Submitted:
23 June 2026
Posted:
24 June 2026
You are already at the latest version
Abstract
Raynaud’s Phenomenon (RP) serves as the clinical sentinel for systemic autoimmunity, frequently predating connective tissue disease (CTD) diagnosis by years. While RP is a largely benign functional vasospasm in the general population, its prevalence in Systemic Sclerosis (SSc) approaches 99%. A fundamental mystery in rheumatology is the divergent fate of the microvasculature across CTDs. While RP can occur in other autoimmune diseases, it is usually not associated with the relentless transition to obliterative vasculopathy. In SSc, the vessel lumen is physically occluded by intimal proliferation and perivascular fibrosis, yet the mechanism triggering this ischemic switch remains poorly understood. Beyond the classic tri-phasic color change, SSc patients frequently report other early distressing sensory symptoms, specifically intense pruritus, neuropathic burning, and finger edema. Historically dismissed as secondary to ischemia, we propose that these sensations are primary markers of the underlying pathological driver. We challenge the traditional B-cell/T-cell-centric view of SSc by introducing the concept of autoallergy. We hypothesize that these sensory symptoms represent chronic, IgE-mediated activation of resident mast cells. This mast cell-driven circuit, triggered by SSc-specific autoantigens, acts as a persistent secretory pump for pro-fibrotic cytokines, fundamentally distinguishing SSc vasculopathy from the purely inflammatory vascular patterns seen in other CTDs. Targeting the IgE-mast cell axis early in the course of the disease may offer a novel therapeutic window to halt the transition from functional vasospasm to permanent structural obliteration.
Keywords:
autoallergy
; mast cells
; IgE autoantibodies
; systemic sclerosis
; vasculopathy
1. Introduction
Systemic Sclerosis (SSc) is a chronic autoimmune disease characterized by a devastating triad of microvascular damage, immune dysregulation with autoantibody development, and progressive interstitial and perivascular fibrosis. SSc is traditionally categorized into limited cutaneous (lcSSc) and diffuse cutaneous (dcSSc) subsets, distinct in their extent of skin involvement and primary IgG autoantibody profiles (anti-centromere vs. anti-topoisomerase and anti-RNA polymerase III). However, the underlying pathogenic trigger for both variants remains unknown, severely limiting effective therapeutic intervention. The traditional model of SSc pathogenesis focuses heavily on Type II (antibody-mediated) and Type IV (T-cell-mediated) autoimmunity. In this paradigm, B-cells produce IgG autoantibodies targeting nuclear antigens, and T-cells release a steady cascade of cytokines that gradually establish a chronic fibrotic tissue state. While this classical framework adequately accounts for long-term tissue scarring and end-organ damage years into the disease, it fundamentally fails to explain the early prodrome, the explosive first few months where the patient is profoundly symptomatic with intense pruritus, neuropathic burning, and non-pitting edema but does not yet exhibit fixed structural fibrosis.
The IgG- and T-cell-mediated mechanisms simply lack the rapid kinetic response required to coordinate these sudden, early vascular and sensory symptoms. Here, I synthesize these divergent early diagnostic features and introduce a new hypothesis regarding the etiology of SSc: that tissue-resident mast cells, driving a localized autoallergy (Type I hypersensitivity), serve as the primary initiating factor in SSc pathogenesis.
2. The Vascular Gatekeeper
Raynaud’s phenomenon (RP) is a relatively common, transient vasospastic condition in the general population, driven by sympathetic overactivity [1,2,3,4,5]. Its clinical presentation in SSc is fundamentally distinct. In the general population, RP represents a mostly benign functional spasm showing seasonal variation; however, the SSc-associated variant serves as an aggressive clinical sentinel that frequently predates an irreversible transition to obliterative, structural vasculopathy [6,7].
2.1. Raynaud’s, the Clinical Paradox in SSc
Although RP typically serves as the clinical sentinel for SSc [8,9,10], it presents a distinct clinical paradox: unlike episodic primary vasospasms, SSc-RP progresses with an aggressive velocity [11,12]. While many patients experience an explosive onset of digital ulcers and cyanotic episodes [10,13], a subset within the GENISOS/CONQUER cohorts exhibits advanced microvascular damage without any formal history of Raynaud's , or reports continuous digital ischemia independent of discrete vasospastic events [14], which has also been reported elsewhere [15]. This underscores that SSc vasculopathy is fundamentally a persistent, structural disease of the microvasculature rather than a transient autonomic failure [16,17]. Nailfold capillaroscopy cleanly illustrates this destructive trajectory, tracking the "Scleroderma pattern" from early giant capillaries to the definitive capillary dropout that marks permanent structural failure [18,19].
Unlike the Type III complement-mediated vasculitis of SLE [20,21,22] and Sjögren's [23] or the membrane attack complex-driven microangiopathy of dermatomyositis [24,25], SSc exhibits a unique structural obliterative signature [26,27,28]. Mixed Connective Tissue Disease (MCTD) remains the primary outlier, mirroring this obliterative vasculopathy in ~40% of patients [29], while an anticentromere antibody (ACA)-positive subset of Sjögren’s exhibits "Scleroderma-pattern" capillary changes in up to 80% of cases without developing definitive skin thickening [30,31,32,33,34]. This distinct divergence across the rheumatological spectrum (Table 1) strongly implies that the SSc/MCTD vascular crisis is governed by a unique immunological mechanism, one that uniquely couples functional spasm with irreversible perivascular remodeling.
Comparative matrix delineating the distinct microvascular signatures, clinical sentinel features, and underlying immunological classifications that separate Systemic Sclerosis and Mixed Connective Tissue Disease from other systemic autoimmune conditions such as Systemic Lupus Erythematosus, Dermatomyositis, and Sjögren’s Syndrome. The parameters contrast the bland, obliterative perivascular remodeling unique to the SSc/MCTD spectrum against the standard complement-mediated or necrotizing vasculitides seen elsewhere across the rheumatological spectrum.
3. Other Early Symptoms of SSc
3.1. Itching (Pruritus)
Intense pruritus is a common, highly debilitating symptom of early SSc [43], affecting 43% to 62% of patients [43,44,45] and severely impacting the quality of life [44,46,47,48]. Early SSc pruritus is broadly associated with winter xerosis [49], and its physical presentation varies significantly by autoimmune subtype: in ACA-positive patients, itching primarily persists over long periods in non-sclerotic regions, whereas in ACA-negative individuals, it emerges concurrently within newly developing sclerotic skin [50].
Importantly, this early sensory prodrome is mechanistically distinct from the pruritus seen across the broader autoimmune spectrum. Unlike the surface itch of dermatomyositis, driven by type I interferons and visible epidermal rashes [51,52,53], the immune complex-mediated vasculitic itch of SLE [52], or the xerosis and direct small-fiber neuropathy characteristic of Sjögren’s syndrome [52], the early, intense neuroimmune itch in SSc points to a primary tissue-resident activation sequence that closely resembles a classic Type I hypersensitivity event.
3.2. Puffy Hands
Diffuse, non-pitting edema of the fingers and dorsum of the hands [54,55,56] is often associated with morning stiffness and the loss of normal knuckle creases, and is a pivotal early feature of SSc that precedes skin hardening and arises directly from acute vascular leakage into the interstitium [57,58]. While other rheumatological conditions present with hand swelling, their temporal progression and mechanisms are distinct. In rheumatoid arthritis, the symmetrical swelling is primarily articular and pitting in nature [59], whereas dermatomyositis-associated hand swelling is rare, and typically limited to anti-synthetase subsets, and is accompanied by hyperkeratotic "Mechanic's hands" [60,61,62]. Conversely, this early puffy phase is an indistinguishable clinical hallmark shared with MCTD [63,64], highlighting a shared, early microvascular insult that strongly mirrors the rapid increase in vascular permeability seen in acute Type I hypersensitivity events.
4. Hypothesis: the Autoallergic Paradigm
The clinical inadequacy of traditional Type II and Type IV hypersensitivity models is most clear in the rapid temporal kinetics of the early SSc prodrome. Classical IgG-mediated autoimmunity and T-cell-driven cytokine cascades lack the rapid response mechanisms required to induce the sudden, massive fluid leakage and vasospastic episodes seen at disease onset. This velocity underscores a fundamental "speed limit" for traditional IgG models, which simply cannot operate fast enough to account for the acute ischemia or the early prodromal symptoms. Conversely, the autoallergic paradigm identifies this non-pitting edema, acute vasospasm, and intense pruritus as definitive hallmarks of a localized Type I hypersensitivity reaction, driven by tissue-resident mast cell activation long before a formal T-cell-coordinated fibrotic response takes hold (Table 2).
A comparative analysis contrasting the rapid temporal kinetics and fluid-phase manifestations of the early SSc prodrome (Type I hypersensitivity autoallergy) against the mechanical limitations of traditional antibody-mediated (Type II) or T-cell-driven (Type IV) autoimmune models. The parameters demonstrate the fundamental biology that distinguishes acute mast cell-driven vascular leakage and neuro-immune flaring from late-stage, progressive tissue fibrosis.
4.1. The Cellular Orchestrator: Mast Cell Dynamics
Within the proposed Type I hypersensitivity autoallergic paradigm, perivascular mast cells function as the central orchestrators of the transition from functional vasospasm to permanent structural obliteration in SSc, serving as key effectors that link innate and adaptive immunity [65,66,67,68,69]. Early clinical data strongly support this model, showing significantly elevated digital and forearm mast cell densities in early-stage SSc that correlate directly with higher modified Rodnan skin thickness scores (mRSS) and Scl-70 positivity [65,69]. Histological evidence confirms that extensive degranulation involving more than half of digital mast cells precedes clinically apparent dermal fibrosis and persists despite tissue hypoxia [65]. Conversely, in late-stage disease, mast cell numbers normalize or transition to histologically undetectable ghost cells [70].
Upon antigen-driven degranulation, these perivascular sentinels release a highly coordinated cocktail of pathogenic signaling molecules that are characteristically elevated in the early SSc prodrome (Figure 1). This secretome includes elevated plasma histamine [71] and elevated tryptase and chymase [66,72] alongside actively synthesized lipid mediators like prostaglandin D2 and leukotrienes [73,74,75,76,77,78]. Simultaneously, mast cells act as a persistent secretory pump for elevated neurovascular growth factors such as vascular endothelial growth factor, nerve growth factor, and platelet-derived growth factor [79,80] and potent pro-fibrotic cytokines, including IL-4, IL-6, IL-13, and TGF-β [81,82,83,84,85,86,87,88], thereby establishing the chronic localized drive required to transform functional spasms into irreversible vascular remodeling.
Within this framework, mast cell degranulation acts as an acute neuro-immune flare. Histamine and leukotrienes rapidly increase vascular permeability to cause non-pitting edema [90,91], while local generation of angiotensin II by mast cell chymase induces the acute vasospasms characteristic of rapid-onset Raynaud’s phenomenon [66,92,93]. Simultaneously, the secretion of tryptase, IL-31, and lysophosphatidic acid directly targets sensory nerves [94,95,96,97], producing the intense pruritus that heralds permanent structural remodeling within a complex neuroimmune landscape further augmented by neuropeptides and endothelin-1 [98,99].
Crucially, these mast cell mediators function as pivotal drivers of tissue fibrosis [79,100,101,102]. Histamine and tryptase directly stimulate fibroblast proliferation and increase type I collagen synthesis [15,103,104,105]. As confirmed by co-culture studies, activated human mast cells directly induce neighboring fibroblasts to differentiate into collagen-producing myofibroblasts, establishing these resident cells as the critical effectors orchestrating the pathological progression from acute inflammatory signaling to permanent, irreversible structural remodeling [105,106].
4.2. Molecular Drivers of Vasculopathy
Unlike the immune-complex-mediated "hot" vasculitis characteristic of SLE, where complement consumption and neutrophil infiltration drive acute vessel wall destruction, the microvascular failure in SSc is a bland, obliterative process. This conspicuous absence of traditional inflammatory markers strongly unmasks an entirely different immunological mechanism, confirming that the SSc ischemic trajectory operates independently of the classic pathways found in SLE.
4.2.1. The IgE Allergic Flare: The Mechanistic Driver of the Ischemia
Although IgE autoantibodies were first identified in SSc sera in 1989 [107], the more recent work by Kramer and colleagues confirmed that IgE targets against centromeric proteins and topoisomerase-1 are highly specific to SSc and CREST compared to other connective tissue diseases [108]. These IgE autoantibodies strongly correlate with their corresponding IgG isotypes, yet they circulate at significantly lower levels, requiring a 1:2 serum dilution for IgE detection compared to a standard 1:1,000 dilution for IgG (Table 3) [108]. This striking disparity in baseline concentration may explain why this critical autoallergic driver has been historically underreported in systemic autoimmunity.
Comparative matrix detailing the clinical specificity and hallmark SSc autoantibody targets (anti-CENP, anti-Topo-I, and anti-U1-RNP) across different connective tissue diseases. The data contrast with the high diagnostic specificity of the IgE autoallergic isotype relative to traditional IgG profiles. *LcSSc is typically associated with the CREST variant and is more likely to have CENP A/B autoantibodies. This may account for the lower prevalence of the IgE isotype reported in this group [108].
The clinical paradox of SSc, where systemic IgE autoantibodies are prevalent despite normal peripheral eosinophil counts [108], unmasks the strictly localized nature of the autoallergic drive (Table 3). This lack of systemic eosinophil recruitment during acute tissue injury confirms that the pathological burden is borne primarily by tissue-resident effectors. Specifically, perivascular mast cells act as the central sentinels, releasing pro-fibrotic and vasospastic mediators upon IgE priming and establishing these cells, rather than eosinophils, as the primary executors of microvascular ischemia. Given that Topo-1 and CENP-A/B are confirmed in the IgE class, we further hypothesize that RNA Polymerase III will follow an identical autoallergic trajectory in dcSSc patients.
Mechanistically, this autoallergic paradigm identifies the IgE-mediated flare as the acute initiating phase of SSc. IgE autoantibodies target nuclear antigens to prime perivascular mast cells via high-affinity FcεRI receptors (Figure 2), triggering the localized degranulation that drives the early prodromal triad of increased vascular permeability, non-pitting edema, and intense neuroimmune pruritus. Crucially, the local generation of angiotensin II by mast cell chymase induces acute vasospasms [93] that clamps the vessel lumen within seconds, permanently flipping a functional ischemic process toward structural remodeling and obliterative vasculopathy.
The antigen-specific nature of the autoallergic drive is clearly demonstrated by the clinical divergence in antibody targets. Patients with MCTD lack SSc-specific IgE autoantibodies against CENP or Topo-I [108], reflecting a distinct antigen mismatch rather than the absence of an autoallergic circuit. MCTD is instead characterized by IgE-isotype autoantibodies targeting U1-RNP, which act as a primary driver of both basophil and mast cell activation [109]. This suggests that while acute ischemia is a universal mechanism across the SSc/MCTD spectrum, the final clinical phenotype is determined by the specific self-antigen that triggers the initial IgE-mediated flare.
4.2.2. IgG: The Chronic Inflammatory Driver
While IgE autoantibodies trigger the initial acute flare, IgG anti-endothelial cell antibodies (AECA) serve as the chronic driver of vascular remodeling. This pro-obliterative stimulus inflicts persistent, day-to-day endothelial damage, serving as the mechanistic bridge that converts transient vasospasms into permanent structural failure. Universally detected in 30% to 80% of SSc patients, depending on the assay [111,112,113,114,115,116,117,118,119], IgG AECAs are strongly biased toward the dcSSc phenotype [112,120,121], where obliteration is most aggressive, compared to a lower prevalence of roughly 30% in lcSSc [122].
Mechanistically, these chronic IgG autoantibodies induce endothelial cell apoptosis and a procoagulant state, stripping the vessel wall of vital vasodilators such as nitric oxide [123,124,125]. Simultaneously, they destroy bone marrow-derived endothelial cell progenitors, stalling the critical cellular recruitment required for vascular repair and neoangiogenesis [123,126]. This sustained dual insult establishes a relentless pro-fibrotic microenvironment that thickens the walls of small arteries and compromises their lumens [127]. Clinically, IgG AECA serves as a high-value biomarker for severe vascular failure, strongly correlating with a 71%–78% risk of secondary pulmonary arterial hypertension [114,117,118] and a twofold to threefold increase in digital infarcts and gangrene [114,128].
4.2.3. Overlap Syndromes (MCTD, SLE, and Sjogren’s): The Functional-to-Structural Bridge
The clinical overlap of MCTD and ACA-positive Sjögren’s syndrome highlights a shared pathogenic basis; in MCTD, AECA prevalence reaches up to 80%, closely corresponding to an SSc-pattern on nailfold capillaroscopy [113,129]. Crucially, the acquisition of IgG AECA in these hybrid cohorts serves as the primary discriminant separating patients who remain in a functional vasospastic state from those who rapidly progress to obliterative digital ischemia or pulmonary arterial hypertension (Table 4).
Comparative summary illustrates how antibody isotype profiles (IgG vs. IgE) dictate the transition from functional vasospasm to structural microvascular remodeling. The matrix highlights how the absence of a Th2-driven IgE isotype switch leaves resident mast cells unprimed in pure SLE and defines the capillaroscopic and biomarker criteria that statistically trigger clinical reclassification into MCTD or SSc overlap syndrome.
Conversely, although anti-U1-RNP antibodies strongly correlate with RP in pure SLE, the manifestation remains strictly episodic and reversible functional vasospasm. Vascular damage in SLE is governed by Type III hypersensitivity and complement consumption [130], an extrinsic inflammatory attack that drives leukocytoclastic or necrotizing lesions without triggering progressive perivascular fibrosis. Because anti-U1-RNP in SLE is predominantly IgG, it lacks the class switch to IgE required to prime tissue-resident mast cells via high-affinity receptors. Consequently, pure SLE lacks the acute secretory flare (histamine, chymase, and tryptase) necessary to generate local angiotensin II or activate latent TGF-β1. Statistically, when an SLE patient exhibits definitive structural changes, such as capillary dropout on capillaroscopy, the clinical phenotype shifts away from pure SLE, and the patient is reclassified as MCTD overlap syndrome (Table 4).
5. Activation of Mast Cells During Wound Healing is Not Identical to IgE Activation
A thorough review by Wilgus et al. on the role of mast cells in wound healing raised pertinent points regarding tissue repair, yet left fundamental questions open about how these cells behave in a chronic fibrotic disorder with no apparent structural wound per se [131]. Investigating this paradox uncovers critical biochemical and kinetic differences that separate physiological tissue restoration from the chronic, self-sustaining loop of IgE-mediated autoallergy (Table 5).
The table highlights the fundamental differences in mast cell behavior, activation triggers, secretion kinetics, and termination signaling between normal homeostatic tissue repair and the chronic, self-perpetuating autoallergic circuit characteristic of early SSc pathogenesis. DAMPs = damage-associated molecular patterns; MRGPRX2 = MAS related GPR family member X2; PIEZO1 = piezo type mechanosensitive ion channel component 1; ST2 = IL-33 receptor; TLR = toll-like receptor; TGF-β = transforming growth factor -beta 1; IL= interleukin; VEGF = vascular endothelial growth factor; TNF-α = tumor necrosis factor alpha; MyD88 = myeloid differentiation primary response protein MyD88; IRAK = interleukin 1 receptor associated kinase.
In classic acute wounding, mast cells are primarily activated by innate immune receptors, such as toll-like receptors (TLRs), or by damage-associated molecular patterns (DAMPs), including alarmins released by ruptured epithelial cells [132,133]. This primitive survival mechanism induces piecemeal degranulation, a slow, highly metered release of specific localized cytokines and growth factors optimized to transition the tissue from acute inflammation to provisional matrix deposition [134]. Crucially, as the structural defect closes and DAMP signaling diminishes, a physiological termination cascade is engaged, allowing local mast cell populations to return to a resting, homeostatic baseline.
Conversely, the SSc autoallergic framework operates via an adaptive immune bypass. Here, tissue-resident perivascular mast cells are primed by antigen-specific IgE autoantibodies structurally cross-linked to high-affinity FcεRI receptors. Upon encountering their specific nuclear autoantigens (such as Topo-1 or CENP), these primed cells undergo degranulation. This sudden exocytosis simultaneously releases high concentrations of preformed chymase, tryptase, histamine, and leukotrienes. Because the underlying targets are intracellular self-antigens that are constantly exposed to or modified by ongoing ischemia-reperfusion cycles, there is no physiological shut-off switch. The termination signal is absent, transforming a temporary tissue-repair response into a permanent, self-perpetuating obliterative driver.
6. Discussion
Microvascular obliteration in SSc is driven by a synergistic, dual-antibody mechanism with a 95–98% co-prevalence [108]. The first hit, an acute, IgE-mediated flare, sensitizes perivascular mast cells to release histamine, tryptase, and chymase, driving vascular permeability and functional vasospasm. Concurrently, the second hit involves chronic IgG AECAs that inflict sustained vascular damage via endothelial cell apoptosis and a procoagulant state [123,135,136]. In this tandem framework, the chronic IgG injury establishes the structural prerequisite for remodeling, while the acute IgE flare provides the secretory stimulus that accelerates perivascular collagen deposition. The definitive terminal effect of this double-hit is the activation of TANGO1-mediated procollagen export in local resident myofibroblasts [127], physically occluding the vessel lumen through intimal hyperplasia and perivascular fibrosis.
Under this framework, Scl-70 and centromeric proteins act as functional allergens (Figure 3), triggering a classic Type I hypersensitivity response via a distinct three-step trajectory.
- Biochemical Modification: Chronic tissue hypoxia and oxidative stress from repeated ischemia-reperfusion injury drive the fragmentation or cross-linking of hallmark antigens like topoisomerase I [137,138]. These chemical modifications alter the protein's physical shape, exposing cryptic epitopes that the immune system perceives as foreign.
- Immunological Misinterpretation (Th2 Bias): Rather than clearing this debris normally, a systemic environment heavily skewed toward Th2 polarization and driven by high estradiol levels misinterprets these modified targets [139]. Estrogen, along with IL-4 and IL-13, promotes IgE class switching in B cells [140,141,142].
- Functional Allergic Behavior: High estradiol levels simultaneously hypersensitize perivascular mast cells, allowing trace autoantigens to trigger degranulation [143]. Once bound to high-affinity FcεRI receptors via specific IgE autoantibodies, the antigen is subsequently cross-linked by tissue exposure, triggering an acute allergic flare. This sudden release of histamine, chymase, and tryptase drives the non-pitting edema, intense pruritus, and aggressive vasospasms that define the early SSc prodrome.
The autoallergic framework effectively resolves several long-standing clinical paradoxes across the spectrum of systemic autoimmunity:
- MCTD: The antigen-specific nature of this mechanism is confirmed by the complete absence of SSc-specific IgE autoantibodies (anti-CENP or anti-Topo-I) in MCTD patients. Instead, the identical obliterative vasculopathy observed in MCTD is driven by a distinct IgE-anti-U1-RNP/basophil axis [109], proving that microvascular failure is determined by the specific self-antigen that triggers the localized allergic circuit. In 81% of MCTD patients, AECAs were identified, further modeling autoantibody similarities with SSc that drives the vasculopathy [144].
- ACA-positive Sjögren’s: This hybrid cohort reinforces the concept that the "Scleroderma-pattern" capillaroscopic signature is an antigen-driven event independent of the primary diagnosis. In this subset, the immune system utilizes the IgE-mast cell circuit (Type I hypersensitivity) rather than the complement-consuming immune complex deposition (Type III) typical of ACA-negative Sjögren’s. Recognizing CENP-A/B as a primary IgE target demonstrates that SSc-like ischemia is a specific, antigen-driven secretory event prioritizing perivascular fibrosis over broad surface inflammation.
- SLE: In pure SLE, while patients may possess IgE autoantibodies against U1-RNP (Table 3), the broader immunological profile is dominated by Th1/Th17 cells and Type I interferons [145]. This environment heavily favors the production of high-affinity IgG autoantibodies [146] that form large, circulating immune complexes. These complexes deposit on vessel walls, activating the complement cascade (Type III hypersensitivity) [147] and recruiting neutrophils and macrophages [148]. These cells release proteolytic enzymes and reactive oxygen species that drive acute destruction (vasculitis and necrotizing lesions) rather than the specific, pro-fibrotic perivascular remodeling seen in SSc. Without a Th2-skewed isotype switch to IgE, resident perivascular mast cells remain unprimed, preventing the acute secretory flare required for localized structural remodeling.
- Female to male predilection in SSc: The striking female predominance of SSc is directly tied to estrogen-mediated amplification, as estradiol promotes Th2-cell polarization and elevates IL-4 and IL-13 [149] to signal B-cell IgE class switching [150]. Crucially, men who develop rapidly progressive dcSSc exhibit a paradoxical hormonal profile, with serum estradiol levels significantly exceeding those of both healthy males and postmenopausal females [151]. This high-estradiol environment drives an identical Th2 bias and a subsequent isotype switch to IgE autoantibodies. In males, this autoallergic switch correlates with a higher risk of visceral myocardial fibrosis, accelerated skin thickening (mRSS), and significantly decreased survival [151,152].
7. Therapeutic Implications: The "Golden Window" and Dual-Pathway Strategy
The current therapeutic failures in SSc likely stem from a fundamental misclassification of the disease as a classic Type II or Type IV autoimmune condition. Repositioning SSc as a localized Type I Autoallergy at the apex of a broader autoallergic gradient unmasks a critical, overlooked therapeutic window that targets early secretory flares rather than late-stage, irreversible fibrosis. Shifting the clinical focus toward the IgE-mast cell axis enables precise pharmacological intervention during the hyper-acute secretory phase of vasculopathy, clinically heralded by distinct neuro-sensory markers such as intense pruritus, neuropathic burning, and non-pitting edema, long before the structural transition to obliterative microangiopathy takes hold.
Within this framework, early-stage serum IgE autoantibody titers and dermal mast cell densities should be prioritized as vital predictive biomarkers. Future diagnostic protocols must use longitudinal screening to track IgE reactivity across an expanded antigenic panel, including predicting the autoallergic trajectory of anti-RNA Polymerase III in rapidly progressive dcSSc. Identifying patients during this transient secretory phase provides an objective tool to start targeted treatments before fixed tissue scarring renders them obsolete. The urgent clinical relevance of this temporal sensitivity is highlighted by historical experiences with mast cell stabilizers. Clinical cohorts treated with Ketotifen demonstrated success in inducing disease remission, but only when administered within a narrow therapeutic "Golden Window" spanning the first 12 to 18 months of symptom onset [153,154]. Beyond this brief boundary, the efficacy of Ketotifen and similar stabilizers abruptly collapses [154]. This failure in patients with established disease durations confirms that the window for purely mast cell-targeted inhibition eventually closes; once persistent ischemia-reperfusion cycles lock the tissue into chronic remodeling, and the pathology transitions from a reversible secretory event into a permanent structural dropout.
Consequently, historical monotherapies utilizing standard mast cell stabilizers (such as cromoglycate) or isolated chymase inhibitors have shown varying or incomplete success [94] because they are typically introduced too late or address only a single component of a multi-isotype insult. To effectively arrest the ischemic switch, future therapeutic strategies must transition toward an aggressive dual-pathway inhibition paradigm: this tandem approach must simultaneously deploy agents targeting the acute autoallergic circuit (e.g., anti-IgE biologics such as omalizumab, high-potency mast cell stabilizers, or localized chymase antagonists) alongside continuous therapies to mitigate chronic IgG-mediated endothelial attrition (e.g., plasma exchange or selective endothelial cell survival factors). By intercepting both circuits during the open secretory window, clinicians can neutralize the dual-isotype synergy required for intimal hyperplasia, protect bone marrow-derived endothelial progenitors, and halt the relentless progression of this devastating microvascular disease.
8. Conclusion
The ischemic switch from benign functional vasospasm to permanent structural obliteration remains a fundamental mystery in SSc. Traditional Type II and Type IV autoimmune models cannot reconcile the hyper-acute temporal kinetics, intense neuroimmune pruritus, and diffuse interstitial edema defining the early prodrome. Repositioning SSc as an antigen-specific Type I autoallergy elegantly solves these anomalies. Chronic ischemia-reperfusion injury and tissue hypoxia induce biochemical cross-linking and fragmentation of hallmark intracellular autoantigens (Topo-1 and CENP), exposing shape-altered cryptic epitopes. Driven by an estrogen-amplified Th2 polarization, B cells undergo an isotype switch to produce highly specific IgE autoantibodies. These molecules bind to high-affinity FcεRI receptors on resident perivascular mast cells, priming them for degranulation upon subsequent antigen exposure. This acute secretory flare releases a wave of histamine, leukotrienes, tryptase, and chymase, rapidly altering vascular permeability, targeting sensory nerves, and generating local angiotensin II to clamp the vessel lumen shut. Crucially, the permanent transition to structural remodeling is governed by a synergistic, dual-antibody two-hit mechanism. Operating in a 95–98% co-prevalence, chronic concurrent IgG AECAs inflict relentless baseline endothelial apoptosis, deplete protective nitric oxide vasodilators, and exhaust bone marrow-derived endothelial progenitors. This continuous vascular attrition ensures that the episodic, pro-fibrotic secretory stimulus delivered by recurrent IgE-mediated mast cell flares cannot be resolved, trapping local microvessels in an irreversible loop of intimal hyperplasia and perivascular remodeling.
Recognizing this dual-isotype axis fundamentally reforms clinical trial architecture. The definitive failure of historical mast cell stabilizer monotherapies outside a narrow Golden Window demonstrates that the opportunity for purely allergic intervention rapidly closes once permanent structural remodeling takes hold. Consequently, future therapeutic strategies must transition toward an aggressive, multi-target dual-pathway paradigm. Simultaneously deploying early anti-IgE biologics or mast cell stabilizers, alongside continuously administered IgG-clearing plasma exchange or endothelial survival factors, provides an objective clinical roadmap. Intercepting the disease during this early, fluid phase provides an unprecedented avenue to arrest the ischemic switch before the pathology locks into irreversible microvascular obliteration.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflicts of interest.
Acknowledgments
I would like to thank Carol Feghali-Bostwick, PhD for her critical comments and valuable insights into the manuscript.
References
- Yeo, R.E.; Eros, F.R.; Demers, P.A.; Sritharan, J. Risk of Raynaud's Phenomenon Among Workers in the Occupational Disease Surveillance System. Am. J. Ind. Med. 2025, 68, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Lemerle, P.; Ganghoffer, J.F. A two scale modeling and computational framework for vibration-induced Raynaud syndrome. J. Mech. Behav. BioMed Mater. 2017, 71, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Khouri, C.; Blaise, S.; Carpentier, P.; Villier, C.; Cracowski, J.L.; Roustit, M. Drug-induced Raynaud's phenomenon: beyond β-adrenoceptor blockers. Br. J. Clin. Pharmacol. 2016, 82, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Fardoun, M.; El Ghawi, O.; Dib, C.; Jaradi, L.; Chaddad, M.T.; Dehaini, H.; Eid, A.H. Cold responses and hormonal echoes: a comprehensive view of Raynaud's vascular dysfunction. Inflammopharmacology 2025, 33, 3637–3651. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Henkin, S. Raynaud's phenomenon and related vasospastic disorders. Vasc. Med. 2021, 26, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.S.; Alcacer-Pitarch, B.; Pauling, J.D.; Flurey, C.A.; Redmond, A.C.; Richards, P.; Herrick, A.L.; Merkel, P.A.; Proudman, S.; Menz, H.B.; et al. Patients' perspectives on systemic sclerosis-related Raynaud's phenomenon in the feet: A qualitative study from the OMERACT Foot and Ankle Working Group. Semin Arthritis Rheum. 2024, 65, 152372. [Google Scholar] [CrossRef] [PubMed]
- Vaportzis, E. Cold Hands, Warm Heart: Quality of life, wellbeing, and mental health in Raynaud's disease - An international survey study. J. Health Psychol. 2026, 13591053251407736. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, P.; Schwager, Z.; Lo Sicco, K.; Franks, A.G., Jr. The clinical effects of l-arginine and asymmetric dimethylarginine: implications for treatment in secondary Raynaud's phenomenon. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Elhai, M.; Avouac, J.; Kahan, A.; Allanore, Y. Systemic sclerosis: Recent insights. Jt. Bone Spine 2015, 82, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Peretti, S.; Bruni, C.; Bonomi, F.; De Angelis, R.; Bajocchi, G.; Giuggioli, D.; Orlandi, M.; Zanframundo, G.; Foti, R.; Visalli, E.; et al. Age and onset timing of Raynaud's phenomenon and first non-Raynaud symptom as prognostic factors in systemic sclerosis: a retrospective analysis from the Italian national multicenter Systemic Sclerosis Progression INvestiGation registry of the Italian Society for Rheumatology (SPRING-SIR). Ther. Adv. Musculoskelet. Dis. 2026, 18, 1759720x251410243. [Google Scholar] [CrossRef] [PubMed]
- Nigro, A. Fast-track capillaroscopic progression in systemic sclerosis: a case-based review of active pattern emerging within 3 months of raynaud's phenomenon onset. Rheumatol. Int. 2025, 45, 203. [Google Scholar] [CrossRef] [PubMed]
- Wirz, E.G.; Jaeger, V.K.; Allanore, Y.; Riemekasten, G.; Hachulla, E.; Distler, O.; Airò, P.; Carreira, P.E.; Tikly, M.; Vettori, S.; et al. Incidence and predictors of cutaneous manifestations during the early course of systemic sclerosis: a 10-year longitudinal study from the EUSTAR database. Ann. Rheum. Dis. 2016, 75, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Hanif, I.; Assassi, S.; Mayes, M.D.; McMahan, Z.H.; Zhang, M.; Charles, J.; VanBuren, J.M.; Alvey, J.S.; Ghaffari, K.; Bernstein, E.J.; et al. Hand Swelling and Other Non-Raynaud Phenomenon Symptoms as the Initial Presentation of Systemic Sclerosis: Prevalence and Clinical Associations in Two US Cohorts. Arthritis Rheum. 2025, 77, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Pauling, J.D.; Reilly, E.; Smith, T.; Frech, T.M. Evolving Symptom Characteristics of Raynaud's Phenomenon in Systemic Sclerosis and Their Association With Physician and Patient-Reported Assessments of Disease Severity. Arthritis Care Res. 2019, 71, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Claman, H.N. Mast cell changes in a case of rapidly progressive scleroderma-ultrastructural analysis. J. Invest Dermatol. 1989, 92, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, A.; Pauling, J.D. Raynaud's phenomenon and digital ulceration in systemic sclerosis. Best Pract. Res. Clin. Rheumatol. 2026, 102121. [Google Scholar] [CrossRef] [PubMed]
- Massay, R.; Zahn, C.; Tsou, P.S. Decoding vascular dysfunction in systemic sclerosis: from endothelial damage to clinical implications. Curr. Opin. Rheumatol. 2025, 37, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Herrick, A.L. Assessing digital vasculopathy in systemic sclerosis. Rheumatology 2026, 65. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, R.; Ferri, C.; Cipolletta, E.; Riccieri, V.; Di Battista, M.; Bajocchi, G.; Bellando-Randone, S.; Bruni, C.; Orlandi, M.; Zanframundo, G.; et al. Prevalence, distribution and associations of the scleroderma capillaroscopic patterns: new insights from the Italian SPRING-SIR registry. Rheumatology 2026, 65. [Google Scholar] [CrossRef] [PubMed]
- Heimovski, F.E.; Simioni, J.A.; Skare, T.L. Systemic lupus erythematosus and Raynaud's phenomenon. An. Bras. Dermatol. 2015, 90, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, P.; Baldini, C.; Rocchi, V.; Bombardieri, S. Anti-Sm and anti-RNP antibodies. Autoimmunity 2005, 38, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Grader-Beck, T.; Wigley, F.M. Raynaud's phenomenon in mixed connective tissue disease. Rheum. Dis. Clin. North Am. 2005, 31, 465–481, vi. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yuan, F. Hypocomplementemia in Primary Sjogren's Syndrome: A Retrospective Study of 120 Treatment-Naive Chinese Patients. Int. J. Gen. Med. 2022, 15, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Lahoria, R.; Selcen, D.; Engel, A.G. Microvascular alterations and the role of complement in dermatomyositis. Brain 2016, 139, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Gumpold, C.; Mueller-Felber, W.; Schoser, B.; Haberler, C.; Loescher, W.N.; Rostásy, K.; Fischer, M.B.; Wanschitz, J.V. Pattern of myogenesis and vascular repair in early and advanced lesions of juvenile dermatomyositis. Neuromuscul. Disord. 2018, 28, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Sato, S. Vasculopathy in scleroderma. Semin Immunopathol. 2015, 37, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Pavlov-Dolijanovic, S.; Damjanov, N.S.; Stojanovic, R.M.; Vujasinovic Stupar, N.Z.; Stanisavljevic, D.M. Scleroderma pattern of nailfold capillary changes as predictive value for the development of a connective tissue disease: a follow-up study of 3,029 patients with primary Raynaud's phenomenon. Rheumatol. Int. 2012, 32, 3039–3045. [Google Scholar] [CrossRef] [PubMed]
- Kasser, C.; Boleto, G.; Allanore, Y.; Avouac, J. Nailfold videocapillaroscopy findings and associations with organ involvement in mixed connective tissue disease. Clin. Exp. Rheumatol. 2025, 43, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y. Is mixed connective tissue disease (MCTD) a subtype of systemic sclerosis? Semin Arthritis Rheum. 2025, 72, 152678. [Google Scholar] [CrossRef] [PubMed]
- Gulati, D.; Kushner, I.; File, E.; Magrey, M. Primary Sjogren's syndrome with anticentromere antibodies--a clinically distinct subset. Clin. Rheumatol. 2010, 29, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Qiao, L.; Duan, X.; Hui, M.; Li, M.; Zhao, Y.; Zeng, X.; Xu, D. Clinical Characteristics and Risk Factors for Progressive Pulmonary Fibrosis in Primary Sjögren's Syndrome: A Case-Control Study. Risk Manag Healthc. Policy 2025, 18, 3017–3028. [Google Scholar] [CrossRef] [PubMed]
- Leehan, K.M.; Pezant, N.P.; Rasmussen, A.; Grundahl, K.; Moore, J.S.; Radfar, L.; Lewis, D.M.; Stone, D.U.; Lessard, C.J.; Rhodus, N.L.; et al. Minor salivary gland fibrosis in Sjögren's syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin. Exp. Rheumatol. 2018, 36 Suppl 112, 80–88. [Google Scholar] [PubMed]
- Androutsakos, T.; Voulgaris, T.A.; Bakasis, A.D.; Koutsompina, M.L.; Chatzis, L.; Argyropoulou, O.D.; Pezoulas, V.; Fotiadis, D.I.; Papatheodoridis, G.; Tzioufas, A.G.; et al. Liver Fibrosis in Primary Sjögren's Syndrome. Front Immunol. 2022, 13, 889021. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, A.; Kobayashi, H.; Ikumi, N.; Kobayashi, Y.; Yokoe, I.; Sugiyama, K.; Matsukawa, Y.; Takei, M.; Kitamura, N. Salivary Gland Focus Score Is Associated With Myocardial Fibrosis in Primary Sjögren Syndrome Assessed by a Cardiac Magnetic Resonance Approach. J. Rheumatol. 2021, 48, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.E. Other manifestations of mixed connective tissue disease. Rheum. Dis. Clin. North Am. 2005, 31, 519–533, vii. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.; Caproni, M.; Marzano, A.V.; De Simone, C.; La Placa, M.; Quaglino, P.; Veller Fornasa, C.; Zane, C.; Vaccaro, M.; Papini, M.; et al. Dermatomyositis in 132 patients with different clinical subtypes: cutaneous signs, constitutional symptoms and circulating antibodies. Acta Derm. Venereol. 2002, 82, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.M.; Graham, D.; Borlenghi, C.E.; Edwards, T.; Schachterle, S.E.; Sile, H. A retrospective natural history study in adult and juvenile patients with incident dermatomyositis and polymyositis using real world data. Clin. Rheumatol. 2025, 44, 4237–4247. [Google Scholar] [CrossRef] [PubMed]
- Lilleker, J.B.; Vencovsky, J.; Wang, G.; Wedderburn, L.R.; Diederichsen, L.P.; Schmidt, J.; Oakley, P.; Benveniste, O.; Danieli, M.G.; Danko, K.; et al. The EuroMyositis registry: an international collaborative tool to facilitate myositis research. Ann. Rheum. Dis. 2018, 77, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, H.; Kimura, T.; Sakate, R.; Maruyama, S.; Isaka, Y.; Narita, I.; Hiromura, K. Characteristics of newly diagnosed systemic lupus erythematosus patients with or without kidney involvement: analysis of the National Database of Designated Intractable Diseases of Japan. Clin. Exp. Nephrol. 2026. [Google Scholar] [CrossRef] [PubMed]
- Barbacki, A.; Rached-d'Astous, N.; Pineau, C.A.; Vinet, E.; Grenier, L.P.; Kalache, F.; Fallavollita, S.; Lukusa, L.; Bernatsky, S. Clinical Significance of Raynaud Phenomenon in Systemic Lupus Erythematosus. J. Clin. Rheumatol. 2022, 28, e488–e490. [Google Scholar] [CrossRef] [PubMed]
- Sargın, G.; Baygin, H.; Cildag, S.; Senturk, T. Interstitial lung disease and associated factors in patients with Sjögren's syndrome. Ir. J. Med. Sci. 2024, 193, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, L.; Lin, W.; Ning, X.; Liu, M.; Cao, J.; Su, Y.; Zheng, X.; Li, S.; Li, F.; et al. Anticentromere antibody positive patients with primary Sjögren's syndrome have distinctive clinical and immunological characteristics. Clin. Exp. Rheumatol. 2023, 41, 2371–2378. [Google Scholar] [CrossRef] [PubMed]
- Razykov, I.; Thombs, B.D.; Hudson, M.; Bassel, M.; Baron, M. Prevalence and clinical correlates of pruritus in patients with systemic sclerosis. Arthritis Rheum. 2009, 61, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Razykov, I.; Levis, B.; Hudson, M.; Baron, M.; Thombs, B.D. Prevalence and clinical correlates of pruritus in patients with systemic sclerosis: an updated analysis of 959 patients. Rheumatology 2013, 52, 2056–2061. [Google Scholar] [CrossRef] [PubMed]
- Théréné, C.; Brenaut, E.; Sonbol, H.; Pasquier, E.; Saraux, A.; Devauchelle, V.; Le Moigne, E.; Misery, L.; Abasq-Thomas, C. Itch and systemic sclerosis: frequency, clinical characteristics and consequences. Br. J. Dermatol. 2017, 176, 1392–1393. [Google Scholar] [CrossRef] [PubMed]
- Meridor, K.; Berookhim, J.; Levy, Y. Low dose naloxone for pruritus in systemic sclerosis: Case series and literature review. Medicine 2022, 101, e28653. [Google Scholar] [CrossRef] [PubMed]
- El-Baalbaki, G.; Razykov, I.; Hudson, M.; Bassel, M.; Baron, M.; Thombs, B.D. Association of pruritus with quality of life and disability in systemic sclerosis. Arthritis Care Res. 2010, 62, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Frech, T.M.; Baron, M. Understanding itch in systemic sclerosis in order to improve patient quality of life. Clin. Exp. Rheumatol. 2013, 31, 81–88. [Google Scholar] [PubMed]
- Stull, C.M.; Weaver, L.A.; Valdes-Rodriguez, R.; Naramala, S.; Lavery, M.J.; Chan, Y.H.; Mendoza, F.A.; Yosipovitch, G. Characteristics of Chronic Itch in Systemic Sclerosis: A Cross-sectional Survey. Acta Derm. Venereol. 2018, 98, 793–794. [Google Scholar] [CrossRef] [PubMed]
- Gourier, G.; Théréné, C.; Mazeas, M.; Abasq-Thomas, C.; Brenaut, E.; Huet, F.; Sonbol, H.; Campillo, E.; Lemerle, J.; Pasquier, E.; et al. Clinical Characteristics of Pruritus in Systemic Sclerosis Vary According to the Autoimmune Subtype. Acta Derm. Venereol. 2018, 98, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, D.; Mangold, A.R.; Werth, V.P.; Christopher-Stine, L.; Femia, A.; Chu, M.; Musiek, A.C.M.; Sluzevich, J.C.; Graham, L.V.; Fernandez, A.P.; et al. Efficacy, safety, and target engagement of dazukibart, an IFNβ specific monoclonal antibody, in adults with dermatomyositis: a multicentre, double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2025, 405, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J. Pruritus in autoimmune connective tissue diseases. Ann. Transl. Med. 2021, 9, 441. [Google Scholar] [CrossRef] [PubMed]
- McKee, S.; Xenakis, J.; Makin, H.; Marshall, C.; Winnette, R.; Aggarwal, R.; Knight, S. Cutaneous Manifestations in Patients with Dermatomyositis, Are They Only Skin Deep? Dermatol. Ther. 2024, 14, 2771–2785. [Google Scholar] [CrossRef] [PubMed]
- Bongi, S.M.; Del Rosso, A.; Passalacqua, M.; Miccio, S.; Cerinic, M.M. Manual lymph drainage improving upper extremity edema and hand function in patients with systemic sclerosis in edematous phase. Arthritis C Res. 2011, 63, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Namas, R.; Dodge, C.; Khanna, D. Hand Impairment in Systemic Sclerosis: Various Manifestations and Currently Available Treatment. Curr. Treatm Opt. Rheumatol. 2016, 2, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Torok, K.S.; Baker, N.A.; Lucas, M.; Domsic, R.T.; Boudreau, R.; Medsger, T.A., Jr. Reliability and validity of the delta finger-to-palm (FTP), a new measure of finger range of motion in systemic sclerosis. Clin. Exp. Rheumatol. 2010, 28, S28–36. [Google Scholar] [PubMed]
- Ture, H.Y.; Lee, N.Y.; Kim, N.R.; Nam, E.J. Raynaud's Phenomenon: A Current Update on Pathogenesis, Diagnostic Workup, and Treatment. Vasc. Spec. Int. 2024, 40, 26. [Google Scholar] [CrossRef] [PubMed]
- Frech, T.M.; Revelo, M.P.; Drakos, S.G.; Murtaugh, M.A.; Markewitz, B.A.; Sawitzke, A.D.; Li, D.Y. Vascular leak is a central feature in the pathogenesis of systemic sclerosis. J. Rheumatol. 2012, 39, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Cui, J.; Weinblatt, M.E.; Solomon, D.H.; Naik, C.; Shadick, N.A. Patterns of Involvement of the Hand Joints in Classical Rheumatoid Arthritis. J. Clin. Rheumatol. 2023, 29, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Kassamali, B.; Mazori, D.R.; LaChance, A.H.; Christopher-Stine, L. Exploring Dermatomyositis through an Interdisciplinary Lens: Pearls from Dermatology and Rheumatology. Int. J. Womens Dermatol. 2021, 7, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Qian, J. The role of nailfold video-capillaroscopy in the assessment of dermatomyositis. Rheumatology 2025, 64, 2987–2994. [Google Scholar] [CrossRef] [PubMed]
- Concha, J.S.S.; Merola, J.F.; Fiorentino, D.; Werth, V.P. Re-examining mechanic's hands as a characteristic skin finding in dermatomyositis. J. Am. Acad. Dermatol. 2018, 78, 769–775.e762. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Sinhamahapatra, P.; Choudhury, S.; Gangopadhyay, A.; Bala, S.; Sircar, G.; Chatterjee, G.; Ghosh, A. Cutaneous manifestations of mixed connective tissue disease: study from a tertiary care hospital in eastern India. Indian J. Dermatol. 2014, 59, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Felis-Giemza, A.; Ornowska, S.; Haładyj, E.; Czuszyńska, Z.; Olesińska, M. Relationship between type of skin lesions and nailfold capillaroscopy pattern in mixed connective tissue disease. Clin. Rheumatol. 2022, 41, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, S.; Yamaoka, K.; Sawamukai, N.; Shimajiri, S.; Kubo, S.; Miyagawa, I.; Sonomoto, K.; Saito, K.; Tanaka, Y. Dermal mast cell density in fingers reflects severity of skin sclerosis in systemic sclerosis. Mod. Rheumatol. 2013, 23, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, S.; Ishikawa, O.; Igarashi, Y.; Kurosawa, M.; Miyachi, Y. Dermal mast cells in scleroderma: their skin density, tryptase/chymase phenotypes and degranulation. Br. J. Dermatol. 1998, 138, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Kobayashi, Y.; Katayama, I.; Takijiri, C. Mast cell numbers in diffuse scleroderma. Arch. Dermatol. 1987, 123, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Prescott, R.J.; Freemont, A.J.; Jones, C.J.; Hoyland, J.; Fielding, P. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J. Pathol. 1992, 166, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A.; Claman, H.N.; Clark, R.A.; Steigerwald, J.C. Increased dermal mast cell populations in progressive systemic sclerosis: a link to chronic fibrosis. Ann. Intern Med. 1985, 102, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Claman, H.N.; Choi, K.L.; Sujansky, W.; Vatter, A.E. Mast cell "disappearance" in chronic murine graft-vs-host disease (GVHD)-ultrastructural demonstration of "phantom mast cells. J. Immunol. 1986, 137, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V.; Soter, N.A.; Altman, R.D.; Kerdel, F.A. Elevated plasma histamine levels in systemic sclerosis (scleroderma). Arch. Dermatol. 1990, 126, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Chanez, P.; Lacoste, J.Y.; Guillot, B.; Giron, J.; Barnéon, G.; Enander, I.; Godard, P.; Michel, F.B.; Bousquet, J. Mast cells' contribution to the fibrosing alveolitis of the scleroderma lung. Am. Rev. Respir. Dis. 1993, 147, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Tontini, C.; Bahri, R.; Higham, A.; Singh, D.; Simpson, A.; Bulfone-Paus, S. Microenvironment-Driven Mast Cell Plasticity: Insights From Cytokine-Activated Gene Signatures in Skin and Respiratory Diseases. Allergy 2025, 80, 3077–3094. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Yuan, X.; Yi, J.; Li, Z.; Li, X.; Guo, W.; Wang, J.; Mu, R. Targeting PTGDS inhibits pro-inflammatory fibroblasts associated with skin fibrosis in systemic sclerosis. Rheumatology 2025, 64, 5551–5561. [Google Scholar] [CrossRef] [PubMed]
- Gilman, K.E.; Limesand, K.H. The complex role of prostaglandin E(2)-EP receptor signaling in wound healing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R287–r296. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Lv, J.; Jiang, Z.; He, H.; Chen, C.; Xiong, Y.; Zhu, X.; Xue, Y.; Yu, Y.; Yang, S.; et al. Promotion of Myofibroblast Differentiation and Tissue Fibrosis by the Leukotriene B(4) -Leukotriene B(4) Receptor Axis in Systemic Sclerosis. Arthritis Rheum. 2020, 72, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Chwieśko-Minarowska, S.; Kowal, K.; Bielecki, M.; Kowal-Bielecka, O. The role of leukotrienes in the pathogenesis of systemic sclerosis. Folia Histochem Cytobiol. 2012, 50, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Dumitraşcu, D. Mast cells as potent inflammatory cells. Rom. J. Intern Med. 1996, 34, 159–172. [Google Scholar] [PubMed]
- Gruber, B.L. Mast cells: accessory cells which potentiate fibrosis. Int. Rev. Immunol. 1995, 12, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Pulito-Cueto, V.; Atienza-Mateo, B.; Batista-Liz, J.C.; Nieto-Nieto, R.; Vaquera-Illescas, C.; Sebastián Mora-Gil, M.; Iturbe-Fernández, D.; Mora-Cuesta, V.M.; Serrano-Combarro, A.; Izquierdo-Cuervo, S.; et al. A New Approach for Achieving Earlier and More Accurate Diagnosis of Connective Tissue Disease-Related Interstitial Lung Disease: TGFB and PDGFA as Novel Promising Biomarkers. Int. J. Molec Sci. 2025, 26. [Google Scholar] [CrossRef] [PubMed]
- Hügle, T.; Hogan, V.; White, K.E.; van Laar, J.M. Mast cells are a source of transforming growth factor β in systemic sclerosis. Arthritis Rheum. 2011, 63, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.I.; Hamburger, J.; Matthews, J.B. Mast cells, extracellular matrix components, TGFbeta isoforms and TGFbeta receptor expression in labial salivary glands in systemic sclerosis. Ann. Rheum. Dis. 2000, 59, 183–189. [Google Scholar] [PubMed]
- Hebbar, M.; Lassalle, P.; Janin, A.; Vanhée, D.; Bisiau, S.; Hatron, P.Y.; Tonnel, A.B.; Gosselin, B. E-selectin expression in salivary endothelial cells and sera from patients with systemic sclerosis. Role of resident mast cell-derived tumor necrosis factor alpha. Arthritis Rheum. 1995, 38, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Fertin, C.; Nicolas, J.F.; Gillery, P.; Kalis, B.; Banchereau, J.; Maquart, F.X. Interleukin-4 stimulates collagen synthesis by normal and scleroderma fibroblasts in dermal equivalents. Cell Mol. Biol. 1991, 37, 823–829. [Google Scholar] [PubMed]
- Wang, X.; Zhang, P.; Tang, Y.; Chen, Y.; Zhou, E.; Gao, K. Mast cells: a double-edged sword in inflammation and fibrosis. Front Cell Dev. Biol. 2024, 12, 1466491. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, K.; Makino, T.; Makino, K.; Kajihara, I.; Fukushima, S.; Ihn, H. IL-16 expression is increased in the skin and sera of patients with systemic sclerosis. Rheumatology 2020, 59, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Sieiro Santos, C.; Antolín, S.C.; Lorenzo, J.C.; Garay, C.L.; Morales, C.M.; de Miguel, E.B.; Guerrero, M.R.; Herránz, L.S.; Álvarez, E.D. KL6 and IL-18 levels are negatively correlated with respiratory function tests and ILD extent assessed on HRCT in patients with systemic sclerosis-related interstitial lung disease (SSc-ILD). Semin Arthritis Rheum. 2024, 65, 152366. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Homer, R.J.; Zhu, Z.; Lanone, S.; Wang, X.; Koteliansky, V.; Shipley, J.M.; Gotwals, P.; Noble, P.; Chen, Q.; et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor B. 1 J. Exp. Med. 2001, 194, 809–821. [Google Scholar] [PubMed]
- Wilgus, T.A.; Wulff, B.C. The Importance of Mast Cells in Dermal Scarring. Adv. Wound Care 2014, 3, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Hennino, A.; Bérard, F.; Guillot, I.; Saad, N.; Rozières, A.; Nicolas, J.F. Pathophysiology of urticaria. Clin. Rev. Allergy Immunol. 2006, 30, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Fireman, P. Understanding asthma pathophysiology. Allergy Asthma Proc. 2003, 24, 79–83. [Google Scholar] [PubMed]
- Hegner, B.; Kretzschmar, T.; Zhu, N.; Kleinau, G.; Zhao, H.; Kamhieh-Milz, J.; Hilger, J.; Schindler, R.; Scheerer, P.; Riemekasten, G.; et al. Autoimmune activation and hypersensitization of the AT1 and ETA receptors contributes to vascular injury in scleroderma renal crisis. Rheumatology 2023, 62, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Miziołek, B.; Bergler-Czop, B.; Kucharz, E.; Kotyla, P.; Kopeć-Mędrek, M.; Widuchowska, M.; Sieńczyk, M.; Brzezińska-Wcisło, L. Significance of the angiotensin I/angiotensin II/angiotensin-(1-7) axis in the pathogenesis of systemic sclerosis. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Tom, K.; Mehta, B.K.; Hoffmann, A.; Aren, K.; Carns, M.; Lee, J.; Martyanov, V.; Popovich, D.; Kosarek, N.; Wood, T.; et al. Mast Cell Activation in the Systemic Sclerosis Esophagus. J. Scleroderma Relat. Disord. 2021, 6, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Aoki, M.; Kawana, S. Urticarial vasculitis appearing in the progression of systemic sclerosis. J. Dermatol. 2006, 33, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Kuzumi, A.; Yoshizaki, A.; Matsuda, K.M.; Kotani, H.; Norimatsu, Y.; Fukayama, M.; Ebata, S.; Fukasawa, T.; Yoshizaki-Ogawa, A.; Asano, Y.; et al. Interleukin-31 promotes fibrosis and T helper 2 polarization in systemic sclerosis. Nat. Commun. 2021, 12, 5947. [Google Scholar] [CrossRef] [PubMed]
- Garcovich, S.; Maurelli, M.; Gisondi, P.; Peris, K.; Yosipovitch, G.; Girolomoni, G. Pruritus as a Distinctive Feature of Type 2 Inflammation. Vaccines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Suzuki, H.; Hara, H.; Hidaka, T.; Ishizuka, T.; Kawagoe, M.; Nakamura, H. Increased endothelin-1 production in fibroblasts derived from patients with systemic sclerosis. Ann. Rheum. Dis. 1994, 53, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Matucci-Cerinic, M.; Giacomelli, R.; Pignone, A.; Cagnoni, M.L.; Generini, S.; Casale, R.; Cipriani, P.; Del Rosso, A.; Tirassa, P.; Konttinen, Y.T.; et al. Nerve growth factor and neuropeptides circulating levels in systemic sclerosis (scleroderma). Ann. Rheum. Dis. 2001, 60, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Lee, C.S.; Park, S.J.; Chung, J.Y.; Jeong, K.S. Kinetics of macrophages, myofibroblasts and mast cells in carbon tetrachloride-induced rat liver cirrhosis. Anticancer Res. 2002, 22, 869–877. [Google Scholar] [PubMed]
- Kondo, S.; Kagami, S.; Kido, H.; Strutz, F.; Müller, G.A.; Kuroda, Y. Role of mast cell tryptase in renal interstitial fibrosis. J. Am. Soc. Nephrol. 2001, 12, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Tóth, T.; Tóth-Jakatics, R.; Jimi, S.; Ihara, M.; Urata, H.; Takebayashi, S. Mast cells in rapidly progressive glomerulonephritis. J. Am. Soc. Nephrol. 1999, 10, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.L. Mast cells in the pathogenesis of fibrosis. Curr. Rheumatol. Rep. 2003, 5, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Jordana, M.; Befus, A.D.; Newhouse, M.T.; Bienenstock, J.; Gauldie, J. Effect of histamine on proliferation of normal human adult lung fibroblasts. Thorax 1988, 43, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Gailit, J.; Marchese, M.J.; Kew, R.R.; Gruber, B.L. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J. Invest Dermatol. 2001, 117, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.L.; Kew, R.R.; Jelaska, A.; Marchese, M.J.; Garlick, J.; Ren, S.; Schwartz, L.B.; Korn, J.H. Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J. Immunol. 1997, 158, 2310–2317. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.D.; Gruber, B.L.; Marchese, M.J.; Seibold, J.R. Anti-IgE autoantibodies in systemic sclerosis (scleroderma). Ann. Rheum. Dis. 1989, 48, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.; Pecher, A.C.; Henes, J.; Klein, R. Detection of IgE-autoantibodies to nuclear antigens in patients with systemic sclerosis and analysis of their clinical relevance. Clin. Exp. Rheumatol. 2024, 42, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Lamri, Y.; Vibhushan, S.; Pacreau, E.; Boedec, E.; Saidoune, F.; Mailleux, A.; Crestani, B.; Blank, U.; Benhamou, M.; Papo, T.; et al. Basophils and IgE contribute to mixed connective tissue disease development. J. Allergy Clin. Immunol. 2021, 147, 1478–1489.e1411. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.; Pecher, A.C.; Henes, J.; Klein, R. IgE autoantibodies to nuclear antigens in patients with different connective tissue diseases: re-evaluation and novel findings. Front Immunol. 2025, 16, 1483815. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.; Savage, C.O.; Black, C.M.; Pearson, J.D. IgG antiendothelial cell autoantibodies from scleroderma patients induce leukocyte adhesion to human vascular endothelial cells in vitro. Induction of adhesion molecule expression and involvement of endothelium-derived cytokines. J. Clin. Invest 1996, 97, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Salojin, K.V.; Le Tonquèze, M.; Saraux, A.; Nassonov, E.L.; Dueymes, M.; Piette, J.C.; Youinou, P.Y. Antiendothelial cell antibodies: useful markers of systemic sclerosis. Am. J. Med. 1997, 102, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, M.; Lassalle, P.; Delneste, Y.; Hatron, P.Y.; Devulder, B.; Tonnel, A.B.; Janin, A. Assessment of anti-endothelial cell antibodies in systemic sclerosis and Sjögren's syndrome. Ann. Rheum. Dis. 1997, 56, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Negi, V.S.; Tripathy, N.K.; Misra, R.; Nityanand, S. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension. J. Rheumatol. 1998, 25, 462–466. [Google Scholar] [PubMed]
- Pignone, A.; Scaletti, C.; Matucci-Cerinic, M.; Vazquez-Abad, D.; Meroni, P.L.; Del Papa, N.; Falcini, F.; Generini, S.; Rothfield, N.; Caqnoni, M. Anti-endothelial cell antibodies in systemic sclerosis: significant association with vascular involvement and alveolo-capillary impairment. Clin. Exp. Rheumatol. 1998, 16, 527–532. [Google Scholar] [PubMed]
- Ihn, H.; Sato, S.; Fujimoto, M.; Igarashi, A.; Yazawa, N.; Kubo, M.; Kikuchi, K.; Takehara, K.; Tamaki, K. Characterization of autoantibodies to endothelial cells in systemic sclerosis (SSc): association with pulmonary fibrosis. Clin. Exp. Immunol. 2000, 119, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Wusirika, R.; Ferri, C.; Marin, M.; Knight, D.A.; Waldman, W.J.; Ross, P., Jr.; Magro, C.M. The assessment of anti-endothelial cell antibodies in scleroderma-associated pulmonary fibrosis. A study of indirect immunofluorescent and western blot analysis in 49 patients with scleroderma. Am. J. Clin. Pathol. 2003, 120, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Tamby, M.C.; Chanseaud, Y.; Humbert, M.; Fermanian, J.; Guilpain, P.; Garcia-de-la-Peña-Lefebvre, P.; Brunet, S.; Servettaz, A.; Weill, B.; Simonneau, G.; et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax 2005, 60, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, K.; Ciurzynski, M.; Gorska, E.; Bienias, P.; Irzyk, K.; Siwicka, M.; Zycinska, K.; Pruszczyk, P.; Demkow, U. Antiendothelial cells antibodies in patients with systemic sclerosis in relation to pulmonary hypertension and lung fibrosis. Adv. Exp. Med. Biol. 2013, 756, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.; Pottinger, B.E.; Woo, P.; Black, C.M.; Loizou, S.; Byron, M.A.; Pearson, J.D. Measurement and characterisation of circulating anti-endothelial cell IgG in connective tissue diseases. Clin. Exp. Immunol. 1988, 72, 450–456. [Google Scholar] [PubMed]
- Hill, M.B.; Phipps, J.L.; Cartwright, R.J.; Milford Ward, A.; Greaves, M.; Hughes, P. Antibodies to membranes of endothelial cells and fibroblasts in scleroderma. Clin. Exp. Immunol. 1996, 106, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Servettaz, A.; Tamby, M.C.; Guilpain, P.; Reinbolt, J.; Garcia de la Penã-Lefebvre, P.; Allanore, Y.; Kahan, A.; Meyer, O.; Guillevin, L.; Mouthon, L. Anti-endothelial cell antibodies from patients with limited cutaneous systemic sclerosis bind to centromeric protein B (CENP-B). Clin. Immunol. 2006, 120, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Michalska-Jakubus, M.M.; Rusek, M.; Kowal, M.; Czop, M.; Kocki, J.; Krasowska, D. Anti-endothelial cell antibodies are associated with apoptotic endothelial microparticles, endothelial sloughing and decrease in angiogenic progenitors in systemic sclerosis. Postep. Dermatol. Alergol. 2020, 37, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Pagkopoulou, E.; Soulaidopoulos, S.; Triantafyllidou, E.; Loutradis, C.; Malliari, A.; Kitas, G.D.; Garyfallos, A.; Dimitroulas, T. Asymmetric dimethylarginine correlates with worsening peripheral microangiopathy in systemic sclerosis. Microvasc. Res. 2023, 145, 104448. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, F.; Ruberto, M.F.; Barcellona, D. Is anticoagulative therapy in systemic sclerosis to be reconsidered? J. Scleroderma Relat. Disord. 2024, 9, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Kramer, B.; Corallo, C.; van den Heuvel, A.; Crawford, J.; Olivier, T.; Elstak, E.; Giordano, N.; Vulto, P.; Lanz, H.L.; Janssen, R.A.J.; et al. High-throughput 3D microvessel-on-a-chip model to study defective angiogenesis in systemic sclerosis. Sci. Rep. 2022, 12, 16930. [Google Scholar] [CrossRef] [PubMed]
- Connolly, L.M.; McFalls, C.M.; McMahon, I.G.; Bhat, A.M.; Artlett, C.M. Caspase 1 Enhances Transport and Golgi Organization Protein 1 Expression to Promote Procollagen Export From the Endoplasmic Reticulum in Systemic Sclerosis Contributing to Fibrosis. Arthritis Rheum. 2023, 75, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Michalska-Jakubus, M.; Kowal, M.; Adamczyk, M.; Krasowska, D. Anti-endothelial cell antibodies do not correlate with disease activity in systemic sclerosis. Postep. Dermatol. Alergol. 2018, 35, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Bodolay, E.; Csipo, I.; Gál, I.; Sipka, S.; Gyimesi, E.; Szekanecz, Z.; Szegedi, G. Anti-endothelial cell antibodies in mixed connective tissue disease: frequency and association with clinical symptoms. Clin. Exp. Rheumatol. 2004, 22, 409–415. [Google Scholar] [PubMed]
- Androulakis, N.; Striligka, O.; Nioti, E.; Dilintas, A.; Darivianaki, A.; Papadopoulou, A.; Nistikaki, F.; Iatridi, T.; Kampa, M.; Kalpadakis, C. Lupus Anticoagulant and Complement C4 Consumption in Antiphospholipid Antibody Testing: Disentangling Inflammation From Autoimmunity. Int. J. Lab Hematol. 2026. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A.; Ud-Din, S.; Bayat, A. A Review of the Evidence for and against a Role for Mast Cells in Cutaneous Scarring and Fibrosis. Int. J. Molec Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Agier, J.; Żelechowska, P.; Kozłowska, E.; Brzezińska-Błaszczyk, E. Expression of surface and intracellular Toll-like receptors by mature mast cells. Cent. Eur. J. Immunol. 2016, 41, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Cho, K.A.; Woo, S.Y. Toll-like receptor 2/6-stimulated HMC-1 mast cells promote keratinocyte migration in wound healing. PLoS ONE 2025, 20, e0317766. [Google Scholar] [CrossRef] [PubMed]
- Vukman, K.V.; Försönits, A.; Oszvald, Á.; Tóth, E.; Buzás, E.I. Mast cell secretome: Soluble and vesicular components. Semin Cell Dev. Biol. 2017, 67, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Pignone, A.; Scaletti, C.; Matucci-Cerinic, M.; Vázquez-Abad, D.; Meroni, P.L.; Del Papa, N.; Falcini, F.; Generini, S.; Rothfield, N.; Cagnoni, M. Anti-endothelial cell antibodies in systemic sclerosis: significant association with vascular involvement and alveolo-capillary impairment. Clin. Exp. Rheumatol. 1998, 16, 527–532. [Google Scholar] [PubMed]
- Mihai, C.; Tervaert, J.W. Anti-endothelial cell antibodies in systemic sclerosis. Ann. Rheum. Dis. 2010, 69, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Abdulle, A.E.; Diercks, G.F.H.; Feelisch, M.; Mulder, D.J.; van Goor, H. The Role of Oxidative Stress in the Development of Systemic Sclerosis Related Vasculopathy. Front Physiol. 2018, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Casciola-Rosen, L.; Wigley, F.; Rosen, A. Scleroderma autoantigens are uniquely fragmented by metal-catalyzed oxidation reactions: implications for pathogenesis. J. Exp. Med. 1997, 185, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Hardman, M.J.; Grencis, R.K. The role of sex hormones in the development of Th2 immunity in a gender-biased model of Trichuris muris infection. Eur. J. Immunol. 2010, 40, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Pauklin, S.; Sernández, I.V.; Bachmann, G.; Ramiro, A.R.; Petersen-Mahrt, S.K. Estrogen directly activates AID transcription and function. J. Exp. Med. 2009, 206, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Kashiwada, M.; Levy, D.M.; McKeag, L.; Murray, K.; Schröder, A.J.; Canfield, S.M.; Traver, G.; Rothman, P.B. IL-4-induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proc. Natl. Acad. Sci. USA 2010, 107, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.K.; Hummelshoj, L. Triggers of IgE class switching and allergy development. Ann. Med. 2007, 39, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Zaitsu, M.; Narita, S.; Lambert, K.C.; Grady, J.J.; Estes, D.M.; Curran, E.M.; Brooks, E.G.; Watson, C.S.; Goldblum, R.M.; Midoro-Horiuti, T. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Molec Immunol. 2007, 44, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Aoun, K.; Yoshida, S.; Kurosawa, Y. Autoantibodies directed against labile epitopes on cell surface proteins in autoimmune disease patients: proposal of a novel ELISA for the detection of anti-endothelial cell antibodies. J. Immunol. Meth 2012, 382, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Y.; Wang, D.C.; Chen, Y.Y.; Xu, W.D.; Huang, A.F. Th1-related transcription factors and cytokines in systemic lupus erythematosus. Front Immunol. 2023, 14, 1305590. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.L.; Szabo, S.J.; Glimcher, L.H. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc. Natl. Acad. Sci. USA 2002, 99, 5545–5550. [Google Scholar] [CrossRef] [PubMed]
- Sunderkötter, C.; Golle, L.; Pillebout, E.; Michl, C. Pathophysiology and clinical manifestations of immune complex vasculitides. Front Med. 2023, 10, 1103065. [Google Scholar] [CrossRef] [PubMed]
- Saavedra Torres, J.S.; Annamaraju, P. Type III Hypersensitivity Reaction. In StatPearls; StatPearls Publishing Copyright © 2026, StatPearls Publishing LLC.: Treasure Island (FL) with ineligible companies. Disclosure: Pavan Annamaraju declares no relevant financial relationships with ineligible companies., 2026.
- Hansda, A.K.; Biswas, B.; Goswami, R. 17-β Estradiol (E2) distinctly regulates the expression of IL-4 and IL-13 in Th2 cells via modulating the interplay between GATA3 and PU.1. Cytokine 2024, 173, 156440. [Google Scholar] [CrossRef] [PubMed]
- Peckham, H.; Radziszewska, A.; Sikora, J.; de Gruijter, N.M.; Restuadi, R.; Kartawinata, M.; Martin-Gutierrez, L.; Robinson, G.A.; Deakin, C.T.; Wedderburn, L.R.; et al. Estrogen influences class-switched memory B cell frequency only in humans with two X chromosomes. J. Exp. Med. 2025, 222. [Google Scholar] [CrossRef] [PubMed]
- Baker Frost, D.; Wolf, B.; Peoples, C.; Fike, J.; Silver, K.; Laffoon, M.; Medsger, T.A., Jr.; Feghali-Bostwick, C. Estradiol levels are elevated in older men with diffuse cutaneous SSc and are associated with decreased survival. Arthritis Res. Ther. 2019, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.; Pauling, J.D.; Armstrong-James, L.; Denton, C.P.; Galdas, P.; Flurey, C. Gender-related differences in systemic sclerosis. Autoimmun. Rev. 2020, 19, 102494. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.L.; Kaufman, L.D. Ketotifen-induced remission in progressive early diffuse scleroderma: evidence for the role of mast cells in disease pathogenesis. Am. J. Med. 1990, 89, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.L.; Kaufman, L.D. A double-blind randomized controlled trial of ketotifen versus placebo in early diffuse scleroderma. Arthritis Rheum. 1991, 34, 362–366. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mast cell degranulation. Mast cells release many different functional mediators that are known to be elevated in early SSc (red bold text). Adapted from Wilgus and Wolff [89]. EFG = Epidermal Growth Factor; FGF-2 = Fibroblast Growth Factor-2; GM-CSF = Granulocyte-Macrophage Colony-Stimulating Factor; IL = Interleukin; KGF = Keratinocyte Growth Factor; LCT4 = Leukotriene C4; MCP-1 = Monocyte Chemoattractant Protein-1; MIP-1 = Macrophage Inflammatory Protein-1; NGF = Nerve Growth Factor; PAF = Platelet-Activating Factor; PGD2 = Prostaglandin D2; PGE2 = Prostaglandin E2; PDGF = Platelet-Derived Growth Factor; TGF-β = Transforming Growth Factor-beta; TNF-α = Tumor Necrosis Factor-alpha; LTB4 = Leukotriene B4; VEGF = Vascular Derived Growth Factor. Created in BioRender.
Figure 1.
Mast cell degranulation. Mast cells release many different functional mediators that are known to be elevated in early SSc (red bold text). Adapted from Wilgus and Wolff [89]. EFG = Epidermal Growth Factor; FGF-2 = Fibroblast Growth Factor-2; GM-CSF = Granulocyte-Macrophage Colony-Stimulating Factor; IL = Interleukin; KGF = Keratinocyte Growth Factor; LCT4 = Leukotriene C4; MCP-1 = Monocyte Chemoattractant Protein-1; MIP-1 = Macrophage Inflammatory Protein-1; NGF = Nerve Growth Factor; PAF = Platelet-Activating Factor; PGD2 = Prostaglandin D2; PGE2 = Prostaglandin E2; PDGF = Platelet-Derived Growth Factor; TGF-β = Transforming Growth Factor-beta; TNF-α = Tumor Necrosis Factor-alpha; LTB4 = Leukotriene B4; VEGF = Vascular Derived Growth Factor. Created in BioRender.

Figure 2.
IgE cross-linking induces mast cell activation and degranulation. The IgE autoantibody to Scl-70 or CEN A/B engages the high-affinity FcεRI receptor, and upon antigen encounter, the mast cell degranulates, releasing signaling molecules that cause vascular leakage, pruritus, and neuropathic pain. Created in Biorender.
Figure 2.
IgE cross-linking induces mast cell activation and degranulation. The IgE autoantibody to Scl-70 or CEN A/B engages the high-affinity FcεRI receptor, and upon antigen encounter, the mast cell degranulates, releasing signaling molecules that cause vascular leakage, pruritus, and neuropathic pain. Created in Biorender.

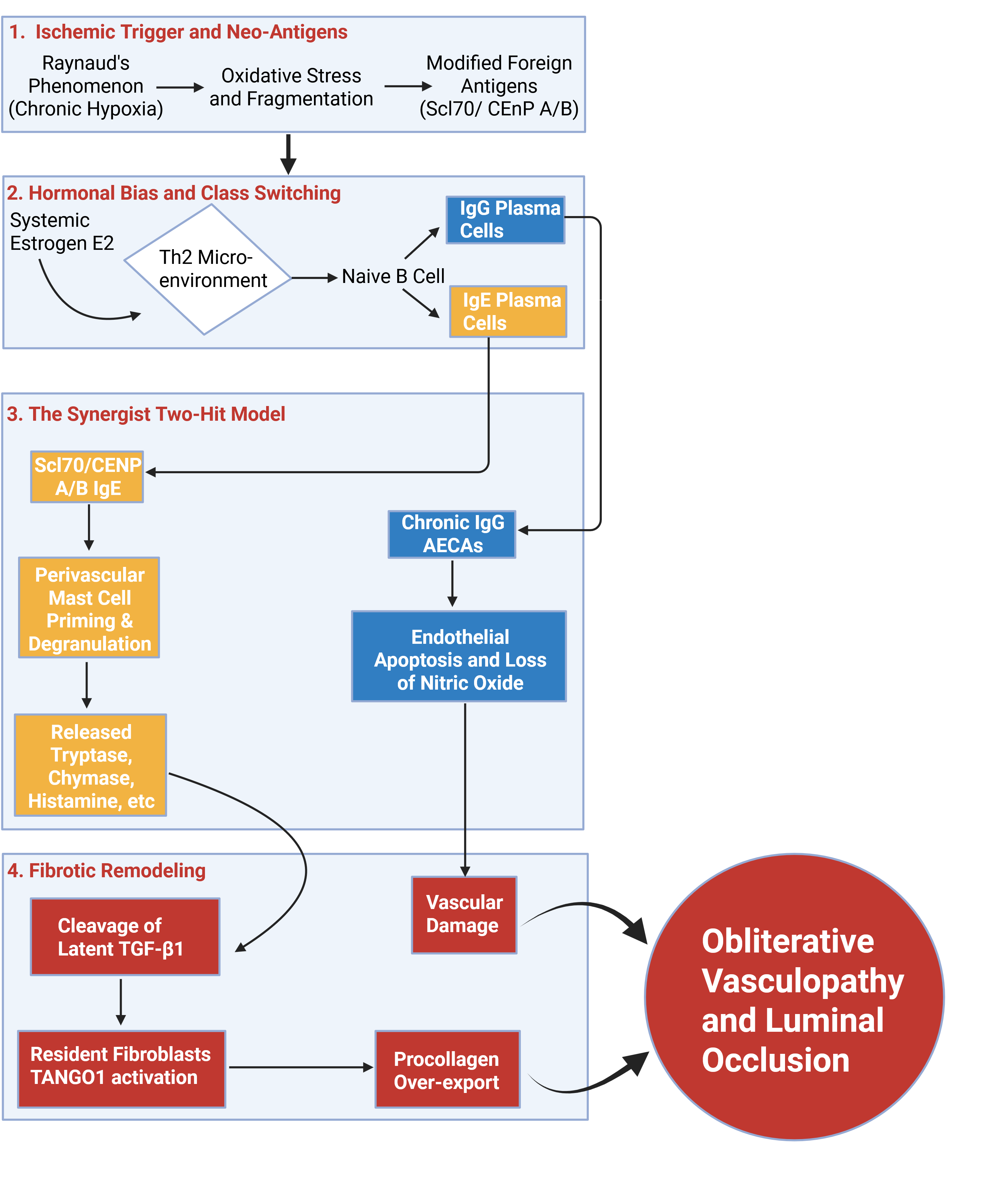
Figure 3.
The Synergistic Dual-Antibody "Two-Hit" Model of Systemic Sclerosis Vasculopathy. The First Hit (Acute IgE Circuit): Estrogen-mediated Th2 polarization promotes a B-cell isotype switch to IgE autoantibodies directed against specific modified nuclear neoantigens (e.g., Topo-1, CENP). These circulating IgE molecules prime tissue-resident perivascular mast cells via high-affinity FcεRI receptors. Upon antigen exposure, the degranulation releases preformed mediators (histamine, leukotrienes, tryptase, and chymase). This acute neuroimmune flare drives increased interstitial permeability (puffy edema), intense sensory nerve stimulation (pruritus), and rapid local generation of angiotensin II, leading to hyperreactive, vasospastic events seen in SSc-RP. The Second Hit (Chronic Concurrent IgG Circuit): Operating in tandem, functional IgG AECAs inflict a relentless, day-to-day structural insult on the vessel lining. This chronic IgG profile drives persistent endothelial cell apoptosis and shifts the microenvironment toward a procoagulant state, permanently removing vital vasodilators such as nitric oxide. Simultaneously, these IgG autoantibodies destroy bone marrow-derived endothelial cell progenitors, inhibiting neoangiogenesis and vascular repair. Pathological Outcome: The chronic endothelial attrition and structural groundwork laid by the concurrent IgG profile ensure that the episodic, pro-fibrotic secretory stimulus delivered by repeated IgE-mediated mast cell flares cannot be homeostatically resolved. This dual signaling permanently activates the TANGO1-mediated secretion in resident vascular myofibroblasts, accelerating intimal hyperplasia and perivascular fibrosis. The ultimate convergence of this dual-isotype cascade physically occludes the vessel lumen, marking the irreversible transition from functional, transient vasospasm to permanent structural obliteration. Created in BioRender. Artlett, C. (2026) https://BioRender.com/mirvpbo.
Figure 3.
The Synergistic Dual-Antibody "Two-Hit" Model of Systemic Sclerosis Vasculopathy. The First Hit (Acute IgE Circuit): Estrogen-mediated Th2 polarization promotes a B-cell isotype switch to IgE autoantibodies directed against specific modified nuclear neoantigens (e.g., Topo-1, CENP). These circulating IgE molecules prime tissue-resident perivascular mast cells via high-affinity FcεRI receptors. Upon antigen exposure, the degranulation releases preformed mediators (histamine, leukotrienes, tryptase, and chymase). This acute neuroimmune flare drives increased interstitial permeability (puffy edema), intense sensory nerve stimulation (pruritus), and rapid local generation of angiotensin II, leading to hyperreactive, vasospastic events seen in SSc-RP. The Second Hit (Chronic Concurrent IgG Circuit): Operating in tandem, functional IgG AECAs inflict a relentless, day-to-day structural insult on the vessel lining. This chronic IgG profile drives persistent endothelial cell apoptosis and shifts the microenvironment toward a procoagulant state, permanently removing vital vasodilators such as nitric oxide. Simultaneously, these IgG autoantibodies destroy bone marrow-derived endothelial cell progenitors, inhibiting neoangiogenesis and vascular repair. Pathological Outcome: The chronic endothelial attrition and structural groundwork laid by the concurrent IgG profile ensure that the episodic, pro-fibrotic secretory stimulus delivered by repeated IgE-mediated mast cell flares cannot be homeostatically resolved. This dual signaling permanently activates the TANGO1-mediated secretion in resident vascular myofibroblasts, accelerating intimal hyperplasia and perivascular fibrosis. The ultimate convergence of this dual-isotype cascade physically occludes the vessel lumen, marking the irreversible transition from functional, transient vasospasm to permanent structural obliteration. Created in BioRender. Artlett, C. (2026) https://BioRender.com/mirvpbo.


Table 1.
Comparison between vascular events in SSc and other autoimmune diseases.
| Disease | Vasculopathy | Primary Vessel Pathology/ Inflammation Type |
Clinical Manifestation |
Raynaud’s Prevalence | Literature |
|---|---|---|---|---|---|
|
Systemic sclerosis |
Yes | Obliterative vasculopathy: intimal proliferation and fibrosis leading to the lumen narrowing. |
Digital ulcers, puffy hands, pulmonary arterial hypertension. |
95-99% | [10,19] |
|
Mixed Connective Tissue Disease |
Yes | Mixed: often shows SSc-like proliferative vasculopathy but can also exhibit SLE-like vasculitis. | Pulmonary arterial hypertension, puffy hands. |
~90% | [29,35] |
|
Dermato- myositis |
No | Microangiopathy: capillary dropout/rarefaction, endothelial swelling (often complement-mediated). | Gottron papules, periungual telangiectasia. |
10-50% | [36,37,38] |
|
Systemic lupus erythematosus |
No | Vasculitis: small vessel leukocytoclastic or necrotizing vasculitis (immune complex-mediated). |
Palpable purpura, glomerulonephritis, skin rashes. |
21-44% | [39,40] |
|
Sjögren’s Syndrome |
Mixed | Small vessel vasculitis: often associated with cryoglobulinemia or hypergammaglobulinemia. A subset shows SSc-like proliferative vasculopathy |
Purpura, urticarial vasculitis, peripheral neuropathy. |
13-30% | [41,42] |
Table 2.
Distinctions between IgG/T-cell vs. IgE/Mast autoallergic paradigm in SSc.
| Feature | Traditional Model (IgG/T-cell) |
Autoallergic Paradigm (IgE/Mast Cell) |
|---|---|---|
|
Early Swelling (Edema) |
Poorly explained, seen as early inflammation | Acute edema due to the release of histamine and leukotrienes. |
|
Pruritus (Itch) |
Viewed as a secondary symptom | A primary symptom due to the release of tryptase and IL-31. |
| Vascular Flare | Structural narrowing (slow) |
Vascular spasm due to the release of chymase and angiotensin II. |
| Biomarker | IgG antinuclear antibodies |
IgE antinuclear antibodies |
Table 3.
Differences in the primary antigen and confirmed IgE isotype between SSc, MCTD, and SLE.
| Disease | Primary Antigen | Confirmed IgE Isotype? | Source |
|---|---|---|---|
| SSc (dcSSc, lcSSc*) | Scl-70 (Topo-1) | Yes (56%) | [108] |
| CREST | CENP A/B | Yes (67-78%) | [108] |
| MCTD | U1-RNP | Yes (High Prevalence) | [109] |
| SLE | U1-RNP | Yes (30%) | [110] |
Table 4.
Association between IgE AECAs and clinical severity of disease.
| SSc Subset | IgG AECA Prevalence |
Primary Vascular Target |
Clinical Severity Signal |
|---|---|---|---|
| dcSSc | High 68-84% | Dermal/renal arterioles | Renal crisis/rapid fibrosis |
| lcSSc | Moderate (~30%) | Pulmonary/digital beds | Pulmonary arterial hypertension/Digital gangrene |
|
Overlap (Hybrid) |
Variable (up to 80%) | Nailfold/Digital loops | Transition to vasculopathy |
Table 5.
Wound healing versus IgE-mediated mast cell activation.
| Mast Cells Activated by IgE | Mast Cells Activated During Wounding | |
|---|---|---|
|
Receptor and Ligand Dynamics |
This is an adaptive immune response requiring prior exposure to a specific antigen. It relies entirely on the cross-linking of allergen molecules by IgE antibodies already bound to the high-affinity FcεRI receptor on the mast cell surface. | This is an innate immune system and a physical response. It does not require antibodies or prior exposure. Instead, it utilizes non-IgE receptors such as TLRs to sense DAMPs (like IL-33 or hyaluronic acid fragments), G-protein coupled receptors (MRGPRX2) to sense neuropeptides like Substance P, and mechanosensitive ion channels (PIEZO1) to sense physical tearing and pressure shifts. |
|
Speed and Mechanics of Release |
When IgE receptors cross-link, it triggers a massive, synchronized influx of intracellular calcium. This causes compounded exocytosis, in which almost all preformed granules fuse with one another and with the plasma membrane simultaneously, rapidly releasing a flood of histamine, tryptase, and leukotrienes into the surrounding tissue within seconds to minutes. |
Activation via DAMPs (like IL-33/ST2 signaling) or TLRs often triggers a slower, controlled, or selective release known as piecemeal degranulation. The mast cell selectively vesiculates and secretes specific cytokines (such as TGF-β1, VEGF, or IL-6) over hours without completely emptying its granules or causing a massive histamine release. |
|
Biological Purpose |
Designed historically to expel macro-parasites like helminths via severe local inflammation, muscle contraction, and fluid flushing (itching, sneezing, swelling). In modern settings, this is the driver of pathological Type I hypersensitivity (allergies and asthma). |
Designed entirely for hemostasis and tissue repair. The goal is to constrict breached vessels (via serotonin and thromboxane), recruit neutrophils to sterilize the tissue (via TNF-α), break down dead matrix (via tryptase/chymase), and initiate angiogenesis and fibroblast recruitment (via VEGF and TGF-β1) to seal the gap. |
|
Downstream Signaling Cascade |
Operates primarily through tyrosine kinase cascades (Lyn/Syk pathways) that rapidly activate phospholipase C gamma (PLCγ), leading to a massive spike in intracellular calcium. |
Operates predominantly through the MyD88/IRAK pathway (for TLRs/IL-33) or direct G-protein subunits (for neuropeptides via MRGPRX2). These pathways preferentially activate transcription factors such as NF-κB and growth kinases such as p38 MAPK, thereby promoting the synthesis of new pro-fibrotic and angiogenic proteins over time. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



