Submitted:
14 July 2026
Posted:
15 July 2026
You are already at the latest version
Abstract
While it remains highly challenging to create oral medicines against so-called “undruggable” targets, progress is accelerating. Using multiple approaches to analyze druggability, we estimate that 17–32% of the genome may now plausibly be considered druggable. This range was derived by extrapolating evidence of tractability from targets with high-quality ligands to related genes within several distinct curated classification schemes. Four distinct scientific advances underlie this progress: more aggressive exploration of chemical space; an improved ability to deliver pharmacologically relevant doses of these medicines; a vastly expanded range of mechanistic drug modalities; and dramatically improved biochemical and cellular screening tools to identify and characterize promising ligands. Underlying these scientific advances is a more optimistic mindset that supports the pursuit of challenging targets of high medical interest. We review recent examples, document progress, and identify remaining bottlenecks. While many challenges undeniably remain, the rapid progress suggests that we need to reconsider the term “undruggable.”
Keywords:
undruggable
; druggability
; ligandability
; drug discovery
; drug targets
1. Introduction
The term “undruggable”[1,2,3] has taken on a wide range of meanings, thereby creating confusion in the world of biomedical research [Box 1]. Often the term refers to the challenge of creating an oral medicine. For example, given the existence of an approved injectable biologic agent that blocks the interaction between FcRn and immunoglobulin G, the statement “FcRn is undruggable” is intended to convey the belief that developing an orally delivered small-molecule FcRn antagonist will not be possible.
| Box 1. Parsing distinct meanings of “undruggable” |
|
Possible meanings of the term “undruggable” when applied to a specific project with the goal of producing an oral medicine Historical fact: no one has made an oral drug against this target (and possibly other related targets). In this sense, a better term would be “undrugged” or “as yet undrugged.” A challenge for future generations: a belief that it is impossible both now and well into the future to make an oral drug against this target. Currently impossible: a more nuanced judgment that a target is unapproachable with the knowledge and technologies available today. There is no implication that the opportunity will remain forever out of reach. Even more narrowly, it may signal that the speaker believes that the problem cannot be solved with the technologies and resources available to them. Very hard: Teams often pursue oral medicines against targets they describe as “undruggable.” However, if those teams literally believed the targets were impossible, pursuing them would be irrational. Generally, such targets are extremely well validated and any resulting therapies directed against such targets would hold the potential to address large patient populations, making them highly attractive despite the extremely high risk of failure. Sometimes these are described as “holy grail” targets. Possible meanings of the phrase “I dislike the term ‘undruggable’” Informed scientific optimism: the speaker believes that most or all targets are druggable now or will be in the near future. This opinion may be based on their own experience or their analysis of the rate of progress in the field. A plea for open-mindedness: the speaker wishes to ensure that all reasonable efforts are made, using all the latest technologies, to pursue medically important but challenging targets that have previously been deemed impossible. Broad confidence in the future: a general and possibly vague belief that future improvements will enable success. Desire to sound optimistic: a wish to avoid appearing negative, defeatist, or even Luddite. |
In this work, we focus on oral drugs of all kinds because they will continue to play an important role in medical practice in the coming decades. Without a doubt, biologics have become an essential and welcome component of the medical armamentarium. More than half of drug revenue now comes from injectable drugs, and in 2021 only 30% of biotech investment was directed at small molecules.[4] Concern about the effect of the Inflation Reduction Act on the pricing for small molecules has further exacerbated funding challenges.[5] However, small molecules still represent the vast majority of drugs overall.[6]
While biologic drugs have revolutionized the treatment of many diseases in recent decades, these medicines often suffer from manufacturing challenges, the high cost of required in-office dosing, the need for cold-chain storage, and the potential for immune-related adverse events. Encouragingly, improvements in the delivery of biologics and large peptides continue to be made. A prominent current example is the at-home, patient-administered weekly subcutaneous injection of GLP-1 agonists.
There remain many advantages to oral small-molecule drugs. These include convenience, broader tissue distribution than is possible for biologics, cellular permeability, the ability to access and modulate diverse functions of biological targets, the ease of treatment cessation to manage adverse events, the availability of synthetic methods to enable the fine-tuning of molecular properties, the ability to create prodrugs to enhance physical or pharmacokinetic properties, long shelf life, no need for cold- chain storage, low manufacturing costs, and the ability to rapidly commercialize inexpensive generics. Of course, there can also be disadvantages to oral medicines, such as the potential for drug-drug interactions, distribution to the wrong tissues, or toxicity resulting from insufficient target selectivity.
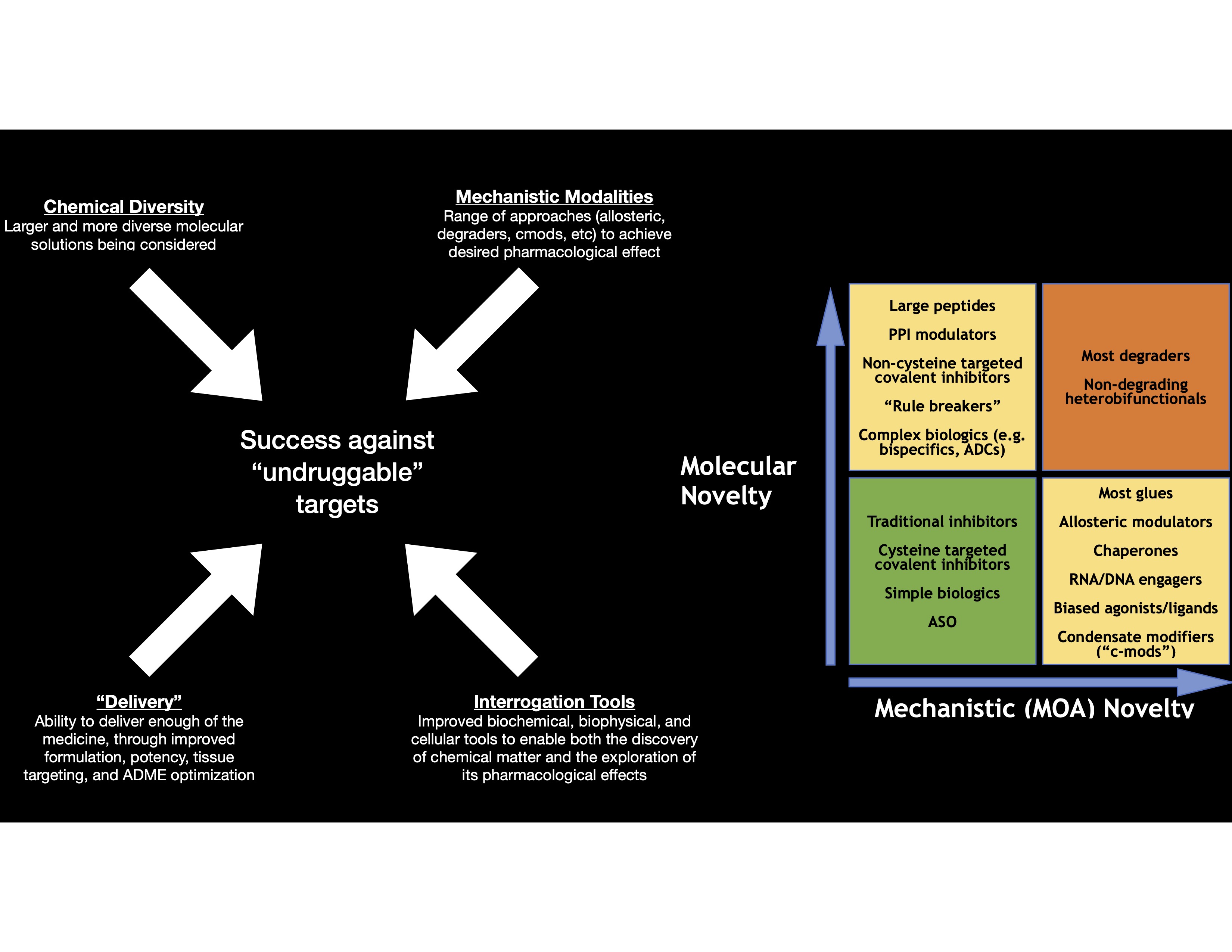
Molecules are getting larger and more complex;[7,8,9,10,11] novel “modalities” are continually being discovered that enable researchers to address an expanding set of biological targets and mechanisms. However, confusion arises from two distinct meanings of the term “modality” [Figure 1]. It may refer to a class of molecules that engage a target and disrupt its function in a fundamentally novel way. In this sense, “modality” refers to mechanism of action. Degraders,[12] biased signaling GPCR agonists,[13,14,15] and condensate-modifying drugs[16] are all examples. Such molecules are found on the right side of the two-by-two matrix in Figure 1. The term “modality” may also refer to molecules that fall outside traditional drug-like property guidelines. In this latter sense, “modality” refers to the chemical structure itself—its molecular novelty or complexity. While the term “modality” is not as widely used in this second sense, and the concept of molecular complexity is still evolving, peptides and other “rule breaker” molecules (molecular mass greater than 500 daltons) are sometimes referred to as a different kind of modality.[11,17,18,19] Molecules exhibiting molecular (chemical) novelty are in the upper half of the two-by-two matrix in Figure 1. Novel modes of administration (e.g., nanoparticles for oral delivery of proteins) may be included in this second, more molecular definition of “modality.” Molecules that exhibit both molecular and mechanistic novelty, such as PROTACs, are in the upper-right quadrant of Figure 1. A molecular glue may operate through a highly novel mechanism of action, but its molecular structure may either be quite conventional and straightforward (lower-right quadrant of Figure 1) or highly complex (upper-right quadrant of Figure 1). Conversely, a large, complex peptide may require novel and creative synthetic work, but its mechanism of action may be that of a conventional enzyme inhibitor or receptor antagonist (upper-left quadrant of Figure 1).
The assessment of the degree of novelty of any modality will be somewhat subjective and will be continually recalibrated in light of each new success, and always against the backdrop of the many specific challenges that may arise during any drug discovery program [Box 2]. Each modality comes with its own set of challenges. For example, with glue discovery projects, it is more difficult to find chemical starting points, but if the molecules are low molecular weight, the optimization process is generally more straightforward; conversely, with heterobifunctionals such as PROTACs, “active leads can usually be found relatively quickly once suitable binary ligands and an E3-ligase system are available, whereas optimization into a drug candidate is often exceptionally difficult.”
| Box 2. Factors that complicate the discovery of oral medicines |
|
Topology. Challenging binding pockets make it more difficult to find a molecule that combines sufficient affinity with desirable pharmacokinetic properties. For example, the pocket may have a small volume, have an extended surface, contain many polar and/or charged amino acids, or form only transiently because of conformational mobility. Competition at the site of action. The natural ligand (e.g., substrate or binding partner) may be present at such a high concentration or bind so tightly that it is difficult to create a molecule that can displace that natural ligand. For example, this often occurs when attempting to disrupt protein-protein interactions, or when attempting to bind to an ATP-binding site of a kinase for which the Kd for ATP is typically in the single-digit micromolar range (since the intracellular concentration of ATP is typically above 1 millimolar). Selectivity. Often targets are members of gene families, making it challenging to discover drugs that bind only to the desired target. In principle, selectivity can be achieved through careful molecular tuning, but in practice this is often quite difficult. Alternatively, if allosteric sites are known or can be discovered, they may provide greater opportunities for selective binding. Mechanism of action. While orthosteric inhibition and antagonism are generally well understood, irreversible or slow-off-rate compounds can be more challenging to interpret; the same is true for molecules that exhibit partial or inverse agonism, bind targets allosterically, or exert their pharmacological impact via alternative signaling pathways. More novel mechanistic modalities such as degraders, glues, and condensate modifiers (“c-mods”) can function in ways that are even more complex to understand. For example, drugs that induce the formation of a ternary complex generally exhibit nonintuitive dose-response behaviors. Toxicology. The target may be essential; it may be pleiotropic and perform multiple functions, many of which are incompletely understood. Tissue targeting. The concentration of drug reaching the desired tissue may be insufficient to exert a therapeutically meaningful effect, or distribution of the drug into other body compartments may cause adverse effects. Cell permeability. For typical small-molecule drugs, achieving cell permeability is generally possible, albeit sometimes with difficulty. However, very large, complex, or highly charged molecules can be far more challenging to deliver into cells. A further complication is delivery to precise locations within the cell—for example, to the nucleus, the mitochondria, or a specific cellular condensate. Pharmacology: exposure and coverage. It may not be possible to achieve a sufficiently high concentration of drug in the target tissue for a long enough period of time. This PK-PD coverage problem is simplified when the required exposure is reduced, e.g., by a higher binding affinity or slower dissociation rate. Pharmacology: incorrect biological or mechanistic hypothesis. The drug may affect a pathway or biological mechanism that is ultimately proven irrelevant to the disease process. A high percentage of Phase II clinical failures result from such errors. Alternatively, the drug may engage the desired target, but in a pharmacologically unproductive manner. The complexity of the voltage-gated sodium channels is instructive in this regard.[178,179] Polypharmacology. To achieve adequate efficacy, we may need to engage multiple targets, but our drug may not accomplish this. Polypharmacology is a deep and fascinating topic but is beyond the scope of this Perspective. Screenability. Designing high-throughput screening (HTS) assays that faithfully report on the biologically critical mechanism—rather than on a convenient surrogate—remains a central challenge in modern drug discovery, particularly for complex, network-level, or conformationally heterogeneous targets. |
In recent years, approximately 25% of newly approved oral medicines have fallen into one or more of these novel modality categories. For some modalities, there are many clinical examples and even approved medicines, while for others the fields are less mature but rapidly evolving. Broadly, macrocyclic peptides,[20,21] allosteric drugs,[22,23,24,25] and protein-protein interaction inhibitors[25] are relatively mature fields, with many approved oral drugs and large clinical pipelines across different target classes (e.g., kinases, GPCRs, and ion channels) and therapeutic areas (e.g., cancer, immunology, and metabolic diseases). Since the emergence of targeted protein degradation through the ubiquitin-proteasome system, PROTACs have become a major drug discovery modality, culminating in the approval of the first PROTAC drug, vepdegestrant, in May 2026 and at least 40 additional PROTACs now being evaluated in clinical trials.[26] The field of molecular glues has deep historical roots, with both stabilizers (such as cyclosporine, FK506, and rapamycin) and degraders (such as the IMiDs). Rationally designed molecular glues are beginning to enter the clinic.[27] Successful examples of non-cysteine covalent inhibitors (other than serine hydrolase inhibitors) remain relatively uncommon, although recent advances in targeting lysine, tyrosine, and other amino acid residues are expanding the scope of covalent drug discovery beyond cysteine.[28,29] Finally, an emerging modality, condensate-modifying drugs (“c-mods”), is just beginning to enter the clinic, with the first clinical candidates now being evaluated.[16]
Largely with the help of these new modalities, both mechanistic and molecular, progress is being rapidly made against targets that were formerly considered “orally undruggable.” We provide several estimates of the fraction of the human genome that may now be considered “druggable.” These estimates reflect the contents of curated compound and gene classification databases at the time of our analysis and the modalities demonstrated to date. They therefore represent a conservative current estimate rather than a fixed property of the genome, and we expect the druggable fraction to grow as new chemistry, mechanisms, and annotations accumulate. We document and discuss the significance of recent successes as well as the remaining challenges that continue to complicate the discovery of new medicines against historically undrugged targets.
2. Analysis
2.1. Defining the Drug Target Space
The definition of “drug target space” (both human targets and those from pathogens) is more complex than it first seems. Two decades ago, Kubinyi[30] pointed out that the phrase “druggable genome”[31] is misleading and suggested instead the terms “druggable proteome” and “druggable targetome” to more accurately reflect the space in which drug discovery operates.
First, many genes produce multiple splice variants, and many proteins occur in multiple post-translationally modified forms. These differences matter: variant “functional alloforms”[32] or “proteoforms” will frequently play divergent roles, contributing to different biological effects on physiology, development, and disease pathology.[33] Historically, it has been reasonable to assume that most drugs interact with multiple proteoforms, although detailed analysis is generally lacking. A recent analysis of ibrutinib demonstrates its promiscuity across ABL proteoforms.[34] Recent studies show that probes can distinguish particular post-translational modification states[35] and other proteoform differences.[36] The ideal proteoform-specific medicine must selectively engage the disease-relevant state of that protein, which may be post-translationally modified in precise ways and reside in specific subcellular compartments in complexes with other proteins and nucleic acids. The tractability of one proteoform does not imply that another proteoform encoded by the same gene will be similarly tractable. For example, consider the discovery of the clinical candidate rezatapopt, which binds selectively to and stabilizes the Y220C mutant form of p53 in a wild-type-like conformation and thereby restores its tumor suppressor functions. This encouraging result has not yet been replicated in any of the other clinically relevant mutant forms of p53.[37]
Second, biological function often depends on heteromeric protein-protein and protein-nucleic acid complexes. Luck et al.[38] have identified approximately 53,000 high-quality human binary protein-protein interactions by screening 90% of the protein-coding genome via three different versions of yeast two-hybrid assays. When combined with previous efforts, their resulting database contains a total of approximately 60,000 interactions involving roughly 9,000 proteins. This interactome map, while admittedly incomplete, is nonetheless vast, and reveals the magnitude of the potential space of drugs that modulate protein-protein interactions. Michaelis et al.[39] have more recently probed the yeast proteome using a highly sensitive high-throughput mass spectrometric approach, leading to a tripling of the number of known interactions in yeast to approximately 31,000.
Third, Kubinyi[30] observed that polypharmacology—drugs that engage combinations of targets and thereby exhibit multiple mechanisms of action—is widespread.
Fourth, not all drug targets are proteins. For example, tens of thousands of long noncoding RNAs have been cataloged, ranging from roughly 36,000 in conservative reference annotations (GENCODE, Ensembl) to nearly 59,000 in comprehensive transcriptome assemblies.[40] The differences are attributable to annotation stringency: conservative curated reference sets versus permissive ab initio assemblies over large, cancer-inclusive compendia. While currently these are properly considered more suitable candidates for biological drugs such as antisense oligonucleotides, emerging evidence suggests that small-molecule oral therapeutics may be possible. This dramatically increases the size of the “targetome” that could potentially be targeted with oral drugs.[41,42,43]
Also of importance is the fact that, although most human proteins now carry some experimental functional annotation, the great majority have not been pharmacologically explored. Of the roughly 20,000 identified human proteins, the Illuminating the Druggable Genome resource classifies about 700 as the target of an approved drug and a further 1,900 as having a well-documented small-molecule ligand.[56] Of the remainder, roughly 12,300 are biologically characterized but lack any chemical probe, while about 5,500 remain essentially unstudied, with minimal functional annotation.[56] As our understanding of disease biology advances, thousands of additional proteins will become targets of interest, and the question then will turn to whether they are “druggable.”
Further, it is becoming clear that the canonical rules for predicting protein-coding open reading frames are too restrictive, and in fact there are many additional proteins of unknown function translated from the genome, including many “microproteins” with fewer than 100 amino acids, many of which may exist in multiple proteoforms. A recent consensus analysis by the TransCODE Consortium found protein-level evidence across 95,520 proteomics experiments for a set of 7,264 candidate non-canonical open reading frames, roughly a quarter of which yield detectable peptides, and introduced the “peptidein” category for translated ORFs whose protein-coding status is not yet established.[45] Many members of this uncharted, non-canonical proteome will no doubt prove to be attractive targets.[46,47]
2.2. The Tractability of the Drug Target Space
Many groups have previously assessed the tractability of target space.[31,48,49,50,51,52,53,54,55,56] Targets may be considered “undruggable” for diverse reasons, generally some combination of the factors in Box 2. Some target classes are generally considered highly druggable, such as GPCRs, kinases, proteases, ion channels, and nuclear hormone receptors. While such targets often still pose considerable challenges, the many past successes within these families engender confidence. This was anticipated in the concept of “chemogenomics,”[55] which posited that within any given gene family, a highly efficient reuse of information, reagents, compounds, assays, intellectual property, and know-how will dramatically increase both speed and quality. This optimism has been borne out for the best-studied gene families. For example, Anderson et al.[53] conclude that essentially all kinases are now tractable. Their conclusion reflects the far greater availability of basic reagents and knowledge (assays, protein structures, tool compounds) as well as novel modalities to address selectivity (allosteric inhibitors, degraders, covalent compounds). Further support for this view comes from the diverse set of kinase chemical probes now available that bind to targets that occupy less-well-studied branches of the kinome tree.[57]
However, “druggable” does not mean “straightforward.” All targets pose one or more of the difficulties listed in Box 2. Specific targets within a class, or entire subsets of that target class, may present special challenges.
The complexity of creating a drug against any specific new target within a previously drugged gene family depends on many variables. How closely related are the active sites of the two targets, and will selectivity be a significant challenge? Do those closely related “anti-targets” present a toxicity concern; that is, how selective does our drug need to be? Are allosteric approaches an option? How potently does the function of the new target need to be inhibited or enhanced to achieve the desired pharmacologic effect? For proteins with multiple functions the difficulty of discovering drugs that block different functions may vary, and of course interfering with different functions will produce different pharmacological or toxicological effects. For example, it was found that the protease activity of PCSK9 was not required for the regulation of the LDL receptor.[58,59]
Even considering these factors, a reasonable default assumption is that pursuing a new target in a previously drugged family, especially if suitable assays, structures, and chemical libraries are available, is more likely to be successful than pursuing a novel target from a previously undrugged gene family. However, the benefits of chemogenomics[55]—the reuse of data and knowledge across a gene family—cannot be assumed to apply equally in every situation. While kinases have been widely enough studied that, in agreement with Anderson et al.,[53] one may be confident that the entire family is tractable, a similar claim cannot be made for other large and complex gene families. Such families often contain subfamilies with substantial structural and functional differences. For example, there are dozens of subfamilies of transcription factors,[54] the intricacies of which remain to be discovered.
2.3. Methods of Estimating Druggability and Results
We infer druggability by similarity: If a product of one gene has been drugged, we assume that products of related genes are also more likely to be tractable. A crucial point is that we use the term “drug” in its broadest sense: a chemical agent that achieves a desired pharmacological effect. These compounds may be marketed drugs, clinical candidates, or preclinically validated ligands. Throughout this analysis, the “druggability” of a target is indicated by the existence of a high-quality ligand for that target, per the Open Targets “high-quality ligand” annotation derived from ChEMBL. Our core assumption is that once a high-quality ligand has been identified against a target, that target becomes potentially far more druggable. As we will discuss, this does not mean that creating a medicine against that target, or closely related targets, will be “easy.”
Inferring similarity requires choosing a metric (sequence similarity, structural similarity, gene function sharing, etc.) and a threshold (percentage of shared residues, RMSD of the best alignment, function assigned by GO classification, etc.). These choices are inherently arbitrary. Therefore, we adopt widely used curated classification systems that encode expert consensus about which targets “belong together.”
A classification system can be visualized as a hierarchical tree characterized by an increasing number of branches at increasing depth. A specified depth of the classification system defines a classification scheme that is characterized by a number of classes or subclasses that each contain one or more genes. Classification schemes trade off coverage and granularity, i.e., the level of detail at which one forms classes. Coarse (low) granularity uses broad thresholds that lump genes together and fine (high) granularity splits sets of genes into smaller groups.
Below, we analyze druggability in two ways: by the ChEMBL class/subclass system and by a hybrid PANTHER class/subclass system. We selected these systems and levels of granularity to bracket a plausible range for the druggable fraction. We chose the two different systems, ChEMBL and PANTHER, because the former covers curated drug targets and their putatively related targets that are a subset of the genome, whereas the latter extends to nearly the entire genome. For each system we carried out the analysis at two different levels of granularity. Coarse (low granularity) groupings allow one to generously infer druggability for many targets based on a single success in the grouping. However, in practice, drug discovery teams usually find that it is only within smaller neighborhoods of more closely related proteins that pocket features and SAR (structure-activity relationships) plausibly transfer. Thus, class (lower granularity) should provide an upper bound on the estimates of druggability, while subclass (higher granularity) analysis should produce a lower bound.
We also considered using a “pocket-ome” analysis to classify target similarity. The pocket-ome[60,61,62] is the complete set of classified binding pockets across proteome structures, characterized by shape and physicochemical properties. If two proteins are classified as having similar binding pockets, it may be comparably difficult to discover ligands that bind those pockets. Therefore, one could imagine classifying and grouping proteins using a pocket-ome analysis. However, most proteins lack experimental 3D structural data, and predicted structures often lack the local accuracy needed for reliable pocket comparison. Furthermore, pocket-ome analysis shares the same inability as our classification approach to account for dynamics, cryptic pockets, selectivity, cellular context, and pharmacological requirements. Accordingly, we concluded that a classification-based approach is more appropriate because of the broader coverage and availability of curated groupings.
We analyze drugged and inferred-druggable genes using ChEMBL at the class and subclass levels. Our findings (Figure 2) are summarized in three additive parts: genes already drugged (cyan); additional genes inferred to be druggable because they share a ChEMBL class or subclass with a drugged member (green), which expands beyond direct targets as expected; and a further increment from the broader ChEMBL class (blue). We show the breakdown by the class label. This stepwise growth makes the trade-off explicit: narrower groupings will preserve specificity and yield conservative propagation, while broader classes increase coverage at the cost of possible over-generalization. Unclassified genes are shown for context and are not used for propagation.
In brief, finer classifications yield more conservative estimates, while broader classifications yield higher but less certain estimates. In greater detail:
- Class level (14 classes): Drugged genes comprise 1,816 classified and 318 unclassified, totaling 2,134 genes (10.6% of the genome). Propagating druggability within classes contributes an additional 1,730 inferred druggable genes, for 3,864 total druggable genes (19.3% of the genome). All 14 ChEMBL classes contain at least one drugged gene (no undrugged classes) (Figure 3 left).
- Subclass level (36 subclasses): Drugged genes comprise 1,574 classified and 560 unclassified, again totaling 2,134. Propagation within subclasses yields 1,268 inferred genes, for 3,402 total druggable genes (17.0%). 34 of 36 subclasses contain at least one drugged gene; 2 subclasses (5.6%) have none.
At both levels, the majority of genes remain outside these druggable sets (80.7% undrugged at Class; 83.0% at subclass) (Figure 3 right).
We repeat the analysis with PANTHER, using (i) Protein Class depth-3 (PC d3) as the class system and (ii) Family (PTHR d2) as the subclass system.
- Class level: PANTHER PC depth-3 (93 classes). Directly drugged genes total 2,134. Propagation within PC d3 classes adds 4,204 inferred druggable genes, for 6,338 total druggable genes (31.7% of the genome). 72/93 classes (77.4%) contain at least one drugged gene, leaving 21 undrugged classes (22.6%). 13,686 genes (68.3%) remain undrugged or share no PC d3 class with a drugged gene (Figure 4 left).
- Subclass level: PANTHER Family (also labeled d2). Of the 7,467 families that contain at least one analyzed protein-coding gene, 932 (12.5%) contain a drugged gene and 6,535 (87.5%) do not. Directly drugged genes total 2,134; propagation within families adds 2,566 inferred genes, for 4,700 total druggable genes (23.5%). 15,324 genes (76.5%) remain outside the druggable set at this level (Figure 4 right).
Figure 4.
Statistics of Druggability by PANTHER. Druggability analysis using PANTHER. Percentage of drugged genes and druggable genes as inferred by shared membership in PANTHER classes (top left) or subclasses (top right) with at least one drugged gene. Percentage of classes with drugged genes in PANTHER classes (bottom left) or in subclasses (bottom right), as defined by having at least one drugged gene in the class or subclass. Counts of genes are given in parentheses.
Figure 4.
Statistics of Druggability by PANTHER. Druggability analysis using PANTHER. Percentage of drugged genes and druggable genes as inferred by shared membership in PANTHER classes (top left) or subclasses (top right) with at least one drugged gene. Percentage of classes with drugged genes in PANTHER classes (bottom left) or in subclasses (bottom right), as defined by having at least one drugged gene in the class or subclass. Counts of genes are given in parentheses.

Despite very different coverage and granularity, the results are consistent: large fractions of the genome remain undrugged and cannot be labeled by similarity-based propagation. At the finer subclass level, the estimated druggable portion ranges from 17% (ChEMBL subclass) to 23% (PANTHER subclass). At the broader class level, estimates range from 19% (ChEMBL Class) to 32% (PANTHER PC d3). We summarize the druggability estimates in Box 3, which also includes a simple flowchart to show the number of targets being analyzed within each system and level of granularity.
| Box 3. Summary of Estimates of Druggability from Analysis of ChEMBL and PANTHER | |||||||||
| (a) | |||||||||
| Drugged or inferred druggable | Undrugged | ||||||||
| Classification System | Drugged Genes (10.6% of genome) | Inferred Druggable Genes | Total Druggable Genes | % of Genome | Druggable Classes | Undrugged Genes | % of Genome Undrugged | Undrugged Classes | % Undrugged Classes |
| ChEMBL Class | 2,134 | 1,730 | 3,864 | 19.3% | 14 | 16,160 | 80.7% | 0 | 0.0% |
|
ChEMBL Subclass |
2,134 | 1,268 | 3,402 | 17.0% | 34 | 16,622 | 83.0% | 2 | 5.6% |
| PANTHER Class | 2,134 | 4,204 | 6,338 | 31.7% | 72 | 13,686 | 68.3% | 21 | 22.6% |
|
PANTHER Subclass |
2,134 | 2,566 | 4,700 | 23.5% | 932 | 15,324 | 76.5% | 6,535 | 87.5% |
| (b) | |||||||||
 A summary of the estimates of currently “druggable” targets. Panel (a) gives the raw data from which the results were derived. Panel (b) shows, at high level, the logical flow of the analysis and the high-level results. PANTHER Family counts are restricted to the 7,467 families containing at least one gene in the analyzed protein-coding universe; see Supplemental Methods. A summary of the estimates of currently “druggable” targets. Panel (a) gives the raw data from which the results were derived. Panel (b) shows, at high level, the logical flow of the analysis and the high-level results. PANTHER Family counts are restricted to the 7,467 families containing at least one gene in the analyzed protein-coding universe; see Supplemental Methods. | |||||||||
Naturally, the druggability estimates depend on the details of the classification system. Grouping genes into a small number of heavily populated sets, as ChEMBL does, gives a more optimistic assessment of druggability because success at one target in the set signals plausibility for the entire set but may over-estimate the ease with which the other targets in that set could be drugged. Conversely, much more fine-grained clusters of very similar genes like PANTHER families give much more confidence that a drugged gene implies others in the same family are druggable but do not leverage the similarity that may exist across classes.
Kinases are among the most intensively pursued of all target families, with 100 FDA-approved small-molecule inhibitors to date and more than 130 worldwide.[23] To continue refining our sense of how best to infer druggability, we turned to this family, conducting an independent analysis using the detailed and comprehensive classification framework of Manning et al.[63] The kinase family is far from representative and should be considered a “best case” scenario for druggability. Results are given in Figure 5.
We examined estimates of druggability using broader and finer kinase classifications as we did with ChEMBL and PANTHER. As before, druggability was inferred based on whether each kinase belongs, at the broad level, to one of the 10 top-level kinase classes (called Groups by Manning et al.) in which there is at least one drugged kinase, or, at the finer level, to one of the 129 subclasses (called Families by Manning et al.) that contain at least one drugged kinase.
The analysis of kinases using the Manning classification demonstrates that as we move from broad group-level classifications (503 inferred druggable kinases) to narrower family-level classifications (467 inferred druggable kinases) and finally to directly drugged kinases (418), the count of druggable kinases decreases. Here ‘drugged’ denotes the OpenTargets/ChEMBL small-molecule high-quality ligand (HQL) tractability criterion, defined as having reasonable activity against the target (pChEMBL ≥ 5.5) and drug-like properties (as indicated by a Property Forecast index ≤7 and ≤2 structural alerts). Compounds meeting these criteria may reasonably be considered valid starting points for a discovery effort but in most cases will not be approved drugs or have been taken into human testing. Of the 503 Manning kinases, 86 are engaged by an approved drug, 167 by an approved or clinical-stage compound, and 418 by a high-quality ligand; 99% of the approved or clinical kinases lie within the high-quality ligand set. This nesting is consistent with the literature: Rudolph et al. report 100 FDA-approved kinase inhibitors (more than 130 worldwide) that target “a small subset of the approximately 500 human kinases,” while noting that “many kinases … remain underexplored pharmacologically.” The larger high-quality ligand count therefore does not conflict with these smaller, program-based counts; it additionally captures kinases that are chemically tractable but have not yet been the focus of a dedicated discovery effort.
This pattern underscores the observation that while broader classifications identify a larger pool of potential targets, finer groupings provide higher confidence that the genes are indeed druggable based on shared characteristics with already drugged kinases. This approach, applied here to kinases, can be extended to other gene families in the broader context of drug discovery.
2.4. Curation of the Discovery of Oral Drugs Against “Undruggable” Targets
The results of our gene-based analysis, summarized in Figure 2 through 5, give a high-level estimate of the current size of the “druggable universe.” To better understand the state of the art, we have assembled a list of recently discovered oral drugs targeting proteins that would previously have been considered “undruggable” [Table 1]. These compounds demonstrate either molecular (chemical) novelty, mechanistic novelty, or both. We generally limited the table to compounds discovered within the past two decades. A handful of trailblazing older compounds, such as cyclosporine, were also included for reference.
For each compound in Table 1, we gathered information related to the target, gene family, therapeutic area, status (approved, clinical, preclinical), and the compound’s degree of molecular and/or mechanistic novelty. We again use our broad definition of “drug”: some compounds have achieved regulatory approval, others are not yet approved but have demonstrated clinical benefit, and a handful of compounds have produced the desired pharmacological effect in validated preclinical models. Unfortunately, for many diseases the available cellular and animal models do not recapitulate the human disease with great fidelity. In such cases, preclinical work cannot satisfy our definition of “drug.”
By contrast, excluded from Table 1 are “tool compounds” at earlier stages of discovery. The mere demonstration of target engagement (biochemical or cellular) is not sufficient for inclusion in Table 1. Neither do we include compounds that affect pharmacological function (in cells, tissues, or animals) but contain unsuitable reactive warheads or known toxicophores or lack adequate target selectivity. Such molecules may engender a sense of optimism, but do not provide sufficient evidence for our purposes that the target will soon be druggable.
As cataloged in Table 1, there has been significant progress in the past 20 years in creating oral medicines against a diverse range of challenging targets. The field of drug discovery would greatly benefit from understanding the transferable lessons that may be drawn from each success. A new target may reside in a precedented gene family, that is, a gene family with prior drug discovery successes. Or there may be reason to believe that the new target could be drugged by using some novel drug modality (molecular and/or mechanistic). Such situations may now be considered tractable, albeit likely still quite challenging. The degree of difficulty will relate to many factors, including the number of prior relevant successes, the similarity (described in more detail below) between the new opportunity and those prior successes, and the modality challenges (whether mechanistic or molecular) relevant to this problem. Naturally, one must also consider the omnipresent complicating factors listed in Box 2.
To make this transferability concrete, each entry in Table 1 reports the number of proteins in its target’s PANTHER protein class—the set of functionally related proteins that frequently share structural and ligandability features with the target of the pioneer molecule. We use the PANTHER protein class rather than the broader ChEMBL classes or the finer PANTHER families because it is the only level at which the grouping is both non-trivial and structurally coherent: ChEMBL classes lump hundreds of functionally diverse proteins together and over-generalize, whereas PANTHER families typically return the target alone and offer no inferential reach. The size of this neighborhood varies widely: sotorasib’s covalent engagement of KRAS G12C extends across a 24-member RAS/RHEB GTPase family, venetoclax across nine BCL-2-related proteins, and the IKZF molecular-glue degraders across a zinc-finger class of 252 members, whereas singleton targets such as MEN1 and EED have no close paralogs at this classification level. Across the table, these classes contain a median of seven proteins (range 1-252), a first-order estimate of the related targets that may now be considered accessible.
Table 1 includes examples of drugs that target proteins from gene families traditionally considered “undruggable,” including phosphatases, helicases, various subfamilies of transcription factors, voltage-gated sodium channels, viral capsids, integrases, and many others.
Even after due consideration of the many potential complications cataloged in Box 2, the significant number of entries in Table 1 suggests there are now many additional drug discovery opportunities of high medical importance that should be considered approachable. For each new target, one’s confidence will depend on the number of relevant related successes and the transferability of lessons from the older to the newer system. None will be “simple” and many will remain impossible for some time to come, but they can no longer be reflexively rejected as “undruggable.” This dramatically widens the opportunity space for oral drug discovery.
3. Discussion and Outlook
Using a high-quality-ligand seed set and several curated gene-grouping systems, we estimate that 17–32% of 20,024 analyzed human protein-coding genes have a known drug-like ligand or belong to a group of targets that includes such a ligand-enabled member. These scenario estimates indicate chemical precedent and a reasonable expectation of “druggability” for these targets.
Building on the preceding analysis, one could posit that a protein family with a higher percentage of targets for which a drug exists should be considered “more druggable” than a protein family with a lower percentage of known agents. In families that have previously been drugged, an important consideration is whether molecules of high molecular complexity and/or a novel mechanism of action were required to overcome efficacy or selectivity challenges. While these scenarios remain challenging, they are increasingly solvable. The most significant hurdles arise when a target requires a truly novel mechanism with no established precedent.
Four technical factors have contributed to the rapid growth in the number of oral medicines against “undruggable” targets: novel molecular modalities, improved ability to deliver the required dose, diverse mechanistic modalities, and improved tools to interrogate these targets.
- Advances in molecular modalities: increasing chemical diversity
The broadening of the chemical space that discovery teams are now willing to explore has enabled much of the progress in the field. Targets such as HCV protease,[64] HCV NS5A,[65] the HIV capsid,[66,67] and PCSK9[68,69,70,71] required larger and more complex molecules that conventional wisdom would have rejected a few decades ago. We envision a further expansion of the chemical space that will be explored in pursuit of oral medicines. Peptide science will continue to advance, leading to peptide-drug conjugates, cell-penetrating peptides, and an increasing number of highly complex macrocyclic compounds with oral bioavailability.[72,73,74] “Rule breaker” drugs such as high-molecular-weight compounds that bind to complex sites (on both proteins and nucleic acids) and heterobifunctional molecules that induce proximity will become increasingly common as we master the complex process of optimizing their bioavailability. New functional groups will provide additional ways to fine-tune molecular interactions and properties.
Table 1 contains multiple examples of drugs that modulate protein-protein interactions (PPIs). As mentioned above, tens of thousands of PPIs have been cataloged in humans, while many remain to be discovered.[38] Considerable progress has been made in discovering oral PPI modulators (both inhibitors of biologically critical protein-protein interactions and glues that enable the stabilization of specific protein-protein interactions).[25,75,76,77,78,79] Often natural products offer useful starting points.[79,80] Researchers have noted the tremendous diversity of the size, shape, and properties of protein-protein interfaces, and the consequence that not all PPI modulators will be equally challenging to develop. Broadly, one can presume that smaller interfaces with specific interactions that contribute significant binding affinity will generally be more straightforward. Arkin, Tang, and Wells suggested that the complexity of the displayed binding epitopes at the protein-protein interface also contributes to the degree of challenge.[75] These considerations reflect the structural requirements for engaging such interfaces.
A trailblazing PPI blocker is venetoclax (ABT-199, Venclexta). Venetoclax is a potent, orally bioavailable BH3-mimetic small molecule that selectively targets BCL-2. BCL-2 is a key anti-apoptotic regulator that is frequently overexpressed or dysregulated in hematologic malignancies, most notably chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML). Venetoclax binds with subnanomolar affinity to the canonical BH3-binding groove of BCL-2 and competitively displaces sequestered pro-apoptotic proteins. The BH3-binding groove is an extended, relatively shallow protein surface, making it challenging to generate ligands with sufficient affinity, selectivity, and drug-like properties.[81,82]
Many highly complex cellular machines exist transiently and often involve highly dynamic hub proteins,[83] which are characterized by a greater-than-average number of partner proteins. While hub proteins and their cellular functions are of great biological interest, their large number of partners means that optimization of a drug that selectively disrupts or enhances the interaction with any specific partner protein is likely to be quite challenging.[84] A further complication is that many hub proteins and their binding partners contain intrinsically disordered regions (IDRs). Many IDR-mediated interactions are transient, context-dependent, and difficult to characterize structurally or biophysically. Hub proteins—and dynamic proteins generally—are also highly regulated via a range of mechanisms including masking of their binding surfaces until needed, post-translational modification, and allosteric crosstalk leading to conformational changes. Finally, high structural plasticity in these hub proteins often enables their many diverse binding interactions. Taken together, these factors greatly complicate the design of potent and selective ligands to modulate such protein complexes. Beyond these design challenges, engaging highly connected proteins can also carry an on-target safety liability, since perturbing a hub may disrupt many partner interactions and physiological functions at once. This risk, however, tracks less with physical connectivity per se than with gene essentiality and phenotypic pleiotropy, the more direct predictors of side-effect burden;[85] the operative question is whether a target’s pleiotropy extends along physiological axes unrelated to the disease being treated, which can be gauged from gene essentiality (for example, using DepMap) together with the breadth of phenotypes linked to the target in human genetics (for example, using GWAS catalog or Open Targets).
A number of heterobifunctional compounds such as PROTAC degraders are included in Table 1. These compounds are defined by their molecular architecture, in which two distinct binding elements are incorporated within a single chemical entity. Such proximity enhancers[86] are part of a rising tide in drug discovery that we will discuss later. The use of large peptides as drugs has grown rapidly in the past two decades,[73] and some of the most dramatic examples from Table 1 demonstrate the progress of this field. Peptides can engage extended and complex binding surfaces that historically were accessible only to biologics, such as the GLP-1 and IL-23 receptors. Their defined yet flexible topologies allow them to achieve high affinity and selectivity across interfaces that are often challenging for conventional small molecules. In addition, advances in peptide design, including macrocyclization and backbone modification, have enabled improved stability and, in some cases, oral bioavailability. The natural product cyclosporine, while unusual, is no longer unique: in the past two decades, many additional complex cyclic peptide drugs have been approved, and the field is undergoing a renaissance.[87]
Several of these novel modalities, such as the PPI modulators, peptides, and heterobifunctionals, occupy a similar region of chemical space in the “millamolecular” size range of 600–6,000 daltons,[18] reflecting an expansion beyond traditional small-molecule space toward larger and more structurally complex compounds. As such, the average molecular weight of compounds synthesized by medicinal chemists has steadily risen.[9] While sometimes time-consuming, optimizing the pharmacokinetic properties of such molecules has proven to be less limiting than previously assumed. Indeed, 32% (35 of 109) of the “small-molecule” drugs approved in 2021–2023 have parent molecular weights (excluding salts) in excess of 500 daltons.[88,89,90]
Finally, selectivity challenges often plague drug discovery. The difficulty of avoiding closely related family members has led to some targets being considered “undruggable.” Similar selectivity challenges apply to the creation of mutant-specific cancer drugs (mutant-specific PI3K inhibitors and G12C KRAS inhibitors being prominent current examples). Often selectivity is achieved via an allosteric strategy or by deploying other novel modalities that avoid the high sequence and structural similarity found at the active site.[91]
- 2.
- Advances in delivery
Drug delivery technologies continue to widen the chemical space of oral medicines. A striking recent example is the 31-mer peptide GLP-1 agonist semaglutide, which can be dosed orally because of two creative choices. First, semaglutide contains a long-chain fatty acid attached to a lysine side chain, resulting in greater than 99% albumin binding and an extended half-life.[92] Second, oral semaglutide is co-formulated with SNAC (salcaprozate sodium) as a permeation enhancer. The formulation enables predominantly transcellular absorption in the stomach by locally increasing pH and facilitating membrane interaction. Even with SNAC, bioavailability remains only ~1%, but this low bioavailability nevertheless provides sufficient systemic exposure for clinical utility.
A second recent example is enlicitide (MK-0616), a potent, once-daily, orally bioavailable macrocyclic peptide inhibitor of PCSK9 designed to lower LDL-C. Formulated with sodium caprate to enhance absorption, it demonstrates high efficacy in reducing unbound plasma PCSK9 by >93% and lowering LDL-C by >60%. The oral bioavailability of enlicitide decanoate formulated with permeation enhancers was estimated to be approximately 2% in humans.[93]
We anticipate that other highly complex molecules such as proteins, antibodies, and RNA therapeutics will also be delivered orally, or via non-traditional routes of administration, including inhaled, sublingual, intranasal, and topical.[94,95,96,97,98,99,100,101,102,103,104,105,106,107] Such delivery approaches may be particularly valuable for challenging targets that require “rule breaker” compounds, whose atypical physicochemical properties can preclude administration by conventional routes.
Of note, reducing the required dose greatly simplifies drug delivery.[108] This may be achieved in various ways, such as higher affinity,[109] enhanced efficacy, tissue targeting, or event-driven degraders that can operate in a catalytic manner, enabling substoichiometric, iterative target knockdown.[12]
- 3.
- Advances in mechanistic modalities.
The dramatic increase in novel, diverse mechanistic modalities enables the pursuit of many previously inaccessible opportunities. Small molecules can disrupt the formation of aberrant supramolecular protein assemblies associated with disease.[110] An important example of such dynamic protein-protein interactions occurs between transcriptional activators and coactivators. An encouraging early result was the demonstration that interactions between the coactivator MED25 and three different transcriptional activators could be modulated by allosteric small molecules that engaged a highly mobile region of MED25. It remains to be seen whether this is a generally exploitable mechanism for the design of drugs to target transcription factors.[111]
3.1 Enzyme activators
While many allosteric agonists of GPCRs are known, in general, the discovery and development of enzyme activators has proven to be quite challenging. Fortunately, progress is being made, and several groups have documented the growing number of cases of synthetic enzyme activators and cataloged the mechanisms by which such compounds operate.[112,113,114] A pioneering paper by Zorn and Wells[112] pointed out that proteins that exist in multiple conformational states are more likely to be stabilized in an activated state, suggesting that for many historically “undruggable” targets or multiprotein complexes, allosteric activation may prove possible. They also noted that low-potency compounds may be sufficient to elicit the desired effect.
Despite the difficulties, more than a dozen enzyme activators have progressed to the clinic, and a handful have reached the market. Soluble guanylate cyclase (sGC) stimulators have been shown to bind directly to sGC at a site independent of nitric oxide (NO) but act synergistically with NO. The approved medicines riociguat and vericiguat are used in the treatment of certain cardiovascular indications.[115,116] Preclinical GCase activators, of interest for the treatment of Gaucher disease and potentially other lysosomal storage disorders, have recently been reported. These compounds bind at several different allosteric pockets and enhance protein dimerization.[117] The ultra-rare disease N-acetylglutamate synthase deficiency (NAGSD) results in low levels of N-acetylglutamate (NAG), which can lead to neonatal-onset, life-threatening hyperammonemia. NAG is an allosteric activator of carbamoyl phosphate synthetase 1 (CPS1), which is essential for the urea cycle. Carglumic acid is an approved therapy for NAGSD; it acts as an analog of N-acetylglutamate and activates CPS1.[118,119]
Many kinase activators have been reported. The approved EGFR inhibitors neratinib and erlotinib have recently been found to stabilize the dimeric form of GCN2, increasing its enzymatic activity.[120] A molecule with a very different chemical structure and kinome selectivity profile, HC-7366, has also progressed to clinical trials.[121] Additional clinical-stage and advanced preclinical kinase activators have been reported; the majority of the published efforts involve kinases such as c-ABL,[122] ULK1,[113] PI3K⍺,[123] and TAK1.[124]
There are several preclinical examples of activators that target other traditionally “undruggable” gene families. Activators of the Ser/Thr phosphatase 2A (PP2A) have been discovered that reduce cell proliferation. These compounds, which promote the assembly of specific heterotrimeric forms of PP2A, include approved antipsychotic medicines from the phenothiazine class, such as perphenazine. While exciting, this finding should not be generalized to the entire Ser/Thr phosphatase family. PP2A exists as a collection of trimeric holoenzymes that carry out diverse biological functions. The design of molecules to inhibit or stabilize any particular PP2A holoenzyme will remain challenging for some time.[125] AAA (ATPases associated with diverse cellular activities) proteins are essential for a wide range of cellular functions; they deploy ATP hydrolysis to drive conformational change in substrate macromolecules. Recently, the Kapoor lab reported the discovery of an activator of valosin-containing protein (VCP/p97) and, crucially, solved cryo-EM structures identifying the druggable allosteric site to which the compound binds. This led to the suggestion of a plausible mechanism by which this site regulates VCP/p97 function.[126] Caseinolytic protease P (ClpP) is a mitochondrial serine protease that, together with its ATP-dependent unfoldase partner ClpX, forms the ClpXP complex responsible for maintaining protein homeostasis. ADEP-28[127] and TR-107[128] are sub-µM binders, activate ClpP in a dose-dependent manner, potently kill cancer cells, and can be modeled to fit into the same hydrophobic pocket of the enzyme. Lysophospholipase-like-1 (LYPLAL1) is a serine hydrolase of possible importance for the development of drugs for metabolic disorders. Activators of LYPLAL1 were identified using a fluorescence polarization assay. The best activators showed sub-µM EC50 values, were reversible, and demonstrated enhanced glucose tolerance and insulin sensitivity in obese mice.[129] Multiple groups have reported activators of NAMPT and demonstrated efficacy in a variety of preclinical models.[130] Potent activators of the mitochondrial zinc metalloprotease neurolysin, implicated in stroke, have been reported; the compounds are described as having excellent drug-like properties, selectivity over related peptidases, and high brain concentrations, making them suitable tools for validating the utility of this mechanism in stroke and perhaps several neurodegenerative diseases.[131] 8-Oxoguanine DNA glycosylase 1 (OGG1) recognizes oxidative DNA damage. A small-molecule activator, TH10785, has been described that enables OGG1 to carry out a novel catalytic lyase reaction, leading to increased repair of DNA damage.[132] Taken together, these early findings suggest areas of future progress and reasons for optimism.
3.2 Proximity enhancers
Molecular glues,[133] PROTACs,[12] related degraders,[134,135,136] and a wide range of heterobifunctional modalities[86] are becoming mainstream. These classes offer a range of potential advantages: greater efficacy via event-driven target turnover; chemically induced proximity-based activation or deactivation; the ability to phosphorylate[136] or dephosphorylate[135] proteins on demand; the opportunity to use molecules with somewhat reduced affinity for one or both binding partners (so long as an optimized ternary complex can be formed); and the opportunity for these molecules to bind to a range of sites on the target proteins, some of which may be more suitable for achieving the requisite binding affinity or selectivity. For example, it has been shown that inducers of proximity, such as PROTACs, can sometimes achieve selectivity—even among kinases that have identical ATP sites—due to differences in surface residues that impact ternary complex formation (and differences in ubiquitination sites in the case of degraders).[137] Also of note, although optimizing the drug-like properties of large heterobifunctional molecules presents considerable challenges beyond those encountered with classical small molecules, an emerging body of literature has begun to delineate the unique physicochemical and biological mechanisms governing their cellular uptake and oral absorption. Deeper mechanistic understanding of these processes is expected to substantially accelerate the rational design of future heterobifunctional therapeutics.[138]
Many challenging targets may be especially appropriate for a heterobifunctional approach. Examples include scaffolding proteins, subunits of larger functional complexes, or non-enzymatic proteins such as transcription factors or RNA-binding proteins. Schneider et al.[139] describe a systematic framework for deciding whether any proposed target is “PROTAC-able” based on the availability of information on that target’s ubiquitination, half-life, known binders, and cellular location. More recently, efforts have been directed at lysosomal induction of extracellular protein degradation.[140,141,142] Lysosomal degradation of extracellular and membrane proteins can expand the range of protein degradation strategies to treat debilitating human diseases.
Glues are particularly promising for scaffolding proteins, subunits of large complexes, or transcription factors, which often lack the well-defined pockets upon which PROTACs depend.[143] By stabilizing otherwise weak interactions, glues can reduce the stringent requirement for high-affinity binary ligands and extend the scope of degradable proteins.
Chemical inducers of proximity may be divided into two categories: heterobifunctional proximity inducers (TACs, including PROTACs, LYTACs, RNATACs, RIPTACs) and molecular glues. Not all glues are degraders—for example, cyclosporine inhibits calcineurin rather than degrading it, and the orthosteric molecular glue CSN5i-3 inhibits the COP9 signalosome by stabilizing the interaction between its catalytic subunit and the enzyme’s own NEDD8 substrate, achieving potent, substrate-dependent inhibition without high affinity for the free enzyme.[144]
The plant hormone auxin (indole-3-acetic acid; 175 daltons) holds a foundational place in the history of molecular glue research. In a landmark 2007 Nature study, Tan et al. solved the crystal structure of the Arabidopsis TIR1–ASK1 complex alone and in ternary complexes with three distinct auxin ligands and an Aux/IAA degron peptide. This revealed that auxin does not act as a classical allosteric regulator; rather, it demonstrated that this remarkably small molecule acts as a molecular glue. Auxin binds in a preformed hydrophobic cavity at the TIR1-Aux/IAA interface and simultaneously contacts both proteins, enhancing the affinity of an E3 ubiquitin ligase for its Aux/IAA transcriptional repressors. The notion that a molecule smaller than aspirin could stabilize a protein–protein interaction and drive substrate ubiquitination and proteasomal degradation was conceptually striking and established the molecular glue paradigm that now underpins modern targeted protein degradation strategies.[145]
Proximity-enhancing drugs take many forms and can have profound effects on biomolecular localization and function.[86] Drugs that stabilize or disrupt the interactions between RNA and RNA-binding proteins may affect diverse functions including transcription, translation, splicing, processing, or degradation.[146] A new class of proximity enhancers can recruit diverse cancer drivers to any number of downstream promoters of cell death to activate the cell death pathway, rewiring the cancer driver circuitry and selectively killing those cancer cells.[147]
Glues as well as heterobifunctional molecules may affect diverse functions such as signaling, target lifetime, condensate properties, gene expression, or localization.
TAC degraders and glue degraders are different in design and mechanism. A limitation of TACs in general is that they require the availability of a ligand that binds with reasonable affinity and selectivity to the protein to be degraded, an obstacle for proteins lacking well-defined binding sites. As such, TACs do not solve the problem of ligandability. Furthermore, many TAC-based ternary complexes exhibit relatively low cooperativity, sometimes described as being “force-fitted.”[148] Another challenge is the presence of the linker connecting the two halves of the molecule, raising molecular weight and conformational lability, both factors that complicate the optimization of drug properties. Another concern is that by degrading a protein, all its functions (including any that may be beneficial) are neutralized rather than just the specific function of the protein that is relevant to the disease being treated. More positively, the ability of a TAC to engage any suitable pocket on the protein being degraded expands the set of proteins that can be drugged by these modalities. For example, RIPTACs form a tight complex between a target protein that is enriched in a tumor cell and a second protein that is essential for the cancer cell’s survival. This complex silences the essential protein, leading to the death of the tumor cell. The first RIPTAC has reportedly entered clinical trials.[149]
Glues, on the other hand, often degrade poorly ligandable targets for which no binary ligands exist; the corresponding ternary complexes are often characterized by a high level of cooperativity, meaning that the recruitment of the effector protein (e.g., cereblon or DCAF15) plays a major role in engaging the target protein and forming the requisite ternary complex. The finding of a ternary complex between RBM39, DCAF15, and the drug indisulam highlights the benefits that glues provide.[143] Glues can stabilize or induce protein-protein interactions without requiring two high-affinity binding moieties, effectively delivering a dual advantage—addressing ligandability and providing novel pharmacology in a single molecule. A glue, in effect, remodels the surface of a protein. This change may induce neomorphic protein-protein interactions, thereby changing the complement of intracellular interactions available to those proteins, or it may increase the affinity or residence time of two proteins that are natural partners. It may also expose a potential degron or alter a post-translational modification.[150] This benefit is exemplified by lenalidomide and pomalidomide in the degradation of IKZF1/3. However, discovering new glues can be highly challenging, as it typically depends on serendipitous screening or advanced chemical biology approaches. A promising avenue is to leverage existing basal affinity between proteins and effectors.[148]
The reach of this modality can be gauged by a less stringent criterion than the validated-ligand criterion used for the genome-wide estimate presented earlier. Rather than requiring an actual compound, one can ask how many proteins carry the recognition features that would make them recruitable to an E3 ligase. Analysis of canonical G-loop degrons identifies more than 1,600 candidate proteins as potentially glueable, and thus potentially degradable, through cereblon,[151] with related work expanding the recognized degron architecture;[152,153] cross-linking proteomics in living cells is beginning to test which of these are genuinely recruited.[154] Such counts identify candidate targets rather than targets with validated compounds and suggest how far the glue-addressable frontier may ultimately extend.
An important feature of mechanistic novelty is the potential for domain-level specificity. Many complex proteins have multiple domains and/or multiple binding partners. To achieve the desired pharmacological response, there may be advantages to engaging certain domains or blocking or enhancing certain protein-protein interactions. This also highlights the alloform/proteoform considerations mentioned earlier.[32,33]
Improved understanding of the obligate intracellular partners of challenging targets enables the adoption of a wider range of intervention strategies.[155,156] For example, transcription factors (TFs) may be targeted in a variety of ways: by inhibiting the interactions between TF and cofactors; by disrupting TF-DNA interactions; by altering levels of ubiquitination, e.g., by inducing non-native protein-protein interactions between the TF and an E3 ligase; by inhibiting targets that regulate TF expression, such as kinases or epigenetic proteins; or by targeting intrinsically disordered regions in the TFs or partner proteins. Perhaps more than any other gene family, TFs will require researchers to employ a broad range of strategies to achieve the desired pharmacological effects. The early TF examples, while largely still preclinical, are encouraging.
- 4.
- Advances in interrogation tools.
Practical experimental difficulties often complicate the exploration of “undruggable” targets. First, it may not be possible to establish an appropriate biochemical or cellular assay—that is, not all targets will be “screenable.”[157] As an example of “screenability” challenges, the history of tau assay development is instructive. Tau research has been hampered by the difficulty of generating in vitro fibrils and aggregation models that faithfully reproduce the polymorphic, cofactor-dependent, post-translationally modified folds observed in human tauopathies. This has led to extensive screening against heparin-induced or otherwise non-disease-relevant aggregates and a large body of misleading preclinical SAR. This mismatch between assay substrates and authentic brain-derived tau strains undermines target engagement claims, confounds mechanistic interpretation, and likely contributes to the repeated clinical failure of tau-directed therapeutics developed using these systems.[158] Second, even a well-run assay using a large and diverse chemical library may fail to yield useful hits—that is, not all targets will be “ligandable” in practice. The industry is replete with stories of high-throughput screening campaigns where testing millions of compounds yielded no useful chemical matter. Third, for the subset of targets that are ligandable, it will not always be possible to optimize the hits into drugs. Fortunately, to address these challenges, an expanding range of powerful biochemical, biophysical, and cellular screening technologies enables more rational exploration of novel drug modalities.[159]
The techniques of chemical biology continue to mature and grow in predictive power, and chemoproteomics can now be applied to identify chemical starting points against a substantially expanded fraction of the proteome.[160,161,162,163] Enantioprobe libraries, composed of highly functionalized enantiomeric fragments, may be used to discover “ligandable” targets in cell-based assays.[164] Applied across the proteome in living cells, such fragment-based approaches have mapped reversible small-molecule binding to more than 2,000 proteins—83% of them without any previously known ligand—spanning proteins with highly diverse functions including scaffolds, adaptors, and transcriptional regulators.[165] These assays yield compounds that bind enantioselectively to a desired target and may elicit a desired phenotype, providing a credible starting point for further chemical exploration. Another approach is disulfide tethering, which was used to discover mutant-selective covalent inhibitors of KRAS G12C. These compounds bind a previously unrecognized switch-II allosteric pocket, thereby shifting the nucleotide preference toward GDP and disrupting effector interactions, providing the first structure-based validation that oncogenic KRAS can be directly and specifically drugged.[166] Another notable application of disulfide tethering was the identification of selective stabilizers (glues) of interactions between the hub protein 14-3-3 and a range of client proteins.[161]
As demonstrated by the current-generation chemical biology tools described above, an important strategy in the pursuit of “undruggable” targets is phenotypic screening. Pioneering examples leading directly to the discovery of breakthrough medicines include the myosin-blocking drug mavacamten for the treatment of hypertrophic cardiomyopathy,[167] the cystic fibrosis (CF) potentiator ivacaftor,[168] the CF corrector lumacaftor,[169] the hepatitis C NS5A replication-complex inhibitor daclatasvir,[170] and the SMN2 splicing modifier risdiplam for the treatment of spinal muscular atrophy.[171] The mechanism of action for compounds discovered in phenotypic programs will, in most cases, initially be unknown or only poorly understood; medicinal chemistry decisions at this early stage must be informed by the cellular phenotype. Mechanistic understanding may emerge later, which will then guide future efforts on related targets.
The importance of phenotypic screening for MOA discovery is clearly exemplified in the discovery and optimization of thalidomide analogs (IMiDs). Robust, unbiased cellular assays drove the identification of immunomodulatory activity long before the underlying molecular target was known. Subsequent mechanism-of-action studies established that IMiDs bind cereblon within the CRL4-CRBN E3 ligase complex and reprogram its substrate specificity to promote neosubstrate degradation, thereby expanding the concept of the druggable proteome to include ligase-modulated targets and laying the groundwork for contemporary targeted protein degradation.[172]
Although phenotypic programs have historically been driven solely by cellular or animal data, the rapidly maturing field of chemical biology will enable target deconvolution in an increasing proportion of cases.[173] Protein structure prediction algorithms, supplemented by advanced biophysical and structural biology techniques, will then be deployed to reveal the precise details of molecular mechanism.[174,175,176,177]
To achieve the desired pharmacological effects or an adequate safety window for complex targets, it will often be necessary to understand and exploit subtle relationships between the target’s structure and function. Examples include selective and state-dependent inhibitors of ion channels such as the voltage-gated sodium channels[178,179] and GPCR agonists that confer biased signaling.[180] Developing such medicines requires a detailed understanding of the precise structural determinants of pharmacological function, which will generally be technically more challenging to measure.
In addition to these four technical factors, there has been a critical shift in mindset toward a more adventurous and open-minded approach to discovery,[181,182] with each demonstrable success on an “undruggable” target emboldening teams to pursue other similarly difficult opportunities. This shift in mindset is exhibited in diverse ways. First, novel targets of all kinds are no longer being reflexively rejected. Second, novel modalities are being aggressively pursued. Third, covalent drugs, formerly verboten, are now embraced.
Fourth and perhaps most importantly, “rule breaker” compounds have become commonplace.[10] While “rule breakers” can be defined simply as any molecule that does not conform with the Rule of Five, there are as yet no clear, established principles by which one can confidently predict which “rule breakers” are likely to have desirable pharmacokinetic and physical properties. There is a steadily growing awareness that it is naïve to believe that “drug-like” molecules must occupy narrow bands of property space. The Medicinal Chemistry Gordon Research Conference, always a harbinger of the evolving practice of the field, featured a session on this topic as early as 2010.[183] Scientists at Bristol-Myers Squibb have referred to cyclic and acyclic compounds with a molecular weight between 600-6000 daltons as “millamolecules” and were among the first to argue that compounds of this size, especially when macrocyclic, are ideal for pursuing “undruggable” targets.[17,18,184] Doak et al., in 2014, analyzed the most recent data available at that time to produce an expanded definition of drug-like space, noting there are no hard-and-fast restrictions on molecular weight or other properties. Further, the use of intramolecular hydrogen bonding, macrocyclization, and alternative formulations are strategies for expanding the chemical space accessible to oral drugs.[10] Wills and Lipkus have documented the acceleration of the discovery of “pioneering” drugs that occupy novel chemical space.[185]
4. Concluding Remarks
Oral medicines remain highly desirable because of their ability to reach different tissues and targets, reduced patient burden, lower cost of administration, and reduced manufacturing and supply chain complexity. It is therefore encouraging to see the many recent successes against challenging targets (Table 1). Some problems are now solvable with approaches that have become mainstream; others require more disruptive approaches.
Increasing knowledge and advancing technologies are somewhat offset by the fact that, as established target classes are increasingly exploited, many if not most of the remaining medically important targets present greater structural, mechanistic, and delivery challenges. In short, the targets we wish to pursue today are becoming steadily more challenging. Many such targets function in highly complex intracellular environments, making it unlikely that a full understanding of the structure and function of such systems is possible outside their native cellular environment. Further, targets may be transiently expressed, operate within cellular condensates, or contain disordered regions. Selective binding of small molecules to specific nucleic acids is rare. Novel modalities, while promising, bring their own challenges. Finally, recall that we have no proven strategies for the majority of the genome. For all these reasons, it would be unwise to extrapolate from the progress made thus far to the conclusion that all targets are now “druggable.”
We anticipate that these “even harder problems” will gradually become more approachable as our toolkit of technologies continues to mature. As recently as three decades ago, target families such as the kinases were once considered “undruggable” because of concerns that, given the similarities of the ATP binding site, it would be impossible to achieve selectivity. Similarly, each modality (Figure 1) will gradually move down and to the left as we gain experience. For example, the application of a range of chemical biology techniques may enable the discovery of molecular glues that engage complex intracellular oligomeric macromolecules to achieve a desired phenotypic response. Crucially, success depends on appropriate assays and subsequent experiments to identify suitable chemical matter.
The two-by-two matrix in Figure 1, therefore, is intended to provide broad guidance on the current degree of challenge for each modality. It cannot account for the specific requirements of each program; some seemingly straightforward problems will long remain unexpectedly difficult, whether because of dosing, efficacy, safety, or other challenges.
Therefore, by avoiding the term “undruggable,” we are not suggesting that all possible targets are now straightforward to drug. Our analysis of the ChEMBL and PANTHER databases, using the high-quality ligand seed set and curated class-wise propagation rules, suggests that up to one-third of the analyzed protein-coding genes have now been drugged or are likely to be druggable. Recall that this is a generous estimate, using our broad definition of “drug,” which includes preclinical compounds. Even with the substantial technological advances and encouraging successes across a broad target space, it would be misleading to say “nothing is undruggable” as if to imply that today all projects are readily solvable. Rather, our mindset has changed, and we understand that even extremely difficult targets are now far more approachable with new thinking and technologies. When pursuing such targets, it is certainly prudent to assume higher risk, longer timelines, the need to consider alternative modalities, and the need for new interrogation tools.
Other researchers strike a similar blend of optimism and realism, referring to hard targets as “yet to be drugged” rather than “undruggable.” Such perspectives acknowledge the many daunting challenges that remain,[186] while celebrating the ingenuity that continually advances the field.[187,188]
In summary, we are witnessing the discovery of remarkable oral medicines against previously inaccessible targets. This stems from four scientific advances: more aggressive exploration of chemical space; an improved ability to deliver pharmacologically relevant doses of these medicines, sometimes through novel delivery technologies; a vastly expanded range of mechanistic modalities; and dramatically improved biochemical and cellular screening tools to increase the chances that we can identify and characterize pharmacologically relevant ligands (Figure 6). Crucially, underlying these technical advances is a more optimistic mindset that supports the pursuit of hard targets of high medical interest. Given the complexities of such groundbreaking work, it is noteworthy that more than half the entries in Table 1 were discovered by relatively small biotechnology companies, often defined as those companies with annual R&D spending below two hundred million dollars.
We are also struck by the convergence between “large” and “small” drugs and between “oral” and “non-oral” medicines. Small molecules are getting bigger; large biologics are getting smaller. It is increasingly possible to deliver peptides and other “millamolecules” orally, or through convenient weekly subcutaneous injections, while evolving delivery technologies hold great promise for further advances.
For all these reasons, while indeed many extremely challenging targets remain, the term “undruggable” is already quite often misleading, and we anticipate that it will increasingly become so in the coming decade. We conclude that “undruggable” no longer serves a useful purpose. Rather than calling a target “undruggable,” investigators should identify the current bottleneck explicitly: not screenable, lacking a validated ligand, lacking a bioavailable ligand, or lacking a demonstrated therapeutic window.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
References
- Moo-Young, Murray. Comprehensive Biotechnology. (Pergamon, 2019).
- Jacoby, E. et al. Key Aspects of the Novartis Compound Collection Enhancement Project for the Compilation of a Comprehensive Chemogenomics Drug Discovery Screening Collection. Curr. Top. Med. Chem. 5, 397–411 (2005).
- Mayer, J. P. & DiMarchi, R. D. Drugging the Undruggable. Chem. Biol. 12, 860–861 (2005).
- Dhawan, N. The future of pharma and health care will come in small (molecule) packages. STAT NEWS (2022).
- Cohen, J. Inflation Reduction Act Favors Biologics Over Small Molecules: In The Long Term, This Could Partly Undermine Bill’s Effort To Contain Costs. Forbes (2023).
- Seoane-Vazquez, E., Rodriguez-Monguio, R. & Powers, J. H. Analysis of US Food and Drug Administration new drug and biologic approvals, regulatory pathways, and review times, 1980–2022. Sci. Rep. 14, 3325 (2024).
- Stegemann, S. et al. Trends in oral small-molecule drug discovery and product development based on product launches before and after the Rule of Five. Drug Discov. Today 28, 103344 (2023).
- Leeson, P. D. Molecular inflation, attrition and the rule of five. Adv. Drug Deliv. Rev. 101, 22–33 (2016).
- Walters, W. P., Green, J., Weiss, J. R. & Murcko, M. A. What Do Medicinal Chemists Actually Make? A 50-Year Retrospective. J. Med. Chem. 54, 6405–6416 (2011).
- Doak, B. C., Over, B., Giordanetto, F. & Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 21, 1115–1142 (2014).
- DeGoey, D. A., Chen, H.-J., Cox, P. B. & Wendt, M. D. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem. 61, 2636–2651 (2018).
- Békés, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
- Tan, L., Yan, W., McCorvy, J. D. & Cheng, J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure–Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 61, 9841–9878 (2018).
- Smith, J. S., Lefkowitz, R. J. & Rajagopal, S. Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 17, 243–260 (2018).
- Wisler, J. W., Rockman, H. A. & Lefkowitz, R. J. Biased G Protein–Coupled Receptor Signaling. Circulation 137, 2315–2317 (2018).
- Mitrea, D. M., Mittasch, M., Gomes, B. F., Klein, I. A. & Murcko, M. A. Modulating biomolecular condensates: a novel approach to drug discovery. Nat. Rev. Drug Discov. 21, 841–862 (2022).
- Bristol-Myers Squibb Annual Report (2010).
- Bristol-Myers Squibb Annual Report (2013).
- Bristol-Myers Squibb Annual Report (2024).
- Xiao, W. et al. Advance in peptide-based drug development: delivery platforms, therapeutics and vaccines. Signal Transduct. Target. Ther. 10, 74 (2025).
- Stein Gold, L. et al. Oral Peptide Therapeutics as an Emerging Treatment Modality in Immune-Mediated Inflammatory Diseases: A Narrative Review. Adv. Ther. 42, 3158–3172 (2025).
- Han, B., Salituro, F. G. & Blanco, M.-J. Impact of Allosteric Modulation in Drug Discovery: Innovation in Emerging Chemical Modalities. ACS Med. Chem. Lett. 11, 1810–1819 (2020).
- Rudolph, J., Hoeflich, K. P. & Dar, A. C. Contemporary design of small-molecule kinase modulators: orthosteric, allosteric and induced-proximity strategies. Nat. Rev. Drug Discov. Online ahead of print – April 2026 - 10.1038/s41573-026-01411-9.
- Ma, F. et al. Allosteric inhibitors for the treatment of cancers (2021-): A review from medicinal chemistry perspectives. J. Mol. Struct. 1344, 142985 (2025).
- Nada, H. et al. New insights into protein–protein interaction modulators in drug discovery and therapeutic advance. Signal Transduct. Target. Ther. 9, 341 (2024).
- Fan, G. et al. Proteolysis-Targeting Chimera (PROTAC): Current Applications and Future Directions. MedComm 6, no. 10 (2025): e70401. [CrossRef]
- Dewey, J. A. et al. Molecular Glue Discovery: Current and Future Approaches. J. Med. Chem. 66, 9278–9296 (2023).
- Csorba, N., Ábrányi-Balogh, P. & Keserű, G. M. Covalent fragment approaches targeting non-cysteine residues. Trends Pharmacol. Sci. 44, 802–816 (2023).
- Kim, G., Grams, R. J. & Hsu, K.-L. Advancing Covalent Ligand and Drug Discovery beyond Cysteine. Chem. Rev. 125, 6653–6684 (2025).
- Kubinyi, H. Drug research: myths, hype and reality. Nat. Rev. Drug Discov. 2, 665–668 (2003).
- Hopkins, A. L. & Groom, C. R. The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 (2002).
- Yang, X. et al. Widespread Expansion of Protein Interaction Capabilities by Alternative Splicing. Cell 164, 805–817 (2016).
- Kjer-Hansen, P., Phan, T. G. & Weatheritt, R. J. Protein isoform-centric therapeutics: expanding targets and increasing specificity. Nat. Rev. Drug Discov. 23, 759–779 (2024).
- Leo, I. R. et al. Functional proteoform group deconvolution reveals a broader spectrum of ibrutinib off-targets. Nat. Commun. 16, 1948 (2025).
- Suraritdechachai, S., Lakkanasirorat, B. & Uttamapinant, C. Molecular probes for cellular imaging of post-translational proteoforms. RSC Chem. Biol. 3, 201–219 (2022).
- Korchak, J. A., Stephen Yi, S., Kelleher, N. L., Sahni, N. & Sheynkman, G. M. Proteoform medicine: characterizing and targeting protein forms in human disease. Nat. Rev. Genet. 27, 271–291 (2026).
- Vu, B. T. et al. Discovery of Rezatapopt (PC14586), a First-in-Class, Small-Molecule Reactivator of p53 Y220C Mutant in Development. ACS Med. Chem. Lett. 16, 34–39 (2025).
- Luck, K. et al. A reference map of the human binary protein interactome. Nature 580, 402–408 (2020).
- Michaelis, A. C. et al. The social and structural architecture of the yeast protein interactome. Nature 624, 192–200 (2023).
- Iyer, M. K. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208 (2015).
- Arun, G., Diermeier, S. D. & Spector, D. L. Therapeutic Targeting of Long Non-Coding RNAs in Cancer. Trends Mol. Med. 24, 257–277 (2018).
- Crooke, S. T., Baker, B. F., Crooke, R. M. & Liang, X. Antisense technology: an overview and prospectus. Nat. Rev. Drug Discov. 20, 427–453 (2021).
- Setten, R. L., Rossi, J. J. & Han, S. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 18, 421–446 (2019).
- Rocha, J. J. et al. Functional unknomics: Systematic screening of conserved genes of unknown function. PLoS Biol. 21, e3002222 (2023).
- Deutsch, E. W. et al. Expanding the human proteome with microproteins and peptideins. Nature 654, 813–825 (2026).
- Wright, B. W., Yi, Z., Weissman, J. S. & Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends Cell Biol. 32, 243–258 (2022).
- Cassidy, L., Kaulich, P. T. & Tholey, A. Proteoforms expand the world of microproteins and short open reading frame-encoded peptides. iScience 26, 106069 (2023).
- Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017).
- Brown, K. K. et al. Approaches to target tractability assessment – a practical perspective. Medchemcomm 9, 606–613 (2018).
- Zdrazil, B., Richter, L., Brown, N. & Guha, R. Moving targets in drug discovery. Sci. Rep. 10, 20213 (2020).
- Carvalho-Silva, D. et al. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 47, D1056–D1065 (2019).
- Klabunde, T. Chemogenomic approaches to drug discovery: similar receptors bind similar ligands. Br. J. Pharmacol. 152, 5–7 (2007).
- Anderson, B. et al. How many kinases are druggable? A review of our current understanding. Biochemical Journal 480, 1331–1363 (2023).
- Lambert, S. A. et al. The Human Transcription Factors. Cell 172, 650–665 (2018).
- Caron, P. R. et al. Chemogenomic approaches to drug discovery. Curr. Opin. Chem. Biol. 5, 464–470 (2001).
- Sheils, T. et al. How to Illuminate the Druggable Genome Using Pharos. Curr. Protoc. Bioinformatics 69, (2020).
- Serafim, R. A. M., Elkins, J. M., Zuercher, W. J., Laufer, S. A. & Gehringer, M. Chemical Probes for Understudied Kinases: Challenges and Opportunities. J. Med. Chem. 65, 1132–1170 (2022).
- Zhang, D.-W. et al. Binding of Proprotein Convertase Subtilisin/Kexin Type 9 to Epidermal Growth Factor-like Repeat A of Low Density Lipoprotein Receptor Decreases Receptor Recycling and Increases Degradation. Journal of Biological Chemistry 282, 18602–18612 (2007).
- McNutt, M. C., Lagace, T. A. & Horton, J. D. Catalytic Activity Is Not Required for Secreted PCSK9 to Reduce Low Density Lipoprotein Receptors in HepG2 Cells. Journal of Biological Chemistry 282, 20799–20803 (2007).
- An, J., Totrov, M. & Abagyan, R. Pocketome via Comprehensive Identification and Classification of Ligand Binding Envelopes. Molecular & Cellular Proteomics 4, 752–761 (2005).
- Bhagavat, R., Sankar, S., Srinivasan, N. & Chandra, N. An Augmented Pocketome: Detection and Analysis of Small-Molecule Binding Pockets in Proteins of Known 3D Structure. Structure 26, 499-512.e2 (2018).
- Zillmer, H. & Walther, D. Towards a comprehensive view of the pocketome universe—biological implications and algorithmic challenges. PLoS Comput. Biol. 21, e1013298 (2025).
- Manning, G., Whyte, D. B., Martinez, R., Hunter, T. & Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science (1979). 298, 1912–1934 (2002).
- Chatel-Chaix, L., Baril, M. & Lamarre, D. Hepatitis C Virus NS3/4A Protease Inhibitors: A Light at the End of the Tunnel. Viruses 2, 1752–1765 (2010).
- Wagner, R.et al. Highlights of the Structure–Activity Relationships of Benzimidazole Linked Pyrrolidines Leading to the Discovery of the Hepatitis C Virus NS5A Inhibitor Pibrentasvir (ABT-530). J. Med. Chem.61, 4052–4066 (2018).
- Campbell, E. M. & Hope, T. J. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 13, 471–483 (2015).
- Link, J. O. et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 584, 614–618 (2020).
- Tucker, T. J. et al. A Series of Novel, Highly Potent, and Orally Bioavailable Next-Generation Tricyclic Peptide PCSK9 Inhibitors. J. Med. Chem. 64, 16770–16800 (2021).
- Evison, B. J. et al. A small molecule inhibitor of PCSK9 that antagonizes LDL receptor binding via interaction with a cryptic PCSK9 binding groove. Bioorg. Med. Chem. 28, 115344 (2020).
- Petersen, D. N. et al. A Small-Molecule Anti-secretagogue of PCSK9 Targets the 80S Ribosome to Inhibit PCSK9 Protein Translation. Cell Chem. Biol. 23, 1362–1371 (2016).
- Alleyne, C. et al. Series of Novel and Highly Potent Cyclic Peptide PCSK9 Inhibitors Derived from an mRNA Display Screen and Optimized via Structure-Based Design. J. Med. Chem. 63, 13796–13824 (2020).
- Nicze, M. et al. The Current and Promising Oral Delivery Methods for Protein- and Peptide-Based Drugs. Int. J. Mol. Sci. 25, 815 (2024).
- Ji, X., Nielsen, A. L. & Heinis, C. Cyclic Peptides for Drug Development. Angew. Chem. Int. Ed. 63, (2024).
- Garcia Jimenez, D., Poongavanam, V. & Kihlberg, J. Macrocycles in Drug Discovery─Learning from the Past for the Future. J. Med. Chem. 66, 5377–5396 (2023).
- Arkin, M. R., Tang, Y. & Wells, J. A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 21, 1102–1114 (2014).
- Gestwicki, J. E. & Shao, H. Inhibitors and chemical probes for molecular chaperone networks. Journal of Biological Chemistry 294, 2151–2161 (2019).
- Ran, X. & Gestwicki, J. E. Inhibitors of protein–protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 44, 75–86 (2018).
- Linhares, B. M., Grembecka, J. & Cierpicki, T. Targeting Epigenetic protein–protein Interactions With small-molecule Inhibitors. Future Med. Chem. 12, 1305–1326 (2020).
- Begnini, F. et al. Mining Natural Products for Macrocycles to Drug Difficult Targets. J. Med. Chem. 64, 1054–1072 (2021).
- Stone, S., Newman, D. J., Colletti, S. L. & Tan, D. S. Cheminformatic analysis of natural product-based drugs and chemical probes. Nat. Prod. Rep. 39, 20–32 (2022).
- Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 (2013).
- Lasica, M. & Anderson, M. A. Review of Venetoclax in CLL, AML and Multiple Myeloma. J. Pers. Med. 11, 463 (2021).
- Hu, G., Wu, Z., Uversky, V. & Kurgan, L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int. J. Mol. Sci. 18, 2761 (2017).
- Garlick, J. M. & Mapp, A. K. Selective Modulation of Dynamic Protein Complexes. Cell Chem. Biol. 27, 986–997 (2020).
- Nguyen, P. A., Born, D. A., Deaton, A. M., Nioi, P. & Ward, L. D. Phenotypes associated with genes encoding drug targets are predictive of clinical trial side effects. Nat. Commun. 10, 1579 (2019).
- Deshaies, R. J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 580, 329–338 (2020).
- Zhang, H. & Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 3, 18–31 (2022).
- Rudolph, J. 2022 Medicinal Chemistry Reviews. vol. 57 (MEDI, Inc. Published by American Chemical Society., Washington, DC, 2022).
- Rudolph, J. 2023 Medicinal Chemistry Reviews. vol. 58 (MEDI, Inc. Published by American Chemical Society., Washington, DC, 2023).
- Rudolph, J. Medicinal Chemistry Reviews. vol. 59 (American Chemical Society, Washington, DC, 2024).
- Huggins, D. J., Sherman, W. & Tidor, B. Rational Approaches to Improving Selectivity in Drug Design. J. Med. Chem. 55, 1424–1444 (2012).
- Overgaard, R. V., Navarria, A., Ingwersen, S. H., Bækdal, T. A. & Kildemoes, R. J. Clinical Pharmacokinetics of Oral Semaglutide: Analyses of Data from Clinical Pharmacology Trials. Clin. Pharmacokinet. 60, 1335–1348 (2021).
- Johns, D. G. et al. Orally Bioavailable Macrocyclic Peptide That Inhibits Binding of PCSK9 to the Low Density Lipoprotein Receptor. Circulation 148, 144–158 (2023).
- Mitchell, M. J. et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20, 101–124 (2021).
- Vargason, A. M., Anselmo, A. C. & Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 5, 951–967 (2021).
- Caffarel-Salvador, E., Abramson, A., Langer, R. & Traverso, G. Oral delivery of biologics using drug-device combinations. Curr. Opin. Pharmacol. 36, 8–13 (2017).
- Abramson, A. et al. An ingestible self-orienting system for oral delivery of macromolecules. Science (1979). 363, 611–615 (2019).
- Abramson, A. et al. Oral delivery of systemic monoclonal antibodies, peptides and small molecules using gastric auto-injectors. Nat. Biotechnol. 40, 103–109 (2022).
- Bellinger, A. M. et al. Oral, ultra–long-lasting drug delivery: Application toward malaria elimination goals. Sci. Transl. Med. 8, (2016).
- Kreitz, J. et al. Programmable protein delivery with a bacterial contractile injection system. Nature 616, 357–364 (2023).
- Han, Y. et al. Biomimetic injectable hydrogel microspheres with enhanced lubrication and controllable drug release for the treatment of osteoarthritis. Bioact. Mater. 6, 3596–3607 (2021).
- Li, D. et al. Nanoparticles for oral delivery: targeted therapy for inflammatory bowel disease. J. Mater. Chem. B 10, 5853–5872 (2022).
- Zhang, G. et al. Glucosylated nanoparticles for the oral delivery of antibiotics to the proximal small intestine protect mice from gut dysbiosis. Nat. Biomed. Eng. 6, 867–881 (2022).
- Nguyen-Trinh, Q. N. et al. A silica-based antioxidant nanoparticle for oral delivery of Camptothecin which reduces intestinal side effects while improving drug efficacy for colon cancer treatment. Acta Biomater. 143, 459–470 (2022).
- Rump, A. et al. In Vivo Evaluation of a Gastro-Resistant HPMC-Based “Next Generation Enteric” Capsule. Pharmaceutics 14, 1999 (2022).
- Morales, J. O. et al. Challenges and Future Prospects for the Delivery of Biologics: Oral Mucosal, Pulmonary, and Transdermal Routes. AAPS J. 19, 652–668 (2017).
- Arrick, G. et al. Cephalopod-inspired jetting devices for gastrointestinal drug delivery. Nature 636, 481–487 (2024).
- Miller, R. R. et al. Integrating the Impact of Lipophilicity on Potency and Pharmacokinetic Parameters Enables the Use of Diverse Chemical Space during Small Molecule Drug Optimization. J. Med. Chem. 63, 12156–12170 (2020).
- Murcko, M. A. The Affinity Advantage. J. Med. Chem. 69, 1963–1969 (2026).
- Fischer, E. S. & Jones, L. H. Small molecule modulation of protein polymerization. Chem. Soc. Rev. 51, 2392–2396 (2022).
- Henderson, A. R. et al. Conservation of coactivator engagement mechanism enables small-molecule allosteric modulators. Proceedings of the National Academy of Sciences 115, 8960–8965 (2018).
- Zorn, J. A. & Wells, J. A. Turning enzymes ON with small molecules. Nat. Chem. Biol. 6, 179–188 (2010).
- Teng, M., Young, D. W. & Tan, Z. The Pursuit of Enzyme Activation: A Snapshot of the Gold Rush. J. Med. Chem. 65, 14289–14304 (2022).
- Dow, L. F. et al. The evolution of small molecule enzyme activators. RSC Med. Chem. 14, 2206–2230 (2023).
- Sandner, P. et al. Soluble Guanylate Cyclase Stimulators and Activators. H. H. H. W. Schmidt et al. (eds.), Reactive Oxygen Species, Handbook of Experimental Pharmacology 264, https://doi.org/10.1007/164_2018_197. Pp 355–394 (2018).
- Mittendorf, J. et al. Discovery of Riociguat (BAY 63-2521): A Potent, Oral Stimulator of Soluble Guanylate Cyclase for the Treatment of Pulmonary Hypertension. ChemMedChem 4, 853–865 (2009).
- Schulze, M. E. D. et al. Identification of β-Glucocerebrosidase Activators for Glucosylceramide hydrolysis. ChemMedChem 19, (2024).
- Häberle, J. Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther. Clin. Risk Manag. 327 (2011). [CrossRef]
- Caldovic, L. et al. Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J. Pediatr. 145, 552–554 (2004).
- Tang, C. P. et al. GCN2 kinase activation by ATP-competitive kinase inhibitors. Nat. Chem. Biol. 18, 207–215 (2022).
- Thomson, C. G. et al. Discovery of HC-7366: An Orally Bioavailable and Efficacious GCN2 Kinase Activator. J. Med. Chem. 67, 5259–5271 (2024).
- Simpson, G. L. et al. Identification and Optimization of Novel Small c-Abl Kinase Activators Using Fragment and HTS Methodologies. J. Med. Chem. 62, 2154–2171 (2019).
- Gong, G. Q. et al. A small-molecule PI3Kα activator for cardioprotection and neuroregeneration. Nature 618, 159–168 (2023).
- Sun, W. et al. Small molecule activators of TAK1 promotes its activity-dependent ubiquitination and TRAIL-mediated tumor cell death. Proceedings of the National Academy of Sciences 120, (2023).
- Brautigan, D. L., Farrington, C. & Narla, G. Targeting protein phosphatase PP2A for cancer therapy: development of allosteric pharmaceutical agents. Clin. Sci. 135, 1545–1556 (2021).
- Jones, N. H. et al. Allosteric activation of VCP, an AAA unfoldase, by small molecule mimicry. Proceedings of the National Academy of Sciences 121, (2024).
- Wong, K. S. & Houry, W. A. Chemical Modulation of Human Mitochondrial ClpP: Potential Application in Cancer Therapeutics. ACS Chem. Biol. 14, 2349–2360 (2019).
- Fennell, E. M. J. et al. Characterization of TR-107, a novel chemical activator of the human mitochondrial protease ClpP. Pharmacol. Res. Perspect. 10, (2022).
- Kok, B. P. et al. Discovery of small-molecule enzyme activators by activity-based protein profiling. Nat. Chem. Biol. 16, 997–1005 (2020).
- Yao, H. et al. Discovery of small-molecule activators of nicotinamide phosphoribosyltransferase (NAMPT) and their preclinical neuroprotective activity. Cell Res. 32, 570–584 (2022).
- Nozohouri, S. et al. In-Vivo and Ex-Vivo Brain Uptake Studies of Peptidomimetic Neurolysin Activators in Healthy and Stroke Animals. Pharm. Res. 39, 1587–1598 (2022).
- Michel, M. et al. Small-molecule activation of OGG1 increases oxidative DNA damage repair by gaining a new function. Science (1979). 376, 1471–1476 (2022).
- Schreiber, S. L. The Rise of Molecular Glues. Cell 184, 3–9 (2021).
- Lai, A. C. & Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 (2017).
- Chen, P.-H. et al. Modulation of Phosphoprotein Activity by Phosphorylation Targeting Chimeras (PhosTACs). ACS Chem. Biol. 16, 2808–2815 (2021).
- Siriwardena, S. U. et al. Phosphorylation-Inducing Chimeric Small Molecules. J. Am. Chem. Soc. 142, 14052–14057 (2020).
- Smith, B. E. et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 10, 131 (2019).
- Tashima, T. Emerging cellular uptake mechanisms of bRo5 PROTACs. RSC Med. Chem. 17, 2850–2859 (2026).
- Schneider, M. et al. The PROTACtable genome. Nat. Rev. Drug Discov. 20, 789–797 (2021).
- Ahn, G., Banik, S. M. & Bertozzi, C. R. Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem. Biol. 28, 1072–1080 (2021).
- Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
- Caianiello, D. et al. Bifunctional Small Molecules That Mediate the Degradation of Extracellular Proteins. Preprint at (2020). [CrossRef]
- Han, T. et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science (1979). 356, (2017).
- Shi, H. et al. CSN5i-3 is an orthosteric molecular glue inhibitor of COP9 signalosome. Nature 652, 1375–1383 (2026).
- Tan, X. et al. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645 (2007).
- Kathman, S. G. et al. Remodeling oncogenic transcriptomes by small molecules targeting NONO. Nat. Chem. Biol. 19, 825–836 (2023).
- Gourisankar, S. et al. Rewiring cancer drivers to activate apoptosis. Nature 620, 417–425 (2023).
- Cao, S. et al. Defining molecular glues with a dual-nanobody cannabidiol sensor. Nat. Commun. 13, 815 (2022).
- Mullard, A. Induced proximity pushes beyond protein degraders, as first RIPTAC moves into the clinic. Nat. Rev. Drug Discov. 24, 235–237 (2025).
- Yoon, H., Rutter, J. C., Li, Y.-D. & Ebert, B. L. Induced protein degradation for therapeutics: past, present, and future. Journal of Clinical Investigation 134, (2024).
- Petzold, G. et al. Mining the CRBN target space redefines rules for molecular glue–induced neosubstrate recognition. Science (1979). 389, eadt6736 (2025).
- Sievers, Q. L. et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science (1979). 362, eaat0572 (2018).
- Słabicki, M. et al. Expanding the druggable zinc-finger proteome defines properties of drug-induced degradation. Mol. Cell 85, 3184-3201.e14 (2025).
- Xu, Y. et al. Cross-Linking Profiling of Molecular Glue Degrader-Induced E3 Ligase Interactome to Expand Target Space. Angew. Chem. Int. Ed. 64, e202505053 (2025).
- Bushweller, J. H. Targeting transcription factors in cancer—from undruggable to reality. Nat. Rev. Cancer 19, 611–624 (2019).
- Henley, M. J. & Koehler, A. N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 20, 669–688 (2021).
- Niphakis, M. J. & Cravatt, B. F. Ligand discovery by activity-based protein profiling. Cell Chem. Biol. 31, 1636–1651 (2024).
- Wu, J. W. et al. Development of a robust, physiologically relevant neuronal tau aggregation assay for tau-related drug discovery. Journal of Biological Chemistry 301, 110853 (2025).
- Pathmanathan, S., Grozavu, I., Lyakisheva, A. & Stagljar, I. Drugging the undruggable proteins in cancer: A systems biology approach. Curr. Opin. Chem. Biol. 66, 102079 (2022).
- Lazear, M. R. et al. Proteomic discovery of chemical probes that perturb protein complexes in human cells. Mol. Cell 83, 1725-1742.e12 (2023).
- Kenanova, D. N. et al. A Systematic Approach to the Discovery of Protein–Protein Interaction Stabilizers. ACS Cent. Sci. 9, 937–946 (2023).
- Offensperger, F. et al. Large-scale chemoproteomics expedites ligand discovery and predicts ligand behavior in cells. Science (1979). 384, (2024).
- Zhang, X. & Cravatt, B. F. Chemical Proteomics–Guided Discovery of Covalent Ligands for Cancer Proteins. Annu. Rev. Cancer Biol. 8, 155–175 (2024).
- Wang, Y. et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem. 11, 1113–1123 (2019).
- Parker, C. G. et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 168, 527-541.e29 (2017).
- Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013).
- Green, E. M. et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science (1979). 351, 617–621 (2016).
- Hadida, S. et al. Discovery of N-(2,4-Di-tert -butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator. J. Med. Chem. 57, 9776–9795 (2014).
- Van Goor, F. et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences 108, 18843–18848 (2011).
- Belema, M. & Meanwell, N. A. Discovery of Daclatasvir, a Pan-Genotypic Hepatitis C Virus NS5A Replication Complex Inhibitor with Potent Clinical Effect. J. Med. Chem. 57, 5057–5071 (2014).
- Ratni, H. et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 61, 6501–6517 (2018).
- Ege, N., Bouguenina, H., Tatari, M. & Chopra, R. Phenotypic screening with target identification and validation in the discovery and development of E3 ligase modulators. Cell Chem. Biol. 28, 283–299 (2021).
- Cravatt, B. F., Wright, A. T. & Kozarich, J. W. Activity-Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu. Rev. Biochem. 77, 383–414 (2008).
- van den Bedem, H. & Fraser, J. S. Integrative, dynamic structural biology at atomic resolution—it’s about time. Nat. Methods 12, 307–318 (2015).
- Fraser, J. S., Lindorff-Larsen, K. & Bonomi, M. What Will Computational Modeling Approaches Have to Say in the Era of Atomistic Cryo-EM Data? J. Chem. Inf. Model. 60, 2410–2412 (2020).
- Lane, T. J. Protein structure prediction has reached the single-structure frontier. Nat. Methods 20, 170–173 (2023).
- Cushing, V. I. et al. High-resolution cryo-EM of the human CDK-activating kinase for structure-based drug design. Nat. Commun. 15, 2265 (2024).
- Catterall, W. A., Lenaeus, M. J. & Gamal El-Din, T. M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 60, 133–154 (2020).
- Wisedchaisri, G. & Gamal El-Din, T. M. Druggability of Voltage-Gated Sodium Channels—Exploring Old and New Drug Receptor Sites. Front. Pharmacol. 13, (2022).
- Slosky, L. M., Caron, M. G. & Barak, L. S. Biased Allosteric Modulators: New Frontiers in GPCR Drug Discovery. Trends Pharmacol. Sci. 42, 283–299 (2021).
- Murcko, M. A. What Makes a Great Medicinal Chemist? A Personal Perspective. J. Med. Chem. 61, 7419–7424 (2018).
- Murcko, M. A. Continually Humbling, Endlessly Fascinating, And Unfailingly Rewarding: Practical Lessons From A Lifelong Quest To Discover Breakthrough Medicines. Med Chem Rev 57, 27–57 (2022).
- Gordon Research Conference on Medicinal Chemistry. www.grc.org/medicinal-chemistry-conference/2010/ (2010).
- Zhang, Y. et al. Dimeric Macrocyclic Antagonists of Inhibitor of Apoptosis Proteins for the Treatment of Cancer. ACS Med. Chem. Lett. 6, 770–775 (2015).
- Wills, T. J. & Lipkus, A. H. Structural Approach to Assessing the Innovativeness of New Drugs Finds Accelerating Rate of Innovation. ACS Med. Chem. Lett. 11, 2114–2119 (2020).
- Dang, C. V., Reddy, E. P., Shokat, K. M. & Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 17, 502–508 (2017).
- Zhang, Z. & Shokat, K. M. Introduction: Drugging the Undruggable. Chem. Rev. 125, 6433–6434 (2025).
- In Drugging the Undruggable: Chemical Reviews Special Issue; 2025; 188. Drugging the Undruggable: Chemical Reviews Special Issue. (2025).
- Czeleń, P. Molecular dynamics study on inhibition mechanism of CDK-2 and GSK-3β by CHEMBL272026 molecule. Struct. Chem. 27, 1807–1818 (2016).
- Fischer, P. M. CDK versus GSK-3 Inhibition. Chem. Biol. 10, 1144–1146 (2003).
- Lanman, B. A. et al. Discovery of a Covalent Inhibitor of KRAS G12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 63, 52–65 (2020).
- LaMarche, M. J. et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 63, 13578–13594 (2020).
- Perni, R. B. et al. Preclinical Profile of VX-950, a Potent, Selective, and Orally Bioavailable Inhibitor of Hepatitis C Virus NS3-4A Serine Protease. Antimicrob. Agents Chemother. 50, 899–909 (2006).
- Lau, J. et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 58, 7370–7380 (2015).
- Kawai, T. et al. Structural basis for GLP-1 receptor activation by LY3502970, an orally active nonpeptide agonist. Proceedings of the National Academy of Sciences 117, 29959–29967 (2020).
- Szlavik, Z. et al. Discovery of S64315, a Potent and Selective Mcl-1 Inhibitor. J. Med. Chem. 63, 13762–13795 (2020).
- Ding, Q. et al. Discovery of RG7388, a Potent and Selective p53–MDM2 Inhibitor in Clinical Development. J. Med. Chem. 56, 5979–5983 (2013).
- Guerlavais, V. et al. Discovery of Sulanemadlin (ALRN-6924), the First Cell-Permeating, Stabilized α-Helical Peptide in Clinical Development. J. Med. Chem. 66, 9401–9417 (2023).
- Zhong, M. et al. Discovery of tetrahydroisoquinoline (THIQ) derivatives as potent and orally bioavailable LFA-1/ICAM-1 antagonists. Bioorg. Med. Chem. Lett. 20, 5269–5273 (2010).
- Wang, T. et al. Discovery of the Human Immunodeficiency Virus Type 1 (HIV-1) Attachment Inhibitor Temsavir and Its Phosphonooxymethyl Prodrug Fostemsavir. J. Med. Chem. 61, 6308–6327 (2018).
- Dorr, P. et al. Maraviroc (UK-427,857), a Potent, Orally Bioavailable, and Selective Small-Molecule Inhibitor of Chemokine Receptor CCR5 with Broad-Spectrum Anti-Human Immunodeficiency Virus Type 1 Activity. Antimicrob. Agents Chemother. 49, 4721–4732 (2005).
- Weisberg, E. et al. Smac mimetics: implications for enhancement of targeted therapies in leukemia. Leukemia 24, 2100–2109 (2010).
- Picaud, S. et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proceedings of the National Academy of Sciences 110, 19754–19759 (2013).
- Xu, R. et al. 3-[(1S, 2S, 3R )-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a Hypoxia-Inducible Factor 2α (HIF-2α) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. J. Med. Chem. 62, 6876–6893 (2019).
- Issa, G. C. et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature 615, 920–924 (2023).
- Illendula, A. et al. A small-molecule inhibitor of the aberrant transcription factor CBFβ-SMMHC delays leukemia in mice. Science (1979). 347, 779–784 (2015).
- Snyder, L. B. et al. Preclinical Evaluation of Bavdegalutamide (ARV-110), a Novel PROteolysis TArgeting Chimera Androgen Receptor Degrader. Mol. Cancer Ther. 24, 511–522 (2025).
- Gough, S. M. et al. Oral Estrogen Receptor PROTAC Vepdegestrant (ARV-471) Is Highly Efficacious as Monotherapy and in Combination with CDK4/6 or PI3K/mTOR Pathway Inhibitors in Preclinical ER+ Breast Cancer Models. Clinical Cancer Research 30, 3549–3563 (2024).
- Kasembeli, M. M. et al. TTI-101: A competitive inhibitor of STAT3 that spares oxidative phosphorylation and reverses mechanical allodynia in mouse models of neuropathic pain. Biochem. Pharmacol. 192, 114688 (2021).
- Lehal, R. et al. Pharmacological disruption of the Notch transcription factor complex. Proceedings of the National Academy of Sciences 117, 16292–16301 (2020).
- Bonazzi, S. et al. Discovery and characterization of a selective IKZF2 glue degrader for cancer immunotherapy. Cell Chem. Biol. 30, 235-247.e12 (2023).
- Kumar, S. & Anderson, K. C. Drug Insight: thalidomide as a treatment for multiple myeloma. Nat. Clin. Pract. Oncol. 2, 262–270 (2005).
- Pasqua, A. E. et al. HSF1 Pathway Inhibitor Clinical Candidate (CCT361814/NXP800) Developed from a Phenotypic Screen as a Potential Treatment for Refractory Ovarian Cancer and Other Malignancies. J. Med. Chem. 66, 5907–5936 (2023).
- Reisman, S. A. et al. Pharmacokinetics and pharmacodynamics of the novel Nrf2 activator omaveloxolone in primates. Drug Des. Devel. Ther. Volume 13, 1259–1270 (2019).
- Hong, C. S. et al. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 16, 821–833 (2015).
- Li, G. et al. A Small Molecule Inhibitor of VE-PTP Activates Tie2 in Schlemm’s Canal Increasing Outflow Facility and Reducing Intraocular Pressure. Investigative Ophthalmology & Visual Science 61, 12 (2020).
- Krishnan, N. et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 10, 558–566 (2014).
- Faulds, D., Goa, K. L. & Benfield, P. Cyclosporin. Drugs 45, 953–1040 (1993).
- Johns, B. A. et al. Carbamoyl Pyridone HIV-1 Integrase Inhibitors 3. A Diastereomeric Approach to Chiral Nonracemic Tricyclic Ring Systems and the Discovery of Dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 56, 5901–5916 (2013).
- Xu, C. et al. Alpha-kinase 1 (ALPK1) agonist DF-006 demonstrates potent efficacy in mouse and primary human hepatocyte (PHH) models of hepatitis B. Hepatology 77, 275–289 (2023).
- Forsyth, S. et al. Safety, Pharmacokinetics, and Pharmacodynamics of Etavopivat (FT-4202), an Allosteric Activator of Pyruvate Kinase-R, in Healthy Adults: A Randomized, Placebo-Controlled, Double-Blind, First-in-Human Phase 1 Trial. Clin. Pharmacol. Drug Dev. 11, 654–665 (2022).
- Knutson, S. K. et al. Selective Inhibition of EZH2 by EPZ-6438 Leads to Potent Antitumor Activity in EZH2-Mutant Non-Hodgkin Lymphoma. Mol. Cancer Ther. 13, 842–854 (2014).
- Osteen, J. D. et al. Pharmacology and Mechanism of Action of Suzetrigine, a Potent and Selective NaV1.8 Pain Signal Inhibitor for the Treatment of Moderate to Severe Pain. Pain Ther. 14, 655–674 (2025).
- Witty, D. R. et al. Discovery of Vixotrigine: A Novel Use-Dependent Sodium Channel Blocker for the Treatment of Trigeminal Neuralgia. ACS Med. Chem. Lett. 11, 1678–1687 (2020).
- Ferretti, S. et al. Discovery of WRN inhibitor HRO761 with synthetic lethality in MSI cancers. Nature 629, 443–449 (2024).
- Metcalf, B. et al. Discovery of GBT440, an Orally Bioavailable R-State Stabilizer of Sickle Cell Hemoglobin. ACS Med. Chem. Lett. 8, 321–326 (2017).
- Yen, K. et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 7, 478–493 (2017).
- Angelaud, R., Sutherlin, D. P., Reynolds, M., Savage, S. & Stumpf, A. The Medicinal Chemistry and Commercial Manufacturing Behind the Discovery and Development of the Hedgehog Inhibitor Vismodegib. in 215–236 (2016). [CrossRef]
- Hansen, J. D. et al. CC-90009: A Cereblon E3 Ligase Modulating Drug That Promotes Selective Degradation of GSPT1 for the Treatment of Acute Myeloid Leukemia. J. Med. Chem. 64, 1835–1843 (2021).
- Collibee, S. E. et al. Discovery of Tirasemtiv, the First Direct Fast Skeletal Muscle Troponin Activator. ACS Med. Chem. Lett. 9, 354–358 (2018).
- Marks, P. A. & Breslow, R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90 (2007).
- Morgan, B. P. et al. Discovery of Omecamtiv Mecarbil the First, Selective, Small Molecule Activator of Cardiac Myosin. ACS Med. Chem. Lett. 1, 472–477 (2010).
- Fog, C. K. et al. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 38, 142–153 (2018).
- Ernst, J. T. et al. Design of Development Candidate eFT226, a First in Class Inhibitor of Eukaryotic Initiation Factor 4A RNA Helicase. J. Med. Chem. 63, 5879–5955 (2020).
- Holden, J. K. et al. Small Molecule Dysregulation of TEAD Lipidation Induces a Dominant-Negative Inhibition of Hippo Pathway Signaling. Cell Rep. 31, 107809 (2020).
- Thurairatnam, S. et al. Brain Penetrable Inhibitors of Ceramide Galactosyltransferase for the Treatment of Lysosomal Storage Disorders. ACS Med. Chem. Lett. 11, 2010–2016 (2020).
- Koralewski, R. et al. Discovery of OATD-01, a First-in-Class Chitinase Inhibitor as Potential New Therapeutics for Idiopathic Pulmonary Fibrosis. J. Med. Chem. 63, 15527–15540 (2020).
- Konteatis, Z. et al. Discovery of AG-270, a First-in-Class Oral MAT2A Inhibitor for the Treatment of Tumors with Homozygous MTAP Deletion. J. Med. Chem. 64, 4430–4449 (2021).
- Christman, J. K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–5495 (2002).
- Chuang, C. et al. Discovery of Aficamten (CK-274), a Next-Generation Cardiac Myosin Inhibitor for the Treatment of Hypertrophic Cardiomyopathy. J. Med. Chem. 64, 14142–14152 (2021).
- Huang, Y. et al. Discovery of the Clinical Candidate MAK683: An EED-Directed, Allosteric, and Selective PRC2 Inhibitor for the Treatment of Advanced Malignancies. J. Med. Chem. 65, 5317–5333 (2022).
- Wobst, H. J. et al. SLC6A19 inhibition facilitates urinary neutral amino acid excretion and lowers plasma phenylalanine. JCI Insight 9, (2024).
- Priestley, E. S. et al. Discovery of Two Novel Antiplatelet Clinical Candidates (BMS-986120 and BMS-986141) That Antagonize Protease-Activated Receptor 4. J. Med. Chem. 65, 8843–8854 (2022).
- Jacobs, J. W. et al. Discovery of Tenapanor: A First-in-Class Minimally Systemic Inhibitor of Intestinal Na+/H+ Exchanger Isoform 3. ACS Med. Chem. Lett. 13, 1043–1051 (2022).
- Vaswani, R. G. et al. Discovery of FHD-286, a First-in-Class, Orally Bioavailable, Allosteric Dual Inhibitor of the Brahma Homologue (BRM) and Brahma-Related Gene 1 (BRG1) ATPase Activity for the Treatment of SWItch/Sucrose Non-Fermentable (SWI/SNF) Dependent Cancers. J. Med. Chem. 68, 1772–1792 (2025).
- Ofir-Rosenfeld, Y. et al. STC-15, an oral small molecule inhibitor of the RNA methyltransferase METTL3, inhibits tumour growth through activation of anti-cancer immune responses associated with increased interferon signalling, and synergises with T cell checkpoint blockade. Eur. J. Cancer 174, S123 (2022).
- Haddach, M. et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 3, 602–606 (2012).
- Fourie, A. M. et al. JNJ-77242113, a highly potent, selective peptide targeting the IL-23 receptor, provides robust IL-23 pathway inhibition upon oral dosing in rats and humans. Sci. Rep. 14, 17515 (2024).
Figure 1.
The two meanings of “novel modality.” A simple two-by-two rubric for understanding the term “modality” is to distinguish between mechanistic and molecular novelty. The x-axis refers to the novelty of the mechanism of action being pursued, while the y-axis describes the degree of chemical complexity or molecular novelty. Compounds on the right side of the matrix have greater mechanistic novelty, while compounds on the upper half of the matrix are chemically more novel. The placement of any modality is intended to give a general perspective on the class as a whole; there will always be exceptions. Also note that some drugs can be defined by more than one modality; for example, an ASO may also disrupt a protein-RNA interaction and be a condensate modifier (c-mod). “Simple biologics” refers to conventional Ab and protein therapeutics. “RNA/DNA engagers” refers to non-nucleic acid molecules, typically low molecular weight, that directly engage RNA or DNA. While all discovery projects are challenging, modalities in the lower-left box (green) will generally present fewer difficulties, while modalities in the upper-right box (orange), which possess both chemical and mechanistic novelty, are generally the most complex. The other two boxes (yellow) represent intermediate cases. Over time, the placement of each modality will be subject to change in light of new discoveries; as experience accumulates, all modalities will be perceived as less novel or difficult and will generally shift towards the lower-left portion of the matrix. As an illustration, consider an example of a “rule breaker” compound with high molecular novelty, such as the PCSK9 macrocyclic peptide (enlicitide or MK-0616, molecular weight 1,616 daltons). Despite its size and structural complexity, this molecule has low mechanistic novelty—it is an inhibitor that disrupts a protein-protein interface and thus belongs in the upper-left quadrant. In contrast, consider a PROTAC degrader such as vepdegestrant (ARV-471), with a molecular weight of 723 daltons. It deploys a novel MOA and is a high-molecular-weight “rule breaker” compound and thus belongs in the upper-right quadrant. Other molecules may be placed in the figure in their appropriate locations based on the current understanding of the degree of challenge associated with that class of oral agents. Note that the choice of placement is to a degree subjective.
Figure 1.
The two meanings of “novel modality.” A simple two-by-two rubric for understanding the term “modality” is to distinguish between mechanistic and molecular novelty. The x-axis refers to the novelty of the mechanism of action being pursued, while the y-axis describes the degree of chemical complexity or molecular novelty. Compounds on the right side of the matrix have greater mechanistic novelty, while compounds on the upper half of the matrix are chemically more novel. The placement of any modality is intended to give a general perspective on the class as a whole; there will always be exceptions. Also note that some drugs can be defined by more than one modality; for example, an ASO may also disrupt a protein-RNA interaction and be a condensate modifier (c-mod). “Simple biologics” refers to conventional Ab and protein therapeutics. “RNA/DNA engagers” refers to non-nucleic acid molecules, typically low molecular weight, that directly engage RNA or DNA. While all discovery projects are challenging, modalities in the lower-left box (green) will generally present fewer difficulties, while modalities in the upper-right box (orange), which possess both chemical and mechanistic novelty, are generally the most complex. The other two boxes (yellow) represent intermediate cases. Over time, the placement of each modality will be subject to change in light of new discoveries; as experience accumulates, all modalities will be perceived as less novel or difficult and will generally shift towards the lower-left portion of the matrix. As an illustration, consider an example of a “rule breaker” compound with high molecular novelty, such as the PCSK9 macrocyclic peptide (enlicitide or MK-0616, molecular weight 1,616 daltons). Despite its size and structural complexity, this molecule has low mechanistic novelty—it is an inhibitor that disrupts a protein-protein interface and thus belongs in the upper-left quadrant. In contrast, consider a PROTAC degrader such as vepdegestrant (ARV-471), with a molecular weight of 723 daltons. It deploys a novel MOA and is a high-molecular-weight “rule breaker” compound and thus belongs in the upper-right quadrant. Other molecules may be placed in the figure in their appropriate locations based on the current understanding of the degree of challenge associated with that class of oral agents. Note that the choice of placement is to a degree subjective.

Figure 2.
Druggability of genes by ChEMBL class and subclass. Drugged genes and druggable genes inferred by ChEMBL class and subclass. Drugged genes are those with the Open Targets “high-quality ligand” annotation, as derived from ChEMBL (light blue). Genes inferred to be druggable based on shared ChEMBL subclass with at least one drugged member are colored green. Genes inferred to be druggable based on shared membership in a ChEMBL class with at least one drugged member are colored dark blue. A set of genes in ChEMBL are given ‘Unclassified’ status which we include here for completeness but do not use to infer druggability (far right).
Figure 2.
Druggability of genes by ChEMBL class and subclass. Drugged genes and druggable genes inferred by ChEMBL class and subclass. Drugged genes are those with the Open Targets “high-quality ligand” annotation, as derived from ChEMBL (light blue). Genes inferred to be druggable based on shared ChEMBL subclass with at least one drugged member are colored green. Genes inferred to be druggable based on shared membership in a ChEMBL class with at least one drugged member are colored dark blue. A set of genes in ChEMBL are given ‘Unclassified’ status which we include here for completeness but do not use to infer druggability (far right).

Figure 3.
Statistics of Druggability by ChEMBL. Druggability analysis using ChEMBL. Percentage of druggable genes as inferred by membership in ChEMBL classes (top left) or in subclasses (top right) with at least one drugged gene. A classified gene is included in the ChEMBL classification system. Percentage of classes with drugged genes in ChEMBL classes (bottom left) or in subclasses (bottom right), as defined by having at least one drugged gene in the class or subclass. Gene counts are given in parentheses.
Figure 3.
Statistics of Druggability by ChEMBL. Druggability analysis using ChEMBL. Percentage of druggable genes as inferred by membership in ChEMBL classes (top left) or in subclasses (top right) with at least one drugged gene. A classified gene is included in the ChEMBL classification system. Percentage of classes with drugged genes in ChEMBL classes (bottom left) or in subclasses (bottom right), as defined by having at least one drugged gene in the class or subclass. Gene counts are given in parentheses.

Figure 5.
Druggability of the Protein Kinase Family. The total number of kinases is 503. Following the classification scheme in Manning et al., the number of kinases with a high-quality ligand (“drugged”) is 418 (cyan). The number inferred to be druggable by sharing subclass membership with a drugged kinase is 467 (green). The number inferred to be druggable by sharing class membership with a drugged kinase is 503 (blue).
Figure 5.
Druggability of the Protein Kinase Family. The total number of kinases is 503. Following the classification scheme in Manning et al., the number of kinases with a high-quality ligand (“drugged”) is 418 (cyan). The number inferred to be druggable by sharing subclass membership with a drugged kinase is 467 (green). The number inferred to be druggable by sharing class membership with a drugged kinase is 503 (blue).

Figure 6.
Scientific Advances That Enable Progress against “Undruggable” Targets. A range of technical advances relating to molecular modalities, delivery, mechanistic modalities and interrogation tools provides powerful new strategies to discover oral medicines against challenging targets. Molecular modalities: larger and chemically more complex molecules are now routinely pursued. Delivery: we are using this term in two distinct ways. Improved methods of formulation and complex excipients enable the delivery of drugs that formerly could not be expected to achieve the required therapeutic concentrations. Further, improved potency and/or PK properties can significantly lower the required dose, greatly simplifying the delivery challenge. Mechanistic modalities: novel approaches such as degraders, glues, c-mods, biased agonists, and allosteric inhibitors provide discovery teams with a wide range of options in cases where traditional approaches cannot achieve adequate potency or selectivity. Interrogation tools: Improved biochemical and cellular screening tools such as chemoproteomics enable both the discovery of chemical matter and the exploration of mechanistic modalities and their relevance to pharmacology.
Figure 6.
Scientific Advances That Enable Progress against “Undruggable” Targets. A range of technical advances relating to molecular modalities, delivery, mechanistic modalities and interrogation tools provides powerful new strategies to discover oral medicines against challenging targets. Molecular modalities: larger and chemically more complex molecules are now routinely pursued. Delivery: we are using this term in two distinct ways. Improved methods of formulation and complex excipients enable the delivery of drugs that formerly could not be expected to achieve the required therapeutic concentrations. Further, improved potency and/or PK properties can significantly lower the required dose, greatly simplifying the delivery challenge. Mechanistic modalities: novel approaches such as degraders, glues, c-mods, biased agonists, and allosteric inhibitors provide discovery teams with a wide range of options in cases where traditional approaches cannot achieve adequate potency or selectivity. Interrogation tools: Improved biochemical and cellular screening tools such as chemoproteomics enable both the discovery of chemical matter and the exploration of mechanistic modalities and their relevance to pharmacology.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



