Submitted:
26 June 2026
Posted:
26 June 2026
You are already at the latest version
Abstract
The taxonomic status of Cipangopaludina chinensis (ZGYTL) and C. cathayensis (ZHYTL) remains controversial due to morphological overlap and intermediate forms. We combined morphometrics (24 landmarks vs. 200 semilandmarks) with multi‑locus phylogenetics (COI, 16S rRNA, H3, 28S rRNA) and haplotype networks on sympatric populations from Liuzhou and GenBank. Morphometrics completely separated ZGYTL from ZHYTL in shell shape; key diagnostic traits include aperture size, spire height, and body whorl inflation. The high‑density semilandmark method outperformed discrete landmarks in dimensionality reduction and fine‑scale resolution, capturing subtle apex and lateral whorl variations. No sexual dimorphism occurred in ZHYTL, with only weak dimorphism in ZGYTL. Molecular data were ambiguous: the species tree and mitochondrial gene tree recovered C. chinensis and C. cathayensis as monophyletic, but the nuclear gene tree showed mixing of ZGYTL/ZHYTL with C. wisseli, C. chinensis, and C. cathayensis. Haplotype networks shared haplotypes between ZGYTL and ZHYTL, yet neither shared haplotypes with GenBank sequences of the two nominal species. Divergence time placed the C. chinensis complex split at ~25.62 million years ago (Ma) and ZGYTL–ZHYTL separation at ~11 Ma. Morphology strongly supports ZGYTL and ZHYTL as distinct species, whereas molecular evidence shows low level differentiation with mitonuclear conflicts. We conclude they are likely valid species, yet mitonuclear discordance warrants further genomic investigation. The distinct shell morphologies suggest possible ecological divergence, supporting separate management under ongoing habitat and harvesting pressures. This study validates geometric morphometrics as an efficient frontline tool for biodiversity assessment in morphologically diverse yet genetically conserved freshwater snails, and reinforces the need for genome‑wide data to resolve species boundaries within the Cipangopaludina complex.
Keywords:
Cipangopaludina
; geometric morphometrics
; phylogenetic analysis
; species delimitation
; sexual dimorphism
1. Introduction
Freshwater gastropods of the family Viviparidae display extraordinary morphological diversity in shell form [1,2,3,4,5,6]. This variation has long confounded taxonomic classification, generating persistent systematic instability across the group [2,4,5,7,8,9,10,11]. For instance, in East Asia, this challenge is epitomized by two widespread and economically important congeneric morphospecies, Cipangopaludina chinensis (Gray, 1834) and C. cathayensis (Heude, 1890), collectively called the Chinese river snails [12]. For over a century, these taxa have been distinguished primarily by shell shape. The key traits used include height-to-width ratio, spire height, body whorl outline, and aperture form. Despite these diagnostic traits, field surveys frequently uncover intermediate forms that resist clear taxonomic assignment. The critical question, therefore, is whether these nominal taxa are genuinely distinct species or synonyms [2,10]. This taxonomic uncertainty carries significant practical consequences, as both species are harvested extensively for human consumption, serve as critical feed sources in aquaculture, accelerate aquatic nutrient cycling, maintain ecosystem stability, and act as sensitive bioindicators of water quality in shallow lentic ecosystems [13,14,15,16]. Resolving their systematic status is therefore essential for effective fisheries management, ecological monitoring, and conservation planning.
Landmark-based geometric morphometrics (GM) has revolutionized the analysis of organismal shape by enabling rigorous, size-independent quantification of morphological variation by eliminating non-shape confounding factors (e.g., position, orientation, and overall size) via Generalized Procrustes Analysis [17,18]. Its utility in dissecting adaptive variation has been demonstrated across diverse systems, including squamate osteoderms, mosquito wings, and anuran skeletal morphology [19,20,21]. Landmark-based geometric morphometrics (GM) has revolutionized the study of mollusk shell form, enabling rigorous, size-independent quantification of morphological variation while preserving the full geometry of shell shape [22]. It provides a robust analytical framework for investigating molluscan shell evolution, adaptation, and phenotypic diversification [23,24,25]. And GM has proven powerful in resolving taxonomic debates, delineating cryptic species, quantifying ecophenotypic plasticity, and detecting subtle sexual dimorphism [26,27,28,29]. Viviparid snails are gonochoristic, and pronounced sexual dimorphism in shell outline, aperture morphology, and body size has been documented in multiple genera, including Viviparus, Filopaludina, and Margarya [30,31,32,33]. For the C. chinensis–C. cathayensis species complex, a key question remains unaddressed. It is unclear whether the morphological gap between them reflects species-level divergence, or simply sexual dimorphism and intraspecific variation misclassified by traditional taxonomy. This possibility, a potential driver of taxonomic inflation, has never been directly tested.
Alongside these morphological advances, the widespread application of DNA barcoding and multi-locus phylogenetics has revealed pervasive discordance between morphological and genetic differentiation across numerous mollusk groups [34,35], with many traditionally recognized morphospecies exhibiting deep mito-nuclear conflicts, incomplete lineage sorting, or even complete genetic homogeneity despite pronounced phenotypic divergence [36,37]. In freshwater gastropods, molecular analyses using standard mitochondrial and nuclear markers have repeatedly shown that morphologically distinct nominal species can be genetically indistinguishable [2,10,23,38,39,40,41]. This pattern is typically attributed to recent divergence with incomplete lineage sorting, ongoing gene flow via hybridization, or phenotypic plasticity, rather than long-term independent evolutionary trajectories [1,23,40,42,43]. Preliminary genetic data indicate remarkably low interspecific distances between C. chinensis and C. cathayensis [11,12], yet no integrative study has combined high-resolution geometric morphometrics with multi-locus genetics to resolve this systematic conundrum.
In this study, we performed the first integrative taxonomic assessment of C. chinensis and C. cathayensis. We combined landmark-based geometric morphometrics of shell shape with phylogenetic and network analyses of four markers (mitochondrial COI and 16S rRNA; nuclear 28S rRNA and H3), incorporating diverse gastropods and appropriate outgroups for robust phylogenetic inference. These results have important implications for the taxonomy, evolutionary biology, and conservation of freshwater viviparids.
2. Materials and Methods
2.1. Sample Collection, Traditional and Geometric Morphometrics Analyses
C. chinensis (ZGYTL) and C. cathayensis (ZHYTL) were both collected from Liuzhou City, Guangxi Zhuang Autonomous Region, with 31 individuals of each species sampled (16 males and 15 females). These specimens were used for traditional morphometrics, geometric morphometric and molecular phylogenetic analyses. In addition, other published Cipangopaludina were obtained from GenBank, as well as other Mollusca species sequences as outgroup (Table S1). To test for morphometric differences between ZGYTL and ZHYTL and to assess intraspecific sexual dimorphism, traditional linear measurements were performed on the same specimens used for geometric morphometric analyses. Seven shell characters—shell height (SH), shell width (SW), spiral height (SpH), body whorl height (BWH), aperture width (AW), operculum height (OH), and operculum width (OW)—were measured using digital calipers (0.01 mm precision, Table S2). To remove size effects, all raw measurements were divided by SH to obtain morphometric ratio indices (SW/SH, SpH/SH, BWH/SH, AW/SH, OH/SH, OW/SH). Principal component analysis (PCA) and principal coordinate analysis (PCoA) based on Euclidean distance matrices were conducted on the ratio data, followed by pairwise Wilcoxon rank-sum tests (wilcox.test) with Bonferroni correction (α = 0.05). All statistical analyses were performed in R 4.2.1.
Furthermore, shell geometric morphometrics was also used to examine shape variation in MorphoJ v. 2.0 [44]. Two complementary landmark-based geometric morphometric approaches were employed to quantify shell shape variation. Each specimen was assigned a unique identification number and photographed in a standardized orientation and at a fixed focal length using a Canon EOS R50 digital camera, ensuring image consistency across all samples. The first method followed the protocol of Minton & Wang [31], who demonstrated sexual shape dimorphism in Viviparus (Gastropoda: Viviparidae) using a set of 24 type II landmarks (homology assumed on relative position only) placed at suture lines or extreme points on the shell surface (as illustrated in Supplemental Figure S1). These landmarks were digitized on each shell image using tpsDig2 [45]. The X and Y coordinates of all landmarks were recorded and subjected to Generalized Procrustes Analysis (GPA) in tpsRelw [45] to remove non-shape variation arising from differences in position, orientation, and scale. The resulting Procrustes coordinates were imported into MorphoJ v2.0 for subsequent multivariate analyses. The second approach employed high-density outline-based semilandmarks to capture fine-scale shell contour variation. Shell images were compiled into a single TPS file using tpsUtil [45]. In tpsDig2, the “draw curves” function was used to trace each shell outline counterclockwise from the apex around the entire shell perimeter. Each curve was then resampled into 200 equidistant semilandmarks using the “resample curves” function with the “by length” option. This method generates points that are geometrically, rather than anatomically, corresponding across specimens, and thus represents a complementary strategy for shape quantification. Because MorphoJ does not recognize curve control lines (“LM=0, CURVES=1, POINT=150/200/250”), the file was opened in a text editor and these lines were batch-replaced with “LM=200” prior to import, retaining 200 equidistant semilandmarks for analysis.
For both landmark datasets, subsequent analyses followed the same workflow in MorphoJ v2.0. Datasets for each species were imported separately and then merged using “Preliminaries → Combine Datasets.” Outliers were identified and removed using the “Find Outliers” function based on the 95th percentile of Procrustes distances (default threshold). Covariance matrices were generated via “Preliminaries → Generate Covariance Matrix.” Principal component analysis (PCA) was performed on both single-species and combined datasets to visualize patterns of shape variation. For the combined dataset, a classifier variable was created with four categories: ZGYTL female (ZGC, n1 = 15), ZGYTL male (ZGX, n2 = 16), ZHYTL female (ZHC, n3 = 15), and ZHYTL male (ZHX, n4 = 16). Canonical variate analysis (CVA) was conducted using both species identity and the combined species–sex classifier, with pairwise permutation tests (10,000 iterations) to assess the significance of group differences. Due to the small sample size relative to the number of shape variables (n < p), Pillai’s trace test is invalid; therefore, only Goodall’s F test and permutation tests were used for group comparison. For both PCA and CVA, results including transformation grids, scatter plots with confidence ellipses, and Procrustes and Mahalanobis distances with associated P-values were exported.
2.2. DNA Extraction, Sequencing and Molecular Analyses
Foot muscle tissue was used for genomic DNA extraction with a QIAGEN DNeasy Blood & Tissue Kit (Shanghai, China) following the manufacturer’s instructions. DNA concentration and purity were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Four molecular markers—two mitochondrial (COI and 16S rRNA) and two nuclear (H3 and 28S rRNA)—were targeted for PCR amplification. Primers for COI (LCO1490/HC02198), 16S rRNA (16sar-L/16sbr-H), and H3 (F/R) were obtained from published sources [46,47,48]. For 28S rRNA gene amplification, we referred to Gu et al. (2019)[23] but introduced minor modifications to the PCR conditions to ensure successful amplification for each sequence. Amplification was performed in 50-μL reactions containing 1 μL of DNA template, 10 μL of 5× PrimeSTAR Buffer (Mg²⁺ plus), 4 μL of dNTP mixture (2.5 mM each), 1 μL of each primer, 0.5 μL of PrimeSTAR HS DNA Polymerase (2.5 U/μL; TaKaRa, Dalian, China), and sterile water to volume. Thermal cycling consisted of an initial denaturation at 94 °C for 4 min, followed by 35 cycles of 98 °C for 10 s, annealing for 5 s, and extension at 72 °C for 45 s, with a final 8-min extension at 72 °C. Annealing temperatures differed among loci: 50 °C for COI, 48 °C for 16S rRNA, 54 °C for H3, and 52 °C for 28S rRNA.
PCR products were purified with a commercial purification kit and sequenced bidirectionally using BigDye Terminator chemistry on an automated sequencer. Sequence reads were aligned using MAFFT as implemented in PhyloSuite v1.2.3 [49], and checked against GenBank entries for verification. All newly obtained sequences were submitted to GenBank under accession numbers, please see Table S1.
2.3. Phylogenetic Analyses
To evaluate the hypothesis that the nominal Cipangopaludina species under study constitute monophyletic lineages, species-tree inference was performed using the multi-locus coalescent method implemented in BEAST v2.6.7 [50]. The analysis encompassed 44 individuals for which sequences from all four targeted markers were available (Table S1). The Cephalopoda, Sepioteuthis lessoniana, were designated as outgroups. Optimal nucleotide substitution models were selected independently for each gene partition using PartitionFinder as implemented in PhyloSuite v1.2.3 [49]. The resulting assignments were as follows: GTR+I+G+X for COI and H3, TRN+G+X for 28S rRNA, and HKY+G+X for 16S rRNA. Prior to multi-locus analysis, a partition homogeneity test was conducted in PAUP; the four gene fragments showed no significant phylogenetic conflict (P < 0.01), supporting their combined use in a simultaneous analysis. Input files for the coalescent analysis were generated with BEAUTi v2 available in the BEAST package. A Yule pure-birth process was specified as the species-tree prior, consistent with recommendations for species-level datasets [51], and a constant population size was assumed. The Markov chain Monte Carlo (MCMC) chain was run for 30 million generations, with parameters and trees sampled at intervals of 3,000 generations. Adequate mixing and convergence were verified in Tracer v1.7 [52] by confirming that effective sample sizes (ESS) for all parameters exceeded 200. The initial 10% of sampled trees (1,000 trees) were discarded as burn-in after visual inspection confirmed that likelihood values had reached a stationary plateau. A maximum-clade-credibility consensus tree was subsequently summarized using TreeAnnotator v2.6.7 available in the BEAST package, again with 10% burn-in removed, and nodes receiving posterior probability support ≥ 95% were considered strongly supported. The resulting species tree was visualized in FigTree v1.4.4 (https://tree.bio.ed.ac.uk/software/figtree/, accessed on 25-May-26). To further assess topological concordance across the posterior sample, all post-burn-in trees were superimposed as transparent lines using DensiTree v2.01 [53]; regions of the resulting plot exhibiting dense coloration indicate high agreement among trees on a particular topology and set of branch lengths.
Additionally, phylogenetic relationships were also reconstructed separately for the mitochondrial (COI + 16S rRNA) and nuclear (H3 + 28S rRNA) concatenated datasets using both maximum likelihood (ML) and Bayesian inference (BI). The mitochondrial dataset encompassed nearly all sequenced individuals, as amplification success was highest for COI and 16S rRNA; the nuclear dataset comprised specimens with complete sequences for both H3 and 28S rRNA. All sequences were aligned using MAFFT as implemented in PhyloSuite v1.2.3. For the protein-coding genes (COI and H3), sequences were partitioned by codon position; the ribosomal genes (H3 and 28S rRNA) were treated as single partitions. The optimal nucleotide substitution model for each partition was selected with ModelFinder, with separate runs conducted for the mitochondrial and nuclear datasets. Base saturation was assessed under the GTR model in DAMBE v7.3.11 (https://dambe.bio.uottawa.ca/DAMBE/dambe.aspx, accessed on 21 March-2026). Maximum likelihood trees for each dataset were inferred with RAxML 8.0 [54], applying the GTR+G model to all partitions and performing 1,000 bootstrap replicates to assess nodal support. Bayesian analyses were run in MrBayes 3.2.2 [55], with each dataset partitioned according to the optimal schemes identified by ModelFinder. Two independent MCMC runs, each with four chains, were executed for approximately 10 million generations, sampling every 1,000 generations. Convergence was evaluated in Tracer v1.7 by verifying that the average standard deviation of split frequencies fell below 0.01 and that effective sample sizes (ESS) exceeded 200 for all parameters. The initial 10% of sampled trees were discarded as burn-in. Final consensus trees were visualized and edited in iTOL (https://itol.embl.de/, accessed on 18 May-2026). To detect potential phylogenetic conflict between mitochondrial and nuclear markers, the topologies of the resulting ML and BI trees from the two datasets were compared manually. Well-supported nodes (bootstrap ≥ 70% in ML; posterior probability ≥ 0.95 in BI) that exhibited discordant relationships between the mitochondrial and nuclear gene trees were identified and recorded.
2.4. Divergence Time Estimation
Divergence times were inferred using BEAST v2.6.7 under an uncorrelated lognormal relaxed molecular clock and a birth–death tree prior, applied to the multilocus (COI + 16S rRNA) dataset. The substitution models assigned to each partition followed those used in the preceding phylogenetic analyses (Table S1). Two secondary calibration points were adopted from TimeTree (http://www.timetree.org/, accessed on 25-May 2026) and implemented as soft-bound priors: (1) the divergence between Lottia gigantea and Pomacea canaliculata, with a mean of 488.7 Ma (95% CI: 468.8–526.6 Ma), and (2) the split between P. canaliculata and Cipangopaludina sp., with a mean of 129.7 Ma (95% CI: 79.0–165.1 Ma). These calibrations were modelled as normal distribution priors centred on the respective mean ages, with standard deviations set to cover the reported 95% confidence intervals, thereby providing soft maximum and minimum constraints.
Four independent MCMC chains were run for 20 million generations each, sampling every 10,000 generations, with substitution and clock models unlinked across partitions. Convergence and adequate mixing were confirmed in Tracer v1.7, ensuring that effective sample sizes for all parameters exceeded 200. The initial 10% of samples were discarded as burn-in, and post-burn-in tree files from the four runs were combined using LogCombiner v2.6.7. A maximum clade credibility tree was summarised in TreeAnnotator v2.6.7, applying a posterior probability limit of 0.5 and reporting mean node heights.
3. Results
3.1. Morphological and Sexual Divergence
3.1.1. Traditional Morphometrics
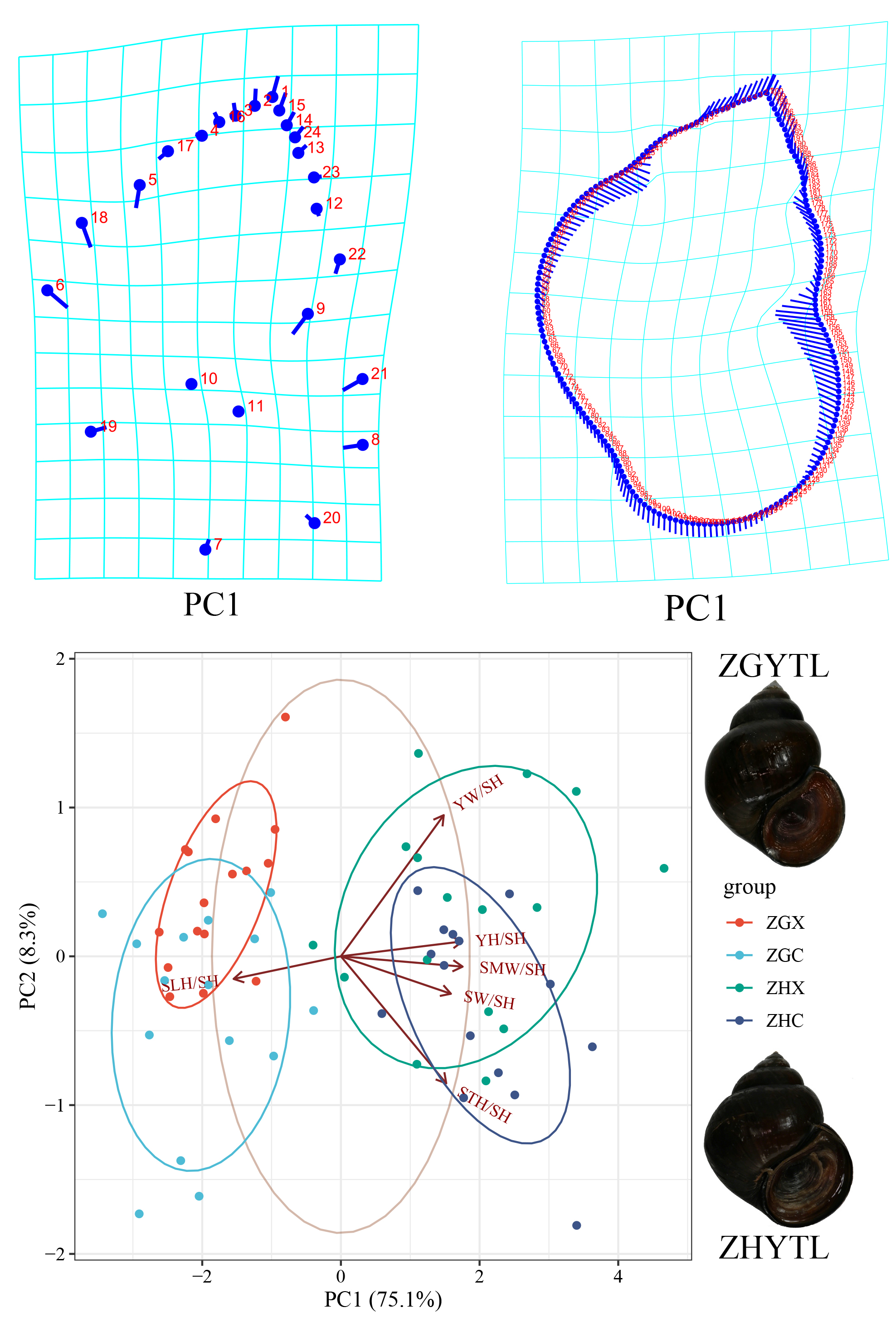
PCA showed that PC1 and PC2 explained 75.1% and 8.3% of total variation, respectively. ZGYTL and ZHYTL were completely separated along PC1 (68% confidence ellipses non-overlapping), indicating significant shell differentiation (Figure 1a). No sex-associated shape variation was detected within either taxon (highly overlapping distributions). Loading analysis identified SpH/SH, (negative PC1 loadings) and AW/SH, SW/SH, OW/SH, OH/SH, BWH/SH (positive PC1 loadings) as core drivers of divergence; OW/SH and BWH/SH also contributed strongly to PC2, reflecting within-group individual variation (Figure 1a). PCoA based on morphological distance matrices showed that PCoA1 and PCoA2 explained 85.45% and 5.46% of variation (cumulative 90.91%, Figure 1b). ZGYTL and ZHYTL were fully separated along PCoA1 (no sample overlap), with sexes interspersed within each group, confirming no sex effect on morphological divergence. Two-way ANOVA followed by Tukey’s HSD confirmed highly significant interspecific differences for all six proportional traits (SW/SH, SpH/SH, BWH/SH, AW/SH, OH/SH, OW/SH; all P < 0.001, Figure 1c), reinforcing the pronounced shell differentiation between the two taxa group.
3.1.2. Geometric Morphometrics
Two geometric morphometric approaches (24 discrete landmarks vs. 200 high-density semilandmarks) were applied. PCA showed that the first nine principal components from discrete landmarks accounted for 90.63% of total shape variation (PC1 alone contributed 56.91%) (Figure 2a), while the first three components from high-density semilandmarks captured 90.91% of variation (PC1 alone contributed 80.80%) (Figure 2b), demonstrating superior dimensionality reduction efficiency. Both methods clearly separated the two species along PC1: ZHYTL clustered on the negative side and ZGYTL on the positive side, with the latter exhibiting a more compact distribution and minimal individual overlap (Figure 2). Thin-plate spline deformation grids identified core interspecific differences in the outer aperture lip, left side of the first whorl near the aperture, and umbilicus: ZHYTL had an expanded aperture and adjacent whorl, a depressed apex, and a flatter overall shell, whereas ZGYTL showed a retracted aperture and whorl, an elongated apex, and a more slender form (Figure 2c,d). The high-density semilandmark method further resolved subtle apex variation and contraction/expansion of the right whorl, significantly improving morphological resolution (Figure 2e,f).
CVA revealed that CV1 explained 100% of interspecific variation for both datasets, with complete separation of the two group along this axis (Figure 3a,b). Interspecific Mahalanobis distances were 15.4901 (discrete landmarks) and 12.3682 (high-density semilandmarks), with corresponding Procrustes distances of 0.0573 and 0.0524, respectively (Table 1). All 10,000-iteration permutation tests and Goodall’s F tests confirmed highly significant interspecific differences (P < 0.0001). For species-sex grouped analysis, the first two CVA axes from high-density data explained 95.56% of among-group variation. Interspecific Mahalanobis distances (12.0114–12.7506) were substantially larger than intraspecific sexual distances (3.8391–4.0049) (Table 2). In contrast, the 24-landmark dataset yielded consistently high Mahalanobis distances across all pairwise group comparisons (interspecific: 20.9306–71.8304; intraspecific sexual: 25.2567–35.7758), with no clear separation between interspecific and intraspecific sexual distances. Permutation tests revealed significant intergroup differences (P < 0.0001); weak but significant sexual dimorphism was detected only in ZGYTL (P = 0.0078) using high-density data (Figure 3c), whereas no significant dimorphism was observed in ZHYTL (P = 0.2740). Discrete landmark data showed similar trends, but intraspecific sexual differences were not statistically significant.
3.2. Phylogenetic Analysis
3.2.1. Species Tree
The species tree was reconstructed for the genus Cipangopaludina using a multispecies coalescent model in BEAST2, based on 44 individual sequences from these four molecular markers (Figure 4a). The MCMC chain ran for 30 million generations and achieved good convergence (ESS > 200). The outgroup, the cephalopod S. lessoniana, was positioned at the base of the tree. Bivalves formed a highly supported monophyletic group with maximal posterior probability (PP = 1.0), and were resolved as sister to the patellogastropod Lottia lineage (PP = 0.99) instead of the entire gastropod clade (Figure 4b). Overlay of the full posterior tree set in DensiTree showed substantial overlap of the major branches, indicating a robust backbone topology. Within Gastropoda, Cipangopaludina was recovered as a strongly supported monophyletic group (PP = 1.0). All seven nominal species included in this study — C. japonica, C. tussurientis, C. suffiensis, C. ziaenensis, C. chinensis, C. compactus, and C. catayensis — formed distinct monophyletic lineages. Most intra- and interspecific divergence nodes had PP ≥ 0.95 or 1.0. The basal node of the C. chinensis complex (including its close relatives) also received high support (Figure 4b). All field-collected samples of the ZGX and ZHX series fell within the C. chinensis complex. DensiTree overlay revealed extensive branch overlap and blurred lineage boundaries between ZGX and ZHX, indicating the absence of significant genetic differentiation and that they do not form reciprocally monophyletic evolutionary lineages. Nevertheless, both ZGX and ZHX exhibited clear genetic differentiation from the seven nominal species of Cipangopaludina, especially from C. chinensis and C. catayensis (Figure 4a).
3.2.2. Gene Tree
Separate maximum likelihood (ML) and Bayesian inference (BI) analyses of concatenated mitochondrial (COI+16S rRNA) and nuclear (H3+28S rRNA) datasets produced congruent topologies. Mitochondrial trees exhibited high nodal support, whereas nuclear trees showed overall low support. Significant mitonuclear phylogenetic conflicts were detected outside Viviparidae (e.g., among bivalves, limpets, Ellobium, hydrothermal vent gastropods, and top shells) (Figure 5).
Within Cipangopaludina, the mitochondrial phylogenetic tree resolved all seven nominal species as monophyletic groups. C. chinensis and C. catayensis were identified as sister taxa, with C. compactus forming a clade that included them. The ZGYTL and ZHYTL samples were nested within the C. chinensis complex, exhibiting differentiation from both C. chinensis and C. catayensis (Figure 5). However, these samples displayed short branch lengths and blurred lineage boundaries among each other. The nuclear tree supported the monophyly of Cipangopaludina and the independence of the seven nominal species, but conflicted with the mitochondrial tree regarding ZGYTL/ZHYTL. These samples did not form a distinct lineage but instead intermixed with individuals of C. chinensis, C. catayensis, and C. wisseli, exhibiting paraphyly or polytomies.
3.3. Haplotype Network
Median-joining haplotype network analyses based on the 16S rRNA and COI genes yielded overall consistent results. The focal populations ZGYTL and ZHYTL formed distinct, independent haplotype lineages (Figure 6a,b). In the 16S rRNA dataset, four haplotypes (Hap_1–4) were detected with sharing between the two populations; in the COI dataset, the two populations shared a single haplotype (Hap_1). No haplotype sharing was observed between these focal populations and the GenBank sequences of C. chinensis and C. cathayensis; they were separated by many mutational steps, indicating clear lineage segregation. Overall, interspecific genetic boundaries were clearly defined among most congeneric species. Haplotype sharing was restricted to the C. chinensis species complex (encompassing C. cathayensis and C. compactus) and between C. zejaensis and C. sujfunensis, suggestive of genetic introgression or ambiguous taxonomic delimitation. The vast majority of remaining congeneric taxa formed discrete haplotype clusters in both genes, with substantial interspecific genetic distances and unambiguous species differentiation. Several undetermined specimens shared haplotypes with their corresponding known species and exhibited high genetic affinity, presumably representing misidentified specimens or recently diverged lineages.
3.4. Divergence Time
Divergence time estimation based on concatenated mitochondrial COI and 16S rRNA sequences yielded a tree topology highly congruent with prior mitochondrial gene trees and the BEAST2 multi-locus coalescent species tree at generic and specific levels. MCMC chains converged well (all ESS > 200), confirming reliable time estimates. Major clade divergences: Cephalopods split from other mollusks at ~494.98 Ma (approx. 500 Ma, Late Cambrian); Patelloidea (limpets) diverged from the clade containing remaining gastropods + all Bivalvia at 483.67 Ma (Early Ordovician); the combined clade of Bivalvia + Heterobranchia diverged from Caenogastropoda at 400.99 Ma (Early Devonian); within this composite Bivalvia+Heterobranchia lineage, Bivalvia separated from Heterobranchia at 361.01 Ma (Late Devonian); inside Bivalvia, Pectinidae diverged from all remaining bivalve taxa at 330.61 Ma (Early Carboniferous); Trochoidea diverged from the rest of Caenogastropoda at 250.07 Ma (Late Permian) (Figure 7). Viviparidae diverged from Ampullariidae (Pomacea canaliculata) at 126.02 Ma (Early Cretaceous), consistent with secondary calibrations. Within Viviparidae, Viviparus split from Cipangopaludina at 88.02 Ma (Late Cretaceous). The MRCA of Cipangopaludina dates to 58.04 Ma (Paleocene), with radiative diversification through the Cenozoic: C. japonica diverged at 34.35 Ma (Oligocene); the C. chinensis complex (including C. compactus, C. catayensis) split at 25.62 Ma (Oligocene-Miocene boundary); C. sujfunensis and C. zejaensis speciated at 14.36 Ma (Middle Miocene); ZG and ZH lineages diverged at 11.07 Ma (Late Miocene).
4. Discussion
Our integrative study provides the first simultaneous evaluation of shell morphology and multi-locus genetics for the two sympatric Chinese river snail populations, ZGYTL and ZHYTL, traditionally assigned to Cipangopaludina chinensis and C. cathayensis, respectively. The results reveal a striking discordance: morphometric evidence strongly supports their status as two distinct species, whereas molecular data show only moderate and partially conflicting genetic differentiation. This pattern underscores the necessity of integrative taxonomy for resolving species boundaries in morphologically plastic freshwater gastropods.
4.1. Morphological Evidence: Clear and Consistent Species Separation
Three multi-scale morphometric approaches all recovered clear shell shape divergence between ZGYTL and ZHYTL. They ranged from traditional linear measurements and 24 landmark geometric morphometrics to 200-point high-density semilandmark outline analysis, and differed substantially in resolving power and trait detection capacity. Traditional linear measurements provided size-adjusted shape discrimination. The complete separation along PC1 and PCoA1 demonstrated that overall shell proportions differ profoundly between the two taxa (Figure 1). The proportional traits driving this separation (SpH/SH, AW/SH, SW/SH, OW/SH, OH/SH, BWH/SH) directly correspond to the classic diagnostic characters described for C. chinensis and C. cathayensis (Figure 1). C. chinensis develops a larger shell with an elongate conical form, characterized by a slender, high-spired outline; its spire is conical, with height exceeding that of the aperture, and each whorl increases rapidly in both height and width [58]. By contrast, C. cathayensis assumes a broadly conical to ovoid shape, possesses a short and broad spire, with whorls expanding rapidly in width and exhibiting convex whorl surfaces. Its body whorl is more inflated and stout, representing a higher proportion of overall shell height [30]. Traditional morphometrics thus validated these classical traits quantitatively, but it could not capture fine whorl surface details or aperture curvature.
Discrete landmark-based geometric morphometrics (24 landmarks) afforded higher-resolution shell shape characterization. Thin-plate spline deformation grids localized interspecific divergence to three key regions, namely the outer aperture lip, and the first whorl flank adjacent to the aperture (Figure 2c,d). These deformation patterns align precisely with established diagnostic traits. ZHYTL exhibits an expanded aperture and inflated adjacent whorl consistent with its broadly conical-ovoid form and convex whorl surfaces, as well as a depressed apex matching its short, broad spire [31]. ZGYTL by contrast shows a retracted aperture and elongated apex corresponding to its slender, high-spired outline. This landmark-based framework reliably captures holistic whorl and aperture geometry. Analogous approaches have been widely adopted to delimit closely related viviparid species [2,59] and detect intraspecific sexual shape dimorphism within the family [31,33]. Nevertheless, limited landmark density precludes resolution of subtle variation in the fine apex curvature.
The high-density semilandmark approach with 200 equidistant points outperformed discrete landmarks in both dimensionality reduction efficiency and fine-scale shape resolution. The first three principal components accounted for nearly all total shape variation, indicating far higher information efficiency per component relative to the discrete landmark dataset [60,61]. This method not only reproduced all major deformation patterns identified by discrete landmarks but also resolved additional subtle shape differences including lateral whorl contraction or expansion and fine-scale apex morphological variation (Figure 2e,f). These fine-scale features align directly with documented morphological disparities between the two species. C. chinensis exhibits coordinated rapid growth in both whorl height and width, while C. cathayensis shows preferential width expansion and strongly convex whorl surfaces [58]. These patterns were only partially captured by discrete landmark analysis. This approach further demonstrated that the characteristically inflated, stout body whorl of C. cathayensis extends across the lateral whorl surface, a morphological detail not emphasized in prior taxonomic descriptions. The improved resolution of subtle shell traits via high-density semilandmarks is consistent with findings from other gastropod morphometric studies [17]. For morphological structures with limited unambiguous homologous landmarks, quantitative comparisons have shown semilandmark-based analyses outperform traditional landmark methods in detecting subtle shape variation [61,62]. Both geometric morphometric approaches reliably discriminate the two species. The high-density semilandmark method captures more shape variation with fewer components and resolves subtle shell features beyond the resolution of discrete landmarks (Figure 2a,b). Core diagnostic traits include aperture size, first whorl contour, apex morphology, and umbilicus position. No significant sexual dimorphism was detected in ZHYTL, whereas ZGYTL showed only weak sex-associated differentiation (Figure 3c, Table 2). The observed shape divergence cannot be attributed to allometric scaling, as all analyses were size-standardized (Procrustes alignment, ratio indices). Moreover, given the sympatric collection from a single locality, ecophenotypic plasticity alone is unlikely to account for such consistent and pronounced shell differentiation [2,24]. The persistent morphological gap is therefore most likely sustained by either reproductive isolation or long-term disruptive selection that maintains discrete phenotypic clusters [63,64,65].
4.2. Molecular Evidence: Mitonuclear Conflict and Limited Genetic Differentiation
In contrast to the clear morphological partition, molecular data exhibit substantial mitonuclear discordance and a more complex phylogenetic pattern. The BEAST2 species tree and mitochondrial gene tree (COI+16S rRNA) consistently recovered C. chinensis and C. cathayensis as monophyletic (Figure 4,5), with divergence time estimation placing their separation in the Miocene (Figure 7). In contrast, the nuclear gene tree (H3+28S rRNA) failed to recover reciprocal monophyly between ZGYTL and ZHYTL, instead showing them intermixed with C. wisseli, C. chinensis and C. cathayensis in paraphyletic or polytomous arrangements (Figure 5). This pronounced mitonuclear discordance—strong mitochondrial differentiation contrasted with unresolved nuclear topology—constitutes the primary molecular incongruence of our study. Such discordance is not uncommon in molluscan systematics, as exemplified by Bivalvia, where mitochondrial markers recover Amarsipobranchia whereas nuclear markers support Heteroconchia [66]; the cone snail Lautoconus ventricosus shows similar patterns with incomplete lineage sorting and introgression [67]; within freshwater gastropods, phylogenomic analyses of East Asian viviparids resolved four mitochondrial clades but only two nuclear clusters [68]. These precedents demonstrate that mitonuclear incongruence is pervasive across Mollusca, underscoring the value of integrating multiple genetic datasets for robust species delimitation.
The haplotype networks provide additional insights. In both COI and 16S rRNA, ZGYTL and ZHYTL shared some haplotypes (Figure 6), indicating shared ancestry or limited ongoing gene flow. Crucially, neither population shared any haplotype with GenBank sequences of C. chinensis or C. cathayensis, suggesting that the Liuzhou populations represent a distinct, previously unsampled genetic lineage within the C. chinensis complex. This finding aligns with the growing awareness that many nominal viviparid species exhibit mitonuclear conflicts or genetic homogeneity despite morphological divergence [2,10]. Such discordance could arise from incomplete lineage sorting, historical gene flow, or a combination of both, but further genomic data are required to disentangle these processes.
4.3. Reconciling the Discordance: Species Validity Despite Genetic Mixing
Given the strong morphological separation but weak and conflicting genetic differentiation, the preponderance of evidence supports recognizing ZGYTL and ZHYTL as two valid morphospecies, while acknowledging that their genetic boundaries are not fully resolved.
Firstly, under a morphological species concept, the shell divergence between the two taxa groups is clear and consistent. This is evidenced by complete non-overlap in multivariate spaces, Mahalanobis distances exceeding 10 (Table 1), and sympatric co-occurrence. Such divergence far exceeds typical intraspecific variation documented in viviparids [2,30,68]. Furthermore, in the absence of evidence for ecophenotypic plasticity—since both populations were collected from the same habitat—the observed morphological gap most likely reflects either reproductive isolation or long-term disruptive selection maintaining discrete phenotypic clusters [24,69].
Secondly, the lack of reciprocal monophyly in nuclear genes and the shared haplotypes in mitochondrial genes do not necessarily invalidate species status. It is well documented in freshwater gastropods that morphologically distinct nominal species can be genetically indistinguishable or show mitonuclear discordance [2,10,23,40,41]. The estimated divergence time of ~11 Ma is sufficiently deep for the accumulation of morphological differences but may still be too recent for complete sorting of nuclear alleles, especially given the typically large effective population sizes of freshwater snails. This scenario is conceptually analogous to the ancient incomplete lineage sorting documented in Mollusca by Song et al. (2023), who demonstrated that phylogenetic discordance at deep nodes (~520 Ma) can persist when rapid diversification outpaces coalescent sorting, reinforcing ILS as a pervasive source of phylogenetic error across timescales [70,71,72,73]. The observed nuclear mixing may reflect incomplete lineage sorting, although limited introgression cannot be excluded. Importantly, the complete absence of haplotype sharing between the Liuzhou populations and GenBank sequences of C. chinensis indicates that ZGYTL and ZHYTL are not misidentified but represent a distinct genetic cluster, further supporting their specific distinctiveness.
Thirdly, the mitonuclear conflict itself does not argue against species validity. In Mollusca, numerous well-documented cases illustrate that such discordance commonly coexists with valid species boundaries. For instance, mitochondrial markers recover the monophyly of Amarsipobranchia (Heterodonta + Pteriomorphia), whereas nuclear markers support the monophyly of Heteroconchia (Heterodonta + Palaeoheterodonta), yet both major clades represent well-established higher-level taxa [66]. In the Octopus vulgaris species complex, genome-wide SNP data revealed significant mitonuclear discordance. Nuclear loci supported O. vulgaris sensu stricto and Type III (South Africa) as distinct species, a relationship not recovered by mtDNA [74]. Despite this discordance, both lineages remain recognized as valid taxonomic units. Similarly, the Mediterranean cone snail Lautoconus ventricosus exhibits pronounced mitonuclear discordance with instances of incomplete lineage sorting and introgression; species delimitation tests nonetheless proposed the existence of at least three valid species [67]. In freshwater gastropods, the New Zealand snail Potamopyrgus antipodarum shows strikingly distinct nuclear versus mitochondrial genomic signatures, yet it is recognized as a cohesive species [75].
Therefore, we conclude that the morphological evidence, combined with the sympatric occurrence and the genetic distinctness from GenBank references, strongly supports treating ZGYTL and ZHYTL as two valid species, while acknowledging that their genetic boundaries are not fully closed.
4.4. Implications for Taxonomy and Conservation
Our results underscore the necessity of integrative taxonomy when morphology and molecules conflict. Relying solely on morphology risks over-splitting by misinterpreting genetic mixing as intraspecific variation, whereas genetics alone risks under-splitting by failing to detect species boundaries masked by incomplete lineage sorting. For the C. chinensis–C. cathayensis complex, we recommend retaining both as valid species, while recognizing that the Liuzhou populations (ZGYTL, ZHYTL) represent distinct evolutionarily significant units (ESUs) that do not fully align with current GenBank reference sequences. This conclusion reinforces the view that species delimitation in viviparids requires integrative evidence combining morphology, multi-locus genetics, and, where feasible, genomic data (Du et al., 2013; Stelbrink et al., 2020; Zhang et al., 2026).
Future work should employ whole-genome sequencing to clarify the extent of nuclear admixture and the relationship of the Liuzhou populations to topotypical material. From a conservation standpoint, both taxa are harvested and play important ecological roles in freshwater ecosystems (Dewi et al., 2017; Ma et al., 2023). We therefore recommend managing ZGYTL and ZHYTL as separate units pending further genomic evidence, to safeguard their genetic diversity and local adaptations.
5. Conclusions
This integrative study reveals a pronounced discordance between morphology and molecules in the two sympatric Chinese river snails, ZGYTL and ZHYTL. Shell shape completely separates the two taxa, whereas molecular data show low level differentiation with mitonuclear conflicts. Despite nuclear mixing and haplotype sharing, the absence of haplotype sharing with GenBank references, combined with clear morphometric separation and sympatric occurrence, supports recognizing them as valid species. The mitonuclear discordance likely reflects incomplete lineage sorting or historical introgression, but genome-wide data are needed to resolve these processes. We conclude that ZGYTL and ZHYTL are valid species, though their genetic boundaries remain partially porous. This study underscores the value of integrative taxonomy for species delimitation in morphologically challenging freshwater gastropods. From a conservation perspective, the two species should be managed as separate units to preserve their genetic diversity and local adaptations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Positions of the 24 landmarks digitized on a representative shell of Cipangopaludina chinensis. Table S1. Taxon names, specimen codes, and GenBank accession numbers for all sequences used in the phylogenetic analyses. Table S2: Shell morphometric measurements (mm) of ZGYTL and ZHYTL: shell height (SH), shell width (SW), spiral height (SpH), body whorl height (BWH), aperture width (AW), operculum height (OH), and operculum width (OW). Table S3: Taxon names, specimen codes, and GenBank accession numbers for sequences used in the haplotype network analyses.
Author Contributions
Y.W.: investigation, methodology, visualization, and writing—original draft. A.L.: investigation, methodology, visualization, and writing—original draft. F.L.: investigation, methodology, visualization. Z.W.: methodology. X.C: methodology. Z.W.: investigation. Q.G.: conceptualization, formal analysis, methodology and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Guangxi Key R&D Program Projects (Guikenong AB2506910042), and the National Key Research and Development Plan of China (No. 2022YFD2400700), and the National Science Foundation of China (No. 31601851).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article, and all genes can be obtained by contacting the author (gqh@hunnu.edu.cn). Newly generated 28S rRNA, 16S rRNA, COI, H3 sequences are available in GenBank (accession number: PZ545870-PZ545924, PZ545927-PZ545980, PZ545982-PZ546036).
Acknowledgments
We thank the High-performance Computing Platform of Yuelushan Laboratory Aquatic Variety Breeding Center for providing computing resources.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sengupta, M.E.; Kristensen, T.K.; Madsen, H.; Jorgensen, A. Molecular phylogenetic investigations of the Viviparidae (Gastropoda: Caenogastropoda) in the lakes of the Rift Valley area of Africa. Mol Phylogenet Evol 2009, 52(3), 797-805. [CrossRef]
- Ye, B.; Hirano, T.; Saito, T.; Dong, Z.Z.; Do, V.T.; Chiba, S. Molecular and morphological evidence for a unified, inclusive Sinotaia quadrata (Caenogastropoda: Viviparidae: Bellamyinae). J Molluscan Stud 2021, 87. [CrossRef]
- Zhang, L.J.; von Rintelen, T. The neglected operculum: a revision of the opercular characters in river snails (Caenogastropoda: Viviparidae). J Molluscan Stud 2021, 87.
- Stelbrink, B.; Richter, R.; Köhler, F.; Riedel, F.; Strong, E.E.; Van Bocxlaer, B.; Albrecht, C.; Hauffe, T.; Page, T.J.; Aldridge, D.C.; et al. Global Diversification Dynamics Since the Jurassic: Low Dispersal and Habitat-Dependent Evolution Explain Hotspots of Diversity and Shell Disparity in River Snails (Viviparidae). Systematic Biology 2020, 69(5), 944-961. [CrossRef]
- Hirano, T.; Saito, T.; Tsunamoto, Y.; Koseki, J.; Prozorova, L.; Tu, D.V.; Matsuoka, K.; Nakai, K.; Suyama, Y.; Chiba, S. Role of ancient lakes in genetic and phenotypic diversification of freshwater snails. Molecular Ecology 2019, 28(23), 5032-5051. [CrossRef]
- Zhang, L.J.; Liu, C.X.; Qin, T.; Oo, T.N.; Ounekham, K.; Lorphengsy, S.; Chen, X.Y. Time-Calibrated Phylogeny of River Snails (Gastropoda: Viviparidae) Reveals a New Viviparid Genus With a "Lazarus Species". Zool Scr 2026, 55(1), 52-61. [CrossRef]
- Hirano, T.; Saito, T.; Ito, S.; Ye, B.; Linscott, T.M.; Do, V.T.; Dong, Z.Z.; Chiba, S. Phylogenomic analyses reveal incongruences between divergence times and fossil records of freshwater snails in East Asia. Mol Phylogenet Evol 2023, 182. [CrossRef]
- Zhang, L.J.; Chen, S.C.; Yang, L.T.; Jin, L.; Köhler, F. Systematic revision of the freshwater snail Margarya Nevill, 1877 (Mollusca: Viviparidae) endemic to the ancient lakes of Yunnan, China, with description of new taxa. Zool J Linn Soc 2015, 174(4), 760-800. [CrossRef]
- Du, L.; Yang, J.; von Rintelen, T.; Chen, X.; Aldridge, D. Molecular phylogenetic evidence that the Chinese viviparid genus Margarya (Gastropoda: Viviparidae) is polyphyletic. Chinese Science Bulletin 2013, 58(18), 2154-2162. [CrossRef]
- Zheng, H.; Zhang, L.J.; Zhang, Y.Y.; Xiang, H.Q.; He, Y.M.; Ouyang, S.; Wu, X.P. Multilocus Phylogeny Reveals New Species From the Ancient Lake Fuxian and Synonymy of Cipangopaludina With Margarya (Gastropoda: Viviparidae). Zoologica Scripta 2026. [CrossRef]
- Wang, J.G.; Zhang, D.; Jakovlic, I.; Wang, W.M. Sequencing of the complete mitochondrial genomes of eight freshwater snail species exposes pervasive paraphyly within the Viviparidae family (Caenogastropoda). Plos One 2017, 12(7). [CrossRef]
- Natural England.; Chinese mystery snail, Cipangopaludina chinensis, phylogenetic analysis and barcoding: Pevensey Levels, Sussex; and Southampton, Hampshire.In: Natural England Commissioned Report. Edition 1 edn; 2024.
- Ma, B.H.; Jin, W.; Fu, H.Y.; Sun, B.; Yang, S.; Ma, X.Y.; Wen, H.B.; Wu, X.P.; Wang, H.H.; Cao, X.J. A High-Quality Chromosome-Level Genome Assembly of a Snail Cipangopaludina cathayensis (Gastropoda: Viviparidae). Genes 2023, 14(7).
- Papes, M.; Havel, J.E.; Vander Zanden, M.J. Using maximum entropy to predict the potential distribution of an invasive freshwater snail. Freshwater Biology 2016, 61(4), 457-471. [CrossRef]
- Dewi, V.K.; Sato, S.; Yasuda, H. Effects of a mud snail Cipangopaludina chinensis laeta (Architaenioglossa: Viviparidae) on the abundance of terrestrial arthropods through rice plant development in a paddy field. Applied Entomology and Zoology 2017, 52(1), 97-106. [CrossRef]
- Van Bocxlaer, B.; Strong, E.E. Anatomy, functional morphology, evolutionary ecology and systematics of the invasive gastropod Cipangopaludina japonica (Viviparidae: Bellamyinae). Contributions to Zoology 2016, 85(2), 235-263.
- Mitteroecker, P.; Schaefer, K. Thirty years of geometric morphometrics: Achievements, challenges, and the ongoing quest for biological meaningfulness. American Journal of Biological Anthropology 2022, 178, 181-210. [CrossRef]
- Adams, D.C.; Rohlf, F.J.; Slice, D.E. A field comes of age: geometric morphometrics in the 21st century. Hystrix-Italian Journal of Mammalogy 2013, 24(1), 7-14. [CrossRef]
- Annear, E.; Van Linden, L.; MacLaren, J.A.; Baeckens, S.; Broeckhoven, C.; Van Damme, R. Intraspecific variation in osteoderm morphology in a cordylid lizard. Zool J Linn Soc 2025, 205(4). [CrossRef]
- Aytekin, S.; Sakaci, Z.; Talay, S.; Alten, B. Effects of High Larval Density on Wing Shape Deformations of Culex pipiens (Culicidae: Diptera) via Geometric Morphometrics. Insects 2025, 16(12). [CrossRef]
- Zhang, W.Y.; Wang, X.Z.; Huang, J.; Wang, X.P.; Wang, B.; Jiang, J.P.; Dong, B.J.; Zhang, M,H, Sexual Differences in Appendages of a Fossorial Narrow-Mouth Frog, Kaloula rugifera (Anura, Microhylidae). Animals 2025, 15(17). [CrossRef]
- Smith, U.E.; Hendricks, J.R. Geometric Morphometric Character Suites as Phylogenetic Data: Extracting Phylogenetic Signal from Gastropod Shells. Systematic Biology 2013, 62(3),366-385. [CrossRef]
- Gu, Q.H.; Husemann, M.; Wu, H.H.; Dong, J.; Zhou, C.J.; Wang, X.F.; Gao, Y.N.; Zhang, M.; Zhu, G.R.; Nie, G.X. Phylogeography of Bellamya (Mollusca: Gastropoda: Viviparidae) snails on different continents: contrasting patterns of diversification in China and East Africa. BMC Evol Biol 2019, 19. [CrossRef]
- Van Bocxlaer, B.; Ortiz-Sepulveda, C.M.; Gurdebeke, P.R.; Vekemans, X. Adaptive divergence in shell morphology in an ongoing gastropod radiation from Lake Malawi. Bmc Evolutionary Biology 2020, 20(1). [CrossRef]
- Morán, G.A.; Martínez, J.J.; Boretto, G.M.; Gordillo, S.; Boidi, F.J. Shell morphometric variation of Ameghinomya antiqua (Mollusca, Bivalvia) during the Late Quaternary reflects environmental changes in North Patagonia, Argentina. Quaternary International 2018, 490, 43-49. [CrossRef]
- Horsakova, V.;Llznarova, E.; Razkin, O.; Nekola, J.C.; Horsak, M. Deciphering "cryptic" nature of European rock-dwelling Pyramidula snails (Gastropoda: Stylommatophora). Contributions to Zoology 2022, 91(4-5), 233-260. [CrossRef]
- Pastorino, G. Sexual dimorphism in shells of the southwestern Atlantic gastropod Olivella plata (Ihering, 1908) (Gastropoda: Olividae). J Molluscan Stud 2007, 73, 283-285. [CrossRef]
- Lai, S.Q.; Shi, L.; Han, Y.D.; Tian, Y.; Hao, Z.L. Adaptation of shell morphology to different tidal zones-insights into phenotypic plasticity of Littorina brevicula. Frontiers in Ecology and Evolution 2025, 12. [CrossRef]
- Van Bocxlaer, B.; Dollion, A.Y.; Ortiz-Sepulveda, C.M.; Calarnou, C.; Habert, R.; Pawindo, G.; Vekemans, X. Adaptive shell-morphological differences and differential fitness in two morphospecies of Lanistes (Gastropoda: Ampullariidae) from the northern region of the Malawi Basin. Evolutionary Journal of the Linnean Society 2024, 3(1). [CrossRef]
- Lu, H-F.; Du, L-N.; Li, Z-Q.; Chen, X-Y.; Yang, J-X. Morphological analysis of the Chinese Cipangopaludina species (Gastropoda; Caenogastropoda: Viviparidae). Dong wu xue yan jiu = Zoological research 2014, 35(6), 510-527. [CrossRef]
- Minton, R.L.; Wang, L.L. Evidence of sexual shape dimorphism in Viviparus (Gastropoda: Viviparidae). Journal of Molluscan Studies 2011, 77, 315-317. [CrossRef]
- Sawangproh, W.; Phaenark, C.; Chunchob, S.; Paejaroen, P. Sexual dimorphism and morphometric analysis of Filopaludina martensi martensi (Gastropoda: Viviparidae). Ruthenica, Russian Malacological Journal 2021, 31, 87-92. [CrossRef]
- Uvayeva, O.; Vakaliuk, T.; Shcherbina, G.; Shimkovich, E. Sexual dimorphism in shell morphology of mollusks of the genus Viviparus – important objects of water resources of Ukraine. E3S Web of Conferences 2021, 280, 10011.
- Chen, Z.; Baeza, J.A.; Chen, C.; Gonzalez, M.T.; González, V.L.; Greve, C.; Kocot, K.M.; Arbizu, P.M.; Moles, J.; Schell, T.; et al. A genome-based phylogeny for Mollusca is concordant with fossils and morphology. Science 2025, 387(6737), 1001-1007. [CrossRef]
- Wanninger, A.; Wollesen, T. The evolution of molluscs. Biol Rev 2019, 94(1), 102-115. [CrossRef]
- Emerson, B.C. Delimiting Species-Prospects and Challenges for DNA Barcoding. Molecular Ecology 2025, 34(5), 8. [CrossRef]
- Gaughran, S.J.; Gray, R.; Ochoa, A.; Jones, M.; Fusco, N.;Miller, J.M.; Poulakakis, N.; de Queiroz, K.; Caccone, A.; Jensen, E.L. Whole-genome sequencing confirms multiple species of Galapagos giant tortoises. Evolution 2024, 79(2), 296-308. [CrossRef]
- Dan, X.Q.; Cheng, G.; Wen, Y.H.; Luo, F.G.; Huang, J.; Wang, W.M. Comparative analyses of morphological characters for three species in genus Bellamya. Freshwater Fisheries 2020, 50(03), 50-55.
- Hobbs, C.S.; Vega, R.; Rahman, F.; Horsburgh, G.J.; Dawson, D.A.; Harvey, C.D. Population genetics and geometric morphometrics of the freshwater snail Segmentina nitida reveal cryptic sympatric species of conservation value in Europe. Conservation Genetics 2021, 22(6), 855-871. [CrossRef]
- Köhler, F.; Deein, G. Hybridisation as potential source of incongruence in the morphological and mitochondrial diversity of a Thai freshwater gastropod (Pachychilidae, Brotia H. Adams, 1866). Zoosystematics and Evolution 2010, 86(2), 301-314. [CrossRef]
- Van Bocxlaer, B.; Clewing, C.; Duputié, A.; Roux, C.; Albrecht, C. Population collapse in viviparid gastropods of the Lake Victoria ecoregion started before the Last Glacial Maximum. Molecular Ecology 2021, 30(2), 364-378. [CrossRef]
- Brönmark, C.; Lakowitz, T.; Hollander, J. Predator-Induced Morphological Plasticity Across Local Populations of a Freshwater Snail. Plos One 2011, 6(7). [CrossRef]
- Schreiber, K.; Hauffe, T.; Albrecht, C.; Wilke, T. The role of barriers and gradients in differentiation processes of pyrgulinid microgastropods of Lake Ohrid. Hydrobiologia 2012, 682(1), 61-73. [CrossRef]
- Klingenberg, C.P. MorphoJ: an integrated software package for geometric morphometrics. Molecular Ecology Resources 2011, 11(2), 353-357.
- Rohlf, F. The Tps series of software. Hystrix 2015, 26, 1-4. [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular marine biology and biotechnology 1994, 3(5), 294-299.
- Palumbi, S.R. The Simple Fool's Guide to PCR. Version 2. 1991.
- Colgan, D.; McLauchlan, A.; Wilson, G.; Livingston, S.; Edgecombe, G.; Macaranas, J.; Cassis, G.; Gray, M. Histone H3 and U2 snRNA DNA sequences and arthropod molecular evolution. Australian Journal of Zoology 1999, 46, 419-437. [CrossRef]
- Xiang, C.Y.; Gao, F.L.; Jakovlic, I.; Lei, H.P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. Imeta 2023, 2(1). [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. Plos Computational Biology 2019, 15(4).
- Drummond, A.J.; Ho, S.Y.W.; Phillips, MJ..; Rambaut A. Relaxed phylogenetics and dating with confidence. Plos Biology 2006, 4(5), 699-710. [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Systematic Biology 2018, 67(5), 901-904.
- Bouckaert, R.R. DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 2010, 26(10), 1372-1373. [CrossRef]
- Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30(9), 1312-1313.
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Systematic Biology 2012, 61(3), 539-542.
- Leigh, J.W.; Bryant, D. POPART: full-feature software for haplotype network construction. Methods in Ecology and Evolution 2015, 6(9), 1110-1116. [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Molecular biology and evolution 1999, 16(1), 37-48. [CrossRef]
- Liu, Y.Y.; Zhang, W.Z.; Wang, Y.X.; Wang, S.Y. Economic Fauna of China (Freshwater Mollusks). Beijing: Science Press 1979.
- Western, L.; Zanatta, D.T. Identifying environmental drivers of shell shape variation in the freshwater gastropod Campeloma decisum (Say, 1817). American Malacological Bulletin 2025, 41(1), 14-14. [CrossRef]
- Goswami, A.; Watanabe, A.; Felice, R.N.; Bardua, C.; Fabre, A.C.; Polly, P.D. High-Density Morphometric Analysis of Shape and Integration: The Good, the Bad, and the Not-Really-a-Problem. Integrative and Comparative Biology 2019, 59(3), 669-683.
- Bardua, C.; Felice, R.N.; Watanabe, A.; Fabre, A.C.; Goswami, A. A Practical Guide to Sliding and Surface Semilandmarks in Morphometric Analyses. Integrative Organismal Biology 2019, 1(1). [CrossRef]
- Cardini, A. Integration and Modularity in Procrustes Shape Data: Is There a Risk of Spurious Results? Evolutionary Biology 2019, 46(1), 90-105. [CrossRef]
- Johannesson, K.; Rolan-Alvarez, E.; Ekendahl, A. Incipient reproductive isolation between two sympatric morphs of the intertidal snail Littorina Saxatilis. Evolution; international journal of organic evolution 1995, 49(6), 1180-1190. [CrossRef]
- Ito, S.; Konuma, J. Disruptive selection of shell colour in land snails: a mark-recapture study of Euhadra peliomphala simodae. Biological Journal of the Linnean Society 2020, 129(2), 323-333.
- Van Bocxlaer, B.; Hunt, G. Morphological stasis in an ongoing gastropod radiation from Lake Malawi. Proceedings of the National Academy of Sciences of the United States of America 2013, 110(34), 13892-13897.
- Formaggioni, A.; Plazzi, F.; Passamonti, M. Mito-nuclear coevolution and phylogenetic artifacts: the case of bivalve mollusks. Scientific Reports 2022, 12(1).
- Abalde, S.; Crocetta, F.; Tenorio, M.J.; D'Aniello, S.; Fassio, G.; Rodríguez-Flores, P.C.; Uribe, J.E.; Afonso, C.M.L.; Oliverio, M.; Zardoya, R. Hidden species diversity and mito-nuclear discordance within the Mediterranean cone snail, Lautoconus ventricosus. Molecular Phylogenetics and Evolution 2023, 186, 107838. [CrossRef]
- Hirano, T.; Saito, T.; Tsunamoto, Y.; Koseki, J.; Ye, B.; Do, V.T.; Miura, O.; Suyama, Y.; Chiba, S. Enigmatic incongruence between mtDNA and nDNA revealed by multi-locus phylogenomic analyses in freshwater snails. Sci Rep 2019, 9(1), 6223. [CrossRef]
- Pickles, G. Mate choice in divergent morphs of the gastropod mollusc Littorina saxatilis (Olivi): speciation in action? Animal behaviour 1999, 58(1), 181-184.
- Neiman, M.; Sharbrough, J. A tale of two genomes: What drives mitonuclear discordance in asexual lineages of a freshwater snail? Bioessays 2023, 45(6). [CrossRef]
- Song, H.; Wang, Y.A.; Shao, H.J.; Li, Z.Q.; Hu, P.L.; Yap-Chiongco, M.K.; Shi, P.; Zhang, T.; Li, C.; Wang, Y.G.; et al. Scaphopoda is the sister taxon to Bivalvia: Evidence of ancient incomplete lineage sorting. Proceedings of the National Academy of Sciences of the United States of America 2023, 120(40). [CrossRef]
- Naciri, Y.; Linder, H.P. Species delimitation and relationships: The dance of the seven veils. Taxon 2015, 64(1), 3-16. [CrossRef]
- Hirano, T.; Saito, T.; Chiba, S. Phylogeny of freshwater viviparid snails in Japan. Journal of Molluscan Studies 2015, 81, 435-441. [CrossRef]
- Amor, M.D.; Doyle, S.R.; Norman, M.D.; Roura, A.; Hall, N.E.; Robinson, A.J.; Leite, T.S.; Strugnell, J.M. Genome-wide sequencing uncovers cryptic diversity and mito-nuclear discordance in the Octopus vulgaris species complex. bioRxiv 2019.
- Paczesniak, D.; Jokela, J.; Larkin, K.; Neiman, M. Discordance between nuclear and mitochondrial genomes in sexual and asexual lineages of the freshwater snail Potamopyrgus antipodarum. Mol Ecol 2013, 22(18), 4695-4710. [CrossRef]
Figure 1.
(a) Principal component analysis (PCA), and (b) Principal coordinates analysis (PCoA) biplot of proportional morphological measurement traits (Table S2) illustrating morphological divergence between ZGYTL and ZHYTL, and (c) the statistical significance of intergroup differences was evaluated by pairwise Wilcoxon rank sum tests (wilcox.test). Specimens are grouped by taxon and sex: ZGX denotes male individuals of the ZGYTL, ZGC denotes female individuals of ZGYTL, ZHX denotes male individuals of the ZHYTL taxon, and ZHC denotes female individuals of ZHYTL; Arrows in the plot correspond to the loading vectors of the input shape ratio indicators: the orientation of each arrow reflects the direction of correlation between the respective morphological trait and the two principal component axes, indicating how the trait value shifts along the PC gradients; the length of an arrow is positively proportional to the trait’s contribution to the total morphological variation captured by PC1 and PC2, with longer arrows indicating a stronger driving effect on the observed morphological differentiation among groups.
Figure 1.
(a) Principal component analysis (PCA), and (b) Principal coordinates analysis (PCoA) biplot of proportional morphological measurement traits (Table S2) illustrating morphological divergence between ZGYTL and ZHYTL, and (c) the statistical significance of intergroup differences was evaluated by pairwise Wilcoxon rank sum tests (wilcox.test). Specimens are grouped by taxon and sex: ZGX denotes male individuals of the ZGYTL, ZGC denotes female individuals of ZGYTL, ZHX denotes male individuals of the ZHYTL taxon, and ZHC denotes female individuals of ZHYTL; Arrows in the plot correspond to the loading vectors of the input shape ratio indicators: the orientation of each arrow reflects the direction of correlation between the respective morphological trait and the two principal component axes, indicating how the trait value shifts along the PC gradients; the length of an arrow is positively proportional to the trait’s contribution to the total morphological variation captured by PC1 and PC2, with longer arrows indicating a stronger driving effect on the observed morphological differentiation among groups.

Figure 2.
Results of principal component analysis (PCA) for two groups ZHYTL and ZGYTL. (a, b) PCA scatter plots constructed from 24 landmarks and 200 dense semilandmarks, respectively; (c, d) Morphological deformation grids of PC1 and PC2 based on 24 landmarks; (e, f) Morphological deformation grids of PC1 and PC2 based on 200 dense semilandmarks.
Figure 2.
Results of principal component analysis (PCA) for two groups ZHYTL and ZGYTL. (a, b) PCA scatter plots constructed from 24 landmarks and 200 dense semilandmarks, respectively; (c, d) Morphological deformation grids of PC1 and PC2 based on 24 landmarks; (e, f) Morphological deformation grids of PC1 and PC2 based on 200 dense semilandmarks.

Figure 3.
Results of canonical variate analysis (CVA) for shell morphological variation. (a, b) Frequency histograms of CV1 scores comparing populations ZHYTL and ZGYTL, constructed using 24 landmarks and 200 dense semilandmarks, respectively; (c) Two-dimensional CVA scatter plot along CV1 and CV2 axes based on 200 dense semilandmarks, illustrating morphological differentiation among four subgroups delineated by sampling population and sex (ZGC, ZGX, ZHC, ZHX).
Figure 3.
Results of canonical variate analysis (CVA) for shell morphological variation. (a, b) Frequency histograms of CV1 scores comparing populations ZHYTL and ZGYTL, constructed using 24 landmarks and 200 dense semilandmarks, respectively; (c) Two-dimensional CVA scatter plot along CV1 and CV2 axes based on 200 dense semilandmarks, illustrating morphological differentiation among four subgroups delineated by sampling population and sex (ZGC, ZGX, ZHC, ZHX).

Figure 4.
BEAST2-reconstructed species trees to resolve interspecific phylogenetic relationships within the genus Cipangopaludina. The phylogenetic inference was built on the concatenated dataset consisting of mitochondrial COI, 16S rRNA and nuclear H3, 28S rRNA gene sequences, and other gastropods, bivalves and cephalopods were included as outgroups. (a) Full collection of posterior trees visualized in DensiTree; (b) Final summarized species tree generated by TreeAnnotator, with posterior probability values labeled on each evolutionary clade.
Figure 4.
BEAST2-reconstructed species trees to resolve interspecific phylogenetic relationships within the genus Cipangopaludina. The phylogenetic inference was built on the concatenated dataset consisting of mitochondrial COI, 16S rRNA and nuclear H3, 28S rRNA gene sequences, and other gastropods, bivalves and cephalopods were included as outgroups. (a) Full collection of posterior trees visualized in DensiTree; (b) Final summarized species tree generated by TreeAnnotator, with posterior probability values labeled on each evolutionary clade.

Figure 5.
Gene trees reconstructed from mitochondrial (COI + 16S rRNA) and nuclear (28S rRNA + H3) genes. The datasets include all samples of ZGC/ZGX and ZHC/ZHX, other taxa of the genus Cipangopaludina, additional gastropods and bivalves, and the cephalopod sequences were retrieved from GenBank (Table S1). Obvious mitonuclear conflicts were observed in both outgroup placement and interspecific relationships within Cipangopaludina between the mitochondrial and nuclear gene trees.
Figure 5.
Gene trees reconstructed from mitochondrial (COI + 16S rRNA) and nuclear (28S rRNA + H3) genes. The datasets include all samples of ZGC/ZGX and ZHC/ZHX, other taxa of the genus Cipangopaludina, additional gastropods and bivalves, and the cephalopod sequences were retrieved from GenBank (Table S1). Obvious mitonuclear conflicts were observed in both outgroup placement and interspecific relationships within Cipangopaludina between the mitochondrial and nuclear gene trees.

Figure 6.
Haplotype networks constructed using 16S rRNA (a) and COI (b) gene sequences of Cipangopaludina retrieved from GenBank (Table S3), together with all samples of ZGYTL and ZHYTL. Conspecific species of the genus Cipangopaludina are marked with unified colors. The circle size corresponds to haplotype frequency; each connecting line indicates a single nucleotide substitution, and each small tick represents a mutational position.
Figure 6.
Haplotype networks constructed using 16S rRNA (a) and COI (b) gene sequences of Cipangopaludina retrieved from GenBank (Table S3), together with all samples of ZGYTL and ZHYTL. Conspecific species of the genus Cipangopaludina are marked with unified colors. The circle size corresponds to haplotype frequency; each connecting line indicates a single nucleotide substitution, and each small tick represents a mutational position.

Figure 7.
Divergence time estimation of the genus Cipangopaludina based on the mitochondrial COI and 16S rRNA datasets (Table S1) derived from the species tree. Two calibration points are applied, which are marked by red dots: (1) the divergence between L. gigantea and P. canaliculata (mean = 488.7 Ma, 95% CI: 468.8–526.6 Ma); (2) the split between P. canaliculata and Cipangopaludina sp. (mean = 129.7 Ma, 95% CI: 79.0–165.1 Ma). Node ages and their corresponding 95% highest posterior density (HPD) bars for age ranges are presented on the tree.
Figure 7.
Divergence time estimation of the genus Cipangopaludina based on the mitochondrial COI and 16S rRNA datasets (Table S1) derived from the species tree. Two calibration points are applied, which are marked by red dots: (1) the divergence between L. gigantea and P. canaliculata (mean = 488.7 Ma, 95% CI: 468.8–526.6 Ma); (2) the split between P. canaliculata and Cipangopaludina sp. (mean = 129.7 Ma, 95% CI: 79.0–165.1 Ma). Node ages and their corresponding 95% highest posterior density (HPD) bars for age ranges are presented on the tree.


Table 1.
Interspecific morphological distances and Goodall’s F-tests between ZGYTL and ZHYTL. Lower-triangular values (ZGYTL vs. ZHYTL) are based on the 24-landmark geometric morphometric dataset, and upper-triangular values (ZHYTL vs. ZGYTL) are based on the 200-point high-density semilandmark dataset. Mahalanobis and Procrustes distances are given together with Goodall’s F-statistics; all permutation tests (10,000 iterations) were significant at P < 0.0001.
Table 1.
Interspecific morphological distances and Goodall’s F-tests between ZGYTL and ZHYTL. Lower-triangular values (ZGYTL vs. ZHYTL) are based on the 24-landmark geometric morphometric dataset, and upper-triangular values (ZHYTL vs. ZGYTL) are based on the 200-point high-density semilandmark dataset. Mahalanobis and Procrustes distances are given together with Goodall’s F-statistics; all permutation tests (10,000 iterations) were significant at P < 0.0001.
| Mahalanobis distances | Procrustes distances | Goodall’s F tests | ||||||
| ZGYTL | ZHYTL | ZGYTL | ZHYTL | ZGYTL | ZHYTL | |||
| ZGYTL | 12.3682 | 0.0524 | 106.1928 | |||||
| ZHYTL | 15.4901 | 0.0573 | 37.0984 | |||||
Table 2.
Pairwise Mahalanobis and Procrustes distances among the four species-sex groups (ZGC, ZGX, ZHC, ZHX). Lower-triangular values are derived from the 24-landmark geometric morphometric dataset, and upper-triangular values from the 200-point high-density semilandmark dataset. All permutation tests (10,000 iterations) were significant at P < 0.0001, except for comparisons involving same-species sexes (see text for details).
Table 2.
Pairwise Mahalanobis and Procrustes distances among the four species-sex groups (ZGC, ZGX, ZHC, ZHX). Lower-triangular values are derived from the 24-landmark geometric morphometric dataset, and upper-triangular values from the 200-point high-density semilandmark dataset. All permutation tests (10,000 iterations) were significant at P < 0.0001, except for comparisons involving same-species sexes (see text for details).
| Mahalanobis distances | Procrustes distances | ||||||||
| ZGC | ZGX | ZHC | ZHX | ZGC | ZGX | ZHC | ZHX | ||
| ZGC | 3.8391 | 12.0114 | 12.3953 | 0.0119 | 0.0514 | 0.0451 | |||
| ZGX | 25.2567 | 12.4203 | 12.7506 | 0.0139 | 0.0605 | 0.0537 | |||
| ZHC | 20.9306 | 42.0034 | 4.0049 | 0.0650 | 0.0666 | 0.0087 | |||
| ZHX | 48.0067 | 71.8304 | 35.7758 | 0.0512 | 0.0529 | 0.0192 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



