
Submitted:
31 January 2023
Posted:
01 February 2023
You are already at the latest version
Abstract
Much evidence suggests a correlation between degeneration and mitochondrial impairment. Typical cases of degeneration can also be observed in physiological phenomena (aging) as well as in neurological neurodegenerative diseases and cancer. All these pathologies have as a common denominator the dyshomeostasis of mitochondrial bioenergy. Even neurodegenerative diseases show a bioenergetics imbalance in their pathogenesis or progression. Huntington's chorea and Parkinson's disease are both neurodegenerative diseases, but while Huntington's disease is a genetic, and progressive disease with early manifestation and severe penetrance, Parkinson's disease is a pathology with a multifactorial aspect. Indeed, there are different types of Parkinson/Parkinsonism. Many forms are early onset diseases linked to gene mutation, others can appear in young adults and senescent only post-injury, and a final group is idiopathic. Huntington's was defined as a hyperkinetic disorder, while Parkinson's is a hypokinetic disorder; but in the middle, there are a lot of similarities as well as neuronal excitability, the loss of striatal function, psychiatric comorbidity, etc. In the review, we would embrace the theories that both diseases start and develop in light of mitochondrial dysfunction. These dysfunctions act on energy metabolism and reduce the vitality of neurons in many different brain areas.
Keywords:
movement disorders
; mitochondria
; energy metabolism
; synaptic plasticity
; basal ganglia
; calcium
Introduction
The Mitochondrion is an evolutionary organelle that originates from alphaproteobacteria that are entered into eukaryotic cells and have established a symbiotic ratio. It led during mitotic cell divisions to stabilize the host as an organelle [1]. In light of this hypothesis, it is possible that mitochondria have enabled the development of life as we know it and represents the common factor through which many organisms function or are dysfunctional [2]. Mitochondria are well-known as energetic units of the eukaryotic cells. They supply energy in the form of ATP to support all cellular functions. They provide for the aerobic respiration, a complex biochemical pathway by which starting from pyruvate and adding different co-factors, mitochondria synthesized ATP molecules. Moreover, mitochondrial proteomes, aggregates of more than 1000 proteins, can realize a wide variety of critical biochemical processes, including amino acid metabolism, nucleotide metabolism, protein synthesis, and fatty-acid catabolism [3]. There is abundant evidence in literature associating mitochondrial disruption with neurological and neurodegenerative diseases [4,5,6,7]. The brain requires high-energy consumption in the form of ATP; it was estimated that a cortical neuron in the human brain could utilize 4.7 billion molecules of ATP/sec. In addition, the ATP used at rest corresponds to about 5.7 kg per day [8]. The brain's highenergy demand supports many important functions, such as neurotransmission, cellular activities required for learning and memory, neural plasticity, and synapse development [9]. Since the mitochondrion, through oxidative phosphorylation and the production of ATP, meets all the energy demands of the nervous system, even with a rapid turnover [10], mitochondrial dysfunction with consequent loss in the energetic capacity and may represent a neurodegenerative trigger. Severe bioenergetic dysfunction makes neurons very vulnerable to oxidative stress and predisposed to neuronal cell death [11]. Additionally, neurodegeneration can be seen as an energy disorder, which manifests indifferent aspects depending on the severity of the process and the area involved [12]. This energetic disorder can lead to different pathologies with sometimes-opposite symptoms, such as hypokinetic and hyperkinetic disorders. The purpose of the present review is to emphasize how mitochondrial dysfunctions, which may be associated with phenomena as inflammation and degeneration, are actually a common element in two major neurodegenerative diseases, one purely genetic and the other resulting from a combination of genetic and environmental factors: Huntington’s disease (HD) and Parkinson’s disease (PD).
Role of Mitochondria in Brain Energy Metabolism, Calcium Homeostasis, and Signal Transduction
Mitochondria are organelles responsible for several critical processes in neuronal function and dysfunction, including energy metabolism, calcium homeostasis, and signal transduction [13]. The brain consumes 20% of total oxygen respired to meet the high metabolic demand of neurons that must maintain ionic gradients across membranes, transport of molecules from the soma along axons and dendrites, as well as for neurotransmission [14]. The metabolic activities to generate ATP rely mainly on mitochondrial oxidative phosphorylation (OXPHOS). During OXPHOS, enzymes of the Krebs cycle utilize acetyl-coenzyme A to reduce the cofactors nicotinamide adenine dinucleotides (NADH) and flavin adenine dinucleotides (FADH2) that serve in energy transfer to the electron transport chain (ETC), embedded within the extensive inner mitochondrial membrane [15,16]. The ETC consists of four complexes which transfer electrons from NADH and FADH2 to O2. Ultimately, the energy released by electron’s transfer is used to allow the thermodynamically unfavorable pumping of protons against their concentration gradient from matrix to the intermembrane space and generate an electrochemical gradient, known as mitochondrial membrane potential (ΔΨm), essential in the process of energy storage. The protons accumulated in the intermembrane space are allowed to move, according to a concentration gradient, back into the matrix by passing through one of the domains of the enzyme ATP synthase (or Complex V), which finally harnesses the released energy to phosphorylate ADP molecules into ATP [15]. Proper neuronal functioning is strongly linked to the retention of mitochondrial membrane potential and ATP levels [17]. Indeed, mitochondrial functions go beyond the primary role of ATP production because these organelles are intimately involved in numerous processes operating within the cell, including calcium homeostasis, generation of free radical species (ROS), steroid synthesis, apoptosis, and participate in cell signaling pathways [18]. The direction of the mitochondrial membrane potential (negative internal) has important implications as it elicits the thermodynamic force supporting the accumulation of metal cations, in particular the calcium (Ca2+), in the mitochondria [19]. Mitochondrial Ca2+ homeostasis undertakes a key role in cellular bioenergetics and signaling [20]. Ca2+ passage across the outer mitochondrial membrane (OMM) is mediated by the VDAC channel, whose different selectivity for cations or anions is voltage-dependent [21]. At low potentials (10 mV), the VDAC channel is highly permeable to anions and able to maintain low Ca2+ flux. Conversely, increases in the membrane potential (20-30 mV) result in the conformational change that allows a 4- to 10-fold increase in Ca2+ influx [22]. In comparison, the inner mitochondrial membrane (IMM) has considerable Ca2+ permeability: the mitochondrial calcium uniporter (MCU) contributes to the potential-dependent Ca2+ influx into the mitochondrial matrix while the mitochondrial Na+/Ca2+ exchanger (NCLX) is one of the main units involved in Ca2+ extrusion [23]. In general, cytosolic calcium uptake by mitochondria will occur only if they are exposed to elevated Ca2+ concentrations [24,25]. Within the cell, however, Ca2+ is compartmentalized and the average resting cytosolic concentration is remarkably low [26,27,28]. One of the main stores of cellular calcium is the endoplasmic reticulum (ER), which contributes to orchestrating cellular Ca2+ homeostasis through its interaction with mitochondria [29]. Notably, Ca2+ uptake in mitochondria probably occurs only at proximity sites between the ER and mitochondria [20]. Neuronal mitochondria, unlike those in non-neuronal cells, have a considerably lower threshold for Ca2+ uptake. In addition, ER-mitochondria contacts are critical for Ca2+ uptake in dendritic mitochondria but not in axonic mitochondria [30]. This property of axonal mitochondria is due to the presence of the brain-specific uniporter, MICU3, which unlocks axons from the constraint of relying on intracellular Ca2+ storage. In the absence of MICU3, synaptic function is impaired [31]. Mitochondrial calcium signaling in neurons regulates metabolism and energy production, which are crucial for neurotransmission and sustaining synaptic plasticity [32]. In addition, mitochondrial Ca2+ also drives the production of reactive oxygen species (ROS) [33]. Mitochondria are the primary sources of ROS in cells and actively participate in cellular redox regulation and ROS signaling [34]. ROS, in the form of superoxide, are natural byproducts of normal mitochondrial activity, naturally converted to H2O2, in turn scavenged, by the enzyme catalase, into water [34]. Under normal conditions, through the generation of ROS and redox signaling, mitochondria can control cell metabolism and physiology, as well as inflammatory response, immune function, autophagy, and stress response [35,36,37]. Indeed, ROS, and in particular hydrogen peroxide, at low concentrations, act as important signaling molecules in the cell, activating several protein kinases, such as PKA, PKC, PI3K, and p38 [38]. Moreover, in immune cells, mitochondrial metabolites and ROS finely regulate signaling pathways and cell fate, orchestrating the immune response [39]. The immune system comprises a diverse family of cells with multiple roles during homeostasis and inflammation, capable of using distinct metabolic programs to undertake their functions. For example, during the immune response, effector T cells promote aerobic glycolysis while, in contrast, memory T cells and regulatory T cells promote fatty acid oxidation [40]. Particularly, mitochondria can modulate the metabolic and physiological states of different types of immune cells [41] and stimulate the innate immune signaling cascade that can intensify inflammation following cytotoxic stimuli or microbial infection [42]. For example, the innate immune receptor NLRX1, a member of the Nod-like Receptor (NLR) family, is located in mitochondria and undertakes an important role in maintaining cellular homeostasis following acute mitochondrial injury [43,44]. In addition, it has recently been discovered that mitochondria play a central role in initiating and regulating the NLRP3 (nucleotide-binding domain, leucine-rich-repeat containing family, pyrin domain-containing 3) inflammasome [45]. This multiprotein complex, activated upon infection or cellular stress, leads to the secretion of proinflammatory cytokines such as interleukin-1β (IL-1β) and IL-18 that trigger an inflammatory form of cell death called pyroptosis [46,47]. Although the mechanisms of NLRP3 inflammasome activation are still debated, it is widely believed that changes in the mitochondrial membrane potential, permeabilization of the outer mitochondrial membrane, and increased formation of mitochondrial ROS are crucial factors in inducing the cytosolic translocation of mitochondrial molecules, such as cardiolipin and mitochondrial DNA, which are capable of activating the inflammasome [45]. Indeed, the overproduction of ROS and dysregulation of the redox signaling system results in oxidative stress that can lead to mitochondrial damage, induce mitochondrial DNA mutations, damage the respiratory chain, alter membrane permeability, and affect the Ca2+ homeostasis and mitochondrial defense systems [48]. Accumulating evidence suggests that oxidative stress, and mitochondrial injury can result in cellular DNA damage and degradation of proteins and lipids as well involved in the pathogenesis of neurodegenerative diseases [49,50]. Nevertheless, cells have an accurate endogenous antioxidant defense system that can maintain cellular redox homeostasis between ROS production and elimination that ensure normal cellular signaling and redox regulation [51]. The nuclear factor erythroid 2–related factor 2 (Nrf2) is an emerging therapeutic target since it is involved in cellular resistance to oxidants. In detail, Nrf2 controls basal expression of genes coding for enzymes and proteins involved in antioxidant and detoxifying action, repair and removal of damaged proteins and organelles, inflammatory response, and mitochondrial bioenergetics [52,53]. Additionally, malfunctioning mitochondria can be selectively removed through a conserved cellular recycling process known as mitochondrial autophagy or mitophagy. Efficient elimination of damaged mitochondria prevents activation of cell death pathways, protects against ROS overproduction, and maintains efficient ATP production [54]. Damaged mitochondria are swallowed into autophagic vesicles that subsequently transport them to lysosomes for destruction. Mitophagy is a strictly regulated process, modulated by mitochondrial fission and fusion proteins, BCL-2 (B-cell lymphoma 2) family proteins [55], and the PINK1/Parkin pathway [56]. As all other defense mechanisms fail, the neuron can orchestrate its self-destruction by activating an intrinsic suicide program otherwise known as apoptosis [57]. The underlying mechanisms of apoptosis are very sophisticated and engage an energy-dependent cascade of molecular events. In addition, it can follow different molecular pathways, one of which, known as the intrinsic pathway, involves the mitochondria [58]. Indeed, these organelles represent the site where antiapoptotic and proapoptotic proteins interact and the origin of signals that initiate the activation of caspases, the cysteine proteases capable of cleaving many cellular substrates to disrupt cellular contents [59].
It is therefore understood that mitochondrial integrity and homeostasis are prerequisites for proper cellular functioning, most notably for neurons, which are polarized, complex cells with high energy demand [60]. Not surprisingly, neurons have the highest content of mitochondria compared to other cell populations. Mitochondrial functionality is essential for ensuring membrane excitability and performing the complex neurotransmission and plasticity processes. First, mitochondria provide the energy needed to undertake a wide range of neuronal functions: the maintenance of resting membrane potential, the restoration of ionic balance after depolarization, the cycling of synaptic vesicles, as well as the transport of proteins and organelles from the soma to distal sites. Interestingly, mitochondria in axons and dendrites have different morphologies, small and sparsely distributed in the former and elongated and densely distributed in the latter. In addition, axonal and dendritic mitochondria differ in movement, metabolism, and responses to neuronal activity [61]. Second, several lines of evidence support the role of mitochondria in the mobilization and recycling of synaptic vesicles [62]. Neurotransmission is underpinned by endocytosis and the local filling of synaptic vesicles in the presynaptic terminal [63]. Third, mitochondria support synaptic activity through cytosolic calcium reabsorption, a critical buffering mechanism for establishing and maintaining synaptic activity and preventing neuronal toxicity and excitotoxicity. Nevertheless, mitochondrial calcium buffering appears necessary at the synapse even when the neuron is at rest: synaptic terminals lacking mitochondria show a higher frequency of spontaneous release of neurotransmitter-containing vesicles [64]. Overall, the importance of mitochondria in neurons is unequivocal, and it is not unexpected that mitochondrial dysfunction has emerged as a critical factor in a plethora of conditions, from impaired neuronal development to various neurodegenerative diseases.
Role of Mitochondria in Degenerative Disease
A great deal of evidence supports the role of mitochondrial dysfunction in degenerative diseases [65] diseases caused by mitochondrial alterations often show a neurodegenerative component involving the nervous system and, similarly, mitochondrial defects are frequently observed in tissue samples taken from patients with a neurodegenerative disorder. This evidence found a common goal in the high-energy requirements for all biological processes. The continuous increase in energy demand, mainly due to excess food consumed daily, the constant increase in energy required for thermoregulation, and the increase in energy needed to counteract the environmental toxicity caused by human them self, require an overwork of the mitochondria resulting in impaired bioenergy efficiency [66,67]. It was demonstrated that the cells exposed to an opulent nutrient environment are inclined to have their mitochondria in a fragmented state. Moreover, mitochondria observed in cells below malnourishment are apt to remain for a longer time in the associated state [68,69]. This is evidence that mitochondria can change their architecture and therefore their bioenergy capacity according to external events. A degenerative disease is defined as a type of un-physiological condition whose origins in a tissue or organ worsen over time. Energy failure was linked to degenerative processes as well as cancer, aging, neuroendocrine disease, neurodegenerative disease, and inflammatory diseases. In agreement, a great deal of evidence supports the involvement of mitochondria in neurodegenerative particularly in PD [70,71,72,73,74]. The story of MPTP toxin first highlighted the role of mitochondrial complex I dysfunction and neurodegeneration in PD [1]. In association with other more used toxins as well as rotenone and Paraquat, MPTP offered a new scenario to define the effective role of mitochondrial bioenergy in neurodegeneration [75]. Indeed, more than five thousand manuscripts dealing with the association between Parkinson's disease and mitochondrial alteration can be found on Pubmed. In addition, due to studies on PD, particularly genetic Parkinsonism, all mitochondrial dysfunction leading to cell death has been well defined [76]. PINK1 gene mutation, responsible for an early onset Parkinsonism, is an example [77]. This gene codifies for the mitochondrial protein phosphatase and tensin homolog serine/threonine-protein kinase 1(PTEN-induced kinase 1) [78]. PTEN- protein can protect the cells against oxidative stress, proton chain dysfunction, and bioenergy failure [77,79] Silvestri et al., 2005). The PINK1 gene has been also identified, the first, as an oncogene with tumor suppressor properties [78,80,81]. Additionally, PINK1 protective role is observed in many disorders characterized by progressive inflammation and neurodegeneration, such as Alzheimer's disease, multiple sclerosis, amyotrophic lateral sclerosis, and HD [78]. In physiological conditions, PINK1 is translocated inside the mitochondria in its mature isoform with the scopes to withstand the activity of the mitochondrial chain and to produce (at the level of complex I) the molecules of ATP showing the maximal bioenergetics efficiency. Moreover, PINK1 is located, also, in the inner mitochondrial membrane and can interact with the chaperone TRAP1 (recognized also, as an interactor of the type 1 tumor necrosis factor receptor), to maintain important bioenergetics and proteostatic functions [78,82]. Clivate PINK1 can interact with other chaperon proteins and when it is located in the cytosol can activate the m-Tork/Atk pathway. Besides, PINK1 can mediate the phosphorylation of another important gene for PD: Parkin. The coupled action of PINK1 and Parkin may induce mitochondrial fusion with the scope to cause the elimination of dysfunctional mitochondria. PINK1 is also involved in the formation of an autophagosome complex by the activation of beclin protein and may regulate the apoptotic process [78]. The role of the PINK1 gene in PD was well investigated by the use of animal models and cellular culture by human fibroblast [83,84,85,86,87,88]. These studies have confirmed the role of bioenergetics efficiency in the maintenance of health state or the progression of neurodegeneration. The absence of PINK1 results in low bioenergetic efficiency and programmed cell death after each minor stress [87,89]. It is very interesting to note that PINK1 has not only a key role in degeneration in general, in oncological degenerative processes, and neurodegenerative PD, but also neurodegeneration found in HD. PD and HD are two different neurological diseases involving the central nervous system. The symptomatology of both is quite similar (cognitive impairment, limb inflexibility, and problems in walking or talking), but while PD results from a combination of genetic and environmental factors, HD is only an inherited genetic disease. Both pathologies have a common denominator: the loss of bioenergetics efficiency [78,90]. HD is an incurable degenerative disorder caused by a mutation in the huntingtin gene where the CAG sequence is excessively repeated. This mutation alters numerous cellular processes and leads the cell to apoptosis. One important alteration is caused by the impairment of mitochondrial metabolism. In a study conducted on the fly model was found the formation of abnormal ring-shaped mitochondria; this particular shape was previously identified in mitophagy-blocked cells in which PINK1 overexpression was able to rescue the regular shape and function of mitochondria. PINK1 over-expression was able to improve bioenergetics efficiency (increasing ATP levels) and rescue neuronal integrity in the adult drosophila model of HD [91]. HD is a degenerative pathology caused by pathological expansion of CAG repeats in the Huntington gene codifying for the Huntingtin protein. The gene is located on chromosome number 4 and is characterized by a high level of polymorphisms. Unlike PD, HD has an age of onset of around 35-44 years and an estimated post-onset life span of 15/18 years; and is characterized by advanced motor disability including chorea [92]. Many pieces of evidence suggest that the mutation may cause alteration in mitochondrial trafficking, increase in oxidative stress, dyshomeostasis in intracellular calcium content, alteration in bioenergetics, and alteration in a respiratory mitochondrial chain [93,94,95,96]. HD, PD, and Alzheimer's diseases are three-neurodegenerative diseases that have 37 common genes and about 40 % of whose product acts at the mitochondrial level [97]. These neurodegenerative diseases are coupled to a physiological, but equally degenerative process called aging or senescence that starts at mitochondrial levels and results in reduced bioenergetics efficiency [98]. Cellular senescence is characterized by heavy changes in cellular metabolism and by an increase in pyruvate utilization that may produce differences in phosphorylation state increasing the activity, of the mitochondrial pyruvate dehydrogenase complex; and the increase of ROS production [99,100]. These features may lead to an impairment of cellular function that induces degeneration equal to cancer or neurodegenerative processes [101]. Moreover, recent data have demonstrated that key oncogenes and tumor suppressors modulate mitochondrial metabolism and dynamics. Indeed, different types of cancer result in more or less sensitivity to the modulation of mitochondrial function in the function of the lesion caused by the tumor [102]. Therefore, mitochondria play a cardinal role in many degenerative processes and it is only in animal models, even before humans, that they can make us understand their importance.
Mitochondria Bioenergy in Parkinson’s Disease, Huntington Disease Rodents Animal Models
In PD and HD are both neurodegenerative diseases with multiple mitochondria bioenergy alterations related to metabolism oxidative stress, dynamics biogenesis transport, and mitophagy. Since there is a close relationship between neurodegeneration and mitochondria bioenergy, one might presume that mitochondria homeostasis changes, with the release of calcium and NO as well as reactive oxygen species (ROS), will affect the unfolded protein response modulating specific signalling molecules and reprogramming mitochondria bioenergy. The mice models of PD e HD provide a valuable tool to define the role of mitochondria bioenergy in pathogenic mechanisms in these diseases.
Parkinson Disease
PD animal models are associated with multiple mitochondrial defects [103,104,105,106]. The current view is that abnormalities in more related mitochondrial factions such as membrane potential, energy production, disruption of mitochondrial-ER Ca2+ homeostasis, inhibition of mitochondrial dynamics, and induced mitochondrial pro-apoptotic protein cytochrome c release led to neurodegeneration [107,108,109]. In fact, the neurotoxin MPTP, rotenone, and paraquat block mitochondrial bioenergetics in dopaminergic neurons and induce Parkinsonian syndrome [110,111,112,113,114,115,116]. However, it is important to consider that overexpression of α-synuclein or agent that generates stress or cytosolic acidification shows translocation of α-synuclein into the mitochondria or in the mitochondrial membrane [117,118,119]. α-synuclein is a presynaptic neuronal protein that is linked to familial and idiopathic PD [120] both over-expression or loss-of-function of SNCA gene contribute to disease manifestation [121] according to the “α-synuclein cascade hypothesis” [122]. Experimental studies in A53T mutant mice, a transgenic animal model that expresses human α- synuclein, develop mitochondrial DNA damage and degeneration with an apoptotic-like death of neocortical, brainstem, and motor neurons [123]. In the same animal model, the mitochondrial complex IV activity was reduced significantly in the spinal cord [123]. Additionally, in vivo human α-synuclein expression is associated with a decrease of Drp1 (Dynamin-related protein 1), the major player in the regulation of mitochondrial dynamics and plays a critical role in the maintenance of their proper function [124]. The dysfunction of Drp1 in the mitochondria is associated with enlarged neuronal mitochondria [124].
Other protein that facilitate mitochondrial fusion are decreased in these models, Mfn1 (Mitofusin1) [124], which correlates with the mitochondria bioenergy dynamic changes. Previous studies in A53T mice suggest that α-synuclein distress the mitochondrial morphology and reduces both Mfn1 and Mfn2 in an age-dependent manner [125]. In vivo study in midbrain dopaminergic neurons suggested that α-synuclein predominantly accumulates in the central mitochondrial membrane and interacts with the complex I resulting in the impaired activity of the mitochondrial electron transport chain [126]. Consistent with these findings other experimental analyses show that after post-inoculation of human α-synuclein in rats, lends its accumulation in striatal dopaminergic terminals from the SNpc and early synapse loss [127]. To explain this early synapse loss a proteomic analysis using a real-time cell metabolic method and isolated synaptosomes was used to investigate functional and molecular mechanisms. Upon injection, in isolated striatal synapses was found multiple dysfunctions of mitochondrial bioenergetics and morphology were with the upregulation of PRKAG2 and TTR, respectively sensor protein of the cell energy status and marker of oxidative stress [127]. Moreover, ultrastructural examination of subsequent human α-synuclein accumulation shows an altered expression of Rab5 endocytic and LC3 autophagic proteins that potentially reflect an accumulation of autophagic vesicles [127]. This study has allowed the evaluation of an interesting aspect such as the contribution of oxidative stress and the mitochondria bioenergy alteration in both dopaminergic neuron death and striatal neurons, which could be responsible together for the induction of Parkinsonian syndrome. The neurons are high- energy consumers and employ most of the energy at the level of the synapse to preserve and restore ionic gradients, and for the uptake and recycling of neurotransmitters [128]. Various mouse lines expressing a different type of α-synuclein present impaired dopamine neurotransmission and striatal synaptic plasticity [129]. In rodents, the injection of α-synuclein with an adeno-associated viral vector blocks the induction of long-term potentiation (LTP) long-term depression (LTD) that is normally expressed in medium spine neuron (SPNs) [116,130,131,132] that are associated to early memory and motor alterations. There is a relatively large body of evidence that α-synuclein misfolding and aggregation lead to mitochondrial stress that appears to be related to the dysfunction of synaptic plasticity. Another interesting aspect is that α-synuclein overexpression promotes by the endoplasmic reticulum (ER) C2+ transfer in the mitochondria, while its silencing impairs mitochondrial function by loosening the ER-mitochondria interface [133].
Although, the precise mechanism by which α-synuclein accomplished pathological characteristics and specific cell death remain to be delineated more clearly. However, it is important to note that α-synuclein plays a role as pro-apoptotic in different neuronal cells [134]. The wild-type α-synuclein can defend neurons from apoptosis by inhibition of caspase-3 and the mutant α-synuclein loses this activity [134]. Caspases-3 exerts its actions trigger general damage and degeneration, aggregates in synapses, and persists in neurons without causing acute cell death [135,136,137]. Furthermore, the discovery of many genes related directly or indirectly with mitochondria to PD underlines the implication of mitochondria bioenergy in its pathogenesis features. For instance, genes linked to early-onset recessive PD include Parkin [84], PINK1 [77] and DJ-1 [138], these genes have been powerfully implicated in the morphology, damaged and mitochondria degradation via mitophagy [139], involved in the mitochondrial activity and quality control mechanisms [78,140,141,142,143]. Parkin encodes a protein localised in the cytoplasm that contains an N-terminal ubiquitin-like domain with a function as E3 ubiquitin-protein ligase [144,145,146]. Parkin knockout (KO) mice show alteration of the levels of different proteins involved in detoxification, stress-related chaperones, without neuronal degeneration [147]. Parkin has neuroprotective effects through the regulation of different cellular processes or pathways such as mitochondrial swelling and cytochrome c release [148,149,150]. Normally, Parkin resides in the cytoplasm but can translocate to depolarized or damaged mitochondria to mediate their removal by mitophagy in cooperation with PINK1 and potentially other factors [151]. In a model mouse of Parkin KO was reported to cause a decrease of subunits of complexes I and IV with redaction of peroxide reductases. In addition, the serum presents a redaction of antioxidant capacity and increased protein of lipid peroxidation [152]. Other studies in Parkin-deficient mice show an increase in the content of dopamine in the striatum and consequently redaction of the excitability of striatal SPNs [153]. Deficits in glutamate neurotransmission and amphetamine-induced dopamine release were also observed in other parkin mutant mice with an increase in the metabolism of dopamine (MAO) [154]. Functional analysis of striatal SPNs showed an impairment of bidirectional corticostriatal synaptic plasticity, with the loss of the LTD and LTP in Parkin KO mice, while synaptic plasticity was not altered in the hippocampus of these animals [155]. The dopaminergic defect [153,154] may explain a selectively enhanced sensitivity to striatal group II metabotropic glutamate receptor in cortically-evoked excitatory postsynaptic potentials recorded from SPNs, this reinforcing effect is an adaptive change [79]. Recent evidence in Parkin KO rats offers the suggestion that lack of Parkin is also necessary for the maintenance of postsynaptic endocytosis of AMPARs, even decreased expression of the postsynaptic protein Homer1, which is essential for coupling AMPA receptor endocytic zones with the postsynaptic density [156]. On the other hand, changes to the numbers or function of ER-mitochondrial contact sites may also affect mitochondrial calcium homeostasis regulated by Parkin [157], calcium uptake into the mitochondrial matrix results in enhanced respiratory function, tuning synaptic activity [158,159]. Recent work showed that conditional knockout of Parkin in adult animals expresses the progressive loss of dopamine neurons. Moreover, overexpression of PGC-1α leads to the selective loss of DA neurons in the substantia nigra [160].
One more gene linked to early-onset recessive PD is PINK1 which encodes a protein with a serine/threonine kinase catalytic domain cytosolic or mitochondrial-associated [78]. Mice models of PINK KO exhibited significant functional impairment of synaptic plasticity and mitochondria morphology, without motor deficits or dopaminergic neuronal loss [84]. Consistent with these findings, PINK KO mice models generated with the silencing of the PINK1 gene did not develop dopaminergic neurodegeneration [161]. PINK1 heterozygous KO mouse show selective impairment of LTP with a normal expression of LTD [162], nevertheless, low doses of rotenone has been sufficient to induce severe alterations of the corticostriatal LTP and LTD [85]. This led to the proposal that the PINK1 homozygous KO model represents a good model to study the effect of gene- environment interaction. While PINK1 KO rats show mitochondrial alteration, locomotor deficits, and α-synuclein aggregates in several brain regions such as the cerebral cortex, dorsal striatum, and degeneration of substantia nigra [163,164,165,166]. However, this model presents clear symptomatic deficits that occur at 9 months old and at 4 months are asymptomatic [165]. In the same model, using magnetic resonance spectroscopy, the mitochondrial metabolomic alterations arise in the cortex and striatum, during the asymptomatic period, which coincides with the metabolic alterations [165]. Functional, bioenergetics experiments revealed that mitochondrial alteration in PINK1 KO rats is due to impaired complex I respiration carrying a dysfunction in ATP synthase and reduced substrate oxidation [164]. New scientific research performed in animal models with the deletion of PINK1 mice provides an indication that lack of PINK1 results in impaired synaptic plasticity caspase-mediated [86]. Caspase-3 presents an additional role in neuronal processes, including the regulation of synaptic function, and pruning [136,167,168]. Previous studies in PINK1 KO show a loss of LTP and LTD with alteration on dopamine release [84]. Indeed, low activation of the caspase-3 is critical to restoring cortical-striatal LTD [86] although chronic activation is involved in degenerative processes [168]. In PINK1 KO mice analysis with electron microscopic and functional studies in the striatum revealed significantly enlarged mitochondria shape and impaired activity [169]. Indeed, functional examination showed impaired mitochondrial respiration and aconitase activity in the striatum but not in the cerebral cortex [169]. Additional independent studies in PINK1 KO mice show a decreased dopamine release in the dorsal striatum in an age-dependent manner with impairment of basal mitochondria respiration [170].
DJ-1 gene mutations cause autosomal recessive early-onset PD. DJ1 encodes a protein of the superfamily which belongs to the ThiJ/PfpI-like. normal conditions, DJ-1 is confined throughout then cytoplasm of neurons with a small amount localised to the mitochondrial matrix and intermembrane space [171] indicating a role in the balance in mitochondrial physiology [19]. DJ-1 KO mice studies fail to express PD features such as substantia nigra degeneration or formation of protein inclusions similar to PINK1 and Parkin KO mice [141]. Recent experiments in DJ-1 KO mice show increasing facilitation of calcium influx into the neuron during its activity, creating basal mitochondrial oxidant stress [172,173]. Functional analysis in isolated mitochondria shows increased ROS and decreased aconitase activity in DJ-1 KO mice [180]. Additionally, DJ-1 gene deletion reveals a deficit in scavenging because the loss DJ-1 pathway loses atypical peroxiredoxin-like activity [174]. In contrast, in an independent study of DJ-1 KO mice present an increase in mitochondrial respiration-dependent H2O2 consumption and increase mitochondrial Trx activity, total glutathione (GSH and GSSG respectively) levels, mitochondrial glutaredoxin (GRX) activity and a decrease in mitochondrial glutathione reductase (GR) activity [175]. Furthermore, new research of the semi-quantitative measurement of cerebral metabolites in DJ-1 knockout mice detect significantly increased glutathione (GSH) level and GSH/glutamate (Glu) ratio in prefrontal cortex [176] (Table 1). It is important to note that the GSH system is the more important antioxidant system in ROS detoxification in the brain [177,178,179]. Accumulated scientific information allowed the hypothesise that the increased sensitivity to oxidative stress in DJ-1 KO mice and dopaminergic neuronal cells is related to a decrease in ROS scavenging arising from deteriorated peroxidase-like scavenging with a deficient Nrf2 transcriptional factors and increased mitochondrial dysfunction due to complex I deficiency [180,181,182,183,184]. Although, DJ-1KO mice had normal corticostriatal LTP but the LTD was absent [185]. Overall, the data from animal models provide a framework that suggests that genes linked to early-onset recessive PD are important for maintaining normal mitochondria bioenergy function. Gradually, the mutation protein described above related to the mitochondrial bioenergy activates the response to mitigate the mitochondrial alteration. Indeed, genetic inactivation PINK1/Parkin/DJ-1 with a triple KO mouse exhibit abnormalities in mitochondrial pathway and morphology or activity without PD-related phenotype [85] (Table 1).
Huntington Disease
For HD several mouse lines of transgenic and knock-in mice have been engineered to address specific pathological characteristics [186]. Accumulation of scientific information implicates that gain and/or loss of function mutant HTT leads to impaired mitochondrial bioenergy with alteration of oxidative stress, dysfunction of mitochondrial trafficking, and mitochondrial calcium dyshomeostasis [187]. In fact, malonate and 3-nitropropionic acid (3-NPA) are considered potent neurotoxins and are used to induce experimental pharmacological models of HD di rodents [188]. These oldest models provided the first piece of evidence that mitochondria bioenergy, targeting the electron transport chain, is involved in the pathophysiology of HD. However, degenerating mitochondria have been identified in different areas, brain, liver, and muscle in genetic mouse models of HD [189,190,191] (Table 1).
The R6/1 and R6/2 both strains express a single copy of a human genomic fragment that contains 116 and 144 CAG repeat HTT under the control of the human HTT promoter [192]. These mice express relatively rapid onset and progression of symptoms that include motor defects and neurodegeneration. Another major contribution to the research was the generation of knockin mice models where CAG is repeated extensively (CAG94 and CAG140), compared to the human trans gene, for developing HD-associated phenotypes [186]. The R6/2 model mice have been the most widely utilised because the disease starts earlier with a rapid progression compared to the YAC, BAC, and other knock-in lines [193]. Transgenic animal models expressing the amino (N)-terminal fragment of the mutant form of HTT (R6/2) show an increase of 8-hydroxy-2-deoxyguanosine OH(8)dG in the late stages of the illness [194], thus, oxidative stress occurs in the striatum before the onset of motor symptoms. Biochemical analysis performed in the striatum of R6/2 mice at 12 weeks shows a significant reduction of mitochondrial complex IV activities and a decrease in aconitase in the cerebral cortex [195]. In accordance with these data, new scientific research using synaptosomes isolated from R6/2 mice shows a decrease in mitochondrial mass with increased ROS production and antioxidant levels in the striatum compared to the cortex [196] indicating oxidative stress. Additionally, in the same model through depolarization conditions the oxygen consumption rates in synaptosomal were significantly amplified, which was accompanied by a clear increase of mitochondrial proton leak of the striatal synaptosomes [196], indicating synaptic mitochondrial stress. Indeed, synaptic dysfunction occurs in various mouse models of HD, appears to involve, with dysfunction of LTD, and displays a physiological LTP but lost synaptic depotentiation. These have been directly connected with the onset of cognitive deficits in HD models [197,198,199,200,201]. Using the brain slices from R6/1 mice, at different ages, presents a significant increase of Ca2+ content after glutamate stimulation, such alterations impact in mitochondrial dysfunction with a decrease in NAD(P)H fluorescence and loss in Mitochondrial membrane potential (ΔΨm) [202]. Several groups characterise the redaction of N-acetyl aspartate in R6/2 mice, similar to symptomatic HD patients [203,204,205,206,207] fluctuations in some metabolite suggest the impairment in cellular metabolism and mitochondrial bioenergy [208]. Another interesting contribution to assessing the deficits of mitochondrial function and redox deregulation is the experimental data performed with PET in YAC128 transgenic mice [209]. PET analysis displays a specific accumulation of [64Cu]-ATSM in the striatum with concomitant alteration of mitochondrial respiration and ATP production, and increased complex II and III activities. In turn, an increase in mitochondrial H2O2 levels in YAC128 mice and defects in Ca2+ handling [209] support an early increased striatal susceptibility of mitochondrial. Furthermore, functional assays revealed impairment of mitochondrial respiratory capacity in the striatum and cortex of R6/1 mice. It is important to note that N-acetylcysteine administration delayed the onset and progression of motor deficits in R6/1 mice [210] which may reduce both excitotoxicity and oxidative stress in the striatum. Recent work established that voluntary wheel running reduces hindlimb clasping in the R6/1 mouse model. Chronic exercise in the R6/1 showed the clasping phenotype with normal mitochondrial respiration in the cortex and striatum, suggesting mitochondrial dysfunction is not necessary for the progression of symptoms [211]. At the molecular level, different studies pointed to considering the contribution of mitochondria in their role in apoptotic cell death processes with an elevated caspase activity in HD mice models [189,212]. HTT has been shown to be involved in vitro for caspases-1,2,3 and [213,214,215]. In fact, immunostaining analysis of YAC72 mice displays an increase of caspase-2 in the SPNs in the striatum, with concomitantly decreased levels of a brain-derived neurotrophic factor in the cortex and striatum at 3 months [213], demonstrating that caspase-2 participates selective on the neurodegeneration of SNPs in the striatum. In YAC128 model mice, to contrast the cognitive dysfunction such as performance on the rotarod, swimming T-maze, the lacking caspase-2 produce a protection from the well-validated motor and cognitive features of HD [216]. Moreover, previous studies in transgenic mice expressing exon 1 of the human HTT demonstrate that intracerebroventricular administration of a caspase inhibitor 1 delays the disease progression of the disease [215] (Table 1).
In conclusion, while HD animal models have not yet provided definitive evidence whether or not mitochondria bioenergy is critically involved in htt-induced disease symptoms compared to PD animals models genes, mitochondrial bioenergy defects are nonetheless a major component for both disease progression. On the other hand, the creation of HD mouse models using different strain backgrounds and expressing only a portion of the htt protein leads to increased difficulty in understanding compelling findings that instead were obtained with the genetic models of PD.
Mitochondria Bioenergy in Parkinson’s Disease, Huntington Disease, Human Evidences
Assessment of mitochondrial activity (or dysfunction) is not easy in CNS of living patients. However, analyzing peripheral tissues and fluids can offer a reliable measure. Albeit the core pathology in PD and HD is central, a number of substantial abnormalities result at the systemic or peripheral level. Peripheral blood mononucleate cells (PBMCs) are one of the most reliable “in vivo PD model”. PBMCs present several alterations in critical metabolic pathways and accumulate α- synuclein pathological forms, well recapitulating PD-related neuropathology [217,218]. In a recent work, PBMCs mitochondrial bioenergetics from PD patients was assessed by the Seahorse Bioscience technology, showing a peculiar pattern of mitochondrial respiration, including normal basal respiration, significant augmentation of the maximal respiratory capacity and spare respiratory capacity, and a tendency to higher ATP production. The increased spare respiratory capacity was found to follow the disease duration and the severity of motor disturbances, revealing some mitochondrial adaptations to the higher bioenergetics requirements occurring at later disease stages [219]. Similar, consistent findings of greater mitochondrial respiration were also observed in different cell lines obtained from PD patients, such as lymphoblasts (immortalized blood lymphocyte-derived cells) [220] and fibroblasts [221,222]. It is thus reasonable that, in PD patients, oppositely from animal models, the mitochondrial activity may increase in a compensatory manner. Indeed, the “nuclear factor erythroid 2-related factor 2” (Nrf2) pathway, a master regulator of the cellular defence and mitochondrial activity, can be overexpressed in PD patients’ PBMCs proportionally to the disease duration, suggesting a systemic defensive response to the PD clinical-pathological progression [218]. Otherwise, changes in respiratory activity could reflect defects in mitochondrial structures, metabolism, or respiratory proteins functioning [223]. Indeed, another study also found in PD patients’ PBMCs increased glycolysis and deficits in superoxide dismutase, together with a peculiar mitochondrial vulnerability in monocyte subpopulation [224]. Substantial evidence also arises from the assay of mitochondrial dysfunction biomarkers in PD patients’ fluids. There are metabolomic studies performed in blood and cerebrospinal fluid (CSF) disclosing various abnormalities in metabolic pathways related to mitochondria (in alanine, branched-chain amino acids, fatty acids, and acylcarnitines levels; in steroidogenesis; in glutathione cycle) [225,226,227]. Likewise, the redox balance-related biomarker levels, such as uric acid [228,229,230,231] catalase and total glutathione [232], nonmercaptalbumin (oxidized form of albumin) [233], oxidized glutathione [218], oxidized DJ-1 [234], α-klotho [235], lactoperoxidase [236], and heme-oxygenase-1 [237] were altered in the same fluids. Genomic studies on peripheral blood showed the down-regulation of genes critical for mitochondrial functions (COX4I1, ATP5A1, and VDAC3) [238], while the analysis of blood-circulating extracellular vesicles demonstrated the reduction of mitochondrial components (i.e., ATP5A, NDUFS3, and SDHB) in PD patients [239]. Analysis of mitochondrial bioenergetics has also been performed in peripheral tissues of HD patients with some controversial findings depending on the matrix or the experimental technique. Indeed, mutant huntingtin is expressed out of the CNS as well, accounting for the substantial impairment in multiple cellular pathways related to mitochondria and redox balance [240]. It has been observed that skin fibroblast may show some energetic, respiratory, redox, and morphological abnormalities [241,242]. Likewise, several functional or structural mitochondrial defects have been found in HD patients-derived lymphobasts [243] (Figure 1). Finally, increased oxidative damage, reduced antioxidant capacity, and mitochondrial abnormalities have been tracked even in HD patients’ blood trough different fluid markers [244].
Discussion
Degenerative diseases show as a common point the mitochondrial impairment. Mitochondria represent the major source of bioenergy for the cell and, as a consequence, their putative failure leads to a lack of energy. Daily energy metabolism for a human and also for an organism is the sum of daily energy expenditure and daily energy intake. Daily Energy Expenditure may be divided into diverse workings: 1) energy spent on basal metabolism; 2) energy by thermic increment following food intake; 3) energy spent on thermoregulation; 4) energy spent during daily activities. In particular, energy metabolism is the process of generating energy (ATP) from nutrients, and mitochondria are the headmaster in the powerhouses of the cell. To produce energy in the form of ATP and GTP, all the cells, and in specific the neurons, devour glucose, amino acids, and fatty acids [245]. These nutrients are processed and transferred into the tricarboxylic acid (TCA) cycle, and electrons are stored in the reducing equivalents NADH and FADH2, through iterative oxidations. NADH and FADH2 represent the carrier molecule that withdraws electrons into the electron transport chain (ETC); protons flow down to generate ATP [246]. The capacity of mitochondria to supply at the request of ATP, defined as bioenergetics efficiency may change, during life for the effect of environmental stress, aging, or pathologies. During aging, for example, mitochondria go through alterations in their capacity to produce ATP. This is due to the release of a great number of reactive oxygen species (ROS). ROS are able to increase spontaneous DNA mutations and can start the processes that lead to cancer [247,248]. Mitochondria from aging people show different features [249]; they swell while their numbers dwindle, unable to replace themselves as quickly in their dysfunctional state [250]. Failure in mitochondria is the starting phase of the neurodegenerative process [251]. It was demonstrated in both PD and HD that the presence of environmental stress can increase the production of oxidative species and also can modify the structure of protein and DNA inside mitochondria and its nucleo [252]. Usually, HD is considered a hyperkinetic disorder, despite the presence of hypokinetic features in the motor symptoms [253]. Conversely, PD is judged a hypokinetic disorder in which the resultant speech motion disease may be classified as hyperkinetic or hypokinetic dysarthria [254,255] (Figure 1). Both are defined as neurodegenerative disorders causing the gradual loss of neurons. In addition, both disorders result in a deficit at the level of the basal ganglia, with peculiar references to striatal [256] and hippocampal areas [257]. These areas are deputized to movement and cognitive ability; for this reason, it is not a surprise that both diseases can affect cognitive or thinking abilities and motor functions. Studies on rodent models of PD show the impairment in bioenergetics pathway and a block of function in mitochondrial complexes I and III. Also, in genetics in phenotypic models induced by toxin exposure, in the early stages of the disease as well as in the developmental stage [4,86,103,105,106,173]. While, study on animal models of HD, show an evident alteration in mitochondrial complexes II and III, and describe a reduction of bioenergetics efficiency that was described essentially in the early pathogenic mechanism [258]. The confirmation of this evidence comes from translational studies in man in which the association between mitochondrial deficit and biomarkers of PD and HD is shown [253]. Actually, there is no cure for both pathologies, and neither exists drugs to modify the development and gravity of the diseases. For all these reasons the knowledge of pathogenetic mechanisms and the similarities in the disease can help us to revise the traditional approach and can help in understanding new scenarios. New pharmacological approach considering mitochondria target agents could be used in the future to cure degenerative and in particular neurodegenerative pathologies that although different show so many common elements.
Author Contributions
Conceptualization: M.M., A.T., T.S. and M.G., bibliography search & collecting: M.M., A.T., T.S. and M.G., resources, P.B.; supervision, P.B.; visualization, G.P.; writing—original draft: M.M., A.T., T.S. and M.G., writing—review and editing, M.M., A.T., T.S. and P.B., table and figure M.M., A.T. and M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work is partially supported by Italian Ministry of Health “Ricerca Finalizzata” grants Nr. RF-2021-12374979 to A.P.; RF-2019-12370182 to P.B. The funding sources had no involvement in the design or writing of the report and in the decision to submit the article for publication.
Data Availability Statement
The data presented in this study are available in Pub-Med library.
Acknowledgments
All the authors wish to thank Massimo Tolu for his excellent technical assistance.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the revision, in the collection, interpretation of data, or in the writing of the manuscript.
References
- Sagan L. On the origin of mitosing cells. J Theor Biol. 1967 Mar;14, 255-74. https://doi.org/10.1016/0022-5193(67)90079-3. PMID: 11541392.
- Pizzorno J. Mitochondria-Fundamental to Life and Health. Integr Med. 2014 Apr;13, 8-15. PMID: 26770084; PMCID: PMC4684129.
- Alberts B, Johnson A, Lewis J, et al. New York: Garland Science; 2002.
- Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci Ther. 2017 Jan;23, 5-22. https://doi.org/10.1111/cns.12655. Epub 2016 Nov 22. PMID: 27873462; PMCID: PMC6492703.
- Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017 Apr 5;6, 25. https://doi.org/10.3390/antiox6020025. PMID: 28379197; PMCID: PMC5488005.
- Wang Y, Xu E, Musich PR, Lin F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther. 2019 Jul;25, 816-824. https://doi.org/10.1111/cns.13116. Epub 2019 Mar 19. PMID: 30889315; PMCID: PMC6566063.
- Jadiya P, Garbincius JF, Elrod JW. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol Commun. 2021 Jul 7;9, 124. https://doi.org/10.1186/s40478-021-01224-4. PMID: 34233766; PMCID: PMC8262011.
- Zhu XH, Qiao H, Du F, Xiong Q, Liu X, Zhang X, Ugurbil K, Chen W. Quantitative imaging of energy expenditure in human brain. Neuroimage. 2012 May 1;60, 2107-17. https://doi.org/10.1016/j.neuroimage.2012.02.013. Epub 2012 Feb 17. PMID: 22487547; PMCID: PMC3325488.
- Vergara RC, Jaramillo-Riveri S, Luarte A, Moënne-Loccoz C, Fuentes R, Couve A, Maldonado PE. The Energy Homeostasis Principle: Neuronal Energy Regulation Drives Local Network Dynamics Generating Behavior. Front Comput Neurosci. 2019 Jul 23;13:49. https://doi.org/10.3389/fncom.2019.00049. Erratum in: Front Comput Neurosci. 2020 Oct 29;14:599670. PMID: 31396067; PMCID: PMC6664078.
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011 Apr 15;435, 297-312. https://doi.org/10.1042/BJ20110162. Erratum in: Biochem J. 2011 Aug 1;437, 575. PMID: 21726199; PMCID: PMC3076726.
- Akbar M, Essa MM, Daradkeh G, Abdelmegeed MA, Choi Y, Mahmood L, Song BJ. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016 Apr 15;1637:34-55. https://doi.org/10.1016/j.brainres.2016.02.016. Epub 2016 Feb 13. PMID: 26883165; PMCID: PMC4821765.
- Jellinger KA. Basic mechanisms of neurodegeneration: a critical update. J Cell Mol Med. 2010 Mar;14, 457-87. https://doi.org/10.1111/j.1582-4934.2010.01010.x. Epub 2010 Jan 11. PMID: 20070435; PMCID: PMC3823450.
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Murali Mahadevan, H.; Hashemiaghdam, A.; Ashrafi, G.; Harbauer, A.B. Mitochondria in neuronal health: from energy metabolism to parkinson’s disease. Advanced Biology 2021, 5, e2100663. [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; da Cruz E Silva, O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: more than just a powerhouse. Curr. Biol. 2006, 16, R551-60. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; Suski, J.M.; Wieckowski, M.R.; Pinton, P. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Bezrukov, S.M.; Hoogerheide, D.P. Regulation of Mitochondrial Respiration by VDAC Is Enhanced by Membrane-Bound Inhibitors with Disordered Polyanionic C-Terminal Domains. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Jung, H.; Kim, S.Y.; Canbakis Cecen, F.S.; Cho, Y.; Kwon, S.-K. Dysfunction of mitochondrial ca2+ regulatory machineries in brain aging and neurodegenerative diseases. Front. Cell Dev. Biol. 2020, 8, 599792. [Google Scholar] [CrossRef] [PubMed]
- Sparagna, G.C.; Gunter, K.K.; Gunter, T.E. A system for producing and monitoring in vitro calcium pulses similar to those observed in vivo. Anal. Biochem. 1994, 219, 96–103. [Google Scholar] [CrossRef]
- Sparagna, G.C.; Gunter, K.K.; Sheu, S.S.; Gunter, T.E. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J. Biol. Chem. 1995, 270, 27510–27515. [Google Scholar] [CrossRef] [PubMed]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef]
- Petersen, O.H. Calcium signal compartmentalization. Biol. Res. 2002, 35, 177–182. [Google Scholar] [CrossRef]
- Laude, A.J.; Simpson, A.W.M. Compartmentalized signalling: Ca2+ compartments, microdomains and the many facets of Ca2+ signalling. FEBS J. 2009, 276, 1800–1816. [Google Scholar] [CrossRef]
- Panda, S.; Behera, S.; Alam, M.F.; Syed, G.H. Endoplasmic reticulum & mitochondrial calcium homeostasis: The interplay with viruses. Mitochondrion 2021, 58, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; Polleux, F. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef]
- Ashrafi, G.; de Juan-Sanz, J.; Farrell, R.J.; Ryan, T.A. Molecular tuning of the axonal mitochondrial ca2+ uniporter ensures metabolic flexibility of neurotransmission. Neuron 2020, 105, 678–687.e5. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The role of mitochondrial calcium homeostasis in alzheimer’s and related diseases. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R. Sources and triggers of oxidative damage in neurodegeneration. Free Radic. Biol. Med. 2021, 173, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of NADPH oxidases: the role of Rac proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.-H.; Yates, C.; You, Z.; Tan, M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front. Immunol. 2018, 9, 1605. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Breda, C.N. de S.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, oxidative stress and innate immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef]
- Chu, X.; Wu, S.; Raju, R. NLRX1 regulation following acute mitochondrial injury. Front. Immunol. 2019, 10, 2431. [Google Scholar] [CrossRef] [PubMed]
- Stokman, G.; Kors, L.; Bakker, P.J.; Rampanelli, E.; Claessen, N.; Teske, G.J.D.; Butter, L.; van Andel, H.; van den Bergh Weerman, M.A.; Larsen, P.W.B.; Dessing, M.C.; Zuurbier, C.J.; Girardin, S.E.; Florquin, S.; Leemans, J.C. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J. Exp. Med. 2017, 214, 2405–2420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Kanneganti, T.-D. Nlrp3: an immune sensor of cellular stress and infection. Int. J. Biochem. Cell Biol. 2010, 42, 792–795. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24. [Google Scholar] [CrossRef] [PubMed]
- Behrouzi, A.; Kelley, M.R.; Fehrenbacher, J.C. Oxidative DNA damage: A role in altering neuronal function. J. Cell Signal. 2022, 3, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.L.; Gustafsson, A.B. Mitochondrial autophagy--an essential quality control mechanism for myocardial homeostasis. Circ. J. 2013, 77, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.-F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.-H. The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 2020, 4, NS20200008. [Google Scholar] [CrossRef]
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2010, 2, 139. [Google Scholar] [CrossRef]
- Chanaday, N.L.; Cousin, M.A.; Milosevic, I.; Watanabe, S.; Morgan, J.R. The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J. Neurosci. 2019, 39, 8209–8216. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Jaiswal, M. Mitochondrial calcium at the synapse. Mitochondrion 2021, 59, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Orth M, Schapira AH. Mitochondria and degenerative disorders. Am J Med Genet. 2001 Spring;106, 27-36. https://doi.org/10.1002/ajmg.1425. PMID: 11579422.
- Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013 Apr 2;17, 491-506. https://doi.org/10.1016/j.cmet.2013.03.002. PMID: 23562075; PMCID: PMC5967396.
- Zhang PN, Zhou MQ, Guo J, Zheng HJ, Tang J, Zhang C, Liu YN, Liu WJ, Wang YX. Mitochondrial Dysfunction and Diabetic Nephropathy: Nontraditional Therapeutic Opportunities. J Diabetes Res. 2021 Dec 9;2021:1010268. https://doi.org/10.1155/2021/1010268. PMID: 34926696; PMCID: PMC8677373.
- Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE, Shirihai OS. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–2315.
- Gomes LC, Di BG, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598.
- Lewin R. Trail of ironies to Parkinson's disease. Science. 1984 Jun 8;224, 1083-5. https://doi.org/10.1126/science.6426059. PMID: 6426059.
- Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol Dis. 2013;51:35–42.
- Surmeier, D. J., Obeso, J. A. & Halliday, G. M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113 (2017).
- Park JS, Davis RL, Sue CM. Mitochondrial Dysfunction in Parkinson's Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr Neurol Neurosci Rep. 2018 Apr 3;18, 21. https://doi.org/10.1007/s11910-018-0829-3. PMID: 29616350; PMCID: PMC5882770.
- Magalhães JD, Cardoso SM. Mitochondrial signaling on innate immunity activation in Parkinson disease. Curr Opin Neurobiol. 2022 Dec 16;78:102664. https://doi.org/10.1016/j.conb.2022.102664. Epub ahead of print. PMID: 36535149.
- Rossi A, Pizzo P. Mitochondrial bioenergetics and neurodegeneration: a paso doble. Neural Regen Res. 2021 Apr;16, 686-687. https://doi.org/10.4103/1673-5374.295331. PMID: 33063726; PMCID: PMC8067919.
- Zhang C, Chen S, Li X, Xu Q, Lin Y, Lin F, Yuan M, Zi Y, Cai J. Progress in Parkinson's disease animal models of genetic defects: Characteristics and application. Biomed Pharmacother. 2022 Nov;155:113768. https://doi.org/10.1016/j.biopha.2022.113768. Epub 2022 Sep 28. PMID: 36182736.
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304: 1158– 1160.
- Arena G, Valente EM. PINK1 in the limelight: multiple functions of an eclectic protein in human health and disease. J Pathol. 2017 Jan;241, 251-263. https://doi.org/10.1002/path.4815. Epub 2016 Nov 12. PMID: 27701735.
- Silvestri L, Caputo V, Bellacchio E, et al. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 2005; 14: 3477– 3492.
- Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001; 20: 4457–4465.
- Garber K. Parkinson's disease and cancer: the unexplored connection. J Natl Cancer Inst. 2010 Mar 17;102, 371-4. https://doi.org/10.1093/jnci/djq081. Epub 2010 Mar 9. PMID: 20215596.
- Masgras I, Laquatra C, Cannino G, Serapian SA, Colombo G, Rasola A. The molecular chaperone TRAP1 in cancer: From the basics of biology to pharmacological targeting. Semin Cancer Biol. 2021 Nov;76:45-53. https://doi.org/10.1016/j.semcancer.2021.07.002. Epub 2021 Jul 6. PMID: 34242740.
- Exner N, Treske B, Paquet D, Holmström K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Krüger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007 Nov 7;27, 12413-8. https://doi.org/10.1523/JNEUROSCI.0719-07.2007. PMID: 17989306; PMCID: PMC6673250.
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A. 2007 Jul 3;104, 11441-6. https://doi.org/10.1073/pnas.0702717104. Epub 2007 Jun 11. PMID: 17563363; PMCID: PMC1890561.
- Martella G, Madeo G, Maltese M, Vanni V, Puglisi F, Ferraro E, Schirinzi T, Valente EM, Bonanni L, Shen J, Mandolesi G, Mercuri NB, Bonsi P, Pisani A. Exposure to low-dose rotenone precipitates synaptic plasticity alterations in PINK1 heterozygous knockout mice. Neurobiol Dis. 2016 Jul;91:21-36. https://doi.org/10.1016/j.nbd.2015.12.020. Epub 2016 Feb 23. PMID: 26916954.
- Imbriani P, Tassone A, Meringolo M, Ponterio G, Madeo G, Pisani A, Bonsi P, Martella G. Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson's Disease. Int J Mol Sci. 2019 Jul 11;20, 3407. https://doi.org/10.3390/ijms20143407. PMID: 31336695; PMCID: PMC6678522.
- Imbriani P, D'Angelo V, Platania P, Di Lazzaro G, Scalise S, Salimei C, El Atiallah I, Colona VL, Mercuri NB, Bonsi P, Pisani A, Schirinzi T, Martella G. Ischemic injury precipitates neuronal vulnerability in Parkinson's disease: Insights from PINK1 mouse model study and clinical retrospective data. Parkinsonism Relat Disord. 2020 May;74:57-63. https://doi.org/10.1016/j.parkreldis.2020.04.004. Epub 2020 Apr 20. PMID: 32335490.
- Brunelli F, Valente EM, Arena G. Mechanisms of neurodegeneration in Parkinson's disease: keep neurons in the PINK1. Mech Ageing Dev. 2020 Jul;189:111277. https://doi.org/10.1016/j.mad.2020.111277. Epub 2020 Jun 3. PMID: 32504621.
- Zhi L, Qin Q, Muqeem T, Seifert EL, Liu W, Zheng S, Li C, Zhang H. Loss of PINK1 causes age-dependent decrease of dopamine release and mitochondrial dysfunction. Neurobiol Aging. 2019 Mar;75:1-10. https://doi.org/10.1016/j.neurobiolaging.2018.10.025. Epub 2018 Nov 2. PMID: 30504091; PMCID: PMC6778692.
- Onyango IG, Bennett JP, Stokin GB. Regulation of neuronal bioenergetics as a therapeutic strategy in neurodegenerative diseases. Neural Regen Res. 2021 Aug;16, 1467-1482. https://doi.org/10.4103/1673-5374.303007. PMID: 33433460; PMCID: PMC8323696.
- Khalil B, El Fissi N, Aouane A, et al. PINK1-induced mitophagy promotes neuroprotection in Huntington's disease. Cell Death Dis 2015; 6: e1617.
- Anderson KE, Marshall FJ. Behavioral symptoms associated with Huntington's disease. Adv Neurol. 2005;96:197-208. PMID: 16383221.
- Caron NS, Wright GEB, Hayden MR. Huntington Disease. 1998 Oct 23 [updated 2020 Jun 11]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023. PMID: 20301482.
- Damiano M, Galvan L, Déglon N, Brouillet E. Mitochondria in Huntington's disease. Biochim Biophys Acta. 2010 Jan;1802, 52-61. https://doi.org/10.1016/j.bbadis.2009.07.012. Epub 2009 Aug 11. PMID: 19682570.
- Rehman MU, Sehar N, Dar NJ, Khan A, Arafah A, Rashid S, Rashid SM, Ganaie MA. Mitochondrial dysfunctions, oxidative stress and neuroinflammation as therapeutic targets for neurodegenerative diseases: An update on current advances and impediments. Neurosci Biobehav Rev. 2023 Jan;144:104961. https://doi.org/10.1016/j.neubiorev.2022.104961. Epub 2022 Nov 14. PMID: 36395982.
- Jurcau A, Jurcau CM. Mitochondria in Huntington's disease: implications in pathogenesis and mitochondrial-targeted therapeutic strategies. Neural Regen Res. 2023 Jul;18, 1472-1477. https://doi.org/10.4103/1673-5374.360289. PMID: 36571344.
- Kawsar, M., Taz, T.A., Paul, B.K. et al. Identification of vital regulatory genes with network pathways among Huntington’s, Parkinson’s, and Alzheimer’s diseases. Netw Model Anal Health Inform Bioinforma 9, 50 (2020). https://doi.org/10.1007/s13721-020-00257-4.
- Li Z, Zhang Z, Ren Y, Wang Y, Fang J, Yue H, Ma S, Guan F. Aging and age-related diseases: from mechanisms to therapeutic strategies. Biogerontology. 2021 Apr;22, 165-187. https://doi.org/10.1007/s10522-021-09910-5. Epub 2021 Jan 27. PMID: 33502634; PMCID: PMC7838467.
- Quijano C, Cao L, Fergusson MM, Romero H, Liu J, Gutkind S, Rovira II, Mohney RP, Karoly ED, Finkel T. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle. 2012;11:1383–1392.
- Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell. 2016 Mar 3;61, 654-666. https://doi.org/10.1016/j.molcel.2016.01.028. PMID: 26942670; PMCID: PMC4779179.
- Moro L. Mitochondrial Dysfunction in Aging and Cancer. J Clin Med. 2019 Nov 15;8, 1983. https://doi.org/10.3390/jcm8111983. PMID: 31731601; PMCID: PMC6912717.
- Boland ML, Chourasia AH, Macleod KF. Mitochondrial dysfunction in cancer. Front Oncol. 2013 Dec 2;3:292. https://doi.org/10.3389/fonc.2013.00292. PMID: 24350057; PMCID: PMC3844930.
- Wang XL, Feng ST, Wang YT, Yuan YH, Li ZP, Chen NH, Wang ZZ, Zhang Y. Mitophagy, a Form of Selective Autophagy, Plays an Essential Role in Mitochondrial Dynamics of Parkinson's Disease. Cell Mol Neurobiol. 2022 Jul;42, 1321-1339. https://doi.org/10.1007/s10571-021-01039-w. Epub 2021 Feb 2. PMID: 33528716.
- Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci. 2015 Apr;40, 200-10. https://doi.org/10.1016/j.tibs.2015.02.003. Epub 2015 Mar 8. PMID: 25757399.
- Büeler H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson's disease. Exp Neurol. 2009 Aug;218, 235-46. https://doi.org/10.1016/j.expneurol.2009.03.006. Epub 2009 Mar 18. PMID: 19303005.
- Chen C, Turnbull DM, Reeve AK. Mitochondrial Dysfunction in Parkinson's Disease-Cause or Consequence? Biology (Basel). 2019 May 11;8, 38. https://doi.org/10.3390/biology8020038. PMID: 31083583; PMCID: PMC6627981.
- Parker WD Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol. 1989 Dec;26, 719-23. https://doi.org/10.1002/ana.410260606. PMID: 2557792.
- Rekha KR, Selvakumar GP. Gene expression regulation of Bcl2, Bax and cytochrome-C by geraniol on chronic MPTP/probenecid induced C57BL/6 mice model of Parkinson's disease. Chem Biol Interact. 2014 Jun 25;217:57-66. https://doi.org/10.1016/j.cbi.2014.04.010. Epub 2014 Apr 24. PMID: 24768735.
- Lee JH, Han JH, Kim H, Park SM, Joe EH, Jou I. Parkinson's disease-associated LRRK2- G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol Commun. 2019 May 2;7, 68. https://doi.org/10.1186/s40478-019- 0716-4. PMID: 31046837; PMCID: PMC6498585.
- Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012 Jun 26;31, 3038-62. https://doi.org/10.1038/emboj.2012.170. PMID: 22735187; PMCID: PMC3400019.
- Meredith GE, Rademacher DJ. MPTP mouse models of Parkinson's disease: an update. J Parkinsons Dis. 2011;1, 19-33. https://doi.org/10.3233/JPD-2011-11023. PMID: 23275799; PMCID: PMC3530193.
- Zeng XS, Geng WS, Jia JJ. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro. 2018 Jan- Dec;10:1759091418777438. https://doi.org/10.1177/1759091418777438. PMID: 29809058; PMCID: PMC5977437.
- Imbriani P, Schirinzi T, Meringolo M, Mercuri NB, Pisani A. Centrality of Early Synaptopathy in Parkinson's Disease. Front Neurol. 2018 Mar 1;9:103. https://doi.org/10.3389/fneur.2018.00103. PMID: 29545770; PMCID: PMC5837972.
- Imbriani P, Sciamanna G, Santoro M, Schirinzi T, Pisani A. Promising rodent models in Parkinson's disease. Parkinsonism Relat Disord. 2018b Jan;46 Suppl 1:S10-S14. https://doi.org/10.1016/j.parkreldis.2017.07.027. Epub 2017 Jul 27. PMID: 28760592.
- Innos J, Hickey MA. Using Rotenone to Model Parkinson's Disease in Mice: A Review of the Role of Pharmacokinetics. Chem Res Toxicol. 2021 May 17;34, 1223-1239. https://doi.org/10.1021/acs.chemrestox.0c00522. Epub 2021 May 7. PMID: 33961406.
- Imbriani P, Martella G, Bonsi P, Pisani A. Oxidative stress and synaptic dysfunction in rodent models of Parkinson's disease. Neurobiol Dis. 2022 Oct 15;173:105851. https://doi.org/10.1016/j.nbd.2022.105851. Epub 2022 Aug 23. PMID: 36007757.
- McLean PJ, Ribich S, Hyman BT. Subcellular localization of alpha-synuclein in primary neuronal cultures: effect of missense mutations. J Neural Transm Suppl. 2000;(58):53-63. https://doi.org/10.1007/978-3-7091-6284-2_5. PMID: 11128613.
- Zhang L, Zhang C, Zhu Y, Cai Q, Chan P, Uéda K, Yu S, Yang H. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 2008 Dec 9;1244:40-52. https://doi.org/10.1016/j.brainres.2008.08.067. Epub 2008 Sep 4. PMID: 18817762.
- Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res. 2008 Jun 10;314, 2076-89. https://doi.org/10.1016/j.yexcr.2008.03.012. Epub 2008 Mar 28. PMID: 18440504; PMCID: PMC2483835.
- Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, Sanges G, Stenroos ES, Pho LT, Schaffer AA, Lazzarini AM, Nussbaum RL, Duvoisin RC. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. 1996 Nov 15;274, 1197-9. https://doi.org/10.1126/science.274.5290.1197. PMID: 8895469.
- Koprich JB, Kalia LV, Brotchie JM. Animal models of α-synucleinopathy for Parkinson disease drug development. Nat Rev Neurosci. 2017 Sep;18, 515-529. https://doi.org/10.1038/nrn.2017.75. Epub 2017 Jul 13. PMID: 28747776.
- Ingelsson M. Alpha-Synuclein Oligomers-Neurotoxic Molecules in Parkinson's Disease and Other Lewy Body Disorders. Front Neurosci. 2016 Sep 5;10:408. https://doi.org/10.3389/fnins.2016.00408. PMID: 27656123; PMCID: PMC5011129.
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006 Jan 4;26, 41-50. https://doi.org/10.1523/JNEUROSCI.4308-05.2006. PMID: 16399671; PMCID: PMC6381830.
- Portz P, Lee MK. Changes in Drp1 Function and Mitochondrial Morphology Are Associated with the α-Synuclein Pathology in a Transgenic Mouse Model of Parkinson's Disease. Cells. 2021 Apr 13;10, 885. https://doi.org/10.3390/cells10040885. PMID: 33924585; PMCID: PMC8070398.
- Xie W, Chung KK. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson's disease. J Neurochem. 2012 Jul;122, 404-14. https://doi.org/10.1111/j.1471-4159.2012.07769.x. Epub 2012 May 23. PMID: 22537068.
- Chinta SJ, Mallajosyula JK, Rane A, Andersen JK. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett. 2010 Dec 17;486, 235-9. https://doi.org/10.1016/j.neulet.2010.09.061. Epub 2010 Sep 29. PMID: 20887775; PMCID: PMC2967673.
- Merino-Galán L, Jimenez-Urbieta H, Zamarbide M, Rodríguez-Chinchilla T, Belloso- Iguerategui A, Santamaria E, Fernández-Irigoyen J, Aiastui A, Doudnikoff E, Bézard E, Ouro A, Knafo S, Gago B, Quiroga-Varela A, Rodríguez-Oroz MC. Striatal synaptic bioenergetic and autophagic decline in premotor experimental Parkinsonism. Brain. 2022 Jun 30;145, 2092-2107. https://doi.org/10.1093/brain/awac087. PMID: 35245368; PMCID: PMC9460676.
- Anandhan A, Jacome MS, Lei S, Hernandez-Franco P, Pappa A, Panayiotidis MI, Powers R, Franco R. Metabolic Dysfunction in Parkinson's Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res Bull. 2017 Jul;133:12-30. https://doi.org/10.1016/j.brainresbull.2017.03.009. Epub 2017 Mar 21. PMID: 28341600; PMCID: PMC5555796.
- Kurz A, Double KL, Lastres-Becker I, Tozzi A, Tantucci M, Bockhart V, Bonin M, García- Arencibia M, Nuber S, Schlaudraff F, Liss B, Fernández-Ruiz J, Gerlach M, Wüllner U, Lüddens H, Calabresi P, Auburger G, Gispert S. A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS One. 2010 Jul 7;5, e11464. https://doi.org/10.1371/journal.pone.0011464. PMID: 20628651; PMCID: PMC2898885.
- Durante V, de Iure A, Loffredo V, Vaikath N, De Risi M, Paciotti S, Quiroga-Varela A, Chiasserini D, Mellone M, Mazzocchetti P, Calabrese V, Campanelli F, Mechelli A, Di Filippo M, Ghiglieri V, Picconi B, El-Agnaf OM, De Leonibus E, Gardoni F, Tozzi A, Calabresi P. Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain. 2019 May 1;142, 1365- 1385. https://doi.org/10.1093/brain/awz065. PMID: 30927362.
- Tozzi A, de Iure A, Bagetta V, Tantucci M, Durante V, Quiroga-Varela A, Costa C, Di Filippo M, Ghiglieri V, Latagliata EC, Wegrzynowicz M, Decressac M, Giampà C, Dalley JW, Xia J, Gardoni F, Mellone M, El-Agnaf OM, Ardah MT, Puglisi-Allegra S, Björklund A, Spillantini MG, Picconi B, Calabresi P. Alpha-Synuclein Produces Early Behavioral Alterations via Striatal Cholinergic Synaptic Dysfunction by Interacting With GluN2D N-Methyl-D-Aspartate Receptor Subunit. Biol Psychiatry. 2016 Mar 1;79, 402-414. https://doi.org/10.1016/j.biopsych.2015.08.013. Epub 2015 Aug 20. PMID: 26392130.
- Tozzi A, Sciaccaluga M, Loffredo V, Megaro A, Ledonne A, Cardinale A, Federici M, Bellingacci L, Paciotti S, Ferrari E, La Rocca A, Martini A, Mercuri NB, Gardoni F, Picconi B, Ghiglieri V, De Leonibus E, Calabresi P. Dopamine-dependent early synaptic and motor dysfunctions induced by α-synuclein in the nigrostriatal circuit. Brain. 2021 Dec 16;144, 3477-3491. https://doi.org/10.1093/brain/awab242. PMID: 34297092; PMCID: PMC8677552.
- Calì T, Ottolini D, Negro A, Brini M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012 May 25;287, 17914-29. https://doi.org/10.1074/jbc.M111.302794. Epub 2012 Mar 27. PMID: 22453917; PMCID: PMC3365710.
- Alves Da Costa C, Paitel E, Vincent B, Checler F. Alpha-synuclein lowers p53-dependent apoptotic response of neuronal cells. Abolishment by 6-hydroxydopamine and implication for Parkinson's disease. J Biol Chem. 2002 Dec 27;277, 50980-4. https://doi.org/10.1074/jbc.M207825200. Epub 2002 Oct 22. PMID: 12397073.
- Chan SL, Mattson MP. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res. 1999 Oct 1;58, 167-90. PMID: 10491581.
- D’Amelio M, Cavallucci V, Cecconi F. Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 2010 Jul;17, 1104-14. https://doi.org/10.1038/cdd.2009.180. Epub 2009 Dec 4. PMID: 19960023.
- Snigdha S, Smith ED, Prieto GA, Cotman CW. Caspase-3 activation as a bifurcation point between plasticity and cell death. Neurosci Bull. 2012 Feb;28, 14-24. https://doi.org/10.1007/s12264-012-1057-5. PMID: 22233886; PMCID: PMC3838299.
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset Parkinsonism. Science. 2003 Jan 10;299, 256-9. https://doi.org/10.1126/science.1077209. Epub 2002 Nov 21. PMID: 12446870.
- Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D, Cedazo-Minguez A, Cookson MR. DJ-1 acts in parallel to the PINK1/Parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet. 2011 Jan 1;20, 40-50. https://doi.org/10.1093/hmg/ddq430. Epub 2010 Oct 11. PMID: 20940149; PMCID: PMC3000675.
- Trancikova A, Tsika E, Moore DJ. Mitochondrial dysfunction in genetic animal models of Parkinson's disease. Antioxid Redox Signal. 2012 May 1;16, 896-919. https://doi.org/10.1089/ars.2011.4200. Epub 2011 Oct 4. PMID: 21848447; PMCID: PMC3292748.
- Dzamko N, Zhou J, Huang Y, Halliday GM. Parkinson's disease-implicated kinases in the brain; insights into disease pathogenesis. Front Mol Neurosci. 2014 Jun 24;7:57. https://doi.org/10.3389/fnmol.2014.00057. PMID: 25009465; PMCID: PMC4068290.
- Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, Wu L, Yan SF, Tieu K, Yan SS. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer's disease. Brain. 2017 Dec 1;140, 3233-3251. https://doi.org/10.1093/brain/awx258. PMID: 29077793; PMCID: PMC5841141.
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015 Aug 20;524, 309-314. https://doi.org/10.1038/nature14893. Epub 2015 Aug 12. PMID: 26266977; PMCID: PMC5018156.
- Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denèfle P, Wood NW, Agid Y, Brice A; French Parkinson's Disease Genetics Study Group; European Consortium on Genetic Susceptibility in Parkinson's Disease. Association between early-onset Parkinson's disease and mutations in the Parkin gene. N Engl J Med. 2000 May 25;342, 1560-7. https://doi.org/10.1056/NEJM200005253422103. PMID: 10824074.
- Moore DJ. Parkin: a multifaceted ubiquitin ligase. Biochem Soc Trans. 2006 Nov;34(Pt 5):749-53. https://doi.org/10.1042/BST0340749. PMID: 17052189.
- Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X, Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009 Mar;119, 650-60. https://doi.org/10.1172/JCI37617. Epub 2009 Feb 23. PMID: 19229105; PMCID: PMC2648688.
- Periquet M, Corti O, Jacquier S, Brice A. Proteomic analysis of Parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J Neurochem. 2005 Dec;95, 1259-76. https://doi.org/10.1111/j.1471-4159.2005.03442.x. Epub 2005 Sep 7. PMID: 16150055.
- Darios F, Corti O, Lücking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003 Mar 1;12, 517-26. https://doi.org/10.1093/hmg/ddg044. PMID: 12588799.
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell. 2011 Mar 4;144, 689-702. https://doi.org/10.1016/j.cell.2011.02.010.
- Jo A, Lee Y, Kam TI, Kang SU, Neifert S, Karuppagounder SS, Khang R, Kang H, Park H, Chou SC, Oh S, Jiang H, Swing DA, Ham S, Pirooznia S, Umanah GKE, Mao X, Kumar M, Ko HS, Kang HC, Lee BD, Lee YI, Andrabi SA, Park CH, Lee JY, Kim H, Kim H, Kim H, Cho JW, Paek SH, Na CH, Tessarollo L, Dawson VL, Dawson TM, Shin JH. PARIS farnesylation prevents neurodegeneration in models of Parkinson's disease. Sci Transl Med. 2021 Jul 28;13, eaax8891. https://doi.org/10.1126/scitranslmed.aax8891. PMID: 34321320.
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. 2009 Jul;5, 706-8. https://doi.org/10.4161/auto.5.5.8505. Epub 2009 Jul 22. PMID: 19377297.
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in Parkin-deficient mice. J Biol Chem. 2004 Apr 30;279, 18614-22. https://doi.org/10.1074/jbc.M401135200. Epub 2004 Feb 24. PMID: 14985362.
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism- linked gene DJ-1. Neuron. 2005 Feb 17;45, 489-96. https://doi.org/10.1016/j.neuron.2005.01.041. PMID: 15721235.
- Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, Ret G, Joubert C, Periquet M, Araujo F, Negroni J, Casarejos MJ, Canals S, Solano R, Serrano A, Gallego E, Sanchez M, Denefle P, Benavides J, Tremp G, Rooney TA, Brice A, Garcia de Yebenes J. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003 Sep 15;12, 2277-91. https://doi.org/10.1093/hmg/ddg239. Epub 2003 Jul 22. PMID: 12915482.
- Kitada T, Pisani A, Karouani M, Haburcak M, Martella G, Tscherter A, Platania P, Wu B, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of parkin-/- mice. J Neurochem. 2009b Jul;110, 613-21. https://doi.org/10.1111/j.1471- 4159.2009.06152.x. Epub 2009 May 5. PMID: 19457102.
- Cortese GP, Zhu M, Williams D, Heath S, Waites CL. Parkin Deficiency Reduces Hippocampal Glutamatergic Neurotransmission by Impairing AMPA Receptor Endocytosis. J Neurosci. 2016 Nov 30;36, 12243-12258. https://doi.org/10.1523/JNEUROSCI.1473-16.2016. PMID: 27903732; PMCID: PMC5148221.
- Calì T, Ottolini D, Negro A, Brini M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta. 2013 Apr;1832, 495-508. https://doi.org/10.1016/j.bbadis.2013.01.004. Epub 2013 Jan 9. PMID: 23313576.
- Bianchi K, Rimessi A, Prandini A, Szabadkai G, Rizzuto R. Calcium and mitochondria: mechanisms and functions of a troubled relationship. Biochim Biophys Acta. 2004 Dec 6;1742(1-3):119-31. https://doi.org/10.1016/j.bbamcr.2004.09.015. PMID: 15590062.
- Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2010 Sep 22;2:139. https://doi.org/10.3389/fnsyn.2010.00139. PMID: 21423525; PMCID: PMC3059669.
- Jo D, Song J. Irisin Acts via the PGC-1α and BDNF Pathway to Improve Depression-like Behavior. Clin Nutr Res. 2021 Oct 20;10, 292-302. https://doi.org/10.7762/cnr.2021.10.4.292. PMID: 34796134; PMCID: PMC8575642.
- Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007 Mar 5;3, 242-50. https://doi.org/10.7150/ijbs.3.242. PMID: 17389931; PMCID: PMC1820878.
- Madeo G, Schirinzi T, Martella G, Latagliata EC, Puglisi F, Shen J, Valente EM, Federici M, Mercuri NB, Puglisi-Allegra S, Bonsi P, Pisani A. PINK1 heterozygous mutations induce subtle alterations in dopamine-dependent synaptic plasticity. Mov Disord. 2014 Jan;29, 41-53. https://doi.org/10.1002/mds.25724. Epub 2013 Oct 25. PMID: 24167038; PMCID: PMC4022284.
- Dave KD, De Silva S, Sheth NP, Ramboz S, Beck MJ, Quang C, Switzer RC 3rd, Ahmad SO, Sunkin SM, Walker D, Cui X, Fisher DA, McCoy AM, Gamber K, Ding X, Goldberg MS, Benkovic SA, Haupt M, Baptista MA, Fiske BK, Sherer TB, Frasier MA. Phenotypic characterization of recessive gene knockout rat models of Parkinson's disease. NeurobiolDis. 2014 Oct;70:190-203. https://doi.org/10.1016/j.nbd.2014.06.009. Epub 2014 Jun 24. PMID:24969022.
- Stauch KL, Villeneuve LM, Purnell PR, Ottemann BM, Emanuel K, Fox HS. Loss of Pink1 modulates synaptic mitochondrial bioenergetics in the rat striatum prior to motor symptoms: concomitant complex I respiratory defects and increased complex II-mediated respiration. Proteomics Clin Appl. 2016 Dec;10, 1205-1217. https://doi.org/10.1002/prca.201600005. Epub 2016 Sep 21. PMID: 27568932; PMCID: PMC5810131.
- Villeneuve LM, Purnell PR, Boska MD, Fox HS. Early Expression of Parkinson's Disease- Related Mitochondrial Abnormalities in PINK1 Knockout Rats. Mol Neurobiol. 2016 Jan;53, 171-186. https://doi.org/10.1007/s12035-014-8927-y. Epub 2014 Nov 25. PMID: 25421206; PMCID: PMC4442772.
- Creed RB, Goldberg MS. Analysis of α-Synuclein Pathology in PINK1 Knockout Rat Brains. Front Neurosci. 2019 Jan 9;12:1034. https://doi.org/10.3389/fnins.2018.01034. PMID: 30686993; PMCID: PMC6333903.
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010 May 28;141, 859-71. https://doi.org/10.1016/j.cell.2010.03.053. PMID: 20510932; PMCID: PMC2909748.
- Snigdha S, Smith ED, Prieto GA, Cotman CW. Caspase-3 activation as a bifurcation point between plasticity and cell death. Neurosci Bull. 2012 Feb;28, 14-24. https://doi.org/10.1007/s12264 012-1057-5. PMID: 22233886; PMCID: PMC3838299.
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008 Aug 12;105, 11364-9. https://doi.org/10.1073/pnas.0802076105. Epub 2008 Aug 7. PMID: 18687901; PMCID: PMC2516271.
- Zhi L, Qin Q, Muqeem T, Seifert EL, Liu W, Zheng S, Li C, Zhang H. Loss of PINK1 causes age-dependent decrease of dopamine release and mitochondrial dysfunction. Neurobiol Aging. 2019 Mar;75:1-10. https://doi.org/10.1016/j.neurobiolaging.2018.10.025. Epub 2018 Nov 2. PMID: 30504091; PMCID: PMC6778692.
- Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI, Torp R, Torgner IA, Ottersen OP, Dawson TM, Dawson VL. Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet. 2005 Jul 15;14, 2063-73. https://doi.org/10.1093/hmg/ddi211. Epub 2005 Jun 8. PMID: 15944198.
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010 Dec 2;468, 696-700. https://doi.org/10.1038/nature09536. Epub 2010 Nov 10. Erratum in: Nature. 2015 May 21;521, 380. PMID: 21068725; PMCID: PMC4465557.
- Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, Surmeier DJ. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci. 2012 Oct;15, 1414-21. https://doi.org/10.1038/nn.3209. Epub 2012 Sep 2. PMID: 22941107; PMCID: PMC3461271.
- Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007 Sep 11;104, 14807-12. https://doi.org/10.1073/pnas.0703219104. Epub 2007 Aug 31. PMID: 17766438; PMCID: PMC1976193.
- Lopert P, Patel M. Brain mitochondria from DJ-1 knockout mice show increased respiration- dependent hydrogen peroxide consumption. Redox Biol. 2014 Apr 24;2:667-72. https://doi.org/10.1016/j.redox.2014.04.010. PMID: 24936441; PMCID: PMC4052521.
- Chen W, Liu H, Liu S, Kang Y, Nie Z, Lei H. Altered prefrontal neurochemistry in the DJ-1 knockout mouse model of Parkinson's disease: complementary semi-quantitative analyses with in vivo magnetic resonance spectroscopy and MALDI-MSI. Anal Bioanal Chem. 2022 Nov;414, 7977-7987. https://doi.org/10.1007/s00216-022-04341-8. Epub 2022 Oct 8. PMID: 36208327.
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003 Apr;384, 505- 16. https://doi.org/10.1515/BC.2003.059. PMID: 12751781.
- Kirkinezos IG, Moraes CT. Reactive oxygen species and mitochondrial diseases. Semin Cell Dev Biol. 2001 Dec;12, 449-57. https://doi.org/10.1006/scdb.2001.0282. PMID: 11735379.
- Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000 Aug;267, 4912-6. https://doi.org/10.1046/j.1432-1327.2000.01597.x. PMID: 10931173.
- Heo JY, Park JH, Kim SJ, Seo KS, Han JS, Lee SH, Kim JM, Park JI, Park SK, Lim K, Hwang BD, Shong M, Kweon GR. DJ-1 null dopaminergic neuronal cells exhibit defects in mitochondrial function and structure: involvement of mitochondrial complex I assembly. PLoS One. 2012;7, e32629. https://doi.org/10.1371/journal.pone.0032629. Epub 2012 Mar 5. PMID: 22403686; PMCID: PMC3293835.
- Junn E, Jang WH, Zhao X, Jeong BS, Mouradian MM. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J Neurosci Res. 2009 Jan;87, 123-9. https://doi.org/10.1002/jnr.21831. PMID: 18711745; PMCID: PMC2752655.
- Irrcher I, Aleyasin H, Seifert EL, Hewitt SJ, Chhabra S, Phillips M, Lutz AK, Rousseaux MW, Bevilacqua L, Jahani-Asl A, Callaghan S, MacLaurin JG, Winklhofer KF, Rizzu P, Rippstein P, Kim RH, Chen CX, Fon EA, Slack RS, Harper ME, McBride HM, Mak TW, Park DS. Loss of the Parkinson's disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. 2010 Oct 1;19, 3734-46. https://doi.org/10.1093/hmg/ddq288. Epub 2010 Jul 16. PMID: 20639397.
- Im JY, Lee KW, Woo JM, Junn E, Mouradian MM. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum Mol Genet. 2012 Jul 1;21, 3013-24. https://doi.org/10.1093/hmg/dds131. Epub 2012 Apr 5. PMID: 22492997; PMCID: PMC3373246.
- Dolgacheva LP, Berezhnov AV, Fedotova EI, Zinchenko VP, Abramov AY. Role of DJ-1 in the mechanism of pathogenesis of Parkinson's disease. J Bioenerg Biomembr. 2019 Jun;51, 175-188. https://doi.org/10.1007/s10863-019-09798-4. Epub 2019 May 3. PMID: 31054074; PMCID: PMC6531411.
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005 Feb 17;45, 489-96. https://doi.org/10.1016/j.neuron.2005.01.041. PMID: 15721235.
- Farshim PP, Bates GP. Mouse Models of Huntington's Disease. Methods Mol Biol. 2018;1780:97-120. https://doi.org/10.1007/978-1-4939-7825-0_6. PMID: 29856016.
- Jurcau A, Jurcau CM. Mitochondria in Huntington's disease: implications in pathogenesis and mitochondrial-targeted therapeutic strategies. Neural Regen Res. 2023 Jul;18, 1472-1477. https://doi.org/10.4103/1673-5374.360289. PMID: 36571344.
- Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington's disease. Nat Rev Neurosci. 2013 Oct;14, 708-21. https://doi.org/10.1038/nrn3570. PMID: 24052178.
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003 Mar 15;23, 2193-202. https://doi.org/10.1523/JNEUROSCI.23-06-02193.2003. PMID: 12657678; PMCID: PMC6742008.
- Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004 Jul 15;13, 1407-20. https://doi.org/10.1093/hmg/ddh162. Epub 2004 May 26. PMID: 15163634.
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002 Aug;5, 731-6. https://doi.org/10.1038/nn884. PMID: 12089530.
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996 Nov 1;87, 493-506. https://doi.org/10.1016/s0092-8674(00)81369-0. PMID: 8898202.
- Menalled L, El-Khodor BF, Patry M, Suárez-Fariñas M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, Morton AJ, Bates G, Leeds J, Park L, Howland D, Signer E, Tobin A, Brunner D. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol Dis. 2009 Sep;35, 319-36. https://doi.org/10.1016/j.nbd.2009.05.007. Epub 2009 May 21. PMID: 19464370; PMCID: PMC2728344.
- Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington's disease. J Neurochem. 2001 Dec;79, 1246-9. https://doi.org/10.1046/j.1471-4159.2001.00689.x. PMID: 11752065.
- Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AH. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000 Jan;47, 80-6. https://doi.org/10.1002/1531-8249(200001)47:1<80::aid-ana13>3.3.co;2-b. PMID: 10632104.
- Petersen MH, Willert CW, Andersen JV, Madsen M, Waagepetersen HS, Skotte NH, Nørremølle A. Progressive Mitochondrial Dysfunction of Striatal Synapses in R6/2 Mouse Model of Huntington's Disease. J Huntingtons Dis. 2022;11, 121-140. https://doi.org/10.3233/JHD-210518. PMID: 35311711.
- Milnerwood AJ, Raymond LA. Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load. J Physiol. 2007 Dec 15;585(Pt 3):817-31. https://doi.org/10.1113/jphysiol.2007.142448. Epub 2007 Oct 18. PMID: 17947312; PMCID: PMC2375504.
- Giralt A, Saavedra A, Alberch J, Pérez-Navarro E. Cognitive Dysfunction in Huntington's Disease: Humans, Mouse Models and Molecular Mechanisms. J Huntingtons Dis. 2012;1, 155-73. https://doi.org/10.3233/JHD-120023. PMID: 25063329.
- Ghiglieri V, Bagetta V, Calabresi P, Picconi B. Functional interactions within striatal microcircuit in animal models of Huntington's disease. Neuroscience. 2012 Jun 1;211:165-84. https://doi.org/10.1016/j.neuroscience.2011.06.075. Epub 2011 Jul 1. PMID: 21756979.
- Nithianantharajah J, Hannan AJ. Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience-dependent mediators of cognitive and psychiatric symptoms in Huntington's disease. Neuroscience. 2013 Oct 22;251:66-74. https://doi.org/10.1016/j.neuroscience.2012.05.043. Epub 2012 May 24. PMID: 22633949.
- Ghiglieri V, Campanelli F, Marino G, Natale G, Picconi B, Calabresi P. Corticostriatal synaptic plasticity alterations in the R6/1 transgenic mouse model of Huntington's disease. J Neurosci Res. 2019 Dec;97, 1655-1664. https://doi.org/10.1002/jnr.24521. Epub 2019 Sep 9. PMID: 31498496.
- Rosenstock TR, Bertoncini CR, Teles AV, Hirata H, Fernandes MJ, Smaili SS. Glutamate-induced alterations in Ca2+ signaling are modulated by mitochondrial Ca2+ handling capacity in brain slices of R6/1 transgenic mice. Eur J Neurosci. 2010 Jul;32, 60-70. https://doi.org/10.1111/j.1460-9568.2010.07268.x. PMID: 20608968.
- Clark JB. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev Neurosci. 1998;20(4-5):271-6. https://doi.org/10.1159/000017321. PMID: 9778562.
- Jenkins BG, Klivenyi P, Kustermann E, Andreassen OA, Ferrante RJ, Rosen BR, Beal MF. Nonlinear decrease over time in N-acetyl aspartate levels in the absence of neuronal loss and increases in glutamine and glucose in transgenic Huntington's disease mice. J Neurochem. 2000 May;74, 2108-19. https://doi.org/10.1046/j.1471-4159.2000.0742108.x. PMID: 10800956.
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF. Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis. 2001 Jun;8, 479-91. https://doi.org/10.1006/nbdi.2001.0406. PMID: 11447996.
- Jenkins BG, Andreassen OA, Dedeoglu A, Leavitt B, Hayden M, Borchelt D, Ross CA, Ferrante RJ, Beal MF. Effects of CAG repeat length, HTT protein length and protein context on cerebral metabolism measured using magnetic resonance spectroscopy in transgenic mouse models of Huntington's disease. J Neurochem. 2005 Oct;95, 553-62. https://doi.org/10.1111/j.1471-4159.2005.03411.x. Epub 2005 Aug 31. PMID: 16135087.
- Tkac I, Dubinsky JM, Keene CD, Gruetter R, Low WC. Neurochemical changes in Huntington R6/2 mouse striatum detected by in vivo 1H NMR spectroscopy. J Neurochem. 2007 Mar;100, 1397-406. https://doi.org/10.1111/j.1471-4159.2006.04323.x. Epub 2007 Jan 8. PMID: 17217418; PMCID: PMC2859960.
- Browne SE. Mitochondria and Huntington's disease pathogenesis: insight from genetic and chemical models. Ann N Y Acad Sci. 2008 Dec;1147:358-82. https://doi.org/10.1196/annals.1427.018. PMID: 19076457.
- Lopes C, Ferreira IL, Maranga C, Beatriz M, Mota SI, Sereno J, Castelhano J, Abrunhosa A, Oliveira F, De Rosa M, Hayden M, Laço MN, Januário C, Castelo Branco M, Rego AC. Mitochondrial and redox modifications in early stages of Huntington's disease. Redox Biol. 2022 Oct;56:102424. https://doi.org/10.1016/j.redox.2022.102424. Epub 2022 Aug 10. PMID: 35988447; PMCID: PMC9420526.
- Wright DJ, Renoir T, Smith ZM, Frazier AE, Francis PS, Thorburn DR, McGee SL, Hannan AJ, Gray LJ. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington's disease. Transl Psychiatry. 2015 Jan 6;5, e492. https://doi.org/10.1038/tp.2014.131. PMID: 25562842; PMCID: PMC4312826.
- Herbst EA, Holloway GP. Exercise training normalizes mitochondrial respiratory capacity within the striatum of the R6/1 model of Huntington's disease. Neuroscience. 2015 Sep 10;303:515-23. https://doi.org/10.1016/j.neuroscience.2015.07.025. Epub 2015 Jul 14. PMID: 26186895.
- Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J Biol Chem. 2005 Jan 7;280, 556-63. https://doi.org/10.1074/jbc.M410210200. Epub 2004 Oct 19. PMID: 15494404.
- Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE, Hackam AS, Logvinova AV, Peel AL, Chen SF, Hook V, Singaraja R, Krajewski S, Goldsmith PC, Ellerby HM, Hayden MR, Bredesen DE, Ellerby LM. Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ. 2004 Apr;11, 424-38. https://doi.org/10.1038/sj.cdd.4401358. PMID: 14713958.
- Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, Hayden MR. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998 Apr 10;273, 9158-67. https://doi.org/10.1074/jbc.273.15.9158. PMID: 9535906.
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999 May 20;399, 263-7. https://doi.org/10.1038/20446. PMID: 10353249.
- Carroll JB, Southwell AL, Graham RK, Lerch JP, Ehrnhoefer DE, Cao LP, Zhang WN, Deng Y, Bissada N, Henkelman RM, Hayden MR. Mice lacking caspase-2 are protected from behavioral changes, but not pathology, in the YAC128 model of Huntington disease. Mol Neurodegener. 2011 Aug 19;6:59. https://doi.org/10.1186/1750-1326-6-59. PMID: 21854568; PMCID: PMC3180273.
- M. Avenali, S. Cerri, G. Ongari, C. Ghezzi, C. Pacchetti, C. Tassorelli, E.M. Valente, F.vBlandini, Profiling the Biochemical Signature of GBA-Related Parkinson’s Disease invPeripheral Blood Mononuclear Cells, Mov. Disord. 36 (2021) 1267–1272. [CrossRef]
- S. Petrillo, T. Schirinzi, G. Di Lazzaro, J. D’Amico, V.L. Colona, E. Bertini, M. Pierantozzi, L. Mari, N.B. Mercuri, F. Piemonte, A. Pisani, Systemic activation of Nrf2 pathway in Parkinson’s disease, Mov. Disord. (2019). https://doi.org/10.1002/mds.27878.
- T. Schirinzi, I. Salvatori, H. Zenuni, P. Grillo, C. Valle, G. Martella, N.B. Mercuri, A. Ferri,vPattern of Mitochondrial Respiration in Peripheral Blood Cells of Patients with Parkinson’s Disease, Int. J. Mol. Sci. 23 (2022) 10863. [CrossRef]
- S.J. Annesley, S.T. Lay, S.W. De Piazza, O. Sanislav, E. Hammersley, C.Y. Allan, L.M. Francione, M.Q. Bui, Z.P. Chen, K.R.W. Ngoei, F. Tassone, B.E. Kemp, E. Storey, A. Evans, D.Z. Loesch, P.R. Fisher, Immortalized Parkinson’s disease lymphocytes have enhanced mitochondrial respiratory activity, Dis. Model. Mech. 9 (2016) 1295. [CrossRef]
- W. Haylett, C. Swart, F. Van Der Westhuizen, H. Van Dyk, L. Van Der Merwe, C. Van Der Merwe, B. Loos, J. Carr, C. Kinnear, S. Bardien, Altered Mitochondrial Respiration and Other Features of Mitochondrial Function in Parkin-Mutant Fibroblasts from Parkinson’s Disease Patients, Parkinsons. Dis. 2016 (2016). https://doi.org/10.1155/2016/1819209.
- P.M.A. Antony, O. Kondratyeva, K. Mommaerts, M. Ostaszewski, K. Sokolowska, A.S. Baumuratov, L. Longhino, J.F. Poulain, D. Grossmann, R. Balling, R. Krüger, N.J. Diederich, Fibroblast mitochondria in idiopathic Parkinson’s disease display morphological changes and enhanced resistance to depolarization, Sci. Rep. 10 (2020). https://doi.org/10.1038/S41598- 020-58505-6.
- M. Fais, A. Dore, M. Galioto, G. Galleri, C. Crosio, C. Iaccarino, Parkinson’s Disease- Related Genes and Lipid Alteration, Int. J. Mol. Sci. 22 (2021). [CrossRef]
- A.M. Smith, C. Depp, B.J. Ryan, G.I. Johnston, J. Alegre-Abarrategui, S. Evetts, M. Rolinski, F. Baig, C. Ruffmann, A.K. Simon, M.T.M. Hu, R. Wade-Martins, Mitochondrial dysfunction and increased glycolysis in prodromal and early Parkinson’s blood cells, Mov. Disord. 33 (2018) 1580–1590. [CrossRef]
- J.F. Havelund, N.H.H. Heegaard, N.J.K. Færgeman, J.B. Gramsbergen, Biomarker researchvin parkinson’s disease using metabolite profiling, Metabolites. 7 (2017). [CrossRef]
- D. Willkommen, M. Lucio, F. Moritz, S. Forcisi, B. Kanawati, K.S. Smirnov, M. Schroeter,A. Sigaroudi, P. Schmitt-Kopplin, B. Michalke, Metabolomic investigations in cerebrospinal fluid of Parkinson’s disease, PLoS One. 13 (2018). [CrossRef]
- S. Saiki, T. Hatano, M. Fujimaki, K.-I. Ishikawa, A. Mori, Y. Oji, A. Okuzumi, T. Fukuhara,T. Koinuma, Y. Imamichi, M. Nagumo, N. Furuya, S. Nojiri, T. Amo, K. Yamashiro, N. Hattori, Decreased long-chain acylcarnitines from insufficient β-oxidation as potential early diagnostic markers for Parkinson’s disease OPEN, (n.d.). [CrossRef]
- Ascherio, M.A. Schwarzschild, The epidemiology of Parkinson’s disease: risk factors and prevention, Lancet Neurol. 15 (2016) 1257–1272. https://doi.org/10.1016/S1474-4422(16)30230-7.
- T. Schirinzi, G. Di Lazzaro, G.M. Sancesario, S. Summa, S. Petrucci, V.L. Colona, S. Bernardini, M. Pierantozzi, A. Stefani, N.B. Mercuri, A. Pisani, Young-onset and late-onset Parkinson’s disease exhibit a different profile of fluid biomarkers and clinical features, Neurobiol. Aging. 90 (2020) 119–124. [CrossRef]
- T. Schirinzi, G. Vasco, G. Zanni, S. Petrillo, F. Piemonte, E. Castelli, E.S. Bertini, Serum uric acid in Friedreich Ataxia, Clin. Biochem. 54 (2018) 139–141. [CrossRef]
- T. Schirinzi, G. Di Lazzaro, V.L. Colona, P. Imbriani, M. Alwardat, G.M. Sancesario, A. Martorana, A. Pisani, Assessment of serum uric acid as risk factor for tauopathies., J. Neural Transm. (2017). https://doi.org/10.1007/s00702-017-1743-6.
- Z. Wei, X. Li, X. Li, Q. Liu, Y. Cheng, Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis, Front. Mol. Neurosci. 11 (2018) 236. [CrossRef]
- S.I. Ueno, T. Hatano, A. Okuzumi, S. Saiki, Y. Oji, A. Mori, T. Koinuma, M. Fujimaki, H. Takeshige-Amano, A. Kondo, N. Yoshikawa, T. Nojiri, M. Kurano, K. Yasukawa, Y.Yatomi, H. Ikeda, N. Hattori, Nonmercaptalbumin as an oxidative stress marker in Parkinson’s and PARK2 disease, Ann. Clin. Transl. Neurol. 7 (2020) 307–317. [CrossRef]
- Y. Yamagishi, K. Saigoh, Y. Saito, I. Ogawa, Y. Mitsui, Y. Hamada, M. Samukawa, H. Suzuki, M. Kuwahara, M. Hirano, N. Noguchi, S. Kusunoki, Diagnosis of Parkinson’s disease and the level of oxidized DJ-1 protein, Neurosci. Res. 128 (2018) 58–62. https://doi.org/10.1016/j.neures.2017.06.008.
- G.M. Sancesario, G. Di Lazzaro, P. Grillo, B. Biticchi, E. Giannella, M. Alwardat, M. Pieri, S. Bernardini, N.B. Mercuri, A. Pisani, T. Schirinzi, Biofluids profile of α-Klotho in patients with Parkinson’s disease, Parkinsonism Relat. Disord. 90 (2021) 62–64. [CrossRef]
- E. Fernández-Espejo, F. Rodriguez de Fonseca, J. Suárez, Á. Martín de Pablos, Cerebrospinal fluid lactoperoxidase level is enhanced in idiopathic Parkinson’s disease, and correlates with levodopa equivalent daily dose, Brain Res. 1761 (2021) 147411. [CrossRef]
- W. Sun, J. Zheng, J. Ma, Z. Wang, X. Shi, M. Li, S. Huang, S. Hu, Z. Zhao, D. Li, Increased Plasma Heme Oxygenase-1 Levels in Patients With Early-Stage Parkinson’s Disease, Front. Aging Neurosci. 13 (2021) 39. [CrossRef]
- R. Shamir, C. Klein, D. Amar, E.J. Vollstedt, M. Bonin, M. Usenovic, Y.C. Wong, A. Maver, S. Poths, H. Safer, J.C. Corvol, S. Lesage, O. Lavi, G. Deuschl, G. Kuhlenbaeumer, H. Pawlack, I. Ulitsky, M. Kasten, O. Riess, A. Brice, B. Peterlin, D. Krainc, Analysis of blood-based gene expression in idiopathic Parkinson disease, Neurology. 89 (2017) 1676–1683. [CrossRef]
- Picca, F. Guerra, R. Calvani, F. Marini, A. Biancolillo, G. Landi, R. Beli, F. Landi, R. Bernabei, A.R. Bentivoglio, M.R. Lo Monaco, C. Bucci, E. Marzetti, Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adult with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study, J. Clin. Med. 2020, Vol. 9, Page 504. 9 (2020) 504. https://doi.org/10.3390/JCM9020504.
- B.D. Paul, S.H. Snyder, Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications, Front. Mol. Neurosci. 12 (2019). https://doi.org/10.3389/FNMOL.2019.00068.
- P. Jędrak, P. Mozolewski, G. Węgrzyn, M.R. Więckowski, Mitochondrial alterations accompanied by oxidative stress conditions in skin fibroblasts of Huntington’s disease patients, Metab. Brain Dis. 33 (2018) 2005–2017. [CrossRef]
- M. Vanisova, H. Stufkova, M. Kohoutova, T. Rakosnikova, J. Krizova, J. Klempir, I. Rysankova, J. Roth, J. Zeman, H. Hansikova, Mitochondrial organization and structure are compromised in fibroblasts from patients with Huntington’s disease, Ultrastruct. Pathol. 46 (2022) 462–475. [CrossRef]
- A. Neueder, M. Orth, Mitochondrial biology and the identification of biomarkers of Huntington’s disease, Neurodegener. Dis. Manag. 10 (2020) 243–255. https://doi.org/10.2217/NMT- 2019-0033.
- C.M. Chen, Y.R. Wu, M.L. Cheng, J.L. Liu, Y.M. Lee, P.W. Lee, B.W. Soong, D.T.Y. Chiu, Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients, Biochem. Biophys. Res. Commun. 359 (2007) 335–340. https://doi.org/10.1016/J.BBRC.2007.05.093.
- Spinelli JB, Zaganjor E. Mitochondrial efficiency directs cell fate. Nat Cell Biol. 2022 Feb;24, 125-126. https://doi.org/10.1038/s41556-021-00834-3. PMID: 35165414.
- Ahmad M, Wolberg A, Kahwaji CI. Biochemistry, Electron Transport Chain. 2022 Sep 5. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 30252361.
- McAdam E, Brem R, Karran P. Oxidative Stress-Induced Protein Damage Inhibits DNA Repair and Determines Mutation Risk and Therapeutic Efficacy. Mol Cancer Res. 2016 Jul;14, 612-22. https://doi.org/10.1158/1541-7786.MCR-16-0053. Epub 2016 Apr 22. PMID: 27106867; PMCID: PMC4955916.
- Korovila I, Hugo M, Castro JP, Weber D, Höhn A, Grune T, Jung T. Proteostasis, oxidative stress and aging. Redox Biol. 2017 Oct;13:550-567. https://doi.org/10.1016/j.redox.2017.07.008. Epub 2017 Jul 12. PMID: 28763764; PMCID: PMC5536880.
- Gerencser AA, Doczi J, Töröcsik B, Bossy-Wetzel E, Adam-Vizi V. Mitochondrial swelling measurement in situ by optimized spatial filtering: astrocyte-neuron differences. Biophys J. 2008 Sep;95, 2583-98. https://doi.org/10.1529/biophysj.107.118620. Epub 2008 Apr 18. PMID: 18424491; PMCID: PMC2517017.
- O'Sullivan JDB, Bullen A, Mann ZF. Mitochondrial form and function in hair cells. Hear Res. 2023 Feb;428:108660. https://doi.org/10.1016/j.heares.2022.108660. Epub 2022 Nov 25. PMID: 36525891.
- Woo J, Cho H, Seol Y, Kim SH, Park C, Yousefian-Jazi A, Hyeon SJ, Lee J, Ryu H. Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants (Basel). 2021 Feb 3;10, 229. https://doi.org/10.3390/antiox10020229. PMID: 33546471; PMCID: PMC7913624.
- Polyzos AA, McMurray CT. The chicken or the egg: mitochondrial dysfunction as a cause or consequence of toxicity in Huntington's disease. Mech Ageing Dev. 2017 Jan;161(Pt A):181-197. https://doi.org/10.1016/j.mad.2016.09.003. Epub 2016 Sep 12. PMID: 27634555; PMCID: PMC5543717.
- Franco-Iborra S, Vila M, Perier C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson's Disease and Huntington's Disease. Front Neurosci. 2018 May 23;12:342. https://doi.org/10.3389/fnins.2018.00342. PMID: 29875626; PMCID: PMC5974257.
- Catherine, E. Lang, Marghuretta D. Bland. Chapter 17 - Impaired Motor Control and Neurologic Rehabilitation in Older Adults, Editor(s): Dale Avers, Rita A. Wong, Guccione's Geriatric Physical Therapy (Fourth Edition), Mosby, 2020, Pages 379-399,ISBN 9780323609128. [CrossRef]
- Lee LN, Huang CS, Chuang HH, Lai HJ, Yang CK, Yang YC, Kuo CC. An electrophysiological perspective on Parkinson's disease: symptomatic pathogenesis and therapeutic approaches. J Biomed Sci. 2021 Dec 9;28, 85. https://doi.org/10.1186/s12929-021-00781-z. PMID: 34886870; PMCID: PMC8656091.
- Jamwal S, Kumar P. Insight Into the Emerging Role of Striatal Neurotransmitters in the Pathophysiology of Parkinson's Disease and Huntington's Disease: A Review. Curr Neuropharmacol. 2019;17, 165-175. https://doi.org/10.2174/1570159X16666180302115032. PMID: 29512464; PMCID: PMC6343208.
- Terreros-Roncal J, Moreno-Jiménez EP, Flor-García M, Rodríguez-Moreno CB, Trinchero MF, Cafini F, Rábano A, Llorens-Martín M. Impact of neurodegenerative diseases on human adult hippocampal neurogenesis. Science 2021 Nov 26;374, 1106-1113. https://doi.org/10.1126/science.abl5163. Epub 2021 Oct 21. PMID: 34672693; PMCID: PMC7613437.
- Carmo C, Naia L, Lopes C, Rego AC. Mitochondrial Dysfunction in Huntington's Disease. Adv Exp Med Biol. 2018, 1049, 59–83. https://doi.org/10.1007/978-3-319-71779-1_3. PMID: 29427098.
Figure 1.
mitochondrial dysfunction in MP and HD. Characteristic features for both disease.
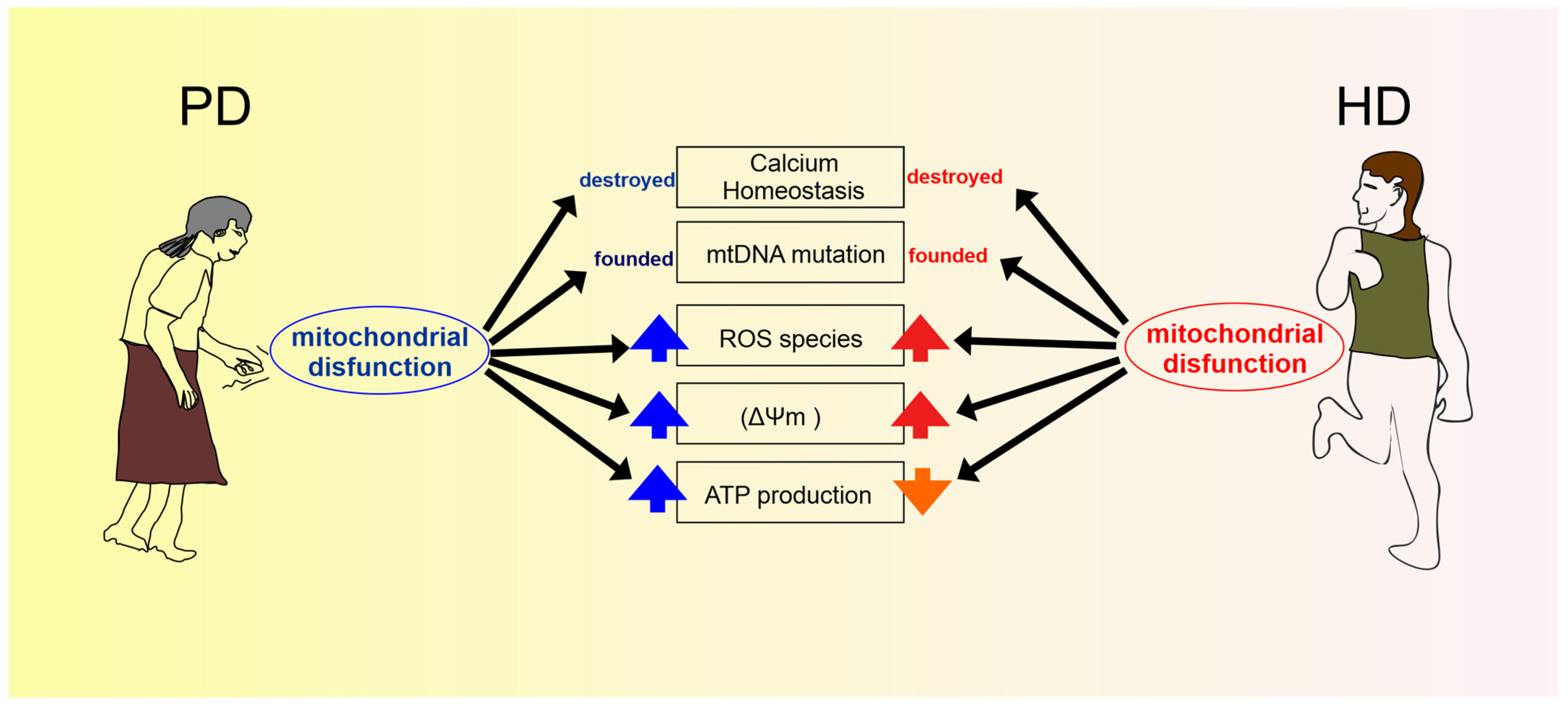
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



