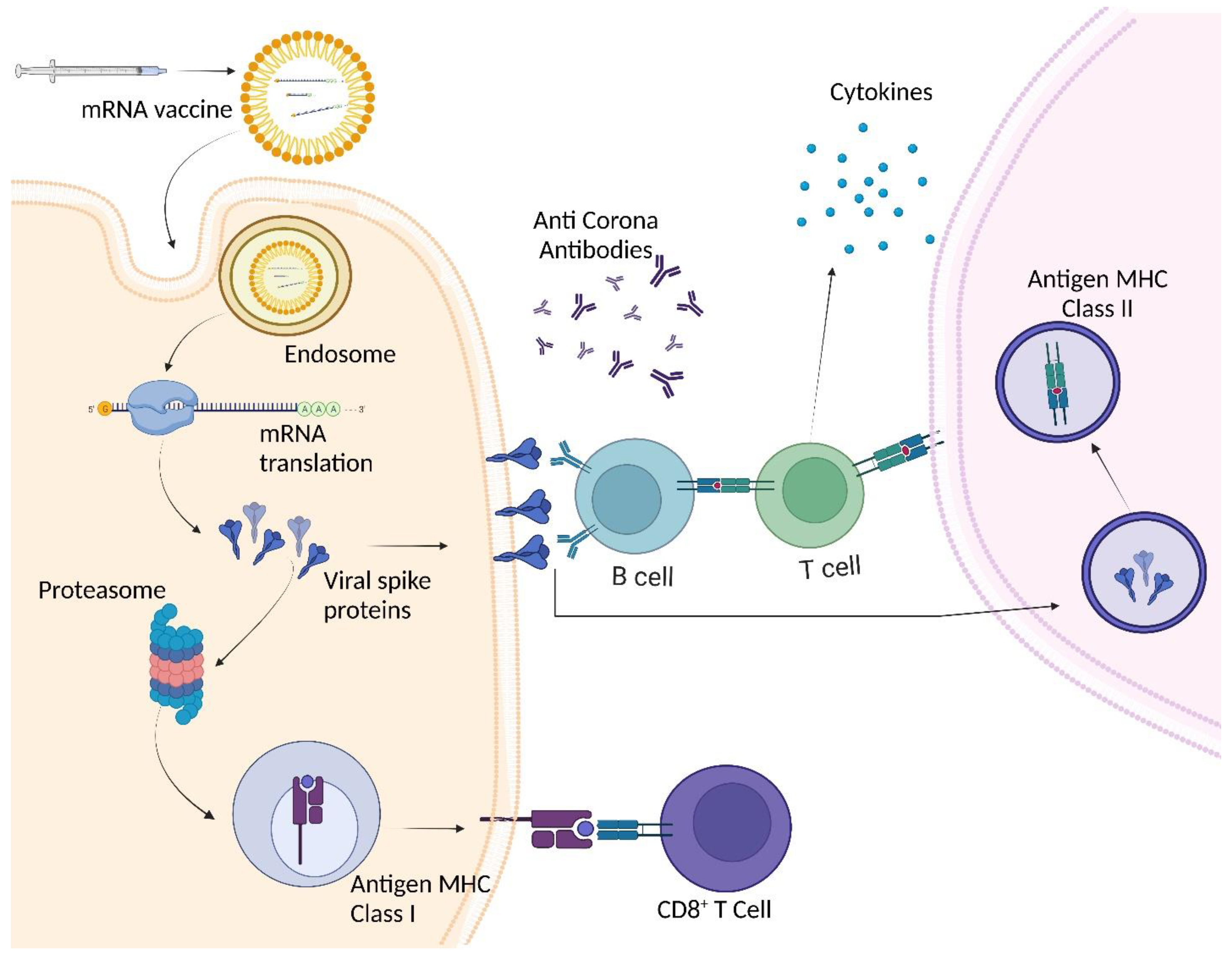
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



