Submitted:
01 July 2026
Posted:
02 July 2026
You are already at the latest version
Abstract
Oleanolic acid is a naturally occurring pentacyclic triterpenoid with broad preclinical interest in inflammation, oxidative stress, liver injury, metabolic disorders, cancer-related models, skin disease, and wound repair. Its further development, however, is constrained by poor aqueous solubility, low and variable bioavailability, limited barrier transport, crystallinity, and strong dependence of biological response on the formulation used. These properties make oleanolic acid a useful example of a hydrophobic natural com-pound whose pharmacological performance is inseparable from delivery design. This review examines polymeric and polymer-assisted nanocarriers developed for oleanolic acid delivery. The systems discussed include biodegradable PLA/PLGA na-noparticles, PEGylated polymeric nanoparticles, polymeric micelles, hyaluron-ic-acid-based nanoprodrugs, polymer-assisted lipid systems, hydrogels, nanogels, polymeric fiber membranes, local depots, and microneedle-compatible platforms. Rather than treating these carriers only as solubility enhancers, the review evaluates how polymer composition, carrier architecture, drug physical state, release behavior, and route of administration affect oleanolic acid exposure. Particular attention is given to controlled release, local retention, disease-oriented de-livery, and critical quality attributes such as particle size, loading, encapsulation effi-ciency, solid-state form, stability, residual solvent, sterility, and batch-to-batch repro-ducibility. The review argues that the most realistic near-term opportunities for polymeric oleanolic acid systems may lie in local and tissue-targeted applications, including in-flammatory skin disease, wound healing, dermal delivery, and osteoarthritis, where sustained exposure at the target site may be more relevant than systemic bioavailability. Future progress will depend less on demonstrating that oleanolic acid can be loaded into another carrier and more on showing that each formulation provides reproducible, safe, and route-appropriate drug exposure.
Keywords:
oleanolic acid
; polymeric nanocarriers
; PLGA nanoparticles
; polymer-assisted delivery
; controlled release
; hydrogels
; microneedles
; local delivery
; wound healing
; psoriasis
1. Introduction
Oleanolic acid (OA) is a naturally occurring pentacyclic triterpenoid present in edible plants, medicinal herbs, olive-derived materials, fruit peels, and other botanical sources [1]. Chemically, it is described as 3β-hydroxy-olean-12-en-28-oic acid, with the molecular formula C30H48O3 and a molecular weight of approximately 456.7 g/mol [2,3,4]. Although OA has attracted broad pharmacological interest, its formulation behavior is difficult: the rigid hydrophobic oleanane scaffold and limited polar functionality result in poor aqueous solubility, slow dissolution, and variable biological exposure [1,5]. These properties make OA less a conventional small-molecule payload and more a formulation-sensitive compound whose apparent activity depends strongly on how it is dispersed, released, and delivered. The natural occurrence, chemical identity, and formulation-relevant limitations of OA are summarized in Figure 1.
The biological literature on OA covers anti-inflammatory, antioxidant, hepatoprotective, metabolic, anticancer, antimicrobial, dermatological, and wound-healing-related effects. These reports are promising, but they are not interchangeable. The measured response depends on dose, route of administration, model, vehicle, and formulation type [1,5]. For a poorly soluble compound, the amount added to an in vitro assay or administered in vivo may differ substantially from the fraction that is actually dissolved, released, absorbed, or retained at the biological target [5].
For this reason, formulation is not a secondary technical detail in OA research. Poor aqueous solubility can limit dissolution and absorption, whereas high lipophilicity can promote partitioning into membranes, proteins, lipid compartments, or delivery matrices [1,5]. Solvents, surfactants, cyclodextrins, emulsions, nanoparticles, and polymeric carriers may therefore change not only OA solubility, but also exposure, cellular uptake, release kinetics, and apparent potency [5,6].
A wide range of OA formulations has been reported, including solid dispersions, cyclodextrin complexes, phospholipid complexes, self-emulsifying systems, liposomes, lipid nanoparticles, polymeric nanoparticles, albumin-based carriers, hydrogels, and local delivery platforms [5,6,7,8,9]. Together, these studies show that OA can be incorporated into many carrier types. They also show that carrier composition, preparation method, physicochemical characterization, and biological model strongly affect how the results should be interpreted [5,9,10].
Polymeric and polymer-assisted nanocarriers are useful in this context because they can address several OA delivery problems simultaneously. Biodegradable polymers such as PLA and PLGA can accommodate hydrophobic OA within nanoparticle or fiber matrices and modulate release through diffusion, relaxation, erosion, or degradation [7,8,9,11,12]. PEGylated systems can improve colloidal stability and alter the biological interface of the carrier [7]. Hyaluronic-acid-based nanoprodrugs add another level of design by combining polymer–drug conjugation, disease-oriented interactions, and stimuli-responsive behavior, as shown in topical psoriasis models [12].
These systems are also relevant for local and transdermal delivery. Many plausible OA applications involve inflammation, skin pathology, tissue repair, osteoarthritis, or localized oxidative stress, where high systemic bioavailability may be less important than controlled exposure at the target site [1,5,8,12]. Polymeric fiber membranes, hydrogels, nanogels, dissolving microneedles, and hydrogel-forming microneedles can therefore be used not only to increase apparent solubility, but also to prolong local retention and reduce unnecessary systemic distribution [6,8,13].
For polymeric OA formulations, the key questions go beyond whether the compound can be loaded into a carrier. A useful formulation must define and control critical quality attributes such as particle size, size distribution, drug loading, encapsulation efficiency, polymer degradation, drug physical state, release profile, residual solvent, colloidal stability, and batch-to-batch reproducibility [6,14]. These parameters determine whether an OA-loaded system can provide reproducible exposure under conditions relevant to the intended route of administration [5,6,13].
This review focuses on polymeric and polymer-assisted nanocarriers for OA delivery. It connects the physicochemical limitations of OA with carrier design, controlled-release principles, local and transdermal delivery, biological evaluation, manufacturing challenges, and clinical perspectives. The review does not aim to summarize all pharmacological effects of OA. Instead, it asks a formulation-centered question: which polymeric design strategies can convert OA from a poorly soluble bioactive compound into a more predictable and route-appropriate delivery system [5,6,7,8,9,11,12,13]?
2. Literature Search Strategy and Review Methodology
This review was prepared as a critical narrative review supported by structured literature mapping. The aim was not to catalogue all reported pharmacological effects of oleanolic acid, but to evaluate how polymeric and polymer-assisted delivery systems may address the formulation barriers that limit its biomedical use [1,5].
The search was organized around four problem-oriented areas: physicochemical and biopharmaceutical limitations of oleanolic acid; polymeric and polymer-assisted carrier systems reported for oleanolic acid; controlled-release and local-delivery strategies; and translational issues related to characterization, manufacturing, safety, and disease relevance [1,5,7,9,11]. Relevant studies were identified through PubMed, Scopus, Web of Science, ScienceDirect, SpringerLink, Wiley Online Library, ACS Publications, MDPI, Taylor & Francis, RSC Publishing, and Google Scholar.
The main search terms combined “oleanolic acid” with “polymeric nanoparticles”, “PLGA”, “PLA”, “PEGylated nanoparticles”, “polymeric micelles”, “nanogels”, “hydrogels”, “hyaluronic acid”, “cyclodextrin”, “polymer-lipid hybrid nanoparticles”, “controlled release”, “transdermal delivery”, “microneedles”, “wound dressing”, “psoriasis”, “osteoarthritis”, “cancer nanomedicine”, and “oral bioavailability”. Priority was given to peer-reviewed articles published from 2000 onward, with particular attention to studies published after 2015 because they better reflect current developments in polymeric nanomedicine, stimuli-responsive carriers, hydrogels, microneedles, and translational nanocarrier design [5,7,8,9,11,12].
Studies were included when they addressed at least one of the following points: OA physicochemical or biopharmaceutical limitations; formulation of OA in polymeric or polymer-assisted systems; controlled release; local, dermal, transdermal, intra-articular, or microneedle-assisted delivery; biological evaluation of OA-loaded carriers; or translational aspects relevant to polymeric nanomedicine [5,7,8,9,11,12,15,16].
Studies focused only on botanical extraction, phytochemical identification, or general pharmacology were used only when they provided necessary background. Non-polymeric systems were discussed selectively, mainly when they served as comparators or when a polymeric component contributed to stabilization, PEGylation, gelation, matrix formation, or route-specific delivery [1,5,11].
The review is structured from formulation barriers to delivery design and translation. First, the intrinsic limitations of OA are defined. Next, polymeric and polymer-assisted carrier types are compared. Controlled-release, local-delivery, and microneedle-compatible strategies are then discussed. The final sections consider characterization, critical quality attributes, manufacturing, safety, clinical relevance, and future development priorities [5,6,9,13].
3. Physicochemical and Biopharmaceutical Barriers in Oleanolic Acid Delivery
3.1. Chemical Identity and Structural Implications
The formulation behavior of OA follows directly from its structure. The molecule contains a rigid pentacyclic oleanane scaffold, one hydroxyl group, and one carboxylic acid group. This combination gives OA a predominantly lipophilic character with only limited capacity for interaction with aqueous media [1,2,3]. As a result, OA is more compatible with hydrophobic or amphiphilic domains than with simple aqueous formulations.
For polymeric delivery, this structural profile is important. OA can partition into hydrophobic polymer matrices, micellar cores, lipid–polymer interfaces, nanogel domains, or other microenvironments that reduce direct contact with water [5,7,9,11].
Biodegradable polymers such as PLA and PLGA are therefore attractive not only because they form nanoparticles, but because their hydrophobic domains can accommodate a compound that is poorly suited to aqueous dispersion. Amphiphilic and PEGylated polymers add a further function by combining hydrophobic drug association with improved colloidal stability in biological media [7,9,13].
The same structural features also create risks. Strong drug–matrix association may slow release, whereas insufficient compatibility between OA and the carrier can lead to crystallization, surface deposition, or precipitation during preparation and dilution [1,5,7,9]. Polymer selection should therefore be guided not only by biodegradability or particle size, but also by polymer–drug compatibility, physical-state control, and the intended route of administration [5,7,9,13].
3.2. Poor Aqueous Solubility as the Primary Formulation Barrier
Poor aqueous solubility remains the central formulation barrier for OA [1,5]. It limits dissolution in gastrointestinal fluids, complicates preparation of parenteral or topical dosage forms, and reduces the reliability of biological testing when nominal concentration is assumed to represent actual exposure [1,5,14]. In practice, OA added to an aqueous assay may exist as dissolved drug, suspended particles, precipitated material, carrier-associated drug, or a mixture of these states.
For polymeric nanocarrier design, poor solubility is both the problem and the rationale. OA may crystallize, precipitate, or remain incompletely incorporated during formulation. At the same time, polymeric nanoparticles, micelles, nanogels, and hydrogels can provide microenvironments in which OA is dispersed, protected, or released gradually rather than presented as a poorly soluble crystalline solid [5,7,9,12].
This distinction matters when biological effects are compared across studies. Cyclodextrin complexation, PEGylated PLA/PLGA nanoparticles, PLGA nanoparticles, micelles, gels, and other carriers do not simply “increase solubility”; they change the form in which OA reaches cells or tissues [7,9,14]. For this reason, OA should be treated as a formulation-dependent compound. Differences in activity between “free” OA and carrier-loaded OA may reflect changes in dissolution, precipitation, uptake, release rate, or tissue deposition rather than a change in intrinsic pharmacological potency [5,7,14].
3.3. Crystallinity, Dissolution, and Physical-State Control
OA is generally handled as a poorly soluble crystalline or semi-crystalline solid, and this solid-state behavior contributes to slow dissolution and variable biological availability [1,5]. In polymeric systems, the physical state of OA can influence drug loading, burst release, long-term release, storage stability, and the risk of precipitation after dilution [5,14].
Encapsulation alone is therefore not enough to define a successful OA formulation. The drug may be molecularly dispersed in the carrier, trapped as an amorphous phase, adsorbed on the particle surface, present as a crystalline domain, or partially separated from the carrier after preparation [5,7,8,9]. These forms can produce very different release profiles even when particle size and encapsulation efficiency appear similar.
However, encapsulation alone does not guarantee physical stability. If the drug is present as a separate crystalline phase inside or outside the carrier, release may become slow, incomplete, or poorly reproducible [5,8,9].
Polymeric carriers can reduce crystallinity or stabilize OA in amorphous or dispersed states, but they can also hide undetected crystalline drug fractions. For this reason, OA-loaded polymeric systems should be characterized not only by particle size, polydispersity, zeta potential, loading, and encapsulation efficiency, but also by drug physical state [5,8,13]. Differential scanning calorimetry, X-ray diffraction, Fourier-transform infrared spectroscopy, Raman spectroscopy, electron microscopy, and validated extraction-based assays can help determine whether OA is crystalline, amorphous, molecularly dispersed, surface-associated, or phase-separated within the formulation [8,13].
3.4. Low Bioavailability and Formulation-Dependent Exposure
Low and variable bioavailability is one of the main reasons OA has attracted formulation interest. Poor dissolution, limited permeability, extensive partitioning into lipid-rich compartments, and possible first-pass metabolism may all reduce systemic exposure after oral administration [1,5]. Polymeric nanocarriers can partly address these problems by improving apparent dispersion, protecting OA during transit, modifying release, and changing interactions with biological barriers [5,7,9]. However, higher apparent exposure is not automatically equivalent to better therapeutic performance. A smaller particle size, higher loading, or improved dispersion is useful only if it produces relevant OA availability at the intended site and remains safe for the selected route [5,6,13]. This distinction is important because OA is being considered for both systemic and local applications.
For skin inflammation, wound healing, and osteoarthritis, systemic bioavailability may be less important than local tissue residence and sustained exposure. Hydrogels, nanogels, fiber membranes, micelles, local depots, and microneedle platforms can therefore be designed to retain OA near the target tissue rather than maximize plasma concentration. In these settings, the most relevant formulation question is not how much OA enters the bloodstream, but how reproducibly the system maintains drug availability at the diseased or injured site [8,12,15,16].
3.5. Permeability Limitations and Biological Barriers
OA delivery is also limited by biological barriers, including the gastrointestinal epithelium, stratum corneum, mucus, extracellular matrix, synovial environment, and cellular membranes. Although OA is lipophilic, lipophilicity alone does not ensure efficient absorption or tissue penetration. Poor aqueous solubility can limit the concentration gradient required for transport, while strong partitioning into carrier or tissue compartments can reduce the amount of freely available OA [1,5].
Polymeric systems can be designed to interact with these barriers in different ways. PEGylated carriers may reduce aggregation and modify protein interactions, hyaluronic-acid-based systems may exploit receptor-mediated uptake in CD44-expressing cells, and hydrogels or microneedles can improve local contact time or bypass the stratum corneum barrier. These mechanisms are route-specific rather than universal. Hydrogels and microneedles can support local tissue delivery by prolonging contact time or bypassing the stratum corneum barrier [7,8,12,13,15,16].
A polymeric OA nanoparticle intended for cancer-cell uptake therefore requires different design criteria from a hydrogel intended for wound treatment or a microneedle patch intended for intradermal delivery [7,8,9,12,15,16]. Carrier selection should begin with the biological barrier and the intended site of action, not simply with the aim of increasing solubility [5,6,13].
3.6. Vehicle-Dependent Biological Activity
A recurring difficulty in OA research is that biological activity depends strongly on the form in which the compound is delivered [5,14]. OA dissolved in an organic solvent, complexed with cyclodextrin, encapsulated in PLGA nanoparticles, incorporated into micelles, or conjugated to hyaluronic acid may produce different exposure patterns even when the nominal OA dose is similar [7,9,12,14].
This vehicle dependence complicates interpretation. A stronger response from an OA-loaded nanocarrier may reflect improved dispersion, reduced precipitation, enhanced cellular uptake, altered intracellular localization, slower release, higher tissue retention, or carrier-associated effects. It should not automatically be interpreted as increased intrinsic potency of OA [5,7,9,12].
Appropriate controls are therefore essential. Studies should compare blank carrier, free OA, solubilized OA, and carrier-loaded OA whenever possible, and the actual OA concentration available under assay conditions should be analytically verified [9,13,14]. Without these controls, it is difficult to determine whether the observed biological effect comes from OA itself, the carrier, the solubilizing environment, or the combined drug–carrier system [5,13,14].
3.7. Why Oleanolic Acid is Suitable for Polymeric Nanocarrier Engineering
Oleanolic acid is a useful payload for polymeric nanocarrier research because it combines biological relevance with difficult formulation behavior [1,5]. Its poor solubility, hydrophobicity, crystallinity, limited bioavailability, barrier-dependent transport, and vehicle-dependent activity create a demanding test case for controlled delivery systems [5,7,9].
The formulation question is no longer whether OA can be incorporated into a carrier. This has already been shown using PEGylated PLA/PLGA nanoparticles, PLGA nanoparticles, hyaluronic-acid-based systems, polymeric micelles, gels, and PLGA fiber membranes [7,8,9,12,15,16]. The more important question is how polymer composition, carrier architecture, drug physical state, release environment, and biological model determine performance [5,6,7,8,9,12,13,15,16].
This shift is important for the whole field. Polymeric and polymer-assisted OA systems should not be judged only by loading efficiency or particle size. They should be judged by whether the carrier creates a reproducible, route-appropriate exposure profile that improves the interpretation and potential usefulness of OA in a defined biological setting [5,6,7,8,9,12,13].
The main formulation-relevant barriers of OA and their implications for polymeric or polymer-assisted carrier design are summarized in Table 1.
4. Current Delivery Strategies for Oleanolic Acid: Positioning Polymeric and Polymer-Assisted Systems
Several formulation strategies have been explored to improve the delivery of oleanolic acid, including solid dispersions, cyclodextrin complexes, phospholipid complexes, self-emulsifying systems, liposomes, lipid nanoparticles, polymeric nanoparticles, protein-based carriers, hydrogels, and local depot systems [1,5,7,8,12]. This broad formulation landscape reflects the central difficulty of OA delivery: the compound is biologically attractive, but its performance depends strongly on how it is dispersed, stabilized, released, and presented to cells or tissues [1,5].
Conventional solubilization approaches, such as solid dispersions and cyclodextrin complexes, are useful because they can improve OA wettability, reduce crystallinity, or increase apparent aqueous compatibility. Their main value is to show that OA activity is not independent of presentation format. However, these systems do not necessarily provide sustained release, tissue retention, or route-specific control over exposure [5,14,19].
Self-emulsifying and self-nanoemulsifying systems are particularly relevant for oral delivery [1,5,20,21]. By maintaining OA in solubilized or lipid-associated states, they may improve dispersion in gastrointestinal fluids and increase the fraction available for absorption [20,21]. Their performance, however, can depend on surfactant concentration, dilution behavior, lipid digestion, precipitation risk, and gastrointestinal variability [1,5,20,21]. These factors make cross-study comparison difficult and show why solubilization alone is not sufficient for translational development.
Lipid-based nanocarriers, including liposomes, solid lipid nanoparticles, nanostructured lipid carriers, and liquid crystalline nanoparticles, are compatible with OA because of its lipophilic triterpenoid structure [15,22,23]. They can improve dispersion, protect OA from direct aqueous exposure, and support local or systemic delivery. At the same time, lipid-based systems may face problems such as drug leakage, lipid oxidation, polymorphic transitions, drug expulsion during storage, and incomplete control over release after dilution in biological media [1,5,15,22,23].
Polymeric and polymer-assisted systems occupy a distinct position because they can combine solubilization with matrix-mediated stabilization, controlled release, surface modification, local retention, and route-specific administration [7,8,9,12,13]. Biodegradable polymeric nanoparticles can incorporate OA into hydrophobic matrices, whereas PEGylated, amphiphilic, hyaluronic-acid-based, hydrogel-forming, or microneedle-compatible systems can add functions that are not limited to dispersion alone [7,8,9,12,13,16].
In this review, polymeric nanocarriers are defined as systems in which the main carrier architecture is formed by polymers, such as PLGA nanoparticles, PLA nanoparticles, polymeric micelles, nanogels, and polymeric fiber membranes [7,8,9,12]. Polymer-assisted systems are broader and include carriers in which polymers provide stabilization, PEGylated surface modification, targeting, gelation, local retention, or administration support, even when another material forms part of the carrier structure. Examples include PEGylated lipid systems, polymer-stabilized lipid nanoparticles, thermosensitive gels, nanocarrier-loaded hydrogels, and polymeric microneedle matrices [10,15,16,22,23].
This distinction is useful because not all OA delivery systems solve the same problem. A self-emulsifying system primarily improves dispersion; a liposome provides lipid-associated delivery; a PLGA nanoparticle offers matrix-based release; a hyaluronic-acid nanoprodrug adds biological interaction and responsive cleavage; and a thermosensitive gel supports local residence after administration [7,8,12,15,22,23]. These differences should guide how each formulation is evaluated.
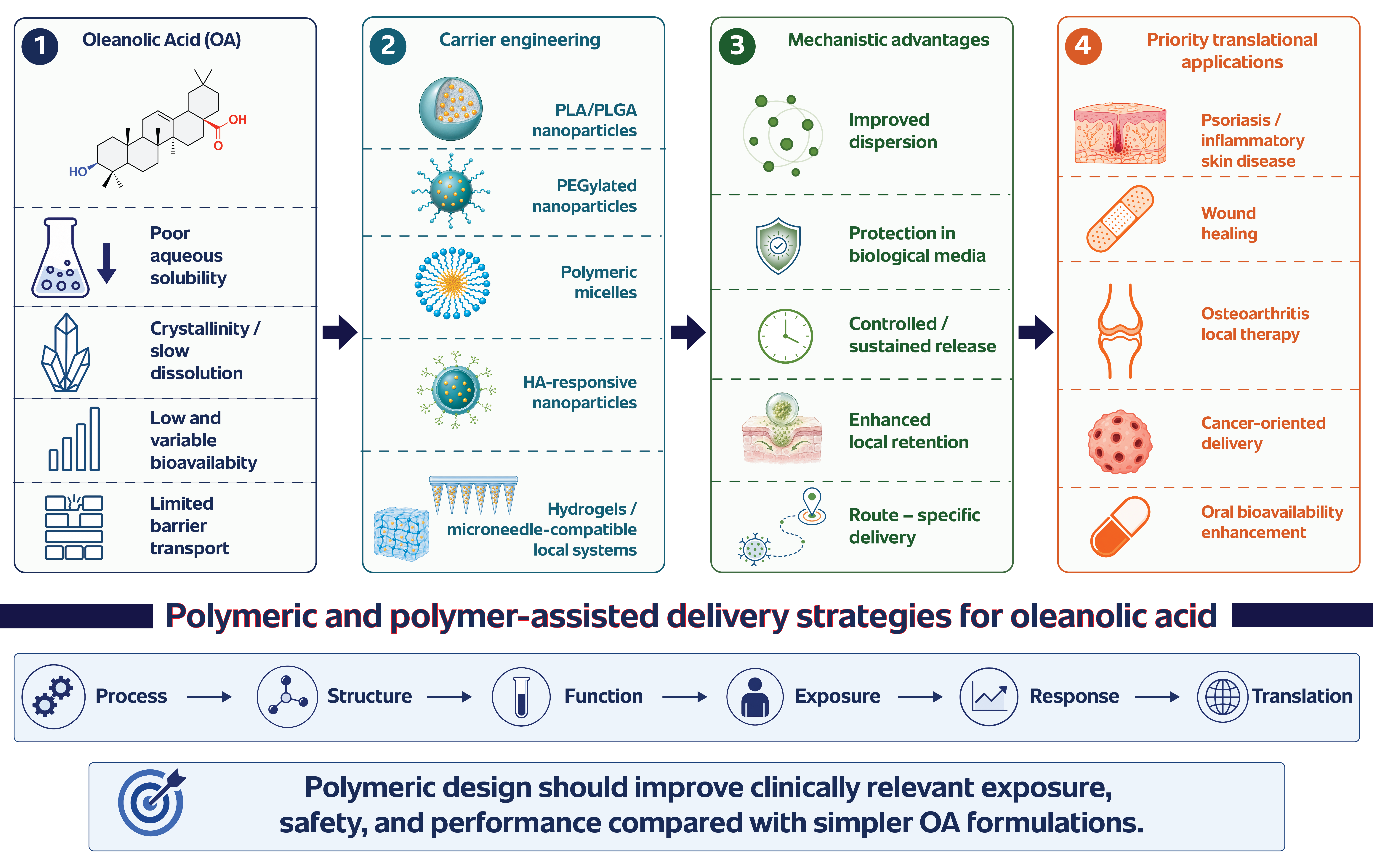
The current evidence supports a focused discussion of polymeric and polymer-assisted OA delivery. The field has moved beyond the simple question of whether OA can be formulated. Multiple carrier types have already demonstrated that it can [1,5,7,8,12]. The more important question is which polymeric design principles improve release control, route suitability, biological interpretation, reproducibility, and development potential [7,8,9,10,12,13,16]. The main polymeric and polymer-assisted carrier architectures discussed in this review are summarized in Figure 2.
This classification helps avoid treating all OA nanocarriers as equivalent. A self-emulsifying system mainly improves dispersion; a liposome may support membrane-associated delivery; a PLGA nanoparticle can provide matrix-based release; a hyaluronic-acid nanoprodrug can introduce receptor-mediated and redox-responsive behavior; and a thermosensitive nanogel can prolong local residence after administration. These systems differ not only in composition but also in how they control OA availability over time [7,8,12,15,22,23].
For translational purposes, polymeric and polymer-assisted carriers are especially relevant because they can be designed around unmet clinical needs rather than only around solubility improvement [7,8,9,10,12,13,16]. In cancer-oriented studies, OA-loaded PEGylated PLA/PLGA nanoparticles and OA/UA-loaded PLGA nanoparticles have been evaluated for cytotoxic activity in cancer cell models [7,9]. In inflammatory skin disease, hyaluronic-acid-based reduction-responsive OA nanoparticles have been developed for topical psoriasis therapy [12]. In local tissue delivery, OA-loaded PLGA fiber membranes and OA-loaded thermosensitive nanogels illustrate the potential of polymeric matrices and polymer-assisted depots for sustained exposure [8,15].
The current delivery landscape therefore supports a focused review on polymeric and polymer-assisted OA systems. The main question is no longer whether OA can be formulated; multiple carrier types have already demonstrated that it can [1,5,7,8,12]. The more important question is which polymeric design principles can improve controlled release, route-specific delivery, biological relevance, reproducibility, and translational feasibility [7,8,9,10,12,13,16].
Representative polymeric and polymer-assisted OA delivery systems are compared in Table 2.
5. Polymeric Nanoparticles for Oleanolic Acid Delivery
5.1. Rationale for Biodegradable Polymeric Nanoparticles
Biodegradable polymeric nanoparticles are among the most relevant platforms for OA delivery because they can accommodate hydrophobic molecules within polymer matrices and release them over time through diffusion, polymer relaxation, erosion, or degradation [7,9,13,24]. For OA, this matrix-based mode of delivery is valuable because the compound is poorly soluble, prone to variable dispersion in aqueous media, and strongly influenced by formulation environment [1,5].
PLA and PLGA are especially important in this context. Their hydrophobicity, molecular weight, lactide ratio, end-group chemistry, and degradation behavior can influence nanoparticle formation, drug loading, release kinetics, and stability [6,13]. These variables make PLA/PLGA systems useful not only as carriers, but also as models for studying how polymer chemistry affects the presentation of OA [7,9,13].
The main advantage of polymeric nanoparticles is that they do not require OA to remain freely dissolved in aqueous medium. Instead, OA can be dispersed within a polymeric matrix and released gradually into the surrounding environment [7,9]. This can improve handling and exposure control, but it also introduces new formulation variables. Residual solvent, incomplete encapsulation, burst release, aggregation, polymer degradation products, crystalline drug domains, and batch-to-batch variability can all affect performance [6,13].
For this reason, OA-loaded polymeric nanoparticles should not be judged only by loading efficiency or particle size. A formulation with high loading may still release OA too slowly, release it too rapidly, contain undetected crystalline drug, or fail to deliver the compound under biologically relevant conditions. The value of a polymeric nanoparticle depends on whether polymer composition, drug physical state, release behavior, and biological response can be linked in a reproducible way [1,5,13].
5.2. PEGylated PLA and PLGA Nanoparticles
One of the most direct examples of polymeric OA nanocarrier design is the study by Man et al., who prepared OA-loaded mPEG–PLA and mPEG–PLGA nanoparticles by nanoprecipitation and evaluated their physicochemical properties, encapsulation efficiency, and cytotoxic activity in cancer-cell models [7]. The study is important because it shows how biodegradable polymer matrices and PEGylated architecture can be combined to improve OA presentation beyond simple solubilization [7].
PEGylation contributes to this design by improving aqueous dispersion, reducing aggregation, and modifying interactions between nanoparticles and biological media [6,7,13]. In OA-loaded mPEG–PLA and mPEG–PLGA systems, the hydrophobic polymeric region can associate with OA, whereas the PEG segment improves colloidal behavior. This architecture directly addresses the poor aqueous compatibility of OA while preserving a nanoscale carrier format [1,5,7].
The biological interpretation of these systems requires caution. Enhanced cytotoxicity of OA-loaded nanoparticles compared with non-encapsulated OA may reflect improved dispersion, altered cellular uptake, reduced precipitation, modified intracellular availability, or changed release kinetics [1,5,7]. It should not be interpreted simply as an increase in the intrinsic potency of OA. The carrier and the drug act as an integrated formulation system [7,13].
From a development perspective, PEGylated PLA/PLGA nanoparticles are attractive because their polymer composition, hydrophobic core, PEG corona, size, and release properties can be tuned. Future OA studies using this approach should connect PEG density, polymer composition, particle size, physical state of the drug, release rate, uptake, and biological response rather than reporting these parameters separately [6,7,13].
5.3. PLGA Nanoparticles for OA and Structurally Related Triterpenoids
PLGA nanoparticles have also been used to deliver OA together with or in comparison to ursolic acid, a structurally related pentacyclic triterpenoid. Silva et al. evaluated OA/UA-loaded PLGA nanoparticles in different cell lines and reported that encapsulation altered cell-specific activity compared with non-encapsulated compounds. This comparative design is useful because OA and ursolic acid share similar formulation challenges but do not necessarily produce identical biological effects [9].
For OA, PLGA nanoparticles provide a hydrophobic matrix that can reduce immediate precipitation, improve nanoscale dispersion, and support gradual release [9,13]. However, their performance depends on preparation method, polymer properties, stabilizer type, drug-to-polymer ratio, solvent history, and post-processing conditions. These variables can influence particle size, encapsulation efficiency, drug physical state, release behavior, and biological response [6,9,13].
The PLGA platform also illustrates a central analytical issue in OA nanocarrier research. Encapsulation efficiency does not reveal whether the drug is molecularly dispersed, amorphous, crystalline, surface-associated, or phase-separated inside the formulation. Since each of these states can influence release and stability, PLGA-based OA systems should be characterized by solid-state and morphological methods in addition to routine size, polydispersity, zeta-potential, and loading measurements [1,9,13].
5.4. Preparation Methods and Process Sensitivity
OA-loaded polymeric nanoparticles have most often been prepared using nanoprecipitation, solvent displacement, or emulsion-based techniques [7,9]. These methods are suitable for hydrophobic compounds, but they are also sensitive to solvent selection, polymer concentration, drug concentration, aqueous-phase composition, stabilizer type, mixing conditions, and solvent-removal procedure [7,9,13].
For OA, this process sensitivity is not a minor technical issue. During nanoparticle formation, uncontrolled supersaturation may cause OA to precipitate outside the polymer matrix, while rapid polymer precipitation may reduce drug incorporation or create heterogeneous particles [1,2,14]. Conversely, strong drug–polymer association may improve encapsulation but slow release to a degree that is unsuitable for the intended biological application [9,13]. The same nominal formulation can therefore produce different exposure profiles if preparation conditions are not controlled.
Nanoprecipitation is attractive because it is simple, scalable in principle, and compatible with hydrophobic drug loading. However, it should not be treated as a fully standardized process. Injection speed, mixing geometry, solvent-to-water ratio, polymer concentration, and purification conditions can alter particle nucleation, growth, residual solvent content, drug loading, and burst release [7,9,13]. Similar process–structure relationships have been reported for biodegradable copolyester nanoparticles loaded with hydrophobic compounds, where polymer composition and nanoprecipitation conditions affected nanoscale morphology and release behavior [24]. Although such systems were not developed specifically for OA, they support the broader point that hydrophobic payload incorporation depends strongly on process history.
For translational development, preparation parameters should be reported in enough detail to allow reproduction and comparison between studies. A polymeric OA formulation cannot be evaluated only as a final particle suspension; it must also be understood as the outcome of a defined process. Batch-to-batch reproducibility, solvent removal, purification, drug recovery, and stability after storage are therefore part of the formulation design rather than secondary methodological details [9,10,13].
The preparation routes and release mechanisms most relevant to OA-loaded polymeric systems are summarized in Figure 3.
5.5. Drug Loading and Encapsulation Efficiency
Drug loading and encapsulation efficiency are central parameters for OA-loaded polymeric nanoparticles, but they are often overinterpreted [7,9,13]. High encapsulation efficiency indicates that a large fraction of the initially added OA is associated with the formulation after preparation. It does not show where the drug is located, whether it is molecularly dispersed, whether it is crystalline, or how much will become available under biological conditions [1,7,9].
For hydrophobic molecules such as OA, apparent encapsulation may include several fractions: drug dissolved in the polymer matrix, drug adsorbed on the particle surface, drug trapped in amorphous domains, and small crystalline aggregates that remain after purification [1,9,13]. These fractions can behave differently during storage, dilution, release testing, and biological exposure. Two formulations with similar encapsulation efficiency may therefore produce different release profiles and biological responses.
Analytical validation is essential. HPLC or LC–MS methods should be used to quantify OA after complete extraction and after separation of free drug from nanoparticles [1,9,12]. Solid-state and morphological methods should complement these assays to detect crystalline OA, phase separation, or drug–polymer interactions that may influence release and stability [1,9,13].
5.6. Release Mechanisms from Polymeric OA Nanoparticles
Controlled release is one of the main reasons to use polymeric carriers for OA. In PLA and PLGA systems, release may involve drug diffusion, polymer swelling, matrix relaxation, hydrolytic degradation, erosion, and redistribution of OA between the polymeric phase and the surrounding medium. The balance between these mechanisms depends on polymer composition, molecular weight, particle size, drug loading, drug physical state, and release environment [6,13].
OA release is expected to be strongly affected by hydrophobic partitioning. The drug may remain associated with the polymer matrix, bind to proteins or surfactants, partition into lipid compartments, or deposit into tissue-like phases [1,5,7]. For this reason, release studies in simple aqueous buffers may underestimate release, whereas surfactant-rich media may overestimate the fraction that would be biologically available [1,5,13]. Release data should therefore be interpreted in relation to the intended route of administration.
The relevant release question differs by application. An oral nanoparticle, an injectable anticancer formulation, a wound-contacting fiber membrane, a microneedle patch, and an intra-articular gel do not require the same release profile [7,25,26,27,28]. For local systems, the key endpoint is often sustained tissue exposure rather than rapid drug liberation into bulk medium. For systemic systems, release must be balanced with circulation, distribution, uptake, and safety.
Release testing should therefore use conditions that reflect the intended biological compartment as closely as possible. Skin-oriented systems may require membrane diffusion studies, ex vivo skin deposition, or hydrogel-based models. Intra-articular systems should consider synovial-fluid-like environments and, where relevant, mechanical conditions. For polymeric nanoparticles intended for systemic delivery, protein-containing media, dilution stability, and uptake-relevant assays may be more informative than buffer release alone [8,13,15,16].
5.7. Biological Evaluation of Polymeric OA Nanoparticles
Biological evaluation of OA-loaded polymeric nanoparticles has often focused on anticancer activity, especially for PLA/PLGA-based systems. These studies demonstrate that polymeric encapsulation can alter cellular response and may increase activity compared with non-encapsulated OA. However, cytotoxicity in monolayer cancer-cell assays is only an early indication of formulation performance and should not be treated as direct evidence of clinical anticancer potential [7,9,13].
Because OA activity is strongly formulation-dependent, biological assays require careful controls. Blank nanoparticles, free OA, solubilized OA, and OA-loaded nanoparticles should be compared under conditions where both OA concentration and carrier concentration are known [7,9,13]. Whenever possible, the available OA fraction should be analytically verified. Without these controls, it remains unclear whether the observed effect reflects OA delivery, solvent effects, carrier effects, altered uptake, or differences in precipitation and release [1,5,7].
The biological model should match the intended use of the formulation. Cancer-oriented systems require more than simple two-dimensional cytotoxicity assays; three-dimensional spheroids, penetration studies, uptake analysis, pharmacokinetics, and biodistribution become important as development progresses [7,9,13]. For skin inflammation, keratinocyte, immune-cell, reconstructed skin, or lesion-relevant models are more informative [12,16]. For joint delivery, cartilage-, synovium-, or osteoarthritis-relevant models are needed [15].
This route-specific evaluation is particularly important for OA because its reported activities include anti-inflammatory, antioxidant, wound-healing-related, metabolic, and anticancer effects [1,5]. A formulation that improves cytotoxicity in cancer cells may not be appropriate for tissue repair, and a formulation designed for local anti-inflammatory activity may not be suitable for systemic exposure. Polymeric carrier design should therefore be guided by the intended biological context, not by the general assumption that increased delivery is always beneficial [7,13,16].
5.8. Formulation Implications
Polymeric nanoparticles provide a rational platform for OA delivery because they can improve dispersion, protect the compound, and modulate release [7,9,13]. PEGylated PLA/PLGA nanoparticles and PLGA nanoparticles have already shown that OA can be incorporated into biodegradable polymeric carriers and that encapsulation can alter biological response [7,9].
The next step is not simply to prepare more OA-loaded nanoparticles. The field needs clearer links between preparation method, polymer chemistry, carrier architecture, drug physical state, release environment, and biological model [6,7,9,13]. In this sense, polymeric OA nanoparticles should be evaluated as process-defined drug–carrier systems rather than as containers that only increase apparent solubility.
6. Polymer-Assisted and Hybrid Nanocarriers for Oleanolic Acid Delivery
6.1. Concept and Classification
Polymer-assisted nanocarriers occupy the space between purely polymeric nanoparticles and non-polymeric delivery systems. In these formulations, the polymer may not form the entire carrier, but it can determine essential functions such as colloidal stabilization, surface protection, solubilization, targeting, gelation, local retention, controlled release, or route-specific administration [1,5,7,8,12]. This category is particularly useful for OA because the compound often requires both hydrophobic accommodation and aqueous stabilization.
For the purposes of this review, polymer-assisted OA systems include PEGylated lipid or polymeric carriers, polymeric micelles, amphiphilic block-copolymer assemblies, hyaluronic-acid-based nanoprodrugs, nanocarrier-loaded hydrogels, thermosensitive gels, polymeric fiber membranes, and microneedle-compatible matrices [7,8,11,12,14,15,18,23]. These systems differ in composition and architecture, but they share a common feature: the polymeric component changes how OA is dispersed, protected, released, retained, or recognized by biological tissues.
This classification avoids treating all OA nanocarriers as equivalent. A lipid vesicle, a PLGA nanoparticle, a hyaluronic-acid nanoprodrug, a polymeric micelle, and a thermosensitive gel do not solve the same delivery problem. Each system controls a different part of the exposure pathway. For OA, that distinction matters because formulation performance depends not only on solubilization, but also on drug physical state, release environment, tissue contact, and route of administration [1,5,7,12,15].
6.2. PEGylated Systems as Polymer-Assisted OA Carriers
PEGylation is one of the most common polymer-assisted strategies used to modify nanocarrier behavior. Polyethylene glycol can improve aqueous dispersion, reduce aggregation, provide steric stabilization, and modify interactions with proteins, cells, and biological fluids [7,13,22]. For OA, PEGylation is relevant because the compound is poorly compatible with aqueous environments and tends to associate with hydrophobic domains [1,5,7].
OA-loaded PEGylated PLA and PLGA nanoparticles illustrate this principle well. In these systems, the biodegradable polymeric region accommodates the hydrophobic payload, whereas the PEG segment improves colloidal behavior and modifies the carrier surface [7]. PEGylated liposomes represent a related polymer-assisted design: the main vesicle is lipidic, but the PEG layer changes its biological interface and potential stability [22].
PEGylation should not be presented as an automatic improvement. PEG molecular weight, surface density, chain distribution, and carrier architecture can influence protein adsorption, cellular uptake, tissue penetration, clearance, and release [7,13,22]. A dense PEG layer may stabilize a formulation but reduce interaction with target cells or delay drug availability. For OA, PEGylated systems should therefore be evaluated through stability, release, uptake, and biological-response data rather than by particle size and encapsulation efficiency alone [7,13].
6.3. Polymeric Micelles for Oleanolic Acid
Polymeric micelles are particularly relevant for OA because they are designed to solubilize hydrophobic molecules in the core of amphiphilic block-copolymer assemblies. Compared with conventional surfactant micelles, polymeric micelles often have lower critical micelle concentrations and improved physical stability, which can be advantageous for poorly soluble compounds such as OA [17,29,30]. OA can be accommodated within the core, while the corona improves dispersion in aqueous or semiaqueous media [29,30]. This makes micelles relevant for dermal and topical applications, where OA must be delivered through or into barrier tissues without relying on systemic exposure.
A direct OA example is the development of polymeric micelles evaluated for particle size, morphology, encapsulation efficiency, skin permeation, three-month stability, and anti-wrinkle performance in a cosmetic formulation. Although the application was cosmetic, the study is useful for drug-delivery discussion because it demonstrates that OA can be converted into a stable nanoscale dispersed form suitable for skin application [17].
The main limitation is dilution stability. Polymeric micelles may dissociate after dilution, exchange unimers with the surrounding medium, bind proteins, release OA prematurely, or retain it too strongly depending on polymer composition and core structure [17,30]. For OA, the relevant question is not only whether the micelle solubilizes the drug, but whether it delivers a usable fraction to the intended skin or tissue compartment. Future micellar systems should therefore be evaluated using release media, dilution conditions, skin deposition models, and biological assays that match their intended route [29,30].
6.4. Hyaluronic-Acid-Based Nanocarriers and Nanoprodrugs
Hyaluronic acid is a particularly attractive polymer for disease-oriented OA delivery because it combines biocompatibility, hydrophilicity, biodegradability, and potential receptor-mediated interactions. Its relevance is strongest in inflammatory or proliferative skin environments, where CD44-related uptake may support local delivery to activated keratinocytes or other target cells [12,29].
A recent OA study used hyaluronic-acid-based reduction-responsive nanoparticles for topical psoriasis treatment [12]. The system combined disulfide-bonded OA–hyaluronic acid nanoprodrugs with an additional encapsulated OA component, producing OA-NPs@OA. This design addressed several barriers at once: poor OA solubility, limited cutaneous delivery, rapid clearance, polymer-mediated cellular interaction, and redox-responsive release [12].
This platform is important because the polymer is not a passive excipient. Hyaluronic acid participates in carrier formation, biological interaction, and disease-oriented delivery, while the disulfide linkage introduces a responsive release mechanism [12]. The system therefore connects polymer chemistry, nanocarrier architecture, topical administration, and a defined inflammatory disease model.
Several issues remain before such systems can be considered mature. Reproducible synthesis of the polymer–drug conjugate, control of substitution degree, nanoprodrug stability, degradation products, skin irritation, long-term safety, dose uniformity, and comparison with standard psoriasis therapies all require attention [12,29]. These questions do not reduce the value of the system; they define the next level of development needed for translational credibility.
6.5. Polymer-Assisted Liquid Crystalline Nanoparticle Gels
Liquid crystalline nanoparticles are not purely polymeric carriers, but they become relevant to this review when incorporated into polymeric or gel-based matrices for local administration. In such systems, the lipidic nanostructure accommodates OA, while the gel or polymeric phase controls residence time, administration properties, and local release [15,18].
Shi et al. developed an OA-loaded cubic liquid crystalline nanoparticle-based topical gel and evaluated rheology, release kinetics, ex vivo permeation, and anti-inflammatory performance [18]. A related study used an OA cubic liquid crystal nanoparticle-based thermosensitive gel for knee osteoarthritis in rats, demonstrating the local-depot logic of polymer-assisted OA delivery [15]. These examples are important because they move the discussion away from general solubility enhancement and toward route-specific local exposure.
For topical inflammation and osteoarthritis, the delivery objective is not necessarily high plasma concentration. The more relevant goal is to keep OA at the target site long enough to support a local biological effect. Gelation, viscosity, injectability, gelation temperature, residence time, release rate, and tissue tolerability are therefore as important as nanoparticle size or encapsulation efficiency [15,18].
6.6. Polymer-Lipid Hybrid Logic in OA Delivery
OA is structurally compatible with lipidic domains, but lipid compatibility alone does not guarantee controlled exposure. Lipid carriers can solubilize or accommodate OA, whereas polymers can improve stabilization, surface properties, release control, or local retention [1,5,7,13,22,23]. This makes polymer–lipid hybrid logic attractive for OA, even when direct OA-specific examples remain limited.
Existing OA formulations already point toward this hybrid approach. PEGylated liposomes use a polymer-modified surface to stabilize a lipid carrier [22]. PEGylated PLA/PLGA nanoparticles combine a hydrophobic polymeric region with a hydrated polymeric corona [7]. Liquid crystalline nanoparticle gels combine lipidic internal structure with a gel-based local delivery matrix [15,18]. These systems differ materially, but each uses more than one functional domain to manage OA delivery.
Future OA research could more explicitly explore PLGA–lipid nanoparticles, lipid-core polymer-shell systems, polymer-coated lipid nanoparticles, and nanocarrier-loaded hydrogels. Such platforms may be suitable for oral, topical, intra-articular, or selected systemic applications, depending on polymer type, lipid composition, carrier architecture, and release requirements [7,15,18,22,23].
6.7. Design Variables in Polymer-Assisted OA Carriers
Polymer-assisted OA systems should be evaluated according to the variables that control both the polymeric and non-polymeric parts of the formulation. Relevant polymer parameters include molecular weight, hydrophilicity, charge, degradability, PEG density, functional groups, crosslinking, stimuli-responsive linkages, and gelation behavior [7,12,15,18,22]. Relevant non-polymeric parameters include lipid composition, surfactant type, internal nanostructure, drug-to-carrier ratio, solvent history, and processing conditions [7,12,13,15,18,22,30].
The interaction between these variables determines whether OA remains dispersed, crystallizes, releases too slowly, partitions into tissue, or becomes biologically available [1,5,18]. A dense hydrophilic shell may improve stability but reduce uptake. A thermosensitive gel may increase residence time but slow diffusion excessively. A micellar core may solubilize OA but lose integrity after dilution [15,17,30].
For this reason, formulation optimization should not rely on a single endpoint. Particle size, encapsulation efficiency, and visual stability are useful but incomplete. A polymer-assisted OA carrier should also be assessed for release kinetics, dilution stability, drug physical state, tissue deposition, biological activity, and route-specific tolerability. The most important quality attributes will differ by application: topical systems require skin deposition and irritation data, intra-articular systems require injectability and joint residence, and systemic systems require uptake, biodistribution, and safety evaluation [7,12,13,15,18].
6.8. Clinical Relevance of Polymer-Assisted OA Systems
The strongest rationale for polymer-assisted OA systems currently lies in local and tissue-targeted delivery. Psoriasis, inflammatory skin disease, wound-related applications, dermal delivery, and osteoarthritis are settings in which sustained local exposure may be more useful than increased systemic bioavailability. Polymeric micelles, hyaluronic-acid nanoprodrugs, gels, nanogels, thermosensitive depots, and polymer-assisted matrices can be designed to prolong contact with the target tissue and reduce unnecessary systemic distribution [12,13,15,17,18].
Cancer-oriented PEGylated and polymeric nanoparticle systems remain scientifically relevant, but they require more cautious interpretation. Increased cytotoxicity in vitro does not by itself establish anticancer potential. For cancer applications, carrier penetration, uptake mechanism, three-dimensional tumor models, pharmacokinetics, biodistribution, safety, and comparison with existing therapies become necessary as the formulation moves beyond proof of concept [7,13,22].
Local inflammatory indications may offer a more realistic near-term path because topical gels, dermal systems, intra-articular depots, and polymeric local-delivery platforms are already familiar dosage-form concepts. However, even local systems must be tested against practical clinical criteria: dose loading, dosing frequency, local tolerability, sterility when needed, shelf-life, ease of administration, and performance compared with standard care [12,15,18,29].
6.9. Design Implications
Polymer-assisted and hybrid nanocarriers broaden the OA delivery landscape beyond conventional polymeric nanoparticles [7,8,12,13,15,17,18,22,23]. Their value lies in combining hydrophobic drug accommodation with functions such as colloidal stabilization, surface modification, responsive release, gelation, tissue retention, or route-specific administration [1,5,7,12].
The most promising systems are not necessarily the most complex. They are the systems in which each material component has a clear purpose: PEG for stabilization, hyaluronic acid for disease-oriented interaction, amphiphilic polymers for micellar solubilization, gels for local residence, or hybrid matrices for controlled tissue exposure. For OA, polymer-assisted design should therefore be judged by functional necessity rather than formulation novelty.
The functional roles, advantages, limitations, and most relevant applications of polymeric materials used in OA delivery are summarized in Table 3.
7. Stimuli-Responsive and Soft Polymeric Systems for Oleanolic Acid Delivery
7.1. Rationale for Soft Polymeric Systems
Soft polymeric systems include polymeric micelles, nanogels, hydrogels, cyclodextrin-containing networks, thermosensitive gels, and stimuli-responsive polymer conjugates. They are relevant to OA delivery because they can combine drug dispersion with mechanisms that regulate release, tissue contact, and local residence [1,5,7,13,15,32,33]. Unlike rigid nanoparticles, these systems often contain hydrated, deformable, or supramolecular domains that respond to dilution, pH, redox conditions, enzymes, temperature, swelling, or the local tissue environment [12,13,15,18,32,33].
For OA, this flexibility is useful because the compound is poorly soluble, lipophilic, and strongly dependent on its formulation environment [1,5]. A rigid polymeric particle may encapsulate OA efficiently but release it too slowly. A simple solubilizing system may improve dispersion but fail to maintain local exposure. Soft polymeric systems occupy the intermediate space between these two extremes: they can keep OA in a dispersed state while allowing release to be influenced by polymer hydration, gel swelling, micelle dissociation, complex dissociation, degradation, or responsive bond cleavage [12,13,15,18,32,33].
This section therefore does not treat soft systems as another list of carrier types. Instead, it focuses on how their dynamic structure can be used to control OA availability. This distinction is important for local and barrier-associated applications, including inflammatory skin disease, psoriasis, wound healing, osteoarthritis, and dermal delivery, where sustained tissue exposure may be more useful than high systemic concentrations [12,15,16,17,18].
7.2. Polymeric Micelles as Solubilizing and Dermal Delivery Systems
Polymeric micelles are formed by amphiphilic polymers that self-assemble into structures with hydrophobic cores and hydrophilic shells. This architecture is compatible with OA because the hydrophobic core can accommodate the triterpenoid, while the shell improves dispersion in aqueous or semiaqueous media [7,17,30].
An et al. developed OA polymeric micelles and evaluated particle size, morphology, encapsulation efficiency, skin permeation, three-month physical stability, and anti-wrinkle performance in a cosmetic formulation [17]. Although the application was cosmetic, the study is relevant to polymeric drug delivery because it shows that OA can be converted into a stable nanoscale dispersed form suitable for skin application [17].
The main limitation of micellar systems is that solubilization does not necessarily equal delivery. Micelles can dissociate after dilution, exchange unimers with the surrounding medium, bind proteins, release OA prematurely, or retain the drug too strongly depending on polymer composition and hydrophobic core structure. For OA, the key question is whether micellar encapsulation increases the fraction that reaches relevant skin or tissue layers. Future micellar OA systems should therefore be evaluated using dilution stability, release kinetics, skin deposition, and biological assays matched to dermal or topical use [17,30].
7.3. Nanogels as Hydrated Polymeric Reservoirs
Nanogels are nanoscale crosslinked polymer networks that can swell in water or biological fluids while maintaining structural integrity. Their hydrated structure makes them attractive for controlled release because drug diffusion can be influenced by mesh size, crosslinking density, polymer composition, swelling behavior, and environmental responsiveness [32,33].
For OA, nanogels are conceptually attractive but technically demanding. Many nanogel networks are hydrophilic, whereas OA is highly lipophilic [1,5,32,33]. Useful OA loading may therefore require hydrophobic domains, inclusion complexes, lipidic substructures, polymer–drug conjugation, or hybrid nanocarrier-in-gel designs [12,15,18]. In other words, the nanogel itself may not be sufficient unless its structure contains a domain capable of accommodating OA.
The OA cubic liquid crystal nanoparticle-based thermosensitive gel developed for knee osteoarthritis illustrates this hybrid logic. The internal nanostructure accommodates OA, whereas the thermosensitive gel provides an injectable local depot that can prolong residence after intra-articular administration [15]. This design is relevant because it links nanoscale drug accommodation with macroscopic gel behavior.
Nanogels and soft gels may be especially useful in local inflammatory conditions, including osteoarthritis, inflamed skin, and wound-associated environments [12,15,18]. Their performance, however, cannot be predicted from drug loading alone. Rheology, swelling, degradation, injectability, gelation temperature, mechanical behavior, release kinetics, and local tolerability may all determine whether the system is useful for a given route [15,18,32,33].
7.4. Cyclodextrin-Containing and Cyclodextrin-Polymer Systems
Cyclodextrins are cyclic oligosaccharides that can form inclusion complexes with hydrophobic molecules and improve the apparent solubility or handling of poorly water-soluble compounds [34,35,38]. For OA, hydroxypropyl-cyclodextrin complexation improved stability, cell compatibility, and biological activity in migration-related assays compared with OA delivered in DMSO [14].
Simple cyclodextrin complexation is not always classified as polymeric nanocarrier delivery in the strict sense [14,34]. However, cyclodextrin-containing polymers, cyclodextrin-modified nanomaterials, cyclodextrin-based nanoparticles, and cyclodextrin nanosponges extend this inclusion-complex principle into polymeric delivery systems. These platforms can combine host–guest complexation with network formation, nanoparticle behavior, local retention, or controlled release [34,35,38].
For OA, cyclodextrin systems have two potential roles. First, they can improve aqueous compatibility and help standardize biological exposure in assays where DMSO-delivered OA may not be an ideal comparator. Second, when incorporated into polymeric networks or nanosponge-like structures, they may support more sustained release or local retention [14,34,35,38,39]. This could be useful for topical, wound-related, or microneedle-compatible formulations in which OA must remain dispersed within a hydrophilic polymeric environment [14,16,18,34].
The main caution is that inclusion complexation does not automatically predict tissue delivery. Complex stability, dissociation rate, competing biological molecules, polymer network structure, and route of administration can all affect OA release [14,34,35,38,39]. Cyclodextrin-polymer OA systems should therefore be compared with reference carriers such as PLGA nanoparticles, micelles, and hydrogels under matched biological conditions [7,14,17,34].
7.5. Redox-Responsive Hyaluronic-Acid Systems
Redox-responsive carriers are designed to release drug in response to differences between extracellular and intracellular redox environments. Disulfide linkages are commonly used for this purpose because they can remain relatively stable outside cells but undergo cleavage in more reductive intracellular compartments [12,32]. This mechanism is directly relevant to hyaluronic-acid-based OA nanoprodrugs developed for topical psoriasis therapy [12].
Han et al. reported disulfide-bonded OA–hyaluronic acid nanoprodrugs with additional OA encapsulation for psoriasis treatment [12]. The formulation was designed to address poor OA solubility, limited cutaneous permeation, and rapid clearance after topical application. The authors reported improved uptake through CD44 receptor-mediated endocytosis in keratinocytes and enhanced anti-psoriasis effects compared with free OA [12].
This system is one of the strongest OA examples because it combines several design elements in one platform: a hydrophilic polymeric framework, receptor-associated cellular interaction, redox-responsive cleavage, nanoscale assembly, and topical disease-oriented delivery [12]. The polymer is not only a stabilizer; it participates in both carrier architecture and biological interaction.
The main translational questions concern reproducibility and safety. The substitution degree of the OA–hyaluronic acid conjugate, disulfide stability, degradation products, batch consistency, skin irritation, repeated-dose safety, and comparison with standard psoriasis therapies all need to be clarified before such a platform can move beyond proof of concept [12,32].
7.6. Thermosensitive, pH-Responsive, and Enzyme-Responsive Systems
Stimuli-responsive polymeric systems can be designed to change swelling, degradation, drug release, or network structure in response to temperature, pH, enzymatic activity, or other local microenvironmental cues [12,15,18,32]. For OA, these mechanisms are attractive because they may help couple drug release to the biological compartment in which local exposure is needed. However, the strength of evidence differs between stimulus types. Thermosensitive OA systems have direct experimental support, whereas pH- and enzyme-responsive approaches remain more conceptual or extrapolated from broader polymeric drug-delivery literature unless OA-specific examples are available.
Thermosensitive polymeric systems are currently the most developed responsive local-depot approach for OA. They can be administered as liquids and form gels at physiological or tissue-relevant temperature, which is useful for topical or intra-articular applications where residence time is a major limitation [15,18,33]. ]. The OA cubic liquid crystal nanoparticle-based thermosensitive gel developed for knee osteoarthritis illustrates this logic: the nanostructured carrier accommodates OA, while the temperature-responsive gel matrix supports local retention and sustained exposure after administration [15].
pH- and enzyme-responsive systems may also be relevant for OA, particularly in inflamed skin, wounds, tumors, or intracellular compartments where local pH, enzyme activity, or redox state may differ from healthy tissue [12,33]. In such systems, the stimulus should not be added as a decorative feature. It should have a clear mechanistic purpose, such as accelerating release in an inflamed microenvironment, promoting carrier degradation after tissue deposition, or reducing premature release before the formulation reaches the target site.
For all responsive OA systems, performance depends on whether the trigger is strong, reproducible, and relevant to the intended route. Gelation temperature, pH sensitivity, enzymatic degradation rate, polymer concentration, injectability, dilution behavior, mechanical strength, release kinetics, and tissue tolerability all need to be matched to the biological setting [15,18,33]. A responsive system is useful only if the stimulus improves local exposure, safety, or dosing practicality compared with a simpler formulation.
7.7. Integration with Microneedles, Hydrogels, and Wound Dressings
Soft polymeric OA systems are especially relevant when they are combined with dosage forms that solve a route-specific barrier. Microneedles can bypass the stratum corneum, hydrogels can maintain moist local contact, fiber membranes can provide tissue-facing matrices, and thermosensitive gels can improve residence after injection or topical placement [15,16,18,36,37].
This integration is important because OA delivery often requires more than one level of control. A nanocarrier may improve OA dispersion, but the dosage form determines where the carrier is placed, how long it remains there, and how the tissue environment affects release [15,16,18]. For example, nanocarrier-loaded microneedles can deposit OA-containing systems into the skin, after which release is governed by microneedle dissolution, carrier stability, tissue fluid interaction, and OA partitioning [16,36,37].
Similarly, hydrogels and wound dressings can combine local retention with controlled release. The polymeric matrix may regulate moisture balance, tissue contact, mechanical compliance, and residence time, while OA-loaded micelles, nanoparticles, or inclusion complexes improve drug dispersion within the matrix [8,15,18]. These combined systems are more complex than simple topical formulations, but they may be better matched to inflammatory skin disease, wounds, and local tissue injury.
Evaluation should reflect this multilevel design. For topical and transdermal OA systems, release data alone are not enough. Skin deposition, tissue distribution, irritation, barrier recovery, inflammatory biomarkers, mechanical performance, and comparison with current therapies are needed to determine whether improved solubilization translates into useful local delivery [12,14,16,17,18].
7.8. Advantages and Limitations of Stimuli-Responsive OA Systems
Stimuli-responsive and soft polymeric systems offer several advantages for OA delivery. They can improve dispersion, support controlled release, prolong local residence, enable disease-oriented interactions, and integrate with topical, intra-articular, or microneedle-based administration [12,15,17,18,32,33]. These properties are most relevant for applications where local tissue exposure matters more than systemic concentration.
Their main weakness is complexity. Performance may depend on polymer chemistry, swelling, degradation, micelle stability, inclusion complex dissociation, gelation behavior, responsive bond cleavage, tissue interaction, and environmental conditions [12,15,18,32]. This complexity increases the need for detailed characterization and may complicate manufacturing, storage, scale-up, and regulatory evaluation [32,33].
A second limitation is the difficulty of separating OA effects from carrier effects. Hyaluronic acid, cyclodextrins, gels, micellar polymers, and responsive linkers may influence tissue interaction or cellular response independently of OA [12,14,17,34]. Blank carrier controls, non-responsive carrier controls, free OA controls, solubilized OA controls, and analytically verified exposure conditions are therefore essential [32].
Finally, many responsive systems remain at proof-of-concept stage. For OA, a soft polymeric system becomes convincing only when it demonstrates reproducible preparation, stable storage, route-specific release, relevant tissue deposition, acceptable safety, and a clear advantage over simpler formulations or existing therapeutic options [12,15,18,32].
7.9. Mechanistic Implications
Soft and stimuli-responsive polymeric systems are valuable for OA delivery because they address more than poor solubility. They can regulate how OA is dispersed, retained, released, and presented to tissue [12,14,15,16,17,18,33]. Polymeric micelles support skin-compatible dispersion, nanogels and hydrogels can act as local reservoirs, cyclodextrin-polymer systems can combine inclusion complexation with controlled release, and hyaluronic-acid nanoprodrugs can introduce receptor-associated and redox-responsive behavior [12,14,17,34,35,38,39].
The most realistic applications are likely to be local or topical, including psoriasis, inflammatory skin disease, wound healing, dermal delivery, and osteoarthritis [12,15,16,17,18]. In these settings, the purpose is not to maximize systemic bioavailability, but to maintain OA exposure at the tissue site for a useful period. The best soft polymeric systems will therefore be those that connect carrier mechanism with route-specific performance, rather than those that add responsiveness without a clear therapeutic reason.
8. Hydrogels, Polymeric Wound Dressings, and Local Depot Systems
8.1. Why Local Polymeric Systems are Important for Oleanolic Acid
Local polymeric delivery systems are particularly relevant for OA because several of its most plausible biomedical applications involve tissue-level inflammation, skin pathology, wound repair, osteoarthritis, or localized oxidative stress rather than immediate systemic therapy [1,5,8,15,18]. In these settings, the main formulation goal is not necessarily to maximize plasma concentration. It is to maintain OA exposure at the target site for a useful period while limiting unnecessary systemic distribution [5,15,18].
This route-specific logic is important because OA has poor aqueous solubility, limited permeability, and formulation-dependent biological activity [1,5]. A formulation that improves oral dispersion may not be suitable for a wound dressing, and a nanoparticle designed for cancer-cell uptake may not be appropriate for intra-articular retention or dermal deposition [5,8,15,18]. Local polymeric matrices allow the formulation to be adapted to the biological environment by controlling residence time, release rate, hydration, tissue contact, mechanical compatibility, and handling [8,12,15,18].
Hydrogels, thermosensitive gels, polymeric fiber membranes, and wound dressings should therefore not be treated as secondary dosage forms. In OA delivery, they represent a distinct strategy: the drug may first be dispersed in nanoparticles, micelles, inclusion complexes, or lipidic nanostructures, and the resulting system can then be placed within a tissue-facing polymeric matrix [8,12,15,18]. In this design, the nanosystem improves OA dispersion, while the macroscopic matrix controls location, contact time, hydration, and the release environment.
The main advantage of local polymeric depots is that they can decouple solubilization from tissue residence. OA can first be incorporated into nanoparticles, micelles, inclusion complexes, or lipidic nanostructures, and the resulting formulation can then be placed in a hydrogel, fiber membrane, or thermosensitive matrix. In this design, the nanosystem addresses OA dispersion, while the polymeric depot controls location, contact time, and release environment [8,12,15,17,18]. In this design, the nanosystem improves OA dispersion, while the macroscopic matrix controls location, contact time, hydration, and the release environment.
8.2. Hydrogels as Soft Matrices for OA Delivery
Hydrogels are useful biomaterials for local drug delivery because they contain high water content, can conform to tissue surfaces, may support moist wound environments, and can be engineered to release drugs over time. Their mechanical softness and tunable chemistry make them suitable for topical formulations, wound dressings, injectable depots, intra-articular systems, and microneedle-compatible platforms [12,39,40,41].
For OA, direct incorporation into a purely hydrophilic hydrogel may be inefficient because the compound is lipophilic and poorly soluble in aqueous media [1,5]. Hydrogel-based OA systems therefore often require an intermediate carrier, such as polymeric nanoparticles, liquid crystalline nanoparticles, polymeric micelles, cyclodextrin complexes, or nanogels, before incorporation into the hydrogel matrix [8,12,15,17,18]. This nanocarrier-in-gel approach is more rational than simply dispersing crystalline OA in a hydrated polymer network.
Hydrogels can regulate release through polymer concentration, crosslinking density, swelling, erosion, mesh size, temperature sensitivity, degradation, and interaction between the embedded carrier and the polymer network. For OA, these variables determine whether the compound is released too rapidly, retained too strongly, or made available at a sustained local concentration. Hydrogel-based OA systems should therefore be characterized not only by drug release, but also by rheology, swelling, mechanical behavior, carrier distribution, and stability during storage and use [8,15,17,18,39].
Hydrogel-based local delivery is most convincing when repeated administration is undesirable or when local residence is central to the therapeutic concept. This applies to chronic wounds, inflamed skin, intra-articular delivery, and post-injury tissue repair, where the desired effect may depend on prolonged anti-inflammatory, antioxidant, or cytoprotective exposure rather than short-term systemic absorption [12,15,17].
8.3. Topical OA Gels and Polymer-Assisted Local Retention
Topical OA gel systems illustrate how a polymer-assisted matrix can convert a nanocarrier into a more practical dosage form. OA-loaded liquid crystalline nanoparticle gels are relevant because the internal nanostructure can accommodate the lipophilic drug, while the gel phase improves topical application, tissue contact, and local residence. In such systems, the gel is not merely a thickening agent. It contributes to administration, retention, and release.
Shi et al. developed an OA-loaded cubic liquid crystalline nanoparticle-based topical gel and evaluated rheological behavior, release kinetics, ex vivo permeation, and anti-inflammatory performance [18]. The topical orientation is important because skin inflammation is a clinically plausible area for OA delivery. Local exposure may be more relevant than systemic bioavailability, and a gel-based format can reduce the need for systemic administration [1,5,18].
The main development questions for topical OA gels are practical rather than only nanoscale. A useful topical system should spread appropriately, remain at the application site, avoid irritation, maintain drug stability, and release OA under skin-relevant conditions [18,40,41]. Evaluation should therefore include rheology, skin deposition, permeation, irritation, barrier compatibility, microbial considerations where relevant, and comparison with established topical treatments [12,18,39].
8.4. Thermosensitive OA Nanogels for Intra-Articular Delivery
Thermosensitive OA nanogels represent one of the most disease-oriented examples of polymer-assisted local delivery. The OA cubic liquid crystal nanoparticle-based thermosensitive gel reported by Shi, Jia, Tang, and Li was designed for intra-articular administration in a rat knee osteoarthritis model [15]. The system combined OA-loaded nanoparticles with a Poloxamer thermosensitive gel base, creating a formulation intended to improve local residence after injection [15].
This approach is relevant because osteoarthritis is a localized degenerative and inflammatory joint disease. For such an indication, sustained intra-articular exposure may be more meaningful than systemic OA administration [15]. OA has anti-inflammatory and cartilage-protective potential, but its poor solubility and limited direct applicability make local depot design attractive [5,15].
The thermosensitive behavior is central to the formulation. It determines whether the system can be administered as a fluid and then form or maintain a depot-like structure under physiological conditions [15]. For intra-articular use, injectability, gelation temperature, viscosity, retention time, release kinetics, sterility, endotoxin control, and joint compatibility are critical quality attributes. These parameters should be considered together with nanoparticle size, OA loading, and drug physical stat [15,39,40].
This example strengthens the clinical orientation of the review because it frames OA delivery around an anatomical and therapeutic problem: local treatment of osteoarthritis. However, translation would require further work on long-term joint safety, repeated dosing, cartilage compatibility, synovial irritation, mechanical joint conditions, comparison with clinically used intra-articular therapies, and manufacturing reproducibility [15,39,40].
8.5. PLGA Fiber Membranes as Polymeric Local Delivery Platforms
OA-loaded PLGA fiber membranes represent a different local delivery strategy. Instead of placing OA in a spherical nanoparticle or soft gel alone, the drug is incorporated into a nonwoven biodegradable polymeric fiber matrix. Aguirre-Chagala et al. developed OA-loaded poly(lactic-co-glycolic acid) fiber membranes and evaluated processing–structure–property relationships relevant to biomedical delivery [8].
This platform is important because polymeric fiber membranes provide a macroscopic tissue-contacting structure. Compared with nanoparticles alone, fiber membranes offer mechanical integrity, surface area, porosity, and a format compatible with wound dressings, local patches, transdermal platforms, or tissue-facing reservoirs [8,39,40,41]. These features are especially relevant when the formulation must remain at the application site and interact with tissue over time.
For OA, PLGA fiber membranes can help avoid direct application of crystalline drug by distributing the compound within a biodegradable polymeric matrix. Fiber processing, fiber diameter, morphology, drug loading, thermal behavior, drug distribution, crystallinity, and degradation can all influence how OA is presented to the local environment [8]. This makes the platform relevant to the broader process–structure–function logic of the review.
The translational value of fiber membranes will depend on more than drug release. Mechanical flexibility, adhesion, tensile strength, porosity, moisture behavior, sterilization, residual solvent, drug stability, degradation rate, and tissue compatibility must be considered before clinical usefulness can be claimed [8,39,40,41]. For wound applications, release testing should also consider wound-like media rather than relying only on simple buffer systems.
8.6. Polymeric Wound Dressings and OA Wound-Healing Relevance
Modern wound dressings are not passive covers. Polymeric dressings can regulate moisture, absorb exudate, protect tissue mechanically, support cell attachment, and deliver therapeutic agents locally [39,40,41]. Hydrogels, electrospun fibers, nanofiber membranes, nanohybrid dressings, and nanoparticle-loaded matrices are therefore relevant platforms for wound-oriented OA delivery [7,39,40,41].
OA is relevant to wound-directed formulations because it has been associated with anti-inflammatory activity, oxidative-stress modulation, and cell migration-related responses [1,5,17,26,28]. Earlier in vivo work also reported wound-healing activity for OA from natural sources, supporting the biological rationale for local wound applications [26,28]. At the same time, wound healing is stage-dependent and involves inflammation, migration, proliferation, extracellular matrix deposition, angiogenesis, and remodeling. OA wound systems should therefore be interpreted carefully and tested in models that reflect the biology of tissue repair [28,39,40,41].
A polymeric wound dressing can support OA delivery by combining local retention with a favorable wound microenvironment. Direct OA loading may be possible in some matrices, but a more robust design may involve OA-loaded nanoparticles, micelles, cyclodextrin complexes, nanogels, or lipidic nanostructures incorporated into the dressing [8,12,17,18]. In this arrangement, the carrier manages OA dispersion and release, while the dressing controls hydration, residence, exudate interaction, tissue contact, and mechanical protection.
Wound-oriented OA systems should include biologically meaningful endpoints in addition to drug release. Relevant readouts include fibroblast viability, keratinocyte migration, inflammatory cytokines, oxidative stress markers, collagen deposition, wound closure, infection risk, exudate interaction, and tissue compatibility [28,39,40,41]. Without these endpoints, it is difficult to determine whether OA release translates into improved wound repair.
8.7. Local Depots for Inflammatory Skin Diseases
Inflammatory skin disease is another plausible direction for local OA polymeric systems. OA has reported anti-inflammatory activity, while topical and intradermal delivery can provide high local exposure with potentially lower systemic burden [1,5,12]. Polymeric micelles, hyaluronic-acid-based nanoparticles, hydrogels, and microneedle-compatible matrices are particularly relevant because they address both OA solubility and skin-barrier limitations [12,16,17].
The hyaluronic-acid-based reduction-responsive OA nanoparticle system for psoriasis is a strong example of disease-oriented local polymeric design. It combines polymeric architecture, receptor-associated cellular interaction, redox-responsive release, and topical application in an inflammatory skin disease model [12]. This type of design is more convincing than a general solubility-enhancing formulation because it is linked to a specific disease context
Local OA systems for inflammatory skin disease should be evaluated in models that reflect barrier function, keratinocyte response, immune signaling, skin deposition, and irritation potential. Simple cytotoxicity assays are not sufficient. A topical anti-inflammatory OA formulation must provide local activity without damaging the barrier, provoking irritation, or relying on unrealistic exposure conditions [12,16,17].
Hydrogels and microneedles may strengthen this direction further. Hydrogels can improve residence and hydration, whereas microneedles can bypass the stratum corneum and deposit OA-loaded systems into viable epidermis or dermis [16,37,42]. For OA, this combination is attractive because poor solubility and limited passive penetration are both major barriers [1,5,16].
8.8. Local Depots for Osteoarthritis and Musculoskeletal Inflammation
Osteoarthritis is a particularly relevant target for polymer-assisted OA depots because it combines inflammation, cartilage degradation, oxidative stress, and chronic pain within an accessible anatomical compartment [15]. Intra-articular delivery can theoretically provide high local exposure while reducing systemic distribution, but rapid clearance from the joint remains a major challenge for small molecules and simple formulations [15,39].
The OA thermosensitive nanogel study directly addresses this problem by placing OA-loaded nanoparticles in a Poloxamer gel base for local knee delivery in a papain-induced osteoarthritis model. The study is therefore more than another solubility-enhancement example. It tests a local depot concept in a disease-relevant anatomical setting [15].
For musculoskeletal applications, the critical formulation question is not only how much OA is loaded, but how long the system remains in the joint and how release relates to biological response [15]. An ideal local depot should be injectable, minimally irritating, mechanically compatible with the joint, stable enough to prolong exposure, and degradable or removable without causing chronic inflammation [15,39].
Future OA osteoarthritis formulations should be compared with clinically relevant controls such as nonsteroidal anti-inflammatory drugs, corticosteroids, hyaluronic acid injections, or other intra-articular therapies where appropriate. They should also evaluate cartilage integrity, synovial inflammation, pain-related behavior, local toxicity, and repeated-dose safety. These endpoints are needed to determine whether polymer-assisted OA depots offer value beyond formulation novelty [15,39].
8.9. Design Criteria for OA Local Polymeric Systems
Local polymeric OA systems require design criteria that differ from oral or systemic nanocarriers [6,7,8,9]. For topical and wound systems, important parameters include tissue adhesion, hydration, breathability, exudate handling, irritation potential, microbial risk, drug release, skin or tissue deposition, and compatibility with damaged or inflamed tissue [8,12,28,39,40,41]. For intra-articular systems, injectability, gelation, residence time, sterility, endotoxin burden, mechanical compatibility, and local toxicity become more important [15,39].
Drug loading and release should be interpreted in relation to the intended tissue [8,12,15,18]. Drug loading and release must be interpreted in relation to the intended tissue. Rapid release may be useful for acute inflammation but insufficient for chronic disease. Very slow release may prolong exposure but fail to reach a useful local level [12,15]. For wound and skin applications, release data should be coupled with deposition or tissue-retention data rather than interpreted only from bulk medium measurements [12,16,17,39,40].
The polymeric matrix must also be compatible with OA physical properties. Because OA is lipophilic and poorly water-soluble, direct dispersion in hydrophilic matrices may lead to poor uniformity, crystallization, or incomplete release [1,5,8]. Nanocarrier-in-matrix systems may therefore be preferable for many local applications [8,12,15,17,18].
Biological controls are essential. Blank matrices, free OA, OA-loaded nanocarriers without matrix, and full nanocarrier-in-matrix systems should be compared where possible [12,16,17,26,28,39,40,41]. These controls help distinguish the effects of OA from the effects of the polymeric matrix, nanocarrier, hydration, occlusion, or local mechanical environment.
8.10. Translational Challenges of Hydrogels, Dressings, and Depots
Hydrogels, polymeric wound dressings, fiber membranes, and local depots are clinically attractive, but they also introduce practical development challenges. These include sterilization, shelf-life, mechanical stability, reproducible drug loading, polymer degradation, residual solvent, microbial contamination, patient handling, packaging, and regulatory classification [15,39,40,41]. For OA, additional concerns arise from poor solubility, crystallization risk, and vehicle-dependent biological activity [1,5].
Wound dressings and topical gels must be evaluated for irritation, sensitization, permeability, microbial compatibility, and performance under realistic skin or wound conditions. In vitro release testing alone is insufficient because wound exudate, enzymes, proteins, salts, variable pH, bacterial contamination, and tissue remodeling can influence polymer swelling, degradation, OA partitioning, and carrier stability. Progressive testing in more realistic models is therefore important. [26,28,39,40,41].
Intra-articular depots have a different translational profile. Joint injections require sterility, low endotoxin burden, appropriate viscosity, injectability through clinically relevant needles, and absence of long-term synovial irritation. A formulation that performs well in a rat model may still require substantial optimization before human translation because joint size, movement, clearance, mechanical loading, and dosing schedules differ substantially [15].
For clinical relevance, local OA systems should be compared not only with free OA, but also with existing standards of care where possible [12,15,40]. In inflammatory skin disease, this may include corticosteroids, calcineurin inhibitors, or approved topical therapies [12]. In osteoarthritis, comparison with hyaluronic acid, corticosteroids, NSAID-based approaches, or other intra-articular therapies may be appropriate [15]. In wound healing, advanced dressings or standard wound-care materials provide more meaningful comparators than free OA alone [39,40,41].
8.11. Local-Delivery Implications
Hydrogels, polymeric wound dressings, fiber membranes, and local depot systems are important for OA delivery because they shift the formulation goal from systemic bioavailability to controlled local exposure [8,12,15,18]. Their value lies in combining OA dispersion with tissue residence, hydration, mechanical contact, and route-specific release.
The most convincing local OA systems are those in which the carrier and matrix have separate but complementary functions. The nanocarrier improves OA dispersion or incorporation, while the hydrogel, dressing, fiber membrane, or thermosensitive depot controls placement, residence, and interaction with the tissue environment. For OA, this local formulation logic is especially relevant to inflammatory skin disease, wound healing, dermal delivery, osteoarthritis, and other tissue-restricted inflammatory conditions [8,12,15,16,17,18,40,41].
9. Polymeric Microneedles and Transdermal Delivery Platforms
9.1. Why Microneedles are Relevant to OA Polymeric Delivery
Polymeric microneedles are relevant to OA delivery because they can bypass the stratum corneum while preserving the advantages of local, minimally invasive administration [16,36,37,42]. This is important for OA because passive topical delivery is limited by poor aqueous solubility, formulation-dependent exposure, and restricted skin penetration [1,5]. By creating transient microchannels or depositing drug-loaded polymeric material into the skin, microneedles can increase intradermal access without requiring systemic administration.
For OA, microneedles are most useful when they are treated as part of an integrated delivery platform rather than as standalone devices [16,36,37,42]. A microneedle array may solve the barrier problem, but it does not automatically solve OA solubility, crystallinity, dose loading, or release control. These formulation barriers may still require polymeric nanoparticles, micelles, nanogels, cyclodextrin complexes, hydrogels, or hyaluronic-acid-based nanocarriers embedded within or applied after the microneedle system [7,9,12,14,17,18].
This carrier-in-microneedle logic is particularly appropriate for OA. The microneedle controls skin access and deposition depth, whereas the embedded carrier controls drug dispersion, physical state, and release after insertion [7,9,12,14,16,17,18,36,37,42]. In this sense, microneedles should be viewed as a route-enabling platform for OA rather than simply as a device for pushing more drug through the skin.
The field should nevertheless be framed cautiously. Direct reports of OA-loaded microneedles remain limited, and much of the rationale comes from the convergence of OA delivery barriers, polymeric OA nanocarrier studies, and broader microneedle literature in transdermal drug delivery [1,5,7,9,12,14,16,17,18,36,37,42]. Microneedles are therefore a plausible future direction for OA, but not yet an established OA formulation class.
9.2. Types of Microneedles and Relevance to OA
Microneedles are commonly classified as solid, coated, hollow, dissolving, and hydrogel-forming systems [16,36,37,42]. Each type creates a different delivery mechanism, and their relevance to OA depends on whether the system can maintain the drug in a dispersed, deliverable, and biologically useful form.
Solid microneedles mainly pretreat the skin by forming microchannels, after which a topical formulation is applied. This strategy could improve OA delivery if the post-treatment formulation is an OA-loaded gel, micelle, nanogel, nanoparticle dispersion, or inclusion-complex system [1,5,9,12,17,18]. However, the approach still depends on diffusion from an external formulation into transient skin pathways, so OA must remain bioavailable during and after application.
Coated microneedles deliver drug from a coating on the microneedle surface. This approach may be less suitable for OA if the coating volume limits dose loading or if OA crystallizes during coating and drying [1,5,42]. Coated systems could still be useful if OA is incorporated into a polymeric coating containing solubilizers, micelles, cyclodextrins, or nanocarriers, but coating uniformity, drying behavior, and dose reproducibility would become critical [9,14,16,36,37,42].
Hollow microneedles can inject liquid formulations through internal channels. They could theoretically deliver OA-loaded nanosuspensions, micelles, or nanoparticle dispersions, but viscosity, clogging, sterility, dose accuracy, and colloidal stability would be major constraints [7,9,12,14,17,18]. For OA, this approach may be more demanding than dissolving or hydrogel-forming systems because the injected formulation must remain stable and free of crystalline precipitates.
Dissolving microneedles are more attractive for OA because the polymeric needle matrix can incorporate drug-loaded carriers and then dissolve after insertion [16,36,37,42]. Rapid dissolution can deposit OA-containing material in the skin, while embedded nanoparticles, micelles, nanogels, or cyclodextrin complexes can provide additional control over dispersion and release [7,9,12,14,17,18]. Hydrogel-forming microneedles offer a related but distinct strategy: the needles swell after insertion and can act as conduits or controlled interfaces between an external reservoir and the skin [16,36,37,42].
9.3. Polymeric Materials for OA-Compatible Microneedles
Polymeric microneedles can be prepared from materials such as hyaluronic acid, polyvinylpyrrolidone, polyvinyl alcohol, carboxymethylcellulose, chitosan, gelatin, alginate, and other water-soluble or hydrogel-forming polymers [16,36,37,42]. Material selection determines mechanical strength, insertion efficiency, dissolution or swelling behavior, drug compatibility, residual moisture, and storage stability.
For OA, polymer choice must account for the poor aqueous compatibility of the payload [1,5]. Hydrophilic microneedle polymers may dissolve or swell well, but they may not solubilize OA efficiently on their own. Direct loading of crystalline OA into a hydrophilic needle matrix could lead to poor dose uniformity, slow or incomplete release, and unpredictable skin deposition. A nanocarrier-loaded design may therefore be more rational than direct drug loading [7,9,12,14,17,18].
Hyaluronic acid is especially relevant because HA-based OA nanoprodrugs have already been explored for topical psoriasis delivery [12]. Incorporating HA-based OA systems into dissolving or hydrogel-forming microneedles could provide a future strategy that combines polymer-mediated skin interaction, microneedle deposition, and responsive release. However, the degree of substitution, molecular weight, mechanical strength, dissolution rate, and repeated-dose skin tolerability would all require careful control [12,16,36,37,42,43].
Chitosan and other cationic polymers may also be useful because they can interact with biological membranes and may provide mucoadhesive or antimicrobial properties. However, cationic polymers can also increase irritation or cytotoxicity, which is important for OA systems intended for inflamed or damaged skin [16,36,37,42]. Polymer selection should therefore balance mechanical performance with skin compatibility and formulation stability.
9.4. Nanocarrier-Loaded Microneedles
Nanocarrier-loaded microneedles are one of the most logical microneedle strategies for OA. The nanocarrier addresses OA solubility, dispersion, physical state, and release, while the microneedle enables deposition into or across the skin barrier [7,9,12,14,16,17,18,42]. This combined approach is more appropriate than attempting to load poorly soluble crystalline OA directly into a dissolving polymeric needle.
Potential OA-compatible carriers include PLGA nanoparticles, PEGylated PLA/PLGA nanoparticles, polymeric micelles, cyclodextrin complexes, hyaluronic-acid-based nanoprodrugs, nanogels, and polymer-assisted lipid systems [5,7,9,12,14,17,18]. Each carrier type would behave differently after microneedle insertion. Polymeric micelles may release OA relatively quickly after dilution, whereas PLGA nanoparticles, nanogels, or depot-forming systems may support slower release [7,9,14,17,18].
Embedding nanocarriers into microneedles adds another processing step and another source of variability. Casting, molding, drying, humidity, polymer concentration, backing-layer formation, and mechanical compression can affect both microneedle geometry and nanocarrier stability [16,36,37,42]. For OA-loaded carriers, these steps may also change drug crystallinity, aggregation, release rate, or carrier recovery [1,5,7,9,12,14,17,18].
Therefore, nanocarrier-loaded OA microneedles should be characterized at several stages: before incorporation into the microneedle matrix, after drying and storage, after insertion or dissolution, and after release from the matrix. Important quality attributes include microneedle geometry, mechanical strength, insertion depth, dissolution or swelling behavior, nanocarrier recovery, OA content uniformity, release kinetics, residual moisture, and storage stability [16,36,37,42].
9.5. Microneedles for Inflammatory Skin Diseases and Psoriasis
Inflammatory skin diseases, including psoriasis, are among the most plausible targets for OA microneedle delivery. OA has reported anti-inflammatory activity, and HA-based OA nanoparticles have already been developed for topical psoriasis treatment [1,5,12]. Microneedles could strengthen this direction by delivering OA-loaded carriers into viable skin layers and reducing reliance on passive penetration through the stratum corneum [12,16,36,37,42].
For psoriasis and related inflammatory skin diseases, microneedles may offer several advantages: barrier bypass, localized deposition, reduced need for chemical penetration enhancers, and the possibility of patient-friendly patch-based administration [16,36,37,42,43]. These features are especially relevant for hydrophobic compounds such as OA, which do not readily cross the skin barrier in free form [1,5,12].
However, inflamed skin is not equivalent to healthy skin. Barrier disruption, immune-cell infiltration, altered cytokine expression, increased vascularity, scaling, and local sensitivity can all influence microneedle insertion, drug deposition, and tolerability. OA microneedle systems for psoriasis should therefore be evaluated in disease-relevant skin models rather than only in healthy excised skin, synthetic membranes, or simple release media [12,16,43].
A realistic development pathway would compare free OA gel, OA micelles, HA-based OA nanoparticles, dissolving microneedles containing OA carriers, and nanocarrier-loaded hydrogel-forming microneedles in the same or comparable skin models. Such comparisons would clarify whether microneedles provide added value beyond improved solubilization [12,16,17,42,43].
9.6. Microneedles for Wound Healing and Tissue Repair
Microneedle systems may also be relevant to wound healing and tissue repair, but this application requires a cautious frame. OA has been associated with wound-healing activity, epithelial cell migration, anti-inflammatory effects, and oxidative-stress modulation [1,5,26,28,40]. However, wounds are dynamic environments, and the timing, dose, depth, and location of delivery are critical.
Direct microneedle application to open wounds may not always be appropriate because damaged tissue can be painful, fragile, infected, highly inflamed, or irregular in structure [40]. OA microneedle systems may be more suitable for periwound skin, chronic wound edges, scar modulation, or controlled intradermal delivery adjacent to damaged tissue [16,40,42]. For open wound surfaces, hydrogels, fiber membranes, and nanocarrier-loaded dressings may be more appropriate than microneedle patches [8,17,18,40].
If OA microneedles are developed for wound-related applications, the polymeric matrix must be designed for biocompatibility, moisture control, minimal irritation, and appropriate mechanical behavior [16,36,37,40,42]. The formulation should also consider whether OA release supports migration, reduces excessive inflammation, or affects remodeling in a stage-specific manner. Excessive or poorly timed delivery could be counterproductive if it interferes with normal inflammatory or proliferative phases of healing.
Relevant evaluation endpoints should include keratinocyte and fibroblast viability, cell migration, inflammatory cytokines, oxidative-stress markers, collagen deposition, wound closure, scar-related markers, infection risk, and tissue histology [26,28,40]. These outcomes are more informative than release data alone because they connect OA deposition to tissue repair.
9.7. Microneedles and OA Penetration-Enhancer Logic
A distinct but relevant part of OA transdermal research concerns OA derivatives as penetration enhancers. Bednarczyk-Cwynar et al. synthesized simple OA amides and evaluated them as potential transdermal penetration enhancers. This work is not a polymeric nanocarrier study, but it shows that OA-based structures can interact with skin permeation processes [44].
This literature supports the broader idea that OA and its derivatives may be relevant to skin delivery not only as therapeutic payloads, but also as molecules capable of modifying barrier interaction. In the context of polymeric microneedles, this raises an interesting possibility: OA derivatives or OA-loaded carriers could be designed to combine therapeutic activity with controlled skin interaction [16,42,44].
This possibility should be treated carefully. Penetration enhancement may increase permeation, but it can also disturb barrier function or increase irritation risk. In inflamed skin, psoriasis, or wound-associated tissue, barrier integrity may already be compromised, making safety evaluation particularly important [16,43,44].
For OA microneedle systems, physical barrier bypass through microneedles may provide a more controllable strategy than aggressive chemical penetration enhancement alone. The most balanced approach may combine shallow, localized microneedle deposition with polymeric control over OA release and carrier stability [16,36,37,42,44].
9.8. Controlled Release from Polymeric Microneedles
Controlled release from OA-loaded microneedles can be engineered through polymer dissolution, hydrogel swelling, nanocarrier release, polymer degradation, drug diffusion, and carrier–matrix interactions. This multilevel control is attractive because OA release can be regulated by both the microneedle matrix and the embedded carrier [7,9,12,14,16,17,18,36,37,42].
For dissolving microneedles, release may occur rapidly after insertion if the polymeric matrix dissolves quickly [16,36,37,42]. This may be useful for short-term local delivery but may not sustain OA exposure. Incorporating PLGA nanoparticles, nanogels, or other sustained-release carriers into dissolving microneedles could create a two-step system: rapid needle dissolution followed by slower OA release from the deposited nanocarrier [7,9,14,17,18].
For hydrogel-forming microneedles, the swollen polymer network can act as a conduit or controlled-release interface. These systems could be paired with OA reservoirs, nanoparticle dispersions, micellar systems, or soft polymeric carriers to support longer delivery periods. The key challenge is ensuring that OA or the OA-loaded carrier partitions effectively from the reservoir through the hydrogel network into the skin.[7,9,12,14,16,17,18,36,37,42].
Release testing should match the microneedle mechanism. Simple immersion of a patch in buffer may not reflect skin insertion, swelling pressure, interstitial-fluid composition, polymer dissolution, or local tissue uptake [16,36,37,42]. OA microneedle studies should therefore include skin-mimicking membranes, ex vivo skin, reconstructed skin, or disease-relevant skin systems where possible [12,16,42,43].
9.9. Manufacturing and Quality Attributes
Polymeric microneedles introduce manufacturing and quality-control requirements that go beyond conventional nanocarrier characterization. Important attributes include needle height, base width, tip sharpness, array density, mechanical strength, insertion efficiency, dissolution or swelling time, dose uniformity, residual moisture, and packaging stability [16,36,37,42].
For OA-loaded microneedles, additional quality attributes include OA loading, drug distribution within the needles, nanocarrier stability after drying, crystallization risk, release profile, and skin deposition [1,5,7,9,12,14,17,18]. If OA-loaded nanoparticles or micelles are embedded in the microneedle matrix, the carrier should be characterized before incorporation, after drying, after storage, and after release from the dissolved or swollen patch [7,9,12,14,17,18].
Manufacturing methods such as micromolding, casting, centrifugation-assisted filling, vacuum-assisted filling, drying, and backing-layer formation can affect payload distribution and mechanical performance [16,36,37,42]. For OA, drying conditions are especially important because solvent removal and water loss may promote drug crystallization, carrier aggregation, or changes in release behavior [1,5,7,9,12,14,17,18].
Scale-up is a major translational issue. Microneedle patches must be produced with reproducible geometry, mechanical strength, microbial control or sterility when required, and dose consistency [16,36,37,42]. For OA systems intended for clinical use, scalable fabrication and packaging are as important as biological activity because inconsistent microneedles may lead to variable dose delivery and tissue exposure.
9.10. Safety and Patient-Oriented Considerations
Microneedle systems are often described as minimally invasive and patient-friendly, but safety and acceptability must still be demonstrated. Potential concerns include local irritation, erythema, incomplete insertion, broken tips, infection risk, polymer residue, allergic reactions, and dose variability. These concerns are especially relevant for chronic skin diseases, where repeated application may be required [16,36,37,42,43].
For OA-loaded microneedles, blank microneedle controls are essential because the polymeric matrix itself can influence skin response [16,36,37,42]. If nanocarriers are included, blank nanocarrier-loaded microneedles should also be tested. This is necessary to distinguish OA effects from polymer effects, nanocarrier effects, hydration, occlusion, mechanical puncture, and local inflammatory responses [7,9,12,14,16,17,18,36,37,42].
Patient-oriented factors also matter. A microneedle OA patch for chronic inflammatory skin disease would need to be easy to apply, stable during storage, acceptable on visible skin areas, minimally painful, non-irritating, and compatible with repeated use [16,36,37,42,43]. If microneedles are intended for periwound or scar-related applications, comfort, dressing compatibility, infection risk, and ease of use become additional design constraints [16,36,37,42].
9.11. Translational Positioning of OA Microneedle Systems
OA microneedle systems should be positioned as an emerging direction rather than as an established formulation class. The rationale is strong: OA is poorly soluble, passive skin delivery is limited, polymeric nanocarriers can improve dispersion and release, and microneedles can bypass the stratum corneum [1,5,7,9,12,14,16,17,18,36,37,42]. However, direct OA-loaded microneedle studies remain needed to confirm feasibility.
The strongest rationale is local treatment of skin inflammation, psoriasis, localized dermatitis, scar-related conditions, and possibly periwound tissue modulation [12,16,40,41,42]. In these settings, microneedles may add value if they improve local deposition, reduce the need for chemical enhancers, provide reproducible dosing, and maintain acceptable skin tolerability [16,36,37,42,43].
Comparative studies will be important. Free OA gel, OA micelles, OA nanoparticles, OA-loaded dissolving microneedles, and OA nanocarrier-loaded microneedles should ideally be compared in the same skin or disease-relevant model [12,16,17,42,43]. Such studies would show whether microneedles provide genuine added value beyond improved solubilization.
9.12. Translational Positioning
Polymeric microneedles provide a compelling but still emerging route for OA delivery. Their value lies in combining physical skin-barrier bypass with polymeric control of drug deposition and release [16,36,37,42]. For a poorly soluble compound such as OA, the most rational strategy is likely to involve nanocarrier-loaded dissolving or hydrogel-forming microneedles rather than direct loading of crystalline drug [1,5,7,9,12,14,17,18].
The most plausible applications are local and skin-oriented: inflammatory skin disease, psoriasis, localized dermatitis, scar-related delivery, and possibly periwound tissue modulation [12,16,26,28,40,43]. OA-loaded microneedles should be evaluated through route-specific criteria, including mechanical insertion, skin deposition, release kinetics, irritation, barrier recovery, inflammatory markers, dose uniformity, patient acceptability, and comparison with existing topical or local therapies [12,16,36,37,42,43].
At present, OA microneedle delivery should be presented as a future-oriented polymeric platform. The field has strong conceptual support from microneedle technology and OA nanocarrier research, but direct OA microneedle formulations still need to demonstrate feasibility, safety, reproducibility, and added value over simpler topical or local systems[1,5,7,9,12,14,16,17,18,36,37,42].
The route-specific positioning of local, transdermal, intra-articular, oral, and systemic OA delivery platforms is summarized in Figure 4.
Local, transdermal, and microneedle-compatible OA delivery platforms are compared in Table 5.
10. Characterization and Critical Quality Attributes of Oleanolic-Acid-Loaded Polymeric Nanocarriers
10.1. Why Characterization is Central for OA Polymeric Nanocarriers
For OA-loaded polymeric and polymer-assisted nanocarriers, characterization is not a supplementary technical step. It determines whether a formulation can be interpreted scientifically and whether it has any realistic development value. OA is poorly soluble, lipophilic, crystalline or semi-crystalline, and strongly formulation-dependent; therefore, small differences in carrier composition, preparation method, drug physical state, and release environment may substantially change biological response [1,5,7,8,9].
A formulation should not be considered successful only because OA can be incorporated into a nanosystem. The more important question is whether the system can be reproduced, stored, characterized, and shown to release OA in a form relevant to the intended route [5,6,7,8,9,13]. This is especially important for polymeric carriers because polymer molecular weight, degradation behavior, surface chemistry, matrix architecture, and drug–polymer compatibility can all affect performance [6,7,8,9,13].
Critical quality attributes should therefore be defined according to the target product profile. An oral polymeric nanoparticle, a topical micellar system, a wound dressing, an intra-articular thermosensitive gel, and a microneedle-loaded nanocarrier do not require identical performance criteria [8,12,18,36,37]. However, all require a rational connection between composition, structure, OA physical state, release behavior, stability, safety, and biological response [6,7,8,9,13].
The characterization workflow and critical quality attributes for OA-loaded polymeric and polymer-assisted systems are summarized in Figure 5.
For OA, characterization must also distinguish total drug content from biologically available drug. A system may contain a high amount of OA but release it too slowly, retain it within a hydrophobic matrix, or contain crystalline drug domains that are poorly available under biological conditions [1,5,7,8,9]. For this reason, OA nanocarrier evaluation should combine conventional nanocarrier characterization with compound-specific assessment of physical state, release, and exposure [1,5,6,7,8,9,13].
10.2. Particle Size, Size Distribution, and Polydispersity
Particle size is one of the most commonly reported parameters for polymeric nanocarriers because it influences colloidal behavior, tissue penetration, cellular uptake, biodistribution, clearance, and release kinetics [6,7,13]. For OA delivery, size can also affect surface area available for release and the likelihood that the compound remains dispersed rather than precipitated [1,5,7,8,9].
Dynamic light scattering is widely used to measure hydrodynamic diameter and polydispersity index, but DLS data require careful interpretation. The method is sensitive to aggregates, large particles, and mixed populations because it provides intensity-weighted measurements that may overrepresent larger species [6,13]. In OA-loaded systems, undetected aggregates, drug crystals, or carrier clusters may distort apparent size and affect biological performance [1,5,7,8,9].
Polydispersity is particularly important for development. A narrow size distribution suggests better process control, whereas broad or multimodal distributions may indicate uncontrolled nucleation, aggregation, mixed carrier populations, or incomplete stabilization [7,13]. For OA, high polydispersity may also reflect coexistence of nanocarriers with free drug aggregates, especially when drug loading exceeds the capacity of the carrier matrix [1,5,7,8,9].
Particle-size analysis should therefore be supported by complementary methods where possible. Nanoparticle tracking analysis, electron microscopy, atomic force microscopy, or field-flow fractionation can help distinguish true nanoscale carriers from aggregates, micelles, fibers, vesicular structures, or gel-associated particles [6,7,12,13,18]. This is particularly important for hybrid OA systems that contain both nanoscale carriers and polymeric matrices [8,15]
10.3. Surface Charge and Zeta Potential
Zeta potential is frequently used as an indirect indicator of colloidal stability and surface properties. For OA-loaded polymeric nanocarriers, surface charge may influence aggregation, protein adsorption, interaction with mucus or skin, cellular uptake, and compatibility with biological fluids [6,7,13].
However, zeta potential should not be treated as a universal predictor of stability or biological performance. Sterically stabilized systems, such as PEGylated nanoparticles or polymeric micelles, may show relatively low absolute zeta-potential values but remain stable because of their hydrated polymeric surface layers [6,7,13,17]. Conversely, a high zeta potential measured in simple buffer does not guarantee stability in serum, wound exudate, synovial-like media, or skin fluids [12,13,15].
Surface properties are especially important when polymers such as PEG, hyaluronic acid, chitosan, poloxamers, or other functional excipients are used. These materials can modify hydration, protein adsorption, receptor interaction, tissue retention, and cellular uptake [7,12,13,15,17]. For this reason, zeta potential should be interpreted together with stability in relevant media, surface composition, release behavior, and biological response.
A route-relevant overview of critical quality attributes and recommended characterization methods is provided in Table 4.
10.4. Morphology and Carrier Architecture
Morphological characterization is essential because carrier architecture can strongly affect OA loading, release, stability, and biological interaction. Microscopy can help determine whether a formulation contains spherical nanoparticles, polymeric micelles, liquid crystalline particles, nanogels, fibers, vesicle-like structures, aggregates, or mixed populations [8,9,12,15,18,37].
Electron microscopy provides direct visual information on shape, surface structure, aggregation, and size distribution. Transmission electron microscopy and scanning electron microscopy are particularly useful for nanoparticles and fiber membranes, whereas cryogenic electron microscopy may be valuable for soft or hydrated systems when available [6,7,13]. For OA-loaded PLGA fiber membranes, morphology is especially important because fiber diameter, porosity, and surface structure can influence drug release and tissue contact [8].
For hydrogels, nanogels, and thermosensitive depots, morphology must be linked with macroscopic behavior. A gel-based OA formulation cannot be characterized only by nanoparticle size because gel network structure, rheology, swelling, and depot integrity also determine release and residence time [15,18,37]. This is particularly relevant for topical and intra-articular systems, where the formulation must remain at the application site.
Morphological analysis can also reveal formulation failure. If OA crystallizes outside the carrier or forms large aggregates, average DLS values may still suggest nanoscale size while the formulation contains poorly controlled drug domains. Morphology should therefore be interpreted together with solid-state analysis, drug loading, and release behavior [1,5,6,7,8,9,13].
10.5. Drug Loading and Encapsulation Efficiency
Drug loading and encapsulation efficiency are central parameters for OA nanocarriers because they describe how much compound is present in the final system and how efficiently the preparation process incorporates OA [1,5,7]. However, these values are not sufficient on their own to establish formulation quality.
For hydrophobic compounds such as OA, apparent encapsulation may include several drug populations: OA molecularly dispersed within the polymer matrix, OA associated with the carrier surface, OA trapped in amorphous domains, OA contained in hydrophobic micellar or lipidic regions, and crystalline or aggregated OA that remains after purification [1,5,7,8,9]. These populations may behave differently during storage, dilution, release testing, and biological exposure.
OA nanocarrier studies should therefore distinguish, where possible, between total OA content, encapsulated OA, surface-associated OA, free or precipitated OA, and released OA [7,8,9,13]. This is especially important when comparing different polymers, preparation methods, stabilizers, or dosage forms. Two systems with similar encapsulation efficiency may have very different release profiles and biological effects if OA is present in different physical states.
Validated analytical methods are essential. HPLC or LC–MS methods should quantify OA after complete extraction and after separation of free drug from the carrier fraction [8,12]. The extraction method should be able to recover OA from polymeric matrices, hydrogels, micelles, fibers, or microneedle materials without degradation or incomplete release from the sample [8,12,16,18]. Drug loading data should also be interpreted alongside solid-state analysis and release testing.
10.6. Physical-State Assessment
Physical-state assessment is particularly important for OA because the compound is poorly soluble and may exist in crystalline, semi-crystalline, amorphous, molecularly dispersed, surface-associated, or phase-separated forms [1,5,7,8,9]. These states can strongly influence release, storage stability, precipitation after dilution, and biological exposure.
A high-loading OA system may be weak from a formulation standpoint if excess drug crystallizes during preparation, drying, storage, or application [1,5,14,20]. This risk is relevant for PLGA nanoparticles, electrospun fiber membranes, polymeric micelles, dried microneedles, nanocarrier-loaded gels, and thermosensitive depots [8,9,15,16,17,18]. Loading must therefore be evaluated together with physical state, not separately from it.
Differential scanning calorimetry, powder X-ray diffraction, Fourier-transform infrared spectroscopy, Raman spectroscopy, polarized-light microscopy, and electron microscopy can help determine whether OA is crystalline, amorphous, molecularly dispersed, or phase-separated [8,9,13]. For hydrated systems such as hydrogels or nanogels, sample preparation may alter the formulation, so complementary methods and careful interpretation are required [18,37].
Physical-state assessment should also be included in stability studies. OA may be initially dispersed within a carrier but recrystallize during drying, lyophilization, humidity exposure, temperature cycling, or long-term storage [1,5,7,8,9]. Monitoring drug state over time is therefore more informative than measuring it only immediately after preparation.
10.7. Release Testing and Exposure Verificatio
Release testing is one of the most important characterization steps for OA-loaded polymeric systems. Because OA is poorly soluble and hydrophobic, release results can vary substantially depending on sink conditions, surfactant concentration, protein content, medium composition, agitation, membrane selection, and separation method [1,5,7,8,9,13]. A release method that is convenient analytically may not reflect the biological environment of the intended route.
Simple aqueous buffer may underestimate OA release if sink conditions are not maintained. In contrast, excessive surfactant or organic cosolvent may overestimate the fraction that would be biologically available in vivo [1,5,7,8,9]. Release media should therefore be selected according to the route and justified mechanistically.
Route relevance is essential. Skin-oriented systems may require skin-compatible receptor media, ex vivo deposition studies, reconstructed skin, or permeation models [12,16,17,18]. Wound systems should consider wound-exudate-like conditions, including proteins, salts, enzymes, and pH variation [7,28,39,40,41]. Intra-articular depots should consider synovial-fluid-like environments and prolonged residence [15]. Oral systems should be tested under biorelevant gastrointestinal conditions [1,5]. Systemic nanoparticles may require protein-containing media and dilution stability testing [7,9,13].
Exposure verification is equally important. Biological activity should not be interpreted only from the nominal OA concentration added to cells, tissues, or animals. Whenever possible, OA should be measured in release medium, cells, skin layers, wound fluid, synovial-like compartments, or tissue extracts [15,16,18,37]. This helps determine whether the carrier changes actual OA availability or only the nominal dose.
10.8. Stability During Storage and Biological Dilution
Stability studies are essential for OA-loaded polymeric systems because both the drug and the carrier can change over time. Relevant instability pathways include aggregation, drug crystallization, polymer degradation, micelle dissociation, gel syneresis, drug leakage, water uptake, drying-related structural changes, and loss of mechanical integrity [1,5,7,8,9,13,15,17,18].
For nanoparticles, stability should include size, PDI, zeta potential, aggregation, drug loading, release profile, and drug physical state after storage [7,8,9,13]. For hydrogels and thermosensitive depots, rheology, gelation behavior, phase separation, water loss, and release should be monitored [15,18]. For fiber membranes and microneedles, mechanical integrity, residual moisture, drug distribution, and dose uniformity are especially important [8,15,16,36].
Stability conditions should reflect the intended product form. A freshly prepared laboratory dispersion, a lyophilized powder, a dried microneedle patch, a hydrated gel, and a packaged wound dressing have different risks [8,12,15,36,37]. For OA, humidity and temperature may be particularly important because they can influence crystallization, polymer relaxation, and release behavior [1,5,8,9].
10.9. Rheology and Mechanical Properties of Gels, Dressings, and Microneedles
Many OA-loaded polymeric nanocarriers are prepared using organic solvents, nanoprecipitation, emulsification, electrospinning, or casting methods [7,8,9,16]. Residual solvents can influence safety, polymer behavior, drug crystallization, and biological response. They should therefore be measured and controlled, especially in systems intended for local, topical, intra-articular, or transdermal use [7,8,9,13,16].
OA raw material quality should also be considered. Purity, polymorphic form, residual solvents, impurity profile, botanical or synthetic origin, and storage conditions may affect loading, crystallization, and release [1,5]. In polymer-assisted systems, excipients such as PEGylated polymers, hyaluronic acid, poloxamers, cyclodextrins, chitosan, surfactants, and hydrogel-forming polymers may also contribute impurities, degradation products, or batch variability [12,15,17,18].
Chemical stability should be monitored for both OA and the carrier. Polymer degradation may change pH, release behavior, and drug stability, while responsive linkages such as disulfide bonds may degrade under storage or biological conditions [12,13,15]. Stability-indicating analytical methods are therefore preferable to simple content assays whenever the formulation is intended for development beyond proof of concept.
10.10. Route-Specific Critical Quality Attributes
Critical quality attributes should be route-specific. For oral polymeric OA systems, relevant attributes include particle size, dispersion after dilution, gastrointestinal stability, precipitation risk, release in biorelevant media, and potential interaction with food, bile salts, or digestive components [1,5]. For systemic injectable systems, sterility, endotoxin burden, colloidal stability in serum, protein interaction, biodistribution, and safety become central [7,9,13].
For topical and dermal systems, the critical attributes include spreadability, skin deposition, permeation, irritation, barrier compatibility, microbial quality, and release under skin-relevant conditions [12,17,18]. For wound dressings, additional attributes include moisture behavior, exudate interaction, mechanical flexibility, porosity, adhesion, microbial risk, and compatibility with damaged tissue [8,28,39,40,41].
For intra-articular depots, the main attributes include injectability, gelation temperature, viscosity, retention time, sterility, endotoxin control, release in synovial-like media, local tolerability, and cartilage or synovial compatibility [15]. For microneedle systems, critical attributes include needle geometry, mechanical strength, insertion efficiency, dose uniformity, dissolution or swelling behavior, residual moisture, skin deposition, and patient acceptability [16,36,37].
This route-specific approach prevents overreliance on generic nanocarrier metrics. Particle size and encapsulation efficiency are useful, but they do not define performance for a gel, dressing, depot, or microneedle patch unless they are connected to the intended route and biological objective [8,12,15,16,18].
10.11. Biological Controls and Assay Interpretation
Biological evaluation is part of characterization for OA systems because the compound is strongly vehicle-dependent [1,5,14]. A stronger response from an OA-loaded carrier may result from improved dispersion, reduced precipitation, altered uptake, carrier-associated effects, or changed release kinetics rather than a change in intrinsic OA activity [7,9,12,14].
Appropriate controls are therefore essential. Studies should include blank carrier, free OA, solubilized OA, OA-loaded carrier, and, where relevant, carrier-in-matrix controls [7,8,9,12,14,15,16,17,18]. For responsive systems, non-responsive analogues or controls without cleavable linkages may help clarify whether the stimulus-responsive mechanism contributes to biological activity [12].
Assays should also match the intended application. Cancer-oriented systems require uptake, penetration, cytotoxic selectivity, and eventually three-dimensional or in vivo models [7,9]. Skin and psoriasis systems require keratinocyte, immune-cell, reconstructed skin, irritation, and deposition endpoints [12,16,17,18]. Wound systems require migration, inflammatory markers, oxidative stress, matrix deposition, and tissue repair endpoints [8,28,39,40,41]. Intra-articular systems require cartilage, synovium, inflammation, pain-related behavior, and local toxicity assessment [15].
Without these controls and application-relevant endpoints, it is difficult to determine whether an OA-loaded polymeric system has meaningful biological value or only produces a formulation-dependent assay effect.
10.12. Reproducibility, QbD Logic, and Minimum Characterization Set
Reproducibility is a major challenge for polymeric OA nanocarriers because preparation methods are sensitive to polymer batch, solvent composition, mixing rate, temperature, concentration, drying, and post-processing conditions. For OA, small process variations may change particle size, loading, drug physical state, release, and apparent biological activity [1,5,6,7,8,9,13].
A quality-by-design-oriented approach would be valuable for OA formulation development. Instead of optimizing one variable at a time, studies should define critical material attributes, critical process parameters, and critical quality attributes, then evaluate how formulation variables affect performance. This is especially relevant for PLGA nanoparticles, electrospun membranes, nanocarrier-loaded hydrogels, thermosensitive depots, and microneedle systems [6,7,8,9,12,13,15,18,26,37].
A minimum characterization set for OA-loaded polymeric nanoparticles should include particle size, size distribution, zeta potential, morphology, drug loading, encapsulation efficiency, physical-state assessment, release kinetics, storage stability, and biological controls [1,5,6,7,8,9,13]. For polymer-assisted gels, dressings, depots, and microneedles, additional attributes should include rheology, mechanical properties, dose uniformity, gelation or dissolution behavior, application performance, and route-specific safety [8,12,15,16,18,40,41].
At least one direct imaging method should complement DLS-based size analysis for nanoparticle systems. For fibers and microneedles, microscopy should confirm morphology, drug distribution, and structural integrity. For gels and depots, rheology and gelation behavior should be connected to release and residence-time expectations [8,15,18,36,37].
Release testing should be justified by the intended application rather than selected only for convenience. A method suitable for a PLGA nanoparticle suspension may not be appropriate for a topical gel, wound dressing, intra-articular depot, or microneedle patch [8,12,15,16,18]. Biological evaluation should similarly include both formulation controls and application-relevant models.
10.13. Characterization Priorities
Characterization of OA-loaded polymeric and polymer-assisted nanocarriers must go beyond routine particle size and encapsulation efficiency. Because OA is poorly soluble, lipophilic, and formulation-dependent, critical quality attributes should include drug physical state, release behavior, stability, exposure verification, carrier safety, and route-specific performance [1,5,6,7,8,9,13].
The central question is whether polymeric design produces reproducible and route-appropriate OA exposure. A high-loading nanoparticle, gel, dressing, depot, or microneedle system has limited value if OA crystallizes, releases unpredictably, destabilizes during storage, or produces biological effects that cannot be separated from carrier artifacts.
For OA, the most useful characterization strategy is a process–structure–function approach. Polymer composition and manufacturing conditions should be linked with carrier architecture, OA physical state, release kinetics, tissue exposure, biological response, and safety. This approach will make OA delivery studies more useful not only for this compound, but also for other poorly soluble natural products developed through polymeric nanocarrier systems [6,7,8,9,12,13,15,18,22,29,37,40].
11. Controlled Release and Mechanistic Delivery Considerations
11.1. Controlled Release as the Central Value of Polymeric OA Systems
Controlled release is one of the main reasons to develop polymeric and polymer-assisted nanocarriers for OA. Improving apparent solubility is necessary, but it is not sufficient. For OA, biological performance depends on how much drug becomes available, where it becomes available, and for how long exposure is maintained. A formulation that disperses OA transiently but releases it too quickly, too slowly, or unpredictably may have limited value despite favorable loading or particle-size data [1,5,7,8,9].
Polymeric systems can regulate OA availability through several mechanisms, including matrix diffusion, polymer swelling, hydrolytic degradation, erosion, micelle dissociation, gel relaxation, prodrug cleavage, responsive linker cleavage, and carrier–tissue interaction [12,14,15,16,17,18]. These mechanisms are especially relevant for a hydrophobic compound such as OA, which may partition into polymeric, lipidic, protein-rich, or tissue-associated environments rather than behave as a freely dissolved drug [1,5,7].
The release objective should be defined by the intended application. For oral delivery, release must support absorption in gastrointestinal conditions. For cancer-oriented nanoparticles, release should support cellular or intracellular exposure. For skin delivery, release should favor deposition in relevant skin layers. For wound dressings, release should match the tissue-repair environment. For intra-articular depots, release should maintain local joint exposure over time [7,8,9,12,14,15,16,17,18,37].
Controlled release should therefore not be treated as a generic formulation advantage. In OA delivery, the same release profile may be useful for one indication and unsuitable for another. The key question is whether the polymeric system produces a route-appropriate exposure pattern for the selected tissue and disease context [7,8,9,12,14,15,16,17,18,37].
11.2. Matrix Diffusion and Degradation in PLA/PLGA-Based OA Nanoparticles
PLA and PLGA nanoparticles release encapsulated drugs through overlapping mechanisms, including diffusion through the polymer matrix, water penetration, polymer swelling, pore formation, hydrolytic degradation, matrix erosion, and drug partitioning into the surrounding medium [1,5,7,9,31,45,46]. For OA-loaded PLA/PLGA systems, these mechanisms are further complicated by the strong hydrophobicity and poor aqueous solubility of the payload [1,5,7,9].
Early release may occur when OA is located near the particle surface, associated with loosely bound domains, or incompletely incorporated into the polymer matrix. A moderate initial release may be useful when rapid local exposure is needed, but excessive burst release can shorten the duration of action and increase local toxicity or irritation risk [31,45,46]. For OA, burst release may also indicate a formulation problem, such as surface-associated crystalline drug or incomplete purification, rather than a deliberate release design [1,5,7,9].
Sustained release is more likely when OA is distributed within the polymeric matrix and diffuses gradually as water penetrates the carrier and the polymer relaxes or degrades [7,9]. Polymer molecular weight, lactide ratio, end-group chemistry, particle size, drug loading, and drug physical state can all influence this process [13,31,45,46]. These variables should be interpreted together rather than as isolated formulation parameters.
For OA, a slow release profile is not automatically better. If the drug remains trapped in a hydrophobic matrix or crystalline domain, sustained release may become incomplete release. A well-designed PLA/PLGA OA system should therefore balance matrix retention with biologically useful drug availability [1,5,7,8,9,13].
11.3. Polymer Variables that Control OA Release
Polymer composition is a major determinant of release. In PLGA systems, a higher glycolide content generally increases hydrophilicity and may accelerate water uptake and degradation, whereas a higher lactide content usually increases hydrophobicity and may prolong release [12,13,31,46]. For OA, this balance is important because the compound itself is highly hydrophobic and may associate strongly with lactide-rich domains [1,5,7,9].
Polymer molecular weight also affects release. Lower-molecular-weight polymers may degrade faster and release drug more rapidly, whereas higher-molecular-weight polymers may form denser or more stable matrices with slower release [12,25,31,35]. The optimal molecular weight depends on whether the intended application requires rapid local exposure, prolonged tissue residence, or gradual systemic availability [14,15,16,18,37].
End-group chemistry can influence hydration, degradation, and drug–polymer interaction. Acid-terminated PLGA may promote water uptake and faster degradation compared with ester-capped PLGA, whereas more hydrophobic end groups may slow hydration and diffusion [12,29,35,39]. For a poorly soluble acidic triterpenoid such as OA, end-group chemistry may also affect the local microenvironment within the carrier and the strength of drug–polymer association [1,5,7,9].
Polymer blending and copolymer modification can provide additional release-control options. Blending PLGA with PEG, poloxamers, hyaluronic acid, chitosan, cyclodextrin-containing polymers, or hydrogel-forming materials may alter hydrophilicity, swelling, degradation, mucoadhesion, skin interaction, and release kinetics [12,13,14,15,17,18,33,34]. Such modifications should have a clear mechanistic purpose. Additional polymers should not be added only to make the formulation appear more advanced.
11.4. Drug Physical State and Burst Release
The physical state of OA inside a polymeric system strongly influences release. OA may be molecularly dispersed, amorphous, crystalline, surface-associated, trapped in hydrophobic domains, or present in phase-separated regions [1,5,7,9]. These states can produce very different release behaviors even when total drug loading appears similar.
Molecularly dispersed or amorphous OA may release more readily than crystalline OA, but it may also be less stable during storage. Crystalline OA may release slowly and incompletely because dissolution becomes the rate-limiting step. Surface-associated OA may produce rapid initial burst release, whereas deeply matrix-associated OA may remain unavailable for extended periods [1,5,7,8,9,13].
This issue is especially important for high-loading formulations. Increasing OA content may improve dose capacity, but once the carrier matrix is saturated, excess OA may crystallize or separate into drug-rich domains [1,5,8,9]. Such systems may show attractive loading values but poor release control and weak reproducibility.
Solid-state characterization should therefore be linked directly to release interpretation. Differential scanning calorimetry, X-ray diffraction, Raman spectroscopy, Fourier-transform infrared spectroscopy, and microscopy can help determine whether OA is crystalline, amorphous, molecularly dispersed, or interacting with the polymer matrix [8,9,13]. Without this information, release profiles may be difficult to interpret mechanistically.
11.5. Release from Soft and Responsive Polymeric Systems
Soft and responsive polymeric systems release OA through mechanisms that differ from rigid nanoparticle matrices. Polymeric micelles may release OA through dilution, unimer exchange, micelle dissociation, diffusion from the hydrophobic core, or partitioning into skin, proteins, or lipidic environments. For dermal OA delivery, micellar release must be balanced: the micelle should retain OA long enough to improve deposition, but release enough drug to support biological activity [17,30].
Nanogels and hydrogels can release OA through swelling, diffusion through hydrated polymer networks, erosion, degradation, or release of embedded nanocarriers [15,18,33]. Because OA is lipophilic, direct release from a hydrophilic gel may be inefficient unless the system contains hydrophobic domains, cyclodextrin complexes, lipidic nanostructures, micelles, or nanoparticles [12,15,17,18,33]. This makes carrier-in-gel systems particularly relevant for OA.
Responsive systems add another layer of control. Redox-responsive hyaluronic-acid-based OA nanoprodrugs can release OA through disulfide cleavage in reductive environments, while thermosensitive gels can regulate local residence through temperature-dependent gelation [12,15]. pH- or enzyme-responsive systems may also be useful in inflamed skin, wounds, tumors, or intracellular environments, but such responsiveness should be mechanistically justified and supported by route-relevant data [12,15,18,33].
11.6. Release from Local Matrices, Depots, and Microneedle-Compatible Systems
Local OA delivery systems often involve more than one release-controlling level. In nanocarrier-in-gel, nanocarrier-in-fiber, nanocarrier-in-dressing, and nanocarrier-in-microneedle systems, both the embedded carrier and the macroscopic polymeric matrix influence release [8,12,15,18,33,36]. This multilevel structure can be valuable, but it also complicates interpretation.
In wound dressings and fiber membranes, OA release may depend on polymer degradation, fiber morphology, porosity, wound fluid penetration, protein binding, pH, enzymes, exudate composition, and tissue contact. The goal is not only to release OA into bulk medium, but to provide local exposure at a tissue surface undergoing inflammation and repair [8,28,39,40,41].
In intra-articular depots, release is linked to gelation, viscosity, joint residence, synovial-fluid interaction, mechanical movement, and clearance from the joint space. The OA thermosensitive nanogel for knee osteoarthritis illustrates this logic because the formulation is designed to maintain local joint exposure rather than maximize systemic absorption [15].
In microneedle-compatible systems, release may involve rapid dissolution of the needle matrix followed by slower release from deposited nanoparticles, micelles, nanogels, or polymeric reservoirs. Release testing for OA microneedles should therefore not rely only on dissolving patches in bulk buffer. More relevant evaluation would include insertion, skin deposition, local retention, recovery of embedded carriers, and release after placement in skin-like environments [16,36,37].
A consolidated comparison of release-control mechanisms across OA polymeric and polymer-assisted systems is provided in Table S1.
11.7. Sink Conditions, Release Media, and Mass Balance
Maintaining sink conditions is a major challenge in OA release testing because the compound is poorly soluble in aqueous media [1,5,7,9]. If the release medium cannot dissolve released OA, release may be underestimated. Conversely, excessive surfactant, ethanol, organic cosolvent, cyclodextrin, albumin, serum protein, lipoprotein, or lipid acceptor may artificially accelerate drug extraction from the carrier and overestimate biologically relevant release [1,5,7,9,14].
Release medium should therefore be chosen according to the intended application. Oral systems require gastrointestinally relevant conditions; topical systems require skin-compatible receptor media, ex vivo skin, reconstructed skin, or deposition models; wound dressings should consider wound-exudate-like media, pH changes, enzymes, salts, and proteins; intra-articular depots require synovial-fluid-like environments and prolonged testing [1,5,8,12,15,16,17,18,28,39,40,41].
For polymeric OA systems, release testing should ideally include mass balance. The amount of OA remaining in the carrier, released into the medium, adsorbed to the apparatus, retained in membranes, precipitated, or deposited in tissue should be quantified when possible [1,5,7,8,9]. This is particularly important because hydrophobic compounds can be lost to plasticware, filters, membranes, proteins, and biological matrices.
11.8. Modeling Release Kinetics
Mathematical modeling can help interpret OA release profiles, but it should be used cautiously. Models such as zero-order, first-order, Higuchi, Korsmeyer–Peppas, Weibull, and diffusion–erosion models can describe release data, but fitting alone does not prove a specific mechanism [7,8,9,31,45,46]. Mechanistic interpretation requires consistency between the model, polymer chemistry, carrier morphology, OA physical state, and experimental conditions.
For OA-loaded PLA or PLGA nanoparticles, release may involve both diffusion and polymer degradation. A Higuchi-type fit may suggest diffusion control, but it may not fully capture systems where water penetration, polymer erosion, matrix swelling, drug dissolution, or crystallinity changes contribute over time [7,8,9,13,31,35,45,46,47]. Modeling should therefore be supported by polymer degradation data, solid-state analysis, morphology, and stability information.
For gels, wound dressings, and microneedles, release modeling is even more complex. Matrix swelling, dissolution, erosion, carrier-in-matrix release, tissue deposition, and changing local fluid conditions can produce multiphase release profiles [12,15,16,18,33,36,37,40]. Empirical models may still be useful for comparing formulations, estimating release half-times, identifying burst release, or supporting quality-by-design studies, but they should not replace mechanistic characterization [7,8,9,13,31,35,45,46,47].
11.9. Connecting Release to Biological Response
For OA polymeric nanocarriers, the most important release question is not simply how much drug leaves the carrier, but whether the released or carrier-associated OA reaches the relevant biological target [1,5,7,9]. OA may act after extracellular release, membrane partitioning, carrier uptake, intracellular release, or local tissue retention. Different carrier designs may therefore produce different exposure pathways [8,12,14,15,16,17,18,36].
In cancer-oriented polymeric nanoparticles, the relevant exposure may involve cellular uptake and intracellular availability [7,9]. In psoriasis-oriented hyaluronic-acid systems, receptor-associated uptake and reduction-responsive release may be central [12]. In topical gels, skin deposition and local anti-inflammatory effects may matter more than systemic absorption [18]. In osteoarthritis depots, joint residence and sustained intra-articular exposure are likely to be key [15]. In wound dressings, local cytokine response, migration, oxidative stress, and tissue repair endpoints may be more informative than cumulative release alone [8,28,39,40,41].
This means that release studies should be paired with biological and analytical exposure measurements. For topical systems, skin deposition and local biomarker changes may be essential. For intra-articular systems, tissue distribution, synovial residence, cartilage protection, and inflammatory markers may be relevant. For wound dressings, migration, cytokines, oxidative-stress markers, and wound closure should be considered [12,15,16,18,39,40,41].
A slower release profile is not automatically better. If release is too slow, OA may remain trapped and biologically inactive. If release is too fast, sustained exposure may be lost. The optimal release profile must be defined by therapeutic objective, tissue environment, dose, safety, and required duration of action [1,5,7,8,9,12,13,14,15,16,17,18].
11.10. Release-Design Implications
Controlled release is the mechanistic bridge between polymeric carrier design and biological performance in OA delivery. Because OA is poorly soluble, lipophilic, and formulation-dependent, release cannot be interpreted only as cumulative percentage released into buffer [1,5,7,8,9]. It must be understood through polymer composition, carrier architecture, OA physical state, release medium, tissue deposition, and biological response [7,8,9,12,13,14,15,16,17,18,37].
Polymeric OA systems can produce release through diffusion, swelling, degradation, erosion, micelle dissociation, gel relaxation, prodrug cleavage, responsive bond cleavage, carrier-in-matrix behavior, or matrix-assisted local retention. These mechanisms are not interchangeable. Their relevance depends on whether the intended application is oral delivery, cancer-oriented delivery, psoriasis, wound healing, osteoarthritis, dermal delivery, or microneedle-assisted administration [7,8,9,12,13,14,15,16,17,18,36,37].
For OA, a controlled-release system should be judged by functional exposure rather than by release duration alone. The best design is not necessarily the slowest or the most complex; it is the one that produces reproducible OA availability at the intended biological site for the intended period of action.
12. Translational and Clinical Perspectives
12.1. Translational Framing of OA Polymeric Nanocarriers
The translational potential of polymeric and polymer-assisted nanocarriers for oleanolic acid should be discussed with caution. Most available evidence remains preclinical and is based on formulation studies, cell models, or animal experiments [1,5,7,9]. OA has broad reported biological activities, but pharmacological breadth alone does not create a clinical product. The compound must be delivered reproducibly, safely, and at concentrations that are meaningful for the intended tissue and disease context.
The main translational limitation of OA is not only pharmacodynamics. It is exposure. Poor aqueous solubility, low and variable bioavailability, crystallinity, limited barrier transport, and vehicle-dependent activity make it difficult to compare studies and to define realistic dosing strategies [1,5]. Polymeric nanocarriers are relevant because they may convert OA from a poorly dispersed hydrophobic compound into a formulation with more controllable release, retention, and tissue exposure [5,7,8,9,12,15,16,17,36,40].
A polymeric OA system should not be considered clinically promising only because it increases apparent solubility or improves activity in vitro [7,8,9,12,15,16,17,36]. Clinical relevance requires a connection between carrier design, release kinetics, tissue exposure, safety, disease biology, patient need, and manufacturability. This is why translational assessment should focus on specific use cases rather than on general enhancement of OA delivery.
The most realistic near-term directions are local and tissue-targeted applications. These include inflammatory skin disease, psoriasis, wound healing, dermal delivery, osteoarthritis, and possibly other localized inflammatory conditions [8,12,15,16,17,36]. In these settings, sustained exposure at the target site may be more important than high systemic bioavailability. Systemic applications such as cancer, liver injury, metabolic disease, and oral bioavailability enhancement remain scientifically relevant, but they require stronger pharmacokinetic, biodistribution, safety, and comparator data before clinical value can be claimed [5,7,9,25,26,28].
12.2. Unmet Medical Needs and Formulation Relevance
Several disease areas associated with OA biology involve chronic inflammation, oxidative stress, tissue remodeling, barrier dysfunction, cartilage degeneration, or abnormal cell proliferation [1,5]. These processes are relevant to inflammatory skin disease, wound repair, osteoarthritis, liver injury, metabolic disorders, and cancer [25,26,28,32]. However, OA’s poor delivery properties limit direct therapeutic use and provide a rationale for formulation-based intervention [1,5].
Polymeric nanocarriers may address these needs by enabling controlled release, sustained local residence, improved tissue deposition, reduced dosing frequency, and better handling of poorly soluble OA. These advantages are most relevant when conventional free-drug administration cannot maintain sufficient exposure or when repeated administration is impractical [5,7,8,9,12,15,16,17,36]. In this sense, polymeric delivery is not merely a technical enhancement. It may make OA biologically testable in settings where unformulated OA is difficult to interpret.
The unmet-need argument must be disease-specific. Psoriasis requires local anti-inflammatory and immunomodulatory effects in skin. Wound healing requires stage-dependent modulation of inflammation, migration, oxidative stress, extracellular matrix deposition, and tissue repair. Osteoarthritis requires joint residence and cartilage- or synovium-relevant activity. Cancer requires selective tumor exposure, penetration, and safety. Liver and metabolic applications require systemic exposure, target-tissue distribution, and long-term tolerability [8,12,15,16,17,25,26,28,32,36]. A single OA formulation is unlikely to be optimal for all these indications.
12.3. Inflammatory Skin Diseases and Psoriasis
Inflammatory skin disease is one of the strongest translational directions for polymeric OA delivery. OA has reported anti-inflammatory activity, and local delivery may provide therapeutic exposure in skin while limiting systemic burden [1,5,12]. This route also matches OA’s formulation limitations: poor solubility and restricted passive penetration can be addressed by polymeric micelles, hyaluronic-acid-based nanoparticles, hydrogels, nanogels, and microneedle-compatible systems [12,16,17,36].
The hyaluronic-acid-based reduction-responsive OA nanoparticle system developed for psoriasis is particularly important because it links polymer chemistry with disease-oriented delivery [12]. The system combines OA–hyaluronic acid conjugation, disulfide-responsive behavior, topical application, and CD44-related cellular uptake in keratinocytes [12]. This makes it more translationally informative than a formulation designed only to increase apparent solubility.
For psoriasis and inflammatory skin disease, the main development questions are local deposition, repeated-dose safety, irritation, barrier recovery, disease-relevant activity, and comparison with current topical therapies [12,16,17,36]. A formulation that improves OA uptake in cells must still show that it can deliver useful exposure in diseased skin without unacceptable irritation or barrier disruption.
Microneedle-assisted systems may become relevant in this area, especially for localized plaques or difficult-to-penetrate lesions [12,16,36,37,40]. However, OA-loaded microneedles should be presented as a future-oriented platform rather than an established OA formulation class. Their value will depend on whether they improve skin deposition, dose reproducibility, patient acceptability, and safety compared with simpler topical gels, micelles, or HA-based nanoparticles [12,16,17,36,37].
12.4. Wound Healing and Tissue Repair
Wound healing is another plausible local application for polymeric OA delivery. OA has been associated with wound-healing-related activity, cell migration, anti-inflammatory effects, and oxidative-stress modulation [1,5,28,41]. However, wound healing is a dynamic process, and the usefulness of OA will depend on timing, dose, tissue context, and formulation design.
Polymeric wound dressings, hydrogels, nanocarrier-loaded dressings, and PLGA fiber membranes are relevant because they can combine OA delivery with tissue contact, moisture control, mechanical protection, and local retention [28,39,40,41]. In these systems, the polymeric matrix is not only a carrier. It also defines how the formulation interacts with wound exudate, damaged tissue, inflammatory cells, and the local repair environment.
The translational challenge is that wound biology is stage-dependent. A formulation may need to reduce excessive inflammation without suppressing necessary immune responses, support migration without causing abnormal proliferation, and maintain moisture without increasing infection risk. OA release data alone are therefore insufficient. Evaluation should include keratinocyte and fibroblast behavior, inflammatory cytokines, oxidative-stress markers, collagen deposition, wound closure, infection risk, exudate interaction, and tissue compatibility [28,39,40,41].
For clinical development, OA wound systems should be compared not only with free OA but also with relevant wound-care materials or advanced dressings. If a nanocarrier-loaded dressing is more complex than a conventional dressing, it must justify that complexity by better tissue response, dosing convenience, safety, or healing performance [8,28,39,40,41].
12.5. Osteoarthritis and Intra-Articular Local Therapy
Osteoarthritis is a strong example of a localized disease where systemic bioavailability may not be the most important formulation endpoint. The disease involves cartilage degeneration, synovial inflammation, oxidative stress, pain, and mechanical joint dysfunction within an accessible anatomical compartment [15]. Intra-articular delivery can provide high local exposure, but rapid clearance from the joint limits the usefulness of simple formulations.
The OA cubic liquid crystal nanoparticle-based thermosensitive gel developed for knee osteoarthritis in rats is therefore one of the most clinically oriented OA delivery examples [15]. It combines OA-loaded nanostructures with a Poloxamer thermosensitive gel base, creating a local depot designed to improve intra-articular residence and sustained exposure [15]. This approach is valuable because it is linked to a specific disease, route, and anatomical site.
For osteoarthritis, the formulation must be assessed through joint-relevant criteria: injectability, gelation temperature, viscosity, residence time, release in synovial-like conditions, sterility, endotoxin control, cartilage compatibility, synovial irritation, and repeated-dose safety [15]. These requirements are more demanding than simple in vitro release or cell-viability assays.
The comparator question is also important. Future OA intra-articular systems should be compared with clinically relevant standards such as hyaluronic acid injections, corticosteroids, nonsteroidal anti-inflammatory approaches, or other local therapies where appropriate [15]. Without such comparisons, it is difficult to judge whether OA depots offer practical value beyond formulation novelty.
12.6. Cancer Nanomedicine
Cancer is relevant to OA polymeric delivery because OA has preclinical anticancer activity and polymeric carriers can alter cellular exposure, uptake, and cytotoxic response [7,9,25,28]. OA-loaded PEGylated PLA/PLGA nanoparticles and OA/ursolic-acid-loaded PLGA nanoparticles provide direct examples of polymeric carrier systems evaluated in cancer-cell models [7,9].
However, cancer nanomedicine requires cautious interpretation. Increased cytotoxicity in monolayer cell culture does not establish clinical anticancer potential [7,9]. A polymeric carrier may increase apparent activity by improving dispersion, reducing precipitation, altering uptake, or changing intracellular availability rather than by enhancing intrinsic OA potency [5,7,9].
For cancer applications, the key development questions include tumor penetration, uptake mechanism, release at the target site, pharmacokinetics, biodistribution, off-target toxicity, immune interaction, and comparison with established anticancer therapies [7,9,25].
Three-dimensional tumor models, spheroids, organoids, and animal biodistribution studies would provide more relevant information than cytotoxicity alone.
Cancer therefore remains scientifically important but translationally difficult. Among the directions discussed in this review, it likely faces a longer development path than local dermatological, wound, or intra-articular applications because systemic exposure, safety margins, and clinical comparators are more demanding [7,9,15,25,28].
12.7. Liver Injury, Metabolic Disorders, and Systemic Applications
OA has a long history of investigation in hepatoprotection, metabolic regulation, antioxidant signaling, and inflammatory pathways [26,28]. These activities are scientifically important, but systemic clinical development is complicated by poor oral bioavailability, variable exposure, and the need to deliver sufficient drug to target tissues without off-target toxicity [1,5].
Polymeric nanocarriers may improve systemic OA delivery by increasing apparent dispersion, protecting the compound, modifying pharmacokinetics, and supporting tissue distribution. However, systemic formulations require more demanding evidence than local formulations because they must demonstrate absorption, circulation stability, biodistribution, metabolism, clearance, and safety [5,7,9,31].
For liver-related applications, carrier design should specify the intended target: hepatocytes, Kupffer cells, hepatic stellate cells, inflammatory pathways, antioxidant response, or metabolic regulation. A formulation that improves plasma exposure may not necessarily improve therapeutic exposure in the relevant liver cell population. Liver-targeted OA polymeric systems therefore require cell-type-specific and pharmacokinetic evaluation [5,26,28,31].
For metabolic disorders, oral or injectable OA systems would need to show sustained exposure, acceptable long-term safety, and disease-relevant outcomes [1,5]. Because metabolic indications usually require chronic administration, polymer safety, degradation products, excipient burden, cost, and patient adherence become central considerations [27]. These requirements make local indications more realistic as initial translational targets [8,12,15,17].
12.8. Oral Delivery and Bioavailability Improvement
Oral delivery is attractive because of patient convenience, but it is one of the most difficult routes for OA because of poor aqueous solubility and low bioavailability [1,5]. Many approaches have been used to improve oral exposure, including solid dispersions, cyclodextrin complexes, phospholipid complexes, self-emulsifying systems, lipid nanoparticles, and polymeric carriers [5,14,20,21].
Polymeric nanocarriers could improve oral OA delivery by enhancing dispersion, protecting the drug in gastrointestinal conditions, modulating release, interacting with mucus, or promoting uptake through intestinal pathways. However, oral nanocarrier translation is difficult. Gastrointestinal dilution, enzymes, bile salts, mucus, food effects, pH variation, transit time, and interindividual variability can all influence performance [5,14,20,21,31].
For OA, the key oral-delivery question is comparative. A polymeric nanocarrier must show an advantage over simpler and often more scalable systems such as self-emulsifying formulations, solid dispersions, or cyclodextrin complexes [5,14,20,21]. If a polymeric system improves in vitro solubility but does not improve in vivo exposure, tissue availability, safety, or therapeutic effect, its translational value may be limited.
Future oral OA studies should include pharmacokinetic evaluation, food-effect considerations, gastrointestinal stability, mucus interaction, release under biorelevant conditions, and comparison with established solubilization systems [5,14,20,21,31]. Oral delivery remains important, but it should be approached as a high-barrier route rather than an automatic consequence of nanosizing.
The process–structure–function–response logic proposed for OA polymeric delivery is summarized in Figure 6.
12.9. Patient-Centered and Product-Development Considerations
Clinical usefulness depends not only on biological activity but also on patient-centered and product-development factors. Topical gels, patches, wound dressings, intra-articular depots, oral formulations, and microneedle systems differ greatly in ease of use, dosing frequency, storage needs, cost, comfort, and acceptability [16,18,35,39]. These factors can determine whether a formulation is realistic even when the biological rationale is strong.
For chronic skin disease, a patient-friendly OA formulation would need to be non-irritating, cosmetically acceptable, stable, easy to apply, and compatible with repeated use [12,16,17,35]. For wound healing, the product would need to fit into dressing changes, moisture management, infection control, and wound-care practice [8,39,40]. For osteoarthritis, injectability, dosing interval, pain during administration, and compatibility with existing intra-articular therapies would be important [15,18].
Polymeric nanocarriers add value only when the added complexity is justified by better performance. If a simple gel, micelle, or cyclodextrin complex provides sufficient local exposure, a complex multi-component nanocarrier may not be necessary [12,16,17,20]. Conversely, if sustained release, targeting, tissue retention, or barrier bypass is required, a polymeric or polymer-assisted system may be justified [7,8,9,15,16,17,35,36].
Cost and scalability also matter. Biodegradable polymeric nanoparticles, microneedles, nanogels, and polymer–drug conjugates may require specialized manufacturing, sterilization, packaging, quality control, and stability programs [16,25,32,36]. Translational development should therefore balance formulation sophistication with manufacturability, regulatory feasibility, patient need, and expected clinical advantage.
12.10. Regulatory and Safety Perspective
Regulatory translation of OA polymeric nanocarriers would require clear definition of the active compound, excipients, polymeric carrier, manufacturing process, critical quality attributes, release profile, safety, and intended clinical use [5,7,8,9,25,31].
Natural origin does not reduce the need for rigorous safety evaluation. OA exposure, impurities, excipients, polymer degradation products, and formulation components can all affect toxicity [1,5,25,31].
For polymeric nanoparticles, safety evaluation should include polymer degradation products, residual solvents, stabilizer or surfactant toxicity, complement activation where relevant, hemocompatibility for injectable systems, off-target tissue distribution, and immune interaction [7,8,9,31]. For topical systems, irritation, sensitization, barrier disruption, photostability if relevant, and repeated-dose local tolerability are critical [12,16,17,25,35]. For intra-articular systems, synovial irritation, cartilage compatibility, sterility, endotoxin burden, viscosity, and repeated injection safety are essential [15,18].
Combination platforms such as microneedles and drug-loaded wound dressings may face additional regulatory complexity because they combine drug delivery with device-like or biomaterial functions [36,40,42]. Dose uniformity, mechanical performance, microbial control, sterility, packaging stability, patient use, and application reproducibility should be considered early in development rather than after biological proof of concept [16,35,39].
For OA specifically, the field would benefit from standardized reporting of dose, formulation composition, free versus carrier-associated drug, release conditions, biological controls, and exposure verification [5,7,8,9]. Without such reporting, comparison between studies will remain difficult and claims of translational potential will remain weak.
12.11. Translational Readiness Across Application Areas
Based on current evidence, local dermatological delivery appears to be one of the most promising directions for OA polymeric systems. This area has direct support from hyaluronic-acid-based OA nanoparticles for psoriasis, polymeric OA micelles for skin application, and broader polymeric or microneedle literature in skin delivery [16,17,35,36]. The main remaining challenges are repeated-dose safety, irritation, skin deposition, barrier recovery, comparison with existing topical therapies, and scalable formulation.
Wound healing is also plausible, especially for hydrogels, PLGA fiber membranes, and nanocarrier-loaded dressings. The main barriers are stage-specific wound biology, infection risk, wound-exudate effects, realistic wound models, and comparison with advanced dressings or standard wound care [8,28,39,40,41].
Osteoarthritis has a strong local-depot rationale because intra-articular delivery can theoretically maintain OA near the diseased joint [15]. The thermosensitive nanogel study provides disease-oriented support, but translation would require stronger evidence on joint residence, cartilage compatibility, repeated dosing, synovial safety, mechanical joint conditions, and comparison with existing intra-articular therapies [15,18].
Cancer, liver injury, metabolic disease, and oral bioavailability enhancement remain important but less immediately translational. These indications require stronger pharmacokinetic, biodistribution, target-tissue exposure, long-term safety, and comparator evidence [5,7,9,20,21,25,26,27,28]. They should be presented as longer-term directions unless direct OA polymeric systems provide more advanced in vivo evidence.
A complementary application-oriented summary of suitable polymeric systems and main translational barriers is provided in Table S2.
12.12. Clinical Positioning
The clinical future of OA polymeric and polymer-assisted nanocarriers will depend on matching formulation design to a realistic use case. Local and tissue-targeted systems appear most promising in the near term because they can use controlled release and tissue residence to compensate for OA’s poor systemic bioavailability [8,15,16,17,36]. Psoriasis, inflammatory skin disease, wound healing, dermal delivery, and osteoarthritis are therefore stronger early translational targets than broad systemic applications.
Systemic and oral applications remain scientifically relevant but require more demanding evidence. For cancer, liver injury, metabolic disease, and oral bioavailability enhancement, polymeric systems must demonstrate not only improved formulation properties but also pharmacokinetic advantage, target-tissue exposure, safety, and superiority over simpler or established delivery approaches [5,7,9,20,21,25,26,27,28].
For OA, translational progress should be judged by functional necessity. A polymeric nanocarrier is justified when it solves a defined clinical delivery problem: local retention, barrier bypass, controlled release, responsive tissue exposure, dose reduction, or improved safety. Formulation complexity should advance only when it improves performance compared with simpler alternatives.
13. Manufacturing, Regulatory, and Scale-Up Challenges
13.1. Why Manufacturability Matters for OA Polymeric Nanocarriers
Manufacturability is a central issue for OA-loaded polymeric and polymer-assisted nanocarriers because formulation performance depends strongly on material attributes, process conditions, and batch reproducibility [7,9,25,27,31]. A system that performs well as a small laboratory batch may not remain stable, reproducible, sterile, or clinically useful after scale-up. This is particularly important for OA because poor solubility, crystallinity, hydrophobicity, and vehicle-dependent activity make the compound sensitive to solvent history, mixing, drying, polymer composition, and storage conditions [1,5,20,21,48].
Many OA formulations are reported as proof-of-concept systems, where the main objective is to demonstrate drug loading, improved dispersion, sustained release, or enhanced biological response [7,9,12,15,17,18]. Translational development requires a different standard. The formulation must be prepared reproducibly, characterized consistently, stored reliably, and manufactured through processes compatible with quality control, sterility or microbial limits where needed, and regulatory expectations [25,27,31].
For polymeric OA systems, manufacturability includes polymer sourcing, raw OA quality, solvent control, process parameters, nanoparticle or matrix formation, drug loading reproducibility, residual solvent removal, drying or lyophilization, sterilization or microbial control, packaging, and stability [7,9,13,25,27,31]. Each of these factors can affect particle size, OA physical state, release profile, biological exposure, and safety. Manufacturing should therefore be considered part of formulation design, not a final technical step.
13.2. Critical Material Attributes
Critical material attributes are especially important for OA polymeric systems because both the drug and the carrier materials influence final performance [1,5,7,20,21,27]. For PLA and PLGA systems, relevant attributes include polymer molecular weight, lactide ratio, end-group chemistry, intrinsic viscosity, crystallinity, residual monomers, water content, degradation behavior, and batch-to-batch variability [13,25,27,31].
OA itself should also be treated as a critical material input rather than a generic active ingredient. Purity, polymorphic form, particle size, residual solvents, impurity profile, botanical or synthetic origin, and storage conditions may influence loading, crystallization, release, and biological response [1,5,20,21,27,48]. For a poorly soluble compound, differences in raw material quality can translate directly into formulation variability.
Excipients such as PEGylated polymers, hyaluronic acid, poloxamers, cyclodextrins, chitosan, surfactants, stabilizers, hydrogel-forming polymers, and microneedle-forming polymers also require control [12,15,16,17,18,34,36,37]. Their molecular weight, substitution degree, charge, purity, moisture content, degradation profile, and biological interaction may influence carrier stability and tissue response. In advanced polymer-assisted systems, an excipient may be part of the therapeutic mechanism rather than a passive ingredient.
Material specifications should therefore be linked to critical quality attributes. Polymer molecular weight may affect particle size and release; hyaluronic-acid molecular weight or substitution degree may affect cellular interaction and degradation; poloxamer concentration may affect gelation temperature and local retention; and cyclodextrin substitution may affect OA complexation and release [12,15,18,30]. This connection between material attributes and formulation performance is essential for quality-by-design-oriented development.
13.3. Critical Process Parameters
Critical process parameters are the manufacturing variables that determine whether the intended formulation can be reproduced. For OA-loaded nanoparticles, these may include solvent type, solvent-to-water ratio, polymer concentration, OA concentration, stabilizer concentration, mixing rate, injection mode, temperature, purification method, and solvent-removal procedure [7,9,13,31].
For hydrogels, thermosensitive depots, and nanocarrier-loaded matrices, process parameters include polymer concentration, mixing order, temperature, hydration time, pH, ionic strength, gelation conditions, carrier distribution, and filling or casting procedure [15,18,31]. For electrospun fiber membranes, polymer solution viscosity, solvent composition, voltage, flow rate, collector distance, humidity, and drying conditions can affect fiber diameter, morphology, drug distribution, and release [8]. For microneedles, molding, casting, drying, backing-layer formation, humidity, and demolding can affect geometry, mechanical strength, dose uniformity, and carrier stability [16,36,37].
OA makes these parameters especially important because uncontrolled supersaturation, solvent evaporation, drying, or concentration changes may promote crystallization or phase separation [1,5,7,8,9]. A process that produces high loading in one batch may produce different OA physical state or release behavior in another batch if mixing, drying, or purification conditions change.
Process descriptions should therefore be detailed enough to allow reproduction. Reporting only the final particle size and encapsulation efficiency is not sufficient. Studies should describe material sources, concentrations, processing sequence, temperature, solvent-removal conditions, purification, drying, storage, and the number of independent batches evaluated [7,9,13,25,27,31].
13.4. Scale-Up of Polymeric Nanoparticles
Scale-up of polymeric nanoparticles is difficult because the processes used at laboratory scale may not translate directly to larger batches. Nanoprecipitation, solvent displacement, and emulsion-based methods depend on mixing time, local concentration gradients, solvent diffusion, interfacial behavior, and energy input [7,9,13,31]. These parameters can shift during scale-up and change particle size, polydispersity, OA loading, and release.
Batch nanoprecipitation may be simple for screening, but it can be sensitive to injection speed, vessel geometry, stirring rate, solvent ratio, and operator technique [7,9,13]. Emulsion-based methods may face additional challenges related to droplet-size control, surfactant removal, residual solvent, and scale-dependent shear [7,9,13,31]. For OA, both approaches must also control drug precipitation and physical state.
Continuous or semi-continuous methods, including microfluidic mixing, controlled impingement, or scalable precipitation platforms, may improve process control for polymeric nanoparticles [25,49,50]. These approaches can provide better control over mixing and nucleation, but they require optimization of flow rate, concentration, solvent compatibility, throughput, fouling, and downstream processing [49,50]. Their value for OA formulations would need to be demonstrated through reproducible loading, physical-state control, release, and stability.
Scale-up should be evaluated through multiple independent batches rather than by increasing batch size once. Important endpoints include particle size, PDI, zeta potential, OA loading, encapsulation efficiency, residual solvent, physical state, release profile, microbial quality, stability, and biological performance [7,9,13,25,27,31]. Without such data, claims of translational potential remain premature.
13.5. Manufacturing of Hydrogels, Depots, Dressings, and Microneedles
Manufacturing challenges differ by dosage form. Hydrogels and thermosensitive depots require control of polymer concentration, mixing order, viscosity, gelation temperature, injectability or spreadability, drug distribution, microbial quality, and filling conditions [15,18,33]. For OA-loaded systems, the distribution of the embedded nanocarrier or drug phase within the gel is especially important because nonuniformity can lead to dose variability and inconsistent release.
Wound dressings and fiber membranes require control of matrix morphology, mechanical strength, porosity, thickness, residual solvent, drug distribution, sterilization compatibility, packaging, and moisture behavior [8,39,40]. If OA is incorporated through nanoparticles, micelles, or cyclodextrin complexes, the stability of those carriers after embedding and drying must also be verified [8,17,18,34].
Microneedles introduce device-like manufacturing requirements. Needle height, base width, tip sharpness, array density, mechanical strength, insertion efficiency, dose uniformity, dissolution or swelling time, residual moisture, and packaging stability must be controlled [16,36,37]. If OA-loaded nanocarriers are embedded in microneedles, the carrier should be characterized before fabrication, after drying, after storage, and after dissolution or swelling [7,8,9,15,29,30,31,32].
These dosage forms are often more clinically practical than nanoparticle suspensions alone, but they are also more complex as products. A nanocarrier-loaded hydrogel, dressing, depot, or microneedle patch must be controlled as the complete formulation, not only as the nanoscale component.
13.6. Sterilization and Microbial Control
Sterilization or microbial control is a major issue for OA polymeric systems intended for topical, wound, intra-articular, transdermal, or injectable use. The required level of control depends on route and product type. Intra-articular depots and injectable systems require strict sterility and endotoxin control, whereas topical gels and wound dressings require microbial limits or sterility depending on intended use and regulatory classification [15,18,39,40].
Common sterilization methods may affect polymeric nanocarriers. Heat can accelerate polymer degradation, alter gelation, or promote OA crystallization. Gamma irradiation or electron-beam sterilization may affect polymer chains, drug stability, or mechanical properties. Filtration may not be feasible for gels, fibers, microneedles, or larger nanocarrier systems. Aseptic manufacturing may be required for sensitive formulations [15,18,31,39,40].
For OA, sterilization should be evaluated not only by microbial outcome but also by formulation performance. Size, PDI, OA loading, physical state, release profile, rheology, mechanical behavior, dose uniformity, and biological activity may change after sterilization [7,9,15,16,18]. This is especially important for thermosensitive gels, wound dressings, and microneedle patches, where polymer structure and mechanical behavior are part of the product function.
13.7. Drying, Lyophilization, and Storage
Drying and storage are critical for OA-loaded polymeric systems because water removal, humidity, temperature, and packaging can alter carrier structure and drug physical state [1,5,7,8,9]. Lyophilization may improve shelf-life for nanoparticle dispersions, but it can also cause aggregation, changes in release, or drug crystallization unless cryoprotectants and reconstitution conditions are optimized [7,9,13].
For microneedles, fiber membranes, and wound dressings, drying is part of the manufacturing process rather than a post-processing option [8,16,36,37]. Drying conditions can influence residual moisture, mechanical strength, OA distribution, carrier stability, and crystallization risk. For hydrogels and thermosensitive depots, storage may involve hydrated or semisolid states, which introduce risks of phase separation, microbial growth, syneresis, viscosity change, and drug redistribution [15,18].
Stability studies should monitor more than appearance. Key parameters include OA content, degradation products, physical state, particle size, release profile, residual moisture, rheology, mechanical strength, dose uniformity, sterility or microbial quality, and biological activity where relevant [7,8,9,15,16,36,37]. Packaging should protect against moisture, oxygen, light, microbial contamination, and mechanical damage according to the dosage form.
13.8. Reproducibility and Batch-to-Batch Variability
Batch-to-batch reproducibility is essential for OA polymeric nanocarriers because small process variations may change particle size, OA physical state, loading, release, and biological response [1,5,7,9,21,25,26,27,48]. This is particularly relevant for poorly soluble compounds whose activity may depend strongly on vehicle and exposure conditions [1,5,21,21,48].
Preclinical studies often report results from optimized batches, but translational development requires multiple independently prepared batches tested against the same critical quality attributes [7,9,25,26,27]. These attributes should include not only size and encapsulation efficiency, but also physical state, release, stability, residual solvent, and biological activity where relevant [1,5,21,25,29].
Reproducibility becomes more difficult for multi-component systems. Hyaluronic-acid nanoprodrugs, nanocarrier-loaded hydrogels, thermosensitive depots, fiber membranes, and microneedle patches involve several material and process steps, each of which may introduce variability [8,12,15,16,18,36,37]. Batch control must therefore cover the full product rather than only the nanoparticle or active-drug component.
A useful approach is to define acceptable ranges for critical quality attributes and link them to biological performance. A range of particle sizes may be acceptable if release and biological activity remain consistent, while a small change in OA physical state may be unacceptable if it alters release or tissue exposure [1,5,7,9,21,25,26,27,29,30,31,48]. This performance-based logic is central to QbD development.
13.9. Quality-by-Design and Risk-Based Development
Quality-by-design provides a useful framework for OA polymeric nanocarrier development because it connects product design, process understanding, risk assessment, and quality control [7,17,21,25,26,27]. Instead of relying only on empirical optimization, QbD encourages definition of the quality target product profile, critical material attributes, critical process parameters, and critical quality attributes.
For OA systems, the target product profile should be indication-specific. A topical psoriasis nanoparticle, a wound dressing, an intra-articular thermosensitive gel, an oral polymeric nanoparticle, and a microneedle patch have different dose, release, safety, sterility, stability, and usability requirements [8,12,15,16,17,18,36,37]. The desired product profile should therefore be defined before selecting polymers, carriers, or manufacturing methods.
Risk assessment can help identify variables most likely to affect performance. For OA, high-risk factors may include drug crystallization, incomplete encapsulation, aggregation, solvent residue, poor release reproducibility, polymer degradation, gelation failure, microneedle fracture, skin irritation, joint irritation, and lack of exposure verification [1,5,7,9,15,16,20,21,25,26,27,28,48].
Design-of-experiments approaches may be especially valuable. DoE can evaluate how polymer concentration, drug-to-polymer ratio, solvent ratio, stabilizer amount, mixing conditions, drying, storage, and sterilization affect size, loading, physical state, release, and stability [25,26,27]. This would help move the field from one-off formulation reports toward more systematic design rules.
13.10. Regulatory Considerations
Regulatory expectations for OA polymeric and polymer-assisted systems would depend on route, composition, dosage form, and intended indication. A topical gel, oral nanoparticle, intra-articular depot, wound dressing, and microneedle patch may fall under different regulatory categories or require different evidence packages [16,25,31,35,39]. Regulatory considerations should therefore be integrated early into formulation design.
Although OA is a natural compound, natural origin does not reduce the need for rigorous quality and safety evaluation [1,5,25,31]. The active compound, excipients, polymeric carrier, manufacturing process, release profile, impurities, degradation products, and intended use all contribute to risk. For polymeric and hybrid systems, the full formulation must be evaluated, not only OA.
For polymer-assisted lipid or hybrid carriers, regulatory principles from complex nanomedicines, liposomal products, and pharmaceutical development provide useful analogies [25,31]. These emphasize composition control, structure, drug loading, release, stability, pharmacokinetics or local exposure, safety, and reproducible manufacturing. OA hybrid systems can draw from these principles even when they are not strictly liposomal products.
Microneedles, wound dressings, and intra-articular depots may require additional consideration because they combine drug delivery with device-like or biomaterial functions [16,35,39]. Dose uniformity, mechanical performance, sterility, biocompatibility, local tolerability, user handling, and packaging stability may be as important as release kinetics. This reinforces the need to design OA polymeric systems as products rather than only experimental formulations.
13.11. Clinical Manufacturing and GMP Considerations
Clinical translation requires manufacturing under controlled quality systems, with defined raw materials, validated processes, validated analytical methods, and documentation [25,26,27,31]. For OA polymeric nanocarriers, GMP-compatible development would require specifications for OA, polymers, excipients, solvents, process parameters, intermediate controls, and final product attributes.
Analytical method validation is particularly important. Methods for OA assay, impurity profiling, residual solvent analysis, release testing, particle size, morphology, physical state, sterility, endotoxin, and stability must be appropriate for the formulation type [7,8,9,12,15,16,17,18,31]. For multi-component systems, separate methods may be needed for the drug, polymer, nanocarrier, gel matrix, dressing, or device component.
Scale-up should also consider equipment compatibility and process robustness. Methods that depend on manual pipetting, small-batch sonication, laboratory-specific drying, or uncontrolled mixing may be difficult to translate. Continuous manufacturing, controlled mixing, spray drying, lyophilization, scalable molding, or aseptic filling may be needed depending on dosage form [16,25,31,49,50,51].
A clinically manufacturable OA product should balance complexity with therapeutic value. If a simple gel, micelle, or cyclodextrin complex provides sufficient local exposure, a highly complex multi-component nanocarrier may not be justified [12,14,17,20]. Conversely, if controlled release, barrier bypass, local retention, or route-specific targeting is necessary, a polymeric or polymer-assisted system may be worth the additional development burden [7,8,9,12,15,16,17,18,36].
13.12. Manufacturing Implications
Manufacturing and scale-up are not peripheral issues for OA-loaded polymeric nanocarriers. Because OA is poorly soluble, hydrophobic, and sensitive to formulation environment, process conditions can strongly affect drug loading, physical state, release, stability, and biological response [1,5,7,8,9,20,21,25,26,27,48].
The most important development need is to connect material attributes and process parameters with critical quality attributes and biological performance. This requires control of OA raw material quality, polymer properties, solvent history, mixing, drying, sterilization, storage, batch reproducibility, and route-specific product performance [7,9,13,15,16,18,25,26,27,31].
For OA, manufacturability should be considered from the beginning of formulation design. The most promising systems will not be those with the greatest complexity, but those that combine a clear clinical purpose with reproducible preparation, stable storage, scalable manufacturing, and measurable performance advantages over simpler alternatives.
14. Future Directions and Research Roadmap
14.1. From Formulation Feasibility to Clinically Oriented Design
Future research on OA-loaded polymeric and polymer-assisted nanocarriers should move beyond demonstrating that OA can be incorporated into another nanosystem [1,5,7,8,9,12]. This feasibility has already been shown for PEGylated PLA/PLGA nanoparticles, PLGA nanoparticles, hyaluronic-acid-based nanoprodrugs, polymeric micelles, PLGA fiber membranes, topical gels, and thermosensitive local depots [7,8,9,12,15,16,17,18]. The next stage is to determine which of these systems can provide reproducible, safe, and route-appropriate OA exposure.
The central shift should be from carrier construction to product logic. Polymer type, carrier architecture, drug loading, release profile, route of administration, and biological model should be selected according to a defined disease problem rather than optimized in isolation. A formulation designed for psoriasis, a wound dressing, an intra-articular depot, an oral delivery system, and a cancer-oriented nanoparticle require different design criteria and different evidence [7,8,9,12,15,16,17,18].
For inflammatory skin disease, the priorities are local deposition, barrier compatibility, controlled release in skin, minimal irritation, and comparison with topical standards [12,16,17]. For wound healing, the formulation must support tissue contact, moisture management, stage-relevant repair biology, and compatibility with wound-care practice [8,39,40,41]. For osteoarthritis, injectability, joint residence, cartilage compatibility, synovial tolerability, and repeated-dose safety are central [15,18]. For cancer, liver, metabolic, or other systemic applications, pharmacokinetics, biodistribution, target-tissue exposure, and safety become much more demanding [8,28,39,40,41].
14.2. Disease-Specific Target Product Profiles
A clear target product profile should be defined before selecting the carrier. For OA, this profile should specify the intended indication, route of administration, target tissue, required exposure duration, dose range, release objective, safety constraints, comparator, and acceptable level of formulation complexity [5,7,8,9,12,15,16,17,18].
This approach would reduce overgeneralization. There is no universal “best” OA nanocarrier. A polymeric micelle may be useful for dermal delivery but insufficient for sustained intra-articular exposure. A PLGA nanoparticle may support sustained release but may not provide enough skin deposition without a topical matrix or microneedle platform. A thermosensitive gel may improve joint residence but would be unnecessary for applications where rapid release is preferred [8,12,15,18].
Target product profiles would also help determine whether polymeric complexity is justified. If a simple gel, cyclodextrin complex, or self-emulsifying formulation provides sufficient exposure, a multi-component nanocarrier may not be needed [14,20,21]. Conversely, if the therapeutic goal requires local retention, barrier bypass, prolonged release, responsive delivery, or reduced systemic distribution, polymeric and polymer-assisted systems become more rational [7,8,9,12,15,16,18].
14.3. Prioritizing Local and Tissue-Targeted Applications
The strongest near-term opportunities for OA polymeric delivery are likely to be local and tissue-targeted applications. These include psoriasis, inflammatory skin disease, wound healing, dermal delivery, osteoarthritis, and other localized inflammatory disorders [8,12,15,16,17,18,28,36,40,41]. These indications match the strengths of polymeric systems because they may benefit from sustained local exposure, controlled release, tissue retention, and reduced systemic burden.
Local delivery also reduces some of the uncertainty associated with poor systemic bioavailability. For OA, a formulation does not always need to maximize plasma concentration if the intended effect is local. A topical gel, nanocarrier-loaded dressing, microneedle patch, or intra-articular depot can be designed around tissue residence rather than systemic absorption [8,12,16,17,18,36].
This does not mean that local systems are simple. They still require dose uniformity, tissue compatibility, release control, irritation testing, sterility or microbial control where relevant, shelf-life stability, and comparison with existing treatments [12,15,16,18,39]. However, the route and endpoint are often more directly connected than in systemic delivery, making local applications a realistic first translational step.
14.4. Better Biological Models and Exposure Verification
A major future priority is the use of biological models that match the intended route and disease. Many OA carrier studies still rely heavily on simplified in vitro assays. These models are useful for early screening, but they cannot fully evaluate skin deposition, wound repair, joint residence, tumor penetration, oral absorption, or systemic biodistribution [7,8,9,12,15,16,17,18].
Skin-oriented systems should be evaluated in keratinocyte, immune-cell, reconstructed skin, ex vivo skin, or disease-relevant inflammatory skin models [12,16,17]. Wound systems should include migration, inflammatory cytokines, oxidative-stress markers, collagen deposition, wound closure, infection-related parameters, and tissue compatibility [8,26,39,40,41]. Intra-articular OA systems should include cartilage, synovium, inflammatory markers, pain-related behavior, repeated dosing, and local toxicity [15,16,17,18]. Cancer-oriented systems require three-dimensional tumor models, penetration, uptake, pharmacokinetics, biodistribution, and safety evaluation [7,8,9].
Exposure verification should become routine. Nominal OA dose is not enough for a formulation-dependent compound [1,5,14]. Future studies should quantify OA in release media, skin layers, wound models, joint compartments, cells, or target tissues where possible. This would help determine whether a polymeric carrier improves actual OA availability or only changes apparent solubility.
14.5. Physical-State Control as a Development Priority
Physical-state control should be treated as a major development priority for OA-loaded polymeric systems. OA may be crystalline, semi-crystalline, amorphous, molecularly dispersed, surface-associated, or phase-separated in a carrier [1,5,7,8,9]. These states can strongly affect release, storage stability, precipitation after dilution, and biological response.
Future studies should connect physical-state analysis with release and biological performance. Differential scanning calorimetry, X-ray diffraction, Raman spectroscopy, Fourier-transform infrared spectroscopy, microscopy, and validated extraction-based assays should be used more consistently to determine how OA is distributed within polymeric nanoparticles, micelles, fibers, hydrogels, depots, and microneedles [8,9,13].
This is particularly important for high-loading systems. Increasing OA content may improve dose capacity, but excessive loading may also promote crystallization, burst release, incomplete release, or batch variability [1,5,8,9]. A formulation with moderate loading and reproducible release may be more useful than a high-loading system with poorly controlled drug state.
14.6. Standardized and Route-Relevant Release Testing
Release testing remains one of the weakest points in many poorly soluble drug-delivery studies. For OA, release profiles are strongly influenced by sink conditions, medium composition, surfactants, proteins, membranes, agitation, and separation methods [1,5,7,8,9,13]. A release method that is analytically convenient may not represent the intended biological environment.
Future OA studies should justify release methods according to route. Oral systems require biorelevant gastrointestinal media. Topical and microneedle systems require skin-compatible receptor media, ex vivo skin, reconstructed skin, or deposition models. Wound dressings should consider exudate-like media, pH, enzymes, proteins, and salts. Intra-articular depots should use synovial-fluid-like conditions where possible [8,12,15,16,17,18,26,28,32,36,39,40,41].
Mass balance should also be improved. Released OA, carrier-retained OA, precipitated OA, tissue-deposited OA, membrane-associated OA, and apparatus-associated OA should be quantified where possible [1,5,7,8,9]. This is especially important because hydrophobic compounds can partition into plastics, proteins, lipids, membranes, and tissue compartments.
14.7. Manufacturing-Aware Innovation
Future OA polymeric systems should be designed with manufacturing in mind from the beginning. Many promising systems fail to progress because they are difficult to reproduce, sterilize, dry, store, scale, or package [25,27,31]. This risk is particularly high for multi-component systems such as polymer–drug conjugates, nanocarrier-loaded hydrogels, thermosensitive depots, fiber membranes, and microneedle patches [8,12,15,16,18,36,37].
Quality-by-design approaches should be used more widely. Critical material attributes, critical process parameters, and critical quality attributes should be defined for each product concept [25,26,27]. For OA, high-risk variables include drug crystallization, polymer molecular weight, solvent history, mixing, drying, residual solvent, release variability, gelation behavior, microneedle mechanics, sterilization effects, and storage stability [1,5,7,8,9,15,16].
Manufacturing-aware innovation does not mean avoiding advanced systems. It means ensuring that each added component has a clear function. PEG should improve stabilization or interface behavior; hyaluronic acid should support biological interaction or responsive delivery; hydrogels should improve residence; microneedles should solve a barrier problem; and thermosensitive gels should improve local depot performance [12,15,16,17,18,36,37]. Complexity is justified only when it improves performance compared with simpler alternatives.
14.8. Comparators and Decision Criteria
Future OA nanocarrier studies should include clinically and formulation-relevant comparators. Free OA is useful but not sufficient. Depending on the route and indication, relevant comparators may include cyclodextrin complexes, self-emulsifying systems, simple gels, polymeric micelles, standard wound dressings, approved topical therapies, hyaluronic acid injections, corticosteroids, NSAID-based approaches, or other established local treatments [9,12,14,15,16,17,18,20,21,39,40].
Comparator selection matters because polymeric systems are often more complex than conventional formulations. A nanocarrier should show added value in exposure, release control, tissue deposition, safety, dosing convenience, or biological performance. If it only improves an in vitro solubility number, its translational value remains uncertain [5,7,8,9,15,16,17].
Go/no-go criteria should also be defined earlier. Examples include minimum drug loading, acceptable release window, absence of drug crystallization after storage, reproducible batch quality, acceptable local tolerability, measurable tissue deposition, and superiority or clear advantage over simpler formulations [5,7,8,9,12,15,16,17,18]. These criteria would help move OA delivery research from formulation screening toward product-oriented development.
14.9. Staged Roadmap for Future OA Polymeric Systems
A staged roadmap may help organize future work. The first stage should define the target product profile: indication, route, target tissue, dosing logic, comparator, and intended clinical advantage. The second stage should select the polymeric or polymer-assisted system according to that profile. The third stage should establish critical quality attributes, including OA physical state, loading, release, stability, sterility or microbial quality where relevant, and route-specific performance.
The fourth stage should use disease-relevant models and exposure verification. At this point, studies should show not only that OA is released, but that it reaches the relevant tissue or cellular compartment and produces a measurable biological response. The fifth stage should address manufacturing, scale-up, storage, packaging, safety, and comparator performance.
14.10. Clinical Prioritization Roadmap
A practical roadmap for OA polymeric nanocarriers should prioritize applications with the strongest combination of biological rationale, delivery feasibility, and clinical relevance. Based on current evidence, local dermatological delivery, wound-oriented systems, and intra-articular osteoarthritis depots appear more immediately realistic than systemic cancer or metabolic-disease applications.
In the first stage, formulation studies should focus on robust polymeric design and characterization. This includes defining polymer role, OA physical state, release kinetics, stability, and appropriate controls. In the second stage, disease-relevant biological models should be used to confirm that the release profile produces meaningful tissue response [8,12,15,16,17,18,26,28,36,40].
In the third stage, translational formulation attributes should be evaluated, including sterilization, scalability, shelf-life, dose uniformity, local tolerability, and comparison with standard therapies [13,16,18,31,32,36,40,45]. In the fourth stage, selected formulations could be advanced into more predictive animal models and, eventually, early clinical feasibility studies if safety and efficacy signals justify progression.
This staged roadmap avoids overclaiming while still identifying a clear path forward [8,12,13,15,16,17,18,26,28,31,32,36,40,45]. OA polymeric nanocarriers are promising, but their future depends on disciplined formulation development, not only on repeated demonstration of improved solubility or in vitro activity [1,5,7,8,9,12,15,16,17,36].
14.11. Research Roadmap
Future progress in OA polymeric and polymer-assisted nanocarriers will depend on moving from formulation feasibility to disease-specific development. The field should prioritize local and tissue-targeted systems, physical-state control, route-relevant release testing, exposure verification, disease-relevant models, QbD-based development, manufacturing-aware innovation, and meaningful comparators [8,13,14,15,16,17,18,26,28,31,36,39,40,45].
The most realistic early opportunities are likely to involve skin inflammation, psoriasis, wound healing, dermal delivery, osteoarthritis, and related localized inflammatory conditions. These applications fit the strengths of polymeric delivery because they can benefit from sustained local exposure, controlled release, tissue retention, and reduced systemic burden [8,12,15,16,17,18,26,28,36,39,40].
The long-term goal should be to establish a process–structure–function framework for OA delivery. This means connecting polymer selection and manufacturing conditions with carrier architecture, OA physical state, release kinetics, tissue exposure, biological response, safety, and patient-relevant performance. For OA, the best future formulations will not be the most complex systems, but those that solve a clearly defined delivery problem better than simpler alternatives.
A staged translational roadmap for OA-loaded polymeric and polymer-assisted nanocarriers is shown in Figure 7.
A narrower comparison of local matrix-based platforms, including hydrogels, fiber dressings, and microneedle-assisted delivery, is shown in Figure 8.
Research priorities and decision criteria for future OA polymeric delivery are summarized in Table 6.
15. Conclusions
Oleanolic acid is a biologically attractive but formulation-limited pentacyclic triterpenoid. Its reported anti-inflammatory, antioxidant, hepatoprotective, anticancer, dermatological, metabolic, and wound-healing-related activities have generated broad preclinical interest, but poor aqueous solubility, low and variable bioavailability, crystallinity, limited barrier transport, and vehicle-dependent biological response continue to restrict its biomedical translation [1,5].
These limitations make OA a clear example of a natural compound whose biological performance cannot be separated from delivery strategy.
Polymeric and polymer-assisted nanocarriers provide a practical way to address several of these barriers at once. PEGylated PLA/PLGA nanoparticles, PLGA nanoparticles, hyaluronic-acid-based responsive nanoprodrugs, polymeric micelles, PLGA fiber membranes, topical gels, hydrogels, thermosensitive depots, and microneedle-compatible systems show that polymeric design can change how OA is dispersed, retained, released, and presented to biological tissues [7,8,9,12,15,16,17,18,36]. The value of these systems lies not only in improved apparent solubility, but in their ability to define exposure more precisely.
The field should now move beyond proof of loading or formulation feasibility. Future OA delivery studies need to connect polymer composition, carrier architecture, drug physical state, release kinetics, tissue deposition, biological response, safety, and manufacturing reproducibility. This process–structure–function logic is essential for distinguishing useful delivery systems from formulations that are technically interesting but not developmentally persuasive [7,8,9,12,15,17,18].
Among the possible clinical directions, local and tissue-targeted applications appear most realistic in the near term. Inflammatory skin disease, psoriasis, wound healing, dermal delivery, osteoarthritis, and related localized inflammatory conditions are well matched to polymeric strategies because they can benefit from sustained local exposure, tissue retention, controlled release, and reduced systemic burden [8,12,13,15,16,17,18,20,21,25,26,27,28,36,40]. In contrast, cancer nanomedicine, liver injury, metabolic disorders, and oral bioavailability enhancement remain scientifically important but require stronger pharmacokinetic, biodistribution, target-tissue exposure, safety, and comparator data before clinical relevance can be claimed [5,7,9,20,21,25,27,31,48,52].
For OA, the most promising formulations will not necessarily be the most complex. They will be those in which each material component has a clear function: improving dispersion, stabilizing the drug, controlling release, increasing local residence, bypassing a biological barrier, or improving safety. Polymeric and polymer-assisted nanocarriers should therefore be advanced only when they solve a defined delivery problem better than simpler alternatives. This disciplined approach may make OA not only a challenging natural-product payload, but also a useful model for the rational development of polymeric delivery systems for poorly soluble bioactive compounds.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/doi/s1, Table S1: Controlled-release mechanisms relevant to OA polymeric and polymer-assisted systems; Table S2: Translational positioning of OA polymeric and polymer-assisted systems by application area.
Author Contributions
Conceptualization, A.G. and B.B.-C.; methodology, A.G.; writing—original draft preparation, A.G.; writing—review and editing, A.G. and B.B.-C.; visualization, A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
The authors would like to thank Mariusz Malinowski, mariusz.malinowski@student.wsb.edu.pl a student of English Philology at WSB University, for his assistance with English-language editing during the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CQA | Critical quality attribute |
| DSC | Differential scanning calorimetry |
| FTIR | Fourier-transform infrared spectroscopy |
| GMP | Good manufacturing practice |
| HA | Hyaluronic acid |
| HPLC | High-performance liquid chromatography |
| LC–MS | Liquid chromatography–mass spectrometry |
| OA | Oleanolic acid |
| PEG | Polyethylene glycol |
| PLA | Poly(lactic acid) |
| PLGA | Poly(lactic-co-glycolic acid) |
| PDI | Polydispersity index |
| PXRD | Powder X-ray diffraction |
| QbD | Quality by design |
| QTPP | Quality target product profile |
References
- Castellano, J.M.; Ramos-Romero, S.; Perona, J.S. Oleanolic Acid: Extraction, Characterization and Biological Activity. Multidiscip. Digit. Publ. Inst. 2022, 14, 623–623. [Google Scholar] [CrossRef] [PubMed]
- Günther, A.; Bednarczyk-Cwynar, B. Oleanolic Acid: A Promising Antioxidant—Sources, Mechanisms of Action, Therapeutic Potential, and Enhancement of Bioactivity. Antioxidants 2025, 14, 598. [Google Scholar] [CrossRef] [PubMed]
- Günther, A.; Bednarczyk-Cwynar, B. Oleanolic Acid in Organelle Stress: Mitochondrial Dysfunction, Endoplasmic Reticulum Stress, Autophagy, and Apoptosis. Stresses 2026, 6, 22. [Google Scholar] [CrossRef]
- Günther, A.; Kulawik, M.; Sip, S.; Zalewski, P.; Jarmołowska-Jurczyszyn, D.; Stawicki, P.; Bednarczyk-Cwynar, B. Targeting Oxidative Stress in Carcinogenesis: Oleanolic Acid and Its Molecular Pathways. Antioxidants 2026, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Wasim, M.; Bergonzi, M.C. Unlocking the Potential of Oleanolic Acid: Integrating Pharmacological Insights and Advancements in Delivery Systems. Pharmaceutics 2024, 16, 692. [Google Scholar] [CrossRef] [PubMed]
- Stevanović, M.M.; Qian, K.; Huang, L.; Vukomanović, M. PLGA-Based Co-Delivery Nanoformulations: Overview, Strategies, and Recent Advances. Pharmaceutics 2025, 17, 1613. [Google Scholar] [CrossRef] [PubMed]
- Man, D.K.W.; Casettari, L.; Cespi, M.; Bonacucina, G.; Palmieri, G.F.; Sze, S.C.W.; Leung, G.P.H.; Lam, J.K.W.; Kwok, P.C.L. Oleanolic Acid Loaded PEGylated PLA and PLGA Nanoparticles with Enhanced Cytotoxic Activity against Cancer Cells. Mol. Pharm. 2015, 12, 2112–2125. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Padilla-Gainza, V.M.; Gilkerson, R.; Narula, A.; Lozano, K. Processing-Structure–Property Relationships of Oleanolic Acid Loaded PLGA Fiber Membranes. J. Mater. Sci. 2023, 58, 4240–4255. [Google Scholar] [CrossRef]
- Silva, A.M.; Alvarado, H.L.; Abrego, G.; Martins-Gomes, C.; Garduño-Ramirez, M.L.; García, M.L.; Calpena, A.C.; Souto, E.B. In Vitro Cytotoxicity of Oleanolic/Ursolic Acids-Loaded in PLGA Nanoparticles in Different Cell Lines. Pharmaceutics 2019, 11, 362. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, H.; Luan, J.; Tian, M.; Zhang, X.; Sohail, A.; Liang, D.; Liu, J.; Tao, F.; Wang, Z.; et al. Study of the Stability and Anti-Inflammatory Activity of Paeonol–Oleanolic Acid Liposomes by Microfluidic Technology. Foods 2025, 14, 2030. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhong, Z.; Tan, W.; Wang, S.; Wang, Y. Recent Advances in Nanoparticle Formulation of Oleanolic Acid. Chin. Med. 2011, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ren, J.; Su, Y.; Zhou, L.; Zhu, X.; Wang, Z.; Kong, F.; Li, G. Hyaluronic Acid-Based Reduction Responsive Nanoparticles for Improved Anti-Psoriasis Effects of Traditional Chinese Herb Monomer Oleanolic Acid via Blocking YAP-AREG Axis. J. Nanobiotechnology 2026, 24, 240. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zeng, H.; Luo, Y.; Chen, Y.; Wang, M.; Wu, C.; Hu, P. Recent Applications of PLGA in Drug Delivery Systems. Polymers 2024, 16, 2606. [Google Scholar] [CrossRef] [PubMed]
- Stelling-Férez, J.; López-Miranda, S.; Gabaldón, J.A.; Nicolás, F.J. Oleanolic Acid Complexation with Cyclodextrins Improves Its Cell Bio-Availability and Biological Activities for Cell Migration. Int. J. Mol. Sci. 2023, 24, 14860. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Jia, F.; Tang, X.; Li, Q. Oleanolic Acid Cubic Liquid Crystal Nanoparticle-Based Thermosensitive Gel Attenuates Knee Osteoarthritis Symptoms in Rats. Front. Pharmacol. 2025, 16, 1730566. [Google Scholar] [CrossRef] [PubMed]
- Qu, F.; Geng, R.; Liu, Y.; Zhu, J. Advanced Nanocarrier- and Microneedle-Based Transdermal Drug Delivery Strategies for Skin Diseases Treatment. Theranostics 2022, 12, 3372–3406. [Google Scholar] [CrossRef] [PubMed]
- An, J.Y.; Yang, H.S.; Park, N.R.; Koo, T.; Shin, B.; Lee, E.H.; Cho, S.H. Development of Polymeric Micelles of Oleanolic Acid and Evaluation of Their Clinical Efficacy. Nanoscale Res. Lett. 2020, 15, 133. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Pan, S.; Wang, L.; Li, S. Topical Gel Based Nanoparticles for the Controlled Release of Oleanolic Acid: Design and in Vivo Characterization of a Cubic Liquid Crystalline Anti-Inflammatory Drug. BMC Complement. Med. Ther. 2021, 21, 224. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, X. Improved Dissolution of Oleanolic Acid with Ternary Solid Dispersions. AAPS PharmSciTech 2007, 8, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Chang, Q.; Chan, C.K.; Meng, Z.Y.; Wang, G.N.; Sun, J.B.; Wang, Y.T.; Tong, H.H.Y.; Zheng, Y. Formulation Development and Bioavailability Evaluation of a Self-Nanoemulsified Drug Delivery System of Oleanolic Acid. AAPS PharmSciTech 2009, 10, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Yang; Huang; Dou; Zhai. Self-Microemulsifying Drug Delivery System for Improved Oral Bioavailability of Oleanolic Acid: Design and Evaluation. Int. J. Nanomed. 2013, 2917. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Tang; Duan; Tong. Oleanolic Acid Liposomes with Polyethylene Glycol Modification: Promising Antitumor Drug Delivery. Int. J. Nanomed. 2012, 3517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Lv; Li; Feng; Li; Liu; Li; Li. Preparation, Characterization, and in Vivo Pharmacokinetics of Nanostructured Lipid Carriers Loaded with Oleanolic Acid and Gentiopicrin. Int. J. Nanomed. 2013, 3227. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.; Gromadzki, D.; Jäger, E.; Giacomelli, F.C.; Kozlowska, A.; Kobera, L.; Brus, J.; Říhová, B.; El Fray, M.; Ulbrich, K.; et al. Novel “Soft” Biodegradable Nanoparticles Prepared from Aliphatic Based Monomers as a Potential Drug Delivery System. Soft Matter 2012, 8, 4343. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Dai, X.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Oleanolic Acid and Its Synthetic Derivatives for the Prevention and Therapy of Cancer: Preclinical and Clinical Evidence. Cancer Lett. 2014, 346, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Stelling-Férez, J.; Gabaldón, J.A.; Nicolás, F.J. Oleanolic Acid Stimulation of Cell Migration Involves a Biphasic Signaling Mechanism. Sci. Rep. 2022, 12, 15065. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Aleksunes, L.M.; Klaassen, C.D. Oleanolic Acid Activates Nrf2 and Protects from Acetaminophen Hepatotoxicity via Nrf2-Dependent and Nrf2-Independent Processes. Biochem. Pharmacol. 2009, 77, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Moura-Letts, G.; Villegas, L.F.; Marçalo, A.; Vaisberg, A.J.; Hammond, G.B. In Vivo Wound-Healing Activity of Oleanolic Acid Derived from the Acid Hydrolysis of Anredera Diffusa. J. Nat. Prod. 2006, 69, 978–979. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, J.M.; Trempus, C.S.; Garantziotis, S. Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int. J. Cell Biol. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kedar, U.; Phutane, P.; Shidhaye, S.; Kadam, V. Advances in Polymeric Micelles for Drug Delivery and Tumor Targeting. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Mauri, E.; Giannitelli, S.M.; Trombetta, M.; Rainer, A. Synthesis of Nanogels: Current Trends and Future Outlook. Gels 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Real, D.A.; Bolaños, K.; Priotti, J.; Yutronic, N.; Kogan, M.J.; Sierpe, R.; Donoso-González, O. Cyclodextrin-Modified Nanomaterials for Drug Delivery: Classification and Advances in Controlled Release and Bioavailability. Pharmaceutics 2021, 13, 2131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, T.; Cheng, C.; Wang, Q.; Lin, S.; Liu, C.; Han, X. Research Progress on Synthesis and Application of Cyclodextrin Polymers. Molecules 2021, 26, 1090. [Google Scholar] [CrossRef] [PubMed]
- Larrañeta, E.; Lutton, R.E.M.; Woolfson, A.D.; Donnelly, R.F. Microneedle Arrays as Transdermal and Intradermal Drug Delivery Systems: Materials Science, Manufacture and Commercial Development. Mater. Sci. Eng. R Rep. 2016, 104, 1–32. [Google Scholar] [CrossRef]
- Aldawood, F.K.; Andar, A.; Desai, S. A Comprehensive Review of Microneedles: Types, Materials, Processes, Characterizations and Applications. Polymers 2021, 13, 2815. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A. Cyclodextrin-Based Nanoparticles for Pharmaceutical Applications: A Review. Environ. Chem. Lett. 2021, 19, 4297–4310. [Google Scholar] [CrossRef]
- Krabicová, I.; Appleton, S.L.; Tannous, M.; Hoti, G.; Caldera, F.; Rubin Pedrazzo, A.; Cecone, C.; Cavalli, R.; Trotta, F. History of Cyclodextrin Nanosponges. Polymers 2020, 12, 1122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Beekman, J.; Hew, J.; Jackson, S.; Issler-Fisher, A.C.; Parungao, R.; Lajevardi, S.S.; Li, Z.; Maitz, P.K.M. Burn Injury: Challenges and Advances in Burn Wound Healing, Infection, Pain and Scarring. Adv. Drug Deliv. Rev. 2018, 123, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-W.; Hou, Y.-T.; Hsu, S. Angiogenic Potential of Co-Spheroids of Neural Stem Cells and Endothelial Cells in Injectable Gelatin-Based Hydrogel. Mater. Sci. Eng. C 2019, 99, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Sheng, T.; Yu, J.; Gu, Z.; Xu, C. Microneedle Biomedical Devices. Nat. Rev. Bioeng. 2023, 2, 324–342. [Google Scholar] [CrossRef]
- Peng, S.; Cheng, L.; Wu, Q.; Li, Y.; Ran, L.; Wang, W.; Huang, K.; Zhu, R.; Xue, S.; Zhou, C.; et al. A Modified Hyaluronic Acid–Based Dissolving Microneedle Loaded With Daphnetin Improved the Treatment of Psoriasis. Front. Bioeng. Biotechnol. 2022, 10, 900274. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk-Cwynar, B.; Partyka, D.; Zaprutko, L. Simple Amides of Oleanolic Acid as Effective Penetration Enhancers. PLoS ONE 2015, 10, e0122857. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Release Mechanisms of PLGA-Based Drug Delivery Systems: A Review. Int. J. Pharm. X 2025, 10, 100440. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, H.; Danede, F.; Siepmann, J.; Siepmann, F. Does PLGA Microparticle Swelling Control Drug Release? New Insight Based on Single Particle Swelling Studies. J. Control. Release 2015, 213, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Stebounova, L.; Ebert, S.M.; Murry, L.T.; Adams, C.M.; Murry, D.J. Rapid and Sensitive Quantification of Ursolic Acid and Oleanolic Acid in Human Plasma Using Ultra-Performance Liquid Chromatography–Mass Spectrometry. J. Chromatogr. Sci. 2018, 56, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, K.; Xia, Y.; Mo, W.; Wang, F.; Dai, W.; Niu, P. The Hepatoprotection by Oleanolic Acid Preconditioning: Focusing on PPAR α Activation. PPAR Res. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, G.; Hui, Y.; Ranaweera, S.; Zhao, C. Microfluidic Nanoparticles for Drug Delivery. Small 2022, 18, 2106580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, J.; Sun, R.; Han, S.; Yang, Z.; Teng, L. Microfluidics for Nano-Drug Delivery Systems: From Fundamentals to Industrialization. Acta Pharm. Sin. B 2023, 13, 3277–3299. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.; Forbes, N.; Roces, C.B.; Anderluzzi, G.; Lou, G.; Abraham, S.; Ingalls, L.; Marshall, K.; Leaver, T.J.; Watts, J.A.; et al. Using Microfluidics for Scalable Manufacturing of Nanomedicines from Bench to GMP: A Case Study Using Protein-Loaded Liposomes. Int. J. Pharm. 2020, 582, 119266. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Cao, Y.; Cao, M.; Wang, Y.; Cao, Y.; Gong, T. Nanomedicine in Cancer Therapy. Signal Transduct. Target. Ther. 2023, 52 8, 293. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Oleanolic acid as a formulation-sensitive payload for polymeric and polymer-assisted delivery. Oleanolic acid (OA) is a naturally occurring pentacyclic triterpenoid found in multiple botanical sources, including triterpene-rich medicinal plants such as mistletoe (Viscum album L.). Its rigid hydrophobic oleanane scaffold, limited polar functionality, poor aqueous solubility, crystallinity, low and variable bioavailability, limited barrier transport, and vehicle-dependent biological response make OA a formulation-sensitive compound. Polymeric and polymer-assisted systems, including PLA/PLGA nanoparticles, PEGylated nanoparticles, polymeric micelles, hyaluronic-acid-based responsive carriers, hydrogels, nanogels, local depots, and microneedle-compatible platforms, can be designed to improve OA dispersion, controlled release, local retention, and clinically relevant exposure.
Figure 1.
Oleanolic acid as a formulation-sensitive payload for polymeric and polymer-assisted delivery. Oleanolic acid (OA) is a naturally occurring pentacyclic triterpenoid found in multiple botanical sources, including triterpene-rich medicinal plants such as mistletoe (Viscum album L.). Its rigid hydrophobic oleanane scaffold, limited polar functionality, poor aqueous solubility, crystallinity, low and variable bioavailability, limited barrier transport, and vehicle-dependent biological response make OA a formulation-sensitive compound. Polymeric and polymer-assisted systems, including PLA/PLGA nanoparticles, PEGylated nanoparticles, polymeric micelles, hyaluronic-acid-based responsive carriers, hydrogels, nanogels, local depots, and microneedle-compatible platforms, can be designed to improve OA dispersion, controlled release, local retention, and clinically relevant exposure.

Figure 2.
Architecture of major polymeric and polymer-assisted carriers for OA delivery. The figure compares representative OA-compatible delivery platforms, including PLGA/PLA nanoparticles, PEGylated nanoparticles, polymeric micelles, hyaluronic-acid-based responsive nanoparticles, hydrogels or nanogels, thermosensitive depots, polymeric fiber membranes, and microneedle-compatible systems. These platforms differ in how they disperse, protect, release, retain, or localize OA.
Figure 2.
Architecture of major polymeric and polymer-assisted carriers for OA delivery. The figure compares representative OA-compatible delivery platforms, including PLGA/PLA nanoparticles, PEGylated nanoparticles, polymeric micelles, hyaluronic-acid-based responsive nanoparticles, hydrogels or nanogels, thermosensitive depots, polymeric fiber membranes, and microneedle-compatible systems. These platforms differ in how they disperse, protect, release, retain, or localize OA.

Figure 3.
Preparation methods and release mechanisms in OA-loaded polymeric systems. OA-loaded polymeric carriers can be prepared by nanoprecipitation, solvent displacement, emulsion-based methods, electrospinning, gelation, or microneedle casting. Their release may involve diffusion, swelling, matrix relaxation, degradation, erosion, micelle dissociation, responsive cleavage, gel-assisted retention, or tissue partitioning. Interpretation of release data should consider OA physical state, sink conditions, release medium, and the intended route of administration.
Figure 3.
Preparation methods and release mechanisms in OA-loaded polymeric systems. OA-loaded polymeric carriers can be prepared by nanoprecipitation, solvent displacement, emulsion-based methods, electrospinning, gelation, or microneedle casting. Their release may involve diffusion, swelling, matrix relaxation, degradation, erosion, micelle dissociation, responsive cleavage, gel-assisted retention, or tissue partitioning. Interpretation of release data should consider OA physical state, sink conditions, release medium, and the intended route of administration.

Figure 4.
Route-specific positioning of polymeric and polymer-assisted OA delivery platforms. The figure compares major administration routes relevant to oleanolic acid delivery, including topical/transdermal, wound/dressing, intra-articular, oral, and systemic applications. For each route, the scheme links the main biological barrier with suitable carrier classes and key evaluation endpoints. This route-specific view helps distinguish general formulation feasibility from application-oriented delivery design.
Figure 4.
Route-specific positioning of polymeric and polymer-assisted OA delivery platforms. The figure compares major administration routes relevant to oleanolic acid delivery, including topical/transdermal, wound/dressing, intra-articular, oral, and systemic applications. For each route, the scheme links the main biological barrier with suitable carrier classes and key evaluation endpoints. This route-specific view helps distinguish general formulation feasibility from application-oriented delivery design.

Figure 5.
Characterization workflow and critical quality attributes for OA-loaded polymeric nanocarriers. OA-loaded polymeric and polymer-assisted systems should be evaluated through an integrated workflow including material attributes, preparation parameters, nanostructure, surface properties, OA loading, physical state, release profile, stability, route-specific performance, biological exposure, and safety. Particle size and encapsulation efficiency alone are insufficient to define translational quality.
Figure 5.
Characterization workflow and critical quality attributes for OA-loaded polymeric nanocarriers. OA-loaded polymeric and polymer-assisted systems should be evaluated through an integrated workflow including material attributes, preparation parameters, nanostructure, surface properties, OA loading, physical state, release profile, stability, route-specific performance, biological exposure, and safety. Particle size and encapsulation efficiency alone are insufficient to define translational quality.

Figure 6.
Process–structure–function–response framework for OA-loaded polymeric and polymer-assisted nanocarriers. The figure illustrates how formulation performance depends on the relationship between process parameters, carrier structure, functional behavior, and biological response. Polymer selection, drug-to-polymer ratio, solvent conditions, drying, and sterilization influence particle or matrix architecture, OA physical state, release kinetics, colloidal stability, tissue interaction, safety, and therapeutic outcome. The framework emphasizes that small size and high loading should not be used as the only development criteria.
Figure 6.
Process–structure–function–response framework for OA-loaded polymeric and polymer-assisted nanocarriers. The figure illustrates how formulation performance depends on the relationship between process parameters, carrier structure, functional behavior, and biological response. Polymer selection, drug-to-polymer ratio, solvent conditions, drying, and sterilization influence particle or matrix architecture, OA physical state, release kinetics, colloidal stability, tissue interaction, safety, and therapeutic outcome. The framework emphasizes that small size and high loading should not be used as the only development criteria.

Figure 7.
Translational roadmap for OA-loaded polymeric and polymer-assisted nanocarriers. A rational development pathway should begin with a disease-specific target product profile, followed by polymer and carrier selection, definition of critical quality attributes, route-relevant release testing, exposure verification, disease-relevant biological models, manufacturing development, safety assessment, and comparison with simpler or standard formulation options. The most realistic near-term opportunities are likely to involve inflammatory skin disease, psoriasis, wound healing, dermal delivery, osteoarthritis, and other localized inflammatory disorders. More demanding systemic applications, including cancer, liver injury, metabolic disease, and oral delivery, require stronger pharmacokinetic, biodistribution, target-tissue exposure, and safety evidence before clinical relevance can be claimed.
Figure 7.
Translational roadmap for OA-loaded polymeric and polymer-assisted nanocarriers. A rational development pathway should begin with a disease-specific target product profile, followed by polymer and carrier selection, definition of critical quality attributes, route-relevant release testing, exposure verification, disease-relevant biological models, manufacturing development, safety assessment, and comparison with simpler or standard formulation options. The most realistic near-term opportunities are likely to involve inflammatory skin disease, psoriasis, wound healing, dermal delivery, osteoarthritis, and other localized inflammatory disorders. More demanding systemic applications, including cancer, liver injury, metabolic disease, and oral delivery, require stronger pharmacokinetic, biodistribution, target-tissue exposure, and safety evidence before clinical relevance can be claimed.

Figure 8.
Local matrix-based polymeric delivery platforms for OA: hydrogels, fiber dressings, and microneedles. The figure compares three local delivery strategies relevant to oleanolic acid: hydrogel and nanogel matrices, electrospun fiber dressings, and microneedle-assisted delivery. These platforms differ in local retention, dose loading, barrier bypass, tissue contact, and practical development challenges. The figure complements the broader route-specific overview in Figure 4 by focusing only on local and tissue-facing polymeric platforms.
Figure 8.
Local matrix-based polymeric delivery platforms for OA: hydrogels, fiber dressings, and microneedles. The figure compares three local delivery strategies relevant to oleanolic acid: hydrogel and nanogel matrices, electrospun fiber dressings, and microneedle-assisted delivery. These platforms differ in local retention, dose loading, barrier bypass, tissue contact, and practical development challenges. The figure complements the broader route-specific overview in Figure 4 by focusing only on local and tissue-facing polymeric platforms.


Table 1.
Formulation-relevant barriers of oleanolic acid and their implications for polymeric delivery.
Table 1.
Formulation-relevant barriers of oleanolic acid and their implications for polymeric delivery.
| Barrier / property | Relevance for OA delivery | Implication for polymeric or polymer-assisted carrier design | Ref. |
|---|---|---|---|
| Hydrophobic pentacyclic triterpenoid structure |
OA contains a rigid hydrophobic scaffold with limited polar functionality, which reduces aqueous compatibility. | Supports incorporation into hydrophobic polymer matrices, micellar cores, lipid–polymer interfaces, and nanogel domains. | [1,2,3] |
| Poor aqueous solubility |
Low solubility limits dissolution, biological availability, and reproducible in vitro exposure. | Polymeric nanoparticles, micelles, cyclodextrin-containing systems, and nanogels can improve apparent dispersion and handling. | [1,5] |
| Crystallinity and solid-state behavior | Crystalline OA may dissolve slowly and show variable release. | Solid-state characterization should be included to determine whether OA is crystalline, amorphous, molecularly dispersed, or phase-separated. | [5,7,9] |
| Low and variable bioavailability | Oral and systemic exposure are limited by dissolution, permeability, and formulation-dependent absorption. | Polymeric systems may improve exposure, but pharmacokinetic and tissue-distribution studies remain necessary. | [1,5,12,17] |
| Limited permeability and barrier transport | Lipophilicity alone does not ensure efficient skin, epithelial, or tissue penetration. | Nanocarriers, hydrogels, local depots, and microneedles may improve tissue deposition or bypass biological barriers. | [5,8,15,18] |
| Vehicle-dependent biological response | OA activity depends strongly on the vehicle, solubilizer, carrier, or matrix used for delivery. | Blank carriers, free OA, solubilized OA, and OA-loaded carrier controls are essential for biological interpretation. | [5,14] |
Table 2.
Representative polymeric and polymer-assisted systems used for oleanolic acid delivery.
| Delivery system |
Polymeric or polymer-assisted component |
Intended relevance |
Main contribution |
Translational significance |
Ref. |
|---|---|---|---|---|---|
| OA-loaded PEGylated PLA/PLGA nanoparticles | mPEG–PLA and mPEG–PLGA |
Cancer-oriented nanocarrier delivery | Demonstrated OA incorporation into biodegradable PEGylated polymeric nanoparticles and enhanced cytotoxic response in cancer-cell models. | Direct example of polymeric OA nanocarrier design using biodegradable PEGylated matrices. | [7] |
| OA/ursolic-acid-loaded PLGA nanoparticles | PLGA | Anticancer / comparative triterpenoid delivery | Encapsulated structurally related triterpenoids into PLGA nanoparticles and evaluated cytotoxicity in different cell lines. | Useful comparative model for hydrophobic pentacyclic triterpenoid delivery. | [9] |
| Hyaluronic-acid-based reduction-responsive OA nanoparticles | HA–OA conjugate with disulfide-responsive design | Topical psoriasis delivery |
Designed to improve topical OA delivery, cellular uptake, and disease-oriented anti-psoriasis activity. |
Strong example of polymeric nanoprodrug design linked to a specific inflammatory skin disease. |
[12] |
| Polymeric micelles of OA | Amphiphilic polymeric micelles | Dermal / skin-compatible delivery | Improved OA dispersion and evaluated skin permeation, stability, and anti-wrinkle efficacy. | Supports the feasibility of soft polymeric micelles for skin-oriented OA delivery. | [17] |
| OA-loaded PLGA fiber membranes | PLGA fiber matrix | Local delivery, wound dressing, transdermal platform | Linked processing, structure, and properties of OA-loaded polymeric membranes. | Relevant for tissue-contacting matrices and sustained local delivery. |
[8] |
| OA-loaded topical gel based on liquid crystalline nanoparticles | Gel-based polymer-assisted matrix | Topical anti-inflammatory delivery | Combined OA-loaded nanostructures with gel formulation for controlled topical release. | Demonstrates polymer-assisted local delivery through gel-based retention. | [18] |
| OA-loaded thermosensitive nanogel for knee osteoarthritis | Poloxamer thermosensitive gel | Intra-articular local delivery | Designed as an injectable local depot in a rat knee osteoarthritis model. | Important example of disease-oriented local sustained delivery. | [15] |
| Cyclodextrin-complexed OA | Cyclodextrin inclusion complex; bridge to cyclodextrin-polymer systems | Improved solubilization and biological exposure | Improved OA cell bioavailability and biological activity compared with DMSO-delivered OA. | Useful comparator and potential building block for cyclodextrin-containing polymeric systems. | [14] |
Table 3.
Functional roles, advantages, and limitations of polymeric materials used in OA delivery.
| Polymeric material / platform | Functional role in OA delivery | Advantages | Main limitations or risks | Most relevant applications | Ref. |
|---|---|---|---|---|---|
| PLGA / PLA | Biodegradable hydrophobic matrix for nanoparticle or fiber-based delivery. | Sustained release, hydrophobic drug accommodation, established use in drug delivery research. | Residual solvent, acidic degradation products, crystallization risk, incomplete release. | Cancer models, local membranes, sustained-release systems. | [7,8,9,13,31] |
| PEGylated polymers | Steric stabilization and improved colloidal dispersion. | Reduced aggregation, improved hydration, altered protein and cell interactions. | Possible reduced cellular uptake; PEG density and surface architecture require control. | PEGylated PLA/PLGA nanoparticles, PEGylated hybrid systems. | [7,32] |
| Hyaluronic acid | Biointeractive polymeric component and possible receptor-mediated delivery element. | Biocompatibility, disease-oriented interaction, potential CD44-related uptake. | Molecular-weight dependence, conjugation variability, degradation control. | Psoriasis and inflammatory skin disease. | [12,29] |
| Poloxamers and thermosensitive polymers | Gelation and local depot formation. | Injectable or topical gel formation, sustained local residence. | Concentration-dependent gelation, dilution sensitivity, local tolerability. | Intra-articular depots, topical gels, thermosensitive systems. | [15,33] |
| Cyclodextrin-containing systems | Inclusion complexation and solubility enhancement. | Improved OA handling and exposure standardization. | Complex dissociation; limited sustained release unless incorporated into polymeric networks. | Solubility enhancement, topical matrices, biological assay standardization. | [14,34,35] |
| Polymeric micelle-forming copolymers | Hydrophobic core formation and aqueous dispersion. | Improved solubilization of hydrophobic OA; dermal delivery potential. | Dilution instability, premature release, micelle dissociation. | Skin delivery and topical formulations. | [17,30] |
| Hydrogel- and microneedle-forming polymers | Local retention, controlled release, and skin-barrier bypass. | Tissue residence, minimally invasive delivery, local exposure. | Mechanical strength, dose uniformity, drying stability, irritation risk. | Wounds, inflammatory skin disease, intradermal delivery. | [15,18,36,37] |
Table 5.
Local, transdermal, and microneedle-compatible OA delivery platforms based on polymeric or polymer-assisted systems.
Table 5.
Local, transdermal, and microneedle-compatible OA delivery platforms based on polymeric or polymer-assisted systems.
| Platform | Main formulation logic |
Potential clinical relevance |
Key evaluation parameters |
Ref. |
|---|---|---|---|---|
| Topical polymeric micelles | OA solubilization in amphiphilic polymeric assemblies. | Dermal delivery, skin-compatible delivery, possible inflammatory skin applications. | Micelle size, dilution stability, skin permeation, deposition, irritation. | [17] |
| HA-based OA nanoparticles | Disease-oriented polymeric nanoprodrug and responsive delivery. | Psoriasis and inflammatory skin disease. | Uptake, skin deposition, cytokine response, irritation, repeated-dose safety. | [8] |
| OA-loaded topical gel | Nanocarrier-in-gel local delivery. | Local anti-inflammatory topical therapy. | Rheology, release, ex vivo permeation, anti-inflammatory response. | [18] |
| OA-loaded thermosensitive nanogel | Injectable local depot with temperature-triggered gelation. | Knee osteoarthritis and intra-articular therapy. | Gelation temperature, injectability, joint residence, cartilage protection, synovial inflammation. | [15] |
| OA-loaded PLGA fiber membrane | Biodegradable tissue-contacting polymeric matrix. | Wound dressing, topical patch, local depot. | Fiber morphology, mechanical strength, drug distribution, release, tissue compatibility. | [8] |
| Nanocarrier-loaded microneedles | Skin-barrier bypass combined with polymeric or nanocarrier-mediated release. | Inflammatory skin disease and intradermal OA delivery. | Needle strength, insertion, dissolution/swelling, OA deposition, carrier stability. | [36,37] |
| Polymeric wound dressing | Moisture-regulating matrix with local OA release. | Wound healing and tissue repair. | Exudate handling, migration assays, cytokines, oxidative stress, wound closure. | [8,26,28] |
Table 4.
Representative polymeric and polymer-assisted delivery systems reported for oleanolic acid.
Table 4.
Representative polymeric and polymer-assisted delivery systems reported for oleanolic acid.
| Critical quality attribute |
Why it matters for OA systems | Recommended methods |
Notes for OA polymeric systems |
Ref. |
|---|---|---|---|---|
| Particle size and polydispersity | Influence colloidal stability, tissue penetration, release, uptake, and reproducibility. | DLS, NTA, TEM, SEM, AFM. | DLS should be supported by imaging when aggregates or mixed populations are possible. | [13,31,32] |
| Surface properties | Affect aggregation, protein adsorption, cell interaction, and tissue retention. | Zeta potential, surface chemistry analysis, stability in biological media. | Especially important for PEGylated, HA-based, and hybrid systems. | [7,12,29,32] |
| Morphology and architecture | Reveal whether the system is a nanoparticle, micelle, fiber, gel, nanogel, or aggregate. | TEM, SEM, cryo-TEM, AFM, optical microscopy. | Essential for distinguishing true nanocarriers from drug-rich aggregates. | [7,8,9,13] |
| Drug loading and encapsulation efficiency | Define OA content and incorporation efficiency. | HPLC, LC-MS, validated extraction methods. | Should include recovery and separation of free vs carrier-associated OA where possible. | [5,7,9,14] |
| OA physical state | Determines release, stability, and crystallization risk. | DSC, PXRD, FTIR, Raman, solid-state NMR, microscopy. | Critical for dried systems, PLGA nanoparticles, fiber membranes, and microneedles. | [5,7,8,9] |
| Release profile | Determines how OA becomes available over time. | Dialysis, sample-and-separate methods, Franz cells, biorelevant release models. | Release medium should maintain sink conditions without artificially overestimating release. | [13,31,45] |
| Stability and shelf-life | Determine reproducibility and product feasibility. | Size/PDI over time, drug content, crystallinity, release profile, rheology. | OA recrystallization and carrier aggregation should be monitored. | [5,7,9,13] |
| Matrix or device performance | Determines clinical usability of gels, membranes, depots, and microneedles. | Rheology, gelation temperature, injectability, tensile testing, insertion tests. | Route-specific performance is essential for topical, intra-articular, wound, and microneedle systems. | [8,15,18,36,37] |
| Biological exposure and safety | Confirms whether OA reaches the target compartment and separates OA effects from carrier effects. | HPLC/LC-MS in media or tissue, blank carrier controls, cytotoxicity, irritation, histology. | Required because nominal OA dose may not equal biologically available OA concentration. | [5,12,14,15] |
Table 6.
Research priorities and translational roadmap for OA-loaded polymeric nanocarriers.
| Research priority |
Current limitation |
Recommended direction |
Expected impact |
|---|---|---|---|
| Disease-specific design | Many systems are optimized for loading rather than clinical use. | Define a target product profile for psoriasis, wound healing, osteoarthritis, cancer, or oral delivery before carrier design. |
Stronger translational relevance. |
| Physical-state control |
OA crystallinity and amorphous/crystalline transitions are often underexplored. | Include DSC, PXRD, Raman/FTIR, microscopy, and stability monitoring. |
More reliable release and shelf-life prediction. |
| Route-relevant release testing |
Simple buffer release may not reflect biological conditions. | Use skin-, wound-, synovial-, gastrointestinal-, or protein-containing media depending on application. |
More meaningful release interpretation. |
| Exposure verification |
Nominal OA dose may not equal biologically available OA. | Quantify OA in media, cells, skin layers, tissue, or local compartments. | Better link between formulation and biological response. |
| Comparative benchmarking | New formulations are often compared only with free OA. | Compare polymeric systems with cyclodextrin complexes, micelles, gels, SEDDS/SNEDDS, and standard therapies when appropriate. | Clarifies whether formulation complexity is justified. |
| Manufacturing-aware development | Many systems are difficult to sterilize, store, or scale. | Consider residual solvent, sterility, drying, packaging, process robustness, QbD, and DoE early. | Higher product-development feasibility. |
| Clinical prioritization |
OA applications are broad but not equally mature. | Prioritize local skin, wound, and osteoarthritis delivery before more demanding systemic indications. | More realistic translational pathway. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



