
Submitted:
07 June 2023
Posted:
07 June 2023
You are already at the latest version
Abstract
3,4-Dihydroquinolin-2(1H)-ones represent a class of valuable bioactive compounds with six-membered nitrogen-containing heterocyclic structures. The development of simple, mild and efficient synthetic methods has been widely concerned by synthetic chemists. In this review, we have summarized a series of different synthetic strategies for the synthesis of dihydroquinoline-2(1H)-ones via catalytic annulation of α,β-unsaturated N-arylamides in the past decade, including covering electrophilic cyclization, radical initiated cyclization and photochemical cyclization reactions. Besides, the substrate scope and mechanistic details are also discussed. This paper will provide a useful reference for the development of diverse synthesis methodologies of 3,4-dihydroquinolin-2(1H)-ones.
Keywords:
3
; 4-dihydroquinolin-2(1H)-one
; α
; β-unsaturated N-arylamides
; radical
; photochemical
; annulation
1. Introduction
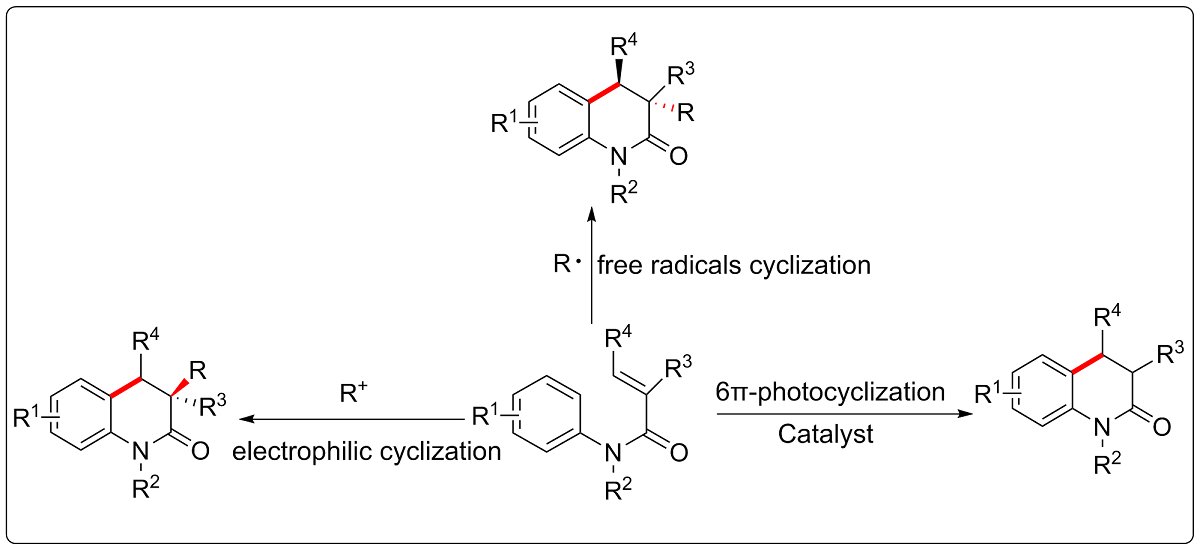
3,4-Dihydroquinolin-2(1H)-one skeleton is an important nitrogen-containing heterocyclic structural unit, which widely exists in natural products, drugs and other bioactive compounds [1-6]. In the past few years, functionalized 3,4-dihydroquinolin-2(1H)-ones have been synthesized and applied to antibiotics, anticancer, antiviral and other important drugs. As shown in Figure 1, selected bioactive compounds containing 3,4-dihydroquinoline-2(1H)-ones have been listed. For example, (+)-Scandine is an alkaloid compound isolated from the twigs and leaves of Melodinus suaveolens, and has been used in the treatment of atherosclerosis [1]. Research also showed that HIV-1 reverse transcriptase inhibitors B could improve antiviral activities against single (K103N) and double (K103N/L100I) mutant viruses [2]. Additionally, (-)-Pinolinone, separated from the roots of boronia pinnata, could strengthen inhibitory effects on Epstein-Barr virus in early antigen (EBV-EA) activation [3]. CYP11B2 (aldosterone synthase) offered possible treatment for breast cancer and the coinstantaneous cardiovascular diseases [4]. Furthermore, Yaequinolones J1, which was extracted from Penicillium sp. FKI-2140, showed toxicity against artemia salina (brine shrimp) [5]. 1-Aryl-3,4-dihydroquinolin-2(1H)-one F was discovered as a potent norepinephrine reuptake inhibitor [6], and so on. Therefore, the synthesis of 3,4-dihydroquinolin-2(1H)-one skeleton compounds will provide more opportunities for the discovery of new bioactive molecules.
The importance of this skeleton compound has stimulated activities of the synthesis community to develop new transformation strategies, which can obtain 3,4-dihydroquinoline-2(1H)-one compounds. In the past decades, some new synthetic methods have been continuously developed, such as Pd-catalyzed Heck reduction-cyclization reaction [7], Pd-catalyzed cyclopropane ring expansion [8], Mn-mediated intramolecular cyclization [9], and Rh-mediated Michael-addition (1,4-additions) of the boronic acid to enone [10], and so on [11,12,13,14]. Among these, using α,β-unsaturated N-arylamides as the key substrates via different cyclization reactions has been shown as a fast, simple and atom economic strategy. Those methodologies include electrophilic cyclization reaction, radical initiated cyclization reaction and photochemical cyclization reaction. This review will focus on the synthesis of 3,4-dihydroquinolin-2(1H)-ones by using α,β-unsaturated N-arylamides as the key substrates in the last decade, and we hope this review will serve as a useful reference for organic chemists to discover more novel strategies for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
2. Synthesis of 3,4-dihydroquinolin-2(1H)-ones via electrophilic cyclization reactions
Synthesis of 3,4-dihydroquinolin-2(1H)-ones via intramolecular Friedel-Crafts alkylation using α,β-unsaturated N-arylamides as the key substrate has been achieved. Some Brønsted acids, such as H2SO4 [15], TsOH [16], CF3COOH [17], polyphosphoric acid (PPA) [18] and Lewis acid AlCl3 [19], were used for the construction of 3,4-dihydroquinolin-2(1H)-ones, in which trifluoroacetic acid (TFA) was the most suitable acid for this reaction (Figure 2).
However, the disadvantages of these reactions are the use of excessively strong acids or high temperature, and the limited range of substrates, especially for the synthesis of 3-substituted dihydroquinolin-2(1H)-ones. Therefore, it is still necessary to develop new synthesis methods under mild conditions.
In 2017, a mild and effective methodology was developed by Zhang’s group for the synthesis of cis-4-aryl-3-arylthio-3,4-dihydroquinolin-2(1H)-ones via electrophilic sulfenylation and cyclization of N-arylcinnamamides with N-arylthiosuccinimides in the presence of BF3·OEt2 [20]. Except for the BF3·OEt2, BBr3 was also effective for the reaction. The reaction showed a broad substrate scope. A series of N-methyl-N-arylcinnamamides were found to be effective in this reaction, and the cis-4-aryl-3-arylthio-3,4-dihydroquinolin-2(1H)-ones 4 were obtained in moderate to excellent yields with high stereoselectivity. N-arylcinnamamides 1 having F, Cl, Br, Me, and MeO groups on the phenyl ring were well tolerated in this reaction. An electrophilic cyclization mechanism was proposed as shown in Figure 3. Firstly, the reaction of N-thiosuccinimides 3 reacted with BF3·OEt2 to generate the electrophilic thio intermediate 5. Then, the thio cation 5 was added to the C=C bond of N-arylcinnamamides 1 via an electrophilic addition process to produce intermediate 6, which further went through intramolecular cyclization to form intermediate 7. Finally, product 4 was formed through the release of a proton.
Reducing the emission of waste to the environment and developing green and atomic economy synthesis technologies are the long-term development goals of synthetic chemists. In 2022, the oxone-mediated direct arylhydroxylation of N-arylcinnamamides was developed by He’s group for the synthesis of hydroxyl-containing 3,4-dihydroquinolin-2-ones [21]. Oxone is a cheap, stable and nontoxic inorganic reagent, which acted as an oxidant for the epoxidation of alkenes and the proton source for the subsequent ring-opening Friedel-Crafts alkylation. This reaction provided an atom-economy strategy for the synthesis of 3-hydroxy-3,4-dihydroquinolin-2-one. The reaction showed a wide functional groups tolerance. The substrates with methyl or n-butyl group on the nitrogen atom worked smoothly, providing the desired products in good to excellent yields. N-arylcinnamamides bearing an electron-donating substituent gave higher yields under the given conditions. Acrylamide substrates with alkyl substituents on the β-position of vinyl group gave the desired products in good yields. However, acrylamide substrates having methyl group on both the α- and β-position of vinyl group afforded a mixture of unidentified products. An epoxidation and subsequent Friedel-Crafts alkylation mechanism was proposed as shown in Figure 4.
3. Synthesis of 3,4-dihydroquinolin-2(1H)-ones via different free radicals initiated cyclization reactions
3.1. Synthesis of 3,4-dihydroquinolin-2(1H)-ones using alkyl radicals.
3.1.1. Carboxylic acids as alkyl radical precursors
Carboxylic acid is an easily obtained chemical raw material, which is widely used in organic synthesis because of its stable nature and easy treatment. More importantly, in the presence of oxidants, carboxylic acids are easy to decarboxylate and produce alkyl radicals [22]. In 2014, Mai and co-workers reported the synthesis of 3,4-disubstituted dihydroquinolin-2(1H)-ones by decarboxylation of N-arylcinnamamides and aliphatic carboxylic acid using AgNO3 as catalyst, K2S2O8 as oxidant and MeCN/H2O (1:1) as solvents [23]. Various primary, secondary and tertiary aliphatic carboxylic acids were tolerated in the reaction, delivering the desired product in moderate to good yields. The mechanism showed that the reaction might undergo a free radical process. Firstly, Ag+ was oxidized by the S2O82− to produce Ag2+, sulfate radical anion and sulfate anion. Then, Ag2+ obtained a single electron from carboxylic acid 14 to produce carboxylic radicals 16, which further underwent a decarboxylation process to provide the alkyl radical 17. Then, the alkyl radical 17 was added to the substrate 1 and further went through intramolecular cyclization to form intermediate 19. Finally, the sulfate radical anion abstracted a hydrogen from 19 to give the final product 15 (Figure 5).
In order to avoid the use of silver salts, a milder method for the synthesis of alkylated dihydroquinolinones was developed by Du’s group [24]. The inexpensive FeCl2·4H2O was used as catalyst and DMF was used as solvent in the reaction, and the peresters (or peroxides) easily prepared from aliphatic acids were used as alkylating reagents and single electron oxidants. A series of alkylated dihydroquinolinone derivatives were obtained in moderate to excellent yields (up to 91%) with excellent diastereoselectivity (dr > 20:1). Peresters prepared from primary acids or secondary acids were effective for this reaction. However, the use of tertiary perester led to only a trace amount of isolated product due to steric hindrance. Peroxides also worked well under the given conditions, delivering the desired alkylated products with good diastereoselectivity. The mechanism showed that the reaction underwent a free radical process. The single-electron transfer (SET) from Fe(II) to perester resulted in the breaking of the O−O bond of perester to produce alkyl radical 22, CO2 and tBuO−. Then alkyl radical 22 reacted with α,β-unsaturated amide 1 via a radical addition/cyclization and re-aromatization process to give the final product (Figure 6).
3.1.2. Alkyl halides as alkyl radical precursors
Alkyl halides are commercially available, accessible substrates and have been widely used in various organic reactions. The alkyl halides containing a,β-hydrogen might be suitable as alkyl radical precursors in the palladium-catalyzed difunctionalization of activated alkenes [25,26,27].
In 2016, Duan’s group developed a palladium-catalyzed alkylarylation of acrylamides with unactivated alkyl halides for the synthesis of dihydroquinolinone and a variety of functionalized oxindoles [28]. PdCl2 was used as catalyst, 1,1'-bis(diphenylphosphino)ferrocene (dppf) was used as ligand, and diglyme was used as solvent. The cinnamamide 1 reacted with cyclohexyl bromide to provide the desired dihydroquinolinone in 39% yield. Mechanistic investigation revealed that this reaction underwent a cascade radical pathway (Figure 7).
Recently, excited-state palladium photocatalysis has become a powerful reagent for inducing single-electron transfer (SET) reactions involving unactivated alkyl C−X bonds (X = Br, I). Photoexcitation of Pd(0) is conducive to forming alkyl radical Pd(I) intermediate by the single-electron oxidative addition of alkyl C−X bonds [29,30]. In this regard, in 2021, Zhang’s group reported visible-light-induced Pd-catalyzed intermolecular radical cascade reaction of N-phenylcinnamamide with cyclohexyl bromide for the preparation of 3,4-dihydroquinolin-2(1H)-ones [31]. The optimization results of test conditions showed that Xantphos was proved to be the most suitable ligand and that Cs2CO3 was the best base for the reaction. N-arylcinnamamides bearing -Cl or -Me group was found to be effective under the given conditions, and the corresponding products were smoothly afforded in moderate yields with good dr values (Figure 8).
A free radical mechanism was proposed in Figure 9. Firstly, Pd (0) complex underwent a single electron transfer process with cyclohexyl bromide under the condition of LEDS irradiation to produce the cyclohexane Pd(I) radical species. Then, cyclohexane radical 28 was added to the double bond of substrate 1 to generate intermediate 29, which underwent an intramolecular cyclization and deprotonation process to obtain the final product 27 and regenerate Pd(0) catalyst.
3.1.3. Hypervalent iodine (III) reagent (HIR) as alkyl radical precursors
In the past decades, the strategic combination of transition-metal catalysts with hypervalent iodine(III) reagent (HIR) has been widely used in the bifunctionalization of olefins [32]. In 2020, Wang and co-workers have described rhenium-catalyzed alkylarylation of cinnamamides with PhI(O2CR)2 via decarboxylation reaction for the synthesis of 3,3-disubstitued dihydroquinolin-2(1H)-ones using Re2(CO)5 as catalyst (Figure 10) [33].
Cinnamamides bearing electron-donating and electron-withdrawing substituents reacted smoothly with PhI(O2CiPr)2 to provide dihydroquinolinones in moderate yields. Different hypervalent iodine(III) reagents were also investigated. When PhI(OAc)2 was used under the given conditions, two different isomers 32f (8:1) were obtained in 40% yield. The decrease of diastereoselectivity might be due to the lower steric hindrance of the methyl group in product. However, when PhI(O2CtBu)2 was used as reaction reagent, only trans-3,4 dihydroquinolin-2(1H)-one 32g was obtained in lower yield due to the larger steric hindrance of tBu group. The mechanism showed that the reaction underwent a free radical process. Firstly, the I–O bond in HIR broke to afford ionic species 33, then the rhenium catalyst transferred an electron to species 33 to produce acyloxy intermediate 34, which further released CO2 to form isopropyl radical 35. Subsequently, the isopropyl radical 35 reacted with the substrate 1 via a radical addition, 6-endo cyclization and re-aromatization processes to produce the product 32.
3.1.4. Aliphatic aldehydes as alkyl radical precursors
Aldehydes are cheap and easily available chemicals. It has great advantages to synthesize various organic compounds through the decarbonylation of aldehydes in the presence of oxidants [34,35]. In this regard, Yang and Pei’s groups developed Fe-catalyzed decarbonylative cascade reaction of N-arylcinnamamides with aliphatic aldehydes for the synthesis of 3,4-dihydroquinolin-2(1H)-ones (Figure 11) [36].
In this reaction, a series of iron salts such as FeCl2, FeCl3, FeSO4, Fe2(SO4)3, Fe(acac)2, Fe(acac)3, FeBr2, and Fe(OAc)2·4H2O were scanned, and Fe(acac)2 was the best choice. DTBP (di-tert-butyl peroxide) was used as oxidant and PhCl was used as solvent. The reaction showed a wide range of substrates. N-arylcinnamamides bearing both electron-withdrawing or electron-donating were well tolerated, and the corresponding 3,4-disubstituted dihydroquinolinones could be obtained in good yields. The steric hindrance and electron effect of substituents were not obvious for this cascade reaction. Interestingly, pyridine units could also be introduced into N-arylcinnamamide substrates, providing the desired product in moderate yields. Different aliphatic aldehydes were also tested, secondary and tertiary alkyl aldehydes worked well in this reaction. However, when a primary alkyl aldehyde was used as reactant, a lower yield was observed due to the self-aldol condensation and the poor stability of primary radicals. Electron-donating groups on the N atom, such as Me, Et, Ph, n-Bu, Bn were effective. However, when the substrate had Ac group on the N atom, the reaction failed to give the desired product. The mechanism study showed that the reaction involved a free radical process. Firstly, the iron catalyst accelerated the homolytic cleavage of DTBP to generate a tert-butoxy radical 40, which abstracted a hydrogen atom from aldehyde 38 to produce the radical intermediate 41. Then, radical 41 released CO to form isopropyl radical 42. Subsequently, the isopropyl radical 42 reacted with the substrate 1 via a radical addition, 6-endo-trig cyclization and re-aromatization process to give the final product 39.
At the same time, Luo and Liu’s groups described a metal-free method for the synthesis of alkylsubstituted 3,4-dihydroquinolin-2(1H)-one derivatives through cascade oxidative decarbonylative radical addition/cyclization of N-arylcinnamamides with aliphatic aldehydes in PhF [37]. The reaction also showed a wide range of substrates. The cinnamamides having electron-donating or electron-withdrawing groups were well tolerated, and the corresponding products were obtained in moderate to good yields. N-Me and N-nBu substituted N-cinnamamides reacted well in this transformation, affording the target products in moderate yields. However, N-H substrate failed to give the expected product. Interestingly, when N-methyl-N-phenyl-3-(pyridin-2-yl) acrylamide was used as substrate, the corresponding product 46f was obtained in 62% yield. However, when oxygen heterocyclic acrylic amide was used, only trace product could be detected. A variety of aliphatic aldehydes were compatible in this reaction (Figure 12).
3.1.5. Cyclohexanone oxime ester as alkyl radical precursors
Recently cyclohexanone oxime ester has been used in organic synthesis as a precursor of cyanoalkyl radicalsand considerable attention has been paid to the selective γ-cyanoalkyl radical functionalization generated from cyclobutanone oxime ester [38]. In 2018, the copper-catalyzed cyclization of N-methyl-N-arylcinnamamides with cyclobutanone O-acyl oximes has been delveloped by Guo’s group for the synthesis of cyanoalkylated dihydroquinolin-2(1H)-ones [39]. N-methyl-N-arylcinnamamides having electron-withdrawing or electron-donating groups on the phenyl rings of the cinnamic acid gave the desired products in moderate to good yields. Cinnamamide bearing a methoxyl group at the para position of the anilide moiety was also compatible in this reaction, delivering the desired product in 51% yield. A reasonable reaction mechanism was proposed in Figure 13. Firstly, Cu(I) catalyst transferred an electron to cyclohexanone oxime ester 47 to give the iminyl radical 49, which underwent β-scission to produce the cyanoalkyl radical 50. Then, the cyanoalkyl radical 50 reacted with substrate 1, producing intermediate 51, which was followed by intramolecular cyclization and deprotonated to give the desired product 48.
3.1.6. Organoboronic acid as alkyl radical precursors
Although trialkylboranes [40] and organotrifluoroborates [41] have been used as the source of alkyl radicals in organic chemistry, the development of directly commercially available alkyl boronic acids was relatively limited. Only a few studies use boronic acid as alkyl radical the precursors mainly because boronic acid has a high oxidation potential [42]. In 2018, Liu’s group described an alkylating method for the synthesis of alkylsubstituted 3,4-dihydroquinolin-2(1H)-one derivatives using cyclohexyl boronic acid as cyclohexyl radical precursor and oxygen as oxidant [43]. The advantage of this reaction was that no transition metal catalyst was used. A series of N-methyl-N-arylcinnamamides were found to be effective substrates, the corresponding alkylated 3,4-dihydroquinolin-2(1H)-ones were obtained in moderate to good yields with excellent chemoselectivity. Only anti-isomers of 3-cyclohexyl-1-methyl-4-aryl-3,4-dihydroquinolin-2(1H)-ones were observed under the given conditions. The mechanism showed that the reaction might undergo a free radical process. The reaction of cyclohexyl boronic acid and molecular oxygen would produce the key cyclohexyl radical 56, which was added to olefin to further form radical intermediate 58 after cyclization. Subsequently, a hydrogen atom transferred (HAT) from 58 to 55 would give product 54 and peroxyboronic acid, which would lead to the formation of boronic acid finally (Figure 14).
3.1.7 Breaking C (sp3)-H bonds as alkyl radical precursors
Breaking various sp3 C-H bonds, such as C-H bonds of alkanes, C-H bonds adjacent to heteroatoms, benzyl and allyl C-H bonds and carbonyl compounds are easy to generate relatively stable free radicals.
In 2014, Mai’s group developed a metal-free protocol for the synthesis of 3,4-disubstituted dihydroquinolin-2(1H)-ones through tandem cyclization of reaction of N-arylcinnamamides with pentane-2,4-dione using K2S2O8 as oxidant and MeCN/H2O (3:3) as solvents [44]. Both electron-withdrawing and electron-donating at the phenyl ring of cinnamic acid were well tolerated, and the corresponding dihydroquinolin-2(1H)-ones could be obtained in moderate yields. Except for methyl group, other groups such as -Bz and -CH2CH2CN at the N atom (R2) were also investigated under the given conditions, and the reaction still proceeded well. Pyridine substituents in substrates 1 were also compatible in this reaction, delivering the desired product in 46% yield. When ethyl 3-oxobutanoate or 1-phenylbutane-1,3-dione were used as substrates, the expected products were not obtained (Figure 15).
In 2016, Duan’s group developed a copper-catalyzed tandem method for the synthesis of dihydroquinolin-2(1H)-ones through cascade radical addition/cyclization of N-arylcinnamamides with benzyl hydrocarbons. (Figure 16).
Cu2O was used as catalyst and tert-butylperoxy benzoate (TBPB) as oxidant [45]. The reaction tolerated a series of substituted N-phenylcinnamamides and toluene derivatives, leading to the desired dihydroquinolinones in moderate to good yields. The mechanism study showed that the reaction underwent a free radical process. The homolytic cleavage of the TBPB in the presence of Cu2O catalyst produced a tert-butoxyl radical 64, which was abstracted a hydrogen from the toluene to generate the key benzyl radical 65.
Further, other types of substrates, such as cyclohexane, cyclopentane, 1,4-dioxane, tetrahydrofuran and isopropanol were all effective under the given conditions, and the corresponding products were obtained in moderate to good yields. However, when a cyclohexene was used as substrate, only a trace amount of the desired product was observed (Figure 17).
Recently, Zhang’s group developed a photo-induced oxidative radical cascade cyclization of N-methyl-N-arylcinnamamides with methanol at room temperature [46]. The reaction avoided the use of expensive photocatalysts and was carried out under very mild conditions. The corresponding hydroxyalkylated dihydroquinolin-2(1H)-one was obtained in 45% yield. A reasonable reaction mechanism was proposed in Figure 18. Under the irradiation of LEDs, (NH4)2S2O8 underwent homolytic cleavage to form sulfate radical anions, which abstracted the α-H from the methanol to give the hydroxyalkyl radical 72. Then, the hydroxyalkyl radical 72 reacted with substrate 1 via an addition/cyclization process to give intermediate 74, which was further oxidized by sulfate radical and deprotonated anion in the presence of Na2CO3 to give the desired product 71.
Liu’s group reported a novel palladium-catalyzed oxidative arylalkylation of alkenes to construct cyano substituted oxindoles and 3,4-dihydroquinolin-2(1H)-one using 2,2'-bipyrimidine 75 as ligand, PhI(OPiv)2 as oxidant and AgF as additive [47]. It was found that the AgF was indispensable to the reaction and AgF played a role to promote the C(sp3)-H bond cleavage in acetonitrile. Only one example was reported for cyano-containing dihydroquinolinone. The desired product 76 was obtained in 77% yield (Figure 19).
In 2018, Luo and Zhang’s groups described a silver-induced tandem radical addition/cyclization of N-arylcinnamamides in CH3CN for the synthesis of cyano-containing 3,4-dihydroquinolin-2(1H)-ones (Figure 20) [48].
The reaction exhibited good functional groups compatibility. The electron-withdrawing groups at the para position of the substrates gave better results compared to that with the electrondonating groups. Different groups at the N atom (R2) were also investigated under the given conditionsand the N-phenyl-substituted substrate gave a yield of 61%. Interestingly, the unprotected or -Ts protected substrate on the N atom was also suitable for this reaction. Although lower yields of products were obtained, it was noteworthy that these products were not easy to obtain through the usual free radical addition/cyclization process. The mechanism indicated that the reaction underwent a free radical process. Firstly, CH3CN reacted with AgOAc to generate AgCH2CN species. There might be two pathways to produce radical 81. One was that AgCH2CN cleaved Ag(0) to form ·CH2CN, which was added to the substrate 1 to produce radical 81. The other was that AgCH2CN was added to the double bond of 1 to provide intermediate 80, followed by a silver-induced formation of radical intermediate 81. Then, the intermediate 81 further went through an intramolecular radical cyclization process to form the intermediate 82. Finally, the intermediate 82 was oxidized by AgOAc to give the product 77.
In 2018, Huang and Hu’s groups developed a Nickel-catalyzed reaction of N-arylcinnamamides with tertiary benzylamines via C-N bond activation for the synthesis of 3,4-dihydroquinolin-2(1H)-ones (Figure 21) [49].
NiI2 and I2 were used as catalysts, 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene 84 was used as ligand and anisole was used as solvent. Tertiary benzylamines 1 having different substituents on the phenyl ring worked well, leading to the six-membered ring dihydroquinolinones 85 in good to excellent yields. Apart from phenylsubstituted amines, naphthyl-substituted amines and thienyl- substituted were also compatible in this reaction, providing the corresponding products in good yields. The reaction went through a free radical reaction process. Firstly, tertiary benzylamines reacted with I2 to form the amine-I2 charge-transfer complex 86, which was extracted a single electron from Ni(0) to generate a benzyl radical 87. Then benzyl radical 87 reacted with substrate 1 underwent an addition/ cyclization process to produce intermediate 89. Finally, intermediate 89 was oxidized by Ni(I) via single-electron transfer (SET) to give product 85 and regenerate Ni(0)-catalyst.
3.2. Synthesis of 3,4-dihydroquinolin-2(1H)-ones using fluoroalkyl radicals
3.2.1. Using CF3SO2Na or HCF2SO2Na as fluorine sources
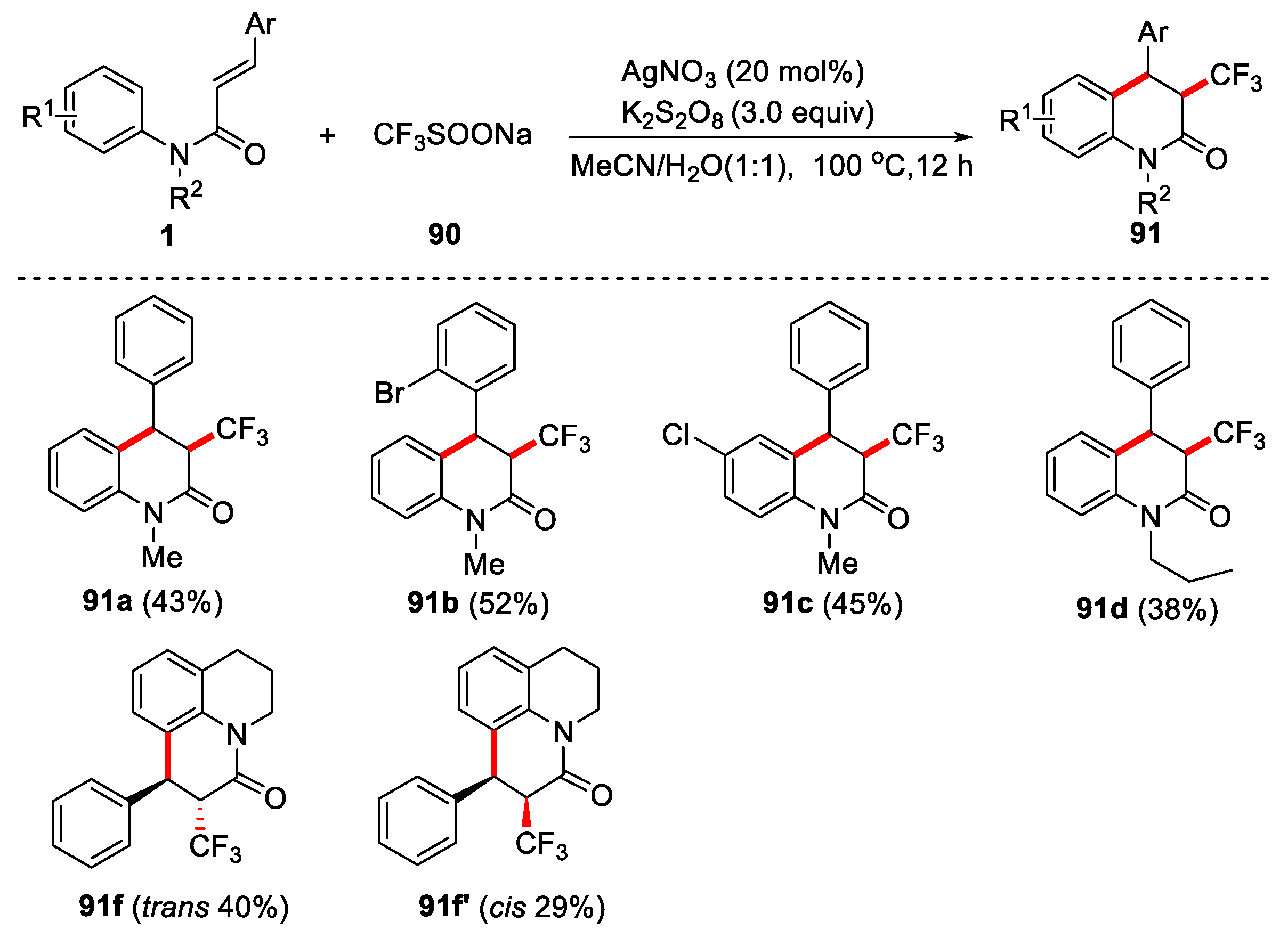
Trifluoromethyl substituted organic compounds have been widely used in medicine, pesticides, materials. Therefore, it is of great research value to develop efficient and practical synthetic methods to prepare trifluoromethyl substituted organic compounds. Langlois’ reagent (CF3SO2Na) is a stable and cheap free radical trifluoromethylation reagent. It has been widely used in the trifluoromethylation reaction of olefins and aromatics in recent years [50,51]. Under oxidation conditions, the reagent usually undergoes single electron transfer, resulting in C-S bond breaking to generate trifluoromethyl radical and SO2. Therefore, in organic synthesis, it can be used as a donor of trifluoromethyl radical to realize diversified trifluoromethylation conversion. In 2014, Mai and co-workers described the silver-catalyzed method for the synthesis of CF3-containing 3,4-dihydroquinolin-2(1H)-ones using the CF3SO2Na as a donor of trifluoromethyl radical sources [23]. AgNO3 was used as catalyst, K2S2O8 was used as oxidant and CH3CN/H2O (1:1) was used as solvents. Various cinnamamides reacted smoothly with CF3SO2Na, affording the desired trifluoromethylated products in moderate yields with excellent diastereoselectivity. However, when (E)-1-(3,4-dihydroquinolin-1(2H)-yl)-3- phenylprop-2-en-1-one was used as substrate, the mixture products 91f and 91f′ were obtained as two stereoisomers under the same reaction conditions (Figure 21).
Figure 22.
Silver-catalyzed method for synthesizing CF3-containing 3,4-dihydroquinolin-2(1H)-ones using CF3SO2Na as CF3 radical source.
Figure 22.
Silver-catalyzed method for synthesizing CF3-containing 3,4-dihydroquinolin-2(1H)-ones using CF3SO2Na as CF3 radical source.
In 2019, Deng and co-workers reported a metal-free, air oxidation difluoromethylation of N-arylacrylamides using Eosin B as photocatalyst and HCF2SO2Na as difluoromethyl sources [52]. Only one example of synthesizing 3,4-dihydroquinolin-2(1H)-ones was reported. The HCF2-containing 3,4-dihydroquinolin-2(1H)-ones 93 were obtained in 33% yield. A plausible photo-induced reaction mechanism was proposed in Figure 23. Firstly, the excited Eosin B* species was formed under the irradiation of white LED light, then an electron transferred from HCF2SO2Na to [Eosin B]* to produce [Eosin B]•- and difluoromethyl radical 95. After that, the difluoromethyl radical 95 reacted with substrate 1 by an addition/ cyclization process to afford intermediate 97. One electron was transferred from [Eosin B]•- to O2 to give peroxide radical anion (O2•–) and regenerate the Eosin B catalyst. The intermediate 97 was oxidized by peroxide radical anion (O2•–) and deprotonated to give product 93 (Figure 23). In ESR experiment, the species of peroxide radical anion was observed, which further proved the rationality of this mechanism.
In 2019, Ruan and coworkers developed an electrochemical di- and trifluoromethylation/cyclization of N-substituted acrylamides for the synthesis of 3,4-dihydroquinolin-2(1H)-ones [53]. The reaction was performed in an undivided cell equipped with a graphite anode and a Ni plate cathode by using nBu4NPF6 as the electrolyte under a constant current of 4 mA, and MeCN as reaction solvent. Control experiments showed the reaction might undergo a free radical process. Firstly, CF3SO2Na lost one electron on the anodic to generate intermediate 101, which further released SO2 to produce the trifluoromethyl radical 102. Then, the trifluoromethyl radical 102 reacted with α, β-unsaturated N-arylamides 1 to generate intermediate 104 through an addition/cyclization process. Then intermediate 104 underwent anodic oxidation to generate intermediate 105, which was deprotonated to give the desired product 106 (Figure 24).
3.2.2. Using Ph3PCH2FI as fluorine sources
Although precious metal catalysts and photocatalysts have been widely used in organic synthesis, it is still necessary to develop protocols using non-photocatalysts and transition metal catalysts due to the fact that those catalysts inevitably produce pollutants. In 2021, Chen and Wang’s groups developed a simple photolysis procedure to realize monofluoromethylation, difluoromethylation and trifluoromethylation of various alkenes [54].
Monofluoromethylated quinolin-one was obtained in 52% yield with 88:12 dr ratio. This strategy avoided the use of oxidants and photocatalysts, promoting SET to generate the corresponding CH2F radicals by using the σ-hole effect of phosphonium salts 107, because of its lower reduction potential and much lower stability than its similar CHF2 and CF3 groups. Hence, this reaction provided a new strategy to produce CH2F radical by a simple method (Figure 25).
3.2.3. Using Togni's Reagents as fluorine sources
Togni's Reagents is a kind of trifluoromethylation reagent with high reactivity. It can not only react with a series of nucleophilic reagents to efficiently realize electrophilic trifluoromethylation, but also be compatible with a variety of sensitive functional groups. It has been widely used to synthesize trifluoromethyl-containing organic compounds [55,56,57]. In 2015, a copper-catalyzed method was developed by Wang and co-workers for the synthesis of trifluoromethylated 3,4-dihydroquinolin-2(1H)-ones by cascade radical addition/cyclization of N-phenylcinnamamides with Togni's Reagents using CuI as catalyst and CHCl3 as solvent (Figure 26) [58].
The reaction showed a wide functional groups tolerance. Different N-protected groups, such as Me, Et, i-Pr, Bn, t-butyloxycarbonyl (Boc) and CH2COOEt were compatible with this reaction, and the desired products were obtained in moderate to good yields. However, when N-Ts(N-para-toluenesulfonyl) protected substrate was tested, almost no target product was obtained. Cinnamamides bearing an electron-withdrawing substituents, such as -F, -CF3, -CN or Br groups in the ortho, meta, or para position worked well under the given conditions, affording the expected products in good yields. However, cinnamamide with electron-donating substituents mainly provided by-products of oxytrifluoromethylation along with low yields of cyclization products. The substrates with two electron-donating substituents in the aniline ring afforded the higher yields compared with those substrates with no substituent or only one methoxy substituent. A reasonable reaction mechanism was proposed in Figure 26. Initially, CuI reacted with Togni's Reagents to generate a trifluoromethyl radical, and then the trifluoromethyl radical was added to the double bond of substrate 1 to produce free radical intermediate 113. Further, 113 was oxidized to carbocation 114 by Cu(II) via a single electron transfer process. Finally, carbocation 114 underwent an intramolecular electrophilic cyclization and deprotonation process to obtain the final product 110. However, the carbocation 114 was attacked by 2-iodobenzoate, giving the oxytrifluoromethylation byproducts 111 via a nucleophilic addition process.
In 2015, Xia and his colleagues developed visible-light induced cascade reaction of N-arylcinnamamides with Togni’s reagent for the synthesis of CF3-containing 3,4-disubstituted dihydroquinolinones and 1-azaspiro [4.5] decanes using fac-Ir(ppy)3 as photocatalyst (Figure 27) [59].
The substituent of N-arylcinnamamides would affect the final product. The N-arylcinnamamides with different groups such as Me, Cl, Br and F on the para-position of anilines were tolerated under the reaction conditions, providing the dihydroquinolinones in moderate yields. However, when the para-position of anilines was substituted by -OH, or -OTBS group, 3,4-disubstituted 1-azaspiro [4.5] decanes were obtained in moderate to good yields. Ortho-Me substituent on the aniline ring gave the relatively lower yield due to steric hindrance. Substrates with different N-protecting groups such as Me, Et, Ph and Bn were effective under the given conditions and provided the desired products in moderate to good yields. A plausible mechanism was described in Figure 27. Firstly, the excited state [fac-IrIII (ppy)3]* was generated under the irradiation of blue LEDs. Then, [fac-IrIII(ppy)3]* transferred an electron to Togni’s reagent led to trifluoromethyl radical and generated the fac-IrIV(ppy)3 species. Subsequently, the trifluoromethyl radical was added to the double bond of the substrate 1 to generate the radical intermediate 118, which was further cyclization by the different processes to produce the intermediate 119 and 121. Finally, 119 or 121 was oxidized by fac-IrIV(ppy)3 and lost H+ or R+ to give the desired product 116 or 117.
3.2.4. Using BrCF2COOEt as a CF2 sources
Except for Togni’s reagent, BrCF2COOEt have been widely used as a CF2 sources in organic synthesis [60,61]. A visible-light induced method was also developed by Zhu’s group for the synthesis of HCF2COOEt-containing 3,4-disubstituted dihydroquinolinones and 1-azaspiro[4.5]decanes [62]. It has been found that the position of the substituent on the cinnamic ring had a great influence on diastereoselectivity. When the cinnamic ring had a meta or ortho substituent, and two pairs of enantiomers could be observed due to steric hindrance. When there was no or only one para substituent on the cinnamic ring, only trans diastereoisomers were obtained. For the para-position substituted anilines with F or CF3 group, the reaction also worked well, providing the desired products in good yields. However, for the para-position of anilines was substituted by OH or OCH3 group, 3,4-disubstituted 1-azaspiro[4.5]decanes 124 were obtained in moderate yields (Figure 28).
3.3. Synthesis of 3,4-dihydroquinolin-2(1H)-ones using chloroalkyl radicals
Chloromethyl groups are found in many bioactive natural products. The introduction of chloromethyl into organic molecules is of great significance for the synthesis of diverse organic molecules. A Cu-catalyzed cross-dehydrogenative coupling of N-arylcinnamamides with chloroform has been developed by Yu and co-workers using tert-butyl peroxybenzoate as oxidant [63]. Trichloromethyl substituted dihydroquinolin-2(1H)-ones were obtained in 78% yield. This mechanism indicated that the trichloromethyl radical might be involved in this cyclization process (Figure 29).
Tang and co-workers developed Mn(OAc)3-mediated radical dichloromethylation of arylacrylamides or N-phenylcinnamamide for the preparation of chloro-containing oxindoles or 3,4-dihydroquinolin-2(1H)-ones [64]. When N-methyl-N-phenylcinnamamide was used, the desired 3,4-dihydroquinolin-2(1H)-one was obtained in 39% yield. The cheap and easily prepared Mn(OAc)3·2H2O was used in this reaction. The reaction mechanism was manganese (III)–o-tolylboronic acid mediated radical oxidative dichloromethylation and cyclization of N-phenylcinnamamide with dichloromethane (Figure 30).
3.4. Synthesis of 3,4-dihydroquinolin-2(1H)-ones using acyl radicals
Carboxylic acids are rich and cheap organic compounds with diverse structures. Synthetic chemists have been trying to apply these compounds to participate in new organic transformation. In 2015, Wallentin and co-workers reported a visible-light photocatalytic tandem acylarylation of olefins by using carboxylic acids as acyl radical precursors for the synthesis of different heterocyclic compounds [65]. When N-phenylcinnamamide reacted with benzoic acid in the presence of the fac-Ir(ppy)3 photocatalyst, 2,6-lutidine and DMDC (dimethyl dicarbonate) under visible-light irradiation, the corresponding dihydroquinolinone was obtained in 32% yield. The mechanism showed that the reaction underwent a free radical process. Benzoic acid reacted with dimethyl dicarbonate (DMDC) to form a mixed anhydride intermediate 138, which could be transferred to key acyl radicals 140 through a single-electron reduction pathway, along with CO2 and methanoate as by products (Figure 31).
α-Keto acids are cheap, easily available and relatively stable organic compounds. Its have been widely used as acylating agents in transition metal catalyzed decarboxylation coupling reactions and photo-induced acyl radical conversion reactions. In 2014, Mai and co-workers developed a silver-catalyzed protocol for the synthesis of 3-acyl-4-aryldihydroquinolin-2(1H)-ones or 3-acyl-4-arylquinolin-2(1H)-ones via intermolecular radical addition/cyclization of N-phenylcinnamamide with α-keto acids in CH3CN/H2O media [66]. It has been found that 3-acyl-4-aryldihydroquinolin-2(1H)-ones 144 or 3-acyl-4-arylquinolin-2(1H)-ones 145 could be selectively obtained by adjusting the amount of K2S2O8. The reaction showed a wide range of substrates. Electron-withdrawing and -donating substituted groups on the phenyl ring of cinnamic acid worked well, affording the corresponding products in moderate yields. Different protective groups on the nitrogen atom, such as -Me, -Bn and -CH2CH2CN were effective in this reaction, providing the final products in moderate yields. Both aliphatic and aromatic keto acids were compatible in this reaction (Figure 32).
At the same time, a similar result was reported by Duan’s group [67]. In this transformation, the decarboxylation coupling/cyclization reaction occurred using acetone/H2O as solvents under much milder condition (Figure 33).
The reaction showed a broad substrate scope and excellent functional groups tolerance. A variety of valuable substituted dihydroquinolinones could be easily obtained in moderate to excellent yields with high stereoselectivity. When free radical scavengers such as 2,2,6,6-tetramethylpiperidine oxide (TEMPO) and butylated hydroxytoluene (BHT) were added to the reaction under the standard conditions, the reaction was significantly inhibited, which indicated that the reaction might undergo a free radical reaction process. Ag (I) transferred one electron to keto acid to obtain free radical intermediate 144, which was further decarboxylated to obtain the key acyl radical 140. The key acyl radical was reacted with substrate 1 via a radical addition/cyclization and re-aromatization process to give the final product 146. This high stereoselectivity could be explained by the intermediate 147, which led to the trans-isomer due to minimizing the strain of benzoyl and phenyl groups on six-membered ring.
In 2015, a metal-free catalyzed method was reported by Mai and co-workers for the synthesis of 3-acyl-4-aryldihydroquinolin-2(1H)-ones by cascade radical addition/cyclization of N-arylcinnamamides with aldehyde [68].
In this reaction, TBAB (tetrabutylammonium bromide) was used as catalyst, K2S2O8 was used as oxidant. Different solvents such as DCE, DMF, CH3CN, EtOAc, dioxane and EtOH were tested for this transformation, and DCE gave the best results. The reaction also showed a wide range of substrates. The substrate scope revealed that the aromatic aldehyde bearing electron-donating groups gave higher yield than those having electron-withdrawing groups on the phenyl ring. When the aromatic aldehydes with strong electron-withdrawing groups, such as -CF3 and -NO2 were used as substrates, no desired products were detected. Except for aromatic aldehydes, aliphatic aldehydes such as acetaldehyde and propyl aldehyde were also compatible in this reaction, affording the target products in moderate yields. The N-arylcinnamamides with electron-donating and electron-withdrawing groups on the various positions were tolerated in this reaction, delivering the corresponding dihydroquinolinones in moderate yields. A possible mechanism was proposed in Figure 34. Firstly, using the catalyst of TBAB, the homolytic cleavage of the S2O82- produced the sulfate radical anions. Then the sulfate radical anion was abstracted a hydrogen atom from aldehyde to generate acyl radical 140. Subsequently, the acyl radical 140 reacted with the substrate 1 via a radical addition, 6-endo-trig cyclization and re-aromatization process to give the final product 149.
Oxime esters have been widely used as a kind of important builing blocks in the synthesis of nitrogen-containing compounds by thermal decomposition, transition metal catalysis or light-induced interruption. [69]. The N-O bond or C-C could be activated to form highly active iminyl radicals or acyl radicals. In 2019, Fan and co-workers developed a photocatalytic method for C-C bond activation of oxime ester to generate acyl radical using fac-Ir(ppy)3 as catalyst under mild conditions [70]. Only one example was reported, in which 3,4-dihydroquinolin-2(1H)-one 153 was obtained in 34% yield. A free radical mechanism was involved in the reaction process. Under the irradiation of blue-LED light, the photocatalyst fac-Ir(ppy)3 was converted to the excited state [fac-Ir(ppy)3] *, which further transferred an electron to oxime ester 151, leading to the N-O bond of oxime ester was dissociated to form iminyl radicals, which underwent fast C-C bond homolysis and elimination of 1 molecule of CH3CN to produce the key acyl radical 154. Next, the addition of the acyl radical 154 to substrate 1 gave the raidical intermediate 155, which was further underwent an intramolecularcyclization process to form intermediate 156. Finally, intermediate 156 was oxidized by the Ir(V)+ species to ggive the product 153 and regenerate the Ir(III) catalyst. (Figure 35).
3.5. Synthesis of 3,4-dihydroquinolin-2(1H)-ones using phosphorus-containing free radicals.
Phosphorus-containing compounds widely exist in organic compounds, such as drugs, natural products and materials [71,72]. In recent years, the development of simple and effective methods has attracted significant attention of organic synthesis chemists for the synthesis of containing phosphorus organic heterocyclic compounds. In 2016, a silver-catalyzed cascade cyclization of cinnamamides with diphenylphosphine oxide was developed by Zhu’s group for the synthesis of dihydroquinolin-2(1H)-ones [73]. The different silver salts, such as AgNO3, Ag2CO3 and AgOAc were screened, and AgNO3 gave the best results. Mg(NO3)2·6H2O was more effective additive, and 0.3 equiv was found to be the ideal amount for the use of additive. MeCN was the most suitable solvent and the optimized reaction temperature was 100 oC. A broad range of cinnamamides bearing electron-donating and electron-withdrawing groups were well tolerated under the given conditions, providing the corresponding dihydroquinolin-2(1H)-ones 158 in moderate to good yields. Different N-protected groups, such as Me, Et, iPr, nBu, and Bn were compatible in this reaction, and the desired products were obtained in moderate to good yields. When radical inhibitor 2,2,6,6-tetramethylpiperidine oxide (TEMPO) was added to the reaction system, the reaction was significantly inhibited, indicating that the reaction experienced a free radical process. The key phosphonyl radical 159 was formed through the reaction of P(O)H compounds 157 with Ag(I) salt (Figure 36). The products could be transferred through the demethylation process, providing free N-protected dihydroquinolin-2(1H)-ones.
3.6. Synthesis of 3,4-dihydroquinolin-2(1H)-ones using sulphur-containing free radicals
In 2015, Wang and Tian reported a tandem sulfenylation/cyclization of N-arylacrylamides with sulfonyl hydrazides in the presence of iodine for the selective synthesis of 3-(sulfenylmethyl)oxindoles and 3- sulfenyl-3,4-dihydroquinolin-2(1H)-ones [74]. In this reaction, iodine played multiple roles as an oxidant, a reductant, and a radical initiator. β-Substituted N-arylacrylamides reacted with sulfonyl hydrazides, followed by 6-endo-trig cyclization to afford 3-sulfenyl-3,4-dihydroquinolin-2(1H)-ones in good yields (Figure 37).
A reasonable reaction mechanism was proposed in Figure 38. At the beginning, sulfonyl hydrazide 162 reacted with I2 to give sulfinic acid 166 and sulfenyl iodide 167. There might be two pathways to produce thiosulfonate 168. One was that sulfinic acid 166 was reduced by I2 to form thiosulfonate 168. The other was that sulfinic acid 166 nucleophilicly attacked sulfenyl iodide 167 to give thiosulfonate 168. Further thiosulfonate 168 was reduced by I2 to give disulfide 169. After that, both thiosulfonate 168 and disulfide 169 were attacked by iodine radical generated from iodine upon heating to give sulfenyl radical 172, which was reacted with α,β-unsaturated amide 1 via a radical addition/cyclization and re-aromatization process to give the final product 163.
Sulfonates and sulfonic acids have been widely used in organic synthesis, material science, natural products and pharmaceutical chemistry. The introduction of sulfonic acid functional groups into the molecule can significantly adjust the water solubility and polarity of the compounds [75]. In this context, in 2022, Mn(OAc)3·2H2O-promoted radical sulfonation from N-phenylcinnamamides and potassium metabisulfite (K2S2O5) was developed by Liang’s group for the synthesis of sulfonyl-containing 3,4-dihydroquinolin-2(1H)-one [76]. The substrate scope revealed that withdrawing groups at para position of the anilines gave excellent yields. The electron-donating or -withdrawing groups on the aromatic ring that was located at the beta-position of double bonds were well tolerated, affording the corresponding products in excellent yields. The sulfonyl-containing 3,4-dihydroquinolin-2(1H)-one could be further converted to aldehyde 177 in DMF. This reaction probably proceeded via the elimination of sulfonyl moiety followed by the in-situ Vilsmeier−Hack formylation (Figure 39).
3.7. Synthesis of 3,4-dihydroquinolin-2(1H)-ones using selenyl free radicals
In the past few years, electrochemistry has become an attractive method in the field of organic synthesis due to its environmental friendliness and practicality. Because of the use of electrons as oxidation or reduction reagents, synthetic electrochemistry can achieve oxidation and reduction reactions without the need for transition metal catalysts or toxic reagents.
In this regard, in 2020, Chen and co-workers successfully developed an electrochemical approach for the synthesis of functionalized 3,4-dihydroquinolin-2(1H)-ones 179 through cascade selenylation/cyclization of N-arylcinnamamides with diphenyl diselenide 178 [77]. The reaction was performed in an undivided cell equipped with a platinum anode and a Fe plate cathode by using nBu4NPF6 as the electrolyte under a constant current of 8 mA, and MeCN as solvent. An evaluation of substrate scope revealed that a series of substituted N-arylcinnamamides were smoothly converted to the selenylated 3,4-dihydroquinolin-2(1H)-ones in moderate to good yields with high diastereoselectivity (Figure 40).
In 2021, Pan’s group also reported an electrochemical protocol for the construction of C-Se bond via tandem cyclization of N-arylacrylamides or N-arylcinnamamide with diselenides [78]. In this transformation, platinum plates was used as anode and graphite rods was used as cathode, nBu4NPF6 was used as the electrolyte under a constant current of 15 mA, and CH3CN/1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as the co-solvents. Only one example was reported, the 1-methyl-4-phenyl-3-(phenylselanyl)- 3,4-dihydroquinolin-2(1H)-one 180 was obtained in 35% yield along with the formation of 1-methyl-4-phenylquinolin-2(1H)-one 181 with a yield of 50%. A possible mechanism was proposed in Figure 41. PhSeSePh lost one electron on the anodic to form selenium free radical 183 and selenium cation 184. The selenium free radical 183 was added to the double bond of substrate 1 to generate radical intermediate 186, which underwent an intramolecular cyclization to lead to radical intermediate 187. Finally, 187 underwent anodic oxidation and deprotonation to produce the final product 180 (path a). In another way selenium cation 184 was added to the double bond of the substrate 1 to generate the cation intermediate 189, which underwent an intramolecular cyclization and deprotonation leading to the formation of product 180 (path b) (Figure 41).
4. Synthesis of 3,4- dihydroquinolin-2(1H)-ones using photoredox cyclization
Photocyclization of acrylanilides 1 to the corresponding 3,4-dihydroquinolin-2-ones was first described by Chapman and co-workers [79]. Later, the research mainly focused on the mechanism of photocyclization using high-energy UV light stimulation with high pressure Hg lamps. These reactions showed a narrow range of substrates [80,81]. To resolve this problem, developing a general and simple system was still desired for the preparation of 3,4-dihydroquinolin-2-ones. In this regard, in 2021, Chi and co-workers developed a photo-induced 6π cyclization of N-arylacrylamides for the synthesis of 3,4- dihydroquinolin-2(1H)-one using luminescent platinum (II) complex Pt-2 as photocatalyst [82]. It was found that additives played a very important role in this reaction. Inorganic bases such as Cs2CO3, Na2CO3, K2CO3, and K3PO4 as additives could dramatically improve the product yield. The reaction showed a broad substrate scope and good functional groups compatibility. Substrates with both electron-withdrawing and electron-donating on the para-position of anilines were well tolerated, providing the corresponding dihydroquinolin-2(1H)-ones in 30-99% yields. Although N-arylamides bearing meta substituents exhibited good reactivity as well, the mixture of products were obtained albeit with poor regioselectivity. Substrates with other alkyl and aromatic substituents on nitrogen atoms were also effective in this transformation. Substrates with a phenyl group at the α and β position of N-arylamides also afforded products in good yields. Heteroaromatic substrates such as quinoline, benzothiazole, 1,2,3,4-tetrahydroquinoline and indoline worked well under the given conditions, providing the desired products in good to excellent yields. Further applying this method to the total synthesis of natural products (+)-SIPI6360 was realized [83] (Figure 42).
Although the use of expensive metal catalysts has achieved great success, there are still strong desire for chemists to develop metal-free method for these important transformations due to people's attention to environmental issues and saving synthetic costs. In 2021, Petersen’s group described a thioxanthone-catalyzed method for the synthesis of 3,4-dihydroquinolin-2-ones via a 6π-photocyclization process [84]. N-methyl-N,2-diphenylacrylamide bearing electron-withdrawing or electron-donating groups on the aniline such as -F, -Cl, -Br, -I, -OMe were well tolerated using 2-CTX as catalyst, providing the target products in good yields. However, substrates with strong electron-withdrawing groups gave poor yields. N-methyl-2-phenyl-N-(pyridin-2-yl) acrylamide also reacted smoothly with a good yield. Variation of the aromatic groups at the 3-position were also well tolerated, affording the desired products in good to excellent yields. Switching the aromatic substitution to the 4- position, only 32% isolated yield was obtained with a 1:2 E/Z ratio. A proposed mechanism was proposed in Figure 43. Under the light irradiation, the ground state 2-TX was transferred to the excited state 12-TX*, which further underwent internal conversion and rapid intersystem crossing (ISC) to produce its triplet state 32-TX*. Then, triplet state 32-TX* transferred the energy to substrate 1 via triplet energy transfer (TET) process to produce the triplet state of substrate 197 and return the catalyst to its ground state 2-TX. The triplet state of substrate 197 went through a 6π-electrocyclization to give intermediate 198, which transferred product 200 either by a direct [1,5]-H shift process or via a two-step re-aromatization−tautomerization process. The mechanism of free radicals might also be involved in the photochemical process [85,86] (Figure 43).
In 2022, Huang and Ji’s groups developed a visible-light induced photoredox cyclization of N-Arylacrylamides for the synthesis of dihydroquinolinones [87]. 4CzIPN was used as a photosensitizer and acetonitrile was used as reaction solvent. Cinnamamides bearing different groups in the para position were tolerated under the given conditions, affording the expected products in good to excellent yields. 2-phenylacrylamides reacted smoothly to give 3-arylquinolinone derivatives in moderate to excellent yields. 2-Methylacrylamides with electron-donating groups gave better results. Substrates derived from 1-naphthylamine and 2-naphthylamine showed high reactivity. Firstly, under the visible-light irradiation, the triplet state of substrate 205 generated via triplet energy transfer (TET) from the photosensitizer. Then the addition to the aniline moiety generated the 1,4-diradical intermediate 206, which underwent an internal conversion and rapid intersystem crossing (ISC) to generate intermediate 207, followed by the process of deprotonation to give the desired product (path a). Alternatively, the [1,3]-H shift of 1,4-diradical 206 proceeds to form the triplet state of intermediate 208, which further underwent an internal conversion and rapid intersystem crossing (ISC) process to give the final product 204 (path b) (Figure 44).
The application of 6π-photocyclization methods were also applied for the synthesis of 3,4-dihydroquinolin-2-ones in the field of asymmetric synthesis. A thiourea/urea catalytic method for the 6π-photocyclization of acrylanilides has been reported for the synthesis of 3,4-dihydroquinolin-2-one [88]. However, poor enantioselectivity was observed in this reaction. Recently, the application of “axial to point chirality transfer” strategy has been shown effectively for the synthesis of 3,4-dihydroquinolin-2-ones with high enantioselectivity [89,90,91]. However, the substrate scope was limited. In 2023, catalytic enantioselective 6π-photocyclization of acrylanilides was developed by Paton and Smith’s groups using Ir((5-CF3)(4′-t-Bu)ppy)3 as photocatalyst in the presence of Sc(OTf)3 and chiral ligand [92]. In this reaction, chiral Lewis acid complexes that was generated through the reaction with Sc(OTf)3 and chiral ligand, played a very important role and could enable selective energy transfer from a photosensitizer to facilitate enantioselective triplet state reactions. A series of substituted dihydroquinolones were obtained in moderate to excellent yields with high diastereo and enantioselectivity. The acrylanilides with various alkyl groups on the N atom, such as Me, Bn, n-Pr, isobutyl, allyl and PMB were well compatible with the reaction. By introducing a 3-substitued group on aniline ring, cyclization onto either ortho position was viable, resulting in the formation of two regioisomers. For the para-position substituted anilines with a variety of functional groups, the substrates bearing electron-donating substituents gave higher yields under the given conditions (Figure 45).
5. Conclusions
The synthetic methods of 3,4-dihydroquinolin-2(1H)-one derivatives have been reviewed in this paper. Using α,β-unsaturated N-arylamides as the key substrate by applying simple and mild reaction conditions to synthesize 3,4-dihydroquinoline-2 (1H)-one is an effective strategy. In these synthesis methods, the application of electrophilic cyclization reaction, free radical initiated cyclization and 6π photochemical cyclization provides a sustainable choice for the synthesis of 3,4-dihydroquinoline-2 (1H)-one.
Despite significant progress has been made in this field and shown its potential application, there are still some key problems to be solved in this field: (1) Developing efficient photochemical catalysts and high regioselective synthesis methods are still in demand. (2) Developing green synthesis protocols by using non-toxic organic reagents is still a challenging area for the synthesis of organic molecules with biological activity. (3) Expanding to the field of asymmetric synthesis to synthesize chiral molecules and natural products with pharmaceutical activity has a broad space. Therefore, it is a long-term development goal in this field to design new and cheap catalytic systems and apply them to the synthesis of natural products, functional materials and drug molecules under mild conditions.
Author Contributions
The manuscript was written by Yan-Ning Niu and Lin-Shuang Tian. Huai-Zhong Lv and Ping-Gui Li gave advices and participated in the modification of the manuscripts. All authors have given approval to the published version of the manuscript.
Funding
The authors gratefully acknowledge the financial support of this work by Science and Technology Plan Projects in Huai’an (HAB202065, HASZ201650); Grain Processing, Storage Inspection Technology Service Platform in Huai’an (HAP202108) and High-level talented person cultivating 333 project of Jiangsu province.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gao, X.-H.; Fan, Y.-Y.; Liu, Q.-F.; Cho, S.-H.; Pauli, G. F.; Chen, S.-N.; Yue, J.-M. Unprecedented dimeric quinoline alkaloids with antimycobacterial activity from melodinus suaveolens. Org. Lett., 2019, 21, 7065–7068. [Google Scholar] [CrossRef]
- Patel, M.; McHugh, R. J.; Cordova, B. C.; Klabe, R. M.; Bacheler, L. T.; Erickson-Viitanen, S.; Rodgers, J. D. Synthesis and evaluation of novel quinolinones as HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett., 2001, 11, 1943–1945. [Google Scholar] [PubMed]
- Ito, C.; Itoigawa, M.; Otsuka, T.; Tokuda, H.; Nishino, H.; Furukawa, H. Constituents of boronia pinnata. J. Nat. Prod., 2000, 63, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Yin, L.; Hartmann, R. W. Selective dual inhibitors of CYP19 and CYP11B2: targeting cardiovascular diseases hiding in the shadow of breast cancer. J. Med. Chem., 2012, 55, 7080–7089. [Google Scholar] [PubMed]
- Uchida, R.; Imasato, R.; Shiomi, K.; Tomoda, H.; Omura, S. Yaequinolones J1 and J2, novel insecticidal antibiotics from penicillium sp. FKI-2140. Org. Lett., 2005, 7, 5701–5704. [Google Scholar]
- Beadle, C. D.; Boot, J.; Camp, N. P.; Dezutter, N.; Findlay, J.; Hayhurst, L.; Masters, J. J.; Penariol, R.; Walter, M. W. 1-Aryl-3,4-dihydro-1H-quinolin-2-one derivatives, novel and selective norepinephrine reuptake inhibitors. Bioorg. Med. Chem. Lett., 2015, 15, 4432–4437. [Google Scholar]
- François, X. F.; Jérôme, C.; Cécile, Z.; Eric, F. Preparation of 2-quinolones by sequential Heck reduction–cyclization (HRC) reactions by using a multitask palladium catalyst. Chem. Eur. J., 2009, 15, 7238–7245. [Google Scholar]
- Tsuritani, T.; Yamamoto, Y.; Kawasaki, M.; Mase, T. Novel approach to 3,4-dihydro-2(1H)-quinolinone derivatives via cyclopropane ring expansion. Org. Lett., 2009, 11, 1043–1045. [Google Scholar] [CrossRef]
- Tsubusaki, T.; Nishino, H. Manganese(III)-mediated facile synthesis of 3,4-dihydro-2(1H)-quinolinones: selectivity of the 6-endo and 5-exo cyclization. Tetrahedron, 2009, 65, 9448–9459. [Google Scholar] [CrossRef]
- Joachim, H.; Ho, Y. L.; Stephen, P. M.; Adam, N.; Rachel, J. S.; Amanda, J. C.; David, H.; Gordon, G. W. Convergent synthesis of dihydroquinolones from o-aminoarylboronates. Tetrahedron, 2009, 65, 9002–9007. [Google Scholar]
- Fujita, K.; Takahashi, Y.; Owaki, M.; Yamamoto, K.; Yamaguchi, R. Synthesis of five-, six-, and seven-membered ring lactams by Cp*Rh complex-catalyzed oxidative N-heterocyclization of amino alcohols. Org. Lett., 2004, 6, 2785–2788. [Google Scholar] [CrossRef]
- Correia, J. T.M.; Santos, M. S.; Pissinati, E. F.; da Silva, G. P.; Paixo, M. W. Recent advances on photoinduced cascade strategies for the synthesis of N-heterocycles. Chem. Rec., 2021, 21, 2666–2687. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-H.; Li,Y. ; Li, J.-H.; Xiang, J.-N.; Deng, W. Recent advances in radical-mediated [2+2+m] annulation of 1,n-enynes. Sci China Chem, 2019, 62, 1463–1475. [Google Scholar] [CrossRef]
- Mieriņa, I.; Jure, M.; Stikute, A. Synthetic approaches to 4-(het)aryl-3,4-dihydroquinolin-2(1H)-ones. Chem. Heterocycl. Com., 2016, 52, 509–523. [Google Scholar] [CrossRef]
- Kraus, J. M.; Tatipaka, H. B.; McGuffin, S. A.; Chennamaneni, N. K.; Karimi, M.; Arif, J.; Verlinde, C. L. M. J.; Buckner, F. S.; Gelb, M. H. Second generation analogues of the cancer drug clinical candidate tipifarnib for anti-chagas disease drug discovery. J. Med. Chem., 2010, 53, 3887–3898. [Google Scholar] [CrossRef]
- Safina, L. Y.; Selivanova, G. A.; Koltunov, K. Y.; Shteingarts, V. D. Synthesis of polyfluorinated 4-phenyl- 3,4-dihydroquinolin-2-ones and quinolin-2-ones via superacidic activation of N-(polyfluorophenyl)cinnamamides. Tetrahedron Lett., 2009, 50, 5245–5247. [Google Scholar] [CrossRef]
- Li, K.; Foresee, L. N.; Tunge, J. A. Trifluoroacetic acid-mediated hydroarylation: synthesis of dihydrocoumarins and dihydroquinolones. J. Org. Chem., 2005, 70, 2881–2883. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Q.; Zhang, D.; Xin, X.; Zhang, R.; Zhou, F.; Dong, D. PPA-Mediated C-C bond formation: a synthetic route to substituted indeno[2,1-c]quinolin-6(7H)-ones. Org. Lett 2013, 15, 776–779. [Google Scholar] [CrossRef]
- Lee, E.; Han, S.; Jin, G. H.; Lee, H. J.; Kim, W.-Y.; Ryu, J.-H.; Jeon, R. Synthesis and anticancer activity of aminodihydroquinoline analogs: identification of novel proapoptotic agents. Bioorg. Med. Chem. Lett., 2013, 23, 3976–3978. [Google Scholar] [CrossRef]
- Ren, H.; Zhang, M.; Zhang, A. Synthesis of 4-aryl-3-arylthio-3,4-dihydroquinolin-2(1H)-ones via cyclization of N-arylcinnamamides with N-thiosuccinimides. Synlett, 2017, 49, 575–582. [Google Scholar]
- Zhang, M.-Z.; Liu, L.; Gou, Q.; Wang, Q.; Li, Y.; Li, W.-T.; Luo, F.; Yuan, M.; Chen, T.; He, W.-M. Synthesis of hydroxyl-containing oxindoles and 3,4-dihydroquinolin-2-ones through oxone-mediated cascade arylhydroxylation of activated alkene. Green Chem., 2020, 22, 8369–8374. [Google Scholar] [CrossRef]
- Xia, X.-F.; Zhu, S.-L.; Chen, C.; Wang, H.; Liang, Y.-M. Silver-catalyzed decarboxylative addition/cyclization of activated alkenes with aliphatic carboxylic acids. J. Org.Chem., 2016, 81, 1277–1284. [Google Scholar] [CrossRef]
- Mai, W.-P.; Wang, J.-T.; Yang, L.-R.; Yuan, J.-W.; Xiao, Y.-M.; Mao, P. and Qu, L.-B. Silver-catalyzed radical tandem cyclization for the synthesis of 3,4-disubstituted dihydroquinolin-2(1H)-ones. Org. Lett., 2014, 16, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; and Du, D.-M. FeCl2-catalyzed decarboxylative radical alkylation/cyclization of cinnamamides: access to dihydroquinolinone and pyrrolo[1,2-a]indole analogues. J. Org. Chem., 2018, 83, 5149–5159. [Google Scholar] [CrossRef] [PubMed]
- Chuentragool, P.; Kurandina, D.; Gevorgyan, V. Catalysis with palladium complexes photoexcited by visible light. Angew. Chem., Int. Ed. 2019, 58, 11586–11598. [Google Scholar] [CrossRef]
- Zhou, W.-J.; Zhang, Y.; Cao, G.; Liu, H.; Yu, D.-G. Palladium-catalyzed radical-type transformations of alkyl halides. Chin. J. Org. Chem., 2017, 37, 1322–1337. [Google Scholar] [CrossRef]
- Fan, J.-H.; Wei, W.-T.; Zhou, M.-B.; Song, R.-J.; Li, J.-H. Palladium-catalyzed oxidative difunctionalization of alkenes with α-carbonyl alkyl bromides initiated through a Heck-type insertion: a route to indolin-2-ones. Angew. Chem., Int. Ed., 2014, 53, 6650−6654. [Google Scholar] [CrossRef]
- Wang, H.; Guo, L.-N.; Duan, X.-H. Palladium-catalyzed alkylarylation of acrylamides with unactivated alkyl halides. J. Org. Chem., 2016, 81, 860–867. [Google Scholar] [CrossRef]
- Zhou, W.-J.; Cao, G.-M.; Zhang, Z.-P.; Yu, D.-G. Visible light-induced palladium-catalysis in organic synthesis. Chem. Lett., 2019, 48, 181–191. [Google Scholar] [CrossRef]
- Fan, J.-H.; Wei, W.-T.; Zhou, M.-B.; Song, R.-J.; Li, J.-H. Palladium-catalyzed oxidative difunctionalization of alkenes with alpha-carbonyl alkyl bromides initiated through a Heck-type insertion: a route to indolin-2-ones. Angew. Chem., Int. Ed., 2014, 53, 6650–6654. [Google Scholar] [CrossRef]
- Du, J.; Wang, X.; Wang, H.; Wei, J.; Huang, X.; Song, J.; Zhang, J. Photoinduced palladium-catalyzed intermolecular radical cascade cyclization of N-arylacrylamides with unactivated alkyl bromides. Org. Lett., 2021, 23, 5635–5635. [Google Scholar] [CrossRef]
- Ciesielski, J.; Dequirez, G.; Retailleau, P.; Gandon, V.; Dauban, P. Rhodium-catalyzed alkene difunctionalization with nitrenes. Chem. – Eur. J., 2016, 22, 9338–9347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Yang, Y.; Zhang, P.; Wang, C. Rhenium-catalyzed alkylarylation of alkenes with PhI(O2CR)2 via decarboxylation to access indolinones and dihydroquinolinon. Org. Chem. Front., 2020, 7, 3234–3241. [Google Scholar] [CrossRef]
- Tang, R.-J.; He, Q.; and Yang, L. Metal-free oxidative decarbonylative coupling of aromatic aldehydes with arenes: direct access to biaryls. Chem. Commun., 2015, 51, 5925–5928. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.-J.; Kang, L.; Yang, L. Metal-free oxidative decarbonylative coupling of aliphatic aldehydes with azaarenes: successful Minisci-type alkylation of various heterocycles. Adv. Synth. Catal., 2015, 357, 2055–2060. [Google Scholar] [CrossRef]
- Gao, R.-X.; Luan, X.-Q.; Xie, Z.-Y.; Yang, L.; Pei, Y. Fe-catalyzed decarbonylative cascade reaction of N-aryl cinnamamides with aliphatic aldehydes to construct 3,4-dihydroquinolin-2(1H)-ones. Org. Biomol. Chem. 2019, 17, 5262–5268. [Google Scholar] [CrossRef]
- Xu, X.; Luo, Z.; Liu, C.-F.; Wang, X. ; Deng, L; Gao. Jie. Metal-free synthesis of 3,4-dihydroquinolin-2(1H)-ones via cascade oxidative decarbonylative radical alkylation/cyclization of cinnamamides with aliphatic aldehydes. Asian J. Org. Chem., 2019, 8, 1903–1906. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Xiao, W.-J.; Chen, J.-R. When light meets nitrogen-centered radicals: from reagents to catalysts. Acc. Chem. Res., 2020, 53, 1066−1083. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, J.-Y.; Gao, P.; Xu, S.-L; Guo, L.-N. Copper-catalyzed redox-neutral cyanoalkylarylation of activated alkenes with cyclobutanone oxime esters. J. Org. Chem., 2018, 83, 1046–1055. [Google Scholar] [CrossRef]
- Sanzone, J. R.; Hu, C. T.; Woerpel, K. A. Uncatalyzed carboboration of seven-membered-ring trans-alkenes: formation of air-stable trialkylboranes. J. Am. Chem. Soc., 2017, 139, 8404–8407. [Google Scholar] [CrossRef]
- Darses, S.; Genet, J. P. Potassium Organotrifluoroborates: new perspectives in organic synthesis. Chem. Rev., 2008, 108, 288–325. [Google Scholar] [CrossRef]
- Seiple, I. B.; Su, S.; Rodriguez, R. A.; Gianatassio, R.; Fujiwara, Y.; Sobel, A. L.; Baran, P. S. Direct C-H arylation of electron deficient heterocycles with arylboronic acids. J. Am. Chem. Soc., 2010, 132, 13194–13196. [Google Scholar] [CrossRef]
- Ling, A.; Zhang, L.; Tan, R. X.; Liu, Z.-Q. Molecular oxygen-promoted general and site-specific alkylation with organoboronic acid. J. Org. Chem. 2018, 83, 14489–14497. [Google Scholar] [CrossRef]
- Wang, H.; Sun, B.; Yang, J.; Wang, J.; Mao, P.; Yang, L.; Mai, W. The synthesis of 3,4-disubstituted dihydroquinolin-2(1H)-one under metal free conditions in aqueous solution. J. Chem. Res., 2014, 38, 542–545. [Google Scholar] [CrossRef]
- Zhou, S.-L.; Guo, L.-Na.; Wang, S.; Duan, X.-H. Copper-catalyzed tandem oxidative cyclization of cinnamamides with benzyl hydrocarbons through cross-dehydrogenative coupling. Chem. Commun., 2014, 50, 3589−3591. [Google Scholar]
- Xu, D.; Yu, Y.; Huang, F.; Zhou, S.; Zhang, W. Photo-induced C(sp3)-H functionalization for the synthesis of 3,3-disubstituted oxindoles. Asian J. Org. Chem., 2022, 11, e202200131 (1-5). [Google Scholar] [CrossRef]
- Wu, T.; Mu, X.; Liu, G. Palladium-catalyzed oxidative arylalkylation of activated alkenes: dual C-H bond cleavage of an arene and acetonitrile. Angew. Chem., Int. Ed., 2011, 50, 12578–12581. [Google Scholar] [CrossRef]
- Wang, K.; Chen, X.; Yuan, M.; Yao, M.; Zhu, H.; Xue, Y.; Luo, Z.; Zhang, Y. Silver-mediated cyanomethylation of cinnamamides by direct C(sp3)−H functionalization of acetonitrile. J. Org. Chem 2018, 83, 1525–1531. [Google Scholar] [CrossRef]
- Yu, H.; Hu, B.; Huang, H. Nickel-catalyzed alkylarylation of activated alkenes with benzylamines via C-N bond activation. Chem. –Eur. J., 2021, 24, 7114−7117. [Google Scholar]
- Lu, M.-J.; Liu, Z.-J.; Zhang, J.-W.; Tian, Y.; Qin, H.-G.; Huang, M.-Q.; Hua, S.-R.; Cai, S.-Y. Synthesis of oxindoles through trifluoromethylation of N-aryl acrylamides by photoredox catalysis. Org.Biomol. Chem., 2018, 16, 6564–6568. [Google Scholar] [CrossRef]
- Wang, S.; Dai, P.; Yan, Z.; Wang, Y.; Shao, J.; Wu, Y.; Deng, C.; Zhang, W. Metal-free, visible-light-induced radical trifluoromethylation/cyclization of N-benzamides with CF3SO2Na to synthesize CF3-containing isoquinoline-1,3-diones. Chemistryselect, 2019, 4, 10329–10333. [Google Scholar] [CrossRef]
- Shao, J.; Wang, Y.; Chen, S.; Wang, S.; Wu, Y.; Li, X.; Zhang, W.; Deng, C. Synthesis of CF2H-containing isoquinoline-1,3-diones through metal-free, visible-light and air-promoted radical difluoromethylation/cyclization of N-benzamides. Tetrahedron 2021, 79, 131822. [Google Scholar] [CrossRef]
- Ruan, Z.; Huang, Z.; Xu, Z.; Mo, G.; Tian, X.; Yu, X.-Y.; Ackermann, L. Catalyst-free, direct electrochemical tri- and difluoroalkylation/cyclization: access to functionalized oxindoles and quinolinones. Org. Lett., 2019, 21, 1237−1240. [Google Scholar] [CrossRef]
- Liu, Q.; Lu, Y.; Sheng, H.; Zhang, C.-S.; Su, X.-D.; Wang, Z.-X.; Chen, X.-Y. Visible-light-induced selective photolysis of phosphonium iodide salts for monofluoromethylations. Angew. Chem., Int. Ed., 2021, 60, 25477–25484. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Y.; Huang, J.; Sun, K.; He, W.-M. Recent advances in transition-metal-free trifluoromethylation with Togni’s reagents. Org. Chem. Front., 2022, 9, 1152–1164. [Google Scholar] [CrossRef]
- Chen, Z.-M.; Bai, W.; Wang, S.-H.; Yang, B.-M.; Tu, Y.-Q.; Zhang, F.-M. Copper-catalyzed tandem trifluoromethylation/ semipinacol rearrangement of allylic alcohols, Angew. Chem., Int. Ed., 2013, 52, 9781–9785. [Google Scholar] [CrossRef]
- Zhang, B.; Mück-Lichtenfeld, C.; Daniliuc, C. G.; Studer, A. 6-Trifluoromethyl-phenanthridines through radical trifluoromethylation of isonitriles. Angew. Chem., Int. Ed., 2013, 52, 10792–10795. [Google Scholar] [CrossRef]
- Wang, Q.; Han, G.; Liu, Y.; Wang, Q. Copper-catalyzed aryltrifluoromethylation of N-phenylcinnamamides: access to trifluoromethylated 3,4-dihydroquinolin-2(1H)-ones. Adv. Synth. Catal., 2015, 357, 2464–2468. [Google Scholar] [CrossRef]
- Gao, F.; Yang, C.; Gao, G.-L.; Zheng, L.; Xia, W. Visble-light induced trifluoromethylation of N-arylcinnamamides for the synthesis of CF3-containing 3,4-disubstituted dihydroquinolinones and 1-azaspiro[4.5]decanes. Org. Lett., 2015, 14, 3478–3481. [Google Scholar] [CrossRef]
- Nguyen, J. D.; Tucker, J. W.; Konieczynska, M. D.; Stephenson, C. R. J. Intermolecular atom transfer radical addition to olefins mediated by oxidative quenching of photoredox catalysts. J. Am. Chem. Soc., 2011, 133, 4160–4163. [Google Scholar] [CrossRef]
- Wallentin, C. J.; Nguyen, J. D.; Finkbeiner, P.; Stephenson, C. R. J. Visible light-mediated atom transfer radical addition via oxidative and reductive quenching of photocatalysts. J. Am. Chem. Soc., 2012, 134, 8875−8884. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Zhang, H.; Xu, P.; Cheng, Y.; Zhu, C. Visible-light-induced radical tandem aryldifluoroacetylation of cinnamamides: access to difluoroacetylated quinolone-2-ones and 1-azaspiro[4.5]decanes. Adv. Synth. Catal. 2015, 357, 3057–3063. [Google Scholar] [CrossRef]
- Chan, C. W.; Lee, P. Y.; and Yu, W. Y. Copper-catalyzed cross-dehydrogenative coupling of N-arylacrylamides with chloroform using tert-butyl peroxybenzoate as oxidant. Tetrahedron Lett., 2015, 56, 2559–2563. [Google Scholar] [CrossRef]
- Li, X.; Xu, J.; Gao, Y.; Fang, H.; Tang, G.; Zhao, Y. Cascade arylalkylation of activated alkenes: synthesis of chloroand cyano-containing oxindole. J. Org. Chem., 2015, 80, 2621−2626. [Google Scholar] [CrossRef] [PubMed]
- Bergonzini, G.; Cassani, C.; Wallentin, C.-J. Acyl radicals from aromatic carboxylic acids by means of visible-light photoredox catalysis. Angew. Chem., Int. Ed., 2015, 54, 14066–14069. [Google Scholar] [CrossRef]
- Mai, W.-P.; Sun, G.-C.; Wang, J.-T.; Song, G.; Mao, P.; Yang, L.-R.; Yuan, J.-W.; Xiao, Y.-M; Qu, L.-B. Silver-catalyzed radical tandem cyclization: an approach to direct synthesis of 3-acyl-4-arylquinolin-2(1H)-ones. J. Org. Chem., 2014, 79, 8094−8102. [Google Scholar] [CrossRef]
- Yang, H.; Guo, L.-N.; Duan, X.-H. Silver-catalyzed tandem radical acylarylation of cinnamamides in aqueous media. RSC Adv., 2014, 4, 52986–52990. [Google Scholar] [CrossRef]
- Mai, W.-P.; Wang, J.-T.; Xiao, Y.-M.; Mao, P.; Lu, K. A novel direct synthesis of 3-acyl-4-aryldihydroquinolin-2(1H)-ones via metal-free radical tandem cyclization between N-arylcinnamamides and aldehydes. Tetrahedron, 2015, 71, 8041–8051. [Google Scholar] [CrossRef]
- Walton, J. C. Functionalised oximes: emergent precursors for carbon-, nitrogen- and oxygen-centred radicals. Molecules, 2016, 21, 63−86. [Google Scholar] [CrossRef]
- Fan, X.; Lei, T.; Chen, B.; Tung, C.-H; Wu, L.-Z. Photocatalytic C−C bond activation of oxime ester for acyl radical generation and application. Org. Lett., 2019, 21, 4153−4158. [Google Scholar] [CrossRef]
- McManus, H. A.; Guiry, P. J. Recent developments in the application of oxazoline-containing ligands in asymmetric catalysis. Chem. Rev., 2004, 104, 4151–4202. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, T.; Réau, R. Organophosphorus π-conjugated materials. Chem. Rev., 2006, 106, 4681–4727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gu, Z.; Li, Z.; Pan, C.; Li, W.; Hu, H.; Zhu, C. Silver-catalyzed cascade radical cyclization: a direct approach to 3,4-disubstituted dihydroquinolin-2(1H)-ones through activation of the P−H bond and functionalization of the C(sp2 )−H bond. J. Org. Chem., 2016, 81, 2122−2127. [Google Scholar]
- Wang, F.-X.; Tian, S.-K. Cyclization of N-arylacrylamides via radical arylsulfenylation of carbon−carbon double bonds with sulfonyl hydrazides. J. Org. Chem., 2015, 80, 12697−12703. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Rakesh, K. P.; Ravidar, L.; Fang, W.-Y.; Qin, H.-L. Pharmaceutical and medicinal significance of sulfur (SVI)-containing motifs for drug discovery: a critical review. Eur. J. Med. Chem., 2019, 162, 679−734. [Google Scholar] [CrossRef]
- Han, B.; Ding, X.; Zhang, Y.; Gu, X.; Qi, Y.; Liang, S. Mn (OAc)3-promoted sulfonation-cyclization cascade via the SO3– radical: the synthesis of heterocyclic sulfonates. Org. Lett. 2022, 24, 8255–8260. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Hasimujiang, B.; Xu, Z.; Cai, H.; Chen, G.; Mo, G.; Ruan, Z. Direct electrochemical selenylation/cyclization of alkenes: access to functionalized benzheterocycles. J. Org. Chem., 2021, 86, 16045−16058. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Y.; Zhong, Y.-F.; Mo, Z.-Y.; Wu, S.-H.; Xu, Y.-L.; Tang, H.-T.; Pan, Y.-M. Synthesis of seleno oxindoles via electrochemical cyclization of N-arylacrylamides with diorganyl diselenides. Adv. Synth. Catal. 2021, 363, 208–214. [Google Scholar] [CrossRef]
- Chapman, O. L.; Adams, W. R. Photochemical transformations. XXII. Photoisomerization of substituted acrylic acids and acrylamides to beta-lactones and beta-lactams. J. Am. Chem. Soc. 1968, 90, 2333–2342. [Google Scholar] [CrossRef]
- Liu, Z.; Zhong, S.; Ji, X.; Deng, G.-J.; Huang, H. Hydroarylation of activated alkenes enabled by proton-coupled electron transfer. ACS Catal., 2021, 11, 4422–4429. [Google Scholar] [CrossRef]
- Li, J.-L.; Yang, X.-L.; Shen, S.; Niu, X. Synthesis of 10-phenanthrenols via photosensitized triplet energy transfer, photoinduced electron transfer, and cobalt catalysis. J. Org. Chem., 2022, 87, 16458−16472. [Google Scholar] [CrossRef]
- Cheng, H.; Lam, T.-L.; Liu, Y.; Tang, Z.; Che, C.-M. Photoinduced hydroarylation and cyclization of alkenes with luminescent platinum(II) complexes. Angew. Chem., Int. Ed., 2021, 60, 1383−1389. [Google Scholar]
- Chen, X.-W.; Liu, Y.; Jin, X.-Q.; Sun, Y.-Y.; Gu, S.-L.; Fu, L.; Li, J.-Q. Development and kilogram-scale synthesis of a D2/5-HT2A receptor dual antagonist (±)-SIPI 6360. Org. Process Res. Dev. 2016, 20, 1662–1667. [Google Scholar] [CrossRef]
- Oddy, M. J.; Kusza, D. A.; Petersen, W. F. Visible-light mediated metal-free 6π-photocyclization of N-acrylamides: thioxanthone triplet energy transfer enables the synthesis of 3,4-dihydroquinolin-2-ones. Org. Lett., 2021, 23, 8963−8967. [Google Scholar] [CrossRef] [PubMed]
- Popescu, M. V.; Mekereeya, A.; Alegre-Requena, J. V.; Paton, R. S.; Smith, M. D. Visible-light-mediated heterocycle functionalization via geometrically interrupted [2 + 2] cycloaddition. Angew. Chem., Int. Ed. 2020, 59, 23020–23024. [Google Scholar] [CrossRef] [PubMed]
- Münster, N.; Parker, N. A.; van Dijk, L.; Paton, R. S.; Smith, M. D. Visible light photocatalysis of 6π heterocyclization. Angew. Chem., Int. Ed., 2017, 56, 9468−9472. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhong, S.; Ji, X. , Deng, G.-J.; Huang, H. Photoredox cyclization of N-arylacrylamides for synthesis of dihydroquinolinones. Org. Lett., 2022, 24, 349−353. [Google Scholar]
- Raghunathan, R.; Jockusch, S.; Sibi, M. P.; Sivaguru, J. Evaluating thiourea/urea catalyst for enantioselective 6π-photocyclization of acrylanilides. J. Photochem. Photobiol. A: Chem., 2016, 331, 84−88. [Google Scholar] [CrossRef]
- Kumarasamy, E.; Ayitou, A. J. L.; Vallavoju, N.; Raghunathan, R.; Iyer, A.; Clay, A.; Kandappa, S. K.; Sivaguru, J. Tale of twisted molecules. Atropselective photoreactions: taming light induced asymmetric transformations through non-biaryl atropisomers. Acc. Chem. Res., 2016, 49, 2713−2724. [Google Scholar] [CrossRef]
- Ayitou, A. J. L.; Sivaguru, J. Light-induced transfer of molecular chirality in solution: enantiospecific photocyclization of molecularly chiral acrylanilides. J. Am. Chem. Soc., 2009, 131, 5036–5037. [Google Scholar] [CrossRef]
- Iyer, A.; Ugrinov, A.; Sivaguru, J. Understanding conformational preferences of atropisomeric hydrazides and its influence on excited state transformations in crystalline media. Molecules, 2019, 24, 3001−3010. [Google Scholar] [CrossRef] [PubMed]
- Jones, B. A.; Solon, P.; Popescu, M.V.; Du, J.-Y.; Paton, R.; Smith, M. D. Catalytic enantioselective 6π photocyclization of Aacrylanilides. J. Am. Chem. Soc., 2023, 145, 171−178. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Examples of medicinally active compounds containing 3,4-dihydro-2(1H)-quinolinones.
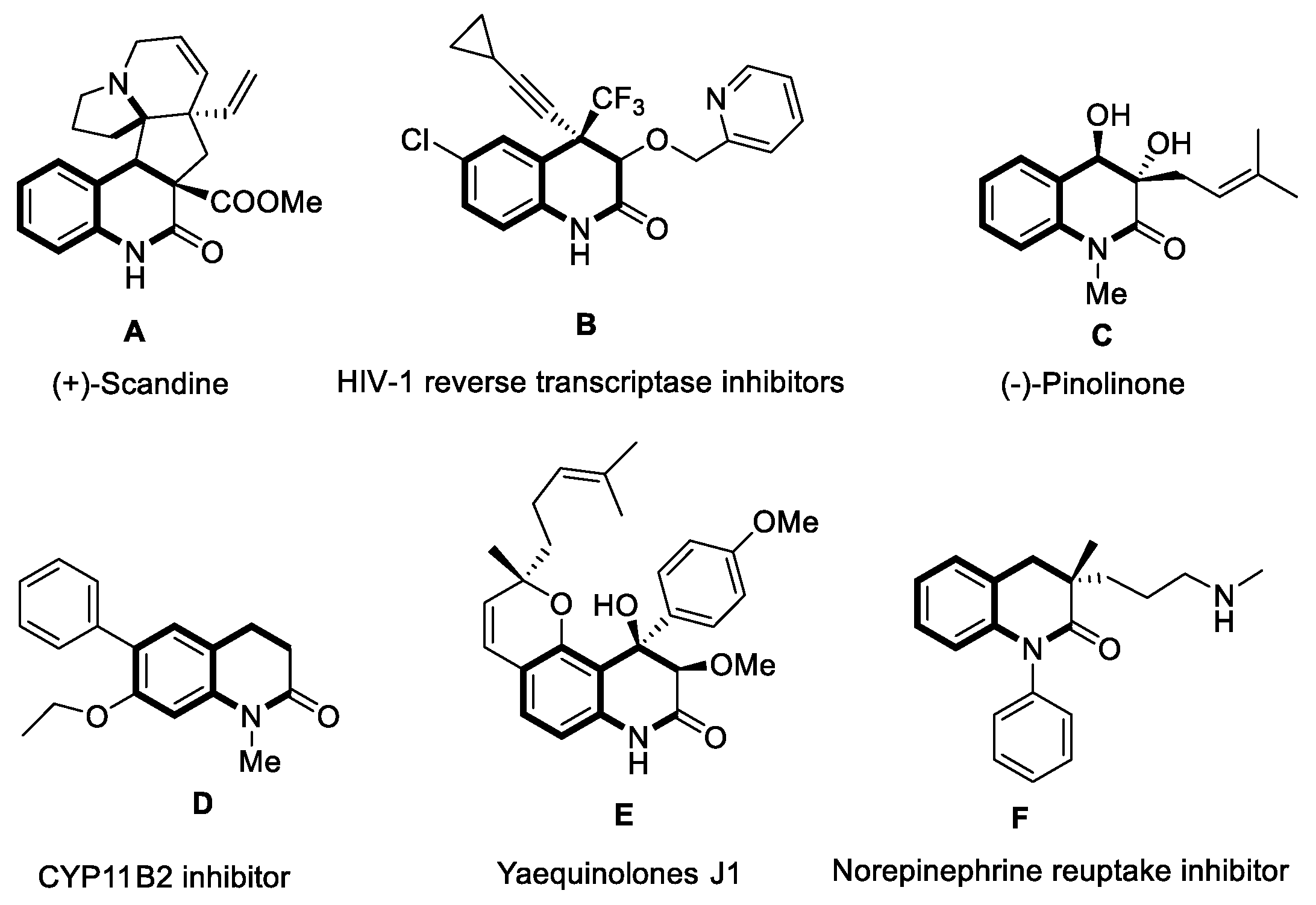
Figure 2.
Trifluoroacetic acid-mediated hydroarylation for synthesis of 3,4-dihydroquinolin-2 (1H)-ones.
Figure 2.
Trifluoroacetic acid-mediated hydroarylation for synthesis of 3,4-dihydroquinolin-2 (1H)-ones.
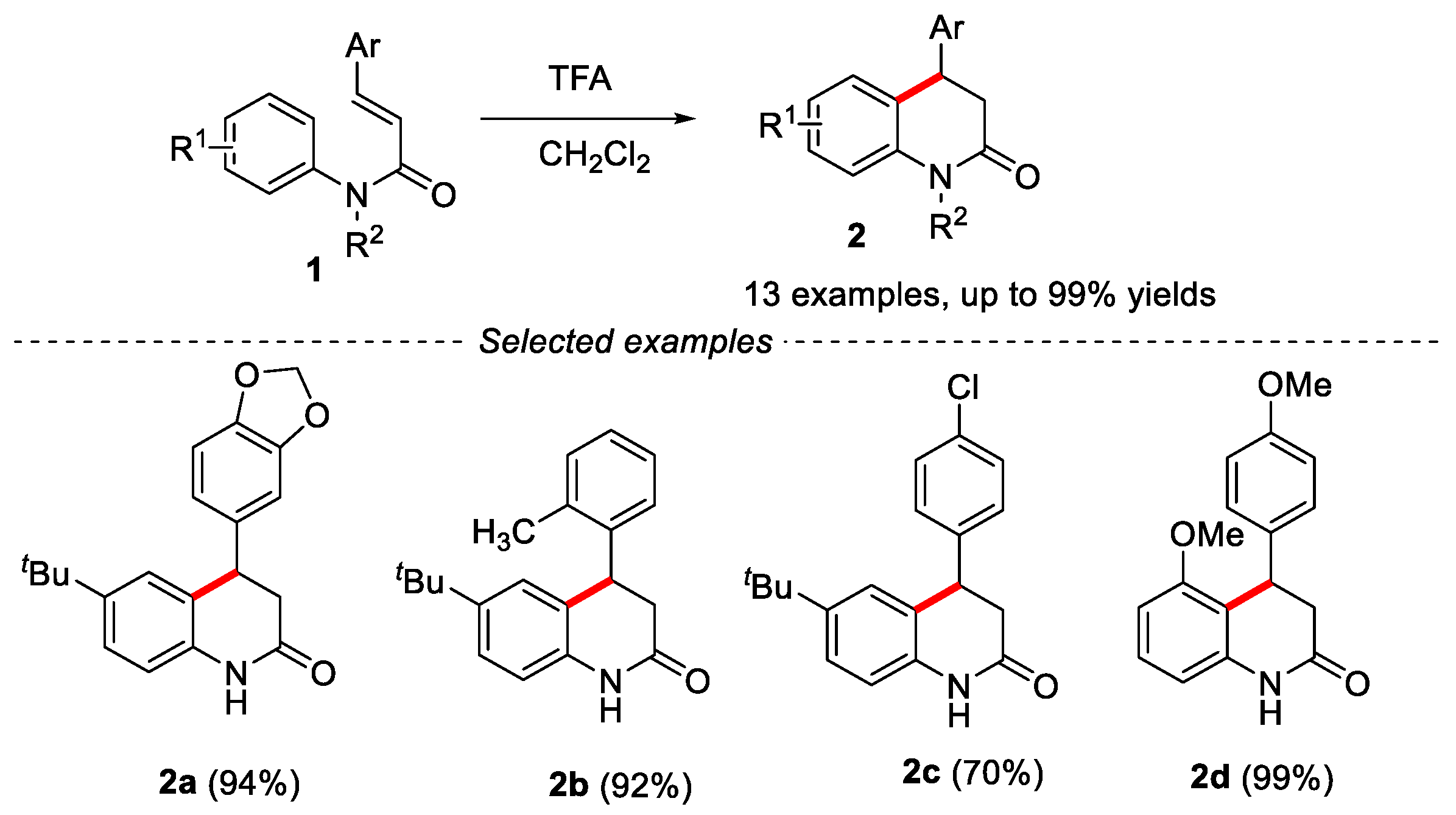
Figure 3.
Synthesis of cis-4-aryl-3-arylthio-3,4-dihydroquinolin-2(1H)-ones via electrophilic sulfenylation and cyclization of N-arylcinnamamides with N-arylthiosuccinimides.
Figure 3.
Synthesis of cis-4-aryl-3-arylthio-3,4-dihydroquinolin-2(1H)-ones via electrophilic sulfenylation and cyclization of N-arylcinnamamides with N-arylthiosuccinimides.
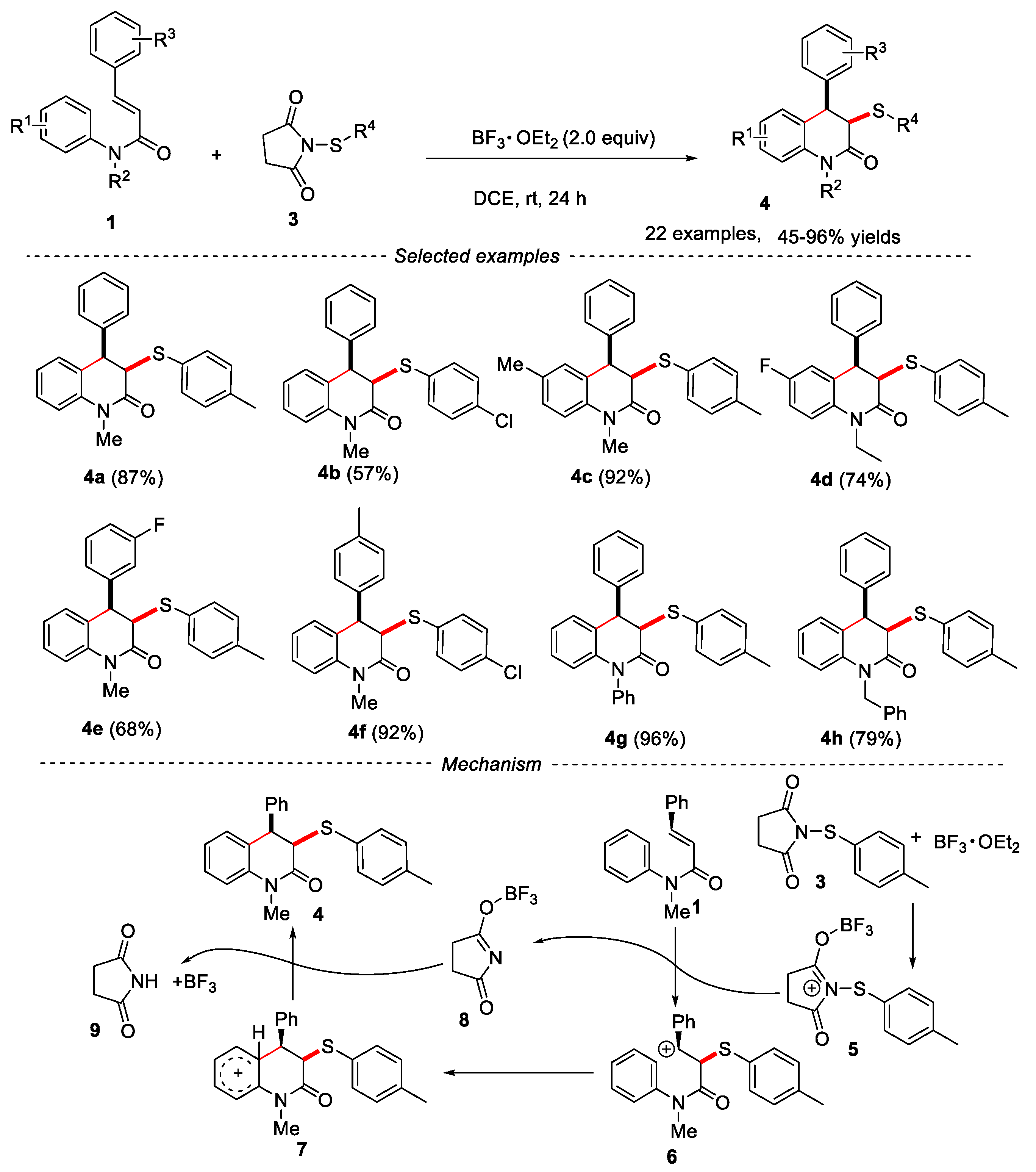
Figure 4.
The oxone-mediated direct arylhydroxylation of N-arylcinnamamides for the synthesis of hydroxyl-containing 3,4-dihydroquinolin-2-ones.
Figure 4.
The oxone-mediated direct arylhydroxylation of N-arylcinnamamides for the synthesis of hydroxyl-containing 3,4-dihydroquinolin-2-ones.
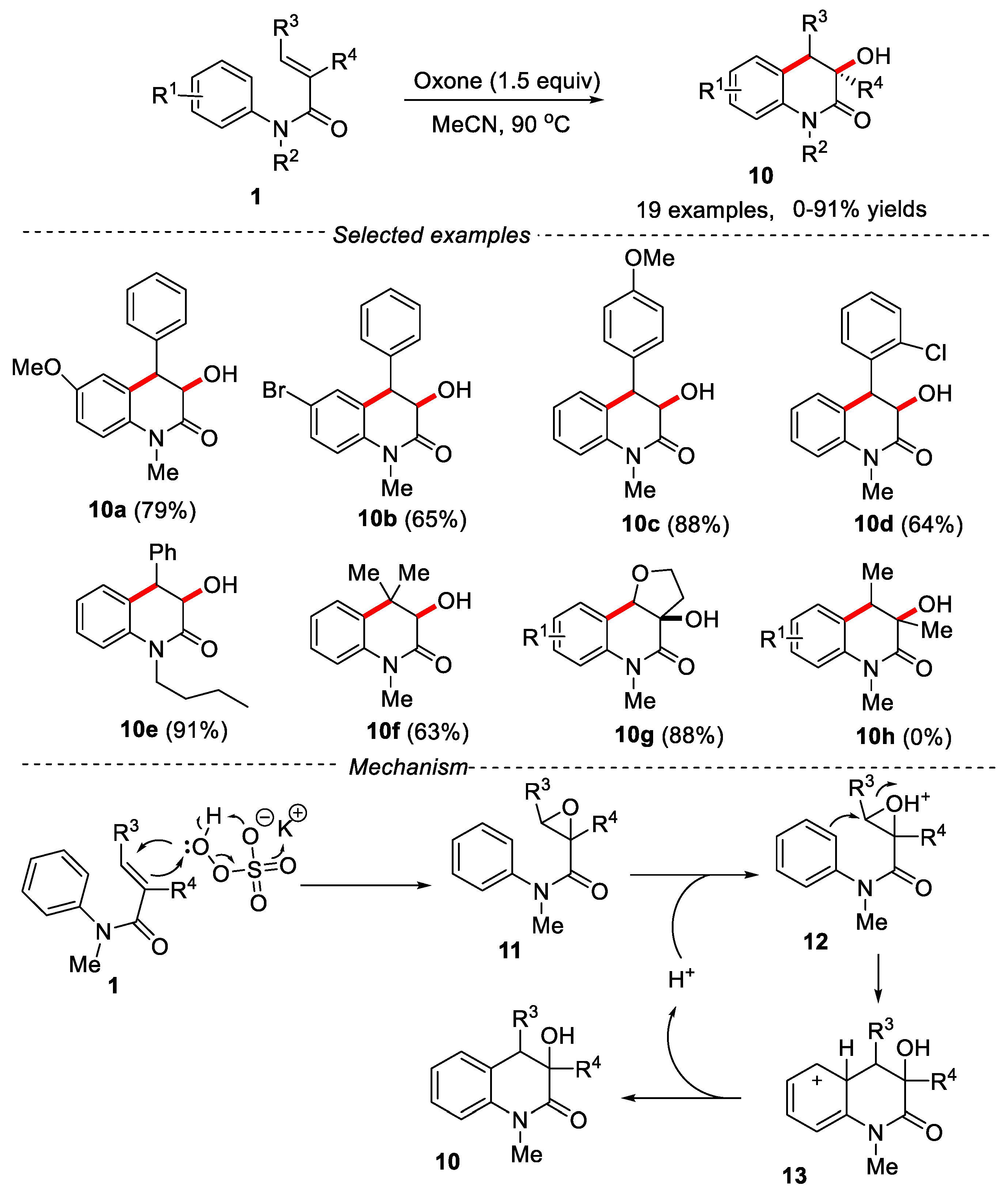
Figure 5.
Synthesis of 3,4-substituted dihydroquinolin-2(1H)-ones by decarboxylation of N-arycinamamides with aliphatic carboxylic acids.
Figure 5.
Synthesis of 3,4-substituted dihydroquinolin-2(1H)-ones by decarboxylation of N-arycinamamides with aliphatic carboxylic acids.
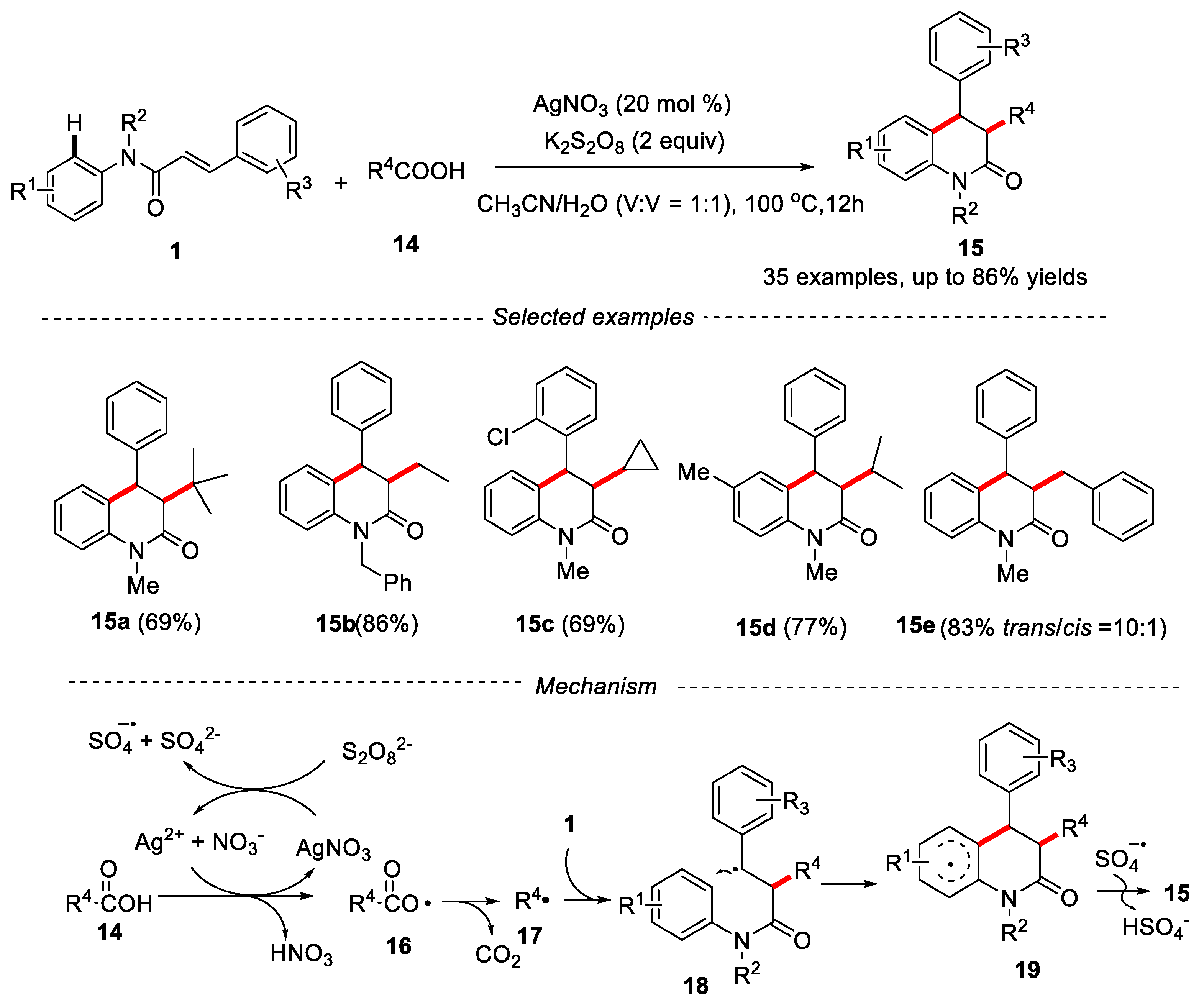
Figure 6.
FeCl2-catalyzed decarboxylative radical alkylation/cyclization of N-phenylcinnamamide for the preparation of dihydroquinolin-2(1H)-ones.
Figure 6.
FeCl2-catalyzed decarboxylative radical alkylation/cyclization of N-phenylcinnamamide for the preparation of dihydroquinolin-2(1H)-ones.
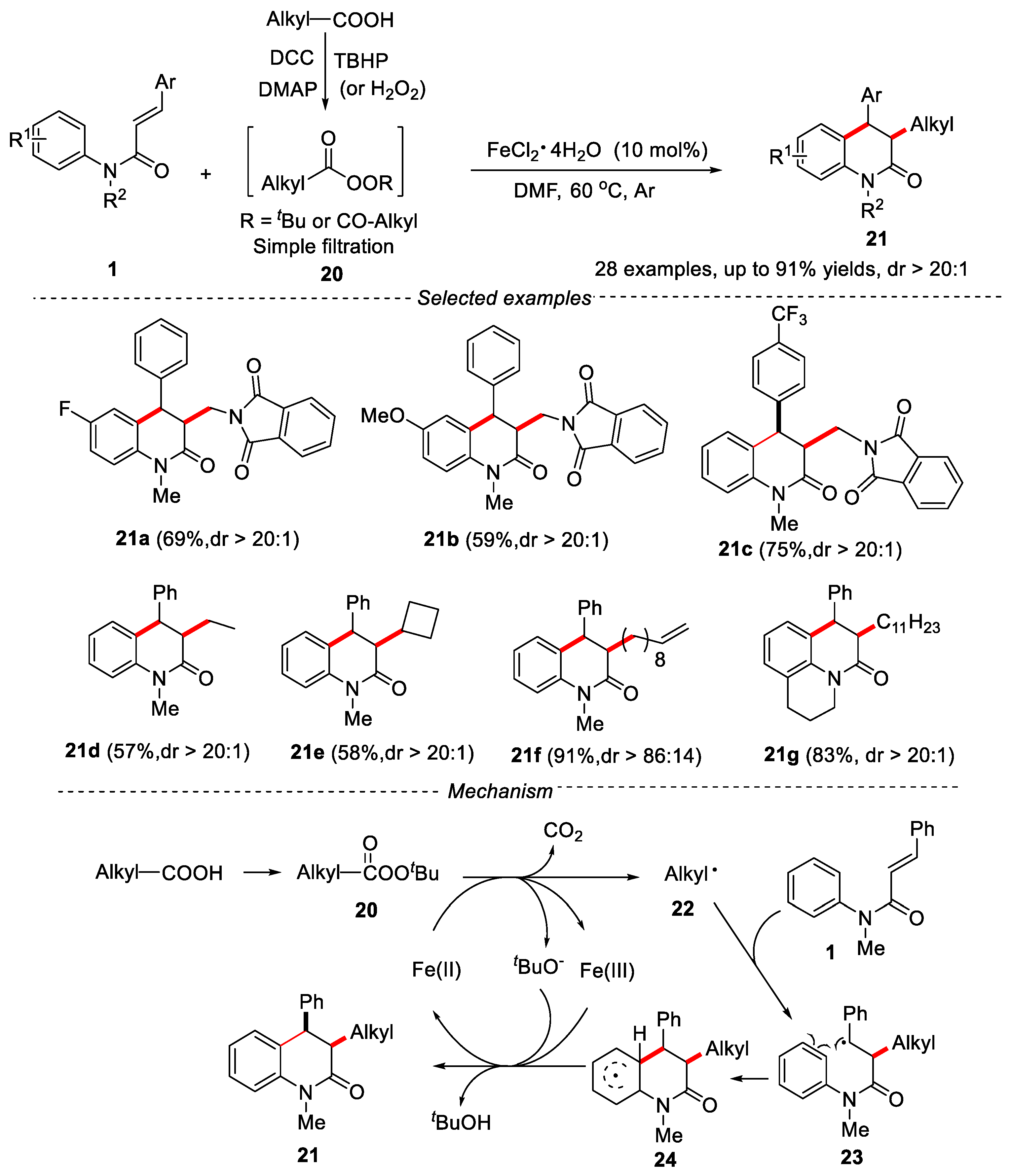
Figure 7.
Pd-catalyzed alkylarylation of acrylamides with unactivated alkyl halides for synthesis dihydroquinolinone 3,4-dihydroquinolin-2(1H)-ones.
Figure 7.
Pd-catalyzed alkylarylation of acrylamides with unactivated alkyl halides for synthesis dihydroquinolinone 3,4-dihydroquinolin-2(1H)-ones.
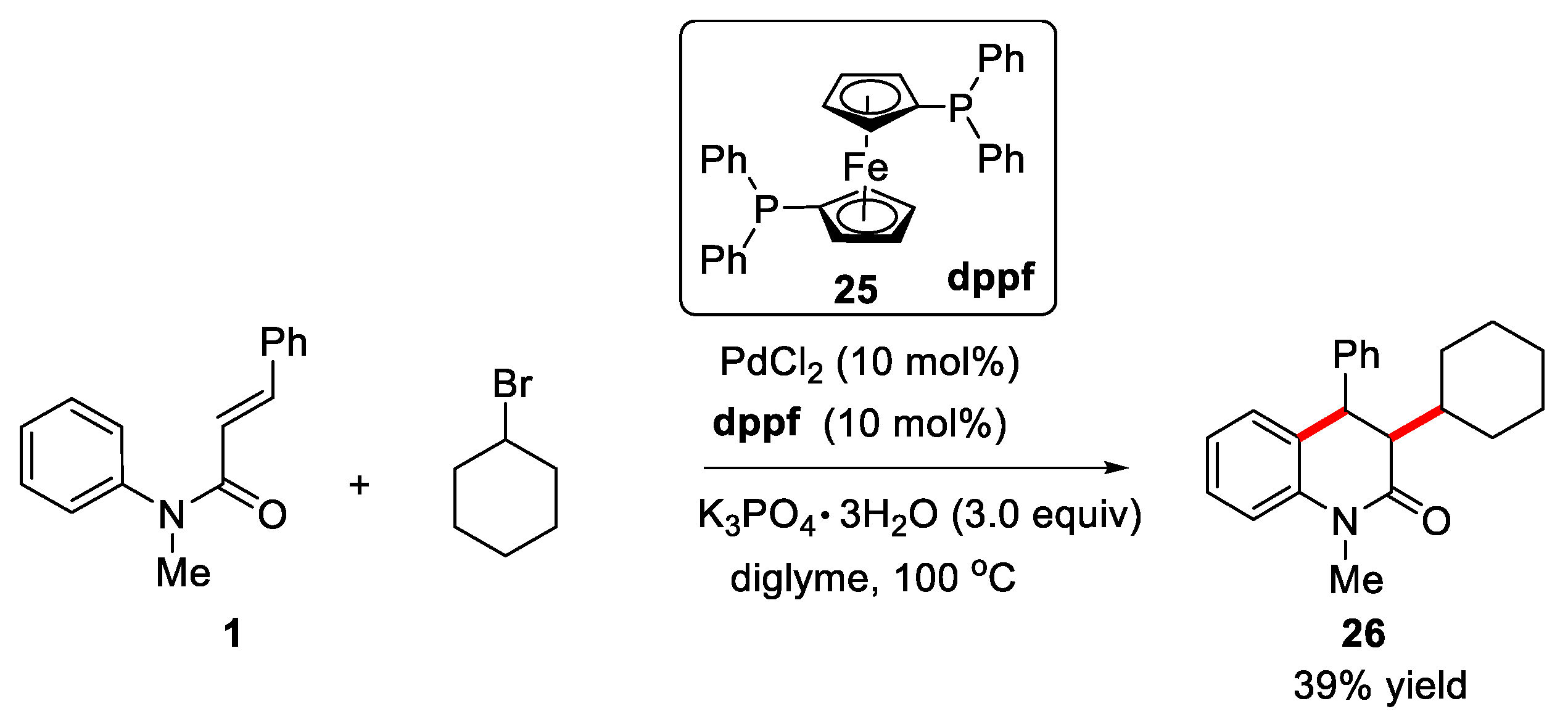
Figure 8.
Visible-light-induced Pd-catalyzed intermolecular radical cascade reaction of N-phenylcinnamamide with cyclohexyl bromide for the preparation of 3,4-dihydroquinolin-2(1H)-ones.
Figure 8.
Visible-light-induced Pd-catalyzed intermolecular radical cascade reaction of N-phenylcinnamamide with cyclohexyl bromide for the preparation of 3,4-dihydroquinolin-2(1H)-ones.
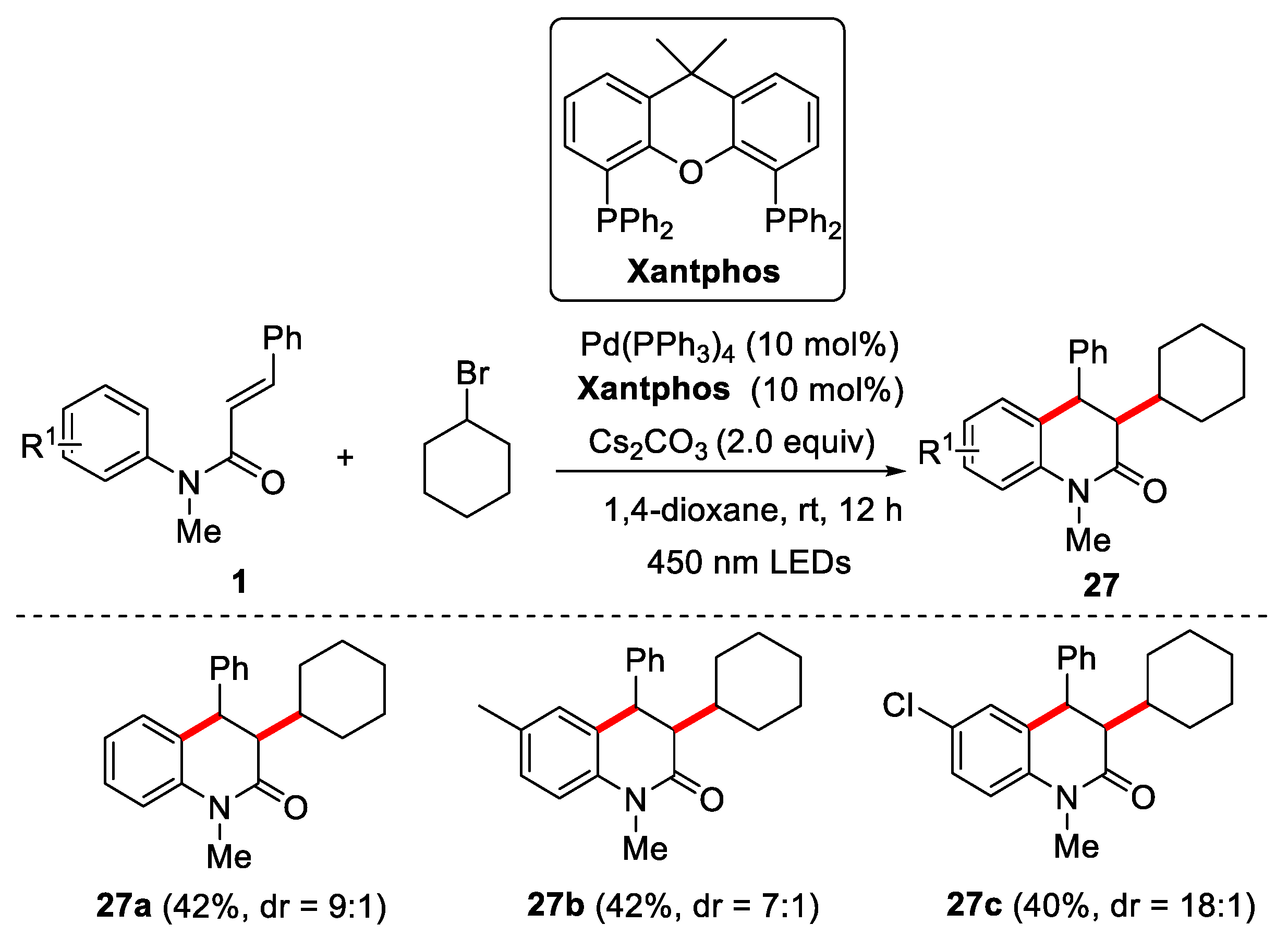
Figure 9.
The mechanism of the Pd-catalyzed intermolecular radical cascade reaction of N-phenylcinnamamide with cyclohexyl bromide.
Figure 9.
The mechanism of the Pd-catalyzed intermolecular radical cascade reaction of N-phenylcinnamamide with cyclohexyl bromide.
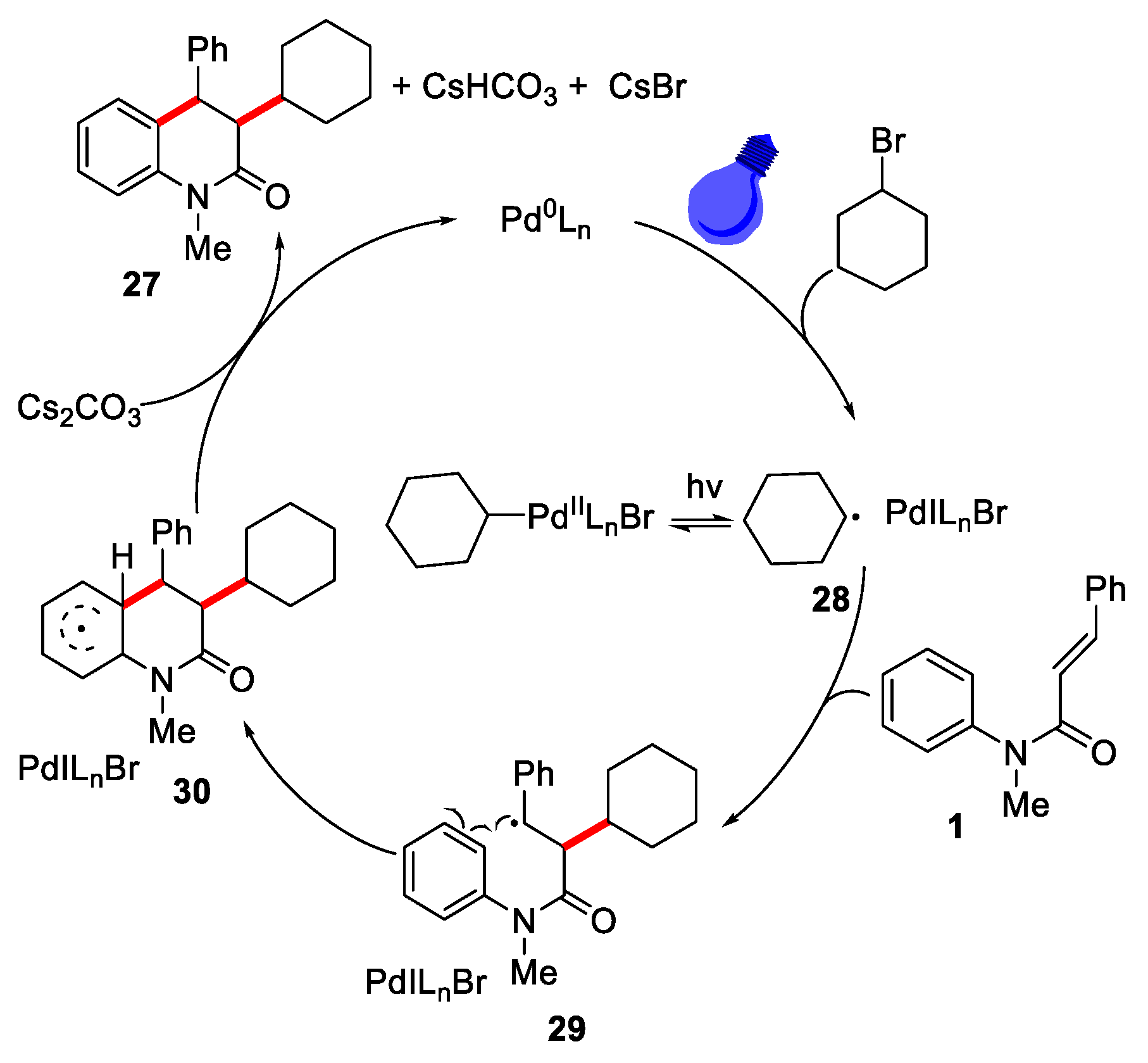
Figure 10.
Rhenium-catalyzed alkylarylation of cinnamamides with PhI(O2CR)2 via decarboxylation for the synthesis of 3,3-disubstitued dihydroquinolin-2(1H)-ones.
Figure 10.
Rhenium-catalyzed alkylarylation of cinnamamides with PhI(O2CR)2 via decarboxylation for the synthesis of 3,3-disubstitued dihydroquinolin-2(1H)-ones.
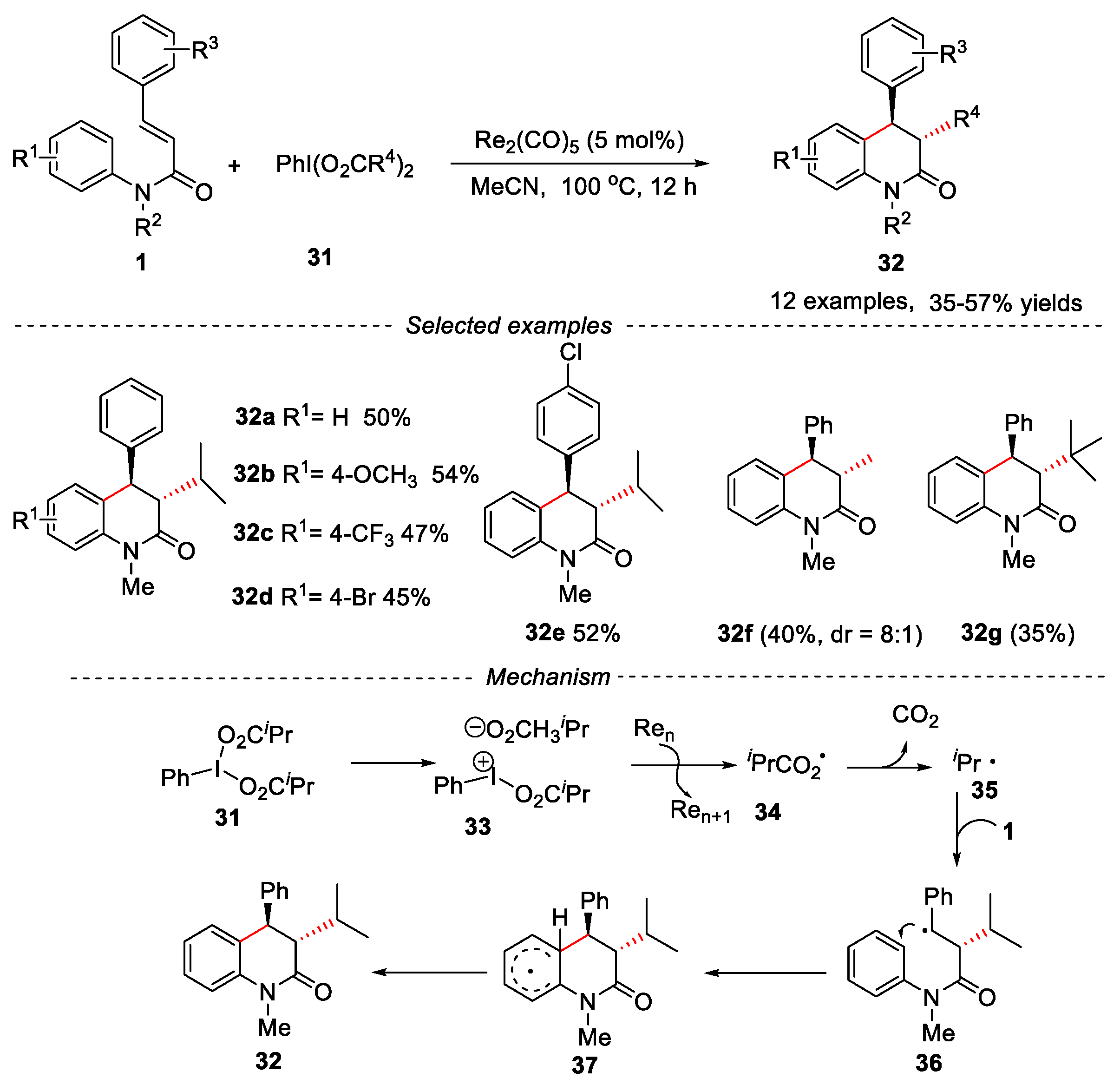
Figure 11.
Fe-catalyzed decarbonylative cascade reaction of N-arylcinnamamides with aliphatic aldehydes for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
Figure 11.
Fe-catalyzed decarbonylative cascade reaction of N-arylcinnamamides with aliphatic aldehydes for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
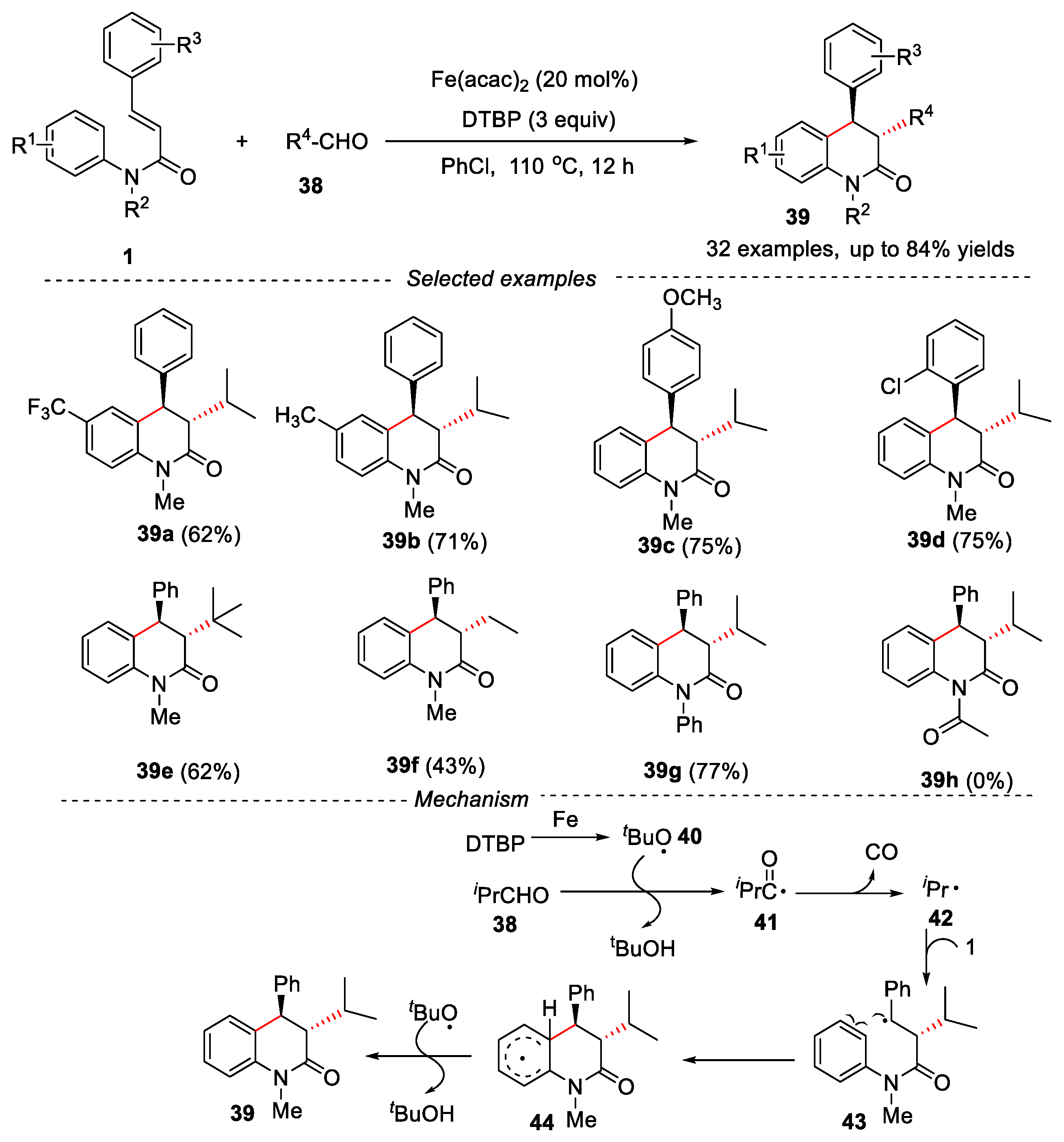
Figure 12.
A metal-free catalyzed method for the synthesis alkylsubstituted 3,4-dihydroquinolin-2(1H)-one derivatives by reactionof N-arylcinnamamides with aliphatic aldehydes in PhF.
Figure 12.
A metal-free catalyzed method for the synthesis alkylsubstituted 3,4-dihydroquinolin-2(1H)-one derivatives by reactionof N-arylcinnamamides with aliphatic aldehydes in PhF.
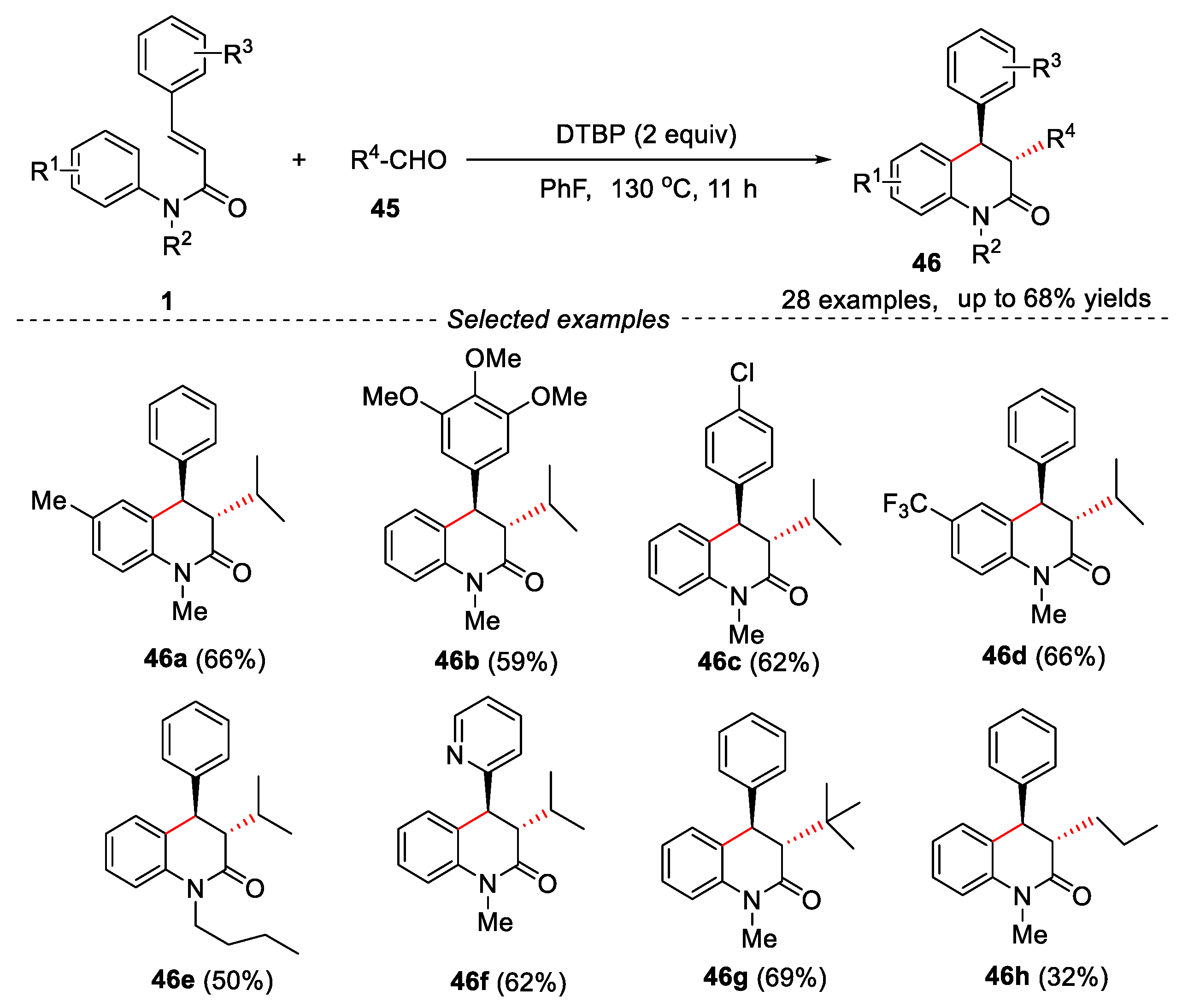
Figure 13.
Cu-catalyzed cyanoalkylarylation of N-arylcinnamamides with cyclobutanone oxime esters for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
Figure 13.
Cu-catalyzed cyanoalkylarylation of N-arylcinnamamides with cyclobutanone oxime esters for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
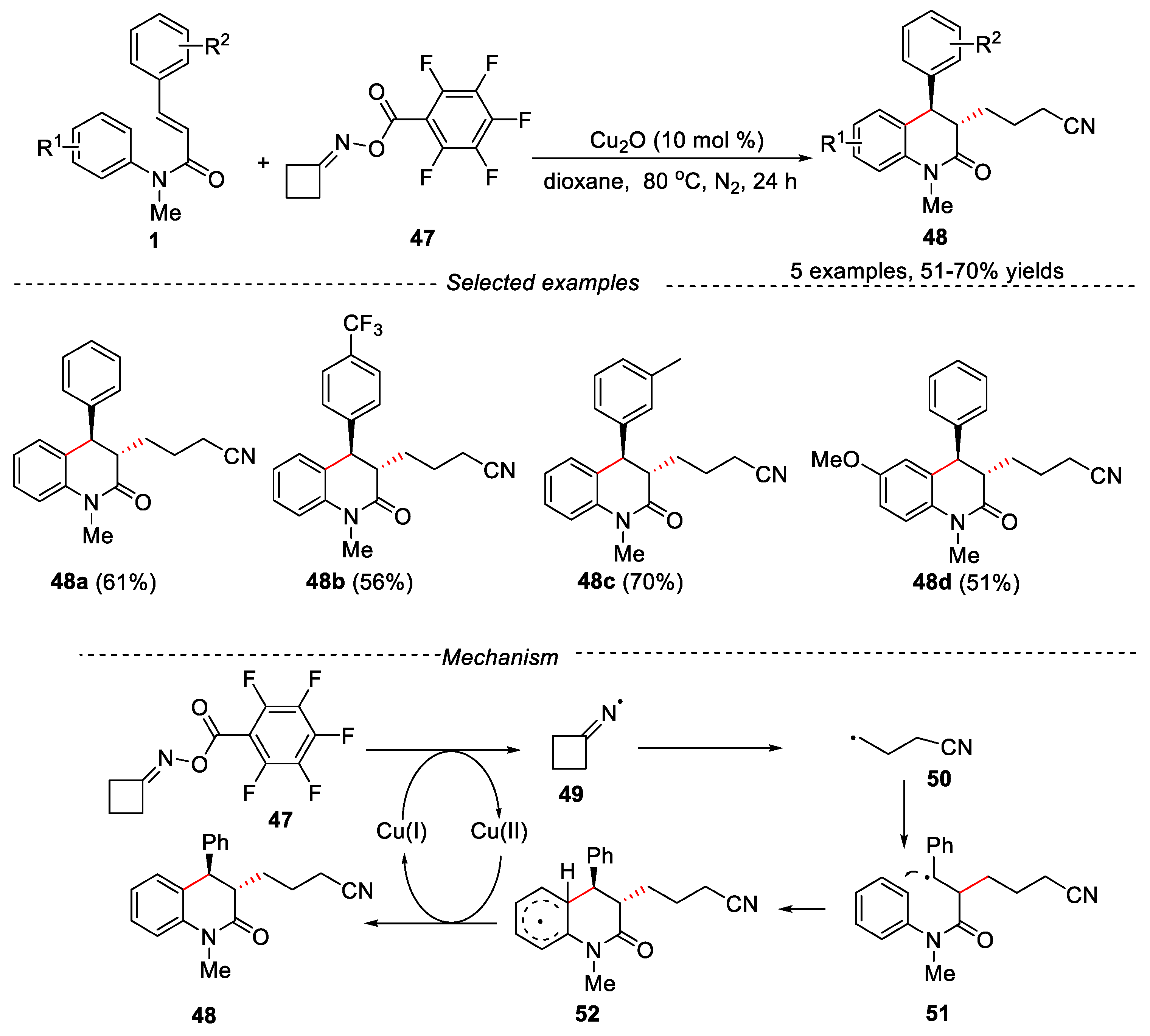
Figure 14.
Synthesis of alkylsubstituted 3,4-dihydroquinolin-2(1H)-one derivatives using cyclohexyl boronic acid as cyclohexyl radical precursor and dioxygen as oxidant.
Figure 14.
Synthesis of alkylsubstituted 3,4-dihydroquinolin-2(1H)-one derivatives using cyclohexyl boronic acid as cyclohexyl radical precursor and dioxygen as oxidant.
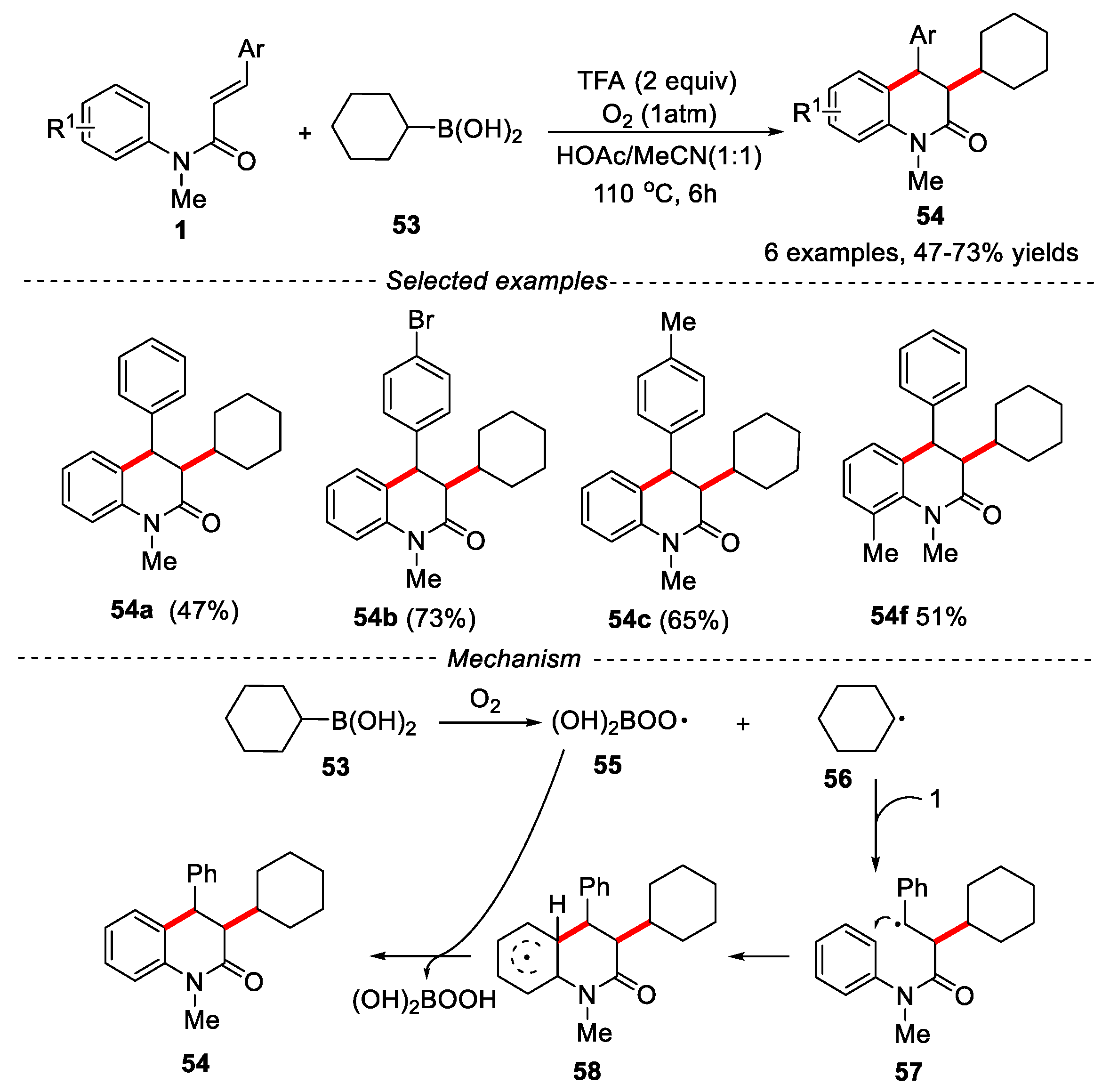
Figure 15.
Tandem cyclization of N-arylcinnamamide with pentane-2,4-diones for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
Figure 15.
Tandem cyclization of N-arylcinnamamide with pentane-2,4-diones for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
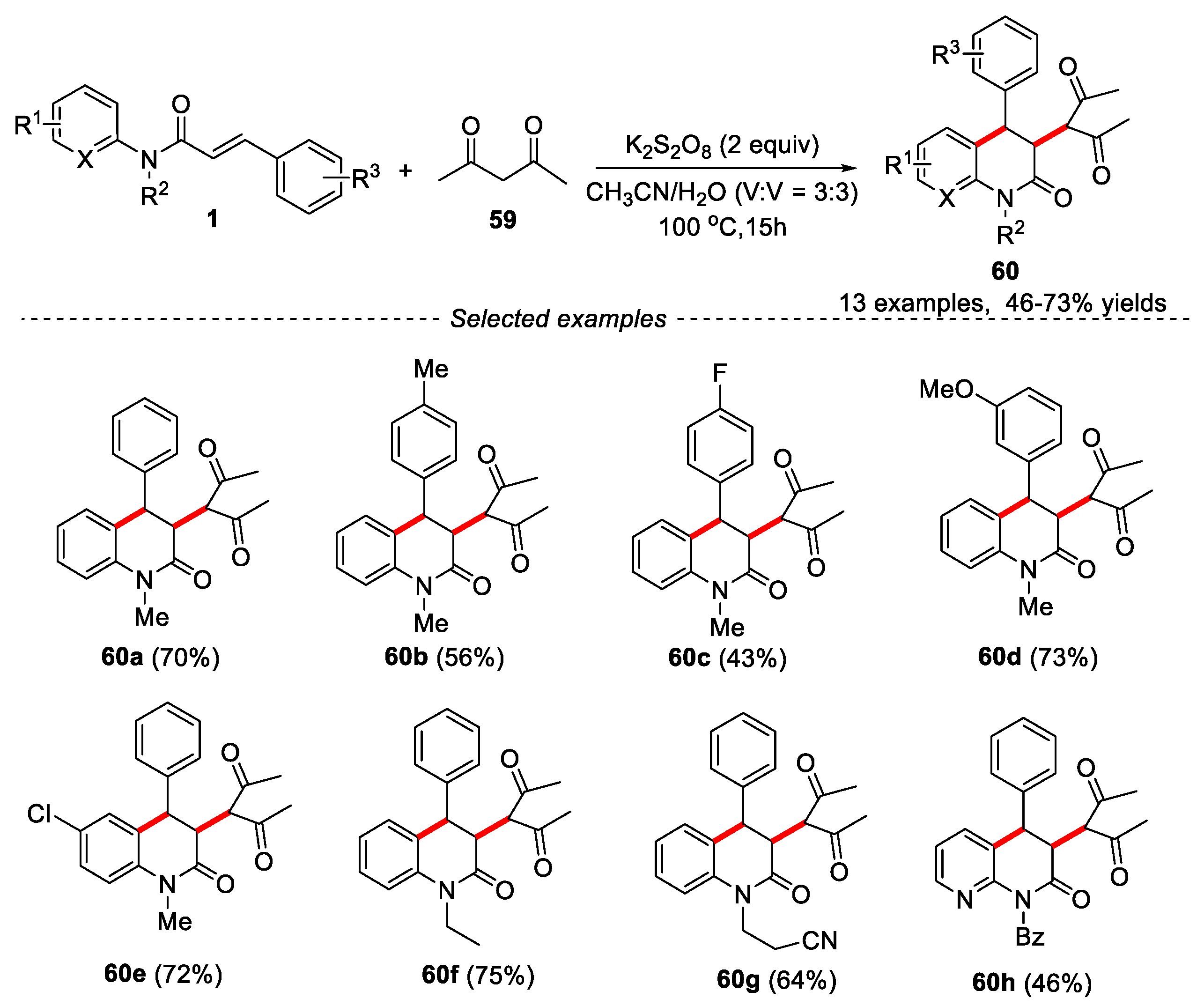
Figure 16.
Copper-catalyzed tandem method for the synthesis dihydroquinolin-2(1H)-ones by cascade radical addition/cyclization of N-arylcinnamamides with benzyl hydrocarbons.
Figure 16.
Copper-catalyzed tandem method for the synthesis dihydroquinolin-2(1H)-ones by cascade radical addition/cyclization of N-arylcinnamamides with benzyl hydrocarbons.
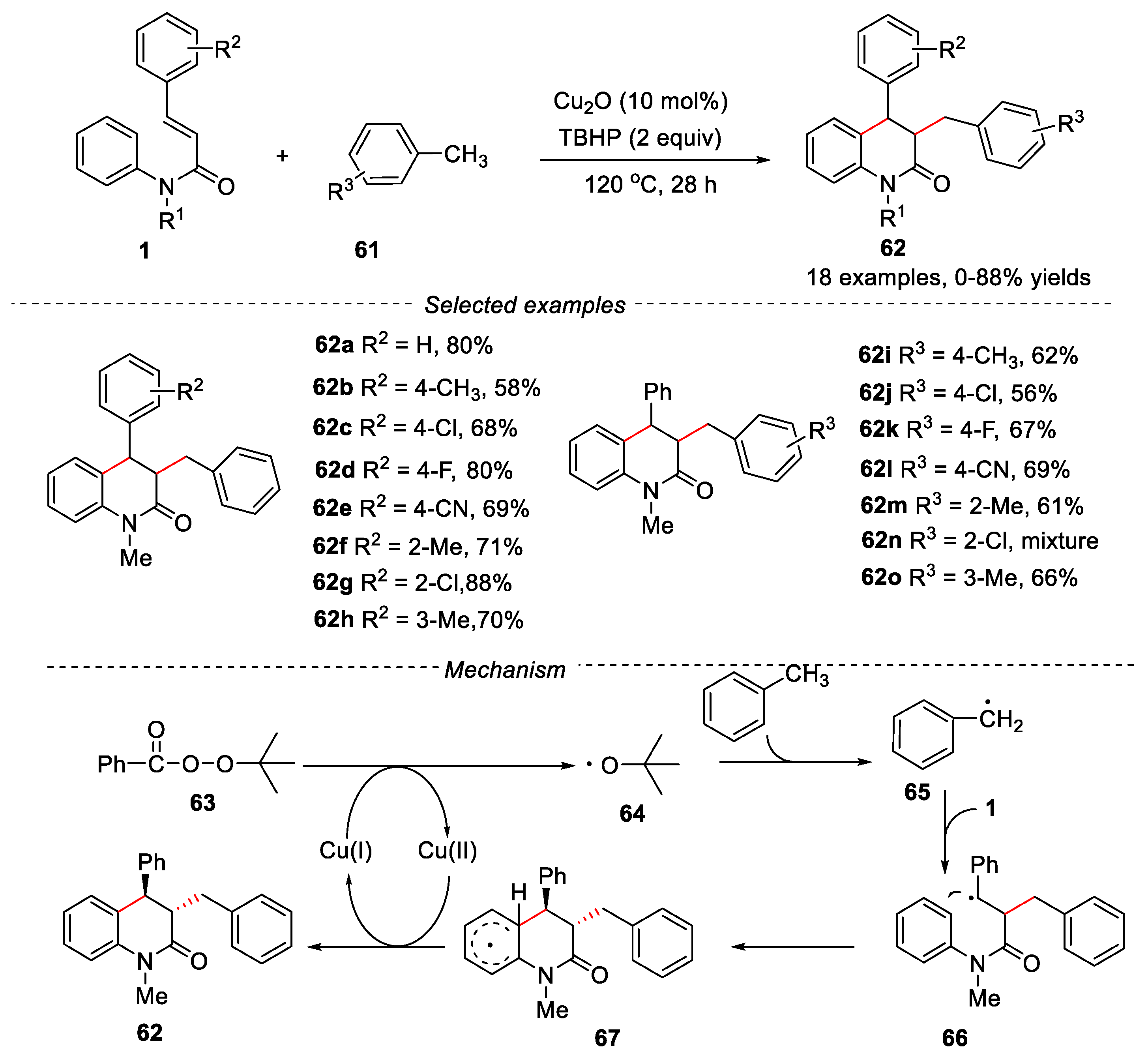
Figure 17.
Copper-catalyzed tandem method for the synthesis of dihydroquinolin-2(1H)-ones by cascade radical addition/cyclization of N-arylcinnamamides with ether, alcohols and alkanes.
Figure 17.
Copper-catalyzed tandem method for the synthesis of dihydroquinolin-2(1H)-ones by cascade radical addition/cyclization of N-arylcinnamamides with ether, alcohols and alkanes.
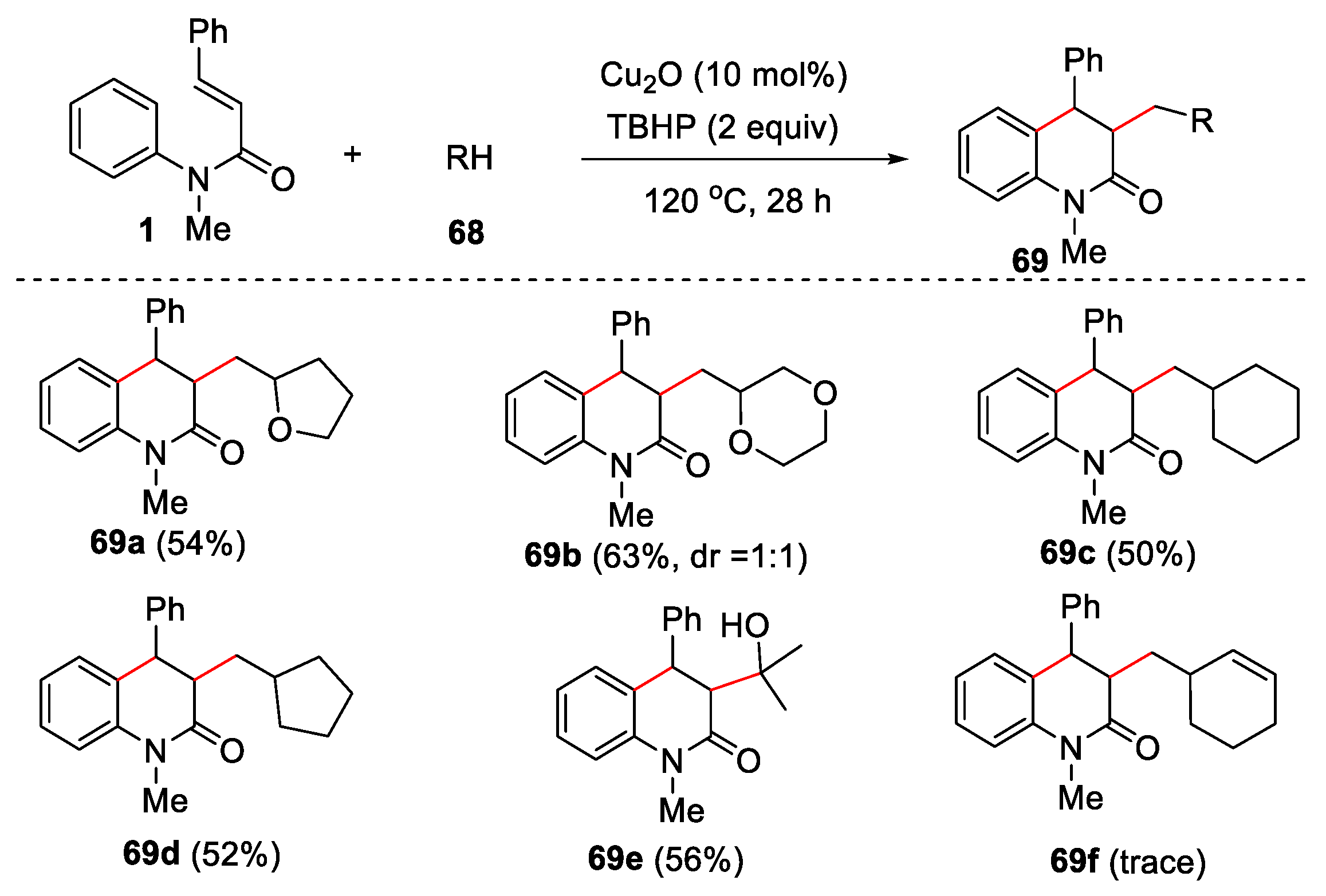
Figure 18.
Photo-induced C(sp3)-H functionalization for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
Figure 18.
Photo-induced C(sp3)-H functionalization for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
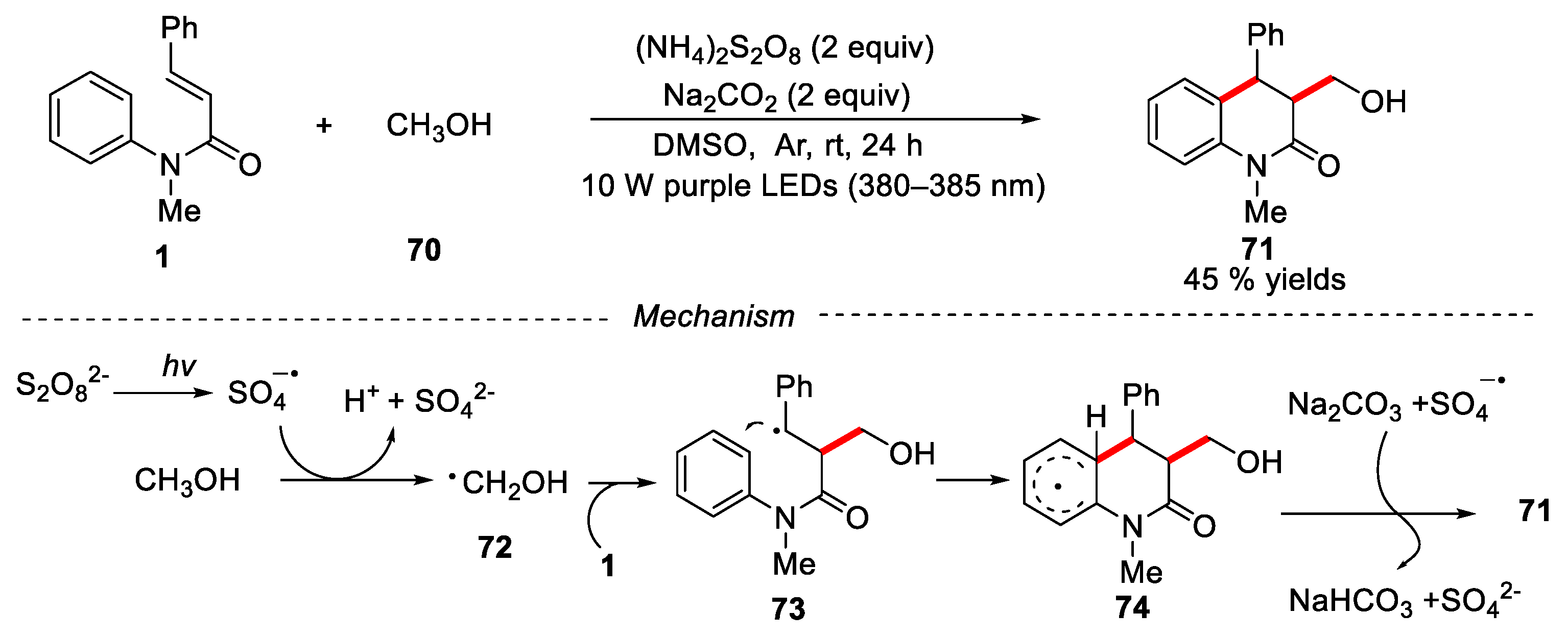
Figure 19.
Palladium-catalyzed oxidative arylalkylation of alkenes to construct cyano substituted oxindoles and 3,4-dihydroquinolin-2(1H)-one.
Figure 19.
Palladium-catalyzed oxidative arylalkylation of alkenes to construct cyano substituted oxindoles and 3,4-dihydroquinolin-2(1H)-one.
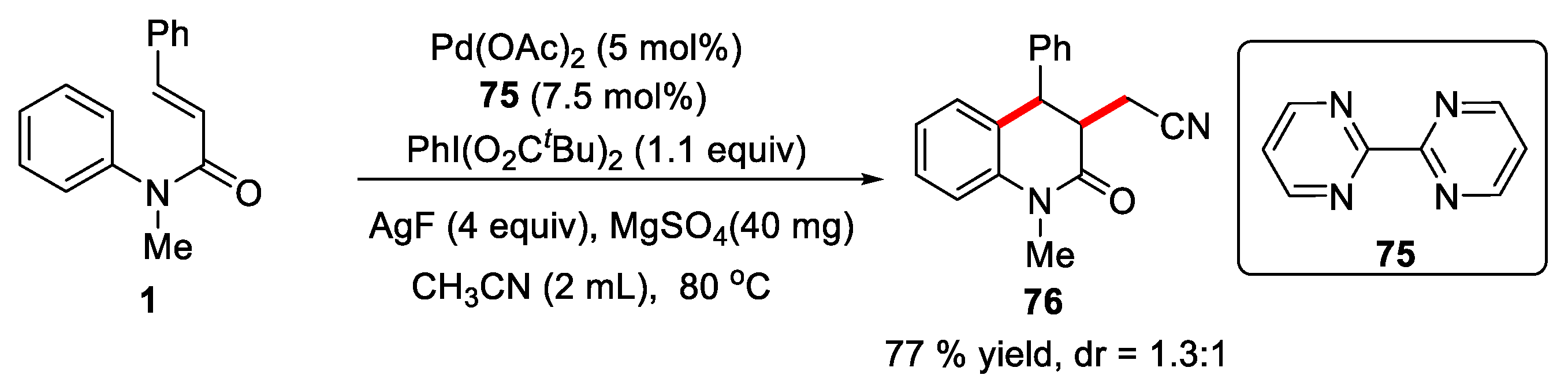
Figure 20.
Silver-induced tandem radical addition/cyclization for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
Figure 20.
Silver-induced tandem radical addition/cyclization for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
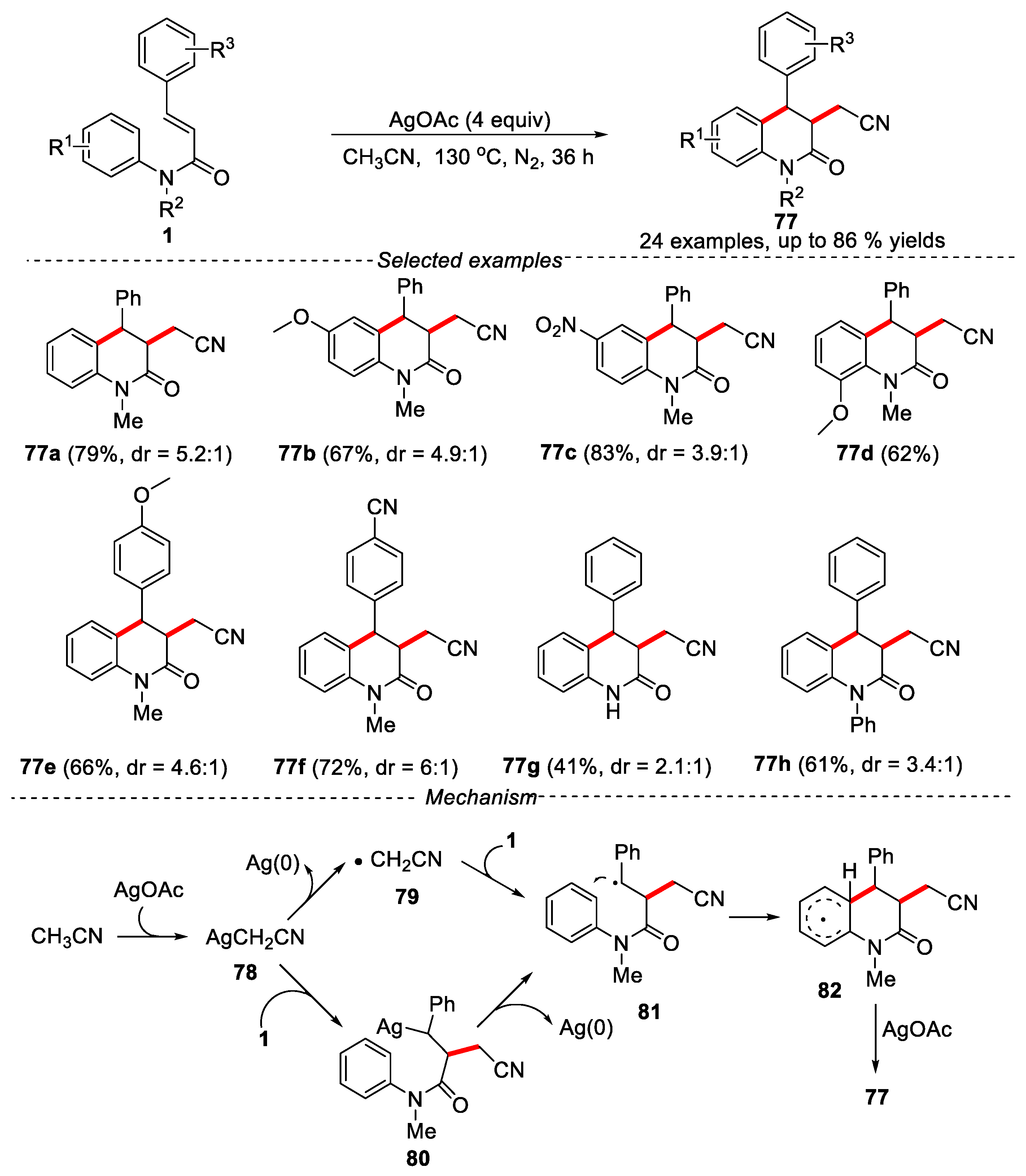
Figure 21.
Nickel-Catalyzed tandem radical addition/cyclization for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
Figure 21.
Nickel-Catalyzed tandem radical addition/cyclization for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
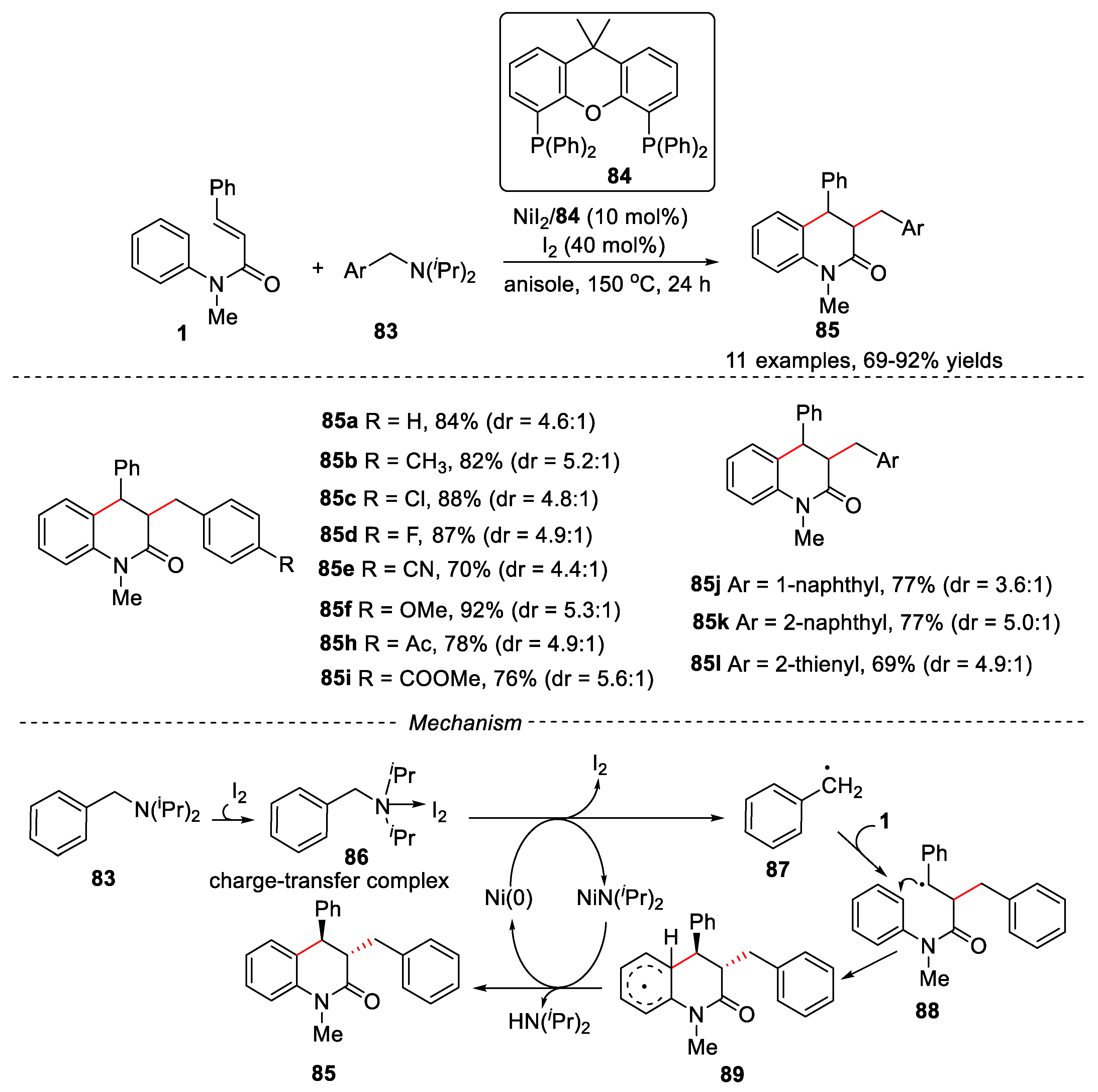
Figure 23.
A white LED light induced reaction of N-arylacrylamides with HCF2SO2Na using the Eosin B as photocatalyst.
Figure 23.
A white LED light induced reaction of N-arylacrylamides with HCF2SO2Na using the Eosin B as photocatalyst.
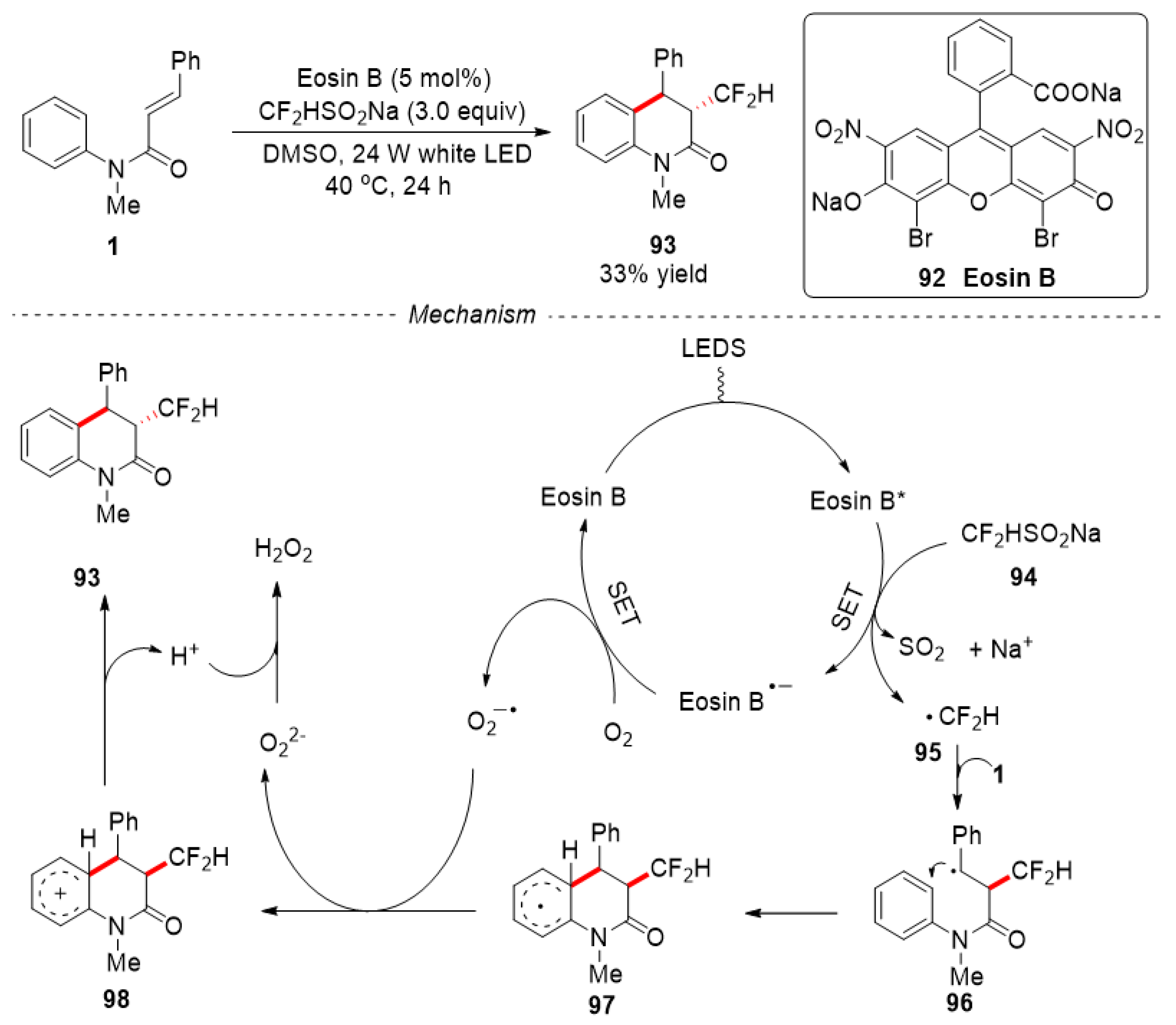
Figure 24.
Di- and trifluoromethylation/cyclization of N-substituted acrylamides for the synthesis of 3,4-dihydroquinolin-2(1H)-ones by an electrochemical method.
Figure 24.
Di- and trifluoromethylation/cyclization of N-substituted acrylamides for the synthesis of 3,4-dihydroquinolin-2(1H)-ones by an electrochemical method.
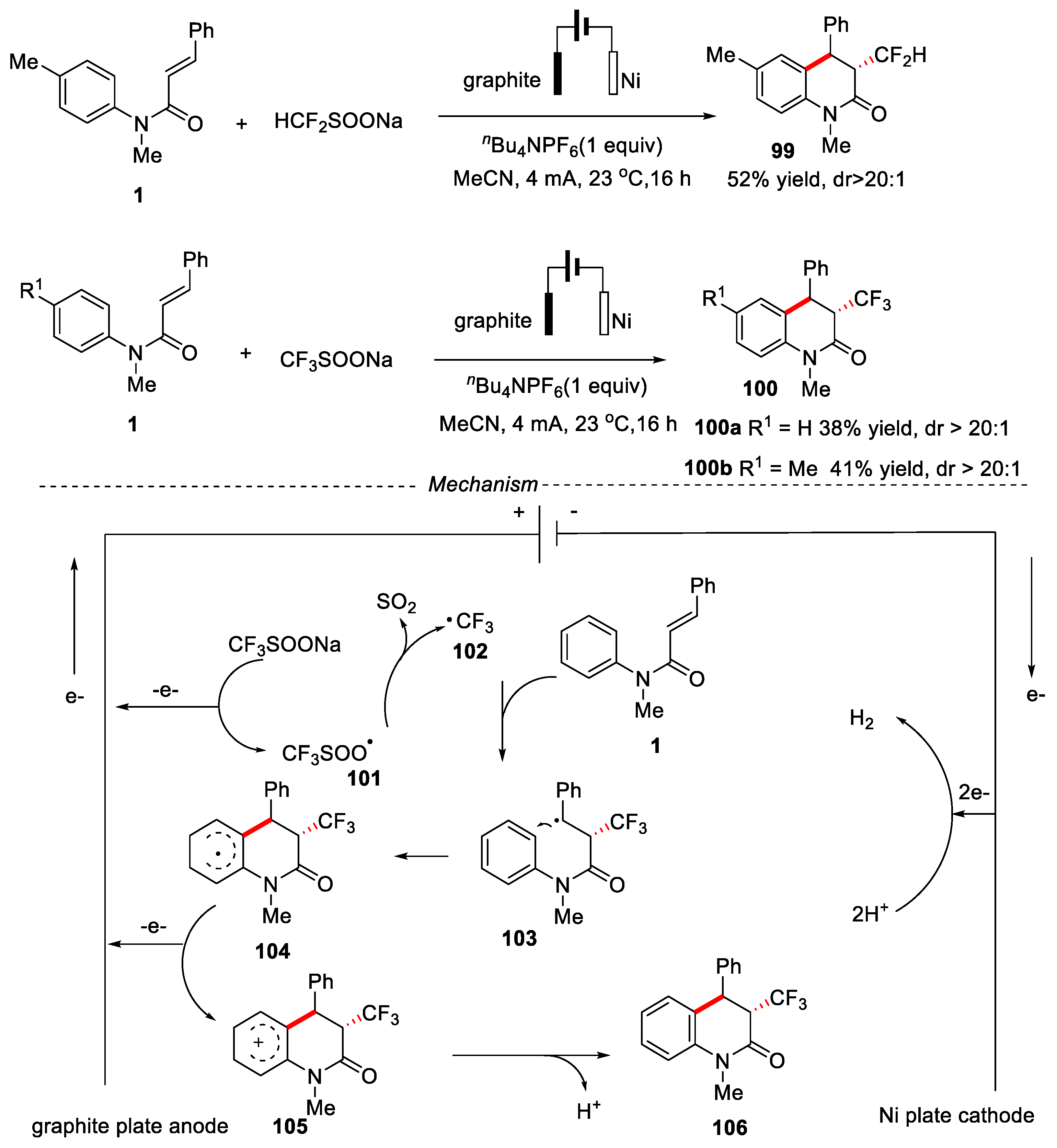
Figure 25.
Blue LED light induced mono-fluoromethylation of phenylcinnamamide using phosphonium salts 107 as fluorine sources.
Figure 25.
Blue LED light induced mono-fluoromethylation of phenylcinnamamide using phosphonium salts 107 as fluorine sources.
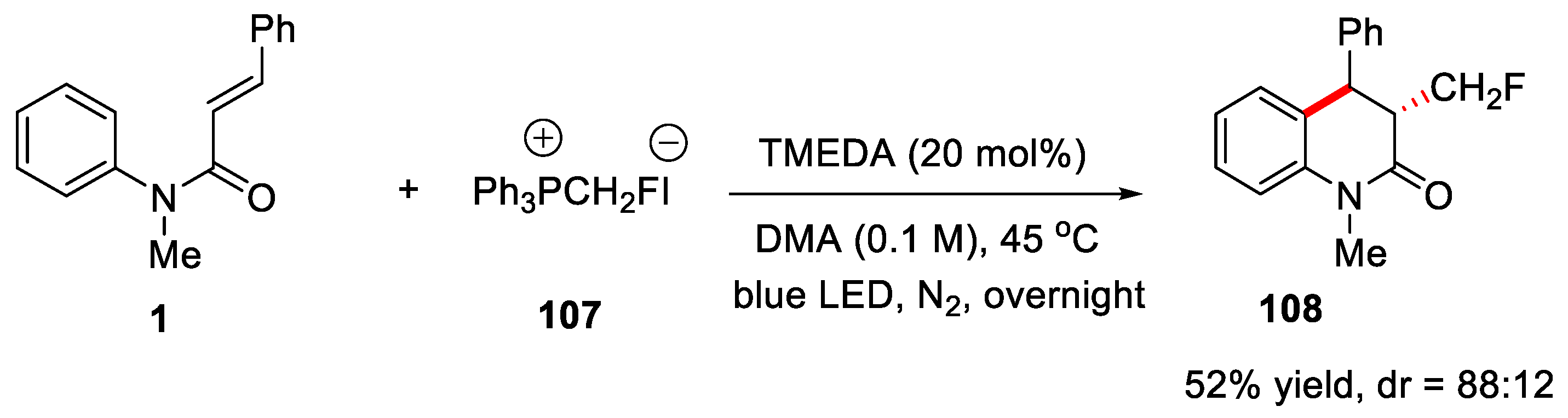
Figure 26.
A CuI-catalyzed protocol for the synthesis of trifluoromethylated 3,4-dihydroquinolin-2(1H)-ones.
Figure 26.
A CuI-catalyzed protocol for the synthesis of trifluoromethylated 3,4-dihydroquinolin-2(1H)-ones.
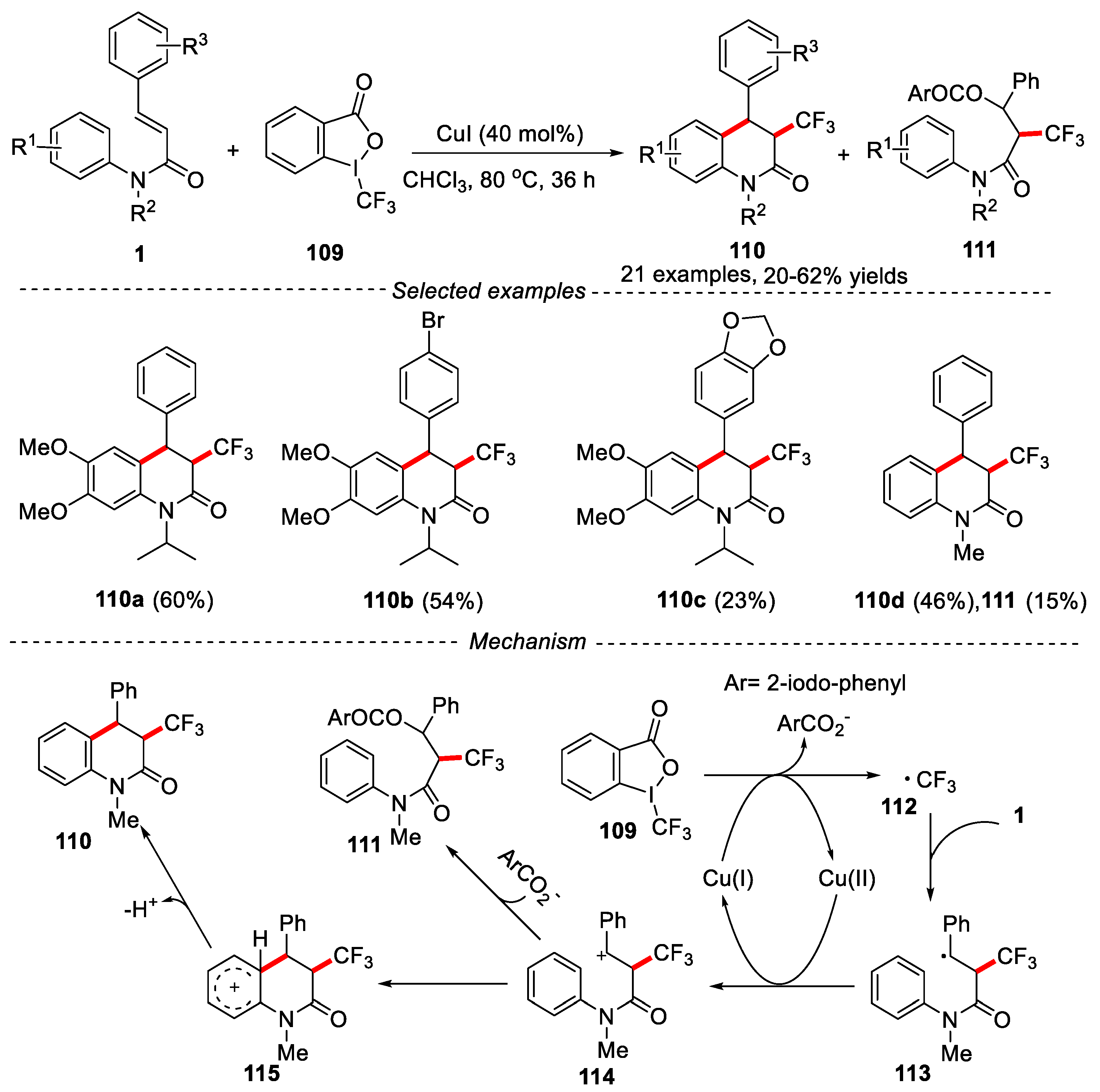
Figure 27.
Visible-light induced reaction of N-arylcinnamamides with Togni’s reagent for the synthesis of CF3-containing 3,4-disubstituted dihydroquinolinones and 1-azaspiro[4.5] decanes.
Figure 27.
Visible-light induced reaction of N-arylcinnamamides with Togni’s reagent for the synthesis of CF3-containing 3,4-disubstituted dihydroquinolinones and 1-azaspiro[4.5] decanes.
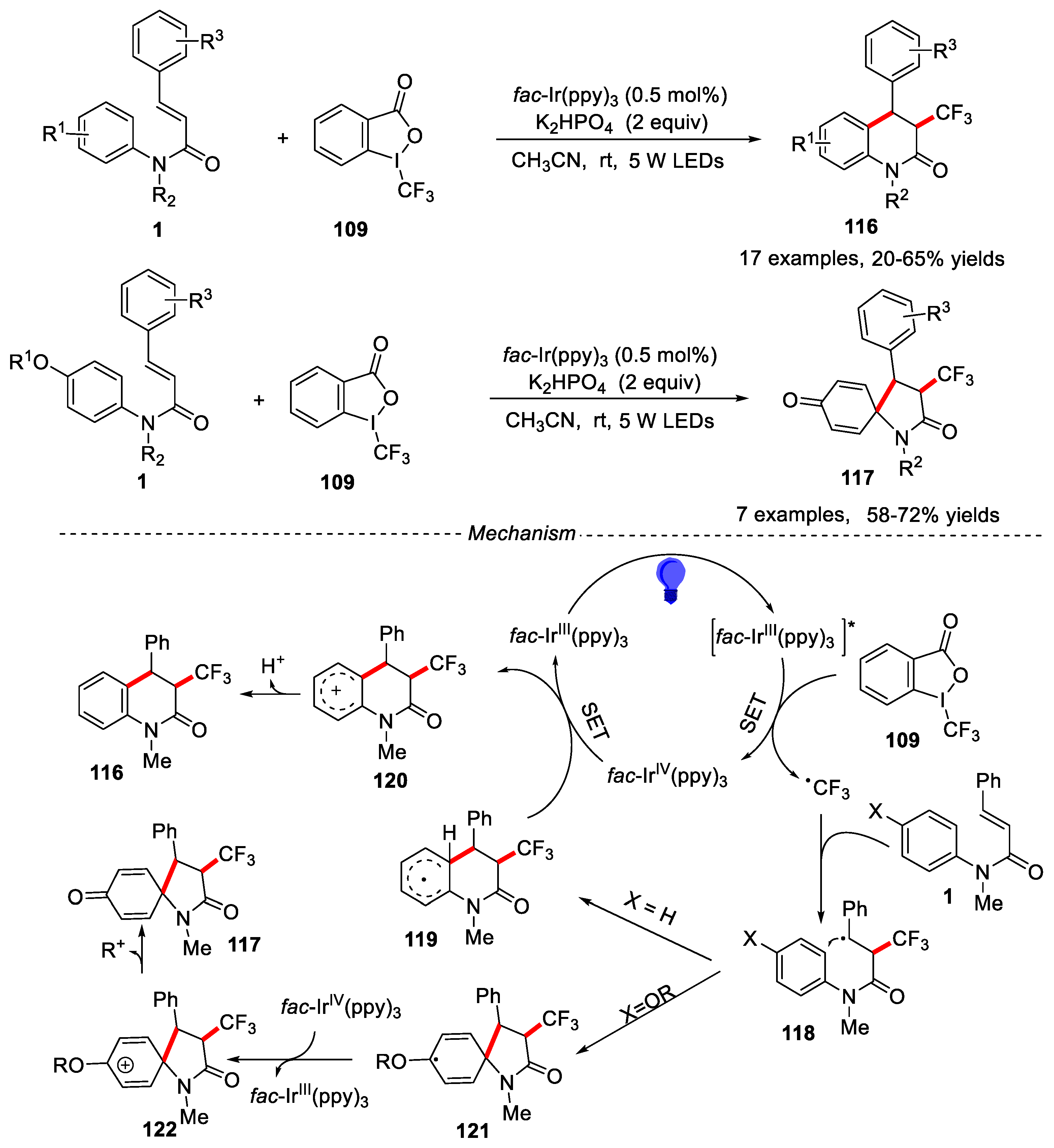
Figure 28.
Visible-light induced cascade reaction of N-arylcinnamamides for the synthesis of HCF2COOEt-containing 3,4-disubstituted dihydroquinolinones and 1-azaspiro[4.5]decanes.
Figure 28.
Visible-light induced cascade reaction of N-arylcinnamamides for the synthesis of HCF2COOEt-containing 3,4-disubstituted dihydroquinolinones and 1-azaspiro[4.5]decanes.
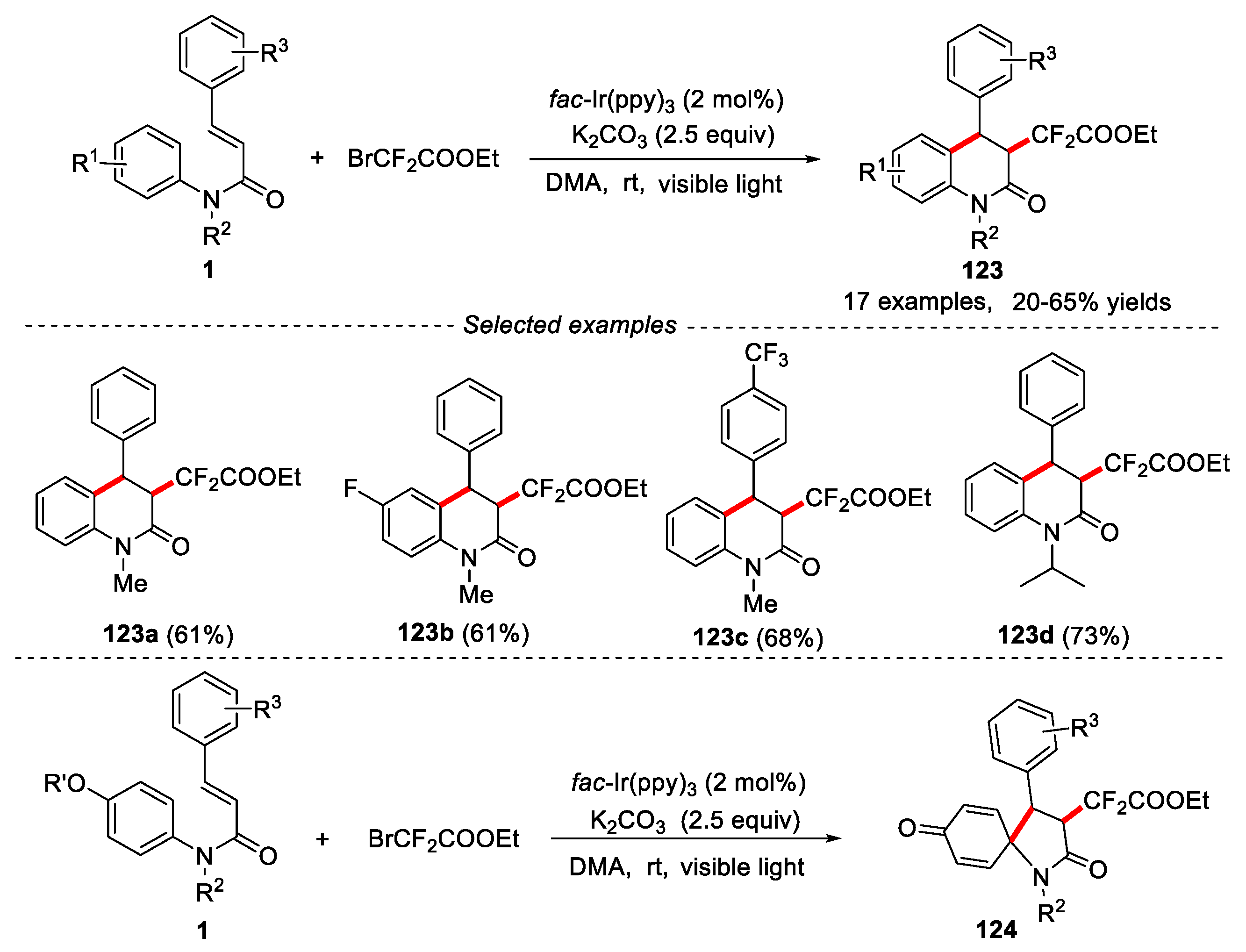
Figure 29.
Copper-catalyzed cross-dehydrogenative coupling of N-arylcinnamamides with chloroform for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
Figure 29.
Copper-catalyzed cross-dehydrogenative coupling of N-arylcinnamamides with chloroform for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
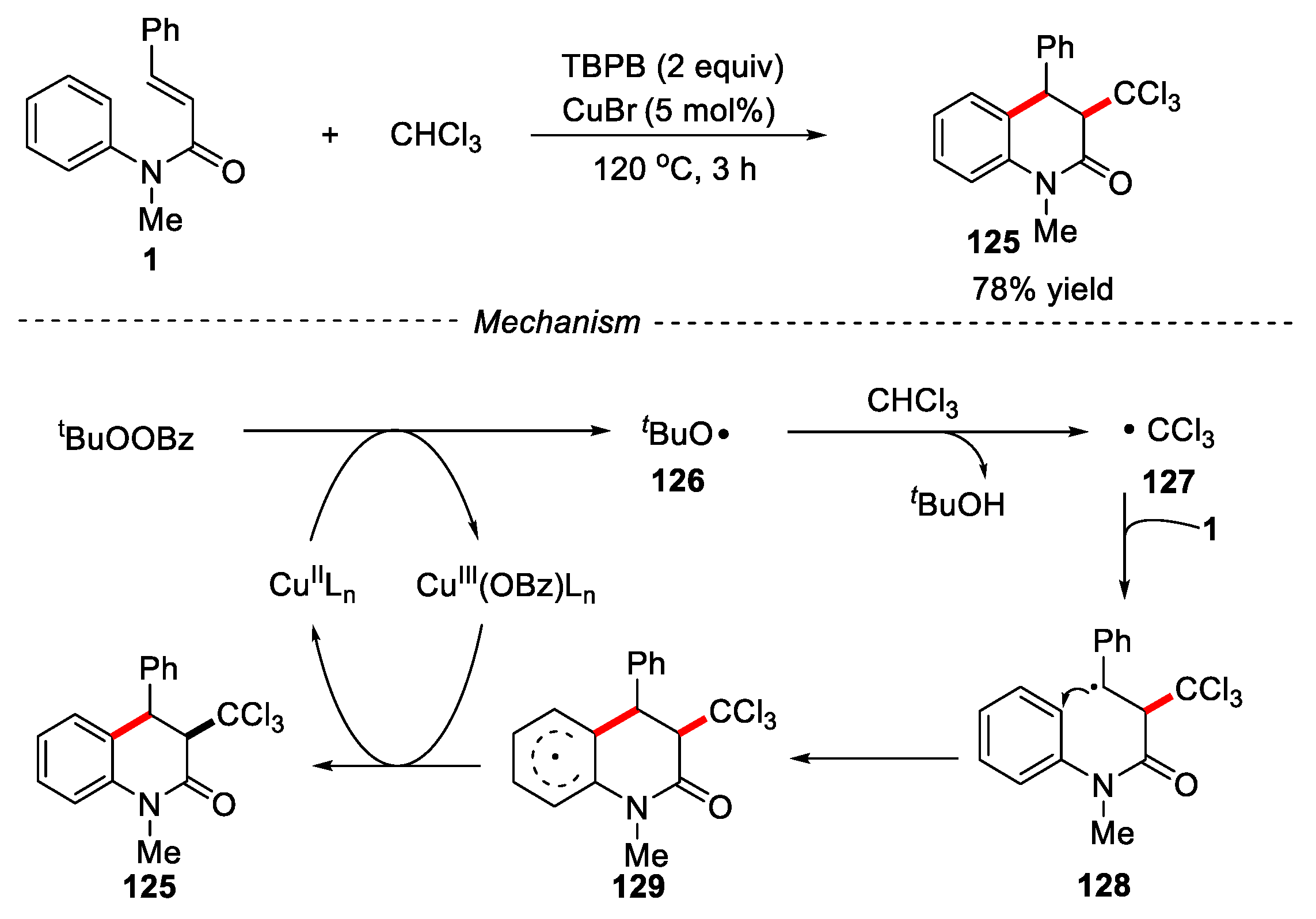
Figure 30.
Mn(OAc)3-mediated radical dichloromethylation of N-phenylcinnamamide for the preparation of chloro-containing 3,4 dihydroquinolin-2(1H)-one.
Figure 30.
Mn(OAc)3-mediated radical dichloromethylation of N-phenylcinnamamide for the preparation of chloro-containing 3,4 dihydroquinolin-2(1H)-one.
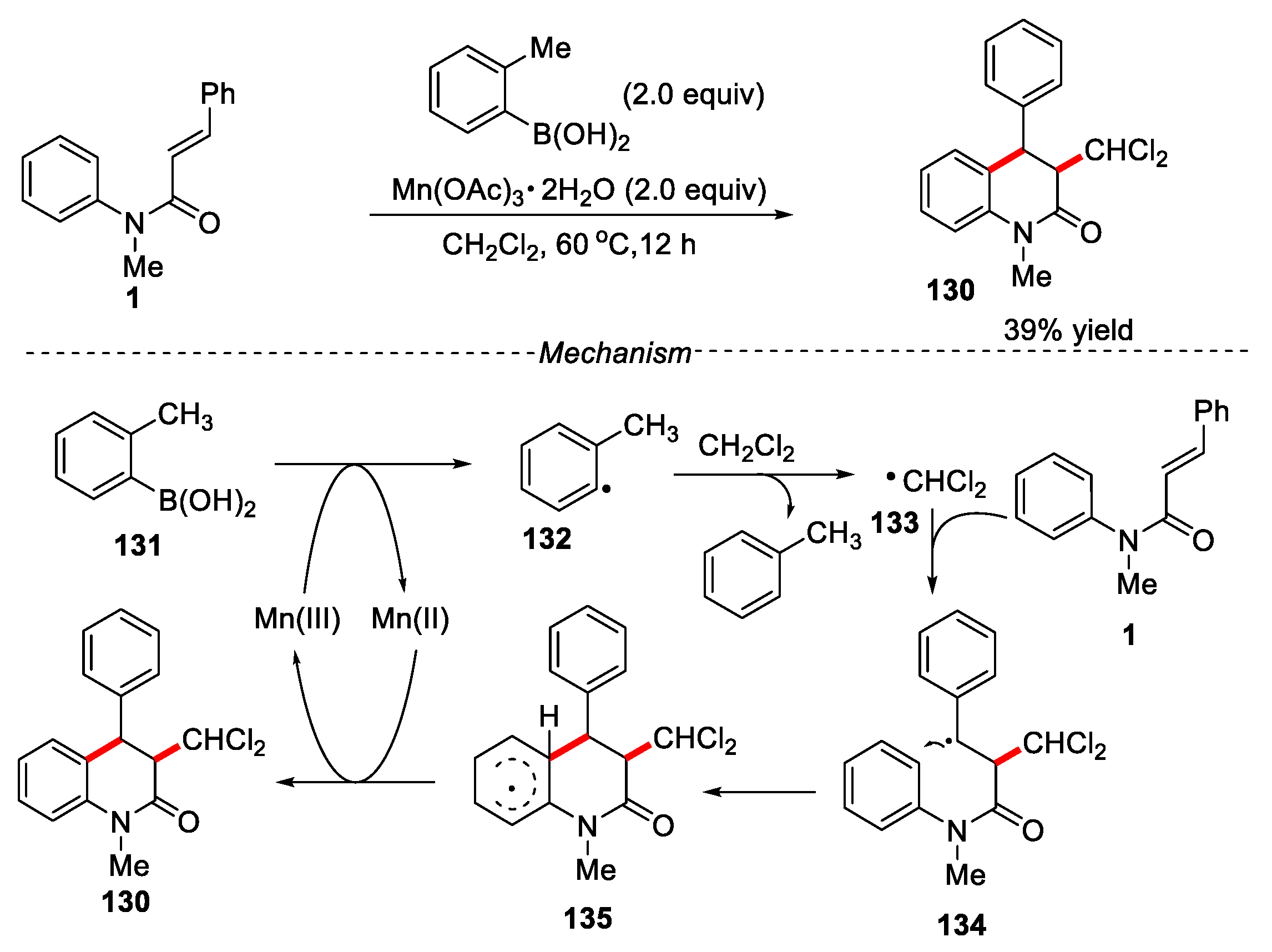
Figure 31.
Photo-induced C(sp3)-H functionalization for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
Figure 31.
Photo-induced C(sp3)-H functionalization for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.
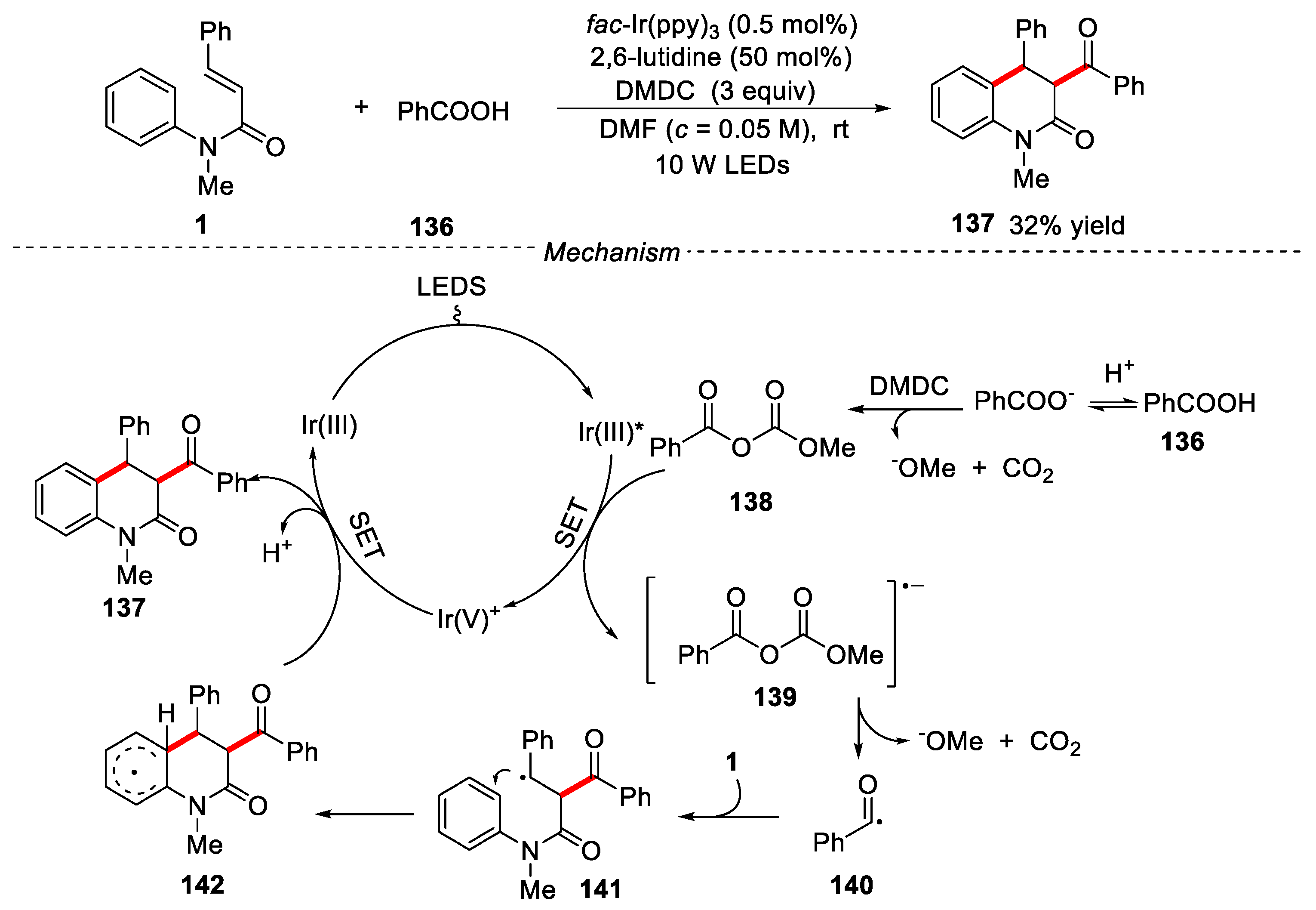
Figure 32.
A silver-catalyzed protocol for the synthesis of dihydroquinolin-2(1H)-ones or quinolin-2(1H)-ones via intermolecular radical addition/cyclization of N-phenylcinnamamide with keto acids.
Figure 32.
A silver-catalyzed protocol for the synthesis of dihydroquinolin-2(1H)-ones or quinolin-2(1H)-ones via intermolecular radical addition/cyclization of N-phenylcinnamamide with keto acids.
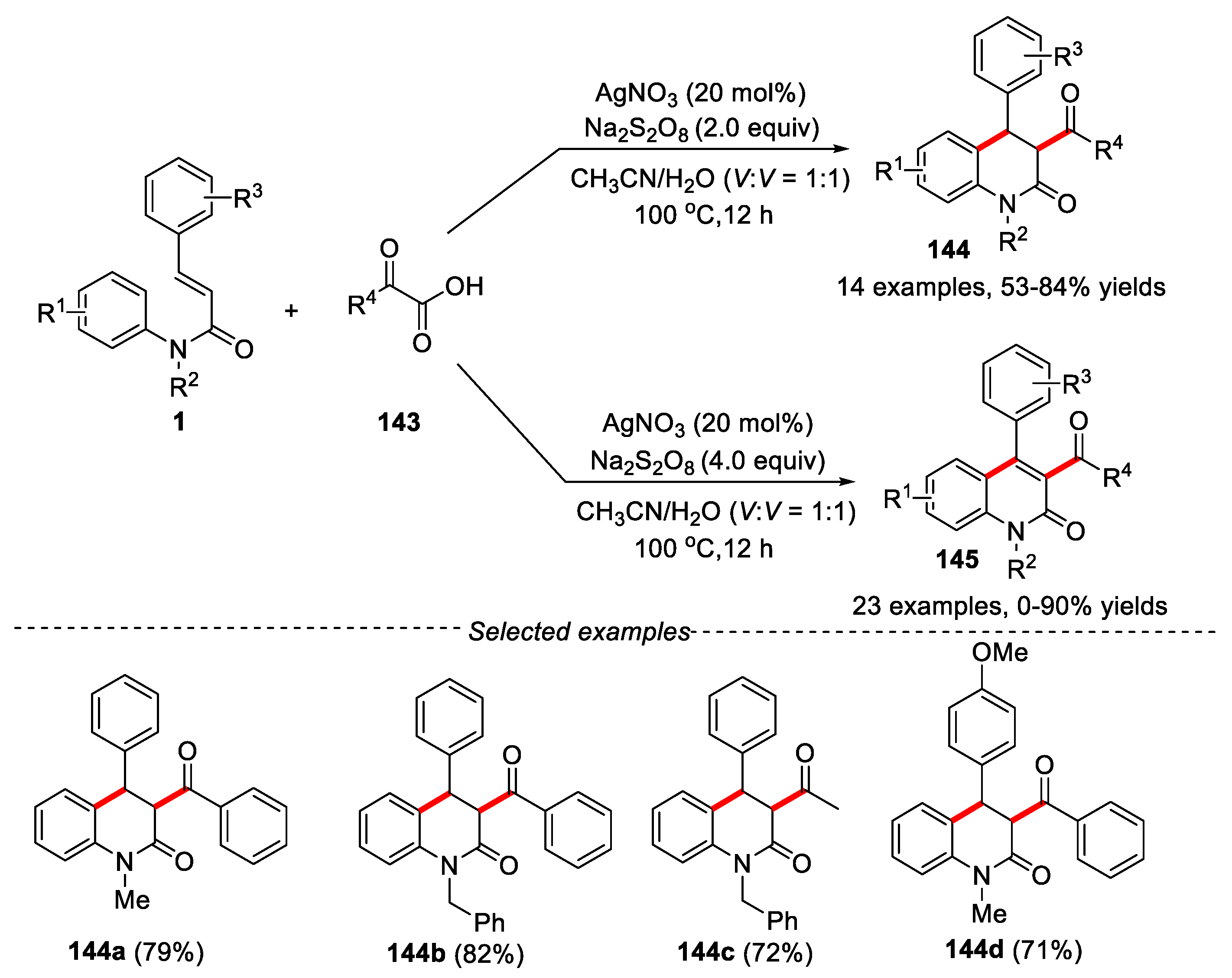
Figure 33.
A silver-catalyzed protocol for the synthesis of dihydroquinolin-2(1H)-ones via radical addition/cyclization of N-phenylcinnamamide with keto acids.
Figure 33.
A silver-catalyzed protocol for the synthesis of dihydroquinolin-2(1H)-ones via radical addition/cyclization of N-phenylcinnamamide with keto acids.
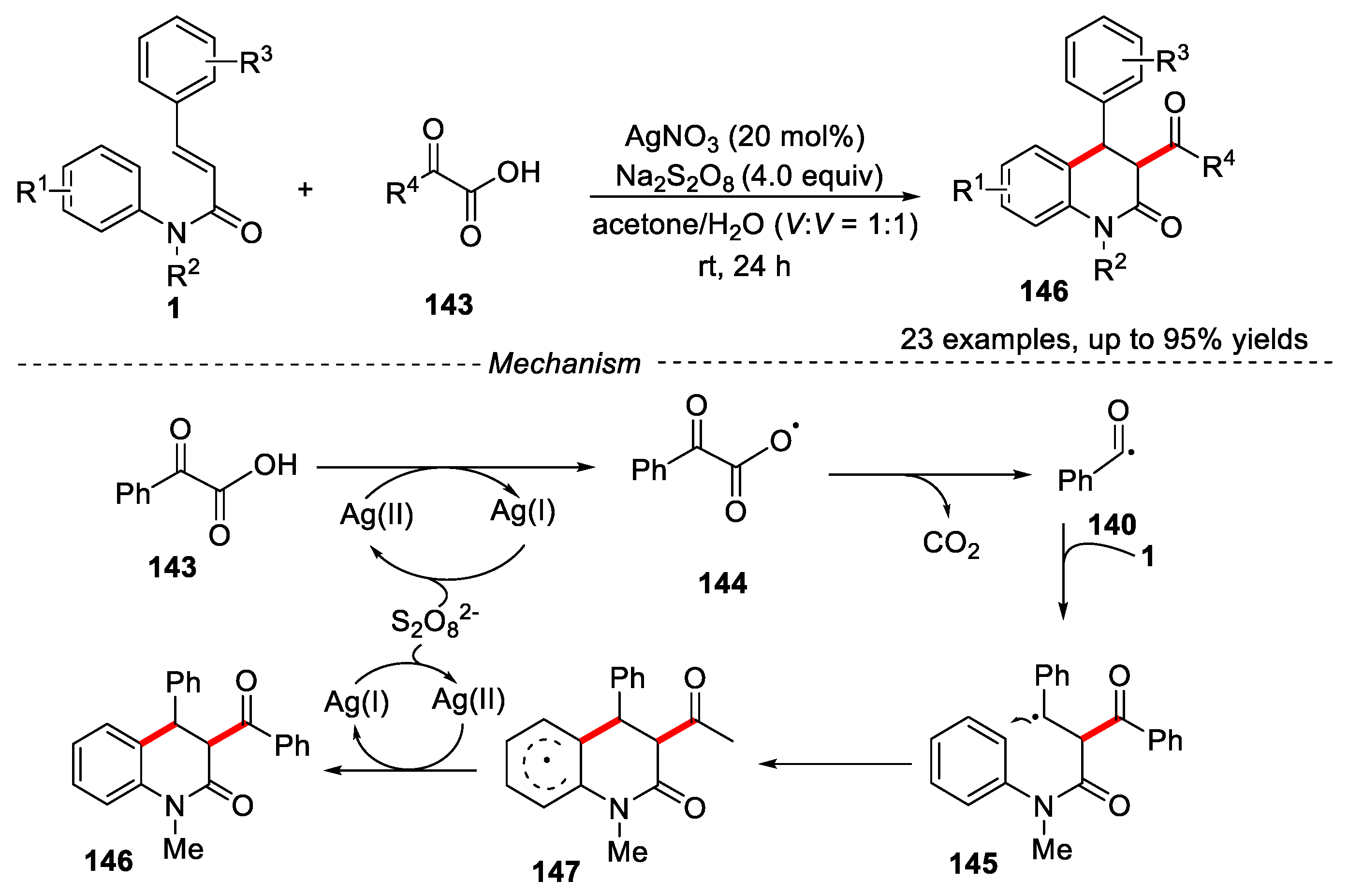
Figure 34.
n-Bu4NBr catalyzed method for the synthesis of 3-acyl-4-aryldihydroquinolin-2(1H)-ones by cascade radical addition/cyclization of N-arylcinnamamides with aldehyde.
Figure 34.
n-Bu4NBr catalyzed method for the synthesis of 3-acyl-4-aryldihydroquinolin-2(1H)-ones by cascade radical addition/cyclization of N-arylcinnamamides with aldehyde.
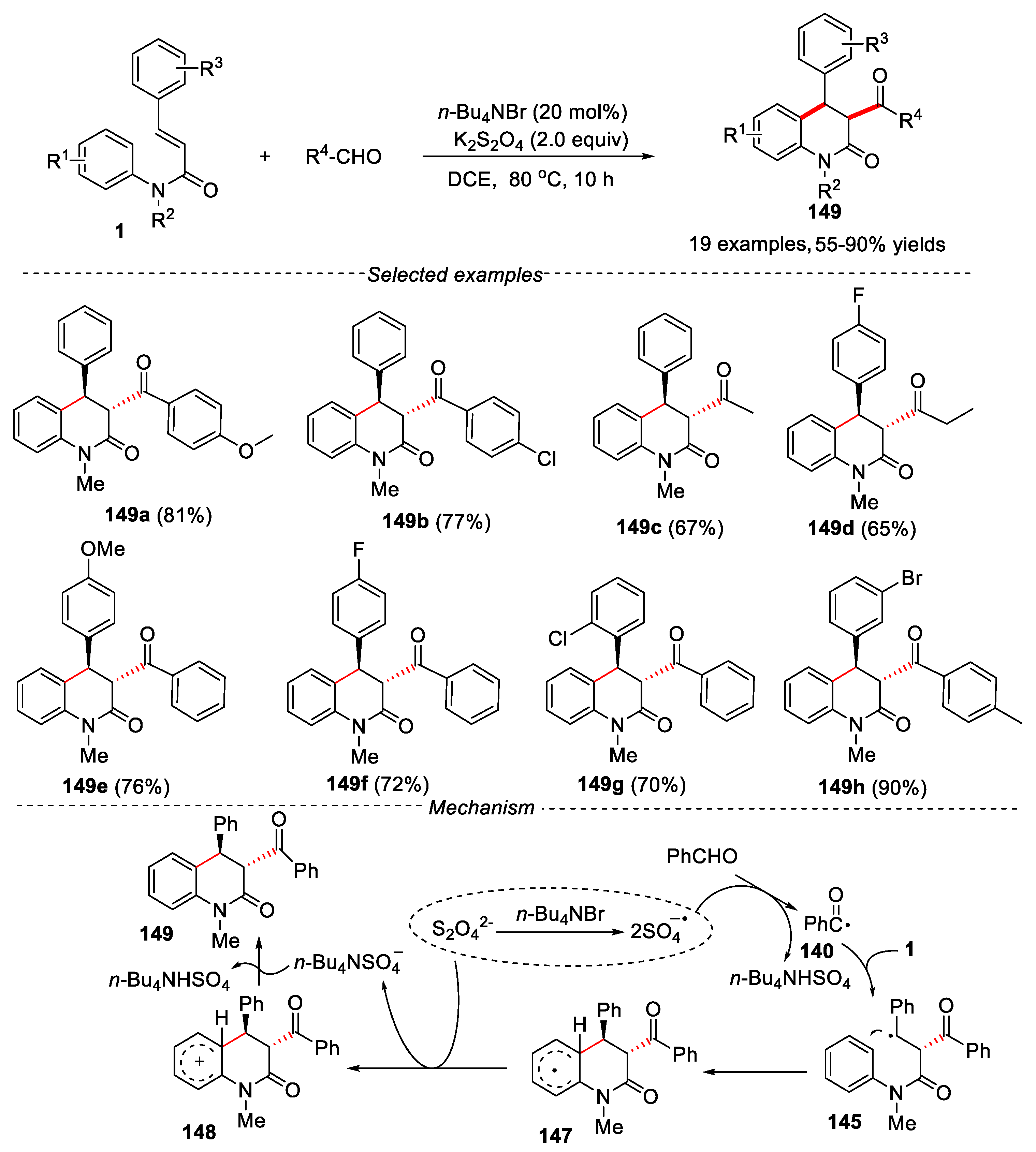
Figure 35.
A photocatalytic strategy for the synthesis of dihydroquinolin-2(1H)-one from N-methyl-N-arylcinnamamides with oxime ester.
Figure 35.
A photocatalytic strategy for the synthesis of dihydroquinolin-2(1H)-one from N-methyl-N-arylcinnamamides with oxime ester.
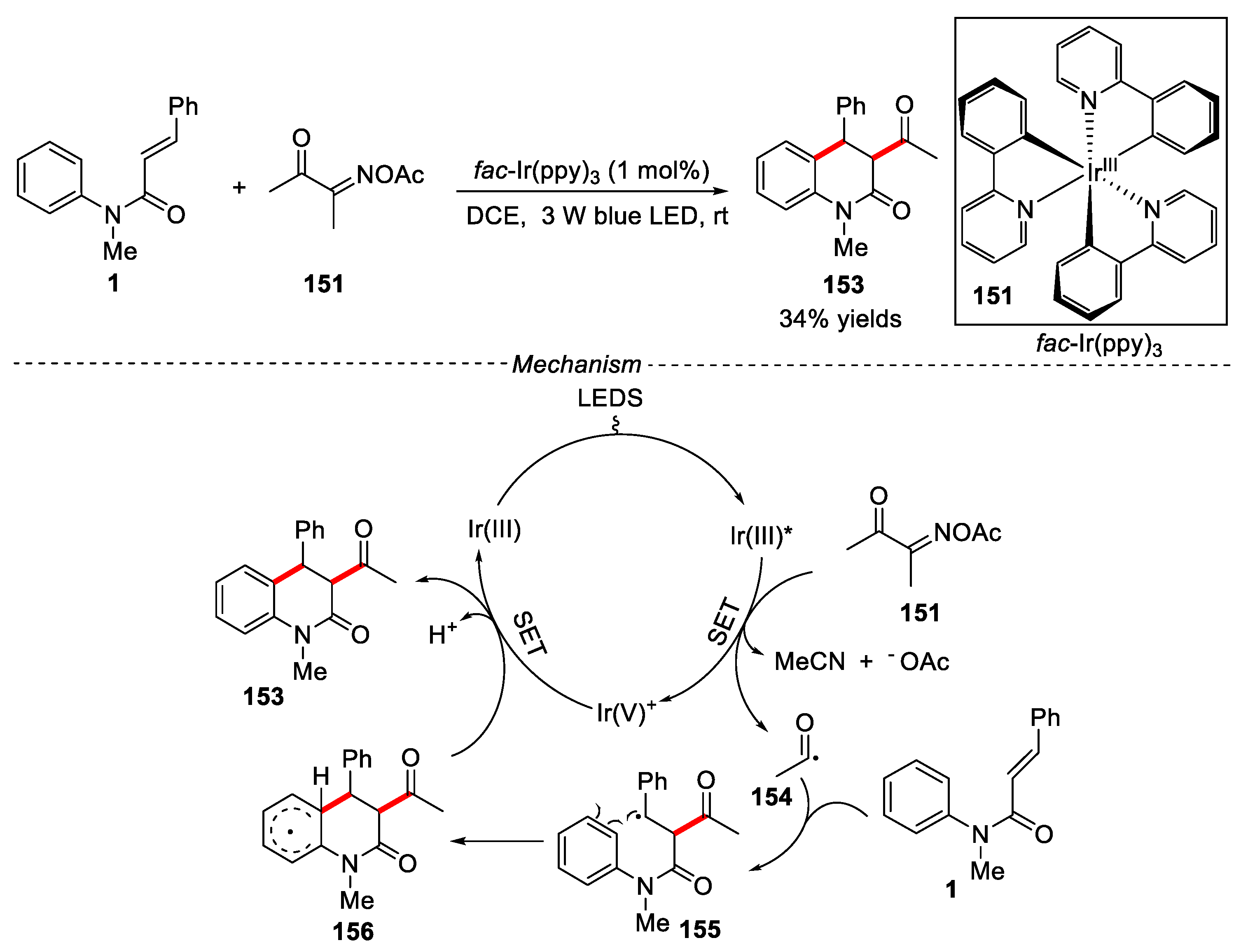
Figure 36.
Silver-catalyzed cascade cyclization of cinnamamides with diphenylphosphine oxide for the synthesis of dihydroquinolin-2(1H)-ones.
Figure 36.
Silver-catalyzed cascade cyclization of cinnamamides with diphenylphosphine oxide for the synthesis of dihydroquinolin-2(1H)-ones.
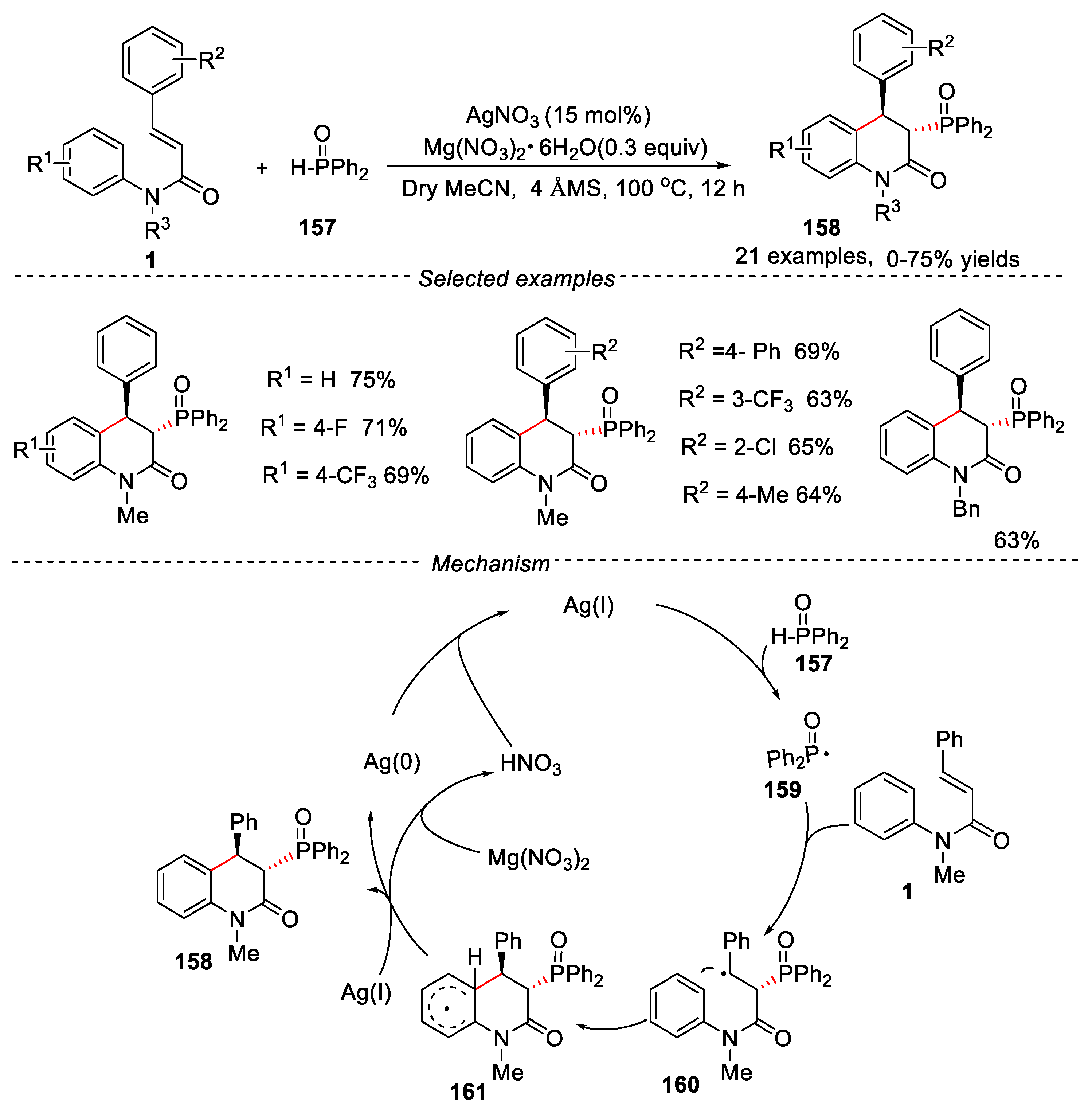
Figure 37.
A tandem sulfenylation/cyclization of N-arylacrylamides with sulfonyl hydrazides for the synthesis of 3-sulfenyl-3,4-dihydroquinolin-2(1H)-ones.
Figure 37.
A tandem sulfenylation/cyclization of N-arylacrylamides with sulfonyl hydrazides for the synthesis of 3-sulfenyl-3,4-dihydroquinolin-2(1H)-ones.
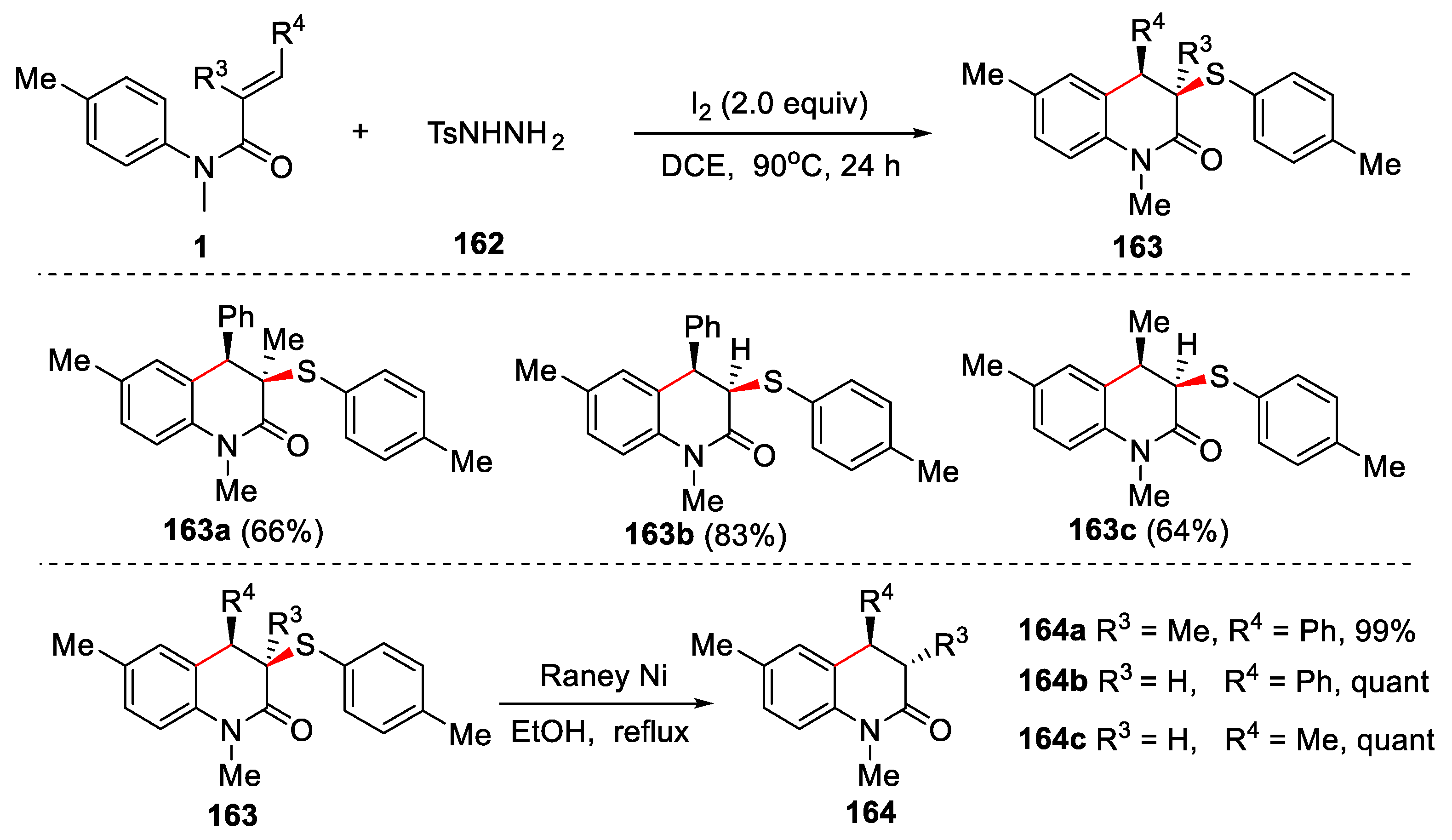
Figure 38.
The mechanism for the synthesis of 3-sulfenyl-3,4-dihydroquinolin-2(1H)-ones.
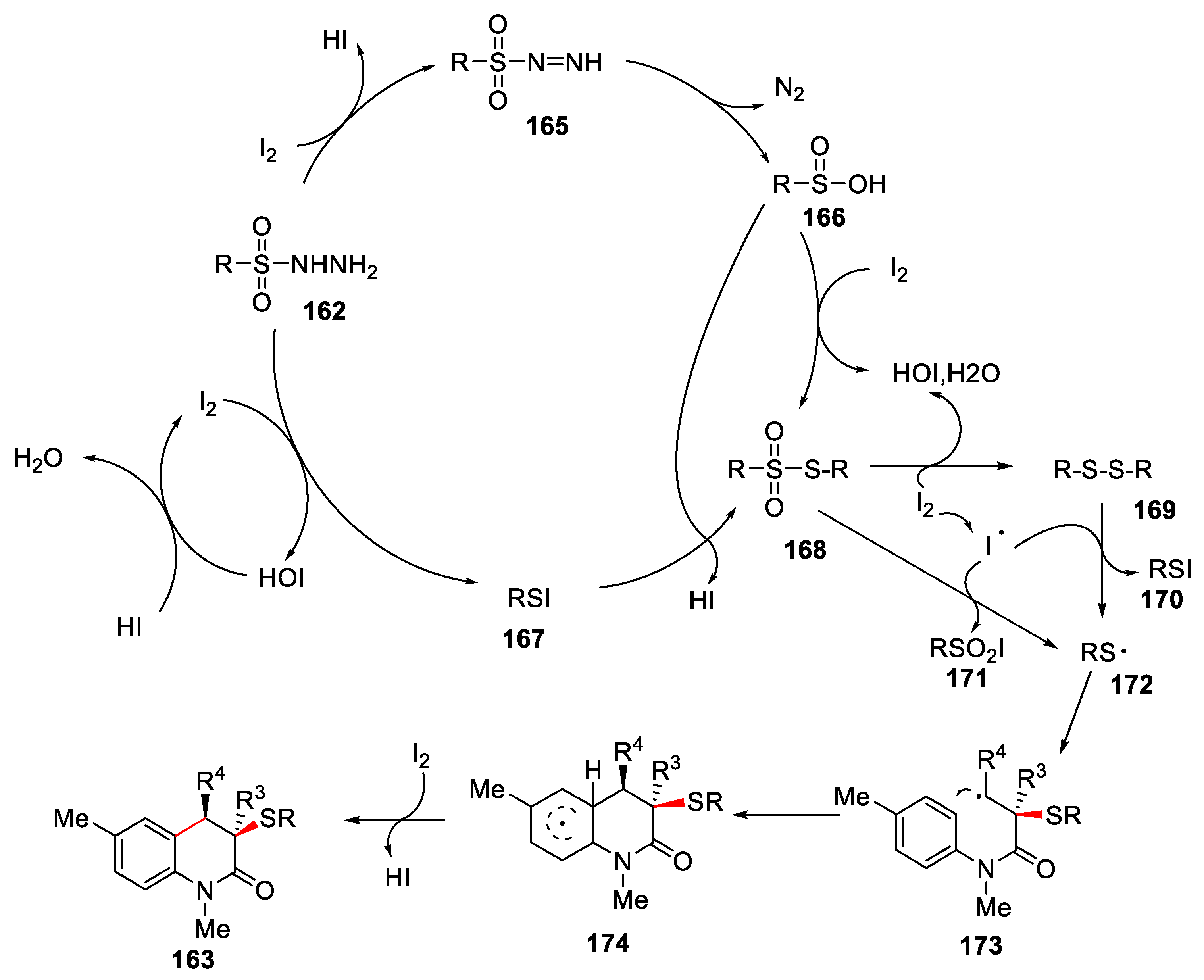
Figure 39.
Mn(OAc)3-promoted radical sulfonation of N-phenylcinnamamide for the preparation of 3,4 dihydroquinolin-2(1H)-ones.
Figure 39.
Mn(OAc)3-promoted radical sulfonation of N-phenylcinnamamide for the preparation of 3,4 dihydroquinolin-2(1H)-ones.
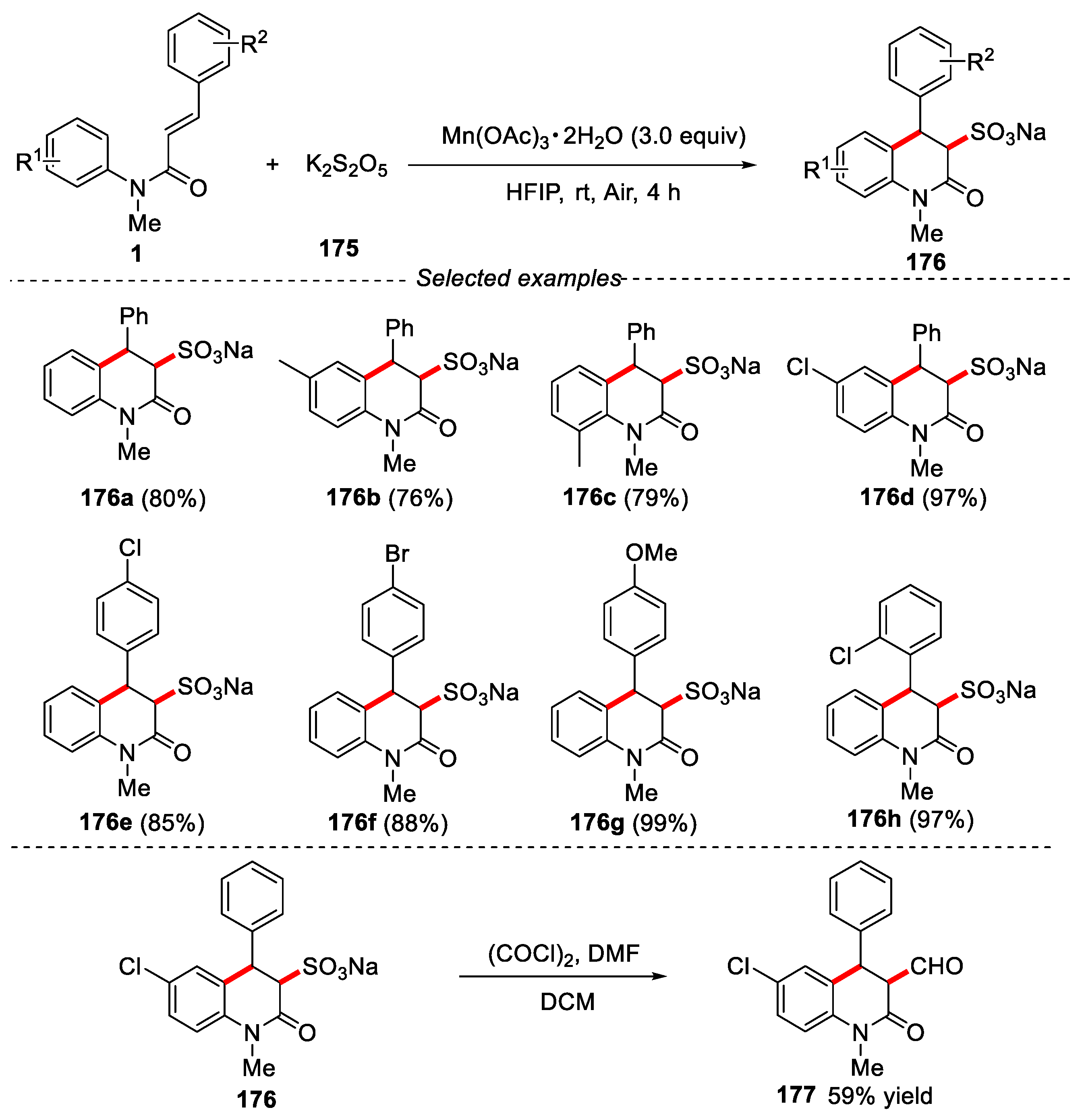
Figure 40.
Cascade selenylation/cyclization of N-arylcinnamamides with diphenyl diselenide for the the synthesis of functionalized 3,4-dihydroquinolin-2(1H)-ones.
Figure 40.
Cascade selenylation/cyclization of N-arylcinnamamides with diphenyl diselenide for the the synthesis of functionalized 3,4-dihydroquinolin-2(1H)-ones.
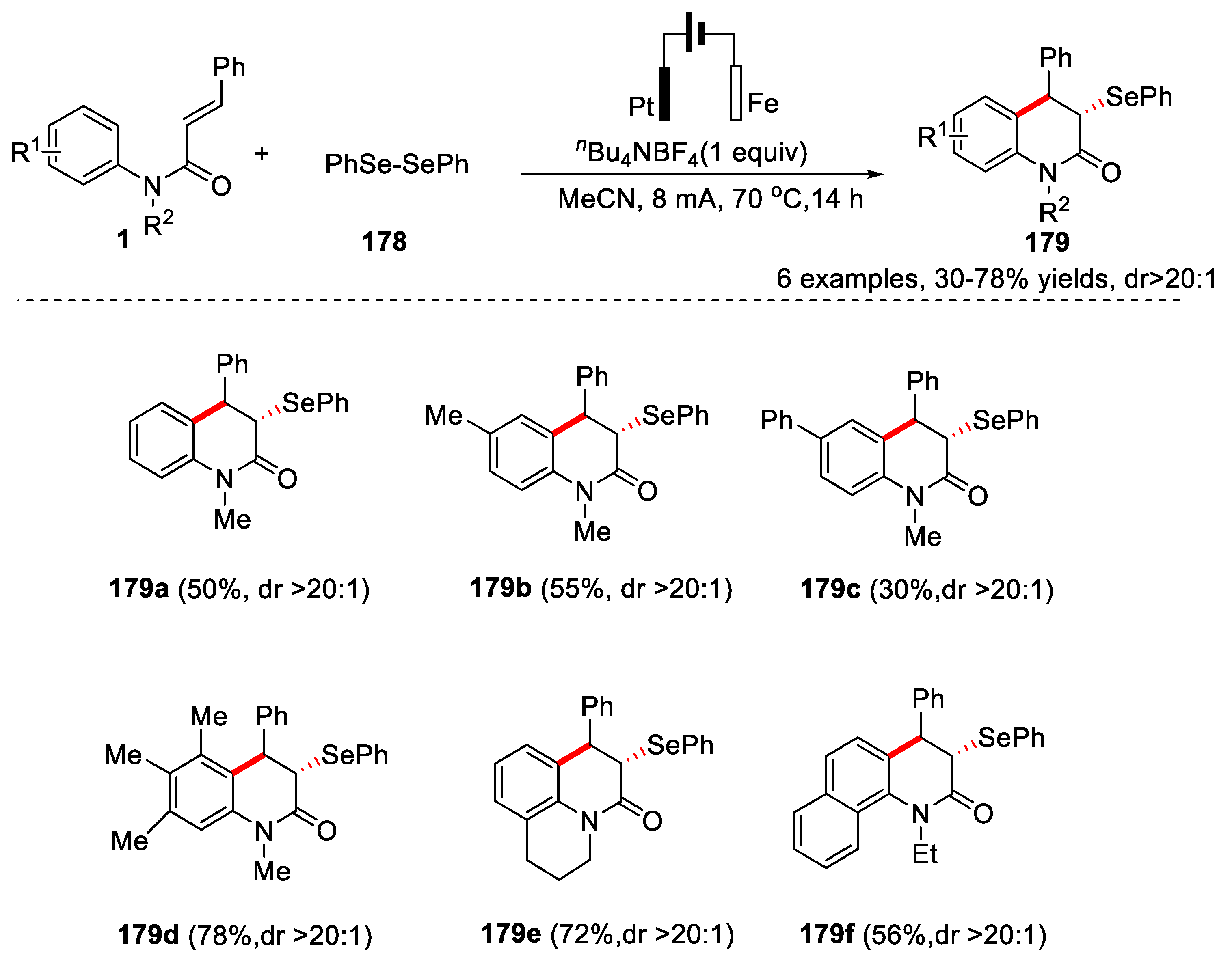
Figure 41.
An electrochemical protocol for the synthesis of 3,4-dihydroquinolin-2(1H)-one via tandem cyclization of N-arylcinnamamide with diselenides.
Figure 41.
An electrochemical protocol for the synthesis of 3,4-dihydroquinolin-2(1H)-one via tandem cyclization of N-arylcinnamamide with diselenides.
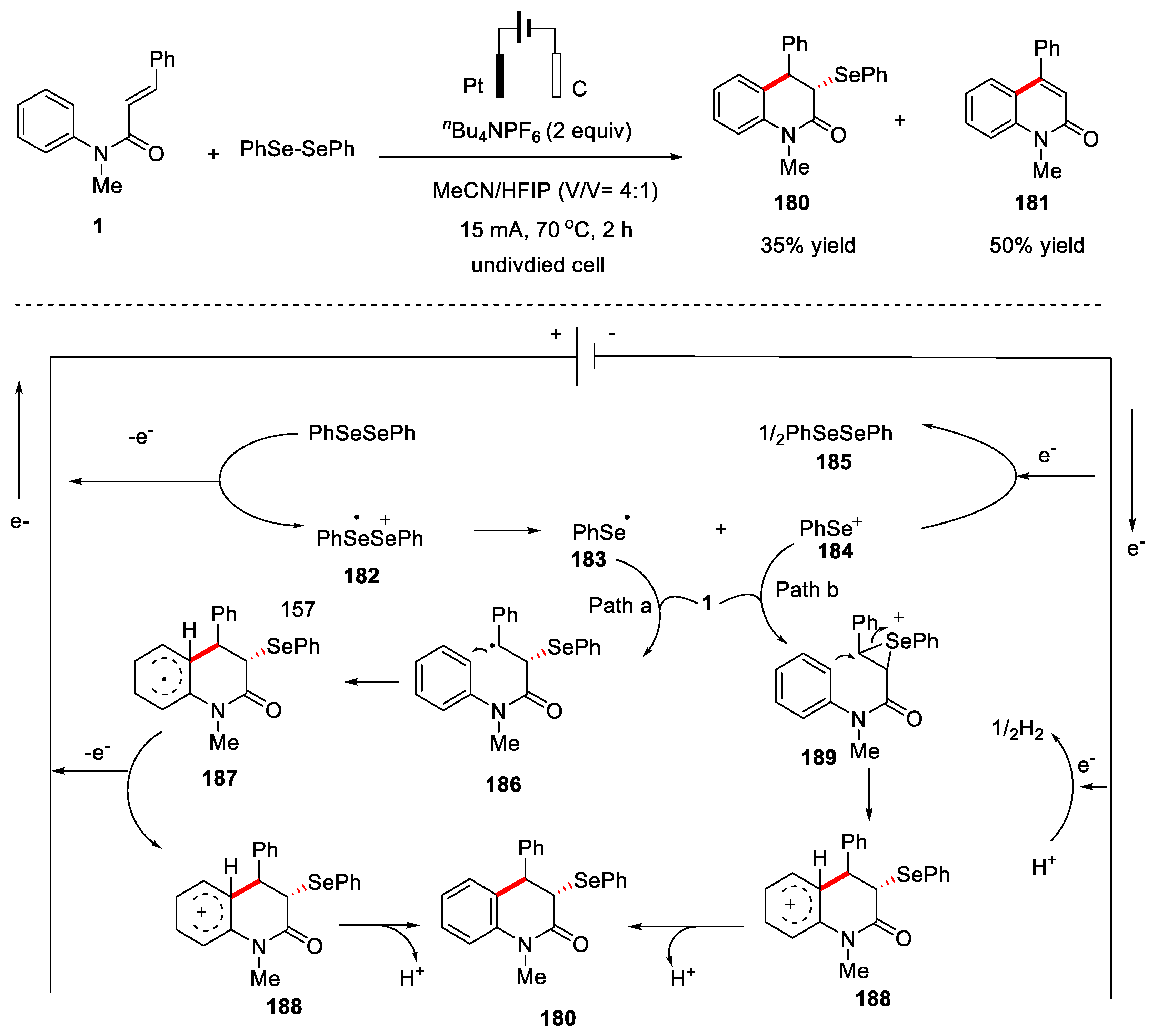
Figure 42.
A photo-induced 6π cyclization of N-arylacrylamides for the synthesis of 3,4-dihydroquinolin-2(1H)-one.
Figure 42.
A photo-induced 6π cyclization of N-arylacrylamides for the synthesis of 3,4-dihydroquinolin-2(1H)-one.
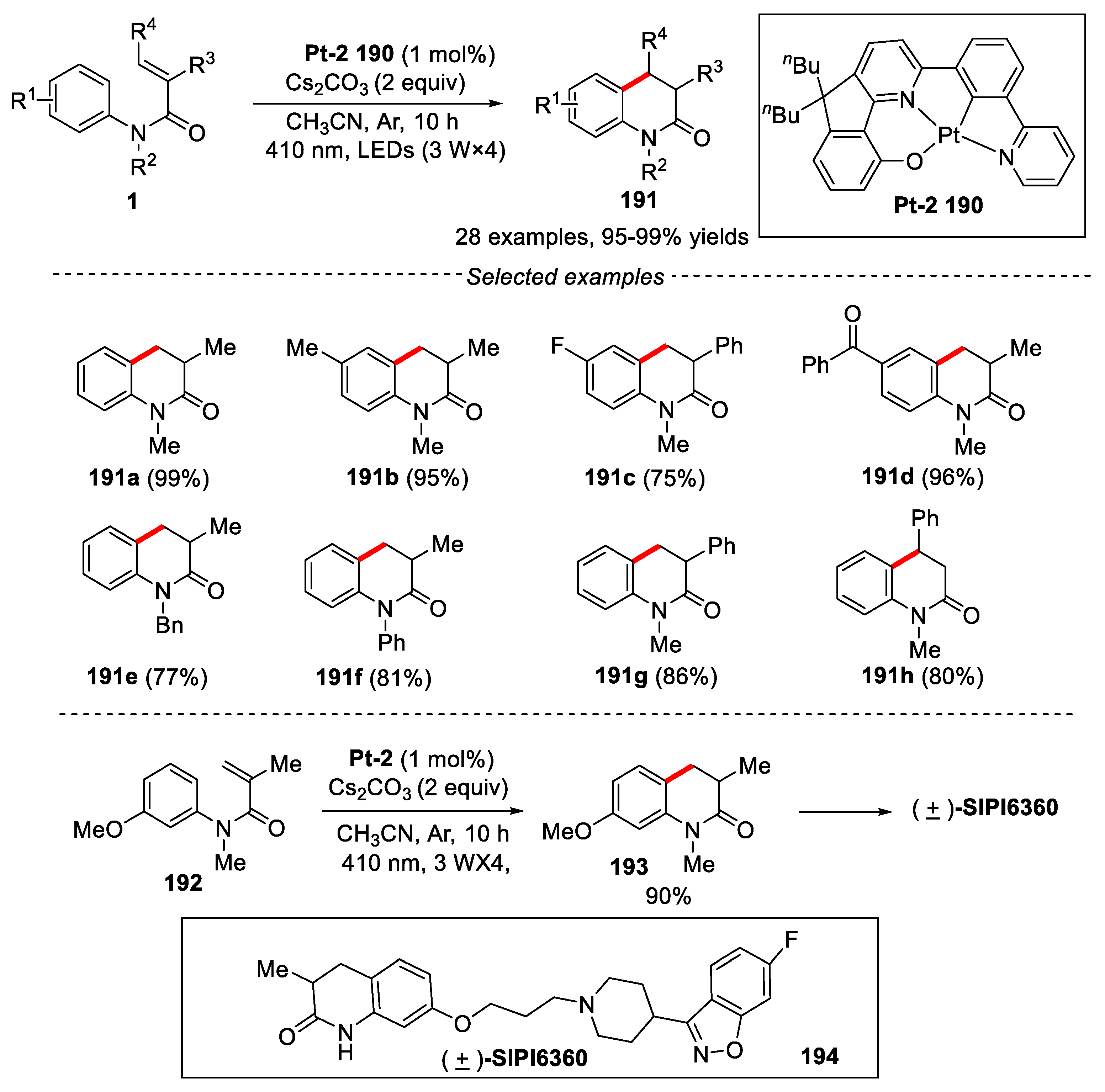
Figure 43.
A thioxanthone-catalyzed method for the synthesis of 3,4-dihydroquinolin-2-ones via a 6π-photocyclization process.
Figure 43.
A thioxanthone-catalyzed method for the synthesis of 3,4-dihydroquinolin-2-ones via a 6π-photocyclization process.
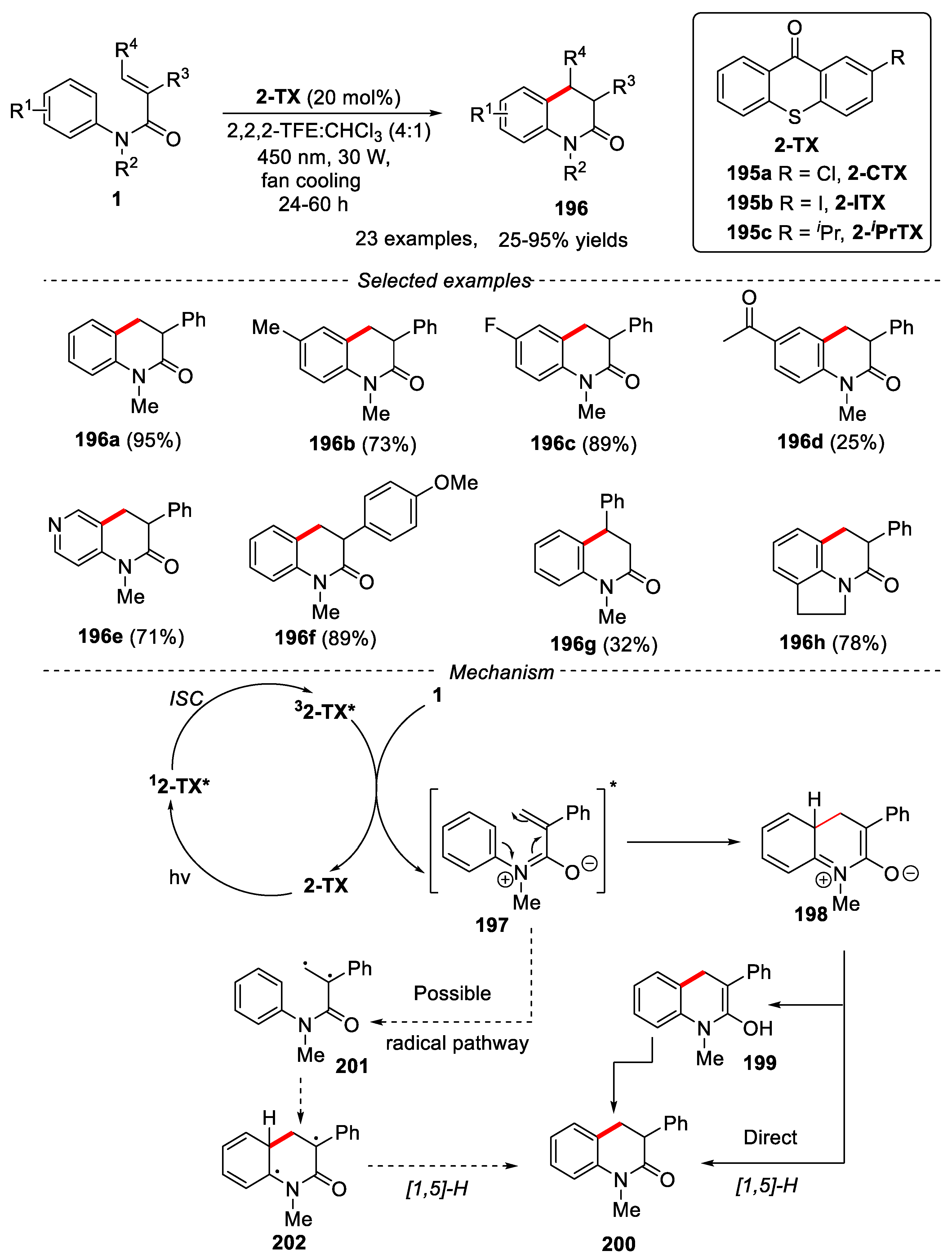
Figure 44.
A visible-light induced photoredox cyclization of N-Arylacrylamides for the synthesis of dihydroquinolinones using 4CzIPN as catalyst.
Figure 44.
A visible-light induced photoredox cyclization of N-Arylacrylamides for the synthesis of dihydroquinolinones using 4CzIPN as catalyst.
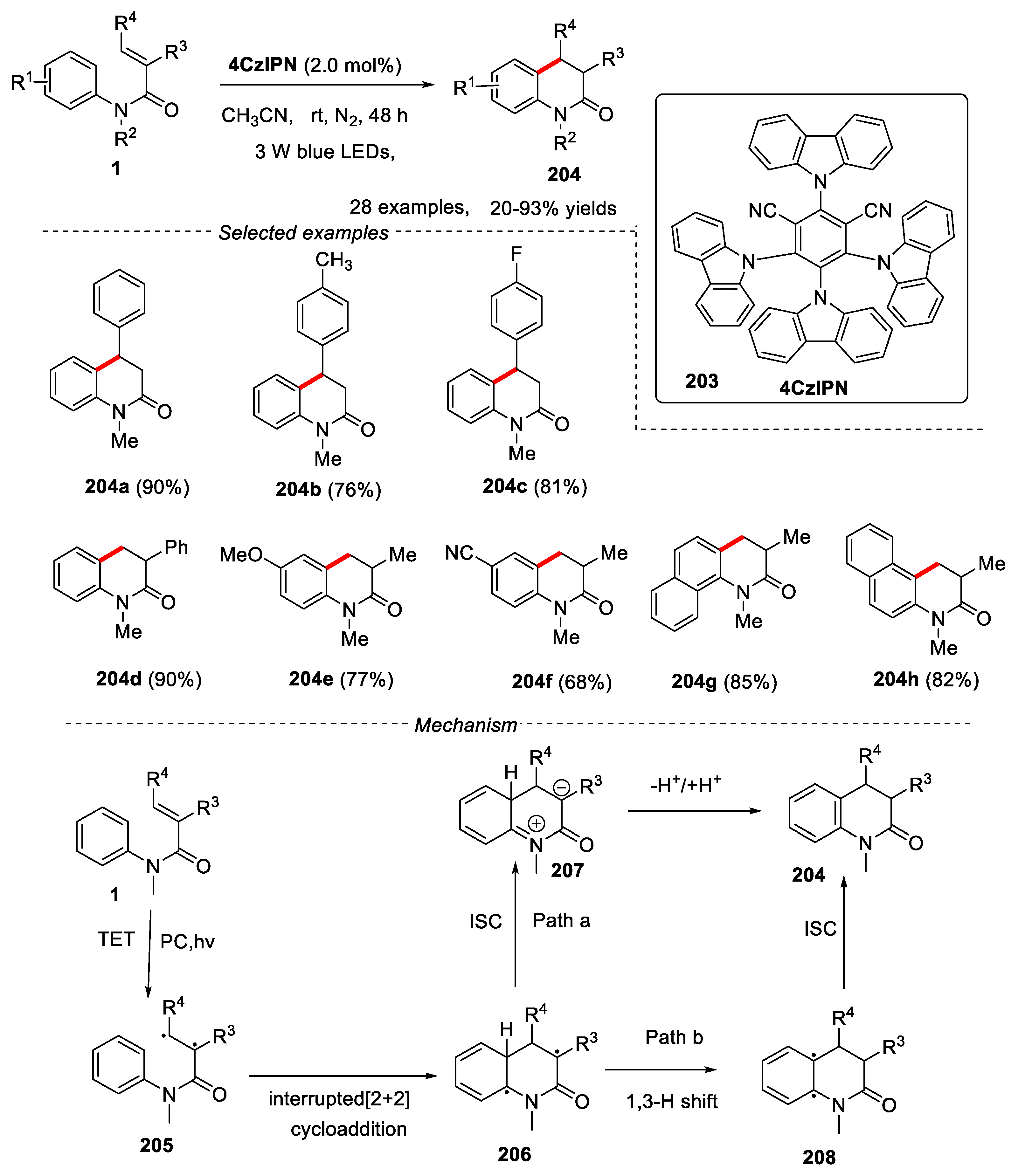
Figure 45.
Catalytic enantioselective 6π-photocyclization of acrylanilides using Ir((5-CF3)(4′-t-Bu)ppy)3 as photocatalyst in the presence of chiral Lewis acid complexes.
Figure 45.
Catalytic enantioselective 6π-photocyclization of acrylanilides using Ir((5-CF3)(4′-t-Bu)ppy)3 as photocatalyst in the presence of chiral Lewis acid complexes.
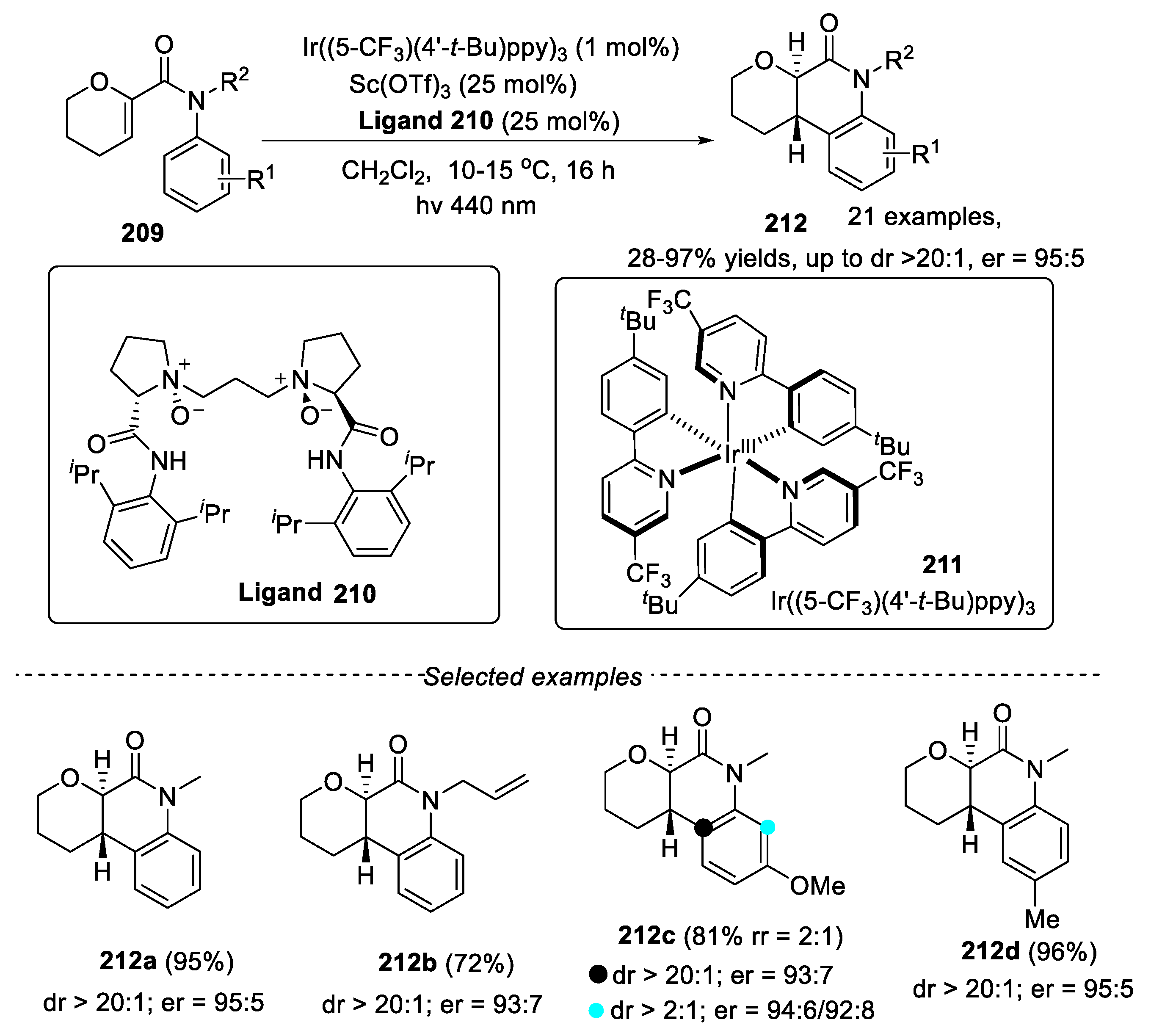
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



