Submitted:
26 June 2026
Posted:
30 June 2026
You are already at the latest version
Abstract
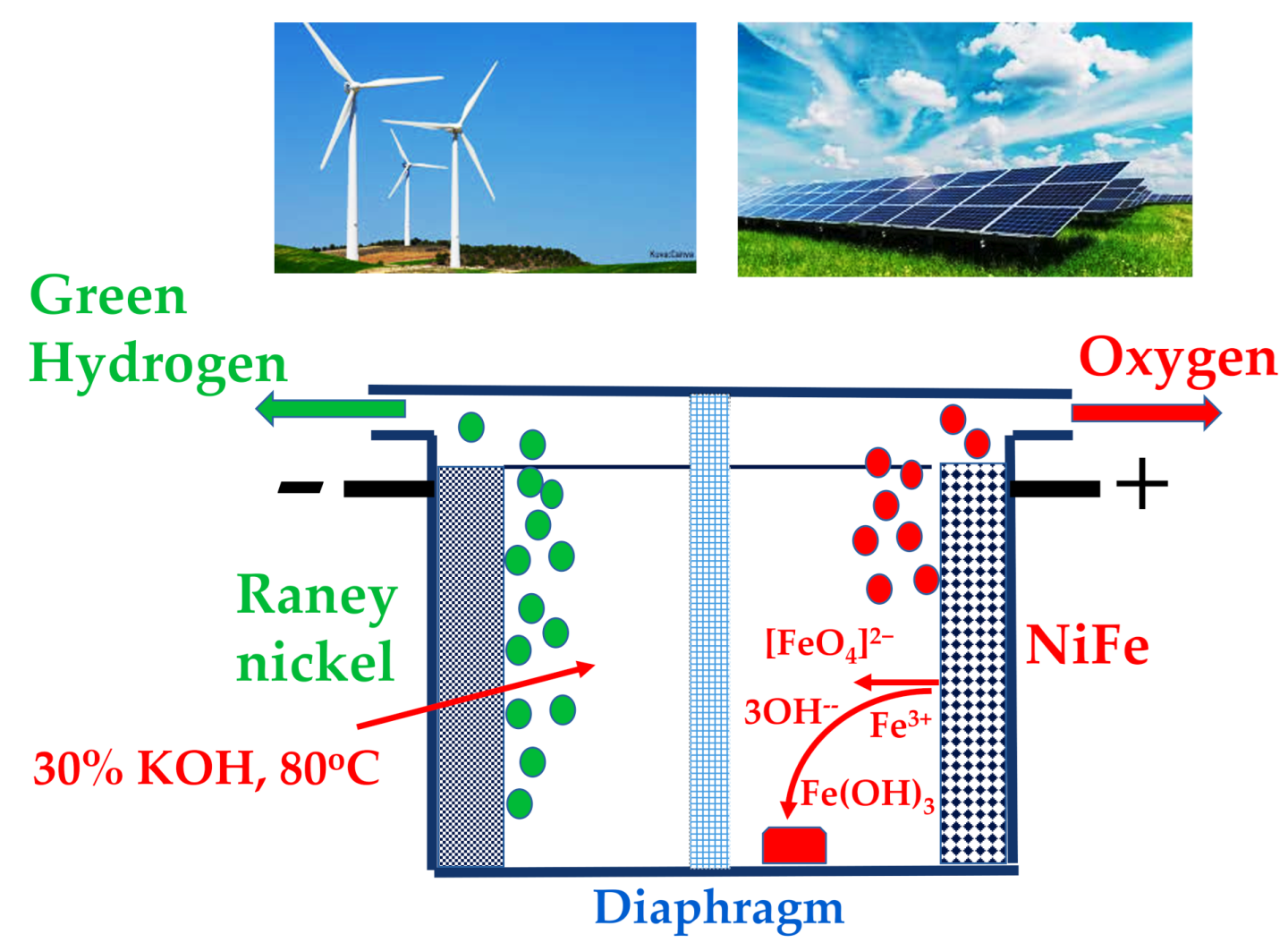
Green hydrogen production via water electrolysis is a cornerstone of the sustainable energy transition. However, the oxygen evolution reaction (OER) remains the kinetic bottleneck, limiting overall efficiency. Nickel-iron (NiFe) based catalysts are among the most promising non precious materials for the OER in alkaline media, offering high activity and low cost. Nevertheless, their practical application at industrially relevant current densities (>100 mA cm–2) is hindered by several challenges: structural degradation, uncontrolled surface reconstruction, metal dissolution (corrosion), particularly Fe leaching, and the ambiguous role of the fundamental mechanisms. This review critically discusses the current understanding of these degradation pathways, the influence of preparation methods, the interplay between Ni and Fe redox chemistry, and strategies for enhancing long-term stability. Future directions for designing durable NiFe OER electrocatalysts are also outlined. The paper also considered a strategy for investigating new catalysts using electrochemical and non-electrochemical techniques, devoted to young scientists interested in this field. In the Outlook and Perspective, the key drawback is present and the possible strategy for improvement is discussed.
Keywords:
1. Introduction
2. Fundamentals of OER and Water Electrolysis










3. The Catalyst Synthesis Methods and Their Influence on the Activity
4. Degradation Mechanisms and Stability Challenges
5. Diagnostic Indicators of Degradation and Guidance to Young Researchers




6. Outlook and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Holechek, J.L.; Geli, H.M.E.; Sawalhah, M.N.; Valdez, R. A Global Assessment: Can Renewable Energy Replace Fossil Fuels by 2050? Sustainability 2022, 14, 4792. [Google Scholar] [CrossRef]
- World Meteorological Organization (WMO). Record carbon emissions highlight urgency of Global Greenhouse Gas Watch. Available online: https://wmo.int/media/news/record-carbon-emissions-highlight-urgency-of-global-greenhouse-gas-watch (accessed on 29 May 2026).
- Elazab, M.A.; Elgohr, A.T.; Bassyouni, M.; Kabeel, A.E.; Attia, M.E.H.; Elshaarawy, M.K.; Hamed, A.K.; Alzahrani, H.A.H. Green Hydrogen: Unleashing the Potential for Sustainable Energy Generation. Results Eng. 2025, 27, 106031. [Google Scholar] [CrossRef]
- Nnabuife, S.G.; Darko, C.K.; Obiako, P.C.; Kuang, B.; Sun, X.; Jenkins, K. A Comparative Analysis of Different Hydrogen Production Methods and Their Environmental Impact. Clean. Technol. 2023, 5(4), 1344–1380. [Google Scholar] [CrossRef]
- Blumberg, T.; Morosuk, T.; Tsatsaronis, G. A Comparative Exergoeconomic Evaluation of the Synthesis Routes for Methanol Production from Natural Gas. Appl. Sci. 2017, 7(12), 1213. [Google Scholar] [CrossRef]
- Franco, A.; Giovannini, C. Recent and Future Advances in Water Electrolysis for Green Hydrogen Generation: Critical Analysis and Perspectives. Sustainability 2023, 15, 16917. [Google Scholar] [CrossRef]
- Tüysüz, H. Alkaline Water Electrolysis for Green Hydrogen Production. Acc. Chem. Res. 2024, 57(4), 558–567. [Google Scholar] [CrossRef] [PubMed]
- Anand, C.; Chandraja, B.; Nithiya, P.; Akshaya, M.; Tamizhdurai, P.; Shoba, G.; Subramani, A.; Kumaran, R.; Yadav, K.K.; Gacem, A.; Bhutto, J.K.; Alreshidi, M.A.; Alam, M.W. Green hydrogen for a sustainable future: A review of production methods, innovations, and applications. Int. J. Hydrogen Energy 2025, 111, 319–341. [Google Scholar] [CrossRef]
- Bhandari, R.; Adhikari, N. A comprehensive review on the role of hydrogen in renewable energy systems. Int. J. Hydrogen Energy 2024, 82, 923–951. [Google Scholar] [CrossRef]
- Zhu, Y.; Keoleian, G.A.; Cooper, D.R. The role of hydrogen in decarbonizing U.S. industry: A review. Renew. Sustain. Energy Rev. 2025, 214, 115392. [Google Scholar] [CrossRef]
- Bhuiyan, M.M.H.; Siddique, Z. Hydrogen as an alternative fuel: A comprehensive review of challenges and opportunities in production, storage, and transportation. Int. J. Hydrogen Energy 2025, 102, 1026–1044. [Google Scholar] [CrossRef]
- Şahin, M.E. An Overview of Different Water Electrolyzer Types for Hydrogen Production. Energies 2024, 17(19), 4944. [Google Scholar] [CrossRef]
- Chatenet, M.; Pollet, B.G.; Dekel, D.R.; Dionigi, F.; Deseure, J.; Millet, P.; Braatz, R.D.; Bazant, M.Z.; Eikerling, M.; Staffell, I.; Balcombe, P.; Shao-Horn, Y.; Schäfer, H. Water electrolysis: from textbook knowledge to the latest scientific strategies and industrial developments. Chem. Soc. Rev. 2022, 51, 4583–4762. [Google Scholar] [CrossRef] [PubMed]
- IRENA. Green hydrogen cost reduction: scaling up electrolysers to meet the 1.5°C climate goal. International Renewable Energy Agency: Abu Dhabi, 2020; Available online: https://www.irena.org/-/media/Files/IRENA/Agency/Publication/2020/Dec/IRENA_Green_hydrogen_cost_2020.pdf (accessed on 29 May 2026).
- Li, Y.-F.; Li, J.-L.; Liu, Z.-P. Structure and catalysis of NiOOH: Recent advances on atomic simulation. J. Phys. Chem. C 2021, 125, 27033–27045. [Google Scholar] [CrossRef]
- Quiroz, S.G.; Cartagena, S.; Calderón, J.A. Enhancement of oxygen evolution performance of water splitting at high current density by novel electrodeposited NiFe-LDH coatings. Electrochim. Acta 2025, 528, 146332. [Google Scholar] [CrossRef]
- Ren, X.; Zhai, Y.; Yang, N.; Wang, B.; Liu, S. Lattice oxygen redox mechanisms in the alkaline oxygen evolution reaction. Adv. Funct. Mater. 2024, 34. [Google Scholar] [CrossRef]
- Xin, S.; Tang, Y.; Jia, B.; Zhang, Z.; Li, C.; Bao, R.; Li, C.; Yi, J.; Wang, J.; Ma, T. Coupling adsorbed evolution and lattice oxygen mechanism in Fe-Co(OH)₂/Fe₂O₃ heterostructure for enhanced electrochemical water oxidation. Adv. Funct. Mater. 2023, 33(45), 2305243. [Google Scholar] [CrossRef]
- Yin, Z.-H.; Liu, H.; Hu, J.-S.; Wang, J.-J. The breakthrough of oxide pathway mechanism in stability and scaling relationship for water oxidation. Natl. Sci. Rev. 2024, 11, nwae362. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Goddard, W.A.; Xiao, H. Potential-dependent transition of reaction mechanisms for oxygen evolution on layered double hydroxides. Nat. Commun. 2023, 14, 4228. [Google Scholar] [CrossRef] [PubMed]
- De Araújo, J.F.; Dionigi, F.; Merzdorf, T.; Oh, H.; Strasser, P. Evidence of Mars-Van-Krevelen mechanism in the electrochemical oxygen evolution on Ni-based catalysts. Angew. Chem. Int. Ed. 2021, 60, 14981–14988. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-Y.; Wu, Z.-Y.; Adler, Z.; Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: From mechanistic understanding to reactor design. Joule 2021, 5, 1704–1731. [Google Scholar] [CrossRef]
- Fabbri, E.; Schmidt, T.J. Oxygen Evolution Reaction—The Enigma in water electrolysis. ACS Catal. 2018, 8, 9765–9774. [Google Scholar] [CrossRef]
- Li, P.; Jang, H.; Zhang, J.; Tian, M.; Chen, S.; Yuan, B.; Wu, Z.; Liu, X.; Cho, J. A Metal-Free N and P-Codoped carbon nanosphere as bifunctional electrocatalyst for rechargeable Zinc-Air batteries. ChemElectroChem 2018, 6(2), 393–397. [Google Scholar] [CrossRef]
- Tachikawa, T.; Beniya, A.; Shigetoh, K.; et al. Relationship Between OER Activity and Annealing Temperature of Sputter-Deposited Flat IrO₂ Thin Films. Catal. Lett. 2020, 150, 1976–1984. [Google Scholar] [CrossRef]
- Palani, R.; Anitha, V.; Karuppiah, C.; Rajalakshmi, S.; Li, Y.-J.J.; Hung, T.-F.; Yang, C.-C. Imidazolatic-Framework Bimetal Electrocatalysts with a Mixed-Valence Surface Anchored on an rGO Matrix for Oxygen Reduction, Water Splitting, and Dye Degradation. ACS Omega 2021, 6(24), 16029–16042. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Huang, X.; Zhou, L.-J.; Li, G.-D.; Fan, M.; Zou, X. Electrospinning Synthesis of Bimetallic Nickel–Iron Oxide/Carbon Composite Nanofibers for Efficient Water Oxidation Electrocatalysis. ChemCatChem 2016, 8, 992. [Google Scholar] [CrossRef]
- Si, S.; Hu, H.-S.; Liu, R.-J.; Xu, Z.-X.; Wang, C.-B.; Feng, Y.-Y. Co–NiFe layered double hydroxide nanosheets as an efficient electrocatalyst for the electrochemical evolution of oxygen. Int. J. Hydrogen Energy 2020, 45(16), 9368–9379. [Google Scholar] [CrossRef]
- Corrigan, D.A. The catalysis of the oxygen evolution reaction by iron impurities in thin-film nickel oxide electrodes. J. Electrochem. Soc. 1987, 134(2), 377–384. [Google Scholar] [CrossRef]
- Trotochaud, L.; Young, S.L.; Ranney, J.K.; Boettcher, S.W. Nickel–iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 2014, 136, 6744–6753. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Li, Y.; Wang, H.; Liang, Y.; Wu, J.Z.; Zhou, J.; Wang, J.; Regier, T.; Wei, F.; Dai, H. An advanced Ni–Fe layered double hydroxide electrocatalyst for water oxidation. J. Am. Chem. Soc. 2013, 135, 8452–8455. [Google Scholar] [CrossRef] [PubMed]
- Mohammed-Ibrahim, J. A review on NiFe-based electrocatalysts for efficient alkaline oxygen evolution reaction. J. Power Sources 2020, 448, 227375. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Khande, G.L.; Gunjakar, J.L.; Koli, V.B. Advances in Layered Double Hydroxide (LDH)-Based Materials for Electrocatalytic Nitrogen Reduction to Ammonia: A Comprehensive Review. Nitrogen 2025, 6, 106. [Google Scholar] [CrossRef]
- Guo, D.; Chi, J.; Yu, H.; Jiang, G.; Shao, Z. Self-Supporting NiFe Layered Double Hydroxide "Nanoflower" Cluster Anode Electrode for an Efficient Alkaline Anion Exchange Membrane Water Electrolyzer. Energies 2022, 15(13), 4645. [Google Scholar] [CrossRef]
- Tyndall, D.; Craig, M.J.; Gannon, L.; McGuinness, C.; McEvoy, N.; Roy, A.; García Melchor, M.; Browne, M.P.; Nicolosi, V. Demonstrating the source of inherent instability in NiFe LDH based OER electrocatalysts. J. Mater. Chem. A 2023, 11, 4067–4077. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shao, M.; An, H.; Wang, Z.; Xu, S.; Wei, M.; Evans, D.G.; Duan, X. Fast electrosynthesis of Fe containing layered double hydroxide arrays toward highly efficient electrocatalytic oxidation reactions. Chem. Sci. 2015, 6, 6624–6631. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, Y.; Lu, X. Construction of NiFe Layered Double Hydroxides Arrays as Robust Electrocatalyst for Oxygen Evolution Reaction. Catalysts 2023, 13(3), 586. [Google Scholar] [CrossRef]
- Li, T.; Liu, W.; Xin, H.; Sha, Q.; Xu, H.; Kuang, Y.; Sun, X. Large-Scale and Simple Synthesis of NiFe(OH)ₓ Electrode Derived from Raney Ni Precursor for Efficient Alkaline Water Electrolyzer. Catalysts 2024, 14(5), 296. [Google Scholar] [CrossRef]
- Li, G.; Zhang, J.; Li, L.; Yuan, C.; Weng, T.-C. Boosting the Electrocatalytic Activity of Nickel-Iron Layered Double Hydroxide for the Oxygen Evolution Reaction by Terephthalic Acid. Catalysts 2022, 12, 258. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, D.; Bao, X.; Yu, H.; Huang, B. Thermal decomposition behavior of nickel-iron hydrotalcite and its electrocatalytic properties of oxygen reduction and oxygen evolution reactions. Int. J. Hydrogen Energy 2018, 43(45), 20734–20738. [Google Scholar] [CrossRef]
- Quiroz, S.G.; Cartagena, S.; Calderón, J.A. Enhancement of oxygen evolution performance of water splitting at high current density by novel electrodeposited NiFe-LDH coatings. Electrochim. Acta 2025, 528, 146332. [Google Scholar] [CrossRef]
- Lu, X.; Zhao, C. Electrodeposition of hierarchically structured three-dimensional nickel–iron electrodes for efficient oxygen evolution at high current densities. Nat. Commun. 2015, 6, 6616. [Google Scholar] [CrossRef] [PubMed]
- Swierk, J.R.; Klaus, S.; Trotochaud, L.; Bell, A.T.; Tilley, T.D. Electrochemical study of the energetics of the oxygen evolution reaction at nickel iron (oxy)hydroxide catalysts. J. Phys. Chem. C 2015, 119(33), 19022–19029. [Google Scholar] [CrossRef]
- Wu, X.; Du, Y.; An, X.; Xie, X. Fabrication of NiFe layered double hydroxides using urea hydrolysis — Control of interlayer anion and investigation on their catalytic performance. Catal. Commun. 2014, 50, 44–48. [Google Scholar] [CrossRef]
- Zuber, A.; Oikonomou, I.M.; Gannon, L.; Chunin, I.; Reith, L.; Can, B.; Lounasvuori, M.; Schultz, T.; Koch, N.; McGuinness, C.; Menezes, P.W.; Nicolosi, V.; Browne, M.P. Effect of the precursor metal salt on the oxygen evolution reaction for NiFe oxide materials. ChemElectroChem 2024, 11(17). [Google Scholar] [CrossRef]
- Hou, C.; Cui, Z.; Zhang, S.; Yang, W.; Gao, H.; Luo, X. Rapid large scale synthesis of ultrathin NiFe LDH nanosheets with tunable structures as robust oxygen evolution electrocatalysts. RSC Adv. 2021, 11, 37624–37630. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Yoshii, H.; Nakamura, H.; Watano, S. Simple and rapid synthesis of Ni–Fe layered double hydroxide by a new mechanochemical method. Appl. Clay Sci. 2012, 58, 120–124. [Google Scholar] [CrossRef]
- Molina-Muriel, M.; Zignani, S.C.; Goberna-Ferrón, S.; Ribera, A.; Aricò, A.S.; García, H. Efficient NiFe-Layered double hydroxide electrocatalyst synthesized via a solvent-free mechanochemical method for oxygen evolution reaction. ACS Omega 2025, 10(22), 22671–22678. [Google Scholar] [CrossRef] [PubMed]
- Del Rosario, J.A.D.; Li, G.; Labata, M.F.M.; Ocon, J.D.; Chuang, P.-Y.A. Unravelling the roles of alkali-metal cations for the enhanced oxygen evolution reaction in alkaline media. Appl. Catal. B 2021, 288, 119981. [Google Scholar] [CrossRef]
- Garcia, A.C.; Touzalin, T.; Nieuwland, C.; Perini, N.; Koper, M.T.M. Enhancement of oxygen evolution activity of nickel oxyhydroxide by electrolyte alkali cations. Angew. Chem. Int. Ed. 2019, 58, 12999–13003. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, D.; Riera, M.; Zhou, R.; Deary, A.; Paesani, F. Hydration structure of Na⁺ and K⁺ ions in solution predicted by data-driven many-body potentials. J. Phys. Chem. B 2022, 126(45), 9349–9360. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Park, E.J.; Zhu, W.; Shi, Q.; Zhou, Y.; Tian, H.; Lin, Y.; Serov, A.; Zulevi, B.; Baca, E.D.; Fujimoto, C.; Chung, H.T.; Kim, Y.S. Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers. Nat. Energy 2020, 5(5), 378–385. [Google Scholar] [CrossRef]
- Kubo, N.M.; Ketter, F.; Palkovits, S.; Palkovits, R. Nickel and Commercially Available Nickel-Containing Alloys as Electrodes for the Electrochemical Oxygen Evolution. ChemElectroChem 2024, 11, e202300460. [Google Scholar] [CrossRef]
- Luo, Y.; He, Y.; Li, J.; Wei, S.; Chen, L. Reinforced lattice oxygen mechanism of NiFe-LDH@Fe₂O₃@NF by optimizing the adsorption of oxygen intermediates for efficient water electrolysis. J. Environ. Chem. Eng. 2025, 13, 115497. [Google Scholar] [CrossRef]
- Li, L.; Bai, L.; She, S.; Chen, G.; Huang, H. Mixed ionic conductor brings extra gain in oxygen-evolving activity of NiFe hydroxide electrocatalyst at practical working temperature. Appl. Catal. B 2025, 371, 125271. [Google Scholar] [CrossRef]
- Pascuzzi, M.E.C.; Man, A.J.W.; Goryachev, A.; Hofmann, J.P.; Hensen, E.J.M. Investigation of the stability of NiFe-(oxy)hydroxide anodes in alkaline water electrolysis under industrially relevant conditions. Catal. Sci. Technol. 2020, 10, 5593–5601. [Google Scholar] [CrossRef]
- Han, Y.; Wang, J.; Liu, Y.; Li, T.; Wang, T.; Li, X.; Ye, X.; Li, G.; Li, J.; Hu, W.; Deng, Y. Stability challenges and opportunities of NiFe-based electrocatalysts for oxygen evolution reaction in alkaline media. Carbon Neutraliz. 2024, 3(2), 172–198. [Google Scholar] [CrossRef]
- Goldsmith, Z.K.; Young, S.L.; Ranney, J.K.; Boettcher, S.W. Characterization of NiFe Oxyhydroxide Electrocatalysts by Integrated Electronic Structure Calculations and Spectroelectrochemistry. Proc. Natl. Acad. Sci. USA 2017, 114(12), 3050–3055. [Google Scholar] [CrossRef] [PubMed]
- El Boumlasy, S.; Pascale, M.; De Luca, O.; Caruso, T.; Mirabella, S.; Terrasi, A.; Aricò, A.S.; Ruffino, F. Highly efficient and stable NiFe oxide-based electrocatalysts for oxygen evolution in alkaline and saline solutions. Appl. Surf. Sci. Adv. 2025, 28, 100809. [Google Scholar] [CrossRef]
- Lin, Z.; Bu, P.; Xiao, Y.; Gao, Q.; Diao, P. β- and γ-NiFeOOH electrocatalysts for an efficient oxygen evolution reaction: an electrochemical activation energy aspect. J. Mater. Chem. A 2022, 10, 20847–20855. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, P.; Cheng, Y.; Mo, Y.; Luo, X.; Liu, P.; Guo, R.; Liu, X. Recent progress on NiFe₂O₄ spinels as electrocatalysts for the oxygen evolution reaction. J. Electroanal. Chem. 2023, 946, 117703. [Google Scholar] [CrossRef]
- He, L.; Zhou, Y.; Wang, M.; Li, S.; Lai, Y. Recent Progress on Stability of Layered Double Hydroxide-Based Catalysts for Oxygen Evolution Reaction. Nanomaterials 2024, 14, 1533. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Ehlers, J.C.; Hussain, I.; Zhang, K.; Chatzichristodoulou, C. Trends and industrial prospects of NiFe-layered double hydroxide for the oxygen evolution reaction. Chem. Eng. J. 2024, 499, 156219. [Google Scholar] [CrossRef]
- Plevová, M.; Hnát, J.; Bouzek, K. Electrocatalysts for the Oxygen Evolution Reaction in Alkaline Media: A Comparative Review. J. Power Sources 2021, 507, 230072. [Google Scholar] [CrossRef]
- Soni, A.; Maurya, S.K.; Malviya, M. Exploring electrocatalysts for oxygen evolution: A comprehensive comparative review in alkaline and acidic medium. J. Power Sources 2025, 636, 236571. [Google Scholar] [CrossRef]
- Li, L.-F.; Li, Y.-F.; Liu, Z.-P. Oxygen Evolution Activity on NiOOH Catalysts: Four-Coordinated Ni Cation as the Active Site and the Hydroperoxide Mechanism. ACS Catal. 2020, 10, 2581–2590. [Google Scholar] [CrossRef]
- Li, X.; Ruan, M.; Shen, Y.; Wen, M.; Li, Z.; Yin, H.; Chen, F.; Cheng, Y.; Lei, P.; Qian, L. Phase locking of NiOOH@β-Ni(Fe)OOH reconstructed simultaneously for robust oxygen evolution at high current density. ACS Appl. Mater. Interfaces 2025, 17(31), 44562–44572. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yoon, J. Strategies for Advancing Electrodeposited Co-Based Oxygen Evolution Catalysts from Alkaline to Pure-Water Conditions. ACS Mater. Lett. 2025, 7(9), 3128–3140. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 2013, 135(45), 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Van Beek Pedersen, T.; Molander, J.; Jakobsen, R.; Tobler, D.J.; Dideriksen, K. Dissolution kinetics for the Fe(II)-Fe(III) layered double hydroxide, green rust. Appl. Clay Sci. 2025, 272, 107814. [Google Scholar] [CrossRef]
- De Koninck, M.; Bélanger, D. The electrochemical generation of ferrate at pressed iron powder electrode: comparison with a foil electrode. Electrochim. Acta 2003, 48, 1435–1442. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Fei, Y.; Zheng, Z. The degradation mechanism of NiFe-LDH catalysts in alkaline oxygen evolution reaction at high temperature. Mater. Lett. 2025, 398, 138901. [Google Scholar] [CrossRef]
- Elsharkawy, S.; Żabiński, P. Effect of Fe/Ni ratio on electrodeposition of Ni-Fe alloys and their bifunctional catalytic performance in hydrogen and oxygen evolution reactions. J. Power Sources 2025, 660, 238516. [Google Scholar] [CrossRef]
- Grgur, B.N.; Popović, A.S. unpublished results. 2025. [CrossRef]
- Yu, J.; Fu, X.; Wang, H.; Lu, S.; Li, B. Rational construction of nano-scaled FeOOH/NiFe-LDH for efficient water splitting. Nanomaterials 2025, 15, 949. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, V.M.; Dimić-Mišić, K.M.; Maslovara, S.L.; Popović, D.P.; Gigov, M.N.; Krstić, S.S.; Kaninski, M.P.M. Advances in Alkaline Water Electrolysis—The role of in situ ionic activation in green hydrogen production. Catalysts 2026, 16, 98. [Google Scholar] [CrossRef]
- Qiao, X.; Zhu, Q.; Hou, G.; Pang, Z.; Kang, H. Pinning effect of lattice Co enhances lattice oxygen regeneration in NiFe-LDH for oxygen evolution reaction. J. Colloid Interface Sci. 2025, 699, 138219. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Du, L.; Yan, L.; Park, S.; Qiu, Y.; Sokolowski, J.; Wang, W.; Shao, Y. Oxygen evolution reaction in alkaline environment: material challenges and solutions. Adv. Funct. Mater. 2022, 32(21). [Google Scholar] [CrossRef]
- Zhang, C.; Wang, J.; Ma, H.; Wang, J.; Xu, R.; Li, G.; Yang, L.; Guo, H. Electronic structure engineering of NiFe hydroxide nanosheets via ion doping for efficient OER electrocatalysis. Chem. Eng. J. 2024, 499, 156430. [Google Scholar] [CrossRef]
- Qu, J.; Dong, Y.; Zhang, T.; Zhao, C.; Wei, L.; Guan, X. Impact of bimetallic synergies on Mo-doping NiFeOOH: Insights into enhanced OER activity and reconstructed electronic structure. Front. Energy 2024, 18, 850–862. [Google Scholar] [CrossRef]
- Assavachin, S.; Ittisanronnachai, S.; Atithep, T.; Chitterisin, N.; Sawangphruk, M. Dynamic ferrous iron regeneration enables stabilization of NiFeCo layered double hydroxide for enhanced alkaline water oxidation. J. Power Sources 2025, 650, 237494. [Google Scholar] [CrossRef]
- Li, J.; Wei, Y.; Zou, L.; Liu, Y.; Li, S.; Luo, Y. Cu dual-site doping: synergistic enhancement of OER activity through LDH and nickel foam interface engineering. New J. Chem. 2025, 49, 17577–17587. [Google Scholar] [CrossRef]
- Zhang, X.; Tong, L.; Shi, X.; Li, Z.; Xiao, Z.; Liu, Y.; Zhang, T.; Lin, S. Tailoring atomically local electric field of NiFe layered double hydroxides with Ag dopants to boost oxygen evolution kinetics. J. Colloid Interface Sci. 2024, 668, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.S.-D.; Hartert, A.; Oestreicher, V.; Romero, J.; Jaramillo-Hernández, C.; Muris, L.J.J.; Thorez, G.; Vieira, B.J.C.; Ducourthial, G.; Fiocco, A.; Legendre, S.; Huck-Iriart, C.; Mizrahi, M.; López-Alcalá, D.; Freiberg, A.T.S.; Mayrhofer, K.J.J.; Waerenborgh, J.C.; Baldoví, J.J.; Cherevko, S.; Varela, M.; Thiele, S.; Lloret, V.; Abellán, G. Scalable synthesis of NiFe-layered double hydroxide for efficient anion exchange membrane electrolysis. Nat. Commun. 2025, 16(1), 6138. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



