
Submitted:
29 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Mucopolysaccharidoses (MPSs) are rare inherited lysosomal storage diseases (LSDs) caused by deficient activity in one of the enzymes responsible for glycosaminoglycans lysosomal degradation. MPS II is caused by pathogenic mutations in the IDS gene, leading to deficient activity of the enzyme iduronate-2-sulfatase, which causes dermatan and heparan sulfate storage in the lysosomes. MPS VI has dermatan sulfate lysosomal storage, owing to a deficiency of arylsulfatase B, due to pathogenic mutations in the ARSB gene. Alterations in the immune system of MPSs mouse models have been described but the data is still scarce concerning MPSs patients. Herein, we study different leukocyte populations in MPS II and VI disease patients. MPS VI, but not MPS II patients, have a decrease percentage of Natural Killer (NK) cells and monocytes when compared with controls. No alterations were identified in the percentage of T, invariant NKT and B cells in both groups of MPS disease patients. However, we uncovered alterations in the naïve versus memory status of both helper and cytotoxic T cells in MPS VI disease patients compared to control group. Indeed, MPS VI disease patients have a higher frequency of naïve T cells and consequently lower frequency of memory T cells than control subjects. Altogether, these results reveal MPS VI disease-specific alterations in some leukocyte populations, suggesting that the type of substrate accumulated and/or enzyme deficient in the lysosome may have a particular effect on the normal cellular composition of the immune system.
Keywords:
Lysosomal storage diseases
; Mucopolysaccharidoses
; Glycosaminoglycans
; Leukocytes
; Lymphocytes
1. Introduction
Mucopolysaccharidoses (MPSs) are a subgroup of lysosomal storage diseases (LSDs) characterized by the progressive intra-lysosomal accumulation of non-degraded or partially degraded glycosaminoglycans (GAGs), which are also increased in urine, blood, and cerebrospinal fluid [1,2].
MPS II (MIM# 309900), or Hunter syndrome, is caused by pathogenic variants in the IDS gene and is transmitted in an X-linked recessive pattern, affecting almost exclusively males. So far, over 800 variants in the IDS gene have been reported as disease-causing mutations: 389 missense/nonsense, 75 splicing mutations, 70 small insertions, 168 small deletions, 20 small indels, 66 gross deletions, 6 gross insertions/duplications, and 22 complex rearrangements [3]. As a result of any of these mutations, the activity of the enzyme iduronate-2-sulfatase (EC 3.1.6.13) is either reduced or absent, which leads to storage of dermatan sulfate (DS) and heparan sulfate (HS) within the lysosomes [1,2,4]. MPS VI (MIM# 253200), or Maroteaux-Lamy syndrome, is an autosomal recessive inherited LSD that results from defects in the ARSB gene which cause the complete or partial loss of enzymatic activity of arylsulfatase B (or N-acetylgalactosamine-4-sulfatase ; EC 3.1.6.12), leading to the accumulation of dermatan sulfate (DS) inside the lysosome [1,2,4,5]. The mutational spectrum of MPS VI includes over 200 variants known to cause the disorder, from missense/nonsense (188, the vast majority) to small deletions, insertions or indels (28, 9 and 2, respectively). A few splicing mutations (16) and some major deletions (8) have also been reported. Unlike MPS II, however, up until now, no major insertions/duplications or complex rearrangements have been reported for MPS VI [3].
The prevalence of MPSs varies among ethnic groups, which will influence which type of MPS dominates a geographic area. When observing the prevalence of all MPS types in 33 countries, it was possible to conclude that Saudi Arabia provided the highest frequency, followed by Portugal, Brazil, Netherlands, and Australia [2,6]. Briefly, Saudi Arabia showed a combined prevalence of 16.9 per 100,000 live births, a value that is probably owed to regional or consanguineous marriages, or even to founder effects. Portugal, on the other hand, has a combined birth prevalence of 4.8 per 100,000 live births, while Netherlands and Australia have 4.5 and 4.46 per 100,000 live births, respectively [2,6]. In 2004, a study about the prevalence of LSDs in Portugal revealed that MPS II comprised 34% and MPS VI corresponded to 16% of all MPSs. In Saudi Arabia, MPS VI has the highest birth prevalence, comprising 46% of all the MPS cases diagnosed. Several other countries reported MPS II as the MPS with highest birth prevalence, as it is the example the Brazilian population, and that accounted for 37% of all MPS cases followed by MPS VI. Since the data from the Portuguese population presented high prevalence values of MPS II and VI these two types of MPSs were chosen to be focused in our study [2,6,7]. It is however worth mentioning that even though there is a relatively uniform distribution of MPSs along the country, MPS I and II are actually exceptions to that pattern. According to that 2004 report, the first one (MPS I) represented 13% of all MPS cases in the North and 35% in the rest of the country; in contrast, MPS II represented 34 and 8% of the MPS in the same respective regions. Therefore, depending on the region, the most prevalent form/type of MPS in Portugal may also MPS I [7]. Unfortunately, because of sample availability, MPS I could not be included in this study.
At the clinical level, MPS II and VI share many features being both characterized by a chronic and progressive course, multisystem involvement, organomegaly, dysostosis multiplex and coarse facial features. Hearing, vision, cardiovascular function and join mobility may be also affected. Both range from severe to attenuated forms that present different onset, rate of progression and degree of clinical symptoms. There is, however, a fundamental difference between both disorders: in the severe forms of MPS II cognitive decline is present whereas in MPS VI patients cognition is usually normal [1,2]. MPS II attenuated form allows the individuals to reach adulthood while with the severe form patients have a life span of approximately two decades. In MPS VI, the slowly progressing forms generally present less pronounced or even atypical symptoms , which make them more difficult to diagnose. Therefore, the age of diagnosis can go from 9 to 42 years old, with an average of 23.5 years [8]. Enzyme replacement therapy (ERT) is available for both diseases. ERT is based on the infusion of a recombinant form of the defective enzyme that leads to a decrease in GAGs storage, and to a somatic improvement [1,9]. Despite their overall success, currently available ERT have some disadvantages, like their high cost and the lifelong dependency on frequent infusions, which is caused by the limited half-life time of the recombinant enzymes. Furthermore, those infusions can elicit adverse reactions, ranging from rash, angioedema and bronchoconstriction to anaphylaxis, all of which have already been reported in MPS II and MPS VI patients [10,11]. In fact, many patients develop antibodies to the recombinant enzyme (anti-ERT antibodies). Altogether, this means that immune system may have an impact on ERT tolerability and on its long-term efficacy. That is why immunological responses to ERTs are still a topic that continues to be studied as it has not yet been definitely clarified if high-titer anti-ERT antibodies may, at least in some cases, reduce the treatment’s efficacy [12,13]. It is also worth mentioning that the currently approved therapies are not able to reach several tissues (including bones, cartilage, eye, heart valves and CNS) and to reduce, for instance, neuroinflammation [14], a process that is now known to play a key role in the overall MPS pathology. In fact, understanding the context, course, and duration of these neuroinflammatory responses has been the focus of numerous teams over the last years, who aim to understand and, ultimately correct their corresponding physiological, biochemical and behavioral consequences [15,16,17,18,19]. Nowadays, promising solutions are being actively studied; they include alternative administration routes for ERT, ERT with fusion proteins that bypass the blood brain-barrier, gene therapy, substrate reduction therapy, anti-inflammatory and pharmacological read-through drugs [1,5,12,20,21].
MPS patients have recurrent upper respiratory infections, pneumonia, bronchitis, and middle ear infections, namely in those affected with the severe forms [5,22,23]. However, it is not well documented if these episodes are caused by the generally poor health status of the severely affected patients, or by specific immune alterations. It is known that GAGs accumulation in MPSs is associated with inflammation, namely in the joints and in the central nervous system [15,24]. The existing body of research on MPSs suggests that GAG fragments may work as an endogenous danger-associated molecular pattern (DAMP), which would initiate an innate immune signaling pathway [5,25]. GAGs can be either exocytosed and bind to Toll-like receptor 4 (TLR4) or released intracellularly due to lysosomal disruption [5,17,20,26]. Indeed, GAGs are structurally similar to the canonical ligand for TLR4, which is lipopolysaccharide (LPS) [26]. Upon binding to TLR4, DAMP GAGs promote myeloid differentiation primary response (88 MyD88) adaptor protein recruitment, which, in turn, leads to the assembly of an interleukin receptor associated kinase (IRAK)4/IRAK1/IRAK2 /TRAF6 complex. This complex then binds to a secondary one, TGF-β activated kinase (TAB) 2/TAK1/TAB1, allowing TRAF6 to dislocate from IRAK1 and move into the cytoplasm. In there, it can activate the NF-Kb pathway, which promotes pro-interleukin-1 β (pro-IL-1β) and NLR family pyrin domain containing 3 (NLRP3) transcription and transcriptional induction of other pro-inflammatory cytokines, culminating in the activation of NLRP3 inflammasome. Through cleavage of caspase-1, mature IL-1β will be secreted, together with the other pro-inflammatory cytokines and, all together, these will cause microglia and astrocyte activation, pyroptosis, and neuronal degeneration [20].
In addition to the alterations in innate immunity, the lysosomal alteration present in MPS can support the investigation of acquired immunity deficiencies since the lysosome has a crucial role in antigen processing, autophagy, and perforin-induced lysis [17,20]. Several studies have revealed that MPS mouse models present irregularities in the function of the immune system. GAGs interact with a wide range of proteins, including chemokines, cytokines, growth factors, enzymes, and adhesion molecules, involved in physiological and pathological processes. Therefore, GAGs might induce inflammation in different cells [27]. Simonaro et al. examined articular chondrocytes from rats with MPS VI and showed an age-progressive increase in the number of apoptotic chondrocytes, also these cells released more nitric oxide (NO) and tumor necrosis factor alpha (TNF-α) when compared to control chondrocytes. It was suggested that apoptosis induction by accumulated GAG fragments might play an important role in cartilage destruction and degenerative joint disease in MPS VI [19]. Tessitore et al studied the relationship between storage and secondary events, such as autophagy, polyubiquitination, mitochondrial function, inflammation, and apoptosis, in cells and tissues of MPS VI affected rats. They detected abundant CD68-positive monocyte/macrophage cells as well as apoptotic cells in liver, spleen, and kidney sections of affected MPS rats compared with controls [28]. In MPS I mouse model, a decrease of circulating CD4+, CD8+ T cells as well as dendritic cells (with decreased expression of cell surface CD123 and CD86) was reported [29]. A murine model for MPS VII showed reduced T cell proliferative response and decreased antibody production after immunization with antigens, possibly due to defective antigen processing [30]. In contrast, in a mouse model of MPS IIIB, there was increased T cell proliferation with mice showing increased numbers of splenic B cells as well as CD4+ and CD8+ T cells [17,31]. Concerning patients with MPS, much less data is available. A recent study reported significantly lower Ig levels, namely IgM and IgA, in the MPS patients comparing with patients with other LSDs [32].
Thus, a wide range of alterations in the immune system were reported in different MPS diseases and although a correlation between innate immunity and MPS has already been established, there is still insufficient data about how the adaptive immune system is affected in MPS patients. Given this, herein, we performed a detailed characterization of lymphocyte populations in MPS II and MPS VI diseases patients. More specifically, we analyzed the B, T, Natural Killer (NK) and invariant natural killer T (iNKT) cell populations.
2. Materials and Methods
Population studied
This study was approved by the Ethical Committees of University Hospital Center São João, Porto, Portugal, that comprises São João Hospital and Valongo Hospital, University Hospital Santo António (Porto, Portugal), Lisbon Academic Medical Center - Santa Maria Hospital – North Lisbon University Hospital Center (Lisbon, Portugal), Pediatric Hospital – University Hospital of Coimbra (Coimbra, Portugal) and Hospital de Clínicas de Porto Alegre (Porto Alegre, Brazil). The informed consent was obtained from participants according to the Helsinki declaration.
Patient Population
Ten male MPS II disease patients (mean age: 15.4 ± 6.6 years; range 7–28 years) and sixteen MPS VI disease patients, comprising eight males and eight females (mean age: 15.4 ± 6.1 years; range 4–27 years), were included in this study. These patients were regularly followed up at São João Hospital, Santo António Hospital, Santa Maria Hospital, Coimbra Pediatric Hospital and Hospital de Clínicas de Porto Alegre (Porto Alegre, Brazil). With the exception of two MPS II disease patients (both of them males with 12 and 17 years old), all individuals in both groups were already under ERT at the time of the analysis.
Control Population
Twenty-seven subjects who were apparently healthy were used as controls, including eighteen males and nine females (mean age: 16.8 ± 7 years; range 6–30 years). The control group was age matched with the patient group. The adult control group was composed by blood donors at the Blood Bank of São João Hospital and the children control group by children undergoing orthopaedic surgery at Valongo Hospital (Valongo, Portugal).
Isolation of Mononuclear Cells from Human Peripheral Blood and Flow Cytometry
Peripheral blood mononuclear cells (PBMCs) were isolated by Histopaque-1077® (Sigma) density gradient centrifugation following the manufacturer’s instructions. Human PBMCs were stained with fluorochrome-conjugated antibody/tetramer mixes diluted in PBS/0.2%BSA/0.1%NaN3 (flow cytometry solution) for 20 minutes, at 4°C, in the dark. To identify and characterize phenotypically the T cell, iNKT cell, NK cell, and B cell populations, the following monoclonal antibodies and tetramer were used: PerCP-Cy5.5-labeled anti-human CD3 (SK7) antibody (eBioscience); PE-Cy7-labeled anti-human CD4 (RPA-T4) antibody (eBioscience); APC-eFluor®780-labeled anti-human CD8 (RPA-T8) antibody (eBioscience); FITC-labeled anti-human CD19 (HIB19) antibody (eBioscience); APC-labeled anti-human CD45RA (MEM-56) antibody (ImmunoTools); eFluor®450-labeled anti-human CD161 (HP-3G10) antibody (ImmunoTools); FITC-labeled anti-human CCR7 (150503) antibody (R&D Systems); and PE-labeled PBS57-loaded hCD1d tetramer (National Institute of Health Tetramer Core Facility, Emory University, Atlanta, GA, USA). After staining, PBMCs were washed with flow cytometry solution and fixed with PBS 1% formaldehyde. In each flow cytometry experiment the controls used were: unstained sample; single stain samples for compensation; unloaded CD1d tetramer (to confirm the specificity of the CD1d PBS57 loaded tetramer staining) and healthy subject control. Sample acquisition was performed in a 3-laser BD FACSCanto™ II (BD Biosciences) flow cytometer, using the BD FACSDiva™ software (BD Biosciences). All flow cytometry analyses were performed using the FlowJo software (Tree Star).
Statistical analysis
All statistical analyses were performed using the GraphPad Prism 6 software. The Shapiro–Wilk test was applied to determine the normal distribution of the variables. For the comparison of a variable between all groups (controls, MPS II and MPS VI disease patients) the parametric one-way ANOVA test was applied when variables were normally distributed, while the non-parametric Kruskal-Wallis test was used for variables that did not follow a normal distribution. For the comparison between two groups (controls versus MPS II disease patients) t-student or Mann-Whitney tests were used. Values of p<0.05 were considered statistically significant.
3. Results
3.1. Major leukocyte populations in MPS II and VI disease patients
We analyzed the peripheral blood mononuclear cells (PBMCs) of MPS II and MPS VI disease patients and compared with control subjects to infer whether GAGs storage could interfere with immune cell composition.
PBMCs consist of monocytes, identified by the expression of CD14, and lymphocytes. Lymphocytes are composed by cells of innate immunity as NK cells, identified by CD56+CD3- expression, and cells of the adaptive immunity, the B and T cells, identified by CD19 and CD3 expression, respectively. The gating strategy to identify these cells population is described in Figure 1A.
When comparing with control subjects, both MPS II and MPS VI disease patient groups showed normal levels of T cells [33] and B cells (Figure 1B). However, we found a significant decrease in the percentage of NK cells (p=0.0107) and monocytes (p=0.0305) in MPS VI disease patients when compared with the control group (Figure 1B). Importantly, this alteration was absent in MPS II disease patients. This suggests that NK cells and monocytes seem to be affected in a disease-specific manner.
3.2. Naïve vs Memory T cell pool in MPS II and VI disease patients
T Lymphocytes are divided into different major functional groups: cytotoxic, identified by the expression of CD8 and helper cells, with a major immune modulatory role, identified by the expression of CD4 (Figure 2A). No alterations in the percentage of helper CD4+ and cytotoxic CD8+ T cells were observed between MPS II and MPS VI disease patients when compared to control subjects [33] (Figure 2B,C). Surprisingly, an increase of T helper cells was found in MPS VI disease patients (p=0.0333) as compared with MPS II disease patients (Figure 2B).
The T helper and cytotoxic T cell populations were further characterized in terms of antigen experience into: naïve T cell (CCR7+CD45RA+), central memory (CCR7+CD45RA-), effector memory (CCR7-) (Figure 1A) in MPS II and MPS VI disease patients. In comparison with the control group, MPS VI disease patients presented a significant increase of naïve CD4+ T cells (p=0.0029) and naïve CD8+ T cells (p=0.0037), with a concomitant decrease of central memory CD4+ T cells (p=0.0004) and effector memory CD8+ T cells (p=0.03) (Figure 2D–I). Importantly, these differences cannot be attributed to age differences between MPS VI disease patients and control subjects, since they were age-matched. On the contrary, MPS II disease patients did not show alterations in the memory state of both helper and cytotoxic T cells as compared with the control population, once again suggesting that these alterations are specific of MPS VI disease patients.
3.3. iNKT cells are normal in number but phenotypically altered in MPS II and MPS VI disease patients
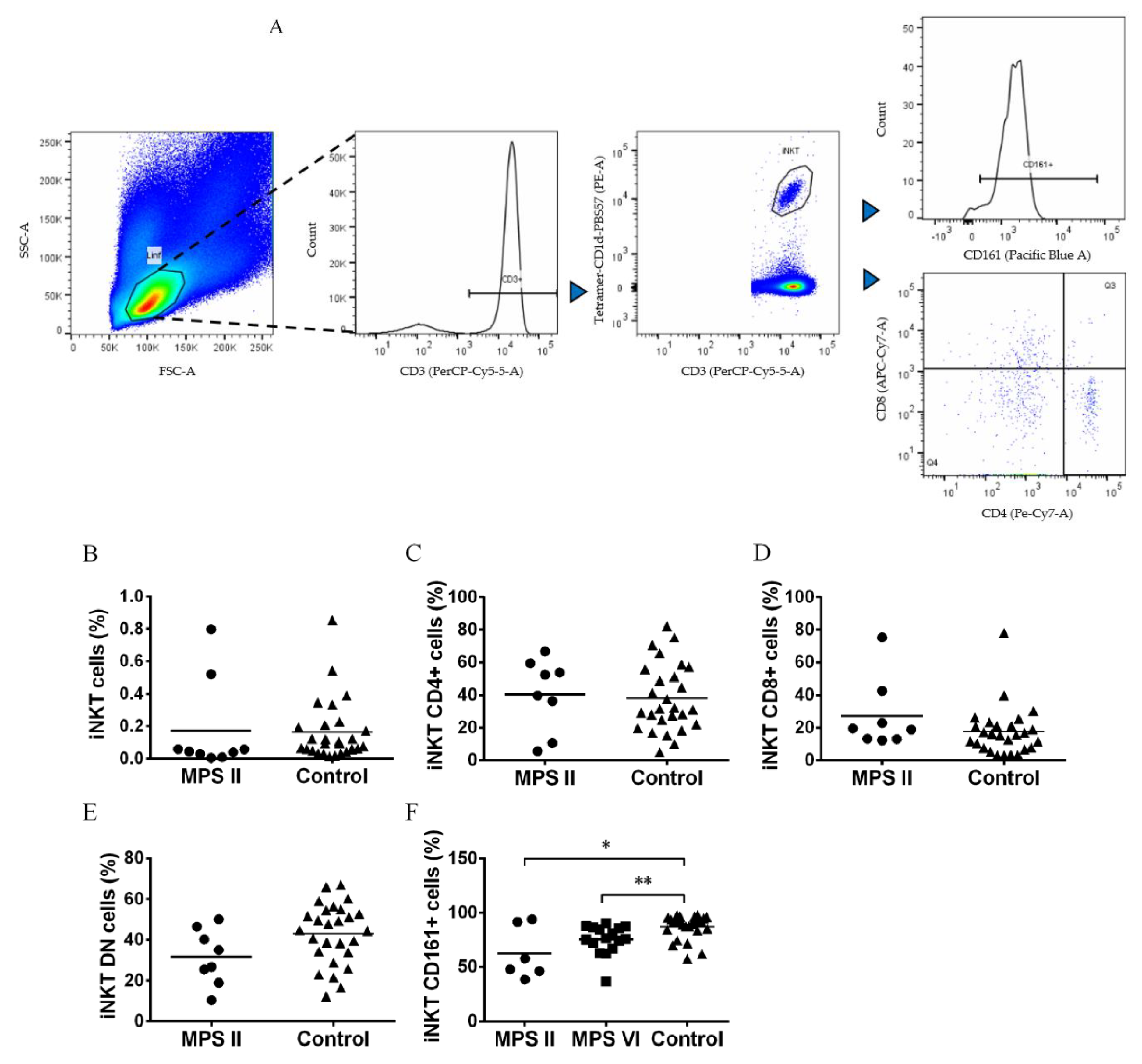
Invariant natural killer T cells (iNKT) are lymphocyte acting in innate and adaptive immunity. They are identified by using CD1d loaded with aGalCer analogous (Figure 3A). iNKT cells are altered in several LSDs animal models and in acid sphingomyelinase deficiency patients [34,35,36,37]. Thus, we also analyzed the percentage of iNKT cells within the lymphocyte population by CD3 expression and reactivity to the PBS57 loaded CD1d tetramer and iNKT cell phenotype by the expression of CD4, CD8 and CD161 (Figure 3A). We previous observed no alterations in the frequency and CD4/CD8 phenotype of iNKT cells in MPS VI disease patients [33]. Here we extended this characterization to MPS II disease patients and went further in the analyses of iNKT cell phenotype by analyzing CD161 expression, an important NK cell marker. In MPS II disease patients we found normal levels of iNKT cells among total T cells (Figure 3B) and a normal iNKT CD4/CD8/DN phenotype (Figure 3C–E). However, MPS II disease patients showed a decrease in the percentage of iNKT cells expressing the NK cell marker CD161 (p=0.0364) as compared with control subjects (Figure 3F). Because of this data it was also investigated the expression of CD161 for MPS VI disease patients, and in accordance with what was seen for MPS II disease patients, iNKT cells from MPS VI disease patients have a lower expression of CD161 when compared to control (p=0.0087, Figure 3F).
4. Discussion
In the present study, we analyzed several immunological parameters in MPS II and MPS VI disease patients to investigate leukocyte modifications in order to understand if immune cells are altered in a disease-specific way and if they could play a role in the clinical course of the disease. We found a decrease in NK cells and monocytes in MPS VI disease patients, as well as alterations in the memory status of both cytotoxic and T helper cells in these patients. Importantly, these alterations were not present in MPS II disease patients. Additionally, we found a decrease in iNKT cells expressing the marker CD161 in both MPS VI and MPS II disease subjects.
A reduction of NK cells had already been described in patients with other LSDs like Gaucher disease [38,39] and Niemann-Pick disease type C1 [40] and could potentially be due to a decrease in shingosine-1-phosphate (S1P) gradients and to defects in calcium storage. Monocytes were also decreased in Fabry disease patients´ blood, which could be related to an increase of extravasation processes with higher migration to peripheral tissues, since monocytes overexpress CD31, a molecule implied in extravasation processes [41]. The reduction of monocytes blood frequencies were also described in Gaucher disease. This reduction could be associated with the decrease in the CXCR4 expression (SDF1α receptor) that consequently leads to an inefficient CXCR4-SDF1α binding capacity and to a decline of monocyte SDF1α-dependent migration [42]. When compared to healthy children, children with Gaucher disease reported increases of activated T cells, T helper cells and cytotoxic T cells [43,44]. These diseases share the characteristic of having sphingolipids as the main storage material. Here, we show for the first time that patients accumulating GAGs also develop defects in the frequency of these populations. The previous demonstration of disease-specific storage material in monocytes from MPS VI disease patients [45] led to the hypothesis that the accumulated GAGs might have a toxic effect in these cells, which may then cause a higher cellular apoptotic rate, thus explaining the decrease found. However, it is also possible that this reduction is mediated by an increased recruitment of monocytes to peripheral sites.
It is known that the lack or deficiencies in NK cells are associated with an increased susceptibility to pulmonary infections, a complication of MPS that often leads to death. This latter information was demonstrated in patients with total absence or functional abnormalities of NK cells [46,47]. This critical role of NK cells was also saw in a study with mice models of IL-15 KO deficient in NK cells and wild type mice depleted of NK cells, that were highly susceptible to pulmonary staphylococcal infection [48]. Consequently, our results may contribute to the explanation of the increased vulnerability to respiratory infections in MPS VI disease patients.
Interestingly, despite a normal percentage of total T cells, we show that MPS VI disease patients have an increase in the percentage of naïve helper and cytotoxic T cells, with concomitant decreases of their memory phenotypes, in comparison with the control group. Of note, these variations are not age-related, given that MPS VI disease patients and control subjects were age-matched. These alterations were not present in MPS II disease patients. Interestingly, in MPS I mouse model, which accumulates both dermatan and heparan sulfate (as MPS II disease patients), a reduction of peripheral blood memory CD4+ T cells was also found [29]. This indicates besides the nature of the accumulated material, the specific enzymatic defect may have an impact on immune cell homeostasis. A reduction in memory T cells might have important consequences for adaptive immune responses. The presence of a lower-number of pathogen specific T cells delays the response, increasing the risk of succumbing to infection. Thus, our results support the idea that the administration of vaccines against respiratory pathogens to increase the pool of pathogen-specific memory T cells in MPS VI disease patients, might be beneficial [49]. Nevertheless, we cannot exclude that T cells may fail to expand even after vaccination with a concomitant decrease in memory cells [50,51]. Indeed patients with alpha-mannosidosis have a reduced response to vaccination when compared to healthy matched controls [50]. Furthermore, in mucolipidosis II (MLII) patients, the specific antibody response to vaccination is poor or not detectable, which is consistent with MLII mouse data. MLII patients have low levels of memory B cells which indicates impaired B cell maturation in response to antigen and defective immunoglobulin class switch, which prevents an effective response against pathogens [51].
Lysosomal integrity is important for iNKT cell development [36]. iNKT cells are immunomodulatory cells previously found to be decreased in several mouse models of LSDs [34,35,36,52,53,54] and in patients with acid sphingomyelinase deficiency [37]. In the present study, we found no differences in the frequency of iNKT cells between MPS II disease patients and control subjects, similar to what we had described for MPS VI disease patients [33]. However, we found a decreased expression of the maturation marker CD161, which is acquired through interactions with CD1d in the periphery [55] in both MPS II and MPS VI disease patients. The presence of alterations in both types of MPS suggests that this population might be particularly sensitive to lysosomal storage of GAGs.
A limitation of this study is the absence of information about patient’s genetic variants of IDS and ARSB genes, that would be important for correlation studies between the immunological findings and genetic variants.
All MPS VI disease patients and eight out of the total ten MPS II disease patients were under ERT at the time of the study. Therefore, we cannot exclude that some immunological alterations can be related to ERT. It would be important to include more untreated patients in this study to clarify this. Nevertheless, since most patients are under treatment, the clinical relevance of our findings persists.
5. Conclusion
In conclusion, with this study we found a reduction in NK cells and monocytes in MPS VI disease patients, as well as alterations in the memory status of both cytotoxic and T helper cells in these patients. But, in MPS II disease patients no alterations were seen in these cells. In addition, we detected a decrease in the expression of the marker CD161 in iNKT cells, both in MPS VI and MPS II disease patients.
These results lead to believe that the type of substrate accumulated or enzyme deficiency in the lysosome may influence the normal cellular composition of the immune system producing MPS VI disease-specific alterations in some leukocyte populations.
Author Contributions
Conceptualization, MFM.; methodology, NL, MLM, CSP, IMR and MFM; validation, NL, MLM, CSP and MFM.; formal analysis, NL, MLM, CSP, IMR and MFM.; investigation, NL, MLM, CSP.; resources, EM, RR, AG, PA, PG, MTC, ER, ELT and RG; writing—original draft preparation, CSP.; writing—review and editing, IMR, MFC, SA and MFM.; supervision, MFM.; project administration, MFM.; funding acquisition, MFM. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by National Funds through FCT—Fundação para a Ciência e a Tecnologia, I.P., under the project UIDB/04293/2020
Institutional Review Board Statement
The study was conducted in accordance with the Declara-tion of Helsinki, and approved by the Institutional Review Boards of: Comitê de Ética em Pesquisa do Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil (protocol code 03-066, date of approval February 14th 2003); Centro Hospitalar de Porto, Porto, Portugal (protocol code 152/13(098-DEFI/124-CES), date of approval May 29th 2013); Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal (protocol code CHUC-48-12, date of approval July 23rd 2013); Centro Hospitalar Lisboa Norte, Lisboa, Portugal (protocol code DIR-CLN-27MAI2013-0179, date of approval May 24th 2013); Centro Hospitalar de São João, Porto, Portugal (protocol code 149/12, date of approval July 27th 2012 and protocol code 212/18, date of approval September 13th 2018).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to confidentially of human subject data.
Acknowledgments
The authors would like to thank the patients and their families for their collaboration, all the medical and nurse staff from all the difference hospitals. The ImmunoHemotherapy department of Hospital de São João (Porto, Portugal) for blood samples of control subjects in special the support of Dr. Cristina Neves, the Tetramer core facility of the National Institute of Health (NIH, USA), for providing the CD1d-PBS57 tetramer. The authors acknowledge the support of the Translational Cytometry Unit i3S Scientific Platform. The authors acknowledge Mariana Miranda for manuscript English editing.
Conflicts of Interest
PA received a Grant / research support from Takeda; honoraria from Takeda, Sanofi-Genzyme, Biomarin, Ultragenyx, Alexion, Amicus and Chiesi. MFM received a grant from Sanofi-Genzyme and honoraria from Takeda. The other authors declare no conflict of interest.
References
- M. F. Coutinho, L. Lacerda, and S. Alves, ‘Glycosaminoglycan Storage Disorders: A Review’, Biochem. Res. Int., vol. 2012, pp. 1–16, 2012. [CrossRef]
- S. A. Khan et al., ‘Epidemiology of mucopolysaccharidoses’, Mol. Genet. Metab., vol. 121, no. 3, pp. 227–240, Jul. 2017. [CrossRef]
- ‘Quiagen Site’, QIAGEN Digital Insights. https://my.qiagendigitalinsights.com/ (accessed Feb. 09, 2023).
- R. Giugliani et al., ‘Mucopolysaccharidosis I, II, and VI: brief review and guidelines for treatment’, Genet. Mol. Biol., vol. 33, no. 4, pp. 589–604, Nov. 2010. [CrossRef]
- H. Parker and B. W. Bigger, ‘The role of innate immunity in mucopolysaccharide diseases’, J. Neurochem., p. jnc.14632, Dec. 2018. [CrossRef]
- B. Celik, S. C. Tomatsu, S. Tomatsu, and S. A. Khan, ‘Epidemiology of Mucopolysaccharidoses Update’, Diagnostics, vol. 11, no. 2, p. 273, Feb. 2021. [CrossRef]
- R. Pinto et al., ‘Prevalence of lysosomal storage diseases in Portugal’, Eur. J. Hum. Genet., vol. 12, no. 2, pp. 87–92, Feb. 2004. [CrossRef]
- F. D’Avanzo, A. Zanetti, C. De Filippis, and R. Tomanin, ‘Mucopolysaccharidosis Type VI, an Updated Overview of the Disease’, Int. J. Mol. Sci., vol. 22, no. 24, p. 13456, Dec. 2021. [CrossRef]
- V. Valayannopoulos and F. A. Wijburg, ‘Therapy for the mucopolysaccharidoses’, Rheumatology, vol. 50, no. suppl 5, pp. v49–v59, Dec. 2011. [CrossRef]
- B. K. Burton and D. A. H. Whiteman, ‘Incidence and timing of infusion-related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real-world setting: A perspective from the Hunter Outcome Survey (HOS)’, Mol. Genet. Metab., vol. 103, no. 2, pp. 113–120, Jun. 2011. [CrossRef]
- P. Harmatz et al., ‘Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase’, Mol. Genet. Metab., vol. 94, no. 4, pp. 469–475, Aug. 2008. [CrossRef]
- A. Broomfield, S. A. Jones, S. M. Hughes, and B. W. Bigger, ‘The impact of the immune system on the safety and efficiency of enzyme replacement therapy in lysosomal storage disorders’, J. Inherit. Metab. Dis., vol. 39, no. 4, pp. 499–512, Jul. 2016. [CrossRef]
- D. Concolino, F. Deodato, and R. Parini, ‘Enzyme replacement therapy: efficacy and limitations’, Ital. J. Pediatr., vol. 44, no. S2, p. 120, Nov. 2018. [CrossRef]
- B. Donida et al., ‘Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy’, Biochim. Biophys. Acta BBA - Mol. Basis Dis., vol. 1852, no. 5, pp. 1012–1019, May 2015. [CrossRef]
- G. M. Viana, D. A. Priestman, F. M. Platt, S. Khan, S. Tomatsu, and A. V. Pshezhetsky, ‘Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms’, J. Clin. Med., vol. 9, no. 2, p. 396, Feb. 2020. [CrossRef]
- B. W. Bigger, D. J. Begley, D. Virgintino, and A. V. Pshezhetsky, ‘Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders’, Mol. Genet. Metab., vol. 125, no. 4, pp. 322–331, Dec. 2018. [CrossRef]
- L. D. Archer, K. J. Langford-Smith, B. W. Bigger, and J. E. Fildes, ‘Mucopolysaccharide diseases: A complex interplay between neuroinflammation, microglial activation and adaptive immunity’, J. Inherit. Metab. Dis., vol. 37, no. 1, pp. 1–12, Jan. 2014. [CrossRef]
- E. Fusar Poli et al., ‘Murine neural stem cells model Hunter disease in vitro: glial cell-mediated neurodegeneration as a possible mechanism involved’, Cell Death Dis., vol. 4, no. 11, pp. e906–e906, Nov. 2013. [CrossRef]
- C. M. Simonaro, M. E. Haskins, and E. H. Schuchman, ‘Articular Chondrocytes from Animals with a Dermatan Sulfate Storage Disease Undergo a High Rate of Apoptosis and Release Nitric Oxide and Inflammatory Cytokines: A Possible Mechanism Underlying Degenerative Joint Disease in the Mucopolysaccharidoses’, Lab. Invest., vol. 81, no. 9, pp. 1319–1328, Sep. 2001. [CrossRef]
- i. Mandolfo, H. Parker, and B. Bigger, ‘Innate Immunity in Mucopolysaccharide Diseases’, Int. J. Mol. Sci., vol. 23, no. 4, p. 1999, Feb. 2022. [CrossRef]
- R. Parini and F. Deodato, ‘Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations’, Int. J. Mol. Sci., vol. 21, no. 8, p. 2975, Apr. 2020. [CrossRef]
- G. M. Campo et al., ‘Glycosaminoglycans modulate inflammation and apoptosis in LPS-treated chondrocytes’, J. Cell. Biochem., vol. 106, no. 1, pp. 83–92, Jan. 2009. [CrossRef]
- i. M. Naur, A. Anggraini, B. W. Indraswari, S. Wandita, T. Wibowo, and E. L. Haksari, ‘Immunisation issues in patient with mucopolysaccharidosis: A case report’, Med. J. Malaysia, vol. 75, no. Suppl 1, pp. 51–52, May 2020.
- V. Opoka-Winiarska, A. Jurecka, A. Emeryk, and A. Tylki-Szymańska, ‘Osteoimmunology in mucopolysaccharidoses type I, II, VI and VII. Immunological regulation of the osteoarticular system in the course of metabolic inflammation’, Osteoarthritis Cartilage, vol. 21, no. 12, pp. 1813–1823, Dec. 2013. [CrossRef]
- D. A. Simon Davis and C. R. Parish, ‘Heparan Sulfate: A Ubiquitous Glycosaminoglycan with Multiple Roles in Immunity’, Front. Immunol., vol. 4, 2013. [CrossRef]
- G. B. Johnson, G. J. Brunn, Y. Kodaira, and J. L. Platt, ‘Receptor-Mediated Monitoring of Tissue Well-Being Via Detection of Soluble Heparan Sulfate by Toll-Like Receptor 4’, J. Immunol., vol. 168, no. 10, pp. 5233–5239, May 2002. [CrossRef]
- N. S. Gandhi and R. L. Mancera, ‘The Structure of Glycosaminoglycans and their Interactions with Proteins’, Chem. Biol. Drug Des., vol. 72, no. 6, pp. 455–482, Dec. 2008. [CrossRef]
- A. Tessitore, M. Pirozzi, and A. Auricchio, ‘Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI’, PathoGenetics, vol. 2, no. 1, p. 4, Dec. 2009. [CrossRef]
- L. D. Archer, K. J. Langford-Smith, W. R. Critchley, B. W. Bigger, and J. E. Fildes, ‘Characterisation of the T cell and dendritic cell repertoire in a murine model of mucopolysaccharidosis I (MPS I)’, J. Inherit. Metab. Dis., vol. 36, no. 2, pp. 257–262, Mar. 2013. [CrossRef]
- T. M. Daly, R. G. Lorenz, and M. S. Sands, ‘Abnormal Immune Function In Vivo in a Murine Model of Lysosomal Storage Disease’, Pediatr. Res., vol. 47, no. 6, pp. 757–762, Jun. 2000. [CrossRef]
- J. DiRosario et al., ‘Innate and adaptive immune activation in the brain of MPS IIIB mouse model’, J. Neurosci. Res., vol. 87, no. 4, pp. 978–990, Mar. 2009. [CrossRef]
- S. Kilavuz et al., ‘Real-world patient data on immunity and COVID-19 status of patients with MPS, Gaucher, and Pompe diseases from Turkey’, Arch. Pédiatrie, vol. 29, no. 6, pp. 415–423, Aug. 2022. [CrossRef]
- C. S. Pereira et al., ‘Lipid Antigen Presentation by CD1b and CD1d in Lysosomal Storage Disease Patients’, Front. Immunol., vol. 10, p. 1264, Jun. 2019. [CrossRef]
- S. D. Gadola et al., ‘Impaired selection of invariant natural killer T cells in diverse mouse models of glycosphingolipid lysosomal storage diseases’, J. Exp. Med., vol. 203, no. 10, pp. 2293–2303, Oct. 2006. [CrossRef]
- M. F. Macedo, R. Quinta, C. S. Pereira, and M. C. Sa Miranda, ‘Enzyme replacement therapy partially prevents invariant Natural Killer T cell deficiency in the Fabry disease mouse model’, Mol. Genet. Metab., vol. 106, no. 1, pp. 83–91, May 2012. [CrossRef]
- C. Pereira, H. Ribeiro, and M. Macedo, ‘From Lysosomal Storage Diseases to NKT Cell Activation and Back’, Int. J. Mol. Sci., vol. 18, no. 3, p. 502, Feb. 2017. [CrossRef]
- E. Melum et al., ‘Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin’, Nat. Immunol., vol. 20, no. 12, pp. 1644–1655, Dec. 2019. [CrossRef]
- A. M. Zahran et al., ‘Upregulation of Cytotoxic T-cells in pediatric patients with Gaucher disease’, Sci. Rep., vol. 12, no. 1, p. 4977, Mar. 2022. [CrossRef]
- Y. Burstein, V. Zakuth, G. Rechavi, and Z. Spirer, ‘Abnormalities of cellular immunity and natural killer cells in Gaucher’s disease’, J. Clin. Lab. Immunol., vol. 23, no. 3, pp. 149–151, Jul. 1987.
- A. O. Speak et al., ‘Altered distribution and function of natural killer cells in murine and human Niemann-Pick disease type C1’, Blood, vol. 123, no. 1, pp. 51–60, Jan. 2014. [CrossRef]
- P. Rozenfeld, E. Agriello, N. De Francesco, P. Martinez, and C. Fossati, ‘Leukocyte perturbation associated with Fabry disease’, J. Inherit. Metab. Dis., vol. 32, no. S1, pp. 67–77, Dec. 2009. [CrossRef]
- N. Bettman, I. Avivi, H. Rosenbaum, L. Bisharat, and T. Katz, ‘Impaired migration capacity in monocytes derived from patients with Gaucher disease’, Blood Cells. Mol. Dis., vol. 55, no. 2, pp. 180–186, Aug. 2015. [CrossRef]
- M. Zahran, A. A. Eltayeb, K. I. Elsayh, K. Saad, F.-A. Ahmad, and A. I. M. Ibrahim, ‘Activated and Memory T Lymphocytes in Children with Gaucher Disease’, Arch. Immunol. Ther. Exp. (Warsz.), vol. 65, no. 3, pp. 263–269, Jun. 2017. [CrossRef]
- D. Rigante, C. Cipolla, U. Basile, F. Gulli, and M. C. Savastano, ‘Overview of immune abnormalities in lysosomal storage disorders’, Immunol. Lett., vol. 188, pp. 79–85, Aug. 2017. [CrossRef]
- B. C. Kieseier, K. E. Wisniewski, and H. H. Goebel, ‘The monocyte-macrophage system is affected in lysosomal storage diseases: an immunoelectron microscopic study’, Acta Neuropathol. (Berl.), vol. 94, no. 4, pp. 359–362, Oct. 1997. [CrossRef]
- J. S. Orange, ‘Natural killer cell deficiency’, J. Allergy Clin. Immunol., vol. 132, no. 3, pp. 515–525, Sep. 2013. [CrossRef]
- J. S. Orange, ‘Human natural killer cell deficiencies’:, Curr. Opin. Allergy Clin. Immunol., vol. 6, no. 6, pp. 399–409, Dec. 2006. [CrossRef]
- C.-L. Small et al., ‘NK Cells Play a Critical Protective Role in Host Defense against Acute Extracellular Staphylococcus aureus Bacterial Infection in the Lung’, J. Immunol., vol. 180, no. 8, pp. 5558–5568, Apr. 2008. [CrossRef]
- V. Valayannopoulos, H. Nicely, P. Harmatz, and S. Turbeville, ‘Mucopolysaccharidosis VI’, Orphanet J. Rare Dis., vol. 5, no. 1, p. 5, Dec. 2010. [CrossRef]
- D. Malm, D. S. Halvorsen, L. Tranebjærg, and H. Sjursen, ‘Immunodeficiency in alpha-mannosidosis: a matched case-control study on immunoglobulins, complement factors, receptor density, phagocytosis and intracellular killing in leucocytes’, Eur. J. Pediatr., vol. 159, no. 9, pp. 699–703, Aug. 2000. [CrossRef]
- T. Otomo et al., ‘Mannose 6 phosphorylation of lysosomal enzymes controls B cell functions’, J. Cell Biol., vol. 208, no. 2, pp. 171–180, Jan. 2015. [CrossRef]
- Y. Sagiv et al., ‘Cutting Edge: Impaired Glycosphingolipid Trafficking and NKT Cell Development in Mice Lacking Niemann-Pick Type C1 Protein’, J. Immunol., vol. 177, no. 1, pp. 26–30, Jul. 2006. [CrossRef]
- N. Schrantz, Y. Sagiv, Y. Liu, P. B. Savage, A. Bendelac, and L. Teyton, ‘The Niemann-Pick type C2 protein loads isoglobotrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells’, J. Exp. Med., vol. 204, no. 4, pp. 841–852, Apr. 2007. [CrossRef]
- J. Schümann et al., ‘Differential alteration of lipid antigen presentation to NKT cells due to imbalances in lipid metabolism’, Eur. J. Immunol., vol. 37, no. 6, pp. 1431–1441, Jun. 2007. [CrossRef]
- F. W. McNab et al., ‘The Influence of CD1d in Postselection NKT Cell Maturation and Homeostasis’, J. Immunol., vol. 175, no. 6, pp. 3762–3768, Sep. 2005. [CrossRef]
Figure 1.
Major peripheral blood mononuclear cells (PBMCs) populations in MPS II, MPS VI disease patients, and control subjects. A) Gating strategy used to identify T cells (CD3+); B cells (CD19+); NK cells (CD56+ CD3-) and monocytes (CD14+) populations. B) Percentage of T cells, B cells and NK cells was determined among lymphocytes and of monocytes among PBMCs. Horizontal bars represent mean values. *p<0.05.
Figure 1.
Major peripheral blood mononuclear cells (PBMCs) populations in MPS II, MPS VI disease patients, and control subjects. A) Gating strategy used to identify T cells (CD3+); B cells (CD19+); NK cells (CD56+ CD3-) and monocytes (CD14+) populations. B) Percentage of T cells, B cells and NK cells was determined among lymphocytes and of monocytes among PBMCs. Horizontal bars represent mean values. *p<0.05.
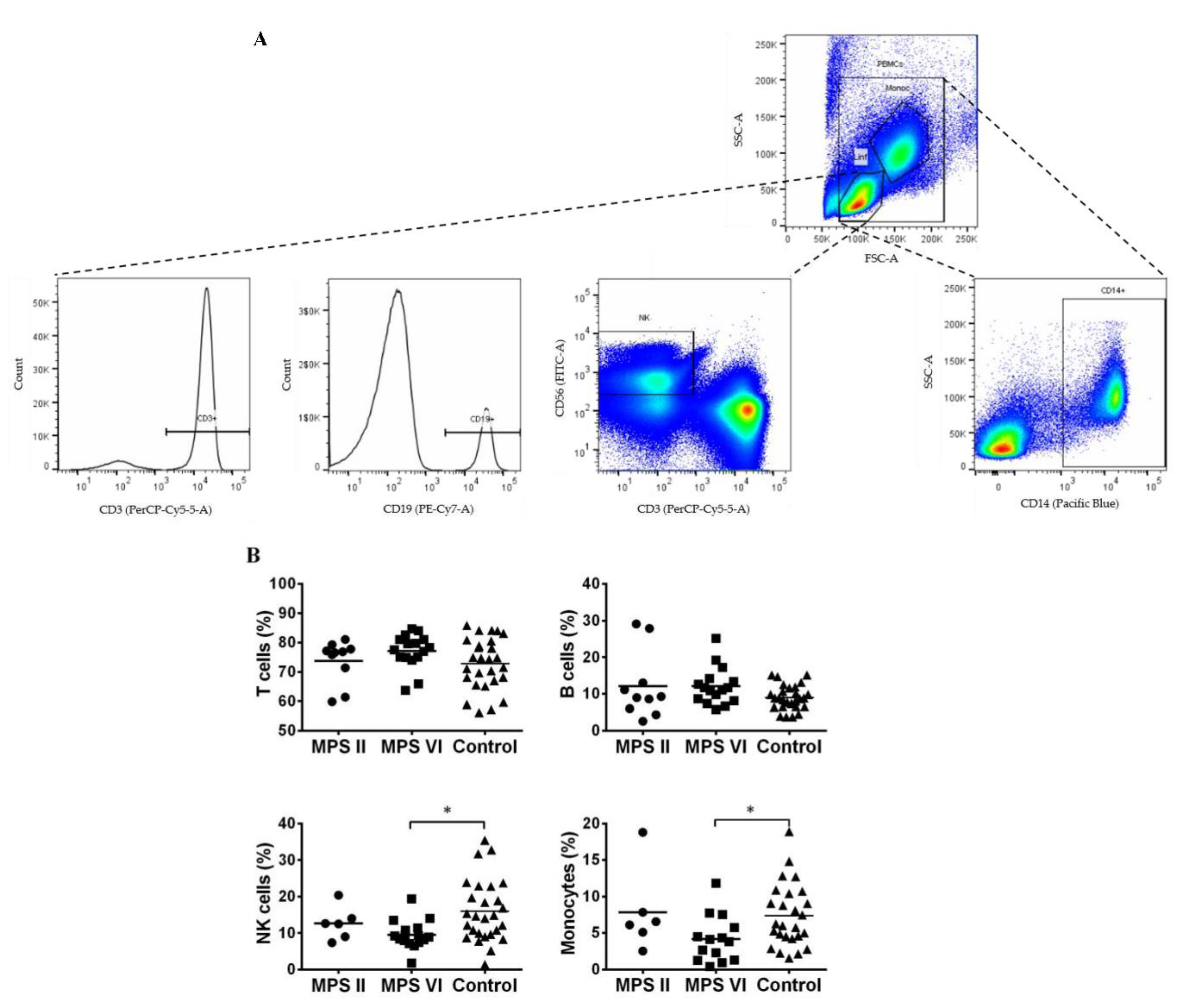
Figure 2.
Frequency of naïve, central memory, effector memory CD4+ and CD8+ T cell subsets in MPS II, MPS VI disease patients, and control subjects. A) Gating strategy applied selecting within the lymphocyte gate the populations of CD4+CD3+ (T helper cells) and CD8+CD3+ (cytotoxic T cells). Within each population: naïve, central memory (CM) and effector memory (EM) cell subset were detailed. B) Percentage of CD4+ T cells in total lymphocytes, (C) Percentage of CD8+ T cells in total lymphocytes, (D) Percentage of naïve cells in CD4+ T cells, (E) Percentage of CM cells in CD4+ T cells, (F) Percentage of EM cells in CD4+ T cells, (G) Percentage of naïve cells in CD8+ T cells, (H) Percentage of CM cells in CD8+ T cells, and (I) Percentage of EM cells in CD8+ T cells. Horizontal bars represent mean values. *p<0.05; **p<0.01; ***p<0.001.
Figure 2.
Frequency of naïve, central memory, effector memory CD4+ and CD8+ T cell subsets in MPS II, MPS VI disease patients, and control subjects. A) Gating strategy applied selecting within the lymphocyte gate the populations of CD4+CD3+ (T helper cells) and CD8+CD3+ (cytotoxic T cells). Within each population: naïve, central memory (CM) and effector memory (EM) cell subset were detailed. B) Percentage of CD4+ T cells in total lymphocytes, (C) Percentage of CD8+ T cells in total lymphocytes, (D) Percentage of naïve cells in CD4+ T cells, (E) Percentage of CM cells in CD4+ T cells, (F) Percentage of EM cells in CD4+ T cells, (G) Percentage of naïve cells in CD8+ T cells, (H) Percentage of CM cells in CD8+ T cells, and (I) Percentage of EM cells in CD8+ T cells. Horizontal bars represent mean values. *p<0.05; **p<0.01; ***p<0.001.
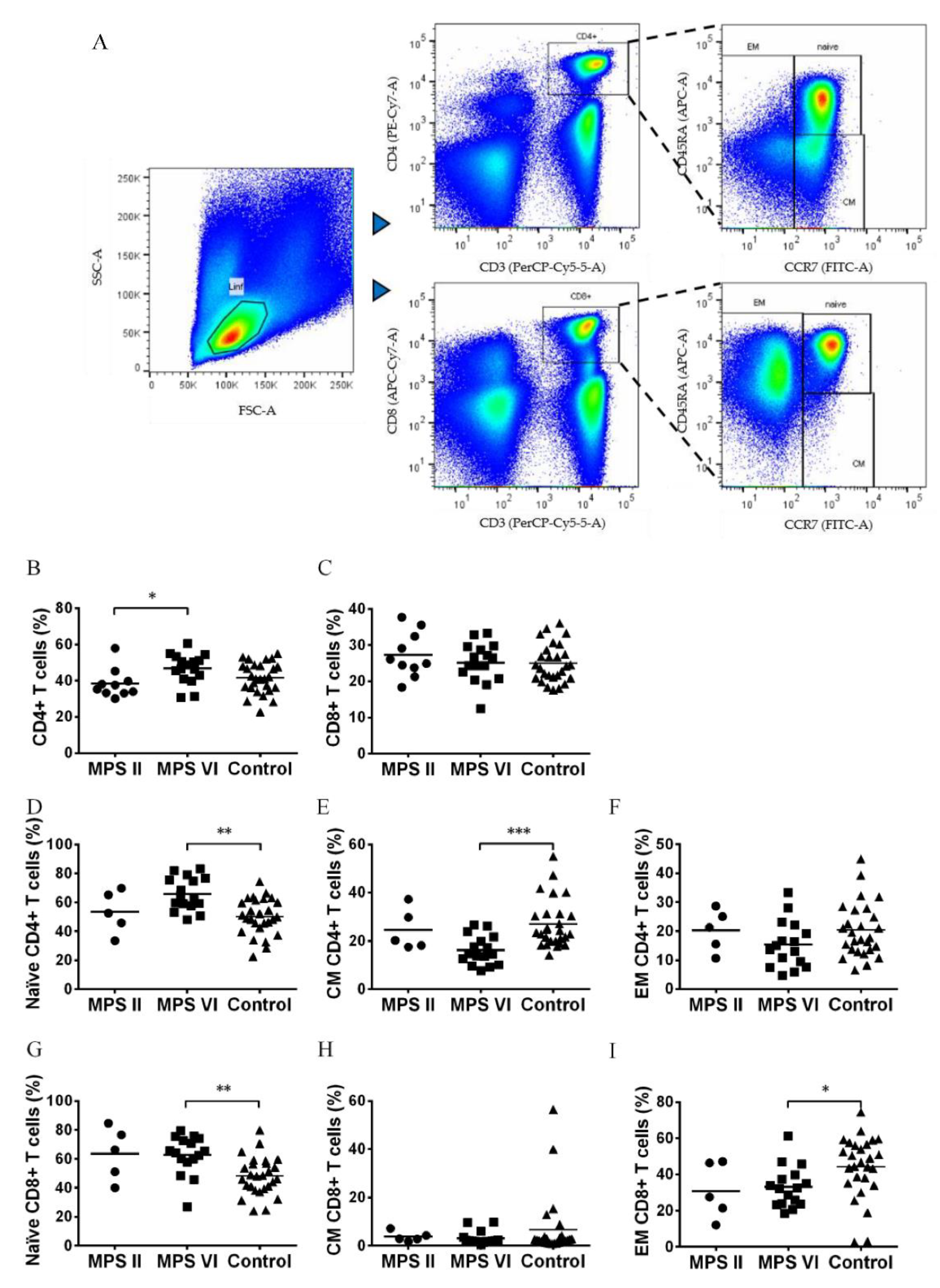
Figure 3.
Percentage and phenotype of iNKT cells in MPS disease patients, and control subjects. A) Gating strategy to characterize iNKT cells by the expression of CD4, CD8 and CD161 among total T cells. B) Percentage of iNKT cells in total T cells of MPS II disease patients and controls, (C) CD4+CD8– cells in iNKT cells of MPS II disease patients and controls, (D) CD4–CD8+ cells in iNKT cells of MPS II disease patients and controls, (E) CD4–CD8–/double negative (DN) cells in iNKT cells of MPS II disease patients and controls and (F) CD161+ cells in iNKT cells of MPS II and VI disease patients and control group. Horizontal bars represent mean values. *p<0.05; **p<0.01.
Figure 3.
Percentage and phenotype of iNKT cells in MPS disease patients, and control subjects. A) Gating strategy to characterize iNKT cells by the expression of CD4, CD8 and CD161 among total T cells. B) Percentage of iNKT cells in total T cells of MPS II disease patients and controls, (C) CD4+CD8– cells in iNKT cells of MPS II disease patients and controls, (D) CD4–CD8+ cells in iNKT cells of MPS II disease patients and controls, (E) CD4–CD8–/double negative (DN) cells in iNKT cells of MPS II disease patients and controls and (F) CD161+ cells in iNKT cells of MPS II and VI disease patients and control group. Horizontal bars represent mean values. *p<0.05; **p<0.01.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or properdty resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



