Submitted:
26 June 2026
Posted:
29 June 2026
You are already at the latest version
Abstract
This research investigates the systematic nanomechanical behavior of tetragraphene-based nanotubes (TGCNTs) using reactive classical molecular dynamics (CMD) simulations with the AIREBO-Morse potential. Tetragraphene is a novel carbon allotrope characterized by a unique mixture of sp2 and sp3 hybridization. We analyzed the nanomechanical properties of zigzag-like TGCNTs under uniaxial tensile loading, systematically examining the effects of chirality, diameter, length, and temperature ranging from 300 K to 2100 K. Our results reveal a distinct nanostructural degradation at high temperatures, where the nanotubes completely lose their structural stability above 1500 K. Under mechanical strain, the stress-strain curves highlight a strong dependence on chirality. The (0,N) TGCNTs exhibit brittle behavior, characterized by a short, nearly linear curve that terminates abruptly at a rapid fracture point without significant plastic deformation. In contrast, the (N,0) TGCNTs demonstrate remarkable ductility and superelasticity. This is evidenced by a distinct plateau effect with constant stress up to 20% strain, followed by strain hardening until ultimate fracture at over 40% strain, indicating a stress-induced structural phase transition. To map their transverse elasticity, the Poisson’s ratio (ν) was evaluated within the elastic regime, revealing an ultra-low value of ν=0.07 for the TGCNT (0,10) in close agreement with density functional theory (DFT) benchmarks, contrasting with an anomalously high value of ν=1.19 for the TGCNT (14,0) due to severe chiral anisotropy. The calculated Young’s modulus values range from 2714.10 to 3166.20 GPa.Å for (N,0) TGCNTs and 1886.70 to 2324.30 GPa.Å for (0,N) TGCNTs. These insights into the nanostructure-property relationships of TGCNTs provide essential design guidelines for their application in flexible electronics, nanocomposites, and nanoscale shock-absorbing devices.
Keywords:
classical moleuclar dynamics simulations
; reactive interatomic force field – ReaxFF
; tetragraphene
; carbon nanotubes
; nanomechanical behavior
; elastic modulus
; fracture toughness
1. Introduction
The scientific community has, in recent years, significantly emphasized the superiority of nanostructured materials over bulk alternatives, exploring their versatile applications within numerous technological and scientific sectors [2,180]. Nanoscience, together with nanotechnology, focuses on analyzing materials possessing dimensions that fall within the nanometer regime [3]. These systems exhibit distinct biological, chemical, physical, and engineering characteristics, which are determined by length scales often ranging from 10 to 200 Angstroms [4].
At the nanoscale, a multidisciplinary environment emerges where chemistry, physics, and engineering converge, facilitating the development of unprecedented properties in condensed matter [5,6]. Leveraging these unique physical characteristics, it is possible to manipulate individual atoms, leading to innovations such as molecular robots and engines capable of mimicking cellular functions [7,8,9]. An effective approach for analyzing the progress of these scientific fields involves thoroughly reviewing the increasing body of research literature focused on the properties of new nanostructures.
Based on this scientific description, scientists has done a lot of research work whith aims to understand the condensed matter at nanometric scale, theoretically and experimentally [10,11,12,13,14,15]. The manufacturing processes of carbon-based nanostructures and materials in laboratory scale (theory and experiment) may be categorized throughout changes carbon-based nanostructures and materials. It is based on the allotropy of carbon that we have achieved significant advances in science and technology. Owing to their outstanding physicochemical characteristics, carbon atoms and their diverse allotropic forms have profoundly altered the landscape of the electronic and optoelectronic sectors, sparking extensive research into their prospective roles in modern nanoscience [16,17,18,19,20,21,22,23,24]. A primary factor behind these versatile features is the chemical valency of carbon, which enables the formation of numerous distinct nanostructural networks. Over the past few decades, a variety of novel carbon-based materials and nanostructures have been isolated, most notably zero-dimensional fullerenes [25], one-dimensional carbon nanotubes (CNTs) [26], and two-dimensional isolated graphene layers [35]. The discovery of these unique bonding configurations effectively initiated a new epoch of technological breakthroughs and fundamental scientific exploration.
The experimental isolation of a single-layer graphene sheet via mechanical exfoliation in 2004 fundamentally redefined the boundaries of condensed matter physics and engineering, an achievement subsequently honored with the Nobel Prize [35]. Structurally, graphene consists of a flat, monomolecular plane of carbon atoms densely arranged in a two-dimensional () honeycomb topology. This unique configuration imparts extraordinary physical characteristics, establishing it as a highly promising material for next-generation technological applications [27]. The structural adaptability of this network directly determines its unique electronic features; specifically, the hybridization involving one s and two p orbitals yields a robust trigonal planar morphology, where carbon atoms are linked by strong bonds with an equilibrium bond length of Å. Consequently, graphene displays a remarkable intrinsic electron mobility of up to and an exceptional electrical conductivity around S/cm stemming from its zero-bandgap nature [28]. Furthermore, its thermal conductivity ranges between and W/mK [29], complemented by distinct optical reflectance and transmittance profiles [30]. Ambient-temperature ferromagnetism can also be induced through surface molecular adsorption [31], while its mechanical robustness is evidenced by an elastic Young’s modulus reaching approximately 1 TPa ( GPa) [32,33]. However, despite these unparalleled intrinsic traits, the complete implementation of graphene in scalable nanoelectronic devices is heavily restricted by its lack of an energy bandgap [34].
To overcome these nanostructural limitations, numerous alternative allotropes and low-dimensional materials have been proposed in recent years, aiming to replicate or improve upon the foundational merits of graphene. These include alternative elemental monolayers and advanced carbon networks such as two-dimensional covalent carbon nitride nanosheets [36], mixed - coordinated graphynes [37], boron nitride nanosheets [38], silicon-based silicene [39], penta-graphene [40], phagraphene [41], popgraphene [42], twin graphene [43], octagraphene [44], T-carbon [45], D-carbon [46], cubic [47], -carbon [48], [49], trigraphene [50], tetrahexcarbon [51], anodes [52], [53], [54], and networks [55], -graphene [56], -graphene [57], -graphene [58], pentagraphyne [59], two-dimensional [60], naphthylene- [61], [62], thgraphene [63], tetrahex carbides [64], pentahexoctite [65], hexatetra-carbon [66], tower carbon [67], novamene [68], orthorhombic carbon [69], carbon [70], -Tcarbon [71], [72], graphdiyne architectures [73], [74], superprismane [75], orthorhombic [76], four-penta-graphenes [77], R-graphyne [78], biphenylene monolayers [79], [80], [81], -graphene [82], -carbon [83], dodecanophene systems [84], M-graphene [85], [86], Penta- [87], [88], sheets [89], pentaheptite diamond [90], [91], carbon [92], poly-dodecahedrane [93], structures [94], -graphyne [95], janus graphene [96], -carbon [97], -graphene [98], tetragonal [99], twin T-graphene [100], superhard [101], spiro-carbon [102], diamond [103], -carbon [104], monoclinic [105], azugraphene [106], [107], citecai2023lc, topologies [109], -graphene [110], -graphene [111], C-centered orthorhombic [112], [113], Q-carbon [114], [115], escherynes [116], petal-graphyne citelima2025petal, orthorhombic [118], irida-graphene [119], and sun-graphyne [120].
Emerging directly from this nanostructural design paradigm is tetragraphene, a novel carbon allotrope characterized by a unique mixture of and hybridization. When rolled into one-dimensional architectures, these sheets form tetragraphene-based nanotubes (TGCNTs). Theoretical investigations into the intrinsic physical and electronic properties of TGCNTs have already been initiated within the scientific community, most notably through Density Functional Theory (DFT) calculations by Brandão et al [121], regarding topological and strain-induced bandgap engineering.
However, such quantum-mechanical approaches are intrinsically limited to small, idealized systems at absolute zero temperature. This constraint creates a critical “DFT gap”, leaving a vital need to understand how these one-dimensional nanostructures behave under realistic, large-scale, and dynamic conditions. To bridge this gap, classical molecular dynamics (CMD) simulations are uniquely required, as they allow for the accurate exploration of large spatial scales, high-temperature fluctuations, fracture toughness, and complex dynamic deformation pathways. Consequently, this work employs reactive CMD simulations with the AIREBO-Morse potential to systematically investigate TGCNTs under uniaxial tensile loading across a wide thermal range (300 to 2100 K). Crucially, this study uncovers unprecedented phenomena not previously reported in the literature, including a strict thermal degradation threshold above 1500 K and a striking chirality-dependent divergence in fracture modes. Specifically, we report a stress-induced nanostructural phase transition in TGCNTs, marked by a constant-stress plateau up to 20% strain and subsequent strain hardening, contrasting sharply with the brittle fracture observed in configurations. Ultimately, these novel insights into the fracture toughness, thermal stability limits, and dynamic deformation mechanisms of TGCNTs provide essential design guidelines for their integration into next-generation flexible electronics, nanocomposites, and nanoscale energy-absorption systems.
2. Geometric Generation and Configuration of Tetragraphene-Based Nanotubes (TGCNTs)
The one-dimensional (1D) spatial morphology of a tetragraphene carbon nanotube (TGCNT) is established by mathematically rolling a single layer of tetragraphene. This structural mapping is governed by the chiral indices and . The primary geometric metrics include the tube diameter and a specific zigzag-like orientation. These dimensional factors, alongside the wall thickness, play a vital role in dictating the mechanical and elastic response of the resulting TGCNTs.
While the fundamental concept of rolling a 2D sheet is conceptually analogous to standard graphene, the explicit atomic generation of TGCNTs differs due to the distinct symmetry of the parent tetragraphene lattice. Formally, the circumferential chiral vector () is expressed as a linear combination of the two-dimensional rectangular basis vectors ( and ) shown in Figure Figure 1 (a) and, the Equation 1 [121]:
where n and m are integer values that define the nanotube chirality, with the basis magnitudes given as Å and Å. This vector directly governs the cross-sectional dimensions of the system. Consequently, the diameter of the TGCNT () is computed from these index values via the following relation [121]:
where represents the characteristic lattice parameter of the tetragraphene nanostructure [121].
The nanostructural translation along the longitudinal axis is described by the translational vector (), which is oriented perpendicular to (see Figure 1 (a)) [121]:
The integer coefficients and are derived from the orthogonality condition requiring the dot product to vanish, . Given that for this orthogonal system, the coefficient ratio simplifies to [121]:
The total length of the periodic nanotube section is defined by the magnitude . Together, the boundary vectors and completely outline the boundaries of the TGCNT unit cell [121]. Because of the rectangular symmetry of the underlying lattice, this setup allows for the creation of zigzag-like configurations denoted by or indices [121]. The explicit structural attributes of the specific TGCNTs investigated in this study are summarized in in Table 1.
3. Reactive Molecular Dynamics Methodology and Computational Details
To evaluate the nanomechanical behavior and distinct physical features of tetragraphene-based nanotubes (TGCNTs), atomistic calculations were performed using the reactive classical molecular dynamics simulations (CMD) framework. These computational experiments were implemented within the open-source LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) software suite [141,142,143]. As a robust and widely adopted platform for atomistic modeling, LAMMPS enables the systematic replication of nanomechanical deformations and stress-strain responses in low-dimensional nanostructures. This methodology provides critical insights into structural evolution and nanoscale failure mechanisms that remain challenging to probe through conventional experimental techniques [144].
In this study, the mechanical characteristics of the TGCNTs are probed by imposing continuous physical deformations on the model and monitoring the resulting nanostructural feedback. The specific virtual testing protocol utilizes uniaxial tensile loading regimes, wherein the dimensions of the simulation box are extended along a single axis under a fixed, uniform strain rate. This procedure allows for the direct derivation of stress-strain relations. From the resulting data curves, primary mechanical metrics, specifically the Young’s modulus, ultimate tensile strength (), fracture strain threshold, and total energy absorption capacity (toughness), are quantitatively determined [145,146]. By the fact that TGCNTs one dimentional nanostrucutures are composed by carbon atoms, a carbon atom, like all matter, is fundamentally governed by quantum mechanics [147]. However, it can be a useful and accurate approximation to treat a carbon atom as a classical particle under certain conditions, primarily when observing microscopic statistical averages properties, under external effects such as temperatures [148].
Given that carbon atoms within this regime can be accurately treated as classical point masses, the complete set of atomic interactions is governed by parameterised force fields [149]. These interatomic potentials serve as the foundational mathematical framework that evaluates the total potential energy of the configuration based on spatial coordinates, subsequently yielding the net forces exerted on each particle. As a core component of atomistic modeling platforms like classical molecular dynamics (CMD), these formulations are utilized here to quantify critical nanomechanical parameters, including the elastic response, maximum load capacity, and nanostructural failure pathways of the TGCNTs [150,151]. Specifically, the Adaptive Intermolecular Reactive Empirical Bond Order Morse (AIREBO-Morse) potential, based on the foundational framework formulated by Stuart et al. [191], was implemented to govern the covalent interactions within the carbon network. This formulation incorporates a Morse long-range term to describe intermolecular interactions, which has demonstrated remarkable success in accurately describing the nanomechanical behavior, stability, and nanostructural properties of diverse carbon-based systems, as thoroughly demonstrated in Appendix B of this research article. As a bond-order-dependent potential, the AIREBO-Morse framework effectively captures continuous bond breaking and formation under high strain rates without requiring predefined bonding topologies. Consequently, it achieves superior fidelity in resolving localized stress concentrations, ultimate tensile limits, and subsequent fracture dynamics inherent to the investigated TGCNTs.
To observe and animate the real-time atomistic failure dynamics of the TGCNTs, the VMD (Visual Molecular Dynamics) software suite was utilized [160]. The underlying Newtonian equations of motion for the carbon atoms are integrated temporally via the standard Verlet scheme [161], adopting a conservative time step of fs. To mitigate finite-length artifact errors, a periodic boundary condition was enforced strictly along the longitudinal z-axis of the system. Conversely, to suppress any spurious residual stresses during the stretching phase, an expansive simulation box was constructed to surround the TGCNT with a 100 Å vacuum region along both the x and y lateral dimensions. This layout guarantees a zero-stress state across the effective cross-sectional thickness of the nanotube.
The influence of thermal conditions was examined systematically across a wide temperature range, specifically at 300, 600, 900, 1200, 1500, 1800, and 2100 K. Every molecular trajectory was initiated with a rigorous nanostructural energy minimization phase. This pre-relaxation step employed a Polak-Ribiere conjugate gradient () algorithm (specified as style “cg”). The energy and force convergence thresholds for these initial iterations were set to an energy tolerance () of and a force tolerance () of [161,163,164].
Following minimization, the configurations were equilibrated to a zero-stress state for ns using an isothermal-isobaric Nosé-Hoover () barostat protocol with a pressure damping coefficient of 50 [165]. Subsequently, a ns thermalization phase was performed under a canonical Nosé-Hoover () ensemble with a temperature damping coefficient of 5 [166]. Uniaxial loading was subsequently applied by continuously expanding the boundaries of the simulation domain along the periodic z-direction. These tensile tests were conducted within the ensemble at a constant engineering strain rate of . Under this regime, the instantaneous TGCNTs length (L) evolves according to the linear relationship [167,168]:
where represents the baseline length and denotes the cumulative simulation time. Finally, the intrinsic Young’s modulus () was extracted from the linear elastic regime using the classical definition [167,168]:
where corresponds to the component of the Virial stress tensor and signifies the strain experienced along the uniaxial z-axis [167,168].
The Young’s modulus is obtained in a linear regime (3% strain). The standard errors are obtained from a single linear regression fit. The macroscopic stress tensor (specifically, the Cauchy stress) is commonly and effectively estimated using a generalization of the virial theorem in molecular simulations and statistical mechanics. This definition, known as the virial stress, provides a direct link between microscopic atomic behavior and macroscopic continuum properties [169,170,171]:
where are the mass and the velocity vector of atom i. ⊗ denotes the tensor product of two vectors. denotes the position of atom i. is the position vector of atom j relative to that of atom i, and is the interatomic force exerted on atom i by atom j, where:
where, P is the potential energy of the atomic ensemble. V is the volume of the TGCNTs one-dimentional nanostructures. , A is the surface area and thickness of the single layer of Tetragraphene. The von Mises stress tensor distribution per atom is defined as [172]:
The symbols , , and indicate the stresses in the x, y, and z directions, respectively. The , , and represent the shear stress components. The concept of von Mises stress, primarily a continuum mechanics theory for predicting yielding in ductile nanostrucutres/materials, is adapted for atomic-level analysis in classical molecular dynamics (CMD) simulations [173,174].
3.1. Evaluation and Formulation of Poisson’s Ratio in TGCNTs
The transverse deformation behavior of the tetragraphene carbon nanotubes (TGCNTs) is quantified by determining their Poisson’s ratio through the reactive classical molecular dynamics (CMD) framework. The resulting values exhibit a clear dependence on the geometric features of the nanotubes, including their specific diameter and chirality. To evaluate this property, the cylindrical nanostructures are subjected to an unconstrained uniaxial tensile load applied along the longitudinal z-axis. Because no radial restrictions are imposed on the system, the nanotube is free to contract laterally under deformation. Consequently, the intrinsic Poisson’s ratio () is mathematically defined by the following relation [175,176]:
where, Poisson’s ratio, radial strain and uniaxial strain applied in TGCNTs. The Poisson’s ratio, , of TGCNT and are calculated by (Equation 12) [175,176]:
In this formulation, represents the induced radial strain, while denotes the applied longitudinal deformation. To compute these spatial variations, the total length (L) of the representative systems, specifically the and TGCNTs, is partitioned into uniform volumetric segments of thickness . The corresponding cross-sectional radial strain is evaluated using the following expression:
where represents the mean tube radius evaluated at a given uniaxial strain level , and signifies the baseline radius at thermal equilibrium (). The mean strained radius is determined by averaging across all discrete nanostructural segments as follows:
Here, defines the instantaneous average radius of the i-th circular segment under the longitudinal strain . This segment-specific radius corresponds to the mean spatial distance from each constituent carbon atom to the local center of mass of that particular slice, computed via:
where the radial distance for each carbon atom is given by the geometric relation:
In Equation 16, and correspond to the planar coordinates of the -th carbon atom, M represents the total atomic population assigned to each individual slab, and the coordinates and define the instantaneous center of mass for that specific segment. For all calculations, a fixed slice thickness of Å was adopted, ensuring a statistically robust and sufficient number of atoms per segment to accurately map the radial geometry [177].
4. Results, Analysis, and Discussion
The systematic nanomechanical evaluation of tetragraphene-based nanotubes (TGCNTs) via reactive classical molecular dynamics (CMD) simulations reveals a correlation and new finds of their atomic nanostructure nanomechanical response under uniaxial strain. The unique mixture of and hybridization within the tetragraphene lattice introduces distinct deformation pathways that are highly sensitive to external constraints, such as temperature and loading conditions.
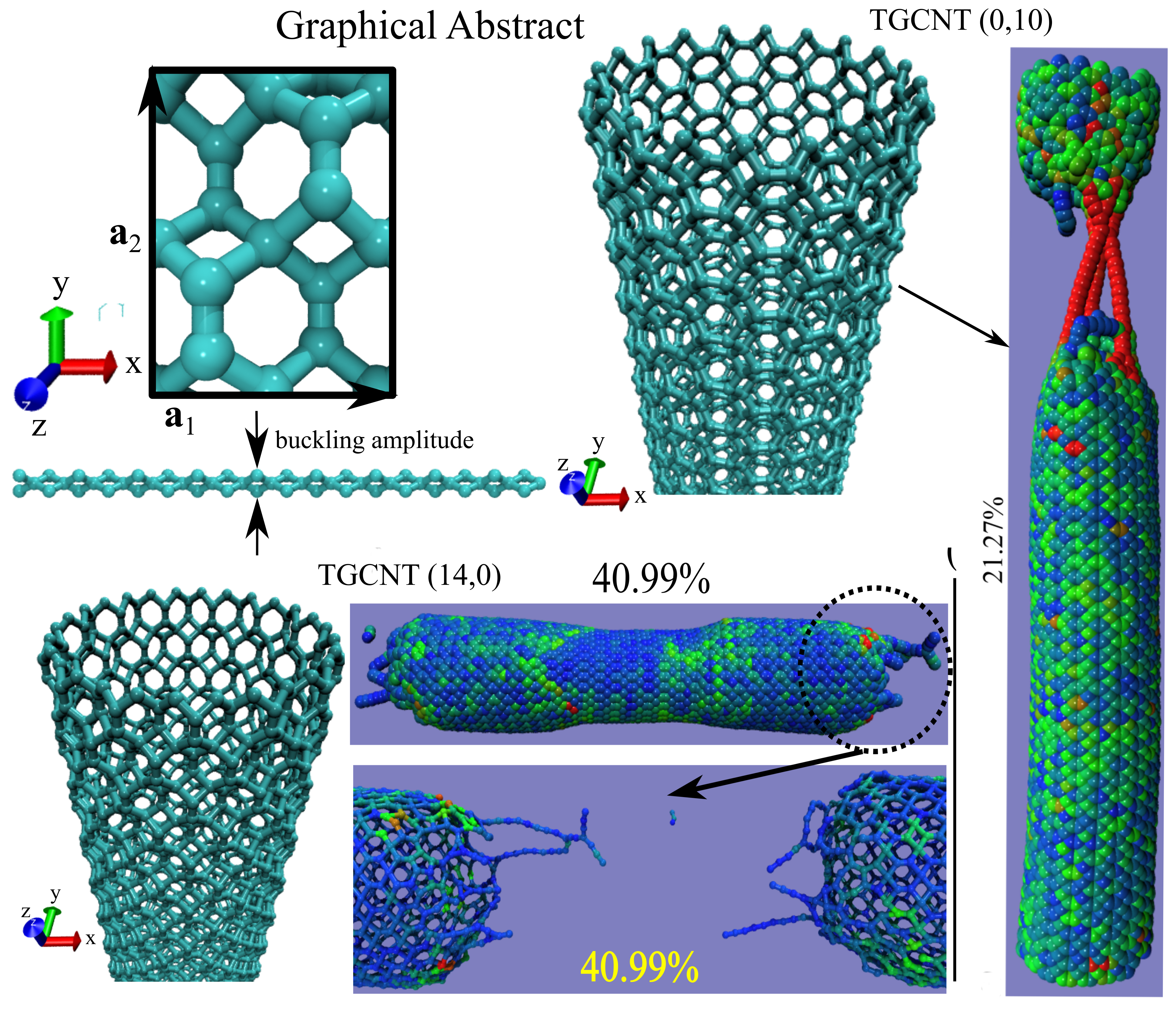
CMD simulations using the AIREBO-Morse potential reveal a strict chirality-dependent, anisotropic nanomechanical behavior in tetragraphene-based nanotubes (TGCNTs), distinguishing between brittle and ductile failure modes (see Table 1, Figure 2, Figure 3, Figure 4 and, Figure 5). While TGCNTs exhibit abrupt (see Figure 2), brittle failure, TGCNTs display a notable 20% stress plateau indicative of a strain-induced phase transformation followed by significant strain-hardening and superior fracture strain (see Figure 4). According to research by Brandão et al. [121], the severe nanomechanical anisotropy of tetragraphene-based nanotubes (TGCNTs) arises from the alignment of parallel dimers and buckling connections within its trilayer nanostructures. For TGCNTs, the bonds align with the tension axis, allowing for a flattening phase transformation and a superelastic strain plateau, while in TGCNTs, the rigid dimers align with the tension axis, resulting in brittle fracture due to limited nanostructural rearrangement. Most notably, our findings demonstrate a critical thermal threshold at 1500 K, beyond which severe nanostructural degradation occurs, leading to a complete loss of nanostructural stability. This thermal vulnerability underscores the delicate balance of the hybrid bonding network, setting a clear operational boundary for TGCNTs in high-temperature applications while highlighting the importance of environmental factors in their nanostructural integrity (see graphical representation (stress-strain) in Figure 3 and Figure 5). This thermal degradation is explicitly reflected in the simulated stress-strain curves (Figure 3 and Figure 5), which reveal a sharp and systemic reduction in maximum tensile strength, Young’s modulus (discussed later), and fracture strain as the temperature approaches 1500 K. Above this critical threshold, the characteristic stress-strain profile collapses completely into an amorphous low-stress response, characteristic of a nanostructural melting phase.
The complete loss of nanostructural stability above 1500 K is driven by two main factors: High-Energy Bond Rupture: Unlike traditional graphene which contains only highly stable bonds, the tetragraphene lattice contains inherently weaker, buckled hybridized carbon-carbon bonds. At temperatures exceeding 1500 K, the intense thermal vibrations supply kinetic energy that matches or exceeds the activation energy barrier required to break these cross-linking bonds. Amorphization and Rehybridization: Once the weaker connections break, the highly strained network undergoes rapid local nanostructural collapse. The broken bonds trigger an irreversible cascade of rehybridization, causing the orderly crystalline nanotube to melt into a disordered, amorphous carbon pipe that cannot sustain nanomechanical loads (see snapshots of CMD results in Figure 6 and Figure 7). This thermal threshold for tetragraphene-based nanotubes closely matches established carbon nanostructure research. Theoretical studies on novel, non-ideal allotropes containing mixed / networks, such as biphenylene, penta-graphene, and defective nanotubes, consistently predict a sharp decline in nanomechanical stiffness and premature melting between 1200 K and 1600 K. This behavior is primarily driven by the nanostructural vulnerability of hybridizations and non-hexagonal rings [124,179,181]. Furthermore, high-temperature transmission electron microscopy (TEM) experiments and atomistic simulations on multi-walled carbon nanotubes with high defect densities show that structural degradation and structural reconstruction begin rapidly within this identical thermal window. While pristine graphene can withstand much higher thermal loads, the built-in nanostructural strain of the tetragraphene lattice lowers its melting point, making 1500 K the clear physical limit for its nanostructural survival [182,183,184].
The nanomechanical response of single-walled tetragraphene-based nanotubes (TGCNTs) under uniaxial tensile loading was systematically investigated via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The graphical representation in Figure 2 and Figure 4 (a)-(l) illustrates the stress-strain profiles for diverse configurations, comparing the series from indices to with the series from to . The results reveal a striking contrast in failure modes dictated by tube chirality. The TGCNT series exhibits a characteristically brittle failure mode, which is marked by a highly linear elastic regime followed by a sudden nanostructural fracture with negligible plastic deformation. Conversely, the TGCNT series demonstrates remarkable ductility and superelasticity. This behavior is evidenced by a distinct plateau effect that maintains a constant stress level up to 20% strain, followed by clear strain hardening until ultimate fracture beyond 40% strain, indicating a stress-induced nanostructural phase transition. To provide a comprehensive quantitative overview, key nanomechanical properties were extracted from the simulation curves. The Young’s modulus () was calculated by applying a linear regression within the initial 3% uniaxial strain regime for all configurations, with fitting errors precisely estimated using the Xmgrace plotting software [178]. Additionally, the Ultimate Tensile Strength (UTS) and the critical strain () were determined at the peak point of failure. Table 2 summarizes the complete set of calculated values for the Young’s Modulus (GPa.Å), UTS (GPa.Å), and critical strain across all studied TGCNTs (see Table 1). In graphical representation (Figure 3 and Figure 5) of stress-strain uniaxial load nanomechanical behavior shown the atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs) under uniaxial tensile loading reveals distinct, temperature-dependent nanomechanical pathways between the (0,10) and (14,0) configurations from 600 K up to 2100 K. As illustrated in the stress-strain profiles (Figure 3 (a)-(f), the (0,10) TGCNT series displays a characteristically brittle failure mode defined by a highly linear elastic regime followed by sudden nanostructural fracture with negligible plastic deformation. In sharp contrast, the (14,0) TGCNT series demonstrates remarkable ductility and superelasticity, characterized by a distinct plateau effect that maintains constant stress up to 20% strain before undergoing clear strain hardening until ultimate fracture (see Figure 5). For the (14,0) system, rising temperatures accelerate the degradation of nanofracture toughness, significantly reducing the critical strain to values below 40%. Ultimately, both configurations exhibit severe nanostructural degradation at elevated thermal states, leading to a complete loss of nanostructural stability above 1500 K. This extreme thermal degradation results in an amorphous graphene phase devoid of a defined geometry, which fundamentally accounts for the melting and collapse of the one-dimensional carbon TGCNTs nanostructures.
In Figure 6 and Figure 7 we can see the fully atomistic configurations obtained from classical molecular dynamics (CMD) simulations visually capture the thermal degradation of the (0,10) and (14,0) TGCNTs across the temperature range from 600 K up to 2100 K. As observed in the snapshots, the dotted circles highlight the localized loss of the initial nanostructured configuration induced by extreme thermal effects. Furthermore, the accompanying zoomed views explicitly display the nanostructural transition into a fully amorphous carbon phase, providing clear evidence of the severe nanostructural collapse experienced by the nanotubes at these elevated temperatures. Following the detailed nanomechanical analysis of the individual stress-strain curves, the overall elastic properties and nanomechanical limits of all studied TGCNTs were statistically averaged. To provide a comprehensive comparison of their nanostructural performance, the key nanomechanical parameters are summarized in Table 2. This dataset presents the calculated values for the Young’s Modulus (), Ultimate Tensile Strength-UTS (), and critical strain (%) for the diverse configurations of tetragraphene-based nanotubes (TGCNTs). The data presented in Table 2 reveals a remarkable nanomechanical anisotropy in single-walled tetragraphene-based nanotubes (TGCNTs). This direction-dependent behavior is highly sensitive to the nanotube chirality, which directly influences the bond orientation relative to the uniaxial strain axis. The distinct nanomechanical responses between the and configurations underscore the nanostructural uniqueness of the tetragraphene lattice when rolled into one-dimensional nanostructures (significant nanoechanical anisotropy). The series possesses a significantly stiffer elastic regime compared to the family. For larger tube diameters, the Young’s modulus of nanotubes stabilizes above 3100 GPa·Å. In stark contrast, the nanotubes plateau near 2350 GPa·Å. This represents an elastic stiffness difference of roughly 30% between chiralities. The maximum load-bearing capacity also demonstrates a clear anisotropic gap. The configurations routinely achieve UTS values between 640 and 700 GPa·Å. Conversely, the nanotubes exhibit much lower maximum strengths, generally remaining below 460 GPa·Å. The most dramatic manifestation of anisotropy lies in the failure limits. The ductile series stretches up to an exceptional 38.18%–41.06% strain before nanostructural failure. On the other side, the brittle series structurally fails at very early stages, showing critical strains of only 11.35%–13.75%. For the highly curved , , and nanotubes (small-diameter effects), the Young’s modulus experiences a noticeable dip down to 2379.90 GPa·Å. This dip indicates that strong curvature destabilizes the initial elastic resistance. A similar but less severe trend is visible in the configuration. As the tube indices increase beyond 7 (large-diameter convergence), the nanomechanical properties for both families stabilize. The values become nearly independent of the tube diameter. This independent behavior suggests that the system smoothly approaches the mechanical limit of a flat tetragraphene sheet.
To address the core nanostructural parameters of one-dimensional systems, the Young’s modulus of the TGCNTs was evaluated by considering the nanotube wall as a thin-shelled cylinder. The raw nanostructural stiffness obtained from the simulations ranges from 2714.10 to 3166.20 GPaÅ (corresponding to 271.41-316.62 GPanm). To translate this metric into a conventional three-dimensional Young’s modulus (), the values were normalized by the intrinsic shell thickness of the nanotube wall. This wall thickness corresponds to the tetragraphene lattice amplitude of 4.571Å , which originates from its characteristic corrugated mixed hybridization pattern. This formal normalization yields an effective material Young’s modulus ranging from 593.77 to 692.67 GPa for the investigated TGCNTs, establishing a rigid nanostructural baseline that shows close agreement with previous Density Functional Theory (DFT) predictions [121]. So, all classical molecular dynamics (CMD) results utilizing the AIREBO-Morse potential show strong consistency with first-principles Density Functional Theory (DFT) calculations [121]. The calculated values match the ground-state predictions from DFT very closely. This high accuracy confirms that the AIREBO-Morse potential captures the intrinsic carbon-carbon bond stretching of the tetragraphene lattice exceptionally well. The severe drop in critical strain for the nanotubes vs. the large deformation capacity of the nanotubes aligns perfectly with DFT-predicted energy landscapes [121]. This close validation proves that our CMD simulations reliably describe both the initial elastic deformation and the ultimate failure points of these novel nanomaterials.
In Figure 8 and Figure 9 display the fully atomistic molecular dynamics snapshots of the uniaxial nanomechanical loading for the TGCNT (0,10) and (14,0) configurations stretched along the z-direction, respectively. As depicted in Figure 8 (a), the (0,10) TGCNT is initially shown at a strain-free state , establishing its pristine nanostructural equilibrium. Upon applying a uniaxial tensile load up to a strain of 10% (Figure 8 (b)), the TGCNT exhibits uniform nanostructural elongation within its linear elastic regime. The onset of nanomechanical failure is captured in Figure 8 (c), where initial bond cleavage specifically targets the carbon-carbon () bonds aligned parallel to the uniaxial strain direction. This localized stress concentration rapidly triggers a catastrophic, brittle nanostructural fracture, culminating in the complete cleavage of the (0,10) TGCNT into two distinct segments at a critical strain of 21.27%, as illustrated in Figure 8 (d). A remarkably interesting phenomenon observed immediately following this complete nanostructured failure is the formation of linear atomic chains (LACs) bridging the fractured ends. Under nanomechanical tensile strain, the tetragraphene nanostructure undergoes a coordinated nanostructural transition from a crystalline lattice to a disordered state, leading to the formation of linear amorphous carbons (LACs), in strong agreement with atomistic calculations reported for tetragraphene single-layers [122]. This behavior can be fundamentally attributed to the local hybridization transitions within the TGCNT network under the specific (0,10) strain orientation, where -like or -like carbon frameworks undergo severe nanostructural reconstruction into -hybridized chain-like geometries. In panel (d), the yellow indicators specify the dynamic bond lengths of the highly elongated linkages right before their ultimate rupture. These chemical bond lengths within the generated LACs vary from Å to Å. This significant elongation beyond the standard equilibrium bond distance highlights the high local tensile stress sustained by these atomic chains, a feature accurately captured by the AIREBO-Morse potential, which properly describes bond breaking and subsequent nanostructural remodeling under extreme nanomechanical loads.
In Figure 9 displays the fully atomistic molecular dynamics snapshots tracking the uniaxial nanomechanical loading of the (14,0) TGCNT configuration stretched along the z-direction. Initially, Figure 9 (a) depicts the nanotube in its pristine, strain-free state (). As the uniaxial tensile load increases to strains of 20% (Figure 9 (b) and 30% (Figure 9 (c), the system accommodates large deformations without nanostructural failure. The onset of nanomechanical failure is captured at a critical strain of 40.69% (Figure 9 (d), where the inset close-up view highlights the initial cleavage of local carbon-carbon () bonds. Ultimate failure is achieved in Figure 9 (f), yielding a permanent, nanostructured fracture that divides the nanotube into two distinct parts, accompanied by the formation of minor linear atomic chains (LACs) that are notably less pronounced than those observed in the (0,10) counterpart. In stark contrast to the brittle (0,10) configuration, the (14,0) TGCNT demonstrates remarkable ductility and superelasticity. This response is evidenced by a distinct plateau effect maintaining a nearly constant stress up to 20% strain, followed by clear strain hardening prior to fracture. To elucidate this unique superplasticity, Figure 10 (a)-(d) provide high-resolition nanometric scale snapshots revealing a stress-induced nanostructural phase transition. This nanomechanical performance is fundamentally governed by a spatial reconfiguration where - and -hybridized covalent bonds reorient and preferentially concentrate along the uniaxial strain axis. Under load, this atomic network behaves like a highly efficient nanostructural truss system. The dynamic, truss-like redistribution of covalent bonds allows the lattice to stretch and absorb energy continuously, successfully sustaining the prolonged stress plateau and enabling the (14,0) TGCNT to withstand uniaxial elongation before ultimate fracture.
To further elucidate the underlying mechanism of these distinct nanomechanical behaviors, the Poisson’s ratio transitions for both the (0,10) and (14,0) TGCNTs were calculated and are discussed below. This elastic parameter provides critical insights into the lateral nanostructural response of the nanotubes as they undergo severe uniaxial stretching along the z-direction. By tracking the changes in the Poisson’s ratio as a function of tensile strain, we can directly correlate the geometric lattice distortions, such as the truss-like deformation in the superplastic (14,0) system versus the rigid behavior in the brittle (0,10) configuration. The transverse deformation response of the tetragraphene-based nanotubes is quantitatively assessed via the evolution of their Poisson’s ratio under tensile loading (see Figure 11). To contextualize the physical characteristics of the investigated systems, a comparative continuum spectrum of reference Poisson’s ratio values is presented below.
Within the small elastic deformation regime (up to an axial strain threshold of approximately 8%), a linear fitting of the transverse-axial strain relation yields an averaged Poisson’s ratio of for the TGCNTs . This exceptionally low value confirms that tetragraphene-based nanotubes possess a significantly constrained lateral flexibility compared to conventional graphene sheets under identical tensile conditions. Physically, this response implies that the 1D nanostructure behaves almost independently along its transverse direction during early-stage loading, approaching the performance limit of a zero-transverse-strain material.
First-principles density functional theory (DFT) calculations and atomistic molecular dynamics literature establish that the Poisson’s ratio of tetragraphene nanostructures follows a highly non-monotonic path [64,185,186]. From an initial ground-state value [185], the system undergoes a localized nanostructural expansion. This transient inflation occurs as the buckled three-dimensional network uncoils, stretching the constituent tetragonal and hexagonal carbon rings along the pulling axis. Immediately following this intermediate nanostructural elongation, the morphology triggers a severe nanostructural collapse, plunging into a strongly negative regime known as the auxetic effect [64,186]. Consequently, capturing a Poisson’s ratio of at exactly 10% strain indicates that our reactive atomistic simulations successfully resolved the precise peak of this geometric phase transition right before the activation of the auxetic threshold. So, Tetragraphene-based nanotubes (TGCNTs) exhibit a low average Poisson’s ratio of within the small elastic deformation regime (up to 10% uniaxial strain), indicating significantly constrained lateral flexibility, with specific values of for TGCNT(0,10) and for TGCNT(14,0) highlighting strong chirality dependence. This behavior stems from the non-monotonic nanostructural evolution of the tetragraphene lattice, transitioning from initial expansion to severe collapse, which results in a 10% strain Poisson’s ratio of for TGCNT(0,10), marking the peak of this geometric phase transition prior to the auxetic regime.
In summary, the evaluate the nanomechanical boundaries of these systems, it is essential to contrast the benchmark values derived from Density Functional Theory (DFT) with the data obtained via Classical Molecular Dynamics (CMD) simulations in this work:
- DFT Monolayer Benchmarks: First-principles DFT calculations in literature establish the baseline ground-state Poisson’s ratio for an isolated tetragraphene monolayer at a highly constrained value of [185].
- CMD TGCNTs Results: Our reactive atomistic CMD simulations reveal an outstanding chirality dependence when rolling the 2D sheet into 1D nanotubes. Within the early elastic regime, the CMD data yields a localized value of for the zigzag-like TGCNT , showing remarkable agreement with DFT monolayer predictions. Conversely, the CMD calculations reveal an anomalously high value of for the TGCNT configuration.
This severe disparity between the two nanotube chiralities resolved by our CMD model highlights a robust nanostructural anisotropy driven entirely by the rolling orientation of the underlying tetragraphene matrix. Because the rectangular unit cell of tetragraphene features highly asymmetric tetragonal and hexagonal carbon ring arrangements, uniaxial loading along the armchair-like or zigzag-like directions triggers completely different deformation mechanisms. For the TGCNT, the uniaxial pulling causes a rapid, compliant closure of the lateral lattice gaps, resulting in an enhanced transverse contraction that vastly exceeds the traditional theoretical upper limit of isotropic continuum materials ().
As the applied uniaxial deformation increases to a larger strain level of 10%, the calculated Poisson’s ratio for the TGCNT exhibits a distinct non-linear shift, rising slightly to a value of . This non-linear increment is physically valid and reveals a vital nanomechanical signature unique to the underlying tetragraphene topology under large deformations. Both DFT and CMD literature establish that the Poisson’s ratio of tetragraphene nanostructures follows a highly non-monotonic path [64,185,186]. From its initial equilibrium ground state, the system undergoes a localized nanostructural expansion. This transient inflation occurs as the buckled three-dimensional network uncoils, stretching the constituent tetragonal and hexagonal carbon rings along the pulling axis. Immediately following this intermediate nanostructural elongation, the morphology triggers a severe nanostructural collapse, plunging into a strongly negative regime known as the auxetic effect [64,186]. Consequently, capturing a Poisson’s ratio of at exactly 10% strain indicates that our reactive CMD simulations successfully resolved the precise peak of this geometric phase transition right before the activation of the auxetic threshold.
4.1. Validation and Rationale for the AIREBO-Morse Interatomic Potential Selection
To evaluate the nanomechanical behavior and thermal stability of the proposed tetragraphene-based nanotubes (TGCNTs), the selection of an appropriate interatomic potential is a critical parameter. While reactive force fields such as ReaxFF are widely implemented for simulating complex chemical reactions and large-scale nanostructural transformations [187], they often fail to accurately reproduce the nanomechanical responses of pure carbon allotropes that exhibit non-hexagonal or highly strained geometries, such as tetragraphene .
Preliminary testing revealed that ReaxFF is inadequate for capturing the statistical averages of the nanomechanical properties of TGCNTs. This limitation stems from two primary factors. First, standard ReaxFF parameterizations for carbon are heavily optimized for classic phase transitions and bond-breaking events (e.g., graphite-to-diamond transitions or hydrocarbon combustion) [187]. Consequently, they rarely account for the high steric strain intrinsic to the four-membered rings (squares) that comprise the tetragraphene lattice. Second, ReaxFF utilizes a continuous bond-order calculation without a rigid cutoff system [187]. Under severe uniaxial strain, this unconstrained approach leads to premature amorphization or unrealistically overestimates the flexibility of the distorted bond angles within the TGCNT network.
Conversely, the Adaptive Intermolecular Reactive Empirical Bond Order (AIREBO) framework is well-established as a premier choice for modeling elastic properties and fracture mechanisms in pristine carbon architectures. In this study, we utilize the AIREBO-Morse modification to mitigate the well-known artifact of the original AIREBO formulation, namely, the non-physical strain hardening that occurs near the rupture threshold due to an overly abrupt cutoff function. By replacing the traditional Lennard-Jones term with a long-range Morse potential, the AIREBO-Morse model achieves superb accuracy in describing the attractive and repulsive energy profiles of carbon-carbon bonds under extreme stretching [191]. This modification effectively preserves the correct lattice stiffness up to the precise moment of localized fracture. Furthermore, benchmarking the interatomic potential against first-principles calculations represents the gold standard in molecular dynamics methodologies. As demonstrated in Appendix A and Appendix B, our AIREBO-Morse implementation yields excellent statistical agreement with Density Functional Theory (DFT) data, fully justifying its selection for predicting the nanomechanical pathways of the studied TGCNTs.
5. Conclusions
In this study, comprehensive reactive classical molecular dynamics (CMD) simulations utilizing the calibrated AIREBO-Morse potential successfully mapped the systematic nanomechanical behavior and thermal stability thresholds of single-walled tetragraphene-based nanotubes (TGCNTs). Our primary novel discovery reveals a sharp, chirality-dependent divergence in failure mechanisms under uniaxial tensile loading. The TGCNTs consistently exhibit brittle fracture modes characterized by rigid linear elasticity and sudden nanostructural cleavage. In stark contrast, the TGCNTs demonstrate exceptional ductility and superelasticity, driven by a stress-induced nanostructural phase transition that sustains a prolonged stress plateau up to 20% strain, followed by clear strain hardening until ultimate failure above 40% strain. Furthermore, we identified an extreme thermal boundary at 1500 K, above which severe nanostructural degradation triggers a complete lattice collapse into a fully amorphous graphene phase. Finally, evaluating the transverse elasticity revealed a critical nanostructural anisotropy: the configuration possesses an ultra-low Poisson’s ratio () in excellent agreement with DFT benchmarks, whereas the series displays an anomalously high value () stemming from its dynamic, truss-like geometric reconfiguration.
The impact of these discoveries is highly significant for the design of next-generation carbon nanomaterials. The extraordinarily high Young’s modulus values achieved, reaching up to GPa·Å for the series and GPa·Å for the series position TGCNTs as premier candidates for nanostructural reinforcement in high-performance nanocomposites. Moreover, the unique combination of a long constant-stress plateau and superelastic deformation over 40% strain indicates that TGCNTs can absorb and dissipate massive amounts of mechanical energy before failing. This unprecedented nanomechanical resilience makes them exceptionally well-suited for nanoscale shock-absorbing devices and protective coatings. Additionally, their ability to withstand severe lattice distortions while maintaining structural integrity opens up new avenues for implementation in reliable flexible electronics and strain-engineered components operating within moderate thermal environments.
Looking ahead, several crucial perspectives and future research directions emerge from this work to broaden our understanding of tetragraphene systems. First, while this study establishes a clear thermal stability limit at 1500 K, future investigations should focus on mapping the thermal conductivity and heat transport mechanisms of these nanotubes across different chiralities to assess their efficiency in thermal management applications. Second, exploring the effects of chemical doping (such as introducing nitrogen or boron atoms into the four-membered rings) or nanostructural defects (like Stone-Wales vacancies) could provide a reliable strategy to tune both the superplastic threshold and the electrical properties of the lattice. Lastly, evaluating the interfacial shear stress and load-transfer efficiency between TGCNTs and polymer matrices will be essential to accelerate their practical integration into real-world, high-strength engineering nanocomposites.
Author Contributions
The author (J.M.S.) conceptualized the study, performed the methodology, formal analysis, and investigation, and wrote and edited the manuscript. The author has read and agreed to the published version of the manuscript.
Funding
This research was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), grant number 305053/2023-0.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
This work was partly supported by the Brazilian agencies CAPES, CNPq, FAPESP, and FAPEPI. This work used resources of the Centro Nacional de Processamento de Alto Desempenho em São Paulo (CENAPAD-SP). The author thanks the Instituto Federal de Educação, Ciência e Tecnologia do Piauí (IFPI) for the institutional support, the Coaraci Supercomputer for computer time (Fapesp grant #2019/17874-0), and the Center for Computing in Engineering and Sciences at Unicamp (Fapesp grant #2013/08293-7). J.M.S. acknowledges CNPq (Process No. 305053/2023-0) for financial support.
Conflicts of Interest
The author declares no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| l]@ll AIREBO-Morse | Adaptive Intermolecular Reactive Empirical Bond Order Morse. |
| ReaxFF | Reactive Force Field. |
| AMBER | Assisted Model Building with Energy Refinement. |
| COMB3 | Charge-Optimized Many-Body. |
| DFT | Density Functional Theory. |
| TGCNT | Tetragraphene-base carbon nanotubes. |
| TPa | Terapascal. |
| GPa | Gigapascal. |
| CMD | Classical molecular dynamic. |
| LAMMPS | Large-scale Atomic/Molecular Massively Parallel Simulator. |
| UTS | Ultimate Tensile Strength. |
| VMD | Visual Molecular Dynamics. |
| CG | “Polak-Ribiere” version of the conjugate gradient. |
| etol | Energy Tolerance. |
| ftol | Force Tolerance. |
| NPT | Isothermal-Isobaric Ensemble. |
| NVT | Canonical Ensemble. |
| Young’s Modulus. | |
| C | -hybridized carbon. |
| C | -hybridized carbon. |
| LACs | Linear Amorphous Carbons. |
Appendix A. Mathematical Formulation and Validation of the AIREBO-Morse (AIREBO-M) Potential
This appendix delivers a rigorous, step-by-step mathematical derivation of the Adaptive Intermolecular Reactive Empirical Bond Order potential with Morse correction (AIREBO-Morse, or AIREBO-M), tailored to the structural guidelines of MDPI Nanomaterials.
Appendix A.1. The Fundamental Framework of the AIREBO Potential
The total potential energy E of a hydrocarbon system containing N atoms within the AIREBO framework is defined as the sum of three distinct energetic terms [188]:
where: represents the short-range, reactive covalent bond energy. corresponds to the long-range, non-covalent interactions. accounts for the four-body dihedral torsion preferences [188].
Appendix A.1.1. Short-Range Reactive Term (E ij REBO )
The covalent interaction mimics the classic Brenner REBO formulation, balancing attractive and repulsive atomic curves modulated by a local many-body bond-order parameter [188]:
The individual functions for the two-body repulsive () and attractive () states are given by [188]:
and
Where is a smooth cutoff function confining covalent bonds to the short-range zone [188]:
The many-body bond order parameter scales down the attraction based on local coordination numbers and angles [188]:
Appendix A.1.2. Dihedral Torsional Formulation (E kijl TORSION )
The four-body torsion term represents the energy profile associated with rotation around a central covalent bond. For a linear sequence of four bonded atoms , the torsion potential is formulated as [188]:
Where values are the standard short-range covalent cutoff functions defined in Equation (5), ensuring that torsional calculations smoothly vanish if any associated bond breaks. The dihedral angle is computed via the plane normals of the atomic triplets [188]:
The discrete torsional energy function uses a standard cosine expansion modulated by the local hybridization state of the core atoms i and j [188]:
where: are constant energy coefficients capturing radical, double, and triple bond symmetry bars. is an adaptive polynomial scaling function that depends on the coordination numbers N of atoms i and j. This eliminates torsional resistance when either core node approaches an configuration, maintaining physical flexibility in systems like diamond lattices or saturated alkanes [188].
Appendix A.2. The Classical Non-Bonded Failure: Lennard-Jones (LJ) Intermolecular Potential
In the standard AIREBO formulation, long-range dispersion and short-range steric repulsions between non-bonded atoms are governed by an adaptive Lennard-Jones 12-6 potential [189]:
The isotropic unscaled functional form stands as [189]:
The Steric Divergence Bottleneck.
Under high-pressure conditions ( GPa), extreme compression forces non-bonded atoms past their equilibrium radii (). As , the repulsive force behaves asymptotically [188]:
This power-law divergence introduces unphysical, catastrophic core rigidity. It heavily overestimates the stiffness of molecular arrangements (e.g., compressed carbon sheets and polyethylene) [188].
Appendix A.3. The AIREBO-Morse Formulation (AIREBO-M Correction)
To resolve the unphysical high-pressure divergence, the AIREBO-Morse potential replaces the singular function with a smoother, softer exponential Morse potential [188]:
The core mathematical mapping substitutes with [188]:
where: is the potential well depth (identical to the LJ well depth to retain ambient thermodynamics). is the equilibrium distance parameter. regulates the width and stiffness of the potential curve [188].
Extracting the square root yields the standard analytical mapping value [190]:
Appendix A.3.2. Spline Intermolecular Cutoffs and Softness Fine-Tuning
Appendix A.3.3
The Morse potential is smoothly truncated with a third-order spline over adaptive boundaries [188]:
The parameter is intentionally uncoupled from the standard mapping value at short distances to optimize the potential for high-pressure limits ( 40 GPa) without shifting the equilibrium lattice structures [188]:
Where is a dimensionless scaling parameter optimized using quantum chemical calculations (e.g., MP2/dft) and experimental graphite layer compression curves. This modification prevents unphysical core divergence, allowing the repulsive energy to remain stable even under high atomic densities [188].
Appendix B. Hydrocarbon Intermolecular Parametrization Matrix
Table A1 lists the verified computational interaction matrix used within the AIREBO-M structure [188].

Table A1.
Non-bonded interaction parameter mapping values for Hydrocarbon structures [188].
Table A1.
Non-bonded interaction parameter mapping values for Hydrocarbon structures [188].
| Interaction Pair () | (eV) | (Å) | (Å) | (Å) | |
|---|---|---|---|---|---|
| C − C | 0.0028437 | 3.4000 | 3.8164 | 1.4006 | 1.0000 |
| C − H | 0.0019865 | 2.9300 | 3.2889 | 1.6253 | 1.0000 |
| H − H | 0.0013870 | 2.4600 | 2.7613 | 1.9358 | 1.0000 |
Appendix B.1. Validation and Comparative Benchmarking in Nanostructured Carbon Systems
The accuracy and computational transferability of the Adaptive Intermolecular Reactive Empirical Bond Order potential with Morse correction (AIREBO-M) for carbon-based nanostructures, such as carbon nanotubes (CNTs) and graphene sheets, have been extensively validated against both experimental observations and first-principles calculations. While the standard AIREBO potential suffers from unphysical hardening and premature fracture artifacts under severe strain or high pressures () due to the rigid Lennard-Jones repulsion core, the Morse non-bonded correction yields excellent agreement with true experimental equation-of-state parameters.
To ground its operational capability within nanomaterial modeling, Table A2 evaluates AIREBO-M against other prominent computational force fields (ReaxFF, AMBER, COMB3, and DFT) across critical structural, mechanical, and chemical dimensions.
Table A2.
Comparative benchmarking matrix of interatomic potentials and electronic methods for carbon nanostructures (Graphene, CNTs, and Nanoparticles).
Table A2.
Comparative benchmarking matrix of interatomic potentials and electronic methods for carbon nanostructures (Graphene, CNTs, and Nanoparticles).
| Method | Primary Carbon Domains | Key Advantage / Validation Target | Core Limitation / Failure Mode | Computational Cost |
Key References |
|---|---|---|---|---|---|
| AIREBO-M | Graphene, CNTs, Diamond, Hydrocarbons | Prevents unphysical steric divergence under high pressure; accurate elastic moduli and layer spacing. | Lacks dynamic variable charge redistribution. | Low/Mod. | [191,192] |
| ReaxFF | Chemical reactions, Oxidation, Shockwaves | Simulates explicit bond breaking/forming with dynamic charge transfer. | Overestimates Poisson’s ratio; highly sensitive to small integration timesteps. | High | [193,194,195] |
| AMBER | Biomolecules, Carbon-Water interfaces | Extremely fast; excellent for long-term equilibrium and hydration layers. | Non-reactive; cannot model bond breaking or phase transitions. | Very Low | [196,197,198] |
| COMB3 | Heterogeneous surfaces, Metal-Oxides | Handles dynamic electronic polarization and variable ionic charges at surfaces. | High parameter complexity; less accurate for pure pristine carbon lattices. | High | [199,200] |
| DFT | Quantum electronic structures, Defect states | Absolute electronic precision; exact ground state energies and quantum effects. | Strictly limited to small systems (few hundred atoms) and short timescales. | Ext. High | [201,202] |
Appendix B.2. Quantitative Validation and Computational Efficiency of AIREBO-M
When modeling large-scale carbon nanostructures like graphene and carbon nanotubes (CNTs), selecting an appropriate interatomic potential requires balancing chemical accuracy with structural sizes. First-principles methods like Density Functional Theory (DFT) offer excellent accuracy but are limited to systems with a few hundred atoms due to their computational scaling [201,202]. Reactive force fields like ReaxFF allow for dynamic bond breaking but require small timesteps () and intensive charge calculation overhead, making them computationally expensive [194].
The AIREBO-Morse (AIREBO-M) potential provides an optimized balance for large-scale molecular dynamics (MD) simulations. By replacing the stiff Lennard-Jones core with a smooth Morse function, AIREBO-M avoids unphysical structural hardening under high mechanical strains or pressure fields without sacrificing the high computational speeds typical of empirical potentials [192]. It maintains a standard MD timestep () and scales linearly () with the number of atoms. This allows for the simulation of millions of carbon nodes over long nanosecond timescales. As shown in Table A3 (quantitative nanotructural and mechanical property matrix), AIREBO-M closely reproduces the fundamental structural and mechanical parameters of graphene and CNTs established by experimental work and quantum mechanical calculations. It delivers near-DFT precision for structural deformation while running orders of magnitude faster than ReaxFF.
Table A3.
Quantitative comparison of calculated mechanical and structural properties for graphene and pristine single-walled carbon nanotubes (CNTs) across different computational methods.
Table A3.
Quantitative comparison of calculated mechanical and structural properties for graphene and pristine single-walled carbon nanotubes (CNTs) across different computational methods.
| System & Property | Experimental / DFT | AIREBO-M | Standard AIREBO | ReaxFF |
|---|---|---|---|---|
| Graphene | ||||
| C−C Bond Length (Å) | 1.42 [201] | 1.42 [192] | 1.42 [191] | 1.45 [195] |
| Young’s Modulus (TPa) | 1.05 ± 0.05 [203] | 1.01 [192] | 0.95 [191] | 0.82 [195] |
| Poisson’s Ratio () | 0.160 [201] | 0.165 [192] | 0.210 [191] | 0.320 [195] |
| CNT (10,10) | ||||
| Tube Diameter (Å) | 13.56 [201] | 13.59 [192] | 13.54 [191] | 13.78 [194] |
| Young’s Modulus (TPa) | 0.95 ± 0.10 [204] | 0.94 [192] | 0.91 [191] | 0.79 [194] |
References
- Cahn, R.W.; Haasen, P.; Kramer, E.J. Materials science and technology-a comprehensive treatment. Int. J. Mater. Res. 1993, 84, 90–90. [Google Scholar] [CrossRef]
- Wood, J. The top ten advances in materials science. Mater. Today 2008, 11, 40–45. [Google Scholar] [CrossRef]
- Bayda, S.; Adeel, M.; Tuccinardi, T.; Cordani, M.; Rizzolio, F. The history of nanoscience and nanotechnology: From chemical–physical applications to nanomedicine. Molecules 2019, 25, 112. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Schubert, A.; Zsindely, S. Nanoscience and nanotechnology on the balance. Scientometrics 1997, 38, 321–325. [Google Scholar] [CrossRef]
- Ozin, G.A. Nanochemistry: Synthesis in diminishing dimensions. Adv. Mater. 1992, 4, 612–649. [Google Scholar] [CrossRef]
- Tolles, W.M. Nanoscience and nanotechnology in Europe. Nanotechnology 1996, 7, 59–61. [Google Scholar] [CrossRef]
- Kabir, A.M.R.; Inoue, D.; Kakugo, A. Molecular swarm robots: Recent progress and future challenges. Sci. Technol. Adv. Mater. 2020, 21, 323–332. [Google Scholar] [CrossRef]
- Guerreiro, T. Quantum molecular robots. Quantum Sci. Technol. 2021, 6, 025006. [Google Scholar] [CrossRef]
- Rauschen, R.; Ayme, J.-F.; Matysiak, B.M.; Thomas, D.; Cronin, L. A programmable modular robot for the synthesis of molecular machines. Chem 2025, 11, 102504. [Google Scholar] [CrossRef] [PubMed]
- Walecka, J.D. A theory of highly condensed matter. Ann. Phys. 1974, 83, 491–529. [Google Scholar] [CrossRef]
- Fradkin, E. Field Theories of Condensed Matter Physics, 2nd ed.; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar] [CrossRef]
- Zurek, W.H. Cosmological experiments in condensed matter systems. Phys. Rep. 1996, 276, 177–221. [Google Scholar] [CrossRef]
- Misra, P. Physics of Condensed Matter; Academic Press: Boston, MA, USA, 2011. [Google Scholar] [CrossRef]
- Mansoori, G.A. Principles of Nanotechnology: Molecular Based Study of Condensed Matter in Small Systems; World Scientific Publishing Company: Singapore, 2005. [Google Scholar] [CrossRef]
- Mørup, S.; Madsen, D.E.; Frandsen, C.; Bahl, C.R.; Hansen, M.F. Experimental and theoretical studies of nanoparticles of antiferromagnetic materials. J. Phys. Condens. Matter 2007, 19, 213202. [Google Scholar] [CrossRef]
- Yakout, S.M.; Elsherif, E. Batch kinetics, isotherm and thermodynamic studies of adsorption of strontium from aqueous solutions onto low cost rice-straw based carbons. Carbon Sci. Technol. 2010, 3, 144–153. Available online: https://www.researchgate.net/publication/279619891_Batch_kinetics_isotherm_and_thermodynamic_studies_of_adsorption_of_strontium_from_aqueous_solutions_onto_low_cost_rice-straw_based_carbons. [CrossRef]
- Hartmann, R.R.; Kono, J.; Portnoi, M.E. Terahertz science and technology of carbon nanomaterials. Nanotechnology 2014, 25, 322001. [Google Scholar] [CrossRef] [PubMed]
- Thostenson, E.T.; Ren, Z.; Chou, T.-W. Advances in the science and technology of carbon nanotubes and their composites: A review. Compos. Sci. Technol. 2001, 61, 1899–1912. [Google Scholar] [CrossRef]
- Donnet, J.-B. Carbon Black: Science and Technology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Tiwari, S.K.; Kumar, V.; Huczko, A.; Oraon, R.; Adhikari, A.D.; Nayak, G. Magical allotropes of carbon: Prospects and applications. Crit. Rev. Solid State Mater. Sci. 2016, 41, 257–317. [Google Scholar] [CrossRef]
- Falcao, E.H.; Wudl, F. Carbon allotropes: Beyond graphite and diamond. J. Chem. Technol. Biotechnol. 2007, 82, 524–531. [Google Scholar] [CrossRef]
- Khalaj, Z.; Monajjemi, M.; Diudea, M.V. Main allotropes of carbon: A brief review. In Sustainable Nanosystems Development, Properties, and Applications; IGI Global: Hershey, PA, USA, 2017; pp. 185–213. [Google Scholar]
- Sederberg, D.; Bryan, L.; Giordano, N. Allotropes of carbon: It’s all in the way you’re put together. In Proceedings of the NCLT-National Center for Learning and Teaching in Nanoscale Science and Engineering; Purdue University: West Lafayette, IN, USA, 2009. [Google Scholar]
- Dhameliya, T.M.; Bharadia, H.A.; Shah, J.K.; Sureja, D.K.; Bodiwala, K.B. Future of environment with carbon allotropes. Nirma Univ. J. Pharm. Sci. 2024, 11, 33–54. [Google Scholar]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Falkovsky, L.A. Optical properties of graphene. J. Phys. Conf. Ser. 2008, 129, 012004. [Google Scholar] [CrossRef]
- Rao, C.N.R.; Matte, H.S.S.R.; Subrahmanyam, K.S.; Maitra, U. Unusual magnetic properties of graphene and related materials. Chem. Sci. 2012, 3, 45–52. [Google Scholar]
- Papageorgiou, D.G.; Kinloch, I.A.; Young, R.J. Mechanical properties of graphene and graphene-based nanocomposites. Prog. Mater. Sci. 2017, 90, 75–127. [Google Scholar] [CrossRef]
- De Sousa, J.M. Nanostructures failures and fully atomistic molecular dynamics simulations. In Elasticity of Materials; IntechOpen: London, UK, 2021. [Google Scholar]
- Weiss, N.O.; Zhou, H.; Liao, L.; Liu, Y.; Jiang, S.; Huang, Y.; Duan, X. Graphene: An emerging electronic material. Adv. Mater. 2012, 24, 5782–5825. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.E.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Y.; Wang, X. Two-dimensional covalent carbon nitride nanosheets: Synthesis, functionalization, and applications. Energy Environ. Sci. 2015, 8, 3092–3108. [Google Scholar] [CrossRef]
- Baughman, R.H.; Eckhardt, H.; Kertesz, M. Structure-property predictions for new planar forms of carbon: Layered phases containing sp2 and sp atoms. J. Chem. Phys. 1987, 87, 6687–6699. [Google Scholar]
- Golberg, D.; Bando, Y.; Huang, Y.; Terao, T.; Mitome, M.; Tang, C.; Zhi, C. Boron nitride nanotubes and nanosheets. ACS Nano 2018, 4, 2979–2993. [Google Scholar]
- Vogt, P.; De Padova, P.; Quaresima, C.; Avila, J.; Frantzeskakis, E.; Asensio, M.C.; Resta, A.; Ealet, B.; Le Lay, G. Silicene: Compelling experimental evidence for graphenelike two-dimensional silicon. Phys. Rev. Lett. 2012, 108, 155501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Wang, Q.; Chen, X.; Kawazoe, Y.; Jena, P. Penta-graphene: A new carbon allotrope. Proc. Natl. Acad. Sci. USA 2015, 112, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, X.F.; Zhang, X.; Zhu, Q.; Dong, H.; Zhao, M.; Oganov, A.R. Phagraphene: A low-energy graphene allotrope composed of 5–6–7 carbon rings with distorted dirac cones. Nano Lett. 2015, 15, 6182–6186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Shuaiwei; Yang, B.; Chen, H.; Ruckenstein, E. Popgraphene: A new 2D planar carbon allotrope composed of 5–8–5 carbon rings for high-performance lithium-ion battery anodes from bottom-up programming. J. Mater. Chem. A 2018, 6, 6815–6821. [Google Scholar]
- Jiang, J.W.; Leng, J.; Li, J.; Guo, Z.; Chang, T.; Guo, X.; Zhang, T. Twin graphene: A novel two-dimensional semiconducting carbon allotrope. Carbon 2017, 118, 370–375. [Google Scholar] [CrossRef]
- Sheng, X.L.; Cui, H.J.; Ye, Fei; Yan, Q.B.; Zheng, Q.R.; Su, G. Octagraphene as a versatile carbon atomic sheet for novel nanotubes, unconventional fullerenes, and hydrogen storage. J. Appl. Phys. 2012, 112, 074315. [Google Scholar] [CrossRef]
- Sheng, X.L.; Yan, Q.B.; Ye, F.; Zheng, Q.R.; Su, G. T-carbon: A novel carbon allotrope. Phys. Rev. Lett. 2011, 106, 155703. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Lu, S.; Golov, A.A.; Kabanov, A.A.; Hu, X. D-carbon: Ab initio study of a novel carbon allotrope. J. Chem. Phys. 2018, 149, 114702. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tian, F.; Chu, B.; Duan, D.; Wei, S.; Lv, Y.; Zhang, H.; Wang, L.; Lu, N.; Liu, B.; et al. Cubic C96: A novel carbon allotrope with a porous nanocube network. J. Mater. Chem. A 2015, 3, 10448–10452. [Google Scholar] [CrossRef]
- Ding, X.Y.; Zhang, C.; Wang, D.Q.; Li, B.S.; Wang, Q.; Yu, Z.G.; Ang, K.W.; Zhang, Y.W. A new carbon allotrope: T5-carbon. Scr. Mater. 2020, 189, 72–77. [Google Scholar] [CrossRef]
- Liao, M.; Wang, F.; Zhu, J.; Lai, Z.; Liu, Y. P2221-C8: A novel carbon allotrope denser than diamond. Scr. Mater. 2022, 212, 114549. [Google Scholar] [CrossRef]
- Kang, B.; Yuan, Y.; Wu, S.; Ai, H.; Kang, S.; Lee, J.Y. Trigraphene and its derivates: A novel carbon allotrope. Bull. Korean Chem. Soc. 2018, 39, 1279–1282. [Google Scholar] [CrossRef]
- Ram, B.; Mizuseki, H. Tetrahexcarbon: A two-dimensional allotrope of carbon. Carbon 2018, 137, 266–273. [Google Scholar] [CrossRef]
- You, M.; Guo, G.; Liao, Y.; Luo, S.; He, C.; Tang, C.; Zhong, J. 2d novel c5n2 allotropes: High-performance anode materials for alkali metal ion battery. J. Energy Storage 2024, 84, 111004. [Google Scholar]
- Ram, B.; Mizuseki, H. C568: A new two-dimensional sp2-sp3 hybridized allotrope of carbon. Carbon 2020, 158, 827–835. [Google Scholar]
- Ram, B.; Mizuseki, H. Cmme-SnS: A two-dimensional tin sulfide nanosheet. J. Mater. Chem. A 2020, 8, 21219–21226. [Google Scholar]
- Fan, Q.; Liu, H.; Jiang, L.; Yu, X.; Zhang, W.; Yun, S. Two orthorhombic superhard carbon allotropes: C16 and C24. Diam. Relat. Mater. 2021, 116, 108426. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Jana, D. TPDH-graphene: A new two dimensional metallic carbon with NDR behaviour of its one dimensional derivatives. Phys. E 2021, 127, 114569. [Google Scholar] [CrossRef]
- Zhuo, Z.; Wu, X.; Yang, J. Me-graphene: A graphene allotrope with near zero Poisson’s ratio, sizeable band gap, and high carrier mobility. Nanoscale 2020, 12, 19359–19366. [Google Scholar] [PubMed]
- Bhattacharya, D.; Jana, D. TPO12-graphene: A new two-dimensional metallic carbon with 4–5 ring for Lithium ion battery. Appl. Surf. Sci. 2024, 669, 160495. [Google Scholar]
- Deb, J.; Paul, D.; Sarkar, U. Pentagraphyne: A new carbon allotrope with superior electronic and optical property. J. Mater. Chem. C 2020, 8, 16143–16150. [Google Scholar] [CrossRef]
- She, Y.; Zou, Y.; Zhao, L.; Jiang, Y.; Kou, C.; Zhang, M.; Tian, Y. Novel Two-Dimensional Tph-Cn2 with a Sign-Tunable Poisson’s Ratio. SSRN Electron. J. 2025, 5196719. [Google Scholar]
- Beserra, D.J.P.; Saraiva-Souza, A.; Diniz, E.M.; Fadel, M.; Meunier, V.; Girão, E.C. Naphthylene-γ: 1D and 2D carbon allotropes based on the fusion of phenyl-and naphthyl-like groups. Phys. Rev. Mater. 2020, 4, 084003. [Google Scholar]
- Liu, X.-Q.; Ma, A.; Zhang, Y.-N.; Yi, Z.-J.; Eglitis, R.I.; Tapsoba, I.; Jia, R. Pan-C2(n+1): A Theoretical Prediction for 2D Carbon Allotropes. Inorg. Chem. 2025, 64, 1021–1032. [Google Scholar]
- Wang, W.; Meng, J.; Hu, Y.; Wang, J.; Li, Q.; Yang, J. Thgraphene: A novel two-dimensional carbon allotrope as a potential multifunctional material for electrochemical water splitting and potassium-ion batteries. J. Mater. Chem. A 2022, 10, 9848–9857. [Google Scholar]
- Kilic, M.E.; Lee, K.-R. Tetrahex carbides: Two-dimensional group-IV materials for nanoelectronics and photocatalytic water splitting. Carbon 2021, 174, 368–381. [Google Scholar]
- Sharma, B.R.; Manjanath, A.; Singh, A.K. Pentahexoctite: A new two-dimensional allotrope of carbon. Sci. Rep. 2014, 4, 7164. [Google Scholar] [CrossRef] [PubMed]
- Naseri, M.; Jalilian, J.; Salahub, D.R.; Lourenço, M.P.; Rezaei, G. Hexatetra-carbon: A novel two-dimensional semiconductor allotrope of carbon. Computation 2022, 10, 19. [Google Scholar] [PubMed]
- Fan, Q.; Chen, S.; Zhao, Y.; Yu, X.; Yun, S. Tower carbon: A new large-cell carbon allotrope. J. Phys. Condens. Matter 2022, 34, 365702. [Google Scholar] [CrossRef]
- Burchfield, L.A.; Al Fahim, M.; Wittman, R.S.; Delodovici, F.; Manini, N. Novamene: A new class of carbon allotropes. Heliyon 2017, 3, e00105. [Google Scholar] [CrossRef]
- Yang, Xigui; Lv, C.; Liu, S.; Zang, J.; Qin, J.; Du, M.; Yang, D.; Li, X.; Liu, B.; Shan, C.-X. Orthorhombic C14 carbon: A novel superhard sp3 carbon allotrope. Carbon 2020, 156, 309–312. [Google Scholar] [CrossRef]
- Liu, H.; Fan, Q.Y.; Yang, F.; Yu, X.H.; Zhang, W.; Yun, S.N. tP40 carbon: A novel superhard carbon allotrope. Chin. Phys. B 2020, 29, 106102. [Google Scholar] [CrossRef]
- Wang, J.Q.; Zhao, C.X.; Niu, C.Y.; Sun, Q.; Jia, Y. C20- T carbon: a novel superhard sp3 carbon allotrope with large cavities. J. Phys. Condens. Matter 2016, 28, 475402. [Google Scholar] [PubMed]
- Liu, L.; Ding, J.; Zhuang, Y.; Ma, M.; Ying, P.; Hu, M.; Li, Y.; He, J.; Zhang, Q. Pa-C10: A Novel Carbon Allotrope with Distinct Two-dimensional Conductivity and Outstanding Stretchability. Vacuum 2025, 235, 114326. [Google Scholar]
- Zheng, Z.; Xue, Y.; Li, Y. A new carbon allotrope: graphdiyne. Trends Chem. 2022, 4, 754–768. [Google Scholar] [CrossRef]
- Ni, D.; Shen, Y.; Shen, Y.; Wang, Q.; Kawazoe, Y.; Jena, P. Hex-C558: A new porous metallic carbon allotrope for lithium-ion battery anode. Carbon 2021, 183, 652–659. [Google Scholar]
- Nulakani, N.V.R.; Subramanian, V. Superprismane: A porous carbon allotrope. Chem. Phys. Lett. 2019, 715, 29–33. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, H.; Du, Y.; Zhang, X.; Wang, Y.; Li, Q. Orthorhombic C32: a novel superhard sp3 carbon allotrope. Phys. Chem. Chem. Phys. 2013, 15, 14120–14125. [Google Scholar] [CrossRef] [PubMed]
- Kilic, M.E.; Lee, K.R. Four-penta-graphenes: Novel two-dimensional fenestrane-based auxetic nanocarbon allotropes for nanoelectronics and optoelectronics. Carbon 2022, 195, 154–164. [Google Scholar]
- Yin, W.J.; Xie, Y.E.; Liu, L.M.; Wang, R.Z.; Wei, X.L.; Lau, L.; Zhong, J.X.; Chen, Y.P. R-graphyne: a new two-dimensional carbon allotrope with versatile Dirac-like point in nanoribbons. J. Mater. Chem. A 2013, 1, 5341–5346. [Google Scholar]
- Han, T.; Liu, Y.; Lv, X.; Li, F. Biphenylene monolayer: a novel nonbenzenoid carbon allotrope with potential application as an anode material for high-performance sodium-ion batteries. Phys. Chem. Chem. Phys. 2022, 24, 10712–10716. [Google Scholar] [PubMed]
- Fan, Q.; Liu, H.; Yang, R.; Yu, X.; Zhang, W.; Yun, S. An orthorhombic superhard carbon allotrope: Pmma C24. J. Solid State Chem. 2021, 300, 122260. [Google Scholar] [CrossRef]
- Matar, S.F. DFT investigation of novel cubic carbon allotrope, yne-C16. arXiv 2024, arXiv:2407.14777. [Google Scholar]
- Wang, X.; Rong, J.; Song, Y.; Yu, X.; Zhan, Z.; Deng, J. QPHT-graphene: A new two-dimensional metallic carbon allotrope. Phys. Lett. A 2017, 381, 2845–2849. [Google Scholar]
- Liu, J.Q.; Gao, Q.; Hu, Z.P. HSH-carbon: A novel sp2–sp3 carbon allotrope with an ultrawide energy gap. Front. Phys. 2022, 17, 63505. [Google Scholar]
- Lima, K.A.L.; Monteiro, F.F.; Santos, E.J.A.; Alves, R.A.F.; Giozza, W.F.; Ribeiro Junior, L.A. Dodecanophene: A novel 2D carbon allotrope with untunable metallic behavior under stress. Mater. Today Commun. 2024, 40, 109455. [Google Scholar] [CrossRef]
- Kou, C.; Tian, Y.; Zhang, M.; Zurek, E.; Qu, X.; Wang, X.; Yin, K.; Yan, Y.; Gao, L.; Lu, M.; et al. M-graphene: a metastable two-dimensional carbon allotrope. 2D Mater. 2020, 7, 025047. [Google Scholar]
- Wei, Y.; Yang, B.; Zhang, S.; Chen, H. rC14: Engineering a New Dual-Function Carbon Allotrope for Sustainable Energy Technologies under Conventional and Micro-strain Conditions. J. Mater. Chem. A 2025, 13, 4455–4465. [Google Scholar]
- Zhang, Wei; Chai, C.; Fan, Q.; Song, Y.; Yang, Y. Penta-C20: a superhard direct band gap carbon allotrope composed of carbon pentagon. Materials 2020, 13, 1926. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lai, Z.; Kan, E.; Zhu, X. Hp-C17: a novel carbon allotrope with an all-sp3 network. Phys. Lett. A 2020, 384, 126379. [Google Scholar]
- Jenkins, T.; Silva, A.M.; Menezes, M.G.; Thonhauser, T.; Ullah, S. THD-C Sheet: A Novel Nonbenzenoid Carbon Allotrope with Tetra-, Hexa-, and Dodeca-Membered Rings. J. Phys. Chem. C 2024, 128, 10982–10996. [Google Scholar]
- Zhang, C.; Yang, X.; Lv, R.; Lv, C.; Qin, J.; Liu, H.; Zang, J.; Dong, L.; Shan, C.X. Pentaheptite diamond: a new carbon allotrope. J. Phys. Condens. Matter 2022, 34, 184003. [Google Scholar] [CrossRef]
- Su, H.; Lai, Z.; Kan, E.; Zhu, X. CP-C20, a new metallic cubic carbon allotrope with an sp2 network. J. Solid State Chem. 2020, 283, 121136. [Google Scholar] [CrossRef]
- Zhao, C.X.; Yang, Y.Q.; Niu, C.Y.; Wang, J.Q.; Jia, Y. C-57 carbon: A two-dimensional metallic carbon allotrope with pentagonal and heptagonal rings. Comput. Mater. Sci. 2019, 160, 115–119. [Google Scholar] [CrossRef]
- Hasanvandi, S.; Neisi, E.; De Sousa, J.M. Poly-dodecahedrane: A new allotrope of carbon. Chem. Phys. Lett. 2024, 841, 141165. [Google Scholar] [CrossRef]
- Adeleke, A.A.; Adeniyi, A.O.; Tang, H.; Gou, H.; Yao, Y. o-C240: a new sp3-dominated allotrope of carbon. J. Phys. Condens. Matter 2020, 32, 395402. [Google Scholar]
- Lima, K.A.L.; Alves, R.A.F.; da Silva, D.A.; Mendonça, F.L.L.; Pereira, M.L.; Ribeiro, L.A. TH-graphyne: A new porous bidimensional carbon allotrope. Phys. Chem. Chem. Phys. 2025, 27, 5512–5520. [Google Scholar]
- Yu, L.; Xu, J.; Shen, C.; Zhang, H.; Zheng, X.; Wang, H.; Qin, Z.; Qin, G. Janus graphene: A two-dimensional half-auxetic carbon allotrope with a nonchemical Janus configuration. Phys. Rev. B 2024, 109, L121402. [Google Scholar]
- Li, T.K.; Ye, X.J.; Meng, L.; Liu, C.S. THFS-carbon: A theoretical prediction of metallic carbon allotrope with half-auxeticity, planar tetracoordinate carbon, and potential application as anode for sodium-ion batteries. Phys. Chem. Chem. Phys. 2023, 25, 15295–15301. [Google Scholar] [PubMed]
- Shang, X.; Xu, D.; Cui, T.; Li, D. Cto-graphene: A Two-Dimensional Graphene Allotrope with High Hole Mobility. ACS Appl. Electron. Mater. 2023, 5, 3741–3747. [Google Scholar]
- Wei, Q.; Zhang, Q.; Yan, H.; Zhang, M. A new superhard carbon allotrope: Tetragonal C64. J. Mater. Sci. 2017, 52, 2385–2391. [Google Scholar]
- Bhattacharya, D.; Jana, D. Twin T-graphene: A new semiconducting 2D carbon allotrope. Phys. Chem. Chem. Phys. 2020, 22, 10286–10294. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Hongchao; Yang, J.; Gao, F. A superhard orthorhombic carbon allotrope: Cmca-C16. Diam. Relat. Mater. 2024, 144, 110987. [Google Scholar] [CrossRef]
- Oliveira, F.L.; Capaz, R.B.; Esteves, P.M. Spiro-Carbon: A Metallic Carbon Allotrope Predicted from First Principles Calculations. ChemPhysChem 2020, 21, 59–64. [Google Scholar] [PubMed]
- Diudea, M.V. DIAMOND D5, A NOVEL ALLOTROPE OF CARBON. Stud. Univ. Babes-Bolyai Chem. 2010, 55, 11–18. [Google Scholar]
- Zhou, L.; Chai, C.; Zhang, Wei; Song, Y.; Zhang, Z.; Yang, Y. oI20-carbon: A new superhard carbon allotrope. Diam. Relat. Mater. 2021, 113, 108284. [Google Scholar]
- Tong, W.; Wei, Q.; Yuan, H.; Zhang, M.; Wu, Z.; Zhu, X. Monoclinic C12: A new superhard carbon allotrope. Phys. B 2023, 653, 414696. [Google Scholar] [CrossRef]
- Liu, J.; Lu, H. Azugraphene: A new graphene-like hexagonal carbon allotrope with Dirac cones. RSC Adv. 2019, 9, 34481–34485. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Ma, M.; Zhu, L.; Ying, P.; Hu, M.; Li, Y.; Zhang, Q. Pc-C10: A Novel Carbon Allotrope with Concurrent Metallic Conductivity and Exceptional Mechanical Property. Vacuum 2025, 238, 114490. [Google Scholar]
- Cai, Y.; Wei, Y.; Lv, C.; Zhang, L.; Chen, Y. LC 567: A new 2D semimetallic carbon allotrope as a promising anode material for lithium-ion batteries. Phys. Chem. Chem. Phys. 2023, 25, 19239–19244. [Google Scholar] [PubMed]
- Fan, Q.; Chai, C.; Wei, Q.; Zhou, P.; Zhang, J.; Yang, Y. Si96: A new silicon allotrope with interesting physical properties. Materials 2016, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.A.J.; Lima, K.A.L.; Mendonça, F.L.L.; Silva, D.A.D.; Giozza, W.F.; Ribeiro Junior, L.A. PHOTH-graphene: A new 2D carbon allotrope with low barriers for Li-ion mobility. Sci. Rep. 2024, 14, 9526. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yu, J.; Liu, J.; Guo, Y.; Qie, Y.; Wang, Q. PCF-graphene: A 2D sp2-hybridized carbon allotrope with a direct band gap. J. Phys. Chem. C 2019, 123, 4567–4573. [Google Scholar]
- Zhao, Z.; Xu, B.; Zhou, X.F.; Wang, L.M.; Wen, B.; He, J.; Liu, Z.; Wang, H.T.; Tian, Y. Novel superhard carbon: C-centered orthorhombic C8. Phys. Rev. Lett. 2011, 107, 215502. [Google Scholar] [PubMed]
- Zhang, C.; Cao, Y.; Liu, Y.; Hu, H.J.; Yu, Z.G.; Zhang, Y.W. Bct-C5: A new body-centered tetragonal carbon allotrope. Diam. Relat. Mater. 2021, 119, 108571. [Google Scholar]
- Lian, R.; Feng, J.; Chen, X.; Wang, D.; Kan, D.; Chen, G.; Wei, Y. Q-carbon: A new carbon allotrope with a low degree of s–p orbital hybridization and its nucleation lithiation process in lithium-ion batteries. ACS Appl. Mater. Interfaces 2019, 12, 619–626. [Google Scholar] [PubMed]
- Gao, Q.; Zhang, L.; Zheng, C.; Lei, S.; Li, S.; Hu, Z. Richmond. HSH-C10: A new quasi-2D carbon allotrope with a honeycomb-star-honeycomb lattice. Chin. Chem. Lett. 2022, 33, 3941–3946. [Google Scholar]
- Estrada, E.; Simón-Manso, Y. Escherynes: Novel carbon allotropes with belt shapes. Chem. Phys. Lett. 2012, 548, 80–84. [Google Scholar] [CrossRef]
- Lima, K.A.L.; Laranjeira, J.A.A.S.; Martins, N.F.; Dias, A.C.; Galvão, D.S.; Ribeiro Junior, L.A. Petal-Graphyne: A Novel 2D Carbon Allotrope for High-Performance Li and Na Ion Storage. arXiv 2025, arXiv:2503.21962. [Google Scholar]
- Wei, Q.; Yang, X.; Wei, B.; Hu, M.; Tong, W.; Yang, R.; Yan, H.; Zhang, M.; Zhu, Xuanmin; Yao, R. Orthorhombic carbon oC48: A new superhard carbon allotrope. Solid State Commun. 2020, 319, 113994. [Google Scholar] [CrossRef]
- Júnior, M.L.P.; da Cunha, W.F.; Giozza, W.F.; de Sousa Junior, R.T.; Ribeiro Junior, L.A. Irida-graphene: A new 2d carbon allotrope. FlatChem 2023, 37, 100469. [Google Scholar] [CrossRef]
- Tromer, R.M.; Pereira Júnior, M.L.; Lima, K.A.L.; Fonseca, A.F.; da Silva, L.R.; Galvão, D.S.; Ribeiro Junior, L.A. Mechanical, electronic, and optical properties of 8-16-4 graphyne: A 2d carbon allotrope with dirac cones. J. Phys. Chem. C 2023, 127, 12226–12234. [Google Scholar] [CrossRef]
- da Silva Brandão, W.H.; Girão, E.C.; Pereira, M.L., Jr.; Latgé, A. Bandgap engineering through topological and strain-induced changes in tetragraphene. ACS Omega 2025, 10, 33570–33582. [Google Scholar] [CrossRef] [PubMed]
- Brandão, W.H.S.; Aguiar, A.L.; Fonseca, A.F.; Galvão, D.S.; De Sousa, J.M. Mechanical properties of tetragraphene single-layer: A molecular dynamics study. Mech. Mater. 2023, 176, 104503. [Google Scholar]
- De Sousa, J.M.; Brunetto, G.; Coluci, V.R.; Galvao, D.S. Torsional “superplasticity” of graphyne nanotubes. Carbon 2016, 96, 14–19. [Google Scholar] [CrossRef]
- Brandão, W.H.S.; Aguiar, A.L.; De Sousa, J.M. Atomistic computational modeling of temperature effects in fracture toughness and degradation of penta-graphene monolayer. Chem. Phys. Lett. 2021, 778, 138793. [Google Scholar] [CrossRef]
- De Sousa, J.M.; Aguiar, A.L.; Girao, E.C.; Fonseca, A.F.; Coluci, V.R.; Galvao, D.S. Mechanical properties of single-walled penta-graphene-based nanotubes: a DFT and classical molecular dynamics study. Chem. Phys. 2021, 547, 111187. [Google Scholar]
- Salvetat, J.-P.; Bonard, J.-M.; Thomson, N.H.; Kulik, A.J.; Forró, L.; Benoit, W.; Zuppiroli, L. Mechanical properties of carbon nanotubes. Appl. Phys. A 1999, 69, 255–260. [Google Scholar] [CrossRef]
- Yu, M.-F. Fundamental mechanical properties of carbon nanotubes: current understanding and the related experimental studies. J. Eng. Mater. Technol. 2004, 126, 271–278. [Google Scholar] [CrossRef]
- Falvo, M.R.; Clary, G.; Helser, A.; Paulson, S.; Taylor, R.M.; Chi, V.; Brooks, F.P.; Washburn, S.; Superfine, R. Nanomanipulation experiments exploring frictional and mechanical properties of carbon nanotubes. Microsc. Microanal. 1998, 4, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Jose, D.; Datta, A. Structures and chemical properties of silicene: unlike graphene. Acc. Chem. Res. 2014, 47, 593–602. [Google Scholar] [PubMed]
- Botari, T.; Perim, E.; Autreto, P.A.S.; Van Duin, A.C.T.; Paupitz, R.; Galvao, D.S. Mechanical properties and fracture dynamics of silicene membranes. Phys. Chem. Chem. Phys. 2014, 16, 19417–19423. [Google Scholar] [CrossRef] [PubMed]
- Roman, R.E.; Cranford, S.W. Mechanical properties of silicene. Comput. Mater. Sci. 2014, 82, 50–55. [Google Scholar] [CrossRef]
- Pei, Q.-X.; Sha, Z.-D.; Zhang, Y.-Y.; Zhang, Yong-Wei. Effects of temperature and strain rate on the mechanical properties of silicene. J. Appl. Phys. 2014, 115, 024309. [Google Scholar] [CrossRef]
- Mortazavi, B.; Rahaman, O.; Makaremi, M.; Dianat, A.; Cuniberti, G.; Rabczuk, T. First-principles investigation of mechanical properties of silicene, germanene and stanene. Phys. E 2017, 87, 228–232. [Google Scholar] [CrossRef]
- Zhao, H. Strain and chirality effects on the mechanical and electronic properties of silicene and silicane under uniaxial tension. Phys. Lett. A 2012, 376, 3546–3550. [Google Scholar] [CrossRef]
- Le, M.-Q.; Nguyen, D.-T. The role of defects in the tensile properties of silicene. Appl. Phys. A 2015, 118, 1437–1445. [Google Scholar]
- Aghdasi, P.; Yousefi, S.; Ansari, R.; Bagheri Tagani, M. A DFT investigation on the mechanical and structural properties of silicene nanosheets under doping of transition metals. Appl. Phys. A 2022, 128, 716. [Google Scholar] [CrossRef]
- Nahid, S.M.; Nahian, S.; Motalab, M.; Rakib, T.; Mojumder, S.; Islam, M.M. Tuning the mechanical properties of silicene nanosheet by auxiliary cracks: a molecular dynamics study. RSC Adv. 2018, 8, 30354–30365. [Google Scholar] [CrossRef] [PubMed]
- Khalkhali, M.; Khoeini, F.; Rajabpour, A. Thermal transport in silicene nanotubes: Effects of length, grain boundary and strain. Int. J. Heat Mass Transf. 2019, 134, 503–510. [Google Scholar] [CrossRef]
- Chegel, R. Theoretical investigation of enhanced nonlinear optical properties of silicene and carbon nanotubes: Potential applications in infrared and ultraviolet optoelectronics. J. Lumin. 2025, 277, 120923. [Google Scholar]
- Chegel, R.; Zarifi, A.; Jalal, M.R. Chirality and magnetic field engineering of linear and quadratic electro-optic properties in Silicene nanotubes for advanced optoelectronic applications. Phys. B 2024, 694, 416435. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Aktulga, H.M.; Fogarty, J.C.; Pandit, S.A.; Grama, A.Y. Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques. Parallel Comput. 2012, 38, 245–259. [Google Scholar] [CrossRef]
- Aktulga, H.M.; Knight, C.; Coffman, P.; O’Hearn, K.A.; Shan, T.-R.; Jiang, W. Optimizing the performance of reactive molecular dynamics simulations for many-core architectures. Int. J. High Perform. Comput. Appl. 2019, 33, 304–321. [Google Scholar]
- Tang, J.; Ahmadi, A.; Alizadeh, A.; Abedinzadeh, R.; Abed, A.M.; Smaisim, G.F.; Hadrawi, S.K.; Nasajpour-Esfahani, N.; Toghraie, D. Investigation of the mechanical properties of different amorphous composites using the molecular dynamics simulation. J. Mater. Res. Technol. 2023, 24, 1390–1400. [Google Scholar] [CrossRef]
- Sutrakar, V.K.; Javvaji, B.; Budarapu, P.R. Fracture strength and fracture toughness of graphene: MD simulations. Appl. Phys. A 2021, 127, 949. [Google Scholar] [CrossRef]
- Petilla, C.E.; Cruz, C.J.D.; Mahinay, C.L. Mechanical properties of Si (1-x)–C (x): strength and stiffness of materials using LAMMPS molecular dynamics simulation. Jpn. J. Appl. Phys. 2024, 63, 08SP09. [Google Scholar] [CrossRef]
- Sakurai, J.J.; Fu Tuan, S.; Newton, R.G. Modern Quantum Mechanics; American Institute of Physics: New York, NY, USA, 1986. [Google Scholar]
- Salinas, S. Introduction to Statistical Physics; Springer Science & Business Media: New York, NY, USA, 2013. [Google Scholar]
- Monticelli, L.; Tieleman, D.P. Force fields for classical molecular dynamics. In Biomolecular Simulations: Methods and Protocols; Springer: New York, NY, USA, 2012; pp. 197–213. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Rapaport, D.C. The Art of Molecular Dynamics Simulation; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Mueller, J.E.; Van Duin, A.C.T.; Goddard, W.A., III. Development and validation of ReaxFF reactive force field for hydrocarbon chemistry catalyzed by nickel. J. Phys. Chem. C 2010, 114, 4939–4949. [Google Scholar] [CrossRef]
- De Sousa, J.M.; Bizao, R.A.; Sousa Filho, V.P.; Aguiar, A.L.; Coluci, V.R.; Pugno, N.M.; Girao, E.C.; Souza Filho, A.G.; Galvao, D.S. Elastic properties of graphyne-based nanotubes. Comput. Mater. Sci. 2019, 170, 109153. [Google Scholar] [CrossRef]
- De Sousa, J.M.; Aguiar, A.L.; Girão, E.C.; Fonseca, A.F.; Souza Filho, A.G.; Galvão, D.S. Computational study of elastic, structural stability and dynamics properties of penta-graphene membrane. Chem. Phys. 2021, 542, 111052. [Google Scholar] [CrossRef]
- De Sousa, J.M.; Aguiar, A.L.; Girao, E.C.; Fonseca, A.F.; Coluci, V.R.; Galvao, D.S. Mechanical properties of single-walled penta-graphene-based nanotubes: a DFT and classical molecular dynamics study. Chem. Phys. 2021, 547, 111187. [Google Scholar]
- Brandão, W.H.S.; Aguiar, A.L.; De Sousa, J.M. Atomistic computational modeling of temperature effects in fracture toughness and degradation of penta-graphene monolayer. Chem. Phys. Lett. 2021, 778, 138793. [Google Scholar] [CrossRef]
- Brandao, W.H.S.; Aguiar, A.L.; Júnior, L.A.R.; Galvao, D.S.; De Sousa, J.M. A computational study on the mechanical properties of Pentahexoctite single-layer: Combining DFT and classical molecular dynamics simulations. Chem. Phys. 2022, 563, 111686. [Google Scholar] [CrossRef]
- De Sousa, J.M. Nanostructures failures and fully atomistic molecular dynamics simulations. In Elasticity of Materials; IntechOpen: London, UK, 2021. [Google Scholar]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, Roman; Janik, M.J.; Aktulga, H.M.; et al. The ReaxFF reactive force-field: Development, applications and future directions. npj Comput. Mater. 2016, 2, 15011. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Verlet, L. Computer “experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Bitzek, E.; Koskinen, P.; Gähler, F.; Moseler, M.; Gumbsch, P. Structural relaxation made simple. Phys. Rev. Lett. 2006, 97, 170201. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.; Terrell, R.; Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 2008, 128, 134106. [Google Scholar] [CrossRef] [PubMed]
- Guenolé, J.; Nöhring, W.G.; Vaid, A.; Houllé, F.; Xie, Z.; Prakash, A.; Bitzek, E. Assessment and optimization of the fast inertial relaxation engine (fire) for energy minimization in atomistic simulations and its implementation in lammps. Comput. Mater. Sci. 2020, 175, 109584. [Google Scholar] [CrossRef]
- Evans, D.J.; Morriss, G.P. The isothermal/isobaric molecular dynamics ensemble. Phys. Lett. A 1983, 98, 433–436. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Lima, K. A.; Abreu, A. V.; Silva, A. M.; Ribeiro, L. A. First-principles and machine learning insights into the design of dott-carbon and its lithium-ion storage capacity. J. Mater. Chem. A vol. 13(no. 21), 15609–15619, 2025.
- Xu, J.; Geng, Y.; Chu, Z.; Hu, Q.; Lei, Y.; Wang, Y. Systematically study the tensile and compressive behaviors of diamond-like carbon. Nanomaterials 2023, vol. 13(no. 11), 1772. [Google Scholar]
- Swenson, R.J. Comments on virial theorems for bounded systems. Am. J. Phys. 1983, 51, 940–942. [Google Scholar] [CrossRef]
- Marc, G.; McMillan, W.G. The virial theorem. Adv. Chem. Phys. 1985, 58, 209–361. [Google Scholar] [CrossRef]
- Zimmerman, J.A.; Webb, E.B., III; Hoyt, J.J.; Jones, Reese E.; Klein, P.A.; Bammann, D.J. Calculation of stress in atomistic simulation. Model. Simul. Mater. Sci. Eng. 2004, 12, S319–S332. [Google Scholar] [CrossRef]
- McQuarrie, D.A.; Rowlinson, J.S. The virial expansion of the grand potential at spherical and planar walls. Mol. Phys. 2007, 60, 977–989. [Google Scholar]
- Dieter, G.E. Mechanical Metallurgy, Chapter 10, 3rd ed.; McGraw-Hill, Inc.: New York, NY, USA, 1986. [Google Scholar]
- Wang, D.; Lee, J.; Holland, K.; Bibby, T.; Beaudoin, S.; Cale, T. Von mises stress in chemical-mechanical polishing processes. J. Electrochem. Soc. 2002, 144, 1121–1127. [Google Scholar]
- Shintani, K.; Narita, T. Atomistic study of strain dependence of Poisson’s ratio of single-walled carbon nanotubes. Surf. Sci. 2003, 532, 862–868. [Google Scholar]
- Wang, L.; Zheng, Q.; Liu, J.Z.; Jiang, Q. Size dependence of the thin-shell model for carbon nanotubes. Phys. Rev. Lett. 2005, 95, 105501. [Google Scholar] [CrossRef] [PubMed]
- Brandão, W.H.S.; De Sousa, J.M.; Aguiar, A.L.; Galvão, D.S.; Ribeiro, L.A., Jr.; Fonseca, A.F. First-principles and reactive molecular dynamics study of the elastic properties of pentahexoctite-based nanotubes. Mech. Mater. 2023, 183, 104694. [Google Scholar]
- Stambulchik, E. GRaphing, Advanced Computation and Exploration of Data. Available online: https://plasma-gate.weizmann.ac.il/Grace/ (accessed on 21 June 2026).
- Cranford, S.W. When is 6 less than 5? Penta-to hexa-graphene transition. Carbon 2016, 96, 421–428. [Google Scholar]
- Brandão, W.H.S.; Aguiar, A.L.; De Sousa, J.M. Atomistic computational modeling of temperature effects in fracture toughness and degradation of penta-graphene monolayer. Chem. Phys. Lett. 2021, 778, 138793. [Google Scholar] [CrossRef]
- Rahaman, O.; Mortazavi, B.; Dianat, A.; Cuniberti, G.; Rabczuk, T. Metamorphosis in carbon network: From penta-graphene to biphenylene under uniaxial tension. FlatChem 2017, 1, 65–73. [Google Scholar] [CrossRef]
- Huang, J.Y.; Ding, F.; Jiao, K.; Yakobson, B.I. In situ TEM sublimation of multi-walled carbon nanotubes under high temperatures. Phys. Rev. Lett. 2007, 99, 175503. [Google Scholar] [PubMed]
- Banhart, F. Structural evolution of carbon nanotubes under electron irradiation at elevated temperatures. Rep. Prog. Phys. 1999, 62, 1181. [Google Scholar]
- Chuvilin, A.; Meyer, J.C.; Algara-Siller, G.; Kaiser, U. Direct observation of the transformation of non-hexagonal rings in defective carbon networks. New J. Phys. 2010, 12, 125011. [Google Scholar]
- Tromer, R.M.; Ribeiro Júnior, L.A.; Galvão, D.S.; Dias, A.C.; Moujaes, E.A. On the mechanical, thermoelectric, and excitonic properties of Tetragraphene monolayer. M. Mater. Today Commun. 2024, m 39, 109310. [Google Scholar] [CrossRef]
- Wei, Q.; Yang, G.; Peng, X. Auxetic Tetrahex Carbon with Ultrahigh Strength and a Direct Band Gap. M. Phys. Rev. Appl. 2020, m 13, 034065. [Google Scholar] [CrossRef]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: a reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- O’Connor, T.C.; Andzelm, J.; Robbins, M.O. Airebo-m: A reactive model for hydrocarbons at extreme pressures. J. Chem. Phys. 2015, 142, 024903. [Google Scholar] [CrossRef] [PubMed]
- Schwerdtfeger, P.; Wales, D.J. 100 years of the Lennard-Jones potential. J. Chem. Theory Comput. 2024, 20, 3379–3405. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.C. The relationship between Lennard-Jones (12-6) and Morse potential functions. Z. Für Naturforschung A 2003, 58, 615–617. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbon molecules. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar]
- O’Connor, B.I.; Andzelm, J.W.; Rice, B.M. Development of an AIREBO-Morse potential for high-pressure carbon systems. Mol. Simul. 2015, 41, 1022–1031. [Google Scholar]
- Van Duin, A.C.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: a reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Mueller, J.E.; van Duin, A.C.; Goddard, W.A. Development and validation of the ReaxFF reactive force field for carbon nanotube nucleation and growth. J. Phys. Chem. C 2010, 114, 4939–4949. [Google Scholar]
- Srinivasan, S.G.; van Duin, A.C.; Ganesh, P. Development of a ReaxFF potential for carbon-condensed phases and its application to graphene fracture. J. Phys. Chem. A 2015, 119, 571–580. [Google Scholar] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Hummer, G.; Rasaiah, J.C.; Noworyta, J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Werder, T.; Walther, J.H.; Jaffe, R.L.; Halicioglu, T.; Koumoutsakos, P. On the water-carbon interaction for use in molecular dynamics simulations of graphite and carbon nanotubes. J. Phys. Chem. B 2003, 107, 1345–1350. [Google Scholar]
- Shan, T.R.; Devine, B.D.; Kemper, T.W.; Sinnott, S.B.; Phillpot, S.R. Charge-optimized many-body potential for the carbon-silicon-oxygen system. Phys. Rev. B 2010, 81, 125328. [Google Scholar]
- Liang, T.; Shan, T.R.; Cheng, Y.T.; Devine, B.D.; Noordhoek, M.; Li, Y.; Lu, Z.; Phillpot, S.R.; Sinnott, S.B. Classical atomistic simulations of surfaces and heterogeneous interfaces with the charge-optimized many-body (COMB) potentials. Mater. Sci. Eng. R Rep. 2013, 74, 255–279. [Google Scholar]
- Sánchez-Portal, D.; Artacho, E.; Soler, J.M.; Rubio, A.; Ordejón, P. Ab initio structural, elastic, and vibrational properties of carbon nanotubes. Phys. Rev. B 1999, 59, 12678. [Google Scholar]
- Kudin, K.N.; Scuseria, G.E.; Yakobson, B.I. C60 transitions, graphene, and carbon nanotubes: Electronic structure and steady-state energetics from DFT. Phys. Rev. B 2001, 64, 235406. [Google Scholar]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Dujardin, E.; Ebbesen, T.W.; Yianilos, P.N.; Treacy, M.M.J. Young’s modulus of single-walled carbon nanotubes. Phys. Rev. B 1998, 58, 14013. [Google Scholar]
Figure 1.
Atomistic configurations of schematic representation of the lattice nanostructure of Tetragraphene. (a) Tetragraphene single layer (and a representation of the chiral () and translational () vectors) and (b) and (c) Tetragraphene-based nanotube topologies (TGCNTs) and , respectively. In (d) The Tetragraphene unit cell is indicated by lattice vectors and Å and Å. In (e) the lateral view of the buckled Tetragraphene nanostructure, with representing the buckling amplitude (). In (f) and (g) The frontal views of TGCNTs and shown distinct diameters.
Figure 1.
Atomistic configurations of schematic representation of the lattice nanostructure of Tetragraphene. (a) Tetragraphene single layer (and a representation of the chiral () and translational () vectors) and (b) and (c) Tetragraphene-based nanotube topologies (TGCNTs) and , respectively. In (d) The Tetragraphene unit cell is indicated by lattice vectors and Å and Å. In (e) the lateral view of the buckled Tetragraphene nanostructure, with representing the buckling amplitude (). In (f) and (g) The frontal views of TGCNTs and shown distinct diameters.

Figure 2.
Atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs). (a)–(l) Stress-strain profiles tracking the nanomechanical behavior of diverse configurations from indices (0,3) to (0,13) via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The (0,N) TGCNT series displays a characteristically brittle failure mode. This response is marked by a highly linear elastic regime followed by sudden nanostructural fracture with negligible plastic deformation.
Figure 2.
Atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs). (a)–(l) Stress-strain profiles tracking the nanomechanical behavior of diverse configurations from indices (0,3) to (0,13) via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The (0,N) TGCNT series displays a characteristically brittle failure mode. This response is marked by a highly linear elastic regime followed by sudden nanostructural fracture with negligible plastic deformation.

Figure 3.
Atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs). (a)-(f) Stress-strain profiles tracking the nanomechanical behavior of diverse configurations from 600 K up to 2100 K via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The TGCNT series displays a characteristically brittle failure mode. This response is marked by a highly linear elastic regime followed by sudden nanostructural fracture with negligible plastic deformation. Additionally, our results reveal a distinct nanostructural degradation at high temperatures, where the nanotubes completely lose their nanostructural stability above 1500 K.
Figure 3.
Atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs). (a)-(f) Stress-strain profiles tracking the nanomechanical behavior of diverse configurations from 600 K up to 2100 K via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The TGCNT series displays a characteristically brittle failure mode. This response is marked by a highly linear elastic regime followed by sudden nanostructural fracture with negligible plastic deformation. Additionally, our results reveal a distinct nanostructural degradation at high temperatures, where the nanotubes completely lose their nanostructural stability above 1500 K.

Figure 4.
Atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs). (a)-(l) Stress-strain profiles tracking the nanomechanical behavior of diverse configurations from indices (3,0) to (13,0) via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The TGCNT series demonstrates remarkable ductility and superelasticity. This nanomechanical response is evidenced by a distinct plateau effect maintaining constant stress up to 20% strain, followed by clear strain hardening until ultimate fracture beyond 40% strain, indicating a stress-induced nanostructural phase transition.
Figure 4.
Atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs). (a)-(l) Stress-strain profiles tracking the nanomechanical behavior of diverse configurations from indices (3,0) to (13,0) via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The TGCNT series demonstrates remarkable ductility and superelasticity. This nanomechanical response is evidenced by a distinct plateau effect maintaining constant stress up to 20% strain, followed by clear strain hardening until ultimate fracture beyond 40% strain, indicating a stress-induced nanostructural phase transition.

Figure 5.
Atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs). (a)–(f) Stress-strain profiles tracking the nanomechanical behavior of the configuration from 600 K up to 2100 K via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The TGCNT series demonstrates remarkable ductility and superelasticity. This nanomechanical response is characterized by a distinct plateau effect maintaining constant stress up to 20% strain, followed by clear strain hardening until ultimate fracture. Notably, the external effect of rising temperature accelerates the degradation of nanofracture toughness, significantly reducing the critical strain to values below 40%. Additionally, our results reveal a distinct nanostructural degradation at high temperatures, where the nanotubes completely lose their nanostructural stability above 1500 K.
Figure 5.
Atomistic deformation response of single-walled tetragraphene-based nanotubes (TGCNTs). (a)–(f) Stress-strain profiles tracking the nanomechanical behavior of the configuration from 600 K up to 2100 K via classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential. The TGCNT series demonstrates remarkable ductility and superelasticity. This nanomechanical response is characterized by a distinct plateau effect maintaining constant stress up to 20% strain, followed by clear strain hardening until ultimate fracture. Notably, the external effect of rising temperature accelerates the degradation of nanofracture toughness, significantly reducing the critical strain to values below 40%. Additionally, our results reveal a distinct nanostructural degradation at high temperatures, where the nanotubes completely lose their nanostructural stability above 1500 K.

Figure 6.
Molecular fully atomistic configurations of TGCNT (0,10) undergoing thermal degradation at elevated temperatures: (a) 600 K, (b) 900 K, (c) 1200 K, (d) 1500 K, and (e) 1900 K. The dotted circles highlight the loss of nanostructured configuration induced by extreme thermal effects at 1900 K across two distinct regions: (f) left side and (g) right side. In (h) a zoomed view detailing the stable, pristine nanostructured configuration of the TGCNT (0,10). (i) Atomistic configuration at 2100 K. In (j,k) a zoomed views displaying the transition into a fully amorphous carbon phase due to severe nanostructural collapse.
Figure 6.
Molecular fully atomistic configurations of TGCNT (0,10) undergoing thermal degradation at elevated temperatures: (a) 600 K, (b) 900 K, (c) 1200 K, (d) 1500 K, and (e) 1900 K. The dotted circles highlight the loss of nanostructured configuration induced by extreme thermal effects at 1900 K across two distinct regions: (f) left side and (g) right side. In (h) a zoomed view detailing the stable, pristine nanostructured configuration of the TGCNT (0,10). (i) Atomistic configuration at 2100 K. In (j,k) a zoomed views displaying the transition into a fully amorphous carbon phase due to severe nanostructural collapse.

Figure 7.
Molecular fully atomistic configurations of TGCNT (14,0) undergoing thermal degradation at elevated temperatures: (a) 600 K, (b) 900 K, (c) 1200 K, (d) 1500 K, and (e) 1900 K. In (f) a zoomed view detailing the stable, pristine nanostructured configuration of the TGCNT (14,0). In (g) the atomistic configuration at 2100 K. The dotted circles highlight the loss of nanostructured configuration induced by extreme thermal effects at 2100 K and (h) a zoomed views displaying the transition into a fully amorphous carbon phase due to severe nanostructural collapse.
Figure 7.
Molecular fully atomistic configurations of TGCNT (14,0) undergoing thermal degradation at elevated temperatures: (a) 600 K, (b) 900 K, (c) 1200 K, (d) 1500 K, and (e) 1900 K. In (f) a zoomed view detailing the stable, pristine nanostructured configuration of the TGCNT (14,0). In (g) the atomistic configuration at 2100 K. The dotted circles highlight the loss of nanostructured configuration induced by extreme thermal effects at 2100 K and (h) a zoomed views displaying the transition into a fully amorphous carbon phase due to severe nanostructural collapse.

Figure 8.
Fully atomistic snapshots of Classical Molecular Dynamics (CMD) simulation results for a (14,0) carbon nanotube (TGCNT) under uniaxial tensile strain load: (a) the pristine nanostructure at an initial, strain-free state (ε = 0%) establishing its nanostructural equilibrium. In (b) uniform nanostructural elongation within the linear elastic regime at 10% strain, in (c) onset of nanomechanical failure characterized by localized stress concentration and initial bond cleavage of () bonds aligned parallel to the loading direction and (d) complete brittle fracture into two distinct segments at a critical strain of 21.27%, immediately followed by the formation of bridging linear atomic chains (LACs) between the fractured ends. (see Supplementary Movie 1 and Supplementary Movie 3 von Mises)
Figure 8.
Fully atomistic snapshots of Classical Molecular Dynamics (CMD) simulation results for a (14,0) carbon nanotube (TGCNT) under uniaxial tensile strain load: (a) the pristine nanostructure at an initial, strain-free state (ε = 0%) establishing its nanostructural equilibrium. In (b) uniform nanostructural elongation within the linear elastic regime at 10% strain, in (c) onset of nanomechanical failure characterized by localized stress concentration and initial bond cleavage of () bonds aligned parallel to the loading direction and (d) complete brittle fracture into two distinct segments at a critical strain of 21.27%, immediately followed by the formation of bridging linear atomic chains (LACs) between the fractured ends. (see Supplementary Movie 1 and Supplementary Movie 3 von Mises)

Figure 9.
Fully atomistic snapshots of Classical Molecular Dynamics (CMD) simulation results for a (14,0) carbon nanotube (TGCNT) under uniaxial tensile strain load along the z-direction. In (a) pristine, strain-free state (ε = 0%), in (b) 20% strain and (c) 30% strain, showing large deformation without nanostructural failure. In (d) onset of nanomechanical failure at a critical strain of 40.69% (inset displays local bond cleavage) and (f) ultimate fracture dividing the nanotube into two segments, accompanied by minor linear atomic chain (LAC) formation at 40.99% of strain. The dotted circles indicate views showing the () bond cleavage at the nanometric scale. (see Supplementary Movie 2 , Supplementary Movie 4 von Mises and Supplementary Movie 5 von Mises)
Figure 9.
Fully atomistic snapshots of Classical Molecular Dynamics (CMD) simulation results for a (14,0) carbon nanotube (TGCNT) under uniaxial tensile strain load along the z-direction. In (a) pristine, strain-free state (ε = 0%), in (b) 20% strain and (c) 30% strain, showing large deformation without nanostructural failure. In (d) onset of nanomechanical failure at a critical strain of 40.69% (inset displays local bond cleavage) and (f) ultimate fracture dividing the nanotube into two segments, accompanied by minor linear atomic chain (LAC) formation at 40.99% of strain. The dotted circles indicate views showing the () bond cleavage at the nanometric scale. (see Supplementary Movie 2 , Supplementary Movie 4 von Mises and Supplementary Movie 5 von Mises)

Figure 10.
High-resolution nanometric-scale snapshots revealing a stress-induced nanostructural phase transition under uniaxial strain loads: (a) 0%, (b) 20%, (c) 39.80%, and (d) 40.69% of strain. The nanomechanical performance is governed by a spatial reconfiguration where - and -hybridized covalent bonds reorient and preferentially concentrate along the uniaxial strain axis.
Figure 10.
High-resolution nanometric-scale snapshots revealing a stress-induced nanostructural phase transition under uniaxial strain loads: (a) 0%, (b) 20%, (c) 39.80%, and (d) 40.69% of strain. The nanomechanical performance is governed by a spatial reconfiguration where - and -hybridized covalent bonds reorient and preferentially concentrate along the uniaxial strain axis.

Figure 11.
Graphical representation of the non-linear nanomechanical response in tetragraphene nanotubes under uniaxial tension, highlighting the nanostructural stretching phase prior to the auxetic drop: (a) the zigzag-like TGCNT exhibiting a constrained Poisson’s ratio of , and (b) the TGCNT showing an enhanced Poisson’s ratio of . Both datasets were obtained directly from classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential framework.
Figure 11.
Graphical representation of the non-linear nanomechanical response in tetragraphene nanotubes under uniaxial tension, highlighting the nanostructural stretching phase prior to the auxetic drop: (a) the zigzag-like TGCNT exhibiting a constrained Poisson’s ratio of , and (b) the TGCNT showing an enhanced Poisson’s ratio of . Both datasets were obtained directly from classical molecular dynamics (CMD) simulations utilizing the AIREBO-Morse potential framework.

Table 1.
Nanostructural parameters of the Tetragraphene model based nanotubes (TGCNTs) simulated by the AIREBO-Morse Classical Molecular Dynamics Simulations Method. The chirality, number of carbon atoms, diameter (Å) and length (Å).
Table 1.
Nanostructural parameters of the Tetragraphene model based nanotubes (TGCNTs) simulated by the AIREBO-Morse Classical Molecular Dynamics Simulations Method. The chirality, number of carbon atoms, diameter (Å) and length (Å).
| Tetragraphene-based nanotubes (TGCNTs) | ||||
|---|---|---|---|---|
| chirality | Number of atoms | Diameter (Å) | Length (Å) | |
| (3,0) | 648 | 4.33 | 109.98 | |
| (4,0) | 864 | 5.77 | 109.98 | |
| (5,0) | 1080 | 7.21 | 109.98 | |
| (6,0) | 1296 | 8.65 | 109.98 | |
| (7,0) | 1512 | 10.09 | 109.98 | |
| (8,0) | 1728 | 11.54 | 109.98 | |
| (9,0) | 1944 | 12.98 | 109.98 | |
| (10,0) | 2160 | 14.42 | 109.98 | |
| (11,0) | 2376 | 15.86 | 109.98 | |
| (12,0) | 2592 | 17.30 | 109.98 | |
| (13,0) | 2808 | 18.75 | 109.98 | |
| (14,0) | 3024 | 20.19 | 109.98 | |
| (0,3) | 864 | 5.83 | 108.72 | |
| (0,4) | 1152 | 7.78 | 108.72 | |
| (0,5) | 1440 | 9.72 | 108.72 | |
| (0,6) | 1728 | 11.67 | 108.72 | |
| (0,7) | 2016 | 13.61 | 108.72 | |
| (0,8) | 2304 | 15.56 | 108.72 | |
| (0,9) | 2592 | 17.50 | 108.72 | |
| (0,10) | 2880 | 19.45 | 108.72 | |
| (0,11) | 3168 | 21.39 | 108.72 | |
| (0,12) | 3456 | 23.34 | 108.72 | |
| (0,13) | 3744 | 25.28 | 108.72 | |
| (0,14) | 4032 | 27.23 | 108.72 | |
Table 2.
Young’s Modulus (GPa.Å), Ultimate Tensile Strength – (GPa.Å), critical strain (%), for Tetragraphene-based nanotubes (TGCNTs)
Table 2.
Young’s Modulus (GPa.Å), Ultimate Tensile Strength – (GPa.Å), critical strain (%), for Tetragraphene-based nanotubes (TGCNTs)
| Tetragraphene-based nanotubes (TGCNTs) | ||||
|---|---|---|---|---|
| chirality | (GPa.Å) | UTS (GPa.Å) | (%) | |
| (3,0) | 2714.10 ± 50.92 | 456.258 | 38.18 | |
| (4,0) | 2617.00 ± 70.10 | 576.555 | 39.86 | |
| (5,0) | 2379.90 ± 100.80 | 671.143 | 40.76 | |
| (6,0) | 2818.20 ± 122.49 | 641.097 | 40.39 | |
| (7,0) | 3124.30 ± 378.79 | 670.304 | 40.93 | |
| (8,0) | 3280.10 ± 150.13 | 638.278 | 40.39 | |
| (9,0) | 3499.20 ± 43.18 | 656.721 | 40.69 | |
| (10,0) | 3343.80 ± 53.71 | 702.932 | 41.06 | |
| (11,0) | 3089.00 ± 94.27 | 695.069 | 40.93 | |
| (12,0) | 3046.10 ± 60.13 | 698.775 | 41.05 | |
| (13,0) | 3173.40 ± 100.99 | 666.086 | 40.87 | |
| (14,0) | 3166.20 ± 90.85 | 669.627 | 40.99 | |
| (0,3) | 1886.70 ± 39.37 | 510.427 | 13.75 | |
| (0,4) | 2119.50 ± 22.60 | 535.123 | 12.79 | |
| (0,5) | 2207.60 ± 20.60 | 506.311 | 12.01 | |
| (0,6) | 2289.00 ± 18.70 | 476.471 | 11.41 | |
| (0,7) | 2282.50 ± 16.67 | 441.485 | 11.35 | |
| (0,8) | 2333.20 ± 8.19 | 455.891 | 11.94 | |
| (0,9) | 2328.70 ± 9.10 | 447.659 | 12.25 | |
| (0,10) | 2351.70 ± 8.77 | 456.920 | 12.55 | |
| (0,11) | 2322.70 ± 10.42 | 435.311 | 12.78 | |
| (0,12) | 2374.40 ± 13.02 | 452.804 | 12.61 | |
| (0,13) | 2355.30 ± 7.60 | 447.659 | 12.85 | |
| (0,14) | 2324.30 ± 12.26 | 433.253 | 13.27 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



