Submitted:
02 July 2026
Posted:
03 July 2026
You are already at the latest version
Abstract
Glaucoma is a leading cause of irreversible vision loss characterized by the pro-gressive degeneration of retinal ganglion cells (RGCs) and structural and biochemical remodeling of the optic nerve head. Although lowering intraocular pressure remains the primary clinical intervention, neurodegeneration often persists, highlighting the com-plexity and multiple mechanisms involved in the disease's pathophysiology. In the healthy retina, astrocytes and Müller cells maintain structural integrity, homeostatic balance, and metabolic support. However, sustained pathological stress triggers reactive gliosis, a phenomenon with a dichotomous phenotype. Initially, the macroglial response is adaptive and neuroprotective. Persistent biomechanical and ischemic insults shift this profile into a typically deleterious one, characterized by extracellular matrix remodeling, complement system activation, and heightened neuroinflammation, factors that inten-sify RGC death. Mechanosensitive pathways, notably Piezo1 and various transient re-ceptor potential (TRP) channels emerge as critical sensors translating physical stress into these reactive cascades within interconnected multicellular networks. This review ex-amines the crucial role of astrocytes and Müller cells in the dynamic modulation of the retinal microenvironment during glaucomatous progression. Finally, it discusses the therapeutic potential of macroglia-directed pharmacological or gene therapies to re-program the retinal environment.
Keywords:
intraocular pressure
; reactive gliosis
; oxidative stress
; neuroinflammation
1. Introduction
Glaucoma comprises a heterogeneous group of progressive optic neuropathies characterized by structural remodeling of the optic nerve head, progressive loss of retinal ganglion cells (RGCs) and their axons, leading to visual deficits [1,2,3]. It is one of the leading causes of irreversible blindness worldwide, and its prevalence is expected to increase substantially with population aging [3,4]. Although elevated intraocular pressure (IOP) remains the most important modifiable risk factor and the main therapeutic target in clinical practice, glaucomatous neurodegeneration may progress despite apparently adequate pressure control [2,5,6]. This indicates that pressure-dependent and pressure-independent mechanisms converge to drive RGC dysfunction and death.
Over the past decades, glaucoma has increasingly been recognized not only as a pressure-related optic neuropathy but also as a complex neurodegenerative condition involving mechanical stress, vascular dysfunction, impaired axonal transport, mitochondrial dysfunction, oxidative stress, excitotoxicity, immune activation, and neuroinflammation [2,6,7,8]. These mechanisms do not act in isolation. Rather, they occur within a highly specialized retinal and optic nerve microenvironment in which neurons, glial cells, vascular elements, extracellular matrix components, and immune mediators continuously interact [5,8,9].
Glial cells are central regulators of this microenvironment. In the healthy retina and optic nerve, macroglial cells, including astrocytes and Müller glia, provide structural and metabolic support, regulate extracellular ion and neurotransmitter homeostasis, contribute to blood-retinal barrier maintenance, modulate synaptic activity, and participate in antioxidant defense [5,10,11]. Astrocytes are particularly abundant, but restricted to the retinal nerve fiber layer, optic nerve head, and optic nerve, where they ensheath RGC axons, support neuronal homeostasis, and contribute to tissue architecture and extracellular matrix organization [9,12,13]. Müller glia span the entire thickness of the retina and are strategically positioned to support neuronal survival, maintain retinal homeostasis, and detect and coordinate responses to injury all over the tissue [5,10]. In glaucoma, however, macroglial cells undergo profound phenotypic and functional changes. Astrocytes and Müller glia become reactive in response to elevated IOP, mechanical deformation of the optic nerve head, ischemic or metabolic stress, oxidative damage, and inflammatory signals [6,8,9,13]. This reactive response may initially represent an adaptive response that helps to preserve tissue integrity and protect neurons. Nevertheless, when sustained or dysregulated, macroglial reactivity can contribute to extracellular matrix remodeling, altered metabolic support, glutamate dysregulation, oxidative stress, complement activation, cytokine release, and amplification of neuroinflammatory signaling [5,8,14].
The dual nature of macroglial activation is particularly relevant in glaucoma. On one hand, reactive astrocytes and Müller cells may limit damage by buffering extracellular glutamate and potassium, producing antioxidant molecules, secreting trophic factors, and preserving the retinal microenvironment [5,10]. On the other hand, chronic macroglial activation may promote a maladaptive state characterized by increased expression of glial fibrillary acidic protein (GFAP), inflammatory mediators, extracellular matrix proteins, and signals that affect RGC soma, dendrites, axons, and synapses [8,9,13,14]. Therefore, macroglia should not be interpreted simply as passive markers of disease progression, but rather as active participants in the balance between neuroprotection and neurodegeneration.
Interactions between macroglia and microglia further increase the complexity of glaucomatous neuroinflammation. Microglial activation is an early event in several experimental models of glaucoma and may influence astrocytic and Müller glial responses through cytokines, complement components, ATP-dependent signaling, and damage-associated molecular patterns [5,6,8]. Conversely, reactive macroglia can shape microglial recruitment, activation state, and inflammatory output, suggesting that the progression of glaucomatous damage may depend, at least in part, on multicellular glial networks rather than on isolated cellular responses [8,14].
In addition to canonical inflammatory and metabolic pathways, transient receptor potential (TRP) channels have emerged as important sensors of mechanical, osmotic, oxidative, and inflammatory stimuli in the retina and optic nerve. Several TRP channels, including TRPV1, TRPV4, TRPC1, and TRPA1, have been associated with retinal homeostasis, mechanotransduction, calcium signaling, and processes relevant to glaucomatous neurodegeneration. Because astrocytes and Müller glia are highly responsive to changes in tissue pressure, oxidative stress, and extracellular signaling, TRP-dependent pathways may contribute to the transition from adaptive glial responses to maladaptive neuroinflammatory signaling in glaucoma [15,16].
Understanding the role of macroglia in glaucoma is therefore essential for identifying new therapeutic strategies beyond IOP lowering. While current treatments are mainly directed toward reducing IOP, they do not directly target glial dysfunction, neuroinflammation, or intrinsic RGC vulnerability [2,3,5]. A better characterization of astrocyte and Müller glial responses may reveal mechanisms that can be selectively modulated to preserve beneficial homeostatic functions while limiting maladaptive inflammatory and neurodegenerative signaling [6,13,14].
In this review, we discuss how macroglial dysfunction contributes to retinal ganglion cell degeneration and optic nerve remodeling in glaucoma, with particular emphasis on astrocytes and Müller glia as active regulators of neuroinflammation, oxidative stress, and the participation of TRP/PIEZO channel. We further examine emerging therapeutic strategies, including gene-based approaches, aimed at modulating macroglial pathways for neuroprotection and disease modification in glaucoma.
2. Glaucoma
Glaucoma comprises a group of progressive and irreversible optic neuropathies characterized by the degeneration of retinal ganglion cells and their axons, ultimately leading to visual loss. This degenerative process results in structural alterations of the optic nerve, including increased optic disc cupping, a morphological hallmark of the disease [17,18,19]. In glaucoma, optic disc cupping develops progressively as a result of axonal loss and lamina cribrosa remodeling, reflecting the underlying structural damage [20]. RGCs transmit visual information to brain visual pathways through their axons, which form the optic nerve. Therefore, their degeneration directly compromises visual function. As the leading cause of irreversible visual loss worldwide, glaucoma exerts a significant impact on the quality of life of affected individuals [21].
From a clinical and anatomical standpoint, glaucoma is classified as open-angle or angle-closure based on the configuration of the anterior chamber angle. Both categories may be further subdivided into primary or secondary forms, depending on the presence or absence of associated ocular or systemic conditions [20]. Primary open-angle glaucoma is the most prevalent subtype and is characterized by an anatomically open drainage angle and altered aqueous humor dynamics. Intraocular pressure is regulated by the balance between aqueous humor production and drainage, and its elevation is most commonly attributed to increased resistance to outflow through the trabecular meshwork, ultimately leading to progressive optic nerve damage. In contrast, angle-closure glaucoma involves partial or complete obstruction of the drainage angle, typically due to mechanical blockage of aqueous humor flow, resulting in acute IOP elevation and subsequent optic neuropathy [3].
From an epidemiological perspective, global estimates indicate that approximately 76 million individuals aged 40 to 80 years were living with glaucoma in 2020, with projections exceeding 110 million by 2040. These figures reflect the impact of population aging and highlight glaucoma as a major public health concern [4]. Furthermore, the disease may remain asymptomatic until advanced stages, which frequently delays diagnosis and treatment initiation [2]. As a result, the true burden of glaucoma is likely underestimated, and a recent systematic review reported that a large proportion of cases worldwide remain undiagnosed [22]. In this context, the identification of modifiable risk factors is essential for the development of effective prevention and management strategies.
Glaucoma is recognized as a multifactorial disease, with several risk factors associated with its development. Elevated intraocular pressure is the main modifiable risk factor and the only proven therapeutic target for slowing disease progression, whereas advanced age constitutes the most important risk factor (non-modifiable) [3]. Reduction of IOP is directly associated with decreased glaucoma progression [23,24,25], and topical ocular hypotensive therapy has been shown to be effective in delaying or preventing the development of primary open-angle glaucoma in, at least, part of patients with elevated IOP [24,26]. Indeed, IOP activates several mechanosensitive ions, especially TRPs and PIEZO, in the retina triggering a number of alterations that generate oxidative stress and inflammation involved in cell death [27].
Currently, the reduction of IOP through pharmacological approaches or surgical interventions constitutes the mainstay of glaucoma treatment. Topical pharmacological therapy is considered the first-line treatment, including different classes of drugs such as prostaglandin analogues, α2-adrenergic agonists, miotic agents, and carbonic anhydrase inhibitors, which act through distinct mechanisms, including reducing aqueous humor production and increasing its outflow [28,29,30]. In addition, fixed combinations of medications have been increasingly used, offering benefits such as greater convenience, improved treatment adherence, reduced exposure to preservatives, and potential cost reduction [31]. In terms of surgical interventions, trabeculectomy is considered the most widely used conventional procedure, aimed at creating an alternative pathway for aqueous humor outflow. Glaucoma may also be treated through laser procedures, such as different modalities of trabeculoplasty (argon laser and selective laser trabeculoplasty), which induce changes in the trabecular meshwork, promoting increased aqueous humor outflow [32,33].
However, a substantial proportion of patients with glaucoma exhibit disease progression despite adequate IOP control [20,34,35], suggesting the contribution of additional pathophysiological mechanisms [24]. Among these mechanisms, neurodegenerative processes, vascular dysfunction, oxidative stress and inflammatory responses may contribute to retinal ganglion cell death independently of intraocular pressure, reinforcing the complexity of glaucoma pathophysiology and the need for complementary therapeutic approaches [2,36].
Accordingly, increasing evidence points to the involvement of glial-mediated mechanisms in glaucoma pathogenesis, since alterations in retinal glial homeostasis have been associated with retinal ganglion cell degeneration and functional visual deficits even under normotensive conditions [14]. In this context, inflammation and oxidative stress have been implicated as key mediators of RGC degeneration. Ischemic events may trigger an inflammatory response characterized by the production of pro-inflammatory mediators and the activation of resident inflammatory cells, contributing to the establishment of a neurotoxic microenvironment that promotes RGC dysfunction and death [2]. Understanding IOP-dependent and -independent mechanisms may complement existing therapeutic strategies or enable the development of novel approaches, contributing to slowing glaucoma progression and preserving visual function.
2.1. Oxidative Stress and Inflammation in Glaucoma
Oxidative stress is a condition that the production of reactive oxygen/nitrogen species (ROS/RNS) overcome the antioxidant cell defenses [37]. Even in physiological conditions, ROS is present, and acts as signals to a number of homeostatic mechanisms [37]. ROS can be derived from oxygen molecular in the mitochondrial electron chain or by the NADPH oxidase (NOX) enzyme, producing superoxide radical (O2∙⁻), a highly reactive oxygen species with one unpaired electron. Superoxide radicals can combine with nitric oxide, generating peroxynitrite, the main RNS. A non-radical ROS, hydrogen peroxide (H2O2), can arise from superoxide radicals by the action of superoxide dismutase (SOD). In an oxidative stress condition, an even highly reactive radical can be produced from H2O2, hydroxyl radicals (OH∙). Cells have a myriad of antioxidant defenses, enzymatic or non-enzymatic, that control the balance of physiological signaling ROS/RNS and oxidative stress state. Among them, it can be highlighted SOD, catalase, peroxiredoxins and glutathione peroxidases, enzymes that detoxifying ROS, and glutathione as the main endogenous non-enzymatic antioxidant of the CNS, working together with melatonin, carotenoids and other [38].
ROS/RNS directly react with proteins, lipids nucleic acids [39,40], altering their function, leading to damage through a number of mechanisms [41]. Elevated ROS levels, a common feature of neurodegenerative diseases, have been linked to mitochondrial dysfunction, protein misfolding, and disruptions in cellular redox homeostasis [40,42,39,42]. Aging increases susceptibility to the accumulation of oxidative damage in cells and tissues, a process that has been linked to cellular senescence and glaucomatous neurodegeneration [43,44]. Additionally, disruption of cellular redox balance may induce a state of microglial hyperactivation, promoting the release of pro-inflammatory mediators such as TNF-α, as well as activation of inflammatory pathways involving NF-κB, nitric oxide synthase, and cyclooxygenase-2 [43]. In glaucoma, alterations in vascular perfusion and reduced oxygen supply may exacerbate oxidative stress, contributing to retinal ganglion cell loss and disease progression [45].
Alterations in ocular vascular autoregulation have been implicated in glaucoma pathophysiology, favoring episodes of reduced ocular perfusion followed by reperfusion, a process associated with the generation of oxidative stress [46,47]. Ocular vascular dysfunction is further characterized by endothelial dysfunction, impaired neurovascular coupling, increased endothelin-1 (ET-1) signaling, and reduced ocular blood flow, all of which contribute to retinal hypoxia and glaucomatous neurodegeneration [47,48]. Persistent hypoxia activates hypoxia-inducible factor-1α (HIF-1α) signaling and promotes the expression of vascular endothelial growth factor (VEGF), while also enhancing the production of inflammatory mediators, including nitric oxide (NO), TNF-α, and interleukins, thereby amplifying oxidative stress, inflammation, and neuronal damage [49,50]. Therefore, vascular dysregulation and reduced oxygen supply may exacerbate oxidative stress, contributing to retinal ganglion cell loss in glaucoma.
The inflammatory response in the CNS exhibits a dual role, initially exerting neuroprotective effects through the removal of cellular debris and maintenance of tissue homeostasis. However, when persistent, this response may become dysfunctional, contributing to the inhibition of reparative processes and progression of neuronal damage [51]. In glaucoma, this transition has been associated with chronic activation of glial cells and the maintenance of a pro-inflammatory environment [52], contributing to neuronal degeneration. There are three types of glial cells in the retina: a) Müller cells, a macroglial and the majoritary retinal glial cell; b) astrocytes, a macroglial with restricted, but strategic localization near ganglion cells; and c) microglial, a myeloid-originated cell, which invade the tissue during development.
3. Astrocytes in the Healthy and Glaucomatous Retina
3.1. Distribution and Physiological Functions of Astrocytes
Astrocytes form a vital support network for the inner retina and the optic nerve head (ONH) [53], offering continuous mechanical and biological support [54]. Embryologically, they derive from neuroepithelial cells in the ventricular zone, where radial glial cells act as progenitors that, following neurogenesis, spatially differentiate to produce astrocyte populations [55]. Early in the glial lineage, tripotent glial-restricted progenitors can also give rise directly to type-1 astrocytes, astrocyte-restricted progenitors, and bipotent O2A cells; the latter can further differentiate into type-2 astrocytes [56].
Within the mature retina, the anatomical distribution of astrocytes is highly localized, strictly confined to the innermost strata comprising the ganglion cell layer (GCL) and the retinal nerve fiber layer (RNFL) [57]. Restricted to this microenvironment, they form a dense plexus that interacts directly with retinal ganglion cell axons and the local vasculature. Here, they orchestrate neurovascular balance, resource distribution, and metabolic support [58]. Classically, these cells are fundamental for maintaining ion and neurotransmitter homeostasis, modulating synapses, and upholding the blood-retinal barrier [59]. Furthermore, some subpopulations play central roles in coordinating immunological and inflammatory responses against tissue adversities [60], while also secreting crucial trophic factors - such as ciliary neurotrophic factor (CNTF) - that protect RGCs from oxidative damage [53,61].
3.2. Biomechanical Stress and Structural Remodeling
Beyond the retinal boundaries, the optic nerve head acts as a critical transition region. Here, RGC axons converge and pass through a dense astrocytic meshwork in an unmyelinated state, heightening their metabolic vulnerability and making them critically dependent on local astrocytic support before acquiring myelin in the retrolaminar optic nerve [62]. This dense astrocytic meshwork at the ONH represents the primary site of initial RGC injury in glaucoma [62]. The transition of astrocytes to a pathological state is primarily driven by early biomechanical stress (usually from elevated IOP), which exerts tension, compression, and shear forces on the lamina cribrosa [63]. Astrocytes detect these continuous loads via mechanosensitive ion channels, such as Piezo1 and TRPs (particularly the TRPC and TRPV subfamilies), translating physical stretch into rapid, calcium-mediated reactive response [27,64,65].
Reactive astrogliosis also induces severe extracellular matrix (ECM) remodeling. Mediated by TGF-β2 signaling, reactive astrocytes increase the deposition of macromolecules such as type I collagen and fibronectin, while upregulating tissue transglutaminase (TGM2) to cross-link this newly remodeled extracellular matrix within the lamina cribrosa [66]. These stressed astrocytes exhibit process retraction, undergo somatic hypertrophy [67] and, eventually, detach from their basal lamina [68]. This remodeling narrows the radial pores of the lamina cribrosa, obstructing axonal transport and resulting in the progressive optic disc cupping, characteristic of glaucomatous progression [69,70].
3.3. Oxidative Stress and Senescence
Alongside this structural remodeling, the transcriptional reprogramming of reactive astrocytes impose a bioenergetic burden. To meet this ATP demand, ONH astrocytes shift from their basal metabolism to a heavy reliance on oxidative phosphorylation (OXPHOS) [71]. While this metabolic shift maximizes energy production needed for their reactive state, the mitochondrial electron transport chain inevitably leaks electrons, generating a secondary wave of mitochondrial reactive oxygen species (mtROS) as a by-product that exacerbates the ongoing oxidative stress [71]. Initially, there is an upregulation of the antioxidant defense system (such as the glutathione and peroxiredoxin systems) in astrocytes, which could help to buffer oxidative stress, as well as an increase in proteasomal activity, that could clear oxidized proteins [71]. However, chronic glaucomatous insult eventually exhausts these defenses, triggering widespread lipid peroxidation and irreversible protein nitration/oxidation. This oxidative collapse destroys the astrocytic cytoskeleton and permanently compromises the metabolic supportive capacity that these cells provide to adjacent RGC axons [[71,72].
The consequences of this oxidative stress permanently alter the astrocytes through genomic instability and cellular senescence. In vivo transcriptomic profiling of ONH astrocytes in experimental glaucoma demonstrates that following the initial oxidative burden, astrocytes significantly upregulate pathways associated with the senescence-associated secretory phenotype (SASP) [71]. The mechanistic basis for this transition has been further mapped utilizing human and murine primary astrocyte cultures exposed to oxidative stressors [73]. These studies confirm that chronic oxidative damage directly induces severe DNA lesions, triggering the DNA damage response (DDR) as evidenced by the accumulation of γH2AX and 53BP1 within the astrocytic nuclei [73]. This genomic stress forces the reactive astrocytes into a permanent cell cycle arrest, characterized by the upregulation of p21 and β-galactosidase, and locks them into the SASP state [73]. These senescent astrocytes become chronic sources of tissue degradation, secreting high levels of matrix metalloproteinases, such as MMP3, and perpetuating the release of inflammatory cytokines like IL-6 and IL-1β [73]. Through this ROS-driven senescence mechanism, astrocytes abandon their supportive role entirely, actively deteriorating the ONH microenvironment and cementing the progression of glaucomatous neuropathy.
3.4. Cytotoxic Shift and Neuroinflammation
Triggered by this oxidative burden and cellular senescence, reactive astrogliosis establishes a self-sustaining neurotoxic shift that perpetuates independently of ongoing IOP elevation, driven by retina-derived chemokine signaling that serves as a primary molecular trigger for a cytotoxic shift [13]. Following early ocular stress, such as elevated IOP, the injured retinal tissue (specifically RGCs and activated resident microglia) locally upregulates specific chemokines, most notably CXCL10 [74]. By binding to the CXCR3 receptor on astrocytes, CXCL10 directly drives their polarization toward a neurotoxic profile, primarily by inducing the expression of complement component 3 (C3) [13,74]. This continuous signaling establishes a localized crosstalk that perpetuates neuropathy in normal-tension glaucoma or even after successful IOP reduction. Ischemia-reperfusion, mitochondrial dysfunction, and cytokine-mediated neuroinflammation serve as potent secondary triggers [75,76]. Regardless of the initial insult, the resulting reactive astrogliosis compromises the vital metabolic support provided to the axons, creating a pro-inflammatory and oxidative microenvironment that dictates irreversible glaucomatous neurodegeneration [62,77].
Ultrastructural analyses from an experimental model of ocular hypertension demonstrate how early these neurodegenerative events can occur. With perilimbal-plexus cauterization, transmission electron microscopy showed progressive axonal disorganization, vacuolization of the myelin sheath, and disruption of the nodes of Ranvier [78]. The mechanical stress triggers a robust reactive glial response within the myelinated portion of the optic nerve, characterized by an upregulation of the astrocytic marker GFAP at 48 hours and a subsequent accumulation of Iba1-positive microglia/macrophages by 72 hours, demonstrating that macroglial and immune remodeling extend rapidly beyond the optic nerve head to propagate the neurotoxic shift [78].
The neuroinflammatory cascade triggers a profound phenotypic shift in astrocytes across a complex and multidimensional spectrum. They exhibit immense heterogeneity, continuously integrating microenvironmental cues to adopt varied transient states before ultimately committing to a pathogenic profile [79]. This polarization towards a cytotoxic state is largely orchestrated by an immune cross-talk with resident microglia, polarized to a pro-inflammatory profile [[80]. These activated microglia secrete mediators (IL-1α, TNF-α, and C1q), which seem to be both necessary and sufficient to drive astrocytes into a cytotoxic state [[81]. At the molecular level, this reactivity is regulated by the transcription factor NF-κB, a master regulator of inflammatory and immune responses. Upon stimulation by the microglial mediators, astrocytic NF-κB translocates to the nucleus and drives the gene regulation of cytotoxic mediators, adhesion molecules and immune receptors[82,83].
While early astrocytic reactivity often serves as an adaptive and protective mechanism, prolonged exposure to this inflammatory microenvironment eventually provokes these cells to lose their neuroprotective functions. In this chronic state, they begin to actively secrete cytotoxic factors and cytokines, such as IL-1β and IL-6, often through NLRP3 inflammasome activation, deeply dependent on the NF-κB signaling [84,85]. This signaling causes RGCs, already weakened by mechanical strain, to become hypersensitive to the astrocyte-derived harmful signals, leading to accelerated apoptosis [81] Furthermore, the aberrant upregulation of MHC-II on reactive astrocytes suggests they act as unconventional antigen-presenting cells, potentially driving a localized autoimmune response that sustains neurodegeneration even after IOP stabilization [86], inducing a cell death cycle driven by chemokines and the complement system.
Driving this cycle, once released by the retinal tissue, CXCL10 binds to the astrocytic CXCR3 receptor, activating the CXCL10/CXCR3 signaling axis which, independent of intraocular pressure, induces astrocytes to upregulate C3 [74]. This astrocyte-derived C3 is cleaved into the C3a anaphylatoxin, which specifically binds to the C3aR receptor that becomes concurrently upregulated on adjacent RGCs, establishing a toxic neuroglial crosstalk that synergistically accelerates neuronal apoptosis [74]. Simultaneously, sustained inflammatory signaling triggers the assembly of the NLRP3 inflammasome within the cytoplasm of reactive astrocytes, activating caspase-1 to cleave Gasdermin D (GSDMD) and initiate pyroptosis, an inflammatory lytic form of programmed cell death that ruptures the glial membrane and spills extensive cytotoxic contents directly onto the unmyelinated axons [74]. Figure 1 compiles the molecular changes undergone by reactive astrocytes in glaucoma, the crosstalk microglia-astrocytes, and the alterations in ONH.
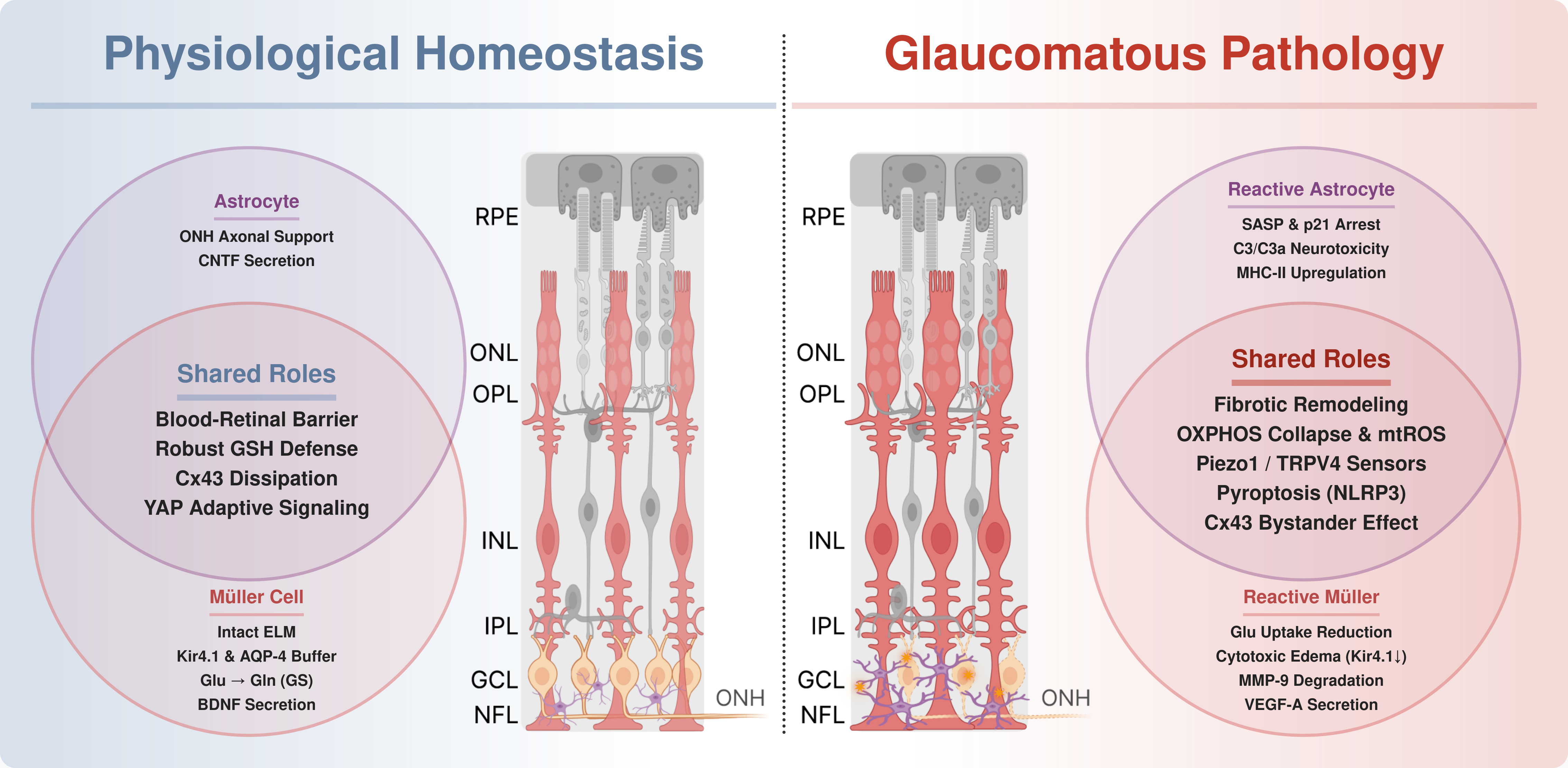
In summary, the convergence of oxidative stress, senescence, and neuroinflammation solidifies the astrocyte as a central executioner in glaucoma. This reactivity is not isolated; astrocytes are physically coupled to Müller cells via Connexin-43 gap junctions [86]. While essential for homeostasis, this connection becomes a pathological conduit that propagates toxic signals, scaling focal optic nerve head injury into a widespread retinal pathology [5]. Therefore, therapeutically decoupling this neurotoxic network, by targeting the SASP, inhibiting specific gap junctions, or modulating the microglial-astrocytic crosstalk, can represent a highly promising path for neuroprotection in the glaucomatous context. The contrast between their essential neuroprotective baseline and the pathogenic shift driven by glaucomatous stress, which dictates the neuropathy progression, is illustrated in Figure 2.
4. Müller Glia in the Healthy and Glaucomatous Retina
4.1. Müller Cells in Physiological Conditions
Müller glia constitute the predominant macroglial population in the retina, accounting for approximately 90% of retinal glial cells, being essential for maintaining retinal architecture and function in physiological conditions [87,88]. They exhibit a unique radial morphology that spans the entire retinal thickness. Due to this peculiar organization, Müller cells interact with all retinal cell types, enabling them to usually be the first glial cell to detect retinal damage [89]. The Müller radial cell processes in the outer retina form critical junctional complexes with the inner segments of photoreceptors to establish the outer limiting membrane (OLM) [90]. This structure acts as an important macromolecular barrier and is intrinsically linked to fluid homeostasis, where Müller cells actively regulate subretinal fluid absorption via aquaporin-4 (AQP-4) water channels [90].
Beyond structural support, these cells play a central role in retinal metabolic compartmentation, acting as the principal retinal source of glutamine synthetase (GS), after rapidly clearing excitatory glutamate from the synaptic cleft, thus preventing excitotoxicity, and converting it into non-toxic glutamine, which is exported back to neurons, allowing continuous neurotransmission [91]. Additionally, they provide metabolic support by supplying lactate to retinal neurons, fueling ATP production via the tricarboxylic acid (TCA) cycle, meeting the retina's fluctuating/high energy demands, also participating in the visual cycle through pigment regeneration [90,91], forming an integrated support network that contributes to RGC survival.
To sustain the intense bioelectric activity of the retina, these cells ensure tight control over ionic and fluid homeostasis. During continuous neuronal firing, they actively buffer extracellular potassium levels, a process dependent predominantly on the inwardly rectifying potassium channel Kir4.1 [92]. The functional coupling of Kir4.1 and AQP4 is critical for coordinating ion and fluid clearance; experimental ablation of this delicate balance (the downregulation of Kir4.1 accompanied by AQP4 upregulation) is a major contributor to cytotoxic Müller cell edema [93].
Complementing this physiological regulation, Müller glia play a central role in the retinal antioxidant response. By utilizing systems such as the cystine/glutamate antiporter (xCT) to import necessary precursors, they sustain the intracellular synthesis of glutathione (GSH), providing a major component of retinal antioxidant defense against ROS continuously generated by the high mitochondrial activity of the outer retina [94,95]. Alongside this redox regulation, Müller cells provide an important neuroprotective mechanism by secreting neurotrophins, most notably brain-derived neurotrophic factor (BDNF). By binding to the TrkB receptor on RGCs, Müller-derived BDNF activates the PI3K/AKT signaling cascade, promoting neuronal survival and mitigating axonal and dendritic degeneration under toxic stress [96]. The disruption of these homeostatic mechanisms marked by Kir4.1 depletion, glutathione depletion, and neurotrophin signaling failure, contributes to the transition from physiological support to reactive gliosis.
4.2. Early Reactive Gliosis and Mechanotransduction
The transition from physiological support to gliosis in glaucoma is not an immediate, but a biphasic response triggered by the biomechanical stress exerted by elevated IOP [10,97]. Initially, Müller cell activation serves as an adaptive and neuroprotective mechanism. A reliable marker of this early phase is the upregulation of GFAP, which transitions from a faint astrocytic immunolabeling in the healthy NFL to robust expression within the radial Müller processes [98]. Importantly, intermediate filaments like GFAP directly enhance retinal stiffness and mechanical shielding against IOP-induced strain [99]. During this compensatory stage, Müller cells amplify their metabolic support capabilities by enhancing the secretion of neurotrophic factors, accelerating the uptake and degradation of glutamate, and increasing the release of antioxidant glutathione to counteract excitotoxic and redox imbalances [10,97,100,101]. Consistent with this protective profile, the upregulation of pigment epithelium-derived factor (PEDF), brain-derived neurotrophic factor (BDNF), and classical antioxidants—such as reduced ascorbate and glutathione—markedly boosts the defensive response against early glaucomatous damage [97,102,103]. Furthermore, this neuroprotective phenotype is characterized by an increased potassium buffering capacity [10], temporarily preserving retinal homeostasis before the chronic mechanical burden triggers pathological remodeling.
Alongside these early biochemical adaptations, the direct biomechanical stress exerted by elevated IOP also regulates the Hippo signaling pathway in Müller glia, primarily through the transcriptional co-activator Yes-associated protein (YAP). Acting as a master mechanosensitive switch, YAP translocates to the nucleus in response to retinal stretch and tissue deformation [104]. In glaucomatous neurodegeneration, YAP appears to have an adaptive and neuroprotective function, rather than a purely pathogenic one. Following excitotoxic retinal injury, YAP activation is markedly upregulated in Müller cells, driving the transcription of survival factors, such as the anti-apoptotic protein Bcl-xL, to preserve mitochondrial integrity and shield adjacent RGCs from cell death [104]. The nature of this mechanosensor for maintaining retinal homeostasis is underscored by the finding that the conditional deletion of YAP specifically in Müller glia precipitates severe functional defects, including impaired water and glutamate clearance, leading to the spontaneous development of uveitic glaucoma-like features [105]. Thus, initial YAP activation serves as a compensatory mechanism against IOP-induced strain. However, its chronic dysregulation or eventual exhaustion under constant mechanical pressure marks a point where Müller cells lose their structural and neuroprotective role.
As this compensatory response fails under persistent stress, Müller glia undergo a pathological transformation. Acting as frontline mechanosensors, they detect physical tissue stretches directly through the transient receptor potential vanilloid 4 (TRPV4) ion channel [106] and PIEZO1 [107]. High pressure induced the increase of both channels levels [107,108]. PIEZO1 activation triggers gliosis and inflammatory response [109,110]. Chronic TRPV4 activation incites a robust intracellular inflammatory cascade, predominantly mediated by the IL-6/STAT3 and the extracellular signal-regulated kinase (ERK 1/2) signaling pathways [111,112]. This cascade drives the classic morphological and molecular hallmarks of reactive gliosis: cellular hypertrophy, proliferation, and the rapid remodeling of constitutive vimentin networks, alongside the robust upregulation of GFAP and de novo expression of nestin [113,114]. Ultimately, chronic gliosis leads to cellular dedifferentiation and the marked downregulation of essential homeostatic enzymes, such as GS [115], functionally uncoupling Müller glia from retinal neurons and shifting their role from neuroprotection to neurotoxicity.
Alongside this early reactivity, the continuous mechanical stretch of Müller cell membranes stabilizes HIF-1α independently of oxygen levels. This mechanotransduction process upregulates HIF-1α expression at both the mRNA and protein levels in a response associated with the activation of transcription factors triggered by extracellular stress, such as NF-κB, driving its intracellular accumulation under normoxic conditions [116]. This mechanically induced HIF-1α stabilization shifts the cellular angiogenic profile by markedly upregulating the secretion of VEGF-A while, simultaneously, downregulating the vessel stabilizing angiopoietin-1 (ANG1) [116]. This reactive transformation is an early, initiating event in glaucoma pathogenesis rather than a late-stage secondary consequence. Histopathological analyses of human post-mortem glaucomatous retinas confirm that widespread Müller cell hypertrophy and GFAP overexpression occur autonomously and independently of the absolute extent of RGC loss [117]. Thus, mechanically activated Müller cells do not merely respond to neuronal death but actively orchestrate the neuroinflammatory microenvironment that accelerates glaucomatous damage. Figure 3 shows the extent of this reactive gliosis, illustrating how Müller cells are overwritten by somatic swelling and fibrotic remodeling.
4.3. Excitotoxicity and Oxidative Stress
Although the early response of reactive Müller glia is beneficial, chronic mechanical stress and severe cytoskeletal remodeling ultimately disrupt their functional polarity [97]. This structural collapse forces spatial mislocalization and downregulation of critical clearance mechanisms, precipitating a robust decrease in potassium buffering capacity. The impaired potassium siphoning directly depolarizes the glial membrane, weakening the driving force for glutamate uptake by GLAST, which forces extracellular glutamate accumulation and induces ganglion cell excitotoxicity [118,119]. Consistent with this, overexpression of Kir4.1 channels in a chronic ocular hypertension model inhibited Müller cell activation, consequent microglial gliosis and inflammatory response, protecting ganglion cells [92]. A cytotoxic profile of Müller glia is also characterized by downregulation of the glutamate recycling enzyme GS, which also occurs in glaucoma models [120,121], accompanied by the reduction of GS activity [122]. This enzymatic failure further elevates extracellular glutamate [123], exacerbating excitotoxic ganglion cell death [118,123]. Supporting this mechanism, one of the animal models of glaucoma used in the literature is the intraocular injection of NMDA receptor agonists [124,125]. The increased activation of ionotropic glutamate receptors, especially NMDA receptors on RGCs, generates a massive intracellular calcium increase that leads to neuronal mitochondrial dysregulation [126], one of the main early sources of reactive oxygen/nitrogen species (ROS/RNS) and a hallmark of glaucoma [127]. This redox imbalance arises when ROS overproduction overwhelms cellular antioxidant defenses. Notably, elevated IOP directly triggers this oxidative burden [128,129,130], leading to pathological protein modifications, such as the oxidation of GS [131], which disrupts important survival mechanisms. In alignment with these findings, total antioxidant status is lower in open-angle glaucoma [132] and exfoliation glaucoma patients [133]. Moreover, this pro-oxidant state downregulates GLAST, crucial to extracellular glutamate concentration control, and xCT, the catalytic subunit of the xc- transporter [134], both important for the synthesis of glutathione [135]. Consequently, there is a marked reduction in glutathione in glaucoma models [136]. Therefore, the accumulation of free radicals establishes a vicious cycle that augments excitotoxicity and accelerates cell death. Furthermore, Müller gliosis induced by glaucoma models is accompanied by cell death, and treatments that prevent the reactivity of the Müller glia protect against neuronal loss [119,137,138]. These data reinforce the crucial role of redox dysregulation in Müller cells in driving the observed ganglion cell death in glaucoma.
4.4. Mitochondrial Collapse and TRP-Driven Oxidative Burden
The intersection between bioenergetic failure and the innate immune activation of Müller glia is a critical point in glaucomatous pathogenesis. This reactive transformation is not spatially uniform, exhibiting a distinct topographical susceptibility: Müller cells located in the peripheral retina are significantly more vulnerable to high intraocular pressure, expressing higher levels of mechanosensitive channels (such as TRPV4 and Piezo1) than their central counterparts [107]. This could predispose the periphery retina to earlier and more severe glaucomatous damage [107]. Under sustained biomechanical strain and a massive intracellular calcium influx, driven primarily by mechanosensitive channels and purinergic signaling rather than canonical excitotoxicity, these susceptible glial cells experience a severe metabolic collapse. A key mechanistic link to this bioenergetic failure is the downregulation of apolipoprotein A-I binding protein (AIBP), a crucial safeguard for mitochondrial dynamics whose expression is repressed by this early cellular stress. AIBP deficiency directly triggers mitochondrial fragmentation via DRP1 phosphorylation, loss of membrane potential, and a massive leakage of mitochondrial ROS (mtROS) [139,140]. Simultaneously, metabolically exhausted Müller cells undergo a pathological reversal of their glutamate transporters, actively flooding the extracellular space with glutamate [141]. The combined mechanosensory and mitochondrial stress also prompts severe extracellular matrix remodeling, driving the robust secretion of matrix metalloproteinases, specifically MMP-9, which degrades the local inner limiting membrane (ILM) architecture[141].
Exacerbating this pro-oxidant state, chronic IOP elevation also triggers additional ROS generation. Müller and other retinal cells detect this massive radical burst through the transient receptor potential ankyrin 1 (TRPA1) channel, a highly sensitive redox sensor that operates as a pathological amplifier of tissue damage [142]. Upon initial activation by ROS, TRPA1 triggers a NADPH oxidase 1 (NOX1)-dependent pathway that further exacerbates the local redox imbalance, evidenced by the severe accumulation of lipid peroxidation end-products such as 4-hydroxynonenal (4-HNE). This TRPA1-driven oxidative response functions as a master switch for retinal inflammatory crosstalk. The amplified oxidative stress disrupts the homeostatic glial-neuronal relationship, recruiting macrophage infiltration and inducing apoptotic cascades in the neural retina via caspase-3 activation. Experimental blockade or genetic deletion of TRPA1 interrupts this axis, preventing NOX1 overactivity, halting immune cell recruitment, and ultimately preserving retinal thickness and RGC viability following this oxidative injury [142]. Consequently, the TRPA1 channel emerges as a central mediator of neuroinflammation and a prime therapeutic target for preventing glaucomatous neurodegeneration [65].
4.5. Müller Glia as Central Drivers of Neuroinflammation
This massive accumulation of mtROS and damage-associated molecular patterns (DAMPs) serves as an endogenous danger signal that forces Müller glia to shift into a neurotoxic, senescence-associated inflammatory phenotype. By generating oxidized phospholipids (OxPLs) and altering membrane dynamics, the severe oxidative burden facilitates the rapid relocalization and membrane association of Toll-like receptor 4 (TLR4) on the Müller cell [143]. This newly membrane-associated TLR4 directly triggers the assembly of the NLRP3 inflammasome (leading to intracellular cleavage of Caspase-1 and -3) while simultaneously activating the NF-κB and ERK signaling pathways [139,144]. This pro-inflammatory cascade initiates a highly coordinated, self-sustaining crosstalk with resident microglia. Acting as primary mechanosensors, reactive Müller cells release early danger signals (such as ATP) that recruit resident microglia to the injury site. Once activated, these early-responding microglia release massive amounts of IL-1β, which potently hyperactivates adjacent Müller glia via the NF-κB and p38 MAPK pathways, prompting a secondary secretory shift that releases chemokines like MCP-1 (CCL2), CXCL1, and CXCL5 [145]. These chemokines act as robust homing signals, binding to CXCR2 receptors on microglia and compelling their rapid migration directly into the ganglion cell layer. Thus, mechanosensitive Müller cells actively dictate the spatial recruitment of immune effectors, driving a toxic positive feedback loop that surrounds RGC axons with concentrated inflammatory signaling and accelerates irreversible visual dysfunction.
This pathogenic transformation of Müller glia is not confined to isolated cells; it propagates across the retinal tissue via Connexin-43 (Cx43) gap junctions and uncoupled hemichannels. While physiological Cx43 networks are vital for ion buffering and metabolic dissipation, elevated IOP triggers a massive pathological upregulation of Cx43 in Müller cells, notably occurring prior to the onset of RGC loss [146]. Under biomechanical and oxidative stress, these connexons frequently operate as uncoupled hemichannels, opening directly into the extracellular space. In an experimental glaucoma model, activated Müller cells utilized these Cx43 hemichannels to release copious amounts of ATP into the microenvironment [147]. The extracellular ATP functions as a danger-associated molecular pattern, binding to purinergic receptors (P2X7R/P2X4R) on resident microglia to forcefully drive their proliferation and migration toward the damaged retinal layers [147]. Through this mechanism, overexpressed Cx43 hemichannels act as conduits for the bystander effect, allowing mechanical injuries to leak inflammatory triggers that cascade into widespread neurodegeneration. Consequently, this interconnected web of macroglial reactivity, spanning from initial mechanosensation and oxidative collapse to Cx43-mediated inflammatory propagation, underscores that retinal glia actively coordinates glaucomatous damage. Figure 4 resumes the molecular mechanisms in reactive Müller cells and the effects on the RGCs and microglia.
Ultimately, the neuroinflammatory crosstalk driven by Müller glia, alongside the previously described astrocyte reactivity, establishes that macroglia are not mere secondary responders in glaucoma, but active drivers of retinal neurodegeneration. This pathogenic cascade, initially triggered by mechanosensors such as TRPV4 and amplified by profound oxidative stress, neuroinflammation, metabolic collapse and glial crosstalk, creates a self-sustaining neurotoxic microenvironment. Because this interconnected glial reactivity can perpetuate neuroinflammation and apoptotic signaling even after the initial mechanical strain is relieved, retinal damage frequently progresses despite intraocular pressure normalization. Therefore, directly interrupting this pathological macroglial network, particularly by targeting primary mechanosensitive and redox sensors, represents a fundamental paradigm shift for neuroprotection, laying the groundwork for advanced pharmacological interventions.
5. Pharmacological Therapies
As mentioned before, current glaucoma treatments primarily aim to normalize IOP. Topical pharmacological therapy remains the first-line treatment and includes prostaglandin analogs, carbonic anhydrase inhibitors, and α2-adrenergic agonists. However, combined pharmacological therapies, as well as surgical interventions and laser procedures, can also be employed [133]. In some cases, the IOP reduction achieved by these treatments does not prevent glaucoma progression. Furthermore, many patients with glaucoma do not exhibit elevated IOP, suggesting that disease development and progression involve additional factors that remain incompletely understood. These findings reinforce the need to explore alternative therapeutic approaches that extend beyond IOP control. In this context, the regulation of retinal glial reactivity has emerged as a promising therapeutic target, together with the modulation of transient receptor potential (TRP) channel activity.
TRP comprises a group of ion channels with similar molecular structures that are permeable to monovalent and divalent ions, with most members exhibiting high calcium permeability. The stimuli responsible for the activation of these receptors are diverse and may include thermal stimuli, plasma membrane stretching, endogenous/exogenous chemical molecules, pathogen-derived fragments, and inflammatory signals [65]. To date, seven TRP subfamilies have been described, including Ankyrin (TRPA), Canonical (TRPC), Melastatin (TRPM), Mucolipin (TRPML), NO-mechano-potential C (TRPN), Polycystin (TRPP), and Vanilloid (TRPV) [134]. Although TRP channels were initially described in somatosensory neurons, it is now well established that they are expressed in different cell types throughout the CNS, including the retina, where they play both physiological and pathological roles [65]. Indeed, in glaucoma, there is evidence demonstrating the involvement of TRPV4 and TRPV1 receptors in retinal ganglion cell loss [135], although the knockout of each of these receptors did not protect retinal cells against cell death induced in IOP-reperfusion model [136]. Intriguingly, only a few studies have investigated the relationship between TRP receptors and glial reactivity in animal models of glaucoma [15,135].
Animal models of glaucoma have demonstrated an important role for TRPs channels in glial reactivity, as well as in the onset and progression of disease-associated symptoms. Among the wide variety of TRP channels, TRPV4 receptors have attracted particular attention. TRPV4 is a mechanosensitive non-selective cation channel that has been described in the retinas of different species and is expressed and functionally active in both neuronal cells and Müller glial cells [94,137]. Chronic elevation of intraocular pressure in mice and rats has been shown to induce Müller glial reactivity, which was associated with increased TRPV4 protein expression in these cells and enhanced retinal ganglion cell death [89,94]. The effects of chronic elevation of intraocular pressure were mimicked by intravitreal administration of GSK101, a TRPV4 agonist, and were prevented by prior treatment with HC-067, a TRPV4 antagonist, or R7050, a soluble TNF-α inhibitor [94]. Pro-inflammatory response-associated genes, such as IL-6, TNF-α, and STAT-3, were also found to be upregulated after 4 weeks of elevated intraocular pressure [89]. Thus, retinal ganglion cell loss in this model appears to depend on Müller glial reactivity and TNF-α release. A similar response has also been observed in in vitro models. Primary cultures and immortalized Müller glial cell lines became reactive following activation of TRPV4 receptors by GSK101 [89,94,138,139]. Activation of TRPV4 receptors in glial cells led to increased TNF-α production and release, which may directly induce retinal ganglion cell death [94,138,140]. Cell death was inhibited by the addition of an anti-TNF-α antibody to the culture medium, indicating that TRPV4 activation-induced gliosis promotes the production and release of TNF-α [138]. The increased production and release of TNF-α are dependent on activation of the JAK/STAT-2 signaling pathway and phosphorylation of the NF-κB p65 subunit [94,138]. Activation of TRPV4 in retinal ganglion cell cultures also reduced cell viability and increased the expression of the TNF-α receptor TNFR1 [94,138]. This scenario supports the hypothesis that elevated intraocular pressure may lead to TRPV4 activation in both Müller glial cells and retinal ganglion cells. In Müller glia, TRPV4 activation induces glial reactivity and promotes the establishment of a pro-inflammatory profile that results in TNF-α release. In retinal ganglion cells, the expression of TNFR1 receptors is increased. In this context, enhanced TNF-α release combined with increased expression of its receptor in retinal ganglion cells appears to be a key factor driving the pro-inflammatory phenotype of Müller glia and the subsequent loss of retinal ganglion cells.
An important question that arises is how elevated intraocular pressure may lead to increased TRPV4 receptor expression in Müller glial cells. A possible clue to this question may come from the relationship between aquaporins and TRPV4 receptors. Aquaporins are among the most abundant water channels in the central nervous system (CNS) and play an important role in water homeostasis in Müller glia, where, together with TRPV4, they regulate membrane permeability and the regulatory volume decrease response induced by cell swelling [140,141]. However, recent studies have shown that aquaporin-4 and TRPV4 can physically interact, and that this relationship may be detrimental in both in vivo and in vitro models of diabetic retinopathy. Interestingly, inhibition of aquaporin-4 expression by shRNA prevents high glucose-induced damage in a manner dependent on the reduction of TRPV4 expression. A similar effect has also been observed in streptozotocin-induced diabetic rats [142]. Thus, Müller glial cell death in diabetic retinopathy models depends on increased aquaporin-4 expression and the associated upregulation of TRPV4. On the other hand, activation of TRPV4 receptors in Müller glial cells by osteopontin, an integral component of the extracellular matrix, in osmotic swelling models reduces membrane permeability and the regulatory volume decrease response by decreasing aquaporin-4 expression [141]. These findings reveal a close interaction between aquaporin-4 and TRPV4 under different pathological conditions. To date, however, this relationship has not been investigated in animal models of glaucoma. It is possible that glial reactivity associated with increased TRPV4 expression may depend on an indirect regulation of aquaporin-4 expression. Therefore, investigating this potential relationship and its implications for glaucoma pathology may be of considerable interest.
Activated Müller glia in glaucoma models may also release pro-survival factors capable of regulating TRPV4 activity and expression. For example, elevated intraocular pressure in animal models has been shown to increase the production and release of lipoxin B4 by Müller glial cells, thereby acting as a neuroprotective agent. Lipoxin B4 is an endogenous eicosanoid produced through the coordinated action of 5-lipoxygenase (5-LOX) and 15-lipoxygenase (15-LOX). Notably, lipoxin B4, as well as 5-LOX and 15-LOX, are expressed in mouse Müller glial cells [89]. Administration of lipoxin B4 was able to prevent the increase in TRPV4 expression, glial reactivity, and the inflammatory response induced in vivo and in vitro by elevated intraocular pressure or TRPV4 activation [89]. Another factor that may regulate TRPV4 activity is the amount of cholesterol present in the membrane. TRPV4 receptor activity induced by GSK101, temperature, and cellular swelling was reduced in Müller glial cells following membrane cholesterol depletion by MβCD [143]. Therefore, reducing TRPV4 receptor activation appears to be a promising therapeutic strategy.
Other members of the TRP family have also been identified as inducers of Müller glial reactivity. A brief 30-minute ischemic event in the rat retina followed by 72 hours of reperfusion led to an increase in TRPM7 immunoreactivity in Müller glial cells, but not in astrocytes. Upregulation of TRPM7 channels following ischemia–reperfusion was accompanied by a reduction in the amplitude of oscillatory potentials in ERG recordings, indicating alterations in the inner retina, as well as mild decreases in b-wave and nSTR amplitudes [144]. TRPV1 activation also plays an important role in retinal ganglion cell loss in glaucoma models [135], however, only a few studies have sought to understand its relationship with Müller glial cell activation. Cultured mouse Müller glial cells proliferate and release ATP in response to increased hydrostatic pressure in a TRPV1- and PLCγ-1-dependent manner [16]. Retinal ganglion cells exposed to conditioned medium from Müller glial cells subjected to increased hydrostatic pressure undergo cell death in a manner dependent on the ATP receptor P2X7. Altogether, the findings suggest that TRPV1-induced ATP release from Müller glia leads to P2X7 activation in retinal ganglion cells, which subsequently undergo autophagy-dependent apoptotic cell death [16]. On the other hand, rat Müller glial cells apparently do not express TRPV1 [145]. Despite this, Müller glial activation in the optic nerve axotomy model appears to depend indirectly on TRPV1 activation, as well as on the production of nitric oxide and peroxynitrite [145]. However, retinas from TRPV1- or TRPV4-knockout animals continued to exhibit damage following ischemia–reperfusion (I/R) injury, indicating that under these experimental conditions these receptors are not essential for the induction of cell death [127]. In contrast, retinas from TRPA1-knockout animals are resistant to I/R injury [127]. Pharmacological blockade of TRPA1 with HC-030031 was also able to inhibit cell death induced by oxygen–glucose deprivation (OGD) in the embryonic chicken retina [136]. Activation of TRPA1 receptors appears to play an important role in retinal ischemic events. However, in these studies, the authors did not evaluate glial reactivity in this context or its dependence on TRPA1 receptor activation. OGD increased TRPA1 protein expression in the retinas of the animals [136]. This increase may be occurring in Müller glial cells and could be responsible for triggering a pro-inflammatory response that promotes cell death.
Although substantial evidence links TRP receptors to Müller glial cell function, there are currently no studies in the literature associating these receptors with astrocyte activation in glaucoma models. Nevertheless, single-cell RT-PCR analyses have demonstrated that several TRP isoforms (TRPC1-2, TRPC6, TRPV2, TRPV4, TRPM2, TRPM4, TRPM6-7, and TRPP1-2), as well as Piezo receptors (Piezo1-2), are expressed in astrocytes of the optic nerve head, with most astrocytes expressing TRPC1 and TRPP1-2 [64]. The lack of information regarding the role of TRP receptors in astrocytes represents an excellent opportunity to further investigate these receptors in glaucoma models.
Taken together, the available evidence reveals a broad involvement of TRP receptors in Müller glial activation and cell loss across different pathological retinal models. To date, however, no studies have evaluated a possible association between TRPM7 expression and glaucoma models, nor the potential involvement of TRPV1 in this context. It is possible that Müller glial activation depends on the coordinated action of TRPV1, TRPV4, and TRPM7. Modulating Müller glial activation therefore represents an attractive strategy for slowing glaucoma progression, with TRP receptors emerging as promising therapeutic targets.
6. Gene Therapy Approaches Targeting Macroglial Dysfunction in Glaucoma
Glaucoma is classically managed by therapies aimed at lowering intraocular pressure; however, disease progression may occur despite adequate pressure control, indicating that additional mechanisms contribute to retinal ganglion cell degeneration. Among these mechanisms, glial activation and neuroinflammation have gained increasing attention. Astrocytes and Müller glia, together with microglia, respond to glaucomatous stress by sensing ATP, reactive oxygen species, damage-associated molecular patterns, mechanical strain, and inflammatory mediators. Although these responses may initially support tissue homeostasis, sustained glial reactivity can amplify neuroinflammatory signaling and contribute to retinal ganglion cell soma, axon, and synapse degeneration [80].
In this context, gene therapy represents an attractive therapeutic approach because it may provide long-lasting modulation of molecular pathways involved in glaucomatous neurodegeneration. Current gene therapy strategies for glaucoma have mainly focused on two broad goals: reduction of intraocular pressure and ganglion cell neuroprotection. Approaches targeting the trabecular meshwork or ciliary body aim to improve aqueous humor outflow or reduce aqueous humor production, whereas retinal approaches seek to protect retinal ganglion cells through delivery of neurotrophic, anti-apoptotic, antioxidant, or regenerative factors. Viral vectors, particularly adeno-associated viral vectors, remain the most widely explored delivery platforms, although non-viral systems and RNA-based strategies are also being developed [146].
Macroglia-directed gene therapy remains less explored but is conceptually highly relevant. Astrocytes and Müller glia are strategically positioned to regulate extracellular glutamate and ion homeostasis, metabolic support, oxidative stress, blood-retinal barrier integrity, cytokine production, complement activation, and neuron-glia communication. Therefore, genetic modulation of these cells could shift the retinal environment from a chronic pro-inflammatory state toward a neuroprotective phenotype. Potential strategies include suppressing NF-κB-dependent inflammatory signaling, reducing TNF-α/IL-1β/C1q-mediated neurotoxic astrocyte conversion, modulating ATP/P2X7R signaling, enhancing antioxidant defenses, or inducing sustained production of neurotrophic factors such as BDNF, CNTF, or NGF [80,147].
Müller glia are particularly attractive targets because they span the entire retinal thickness and provide structural, metabolic, and homeostatic support to neurons. Experimental work in retinal gene therapy has identified viral capsids and promoter systems capable of improving Müller glia targeting, including ShH10/ShH10Y AAV variants and Müller-associated promoters such as RLBP1 or GFAP. These tools suggest that selective or enriched macroglial transduction is feasible, although their application to glaucomatous neurodegeneration remains at an early stage [148].
Despite this promise, several challenges must be addressed before macroglia-targeted gene therapy can be translated to glaucoma. These include achieving cell-type specificity, avoiding excessive or uncontrolled transgene expression, defining the optimal disease stage for intervention, minimizing immune responses to viral vectors, and determining whether long-term suppression of glial reactivity could interfere with beneficial reparative functions. Furthermore, because macroglial activation may be protective in early disease but detrimental when chronic, future studies should consider stage-specific and phenotype-specific modulation rather than complete inhibition of gliosis [80].
Overall, gene therapy provides a promising framework to move beyond pressure-lowering strategies and target the neuroinflammatory component of glaucoma. While most current approaches focus on intraocular pressure regulation or direct retinal ganglion cell protection, macroglia-directed gene therapy may represent a future strategy to reprogram the retinal microenvironment, reduce chronic inflammatory amplification, and preserve retinal ganglion cell function.
7. Conclusions
Macroglial cells, comprising astrocytes and Müller cells, function as central regulators of the healthy retinal microenvironment by providing essential mechanical, structural, and metabolic support. However, under pathological conditions such as glaucoma, mechanical deformation, ischemic/metabolic stress, oxidative damage, and inflammatory signaling trigger profound phenotypic and functional alterations in glial cells that contribute to disease onset and progression. Therefore, it is important to determine whether the prolonged suppression or inhibition of macroglial reactivity may adversely affect their intrinsic reparative and homeostatic functions. Given that glial activation is neuroprotective during the early stages of the disease but becomes detrimental when chronic, future studies should focus on strategies that modulate specific glial phenotypes according to the stage of disease progression, rather than aiming for the complete inhibition of gliosis. Furthermore, the optimal stage of glaucoma progression for therapeutic intervention targeting macroglial pathways remains poorly defined. Therefore, identifying the appropriate therapeutic window will be crucial for maximizing treatment efficacy while preserving the beneficial functions of macroglia.
The transition of the glial response from an adaptive state to maladaptive inflammatory signaling involves mechanosensitive pathways associated with Piezo1 and members of the TRP channel family, including TRPV1, TRPV4, TRPC1, and TRPA1. Activation of these channels converts mechanical stretch into rapid reactive responses through calcium-dependent signaling. Furthermore, the progression of glaucomatous damage depends on multicellular glial networks, in which early macroglial activation modulates microglial responses through the release of cytokines and complement components, while reactive microglia further amplify the inflammatory state of macroglia. In this context, a more comprehensive understanding of the relationship between mechanosensitive channels and gliosis is needed. For example, are specific mechanosensitive channels selectively involved in the adaptive or maladaptive responses of astrocytes and/or Müller cells? Likewise, defining the optimal therapeutic window for targeting each mechanosensitive channel will be essential.
In addition, it will be important to investigate whether these channels contribute to the progression of neurodegeneration in glaucoma patients without elevated intraocular pressure. In this regard, TRPA1 deserves particular attention, as it is activated by ROS, functioning as an oxidative stress sensor while establishing a positive feedback loop that further exacerbates oxidative damage. Therefore, it is conceivable that certain classical TRP channels contribute to neurodegeneration independently of elevated IOP and may represent promising therapeutic targets for the treatment of normal-tension glaucoma. Overall, TRP channels and, in certain contexts, Piezo channels may represent key molecular regulators linking mechanotransduction, gliosis, and glaucoma progression, making them attractive candidates for the development of novel therapeutic strategies.
Finally, regarding the translational challenges associated with macroglia-targeted gene therapy, several important technical gaps remain to be addressed. These include: (a) achieving strict cell type-specific targeting within the retina; (b) preventing excessive or uncontrolled expression of the introduced transgenes; and (c) minimizing host immune responses against viral vectors. Nevertheless, the modulation of macroglial reactivity appears to be a promising therapeutic strategy in experimental models of glaucoma and may provide new opportunities to prevent disease progression and improve treatment outcomes.
Funding
This research was funded by This work was supported by grants from National Council of Scientific and Technological Development (CNPq), Universal grant number: 403325/2023-4, National Institute of Science and Technology— INCT da GLIA (INCT/IGlia) (409204/2024-2); Fellowships from Coordination of Superior Level Staff Improvement (CAPES); Foundation for Research Support of the State of Rio de Janeiro (FAPERJ)– K.C. Calaza (Grant E-26/211.289/2021); R.B. (Grant E-26/211.462/2019, E-26/210.410/2022). K.C. Calaza is supported by a special scholarship for senior scientists of the Rio de Janeiro State (CNE/FAPERJ, Grant E-26/202.977/2017 and E-26/201.025/2021) and CNPq (304410/2019-5, 316553/2023-9). D.S.M.A is supported by a scholarship from the National Council of Scientific and Technological Development (CNPq) number 315139/2025-0.
Acknowledgments
Teixeira, GR, Costa, AGA and Mattos ACL thank the Coordination of Superior Level Staff Improvement (CAPES) for fellowships. KCC is supported by a special scholarship for senior scientists of the Rio de Janeiro State (CNE/FAPERJ, grant number E-26/201.025/2021, E-26/204.180/2024) and CNPq (KCC: 304410/2019-5, 316553/2023-9). D.S.M.A is supported by a scholarship from the National Council of Scientific and Technological Development (CNPq) number 315139/2025-0.
Conflicts of Interest
The authors declare no conflicts of interest
Abbreviations
The following abbreviations are used in this manuscript:
| ANG1 | Angiopoietin-1 |
| TRPA | Ankyrin |
| AIBP | Apolipoprotein A-I binding protein |
| AQP-4 | Aquaporin-4 |
| BRB | Blood–retinal barrier |
| BDNF | Brain-derived neurotrophic factor |
| CNTF | Ciliary neurotrophic factor |
| CNS | Central nervous system |
| C3 | Complement component 3 |
| Cx43 | Connexin-43 |
| DAMP | Damage-associated molecular patterns |
| DDR | |
| ET-1 | DNA damage response |
| ERG | |
| ECM | Endothelin-1 |
| ERK | Electroretinogram |
| GCL | Extracellular matrix |
| GSDMD | Extracellular signal-regulated kinase |
| GLAST | Ganglion cell layer |
| GFAP | Gasdermin D |
| GS | |
| GSH | Glutamate/Aspartate Transporter |
| H2O2 | Glial fibrillary acidic protein |
| HIF-1α | Glutamine synthetase |
| 4-HNE | Glutathione |
| IOP | Hydrogen peroxide |
| IL | Hypoxia-inducible factor-1 alpha |
| ILM | 4-hydroxynonenal |
| LOX | Intraocular pressure |
| MMP | Interleukine |
| MHC | Inner limiting membrane |
| mtROS | Lipoxygenase |
| NOX | Matrix metalloproteinases |
| NF-κB | Major histocompatibility complex |
| NO | Mitochondrial reactive oxygen species |
| NLRP3 | NADPH oxidase |
| O2 | Nuclear factor kappaB |
| ONH | Nitric oxide |
| OLM | Inflammasome |
| OXPHOS | Superoxide radical |
| OGD | Optic nerve head |
| PEDF | Outer limiting membrane |
| ROS | Oxidative phosphorylation |
| RNS | Oxygen–glucose deprivation |
| RGCs | Pigment epithelium-derived factor |
| RNFL | Reactive oxygen species |
| SASP | Reactive nitrogen species |
| SOD | Retinal ganglion cells |
| TCA | Retinal nerve fiber layer |
| TGF-β2 | Senescence-associated secretory phenotype |
| TNF-α | Superoxide dismutase |
| TLR4 | Tricarboxylic acid |
| TGM2 | Transforming Growth Factor-β2 |
| TRP | Tumor necrosis factor alpha |
| VEGF | Toll-like receptor 4 |
| xCT | Transglutaminase |
| YAP | Transient receptor potential |
| Vascular endothelial growth factor | |
| Cystine/glutamate antiporter | |
| Yes-associated protein |
References
- Quigley, H.A.; Broman, A.T. The Number of People with Glaucoma Worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The Pathophysiology and Treatment of Glaucoma: A Review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef]
- Jonas, J.B.; Aung, T.; Bourne, R.R.; Bron, A.M.; Ritch, R.; Panda-Jonas, S. Glaucoma. Lancet 2017, 390, 2183–2193. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.-C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.-Y. Global Prevalence of Glaucoma and Projections of Glaucoma Burden through 2040: A Systematic Review and Meta-Analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- García-Bermúdez, M.Y.; Freude, K.K.; Mouhammad, Z.A.; van Wijngaarden, P.; Martin, K.K.; Kolko, M. Glial Cells in Glaucoma: Friends, Foes, and Potential Therapeutic Targets. Front. Neurol. 2021, 12, 624983. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Albarral, J.A.; Ramírez, A.I.; de Hoz, R.; Matamoros, J.A.; Salobrar-García, E.; Elvira-Hurtado, L.; López-Cuenca, I.; Sánchez-Puebla, L.; Salazar, J.J.; Ramírez, J.M. Glaucoma: From Pathogenic Mechanisms to Retinal Glial Cell Response to Damage. Front. Cell. Neurosci. 2024, 18, 1354569. [Google Scholar] [CrossRef] [PubMed]
- Calkins, D.J. Critical Pathogenic Events Underlying Progression of Neurodegeneration in Glaucoma. Prog. Retin. Eye Res. 2012, 31, 702–719. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects. Cells 2021, 10, 1372. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.R. The Optic Nerve Head in Glaucoma: Role of Astrocytes in Tissue Remodeling. Prog. Retin. Eye Res. 2000, 19, 297–321. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller Cells in the Healthy and Diseased Retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-Neuron Interactions in the Mammalian Retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.E. Optic Nerve Head Structure in Glaucoma: Astrocytes as Mediators of Axonal Damage. Eye (Lond.) 2000, 14 Pt 3B, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, Y.; Chen, D. The Heterogeneity of Astrocytes in Glaucoma. Front. Neuroanat. 2022, 16, 995369. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, Y.; Namekata, K.; Guo, X.; Harada, T. Glial Cells as a Promising Therapeutic Target of Glaucoma: Beyond the IOP. Front. Ophthalmol. 2023, 3, 1310226. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, X.; Liu, Q.; Jin, S.; Chang, H.; Liu, H. The Roles of Transient Receptor Potential Ion Channels in Pathologies of Glaucoma. Front. Physiol. 2022, 13, 806786. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Nie, D.; Fang, M.; He, W.; Zhang, J.; Liu, X.; Zhang, G. Müller Cells under Hydrostatic Pressure Modulate Retinal Cell Survival via TRPV1/PLCγ1 Complex-Mediated Calcium Influx in Experimental Glaucoma. FEBS J. 2024, 291, 2703–2714. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Khaw, P.T. Primary Open-Angle Glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, C. The Morphological Difference between Glaucoma and Other Optic Neuropathies. J. Neuroophthalmol. 2015, 35 Suppl 1, S8–S21. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglio, F.; Pfeiffer, N.; Gericke, A. Immunomodulatory and Antioxidant Drugs in Glaucoma Treatment. Pharmaceuticals 2023, 16, 1193. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.M.; Tanna, A.P. Glaucoma. Med. Clin. North Am. 2021, 105, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Hodge, W.; Malvankar-Mehta, M. The Cost-Effectiveness Analysis of Teleglaucoma Screening Device. PLoS ONE 2015, 10, e0137913. [Google Scholar] [CrossRef] [PubMed]
- Soh, Z.; Yu, M.; Betzler, B.K.; Majithia, S.; Thakur, S.; Tham, Y.C.; Wong, T.Y.; Aung, T.; Friedman, D.S.; Cheng, C.-Y. The Global Extent of Undetected Glaucoma in Adults: A Systematic Review and Meta-Analysis. Ophthalmology 2021, 128, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Heijl, A.; Leske, M.C.; Bengtsson, B.; Hyman, L.; Bengtsson, B.; Hussein, M. Early Manifest Glaucoma Trial Group Reduction of Intraocular Pressure and Glaucoma Progression: Results from the Early Manifest Glaucoma Trial. Arch. Ophthalmol. 2002, 120, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Asrani, S.G.; McGlumphy, E.J.; Al-Aswad, L.A.; Chaya, C.J.; Lin, S.; Musch, D.C.; Pitha, I.; Robin, A.L.; Wirostko, B.; Johnson, T.V. The Relationship between Intraocular Pressure and Glaucoma: An Evolving Concept. Prog. Retin. Eye Res. 2024, 103, 101303. [Google Scholar] [CrossRef] [PubMed]
- Naik, V.; Ohri, S.; Fernandez, E.; Mwanza, J.-C.; Fleischman, D. Changes in Individuals’ Glaucoma Progression Velocity after IOP-Lowering Therapy: A Systematic Review. PLoS ONE 2025, 20, e0324806. [Google Scholar] [CrossRef] [PubMed]
- Kass, M.A.; Heuer, D.K.; Higginbotham, E.J.; Johnson, C.A.; Keltner, J.L.; Miller, J.P.; Parrish, R.K., 2nd; Wilson, M.R.; Gordon, M.O. The Ocular Hypertension Treatment Study: A Randomized Trial Determines That Topical Ocular Hypotensive Medication Delays or Prevents the Onset of Primary Open-Angle Glaucoma. Arch. Ophthalmol. 2002, 120, 701–713; discussion 829–830. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sanchez, J.; Lin, D.; Liu, W.W. Mechanosensitive Ion Channels in Glaucoma Pathophysiology. Vis. Res. 2024, 223, 108473. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Gryciuk, A.; Skup, M.; Waleszczyk, W.J. Glaucoma -State of the Art and Perspectives on Treatment. Restor. Neurol. Neurosci. 2016, 34, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Subbulakshmi, S.; Kavitha, S.; Venkatesh, R. Prostaglandin Analogs in Ophthalmology. Indian J. Ophthalmol. 2023, 71, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.; Rewri, P. A Narrative Review of Pharmacotherapy of Glaucoma. Future Pharmacol. 2024, 4, 395–419. [Google Scholar] [CrossRef]
- Lee, H.-P.; Tsung, T.-H.; Tsai, Y.-C.; Chen, Y.-H.; Lu, D.-W. Glaucoma: Current and New Therapeutic Approaches. Biomedicines 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- European Glaucoma Society Terminology and Guidelines for Glaucoma, 5th Edition. Br. J. Ophthalmol. 2021, 105, 1–169. [CrossRef] [PubMed]
- Takusagawa, H.L.; Hoguet, A.; Sit, A.J.; Rosdahl, J.A.; Chopra, V.; Ou, Y.; Richter, G.; Kim, S.J.; WuDunn, D. Selective Laser Trabeculoplasty for the Treatment of Glaucoma: A Report by the American Academy of Ophthalmology. Ophthalmology 2024, 131, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, D.M. Does Reduction of Intraocular Pressure (IOP) Prevent Visual Field Loss in Glaucoma? Am. J. Optom. Physiol. Opt. 1983, 60, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Killer, H.E.; Pircher, A. Normal Tension Glaucoma: Review of Current Understanding and Mechanisms of the Pathogenesis. Eye (Lond.) 2018, 32, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Calkins, D.J. Adaptive Responses to Neurodegenerative Stress in Glaucoma. Prog. Retin. Eye Res. 2021, 84, 100953. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, J.; Wang, Y.; Deng, F.; Deng, Z. Oxidative Stress: Signaling Pathways, Biological Functions, and Disease. MedComm 2025, 6, e70268. [Google Scholar] [CrossRef] [PubMed]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous Non-Enzymatic Antioxidants in the Human Body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The Molecular Basis of Retinal Ganglion Cell Death in Glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- Iorga, R.E.; Moraru, A.D.; Costin, D.; Munteanu-Dănulescu, R.S.; Brănișteanu, D.C. Current Trends in Targeting the Oxidative Stress in Glaucoma (Review). Eur. J. Ophthalmol. 2024, 34, 328–337. [Google Scholar] [CrossRef] [PubMed]
- de Freitas Azevedo-Repossi, R.; Brito, R.; Cossenza, M.; Dos Santos-Rodrigues, A.; Ferreira, G.C.; Petrs-Silva, H.; Calaza, K.C.; Fragel-Madeira, L. Reactive Oxygen Species Regulation across Retinitis Pigmentosa Animal Models: A 25-Year Systematized Review. Mol. Neurobiol. 2025, 62, 16015–16044. [Google Scholar] [CrossRef] [PubMed]
- Goldsteins, G.; Hakosalo, V.; Jaronen, M.; Keuters, M.H.; Lehtonen, Š.; Koistinaho, J. CNS Redox Homeostasis and Dysfunction in Neurodegenerative Diseases. Antioxidants 2022, 11, 405. [Google Scholar] [CrossRef] [PubMed]
- Vernazza, S.; Tirendi, S.; Bassi, A.M.; Traverso, C.E.; Saccà, S.C. Neuroinflammation in Primary Open-Angle Glaucoma. J. Clin. Med. 2020, 9, E3172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, S.; Xie, B.; Zhong, Y. Aging, Cellular Senescence, and Glaucoma. Aging Dis. 2024, 15, 546–564. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.T.H.; Pattamatta, U.; White, A. Role of Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of Glaucoma. Med. Hypothesis Discov. Innov. Ophthalmol. 2025, 14, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.M.; Chidlow, G.; Graham, M.; Melena, J. Retinal Ischemia: Mechanisms of Damage and Potential Therapeutic Strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.H.; Wang, Z.; Chong, R.S. Caveolin-1 in Vascular Health and Glaucoma: A Critical Vascular Regulator and Potential Therapeutic Target. Front. Med. 2023, 10, 1087123. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Kagemann, L.; Ehrlich, R.; Rospigliosi, C.; Moore, D.; Siesky, B. Measuring and Interpreting Ocular Blood Flow and Metabolism in Glaucoma. Can. J. Ophthalmol. 2008, 43, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Vohra, R.; Tsai, J.C.; Kolko, M. The Role of Inflammation in the Pathogenesis of Glaucoma. Surv. Ophthalmol. 2013, 58, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Tomita, Y.; Miwa, Y.; Kunimi, H.; Nakai, A.; Shoda, C.; Negishi, K.; Kurihara, T. Recent Insights into Roles of Hypoxia-Inducible Factors in Retinal Diseases. Int. J. Mol. Sci. 2024, 25, 10140. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Bariş, M.; Tezel, G. Immunomodulation as a Neuroprotective Strategy for Glaucoma Treatment. Curr. Ophthalmol. Rep. 2019, 7, 160–169. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Sun, Q.-F.; Bai, W.; Yao, J. Retinal Astrocytes: Key Coordinators of Developmental Angiogenesis and Neurovascular Homeostasis in Health and Disease. Biology 2026, 15. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Ren, J.; Qian, X. Study on the Polarization of Astrocytes in the Optic Nerve Head of Rats under High Intraocular Pressure: In Vitro. Bioengineering 2025, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Gong, P.; Pan, X.; Ren, Z.; Liu, Y.; Qi, G.; Li, J.-L.; Sun, W.; Ge, W.-P.; Zhang, C.-L.; et al. Temporal-Spatial Generation of Astrocytes in the Developing Diencephalon. Neurosci. Bull. 2024, 40, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, G.J.; Piscopo, V.E.C.; Goldsmith, T.M.; Chapleau, A.; Sirois, J.; Bernard, G.; Antel, J.P.; Durcan, T.M. Single Cell RNAseq to Identify Subpopulations of Glial Progenitors in iPSC-Derived Oligodendroglial Lineage Cultures. npj Syst. Biol. Appl. 2025, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.M.; Wareham, L.K.; Calkins, D.J. Retinal Astrocyte Morphology Predicts Integration of Vascular and Neuronal Architecture. Front. Neurosci. 2023, 17, 1244679. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.M.; Wareham, L.K.; Calkins, D.J. Morphological and Electrophysiological Characterization of a Novel Displaced Astrocyte in the Mouse Retina. Glia 2024, 72, 1356–1370. [Google Scholar] [CrossRef] [PubMed]
- Endo, F.; Kasai, A.; Soto, J.S.; Yu, X.; Qu, Z.; Hashimoto, H.; Gradinaru, V.; Kawaguchi, R.; Khakh, B.S. Molecular Basis of Astrocyte Diversity and Morphology across the CNS in Health and Disease. Science 2022, 378, eadc9020. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-G.; Rone, J.M.; Li, Z.; Akl, C.F.; Shin, S.W.; Lee, J.-H.; Flausino, L.E.; Pernin, F.; Chao, C.-C.; Kleemann, K.L.; et al. Disease-Associated Astrocyte Epigenetic Memory Promotes CNS Pathology. Nature 2024, 627, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Choi, J.-O.; Hwang, A.; Bae, H.W.; Kim, C.Y. Ciliary Neurotrophic Factor Derived from Astrocytes Protects Retinal Ganglion Cells through PI3K/AKT, JAK/STAT, and MAPK/ERK Pathways. Invest. Ophthalmol. Vis. Sci. 2022, 63, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, R.; Pappas, A.C.; Seifert, P.; Savol, A.; Sadreyev, R.I.; Sun, D.; Jakobs, T.C. Astrocytes in the Optic Nerve Are Heterogeneous in Their Reactivity to Glaucomatous Injury. Cells 2023, 12, 2131. [Google Scholar] [CrossRef] [PubMed]
- Strat, A.N.; Ganapathy, P.S. Considerations and Implications of Current in Vitro Model Systems to Study Optic Nerve Head Cellular Mechanobiology. Front. Cell Dev. Biol. 2025, 13, 1699793. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Sun, D.; Jakobs, T.C. Astrocytes in the Optic Nerve Head Express Putative Mechanosensitive Channels. Mol. Vis. 2015, 21, 749–766. [Google Scholar] [PubMed]
- do Nascimento, T.H.O.; Pereira-Figueiredo, D.; Veroneze, L.; Nascimento, A.A.; De Logu, F.; Nassini, R.; Campello-Costa, P.; Faria-Melibeu, A. da C.; Souza Monteiro de Araújo, D.; Calaza, K.C. Functions of TRPs in Retinal Tissue in Physiological and Pathological Conditions. Front. Mol. Neurosci. 2024, 17, 1459083. [Google Scholar] [CrossRef] [PubMed]
- Fuchshofer, R.; Birke, M.; Welge-Lussen, U.; Kook, D.; Lütjen-Drecoll, E. Transforming Growth Factor-Beta 2 Modulated Extracellular Matrix Component Expression in Cultured Human Optic Nerve Head Astrocytes. Invest. Ophthalmol. Vis. Sci. 2005, 46, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Jakobs, T.C. The Time Course of Gene Expression during Reactive Gliosis in the Optic Nerve. PLoS ONE 2013, 8, e67094. [Google Scholar] [CrossRef] [PubMed]
- Quillen, S.; Schaub, J.; Quigley, H.; Pease, M.; Korneva, A.; Kimball, E. Astrocyte Responses to Experimental Glaucoma in Mouse Optic Nerve Head. PLoS ONE 2020, 15, e0238104. [Google Scholar] [CrossRef] [PubMed]
- Macanian, J.; Sharma, S.C. Pathogenesis of Glaucoma. Encyclopedia 2022, 2, 1803–1810. [Google Scholar] [CrossRef]
- Waisberg, E.; Micieli, J.A. Neuro-Ophthalmological Optic Nerve Cupping: An Overview. Eye Brain 2021, 13, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.G.; Julé, A.M.; Sun, D. Astrocytes of the Optic Nerve Exhibit a Region-Specific and Temporally Distinct Response to Elevated Intraocular Pressure. Mol. Neurodegener. 2023, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, A.H. Nitric Oxide: A Potential Mediator of Retinal Ganglion Cell Damage in Glaucoma. Surv. Ophthalmol. 1999, 43 Suppl 1, S129–S135. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.; Hayashide, L. de S.; Amorim, P.; Fernandes, B.M.; Araujo, A.P.B.; Messor, D.F.; Leocadio, V.E.; Pessoa, B.; Corrêa, J.B.L.P.; Villablanca, C.; et al. Doxorubicin Induces a Senescent Phenotype in Murine and Human Astrocytes. J. Neurochem. 2025, 169, e70177. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Shi, S.; Palacios, E.; Ju, W.-K.; Liu, H.; Zhang, W. CXCR3 Deficiency Alleviates Retinal Ganglion Cell Loss by Regulating Neuron-Astrocyte Communication in a Mouse Model of Glaucoma. Invest. Ophthalmol. Vis. Sci. 2025, 66, 42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-X.; Sun, H.; Guo, W.-Y. Astrocyte Polarization in Glaucoma: A New Opportunity. Neural Regen. Res. 2022, 17, 2582–2588. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, C.; Ren, C.; Zhao, H.; Zhang, X. Immunological Landscape of Retinal Ischemia-Reperfusion Injury: Insights into Resident and Peripheral Immune Cell Responses. Aging Dis. 2024, 16, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Coviltir, V.; Burcel, M.G.; Baltă, G.; Marinescu, M.C. Interplay between Ocular Ischemia and Glaucoma: An Update. Int. J. Mol. Sci. 2024, 25, 12400. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.A.; Ferraz, G.; Dos Santos, R.C.; Conde, L.; Dantas, D.P.; Archanjo, B.S.; Linden, R.; Pimentel-Coelho, P.M.; Allodi, S. Early Ultrastructural Damage in Retina and Optic Nerve Following Intraocular Pressure Elevation. Vis. Res. 2025, 227, 108544. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Molecular Regulation of Neuroinflammation in Glaucoma: Current Knowledge and the Ongoing Search for New Treatment Targets. Prog. Retin. Eye Res. 2022, 87, 100998. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, K.A.; Stafford, B.K.; El-Danaf, R.N.; Adler, D.I.; Münch, A.E.; Weigel, M.K.; Huberman, A.D.; Liddelow, S.A. Neurotoxic Reactive Astrocytes Drive Neuronal Death after Retinal Injury. Cell Rep. 2020, 31, 107776. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Dvoriantchikova, G.; Barakat, D.; Ivanov, D.; Bethea, J.R.; Shestopalov, V.I. Transgenic Inhibition of Astroglial NF-κB Protects from Optic Nerve Damage and Retinal Ganglion Cell Loss in Experimental Optic Neuritis. J. Neuroinflammation 2012, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-κB Function in the Nervous System. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zeng, Q.; Barış, M.; Tezel, G. Transgenic Inhibition of Astroglial NF-κB Restrains the Neuroinflammatory and Neurodegenerative Outcomes of Experimental Mouse Glaucoma. J. Neuroinflammation 2020, 17, 252. [Google Scholar] [CrossRef] [PubMed]
- Chong, R.S.; Martin, K.R. Glial Cell Interactions and Glaucoma. Curr. Opin. Ophthalmol. 2015, 26, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Albarral, J.A.; de Hoz, R.; Matamoros, J.A.; Chen, L.; López-Cuenca, I.; Salobrar-García, E.; Sánchez-Puebla, L.; Ramírez, J.M.; Triviño, A.; Salazar, J.J.; et al. Retinal Changes in Astrocytes and Müller Glia in a Mouse Model of Laser-Induced Glaucoma: A Time-Course Study. Biomedicines 2022, 10, 939. [Google Scholar] [CrossRef] [PubMed]
- Kashihara, T.; Morita, Y.; Hatta, M.; Inoue, S.; Suzuki, Y.; Morita, A.; Nakahara, T. YAP Activation in Müller Cells Protects against NMDA-Induced Retinal Ganglion Cell Injury by Regulating Bcl-xL Expression. Front. Pharmacol. 2024, 15, 1446521. [Google Scholar] [CrossRef] [PubMed]
- Bitard, J.; Grellier, E.-K.; Lourdel, S.; Filipe, H.P.; Hamon, A.; Fenaille, F.; Castelli, F.A.; Chu-Van, E.; Roger, J.E.; Locker, M.; et al. Uveitic Glaucoma-like Features in Yap Conditional Knockout Mice. Cell Death Discov. 2024, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Karnam, S.; Maurya, S.; Nagireddy, R.; Flanagan, J.G.; Gronert, K. Lipoxin B4 Mitigates TRPV4-Activated Müller Cell Gliosis during Ocular Hypertension. Invest. Ophthalmol. Vis. Sci. 2026, 67, 2. [Google Scholar] [CrossRef] [PubMed]
- Pereiro, X.; Ruzafa, N.; Azkargorta, M.; Elortza, F.; Acera, A.; Ambrósio, A.F.; Santiago, A.R.; Vecino, E. Müller Glial Cells Located in the Peripheral Retina Are More Susceptible to High Pressure: Implications for Glaucoma. Cell Biosci. 2024, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Morozumi, W.; Inagaki, S.; Iwata, Y.; Nakamura, S.; Hara, H.; Shimazawa, M. Piezo Channel Plays a Part in Retinal Ganglion Cell Damage. Exp. Eye Res. 2020, 191, 107900. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Chen, G.; Yang, L.; Deng, J.; Pan, Y. Piezo1 Inhibitor Isoquercitrin Rescues Neural Impairment Mediated by NLRP3 after Intracerebral Hemorrhage. Exp. Neurol. 2024, 379, 114852. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bian, W.; Yang, D.; Yang, M.; Luo, H. Inhibiting the Piezo1 Channel Protects Microglia from Acute Hyperglycaemia Damage through the JNK1 and mTOR Signalling Pathways. Life Sci. 2021, 264, 118667. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Y.; Zhang, S.; Sun, X.; Wu, J. TRPV4-Induced Müller Cell Gliosis and TNF-α Elevation-Mediated Retinal Ganglion Cell Apoptosis in Glaucomatous Rats via JAK2/STAT3/NF-κB Pathway. J. Neuroinflammation 2021, 18, 271. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Nakazawa, T.; Shimura, M.; Tomita, H.; Tamai, M. Presence of Mitogen-Activated Protein Kinase in Retinal Müller Cells and Its Neuroprotective Effect Ischemia-Reperfusion Injury. Neuroreport 2002, 13, 2103–2107. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Reichenbach, A. Role of Muller Cells in Retinal Degenerations. Front. Biosci. 2001, 6, E72–E92. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.P.; Fisher, S.K. Up-Regulation of Glial Fibrillary Acidic Protein in Response to Retinal Injury: Its Potential Role in Glial Remodeling and a Comparison to Vimentin Expression. Int. Rev. Cytol. 2003, 230, 263–290. [Google Scholar] [CrossRef] [PubMed]
- Iandiev, I.; Tenckhoff, S.; Pannicke, T.; Biedermann, B.; Hollborn, M.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Differential Regulation of Kir4.1 and Kir2.1 Expression in the Ischemic Rat Retina. Neurosci. Lett. 2006, 396, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Ashimori, A.; Higashijima, F.; Sakuma, A.; Hamada, W.; Sunada, J.; Aoki, R.; Mikuni, M.; Hayashi, K.; Yoshimoto, T.; et al. HIF-1α-Dependent Regulation of Angiogenic Factor Expression in Müller Cells by Mechanical Stimulation. Exp. Eye Res. 2024, 247, 110051. [Google Scholar] [CrossRef] [PubMed]
- Salkar, A.; Palanivel, V.; Basavarajappa, D.; Mirzaei, M.; Schulz, A.; Yan, P.; Gupta, V.; Graham, S.; You, Y. Glial and Immune Dysregulation in Glaucoma Independent of Retinal Ganglion Cell Loss: A Human Post-Mortem Histopathology Study. Acta Neuropathol. Commun. 2025, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular Signaling and Factors Involved in Müller Cell Gliosis: Neuroprotective and Detrimental Effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef] [PubMed]
- Derouiche, A.; Rauen, T. Coincidence of L-glutamate/L-Aspartate Transporter (GLAST) and Glutamine Synthetase (GS) Immunoreactions in Retinal Glia: Evidence for Coupling of GLAST and GS in Transmitter Clearance. J. Neurosci. Res. 1995, 42, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Zhao, G.-L.; Cheng, S.; Wang, Z.; Yang, X.-L. Activation of Retinal Glial Cells Contributes to the Degeneration of Ganglion Cells in Experimental Glaucoma. Prog. Retin. Eye Res. 2023, 93, 101169. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, Z.; Li, S.; Zhou, H.; Guo, Y.; Wang, Y.; Lei, B.; Miao, Y.; Wang, Z. Overexpression of the Inwardly Rectifying Potassium Channel Kir4.1 or Kir4.1 Tyr 9 Asp in Müller Cells Exerts Neuroprotective Effects in an Experimental Glaucoma Model. Neural Regen. Res. 2026, 21, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Gionfriddo, J.R.; Freeman, K.S.; Groth, A.; Scofield, V.L.; Alyahya, K.; Madl, J.E. Alpha-Luminol Prevents Decreases in Glutamate, Glutathione, and Glutamine Synthetase in the Retinas of Glaucomatous DBA/2J Mice. Vet. Ophthalmol. 2009, 12, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-T.; Alyahya, K.; Gionfriddo, J.R.; Dubielzig, R.R.; Madl, J.E. Loss of Glutamine Synthetase Immunoreactivity from the Retina in Canine Primary Glaucoma. Vet. Ophthalmol. 2008, 11, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.C.; Sande, P.; Marcos, H.A.; de Zavalía, N.; Keller Sarmiento, M.I.; Rosenstein, R.E. Effect of Glaucoma on the Retinal Glutamate/glutamine Cycle Activity. FASEB J. 2005, 19, 1161–1162. [Google Scholar] [CrossRef] [PubMed]
- Nucci, C.; Tartaglione, R.; Rombolà, L.; Morrone, L.A.; Fazzi, E.; Bagetta, G. Neurochemical Evidence to Implicate Elevated Glutamate in the Mechanisms of High Intraocular Pressure (IOP)-Induced Retinal Ganglion Cell Death in Rat. Neurotoxicology 2005, 26, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, C.; Zhang, C.; Zhang, W.; Zhu, W.; He, Y.; Xia, Z.; Song, W. p38 MAPK Inhibitor SB202190 Suppresses Ferroptosis in the Glutamate-Induced Retinal Excitotoxicity Glaucoma Model. Neural Regen. Res. 2024, 19, 2299–2309. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, H.; Yang, R.; Ji, D.; Xia, X. GSK872 and Necrostatin-1 Protect Retinal Ganglion Cells against Necroptosis through Inhibition of RIP1/RIP3/MLKL Pathway in Glutamate-Induced Retinal Excitotoxic Model of Glaucoma. J. Neuroinflammation 2022, 19, 262. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Pace, L.; Bedse, G.; Lavecchia, A.M.; De Marco, F.; Gaetani, S.; Serviddio, G. Glutamate and Mitochondria: Two Prominent Players in the Oxidative Stress-Induced Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.-K.; Perkins, G.A.; Kim, K.-Y.; Bastola, T.; Choi, W.-Y.; Choi, S.-H. Glaucomatous Optic Neuropathy: Mitochondrial Dynamics, Dysfunction and Protection in Retinal Ganglion Cells. Prog. Retin. Eye Res. 2023, 95, 101136. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.C.; Campanelli, J.; Sande, P.; Sánez, D.A.; Keller Sarmiento, M.I.; Rosenstein, R.E. Retinal Oxidative Stress Induced by High Intraocular Pressure. Free Radic. Biol. Med. 2004, 37, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.-L.; Peng, P.-H.; Ma, M.-C.; Ritch, R.; Chen, C.-F. Dynamic Changes in Reactive Oxygen Species and Antioxidant Levels in Retinas in Experimental Glaucoma. Free Radic. Biol. Med. 2005, 39, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Reides, C.G.; Evelson, P.A.; Llesuy, S.F. Time Course Changes of Oxidative Stress Markers in a Rat Experimental Glaucoma Model. Invest. Ophthalmol. Vis. Sci. 2010, 51, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Cai, J. Proteomic Identification of Oxidatively Modified Retinal Proteins in a Chronic Pressure-Induced Rat Model of Glaucoma. Invest. Ophthalmol. Vis. Sci. 2005, 46, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Seven, E.; Tekin, S.; Demir, C.; Demir, H.; Batur, M.; Ozer, M.D.; Yasar, T. Altered Serum Biomarkers of Oxidative Stress and Antioxidant Capacity in Primary Open-Angle Glaucoma. Sci. Rep. 2026, 16. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, S.; Cao, W.; Sun, X. The Association of Oxidative Stress Status with Open-Angle Glaucoma and Exfoliation Glaucoma: A Systematic Review and Meta-Analysis. J. Ophthalmol. 2019, 2019, 1803619. [Google Scholar] [CrossRef] [PubMed]
- Mysona, B.; Dun, Y.; Duplantier, J.; Ganapathy, V.; Smith, S.B. Effects of Hyperglycemia and Oxidative Stress on the Glutamate Transporters GLAST and System Xc- in Mouse Retinal Müller Glial Cells. Cell Tissue Res. 2009, 335, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S.; Tateishi, N. Role of Membrane Transport in Metabolism and Function of Glutathione in Mammals. J. Membr. Biol. 1986, 89, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jassim, A.H.; Inman, D.M. Evidence of Hypoxic Glial Cells in a Model of Ocular Hypertension. Invest. Ophthalmol. Vis. Sci. 2019, 60, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Shim, M.S.; Kim, K.-Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.-K. Coenzyme Q10 Inhibits Glutamate Excitotoxicity and Oxidative Stress-Mediated Mitochondrial Alteration in a Mouse Model of Glaucoma. Invest. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.-M.; Chuang, M.-J.; Liu, J.-H.; Liu, X.-Q.; Ho, L.-K.; Pan, W.H.T.; Zhang, X.-M.; Liu, C.-M.; Tsai, S.-K.; Kong, C.-W.; et al. Baicalein Protects against Retinal Ischemia by Antioxidation, Antiapoptosis, Downregulation of HIF-1α, VEGF, and MMP-9 and Upregulation of HO-1. J. Ocul. Pharmacol. Ther. 2013, 29, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Choi, S.-H.; Bastola, T.; Kim, K.-Y.; Park, S.; Weinreb, R.N.; Miller, Y.I.; Ju, W.-K. AIBP Protects Müller Glial Cells against Oxidative Stress-Induced Mitochondrial Dysfunction and Reduces Retinal Neuroinflammation. Antioxidants 2024, 13, 1252. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Choi, S.-H.; Bastola, T.; Park, Y.; Oh, J.; Kim, K.-Y.; Hwang, S.; Miller, Y.I.; Ju, W.-K. AIBP: A New Safeguard against Glaucomatous Neuroinflammation. Cells 2024, 13, 198. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-H.; Cheng, Y.-W.; Lin, F.-L.; Hsu, K.-C.; Wang, M.-H.; Yen, J.-L.; Wang, T.-J.; Lin, T.E.; Liu, Y.-C.; Huang, W.-J.; et al. A Novel HDAC8 Inhibitor H7E Exerts Retinoprotective Effects against Glaucomatous Injury via Ameliorating Aberrant Müller Glia Activation and Oxidative Stress. Biomed. Pharmacother. 2024, 174, 116538. [Google Scholar] [CrossRef] [PubMed]
- Souza Monteiro de Araújo, D.; De Logu, F.; Adembri, C.; Rizzo, S.; Janal, M.N.; Landini, L.; Magi, A.; Mattei, G.; Cini, N.; Pandolfo, P.; et al. TRPA1 Mediates Damage of the Retina Induced by Ischemia and Reperfusion in Mice. Cell Death Dis. 2020, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.C.; Wang, H.; et al. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Veglianti, F.; Di Vito Nolfi, M.; Flati, I.; Dall’Aglio, F.; Di Giovanni, F.; Verzella, D.; Capece, D.; Angelucci, A.; Alesse, E.; Zazzeroni, F.; et al. NF-κB Involvement in Glaucoma-Associated Neuroinflammation: Focus on Glial Cells. Front. Biosci. (Landmark Ed.) 2026, 31, 45644. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Han, L.; Qu, Y.; Zhang, L.; Zhang, X.; Li, J.; Lei, B.; Wang, Y.-C.; Wang, Z. IL-1β-Mediated Interaction between Müller Cells and Microglia through CXCL1/5-CXCR2 Aggravates Visual Dysfunction in Experimental Glaucoma. J. Neuroinflammation 2026. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.M.; Johnson, C.S.; Zhang, J.; Eady, E.K.; Green, C.R.; Danesh-Meyer, H.V. High Pressure-Induced Retinal Ischaemia Reperfusion Causes Upregulation of Gap Junction Protein connexin43 prior to Retinal Ganglion Cell Loss. Exp. Neurol. 2012, 234, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-X.; Zhao, G.-L.; Hu, X.; Zhou, H.; Li, S.-Y.; Li, F.; Miao, Y.; Lei, B.; Wang, Z. P2X7/P2X4 Receptors Mediate Proliferation and Migration of Retinal Microglia in Experimental Glaucoma in Mice. Neurosci. Bull. 2022, 38, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Bilal, A.; Constantin, F.; Chirila, S.; Hangan, T. New Trends in the Treatment of Open-Angle Glaucoma: A Critical Review. Int. Ophthalmol. 2025, 45, 381. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Hughes, T.E.T.; Moiseenkova-Bell, V.Y. Transient Receptor Potential (TRP) Channels. Subcell. Biochem. 2018, 87, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, F.; Su, Y. TRPV: An Emerging Target in Glaucoma and Optic Nerve Damage. Exp. Eye Res. 2024, 239, 109784. [Google Scholar] [CrossRef] [PubMed]
- Araújo, D.S.M.; Miya-Coreixas, V.S.; Pandolfo, P.; Calaza, K.C. Cannabinoid Receptors and TRPA1 on Neuroprotection in a Model of Retinal Ischemia. Exp. Eye Res. 2017, 154, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Greferath, U.; Zhao, D.; Huang, J.Y.; Wang, A.Y.M.; Vessey, K.A.; Chrysostomou, V.; Fletcher, E.L.; Crowston, J.G.; Bui, B.V. Systemic TRPV4 Inhibition Worsens Retinal Response to Acute Intraocular Pressure Elevation in Older but Not Younger Mice. Optom. Vis. Sci. 2025, 102, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.W.; Kim, M.-L.; Sung, K.R. Modulation of TRPV4-Mediated TNF-α Expression in Müller Glia and Subsequent RGC Apoptosis by Statins. Exp. Eye Res. 2024, 239, 109781. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.H.; Choi, G.W.; Kim, M.-L.; Sung, K.R. Statins Inhibit the Gliosis of MIO-M1, a Müller Glial Cell Line Induced by TRPV4 Activation. Int. J. Mol. Sci. 2022, 23, 5190. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.O.; Ryskamp, D.A.; Phuong, T.T.T.; Verkman, A.S.; Yarishkin, O.; MacAulay, N.; Križaj, D. TRPV4 and AQP4 Channels Synergistically Regulate Cell Volume and Calcium Homeostasis in Retinal Müller Glia. J. Neurosci. 2015, 35, 13525–13537. [Google Scholar] [CrossRef] [PubMed]
- Netti, V.; Cocca, M.A.; Cutrera, N.; Molina Ponce, T.; Ford, P.; Di Giusto, G.; Capurro, C. Osteopontin Regulates AQP4 Expression by TRPV4 Activation in Müller Cells: Implications for Retinal Homeostasis. Mol. Neurobiol. 2025, 62, 4769–4784. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, B.; Zhou, D.; Lei, M.; Yang, J.; Hu, Z.; Duan, W. AQP4 Regulates Ferroptosis and Oxidative Stress of Muller Cells in Diabetic Retinopathy by Regulating TRPV4. Exp. Cell Res. 2024, 439, 114087. [Google Scholar] [CrossRef] [PubMed]
- Lakk, M.; Yarishkin, O.; Baumann, J.M.; Iuso, A.; Križaj, D. Cholesterol Regulates Polymodal Sensory Transduction in Müller Glia. Glia 2017, 65, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Gil, N.; Kutsyr, O.; Fernández-Sánchez, L.; Sánchez-Sáez, X.; Albertos-Arranz, H.; Sánchez-Castillo, C.; Vidal-Gil, L.; Cuenca, N.; Lax, P.; Maneu, V. Ischemia-Reperfusion Increases TRPM7 Expression in Mouse Retinas. Int. J. Mol. Sci. 2023, 24, 16068. [Google Scholar] [CrossRef] [PubMed]
- Leonelli, M.; Martins, D.O.; Britto, L.R.G. TRPV1 Receptors Are Involved in Protein Nitration and Müller Cell Reaction in the Acutely Axotomized Rat Retina. Exp. Eye Res. 2010, 91, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Hakim, A.; Guido, B.; Narsineni, L.; Chen, D.-W.; Foldvari, M. Gene Therapy Strategies for Glaucoma from IOP Reduction to Retinal Neuroprotection: Progress towards Non-Viral Systems. Adv. Drug Deliv. Rev. 2023, 196, 114781. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Qi, B.; Ren, Z.; Cong, L.; Pan, X.; Zhou, Q.; Zhang, B.N.; Xie, L. Targeted Neuroprotection of Retinal Ganglion Cells via AAV2-hSyn-NGF Gene Therapy in Glaucoma Models. Invest. Ophthalmol. Vis. Sci. 2025, 66, 48. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, L.P.; Hoek, R.M.; Vos, R.M.; Aartsen, W.M.; Klimczak, R.R.; Hoyng, S.A.; Flannery, J.G.; Wijnholds, J. Specific Tools for Targeting and Expression in Müller Glial Cells. Mol. Ther. Methods Clin. Dev. 2014, 1, 14009. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Microglial cells, in a pro-inflammatory profile, release molecules such as IL-1β, TNF-α, and C1q, via cytokines receptors and NF-ΚB activation, induce the shift of astrocytes to a neurotoxic phenotype. In this scenario, and also in an NF-Κβ-dependent manner, astrocytes begin secreting inflammatory mediators into the retinal microenvironment, thereby reinforcing and sustaining the local inflammatory state. The chemokine CXCL10 is also elevated in glaucomatous tissue; by interacting with the CXCR3 receptor, it stimulates astrocytic expression of C3, contributing to the chronicity of the injury and subsequent neuronal loss. The convergence of these pathways culminates in the activation of the NLRP3 inflammasome, which recruits caspase-1. This enzyme cleaves gasdermin D (GSDMD), triggering pyroptosis, resulting in astrocyte lysis and the release of cytoplasmic contents into the nerve fiber layer and optic nerve head (ONH), thereby exacerbating neurodegeneration. Concurrently, the chronic oxidative stress inherent to the pathology causes severe DNA damage, triggering the DNA damage response (DDR) and upregulating the expression of p21 and β-galactosidase. This genotoxic stress drives astrocytes into a state of cellular senescence and the associated secretory phenotype (SASP); the continuous release of cytokines, metalloproteinases and remodeling molecules from this state directly contributes to tissue deterioration of the microenvironment in which these cells are present. Created in BioRender. https://BioRender.com/eeqnhyd.
Figure 1.
Microglial cells, in a pro-inflammatory profile, release molecules such as IL-1β, TNF-α, and C1q, via cytokines receptors and NF-ΚB activation, induce the shift of astrocytes to a neurotoxic phenotype. In this scenario, and also in an NF-Κβ-dependent manner, astrocytes begin secreting inflammatory mediators into the retinal microenvironment, thereby reinforcing and sustaining the local inflammatory state. The chemokine CXCL10 is also elevated in glaucomatous tissue; by interacting with the CXCR3 receptor, it stimulates astrocytic expression of C3, contributing to the chronicity of the injury and subsequent neuronal loss. The convergence of these pathways culminates in the activation of the NLRP3 inflammasome, which recruits caspase-1. This enzyme cleaves gasdermin D (GSDMD), triggering pyroptosis, resulting in astrocyte lysis and the release of cytoplasmic contents into the nerve fiber layer and optic nerve head (ONH), thereby exacerbating neurodegeneration. Concurrently, the chronic oxidative stress inherent to the pathology causes severe DNA damage, triggering the DNA damage response (DDR) and upregulating the expression of p21 and β-galactosidase. This genotoxic stress drives astrocytes into a state of cellular senescence and the associated secretory phenotype (SASP); the continuous release of cytokines, metalloproteinases and remodeling molecules from this state directly contributes to tissue deterioration of the microenvironment in which these cells are present. Created in BioRender. https://BioRender.com/eeqnhyd.

Figure 2.
Phenotypic transition of astrocytes in glaucoma. The central conceptual graphic employs a split-cell rendering to directly contrast the two functional states. Under physiological homeostasis (left side), astrocytes provide essential neurovascular support, early mechanosensation, and neuroprotection to unmyelinated retinal ganglion cell axons. Sustained biomechanical and oxidative stress (right side) drives a pathological collapse characterized by a senescence-associated secretory phenotype (SASP), fibrotic remodeling of the lamina cribrosa, and the activation of cytotoxic neuroinflammatory cascades, including the C3/C3a neurotoxic axis. Created with BioRender. https://BioRender.com/eeqnhyd.
Figure 2.
Phenotypic transition of astrocytes in glaucoma. The central conceptual graphic employs a split-cell rendering to directly contrast the two functional states. Under physiological homeostasis (left side), astrocytes provide essential neurovascular support, early mechanosensation, and neuroprotection to unmyelinated retinal ganglion cell axons. Sustained biomechanical and oxidative stress (right side) drives a pathological collapse characterized by a senescence-associated secretory phenotype (SASP), fibrotic remodeling of the lamina cribrosa, and the activation of cytotoxic neuroinflammatory cascades, including the C3/C3a neurotoxic axis. Created with BioRender. https://BioRender.com/eeqnhyd.

Figure 3.
Pathological changes of Müller glia under stress. Using a mirrored split-cell illustration, the figure contrasts with the healthy and reactive profiles of a single cell. In a healthy retina (left side), Müller cells maintain structural architecture (forming the outer limiting membrane, OLM), offer metabolic support, and prevent excitotoxicity through rapid glutamate and potassium clearance (via GLAST, GS, and Kir4.1 channels). Glaucomatous stress (right side) triggers a reactive gliosis marked by profound cellular hypertrophy, GFAP overexpression, elongation of radial processes with soma swelling, and the formation of fibrotic glial scars that disrupt retinal integrity. Created with BioRender. https://BioRender.com/eeqnhyd.
Figure 3.
Pathological changes of Müller glia under stress. Using a mirrored split-cell illustration, the figure contrasts with the healthy and reactive profiles of a single cell. In a healthy retina (left side), Müller cells maintain structural architecture (forming the outer limiting membrane, OLM), offer metabolic support, and prevent excitotoxicity through rapid glutamate and potassium clearance (via GLAST, GS, and Kir4.1 channels). Glaucomatous stress (right side) triggers a reactive gliosis marked by profound cellular hypertrophy, GFAP overexpression, elongation of radial processes with soma swelling, and the formation of fibrotic glial scars that disrupt retinal integrity. Created with BioRender. https://BioRender.com/eeqnhyd.

Figure 4.
Signaling pathways and neurotoxic crosstalk of Müller cells in a glaucomatous context. Biomechanical stress induces the activation of mechanosensitive channels (Piezo1, TRPV4), promoting the upregulation of HIF-1α (1). This process leads to the inhibition of AIBP, inducing mitochondrial dysfunction and ROS accumulation (2), reinforced by the decrease of system xc-, and consequent reduction of glutathione (GSH), the main neuronal antioxidant. The activation of ERK and STAT3 by increased ROS levels leads to the upregulation of VEGF and GFAP, characterizing reactive gliosis (3). Concurrently, there is a downregulation of glutamine synthetase (GS), glutamate-aspartate transporter (GLAST) and Kir4.1 potassium channels, leading to the extracellular accumulation of glutamate and overstimulation of NMDA receptors, and excitotoxicity of retinal ganglion cells (RGC)(4). In response to the microenvironment, activated microglia secrete IL-1β, stimulating Müller cell receptors and activating the p38 and NF-κB pathways (5). The translocation of NF-κB triggers the release of pro-inflammatory chemokines (MCP-1, CXCL5, CXCL1) and cytokines (IL-1β, TNF-α) (6). Increased oxidative stress activates TRPA1 channels and induces the formation of inflammasomes, with the subsequent activation of caspases 1 and 3 (7). ATP release through hemichannels stimulates microglia via P2X7 receptors, while excess glutamate hyperactivates NMDA receptors on RGCs. This promotes calcium influx and excitotoxicity, which, together with inflammation and oxidative stress, culminates in neurodegeneration. Created in BioRender. https://BioRender.com/eeqnhyd.
Figure 4.
Signaling pathways and neurotoxic crosstalk of Müller cells in a glaucomatous context. Biomechanical stress induces the activation of mechanosensitive channels (Piezo1, TRPV4), promoting the upregulation of HIF-1α (1). This process leads to the inhibition of AIBP, inducing mitochondrial dysfunction and ROS accumulation (2), reinforced by the decrease of system xc-, and consequent reduction of glutathione (GSH), the main neuronal antioxidant. The activation of ERK and STAT3 by increased ROS levels leads to the upregulation of VEGF and GFAP, characterizing reactive gliosis (3). Concurrently, there is a downregulation of glutamine synthetase (GS), glutamate-aspartate transporter (GLAST) and Kir4.1 potassium channels, leading to the extracellular accumulation of glutamate and overstimulation of NMDA receptors, and excitotoxicity of retinal ganglion cells (RGC)(4). In response to the microenvironment, activated microglia secrete IL-1β, stimulating Müller cell receptors and activating the p38 and NF-κB pathways (5). The translocation of NF-κB triggers the release of pro-inflammatory chemokines (MCP-1, CXCL5, CXCL1) and cytokines (IL-1β, TNF-α) (6). Increased oxidative stress activates TRPA1 channels and induces the formation of inflammasomes, with the subsequent activation of caspases 1 and 3 (7). ATP release through hemichannels stimulates microglia via P2X7 receptors, while excess glutamate hyperactivates NMDA receptors on RGCs. This promotes calcium influx and excitotoxicity, which, together with inflammation and oxidative stress, culminates in neurodegeneration. Created in BioRender. https://BioRender.com/eeqnhyd.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



