Submitted:
25 May 2023
Posted:
26 May 2023
You are already at the latest version
Abstract
DNA double-strand breakage is the most lethal damage to chromosomal DNA. It activates a series of cellular DNA damage response pathways, including DNA damage sensing, control of cell cycle arrest and apoptosis, and DNA repair. DNA damage response pathways are regulated by complex signaling machineries. Of the intracellular signaling cascades, diacylglycerol kinase (DGK) phosphorylates diacylglycerol (DG) to generate phosphatidic acid (PA). Because both DG and PA serve as second messengers, DGK activity induces a shift of signaling pathways from DG-mediated to PA-mediated cascades, thereby implicating DGK in the regulation of widely various functions. Reportedly, one member of the DGK family, DGKζ, is intimately involved in the regulation of stress responses through p53 and NF-κB. Stresses such as ischemia and infarction cause DGKζ downregulation. Experimental DGKζ depletion renders cells and mice vulnerable to various stressors such as chemotherapeutic agents and ionizing irradiation. Nevertheless, how DGKζ is involved in DNA repair, a critical event of DNA damage response for survival remains unknown. For this study, we examined how DGKζ depletion affects DNA repair mechanisms. We demonstrated that DGKζ depletion causes attenuation of Akt activation and DNA-PK protein expression upon DNA damage, which might engender downregulated BRCA1 protein synthesis and stability. Results suggest that DGKζ depletion attenuates BRCA1-mediated DNA repair machinery, thereby conferring vulnerability to DNA damage.
Keywords:
γH2AX
; Actinomycin D
; Akt
; DNA damage response
; DNA double-strand break
; DNA-PK
; Etoposide
; p95/NBS1
1. Introduction
Ionizing radiation and chemotherapeutic agents can cause a DNA double-strand break (DSB), which represents the most lethal type of DNA damage and which can trigger a series of cellular DNA damage response pathways, including transcription factors such as p53 and NF-κB [1]. p53 induction activates an apoptotic program, whereas NF-κB prevents apoptosis [2]. Of the intracellular signaling cascades, diacylglycerol kinase (DGK) phosphorylates a second messenger diacylglycerol (DG) to generate another second messenger phosphatidic acid (PA), thereby regulating distinct downstream cascades and different cellular responses [3,4,5,6]. Therefore, DGK activity induces a phase transition of signaling pathways. Reports of some earlier studies have described one member of the DGK family, DGKζ, as closely involved in stress responses through p53 and NF-κB regulation [7].
DGKζ localizes predominantly to the nucleus and shuttles between the nucleus and the cytoplasm [8,9]. It is particularly interesting that DGKζ exerts distinct effects on these transcription factors in these cellular compartments, catalytic activity-dependently and activity-independently. At the cellular level, DGKζ promotes nuclear p53 transcriptional activity in a catalytic activity-dependent manner and its degradation in the cytoplasm via the ubiquitin–proteasome system (UPS) independently of catalytic activity [7,10,11]. It is particularly interesting that DGKζ itself is subject to degradative breakdown by UPS [12]. Therefore, decreasing DGKζ results in accumulation of p53, which induces cytotoxicity in the cytoplasm because cytoplasmic p53 associates with the proapoptotic protein Bak at the mitochondria to trigger apoptosis in a transcription-independent manner [13,14].
The regulatory mechanism of p53 by DGKζ observed in cell-based experiments is also functional at the animal level. Whole body ionizing radiation strongly induces p53 protein in DGKζ-deficient spleen tissues and renders those mice vulnerable to X-ray irradiation [15]. In this regard, X-ray irradiation-induced DSB is regulated by p53; it represents an initiator of radiation-induced genomic instability [16,17]. DSB is recognized by ataxia telangiectasia mutated kinase (ATM)[18], which promotes the accumulation and activation of p53, CHK2/Cds1, BRCA1, p95/NBS1, and histone H2X. Especially, p53 induces transactivation of various downstream genes such as p21, GAD45, Bax, and p53AIP1, thereby leading to cell cycle arrest, apoptosis, and DNA repair in a context-dependent manner [19].
Regarding the NF-κB system, DGKζ depletion facilitates IκB degradation through specific IKKβ activation, thereby accelerating nuclear translocation of NF-κB p65 subunit [20]. Because DGKζ deficiency also enhances p65 subunit phosphorylation, DGKζ depletion altogether upregulates NF-κB transcription activity. Reportedly, NF-κB activation is favorable to cell survival after DNA damaging agents and ionizing irradiation treatment, although the function of NF-κB can be either pro-apoptotic or anti-apoptotic depending on characteristics of the initial genotoxic signals and the cell type [21].
Reportedly, the cellular responses to DSB are finely tuned by numerous cascades. The cell fate depends on a delicate balance between pro-apoptotic and anti-apoptotic signals [21]. In this sense, DGKζ depletion engenders attenuated p53 transcriptional activity together with enhanced NF-κB activity, the total effects of which confer stress-vulnerability to the cells. However, functional implication of DGKζ in DNA repair, a critical event of DNA damage response, remains unclear. This study was conducted to investigate how the regulatory mechanism of DNA repair is affected by DGKζ depletion.
Results show that depletion of DGKζ engenders decreased Akt activation, together with an attenuated DNA-PKcs protein level, upon DNA damage, those phenomena might downregulate the induction of protein synthesis and stability of BRCA1. Results suggest that BRCA1-mediated DNA repair mechanism is attenuated in DGKζ-depleted cells. Additionally, DGKζ depletion apparently suppresses functional DNA repair protein complex formation in the nucleus.
2. Materials and Methods
2.1. Reagents
Cell culture reagents were obtained from Wako Pure Chemical Industries. DNA damage reagents including Actinomycin D (Act D) and Etoposide (Eto) were purchased from Sigma.
2.2. Cell lines and cell culture
U2OS (p53 wild-type) human osteosarcoma cells and mouse embryonic fibroblasts (MEFs) were used in this study. MEFs obtained from wild-type (C57BL6) and DGKζ-KO mice were immortalized by transfection of SV40 plasmid DNA [20]. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% heat-inactivated fetal bovine serum, penicillin (100 U/ml) and streptomycin (100 U/ml). Cells were incubated at 37C in a humidified atmosphere of 95% air and 5% CO2.
2.3. RNAi and transfection
Human DGKζ-specific small interfering RNAs (siRNAs) including siDGKζ#7 (HSS112447), siDGKζ#8 (HSS122448), and siDGKζ#9 (HSS122449) were purchased from Life technology. Control siRNA (siCont, All stars negative control) were from Qiagen. Cells were transfected with siRNAs using LipofectAMINE RNAiMAX reagent (Invitrogen) according to the manufacturer’s instructions.
2.4. Immunoblotting and antibodies
Cells treated with or without reagents were lysed in lysis buffer consisting 20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 1 mM Na3VO4, 50 mM NaF, 1% Triton X-100, and protease inhibitors cocktail (Sigma). Protein concentration was determined using BCA Protein Assay Reagent (Piece, Rockford, IL) according to the instruction manual. Equal amounts of protein lysate were applied in SDS-PAGE and electrophoretically transferred on PVDF membrane (Millipore). After blocking with 5% skim milk in PBS-T, the membrane were immunoblotted with primary antibodies against DGKζ g/ml; rabbit [9], guinea pig [22]), β-actin (1:10000; #3700; Cell Signaling), Sirt1 (1:1000; #9475; Cell Signaling), p53 (1:1000; DO-1; Santa Cruz), acetyl-p53 Lys382 (human protein) or Lys379 (mouse protein), (1:1000; #2570; Cell Signaling), H2AX (1:1000; #2595; Cell Signaling), γH2AX (1:1000; #9718; Cell Signaling), Akt (1:1000; #9272; Cell Signaling), phospho-Akt Ser473 (1:1000; #4060; Cell Signaling), BRCA1 (1:1000; #9019; Cell Signaling), phospho-BRCA1 Ser1524 (1:1000; #9009; Cell Signaling), DNA-PKcs (1:1000; #4602; Cell Signaling), phospho-DNA-PKcs Ser2056 (1:1000; #68716; Cell Signaling), p95/NBS1 (1:1000; #14956; Cell Signaling), Rad50 (1:1000; #3427; Cell Signaling), Ku80 (1:1000; #2180; Cell Signaling), MERIT40 (1:1000; #12711; Cell Signaling), BRE (1:1000; #12457; Cell Signaling), BRCC36 (1:1000; #18215; Cell Signaling), XLF (1:1000; #2854; Cell Signaling). Immunoreactive complexes were visualized using the chemiluminescent HRP Substrate ImmobilonTM Western (Millipore).
2.5. Immunofluorescence microscopy
Cells seeded on the coverslip were transfected with negative control siRNA or siDGKζ. Forty-eight hours after transfection, cells were fixed with 4% paraformaldehyde, washed with PBS three times, and permeabilized with 0.1% Triton/PBS. Cells were incubated with 10% normal donkey serum for blocking and incubated with primary antibody overnight at room temperature in a moist chamber. For immunofluorescence examination, cells were washed with PBS three times and incubated with fluorescence-conjugated secondary antibody for 1 h. TO-PRO-3 was used for nuclear staining. The images were observed under a confocal laser scanning microscope (LSM510META, Carl Zeiss, Jena, Germany).
2.6. Immunoprecipitation analysis
All cells were dissolved in lysis buffer (20 mM Tris-HCl, pH 7.4, 50 mM NaCl, 1 mM Na3VO4, 50 mM NaF, 1% Triton X-100, protease inhibitors cocktail). Each 8,000 g of cell lysates was precleaned with protein A Sepharose beads (GE Healthcare, Piscataway, NJ, USA) for 2 h at 4 C, incubated with antibodies (anti-DGKζ, anti-Akt, anti-p95/NBS1) in U2OS cells overnight at 4C, and reacted with protein A Sepharose beads for 6 h. After the immunoprecipitated complexes were washed four times with lysis buffer, they were boiled for 15 min in SDS sample buffer (New England Biolabs, Inc. Beverly, MA, USA). Immunoprecipitated complexes were subjected to SDS-PAGE, transferred to PVDF membranes (Millipore), and analyzed with immunoblotting.
3. Results
3.1. Depletion of DGK enhances DNA double strand break
Earlier reports have described that DSB is induced by higher concentrations of chemical agents such as DNA-intercalating actinomycin D (ActD) and topoisomerase II inhibitor etoposide (Eto), as well as X-ray irradiation [23,24,25]. Because p53 is induced strongly upon DSB, we first used U2OS cells that express wild-type p53 and examined the time course of cellular response to ActD. The phosphorylated form of H2AX, designated γH2AX, is well known to serve as a marker for DSB and to accumulate at DSB sites [26]. Immunoblot analysis showed that γH2AX level is increased gradually after 4 h of treatment (Figure 1A). The increased γH2AX level coincided with increased p53 protein level. Furthermore, acetylation level of p53 at Lys382 was increased, which is consistent with previous findings [27]. Through the time course of increasing levels of these DNA damage markers, we found that protein levels of DGKζ and histone deacetylase Sirt1 are inversely decreased, especially at 16 h of treatment. In this regard, we earlier reported that DGKζ knockdown leads to a decreased Sirt1 level [28].
Because an earlier report showed that DGK-KO mice are vulnerable to DNA damage inflicted by whole-body irradiation [15], we next checked whether depletion of DGK promotes DNA damage response upon ActD treatment. To examine the causal relation between the degree of DNA damage and DGKζ level, we performed experimental downregulation of DGKζ via siRNA. Figure 1B shows that DGKζ knockdown caused increased γH2AX level upon ActD treatment. Similar results were obtained using Eto treatment (data not shown). Furthermore, p53-Lys382 acetylation level was significantly enhanced in siDGKζ-transfected cells. Since Sirt1 is shown to deacetylate p53-Lys382 [29], decreased Sirt1 level may lead to enhanced p53 acetylation at this site. This finding suggests that DGKζ depletion enhances DSB and DNA damage signaling.
Results of an earlier study indicated that the quantities of γH2AX foci in the nucleus are comparable to the quantities of induced DSB [30]. Therefore we next performed immunofluorescence staining of U2OS cells transfected with siCont or siDGKζ. Figure 1C shows immunofluorescence analysis results: γH2AX foci abound in the nucleus of siDGKζ-transfected cells after treatment with ActD compared with the controls. Similar results were obtained using Eto treatment (data not shown). These morphological data confirm that depletion of DGKζ facilitates DSB upon DNA damage.
3.2. DNA repair is delayed in DGK-depleted cells
As a universal process in living cells, DNA repair maintains the structural integrity of chromosomal DNA upon damage arising from environmental insult, and from normal metabolic processes [1,31]. Nonhomologous end-joining (NHEJ) and homologous recombination (HR) are the two primary processes used to repair DSB [32].
To investigate the role of DGKζ in DNA repair, we next examined how the γH2AX expression level is changed in DGKζ-depleted cells after washing out DNA-damaging agents. As presented in Figure 2A, in siCont-transfected U2OS cells, the γH2AX level was slightly increased on ActD treatment, but it returned to the basal level after washing out. By contrast, in siDGKζ-transfected U2OS cells, the γH2AX level was robustly increased on ActD treatment. Moreover, it remained high, even after washing out. Similar results were obtained for WT and DGKζ MEFs (Figure 2B). Immunofluorescence analysis confirmed the results obtained from immunoblot analysis: In the nucleus of WT MEFs, γH2AX-immunofluorescence returned to the basal level 2 h after washing out ActD (Figure 2C). In the nucleus of DGKζ-KO MEFs, however, γH2AX-immunofluorescence remained high at this time point of washing out. Together, results suggest that DGKζ depletion leads not only to facilitated DSB but also to delayed DNA repair, thereby enhancing vulnerability to DNA damage.
Earlier reports have described that Akt is involved in the NHEJ repair pathway of DNA damage [32]. Akt activated by phosphorylation facilitates ionizing radiation-induced DNA-PK complex formation and accumulation of the catalytic subunit of DNA-PK (DNA-PKcs) to DNA damage sites [33]. In this regard, reagent-induced DSB also promotes the phosphorylation of Akt at Ser473 [34,35]. Therefore, we next checked whether depletion of DGKζ affects the phosphorylation status of Akt-Ser473 after washing out DNA damaging agents. As presented in Figure 2A, the phosphorylation level of Akt at Ser473 was increased in siCont-transfected U2OS cells upon ActD treatment and was further upregulated after washing out. However, it remained mostly unchanged or was increased slightly in siDGKζ-transfected U2OS cells before and upon ActD treatment, and after washing out. Similar results were obtained for WT and DGKζ MEFs (Figure 2B). Results suggest that the Akt-regulated DNA repair mechanism upon DNA damage is suppressed in DGKζ-depleted cells.
One of the enzymes responsible for Akt phosphorylation at Ser473 upon DNA damage is DNA-PK [34]. Because DNA-PK consists of the DNA-PKcs and Ku antigen complex (Ku70/80) [36], we next examined the DNA-PKcs level upon ActD treatment. As presented in Figure 2A, DNA-PKcs level was decreased in siDGKζ-transfected U2OS cells at 1 or 2 h after washing out.
3.3. DGK depletion attenuates BRCA1-mediated DNA repair mechanism.
BRCA1 plays pivotal roles in the maintenance of genome stability. Reportedly, Akt regulates BRCA1 protein stability and function through direct phosphorylation of BRCA1; also, the phosphorylation status of Akt-Ser473 consistently correlates with the BRCA1 protein stability [37]. Therefore, we further investigated whether depletion of DGKζ affects the total and phosphorylated levels of BRCA1 after washing out DNA damaging agents. Immunoblot analysis revealed that total and phosphorylated (Ser1524) levels of BRCA1 are increased in siCont-transfected U2OS cells upon ActD treatment; furthermore, those levels remained high after washing out (Figure 2A). In siDGKζ-transfected U2OS cells, however, the BRCA1 level remained at the basal level during the course of ActD treatment and washing out. Results suggest that phosphorylation of BRCA1 at S1542 is necessary for DNA repair. This inference is supported by results obtained from an earlier study showing that phosphorylation of BRCA1 at this site is crucially important in the radioresistant cell phenotype [38].
3.4. DGKζ co-immunoprecipitates with DNA damage-repair proteins.
Having demonstrated that depletion of DGKζ attenuates the phosphorylation level of Akt at Ser473 and protein levels of DNA-PKcs and BRCA1 (Figure 2A,B), we next examined whether DGKζ interacts with Akt and DNA damage-repair proteins (Figure 3A). Results showed that DGKζ interacts with total Akt and phospho-Akt (Ser473). It is particularly interesting that DGK was found to interact with BRCA1, p95/NBS1, and Rad50 of HR-related proteins, and Ku80 and DNA-PKcs of NHEJ-related proteins, but not with MERIT40, BRE, BRCC36, and XLF. Results indicate that DGKζ participates in the DNA repair complexes of both HR and NHEJ.
Therefore, we next investigated whether DGKζ depletion affects the binding affinities between proteins in DNA repair protein complexes. Results demonstrated that the binding affinities of Akt with p95/NBS1, BRCC36, BRE, or MERIT40 are decreased in DGKζ-KO MEFs (Figure 3B). Furthermore, the binding affinities of p95/NBS1 with Akt, MERIT40, or Rad50 were decreased in the absence of DGKζ (Figure 3C). These results suggest that DGKζ serves as a scaffold protein that tethers the DNA repair protein complex together. Consequently, DGKζ depletion might loosen DNA repair protein complex binding affinity, thereby attenuating effective DNA repair mechanism.
3.5. Depletion of DGK induces cytoplasmic retention of p95/NBS1 and DNA-PKcs during DNA repair
Repair of radiation-induced DSB is dependent on the multifunctional MRN complex containing Mre11, Rad50, and the NBS1 gene product p95/NBS1 [39]. Of those, p95/NBS1 is necessary for proper G2/M DNA-damage checkpoint function [40] and is crucially important for HR following DSB.
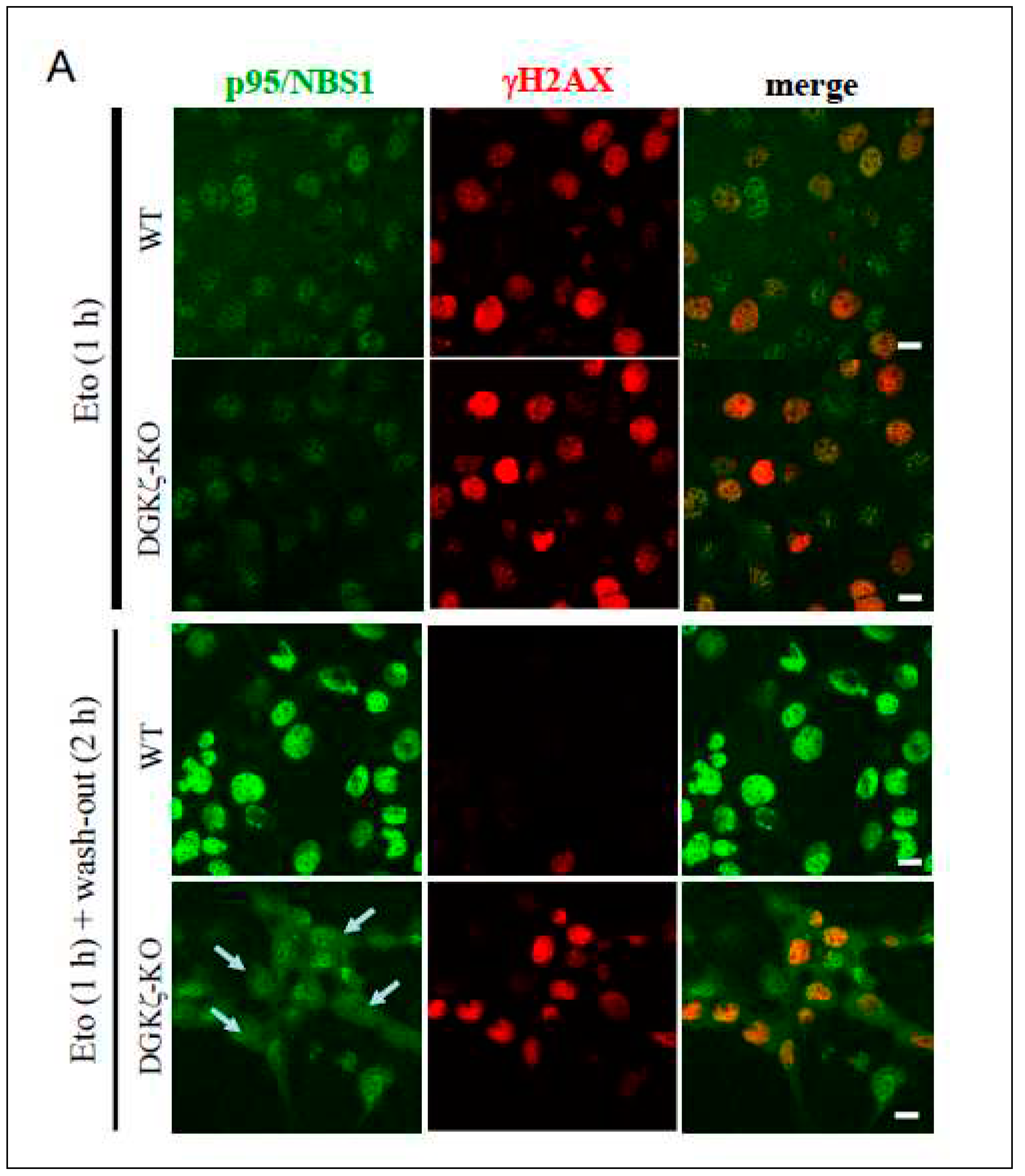
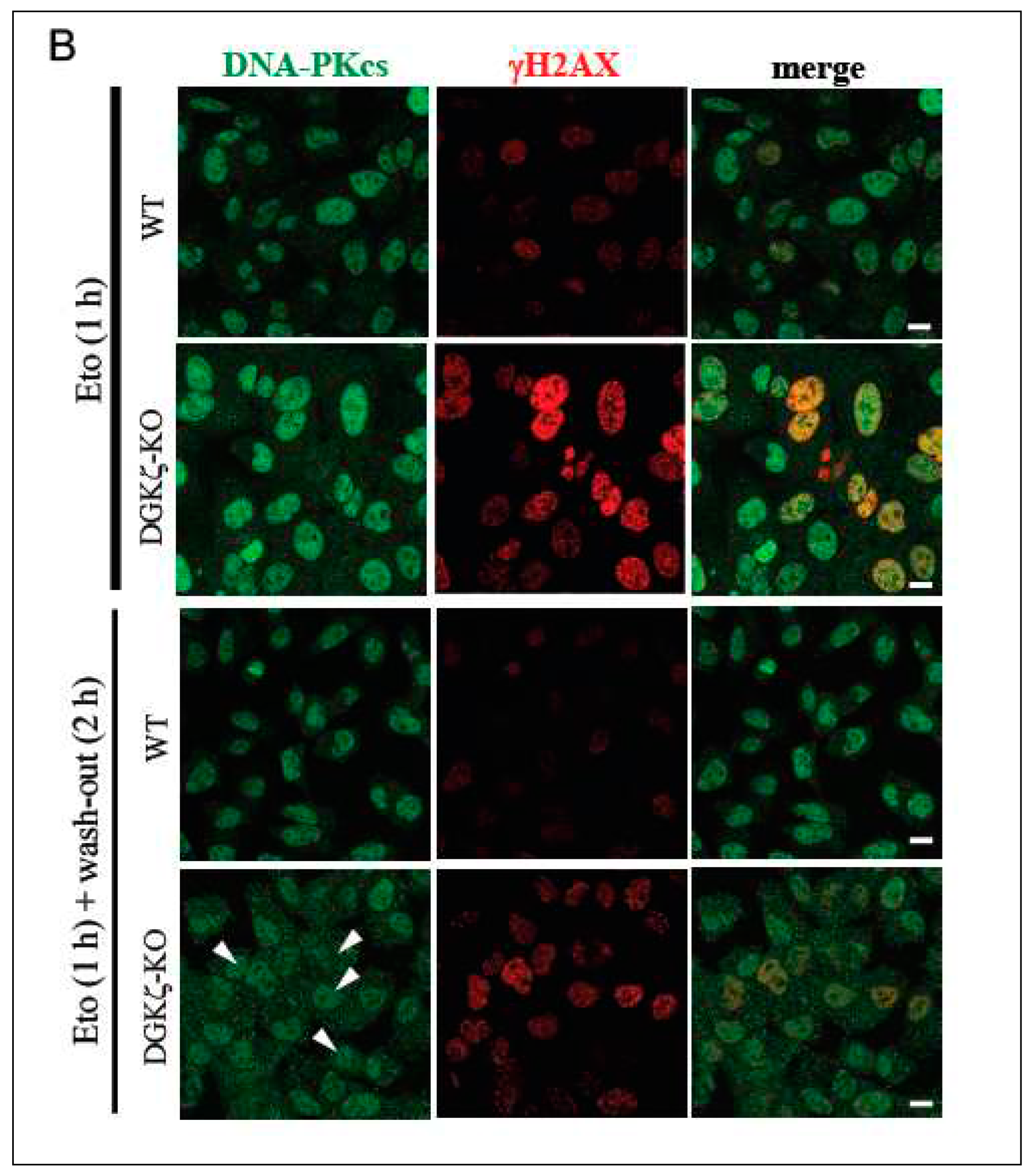
Because DNA repair was delayed in DGK-depleted cells (Figure 2), we next investigated whether depletion of DGKζ exerts some effects on p95/NBS1 and DNA-PKcs. These proteins represent the major components of HR and NHEJ repair mechanisms, respectively. Because DNA repair proteins accumulate to the nucleus upon DNA damage, we specifically examined subcellular localization of these molecules upon DNA damage (Figure 4). Immunofluorescence analysis revealed that, in WT MEFs, p95/NBS1 and DNA-PKcs clearly accumulate to the nucleus 2 h upon Eto treatment and after washing out Eto, whereas in DGKζ-KO MEFs, p95/NBS1 (arrows in Figure 4A) and DNA-PKcs (arrowheads in Figure 4B) partially localize to the cytoplasm. This finding suggests that DGKζ depletion partially induces cytoplasmic retention of p95/NBS1 and DNA-PKcs, thereby attenuating functional DNA repair complex formation in the nucleus.
4. Discussion
Our earlier studies show that DGKζ deficiency renders cells and mice vulnerable to various stressors such as doxorubicin, hypoxia, and X-ray irradiation [15]. Of those, ionizing radiation and chemotherapeutic agents cause DSB, the severest type of DNA damage [1]. Actually, DSB activates a series of cellular DNA damage response pathways, in which tumor suppressor p53 plays a considerably important role. The DNA damage response pathways regulate multiple processes, including DNA damage sensing, control of cell cycle arrest and apoptosis, and DNA repair [41,42]. In this regard, DGKζ depletion leads to an increased level of p53 protein, but attenuates p53 transactivation activity. However, increased p53 proteins cause cytotoxicity in the cytoplasm independently of DGK catalytic activity [7]. The present study specifically addressed DNA repair, a critical event of recovery from DNA damage, in DGKζ-depleted cells. Results demonstrated that DGKζ depletion attenuates the BRCA1-mediated DNA repair mechanism. In light of findings of the earlier and present studies together, vulnerability of DGKζ-depleted cells to DSB can be ascribed to enhanced p53 cytotoxicity and attenuated DNA repair mechanism.
How does DGKζ depletion affect the DNA damage response pathways? First, we found that in DGKζ-depleted cells DSB marker γH2AX expression is increased on DNA damaging agent treatment and that it remains high after washing out. This finding suggests that DGKζ depletion results in facilitated DSB and delayed DNA repair, thereby enhancing vulnerability to DNA damage.
DSB is a life threatening lesion, the repair of which is promoted by an intricate network of multiple DNA repair pathways [41]. It includes NHEJ and HR. Also, NHEJ promotes the potentially inaccurate re-ligation of DSBs, whereas HR precisely restores the genomic sequence of the broken DNA ends using sister chromatids as a template for repair [43]. Also, DSB is bound rapidly by the Ku70/Ku80 heterodimer, which activates the catalytic subunit of DNA-PK to initiated NHEJ [43]. Reportedly, Sirt1 interacts with distinct DNA damage repair proteins including Ku70, which is deacetylated and activated by Sirt1 upon DNA damage [44]. Thus, decreased level of Sirt1 should result in dysregulated Ku70-mediated NHEJ repair pathway under DGKζ-depleted conditions. In addition, Akt, through an immediate complex formation with DNA-PKcs, stimulates the accumulation of DNA-PKcs at DSB sites and promotes DNA-PKcs activity for efficient NHEJ DSB repair. [33].
In this regard, the phosphorylation level of Akt at Ser473 is increased in control U2OS cells upon ActD treatment. Moreover, it is further upregulated after washing out, suggesting that a proper DNA repair mechanism functions. However, Akt phosphorylation level is not increased in DGKζ-depleted cells before and upon ActD treatment, and after washing out. This finding suggests that DGKζ depletion suppresses Akt-mediated NHEJ DNA repair. Consistent with this inference, the protein level of DNA-PKcs, which is responsible for Akt-Ser473 phosphorylation upon DNA damage [34], is not upregulated, but is rather downregulated in DGKζ-depleted cells upon ActD treatment and after washing out.
Actually, BRCA1 is well-known to play a major role in DNA damage response, including both HR and NHEJ repair systems, by participating in cellular pathways for DNA repair, mRNA transcription, cell cycle regulation, and protein ubiquitination [45,46]. In addition to the regulation of DNA-PK, Akt participates in the regulatory mechanism of BRCA1 protein stability and function through direct phosphorylation of BRCA1. Furthermore, the Akt-Ser473 phosphorylation status consistently correlates with stability of the BRCA1 protein [37]. In this regard, BRCA1 protein and phosphorylation levels are upregulated significantly in control cells, but those levels remain quite low in DGKζ-depleted cells upon DNA damaging agent treatment and after washing out.
Collectively, the results presented herein suggest that depletion of DGKζ engenders decreased Akt activation, together with the attenuated DNA-PKcs protein level. As a result, induction of protein synthesis and stability of BRCA1 are downregulated, which attenuates BRCA1-mediated DNA repair mechanism in DGKζ-depleted cells.
Furthermore, we showed that the DNA damage response pathway dysfunction results in destabilization of DNA repair complex in DGKζ-depleted cells. Under normal conditions, DGKζ interacts with Akt and BRCA1, together with HR-related proteins (p95/NBS1, and Rad50) and NHEJ-related proteins (Ku80 and DNA-PKcs). In the absence of DGKζthe binding affinities of Akt with p95/NBS1, MERIT40, BRE, or BRCC36 are decreased. Also, interaction of p95/NBS1 with Akt, MERIT40, or Rad50 is attenuated. These findings suggest that DGKζ functions as a scaffold protein for proper integration of DNA repair proteins. Therefore, DGKζ depletion might suppress effective complex formation of DNA repair proteins. It is also noteworthy that DGKζ depletion partially induces cytoplasmic retention of the major DNA repair proteins, such as NMRN complex protein p95/NBS1 and DNA-PKcs, which might further attenuate functional DNA repair complex formation of both HR and NHEJ mechanisms in the nucleus.
The present study suggests that the critical roles of DGKζ in DNA repair mechanism include the regulation of Sirt1 and Akt, which initiate and regulate downstream pathways for DNA repair program. Several points remain to be elucidated: Do these effects of DGKζ on DNA repair proteins depend on DGK enzymatic activity? Where does DGKζ act? As shown for p53 regulation, DGKζ supports nuclear p53 transcriptional activity in a catalytic activity-dependent manner and facilitates its cytoplasmic degradation via UPS, independently of catalytic activity [7,10,11]. Therefore, further study is warranted to assess the regulatory mechanisms of DGKζ on Sirt1 and Akt, including interaction, activity-dependency, and subcellular localization.
Author Contributions
TT, MI, and KG conceived the study. TT performed experiments and analyzed data. MI helped writing the manuscript. TT and KG wrote the manuscript.
Acknowledgments
This work was supported by Grants-in-Aid from The Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (18K06815 and 22K06804 to TT).
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Chang, H. H. Y.; Pannunzio, N. R.; Adachi, N.; Lieber, M. R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Tergaonkar, V.; Pando, M.; Vafa, O.; Wahl, G.; Verma, I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell 2002, 1, 493–503. [Google Scholar] [CrossRef]
- Kanoh, H.; Yamada, K.; Sakane, F. Diacylglycerol kinase: a key modulator of signal transduction? Trends Biochem. Sci. 1990, 15, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Sakane, F.; Imai, S.; Kai, M.; Yasuda, S.; Kanoh, H. Diacylglycerol kinases: why so many of them? Biochim. Biophys. Acta 2007, 1771, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Hozumi, Y.; Nakano, T.; Saino, S. S.; Kondo, H. Cell biology and pathophysiology of the diacylglycerol kinase family: morphological aspects in tissues and organs. Int. Rev. Cytol. 2007, 264, 25–63. [Google Scholar] [PubMed]
- Topham, M. K.; Epand, R. M. Mammalian diacylglycerol kinases: molecular interactions and biological functions of selected isoforms. Biochim. Biophys. Acta 2009, 1790, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nakano, T.; Hozumi, Y.; Martelli, A. M.; Goto, K. Regulation of p53 and NF-κB transactivation activities by DGKζ in catalytic activity-dependent and -independent manners. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118953. [Google Scholar] [CrossRef]
- Goto, K.; Kondo, H. A 104-kDa diacylglycerol kinase containing ankyrin-like repeats localizes in the cell nucleus. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 11196–11201. [Google Scholar] [CrossRef]
- Hozumi, Y.; Ito, T.; Nakano, T.; Nakagawa, T.; Aoyagi, M.; Kondo, H.; Goto, K. Nuclear localization of diacylglycerol kinase zeta in neurons. Eur. J. Neurosci. 2003, 18, 1448–1457. [Google Scholar] [CrossRef]
- Goto, K.; Tanaka, T.; Nakano, T.; Okada, M.; Hozumi, Y.; Topham, M. K.; Martelli, A. M. DGKζ under stress conditions: “to be nuclear or cytoplasmic, that is the question”. Adv. Biol. Regul. 2014, 54, 242–253. [Google Scholar] [CrossRef]
- Tanaka, T.; Okada, M.; Hozumi, Y.; Tachibana, K.; Kitanaka, C.; Hamamoto, Y.; Martelli, A. M.; Topham, M. K.; Iino, M.; Goto, K. Cytoplasmic localization of DGKζ exerts a protective effect against p53-mediated cytotoxicity. J. Cell Sci. 2013, 126 Pt 13, 2785–2797. [Google Scholar] [CrossRef]
- Okada, M.; Hozumi, Y.; Tanaka, T.; Suzuki, Y.; Yanagida, M.; Araki, Y.; Evangelisti, C.; Yagisawa, H.; Topham, M. K.; Martelli, A. M.; et al. DGKζ is degraded through the cytoplasmic ubiquitin-proteasome system under excitotoxic conditions, which causes neuronal apoptosis because of aberrant cell cycle reentry. Cell. Signal. 2012, 24, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Green, D. R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, N. D.; Wolff, S.; Erster, S.; Becker, K.; Moll, U. M. Monoubiquitylation promotes mitochondrial p53 translocation. Embo J. 2007, 26, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Iseki, K.; Tanaka, K.; Nakano, T.; Iino, M.; Goto, K. DGKζ ablation engenders upregulation of p53 level in the spleen upon whole-body ionizing radiation. Adv. Biol. Regul. 2018, 67, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Fei, P.; El-Deiry, W. S. P53 and radiation responses. Oncogene 2003, 22, 5774–5783. [Google Scholar] [CrossRef]
- Suzuki, K.; Ojima, M.; Kodama, S.; Watanabe, M. Radiation-induced DNA damage and delayed induced genomic instability. Oncogene 2003, 22, 6988–6993. [Google Scholar] [CrossRef]
- Nelson, W. G.; Kastan, M. B. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol. Cell. Biol. 1994, 14, 1815–1823. [Google Scholar]
- Vousden, K. H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Tsuchiya, R.; Tanaka, T.; Hozumi, Y.; Nakano, T.; Okada, M.; Topham, M. K.; Iino, M.; Goto, K. Downregulation of diacylglycerol kinase ζ enhances activation of cytokine-induced NF-κB signaling pathway. Biochim. Biophys. Acta 2015, 1853, 361–369. [Google Scholar] [CrossRef]
- Habraken, Y.; Piette, J. NF-kappaB activation by double-strand breaks. Biochem. Pharmacol. 2006, 72, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Hozumi, Y.; Ichimura, T.; Tanaka, T.; Hasegawa, H.; Yamamoto, M.; Takahashi, N.; Iseki, K.; Yagisawa, H.; Shinkawa, T.; et al. Interaction of nucleosome assembly proteins abolishes nuclear localization of DGKζ by attenuating its association with importins. Exp. Cell Res. 2011, 317, 2853–2863. [Google Scholar] [CrossRef] [PubMed]
- Karpinich, N. O.; Tafani, M.; Rothman, R. J.; Russo, M. A.; Farber, J. L. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J. Biol. Chem. 2002, 277, 16547–16552. [Google Scholar] [CrossRef] [PubMed]
- Porcedda, P.; Turinetto, V.; Lantelme, E.; Fontanella, E.; Chrzanowska, K.; Ragona, R.; De Marchi, M.; Delia, D.; Giachino, C. Impaired elimination of DNA double-strand break-containing lymphocytes in ataxia telangiectasia and Nijmegen breakage syndrome. DNA Repair (Amst) 2006, 5, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Choong, M. L.; Yang, H.; Lee, M. A.; Lane, D. P. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle 2009, 8, 2810–2818. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O. A.; Rogakou, E. P.; Panyutin, I. G.; Bonner, W. M. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat. Res. 2002, 158, 486–492. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Herrera, J. E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C. W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef]
- Akimoto, R.; Tanaka, T.; Nakano, T.; Hozumi, Y.; Kawamae, K.; Goto, K. DGKζ depletion attenuates HIF-1α induction and SIRT1 expression, but enhances TAK1-mediated AMPKα phosphorylation under hypoxia. Cell. Signal. 2020, 71, 109618. [Google Scholar] [CrossRef]
- Cheng, H. L.; Mostoslavsky, R.; Saito, S.; Manis, J. P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F. W.; Chua, K. F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sc.i U.S.A. 2003, 100, 10794–10799. [Google Scholar] [CrossRef]
- Rogakou, E. P.; Boon, C.; Redon, C.; Bonner, W. M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Tebbs, R. S.; Zhao, Y.; Tucker, J. D.; Scheerer, J. B.; Siciliano, M. J.; Hwang, M.; Liu, N.; Legerski, R. J.; Thompson, L. H. Correction of chromosomal instability and sensitivity to diverse mutagens by a cloned cDNA of the XRCC3 DNA repair gene. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 6354–6358. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Wang, H.; Perrault, A. R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Pantelias, G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004, 104, 14–20. [Google Scholar] [CrossRef]
- Toulany, M.; Lee, K. J.; Fattah, K. R.; Lin, Y. F.; Fehrenbacher, B.; Schaller, M.; Chen, B. P.; Chen, D. J.; Rodemann, H. P. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B. A. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef]
- Winograd-Katz, S. E.; Levitzki, A. Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene 2006, 25, 7381–7390. [Google Scholar] [CrossRef] [PubMed]
- Suwa, A.; Hirakata, M.; Takeda, Y.; Jesch, S. A.; Mimori, T.; Hardin, J. A. DNA-dependent protein kinase (Ku protein-p350 complex) assembles on double-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 6904–6908. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A. C.; Lyons, T. R.; Young, C. D.; Hansen, K. C.; Anderson, S. M.; Holt, J. T. AKT regulates BRCA1 stability in response to hormone signaling. Mol. Cell. Endocrinol. 2010, 319, 129–142. [Google Scholar] [CrossRef]
- Ouchi, T. BRCA1 phosphorylation: biological consequences. Cancer Biol. Ther. 2006, 5, 470–475. [Google Scholar] [CrossRef]
- Lee, J. H.; Mand, M. R.; Deshpande, R. A.; Kinoshita, E.; Yang, S. H.; Wyman, C.; Paull, T. T. Ataxia telangiectasia-mutated (ATM) kinase activity is regulated by ATP-driven conformational changes in the Mre11/Rad50/Nbs1 (MRN) complex. J. Biol. Chem. 2013, 288, 12840–12851. [Google Scholar] [CrossRef]
- Hari, F. J.; Spycher, C.; Jungmichel, S.; Pavic, L.; Stucki, M. A divalent FHA/BRCT-binding mechanism couples the MRE11-RAD50-NBS1 complex to damaged chromatin. EMBO Rep. 2010, 11, 387–392. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S. J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Zhou, B. B.; Elledge, S. J. The DNA damage response: putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K. W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S. H.; Min, B. H.; Lee, K. M.; Cho, M. H.; Park, G. H.; Lee, K. H. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R. D.; Quinn, J. E.; Johnston, P. G.; Harkin, D. P. BRCA1: mechanisms of inactivation and implications for management of patients. Lancet 2002, 360, 1007–1014. [Google Scholar] [CrossRef]
- Kennedy, R. D.; Quinn, J. E.; Mullan, P. B.; Johnston, P. G.; Harkin, D. P. The role of BRCA1 in the cellular response to chemotherapy. J. Natl. Cancer Inst. 2004, 96, 1659–1668. [Google Scholar] [CrossRef]
Figure 1.
Depletion of DGKζ promotes DNA double strand break upon ActD treatment (A) U2OS cells were treated with ActD (400 nM) for varying times (~16 h) and subjected to immunoblotting using antibodies against DGKζ, Sirt1, H2AX, γH2AX, p53, and acetyl-p53 K382. (B) U2OS cells were transfected with siRNA for control (siCont), siDGK#7, siDGKζ#8, or siDGKζ#9. After 48 h of transfection, cells were treated with ActD (400 nM) for 4 h and subjected to immunoblotting using indicated antibodies. Anti-β-actin antibody was used as a loading control. (C) Cells transfected with siCont or siDGKζ were treated with ActD (400 nM) for 4 h. After fixation with 4% paraformaldehyde, cells were stained with anti-γH2AX antibody followed by anti-rabbit IgG-conjugated Alexa 488 (green). TO-PRO-3 (blue) was used for nuclear staining. Scale bars = 20 m.
Figure 1.
Depletion of DGKζ promotes DNA double strand break upon ActD treatment (A) U2OS cells were treated with ActD (400 nM) for varying times (~16 h) and subjected to immunoblotting using antibodies against DGKζ, Sirt1, H2AX, γH2AX, p53, and acetyl-p53 K382. (B) U2OS cells were transfected with siRNA for control (siCont), siDGK#7, siDGKζ#8, or siDGKζ#9. After 48 h of transfection, cells were treated with ActD (400 nM) for 4 h and subjected to immunoblotting using indicated antibodies. Anti-β-actin antibody was used as a loading control. (C) Cells transfected with siCont or siDGKζ were treated with ActD (400 nM) for 4 h. After fixation with 4% paraformaldehyde, cells were stained with anti-γH2AX antibody followed by anti-rabbit IgG-conjugated Alexa 488 (green). TO-PRO-3 (blue) was used for nuclear staining. Scale bars = 20 m.
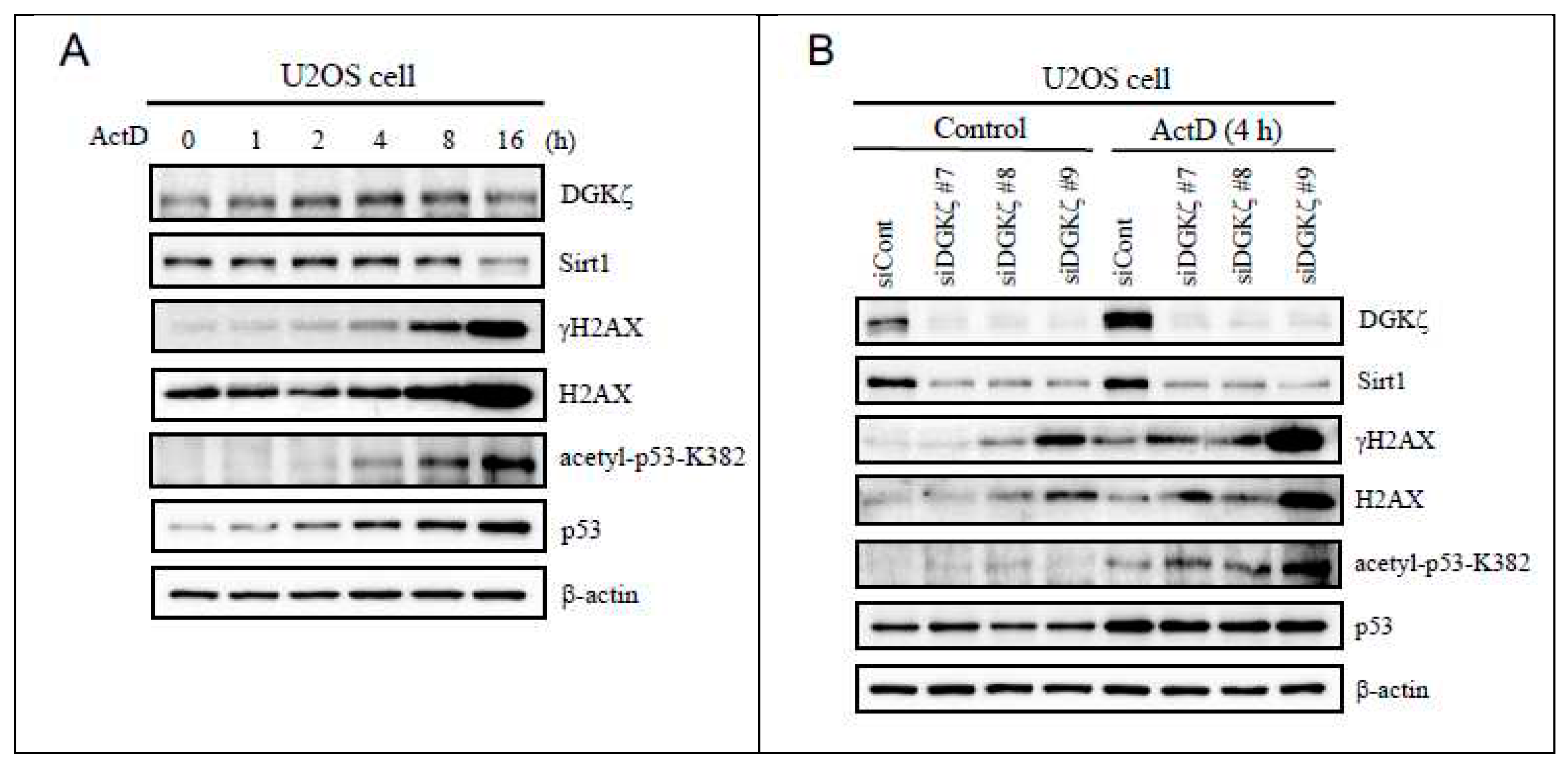
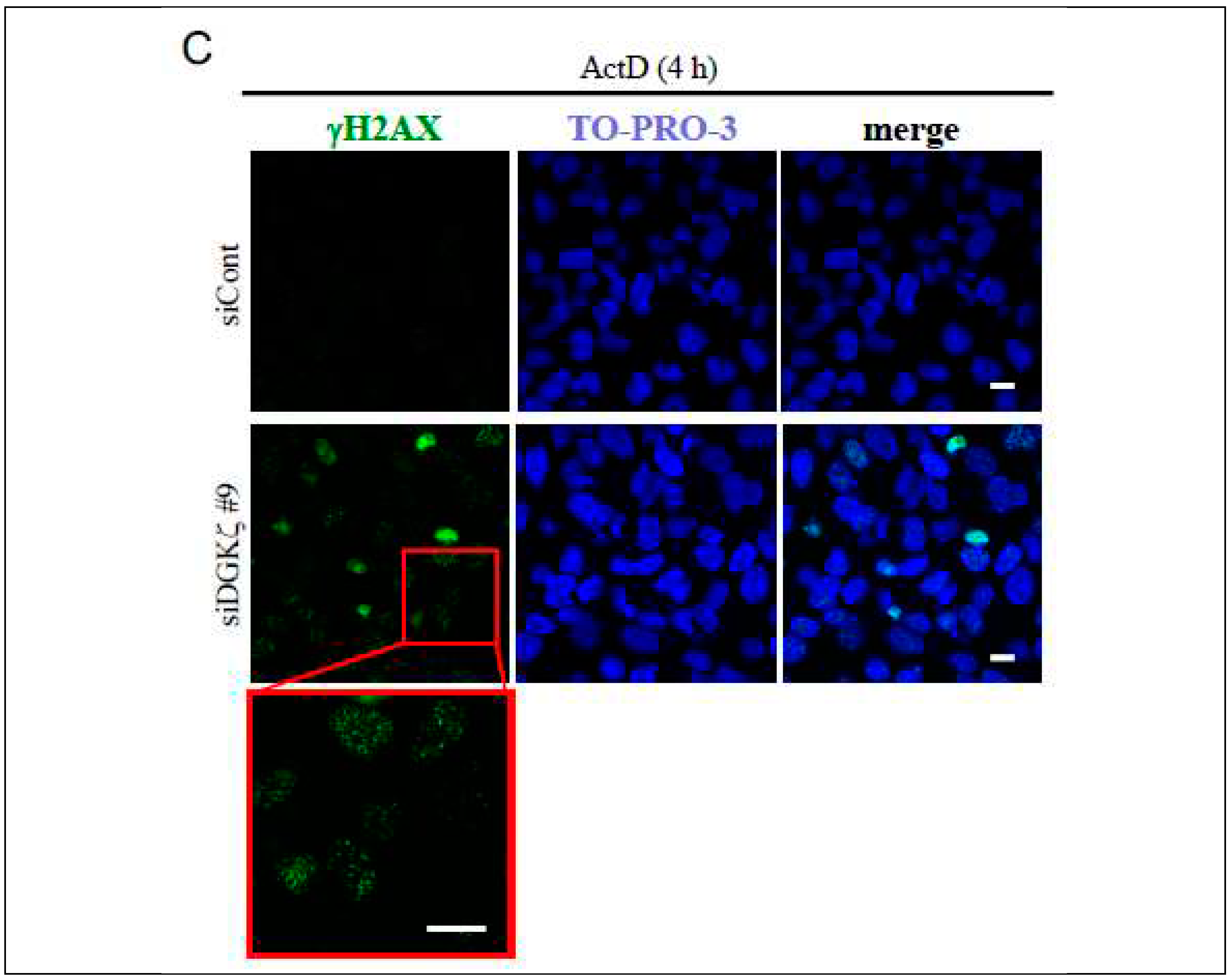
Figure 2.
DNA repair is delated in DGKζ-depleted cells. Because of the most effective induction of acetyl-p53 K382 and γH2AX on ActD treatment in Figure 1B, siDGKζ#9 was used for the subsequent knockdown experiments. U2OS cells transfected with siCont or siDGKζ#9 (A) were treated with or without ActD (400 nM) for 4 h. WT and DGKζ-KO MEFs (B) were also treated similarly. After thoroughly washing out ActD, cells were further incubated with the regular DMEM and subjected to immunoblotting using antibodies against DGKζ, γH2AX, Sirt1, Akt, phopho-Akt S473, DNA-PKcs, BRCA1, and hosphor-BRCA1 S1524. Anti-β-actin antibody was used as a loading control. (C) WT and DGKζ-KO MEFs were treated with Eto (10 μM) for 1 h. After thoroughly washing out Eto, cells were fixed with 4% paraformaldehyde and stained with anti-γH2AX antibody followed by anti-rabbit IgG-conjugated Alexa 488 (green). TO-PRO-3 (blue) was used for nuclear staining. Scale bars = 20 m.
Figure 2.
DNA repair is delated in DGKζ-depleted cells. Because of the most effective induction of acetyl-p53 K382 and γH2AX on ActD treatment in Figure 1B, siDGKζ#9 was used for the subsequent knockdown experiments. U2OS cells transfected with siCont or siDGKζ#9 (A) were treated with or without ActD (400 nM) for 4 h. WT and DGKζ-KO MEFs (B) were also treated similarly. After thoroughly washing out ActD, cells were further incubated with the regular DMEM and subjected to immunoblotting using antibodies against DGKζ, γH2AX, Sirt1, Akt, phopho-Akt S473, DNA-PKcs, BRCA1, and hosphor-BRCA1 S1524. Anti-β-actin antibody was used as a loading control. (C) WT and DGKζ-KO MEFs were treated with Eto (10 μM) for 1 h. After thoroughly washing out Eto, cells were fixed with 4% paraformaldehyde and stained with anti-γH2AX antibody followed by anti-rabbit IgG-conjugated Alexa 488 (green). TO-PRO-3 (blue) was used for nuclear staining. Scale bars = 20 m.
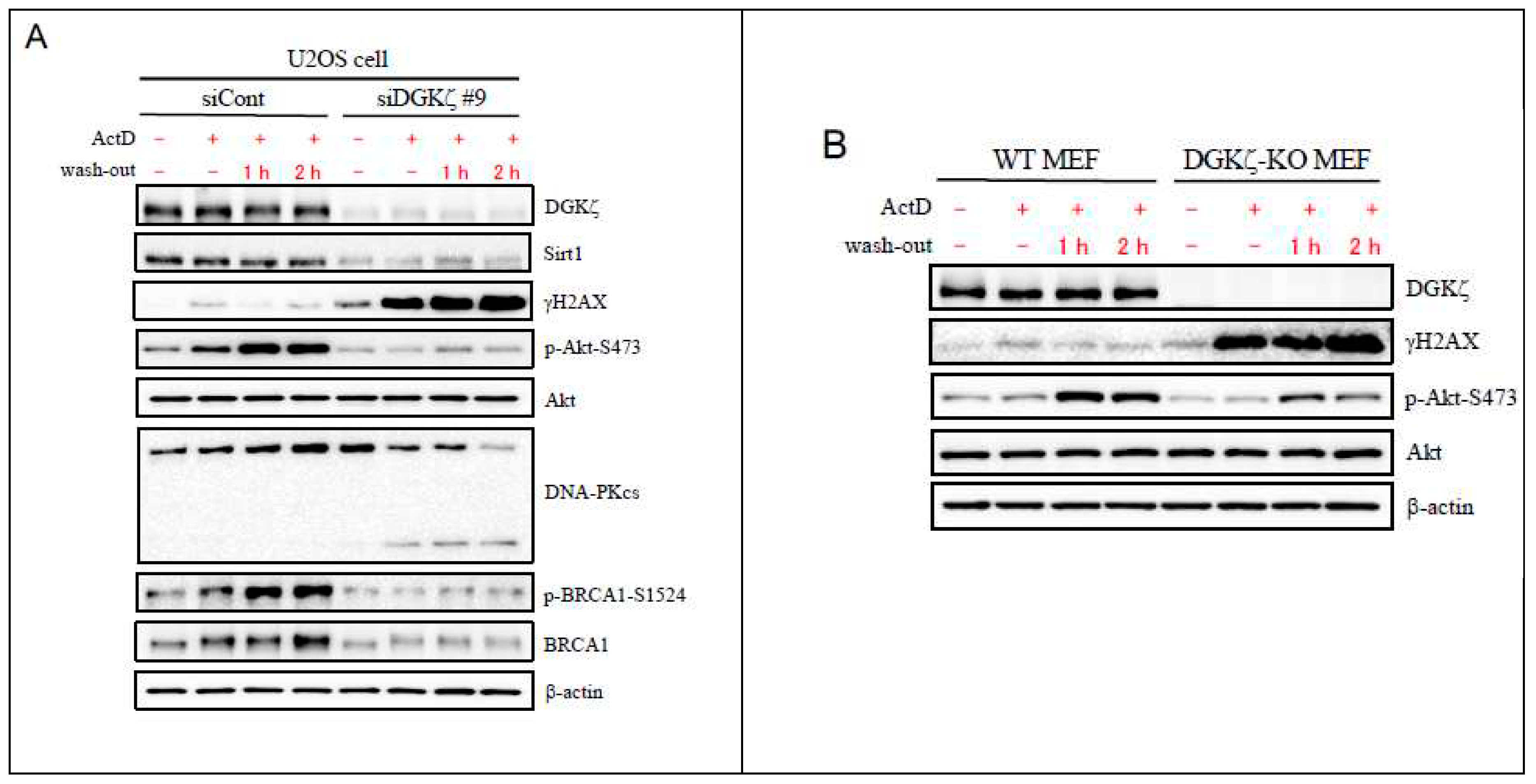
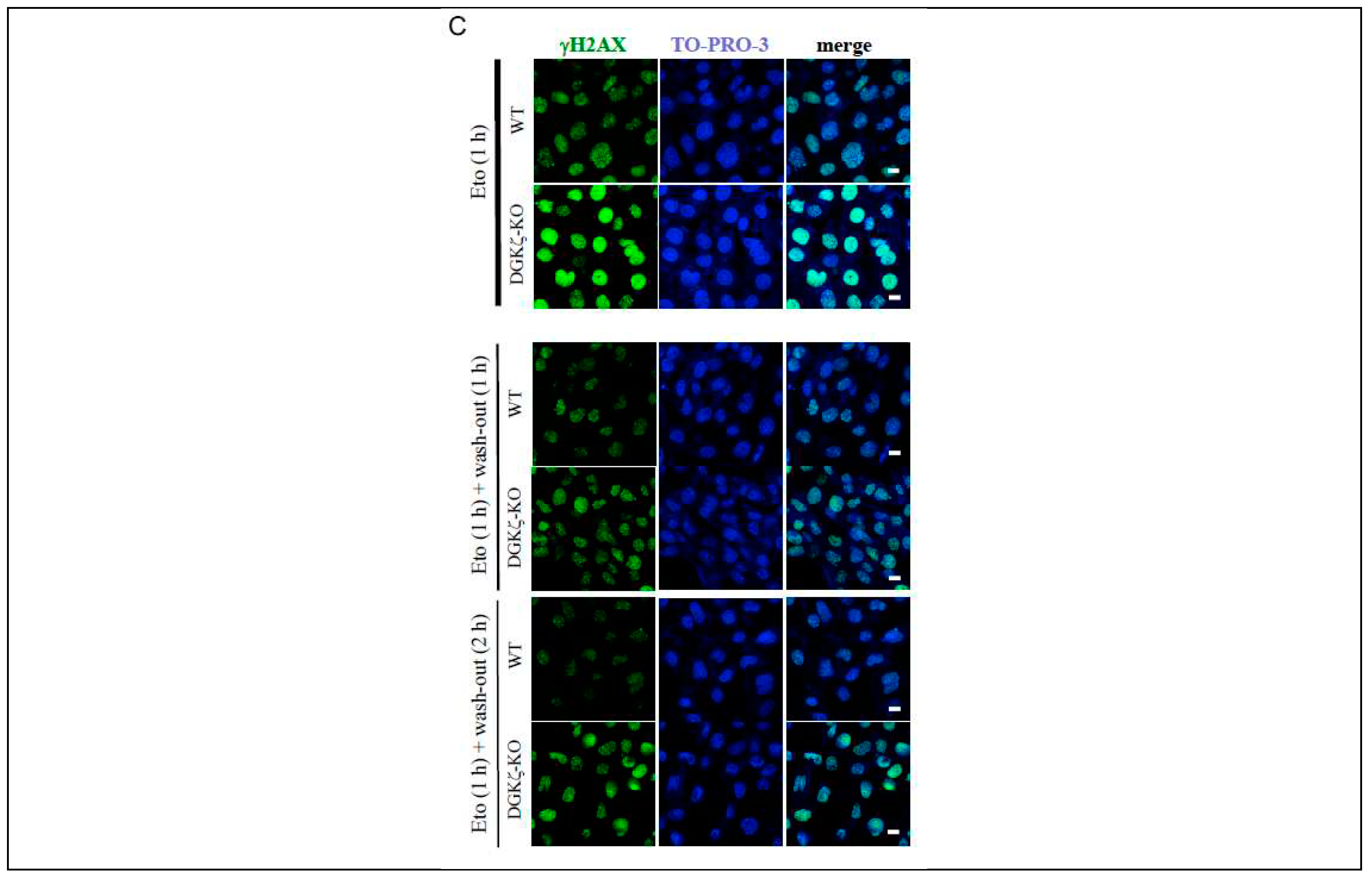
Figure 3.
DGKζ co-immunoprecipitates with DNA damage-repair proteins. (A) U2OS cells were treated with ActD (400 nM) for 4 h or Eto (10 μM) for 1 h. Cell lysates were immunoprecipitated using control guinea pig IgG or anti-DGKζ antibody. The immunoprecipitates were subjected to immunoblotting using antibodies against several DNA damage-repair proteins for homologous recombination (HR) and for non-homologous end-joining (NHEJ) mechanisms. (B, C) The effects of DGKζ depletion on the binding affinities of Akt (B) and of p95/NBS1 (C) with DNA damage-repair proteins. Cell lysates of WT or DGKζ-KO MEFs were immunoprecipitated using anti-Akt or anti-p95/NBS1 antibody. The immunoprecipitates were subjected to immunoblotting using indicated antibodies.
Figure 3.
DGKζ co-immunoprecipitates with DNA damage-repair proteins. (A) U2OS cells were treated with ActD (400 nM) for 4 h or Eto (10 μM) for 1 h. Cell lysates were immunoprecipitated using control guinea pig IgG or anti-DGKζ antibody. The immunoprecipitates were subjected to immunoblotting using antibodies against several DNA damage-repair proteins for homologous recombination (HR) and for non-homologous end-joining (NHEJ) mechanisms. (B, C) The effects of DGKζ depletion on the binding affinities of Akt (B) and of p95/NBS1 (C) with DNA damage-repair proteins. Cell lysates of WT or DGKζ-KO MEFs were immunoprecipitated using anti-Akt or anti-p95/NBS1 antibody. The immunoprecipitates were subjected to immunoblotting using indicated antibodies.
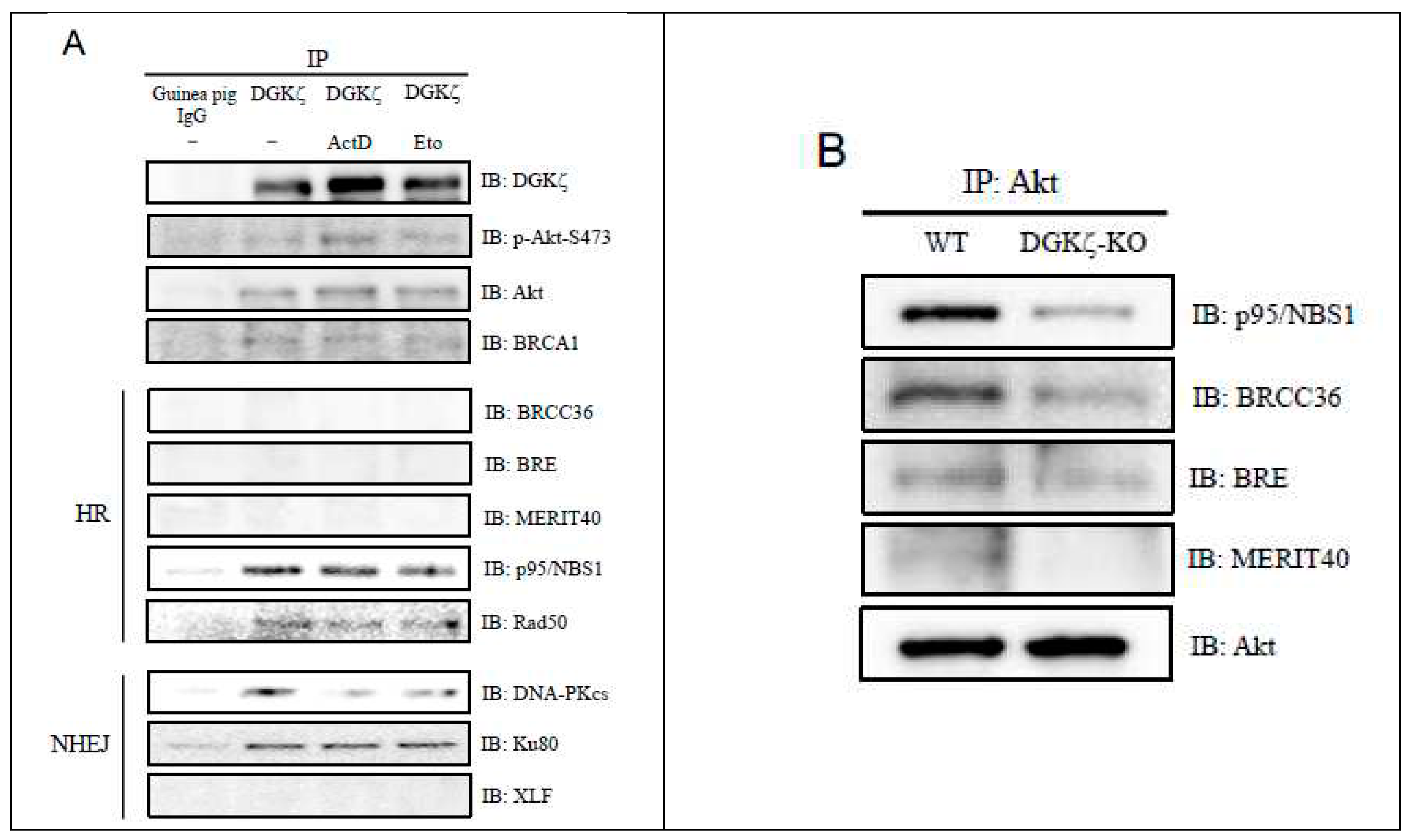
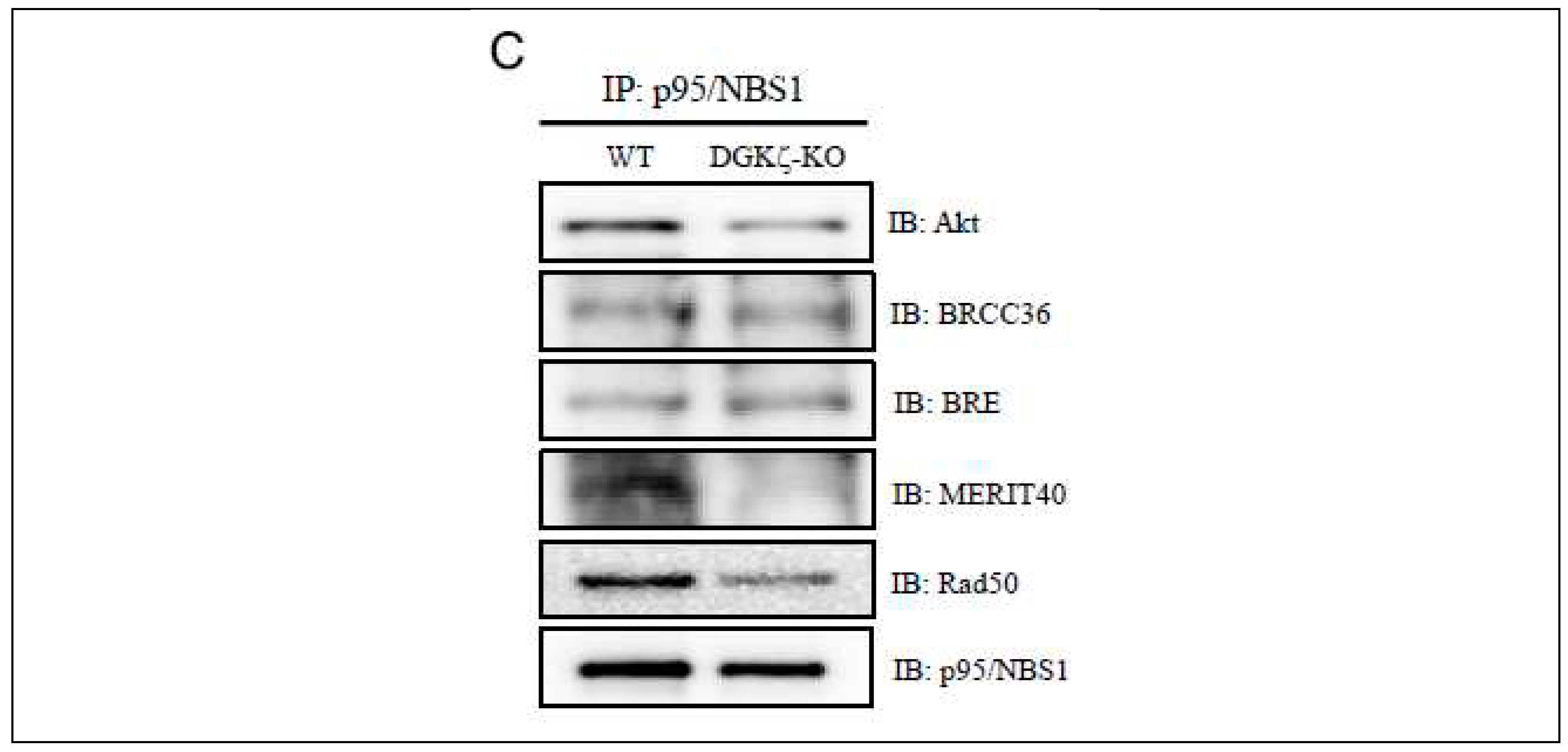
Figure 4.
Depletion of DGKζ induces cytoplasmic retention of DNA damage-repair proteins. WT and DGKζ-KO MEFs were treated with Eto (10 μM) for 1 h. After thoroughly washing out Eto, cells were further incubated with the regular DMEM for 2 h. Cells were fixed with 4% paraformaldehyde and stained with anti-p95/NBS1 (A) or anti-DNA-PKcs (B) antibody followed by anti-rabbit IgG-conjugated Alexa 488 (green). Cells were co-stained and anti-γH2AX antibody followed by anti-mouse IgG-conjugated Alexa 594 (red). The immunoreactivity for γH2AX almost disappears in WT MEFs after washing out for 2 h. Note that the immunoreactivities for p95/NBS1 (arrows in A) and DNA-PKcs (arrowheads in B) are partially recognized to the cytoplasm in DGKζ-KO MEFs after washing out for 2 h.
Figure 4.
Depletion of DGKζ induces cytoplasmic retention of DNA damage-repair proteins. WT and DGKζ-KO MEFs were treated with Eto (10 μM) for 1 h. After thoroughly washing out Eto, cells were further incubated with the regular DMEM for 2 h. Cells were fixed with 4% paraformaldehyde and stained with anti-p95/NBS1 (A) or anti-DNA-PKcs (B) antibody followed by anti-rabbit IgG-conjugated Alexa 488 (green). Cells were co-stained and anti-γH2AX antibody followed by anti-mouse IgG-conjugated Alexa 594 (red). The immunoreactivity for γH2AX almost disappears in WT MEFs after washing out for 2 h. Note that the immunoreactivities for p95/NBS1 (arrows in A) and DNA-PKcs (arrowheads in B) are partially recognized to the cytoplasm in DGKζ-KO MEFs after washing out for 2 h.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



