Submitted:
10 July 2026
Posted:
13 July 2026
You are already at the latest version
Abstract
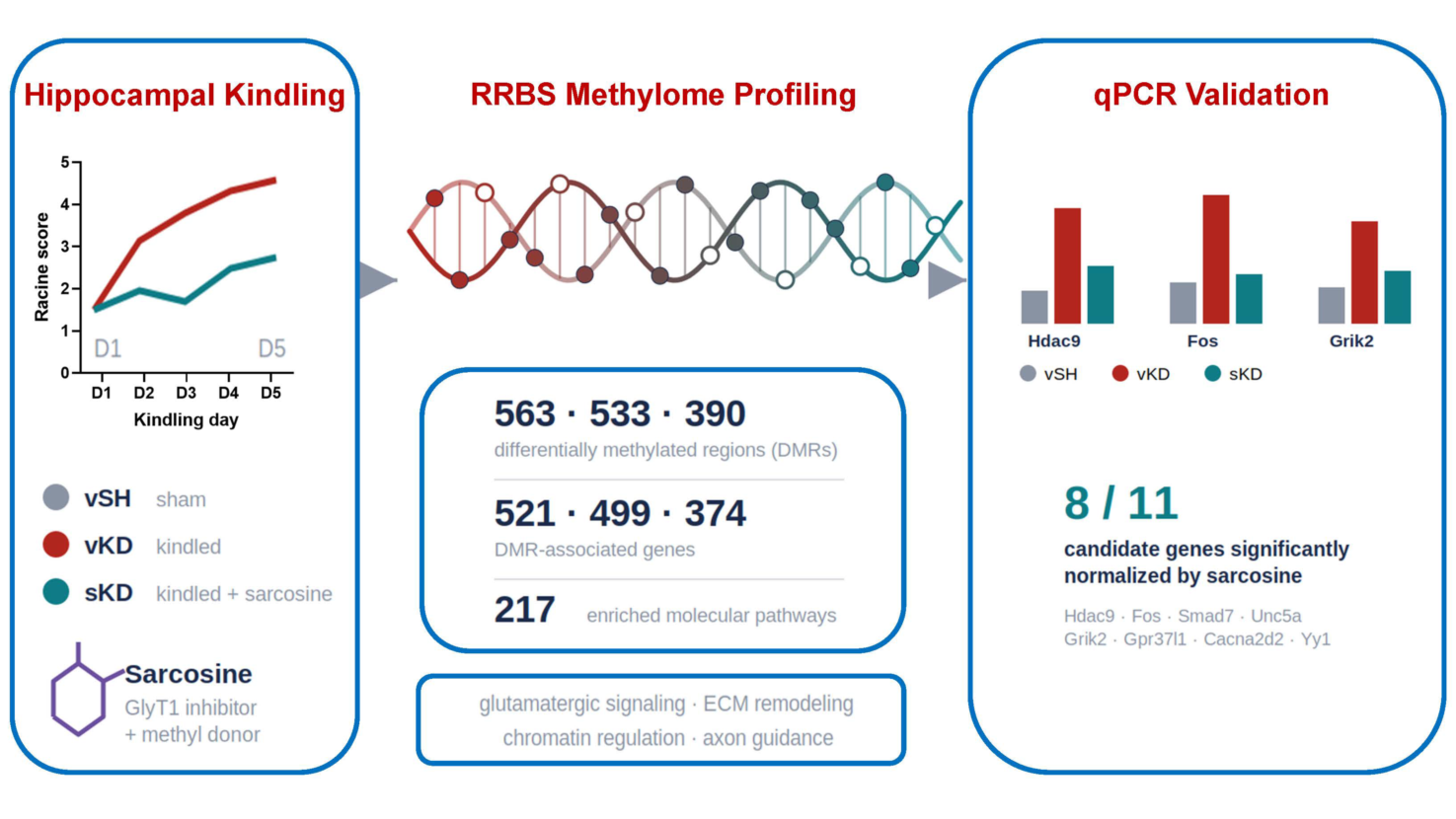
DNA methylation is a key regulator of epileptogenesis. Sarcosine, a glycine transporter 1 (GlyT1) inhibitor and methyl donor, suppresses kindling-induced epileptogenesis and alters hippocampal DNA methylation, but its locus-specific epigenetic effects remain poorly understood. Here, reduced representation bisulfite sequencing (RRBS) was combined with targeted gene expression analysis in the hippocampus of sarcosine-treated kindled rats. RRBS identified 563, 533, and 390 differentially methylated regions (DMRs), corresponding to 521, 499, and 374 DMR-associated genes, in vehicle-kindled versus sham (vKD vs vSH), sarcosine-kindled versus sham (sKD vs vSH), and sarcosine-kindled versus vehicle-kindled (sKD vs vKD) comparisons, respectively. Pathway enrichment analysis identified 217 significantly affected pathways, including glutamatergic signaling, extracellular matrix (ECM) organization, chromatin regulation, axon guidance, and apoptotic processes. Eleven candidate genes involved in epigenetic regulation, excitatory neurotransmission, and ECM remodeling were selected for transcriptional validation. All 11 genes were significantly upregulated in kindled hippocampi, whereas sarcosine normalized expression of eight genes (Hdac9, Fos, Smad7, Unc5a, Grik2, Gpr37l1, Cacna2d2, and Yy1) toward control levels. Collectively, these findings indicate that sarcosine remodels DNA methylation-associated transcriptional networks during epileptogenesis and support GlyT1 inhibition as a potential disease-modifying strategy for epilepsy.
Keywords:
DNA methylation
; epileptogenesis
; sarcosine
; GlyT1 inhibition
; differentially methylated regions (DMRs)
; epigenetic regulation
; hippocampus
1. Introduction
Epileptogenesis refers to the progressive transformation of a functionally normal brain into an epileptic state, culminating in the emergence of spontaneous, recurrent seizures. This pathological process is often initiated by a neurological insult, such as traumatic brain injury, stroke, central nervous system infection, or status epilepticus [1,2]. Although antiepileptic drugs (AEDs) are effective in suppressing acute and chronic seizures, they do not prevent or reverse the underlying development of epilepsy [3]. These limitations underscore a significant unmet need for disease-modifying therapies that can intervene during the latent period and halt progression to chronic epilepsy. Here, we investigate how a disease-modifying intervention, glycine transporter 1 (GlyT1) inhibition by sarcosine, is associated with locus-specific DNA methylation and transcriptional changes during epileptogenesis.
Genome-wide DNA methylation alterations, often accompanied by increased DNA methyltransferase (DNMT) activity, have been reported in patients with temporal lobe epilepsy (TLE), pediatric epilepsy, and multiple experimental epilepsy models [4,5,6,7,8]. These findings suggest that epigenetic dysregulation represents a common pathomechanism across diverse forms of epilepsy [3,4,9,10,11,12]. Importantly, DNA methylation changes are highly context-dependent, with distinct epilepsy models exhibiting unique methylation signatures [13]. Both hypermethylation and hypomethylation occur during epileptogenesis and affect a wide range of genomic regions, including CpG islands, promoters, gene bodies, and intergenic loci [9,12]. These epigenetic alterations are thought to contribute to dysregulation of gene networks involved in inflammation, synaptic remodeling, neuroplasticity, and neuronal survival.
DNA methylation changes during epileptogenesis are also highly cell-type specific, differentially affecting neuronal and glial populations [3,11]. Several epilepsy-associated genes exhibit coordinated changes in DNA methylation and gene expression, suggesting a potential regulatory relationship, although the connection between methylation status and transcriptional output is often complex and context-dependent [3,7]. Despite increasing recognition of the role of epigenetic mechanisms in epilepsy, no currently available therapy has been shown to halt or reverse the epigenetic processes that drive epileptogenesis. While certain AEDs, such as valproic acid, influence DNA methylation and histone acetylation, their impact on long-term disease progression remains limited [14]. Likewise, non-pharmacological interventions such as the ketogenic diet can partially modify methylation profiles in experimental models but have not consistently prevented epileptogenesis in at-risk populations [10,15]. Identifying methylation-associated genes and pathways that contribute to epileptogenesis, and determining whether these alterations can be therapeutically reversed, remain important challenges in the development of disease-modifying therapies [3,16].
We previously demonstrated that sarcosine (N-methylglycine), a GlyT1 inhibitor and methyl donor, significantly attenuates epileptogenesis in a rat kindling model [17]. In addition to inhibiting glycine reuptake, sarcosine influences one-carbon metabolism by affecting the conversion of S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH), thereby modulating cellular methylation potential [18]. Our prior work further showed that sarcosine decreases hippocampal 5-methylcytosine (5mC) levels while increasing 5-hydroxymethylcytosine (5hmC), suggesting activation of TET-mediated DNA demethylation pathways during epileptogenesis [17]. These findings raise the possibility that sarcosine exerts disease-modifying effects through epigenetic remodeling of seizure-responsive gene networks.
To further define the epigenetic mechanisms underlying sarcosine’s anti-epileptogenic actions, we employed our established hippocampal kindling model and performed reduced representation bisulfite sequencing (RRBS) together with targeted gene expression analysis of hippocampal tissue from sham, kindled, and sarcosine-treated animals. Our objectives were to identify gene- and pathway-level methylation changes associated with epileptogenesis and determine whether sarcosine reverses or reshapes these epigenetic alterations. We hypothesized that kindling-induced DNA methylation changes contribute to epileptogenic network remodeling and that sarcosine suppresses epileptogenesis, at least in part, through reprogramming of DNA methylation-associated transcriptional networks involved in neuronal excitability, synaptic plasticity, and structural remodeling. While previous studies have largely cataloged methylation changes associated with epilepsy, few have directly linked genome-wide methylation remodeling to a candidate disease-modifying intervention and corresponding transcriptional rescue. By integrating RRBS, pathway enrichment analyses, and targeted gene expression profiling, the present study defines how GlyT1 inhibition by sarcosine reshapes hippocampal DNA methylation-associated transcriptional networks during epileptogenesis.
2. Materials and Methods
2.1. Ethical Approval and Animal Care
All animal procedures were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-accredited facility and were approved by the Institutional Animal Care and Use Committee of the Legacy Research Institute. Male Sprague–Dawley rats (280-300 g) were obtained from Jackson Laboratory (Sacramento, CA, USA). Animals were acclimated for one week before experimentation and housed under controlled temperature and humidity conditions on a 12 h light/dark cycle (lights on at 07:00 h) with ad libitum access to food and water throughout the study.
2.2. Rat Kindling and Treatment
The hippocampal kindling model was performed as previously described [17]. Eight-week-old male Sprague–Dawley rats were surgically implanted with bipolar electrodes (Plastics One, Roanoke, VA, USA) targeting the left dorsal hippocampus (coordinates relative to bregma: AP −5.0 mm, ML +5.0 mm, DV −7.5 mm). Following a 10-day postoperative recovery period, animals underwent a rapid hippocampal kindling protocol over five consecutive days [17].
Rats were randomly assigned to one of three experimental groups (n = 6 per group): (i) vehicle-sham (vSH), which received sham stimulation and vehicle treatment; (ii) vehicle-kindled (vKD), which received vehicle treatment during the kindling protocol; and (iii) sarcosine-kindled (sKD), which received sarcosine (3 g/kg, i.p.; Cat. No. 131776, Sigma-Millipore, Burlington, MA, USA) during the kindling protocol. Sarcosine or vehicle was administered intraperitoneally 30 min before each daily stimulation session.
Each kindling session consisted of six electrical stimulations delivered using a Grass S-88 stimulator (1 ms biphasic square-wave pulses, 200 μA, 50 Hz, 10 s duration) separated by 30 min intervals. Behavioral seizure severity was scored according to the Racine scale [17,19], and electrographic seizure activity was recorded using the PowerLab data acquisition system (ADInstruments, Colorado Springs, CO, USA) as previously described [17]. The vSH group served as the baseline control for comparisons of seizure susceptibility and gene expression.
2.3. Tissue Collection
Hippocampal tissue from sarcosine-treated (sKD) and vehicle-treated (vKD) kindled rats was collected 24 hours after the final stimulation session. Age-matched vehicle-treated sham animals (vSH) served as baseline controls. Rats were deeply anesthetized with isoflurane and euthanized by rapid decapitation. The hippocampus was rapidly dissected, with tissue from the right hemisphere allocated for DNA extraction and reduced representation bisulfite sequencing (RRBS), and tissue from the left hemisphere used for RNA isolation and quantitative real-time PCR (qPCR) analyses.
2.4. Reduced Representation Bisulfite Sequencing (RRBS)
Genomic DNA was extracted from hippocampal tissue according to Qiagen protocols by the Gene Profiling Shared Resource Core at Oregon Health & Science University (OHSU, Portland, OR, USA). DNA concentration and quality were assessed by UV spectrophotometry and fragment analysis using a TapeStation system. RRBS library preparation and methylation profiling were performed by the KCVI Epigenetics Core at OHSU, and next-generation sequencing was conducted by the OHSU Massively Parallel Sequencing Shared Resource Core.
RRBS libraries were generated from 200 ng genomic DNA per sample. Six animals were initially included in each experimental group; however, one sKD sample failed RRBS quality control and was excluded from methylation analyses. Consequently, RRBS was performed using n = 6 for vSH, n = 6 for vKD, and n = 5 for sKD, following established protocols [20]. Genomic DNA was digested overnight with MspI (New England Biolabs, Ipswich, MA, USA), and libraries were prepared using the NEXTflex Bisulfite-Seq Kit (Bioo Scientific, Austin, TX, USA). Bisulfite conversion was performed using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA), followed by PCR amplification using NEBNext Multiplex Oligos (New England Biolabs). Libraries were sequenced on an Illumina NextSeq 500 platform using a 75 bp paired-end high-output protocol (Illumina, San Diego, CA, USA), generating approximately 40 million reads per sample.
Library quality and bisulfite conversion efficiency were assessed prior to downstream analyses. Bisulfite conversion efficiency exceeded 99% in all samples. Mean CpG coverage depth was approximately 10×, with each sample achieving ≥10× coverage at 716,512-1,337,815 CpG sites. Alignment rates to the Rnor_6.0 reference genome ranged from 70.7% to 75.3% (mean 72.9%), indicating high-quality sequencing data suitable for differential methylation analysis.
2.5. Differential Methylation Analysis and Pathway Enrichment
Differentially methylated cytosines (DMCs) and differentially methylated regions (DMRs) were identified using established bioinformatic pipelines [20]. DMC analysis was performed using the Limma package (v3.34.9). Only CpG sites with a minimum coverage of 10× in at least three biological replicates per group were retained for analysis, yielding a total of 644,003 CpGs. Sequencing reads were aligned to the Rattus norvegicus reference genome (Rnor_6.0) using Ensembl annotations. Methylation values were arcsine-transformed prior to statistical analysis to improve distributional normality.
DMRs were identified using the Comb-p algorithm. Regions containing at least two adjacent CpGs with FDR-corrected p values < 0.05 and located within 300 bp of one another were defined as DMRs. Hypermethylated and hypomethylated DMRs were classified using a methylation difference threshold of ≥10% or ≤−10%, respectively.
To investigate the biological significance of DMR-associated genes, functional enrichment analyses were performed using g:Profiler (https://biit.cs.ut.ee/gprofiler/gost) and PANTHER (http://pantherdb.org), followed by pathway and network analyses using Metascape. Enriched Gene Ontology (GO) terms and biological pathways were used to identify molecular processes affected by kindling-induced epileptogenesis and sarcosine treatment.
Complete DMR lists for all comparison groups are provided in Supplementary Tables S1–S3. DMRs were assigned to their nearest genes using gene-centric mapping based on the nearest transcription start site (TSS). Candidate genes selected for transcriptional validation were identified from these DMR-associated gene lists and pathway enrichment analyses. Associations between DNA methylation changes and gene expression were therefore inferred through DMR-to-gene mapping and functional pathway relationships rather than direct sample-wise methylation–expression correlation analyses.
2.6. Quantitative Real-Time PCR (qPCR)
Total RNA was extracted from hippocampal tissue using the RNeasy Mini Kit (Cat. No. 74104, Qiagen, Germantown, MD, USA). RNA integrity and concentration were assessed using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). First-strand cDNA synthesis was performed using the Promega Reverse Transcription System (Cat. No. A3500, Promega, Leiden, The Netherlands) in a 20 μL reaction (42 °C for 40 min, followed by 85 °C for 5 min). cDNA was stored at 4 °C until qPCR analysis.
Quantitative PCR was performed using SYBR Green Universal Master Mix (Cat. No. 4309155, Thermo Fisher Scientific) and primers targeting genes involved in extracellular matrix remodeling (Antxr2, Spock2), excitatory synaptic signaling (Grik2, Gpr37l1, Cacna2d2), and transcriptional/epigenetic regulation (Hdac9, Fos, Smad7, Unc5a, Dzip1, Yy1) (Table 1). Gapdh served as the reference gene for normalization. Each 20 μL reaction contained 10 μL SYBR Green Master Mix, 0.8 μM forward and reverse primers, and 5 μL cDNA template. No-template controls were included in each run to assess reagent contamination.
Amplification was performed on a QuantStudio™ 5 Real-Time PCR System under the following conditions: 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Relative gene expression was calculated using the 2–ΔΔCt method, with expression normalized to Gapdh.
2.7. Statistical Analysis
Relative gene expression was calculated using the 2–ΔΔCt method. Expression data were normalized to Gapdh, whose stability was verified using the geNorm and NormFinder algorithms (GenEx, MultiD). Accumulated standard deviation (Acc.SD) values were used to determine the optimal number of reference genes.
All data are presented as mean ± SEM. Normality was assessed prior to statistical testing. For comparisons between two groups, either Student’s t-test or the Mann–Whitney U test was used as appropriate. For comparisons among multiple groups, one-way or two-way ANOVA followed by Tukey’s post hoc test was performed. Statistical significance was defined as p < 0.05. All analyses were conducted using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
3. Results
3.1. Sarcosine Suppresses Hippocampal Kindling-Induced Epileptogenesis
To confirm the anti-epileptogenic effects of sarcosine in the cohort used for methylation analyses, we employed our previously established rapid hippocampal kindling model [17] (Figure 1A). Vehicle-treated kindled rats (vKD) developed fully kindled seizures after five days of stimulation, reaching Racine stage 4–5 seizures by the first stimulation on day 5 and exhibiting an average Racine score of 4.6 ± 0.3 (Figure 1B). In contrast, rats receiving daily sarcosine treatment (3 g/kg, i.p.; sKD) displayed significantly reduced seizure severity, with an average Racine score of 2.5 ± 0.8 on day 5 (p < 0.05 vs. vKD; Figure 1B). These findings confirm that sarcosine attenuates hippocampal kindling-induced epileptogenesis in this experimental cohort, consistent with our previous report [17], and provide the foundation for subsequent methylome and gene expression analyses.
Rats underwent rapid hippocampal kindling with daily vehicle (vKD) or sarcosine (sKD; 3 g/kg, i.p.) treatment. Vehicle-treated sham animals (vSH) served as non-kindled controls. (A) Experimental timeline and study design. Sarcosine or vehicle was administered 30 min before each daily kindling session. Hippocampal tissue was collected 24 h after the final stimulation. (B) Racine seizure scores during the 5-day kindling protocol. Vehicle-treated rats developed fully kindled seizures by day 5, whereas sarcosine-treated rats exhibited significantly reduced seizure severity. Data are presented as mean ± SEM (n = 6 per group). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. vKD at the corresponding time point.
3.2. Genome-Wide DNA Methylation Changes Associated with Kindling and Sarcosine Treatment
To define the epigenetic landscape associated with kindling-induced epileptogenesis and sarcosine treatment, we characterized genome-wide differentially methylated regions (DMRs) across experimental groups. Compared with non-kindled sham controls (vSH), vehicle-treated kindled rats (vKD) exhibited 264 hypermethylated and 299 hypomethylated DMRs (|Δmethylation| ≥ 10%, q < 0.05), indicating extensive remodeling of the hippocampal methylome during kindling (Figure 2A). Sarcosine-treated kindled rats (sKD) exhibited 387 hypermethylated and 146 hypomethylated DMRs relative to vSH, with 33 hypermethylated and 15 hypomethylated DMRs shared between vKD and sKD, indicating a subset of methylation changes common to both kindled groups. In the sKD vs vKD contrast, 304 hypermethylated and 86 hypomethylated DMRs were identified, yielding a predominantly hypermethylated profile in sarcosine-treated animals (Figure 2A).
To characterize shared and distinct methylation signatures, overlap analyses were performed separately for hypomethylated and hypermethylated DMRs. Among hypomethylated DMRs, 284 were unique to vKD vs vSH, 131 were unique to sKD vs vSH, and 15 were shared between the two contrasts (Figure 2B, left panel). Among hypermethylated DMRs, 231 were unique to vKD vs vSH, 354 were unique to sKD vs vSH, and 33 were shared (Figure 2C, left panel). The genomic distributions of hypo- and hypermethylated DMRs relative to transcription start sites are shown in Figure 2B and Figure 2C, respectively. Notably, sarcosine-treated animals exhibited a greater proportion of hypermethylated DMRs located within promoter regions 1–3 kb upstream of transcription start sites than vehicle-treated kindled animals.
DMRs were subsequently assigned to their nearest genes to generate DMR-associated gene sets for downstream pathway and transcriptional analyses. Across all contrasts, 563, 533, and 390 DMRs mapped to 521, 499, and 374 unique DMR-associated genes in the vKD vs vSH, sKD vs vSH, and sKD vs vKD groups, respectively (Figure 2D). Venn analysis identified three pairwise overlap gene sets: 47 genes shared between vKD vs vSH and sKD vs vSH, 17 genes shared between vKD vs vSH and sKD vs vKD, and 25 genes shared between sKD vs vSH and sKD vs vKD. No genes were shared across all three contrasts. The 25 DMR-associated genes shared between the sKD vs vSH and sKD vs vKD contrasts were considered a core sarcosine-responsive gene set because methylation changes were consistently detected in both sham-referenced and kindling-referenced analyses. Eleven candidate genes were selected for transcriptional validation from a broader sarcosine-responsive DMR-associated gene set, defined by the union of genes appearing in DMR Venn segments involving sKD comparisons (sKD vs vSH and/or sKD vs vKD), sarcosine-responsive differential expression lists, and Metascape pathway enrichment results. Selection was based on consistent methylation changes across sarcosine-relevant comparisons and established functional roles in epigenetic regulation, excitatory neurotransmission, or ECM remodeling (Figure 2D).
3.3. Comparative Pathway Enrichment Signatures Associated with Epileptogenesis and Sarcosine Treatment
To investigate whether DMR-associated genes participate in coordinated biological processes relevant to epileptogenesis and sarcosine treatment, pathway enrichment analyses were performed using epiPathway, Gene Ontology (GO), and Metascape databases. Comparison of the vKD vs vSH and sKD vs vSH DMR-associated gene sets identified 217 significantly enriched pathways, including 13 shared epiPathway terms and 7 shared GO biological process categories. Representative GO biological process enrichment heatmaps across all three pairwise contrasts are shown in Figure 3. Among hypomethylated DMR-associated genes in the vKD vs vSH and sKD vs vSH comparisons, top enriched terms included myeloid leukocyte migration, chemotaxis, glutamate receptor signaling, Notch signaling, gliogenesis, and tissue morphogenesis (Figure 3A), reflecting neuroinflammatory and structural remodeling programs activated in both kindled groups relative to sham. Among hypermethylated DMR-associated genes in the same comparisons, top enriched terms included positive regulation of kinase activity, modulation of chemical synaptic transmission, regulation of plasma membrane-bounded cell projection organization, postsynapse assembly, and regulation of neurotransmitter receptor activity (Figure 3B). In the pharmacologically direct sKD vs vKD contrast, hypermethylated genes were enriched for developmental growth, synapse organization, myelination, and TGF-β response, whereas hypomethylated genes were enriched for ECM organization, endomembrane system organization, and O-glycan biosynthesis (Figure 3C). Together, these analyses identify both shared and distinct methylation-associated pathway signatures across experimental contrasts, highlighting coordinated alterations in synaptic signaling, neuroinflammatory responses, developmental programs, and extracellular matrix remodeling during epileptogenesis and following sarcosine treatment. These pathway signatures provided the framework for subsequent Metascape analyses focusing separately on kindling-associated and sarcosine-associated methylation programs.
3.4. Kindling-Associated Methylation Pathways Identified by Metascape Enrichment Analysis
To further characterize methylation-associated biological processes during epileptogenesis, Metascape enrichment analyses were performed separately on hypermethylated and hypomethylated DMR-associated genes identified in the vKD vs vSH comparison (Figure 4). The hypermethylated gene set (n = 251) was significantly enriched for pathways related to glutamatergic synapse (ko04724), neuronal system (R-HSA-112316), and regulation of plasma membrane-bounded cell projection organization (GO:0120035) (Figure 4A-C). Additional enriched terms included regulation of neurotransmitter receptor activity, behavior, HDAC Class I pathway signaling, and neuronal depolarization-related processes. Pathway network analysis identified interconnected clusters centered on glutamatergic signaling, neuronal function, and cellular morphogenesis (Figure 4C).
The hypomethylated gene set (n = 284) was enriched for pathways associated with neuroinflammatory and structural remodeling processes, including IL-6 signaling, chemokine signaling, leukocyte differentiation, positive regulation of gliogenesis, regulation of membrane depolarization, small GTPase-mediated signaling, and lamellipodium organization (Figure 4D-F). Network analysis revealed a densely interconnected pathway architecture linking inflammatory signaling, developmental processes, and cellular remodeling pathways (Figure 4F). Collectively, these analyses indicate that kindling-induced DNA methylation changes are associated with coordinated alterations in synaptic signaling, chromatin regulation, neuroinflammatory pathways, and structural remodeling networks relevant to epileptogenesis.
3.5. Sarcosine-Associated Methylation Pathways Identified by Metascape Enrichment Analysis
To characterize methylation-associated pathways linked to sarcosine treatment, Metascape enrichment analysis was performed on DMR-associated genes from the sKD vs vKD comparison, the most pharmacologically informative and mechanistically interpretable contrast in the dataset, stratified by direction of methylation change (Figure 5).
The hypermethylated gene set (n = 295) was significantly enriched for developmental growth (GO:0048589), brain development (GO:0007420), chloride transmembrane transport (GO:1902476), response to transforming growth factor-β (GO:0071559), trans-synaptic signaling (GO:0099537), and positive regulation of synapse assembly (GO:0051965) (Figure 5A-C). Parent Gene Ontology (GO) analysis identified growth, developmental process, and negative regulation of biological process as the most enriched higher-level categories (Figure 5B). Network analysis revealed interconnected pathway clusters associated with developmental regulation, synaptic organization, and growth-related signaling (Figure 5C).
In contrast, the hypomethylated gene set (n = 85) was smaller and enriched for extracellular matrix organization (R-HSA-1474244), sodium ion transport (GO:0006814), regulation of small GTPase-mediated signal transduction (GO:0051056), and regulation of ion transport (GO:0043269) (Figure 5D-F). Parent GO analysis identified biological adhesion and localization as the most enriched higher-level categories (Figure 5E). Network analysis demonstrated two relatively discrete subnetworks dominated by extracellular matrix organization and ion transport pathways (Figure 5F). Collectively, these findings indicate that sarcosine treatment is associated with a distinct methylation signature in the kindled hippocampus characterized by coordinated alterations in developmental regulation, synaptic organization, ion transport, and extracellular matrix remodeling pathways.
3.6. Transcriptional Validation of Methylation-Associated Candidate Genes
To determine whether methylation-associated candidate genes exhibited corresponding transcriptional alterations during epileptogenesis, quantitative PCR was performed on 11 genes selected from the DMR-associated gene sets and pathway enrichment analyses. These candidates represented three functional categories: transcriptional and epigenetic regulation (Hdac9, Fos, Smad7, Unc5a, Dzip1, and Yy1), excitatory synaptic signaling (Grik2, Gpr37l1, and Cacna2d2), and extracellular matrix remodeling (Antxr2 and Spock2) (Figure 6). These genes were prioritized based on overlap within the broader sarcosine-responsive DMR-associated gene set, pathway enrichment significance, and established biological relevance to epigenetic regulation, excitatory neurotransmission, and extracellular matrix remodeling.
All 11 genes exhibited significantly increased expression in vKD hippocampi compared with vSH controls, indicating activation of these molecular pathways during kindling-induced epileptogenesis (Figure 6). Sarcosine treatment during kindling (sKD) significantly attenuated the kindling-induced transcriptional response. Expression of Hdac9, Fos, Smad7, Unc5a, Grik2, Gpr37l1, Cacna2d2, and Yy1 was significantly reduced relative to vKD and restored to levels comparable to those observed in vSH controls. In contrast, Dzip1, Antxr2, and Spock2 displayed partial, non-significant trends toward normalization (Figure 6).
Together, these findings demonstrate that genes embedded within kindling- and sarcosine-associated methylation networks also undergo corresponding transcriptional alterations. The concordant changes observed across methylation, pathway enrichment, and gene expression analyses support the involvement of these molecular pathways in epileptogenesis and their modulation by sarcosine treatment.
4. Discussion
4.1. Mechanistic Insight Into Kindling-Induced Epileptogenesis
In the present study, we combined genome-wide reduced representation bisulfite sequencing (RRBS), pathway enrichment analyses, and targeted transcriptional profiling to investigate DNA methylation-associated molecular networks during hippocampal kindling-induced epileptogenesis and their modulation by sarcosine. Consistent with our previous work, sarcosine significantly attenuated seizure progression in the rapid hippocampal kindling model, confirming its anti-epileptogenic effects in the experimental cohort used for methylation analyses [17]. Beyond seizure suppression, RRBS revealed extensive remodeling of the hippocampal methylome during epileptogenesis, accompanied by coordinated transcriptional alterations in genes involved in epigenetic regulation, excitatory synaptic signaling, and ECM remodeling.
Among the validated genes, several transcriptional and epigenetic regulators - including Hdac9, Fos, Smad7, Unc5a, and Yy1 - were significantly upregulated during kindling and largely normalized by sarcosine treatment. HDAC9 has emerged as an important regulator of neuronal plasticity and chromatin remodeling and has been implicated in activity-dependent transcriptional responses in neurological disorders [3,11,12,21,22,23]. Likewise, Fos is a canonical immediate-early gene activated by neuronal excitation and seizure activity [4,9,24,25], whereas Smad7 serves as a critical modulator of transforming growth factor-β (TGF-β) signaling, a pathway increasingly recognized for its role in neuroinflammation and epileptogenesis [3,7,26]. The coordinated dysregulation of these genes suggests activation of a broader transcriptional program linking neuronal activity, chromatin remodeling, and inflammatory signaling during epileptogenesis.
In addition to transcriptional regulators, we identified significant alterations in genes associated with excitatory synaptic signaling, including Grik2, Gpr37l1, and Cacna2d2. These findings are consistent with extensive evidence implicating glutamatergic dysfunction and aberrant synaptic plasticity in seizure development and progression [4,12,27,28]. The normalization of these genes by sarcosine suggests that GlyT1 inhibition may influence molecular pathways governing excitatory network remodeling during epileptogenesis [17]. Finally, ECM-associated genes, including Antxr2 and Spock2, also exhibited kindling-associated transcriptional changes, supporting the concept that extracellular matrix remodeling contributes to structural plasticity and network reorganization in the epileptic hippocampus [3,29].
Collectively, these findings indicate that epileptogenesis is associated with coordinated activation of transcriptional, synaptic, and extracellular matrix regulatory networks, and that sarcosine attenuates many of these molecular changes. The convergence of methylation-associated pathway enrichment and transcriptional normalization further suggests that epigenetic remodeling may contribute to the anti-epileptogenic actions of GlyT1 inhibition. Although causality cannot be inferred from the present data, these observations support the hypothesis that epigenetic remodeling contributes to the anti-epileptogenic effects of sarcosine.
4.2. Epigenetic Context of the qPCR-Validated Genes
RRBS analysis revealed extensive methylation remodeling across all experimental comparisons. The 11 genes selected for transcriptional validation were derived from a broader sarcosine-responsive DMR-associated gene set, defined by the union of genes across DMR Venn segments involving sKD comparisons, sarcosine-responsive differential expression lists, and Metascape pathway enrichment results, and represented multiple biological pathways identified through enrichment analyses. Importantly, DMR-to-gene relationships were inferred using nearest-transcription-start-site mapping rather than direct methylation-expression correlation analyses. Therefore, the observed associations between methylation changes and gene expression should be interpreted as candidate regulatory relationships rather than definitive causal interactions [3,11,13].
Despite this limitation, the convergence of DMR mapping, pathway enrichment, and transcriptional validation provides evidence that the identified genes occupy biologically relevant positions within methylation-associated regulatory networks. Notably, multiple genes involved in transcriptional regulation (Hdac9, Fos, Smad7, Unc5a, and Yy1), excitatory synaptic signaling (Grik2, Gpr37l1, and Cacna2d2), and extracellular matrix remodeling (Antxr2 and Spock2) exhibited coordinated transcriptional responses that paralleled methylation-associated pathway changes. The normalization of many of these genes following sarcosine treatment further supports the concept that GlyT1 inhibition influences broader epigenetically regulated molecular programs involved in epileptogenesis rather than isolated downstream targets.
4.3. Kindling-Associated Methylation Pathways
Metascape analyses demonstrated that kindling-induced methylation changes were associated with distinct hypermethylated and hypomethylated biological programs. Hypermethylated DMR-associated genes were enriched for glutamatergic synapse, neuronal system, neurotransmitter receptor activity, and HDAC-related pathways, whereas hypomethylated genes were enriched for chemokine signaling, leukocyte differentiation, gliogenesis, and structural remodeling processes. These findings are consistent with previous reports demonstrating that epileptogenesis is accompanied by widespread epigenetic remodeling affecting neuronal signaling and inflammatory pathways [9,11,12,13].
The enrichment of glutamatergic and broader neuronal system pathways within the hypermethylated gene set highlights the central role of excitatory circuit remodeling during epileptogenesis. Dysregulation of glutamatergic signaling is a well-established driver of seizure initiation and propagation, and abnormal plasticity within excitatory networks is thought to contribute to the progressive lowering of seizure threshold during kindling. Interestingly, the coexistence of glutamatergic pathway enrichment within the hypermethylated gene set and transcriptional activation of several excitatory signaling genes suggests that methylation changes during epileptogenesis may not uniformly suppress gene expression. Instead, these findings are consistent with increasingly recognized context-dependent relationships between DNA methylation and transcription, whereby methylation may affect distinct regulatory regions, chromatin states, or feedback networks rather than directly controlling expression of individual genes. Similar complexity has been reported in both experimental and human epilepsy studies, highlighting the multifaceted nature of epigenetic regulation during epileptogenesis [3,11,13].
The enrichment of HDAC-related pathways is particularly noteworthy given the robust upregulation of Hdac9 observed in kindled animals. Histone deacetylases are increasingly recognized as important regulators of activity-dependent gene expression and neuronal plasticity, and dysregulated HDAC signaling has been implicated in several neurological disorders, including epilepsy [3,12,22]. Together, these findings support a potential role for chromatin remodeling in the maintenance of epileptogenic gene expression programs and suggest that epigenetic regulation during epileptogenesis extends beyond DNA methylation alone.
In contrast, hypomethylated DMR-associated genes were enriched for pathways related to chemokine signaling, leukocyte differentiation, gliogenesis, and structural remodeling. The simultaneous enrichment of chemokine signaling, leukocyte differentiation, gliogenesis, and membrane depolarization pathways further supports the concept that epileptogenesis involves coordinated interactions between immune activation and neuronal excitability. These findings are consistent with emerging evidence that neuroinflammatory responses contribute not only to seizure generation but also to long-term circuit remodeling, gliosis, and the establishment of chronic epileptic networks [1,3,12,30].
Together, these results support a model in which epileptogenesis is accompanied by a bidirectional epigenetic landscape involving coordinated remodeling of excitatory synaptic signaling, chromatin regulatory mechanisms, neuroinflammatory pathways, and structural plasticity. Such methylation-associated network alterations may contribute to the progressive transition from a physiologically normal hippocampus to a hyperexcitable epileptic state.
4.4. Sarcosine-Associated Epigenetic Remodeling
The most pharmacologically informative comparison in this study was the direct contrast between sarcosine-treated and vehicle-treated kindled animals. In this comparison, hypermethylated DMR-associated genes were enriched for developmental growth, brain development, chloride transmembrane transport, synaptic organization, and TGF-β signaling pathways, whereas hypomethylated genes were enriched for extracellular matrix organization, ion transport, and small GTPase-mediated signaling pathways. These findings suggest that sarcosine is associated with selective remodeling of biological pathways implicated in neuronal plasticity, circuit reorganization, and epileptogenesis.
Several observations are noteworthy. Developmental growth and brain development emerged among the most significantly enriched pathways in the hypermethylated gene set, suggesting that sarcosine may influence epigenetic programs associated with structural remodeling during epileptogenesis. This observation is particularly intriguing because developmental signaling pathways that are largely quiescent in the mature brain can become reactivated following seizures and brain injury, contributing to aberrant synaptic reorganization, axonal sprouting, and network instability. The enrichment of developmental growth and brain development pathways therefore raises the possibility that sarcosine-associated methylation changes may partially oppose maladaptive developmental reprogramming occurring during epileptogenesis [31,32].
Likewise, enrichment of chloride transmembrane transport pathways raises the possibility that sarcosine-associated methylation changes may influence molecular mechanisms involved in chloride homeostasis and inhibitory neurotransmission. Because chloride homeostasis is a major determinant of GABAergic inhibitory efficacy during epileptogenesis, disruption of chloride transport can promote network hyperexcitability and seizure susceptibility [33,34]. Future studies will be required to determine whether chloride transporters such as KCC2 and NKCC1 are directly affected by GlyT1 inhibition.
The enrichment of TGF-β-responsive pathways is also noteworthy given the normalization of Smad7 expression observed in the present study. TGF-β signaling has emerged as an important contributor to neuroinflammation, blood–brain barrier dysfunction, and epileptogenesis [3,7]. Although causality cannot be established from the current data, these findings raise the possibility that modulation of TGF-β-associated signaling contributes to the anti-epileptogenic actions of sarcosine.
Similarly, enrichment of ECM-associated pathways in the hypomethylated gene set is consistent with the transcriptional changes observed in Antxr2 and Spock2 and further supports a role for ECM remodeling in sarcosine-responsive molecular networks. The prominence of ECM-associated pathways is particularly notable given growing evidence that perineuronal nets (PNNs) regulate neuronal plasticity, inhibitory interneuron stability, and seizure susceptibility. Epileptogenic insults frequently disrupt PNN integrity, leading to altered circuit excitability and impaired inhibitory control. Although PNNs were not directly assessed in the present study, the enrichment of ECM organizational pathways together with sarcosine-associated modulation of Antxr2 and Spock2 suggests that ECM remodeling may represent an underappreciated component of the anti-epileptogenic response [27,35,36,37] . Future studies examining PNN structure and function following GlyT1 inhibition will be important for testing this hypothesis.
Taken together, these findings indicate that sarcosine is associated with a distinct methylation signature involving developmental regulation, synaptic organization, ion transport, TGF-β signaling, and extracellular matrix remodeling. Notably, the sarcosine-associated methylation profile was not simply the inverse of the kindling-associated methylation landscape. While certain pathways appeared to move in opposing directions between comparisons, many sarcosine-associated changes involved biological processes that were not prominently identified in the kindling dataset. This observation suggests that sarcosine may not function as a passive restorer of baseline methylation states but rather as an active remodeler of molecular networks involved in epileptogenesis.
Such a model is consistent with sarcosine’s dual pharmacological properties as both a GlyT1 inhibitor and a participant in one-carbon metabolism. In addition to regulating glycinergic and glutamatergic neurotransmission, sarcosine can influence methyl donor availability through the S-adenosylmethionine/S-adenosylhomocysteine metabolic axis [18]. Together with our previous observation that sarcosine alters hippocampal 5-methylcytosine and 5-hydroxymethylcytosine levels during epileptogenesis [17], the present findings support the hypothesis that sarcosine acts as an active epigenetic remodeler rather than merely suppressing seizure activity.
4.5. Epigenetic Impact of Anti-Epileptogenic Interventions
A growing body of evidence indicates that epigenetic mechanisms contribute to epileptogenesis and may represent promising therapeutic targets [3,10,12]. Several antiepileptic and metabolic interventions, including valproic acid and ketogenic dietary approaches, have been reported to influence DNA methylation, histone modification, and chromatin-associated signaling pathways [14,38]. However, relatively few studies have examined therapeutic interventions at the level of genome-wide methylation remodeling during epileptogenesis while simultaneously linking epigenetic alterations to downstream transcriptional responses.
Our findings place sarcosine within a emerging class of epigenetically active anti-epileptogenic interventions while providing gene- and pathway-level evidence linking methylation-associated remodeling to transcriptional regulation. Recent evidence from pharmacological GlyT1 inhibition studies further supports the emerging role of glycine transport regulation in seizure susceptibility and epileptogenesis [39]. Given its dual role as a GlyT1 inhibitor and participant in one-carbon metabolism, sarcosine represents a unique pharmacological tool for investigating interactions between neurotransmitter regulation and epigenetic plasticity. More broadly, these findings support the concept that modulation of methylation-associated molecular networks may represent a viable strategy for the development of disease-modifying therapies for epilepsy.
4.6. Clinical Implications and Future Directions
The present findings suggest that epigenetically regulated molecular networks may represent viable targets for disease-modifying epilepsy therapies. Unlike conventional antiseizure medications, which primarily suppress seizure activity without preventing disease progression, epigenetic interventions have the potential to modify the molecular pathways that drive epileptogenesis itself [2,3]. The identification of sarcosine-associated methylation changes in pathways related to synaptic organization, developmental regulation, neuroinflammatory signaling, and extracellular matrix remodeling highlights several candidate mechanisms that may be therapeutically exploitable.
Future studies should employ cell-type-specific methylome and transcriptome profiling to define the neuronal and glial populations most responsive to GlyT1 inhibition. Longitudinal analyses spanning multiple stages of epileptogenesis will also be important for distinguishing early epigenetic events that contribute to disease initiation from later adaptations associated with chronic epilepsy. Integration of methylation profiling with transcriptomic, proteomic, and physiological analyses will further help establish causal relationships between epigenetic remodeling and seizure susceptibility.
In addition, emerging locus-specific epigenetic editing technologies offer a powerful framework for mechanistic validation of candidate pathways identified in this study. Future investigations combining GlyT1 inhibition with CRISPR-dCas9-based methylation and demethylation approaches may help determine whether specific regulatory loci, including Hdac9, Fos, and Smad7, play causal roles in epileptogenesis and therapeutic response. Such studies may ultimately facilitate the development of precision epigenetic therapies aimed at modifying disease progression rather than simply suppressing seizures.
4.7. Study Limitations
Several limitations should be considered when interpreting the present findings. First, DMR-associated genes were assigned using nearest-transcription-start-site mapping and were not validated through direct methylation-expression correlation analyses. Consequently, the relationships between methylation changes and transcriptional regulation reported here should be interpreted as candidate regulatory associations rather than definitive causal interactions. Second, transcriptional validation was performed using a targeted qPCR approach rather than unbiased transcriptomic profiling. Although the selected genes were chosen based on DMR mapping and pathway enrichment analyses, this strategy may not fully capture the broader transcriptional consequences of the observed methylation changes. Third, RRBS interrogates only a subset of the methylome enriched for CpG-dense regions and therefore may not detect methylation changes occurring outside the genomic regions sampled by this approach. More comprehensive methylome profiling strategies may reveal additional epigenetic alterations associated with epileptogenesis and sarcosine treatment. Fourth, all analyses were performed using bulk hippocampal tissue, which does not resolve cell-type-specific methylation and transcriptional changes. Given the distinct contributions of neurons, astrocytes, microglia, and oligodendrocytes to epileptogenesis, future studies employing single-cell or cell-type-resolved multi-omic approaches will be important for defining the cellular origin of the molecular changes identified here. Finally, the present study establishes associations among methylation changes, pathway enrichment, and transcriptional regulation but does not directly demonstrate causality. Future investigations incorporating longitudinal analyses, targeted epigenetic manipulation, and functional validation of candidate loci will be necessary to define the mechanistic contribution of individual methylation events to epileptogenesis and therapeutic response.
5. Conclusions
Our findings demonstrate that hippocampal DNA methylation is extensively remodeled during kindling-induced epileptogenesis and that sarcosine treatment is associated with selective, bidirectional, remodeling of methylation-associated molecular pathways together with normalization of key transcriptional networks. By integrating genome-wide reduced representation bisulfite sequencing (RRBS) with targeted gene expression analysis, we identified methylation-associated genes involved in transcriptional regulation, synaptic signaling, and extracellular matrix remodeling that were consistently dysregulated during epileptogenesis and largely normalized by sarcosine treatment.
These results support the concept that GlyT1 inhibition influences epigenetically regulated molecular pathways associated with epileptogenesis and highlight sarcosine-associated methylation changes as a potential mechanism underlying its previously reported anti-epileptogenic effects. More broadly, this study provides evidence that modulation of methylation-associated gene networks may represent a promising strategy for the development of disease-modifying therapies for epilepsy and establishes a framework for future studies investigating epigenetic mechanisms of epileptogenesis and potential disease modification.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: RRBS sequencing quality metrics and sample-level methylation profiles. Table S1: Differentially methylated genes, vKD vs vSH; Table S2: Differentially methylated genes, sKD vs vSH; Table S3: Differentially methylated genes, sKD vs vKD.
Author Contributions
Conceptualization: H-YS. Methodology: GH, NF, and H-YS. Formal analysis: GH, NF, and H-YS. Investigation: NF, LP, WO, GH, and H-YS. Writing – original draft preparation: H-YS and GH. Writing, review and editing: H-YS. Supervision: H-YS. Funding acquisition: H-YS. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Good Samaritan Research Foundation, the NIH (R01 NS084920), the Children’s Health Research Institute (CHRI) of UNMC, and the Cognitive Neuroscience of Development and Aging Center (P20GM130447).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Legacy Research Institute (protocol code #114-2020).
Data Availability Statement
All relevant data are within the manuscript and its Supplementary Materials. The datasets generated for this study are available on request to the corresponding author.
Acknowledgments
The authors thank the Gene Profiling Shared Resource Core, the KCVI Epigenetics Core, and the Massively Parallel Sequencing Shared Resource Core at Oregon Health & Science University (OHSU) for technical support with RRBS library preparation and sequencing. The authors also wish to thank Meng Niu, Ph.D., at UNMC for discussions on bioinformatic analysis.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15(8), 459–472. [Google Scholar] [CrossRef] [PubMed]
- Younus, I.; Reddy, D.S. Epigenetic interventions for epileptogenesis: A new frontier for curing epilepsy. Pharmacol. Ther. 2017, 177, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.C.; Tauboll, E.; Heuser, K. The potential role of DNA methylation as preventive treatment target of epileptogenesis. Front Cell Neurosci. 2022, 16, 931356. [Google Scholar] [CrossRef] [PubMed]
- Miller-Delaney, S.F.; et al. Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain 2015, 138 Pt 3, 616–31. [Google Scholar] [PubMed]
- Caramaschi, D.; et al. Epigenome-wide association study of seizures in childhood and adolescence. Clin. Epigenet. 2020, 12(1), 8. [Google Scholar] [CrossRef]
- Zhu, Q.; et al. Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy. J. Mol. Neurosci. 2012, 46(2), 420–6. [Google Scholar] [PubMed]
- Pedersen, S.; et al. Genome-wide decrease in DNA methylation in adults with epilepsy treated with modified ketogenic diet: A prospective study. Epilepsia 2022, 63(9), 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; et al. DNA Methylation Signature of Epileptic Encephalopathy-Related Pathogenic Genes Encoding Ion Channels in Temporal Lobe Epilepsy. Front Neurol. 2021, 12, 692412. [Google Scholar] [CrossRef] [PubMed]
- Miller-Delaney, S.F.; et al. Differential DNA methylation patterns define status epilepticus and epileptic tolerance. J. Neurosci. 2012, 32(5), 1577–88. [Google Scholar] [CrossRef] [PubMed]
- Kobow, K.; Blumcke, I. The emerging role of DNA methylation in epileptogenesis. Epilepsia 2012, 53 Suppl 9, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.C.; et al. Neuronal and glial DNA methylation and gene expression changes in early epileptogenesis. PLoS ONE 2019, 14(12), e0226575. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.M.; Henshall, D.C.; Lubin, F.D. The Epigenetics of Epilepsy and Its Progression. Neuroscientist 2018, 24(2), 186–200. [Google Scholar] [PubMed]
- Debski, K.J.; et al. Etiology matters - Genomic DNA Methylation Patterns in Three Rat Models of Acquired Epilepsy. Sci. Rep. 2016, 6, 25668. [Google Scholar] [PubMed]
- Navarrete-Modesto, V.; et al. The molecular hallmarks of epigenetic effects mediated by antiepileptic drugs. Epilepsy Res. 2019, 149, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-resistant epilepsy. N Engl. J. Med. 2011, 365(10), 919–26. [Google Scholar] [CrossRef] [PubMed]
- Purnell, B.S.; et al. Commonalities in gene expression and methylation changes across two rat models of acquired epilepsy. Sci. Rep. 2026, 16(1), 5095. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.Y.; et al. Sarcosine Suppresses Epileptogenesis in Rats With Effects on Hippocampal DNA Methylation. Front Mol. Neurosci. 2020, 13, 97. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; et al. Effects of sarcosine and N, N-dimethylglycine on NMDA receptor-mediated excitatory field potentials. J. BioMed Sci. 2017, 24(1), 18. [Google Scholar] [CrossRef] [PubMed]
- Racine, R. Kindling: the first decade. Neurosurgery 1978, 3(2), 234–52. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.; et al. Synergistic Effects of Hyperandrogenemia and Obesogenic Western-style Diet on Transcription and DNA Methylation in Visceral Adipose Tissue of Nonhuman Primates. Sci. Rep. 2019, 9(1), 19232. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; et al. HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells. Int. J. Mol. Sci. 2022, 23(15). [Google Scholar] [CrossRef] [PubMed]
- Citraro, R.; et al. Perampanel effects in the WAG/Rij rat model of epileptogenesis, absence epilepsy, and comorbid depressive-like behavior. Epilepsia 2017, 58(2), 231–238. [Google Scholar] [PubMed]
- de Nijs, L.; et al. DNA methyltransferase isoforms expression in the temporal lobe of epilepsy patients with a history of febrile seizures. Clin. Epigenet. 2019, 11(1), 118. [Google Scholar] [CrossRef]
- Dragunow, M.; Robertson, H.A. Kindling stimulation induces c-fos protein(s) in granule cells of the rat dentate gyrus. Nature 1987, 329(6138), 441–2. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.I.; et al. Mapping patterns of c-fos expression in the central nervous system after seizure. Science 1987, 237(4811), 192–7. [Google Scholar] [CrossRef] [PubMed]
- Cacheaux, L.P.; et al. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J. Neurosci. 2009, 29(28), 8927–35. [Google Scholar] [CrossRef] [PubMed]
- Bortoletto, R.; et al. Is It Time to Test the Antiseizure Potential of Palmitoylethanolamide in Human Studies? A Systematic Review of Preclinical Evidence. Brain Sci. 2022, 12(1). [Google Scholar] [CrossRef] [PubMed]
- Coulter, D.A.; Steinhauser, C. Role of astrocytes in epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5(3), a022434. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, E.; Kurkowska-Jastrzebska, I. Matrix Metalloproteinase 9 in Epilepsy: The Role of Neuroinflammation in Seizure Development. In Mediators Inflamm; 2016; p. 7369020. [Google Scholar]
- Maroso, M.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16(4), 413–9. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Holmes, G.L. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006, 5(12), 1055–63. [Google Scholar] [CrossRef] [PubMed]
- Dudek, F.E.; Sutula, T.P. Epileptogenesis in the dentate gyrus: a critical perspective. Prog. Brain Res. 2007, 163, 755–73. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Staley, K.J. The bumetanide-sensitive Na-K-2Cl cotransporter NKCC1 as a potential target of a novel mechanism-based treatment strategy for neonatal seizures. Neurosurg. Focus 2008, 25(3), E22. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.F.; et al. Knockdown of SIK3 in the CA1 Region can Reduce Seizure Susceptibility in Mice by Inhibiting Decreases in GABA(A)R alpha1 Expression. Mol. Neurobiol. 2024, 61(3), 1404–1416. [Google Scholar] [PubMed]
- Broekaart, D.W.; et al. The matrix metalloproteinase inhibitor IPR-179 has antiseizure and antiepileptogenic effects. J. Clin. Invest 2021, 131(1). [Google Scholar] [CrossRef] [PubMed]
- Chaunsali, L.; Tewari, B.P.; Sontheimer, H. Perineuronal Net Dynamics in the Pathophysiology of Epilepsy. Epilepsy Curr. 2021, 21(4), 273–281. [Google Scholar] [CrossRef] [PubMed]
- Lensjo, K.K.; et al. Differential Expression and Cell-Type Specificity of Perineuronal Nets in Hippocampus, Medial Entorhinal Cortex, and Visual Cortex Examined in the Rat and Mouse. eNeuro 2017, 4(3). [Google Scholar] [CrossRef] [PubMed]
- Kobow, K.; et al. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 2013, 126(5), 741–56. [Google Scholar] [CrossRef] [PubMed]
- Gapinska, N.; et al. Effect of SSR504734, a Selective Glycine Transporter Type 1 Inhibitor, on Seizure Thresholds, Neurotransmitter Levels, and Inflammatory Markers in Mice. ACS Chem. Neurosci. 2025, 16(6), 1210–1226. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Sarcosine suppresses hippocampal kindling-induced epileptogenesis.

Figure 2.
Genome-wide hippocampal DNA methylation landscape in kindled and sarcosine-treated rats. (A) Numbers of hypermethylated and hypomethylated differentially methylated regions (DMRs) identified in hippocampal tissue from the vKD vs vSH, sKD vs vSH, and sKD vs vKD comparisons (|Δmethylation| ≥ 10%, q < 0.05). (B) Overlap and genomic distribution of hypomethylated DMRs in the vKD vs vSH and sKD vs vSH comparisons. Left panel: Venn diagram showing shared and unique hypomethylated DMRs. Right panel: genomic annotation of hypomethylated DMRs relative to transcription start sites (TSSs). (C) Overlap and genomic distribution of hypermethylated DMRs in the vKD vs vSH and sKD vs vSH comparisons. Left panel: Venn diagram showing shared and unique hypermethylated DMRs. Right panel: genomic annotation of hypermethylated DMRs relative to TSSs. (D) Venn diagram showing overlap of DMR-associated genes among the three comparisons. A total of 521, 499, and 374 DMR-associated genes were identified in the vKD vs vSH, sKD vs vSH, and sKD vs vKD comparisons, respectively. Pairwise overlaps included 47 genes overlap between vKD vs vSH and sKD vs vSH, 17 genes overlap between vKD vs vSH and sKD vs vKD, and 25 genes overlap between sKD vs vSH and sKD vs vKD. No DMR-associated genes were overlap among all three comparisons. Eleven candidate genes selected for transcriptional validation were derived from a broader sarcosine-responsive DMR-associated gene set, defined by the union of genes across DMR Venn segments involving sKD comparisons, sarcosine-responsive differential expression lists, and Metascape pathway enrichment results, including the 25-gene overlap shown here.
Figure 2.
Genome-wide hippocampal DNA methylation landscape in kindled and sarcosine-treated rats. (A) Numbers of hypermethylated and hypomethylated differentially methylated regions (DMRs) identified in hippocampal tissue from the vKD vs vSH, sKD vs vSH, and sKD vs vKD comparisons (|Δmethylation| ≥ 10%, q < 0.05). (B) Overlap and genomic distribution of hypomethylated DMRs in the vKD vs vSH and sKD vs vSH comparisons. Left panel: Venn diagram showing shared and unique hypomethylated DMRs. Right panel: genomic annotation of hypomethylated DMRs relative to transcription start sites (TSSs). (C) Overlap and genomic distribution of hypermethylated DMRs in the vKD vs vSH and sKD vs vSH comparisons. Left panel: Venn diagram showing shared and unique hypermethylated DMRs. Right panel: genomic annotation of hypermethylated DMRs relative to TSSs. (D) Venn diagram showing overlap of DMR-associated genes among the three comparisons. A total of 521, 499, and 374 DMR-associated genes were identified in the vKD vs vSH, sKD vs vSH, and sKD vs vKD comparisons, respectively. Pairwise overlaps included 47 genes overlap between vKD vs vSH and sKD vs vSH, 17 genes overlap between vKD vs vSH and sKD vs vKD, and 25 genes overlap between sKD vs vSH and sKD vs vKD. No DMR-associated genes were overlap among all three comparisons. Eleven candidate genes selected for transcriptional validation were derived from a broader sarcosine-responsive DMR-associated gene set, defined by the union of genes across DMR Venn segments involving sKD comparisons, sarcosine-responsive differential expression lists, and Metascape pathway enrichment results, including the 25-gene overlap shown here.

Figure 3.
Comparative pathway enrichment signatures associated with epileptogenesis and sarcosine treatment. Heatmap color intensity reflects −log10(P-value), and gray cells indicate no significant enrichment (P > 0.05). (A) Representative enriched biological processes and pathways among hypomethylated DMR-associated genes in the sKD vs vSH and vKD vs vSH comparisons. Prominent enriched terms include myeloid leukocyte migration, chemotaxis, glutamate receptor signaling pathway, Notch signaling, gliogenesis, and tissue morphogenesis. (B) Representative enriched biological processes and pathways among hypermethylated DMR-associated genes in the same comparisons. Enriched terms include positive regulation of kinase activity, modulation of chemical synaptic transmission, homophilic cell adhesion via plasma membrane adhesion molecules, regulation of plasma membrane-bounded cell projection organization, postsynapse assembly, and regulation of neurotransmitter receptor activity. (C) Representative enriched biological processes and pathways in the sKD vs vKD comparison. The left column shows hypomethylated DMR-associated genes, and the right column shows hypermethylated DMR-associated genes. Enriched hypermethylated terms include actin filament-based process, synapse organization, developmental growth, myelination, and response to transforming growth factor-β (TGF-β), whereas enriched hypomethylated terms include extracellular matrix organization, endomembrane system organization, and O-glycan biosynthesis. Terms were selected using Metascape representative-term filtering. Abbreviations: sKD, sarcosine-kindled; vKD, vehicle-kindled; vSH, vehicle-sham.
Figure 3.
Comparative pathway enrichment signatures associated with epileptogenesis and sarcosine treatment. Heatmap color intensity reflects −log10(P-value), and gray cells indicate no significant enrichment (P > 0.05). (A) Representative enriched biological processes and pathways among hypomethylated DMR-associated genes in the sKD vs vSH and vKD vs vSH comparisons. Prominent enriched terms include myeloid leukocyte migration, chemotaxis, glutamate receptor signaling pathway, Notch signaling, gliogenesis, and tissue morphogenesis. (B) Representative enriched biological processes and pathways among hypermethylated DMR-associated genes in the same comparisons. Enriched terms include positive regulation of kinase activity, modulation of chemical synaptic transmission, homophilic cell adhesion via plasma membrane adhesion molecules, regulation of plasma membrane-bounded cell projection organization, postsynapse assembly, and regulation of neurotransmitter receptor activity. (C) Representative enriched biological processes and pathways in the sKD vs vKD comparison. The left column shows hypomethylated DMR-associated genes, and the right column shows hypermethylated DMR-associated genes. Enriched hypermethylated terms include actin filament-based process, synapse organization, developmental growth, myelination, and response to transforming growth factor-β (TGF-β), whereas enriched hypomethylated terms include extracellular matrix organization, endomembrane system organization, and O-glycan biosynthesis. Terms were selected using Metascape representative-term filtering. Abbreviations: sKD, sarcosine-kindled; vKD, vehicle-kindled; vSH, vehicle-sham.

Figure 4.
Kindling-associated methylation pathways identified by Metascape enrichment analysis. Metascape enrichment analysis of DMR-associated genes from the vKD vs vSH comparison, stratified by direction of methylation change. (A) Top enriched biological processes and pathways among hypermethylated DMR-associated genes, ranked by −log10(P-value). Prominent enriched terms include glutamatergic synapse, neuronal system, regulation of plasma membrane-bounded cell projection organization, and the PID HDAC Class I pathway. (B) Parent Gene Ontology (GO) term enrichment of the hypermethylated gene set. (C) Pathway network (ColorByCluster) of the hypermethylated gene set, demonstrating interconnected modules related to glutamatergic signaling, neuronal function, and cell morphogenesis. (D) Top enriched biological processes and pathways among hypomethylated DMR-associated genes, ranked by −log10(P-value). Prominent enriched terms include tissue morphogenesis, positive regulation of gliogenesis, chemokine signaling pathway, leukocyte differentiation, and regulation of membrane depolarization. (E) Parent GO term enrichment of the hypomethylated gene set. (F) Pathway network (ColorByCluster) of the hypomethylated gene set, revealing interconnected modules associated with neuroinflammatory signaling, developmental processes, and structural remodeling. Together, these analyses reveal coordinated methylation-associated pathway changes involving synaptic signaling, chromatin regulation, neuroinflammatory processes, and structural remodeling during epileptogenesis. Abbreviations: vKD, vehicle-kindled; vSH, vehicle-sham.
Figure 4.
Kindling-associated methylation pathways identified by Metascape enrichment analysis. Metascape enrichment analysis of DMR-associated genes from the vKD vs vSH comparison, stratified by direction of methylation change. (A) Top enriched biological processes and pathways among hypermethylated DMR-associated genes, ranked by −log10(P-value). Prominent enriched terms include glutamatergic synapse, neuronal system, regulation of plasma membrane-bounded cell projection organization, and the PID HDAC Class I pathway. (B) Parent Gene Ontology (GO) term enrichment of the hypermethylated gene set. (C) Pathway network (ColorByCluster) of the hypermethylated gene set, demonstrating interconnected modules related to glutamatergic signaling, neuronal function, and cell morphogenesis. (D) Top enriched biological processes and pathways among hypomethylated DMR-associated genes, ranked by −log10(P-value). Prominent enriched terms include tissue morphogenesis, positive regulation of gliogenesis, chemokine signaling pathway, leukocyte differentiation, and regulation of membrane depolarization. (E) Parent GO term enrichment of the hypomethylated gene set. (F) Pathway network (ColorByCluster) of the hypomethylated gene set, revealing interconnected modules associated with neuroinflammatory signaling, developmental processes, and structural remodeling. Together, these analyses reveal coordinated methylation-associated pathway changes involving synaptic signaling, chromatin regulation, neuroinflammatory processes, and structural remodeling during epileptogenesis. Abbreviations: vKD, vehicle-kindled; vSH, vehicle-sham.

Figure 5.
Sarcosine-associated epigenetic remodeling pathways identified by Metascape enrichment analysis. Metascape enrichment analysis of DMR-associated genes from the sKD vs vKD comparison, stratified by direction of methylation change. (A) Top enriched biological processes and pathways among hypermethylated DMR-associated genes, ranked by −log10(P-value). Prominent enriched terms include developmental growth, brain development, chloride transmembrane transport, response to transforming growth factor-β (TGF-β), trans-synaptic signaling, and positive regulation of synapse assembly. (B) Parent Gene Ontology (GO) term enrichment of the hypermethylated gene set. (C) Pathway network (ColorByCluster) of the hypermethylated gene set, illustrating interconnected modules associated with developmental regulation, synaptic organization, chloride homeostasis, and growth-related signaling pathways. (D) Top enriched biological processes and pathways among hypomethylated DMR-associated genes, ranked by −log10(P-value). Prominent enriched terms include extracellular matrix organization, sodium ion transport, regulation of small GTPase-mediated signal transduction, and regulation of ion transport. (E) Parent GO term enrichment of the hypomethylated gene set. (F) Pathway network (ColorByCluster) of the hypomethylated gene set, demonstrating a small number of functionally distinct modules dominated by extracellular matrix organization and ion transport pathways. Together, these analyses indicate that sarcosine induces a targeted bidirectional methylation program involving developmental regulation, synaptic organization, chloride homeostasis, ion transport, and extracellular matrix remodeling. Abbreviations: sKD, sarcosine-kindled; vKD, vehicle-kindled.
Figure 5.
Sarcosine-associated epigenetic remodeling pathways identified by Metascape enrichment analysis. Metascape enrichment analysis of DMR-associated genes from the sKD vs vKD comparison, stratified by direction of methylation change. (A) Top enriched biological processes and pathways among hypermethylated DMR-associated genes, ranked by −log10(P-value). Prominent enriched terms include developmental growth, brain development, chloride transmembrane transport, response to transforming growth factor-β (TGF-β), trans-synaptic signaling, and positive regulation of synapse assembly. (B) Parent Gene Ontology (GO) term enrichment of the hypermethylated gene set. (C) Pathway network (ColorByCluster) of the hypermethylated gene set, illustrating interconnected modules associated with developmental regulation, synaptic organization, chloride homeostasis, and growth-related signaling pathways. (D) Top enriched biological processes and pathways among hypomethylated DMR-associated genes, ranked by −log10(P-value). Prominent enriched terms include extracellular matrix organization, sodium ion transport, regulation of small GTPase-mediated signal transduction, and regulation of ion transport. (E) Parent GO term enrichment of the hypomethylated gene set. (F) Pathway network (ColorByCluster) of the hypomethylated gene set, demonstrating a small number of functionally distinct modules dominated by extracellular matrix organization and ion transport pathways. Together, these analyses indicate that sarcosine induces a targeted bidirectional methylation program involving developmental regulation, synaptic organization, chloride homeostasis, ion transport, and extracellular matrix remodeling. Abbreviations: sKD, sarcosine-kindled; vKD, vehicle-kindled.

Figure 6.
Transcriptional validation of methylation-associated candidate genes. Quantitative PCR analysis of hippocampal tissues from vehicle-sham (vSH), vehicle-kindled (vKD), and sarcosine-kindled (sKD) rats (n = 6 per group) for 11 candidate genes selected from DMR-associated gene sets and pathway enrichment analyses. (A) Transcriptional and epigenetic regulators (Hdac9, Fos, Smad7, Unc5a, Dzip1, and Yy1). (B) Excitatory synaptic signaling genes (Grik2, Gpr37l1, and Cacna2d2). (C) Extracellular matrix-related genes (Antxr2 and Spock2). All 11 genes exhibited increased expression in vKD relative to vSH, with significant upregulation observed for each candidate gene. Sarcosine treatment significantly reduced expression of Hdac9, Fos, Smad7, Unc5a, Grik2, Gpr37l1, Cacna2d2, and Yy1 compared with vKD, whereas Antxr2, Spock2, and Dzip1 showed partial normalization trends. Data are presented as mean ± SEM. Statistical analyses were performed using one-way ANOVA followed by Tukey’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant.
Figure 6.
Transcriptional validation of methylation-associated candidate genes. Quantitative PCR analysis of hippocampal tissues from vehicle-sham (vSH), vehicle-kindled (vKD), and sarcosine-kindled (sKD) rats (n = 6 per group) for 11 candidate genes selected from DMR-associated gene sets and pathway enrichment analyses. (A) Transcriptional and epigenetic regulators (Hdac9, Fos, Smad7, Unc5a, Dzip1, and Yy1). (B) Excitatory synaptic signaling genes (Grik2, Gpr37l1, and Cacna2d2). (C) Extracellular matrix-related genes (Antxr2 and Spock2). All 11 genes exhibited increased expression in vKD relative to vSH, with significant upregulation observed for each candidate gene. Sarcosine treatment significantly reduced expression of Hdac9, Fos, Smad7, Unc5a, Grik2, Gpr37l1, Cacna2d2, and Yy1 compared with vKD, whereas Antxr2, Spock2, and Dzip1 showed partial normalization trends. Data are presented as mean ± SEM. Statistical analyses were performed using one-way ANOVA followed by Tukey’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant.


Table 1.
Primers used for the qPCR assay.
| Gene | Forward Primer (5’-3’) | Reverse Primer (5’-3’) |
|---|---|---|
| Antxr2 | cgccaattaagggtcgtctg | cgggagaagtttatgcaccg |
| Cacna2d2 | gcgaggtaaacaacgaggac | cactgaagaacctgccaacc |
| Dzip1 | ttccacccgaagaagtaccc | tttcttgagttttggggcgg |
| Fos | tgcaagatccccaatgacct | tgagaagaggcagggtgaag |
| Gpr37l1 | agcagtgtgagagtcagctt | tgttgcagacgttttcaggg |
| Grik2 | actcaggtttgctggatgga | cagggtttgtgtcgattgca |
| Hdac9 | tgtgaatacgaatgcagccg | cttgcaacagccaccatctt |
| Smad7 | ggcttcaccgtgcagattag | tgaagatgacctccagccag |
| Spock2 | actgtgatgacatcgtgggt | ccagatgtagcctccgtcat |
| Unc5a | aagaaggaagggctggactc | ctggtaggtggtagtggtgg |
| Yy1 | gcaagaagagttacctgggc | acctgcttctgttcccactt |
| Gapdh | ggatactgagagcaagagaga | ttatggggtctgggatggaa |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



